


Abstract
Oligonucleotide-based therapeutics have the capacity to engage with nucleic acid immune sensors to activate or block their response, but a detailed understanding of these immunomodulatory effects is currently lacking. We recently showed that 2′-O-methyl (2′OMe) gapmer antisense oligonucleotides (ASOs) exhibited sequence-dependent inhibition of sensing by the RNA sensor Toll-Like Receptor (TLR) 7. Here we discovered that 2′OMe ASOs can also display sequence-dependent inhibitory effects on two major sensors of DNA, namely cyclic GMP-AMP synthase (cGAS) and TLR9. Through a screen of 80 2′OMe ASOs and sequence mutants, we characterized key features within the 20-mer ASOs regulating cGAS and TLR9 inhibition, and identified a highly potent cGAS inhibitor. Importantly, we show that the features of ASOs inhibiting TLR9 differ from those inhibiting cGAS, with only a few sequences inhibiting both pathways. Together with our previous studies, our work reveals a complex pattern of immunomodulation where 95% of the ASOs tested inhibited at least one of TLR7, TLR9 or cGAS by ≥30%, which may confound interpretation of their in vivo functions. Our studies constitute the broadest analysis of the immunomodulatory effect of 2′OMe ASOs on nucleic acid sensing to date and will support refinement of their therapeutic development.
INTRODUCTION
RNA-targeting therapeutics based on synthetic oligonucleotides have been gaining interest, with several regulatory approvals in the US and European Union (1), and multi-billion license deals from big pharma in recent years (2). To permit their essential functions related to gene targeting activities, oligonucleotides-based therapeutics require both increased affinity for their targets and stabilisation against nuclease activities; this can be achieved through the incorporation of modified nucleotides (e.g. with 2′-O-methyl [2′OMe], 2′-methoxyethyl [2′MOE], 2′-fluoro [2′F], or locked nucleic acid [LNA]) as well as modified internucleotide linkages (e.g. phosphorothioate [PS]).
The intricate relationship between synthetic oligonucleotides and nucleic acids sensors of the innate immune system, which are involved in the early detection of pathogens, has been known for two decades. For instance, PS-modified unmethylated ‘CG’ (CpG) containing DNA oligonucleotides have the potential to activate the DNA sensor Toll-Like Receptor (TLR) 9 (3,4), but can also block it in a sequence- and length-dependent manner (5–8). Similarly, 2′OMe modified RNAs block RNA sensing by TLR7 and TLR8 (9,10) and Retinoic Acid Inducible Gene-I (RIG-I) (11). This knowledge has been important for the design of oligonucleotide therapeutics that can evade activation of innate immune sensors, otherwise leading to strong off-target pro-inflammatory immune responses in patients (3,12–14). From this perspective, chemical modifications can have the dual benefit of increasing the targeting efficacy of the oligonucleotides, while decreasing their immunostimulatory effects.
Nonetheless, it has also been clear for some time that select PS-modified DNA oligonucleotides (ODN) have broad immunosuppressive effects (15). This is best exemplified with the ‘TTAGGG’ containing PS-ODN A151, involved in the inhibition of TLR9 (6), TLR7 (16), Absent In Melanoma 2 (AIM2) (17) and cyclic-GMP-AMP synthase (cGAS) (18). These effects are sequence-dependent, with some PS-DNA ODNs displaying limited immunosuppressive activities on individual immune sensors (7,15). Similarly, 2′OMe oligonucleotides can exhibit sequence-dependent inhibitory effects on TLR7 and TLR8 sensing (19). These observations suggest a complex picture of immunosuppression by chemically modified oligonucleotides where sequences dictate their activities on nucleic acid sensors. Since only a handful of ODNs have been studied to date across different receptors (15,18), a detailed understanding of the immunosuppressive sequence determinants of ODNs is currently lacking. Further, our understanding of the immunosuppressive effects of oligonucleotides combining base and/or backbone modifications, as is seen in most oligonucleotide therapeutics approved and in development, is non-existent. While it is potentially useful to generate anti-inflammatory ODNs (20), characterizing the immunosuppressive effects of therapeutic oligonucleotides is becoming important to avoid the potential for increased susceptibility to infection in the large patient populations beginning to receive ODN therapies (2).
We recently investigated the TLR7 and TLR8 immunomodulatory effect of RNase-H1-recruiting gapmer 2′OMe ASOs, which have a central DNA core of 10 nt flanked by 5 nt of 2′OMe, on a PS backbone (21). Unexpectedly, we demonstrated that the majority of the 2′OMe gapmer ASOs tested were potent inhibitors of TLR7, but that selected sequences harbouring 2′OMe ‘CUU’ motifs did not block TLR7 activation (21). Conversely, we found that TLR8 sensing of the low molecular weight synthetic ligand Resiquimod (referred to as R848 hereafter), was potentiated by select 2′OMe ASOs, in a sequence-dependent manner, an observation that aligns with previous findings with poly-dT PS ODNs (21–23). These findings prompted us to investigate the broader immunomodulatory effects of 2′OMe ASOs on nucleic sensing, with the aim of defining the molecular patterns controlling these activities.
In the present study, we focused on the effect of 2′OMe ASOs on two key innate immune sensors of pathogenic DNA: cGAS, which detects cytoplasmic DNA in most tissues, and TLR9, which detects endosomal DNA in select immune cells (20). We demonstrated the sequence-dependent modulation of both receptors by the ASOs and identified specific motifs involved in these activities, based on a panel of 80 2′OMe ASOs. Integration of these results with those from our previous report allowed us to generate a broad overview of immunomodulation by 2′OMe ASOs on RNA (TLR7 or 8) and DNA (cGAS or TLR9) sensing. Specifically, for instance, we have identified a significant correlation between cGAS and TLR7 immunosuppressive effects. Collectively, our studies present the most comprehensive analysis of the modulation of nucleic acid sensing by PS ASOs, confirming their prevalent immunosuppressive activity. Our studies highlight the need to consider these sequence-dependent activities to avoid misinterpreting results and clinical outcomes and to avoid increasing the susceptibility to infection for patients receiving oligonucleotide-based therapeutics.
MATERIALS AND METHODS
Cell isolation, culture and stimulation
Human primary bone marrow-derived mesenchymal stem cell (MSCs) from two healthy adult donors (#1129 and #1980) were purchased from Lonza (#PT-2501) and were cultured in Dulbecco's modified Eagle's medium plus l-glutamine supplemented with 1× antibiotic/antimycotic (Thermo Fisher Scientific) and 10% heat-inactivated foetal bovine serum (referred to as complete DMEM). Culture media was replaced twice a week and cells were passaged at 80% confluency and seeded at a density of 2.5 × 103 cells/cm2. These cells were confirmed free from pathogens and certified to meet MSC criteria as defined by the International Society of Cell and Gene Therapy. MSCs used herein were plated at passage 6–7. Synovial cells were obtained from rheumatoid arthritis (RA) patients who fulfilled the American College of Rheumatology (ACR) criteria for the classification of RA (24). RA and control primary fibroblast-like synoviocytes (FLS) were obtained from surgical specimens of synovial tissue and cultured as previously described (25). FLS were grown in RPMI 1640 plus l-glutamine medium (Life Technologies) complemented with 1x antibiotic/antimycotic and 10% heat inactivated foetal bovine serum (referred to as complete RPMI).
Trex1-mutant mice (used under ANU animal ethics reference A2018/38) have a single-base mutation in Trex1 leading to a premature stop codon (Q98X). Homozygous mutant mice have aberrant accumulation of cytoplasmic DNA, resulting in basal engagement of the cGAS-STING pathway (Ellyard J.I. and Vinuesa C.G., manuscript in preparation), similar to that reported in Trex1-deficient mice (26). Primary bone marrow derived macrophages (BMDMs) from 3 Trex1-mutant mice were extracted and differentiated for 6 days in complete DMEM supplemented with L929 conditioned medium as previously reported (27). 293XL-hTLR7-HA, 293XL-hTLR9-HA and HEK-Blue™ hTLR3 stably expressing human TLR7, TLR9 or TLR3 were purchased from Invivogen, and were maintained in complete DMEM supplemented with either 10 μg/ml and 30 μg/ml Blasticidin (Invivogen), for TLR7 or 9 and TLR3 cells, respectively. HeLa cells, human colorectal cancer HT-29 (kind gift from R. Firestein), human fibroblasts expressing SV40 (large and small T antigens) with HRASG12V (kind gift from E. Sanij (28); referred to as BJ hTERT SV40T herein), LL171 cells (mouse L929 cells expressing an IFN-stimulated response element (ISRE)-Luciferase - kind gift from V. Hornung (29)), and immortalized wild-type mouse bone marrow macrophages (BMDMs) (30) were grown in complete DMEM. Human osteosarcoma MG-63 cells were purchased from ATCC (#CRL-1427) and grown in ATCC-formulated Eagle's Minimum Essential Medium, supplemented with 10% heat-inactivated foetal bovine serum (Thermo Fisher Scientific) and 1× antibiotic/antimycotic (Thermo Fisher Scientific). Human acute myeloid leukemia THP-1 and their CRISPR-Cas9 derivatives (cGAS-/- (31), UNC93B1–/– and UNC93B1–/– reconstituted with UNC93B1 (32)) cells were grown in complete RPMI. THP-1 cells were not differentiated with phorbol 12-myristate 13-acetate (PMA) in any experiments, and rather used in suspension. All the cells were cultured at 37°C with 5% CO2. Cell lines were passaged 2–3 times a week and tested for mycoplasma contamination on a routine basis by PCR.
For cGAS stimulations, cells were treated for the indicated duration with ASOs, prior to transfection with ISD70 (human cells) or ISD45 (mouse cells) (the ASOs were not washed off prior to ISD transfection unless otherwise indicated). The cGAS ligands ISD45 (33) and ISD70 (also known as VACV-70 (34)) (Supplementary Table S1) were resuspended as follows: 5 μl of sense and 5 μl of antisense strands at 10 μg/μl were added to 90 μl PBS under sterile conditions, heated at 75°C for 30 min, prior to cooling to room temperature and aliquoting (stock at 1 μg/1 μl). The ISDs were transfected at a concentration of 2.5 μg/ml at a ratio of 1 μg:1 μl with Lipofectamine 2000 in Opti-MEM (Thermo Fisher Scientific). HEK-TLR3, HEK-TLR7 and HEK-TLR9 were treated with indicated concentrations of ASOs for 20–50 min, prior to stimulation with poly(I:C) (Invivogen), R848 (Invivogen), and the Class B CpG oligonucleotides ODN 2006 (synthesised by IDT and resuspended in RNase-free TE buffer [Supplementary Table S1]), respectively. The TLR2/1 agonist, PAM3CSK4 (Invivogen); the TLR4 agonist, lipopolysaccharide (Invivogen); the mouse Sting agonist, DMXAA (Cayman); and the human STING agonist, (compound #3 from (35), referred to as GSK herein – kind gift from Cancer Therapeutics CRC, Australia), were used at indicated concentrations. Aspirin (Sigma-A2093) was resuspended in pure DMEM to 10 mM, filter sterilised and added to the cells for a final concentration of 2 mM. All ASOs were synthesised by Integrated DNA Technologies (IDT), and resuspended in RNase-free TE buffer, pH 8.0 (Thermo Fisher Scientific). ASO sequences and modifications are provided in Supplementary Tables S1 and S2. Cell viability was assessed by adding 1× fresh resazurin solution to the wells (10X solution made up with 7 mg resazurin [Sigma] dissolved in 3.5 ml PBS, filter sterilised at 0.2 μM) for 4 h, prior to reading with a Fluostar OPTIMA (BMG LABTECH) plate-reader (fluorescence: excitation 535 nm, emission 590); wells with medium only and 1X resazurin were used as blanks.
Luciferase assays
HEK293 cells stably expressing TLR3, 7 or 9 were reverse-transfected with pNF-κB-Luc4 reporter (Clontech), with Lipofectamine 2000 (Thermo Fisher Scientific), according to the manufacturer's protocol. Briefly, 500 000–700 000 cells were reverse-transfected with 400 ng of reporter with 1.2 μl of Lipofectamine 2000 per well of a six-well plate, and incubated for 3–24 h at 37°C with 5% CO2. Following transfection, the cells were collected from the six-wells and aliquoted into 96-wells, just before ASO and overnight TLR stimulation (as above described). Similarly, the LL171 cells expressing an ISRE-Luc reporter were treated overnight. The next day, the cells were lysed in 40 μl (for a 96-well plate) of 1× Glo Lysis buffer (Promega) for 10 min at room temperature. 15 μl of the lysate was then subjected to firefly luciferase assay using 40 μl of Luciferase Assay Reagent (Promega). Luminescence was quantified with a Fluostar OPTIMA (BMG LABTECH) luminometer.
Beta-galactosidase staining
A β-Galactosidase staining assay was performed on FLS and MSCs treated with ASOs using the Senescence β-Galactosidase Staining kit (New England Biolabs). Briefly, FLS and MSCs were washed with PBS, fixed and stained over 24–48 h with X-Gal solution at 37°C according to the manufacturer's protocol. The cells were imaged using inverted phase microscopy. Three to 6 images were taken per condition and analysed with ImageJ, counting the number of β-Galactosidase positive cells (blue) per image (totalling >100 cells for each condition). The relative proportion of blue cells per field was calculated for each image.
