

Abstract
Psychoactive natural products play an integral role in the modern world. The tremendous structural complexity displayed by such molecules confers diverse biological activities of significant medicinal value and sociocultural impact. Accordingly, in the last two centuries, immense effort has been devoted towards establishing how plants, animals, and fungi synthesize complex natural products from simple metabolic precursors. The recent explosion of genomics data and molecular biology tools has enabled the identification of genes encoding proteins that catalyze individual biosynthetic steps. Once fully elucidated, the “biosynthetic pathways” are often comparable to organic syntheses in elegance and yield. Additionally, the discovery of biosynthetic enzymes provides powerful catalysts which may be repurposed for synthetic biology applications, or implemented with chemoenzymatic synthetic approaches. In this review, we discuss the progress that has been made toward biosynthetic pathway elucidation amongst four classes of psychoactive natural products: hallucinogens, stimulants, cannabinoids, and opioids. Compounds of diverse biosynthetic origin – terpene, amino acid, polyketide – are identified, and notable mechanisms of key scaffold transforming steps are highlighted. We also provide a description of subsequent applications of the biosynthetic machinery, with an emphasis placed on the synthetic biology and metabolic engineering strategies enabling heterologous production.
Graphical Abstract

1. Introduction
The consumption of psychoactive natural products predates recorded history.1 For millennia, our ancestors subsisted by consuming materials foraged from the natural world. Over time, innumerable person-hours of trial and error resulted in a keen understanding of the expected physiological and psychological effects upon ingestion of specific plants, animals, and fungi.2 This information propagated initially as traditional knowledge, forming the basis of valuable cultural practices and efficacious traditional medicine.3 The myriad of ethical concerns around the appropriation of indigenous knowledge, exploitation of slave labor, as well as inequitable access to natural product cultivation, sale, and use, typically go unanswered by mainstream science, and we encourage the reader to consult a selection of responsibly written articles on these subject matters.4–8 Scientists are beginning to recognize that natural products have mediated intimate evolutionary relationships between plants, animals, and fungi.9 For instance, over centuries, winemakers selected grapes harboring high-alcohol producing Crabtree-positive yeast, enabling the co-domestication of a plant-fungal symbiont pair.10 An additional, highly speculative example known as the “Stoned Ape Hypothesis” posits that the consumption of psychedelic mushrooms may have played a role in rapid increase of brain size in early hominids.11
This push-pull relationship of humans with natural products continues to this day, as the adoption of single molecule constituents by Western culture has triggered the expansion of traditional cultivation practices to meet global demands.12 Isolation of and characterization of organic plant extracts marked the beginnings of both organic chemistry and Western medicine. Prior to 20th century prohibition, efforts towards the total synthesis of commodified natural products provided a foundation for generations of organic chemists. Sir Robert Robinson’s 1917 route to the cocaine precursor tropinone is widely lauded as a classic in total synthesis,13 while Woodward’s innumerable contributions to the field of natural product total synthesis included a route to the lysergic acid diethylamide (LSD) precursor lysergic acid.14 Incorporation of this knowledge into semi-syntheses prompted researchers to think of biological materials as chemical factories, and beg the question: how do organisms synthesize natural products? Extraordinary progress has been made in the elucidation of the metabolic pathways underpinning the chemical composition of psychoactive substances. In the field of natural product biosynthesis, scientists investigate the biosynthetic logic that enables Nature to synthesize psychoactive natural products with high efficiencies and selectivities.15 Identification and reconstitution of key enzymatic steps uncovers Nature’s synthetic schema towards complex molecular scaffolds from simple metabolic precursors. The accumulation of such biosynthetic information is driven in part by advancements in synthetic biology; emerging biotechnologies promise to outperform traditional synthetic methods in cost, safety, efficiency, and sustainability. Thus, significant achievements have been made in the heterologous expression of natural product pathways towards consumer products.
1.1. Four categories of psychoactive natural products
Only in the last half of a century have scientists begun to investigate the molecular mechanisms of psychoactivity – the alterations in perception, consciousness, and behavior, associated with such small molecules.16 Prior to the 1950s, most scientists believed that synaptic activity was dictated entirely through electrical impulses, and little evidence existed on the role of chemical signaling.17 Our current understanding of psychopharmacology has been directly facilitated by the use of natural products. The extraordinary protein receptor binding affinities of psychoactive natural products allowed scientists to deduce the role of neurotransmitters in the central nervous system.18 We now know that neuroreceptors are the key signal transducers able to integrate chemical signals into biological systems. It is the selective receptor binding and activation by native and non-native chemical ligands that causes modulation of neural pathways, resulting in altered perception.19 These receptors are differentially expressed in different populations of neurons, and may exist as splice variants or exhibit single-nucleotide polymorphisms between individuals.20 Further, differential activation of receptor subtypes by a given ligand makes it difficult to categorize psychoactive drugs based strictly on the physiological target. For example, activation of μ-opioid receptors (MORs) by agonists like morphine (Section 5.2) results in analgesia and sedation,21 whereas activation of κ-opioid receptors (KORs) by the potent ligand salvinorin A (Section 2.9) results in dissociation.22 Thus, while formally an opioid, the consumer of Salvia divinorum would classify the shrub as a bona fide hallucinogen based on perceived psychological effect. As a result, psychoactive drugs have traditionally been categorized based simply on the experience of the user, as opposed to complex molecular mechanisms of psychoactivity. The natural products discussed herein fall within one of four well-recognized classes: hallucinogens, stimulants, cannabinoids, and opioids (Fig. 1).
Fig. 1.
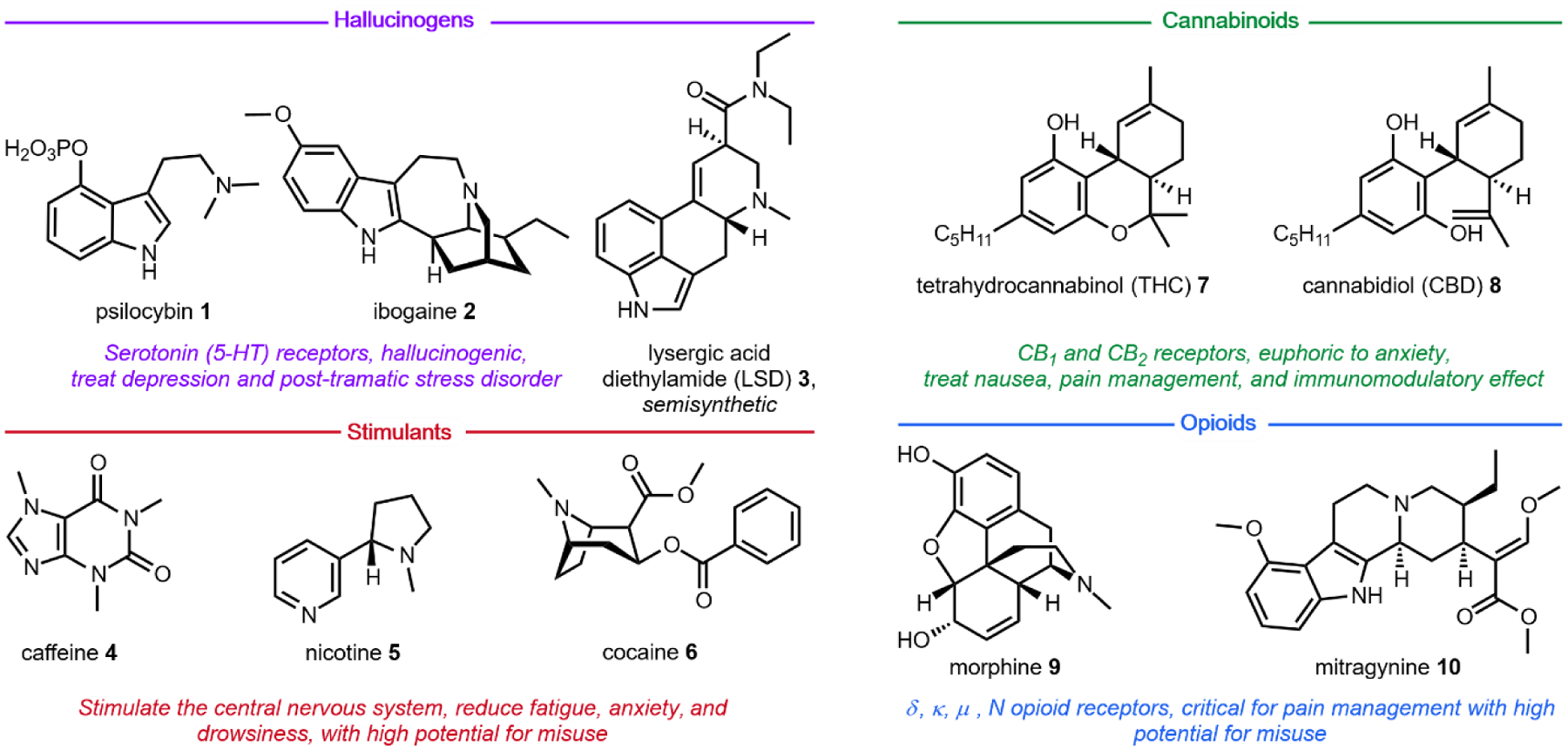
Four categories of psychoactive natural products or derivatives described in this review.
The utility of psychoactive natural products, if used safely, cannot be questioned. Selective, potent binding of a ligand to a target is a hallmark feature of a pharmaceutical agent. While immense pharmaceutical potential has been ascribed to many psychoactive natural products, evidence-based drug development campaigns are largely hindered by regulatory status.23 Natural products in the Schedule I Controlled Substance category have been designated as having no accepted medical use, hindering clinical trials, even though many compounds on the list exhibit great potential for clinical success. For example, evidence implicates psilocybin 1 as a promising candidate for treatment-resistant depression24 and post-traumatic stress disorder,25 whereas the alkaloid ibogaine 2 has undergone development as anti-addictive agent.26 Meanwhile, a recent meta-analysis concluded that the natural product derivative lysergic acid diethylamide (LSD) 3 has strong potential in the treatment of alcoholism.27 These three compounds fall into the category of hallucinogenic natural products, invoking psychedelic, introspective effects. Alkaloidal stimulants are also of great societal value, and include the world’s most widely consumed psychoactive drug, caffeine 4.28 Nicotine 5 and cocaine 6, two other well-known alkaloidal stimulants, exhibit high potential for dependence, but are each approved for specific medicinal indications.29,30 While the legal status of Cannabis is currently in flux, the primary constituents tetrahydrocannabinol (THC) 7 and cannabidiol (CBD) 8 are FDA approved medications.31 State-by-state deregulation has resulted in the ongoing cannabinoid boon driving academia and industry to discover additional applications for THC, CBD, and other rare cannabinoids. Finally, opioid analgesics are included on the World Health Organization’s List of Essential Medicines. Despite the ongoing opioid crisis, morphine 9 plays a critical role in pain management and palliative care.32 Kratom, which contains the potent MOR agonist mitragynine 10, has emerged recently as an alternative to opium-derived substances. Given its potential for abuse, additional epidemiological studies of kratom are warranted.33 As opioid dependence soars, public health organizations have described the importance of research into pain management and addiction. We advocate for an unbiased, evidence-based evaluation of the risks and benefits of psychoactive natural product use in order to maximize societal value.
1.2. Overview of biosynthesis of psychoactive compounds
As with most natural products isolated from microorganisms and plants, the psychoactive compounds discussed in this review are biosynthesized from simple, primary metabolites such as acetate, isoprene, and amino acids.15 With the exception of cannabinoids and a few others, most of the compounds covered are alkaloids derived from the decarboxylation of a small set of amino acids. For example, l-tryptophan 11 is the precursor to ibogaine 2 and psilocybin 3; l-tyrosine 12 is the precursor to mescaline (Section 2.6) and morphine 10; while the nonproteinogenic amino acid l-ornithine 13 is the precursor to nicotine 5 and cocaine 6. The decarboxylation of amino acids is catalyzed by an enzyme family known as amino acid decarboxylase (AADC), which uses pyridoxal-5’-phosphate (PLP) as a cofactor. A few of the compounds contain isoprenoid building blocks, such as the C5 prenyl unit in lysergic acid (Section 2.5) and the C10 geranyl unit in cannabinoids (Section 4.2). The C–C bonds between the isoprenes and the rest of the molecules in these compounds are catalyzed by a group of enzymes known as prenyltransferases. Prenyltransferases are one type of group transfer enzyme used by nature to transfer functional groups from thermodynamically activated carriers to natural product biosynthetic intermediates. Other group transfer enzymes include acyltransferases and S-adenosylmethionine (SAM) dependent methyltransferases, which are frequently found in biosynthetic pathways. Nature also uses redox reactions extensively to modify the natural products to their final, bioactive forms. The enzymes catalyzing these reactions are collectively referred to as oxidoreductases, and include examples such as cytochrome P450s, ketoreductases and amine oxidases.34 The enzymology of these enzymes has been well-studied and the reader can refer to other reviews for more information.35,36 Here we will briefly summarize a few enzyme-catalyzed or enzyme-mediated reactions that will be found throughout the review.
1.2.1. Decarboxylation of amino acids
The aromatic amino acids l-tryptophan 12, l-tyrosine 13 and to a less extent, l-phenylalanine, are commonly used precursors for alkaloid natural product biosynthesis. For example, the indole ring in l-tryptophan 11 is preserved in compounds such as psilocybin 1 and ibogaine 2; while the para-hydroxybenzene side chain in l-tyrosine 12 can be found in mescaline (Section 2.6) and morphine 9. The terminal amine-containing l-lysine and l-ornithine 13 are also used as precursors. Relevant to this review, the four-carbon side chain of l-ornithine 13 is required for the formation of pyrrolidines and tropanes. The first step in the utilization of these amino acids for alkaloid biosynthesis is decarboxylation to give the corresponding primary amines, although in lysergic acid biosynthesis l-tryptophan is used without decarboxylation. The decarboxylation products of l-tryptophan, l-tyrosine and l-ornithine are tryptamine 14, tyramine 15, and putrescine 16, respectively (Fig. 2A). In the case of tyramine 14, hydroxylation of one of the meta positions in the para-phenol ring gives the metabolite dopamine 17. Dopamine 17 is a natural product building block, but also a neurotransmitter in mammals. The chemical logic for the early decarboxylation is straightforward: to facilitate intra- and intermolecular Mannich reactions with aldehydes and ketones using the nucleophilic amine (see section 1.2.2). This decarboxylation-Mannich two step rapidly sets up the (poly)-heterocyclic scaffold of many alkaloidal natural products.
Fig. 2: PLP-Dependent amino acid decarboxylase.
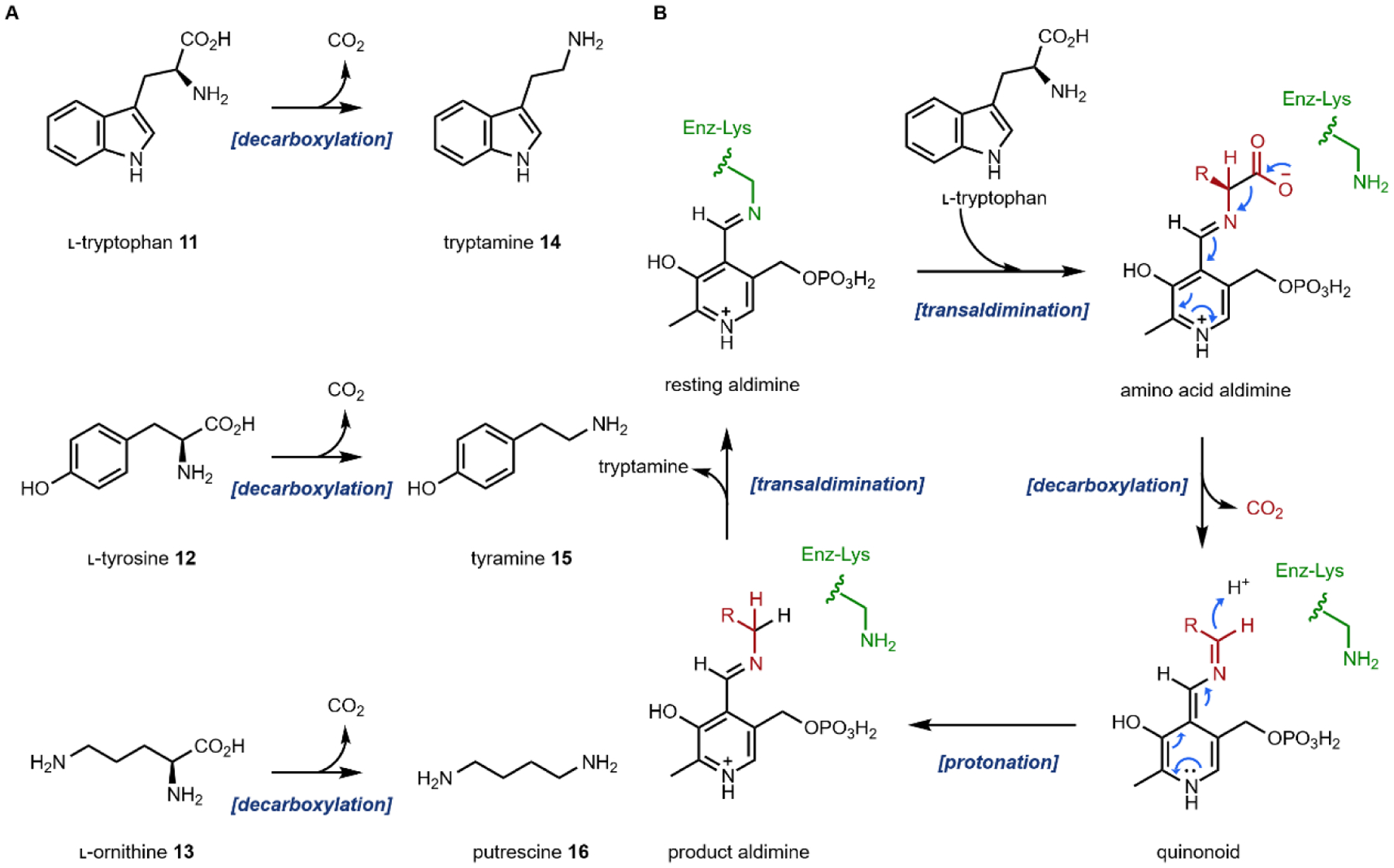
(A) three amino acids are decarboxylated to give primary amines that are building blocks for alkaloids; (B) mechanism of the PLP-dependent tryptophan decarboxylase
The decarboxylation reactions are catalyzed by dedicated amino acid decarboxylases. For example, in the case of l-tryptophan, a tryptophan decarboxylase is involved. These enzymes typically use the PLP cofactor, as expected for many enzymes that perform Cα, Cβ and Cγ modifications on amino acids.37 The mechanism of the reaction is shown in Fig. 2B. The aldehyde of PLP modifies an active site lysine to form the resting aldimine in the decarboxylase active site. A transaldimination step takes place next in which the amine of the substrate amino acid attacks the aldimine and forms the amino acid–PLP aldimine. The PLP then serves as an electron sink in the enzyme-catalyzed cleavage of the Cα-COO− bond via a quinonoid species. Reprotonation of the Cα then generates the product aldimine, which can undergo another transaldimination with the active site lysine to release the product amine and regenerate the resting aldimine.
1.2.2. Mannich/Pictet-Spengler reactions
Following decarboxylation of the amino acids to the corresponding primary amines, a common next step is the Mannich reaction involving the primary amine. The Mannich reaction is a two-step reaction that yields an alkylated amine.38 In the first step, the primary amine reacts with either an aldehyde or a ketone to form the Schiff base. The C=N double bond is then attacked by a carbon nucleophile, such as the acidic Cα of a carbonyl to form the β-amino-carbonyl product. Two examples of an intramolecular Mannich reaction can be found in the formation of the tropane unit in cocaine 6 (Section 3.4).39,40 Starting from putrescine 16, methylation of one of the primary amines gives the intermediate N-methylputrescine 18; oxidation and hydrolysis of the other amine yields N-methylaminobutanal 19, which is in equilibrium with the cyclic N-methylpyrrolinium 20. Attack of the imine by the enolized 3-oxo-glutaric acid 21 yields the adduct pyrrolidine tropane scaffold precursor (Fig. 3A). A subsequent dehydrogenation generates a new pyrrolinium species that can be attacked with Cα of the 1,3-diketo unit in a second Mannich reaction (Section 3.4).
Fig. 3: Mannich reactions in alkaloid biosynthesis.
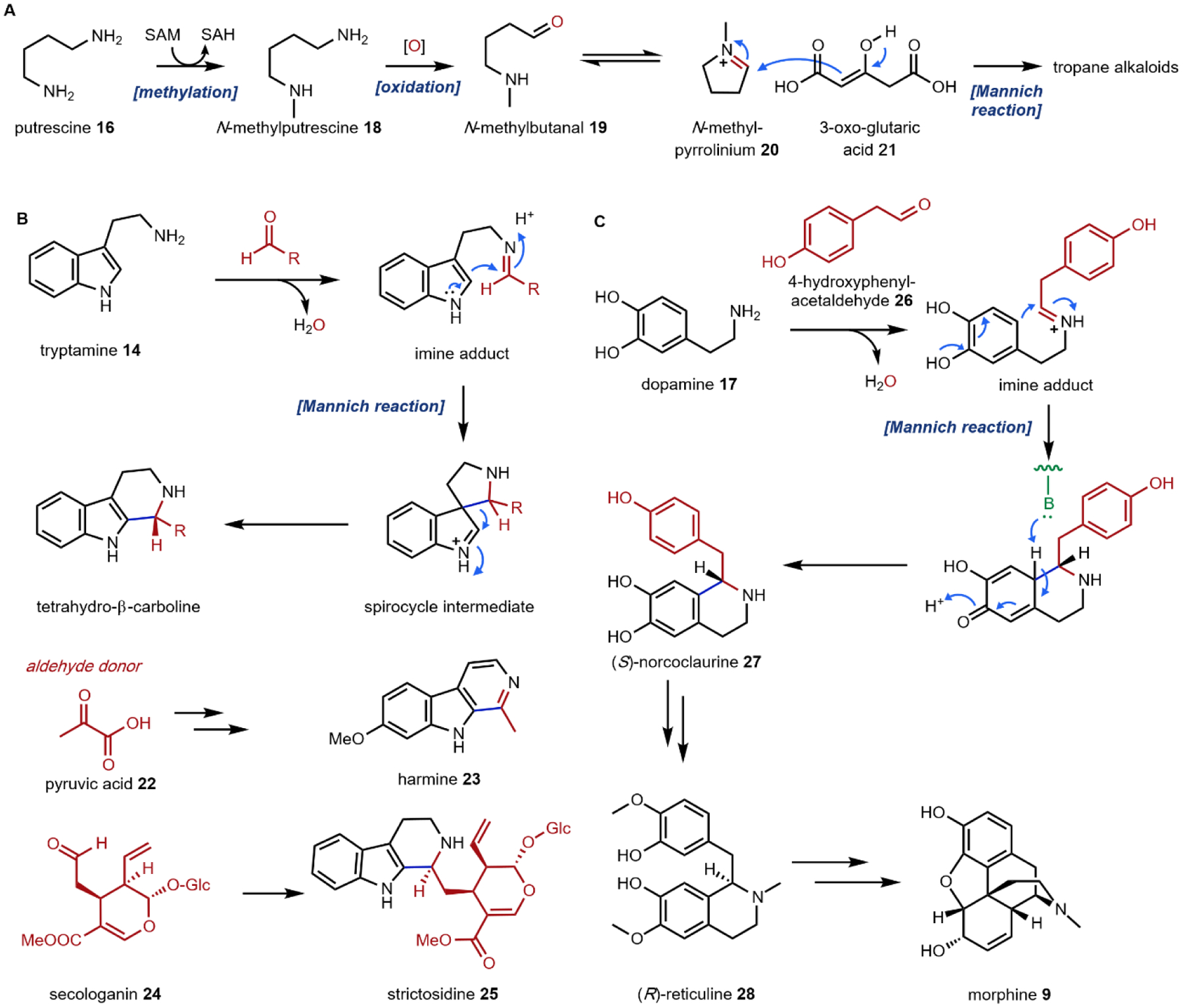
(A) formation of the pyrrolidine intermediate on pathway to tropane alkaloids; (B) the Pictet-Spengler reaction involving tryptamine to form tetrahydro-β-carboline intermediates; (C) the Pictet-Spengler reaction involving dopamine to form tetrahydroisoquinoline on pathway to morphine.