ASO reverse-transfections
For Figures 1A, 3F and G, ASOs were reverse-transfected with Lipofectamine 2000. Briefly, a solution of 1.125 μl Lipofectamine 2000 in 25 μl Opti-MEM was mixed to a solution of 1 μl of 10 μM ASO in 25 μl Opti-MEM. Following 20–25 min incubation at room temperature, the resulting 50 μl solution was dispensed into a 24-well plate, and 50 000 to 80 000 cells in 450 μl antibiotic-free medium were added, giving a final ASO concentration of 20 nM per well. For conditions with 50 and 100 nM ASOs (Figure 3F), the amount of Lipofectamine was kept constant. Cells were lysed in 150 μl RNA lysis Buffer (ISOLATE II RNA Mini Kit (Bioline)) after 24 h incubation.
Figure 1.

Sequence-dependent inhibition of cGAS sensing by ASOs. (A) HeLa and HT-29 cells were transfected for 24 h with 20 nM of indicated ASOs targeted to cGAS (Supplementary Table S1), prior to RNA purification and RT-qPCR analyses. cGAS levels were reported relative to 18S, and normalised to Mock condition. Data shown represent the median of two independent experiments for each cell line. (B) THP-1 pre-treated overnight with 100 nM of the indicated ASO, were transfected or not (non-treated [NT]) with 2.5 μg/ml ISD70 for 8.5 h and IP-10 levels in supernatants were determined by ELISA. Data shown are averaged from two independent experiments in biological triplicate (± s.e.m and ordinary one-way ANOVA with Tukey's multiple comparison tests to the ‘ISD70 only’ condition, or otherwise indicated pairs of conditions are shown). There was no basal effect of the ASOs on NT cells (21). (C) HT-29 cells pre-treated overnight with 125, 250 or 500 nM of indicated ASOs, were transfected or not (non-treated [NT]) with 2.5 μg/ml of ISD70 for 24h, and IP-10 levels in supernatants were determined by ELISA. IP-10 levels were normalised to the ‘ISD70 only’ condition, after background correction with the NT condition. Data shown are averaged from two independent experiments in biological triplicate (± s.e.m. and Mann–Whitney U tests to the ‘ISD70 only’ condition are shown). (D, E) THP-1 pre-treated overnight with 100 nM of the indicated ASOs, were transfected with 2.5 μg/ml of ISD70 for 7–8 h, and IP-10 levels in supernatants were determined by ELISA. (D) Stimulations and ELISAs were carried out in two independent plates and the results presented on each axis (with a correlation r = 0.7716, P < 0.0001). Averaged values from both plates are given in Supplementary Table S2. ISD70 only condition is shown in blue. (E) IP-10 levels were normalised to the ‘ISD70 only’ condition, after background correction with NT condition. Data shown are averaged from three independent experiments in biological triplicate (± s.e.m. and ordinary one-way ANOVA with Tukey's multiple comparison tests to the condition ‘ISD70 only’, or otherwise indicated pairs of conditions, which are shown). (F) HT-29 cells pre-treated overnight with 187.5 nM of indicated ASOs were transfected or not with 2.5 μg/ml of ISD70 for 24 h, and IP-10 levels in supernatants determined by ELISA. IP-10 levels were normalised to the ‘ISD70 only’ condition, after background correction with NT condition. Data shown are averaged from three independent experiments in biological triplicate (± s.e.m. and ordinary one-way ANOVA with Dunnett's multiple comparison tests to the condition ‘ISD70 only’ condition are shown). (G) cGAS–/–, UNC93B1–/– and matched controls with rescued UNC93B1 expression (UNC93B1 WT) THP-1 were pre-treated 6 h with 100 or 250 nM ASOs, and transfected with 2.5 μg/ml of ISD70 overnight. GSK (100 nM) and ODN2006 (500 nM) were used as human STING and TLR9 agonists, respectively. IP-10 levels in supernatants were determined by ELISA and normalised to the ‘GSK’ condition, after background correction with NT condition. Data shown are averaged from three independent experiments in biological triplicate (± s.e.m and ordinary two-way ANOVA with Tukey's multiple comparison tests relative to ‘ISD70 only’ condition are shown). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001, ns: non-significant.
Figure 3.

Sequence-dependent inhibition of cGAS function. (A, B) THP-1 pre-treated 6 h with indicated doses (500, 250, 125, 62.5, 31.25 nM) (A) or 125 nM (B) of C2-Mut1 or A151 oligonucleotides, were transfected with 2.5 μg/ml of ISD70 overnight, and IP-10 (A) or IFN-β (B) levels in supernatants were determined by ELISA. IP-10 (A) and IFN-β (B) levels were normalised to the ‘ISD70 only’ condition, after background correction with the NT condition. (A, B) Data shown are averaged from three independent experiments in biological triplicate (± s.e.m. and ordinary two-way ANOVA with Sidak's multiple comparison tests relative to A151 are shown [A] or ordinary one-way ANOVA with Tukey's multiple comparison tests relative to condition ‘NT’, or otherwise indicated pairs of conditions are shown [B]). MG-63 (C) or mouse immortalised BMDMs (D) were pre-treated or not with 500 nM of C2-Mut1 for 6 h prior to stimulation with 2.5 μg/ml ISD (ISD70 for MG-63, ISD45 for BMDMs), LPS (1 μg/ml), GSK (100 nM), PAM3C (100 ng/ml), ODN1826 (500 nM), or DMXAA (50 μg/ml) overnight. IP-10 (C and D) and TNF-α (D) levels in supernatants were determined by ELISA. (C) Data were normalised to the ‘GSK’ condition, after background correction to the NT condition. Data shown are averaged from two independent experiments in biological triplicate (± s.e.m and Mann-Whitney U tests are shown). (D) IP-10 levels were normalised to the DMXAA condition, while TNF-α levels were normalised to the LPS condition. Data shown are averaged from three independent experiments in biological triplicate (± s.e.m and Mann-Whitney U tests are shown). (E) Recombinant cGAS protein was incubated in vitro with 2.3 μM ISD70 with or without (NT) increasing concentrations of ASOs (0.5, 2 and 10 μM) for 40 min. The reaction was stopped with EDTA and cGAMP levels were analysed by ELISA. Data were normalised to the condition A151 2 μM. Data shown are averaged from three independent experiments run in technical duplicate on the ELISA (± s.e.m and a Mann-Whitney U test is shown). (F) BJ hTERT SV40T cells were transfected overnight with an increasing amount of indicated ASO (20, 50, 100 nM), or 2 mM aspirin (ASP), prior to RNA purification. Expression of the panel of 3 human IFN-driven genes was analysed by RT-qPCR. Expression of the indicated genes was reported against to 18S expression and further normalised to the average of the ‘Mock’ condition. Data shown represent the average of three independent experiments conducted in biological duplicate (± s.e.m and Mann-Whitney U tests are shown). (G) Trex1-mutant primary BMDMs from three individual homozygous mice were transfected with 50 nM ASOs (or lipofectamine only, ‘Mock’) for 20 h, prior to RNA purification. Expression of the panel of three mouse IFN-driven genes was analysed by RT-qPCR. Expression of the indicated genes was reported against to 18s expression and further normalised to the average of the ‘Mock’ condition. Data shown represent the average of three mice conducted in biological duplicate (± s.e.m. and Mann–Whitney U tests are shown). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001, ns: non-significant.
mRNA reverse transcription quantitative real-time PCR (RT-qPCR)
Total RNA was purified from cells using the ISOLATE II RNA Mini Kit (Bioline). Random hexamer cDNA was synthesized from isolated RNA using the High-Capacity cDNA Archive kit (Thermo Fisher Scientific) according to the manufacturer's instructions. RT-qPCR was carried out with the Power SYBR Green Master Mix (Thermo Fisher Scientific) on the HT7900 and QuantStudio 6 RT-PCR system (Thermo Fisher Scientific). Each PCR was carried out in technical duplicate and human or mouse 18S was used as the reference gene. Each amplicon was gel-purified and used to generate a standard curve for the quantification of gene expression (used in each run). Melting curves were used in each run to confirm specificity of amplification. The primers used were the following: Human IFIT1: IFIT1-FWD TCACCAGATAGGGCTTTGCT; hIFIT1-REV CACCTCAAATGTGGGCTTTT; Human 18S: h18S-FWD CGGCTACCACATCCAAGGAA; h18S-REV GCTGGAATTACCGCGGCT; IFI44: IFI44-FWD ATGGCAGTGACAACTCGTTTG; IFI44-REV: TCCTGGTAACTCTCTTCTGCATA; Human IFIT2: IFIT2-RT-FWD TTATTGGTGGCAGAAGAGGAAG; IFIT2-RT-REV CCTCCATCAAGTTCCAGGTG; Human cGAS: hcGAS-FWD CACGTATGTACCCAGAACCC; hcGAS-REV GTCCTGAGGCACTGAAGAAAG; Mouse Rsad2: Rsad2-FWD CTGTGCGCTGGAAGGTTT; Rsad2-REV ATTCAGGCACCAAACAGGAC; Mouse 18s: Rn18s- FWD GTAACCCGTTGAACCCCATT; Rn18s-REV CCATCCAATCGGTAGTAGCG; Mouse Ifih1: Ifih1-FWD TCTTGGACACTTGCTTCGAG; Ifih1-REV TCCTTCTGCACAATCCTTCTC; Mouse Ifit1: Ifit1-RT-FWD GAGAGTCAAGGCAGGTTTCT; Ifit1-RT-REV TCTCACTTCCAAATCAGGTATGT.
Detection of cytokines
Human IP-10 and IFN-β levels were measured using supernatants from the different cultures and were quantified using IP-10 (BD Biosciences, #550926) or IFN-β (PBL assay science, #41415-1) ELISA kits respectively, according to the manufacturers’ protocols. Tetramethylbenzidine substrate (Thermo Fisher Scientific) was used for quantification of the cytokines on a Fluostar OPTIMA (BMG LABTECH) plate-reader.
cGAS in vitro assay and cGAMP ELISA
0.8 μg of recombinant full-length human cGAS (Cayman, #22810) was used per single reaction in a 200 μl volume with 80 mM Tris–HCl (pH 7.5), 200 mM NaCl, 20 μM ZnCl2, 20 mM MgCl2, 0.25 mM GTP (Thermo Fisher Scientific #R0441) and 0.25 mM ATP (Thermo Fisher Scientific #R0461), with 20 μg of ISD70 (freshly annealed at 1 μg/μl in PBS), and 2 μl C2-Mut1/A151 diluted in TE buffer (to get 0.5, 2 or 10 μM ODN concentration in 200 μl). After 40 min at 37°C, 2.5 mM EDTA was added to each tube to stop the enzymatic reaction and the samples were either snap frozen and stored at –80°C or directly processed for cGAMP ELISA. cGAMP levels were measured with the DetectX Direct 2′,3′-Cyclic GAMP Enzyme Immunoassay Kit (Arbor Assays), according to the manufacturer's protocol. Briefly, 50 μl of in vitro reactions were added per well to the kit microplate with 50 μl Assay buffer, 25 μl conjugate, and 25 μl Antibody per well, prior to a minimum of 2 h incubation. The standards were prepared with serial-dilution in Assay buffer. Quantification of cGAMP levels was performed on a Fluostar OPTIMA (BMG LABTECH) plate-reader at 450 nm.
Statistical analyses
Statistical analyses were carried out using Prism 8 (GraphPad Software Inc.). Every experiment was repeated a minimum of two independent times (except the ASO screens – Figures 1D and 4D, conducted in single experiments, but for which key ASOs were validated in a minimum of two independent experiments). One-way and two-way analyses of variance (ANOVA) were used when comparing groups of conditions, while two-tailed unpaired non-parametric Mann-Whitney U tests or unpaired two-tailed t-tests were used when comparing pairs of conditions. Symbols used: * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001, ****P ≤ 0.0001 and ‘ns’ is non-significant.
Figure 4.