One variation of the Mannich reaction that is central to the biosynthesis of plant alkaloids is the Pictet-Spengler (PS) reaction involving β-arylethylamines such as tryptamine 14 and dopamine 17. In the PS reaction, after the amine reacts with an aldehyde or ketone to form the Schiff base, a carbanion resonance structure of the indole in tryptamine or the para-hydroxy phenol ring in dopamine can attack the imine to form the new C–C bond. This can be followed by rearrangements to form the stable tricyclic tetrahydro-β-carboline or bicyclic tetrahydroisoquinoline, respectively. The tryptamine-derived tetrahydro-β-carboline is found in harmala alkaloids (Section 2.4) and iboga alkaloids (Section 2.8). To generate the harmala family of compounds, tryptamine 14 is condensed with pyruvic acid 22, followed by attack of the imine by C3 from the indole ring to form a spirocycle, which collapses via single bond migration to complete the PS reaction (Fig. 3B).41 Similarly, the condensation between the aldehyde donor secologanin 24 and tryptamine 14 is catalyzed by a dedicated Pictet-Spenglerase, yielding strictosidine, the universal precursor to monoterpene indole alkaloids (MIAs) including ibogaine.42 In the biosynthesis of benzylisoquinoline alkaloids (BIAs) such as morphine 9, the PS reaction takes place between dopamine 17 and 4-hydroxyphenylacetaldehyde 26, both oxidation products of tyramine 15, to form the key intermediate S-norcoclaurine 27, precursor to R-reticuline 28 and morphine 9. (Fig. 3C).43
1.2.3. Common group transfer reactions
Group transfer reactions are widely used by Nature in the biosynthesis of natural products. Functional groups that are frequently transferred from donor molecules to biosynthetic intermediates include methyl, acetyl, small, medium and long alkyl-substituted acyl chains, isoprenyl, glucosyl, etc. These reactions serve a multitude of purposes, including i) increasing the size and complexity of the molecules; ii) changing the lipophilicity of molecules; iii) altering the reactivity of functional groups; iv) serving as a transient chemical protection group for downstream modifications; v) acting as leaving groups in elimination reactions; and vi) changing the biological properties of the natural product. Hence, these reactions are indispensable to the structural diversity of natural products that have been isolated to date.
The donor molecules, those that “carry” the groups to be transferred, are kinetically stable and thermodynamically activated: the molecules are high in energy and therefore releasing the groups is a highly exergonic reaction; yet the molecules are stable under cellular conditions and enzyme catalysis is required to overcome the kinetic barriers. We recently reviewed eight such molecules that power cellular metabolism, which include ATP, NAD(P)H, acetyl-CoA, SAM, carbamoyl phosphate, isoprenyl pyrophosphate, UDP-glucose and molecular oxygen.44 NAD(P)H and molecular oxygen are involved in the redox reactions and will be summarized in the next section. Among the remaining six, carbamoyl-phosphate is involved in nitrogen metabolism and is not directly involved in natural product biosynthesis.
The remaining five, however, are frequently used group transfer donor molecules, and examples can be found throughout the review. ATP, the universal cellular energy currency, is the donor in the transferring of phosphate groups to nucleophilic oxygen in the presence of a phosphotransferase. This reaction is ubiquitous in primary metabolism but is quite rare in natural product biosynthesis (or secondary metabolism). One such example can be found in the psilocybin pathway (see section 2.3). Acetyltransferases catalyze the transfer of acetyl groups from the acetyl-CoA thioester to a variety of O and N nucleophiles (Fig. 4A). SAM-dependent methyltransferases use S-adenosylmethionine to transfer a methyl group from the trivalent sulfonium group to C, O, N, and S nucleophiles in an SN2 type substitution reaction (Fig. 4B). This reaction can be found in the majority of biosynthetic pathways described herein. For example, iterative N-methylation of tryptamine yields the psychoactive molecule N,N-dimethyltryptamine 29 (DMT, see Section 2.2). UDP-glucose is an activated glucose donor in cells for the assembly of oligosaccharides and polysaccharides. UDP-glucose is thermodynamically activated but kinetically stable in the absence of glucosyltransferases.44 In the presence of glucosylating enzymes, UDP dissociates via cleavage of the C–O bond in an SN1 fashion to yield a C1 oxocarbonium ion, which can be attacked by incoming nucleophiles (Fig. 4C). A notable example of substrate glucosylation is in the biosynthetic pathway of strictosidine 25, the precursor to ibogaine (Section 2.8). The enzyme 7DLGT glucosylates the hemiacetal in 7-deoxyloganetic acid 30 to give 7-deoxyloganic acid 31.45 The glucose moiety serves as a protecting group to prevent formation of the aldehyde, and remains in strictosidine 25. In order to transform strictosidine 25 into different scaffolds, a glucosidase removes the glucose moiety, unmasking the aldehyde and leading to subsequent rearrangements towards structurally diverse monoterpene indole alkaloids.
Fig. 4: Enzyme catalyzed group transfer reactions in biosynthesis.
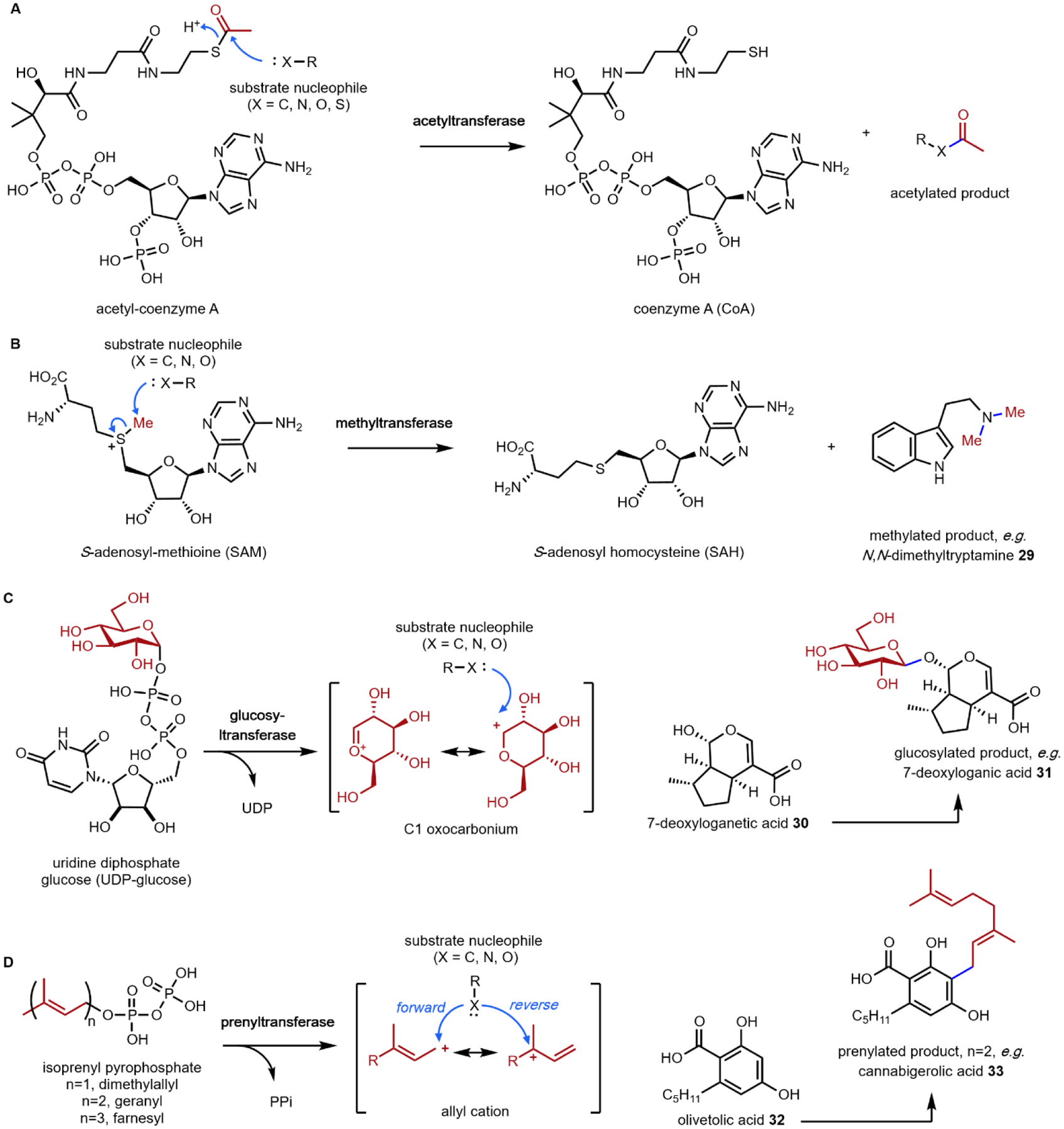
(A) acetyltransferase-catalyzed acetyltransfer; (B) methyltransferase-catalyzed methyl transfer; (C) glucosyltransferase-catalyzed glucosyl transfer.; and (D) prenyltransferase-catalyzed prenyl transfer.
The final group transfer reaction that is relevant to this review is the transfer of prenyl groups from isoprenyl pyrophosphate to different nucleophiles in small molecules. These reactions are catalyzed by a family of enzymes known as prenyltransferases. The prenyl unit that is transferred from the pyrophosphorylated donor to the substrate can be as small, as in the five-carbon dimethylallyl (most common), or the more elongated oligoprenyl groups such as the ten-carbon geranyl, fifteen-carbon farnesyl, etc. In the enzyme active site, the Δ2-prenyl pyrophosphate donors can undergo C–O bond cleavage to yield the C1 carbocation, which is stabilized by delocalization of the positive charge. Attack of the carbocation by a nucleophile carbon forges the new bond and completes the prenyl transfer reaction (Fig. 4D). Electron rich aromatic rings, such as hydroxybenzenes and indoles can serve as nucleophiles to attack the allyl cation to perform in essence an electrophilic aromatic substitution. Two examples in this review illustrate this reaction. The first is the dimethylallyl tryptophan synthase (DMATS) in lysergic acid biosynthesis, which prenylates the C4 position in l-tryptophan 11 to give 4-dimethylallyl-l-tryptophan (4-DMAT, Section 2.4).46 This modification introduces an olefin-containing five carbon unit into l-tryptophan, which can be further oxidized and cyclized into the hallucinogenic lysergic acid. The mechanism of this reaction has been thoroughly studied, and is likely a two-step reaction.47 The C3 position of the indole ring is the most nucleophilic due to resonance with the indole nitrogen lone pair. Attack on the allyl cation can occur at either C1 or C3; this attack is proposed to take place at the more stable C3 position of the allyl cation. This generates a “reverse”-prenylated product that is proposed to undergo a nonenzymatic sigmatropic Cope rearrangement to yield the “forward”-prenylated 4-DMAT. In addition to serving as the starting point for lysergic acid (Section 2.5), indole prenylation of early pathway intermediates is commonly observed in the biosynthesis of other fungal indole alkaloids.48–52
The second notable pathway that involves prenyl transfer is in cannabinoid biosynthesis (Section 4.2).53 Starting with the first intermediate in the pathway, olivetolic acid 32 which is a resorcinol derivative, the aromatic prenyltransferase transfers the ten-carbon geranyl group from geranyl pyrophosphate to the C3 position in the ring to give cannabigerolic acid (CBGA, 33). As in the lysergic acid example, the introduced ten-carbon unit can undergo oxidative intramolecular cyclization, providing a variety of cannabinoids (Section 4.2).
1.2.4. Oxidative and reductive reactions
Natural product biosynthetic pathways employ powerful redox enzymes to modify the intermediates en route to the final product. The redox modification can directly modify the molecular scaffolds, or trigger rearrangement cascades, to introduce considerable structural complexities.34 On the reductive side, the NAD(P)H utilizing enzymes dominate as one would expect. These include ketoreductases, short-chain dehydrogenase/reductases (SDRs), ene-reductases, and imine reductases, etc. The two-electron reduction of C=C, C=O or C=N bonds are initiated through the attack by a hydride equivalent from either the dihydropyridine ring of NAD(P)H or the hydroquinone form of flavin adenine dinucleotide (FADH2). On the oxidative side, aerobic organisms use an assortment of enzymes and molecular oxygen as the oxidant to perform a dazzling array of chemical modifications.15 Both single electron (radical) and two electron manifolds are used by enzymes. These enzymes include the large family of heme-dependent cytochrome P450 monooxygenases that are abundant in plants and fungi; nonheme, iron and α-ketoglutarate dependent oxygenases, copper-dependent oxidases (such as the amine oxidase mentioned above), and flavin-dependent monooxygenases and oxidases. In two-electron oxidation of substrates catalyzed by oxidases, molecular oxygen is reduced to hydrogen peroxide. In monooxygenases where oxygen is reduced fully to water (four electron reduction), the substrate undergoes a two-electron oxidation, while NADPH is oxidized to NADP+. Here, the substrate can incorporate one of the oxygen atoms via hydroxylation or epoxidation, or alternatively the substrate can be oxidized without incorporation of oxygen atoms. Hence, depending on the mechanism of the redox enzyme, the outcome of the reaction can be very different. This topic has been extensively reviewed in the literature,15,34,54 and will not be discussed in detail here. However, we will highlight two reactions to illustrate the enzymatic prowess of the P450s, a staple of the plant biosynthetic pathways.
P450 enzymes use heme as a coenzyme to bind molecular oxygen. The coordinated iron is reduced to the Fe(II) state by an associated cytochrome P450 reductase (CPR). Binding of molecular oxygen and electron transfer from the Fe(II) and CPR leads to a hydroperoxy Fe(III)–O–O–H species. Cleavage of the O–O bond and the loss of water generates the high valent Fe(IV)=O porphyrin cation radical, which is also referred to as Compound I. This is a highly oxidizing species that can abstract hydrogen from substrate C, O, and N atoms to generate substrate radicals, including “unactivated” sp3 carbons. This generates the Fe(IV)–OH species also known as Compound II. Radical OH transfer to the substrate carbon radical produces the hydroxylated product in a process known as oxygen rebound. In many P450-catalyzed reactions in biosynthesis, the substrate radical can migrate to other atoms in the molecule through internal reactions and delocalization through π-bonds. This can lead to rearrangement of the carbon skeleton, as well as oxygen atom incorporation at distal positions from the initial abstraction site. In some cases, the Fe(IV)–OH can abstract a second hydrogen atom from the substrate to generate a second radical in the substrate that can recombine with the first one to terminate the reaction cycle. In this scenario, no oxygen atom is incorporated yet molecular oxygen is consumed. An additional feature of some biosynthetic P450s is the ability to iteratively oxidize a substrate, either at a single carbon or at nearby atoms. For example, it is not uncommon to find a single P450 that can perform the six-electron oxidation of a methyl group into a carboxylic acid in both fungal and plant biosynthetic pathways.
One notable example of P450 catalysis in this review is the secologanin synthase (SLS) found in the strictosidine biosynthetic pathway that ultimately leads to ibogaine (Section 2.8).55,56 The substrate is loganin 34 which contains the iridoid core. SLS performs hydrogen abstraction followed by oxygen rebound at the methyl group on the cyclopentanol ring to give a primary hydroxyl group. This species then undergoes a Grob fragmentation-like reaction to cleave the C–C bond which reveals both an aldehyde and a terminal olefin in the product secologanin 24 (Fig. 5A).57 This aldehyde then participates in the aforementioned Pictet-Spengler reaction with tryptamine 14 to give strictosidine 25. Hence, although this example illustrates a “standard” P450 reaction, the hydroxylation modification triggers a significant skeletal rearrangement.
Fig. 5: Two examples of P450 catalyzed oxidative modifications in biosynthesis of plant natural products.
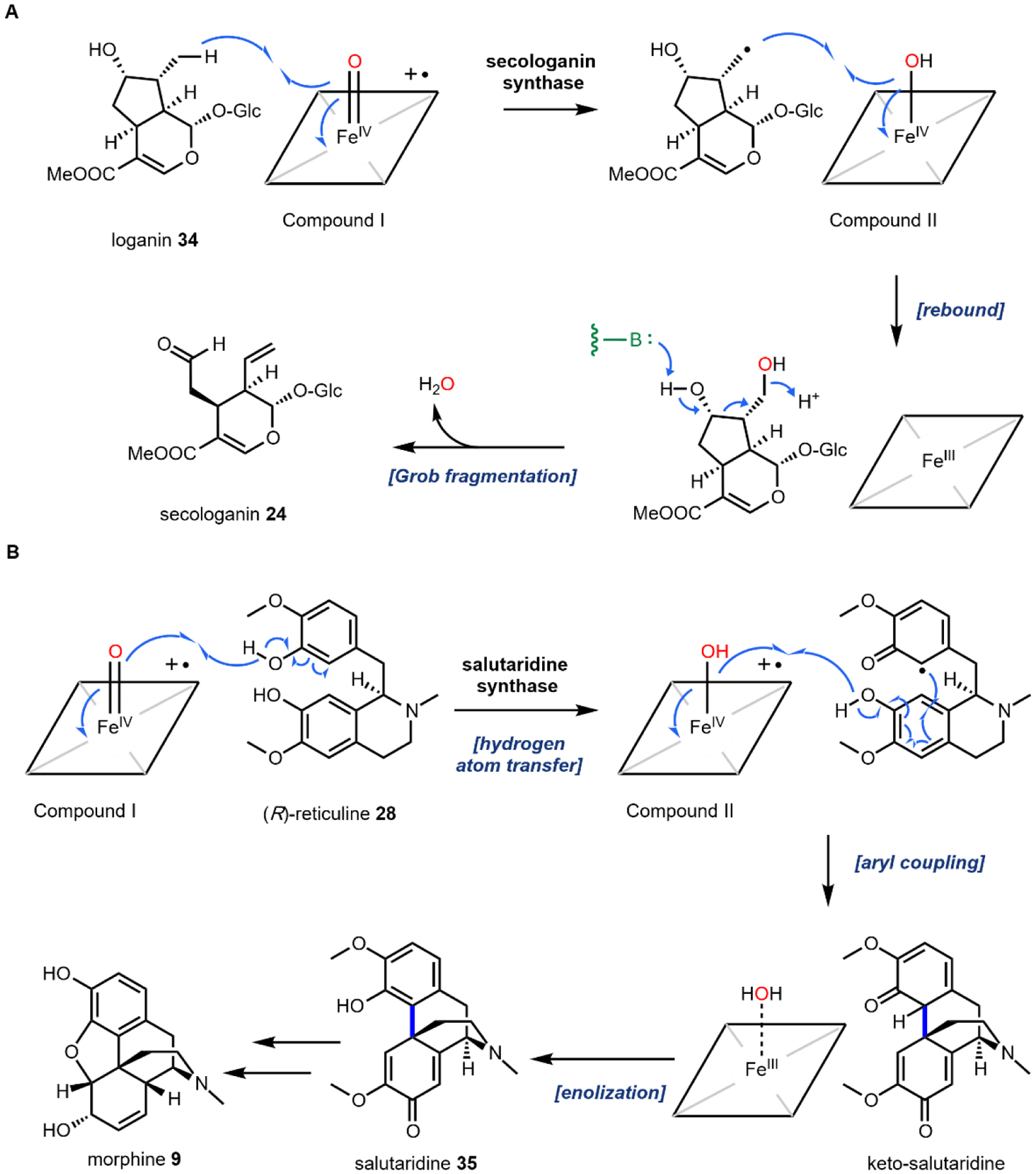
(A) secologanin synthase in biosynthesis of monoterpene indole alkaloids; (B) salutaridine synthase in biosynthesis of morphine family of opioids.
A second example that illustrates oxidation without oxygen incorporation is found in the morphine biosynthetic pathway, in which the salutaridine synthase catalyzes the phenyl coupling in R-reticuline 28 to yield salutaridine 35 (Fig. 5B).58 A radical addition mechanism is currently favored for this reaction: hydrogen abstraction from one of the phenol group generates an oxygen radical that is delocalized throughout the aromatic ring. The carbon radical then adds into the isoquinoline ring and recombines with the second radical that is generated by the P450 through the second hydrogen abstraction step. This forms a C–C bond that couples the two phenolic rings and gives rise to the rigidified morphinan scaffold of salutaridine 35 that is found in morphine 9 and related opioids.
1.3. Synthetic biology of psychoactive natural products
The psychoactivity of a given plant or fungi is often attributed to a short list of molecules. In reality, psychoactive natural products are produced as complex mixtures of metabolites and frequently have partially undefined compositions.59 Variability in growth conditions, in addition to pests, disease, agrochemicals, and climate may introduce further inconsistencies in product composition.60 In the event that a single psychoactive constituent is desired by the consumer and isolation from the native host is costly, total synthesis may be one strategy to establish a robust supply chain. In the last two decades, advances in DNA technologies have resulted in the development of an alternative production strategy: synthetic biology.61,62 Synthetic biologists use genetic tools to build designed biological systems with useful functionality. Whether or not synthetic biology can produce a viable process depends on the economic, environmental, and societal cost of alternative production strategies. However, as novel DNA-related technologies continue to arise, capabilities of molecular biologists are expected to expand. In 2010, Gibson assembly,63 DNA microarrays,64 and zinc-finger nucleases65 were considered state-of-the-art. A PhD student that graduated in 2020, however, would have witnessed cost-efficient gene synthesis,66 RNA-seq,67 and CRISPR/Cas968 emerge as routine. The substantial unrealized potential of synthetic biology is evidenced by continued investments across industry and academia.
As these technologies expand, successful refactoring of a biosynthetic pathway relies on the use of well-characterized “genetic parts” – these DNA-based elements permit coordinated expression of genes of interest in a heterologous host.69 Following the standardization of genetic engineering protocols and genetic parts, reliable metabolic engineering techniques have been established that enable improvements in engineered systems. The general methodology for synthetic biology-based heterologous production of natural products is outlined in Fig. 6. First, a biosynthetic pathway must be elucidated such that a heterologous production strategy can be envisaged. Second, an appropriate biosynthetic chassis must be selected. Finally, the engineer must iterate through the design, build, test, learn (DBTL) cycle until sufficiently high titers, production rates, and yields are reached.
Fig. 6.
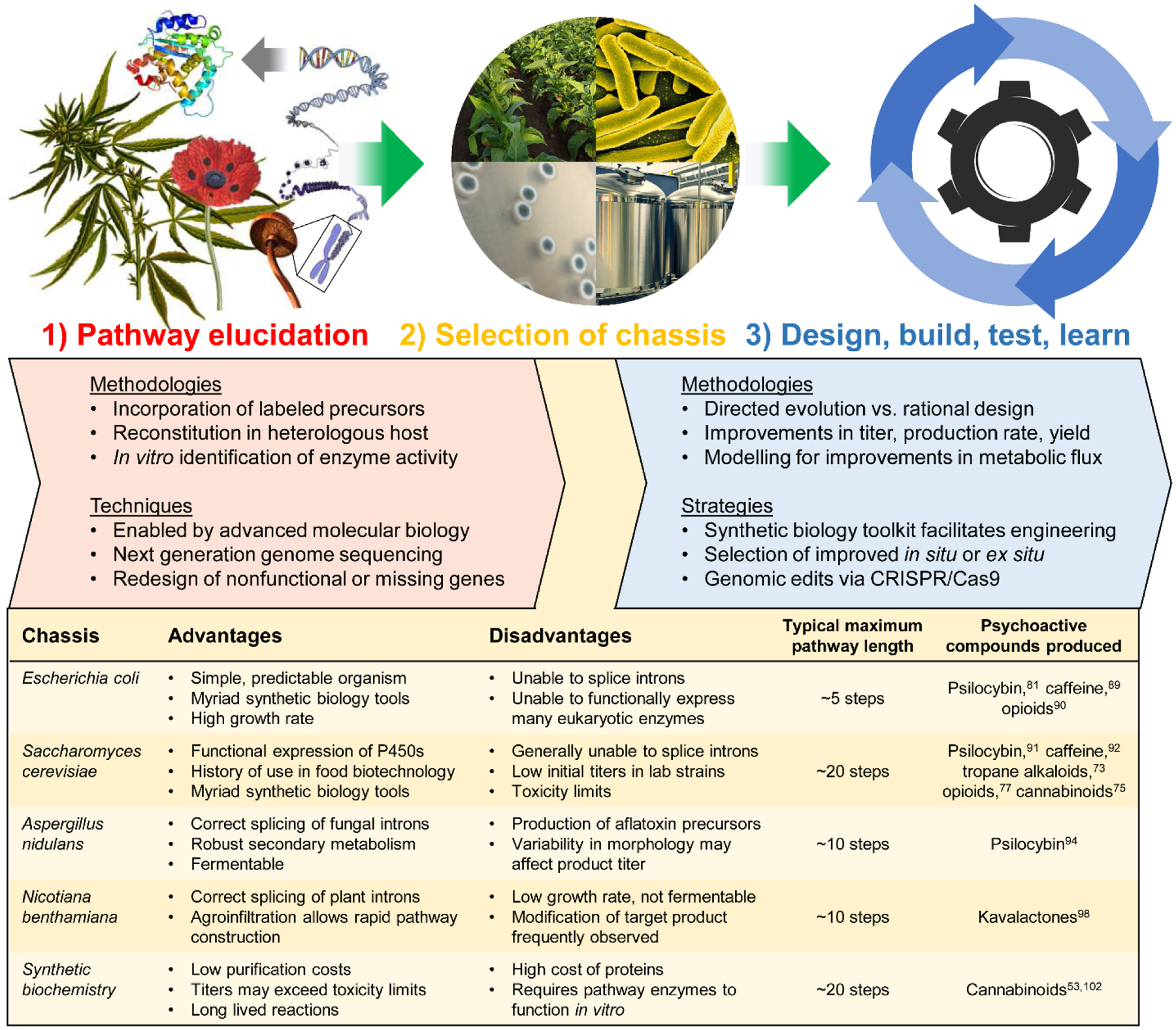
Strategies in synthetic biology.
1.3.1. Pathway elucidation and design
Biocatalytic production methods benefit greatly from fully elucidated biosynthetic pathways; a single missing biosynthetic step may completely derail heterologous production efforts. Identification of natural product biosynthetic logic is the primary focus of Sections 2 – 5. Early biosynthetic investigations involved demonstrating that isotope labeled precursors could be site-specifically incorporated into final products, which provided connections between primary metabolism and natural product biogenesis. Now, genomic sequencing and synthetic biology toolkits permit gene knockouts in the native host or expression in a heterologous host for functional analysis. “Reconstitution” of the activity of a recombinantly expressed enzyme activity in vitro affords the most unequivocal evidence of a biosynthetic sequence. It should be mentioned that availability of transcriptomics data has provided a quantum leap in the ability to identify candidate enzymes, particularly in unclustered plant pathways. Whereas bacterial and fungal biosynthetic pathways are frequently colocalized in a “gene cluster,” examples of clustered plant pathways are scarce.70–72 Meanwhile, the differential abundance of RNA across plant tissues and cultivars gives metabolic engineers precise spatiotemporal gene expression data, which can be mined for information about biosynthetic pathways. In recent years, RNA-Seq has been used to identify a wide range of plant natural product biosyntheses, including a number of key conversions in psychoactive natural product pathways.45,73 For instance, Facchini and coworkers utilized RNA-Seq to discover neopinone isomerase, which catalyzes a reaction previously believed to occur spontaneously in morphine biosynthesis.74 As an additional example, Luo et al. identified a functional prenyltransferase enabling cannabinoid production in S. cerevisiae by interrogating Cannabis sativa transcriptome data.75
In some cases, a biosynthetic step from the native organism cannot be identified, or functional expression of a known pathway gene may not be feasible in a given organism. In this event, bioprospecting or mining the genomes of alternative organisms to identify functional proteins that carry out key reactions has been successfully applied. For example, incorporation of genes from Gallus gallus (chicken) and Rattus norvegicus (rat) in place of missing or non-functional yeast metabolic steps was a crucial advancement in the development of MIA and BIA producing strains.76,77 Alternatively, protein engineering strategies may be employed to alter the regiospecificity or substrate specificity of other well-characterized proteins in order to generate de novo suitable replacements for missing or nonfunctional steps. Dueber and coworkers employed this method to engineer a l-tyrosine hydroxylase, which normally requires a cofactor not produced in yeast, and used the evolved enzyme to produce a morphine precursor.78 The field of directed evolution is now well established,79 which can be implemented prior to DBTL or integrated into the DBTL pipeline.
Following partial or complete pathway elucidation, a biosynthetic strategy may be designed. For many psychoactive natural products, especially those which can be easily constructed from primary metabolites, de novo production from minimal media will provide the most cost-efficient route to a final product. Stephanopoulos and coworkers recently highlighted an alternative approach: the use of a late-stage pathway entry point to circumvent troublesome early biosynthetic steps.80 Such “mixed carbon” feeding strategies may prove useful if an intermediate is commercially available or accessible via facile chemical synthesis. Efficient uptake of the late-stage entry point is another requirement, as transport limitations may prevent efficient substrate incorporation. The terms biotransformation (single step) and bioconversion (multistep) are commonly used to refer to this type of hybrid synthetic approach, which has been leveraged in the biosynthesis of psilocybin81 and an ibogaine precursor.82 Lastly, many in silico pathway design algorithms have been described in recent years, which perform automated retrobiosynthetic analyses to predict novel or optimized pathways.83,84 This approach has been successfully applied to primary metabolic products, highlighting the demand for continued investigation of secondary metabolic pathways.85,86,87 Machine-learning technologies linked to databases of reactions using automated DBTL are predicted to play a role in the future of natural product biomanufacturing.88
1.3.2. Chassis selection
A critical parameter in the successful refactoring of a natural product pathway is the selection of a suitable biosynthetic chassis. Five representative biosynthetic chasses are shown in Fig. 6. The model bacterium Escherichia coli has become a foundation of biotechnology as a DNA bearing model organism. E. coli laboratory strains have been customized for plasmid propagation and protein expression. Production of drugs with relatively short biosynthetic pathways has been shown,81,89 with stepwise mixed-strain cultures leveraged for longer pathways.90 Saccharomyces cerevisiae (brewer’s yeast) was initially the subject of genetic studies, but has become a favorite organism in academia to demonstrate heterologous production of an impressive variety of plant or fungus-derived psychoactive drugs.73,75,77,91,92 The model ascomycete Aspergillus nidulans has also been used for the production of bioactive molecules due to its robust secondary metabolism and ability to splice fungal introns.93–95 Nicotiana benthamiana has proven useful in characterizing and reconstituting difficult plant pathways, and is particularly attractive due to the well-established and modular transient gene expression technologies.96–99 The fifth chassis is synthetic biochemistry, wherein long-lived “cell-free” enzymatic reactions have enabled high-titer flux through lengthy biosynthetic pathways.53,100–102
One must carefully consider the features of a given pathway before deciding if a particular chassis meets the biosynthetic requirements. Many natural product pathways evolved in the context of highly specialized organelles, cells, or tissues.103 In this case, pathway compartmentalization may be required in order to sequester reactive biosynthetic intermediates from endogenous metabolism. Currently, sub-cellular localization is possible through the use of organelle-targeting peptide signals fused to the N-terminus of pathway enzymes, or the use of intracellular protein scaffolds.104,105 The recent production of tropane alkaloids in yeast required extensive localization across six sub-cellular locations.73 Tissue specific pathway localization in multicellular model organisms has yet to be employed but will require the implementation of intercellular metabolite transport. Special attention must be given to enzymes that are membrane associated, including the cytochrome P450s.106 Even in the most appropriate chassis, functional expression of trafficked proteins may require extensive engineering. Galanie et al. employed a protein chimera strategy to ameliorate improper processing of a P450 for opioid biosynthesis in yeast.77 Solubilization of membrane anchored P450s has been successfully demonstrated, but a general strategy guaranteeing functional soluble expression of P450s is still a major technological hurdle.107 It is also important to consider the primary metabolite building blocks required for construction of the secondary metabolite to be produced. Individual organisms exhibit variable fluxes towards given metabolic pools, dictating initial maximum titers prior to strain engineering. To address this limitation, “metabolic chassis strains” – strains with increased flux towards dedicated natural product building blocks – have been developed. Microbial chasses for the production of N-methylpyrrolinium 20,108 strictosidine 25,76 (R)-reticuline 28,90,109 and a number of other psychoactive natural product precursors have been established in the last decade.
The availability of a robust synthetic biology toolkit is another important factor to consider when selecting a production host. An ideal suite of molecular biology tools permits accurate and rapid genomic edits, precisely controlled gene expression, and diversity generation using libraries of genetic parts. More industrially “robust” organisms may also be utilized. These may be proprietary strains that outperform laboratory strains, but oftentimes lack the synthetic biology toolkit characteristic of the previously described model organisms. Proprietary methods may be developed for rational engineering, or random mutagenesis may be employed for nonrational diversity generation. Additional properties of robust chasses are faster growth, resistance to contamination, and a tailored metabolic profile. Predictable scalability and ease of downstream purification costs should also be considered when assessing platform commercialization.110 For academic purposes, however, it is most common to recapitulate biosynthetic pathways in model organisms as a proof-of-concept.
1.3.3. Design, Build, Test, Learn
Iterative design methodologies are now commonplace in deploying synthetic biology-based engineering. In natural product production chasses, first generation strain prototypes almost never produce compounds in sufficient quantities to compete with alternative production strategies. As a result, many iterations of design, build, test, and learn (DBTL) are required before a process is cost competitive. The industrial feasibility of bioprocess is often measured by titer (mass per volume), rate (mass per volume per time), and yield (mass product per mass substrate) as these metrics relate to cost of goods sold (COGS).111 In addition to improving titers on the strain engineering front, large improvements in productivity can be made through bioprocess engineering, which has benefitted immensely from automated design of experiment methodologies. The ability to iterate through the DBTL process is dependent on the biosynthetic chassis, engineering strategy, and screening strategy, among other factors. Novel metabolic engineering approaches aim to reduce the cost or duration of some aspect of the DBTL cycle.112,113 As previously mentioned, “automated design” and “machine learning” technologies have only recently been deployed in metabolic engineering studies. Thus, we focus below on methodologies which streamline the “build” and “test” phases of iterative design.
Within the DBTL cycle, synthetic biology toolkits have had the greatest impact on the “build” phase. Rapid and precise diversity generation, including the construction and integration of expression assemblies into a platform, is a vital prerequisite to screening. Libraries of well characterized genetic parts provide metabolic engineers with a set of elements that can precisely control the expression of a pathway gene. To this end, vector sets, promoter sets, terminator sets, and signal peptide sets are the most common control elements used. A vector is a circular fragment of DNA that harbors pathway genes, a selection marker, and an origin of replication which dictates copy number and plasmid stability. Integration of synthetic biology constructs directly into the genome may obfuscate the use vectors, however shuttle vectors for cloning of constructs are generally still employed. Promoters are regulatory elements directly upstream of a gene of interest, which recruit transcriptional elements for gene expression. Promoters may be constitutive (always on) or inducible (turned on by a condition). The promoter “strength” correlates to the copy number of mRNA upon induction; promoters are often referred to as tight (no basal expression) or leaky (measurable basal expression). Terminators are the regulatory elements downstream of the protein coding sequence, signaling transcriptional termination, and impact the half-life of mRNA. Signal peptides may be employed to direct expression to an organelle for localization or secretion. Prior to use, these genetic parts must be assembled into a single contiguous DNA fragment. Sequence independent cloning techniques such as Gibson assembly and yeast homologous recombination have replaced traditional methods such as digestion-ligation.63 Furthermore, gene fragments can now be affordably synthesized, circumventing strain procurement and DNA isolation.66 A once tedious and unpredictable process, heterologous gene expression has been streamlined using reliably functional elements; gene expression is now definitively “engineerable”. As we gain a more comprehensive understanding of sophisticated cellular programs, we will be able to assemble even more robust and dynamic synthetic biology circuits. Once such systems are constructed, integration into the heterologous host is the final hurdle in the “build” phase. The recent discovery of CRISPR/Cas9 has ameliorated this challenge. Cas9, an RNA-guided DNA endonuclease, enables genomic modifications with unprecedented precision, greatly accelerating strain construction.68
Following the “build” phase, a screening approach is required in order to “test” the performance of synthetic constructs. Screening throughput is dependent on the strategy used to quantify production of a natural product. Direct measurement of product titer using chromatography, mass spectrometry, and spectrophotometry and comparison to an authentic standard is the most accurate quantification method. Advancements in instrumentation have increased the throughput and accuracy while decreasing costs, however these methods are still considered low-to-medium throughput, requiring 1 minute – 1 hour per sample. Meanwhile, indirect measurements of product titer employing biological readouts have enabled high-throughput testing of strains. So called “biosensors” transduce chemical inputs into physiological outputs in order to establish a correlation between a titer and a selectable phenotype. Biosensors enable screening of constructs on the order of seconds or less per sample. In rare circumstances, a natural product is produced in sufficient quantities and has a unique enough absorbance spectrum to function directly as the selectable chromophore. More typically, a genetically-encoded biosensor must be engineered that robustly actuates a signal that can be correlated to the metabolite’s concentration. Biosensors consist of a sensor-actuator pair and are either RNA-based or protein-based. The sensor-input consists of binding of the biosensor to the secondary metabolite. Then, an actuator-output is generated resulting in modulation of transcription or translation of a selectable protein. The genetic circuit may also encode Boolean logic in order to improve biosensor properties such as dynamic range or sensitivity.114 Selection is then performed either in situ (cell viability) or ex situ (high-throughput cell sorting). For example, a cell viability screen can be established by tying a biosensor output to expression of an antibiotic resistance gene or complementation of an auxotroph. On the other hand, biosensor-dependent expression of a fluorescent protein enables high-throughput fluorescence-activated cell sorting (FACS) for rapid analysis of entire populations of cells. Microbial opioid production has benefited greatly from the use of biosensors, as both RNA and protein based metabolite sensors have been reported for benzylisoquinoline alkaloid pathway intermediates.78,115 Adaptive laboratory evolution (ALE) has also emerged as an efficient method to circumvent traditional DBTL strain construction. ALE employs natural selection and in vivo diversity generation for population-wide engineering, and has been primarily applied to primary metabolic products.116 Although several generalizable biosensor development platforms have been proposed, research towards rapid expansion of the variety of sensed metabolites is ongoing.
Compared to organic synthesis and biochemical engineering, synthetic biology is a relatively nascent applied science. Despite this, immense progress has been made in the last 20 years, and a number of recent success stories illustrate the field’s potential. Research groups now routinely refactor pathways with more than 10 steps in A. nidulans and N. benthamiana, and pathways with more than 20 steps have been reconstituted using both S. cerevisiae and synthetic biochemistry. The ongoing challenge for these platforms is to improve titers and reduce costs sufficiently to compete with traditional production methods. General strategies range from improving flux through pathway bottlenecks to ameliorating growth defects from metabolic burden or toxicity, however a more nuanced engineering approach is often required. In depth discussions of the engineering strategies enabling benchmark production of the psychoactive natural products described in this review accompany the biosynthetic pathway descriptions.
2. Hallucinogenic natural products
Of all the psychoactive compounds that are either isolated as natural products or produced synthetically, hallucinogens may impart the most dramatic shifts in one’s psyche. This broad class of substances can induce potent alterations to consciousness, mood, and perception resulting in vivid visual hallucinations, synesthesia, and a warped sense of time and space.117 The precise mixture of perceptual and somatic effects of hallucinogens is highly compound specific and thus has led to many debates on accurate nomenclature. There is yet to be a consensus with terms such as “psychedelic” and “entheogen” often used interchangeably with “hallucinogen” in different contexts.
Natural sources of hallucinogens famously include “magic mushrooms” of the Psilocybe genus and other fungi such as ergot and fly agaric. Other well-known sources of hallucinogens are from the spineless cactus, peyote, the psychoactive brew, ayahuasca, and with a recent resurgence, nutmeg.118 Most natural hallucinogens are alkaloids derived from amino acids such a l-tryptophan 11, l-tyrosine 12, and l-glutamic acid 36 (Fig. 7), with one notable exception being the terpenoid salvinorin A 37. Numerous extensive reviews exist on the history, pharmacology, and potential as therapeutics of hallucinogens which we recommend.117,119,120
Fig. 7.
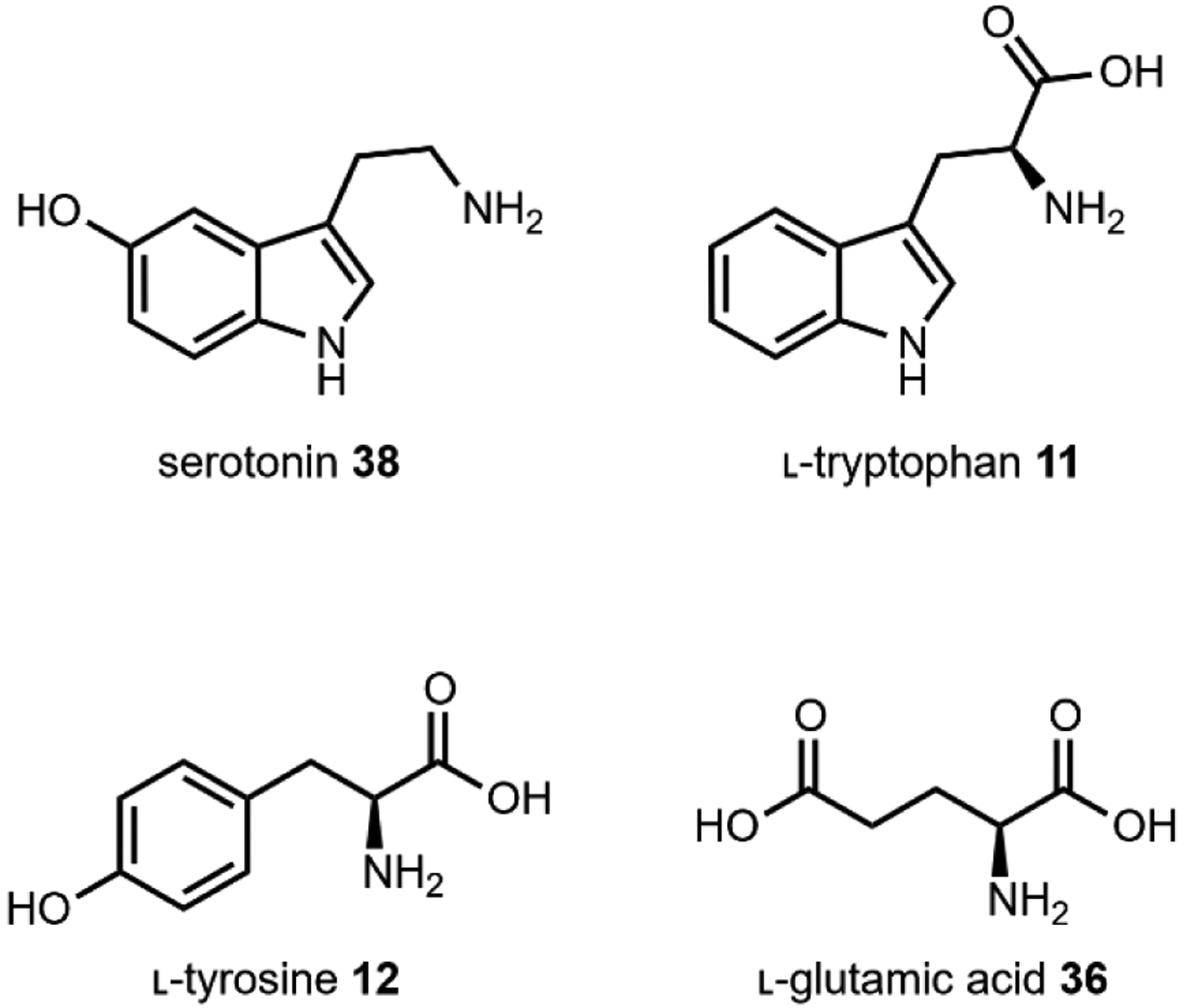
Amino acid building blocks for hallucinogens that target serotonin receptors.
2.1. Serotonin Receptors
The serotonin or 5-hydroxytryptamine (5-HT) receptors, named for their native ligand, serotonin 38, have been implicated in the modulation of sensory perception, mood, cognition, memory, and more through the peripheral and central nervous systems (Fig. 7).121 There are many subtypes, and with the exception of 5-HT3 which is a ligand-gated ion channel, the rest are G-protein-coupled receptors, each with unique spatial distribution and localization in the brain.122 Phylogenetic analysis and low sequence identity (~25% between the major subtypes) demonstrates early divergence, implicating 5-HT receptors as one of the oldest receptor systems.121
The relationship between 5-HT receptors was first determined through testing of LSD 3. While hallucinogenic compounds like 3 (Fig. 8) have been shown to target multiple 5-HT receptors, the 5-HT2A receptor is most commonly associated with the majority of psychotropic effects.123 Previously, structure-activity relationship studies between 5-HT2A and numerous psychoactive compound scaffolds have demonstrated that hallucinogenic potency is not necessarily a function of affinity, likely due to more nuanced mechanisms of functional selectivity.124 However, a recent crystal structure of 3 complexed with 5-HT2B (a model system for 5-HT2A) was reported and combined with molecular dynamic simulations, identified a molecular basis for the particular potency of 3.125 The authors demonstrate that the diethylamide side chain of 3 adopts a restrictive conformation when bound to 5-HT2B that increases residence time and improves β-arrestin translocation to the cell membrane. This enhanced β-arrestin translocation results in desensitization of the cell to stimuli by uncoupling G-proteins from receptors and could explain the long duration of action of 3.
Fig. 8. Overview of hallucinogenic natural products.