Sequence-dependent TLR9 inhibition. (A, C) HEK-TLR9 cells expressing a NF-κB-luciferase reporter were treated with 500 nM indicated ASOs for 30 min prior to stimulation or not (non-treated [NT]) with 200 nM ODN2006. NF-κB-luciferase levels were measured after overnight incubation. NF-κB-luciferase levels were normalised to the ‘ODN2006 only’ condition, after background correction with the NT condition. Data shown are averaged from two (A) or three (C) independent experiments in biological triplicate (± s.e.m. and ordinary one-way ANOVA with Dunnett's multiple comparison tests to the ‘ODN2006 only’ condition [A] or Mann–Whitney U tests [C] are shown). (B) ASO2 and ASO11 sequence mutants. ASO11Mut1 and ASO11Mut2 contain the 3′ and 5′ end of ASO2, respectively. (D) HEK-TLR9 cells expressing a NF-κB-luciferase reporter were treated with 500 nM indicated ASOs for 45 min prior to stimulation or not with 200 nM ODN2006. NF-κB-luciferase levels were measured after overnight incubation. NF-κB-luciferase levels were normalised to the ‘ODN2006 only’ condition, after background correction with the NT condition. Stimulations and luciferase assays were carried out in two independent plates and the results are presented on each axis (with a correlation r = 0.7909, P < 0.0001) (averaged data are provided in Supplementary Table S2). The 10 most potent ASOs (reference position in the plate is given as per Supplementary Table S2) are highlighted on the plot. ASOs with ≤50% reduction of TLR9 activity at 500 nM are highlighted with blue shading. (E) Bottom: MEME pictogram of the relative frequency of bases constituting the TLR9 inhibitory motif. Top: alignment of the sequences enriched with the motif identified as per Supplementary Figure S1D. The reference position in the plate is given as per Supplementary Table S2. (F) Alignment of the sequences enriched with ASO2 motif (Supplementary Figure S1C). (G) The central 10 DNA bases of the top and bottom 16 TLR9 inhibitors from the 80 ASOs screened (see Supplementary Table S2) were analysed for base content. The violin plots show the distribution of the cumulative number of each central base for both ASO populations. Ordinary two-way ANOVA with Sidak's multiple comparison tests (between top and bottom populations) are shown. (H, J, L) HEK-TLR9 cells expressing a NF-κB-luciferase reporter were treated with 500 nM indicated ASOs for 45 min prior to stimulation or not (non-treated [NT]) with 200 nM ODN2006. NF-κB-luciferase levels were measured after overnight incubation. NF-κB-luciferase levels were normalised to the ‘ODN2006 only’ condition, after background correction with the NT condition. Data shown are averaged from three independent experiments in biological triplicate (± s.e.m). One-way ANOVA with Dunnett's multiple comparison tests to the ‘ASO2138’ condition (H), or the ‘ASO108’ condition (J), are shown. (L) One-way ANOVA with Tukey's multiple comparison tests relative to the condition ‘661’, or otherwise indicated pairs of conditions are shown. (I, K) Sequence alignments showing the ‘CUU’ motifs in families of closely related ASOs; the yellow region highlights the DNA moiety of the gapmers. (M) Correlation of TLR7 and TLR9 inhibition based on 80 ASOs (used at 100 nM for TLR7, and 500 nM for TLR9). Percentages of NF-κB-luciferase levels relative to the conditions ‘R848 without ASO’ (for TLR7) or ‘ODN2006 without ASO’ (for TLR9) are averaged from biological duplicate (averaged data are provided in Supplementary Table S2) (with a correlation r = 0.05256, P = 0.6433). Selected ASOs are indicated. *P ≤ 0.05, **P ≤ 0.01, ****P ≤ 0.0001, ns: non-significant.
RESULTS
Sequence-dependent effects of 2′OMe gapmer ASOs on cGAS sensing
cGAS has recently emerged as an essential sensor of cytosolic DNA deriving from pathogens and damaged endogenous nucleic acids (20). Upon activation by DNA, cGAS drives the formation of cyclic GMP-AMP (cGAMP), which binds to stimulator of interferon (IFN) genes (STING) and promotes transcriptional induction of IRF3 responsive genes, including CXCL10 (IP-10) and IFNB1. Since it instigates deleterious immune responses linked to a wide range of diseases, various approaches are being investigated currently to therapeutically target cGAS (36–40). We originally hypothesised that strategies relying on the down-regulation of cGAS mRNA could provide therapeutic alternatives to chemical inhibitors of cGAS, which have been pursued in many other studies (36–40). To study this possibility, we designed a panel of 11 2′OMe gapmer ASOs (ASO1 to ASO11) that targeted the mRNA of human cGAS and tested their effects in HeLa cells and HT-29 cells, which endogenously express cGAS (Figure 1A and Supplementary Table S1). Transfection of the ASOs led to a sequence-dependent decrease of cGAS mRNA, although variable between the two cell lines, with ASO2 having a 30–50% impact, while ASO3/ASO4 robustly failed to decrease cGAS expression (Figure 1A).
We next tested the functional effect of our panel of ASOs on undifferentiated monocytic THP-1 cells, upon overnight uptake of the naked ASOs (Figure 1B). In contrast to the down-regulation signature seen in HeLa and HT-29 cells, ASO2 was the most potent oligonucleotide blocking IP-10 production after transfection of the synthetic 70-bp interferon stimulating DNA (ISD70), which acted as the cGAS ligand (34). This observation was reproduced in cGAS/STING-competent epithelial HT-29 cells (41) where overnight incubation with 125 nM of ASO2 (but not ASO4, ASO10 or ASO11) blunted ISD70-induced IP-10 production (Figure 1C). Critically, however, increased doses of ASO4, ASO10 and ASO11 also blocked ISD70-induced IP-10 production, when used at 250 (ASO4/ASO10) or 500 (ASO11) nM in HT-29 (Figure 1C). Taking into account their differing effects on cGAS mRNA targeting (Figure 1A), the dose-dependent inhibitory effect of ASO4/ASO10/ASO11 on ISD70-sensing strongly suggests it was independent of cGAS mRNA targeting, but rather related to a competitive effect on cGAS sensing of ISD70; this aligns with the effect observed for PS-ODN A151 on cGAS sensing (18). Our hypothesis that ASOs have a direct effect on cGAS function was consistent with the observation that ASO11 increased ISD70-sensing at low doses, but was also inhibitory at higher doses, which may relate to the binding of the ASO to the third DNA binding domain of cGAS, increasing its enzymatic activity at low dose (42) (Figure 1C).
Since ASO2 inhibited ISD70-sensing at a lower concentration than the other ASOs, we posited that select motifs may increase the inhibitory effect of 2′OMe gapmer ASOs on ISD70-sensing of cGAS. To define this further, we screened a library of 80 2′OMe ASOs, designed to target the CDKN2B-AS1 and LINC-PINT transcripts (Supplementary Table S2 and (21)), in THP-1 transfected with ISD70 (Figure 1D). While none of these ASOs targeted the cGAS mRNA, 23 out of 80 decreased ISD70-driven IP-10 production by >40% (Figure 1D and Supplementary Table S2). A few highly inhibitory ASOs were selected from the screen for validation in THP-1 (C2, E10 and F2 ASOs) and HT-29 cells (C2 and E10 ASOs) transfected with ISD70 (Figure 1E, F). Overnight pre-treatment with C2 robustly inhibited ISD70-dependent IP-10 production in both cell models, while a significant effect of E10 was seen only in THP-1 cells (noting a reduction of 25% with this sequence in HT-29 cells). Importantly, C2 was a more potent inhibitor than ASO2 (Figure 1E) and A151 (Figure 1E, F).
We have recently shown that 2′OMe gapmer ASOs are spontaneously taken up by undifferentiated THP-1 and can modulate endosomal TLR7/8 sensing (21). To exclude a putative contribution of endosomal TLRs upon ISD70 transfection, we next tested the effect of C2 and ASO11 in THP-1 cells lacking cGAS (31) or UNC93B1 (32), the latter being devoid of TLR7/8 and 9 sensing. While cGAS-deficient and UNC93B1-deficient cells similarly produced IP-10 upon stimulation with a synthetic human STING agonist (35), cGAS-deficient THP-1 cells also lacked responsiveness to ISD70 transfection (Figure 1G). Conversely, we observed a dose-dependent inhibitory effect of C2 on ISD70 sensing in UNC93B1-deficient and matched wild-type cells, not seen with ASO11 (Figure 1G), ruling out a contribution of endosomal TLRs in the effects of C2 on ISD70 sensing. Collectively these results demonstrate a capacity for 2′OMe gapmer ASOs to inhibit cGAS sensing of transfected DNA, in a sequence-dependent manner.
Identification of a 2′OMe motif controlling cGAS inhibition
Having shown that ASO2, C2 and E10 were potent inhibitors of ISD70 sensing in THP-1, we next sought to define whether select motifs in these ASOs were involved in their inhibitory activities. MEME motif discovery analysis (43) performed on the three sequences identified a putative enriched motif in the 5′ half of C2 and 3′ half of both ASO2 and E10, with four highly conserved bases (Figure 2A and Supplementary Figure S1A). To test the importance of these four bases, we first designed two variants of C2, substituting two (C2-Mut1) or four (C2-Mut2) of these bases (Figure 2A). The bases used to replace those in the motif were arbitrarily chosen, and could equally decrease or increase the inhibitory activity. In accordance with our hypothesis that these specific bases made a direct contribution to inhibition, dose-response analyses of the C2 mutants on ISD70-driven IP-10 production in HT-29 cells showed increased inhibitory activity for both ASOs at 125 and 250 nM compared to the parent C2 ASO (this decrease being significant for C2-Mut1 at 125 nM; Figure 2B). Similarly, C2-Mut1 was significantly more inhibitory on the ISD45-driven interferon stimulated response element (ISRE)-luciferase than was C2 and ASO2 in mouse LL171 cells (Figure 2C). Conversely, in LL171 cells the 20-mer PS oligonucleotide (dT20) did not inhibit ISD45 sensing even at 600 nM, confirming the sequence-specific nature of the ASO inhibition in mouse cells (Figure 2C). In parallel, having shown that C2 was a more potent inhibitor than ASO2 (Figure 1E), we designed mutants of ASO2 harbouring the 5′ region of C2 (ASO2up), or a mutant of the four conserved bases (ASO2down – Figure 2A). While not significant in HT-29 cells, the trend was that ASO2up was a more potent inhibitor of ISD70-driven IP-10 production than ASO2 (at 125 nM). Conversely, ASO2down significantly increased IP-10 production at 62.5 and 125 nM, aligning with what was seen with ASO11 in HT-29 cells (Figure 2D).
Figure 2.

Identification of a highly potent inhibitor of cGAS sensing. (A) Top: sequence alignments of cGAS inhibitors and identification of important bases predicted by MEME analysis, highlighted with arrows (Supplementary Figure S1A). Bottom: design of C2 and ASO2 mutants incorporating point mutations at selected positions of the MEME motif (mutations are highlighted in yellow). (B, D) HT-29 cells pre-treated overnight with indicated doses of ASOs (500, 250, 125, 62.5 nM), were transfected or not with 2.5 μg/ml of ISD70 for 24 h and IP-10 levels in supernatants were determined by ELISA. IP-10 levels were normalised to the ‘ISD70 only’ condition, after background correction to the NT condition. Data shown are averaged from two (D) or three (B) independent experiments in biological triplicate (± s.e.m and ordinary two-way ANOVA with Tukey's multiple comparison tests relative to C2 [B] or ASO2 [D] conditions, are shown; comparisons were not significant between the ASOs for 250 and 500 nM). (C) Mouse LL171 cells were treated for 6 h with indicated amount of ASOs (200, 400, 600 nM) prior to overnight stimulation with 2.5 μg/ml ISD45. Cells were lysed and ISRE-Luc levels were analysed by luciferase assay the next day. ISRE-luciferase levels were normalised to the ‘ISD45 only’ condition, after background correction with NT condition. Data shown are averaged from two independent experiments in biological triplicate (± s.e.m and Mann-Whitney U tests are shown). (E) Top: sequence alignments of C2-Mut1 variants; C2-ASO2-A and C2-ASO2-B have the 3′ ends from ASO2up and ASO2down underlined. Bottom: variants of the homopolymer dC20; 2′OMe bases are in pink and the cGAS inhibitory motif underlined. (F) HT-29 cells pre-treated overnight with 187.5 nM indicated ASOs were transfected or not with 2.5 μg/ml of ISD70 for 24 h and IP-10 levels in supernatants were determined by ELISA. (G) THP-1 pre-treated overnight with 100 nM indicated ASOs were transfected with 2.5 μg/ml of ISD70 for 24 h and IP-10 levels in supernatants determined by ELISA. (F, G) IP-10 levels were normalised to the ‘ISD70 only’ condition, after background correction with the NT condition. Data shown are averaged from three independent experiments in biological triplicate (± s.e.m. and ordinary one-way ANOVA with Tukey's multiple comparison tests relative to condition ‘ISD70 only’, or otherwise indicated pairs of conditions are shown). (H) LL171 cells were treated for 6 h with 200 nM ASOs prior to overnight stimulation with 2.5 μg/ml ISD45. Cells were lysed and ISRE-Luc levels were analysed by luciferase assay the next day. ISRE-luciferase levels were normalised to the ‘ISD45 only’ condition, after background correction with the NT condition. Data shown are averaged from three independent experiments in biological triplicate (± s.e.m. and ordinary one-way ANOVA with Dunnett's multiple comparison tests to the ‘ISD70 only’ condition are shown). (I) THP-1 cells treated for 6 h with 100 nM C2-Mut1, or 100, 250, 500 nM of C2-Mut1-PS were transfected with 2.5 μg/ml of ISD70 overnight. IP-10 levels were normalised to the ‘ISD70 only’ condition, after background correction with the NT condition. Data shown are averaged from two independent experiments in biological triplicate (± s.e.m and Mann-Whitney U tests to the ‘ISD70 only’ condition are shown). (J) Sequences of [LINC-PINT] ASOs 101–116, showing the location of the inhibitory GGUCCC motif (in pink) from ASO103 identified by MEME (see E and Supplementary Figure S1B). The yellow region highlights the DNA moiety of the gapmers. (K) THP-1 pre-treated overnight with 100 nM indicated [LINC-PINT] ASOs, were transfected with 2.5 μg/ml of ISD70 for 7.5 h and IP-10 levels in supernatants were determined by ELISA. IP-10 levels were normalised to the ‘ISD70 only’ condition, after background correction with the NT condition. Data shown are averaged from three independent experiments in biological triplicate (± s.e.m and one-way ANOVA with Dunnett's multiple comparison tests to the ‘ISD70 only’ condition are shown). (L, M) LL171 cells were treated for 6 h or 20 min with 200 nM ASOs prior to being ‘washed’ or not (L), or treated with 50, 100 or 200 nM ASOs for 20 min (M), and stimulated overnight with 2.5 μg/ml ISD45. Cells were lysed and ISRE-Luc levels were analysed by luciferase assay the next day. ISRE-luciferase levels were normalised to the ‘ISD45 only’ condition, after background correction with the NT condition. Data shown are averaged from two (L) or three (M) independent experiments in biological triplicate (± s.e.m. and one-way ANOVA with Tukey's multiple comparison tests to the ‘ISD45 only’ condition (L), or Mann-Whitney U tests to the ‘ISD45 only’ condition (M), or otherwise indicated pairs of conditions, are shown). (N) THP-1 pre-treated for 40 min with 250 nM indicated ASOs, were transfected overnight with 2.5 μg/ml of ISD70 and IP-10 levels in supernatants were determined by ELISA. Data shown are averaged from three independent experiments in biological triplicate (±s.e.m. and ordinary one-way ANOVA with Dunnett's multiple comparison tests to the ‘ISD70 only’ condition, and a Mann-Whitney U test comparing Mut1-dC and dC20 are shown). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001, ns: non-significant.
Since two base mutations of C2 in C2-Mut1 significantly increased ISD70-driven IP-10 inhibition, we next designed a series of four C2-Mut1 mutants (C2-Mut1v1 to C2-Mut1v4) with base permutations to define whether the inhibition seen with C2-Mut1 could be enhanced further (Figure 2E). We also designed hybrid ASOs fusing the 5′ half of C2-Mut1 to the 3′ half of ASO2up (C2-ASO2-A) or ASO2down (C2-ASO2-B). Analyses of these sequences in HT-29 (Figure 2F) and THP-1 (Figure 2G) revealed that C2-Mut1 was robustly the most inhibitory sequence of ISD70 sensing and was significantly more potent than its mutant C2-Mut1v3. Interestingly, C2-ASO2-A, which contains two fused inhibitory motifs (that of C2-Mut1 in the 5′ half, and that of ASO2up/C2 in the 3′ half), had a similar potency to that of C2-Mut1, indicating that duplicating the inhibitory motif in the 3′ end region of the ASO did not significantly enhance the inhibition (Figure 2F, G). Nonetheless, in THP-1 cells, C2-ASO2-B was significantly less inhibitory than C2-ASO2-A and ASO2 down was significantly less inhibitory than ASO2up, confirming that 3′ end regions could also play a role in the inhibition of ISD70 sensing (Figure 2G).
cGAS inhibition by a minimal mGmGmUATC motif
We next asked whether the 2′OMe chemical modification of gapmer ASOs was at play in their effect on cGAS. In both mouse LL171 and human THP-1 cells, the inhibitory effect of C2-Mut1 was significantly decreased in a C2-Mut1 analogue in which the 2′OMe ends were replaced by DNA bases on a PS-backbone (referred to as C2-Mut1-PS) (Figure 2H, I and Supplementary Table S1). In addition, MEME motif discovery on the 10 most inhibitory ASOs from our screen (Figure 1D and Supplementary Table S2) revealed that five ASOs harboured a conserved [A/G]GUC[U/C]C motif aligned with that of C2 and C2-Mut1, which was predominantly overlapping with their 2′OMe 5′ end (Supplementary Figure S1B). Interestingly, one of these sequences, ‘[LINC-PINT]103′ (referred to as ASO103), was part of a family of sequences with single base increments (Supplementary Table S2 and Figure 2J). Within this series, ASO103 was the only sequence significantly inhibiting ISD70-driven IP-10 production in THP-1 cells, further supporting the idea that the 5′-end location and 2′OMe modifications were likely essential to the ASO inhibitory function (Figure 2K).
The data collected so far demonstrated that long (i.e. 6–16 h) pre-incubation with 2′OMe gapmer led to inhibition of ISD sensing by cGAS. To tease out the role of the pre-incubation on the inhibitory activity of the ASOs, we compared the effect of C2-Mut1 pre-incubated for 6 h in LL171 cells, with or without a wash prior to the transfection of ISD45. We also tested the effect of ASOs that were added a short time prior to the transfection of ISD45 (∼20 min). While pre-incubation of the ASOs for a shorter duration did not impact their inhibitory effect on ISD45-driven ISRE-Luc expression, the addition of a washing step significantly blunted inhibition (Figure 2L). These findings suggested that the ASOs were somewhat co-transfected with the liposome containing ISD. Accordingly, increased intracellular fluorescent puncta of ASO2-Cy3 were observed following ISD70 transfection of HT-29 cells (Supplementary Figure S2), altogether suggesting that the ASOs competed with ISD for cGAS sensing intracellularly.
Relying on this shorter ASO pre-incubation, we next sought to confirm the core motif modulating cGAS inhibition in C2-Mut1. We started from a 20-mer homopolymeric sequence of dCs on a PS backbone (dC20), to which the minimal 2′OMe containing terminal 5′ mGmGmUATC motif of C2-Mut1 was added (referred to Mut1-dC) (Figure 2E). Mut1-dC was significantly more inhibitory than its precursor dC20 on ISD sensing in LL171 and THP-1 cells (Figure 2M, N). In addition, the two base-mutations (mCmGmUTTC) in Mut1v3-dC lacked significant inhibitory activity in both cell models (Figure 2M, N). Together with our previous observations, these results establish that the minimal mGmGmUATC motif is sufficient to confer cGAS inhibition to a PS-modified homopolymeric sequence, with a predominant role for the two bases previously identified.
Sequence-specific inhibition of cGAS activity by C2-Mut1
Having identified C2-Mut1 as our most potent 2′OMe ASO inhibitor of cGAS, we performed dose-response analyses to determine its IC50 in our THP-1 model and to compare it to that of A151 (18). Based on IP-10 driven production after ISD70 transfection, we determined that the IC50 of C2-Mut1 was 56 nM compared to 165 nM for A151 (Figure 3A). This greater inhibitory activity of C2-Mut1 over that of A151 on DNA sensing was confirmed looking at the production of IFN-β, which was blocked with C2-Mut1 at 125 nM, while only reduced by ∼50% for A151 at that dose (Figure 3B). It is also noteworthy that treatment of the cells with C2-Mut1 did not significantly impact cell viability in our assays in THP-1 and HT-29 cells (Supplementary Figure S3).
In human MG-63 cells and immortalised mouse bone marrow derived macrophages (BMDMs), a 6 h pre-incubation with high dose C2-Mut1 (500 nM) did not significantly affect IP-10 production driven by lipopolysaccharide (LPS–TLR4 ligand) or STING synthetic agonists, although the pre-incubation significantly impacted ISD sensing (Figure 3C, D). Similarly, C2-Mut1 did not impact production of TNF-α upon treatment with LPS, PAM3CSK4 (TLR2/1 ligand) or DMXAA (mouse Sting agonist) (Figure 3D). Nonetheless, C2-Mut1 pre-incubation significantly decreased sensing of the CpG ODN 1826, which activates mouse Tlr9, as revealed by dampened TNF-α production in immortalised BMDMs (Figure 3D). This is consistent with our previous report that PS-2′OMe ASO can impact endosomal sensing by TLRs such as TLR7/8 in phagocytes (21), but indicates that C2-Mut1 is not broadly inhibiting non-nucleic acid sensing pathways such as TLR1/2/4, nor acting at the level of downstream signalling cascades.
In vitro, cGAS has previously been shown to be bound and weakly activated by single stranded ISD45 (44), leading us to posit that single stranded PS-ASOs could act as ‘inactive’ competitors of double stranded ISD. To directly assess this, recombinant cGAS was incubated in vitro with 2.3 μM (0.1 μg/μl) of ISD70 in the presence of increasing amounts of C2-Mut1 or A151 (0.5, 2 and 10 μM). Critically, while both PS-ODNs similarly reduced cGAMP production driven by ISD70 at 0.5 μM, the inhibitory activity of C2-Mut1 was significantly greater than that of A151 (about 2-fold more) at 2 μM (Figure 3E). Under these specific conditions in vitro, C2-Mut1 was therefore able to displace more molecules of ISD70 than A151, suggesting C2-Mut1 had stronger affinity for cGAS than did A151. Together with our cell-based assays, these observations support that C2-Mut1 acts as an inactive competitor of ISD70 binding to cGAS.
C2-Mut1 inhibits constitutively active cGAS signalling
Since our experiments to this point strictly relied on cGAS sensing of exogenously transfected ISD, we next investigated the effects of our ASOs on cell models with constitutive cGAS activation. We first compared the dose-dependent effect of C2-Mut1 and C2-Mut1v3 transfected in human BJ hTERT fibroblasts stably expressing SV40T and RASG12V (28), which display a basal level of constitutive cGAS activation (45). Transfected C2-Mut1 was significantly more potent than its two nucleotide variant C2-Mut1v3 in inhibiting expression of several interferon stimulated genes (ISGs) (IFIT1, IFIT2 and IFI44) constitutively expressed in these cells (46) and had an effect comparable to aspirin treatment that blocks cGAS activity (47) (Figure 3F). Secondly, overnight transfection of C2-Mut1 and C2-Mut1-v3 significantly decreased the basal expression of two out or three ISGs (Rsad2 and Ifit1) in primary mouse bone marrow derived macrophages (BMDMs) from Trex1-mutant mice (Q98X – see Material and Methods), which exhibit accumulated cytoplasmic DNA and constitutive cGAS-STING signalling (Figure 3G) (20). Conversely, transfection of a similar dose of A151 failed to inhibit these ISGs (Figure 3G).
cGAS activation has been shown recently to play an important role in the paracrine propagation of senescence between cells (48,49). As such, senescence-associated β-galactosidase (SAB) was strongly decreased during the expansion of primary mouse fibroblasts from cGas-deficient mice (48). We therefore decided to investigate the capacity of C2-Mut1 to inhibit spontaneous engagement of cGAS-STING signalling, in pre-senescent primary fibroblast-like synoviocytes (FLS) and primary bone marrow-derived mesenchymal stem cells (MSCs). For this purpose, we grew the primary cells in the presence of 1–5 μM C2-Mut1 ASO (passively taken up by the cells by gymnosis) for one to two weeks. While a large proportion of cells were senescent in non-ASO treated cells after this period (based on β-galactosidase positive staining, seen in up to ∼40–50% of the cells), senescence was strongly reduced in C2-Mut1-treated FLS cells and MSCs (Supplementary Figure S4A, S4B). Further experiments in another set of FLS cells showed that C2-Mut1 was more potent in inhibiting SAB than its PS only variant C2-Mut1-PS, or its mutant C2-Mut1v3. Similarly, C2-Mut1 was a more potent inhibitor of SAB than A151 (Supplementary Figure S4C), aligning with the concept that this activity pertained to its increased inhibition of cGAS.
Collectively, these results demonstrate that C2-Mut1 significantly inhibits constitutive cGAS sensing of endogenous cytoplasmic DNA, in a sequence-dependent manner.
Sequence-dependent TLR9 inhibition by 2′OMe gapmer ASOs
Having demonstrated the sequence-dependent modulation of cGAS sensing of DNA by 2′OMe gapmer ASOs, we next turned to their effect on DNA sensing by TLR9, with the aim of providing a broad picture of their impact on DNA sensing in human cells. As such, while there is ample evidence for sequence-dependent TLR9 modulation with select PS-ASOs (3,7), including A151 (6), the impact of 2′OMe moieties in the context of PS-gapmer ASOs, is not defined. We recently demonstrated that 2′OMe gapmer ASOs did not frequently activate TLR9, with the exception of ‘T’ rich sequences (21). To characterise whether 2′OMe gapmer ASOs instead led to inhibition of DNA sensing by TLR9, as indicated with our results in BMDMs with C2-Mut1 (Figure 3D), we tested our panel of 11 cGAS ASOs on TLR9 sensing of CpG ODN2006 in HEK cells expressing human TLR9 (HEK-TLR9 hereafter) and a NF-κB luciferase reporter. We included PS-ODN A151 and IRS957, which have been reported to inhibit TLR9, along with their respective controls, C151 and IRS661 (6,7)). Several ASOs significantly inhibited TLR9 sensing of ODN2006, including ASO2, 4, 5, 6, 7, 8, 9, 10 (Figure 4A). However, ASO2, IRS957 and A151 were the only oligonucleotides reducing NF-κB luciferase induction by more than 80%. Our previous observation that ASO11, which lacked the capacity to inhibit TLR9 but strongly potentiated TLR8 in HEK cells (21), indicated that the sequence-specific effects seen here were not related to uptake of the ASOs, but rather due a competitive effect of specific motifs on ODN2006 sensing by TLR9.