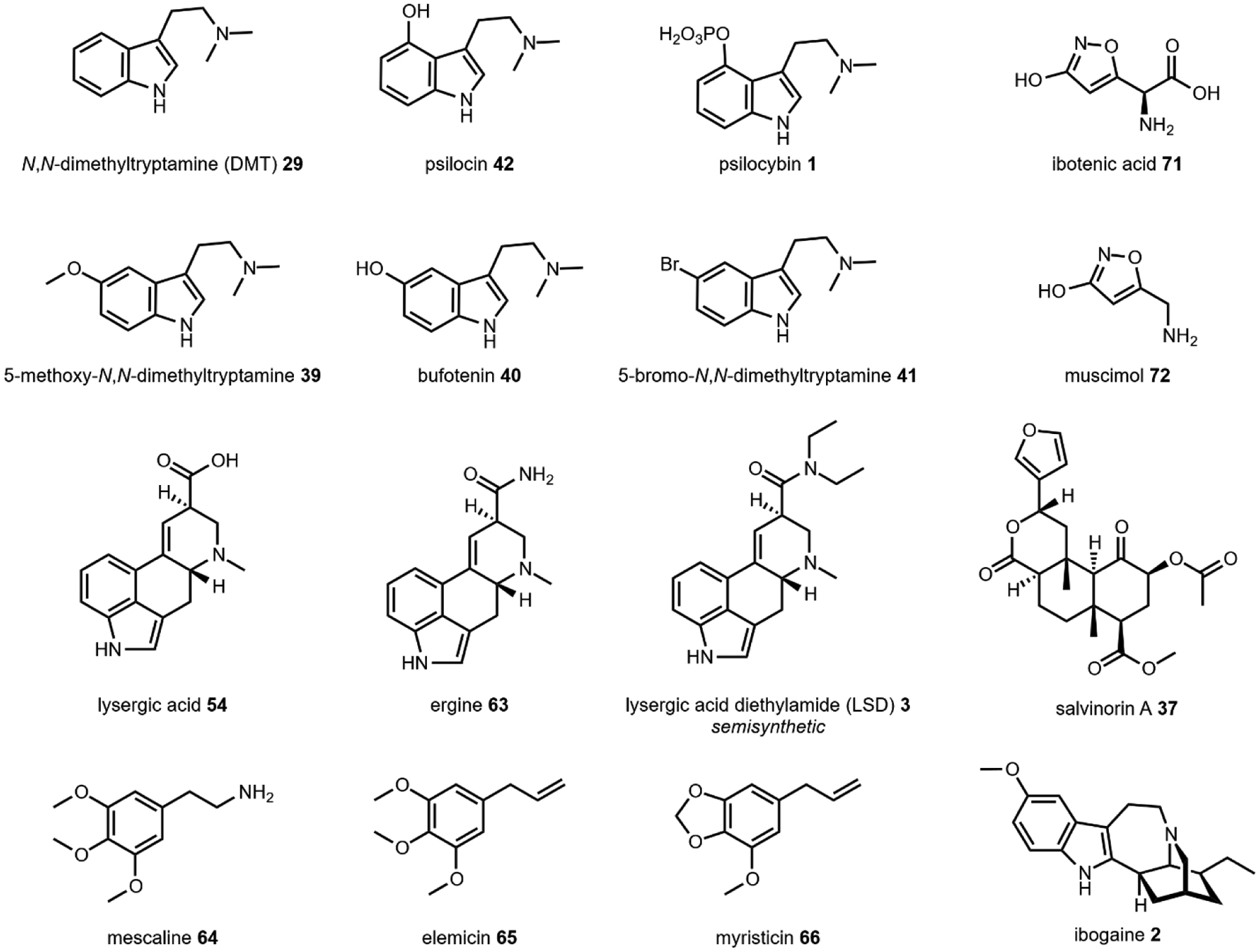
*Note that LSD 3 is a semisynthetic compound derived from lysergic acid (Section 2.5).
2.2. N,N-Dimethyltryptamine
N,N-dimethyltryptamine (DMT) 29 (Fig. 9) is likely the most pervasive psychoactive compound across species and is found in dozens of plant and animal species, including humans.126 Root, bark, and leaf preparations from plants such as Psychotria viridis, containing DMT and its structural analogs (Fig. 9) have been used in shamanic ritual practices for at least 1000 years.127 Interestingly, in addition to plants, structural analogs 5-methoxy-N,N-dimethyltryptamine 39 and bufotenin 40, are also found in the toxin of the Colorado River toad Incilius alvarius, formerly known as Bufo alvarius, whose remains have been found as a part of Olmec ritual ceremonies dating back to pre-Columbian Mesoamerica (Fig. 10).128,129 Referred to colloquially as the “Psychedelic Toad of the Sonoran Desert,” exudates from the amphibian’s specialized glands may contain up to fifteen percentage dry weight 39, representing the most notable example of a psychoactive natural product of animal origin.130 DMT 29 was first isolated from the shrub Mimosa tenuiflora in 1946 by Oswaldo Gonçalves de Lima,131 but its hallucinogenic effects were not discovered for another decade.132 29, like all l-tryptophan derived hallucinogens, is a serotonin receptor agonist. While the functional selectivity of 29 towards the 5HT2A receptor is believed to be necessary for its effects, 29 can bind to many serotonin receptors that may also contribute to its psychoactivity.126
Fig. 9. Psychotria viridis is one of the common sources of DMT for ritual purposes.
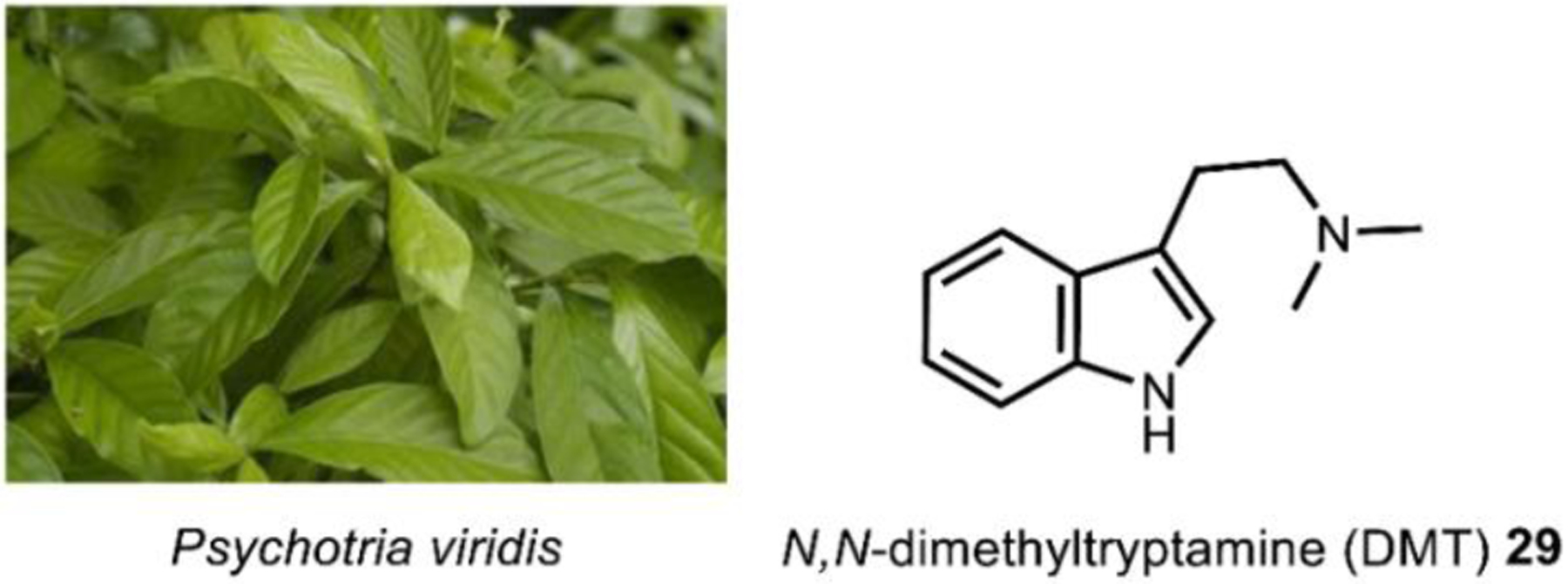
Image on the left courtesy of Paulo Pedro P. R. Costa via. CC-4.0.
https://upload.wikimedia.org/wikipedia/commons/0/01/PsychotriaviridisFrutoDSC75.jpg
Fig. 10. Incilius alvarius’s skin and exudates contain 5-methoxy-N,N-ditryptamine and bufotenin.

Image on top courtesy of Wildfeurer via. CC-3.0. https://upload.wikimedia.org/wikipedia/commons/4/4f/2009-03-13Bufo_alvarius067.jpg
While the precise role of endogenous 29 in humans has yet to be ascertained,133 one study speculates it may have a role in protecting from hypoxia.134 Further, 29 has shown promise as a therapeutic anti-depressive agent and is known to promote neural plasticity.135,136 Interestingly, brominated forms of DMT such as, 5-bromo-N,N-dimethyltryptamine 41, have been isolated from the marine sponges137,138 and show particular promise as anti-depressives.139 Finally, 29 has limited neurotoxicity and only exhibits cardiovascular effects when taken intravenously in large doses, furthering its therapeutic potential.126
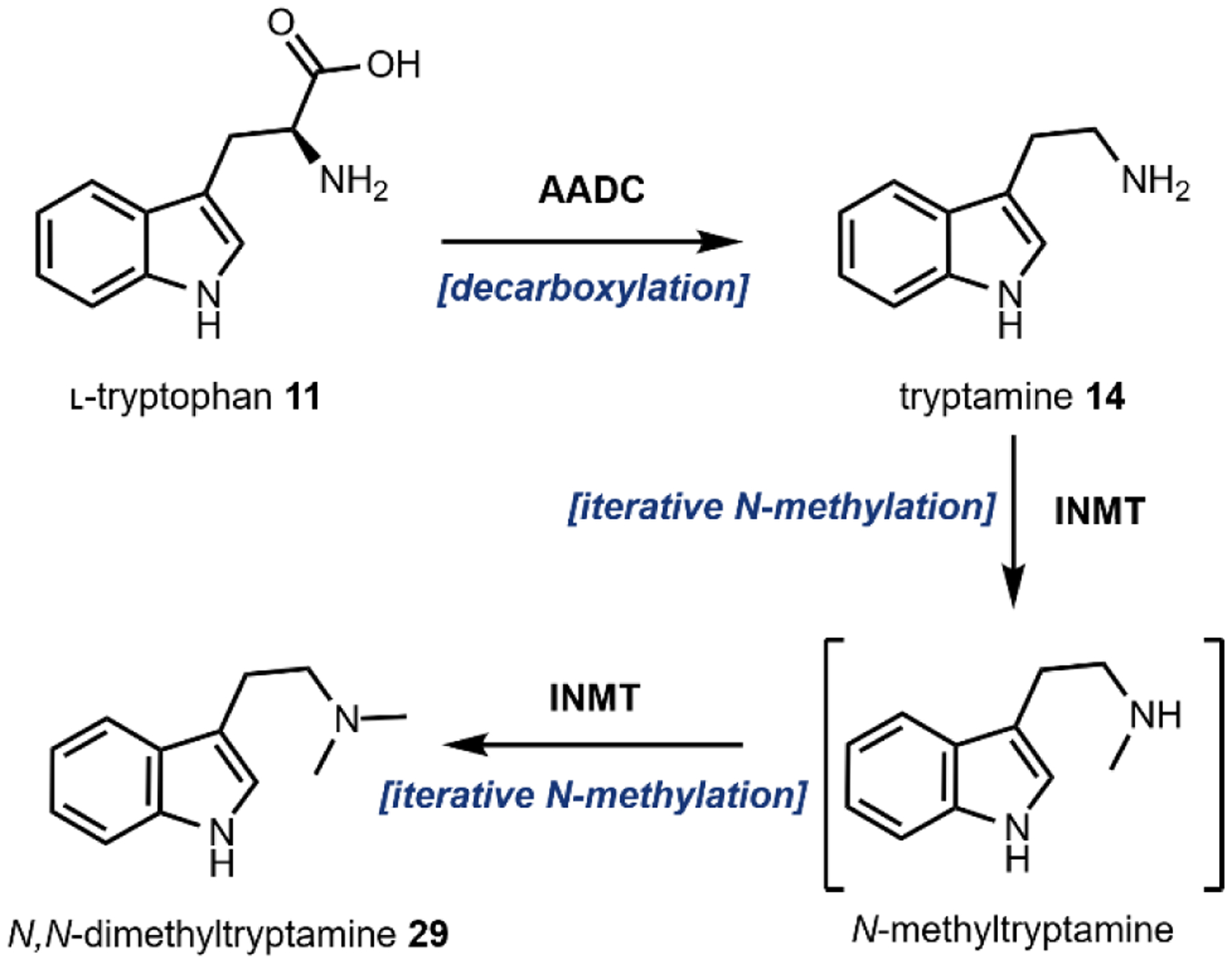
2.2.1. Biosynthesis of DMT
The biosynthesis of DMT 29 is the shortest pathway described in this review, requiring just two enzymes. Biogenesis begins with the decarboxylation of the proteinogenic amino acid l-tryptophan 11 to form tryptamine 14 by an aromatic amino acid decarboxylase (AADC) (Fig. 11, and Fig. 2).140 The PLP-dependent AADCs in most species display a broad substrate scope, operating on multiple aromatic amino acids and derivatives.140 Tryptamine 14 is then methylated sequentially by an iterative N-methyltransferase (INMT) to first form the secondary amine, then 29, using SAM (Fig. 2B) as a methyl donor.141,142
Fig. 11.
Biosynthesis of DMT.
2.3. Psilocybin
Psilocybin (4-phosphoryloxy-N,N-dimethyltryptamine) 1, one of the major natural products from hallucinogenic Psilocybe sp. (“magic mushrooms”), was first isolated from Psilocybe mexicana by Albert Hofmann in 1958 (Fig. 12).143 The description of “magic mushrooms” in scientific literature and the subsequent isolation and characterization of their psychoactive metabolites was the culmination of decades of effort to identify the sacred mushroom that the South American Aztecs referred to as teonanacatl, meaning “god’s flesh.”144 Psilocybin 1 itself is not psychoactive, but rather exists as a prodrug. After ingestion, psilocybin 1 is metabolized through dephosphorylation and becomes psilocin (4-hydroxy-N,N-dimethyltryptamine) 42, a potent psychotropic 5HT2A receptor agonist.145,146 In addition to its psychoactivity, 1 has shown some promise as a therapeutic for treating depression, anxiety and tobacco addiction.147–149
Fig. 12. Psilocybe mexicana contains ~1% psilocybin.
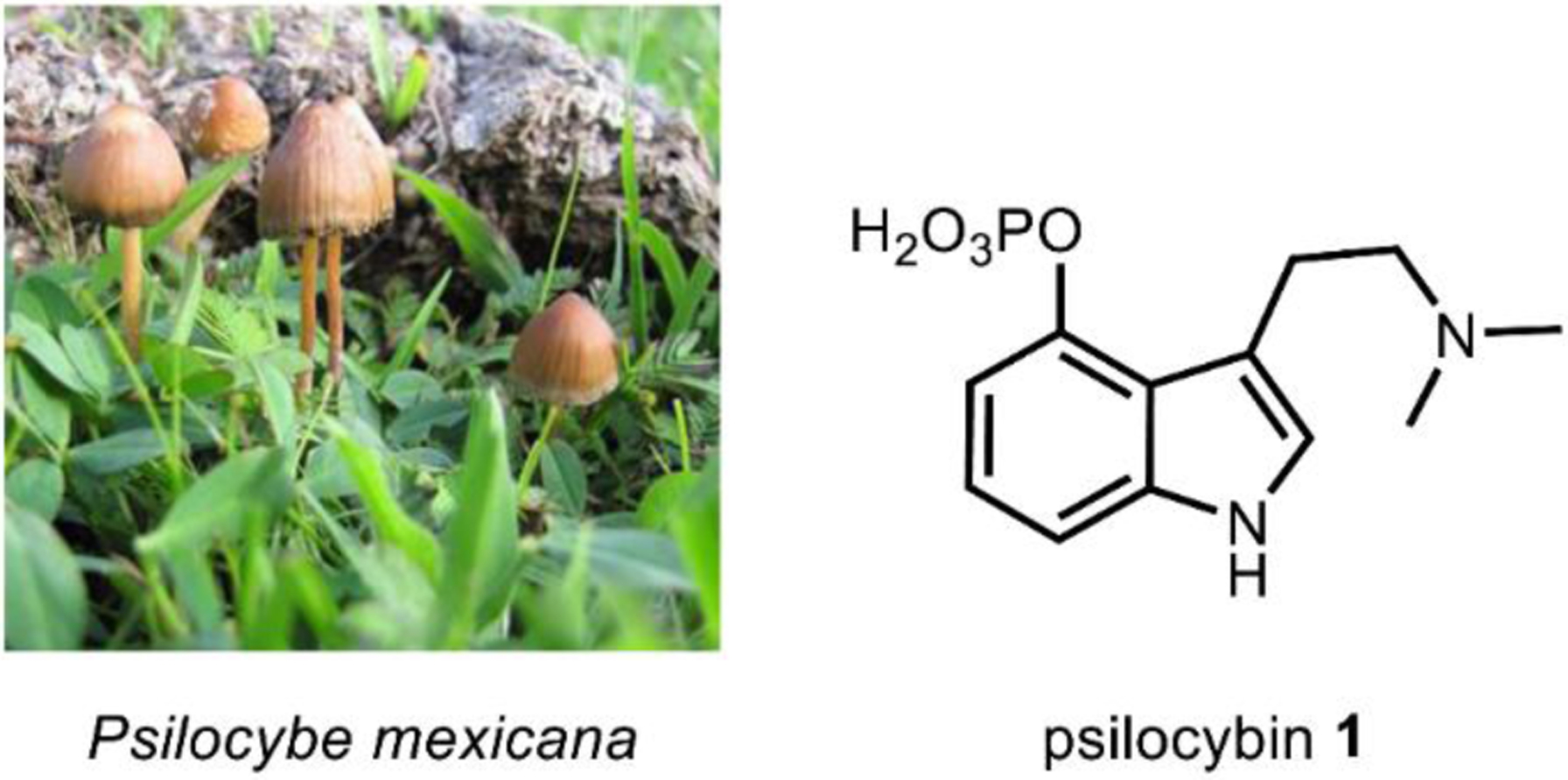
Image on left courtesy of Alan Rockefeller via CC-3.0.
https://upload.wikimedia.org/wikipedia/commons/4/46/Psilocybe_mexicana_53960.jpg
2.3.1. Biosynthesis of psilocybin
A biosynthetic pathway for psilocybin was proposed based on isotope feeding studies as early as 1968.150 Agurell et al. hypothesized that following decarboxylation, l-tryptophan 11, now tryptamine 14, would be methylated iteratively to form the psychoactive dimethyltryptamine 29. This was a reasonable hypothesis because indolethylamine(tryptamine)-N-methyltransferases were a popular enzyme for study at the time following their discovery rat, rabbit, and human tissues.
Recently, a psilocybin biosynthetic cluster from Psilocybe cubensis and Psilocybe cyanescens was identified and characterized by Fricke et al. (Fig. 13).151 The authors first sequenced the genomes of both Psilocybe sp. Then, using a combination of a methyltransferase, a hydroxylase, and a kinase as queries, a putative biosynthetic cluster present in both species was identified and characterized. Fricke et al. determined that the iterative N-methylation was the terminal step of psilocybin biosynthesis by enzymatic action of PsiM whose sequence is unrelated to the well-characterized mammalian indolethylamine-N-methyltransferases, and thus revised the hypothesis that DMT 29 is an intermediate in psilocybin biosynthesis. Starting from l-tryptophan 11, PsiD catalyzes a decarboxylation reaction to yield 14. The amino acid sequence for PsiD diverges from the more common PLP-dependent aromatic amino acid decarboxylases and instead shares similarity with the PLP-independent phosphatidylserine decarboxylases. PsiH, a P450 monooxygenase, then hydroxylates the indole C4 to yield 4-hydroxytryptamine 43.
Fig. 13.
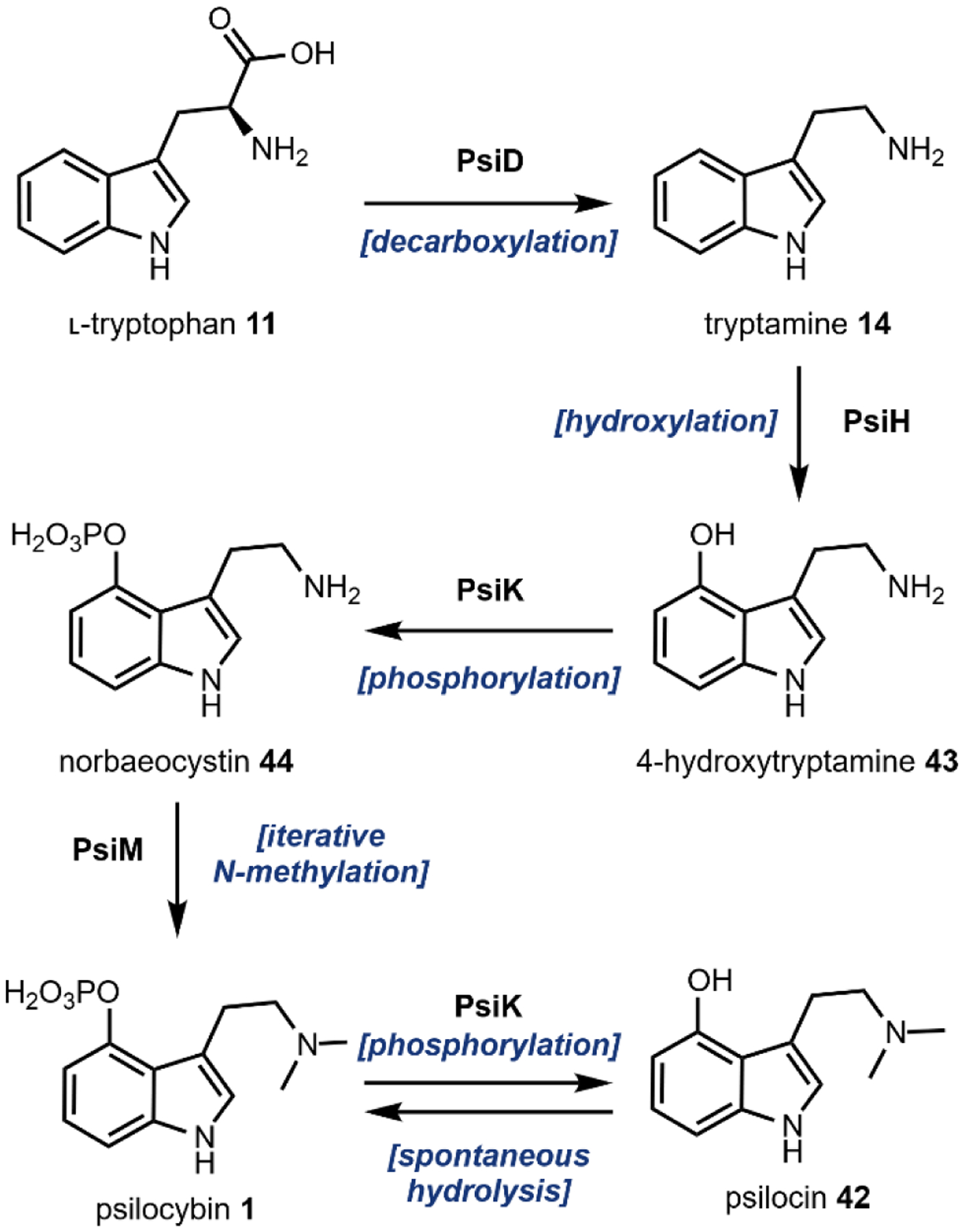
Biosynthetic pathway of psilocybin and psilocin from l-tryptophan.
Next PsiK, a predicted kinase, catalyzes the phosphorylation of 4-hydroxytryptamine 43 into norbaeocystin 44 using ATP as the phosphate donor. Phosphoryltransferase (or kinases) are relatively uncommon in natural product biosynthesis. Recent examples include the biosynthesis of calyculin protoxins and the lasso peptide paeninodin, in which phosphorylation plays a role in self-immunity which could highlight the importance of dedicated kinases.152,153 Lastly, PsiM methylates the terminal amine in 44 in an iterative fashion using SAM as a methyl donor to give 1. PsiM only methylates phosphorylated tryptamine 44, indicating that psilocybin biosynthesis is nearly linear. In water, 1 undergoes spontaneous hydrolysis of the phosphate group to form 42, but PsiK accepts psilocin as a substrate and readily phosphorylates to reform psilocybin 1. As previously mentioned, this hydrolysis results in the psychoactive form, 42, upon ingestion by vertebrates.