To investigate further the sequence determinants of TLR9 inhibition by 2′OMe gapmer ASOs, we initially assessed the effect of ASO2 3′-end mutants (ASO2up and ASO2down) and ASO11 mutants where the 5′ or 3′ 2′OMe regions had been swapped with those of ASO2 (ASO11Mut1 and ASO11Mut2) in HEK-TLR9 cells (Figure 4B, C). The four-base substitutions in the 3′-end of ASO2down significantly reduced TLR9 inhibition, unlike those in ASO2up, indicating the inhibitory effect was partially dependent on the sequence of the 3′ half of ASO2. Critically, substitution of the ASO11 5′-end 2′OMe region with that of ASO2 (ASO11Mut2), but not its 3′-end region (ASO11Mut1), conferred significant TLR9 inhibition to ASO11, suggesting that the 5′-end of ASO2 is also at play in its regulatory effect on TLR9 (Figure 4C).
To gain further insights into the sequence-dependent inhibition of TLR9 sensing of DNA by 2′OMe gapmer ASOs, we next tested our panel of 80 2′OMe ASOs in HEK-TLR9 cells, (Figure 4D and Supplementary Table S2). We observed that only 8 out of the 80 ASOs inhibited TLR9 by more than 50% (Supplementary Table S2), and ASO2 remained the most potent ASO tested. MEME motif discovery on the top 10 ASOs from this screen revealed significant enrichment in three ASOs of a ‘GGCCTC’ motif also present in ASO2, and a ‘A’-rich central region in 5 out of the 10 ASOs (Figure 4E, F and Supplementary Figures S1C, S1D). Accordingly, when we compared the central DNA regions of the 16 most and least inhibitory ASOs in the TLR9 screen, we observed a significantly higher proportion of central ‘As’ in the more potent TLR9 inhibitory sequences (Figure 4G and Supplementary Table S2).
Closer analyses of two ASOs families from the screen with single base increments also pointed to an important role for 5′ 2′OMe ‘CUU’ motifs in TLR9 inhibition (Supplementary Table S2). Validation of the first family, referred to as the ‘CDKN2B-AS1’ series (ASO2133-2139), suggested that addition of a 5′-end terminal ‘CUU’ motif in ASO2139 significantly increased TLR9 inhibition (Figure 4H, I). Aligning with this observation, analyses of the ‘LINC-PINT’ series of ASOs (ASO108-ASO116), also harbouring 5′ or 3′ ‘CUU’ motifs, demonstrated that ‘CUU/CUT’ motifs present in the 5′ 2′OMe region of the ASOs (ASO112-115) were associated with a significant increase in TLR9 inhibition compared to ASO108-110 containing such 2′OMe ‘CUU’ motifs in their 3′ end (Figure 4I, J).
To define further the role of 2′OMe ‘CUU’ motifs on the modulation of TLR9 sensing, we next used an additional panel of 2′OMe ASOs targeting the mRNA of HPRT (Figure 4K and (21)). While 5′-end terminal ‘CUU’ motifs in ASO [HPRT]551 and 660 were associated with >60% inhibition of TLR9, addition of a single 5′-end terminal base to the ‘CUU’ motif (‘ACUU’) in ASO661 significantly limited inhibition (Figure 4K, L). Conversely, ASO662, with its ‘CACUUU’ 5′-end 2′OMe region displayed the strongest inhibition of this series of ASOs, although this may also be related to a contribution of its ‘UUUUC’ 3′-end (uniquely present in this sequence in this series). Interestingly, ASO666 also strongly inhibited TLR9, while not displaying any ‘CUU’ motif and only differing by two bases from ASO665, which was only poorly inhibitory (Figure 4K, L). Nonetheless, these results are only suggestive of a role for selected motifs in the modulation of TLR9 inhibition, and need to be taken with caution given the putative impact of base increments on 2′OMe regions, which could confound the interpretation of results. Taken together, these findings demonstrate a complex regulation of TLR9 inhibition by 2′OMe gapmer ASOs, including preferential contributions from central (preferentially ‘A’ rich) and 5′-end 2′OMe regions.
In contrast with these findings, we have recently reported that 2′OMe ‘CUU’ motifs could help mitigate TLR7 inhibition by 2′OMe gapmer ASOs (21), suggesting an inverse relationship between the modulation of these two sensors. We therefore analysed the correlation of the effect of the 80 ASOs on TLR9 and TLR7 inhibition (21), presented in Figure 4M. There was no correlation between the activities of the ASOs on TLR7 and TLR9, exemplified with ASO[LINC-PINT]108, ASO109, ASO116 and A10 which had limited effect on TLR9, and TLR7, unlike ASO[CDKN2B-AS1]2139, which did not inhibit TLR7 but strongly inhibited TLR9.
C2-Mut1 is a potent inhibitor of human cGAS, TLR7 and TLR3 but not TLR9
Having generated immunomodulatory profiles for our panel of 80 2′OMe ASOs on TLR7/TLR8 (21), TLR9 and cGAS, we generated a bubble chart to visualise the correlation of inhibition of DNA sensing by cGAS and TLR9, while also incorporating data on TLR7 inhibition and TLR8 potentiation (Figure 5A and Supplementary Table S2). There was no significant correlation between TLR9 and cGAS inhibition, illustrated by the example that C2, the most potent cGAS inhibitor in our screen did not inhibit TLR9 (Figure 5A). This observation was validated in HEK-TLR9 cells with C2 and its mutants C2-Mut1 and C2-ASO2-A. While the C2 mutants were significantly better inhibitors of TLR9 than their parent ASO, their inhibitory effect on TLR9 sensing was limited compared to that of ASO2 or A151 (Figure 5B).
Figure 5.
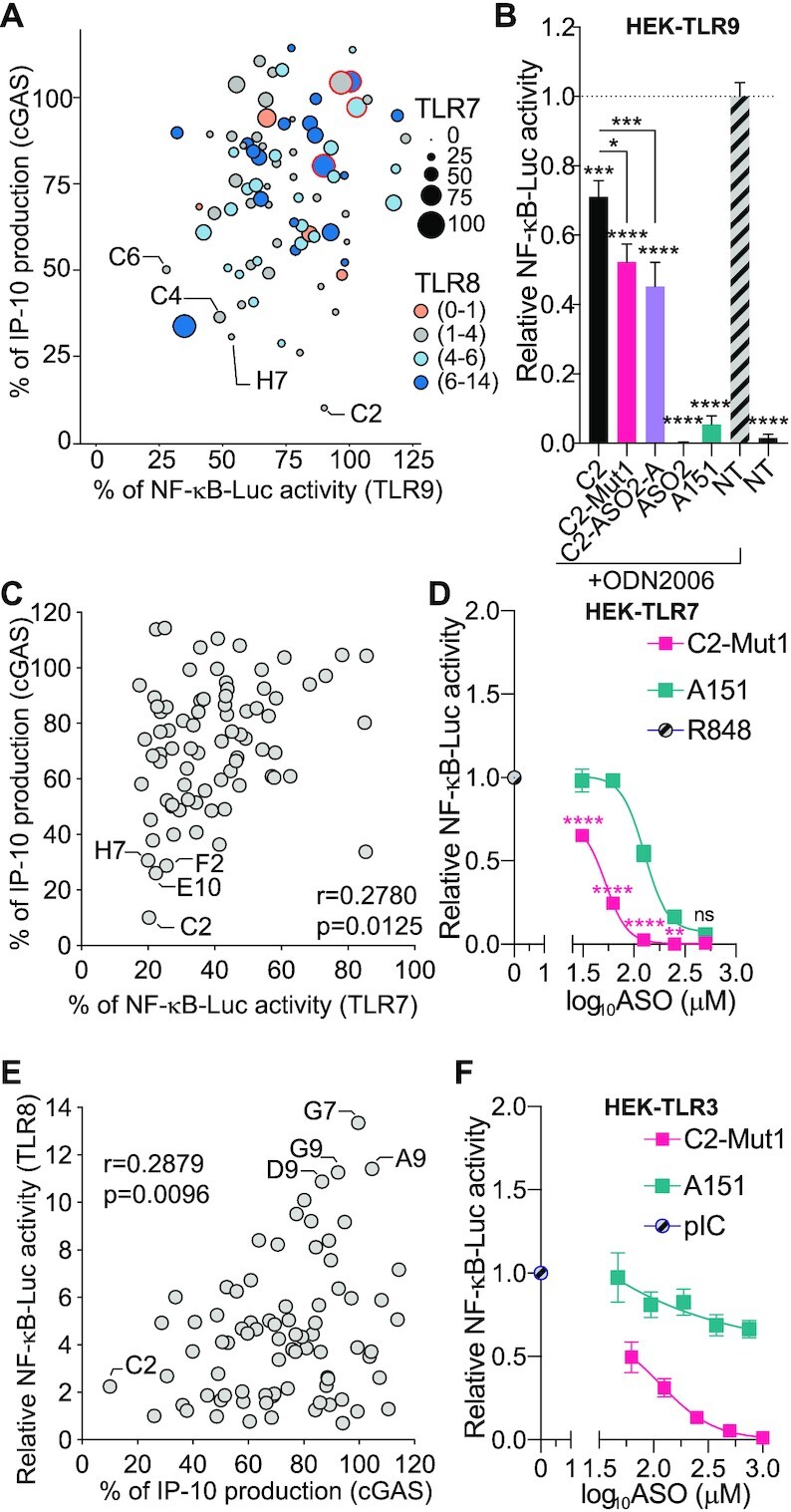
The broad immunosuppressive effects of 2′OMe ASOs. (A) Bubble graph showing the relationship between cGAS, TLR9, TLR7 inhibition, and TLR8 potentiation (based on the data from Supplementary Table S2 and (21)), for 80 ASOs. For TLR7 and TLR9, percentages of NF-κB-luciferase levels relative to the conditions ‘R848 without ASO’ (for TLR7) or ‘ODN2006 without ASO’ (TLR9) are shown (the size of the bubbles reflects TLR7 signalling strength). For TLR8, fold increases relative to the condition ‘R848 without ASO’ are shown, using a 4 colour-scale. For cGAS, percentages of IP-10 production relative to the condition ‘ISD70 only’ are shown. Selected ASOs are indicated: the reference position in the plate is given as per Supplementary Table S2. (B) HEK-TLR9 cells expressing a NF-κB-luciferase reporter were treated with 500 nM indicated ASOs for 30–45 min prior to stimulation or not (non-treated [NT]) with 200 nM ODN2006. NF-κB-luciferase levels were measured after overnight incubation. NF-κB-luciferase levels were normalised to the ‘ODN2006 only’ condition, after background correction with NT condition. Data shown are averaged from three independent experiments in biological triplicate (± s.e.m and one-way ANOVA with Tukey's multiple comparison tests relative to condition ‘ODN2006 only’, or otherwise indicated pairs of conditions are shown). (C) Correlation of TLR7 and cGAS inhibition based on 80 ASOs (used at 100 nM for TLR7 and cGAS). Percentages of NF-κB-luciferase levels relative to the conditions ‘R848 without ASO’ (for TLR7) or percentages of IP-10 production relative to the condition ‘ISD70 only’ are shown (with a significant correlation r = 0.2780, P = 0.0125). Selected ASOs are indicated; the reference position in the plate is given as per Supplementary Table S2. (D) HEK-TLR7 cells expressing a NF-κB-luciferase reporter were treated with the indicated concentration of ASOs (500, 250, 125, 62.5, 31.25 nM) for 30–50 min prior to stimulation with 1 μg/ml R848. NF-κB-luciferase levels were measured after overnight incubation. NF-κB-luciferase levels were normalised to the ‘R848 only’ condition, after background correction with the NT condition. Data shown are averaged from three independent experiments in biological triplicate (± s.e.m. and ordinary two-way ANOVA with Sidak's multiple comparison tests relative to A151 are shown). (E) Correlation of TLR8 potentiation and cGAS inhibition based on 80 ASOs (used at 500 nM for TLR8 and 100 nM cGAS). Fold increases of NF-κB-luciferase levels relative to the conditions ‘R848 without ASO’ (for TLR8) or percentages of IP-10 production relative to the condition ‘ISD70 only’ are shown (with a significant correlation r = 0.2879, P = 0.0096). Selected ASOs are indicated; the reference position in the plate is given as per Supplementary Table S2. (F) HEK-TLR3 cells expressing a NF-κB-luciferase reporter were treated with indicated concentrations of C2-Mut1 (1000, 500, 250, 125, 62.5, nM) or A151 (753, 376.5, 188.25, 94.125 or 47.0625 nM) for 30–50 min prior to stimulation with 0.5 μg/ml pIC. NF-κB-luciferase levels were measured after overnight incubation. NF-κB-luciferase levels were normalised to the ‘pIC only’ condition, after background correction with NT condition. Data shown are averaged from three independent experiments in biological triplicate (± s.e.m.). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001, ns: non-significant.