Subsequently, additional psilocybin biosynthetic clusters were found in distant fungal species and provide some evolutionary evidence of the ecological role of psilocybin in influencing mycophagy in animals, which is to reduce their consumption from invertebrate predators.154 Thus, the bioactivity of 1 may provide a fitness advantage to natural producers over their competitors. Further, a recent preprint presents evidence of a new, diverged psilocybin cluster in Inocybe corydalina that contains a second methyltransferase that may produce the trimethylated, quaternary ammonium salt analogue of 1, aeruginascin.155
2.3.2. Heterologous production of psilocybin
Since the elucidation of the psilocybin biosynthetic pathway, engineering efforts for high-titer production of psilocybin 1 in various microbial hosts, such as the filamentous fungus A. nidulans, Baker’s yeast S. cerevisiae, and the model bacterium E. coli have been reported.81,91,94 Hoefgen et al. developed a polycistronic expression system in A. nidulans and used the psilocybin pathway as a proof-of-concept. They obtained 110 mg/L of 1 at 1.5% dry mycelial weight which is a titer comparable to native psilocybin producers.
Adams et al. were able to combine heterologous expression and metabolic engineering strategies to achieve a titer of 1.16 g/L psilocybin 1 in E. coli in a 1.5-L bioreactor from 3.05 g/L of gradually supplied 4-hydroxyindole 45 (Fig. 14) over several days. The exogenously supplied 45 is first converted into 4-hydroxy-l-tryptophan 46 by TrpB, an endogenous bacterial enzyme in the l-tryptophan biosynthetic pathway that catalyzes the condensation of indole with serine to form 46. PsiD, PsiK, and PsiM from P. cubensis were heterologously expressed under a single T7 promoter on a high copy plasmid, which facilitated the conversion of 44 formed in situ into psilocybin 1. Endogenous levels of serine and SAM, required by TrpB and PsiM, respectively, were not sufficient for high-titer production and thus the media was supplemented with excess amounts of serine and methionine. The native E. coli enzyme MetK is able to anabolize the exogenous methionine into SAM while the E. coli enzymes Mtn, LuxS, and MetE are able to recycle the by-product S-adenosylhomocysteine (SAH) into more methionine.
Fig. 14.
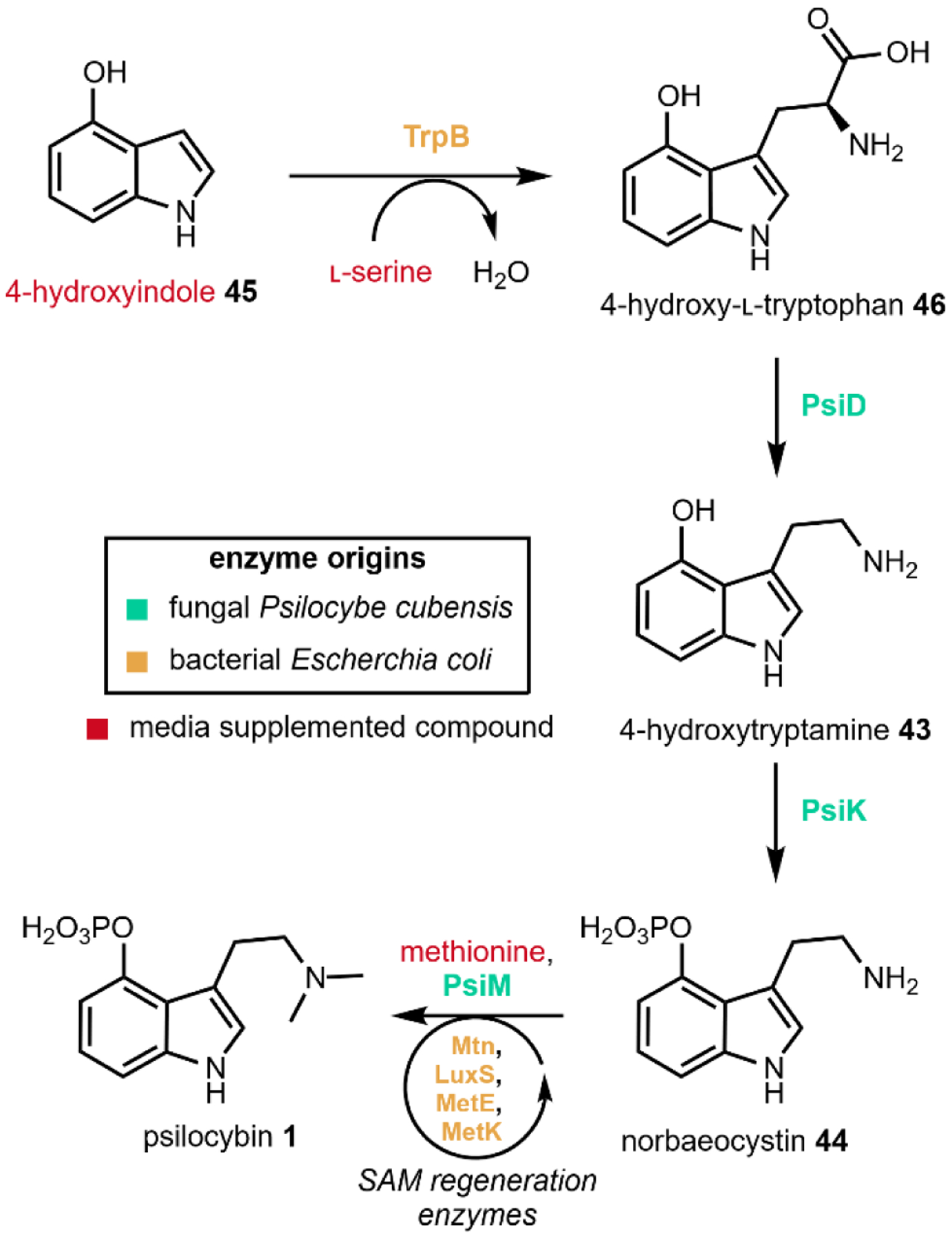
Engineered production of psilocybin in E. coli.81
Engineered de novo production of psilocybin 1 was recently reported in a fully integrated S. cerevisiae strain with a titer of 627 ± 140 mg/L of psilocybin 1 and 580 ± 276 mg/L of psilocin 1 from 1L scale fed-batch fermentation over ~9 days (Fig. 15). Psilocybin pathway genes from P. cubensis were expressed under strong, constitutive promoters. Instead of expressing the pathway decarboxylase PsiD, Milne et al. expressed a tryptophan decarboxylase from Catharanthus roseus, CrTDC, which was previously shown to have high catalytic efficiency when expressed in yeast.76 Additionally, the authors expressed a cytochrome P450 reductase (CPR) from P. cubensis to improve activity of PsiH. Matching a P450 with its cognate reductase partner has been demonstrated to be important for functional heterologous expression and is an effective technique to improve heterologous expression of P450 enzymes.106,156 To increase endogenous l-tryptophan levels, the authors overexpressed ARO1 and ARO2, which are involved in combining erythrose 4-phosphate 47 and phosphoenolpyruvic acid 48 to form chorismic acid 49 in the shikimate pathway leading to l-tryptophan biosynthesis. This, combined with knockout of a shikimate pathway regulator, RIC1, were effective towards elevating l-tryptophan supply.
Fig. 15.
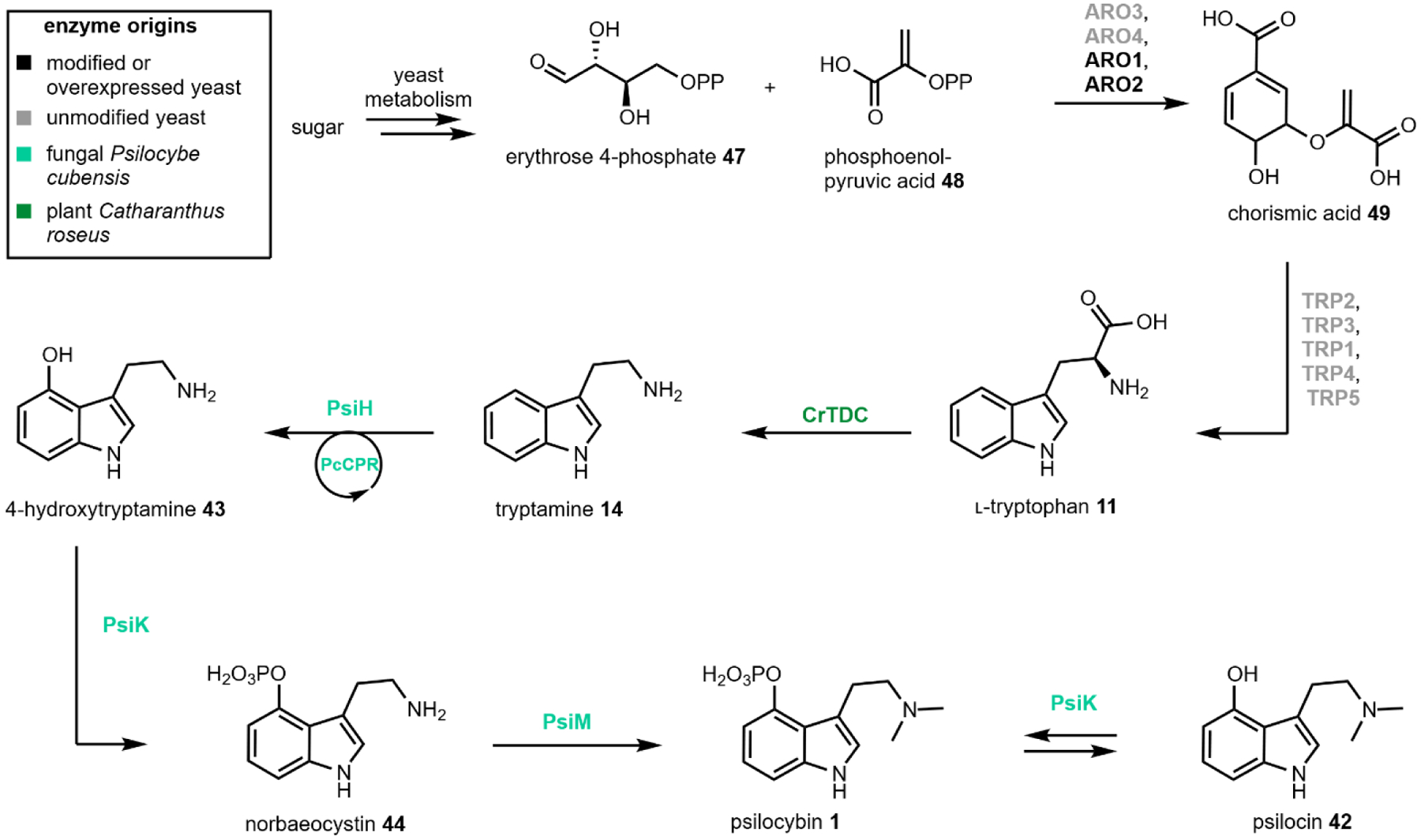
Engineered production of psilocybin and psilocin in yeast.91
2.4. Ayahuasca
Most hallucinogens are rapidly metabolized in vivo following ingestion by the action of monoamine oxidases (MAO) for eventual renal elimination, resulting in many hallucinogens being orally inactive. MAOs, as the name suggest, catalyze the oxidative deamination of neurotransmitters and structurally similar compounds.157 It follows that ingestion of MAO inhibitors (MAOIs) concurrently with hallucinogens can increase their bioavailability. This synergy is best demonstrated by ayahuasca, a psychoactive decoction commonly prepared from the vine Banisteriopsis caapi, containing MAO inhibiting harmala alkaloids, and some DMT 29 containing species such as the shrub Psychotria viridis.158,159 Ayahuasca, derived from a Quechua term meaning “vine of the soul,” has been used as a spiritual medicine by indigenous groups in South America’s Amazon basin for at least one thousand years.127 During a ceremony in which the brew is ingested, practitioners experience several stages of visual and purgative experiences in order to heal physical, emotional, and spiritual imbalances.160 While there are no currently approved therapeutic uses for ayahuasca or its active metabolites, harmala alkaloids have shown promise as an antidepressant through brain plasticity modulation.161
The harmala alkaloids are compounds that contain a β-carboline scaffold with various methyl or methoxy substitutions and different degrees of unsaturation. The β-carboline scaffold itself is characterized by a pyridine ring ortho-fused to indole resulting in a 6-5-6 tricycle with possible substitutions at the ortho position to the pyridine nitrogen. The major harmala alkaloids that contribute to the MAOI activity are harmine 23, harmaline 50, and tetrahydroharmine 51. These compounds are abundant at ~ 0.05 – 0.1% of dried plant material in B. caapi (Fig. 16).162 Thorough pharmacokinetic data is scarce, but psychotropic action of harmala alkaloids is expected to occur around 20–50 mg with a typical 100 mL ayahuasca brew containing between ~300–600 mg of harmala alkaloids and 20–60 mg of 29.163
Fig. 16. Banisteriopsis caapi contains many compounds with the β-carboline scaffold, including harmine.
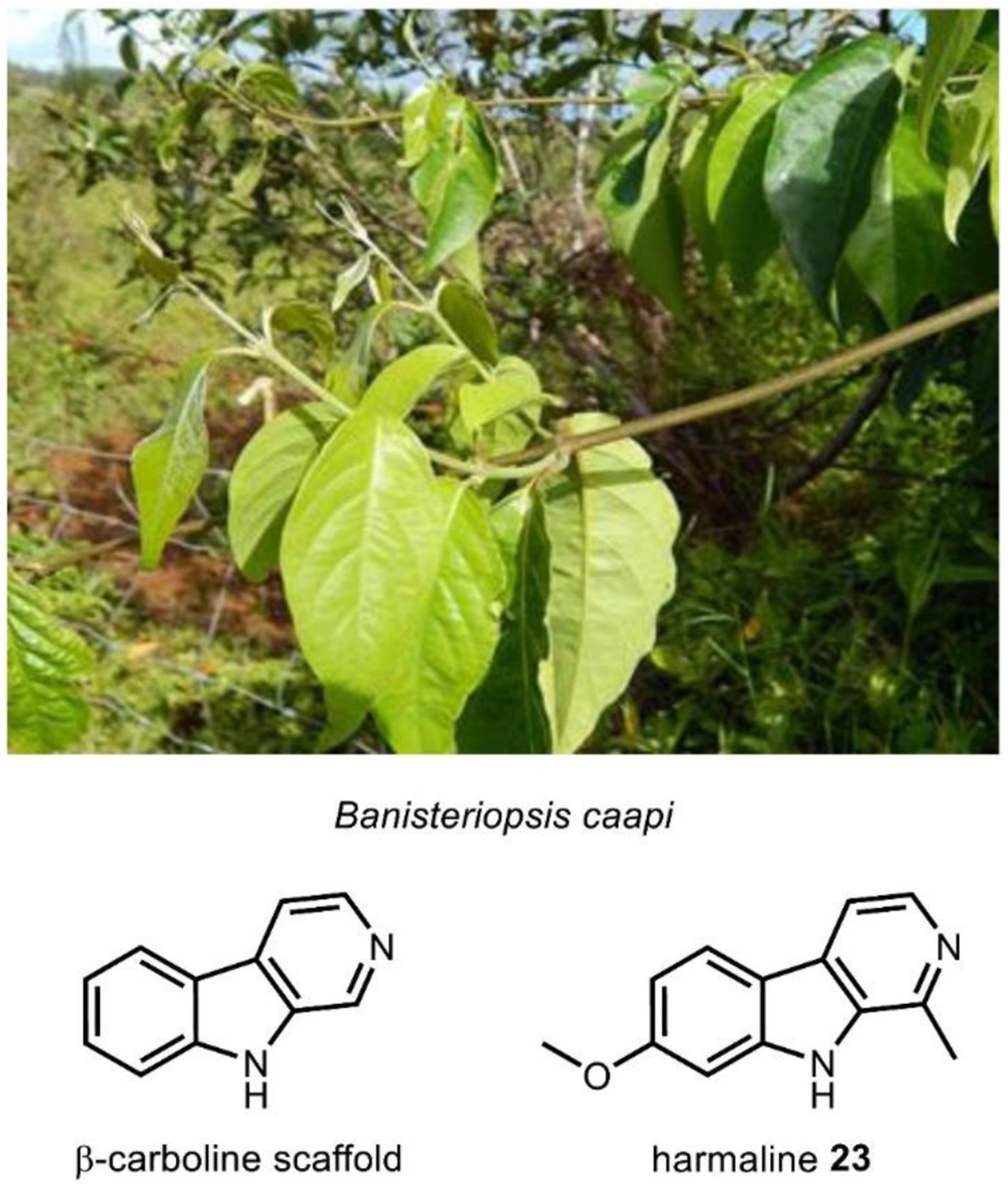
Image on left courtesy Forest and Kim Starr via CC-2.0. https://upload.wikimedia.org/wikipedia/commons/1/17/Starr-140222-0335-Banisteriopsis_caapi-leaves-Haiku-Maui_%2825240510635%29.jpg
Another example of a MAOI natural product cocktail is the recent isolation of 23 and related β-carbolines from numerous hallucinogen-producing Psilocybe sp. as known as “magic mushrooms”164 This serves as an interesting example of a single organism with diverged secondary metabolite scaffolds, where the biosynthetic pathways of both compounds diverge at tryptamine 14 but contribute to the same psychoactive effect.
2.4.1. Biosynthesis of harmala alkaloids
Initial feeding studies with radioactively labelled substrates into seedlings of the known harmala alkaloid producer, Peganum harmala, demonstrated that l-tryptophan and l-methionine are precursors in biosynthesis of harmala alkaloids.165 A later study demonstrated that radiolabeled 26 could be converted into its dehydrogenated form, 50, and that harmala alkaloid biosynthesis is likely compartmentalized across different tissues.166
While the complete set of biosynthetic genes implicated in harmala alkaloid formation have yet to be determined, one proposal167 postulates the sequence shown in Fig. 17. As in the case of the other indole containing compounds described, 11 is first decarboxylated to form 14 (Fig. 17). Next, pyruvic acid 22 is incorporated by a Mannich or Pictet-Spengler type reaction to form the β-carboline carboxylic acid 52 (also see Fig. 2). To determine the biosynthetic origin of the C-1 β-carboline methyl, radiolabeled feeding of acetic acid and pyruvic acid was performed.165 Stolle et al. observed specific incorporation of the radiolabeled C-2 and C-3 carbons of pyruvic acid 22 into the pyridine ring of the β-carboline scaffold, while radiolabeled acetic acid carbons were non-specifically incorporated throughout as a result of primary metabolism. 1-methyl β-carboline 53 is then formed by oxidative decarboxylation, followed by subsequent hydroxylation and O-methylation reactions to form harmaline 50. Formation of harmine 23 or tetrahydroharmine 51 takes places through either oxidation, or reduction of 50, respectively.
Fig. 17.
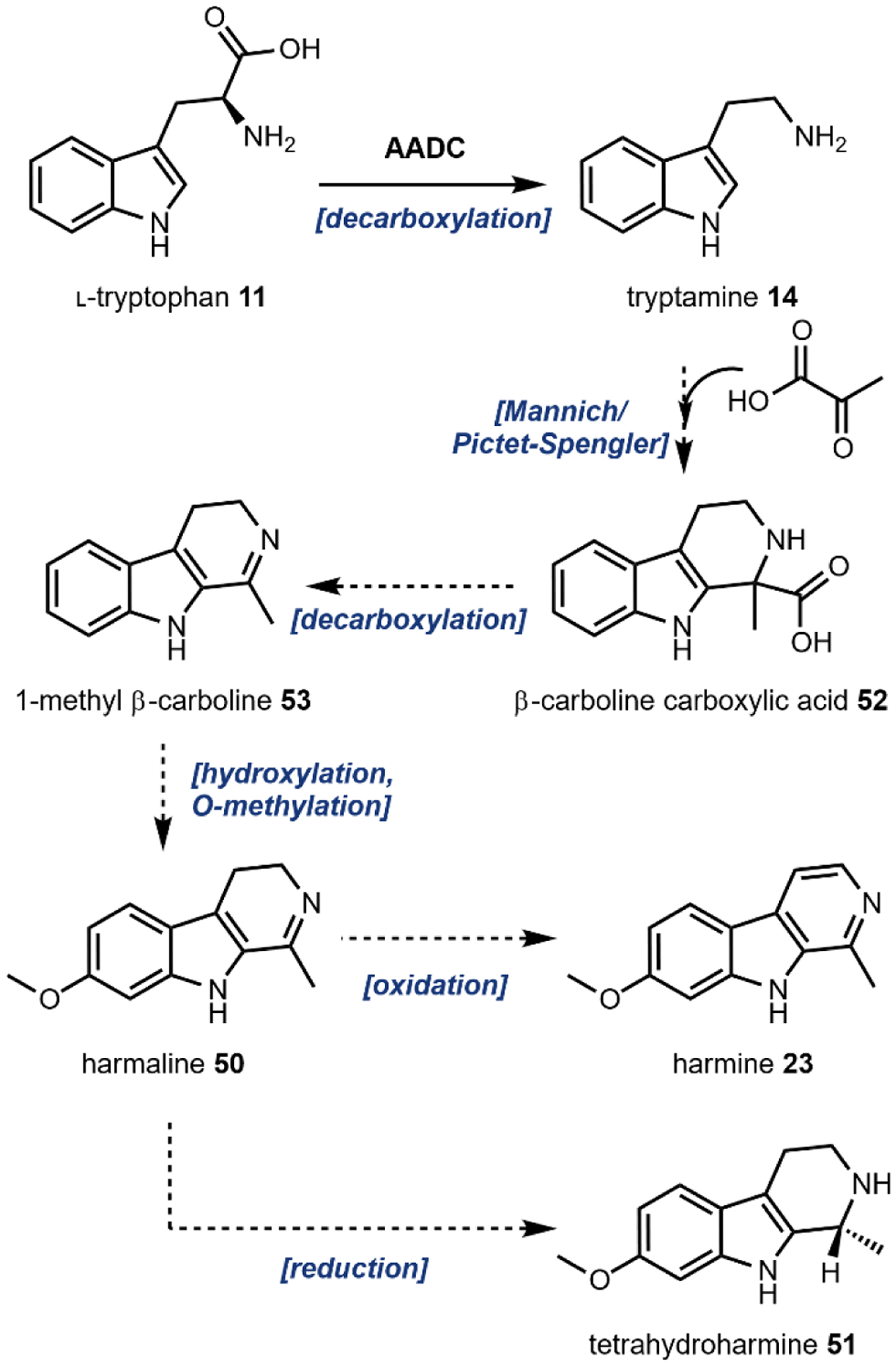
Proposed biosynthesis of harmala alkaloids.167
2.5. Lysergic acid and LSD
Lysergic acid diethylamide (LSD) 3 was first synthesized from lysergic acid 54 by Albert Hofmann in 1938. Like other 5HT2A receptor agonists, ingestion of 3 results in altered states of consciousness and visual hallucinations.168 While 3 has not been observed to occur naturally, its precursor, 54, is a natural product belonging to a class of diverse molecules broadly known as ergot alkaloids. 54 is isolated from many fungi with the ergot fungus, Claviceps purpurea (Fig. 18) being the most notable.169,170 Ergot alkaloids are commonly associated with the disease ergotism, known colloquially as Saint Anthony’s Fire, caused by eating rye or other cereal crops contaminated with ergot fungi.171 In addition to the vasoconstrictive and convulsive symptoms of the disease, mania and psychosis have been observed, underlining the psychoactivity of ergot alkaloids.171
Fig. 18. Claviceps purpurea (ergot fungus) infecting Dactylis glomerata (cat grass).