As shown in Figure 5A, the patterns of immunomodulation on these sensors was highly variable with only a few ASOs inhibiting TLR7, TLR9 and cGAS (e.g. C4, C6 and H7) (Supplementary Table S2). Nonetheless, there was a significant correlation between cGAS and TLR7 inhibition (Figure 5C), with the strongest inhibitors of cGAS also displaying strong TLR7 repression (e.g. C2, E10, F2, H7; Figure 5C). Consistent with this, the most potent cGAS inhibitor, C2-Mut1, was also a strong inhibitor of R848 sensing by TLR7, with an IC50 of 44 nM, against 132 nM for A151 (Figure 5D). Critically, only 4 ASOs in our screen (i.e. 5%) did not inhibit any of TLR7, TLR9 or cGAS by >30% (highlighted in red in Figure 5A, and in yellow in Supplementary Table S2), demonstrating that most 2′OMe ASOs have immunosuppressive effects.
Unlike their inhibitory effect on TLR7, we recently discovered that select 2′OMe gapmer ASOs could potentiate TLR8 sensing of R848, presenting potential therapeutic opportunities in immuno-oncology (21). Interestingly, TLR8 potentiation was also inversely correlated with cGAS inhibition, with the best TLR8 potentiators lacking cGAS inhibition (e.g. G7, A9, D9, G9) (Figure 5E). Accordingly C2 was a weak potentiator of TLR8, but select ASOs may be able to inhibit cGAS while potentiating TLR8 sensing, as seen with the example of ASO2 (Figure 1 and (21)).
Finally, we tested the effect of our panel of 11 cGAS ASOs (including IRS661, IRS957, A151 and its mutant C151) on the sensing of untransfected double stranded RNA (polyI:C) by human TLR3 sensing in HEK 293 cells stably expressing human TLR3 (HEK-TLR3) and a NF-κB luciferase reporter (it should be noted that these cells are not responsive to the amount of untransfected polyI:C we used, through RIG-I or MDA5). While all 2′OMe gapmer ASOs used at 500 nM along with IRS661 and IRS957 blocked polyI:C sensing, A151 and C151 only partially reduced polyI:C-dependent TLR3 activation in these cells (Supplementary Figure S5A). Sequence-specific inhibition of TLR3 was more visible with 100 nM ASOs, where only ASO5 retained significant inhibition of polyI:C sensing (Supplementary Figure S5B). Direct comparison of the inhibitory effect of C2-Mut1 and A151 on TLR3 sensing in HEK-TLR3 cells confirmed the stronger inhibition of C2-Mut1, with an IC50 of 62 nM for C2-Mut1 and an IC50 greater than 750 nM for A151 (Figure 5F). Collectively these results demonstrate that C2-Mut1 is a potent inhibitor of human cGAS, TLR7 and TLR3, but a weaker inhibitor of TLR9.
DISCUSSION
There is growing evidence that the cGAS-STING pathway not only has roles in detecting viral and bacterial cytosolic DNA (50,51), but also acts a sensor of cellular integrity, activated by damaged nuclear DNA (45,48,49,52), and cytoplasmic mitochondrial DNA (53), to facilitate clearance of damaged cells by immune cells (48,49). Critically, cGas/Sting-deficiency has been shown to be protective against many diseases in pre-clinical animal models (including in Parkinson's disease, amyotrophic lateral sclerosis, acute pancreatitis, macular degeneration, myocardial infarction, alcohol-related liver disease, and polyarthritis (54–56)), providing strong rationale for the development of cGAS inhibitors as new therapeutic avenues for these diseases.
Most of the strategies reported to date to inhibit cGAS-STING signalling have focussed on the development of small molecules inhibiting cGAS enzymatic activity (36–40,47,57,58), with a propensity to target cGAS systemically rather than in specific tissues. Conversely, our approach has been to develop therapeutic ASOs that could be directed to modulate cGAS levels in selected tissues. While initially screening 2′OMe ASOs targeted to the mRNA of human cGAS, we discovered a disconnect between the ASOs that decreased cGAS mRNA expression and those inhibiting cGAS activation by transfected immunostimulatory DNA. Hypothesising that some of our PS-2′OMe ASOs were acting as competitive inactive ligands for cGAS, as was the case with the PS-ODN A151 (18), we screened a panel of 80 2′OMe ASOs targeted to two long non-coding RNAs, unrelated to cGAS signalling. These analyses confirmed that inhibition of cGAS relied on sequence-dependent features of the ASOs, independent of cGAS mRNA targeting.
In vitro, we showed that our most potent ASO, C2-Mut1, competes with double stranded DNA to restrict cGAMP synthesis by cGAS, aligning with the proposed mechanism of action of A151 (18). However, since C2-Mut1 was a more potent inhibitor of cGAMP synthesis than A151, we propose that C2-Mut1 has a stronger affinity for cGAS; although this warrants confirmatory biochemical analyses. Importantly, C2-Mut1 was also a strong inhibitor of mouse cGas, unlike other small molecule antagonists that display species-specific activities (39). However C2-Mut1 did not impact activation of the downstream cGAMP sensor STING, upon stimulation with its specific agonists (DMXAA in mouse cells (59), and GSK#3 in human cells (35)). In addition, C2-Mut1 did not impact sensing of non-nucleic acid sensors TLR2/1 and TLR4. This confirms further that it does not act at the level of downstream signalling adaptors shared between these pathways, which control the production of cytokines such as IP-10. We also extended our findings to models where cGAS is spontaneously engaged by endogenous cytosolic DNA, rather than transfected ISD. As such, we demonstrated that transfection of C2-Mut1 in BJ hTERT SV40T fibroblasts or Trex1 mutant primary macrophages, causing basal engagement of the pathway (45), reduced basal expression of interferon-stimulated genes (ISGs); this occurred in a sequence-dependent manner, comparable to what is observed with the cGAS inhibitor, aspirin (47). Finally, we showed that culture of primary human cells with high concentrations of C2-Mut1 for 1 to 2 weeks, robustly reduced the number of senescent positive cells, in a sequence-specific manner. Recent evidence suggests that cGAS is engaged upon destabilisation of the nuclear membrane in senescent cells (48,49), resulting in a paracrine amplification of senescence (48), consistent with our prior observation of cGAMP-like activity in senescent joint fibroblast-like synoviocytes (60). The present results indicate that spontaneous uptake of the ASOs, also known as gymnotic delivery, is able to block engagement of cGAS in senescent cells. These findings are in agreement with the previous demonstration that gymnotic delivery of the PS-ODN A151 reduced basal ISG signature in TREX1-deficient macrophages (18). Although further studies would be warranted to confirm direct involvement of cGAS inhibition by C2-Mut1 in the decreased senescence observed here, these observations support the idea that spontaneous uptake of C2-Mut1 is biologically relevant to limit senescence.
Direct comparison between our most potent cGAS inhibitor ASO (C2-Mut1) and A151, confirmed that pre-treatment with both molecules inhibited sensing of transfected immunostimulatory DNA, to significantly reduce IP-10 production in a dose-dependent manner, along with IFN-β levels. However, C2-Mut1 was more potent than A151 in cells and in vitro, blocking IP-10 and IFN-β production at a concentration of ∼125 nM in our assays. Steinhagen et al. recently demonstrated that A151 competed against double stranded DNA for binding cGAS, in cells and cellular extracts (18), but did not provide a detailed study of the sequence determinants involved in this effect, beyond the demonstration that the C151 mutant (which contains 16 mutations compared to A151) did not inhibit cGAS. In this manuscript we demonstrate that cGAS inhibition by DNA-containing PS-ASOs is intrinsically dependent on their sequence. Most importantly, we have identified a minimal 5′-end [mGmGmUATC] highly potent cGAS inhibitory motif, the activity of which can be impacted by as few as two nucleotide substitutions (as seen with Mut1v3 sequences). Our studies therefore provide proof-of concept that 2′OMe ODNs can be rationally designed to inhibit cGAS. Although duplication of the inhibitory motif in C2-ASO2-A did not robustly increase cGAS inhibition in our assays, we note that the duplicated motif did not contain the correct 2′OMe pattern, which is essential to the activity of C2-Mut1 (as shown with C2-Mut1-PS). Further studies will help define whether other sequence variations could help improve the cGAS inhibitory activity of C2-Mut1. As recently reported with the example of A151, which protects against brain injury, oligonucleotides-based inhibition of cGAS can present novel therapeutic avenues, by leveraging their unique capacity to inhibit additional immune pathways (61). Importantly, A151 used at 15 mg/kg intraperitoneally (i.p.) led to cGAS inhibition in vivo (61). Given that select ASOs such as C2-Mut1 are more potent inhibitors of cGAS than A151, and that a dosage of 5–15 mg/kg ASO i.p. is commonly used in pre-clinical studies, it is therefore possible that gene targeting ASOs used in vivo trigger unintended cGAS inhibition.
When broadly considering the off-target inhibition of cGAS by 2′OMe ASOs as a class of molecules, we note that only 13 out of 80 ASOs decreased cGAS activity by >50% at the dose used in our screen, and we found several inhibitory sequences harbouring a [A/G]GUC[U/C]C motif overlapping with their 5′ 2′OMe end. Although this motif can be useful to identify cGAS inhibitory ASOs, it is clear that some sequences are inhibiting cGAS independent of this motif (e.g. C4, E6 or H7 – Supplementary Table S2 and Supplementary Figure S1B). Nevertheless, we found that the least inhibitory sequences in our screen exhibited an enriched 5′ or 3′ end ‘CCUUCU’ motif, which may help selecting ASOs with limited cGAS inhibitory effects (Supplementary Figure S1E). This approach warrants further validations. Based on the data currently at hand, we recommend avoiding 2′OMe ASOs exhibiting a 5′ [A/G]GUC[U/C]C motif, and rather favour those with a 3′ end ‘CCUUCU’ to avoid unintended cGAS inhibition. Interestingly, a few ASOs, such as ASO11, potentiated cGAS sensing when used at low concentrations in HT-29 and LL171 cells. While this observation could be due to a potentiation of transfection efficiency of liposome-encapsulated ISD70 by select ASOs, it could also be due to the binding of such ASOs to the third DNA binding domain of cGAS, which facilitates oligomer formation and increases its enzymatic activity (42). While additional studies would be required to define their mode of action, it is clear that select 2′OMe ASOs can potentiate cGAS sensing when used at low doses, raising the possibility that these molecules instigate off-target inflammation in cells where the pathway is basally active.
In addition to their effect on cytoplasmic cGAS, we present here the first detailed analysis of the impact of 2′OMe gapmer ASOs on endosomal TLR9 inhibition, based on over 100 ASOs. Since A151 is a potent TLR9 inhibitor, we were interested to define whether cGAS and TLR9 modulation by 2′OMe ASOs correlated. Nonetheless, our analyses of 80 2′OMe ASOs did not find any significant correlation between the effect of the ASOs on TLR9 and cGAS inhibition. Based on our best human TLR9 inhibitor, ASO2, and its associated mutants, we demonstrated a direct involvement of its 5′ 2′OMe end in sequence-specific TLR9 inhibition, which conferred inhibition to the otherwise non-inhibitory ASO11. We note, however, that mutations of 4 bases over the 3′-half of ASO2 could also reduce inhibition. Motif enrichment analyses from our screen also revealed a potential role for ‘A’-rich central DNA regions in TLR9 inhibition. Given the importance of 5′ 2′OMe regions, we sought to identify motifs that condition TLR9 inhibition and discovered that ASOs with a terminal 5′ ‘CUU/CUT’ motif were often more inhibitory than closely related sequences. Yet, as shown with the case of ASO[HPRT]661, other sequence determinants can impact the activity of such 5′ ‘CUU’ motifs on TLR9 sensing, underlining the complexity of the ASO determinants affecting TLR9 function.
Overall, human TLR9 sequence-dependent inhibition by 2′OMe ASOs was mostly limited, as only 20 out of 80 ASOs inhibited TLR9 by more than 40% in our screen. However, dose-response analyses of ASO2 demonstrated it could still block TLR9 activity when using half the concentration of molecules of the TLR9 agonist ODN2006, indicating a strong potential antagonist effect of some 2′OMe ASOs (Supplementary Figure S6). CpG ODN are generally used at around 1–10 mg in humans, suggesting the capacity of select 2′OMe ASOs to block TLR9 in vivo when used at a therapeutic dose greater than 1 mg.