Image on the left courtesy of Bildoj via CC-3.0.
https://upload.wikimedia.org/wikipedia/commons/c/c4/Dactylis_026.JPG
Ergot alkaloids, derived from l-tryptophan 11, are characterized by a unique tetracyclic ergoline skeleton where the indole comprises the A and B rings. The C and D rings of the ergoline scaffold are derived from a cyclization of dimethylallyl pyrophosphate with the l-tryptophan amino group.172 There are three main ergot alkaloid classes, clavines, ergoamides (lysergamides), and ergopeptides, with 3 belonging to the ergoamide class.173 Ergoamides contain a C8-amide linkage on the D ring of the ergoline scaffold and is a common point of derivatization for drug development.174 Modifications on the amide can greatly affect bioactivity and in the case of 3, the diethylamide moiety is crucial for its prolonged psychoactivity.125
2.5.1. Biosynthesis of lysergic acid
Isotope labeling studies during the 1950s and 1960s determined that a mevalonate acid-derived isoprenoid, a methionine-derived methyl group and l-tryptophan 11 were key precursors to ergot alkaloid biosynthesis.175 The first enzymatic study in Claviceps sp. was the purification and characterization of 4-dimethylallyl- l-tryptophan synthetase (DMATS) that catalyzes the first committed step in ergot alkaloid biosynthesis: C-prenylation of l-tryptophan 11 with dimethylallylpyrophosphate at the indole C4 position to form 4-dimethylallyl-l-tryptophan 55 (Fig. 19, also see Fig. 4D).176 Recently, many laboratories have focused on characterizing prenyltransferases, of which DMATS is the original member of a new superfamily of prenyltransferase enzymes. Since the discovery of the DMATS, prenyltransferases that can regioselectively transfer allylic prenyl groups to almost every position on the indole ring have been identified.48,177–181 Members of the DMATS superfamily also have broad substrate scopes while maintaining regioselectivity which has aided in their development as tools for chemoenzymatic syntheses of natural and unnatural prenylated compounds, including the cannabinoid family (see 4.2.2).47,53,182,183
Fig. 19.
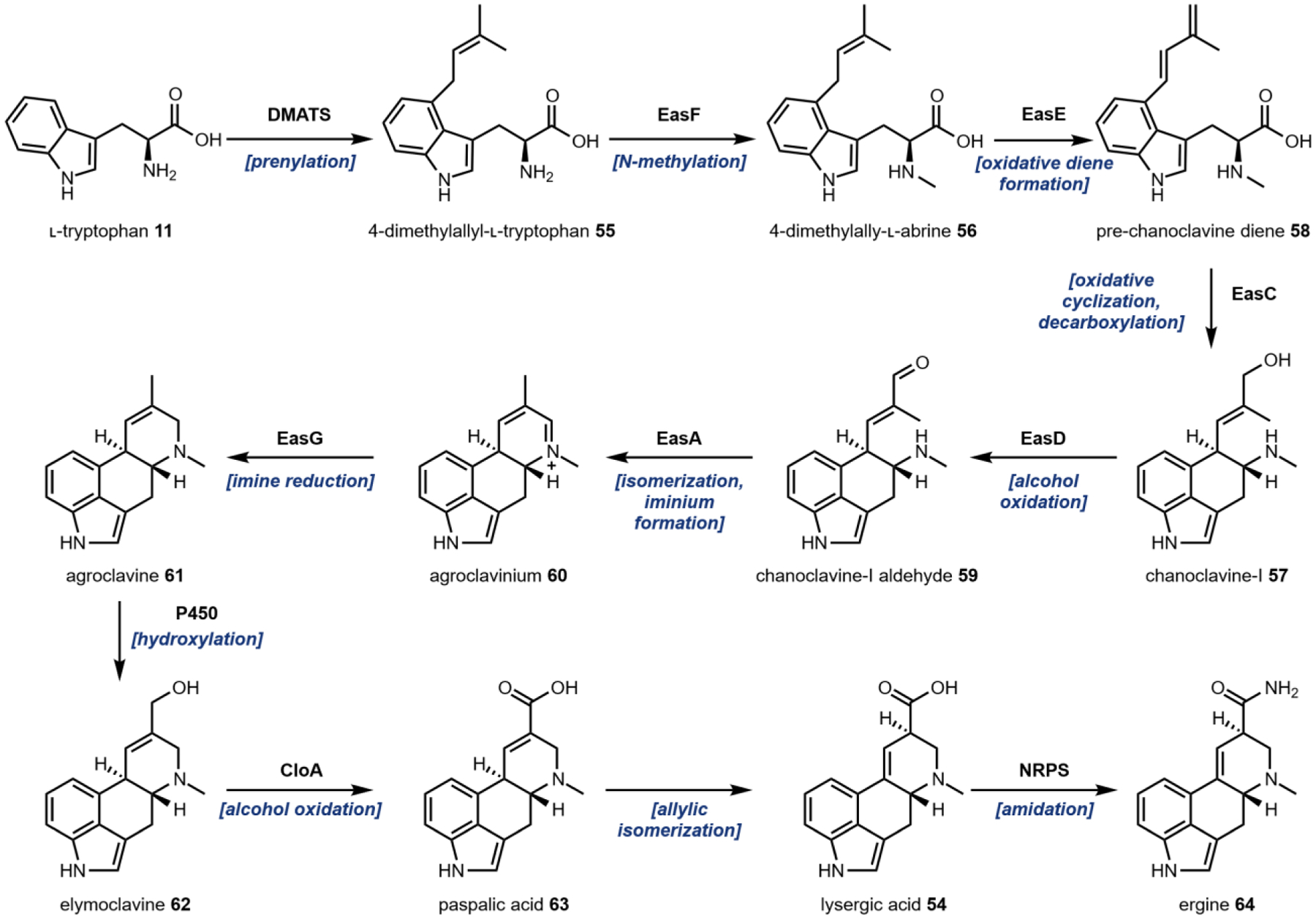
Biosynthesis of lysergic acid from l-tryptophan.
Chromosome walking using the gene encoding DMATS as a step-off point led to the identification of an ergot alkaloid biosynthetic gene cluster in the fungus C. purpurea.184,185 Sequence alignment revealed an N-methyltransferase, EasF which was proposed to convert 4-dimethylallyl-l-tryptophan 55 into 4-dimethylallyl-l-abrine 56 using SAM as a methyl donor. Thorough characterization of a homologous enzyme in an Aspergillus fumigatus ergot gene cluster, FgaMT, supported this hypothesis.186
Conversion of 56 into the cyclized chanoclavine-I 57 is facilitated by the FAD-linked oxidoreductase EasE and EasC, which was initially annotated as a catalase. Knock-out studies in both C. purpurea and the homologous cluster in A. fumigatus confirmed that both enzymes are necessary for production of 57.187,188 Subsequent pathway reconstitution studies in Aspergillus nidulans and Saccharomyces cerevisiae further supported the essential roles of EasE and EasC in biosynthesis.189,190 Until recently, however, the precise mechanisms of EasE and EasC were not resolved. Lorenz et al. initially postulated that EasE catalyzes the oxidative diene formation from 56 followed by decarboxylation through an epoxide intermediate to yield chanoclvaine-I 57, with EasC serving as a scavenger of hydrogen peroxide generated from EasE.188 A recent pathway reconstitution in A. nidulans enabled isolation of the a previously unknown intermediate, pre-chanoclavine diene 58, which verified the diene formation activity of EasE.191,192 Subsequent incubation of 58 with EasC recombinantly purified from E. coli led to the formation of 57 via a proposed radical addition mechanism using O2 as an oxidant.192 Hence, EasC is an essential redox enzyme in the main pathway to 54.
A short-chain reductase (SDR), FgaDH, was identified in an A. fumigatus gene cluster that produces a related ergot alkaloid fumigaclavine C.193 In vitro assays using recombinantly expressed enzyme determined that FgaDH catalyzes the oxidation of the allylic alcohol on 57 to an aldehyde to form chanoclavine-I aldehyde 59, strictly using NAD+ as the electron acceptor.193 A homologous SDR was subsequently identified in the lysergic acid biosynthetic gene cluster in C. purpurea and named EasD.194
The next steps in the pathway represent a branching point for ergot alkaloids. Functional differences in a conserved flavin-dependent old yellow enzyme known as EasA (an isomerase) from C. purpurea and FgaOx3 (a reductase) from A. fumigatus and P. commune represent a mechanistic branching point in D-ring formation.195–197 Here we will focus on the formation of agroclavine 61 from 59 towards the psychoactive lysergic acid amides in C. purpurea. EasA performs a hydride mediated isomerization of the α,β-unsaturated carbonyl from the E-alkene geometry to the Z-configuration through an enolate intermediate.196 This rearrangement positions the carbonyl for an intramolecular cyclization with the secondary amine resulting in the formation of the D-ring.196 Following ring closure, the iminium intermediate agroclavinium 60 then undergoes NADPH-dependent reduction by the oxidoreductase EasG to form 61.198
Assays of microsomal fractions from C. purpurea determined that 61 undergoes a 2-electron oxidation of the methyl group to an alcohol to form elymoclavine 62 by an unidentified cytochrome P450 monooxygenase.199 The only P450 enzyme in the biosynthetic gene cluster, CloA, does not catalyze this transformation and instead performs the 4-electron oxidation of 62 to paspalic acid 63 as suggested from two knock-out studies.200,201In ΔcloA mutants, 62 was still detected and supports the likelihood of an additional P450 enzyme in the host that can perform the first 2-electron oxidation. Finally, allylic isomerization of 63 forms the product lysergic acid 54. This transformation can occur spontaneously, but it remains possible that that an unidentified isomerase can catalyze this reaction as enzyme-catalyzed allylic rearrangements have been observed in other pathways.202,203
54 itself serves as a branching point for the formation of many ergopeptines or ergoamides. These derivatives are formed by a non-ribosomal peptide synthase (NRPS) enzyme complex of two synthetases, LPS1 and LPS2.173 One of these lysergic acid derivatives from Ipomoea purpurea (Morning Glory), ergine 64 (lysergic acid amide, LSA) is psychoactive. The pathway leading to formation of 64, while unconfirmed, could involve amidation by an NRPS or degradation of another NRPS product.204
2.6. Peyote
Peoples indigenous to North America have consumed the cactus, peyote, for over one thousand years as a part of their religious practices.205 Peyote, Lophophora williamsii (Fig. 20), is a small, spineless cactus with a crown consisting of round buttons that, among other cacti species, contain the hallucinogen, mescaline 65.205 The psychoactive effects have been described to be similar to LSD, but with a significantly lower potency at a ratio of about 1:2500 mescaline:LSD.117 Despite peyote’s status as a Schedule I controlled substance in the United States, it remains legal as an important part of religious practices by the Native American Church and other religious organizations who are protected by the American Indian Religious Freedom Act.
Fig. 20. Lophophora williamsii, one of the many cacti species that contain mescaline.

Image on the left courtesy of Peter A. Mansfeld via CC-3.0.
https://upload.wikimedia.org/wikipedia/commons/6/69/Lophophora_williamsii_pm.jpg
The natural products, elemicin 66 and myristicin 67 (Fig. 8) from nutmeg, or Myristica fragrans, are tetrasubstituted benzenes and structurally related to 65. Despite not being psychoactive, 66 and 67 are believed to be prodrugs as they are metabolized in the liver into 3-methoxy-4,5-methylenedioxyamphetamine, also known as MMDA.206,207 MMDA and its analogs were first synthesized from 65 by Alexander Shulgin, and similar to 65, MMDA is a 5HT2A receptor agonist, but with almost double the potency.208 Shulgin would later detail his extensive clandestine investigations into the syntheses and effects of substituted phenethylamines and tryptamines, earning him the title “godfather of psychedelics.”209,210
2.6.1. Biosynthesis of mescaline
Before the discovery of the mammalian iterative methyltransferase that catalyzes N-methylation of tryptamine 14 and serotonin 38 into hallucinogenic compounds,141 Axelrod and Tomchick identified another neurotransmitter methyltransferase, catechol O-methyltransferase (COMT).211 COMT, along with monoamine oxidase, modified the l-tyrosine-derived catecholamine neurotransmitter dopamine 17 (Fig. 21) for excretion in the urine.212. In the years following, similar to the case of endogenous DMT biosynthesis, several studies identified enzymes in mammalian tissues that could catalyze the chemical transformations of dopamine-related metabolites 3-methoxytyramine 68 into 3-methoxy-5-hydroxytyramine 69 and 3,5-dimethoxytyramine 70 into 65, although no endogenous 65 could be identified from mammalian organisms.213,214 Several mechanisms for 65 biosynthesis in peyote and related cacti have been proposed by metabolite isolation and radiolabeled feeding studies.215–219 One proposed pathway by Lundström is shown in Fig. 21.219
Fig. 21.
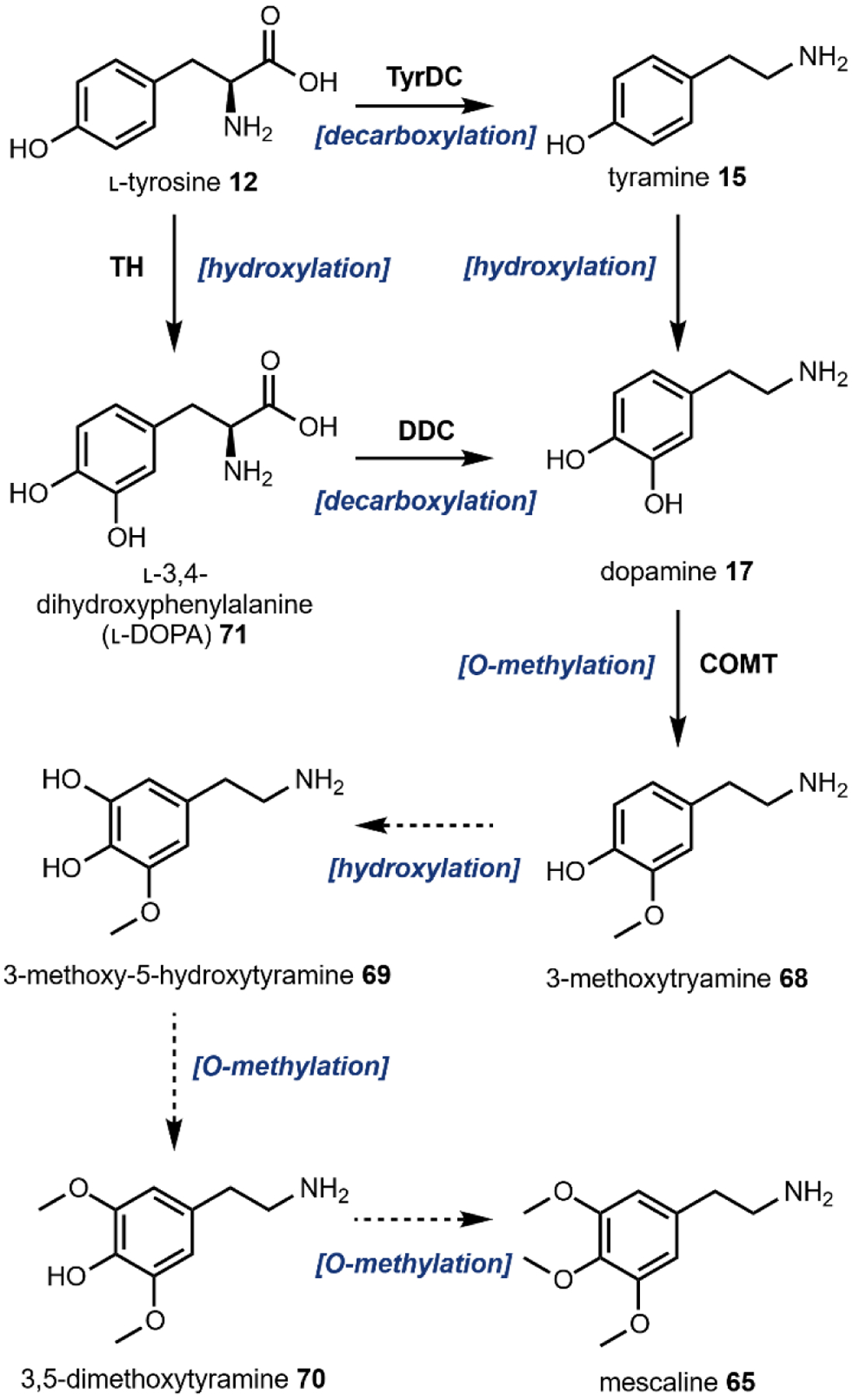
Proposed biosynthesis of mescaline.221
The proposed biosynthesis begins with hydroxylation of l-tyrosine 12 to 3-hydroxy-L-tyrosine (l-DOPA, 71) by tyrosine hydroxylase (TH), followed by decarboxylation catalyzed by DOPA decarboxylase (DDC) to yield 17. Alternatively, 12 may also be converted to tyramine 15 through a decarboxylation catalyzed by tyrosine decarboxylase (TyrDC), followed by aromatic hydroxylation to 17 by an unknown enzyme. From either route, 17 can be converted into 3-methoxytyramine 68, which has been isolated from mescaline producing plants, by the enzyme catechol O-methyltransferase (COMT) using SAM as the methyl donor. The final intermediates towards mescaline production 3-methoxy-5-hydroxytyramine 69 and 3,5-dimethoxytyramine 70 have been determined to be naturally occurring in mescaline producing plants by inverse isotope dilution, but neither have been isolated from plants. These are likely to be on pathway intermediates since they are incorporated into mescaline to a greater extent than other possible intermediates.219
While the biosynthesis of 65 in peyote has yet to be elucidated, Ibarra-Laclette et al. recently generated two cDNA libraries of the L. williamsii transcriptome, one for button and one for root, using RNA-seq.220 From this data set, the authors identified putative genes that may encode biosynthetic enzymes for mescaline production including DOPA decarboxylases, hydroxylases, and O-methyltransferases based on phylogenetic analysis.220 Careful in vitro experiments will be required to finally ascertain the mescaline biosynthetic pathway.
2.7. Fly agaric
Ibotenic acid 72, a nonproteinogenic amino acid with a hydroxylated isoxazole ring, and its decarboxylated form, muscimol 73, are the main psychoactive constituents of the toadstool, Amanita muscaria, commonly known as fly agaric (Fig. 22).164 Similar to Psilocybe sp., recreational consumption of Amanita sp. rose in popularity in the 1960s. However, contrary to other fungal psychoactives that target the serotonin receptor, these compounds are γ-aminobutyric acid type A (GABAA) receptor agonists.222 GABAA receptors are found in multiple regions of the brain and thus 72 and 73 can alter the activity of the cerebral cortex and cerebellum leading to alterations in sensory processing and motor function, respectively.223 A. muscaria is classified as poisonous, which can in part be attributed to the neurotoxicity of 72. Its structural similarity to l-glutamic acid 36 allows 72 to act as an agonist towards the N-methyl-d-aspartate (NMDA) receptor resulting in electrolytic lesions in the brain.224
Fig. 22.

Amanita muscaria contains about ~100–1000 ppm of ibotenic acid and muscimol.
72 and 73 naturally occur in low concentrations (~100 – 1000 ppm) in the cap and stem of A. muscaria.225 Minimal dosage for psychedelic effects are estimated as low as 6 mg for 46 and 30–60 mg for 72.226 Interestingly, A. muscaria and its constituents are not regulated by the United States federal government, in contrast to 1 and 42 from Psilocybe sp.
While 72 was first isolated over 50 years ago, its biosynthesis remained elusive.227 Recently, Obermaier and Muller identified a gene cluster encoding 72 and 73 biosynthesis in A. muscaria.228 The key to locating this cluster was the identification of a glutamate hydroxylase, an enzyme first implicated in 72 biosynthesis over 50 years ago, but never found. This enzyme, a nonheme, iron and α-ketoglutarate-dependent dioxygenase named IboH, hydroxylates l-glutamate 36 at the C3 position resulting in the formation of 3-hydroxy-l-glutamic acid 74.
2.7.1. Biosynthesis of ibotenic acid
Obermaier and Muller proposed two pathways (A and B) for ibotenic acid biosynthesis diverging from 74 (Fig. 23). One proposal (Pathway A) is that 74 undergoes a condensation reaction catalyzed by IboA, an adenylating enzyme, with ammonia from an unidentified donor to form 3-hydroxyglutamine 75. A likely amine source is glutamine which is the amine donor in various metabolic reactions. IboF, a flavin-dependent monooxygenase, would then catalyze N-oxidation of the terminal amide to form 3-hydroxyglutamine hydroxamic acid 76. Next, either IboG1 or IboG2, PLP-dependent paralogs found in the biosynthetic gene cluster, catalyzes the intramolecular cyclization of the hydroxamic acid with the hydroxyl group at the C3 position to form the five-membered heterocycle tricholomic acid 77. Alternatively, Pathway B involves N–O bond formation between an unidentified, hydroxylamine 78 with the C3 hydroxyl group on 74 by IboG1/G2 to form a 3-hydroxy-l-glutamic acid derivative 79. In this pathway, the external hydroxylamine could derive from hydroxylation of an external amine 80 catalyzed by IboF. IboA would then facilitate cyclization of the hydroxylamine with the C-5 carbonyl of the 3-hydroxy-l-glutamic acid derivative 79 to form 50. From tricholomic acid 77, IboC, a cytochrome P450, catalyzes the desaturation of the 3-oxoisoxazolidine ring to form ibotenic acid 72. IboD, a PLP-dependent decarboxylase can catalyze the further decarboxylation of 72 to form the other major psychoactive compound, muscimol 73.
Fig. 23.
Biosynthesis of ibotenic acid and muscimol from l-glutamic acid.228
2.8. Iboga alkaloids
Root and bark from the iboga tree, Tabernanthe iboga, has been used for both therapeutic and spiritual ritual purposes in West Central Africa for hundreds of years.229 T. iboga is rich in l-tryptophan derived-monoterpene indole alkaloids (MIAs), an expansive class of over 3000 plant natural products starting from the universal MIA precursor, strictosidine 25.230,231 Many molecules of this class have broad bioactivities that include anti-cancer221, anti-malarial232, anti-addiction233 and more.234 The potent MIA cancer therapeutics vincristine and vinblastine from Catharanthus roseus are listed on the World Health Organization’s List of Essential Medicines, underlining the value of MIAs as human therapeutics. One of the MIAs from iboga roots is the psychedelic (–)-ibogaine 2 that has multiple neurotransmitter interactions including the κ- and μ-opioid receptors and the serotonin transporter, which collectively results in a feeling of a dream-like state of consciousness.229 Additionally, 2 and some of its derivatives have shown promise as anti-addictive agents.233,235
The iboga alkaloid scaffold is characterized by a 6-5-7 ring system comprised of indole and tertrahydroazepine fused to an isoquinuclidine ring to form a pentacyclic skeleton with a tertiary amine serving as the bridgehead (Fig. 24). The addition of a C5 methoxy group on the indole ring within the iboga scaffold provides 3. Variable substitutions on the indole ring and the presence of a carbomethoxy group at the indoloazepine-isoquinuclidine junction lead to different family members within this class. Interestingly, 2 is the only known compound with the iboga scaffold to have hallucinogenic properties, which raises questions about the structure-activity relationship between 2 and 5-HT receptors. According to a recent study, iboga scaffolds lacking the isoquinuclidine ring resulting in an indole-tetrahydroazepine tricycle lost their hallucinogenic properties but maintained their ability to promote neural plasticity, the mechanism that may be the key to its anti-addiction properties.26
Fig. 24. Tabernanthe iboga in fruit.