We have previously recommended the selection of 2′OMe ‘CUU’ motifs to limit TLR7 inhibition, when designing ASOs (21). In light of the inhibitory effects of such 5′ ‘CUU’ motifs on TLR9, it appears that selection of ASOs with 3′ ‘CUU’ motifs may be preferable to limit TLR7 inhibition, without increasing TLR9 inhibition. Nevertheless, the stark differences in TLR9 inhibition seen between very closely related sequences with 5′ ‘CUU/CUT’ motifs (e.g. with ASO661 and ASO662 or ASO115 and ASO116), suggest that mitigation of TLR9 inhibition could be achieved by limited sequence modification of the ASOs – for instance, by shifting the ASO by one base. We also note that potential discrepancies may exist between species regarding TLR9 inhibition by 2′OMe ASOs, since C2-Mut1 was a much stronger inhibitor in mouse macrophages. Mechanistically, we speculate that 2′OMe ASOs inhibit TLR9 by binding to it directly and competing with other agonistic DNA sequences. Although warranting further validation, this mode of action is supported by our observation that select 2′OMe ASOs were able to elicit a mild TLR9 activation on their own (21). However, it is also possible 2′OMe ASOs indirectly modulate cGAS/TLR9 sensing through other mechanisms such as an inhibitory effect on nucleases (e.g. DNase II), which should be investigated in further studies.
Finally, relying on our previous results regarding TLR7 and TLR8 modulation by 2′OMe ASOs, we performed a comprehensive comparison of the regulatory effects of 2′OMe ASOs on RNA and DNA sensing in human cells on our panel of 80 ASOs. Interestingly, TLR7 inhibition correlated with that of cGAS, as exemplified with C2-Mut1, which was also a very potent TLR7 inhibitor (with an IC50 3 times lower than that of A151). TLR8 potentiation was inversely correlated to cGAS inhibition, with high potentiators of TLR8 often displaying low cGAS inhibition (as seen with ASO [LINC-PINT]108 in Supplementary Table S2), suggesting that the rational design of gene targeting ASOs that can potentiate TLR8 sensing without inhibiting TLR7 and cGAS signalling may be achievable (21).
Critically, our findings suggest that it may be extremely challenging to generate 2′OMe gapmer ASOs that do not inhibit any of these three nucleic acids sensors (i.e. TLR7, TLR9 and cGAS). Based on our screens, we found that 95% of the ASOs decreased sensing by at least one of these three sensors by more than 30% and 2′OMe ASOs may have further sequence-specific suppressive effects on other nucleic acids sensors, as seen with the example of TLR3.
In conclusion, we demonstrate here that 2′OMe gapmer ASOs can be potent inhibitors of DNA sensing by human cGAS and TLR9, complementing our recent findings that these molecules can inhibit TLR7 sensing (21). Paving the way for the design of further highly potent oligonucleotide-based cGAS inhibitors, we identified the most potent cGAS-inhibiting oligonucleotide reported to date, while defining the motif regulating its activity. Although our detailed analyses of the sequence-dependent effects of 2′OMe ASOs on TLR7, TLR9 and cGAS sensing may help design ASOs that circumvent inhibition by these receptors, our findings indicate that only a limited number of molecules have the features to achieve this. This has important implications for the functional characterization and therapeutic development of ASOs, for which different immunosuppressive effects between tested and control ASO sequences may lead to the misinterpretation of results. Whether similar immunosuppressive effects can be seen for gapmer ASOs relying on different chemistries remains to be defined, but our previous analyses on TLR7 sensing with LNA and 2′MOE (21), along with the broad immunosuppressive effects of the PS-ODN A151, suggest that the PS-DNA moiety found in all gapmer ASOs plays a critical role in immune inhibition - independent of the chemistries used in the 5′ and 3′ regions of the gapmers. Finally whether other key nucleic acid sensors such as AIM2 or Retinoic Acid Inducible Gene-I (RIG-I) can also be impacted by 2′OMe ASOs remains to be defined. There is, however, prior evidence that PS-ODN can inhibit AIM2 (17) and that 2′OMe moieties can modulate RIG-I sensing (11), indicating that the immunosuppressive effects of ASOs are even broader than considered here.
DATA AVAILABILITY
Additional numerical data used to generate published graphs that support the findings of this study are available on request from the corresponding author.
Supplementary Material
ACKNOWLEDGEMENTS
We thank R. Firestein for HT-29 cells; V. Hornung for LL171 and cGAS–/– THP-1 cells; E. Latz for UNC93B1–/– and matched reconstituted cells; E. Sanij for BJ hTERT SV40T cells; The Cancer Therapeutics CRC for the GSK human STING agonist; J. Gearing for help with generating Figure 5; G. Ballerin (Monash Micro-Imaging at MHTP) for help with imaging of senescent cells; B.R.G. Williams for comments on the manuscript and R.E. Smith for helping with the preparation of the manuscript.
Notes
Present address: Geneviève Pépin, Department of Medical Biology, Université du Québec à Trois-Rivières, Quebec G8Z 4M3, Canada.
Contributor Information
Roxane Valentin, Centre for Innate Immunity and Infectious Diseases, Hudson Institute of Medical Research, Clayton, Victoria 3168, Australia; Department of Molecular and Translational Science, Monash University, Clayton, Victoria 3800, Australia.
Christophe Wong, Centre for Innate Immunity and Infectious Diseases, Hudson Institute of Medical Research, Clayton, Victoria 3168, Australia; Department of Molecular and Translational Science, Monash University, Clayton, Victoria 3800, Australia.
Arwaf S Alharbi, Centre for Innate Immunity and Infectious Diseases, Hudson Institute of Medical Research, Clayton, Victoria 3168, Australia; Department of Molecular and Translational Science, Monash University, Clayton, Victoria 3800, Australia; The Department of Clinical Laboratory Sciences, College of Applied Medical Sciences, Taif University, Turabah 29179, Saudi Arabia.
Solène Pradeloux, Centre for Innate Immunity and Infectious Diseases, Hudson Institute of Medical Research, Clayton, Victoria 3168, Australia; Department of Molecular and Translational Science, Monash University, Clayton, Victoria 3800, Australia.
Makala P Morros, Centre for Innate Immunity and Infectious Diseases, Hudson Institute of Medical Research, Clayton, Victoria 3168, Australia; Department of Molecular and Translational Science, Monash University, Clayton, Victoria 3800, Australia.
Kim A Lennox, Integrated DNA Technologies Inc., Coralville, IA 52241, USA.
Julia I Ellyard, Department of Immunology and Infectious Diseases, John Curtin School of Medical Research, Australian National University, Canberra, ACT 2601, Australia; Centre for Personalised Immunology, John Curtin School of Medical Research, Australian National University, Canberra, ACT, 2601, Australia.
Aurélie J Garcin, Centre for Innate Immunity and Infectious Diseases, Hudson Institute of Medical Research, Clayton, Victoria 3168, Australia; Department of Molecular and Translational Science, Monash University, Clayton, Victoria 3800, Australia.
Tomalika R Ullah, Centre for Innate Immunity and Infectious Diseases, Hudson Institute of Medical Research, Clayton, Victoria 3168, Australia; Department of Molecular and Translational Science, Monash University, Clayton, Victoria 3800, Australia.
Gina D Kusuma, Ritchie Centre, Hudson Institute of Medical Research, Clayton, Victoria 3168, Australia; Department of Obstetrics and Gynaecology, Monash University, Clayton, Victoria 3800, Australia.
Geneviève Pépin, Centre for Innate Immunity and Infectious Diseases, Hudson Institute of Medical Research, Clayton, Victoria 3168, Australia; Department of Molecular and Translational Science, Monash University, Clayton, Victoria 3800, Australia.
Hong-Mei Li, Centre for Innate Immunity and Infectious Diseases, Hudson Institute of Medical Research, Clayton, Victoria 3168, Australia; Department of Molecular and Translational Science, Monash University, Clayton, Victoria 3800, Australia.
Jaclyn S Pearson, Centre for Innate Immunity and Infectious Diseases, Hudson Institute of Medical Research, Clayton, Victoria 3168, Australia; Department of Molecular and Translational Science, Monash University, Clayton, Victoria 3800, Australia.
Jonathan Ferrand, Centre for Innate Immunity and Infectious Diseases, Hudson Institute of Medical Research, Clayton, Victoria 3168, Australia; Department of Molecular and Translational Science, Monash University, Clayton, Victoria 3800, Australia.
Rebecca Lim, Ritchie Centre, Hudson Institute of Medical Research, Clayton, Victoria 3168, Australia; Department of Obstetrics and Gynaecology, Monash University, Clayton, Victoria 3800, Australia; Australian Regenerative Medicine Institute, Monash University, Clayton, Victoria 3800, Australia.
Rakesh N Veedu, Centre for Molecular Medicine and Innovative Therapeutics, Murdoch University, Perth, Western Australia 6150, Australia; Perron Institute for Neurological and Translational Science, Perth, Western Australia 6150, Australia.
Eric F Morand, School of Clinical Sciences at Monash Health, Monash University, Clayton, Victoria 3168, Australia.
Carola G Vinuesa, Department of Immunology and Infectious Diseases, John Curtin School of Medical Research, Australian National University, Canberra, ACT 2601, Australia; Centre for Personalised Immunology, John Curtin School of Medical Research, Australian National University, Canberra, ACT, 2601, Australia.
Mark A Behlke, Integrated DNA Technologies Inc., Coralville, IA 52241, USA.
Michael P Gantier, Centre for Innate Immunity and Infectious Diseases, Hudson Institute of Medical Research, Clayton, Victoria 3168, Australia; Department of Molecular and Translational Science, Monash University, Clayton, Victoria 3800, Australia.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Australian National Health and Medical Research Council [1081167, 1124485 to M.P.G.]; Australian Research Council [140100594 Future Fellowship to M.P.G.]; Quebec Fonds de Recherche du Québec (FRSQ) – Santé [35071 to G.P.]; Fielding Foundation Innovation Award [to M.P.G.]; Victorian Government's Operational Infrastructure Support Program. Funding for open access charge: Hudson Institute of Medical Research.
Conflict of interest statement. Mark A. Behlke and Kim A. Lennox are employed by Integrated DNA Technologies Inc. (IDT), a Danaher company, which offers oligonucleotides for sale similar to some of the compounds described in the manuscript. Mark A. Behlke owns equity in DHR, the parent company of IDT. Michael Gantier is a named inventor of a provisional patent relating to immunomodulatory oligonucleotides.