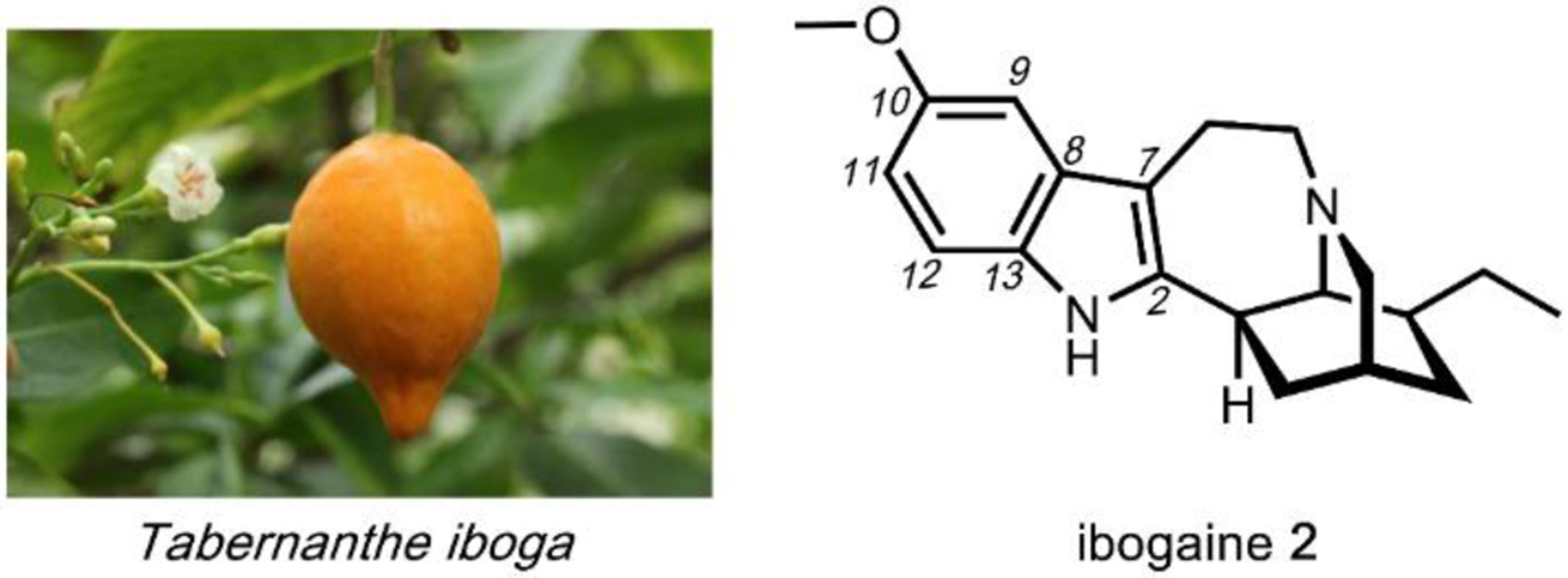
Image courtesy of Christian Kunath via CC-3.0.
https://twitter.com/sesamothamnus/status/1031998713760231424
2.8.1. Biosynthesis of iboga alkaloids
Given that strictosidine 25 is the central metabolite in the MIA biosynthetic pathways in plants, there has been intense efforts to understand how nature transforms the simple geranyl (C10) precursor that combines with tryptamine 14 to yield the complex 25. These efforts from different labs have fully elucidated the pathway to 25. In recent years, further efforts have led to the complete mapping of the downstream enzymatic transformation to vinblastine in C. roseus, which comprise of over 30 enzymes starting from primary metabolites.45,236–243 Shortly after, the (−)-ibogaine biosynthetic pathway from 25 was also elucidated, as well as other complex MIA compounds.244
The first committed step in the seco-iridoid pathway towards the monoterpene scaffold in 25 is the formation of geraniol 81 (Fig. 25). While it was predicted that 81 was hydrolyzed from the mevalonate pathway intermediate, geranyl pyrophosphate (GPP) 82,245,246 the enzymatic basis of its formation was unknown until the discovery of geraniol synthase (GES) from sweet basil (Ocimum basilicum) decades later.193 Since then, many GES homologs have been discovered from various plants. The activity of GES, which is to hydrolyze 82 to 81, represents a divergence point between primary and secondary terpene metabolism in plants. In primary metabolism, GPP is further elongated to farnesyl pyrophosphate (FPP), which is central towards the synthesis of steroids and coenzyme Q. By hydrolyzing the pyrophosphate in GPP, GES commits the geraniol group for MIA biosynthesis and siphons GPP away from primary metabolism. In the MIA pathway, geraniol 81 is then hydroxylated by the P450 enzyme geraniol 8-hydroxylase (G8H) to form 8-hydroxygeraniol 83.247
Fig. 25.
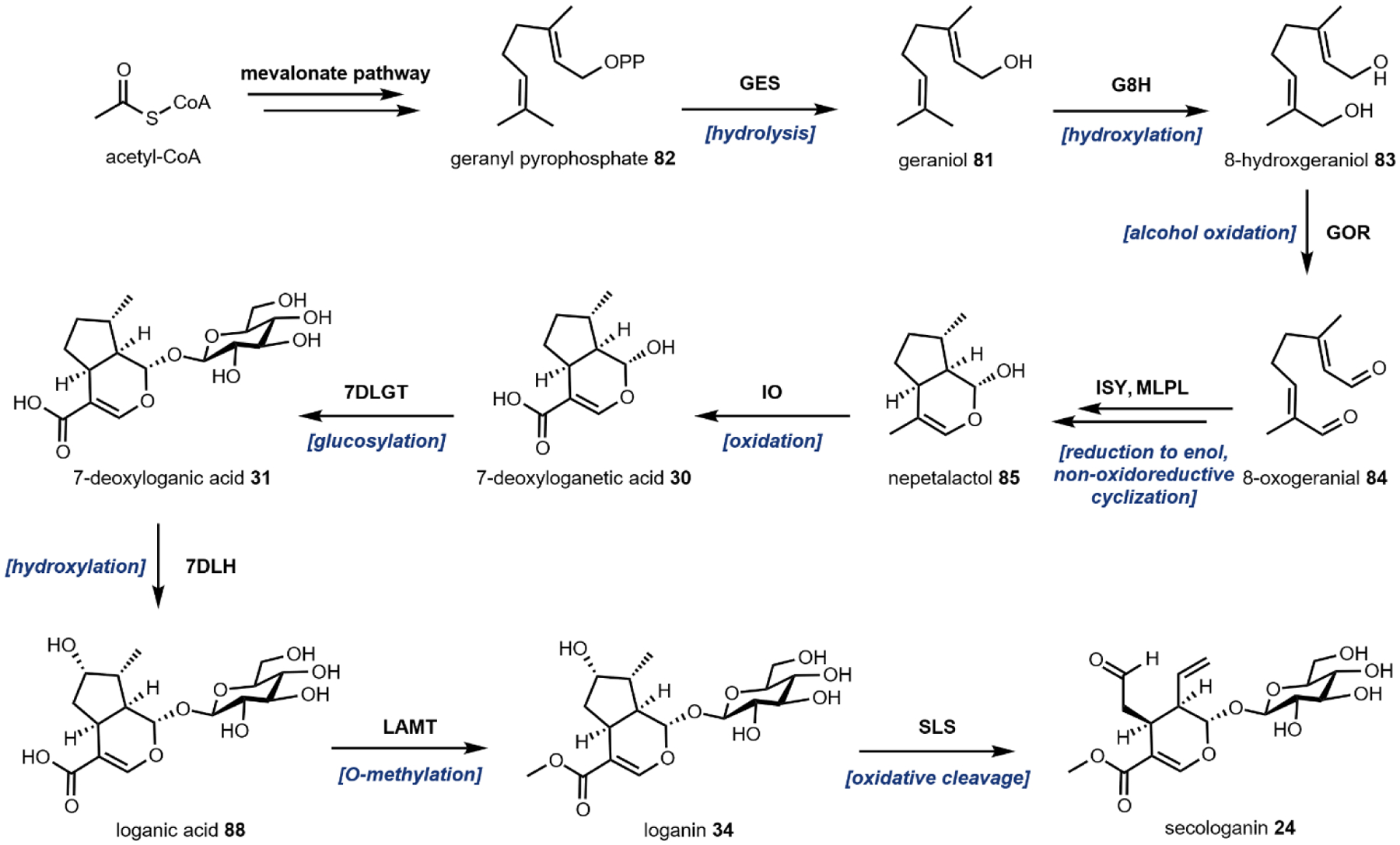
Biosynthesis of secologanin from geranyl pyrophosphate (GPP).
The next four biosynthetic steps were all discovered from analysis of the C. roseus transcriptome.45 8-hydroxygeraniol oxidoreductase (GOR) iteratively oxidizes the two alcohols in 83 to yield 8-oxogeranial 84, a dialdehyde that is poised for intramolecular cyclization. It was initially believed that iridoid synthase (ISY) was an NAD(P)H-dependent cyclase.248 However, a recent report demonstrated that ISY is a reductase that can reduce 84 to an enol intermediate.249 A previously undiscovered cyclase, major latex protein-like (MLPL), then facilitates the cyclization of the reduced enol to form cis-trans nepetalactol 85 in a non-cofactor dependent mechanism.243 85 is the first molecule in the pathway that has the iridoid structure. In plants such as Nepeta, 85 can be oxidized to neptalactone, which is the cat attractant produced by these plants.249 In the MIA pathway 85 undergoes a 4-electron oxidation catalyzed by the P450 iridoid oxidase (IO) to install an α,β-unsaturated carboxylic acid in 7-deoxyloganetic acid 30. The next step is glucosylation by 7-deoxyloganetic acid glucosyl transferase (7DLGT) with UDP-glucose to form 7-deoxyloganic acid 31 (See Fig. 3C). Glucosylation of the hemiacetal presumably stabilizes the compound and prevents spontaneous ring opening. The third P450 in the pathway, 7-deoxyloganic acid hydroxylase (7DLH), catalyzes hydroxylation of the cyclopentane ring in 31 to form loganic acid 86.
Expression data revealed that the next two genes in the seco-iridoid pathway encoding for loganic acid O-methyltransferase (LAMT) and secologanin synthase (SLS) are part of a separate regulon from the early pathway.250,251 The seco-iridoid pathway is also spatially segmented between the internal phloem associated parenchyma (IPAP) cells for iridoid production and leaf epidermis cells for the remaining steps towards production of strictosidine 25.252 86 is first transported from the cytosol of the IPAP cells into the cytosol of epidermic cells by a nitrate/peptide family (NPF) transporter.253 The cytosolic LAMT subsequently converts 86 into loganin 34.250 The fourth P450 in the pathway, SLS then catalyzes oxidative cleavage of the cyclopentanol ring of 34 to unveil the reactive aldehyde handle in secologanin 24 (See Fig. 5A).56
To form strictosidine 25, 24 and 14 are condensed through a stereospecific Pictet-Spengler reaction catalyzed by strictosidine synthase (STR) (Fig. 26, and see Fig. 3).254 This mechanism had been long proposed before the discovery of STR, modeled after the formation of l-benzylisoquinolines alkaloids.255 Considering the synthetic challenges associated with accessing 25, STR has become an attractive enzyme for the chemoenzymatic and biotransformative syntheses of analogs of 25.256–258 The regulation and complexity of MIA biosynthesis is further highlighted by the transient sub-cellular compartmentalization of 25 formation in the vacuole of epidermis cells followed by immediate export towards the nucleus.259 It is believed that the spatial isolation of STR and its substrates prevents accumulation of the highly-reactive strictosidine aglycone intermediate, 4,21-dehydrogeissoschzine 87 (vide infra), a dialdehyde which leads to toxic protein cross-linking.260 It is hypothesized that this is a plant defense mechanism from herbivores mirroring the activation of the related phenolic secoiridoid glycoside, oleuropein, from the privet tree, Ligustrum obstusifolium following tissue damage.261
Fig. 26.
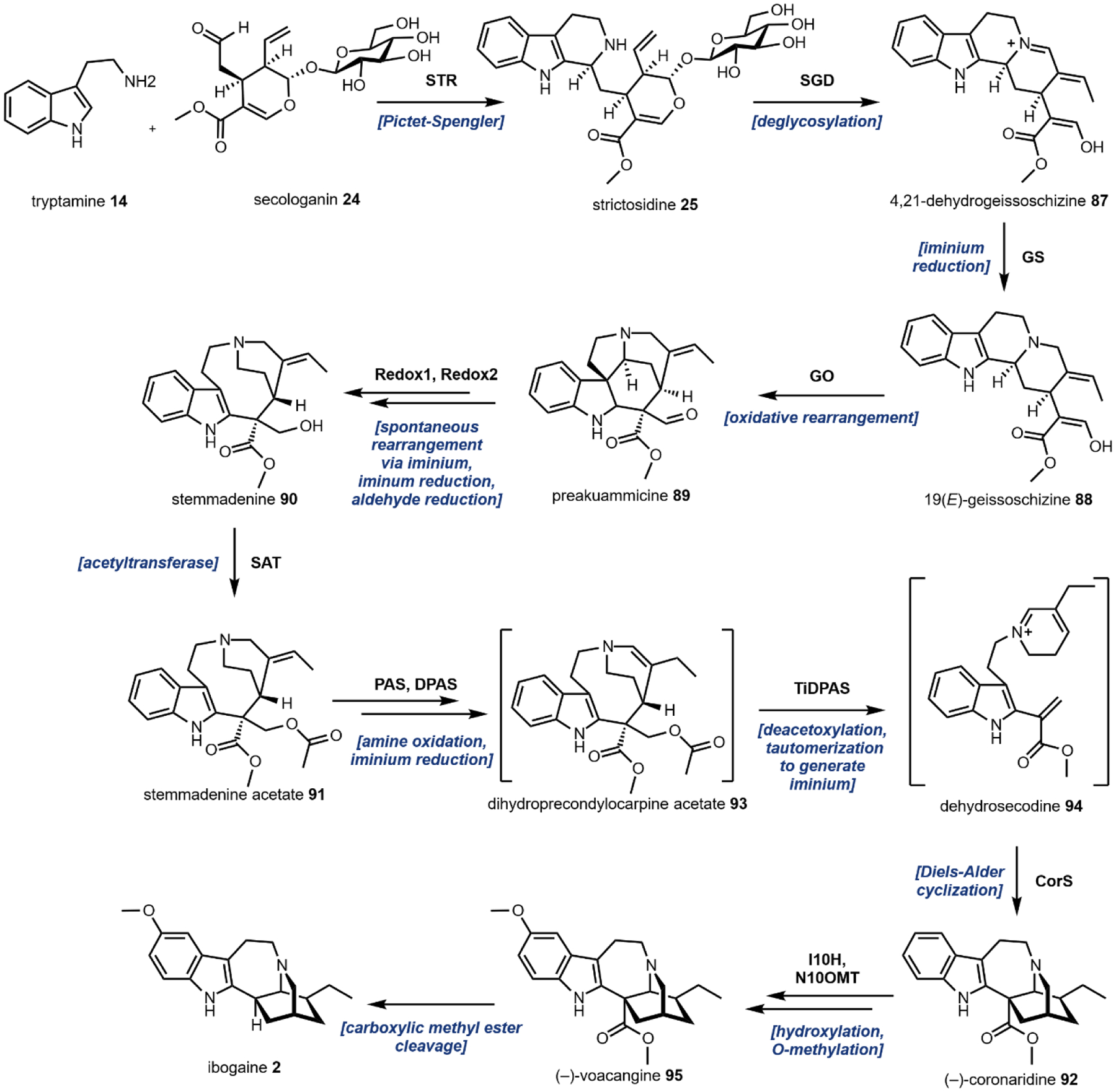
Biosynthesis of ibogaine from tryptamine and secologanin.
From 25, different branches of the MIA family can be accessed. The first step is the deglucosylation of 25 by the enzyme strictosidine-O-β-glucosidase (SGD).262 Whereas 25 is relatively stable and benign to the host, removal of the glucose group which essentially serves to mask the hemiacetal, leads to the dialdehyde 4,21-dehydrogeissoschizine 87 that is prone to cross-linking. 87 can exists in equilibrium with the more stable epimers cathenamine and epicathenamine.263 Each of these aglycone intermediates represents a divergence point towards different terminal alkaloids.241,264 From 87, the next two transformations to form 19(E)-geissoschizine 88 and preakuammicine 89 catalyzed by geissoscizine synthase (GS) and geissoschizine oxidase (GO), respectively, were characterized by Tatsis et al.242 87 is converted into 88 through iminium reduction catalyzed by GS.241 88 then undergoes an oxidative rearrangement catalyzed by the P450 GO to yield an unstable intermediate, preakuammicine 89, which can undergo spontaneous rearrangement and tandem enzyme-catalyzed reductions to form the stable stemmadenine 90. Reactive intermediates that form between 88 and 90 exist transiently can spontaneously undergo chemical transformations that diverge towards different MIAs including corynanthean, strychnos, iboga, and aspidosperma skeletons.236 From 90, stemmadenine O-acetyltransferase (SAT) catalyzed acetylation forms stemmadenine acetate 91.
A series of redox transformations and divergent cycloaddition reactions take place to transform 91 into catharanthine, tabersonine, and (−)-coronardine 92. Catharanthine and tabersonine are both on-pathway intermediates to vinblastine, while 92 has essentially the same carbon skeleton as ibogaine 2. These transformations have recently been characterized through analysis of transcriptome datasets from T. iboga and subsequent biochemical characterizations.244,265 First, a tandem amine oxidation-iminium reduction cascade catalyzed by precondylocarpine acetate (PAS) and dihydroprecondylocarpine acetate synthase (DPAS), respectively, would generate the enamine dihydroprecondylocarpine acetate 93. The net outcome from 92 to 93 is migration of the olefin to set up the subsequent [4 + 2]-Diels–Alder reactions.237 In ibogaine biosynthesis, TiDPAS would promote the deacetoxylation with concomitant carbon-carbon bond cleavage, and NADPH-dependent tautomerization to generate the iminium dehydrosecodine 94. The enzyme coronaridine synthase (CS) would then catalyze a formal [4 + 2]-Diels–Alder to form (–)-coronaridine 92. In the biosynthesis of catharanthine and tabersonine, a corresponding pair of DPAS and cyclization enzyme (catharanthine synthase and tabersonine synthase, respectively) are involved to forge the different connectivities via cycloadditions. A recent study by the O’Connor group reports the structural basis for the divergence in regio- and stereo-selectivity of the Diels-Alderases found in iboga and aspidosperma alkaloid biosynthesis.266 From 92, the P450 enzyme ibogaine 10-hydroxylase (I10H) catalyzes hydroxylation at the C-5 position of the indole ring, followed by noribogaine 10-O-methyltransferase (N10OMT)-catalyzed O-methylation to yield (−)-voacangine 95.265 Both 92 and 95 have shown promise as acetylcholinesterase inhibitors.267 In the last step, 92 undergoes decarboxylation to form (−)-ibogaine 2. This process can occur nonenzymatically under heat, but it is likely there is an unidentified decarboxylase that facilitates this step in planta.
2.8.2. Heterologous production of iboga alkaloids
De novo production of strictosidine 25 in S. cerevisiae was demonstrated by Brown et al. in a landmark achievement of synthetic biology in 2015 (Fig. 27). The authors’ engineered yeast strain comprised of twenty-one genome integrated genes, three genome-deletions and expression of a high-copy plasmid encoding a codon-optimized G8H gene. The host produced ~ 0.5 mg/L of extracellular strictosidine after 6 days. Since simple expression of the required pathway genes did not result in detectable production of pathway intermediates, the authors employed a series of metabolic engineering steps to boost precursor titers, reduce nonproductive shunt product formation, and increase P450 activity.
Fig. 27.
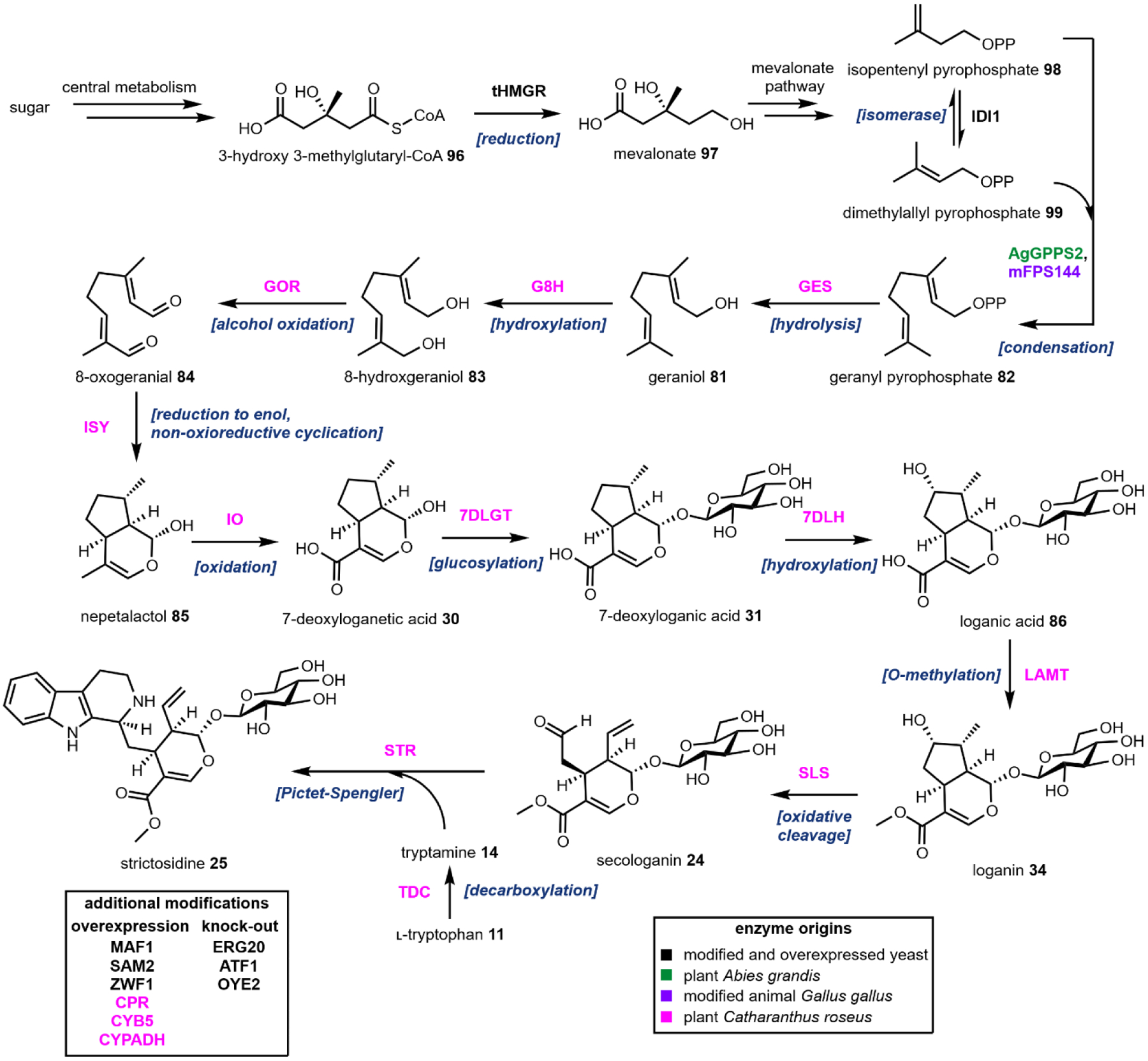
Heterologous production of strictosidine in S. cerevisiae.76
Towards increasing precursor titers, a truncated yeast 3-hydroxy-3-methylglutaryl-CoA reductase (tHMGR) was expressed to increase the reduction of 3-hydroxy-3-methylglutaryl-CoA 96 to form mevalonate 97. Since GPP 82 is not a native yeast metabolite, expression of a GPP synthase (AgGPPS1) from Abies grandis combined with expression of a mutated farnesyl pyrophosphate synthase (mFPS144) with partial GPP synthase activity from the avian Gallus gallus resulted in 82 biosynthesis. Maintaining some level of essential yeast metabolite farnesyl pyrophosphate (FPP) biosynthesis with mFPS144 was required since yeast FPP synthase, ERG20, was knocked-out to shift mevalonate pathway flux from FPP to 82. Balancing concentrations of isomers isopentenyl pyrophosphate 98 and dimethylallyl pyrophosphate 99 required for 82 formation was achieved through expression of a second copy of yeast isomerase IDI1. To further increase 81 titers, the authors overexpressed MAF1, a negative tRNA biosynthesis regulator, which reduced the amount of 98 utilized for tRNA formation.