REFERENCES
- 1. Yin W., Rogge M.. Targeting RNA: a transformative therapeutic strategy. Clin. Transl. Sci. 2019; 12:98–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Byrne P., Cullinan J., Mintzes B., Smith S.M.. UK deal over inclisiran. BMJ. 2020; 368:m579. [DOI] [PubMed] [Google Scholar]
- 3. Krieg A.M., Yi A.K., Matson S., Waldschmidt T.J., Bishop G.A., Teasdale R., Koretzky G.A., Klinman D.M.. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995; 374:546–549. [DOI] [PubMed] [Google Scholar]
- 4. Hemmi H., Takeuchi O., Kawai T., Kaisho T., Sato S., Sanjo H., Matsumoto M., Hoshino K., Wagner H., Takeda K.et al.. A Toll-like receptor recognizes bacterial DNA. Nature. 2000; 408:740–745. [DOI] [PubMed] [Google Scholar]
- 5. Krieg A.M., Wu T., Weeratna R., Efler S.M., Love-Homan L., Yang L., Yi A.K., Short D., Davis H.L.. Sequence motifs in adenoviral DNA block immune activation by stimulatory CpG motifs. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:12631–12636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gursel I., Gursel M., Yamada H., Ishii K.J., Takeshita F., Klinman D.M.. Repetitive elements in mammalian telomeres suppress bacterial DNA-induced immune activation. J. Immunol. 2003; 171:1393–1400. [DOI] [PubMed] [Google Scholar]
- 7. Barrat F.J., Meeker T., Gregorio J., Chan J.H., Uematsu S., Akira S., Chang B., Duramad O., Coffman R.L.. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J. Exp. Med. 2005; 202:1131–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Trieu A., Roberts T.L., Dunn J.A., Sweet M.J., Stacey K.J.. DNA motifs suppressing TLR9 responses. Crit. Rev. Immunol. 2006; 26:527–544. [DOI] [PubMed] [Google Scholar]
- 9. Robbins M., Judge A., Liang L., McClintock K., Yaworski E., MacLachlan I.. 2′-O-methyl-modified RNAs act as TLR7 antagonists. Mol. Ther. 2007; 15:1663–1669. [DOI] [PubMed] [Google Scholar]
- 10. Sioud M., Furset G., Cekaite L.. Suppression of immunostimulatory siRNA-driven innate immune activation by 2′-modified RNAs. Biochem. Biophys. Res. Commun. 2007; 361:122–126. [DOI] [PubMed] [Google Scholar]
- 11. Devarkar S.C., Wang C., Miller M.T., Ramanathan A., Jiang F., Khan A.G., Patel S.S., Marcotrigiano J.. Structural basis for m7G recognition and 2′-O-methyl discrimination in capped RNAs by the innate immune receptor RIG-I. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Judge A.D., Sood V., Shaw J.R., Fang D., McClintock K., MacLachlan I.. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat. Biotechnol. 2005; 23:457–462. [DOI] [PubMed] [Google Scholar]
- 13. Judge A.D., Bola G., Lee A.C., MacLachlan I.. Design of noninflammatory synthetic siRNA mediating potent gene silencing in vivo. Mol. Ther. 2006; 13:494–505. [DOI] [PubMed] [Google Scholar]
- 14. Frazier K.S. Antisense oligonucleotide therapies: the promise and the challenges from a toxicologic pathologist's perspective. Toxicol. Pathol. 2015; 43:78–89. [DOI] [PubMed] [Google Scholar]
- 15. Bayik D., Gursel I., Klinman D.M.. Structure, mechanism and therapeutic utility of immunosuppressive oligonucleotides. Pharmacol. Res. 2016; 105:216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Beignon A.S., McKenna K., Skoberne M., Manches O., DaSilva I., Kavanagh D.G., Larsson M., Gorelick R.J., Lifson J.D., Bhardwaj N.. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J. Clin. Invest. 2005; 115:3265–3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kaminski J.J., Schattgen S.A., Tzeng T.C., Bode C., Klinman D.M., Fitzgerald K.A.. Synthetic oligodeoxynucleotides containing suppressive TTAGGG motifs inhibit AIM2 inflammasome activation. J. Immunol. 2013; 191:3876–3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Steinhagen F., Zillinger T., Peukert K., Fox M., Thudium M., Barchet W., Putensen C., Klinman D., Latz E., Bode C.. Suppressive oligodeoxynucleotides containing TTAGGG motifs inhibit cGAS activation in human monocytes. Eur. J. Immunol. 2018; 48:605–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sarvestani S.T., Stunden H.J., Behlke M.A., Forster S.C., McCoy C.E., Tate M.D., Ferrand J., Lennox K.A., Latz E., Williams B.R.et al.. Sequence-dependent off-target inhibition of TLR7/8 sensing by synthetic microRNA inhibitors. Nucleic Acids Res. 2015; 43:1177–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McWhirter S.M., Jefferies C.A.. Nucleic acid sensors as therapeutic targets for human disease. Immunity. 2020; 53:78–97. [DOI] [PubMed] [Google Scholar]
- 21. Alharbi A.S., Garcin A.J., Lennox K.A., Pradeloux S., Wong C., Straub S., Valentin R., Pepin G., Li H.M., Nold M.F.et al.. Rational design of antisense oligonucleotides modulating the activity of TLR7/8 agonists. Nucleic Acids Res. 2020; 48:7052–7065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gorden K.K., Qiu X., Battiste J.J., Wightman P.P., Vasilakos J.P., Alkan S.S.. Oligodeoxynucleotides differentially modulate activation of TLR7 and TLR8 by imidazoquinolines. J. Immunol. 2006; 177:8164–8170. [DOI] [PubMed] [Google Scholar]
- 23. Jurk M., Kritzler A., Schulte B., Tluk S., Schetter C., Krieg A.M., Vollmer J.. Modulating responsiveness of human TLR7 and 8 to small molecule ligands with T-rich phosphorothiate oligodeoxynucleotides. Eur. J. Immunol. 2006; 36:1815–1826. [DOI] [PubMed] [Google Scholar]
- 24. Arnett F.C., Edworthy S.M., Bloch D.A., McShane D.J., Fries J.F., Cooper N.S., Healey L.A., Kaplan S.R., Liang M.H., Luthra H.S.et al.. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis & Rheumatism. 1988; 31:315–324. [DOI] [PubMed] [Google Scholar]
- 25. Leech M., Metz C., Hall P., Hutchinson P., Gianis K., Smith M., Weedon H., Holdsworth S.R., Bucala R., Morand E.F.. Macrophage migration inhibitory factor in rheumatoid arthritis: evidence of proinflammatory function and regulation by glucocorticoids. Arthritis & Rheumatism. 1999; 42:1601–1608. [DOI] [PubMed] [Google Scholar]
- 26. Gray E.E., Treuting P.M., Woodward J.J., Stetson D.B.. Cutting Edge: cGAS is required for lethal autoimmune disease in the Trex1-deficient mouse model of Aicardi–Goutières syndrome. J. Immunol. 2015; 195:1939–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ferrand J., Gantier M.P.. Assessing the inhibitory activity of oligonucleotides on TLR7 sensing. Methods Mol. Biol. 2016; 1390:79–90. [DOI] [PubMed] [Google Scholar]
- 28. Quin J., Chan K.T., Devlin J.R., Cameron D.P., Diesch J., Cullinane C., Ahern J., Khot A., Hein N., George A.J.et al.. Inhibition of RNA polymerase I transcription initiation by CX-5461 activates non-canonical ATM/ATR signaling. Oncotarget. 2016; 7:49800–49818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ablasser A., Schmid-Burgk J.L., Hemmerling I., Horvath G.L., Schmidt T., Latz E., Hornung V.. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature. 2013; 503:530–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ferrand J., Croft N.P., Pépin G., Diener K.R., Wu D., Mangan N.E., Pedersen J., Behlke M.A., Hayball J.D., Purcell A.W.et al.. The use of CRISPR/Cas9 gene editing to confirm congenic contaminations in host-pathogen interaction studies. Front. Cell. Infect. Microbiol. 2018; 8:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mankan A.K., Schmidt T., Chauhan D., Goldeck M., Höning K., Gaidt M., Kubarenko A.V., Andreeva L., Hopfner K.P., Hornung V.. Cytosolic RNA:DNA hybrids activate the cGAS –STING axis. The EMBO Journal. 2014; 33:2937–2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pelka K., Phulphagar K., Zimmermann J., Stahl R., Schmid-Burgk J.L., Schmidt T., Spille J.H., Labzin L.I., Agrawal S., Kandimalla E.R.et al.. Cutting edge: the UNC93B1 tyrosine-based motif regulates trafficking and TLR responses via separate mechanisms. J. Immunol. 2014; 193:3257–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stetson D.B., Medzhitov R.. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006; 24:93–103. [DOI] [PubMed] [Google Scholar]
- 34. Unterholzner L., Keating S.E., Baran M., Horan K.A., Jensen S.B., Sharma S., Sirois C.M., Jin T., Latz E., Xiao T.S.et al.. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010; 11:997–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ramanjulu J.M., Pesiridis G.S., Yang J., Concha N., Singhaus R., Zhang S.-Y., Tran J.-L., Moore P., Lehmann S., Eberl H.C.et al.. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature. 2018; 564:439–443. [DOI] [PubMed] [Google Scholar]
- 36. An J., Woodward J.J., Lai W., Minie M., Sun X., Tanaka L., Snyder J.M., Sasaki T., Elkon K.B.. Inhibition of Cyclic GMP-AMP synthase using a novel antimalarial drug derivative in Trex1-deficient mice. Arthritis Rheumatol. 2018; 70:1807–1819. [DOI] [PubMed] [Google Scholar]
- 37. Lama L., Adura C., Xie W., Tomita D., Kamei T., Kuryavyi V., Gogakos T., Steinberg J.I., Miller M., Ramos-Espiritu L.et al.. Development of human cGAS-specific small-molecule inhibitors for repression of dsDNA-triggered interferon expression. Nat. Commun. 2019; 10:2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Padilla-Salinas R., Sun L., Anderson R., Yang X., Zhang S., Chen Z.J., Yin H.. Discovery of small-molecule cyclic GMP-AMP synthase inhibitors. J. Org. Chem. 2020; 85:1579–1600. [DOI] [PubMed] [Google Scholar]
- 39. Vincent J., Adura C., Gao P., Luz A., Lama L., Asano Y., Okamoto R., Imaeda T., Aida J., Rothamel K.et al.. Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat. Commun. 2017; 8:750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhao W., Xiong M., Yuan X., Li M., Sun H., Xu Y.. In silico screening-based discovery of novel inhibitors of human cyclic GMP-AMP synthase: A cross-validation study of molecular docking and experimental testing. J. Chem. Inf. Model. 2020; 60:3265–3276. [DOI] [PubMed] [Google Scholar]
- 41. Xia T., Konno H., Ahn J., Barber G.N.. Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. 2016; 14:282–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xie W., Lama L., Adura C., Tomita D., Glickman J.F., Tuschl T., Patel D.J.. Human cGAS catalytic domain has an additional DNA-binding interface that enhances enzymatic activity and liquid-phase condensation. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:11946–11955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bailey T.L., Elkan C.. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994; 2:28–36. [PubMed] [Google Scholar]
- 44. Kranzusch Philip J., Lee Amy S.-Y., Berger James M., Doudna Jennifer A.. Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Reports. 2013; 3:1362–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pepin G., Nejad C., Ferrand J., Thomas B.J., Stunden H.J., Sanij E., Foo C.H., Stewart C.R., Cain J.E., Bardin P.G.et al.. Topoisomerase 1 inhibition promotes cyclic GMP-AMP synthase-dependent antiviral responses. mBio. 2017; 8:e01611-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Uhlen M., Zhang C., Lee S., Sjostedt E., Fagerberg L., Bidkhori G., Benfeitas R., Arif M., Liu Z., Edfors F.et al.. A pathology atlas of the human cancer transcriptome. Science. 2017; 357:eaan2507. [DOI] [PubMed] [Google Scholar]
- 47. Dai J., Huang Y.J., He X., Zhao M., Wang X., Liu Z.S., Xue W., Cai H., Zhan X.Y., Huang S.Y.et al.. Acetylation blocks cGAS activity and inhibits Self-DNA-Induced autoimmunity. Cell. 2019; 176:1447–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gluck S., Guey B., Gulen M.F., Wolter K., Kang T.W., Schmacke N.A., Bridgeman A., Rehwinkel J., Zender L., Ablasser A.. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017; 19:1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dou Z., Ghosh K., Vizioli M.G., Zhu J., Sen P., Wangensteen K.J., Simithy J., Lan Y., Lin Y., Zhou Z.et al.. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017; 550:402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ni G., Ma Z., Damania B.. cGAS and STING: At the intersection of DNA and RNA virus-sensing networks. PLoS Pathog. 2018; 14:e1007148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Watson R.O., Bell S.L., MacDuff D.A., Kimmey J.M., Diner E.J., Olivas J., Vance R.E., Stallings C.L., Virgin H.W., Cox J.S.. The cytosolic sensor cGAS detects Mycobacterium tuberculosis DNA to induce type I interferons and activate autophagy. Cell Host Microbe. 2015; 17:811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pepin G., Nejad C., Thomas B.J., Ferrand J., McArthur K., Bardin P.G., Williams B.R., Gantier M.P.. Activation of cGAS-dependent antiviral responses by DNA intercalating agents. Nucleic Acids Res. 2017; 45:198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Maekawa H., Inoue T., Ouchi H., Jao T.M., Inoue R., Nishi H., Fujii R., Ishidate F., Tanaka T., Tanaka Y.et al.. Mitochondrial damage causes inflammation via cGAS-STING signaling in acute kidney injury. Cell Rep. 2019; 29:1261–1273. [DOI] [PubMed] [Google Scholar]
- 54. Ablasser A., Chen Z.J.. cGAS in action: expanding roles in immunity and inflammation. Science. 2019; 363:eaat8657. [DOI] [PubMed] [Google Scholar]
- 55. Luther J., Khan S., Gala M.K., Kedrin D., Sridharan G., Goodman R.P., Garber J.J., Masia R., Diagacomo E., Adams D.et al.. Hepatic gap junctions amplify alcohol liver injury by propagating cGAS-mediated IRF3 activation. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:11667–11673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yu C.-H., Davidson S., Harapas C.R., Hilton J.B., Mlodzianoski M.J., Laohamonthonkul P., Louis C., Low R.R.J., Moecking J., De Nardo D.et al.. TDP-43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell. 2020; 183:636–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hall J., Brault A., Vincent F., Weng S., Wang H., Dumlao D., Aulabaugh A., Aivazian D., Castro D., Chen M.et al.. Discovery of PF-06928215 as a high affinity inhibitor of cGAS enabled by a novel fluorescence polarization assay. PLoS One. 2017; 12:e0184843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang M., Sooreshjani M.A., Mikek C., Opoku-Temeng C., Sintim H.O.. Suramin potently inhibits cGAMP synthase, cGAS, in THP1 cells to modulate IFN-beta levels. Future Med. Chem. 2018; 10:1301–1317. [DOI] [PubMed] [Google Scholar]
- 59. Conlon J., Burdette D.L., Sharma S., Bhat N., Thompson M., Jiang Z., Rathinam V.A., Monks B., Jin T., Xiao T.S.et al.. Mouse, but not human STING, binds and signals in response to the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid. J. Immunol. 2013; 190:5216–5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pepin G., De Nardo D., Rootes C.L., Ullah T.R., Al-Asmari S.S., Balka K.R., Li H.M., Quinn K.M., Moghaddas F., Chappaz S.et al.. Connexin-dependent transfer of cGAMP to phagocytes modulates antiviral responses. mBio. 2020; 11:e03187-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li Q., Cao Y., Dang C., Han B., Han R., Ma H., Hao J., Wang L.. Inhibition of double-strand DNA-sensing cGAS ameliorates brain injury after ischemic stroke. EMBO Mol. Med. 2020; 12:e11002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Additional numerical data used to generate published graphs that support the findings of this study are available on request from the corresponding author.