Geraniol 81 can be rapidly metabolized by yeast enzymes to form esterified and reduced shunt products.268 ATF1, an alcohol acetyltransferase, and OYE2, an NADPH-dependent oxidoreductase, were knocked-out to reduce nonproductive shunt product formation. A later study by Billingsley et al. identified more yeast enzymes that when knocked-out, further attenuate shunt product formation from 8-hydroxygeraniol 83 and channel additional flux of 83 towards iridoid biosynthesis.82
The strictosidine biosynthetic pathway contains four P450 enzymes which require reductase partners to facilitate the electron shuttling during catalysis. The C. roseus P450 reductase partners, cytochrome P450 reductase (CPR) and cytochrome b5 (CYB5), were expressed in yeast along with a putative alcohol dehydrogenase that was identified from MIA biosynthesis coexpression profiles (CYPADH) to increase P450 activity. Since these P450s also all require NADPH as an electron donor, ZWF1 which is yeast glucose-6-phosphate dehydrogenase, was overexpressed to increase intracellular NADPH concentrations. SAM2, yeast SAM synthetase, was also overexpressed to increase SAM availability for LAMT.
Overall, the metabolic engineering and synthetic biology strategies employed to create this yeast platform illustrates the possibility of an alternative pipeline for MIA production. Biotransformation of 25 into ibogaine 2 is another twelve enzymatic steps. While a de novo ibogaine biosynthesis yeast platform has yet to be published, many of the enzymes downstream of 53 biosynthesis have been demonstrated to be functional in yeast which is promising for providing sustainable access to 2 and potential derivatives.236,244
2.9. Salvia
Salvia divinorum colloquially known as the “sage of the diviners”was introduced to western academics by the indigenous people of the Sierra Mazateca in Mexico. While the botanical history of this plant has remained elusive, the hallucinogenic properties of salvia are currently employed by Mazatec shamans to facilitate visions of curing and divination. The active constituent, salvorin A 37, was first isolated by Ortega and coworkers in 1982 and further characterized by Valdes et al. two years later (Fig. 28).269,270 The potent hallucinogenic properties of the isolated 37 were confirmed by ethnobotanist Daniel Siebert, who noted that a dose of 200 μg could product effects identical to that of whole herb ingestion.271 Unlike other known hallucinogenic natural products, salvia does not function as a serotonin receptor agonist. Instead, 37 is a selective opioid agonist, binding strongly to κ−opioid receptors (KORs), and was identified as the first non-nitrogenous opioid receptor ligand.22,272 Given these unique properties, numerous therapeutic uses for salvinorins have been proposed, including anti-inflammatory, analgesic, and anti-addiction treatments.273,274 Salvia is consumed primarily as a recreational drug inducing powerful, sometimes disorienting hallucinations and has a legal status that is highly contested.
Fig. 28.
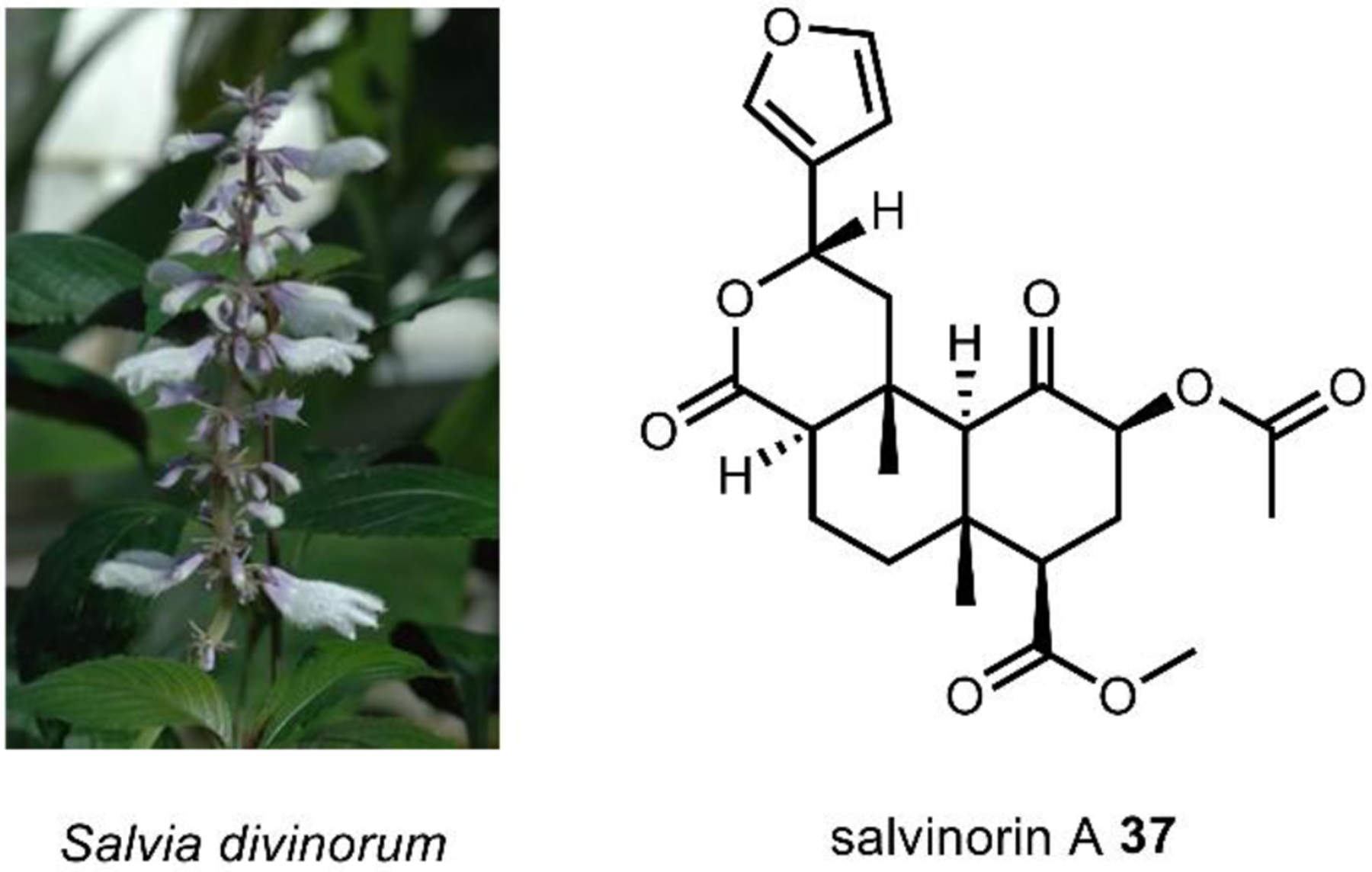
Salvia divinorum contains salvinorin A, a structurally unique terpene hallucinogen. Image on the left courtesy of Eric Hunt via CC-2.5.
https://upload.wikimedia.org/wikipedia/commons/3/35/Salvia_divinorum_-1.jpg
2.9.1. Biosynthesis of salvinorins
37 is a modified neo-clerodane type diterpenoid featuring a unique furyl-δ-lactone fragment. Structural-activities relationship studies of 37 analogues with modifications to the of furanyl group, as well as molecular modelling have implicated the furan ring in selective KOR binding.275 In 2015, Gupta et al. reported collybolide, a fungal sesquiterpene bearing a similar furyl-δ-lactone, exhibiting KOR agonism similar to salvorin.276 Investigations into the biosynthetic route to 37 are still in their infancy. Produced and stored in the leaf trichomes,277 tissue culture of S. divinorum grown on isotopically labelled substrate confirmed that the diterpene core of salvorins arises via the deoxyxylulose phosphate (DXP) pathway.278 This information aided the trichome-specific transcriptomics studies that investigators have used to identify pathway genes. In 2016, two research groups simultaneously identified and characterized the first enzyme involved in biosynthesis of 37, the (–)-kolavenyl diphosphate synthase (KPS) (Fig. 29). KPS is a class II diterpene synthase, performing cycloisomerization of geranylgeranyl pyrophosphate 100 through a carbocation intermediate to form (–)-kolavenyl pyrophosphate 101.279,280 Hardwickiic acid 102 has been proposed as an on-pathway intermediate based on co-localization and structural similarity to the salvorins. Based on more than a dozen salvorin-like molecules that have been isolated, a hypothetical downstream biosynthetic pathway has been proposed.279 However, the exact series of oxidative decorations and cyclizations leading formation of the rare furyl-δ-lactone moiety will be of interest to biosynthetic chemists and metabolic engineers alike.
Fig. 29.
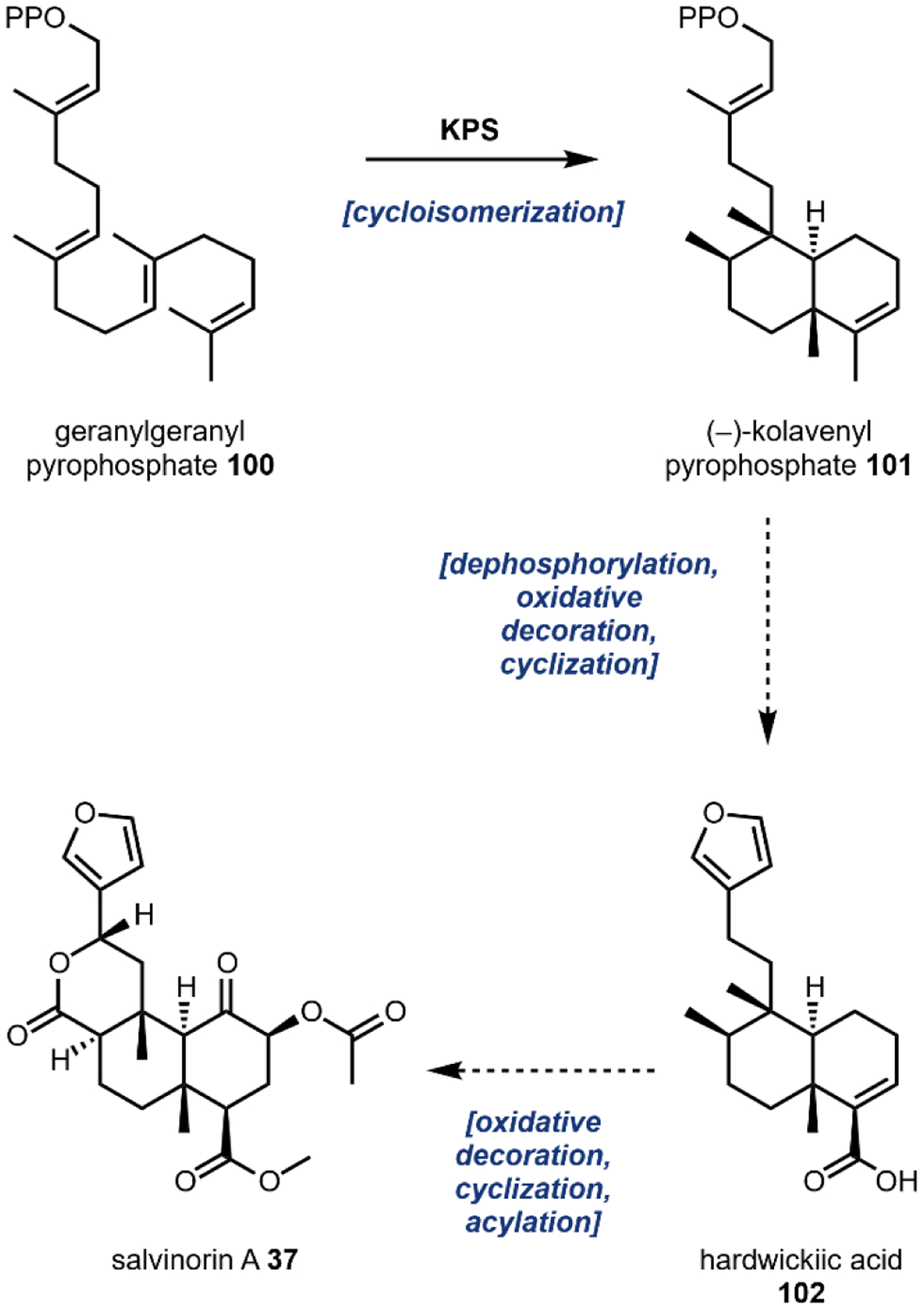
Proposed biosynthetic pathway for salvinorin A.279
3. Alkaloidal Stimulants
Alkaloidal stimulants may be regarded as the most culturally pervasive secondary metabolites; consumption of plants containing the alkaloidal stimulant caffeine 4 may have occurred as early as 2500 BC in China. By the 1600s, alkaloid containing plants were distributed as luxury commodities along every major trade route. Alkaloid consumption was a key driver of the Euro-American slave trade, which occurred from the 16th to the 19th centuries and enabled early efforts to characterize active constituents.281 Indeed, the alkaloid caffeine is currently world’s most widely consumed psychoactive drug; although global consumption statistics have been difficult to estimate, more than 85% of adults in the U.S. regularly consume caffeine at an average rate of 0.2 grams per person per day.28 While an exhaustive list of natural product stimulants would encompass molecules that are of diverse biosynthetic origins, the well-known members covered in this review fall within three major categories – the purine alkaloids, pyridine alkaloids, and tropane alkaloids. In addition to these alkaloids for which the biosynthesis has been well-studied, stimulants from a number of other plants including khat, areca, and ephedra are increasing in notoriety. Investigations into the biosynthesis, safety, and efficacy of these alkaloidal stimulants remain in their infancy.
3.1. Catecholamine neurotransmitters
Generally speaking, a stimulant may be defined as a substance that increases the activity of the central nervous system (CNS). Most stimulants function by increasing the synaptic concentrations of catecholamine neurotransmitters – namely dopamine 17, epinephrine 103, and norepinephrine 104.282 Produced by adrenal glands, catecholamines act as signaling molecules to activate the sympathetic nervous system. Increases in synaptic catecholamine levels are primarily achieved via blocking their reuptake or stimulating their efflux, however there are notable examples of stimulants with more indirect modes of CNS activation. As a result, a description of the physiological targets of natural products described in this section is provided alongside the individual stimulant. Despite disparate mechanisms of achieving their effects, all known natural product stimulants are alkaloidal in nature (Fig. 30). Alkaloids are commonly defined as molecules possessing one or more basic nitrogen atoms; this chemical property facilitated early isolation via acid–base extraction, making alkaloidal stimulants some of the first natural products to undergo biosynthetic investigation.
Fig. 30.
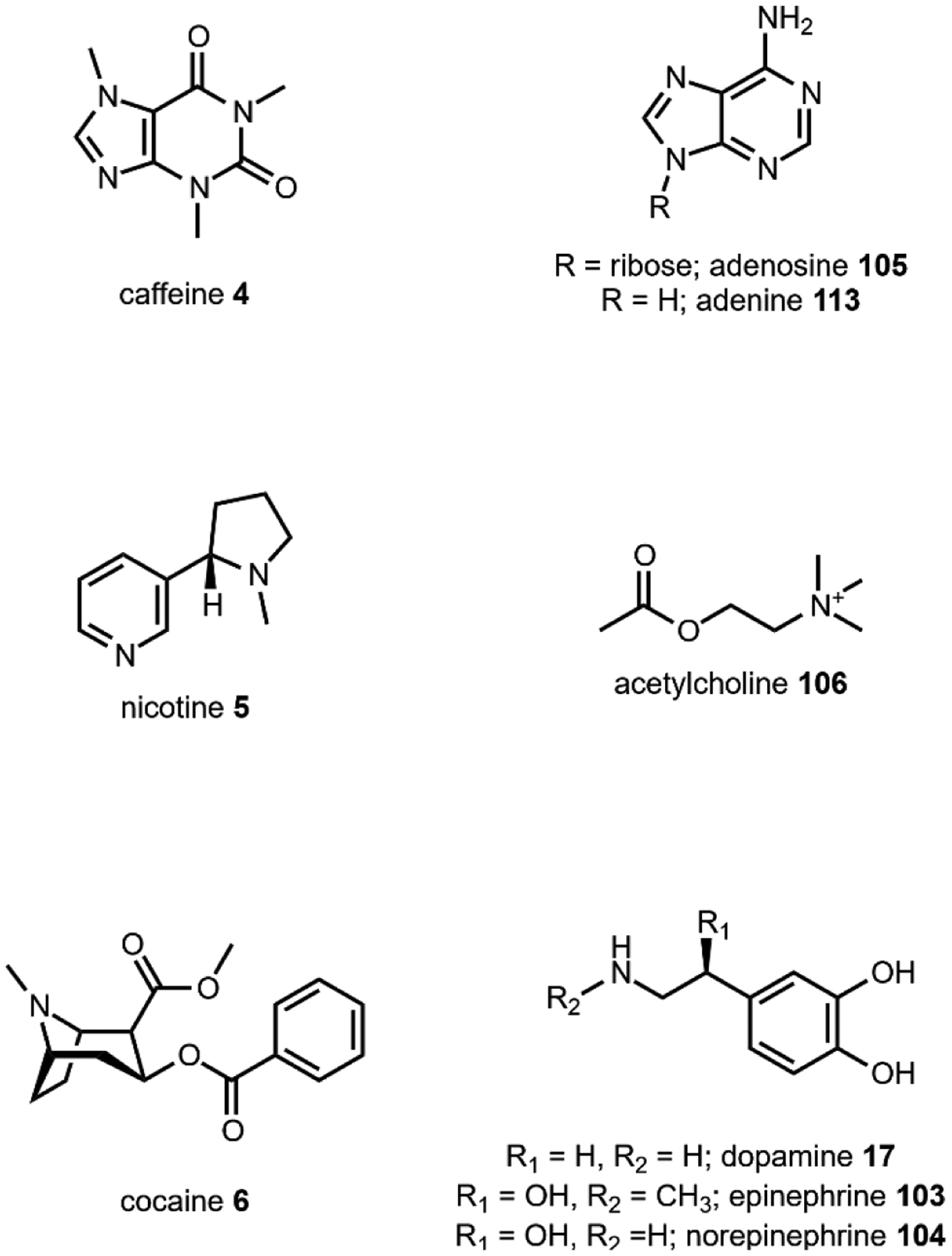
Alkaloidal stimulants as structural mimics of neurotransmitters.
3.2. Caffeine
In addition to early reports of human consumption of caffeine 4 in the Yunnan Province of China, caffeine containing plants were independently discovered in Africa and South America, where they were consumed for their energizing, anti-fatigue effects. Caffeine belongs to the purine alkaloid (PuA) family of natural products, which are defined by their 3,7-dihydropurine-2,6-dione core. Despite their structurally simplicity, at least 80 plant species in 13 orders of the kingdom are known to produce PuAs, indicating important biological function.283 Bitter in taste PuAs are primarily involved in plant defense as an antifeedant, comprising between 1–3 percent dry weight in most producing organisms.284,60 Additional research suggests that caffeine may function as an allelopathic signaling molecule,285 or even a conditioning molecule to train plant pollinators.286 In humans, PuAs work as antagonists of adenosine A2AG protein-coupled receptors. During the course of the day, adenosine 77 accumulates in the neuronal synapse; subsequent binding results a negative regulation of CNS activity causing drowsiness. As structural analogues of adenosine, PuAs bind tightly (caffeine KD = 2.4 μM) but do not activate adenosine receptors.287 The resulting activation of specific regions of the brain causes accumulation of stimulatory dopamine 17 and acetylcholine 106, facilitating wakefulness. Other purine alkaloids with varying methylation patterns of the purine heterocycle exhibit variable potencies and include theobromine 107, theophylline 108, and xanthine 109.
3.2.1. Biosynthesis of purine alkaloids
Caffeine 4 was first isolated in 1819 by the German chemist Friedlieb Ferdinand Runge.288 By the end of the century, Fisher devised a synthesis from theobromine 107 (Fig. 32) which employed methyl iodide for base-catalyzed N-alkylation, thus establishing caffeine’s structure and formula.289 Given the widespread occurrence of caffeine across the plant kingdom, the biosynthesis of caffeine and related PuAs has been of interest from both a secondary metabolism and evolutionary perspectives. The biosynthesis has been studied primarily in Camellia sinensis (tea plant) and Coffea arabica (coffee plant). As early as 1962, the feeding of 14C-labeled precursors confirmed that PuAs originate from the primary purine metabolite xanthosine in Coffea.290 Direct evidence for the conversion of xanthosine 110 to 7-methylxanthosine 111 was first shown by Negishi et al. using plant extracts.291 Elucidation of the subsequent hydrolysis step by a nonspecific N-methyl nucleosidase was frustrated by contaminating nucleosidase activity in crude enzyme extracts, but eventually confirmed using advanced chromatography methods.292 Finally, tedious preparation of tea leaf enzymatic extracts in 1975 provided direct evidence for the transfer of methyl groups from SAM in the conversion of 7-methylxanthine 111 via theobromine 107 to caffeine 4.293 Development of methods for recombinant protein production enabled Ashihara, Fujimura, and others to provide conclusive in vitro evidence for the biosynthetic route from xanthosine shown in Fig. 32A, with the genes encoding the responsible enzymes identified in both coffee and tea.294,295
Fig. 32. Caffeine biosynthesis and microbial engineering strategies.
(A) Major caffeine biosynthetic route identified in Camellia sinensis and Coffea arabica. (B) SAH-derived adenosine may be funneled into purine metabolism in tea leaves following methyl transfer. (C) Xanthine recycle pathway utilized during heterologous production in yeast. (D) Novel xanthine-to-caffeine conversion pathway leveraged for caffeine production in E. coli.
Several routes to the primary metabolite xanthosine 110 have been elucidated, however efficient incorporation of adenine 113 implicated adenosine monophosphate (AMP) 114 as a prominent source of purine equivalents.296 Caffeine production from AMP 114 begins with deamination to inosine monophosphate 115, oxidation to xanthosine monophosphate 116, and hydrolysis to xanthosine 110 by AMP deaminase (AMPD), IMP dehydrogenase (IMPDH), and 5ʹ-nucleotidase (XMPN), respectively.297 The resulting xanthosine 110 is methylated by a xanthosine methyltransferase (XMT) and hydrolyzed by N-methylnucleosidase (NS) to give 7-methylxanthine 112. Iterative methylation of 112 in tea has been confirmed by isolation of a caffeine synthase (CsTCS1) exhibiting both N3 and N1 methylation activity.294 Orthologous genes in coffee have been identified which exhibit either theobromine synthase (CaMXMT1) or caffeine synthase (CaDXMT1) activity, using 112 and 107 as a substrates.298,299
In addition to the major pathway described above, caffeine biosynthesis evolved independently at least five times during flowering plant history, a striking example of convergent evolution towards a secondary metabolite.300 Analysis of the enzymes recruited by distantly related plants to carry out identical reactions has provided strong evidence for the “patchwork hypothesis” as a model to describe pathway evolution. Additional studies aimed at unravelling pathway regulation in the plant have given further insight into the “provider pathways” used by plants to increase xanthosine 110 pools. In 2001, Koshiishi et al. unexpectedly observed incorporation of SAM-derived adenosine 105 into the purine ring using cell free extracts of tea leaves.301 As shown in Fig. 32B, SAH-equivalents released upon substrate methylation with SAM could be funneled into purine metabolism, providing an alternative pathway to the well-established de novo adenosine production routes. Alternative guanosine recycling pathways have also been identified via incorporation of [8-14C]guanosine.297 Sub-cellular localization of the caffeine biosynthetic pathway has also been examined. Like many plant secondary metabolites, caffeine accumulates in the vacuole,302 whereas several enzymes involved in the biosynthesis associate with the chloroplasts303 or cytosol.301
3.2.2. Heterologous production of purine alkaloids
Extensive biosynthetic investigations provided a foundation for numerous efforts in plant and microbial engineering, facilitating the creation of caffeine (and caffeine-free) biotechnologies. Knockdown of the CaMXMT1 encoding theobromine synthase using RNA interference resulted in a 70% reduction of caffeine content, highlighting the possibility to circumvent costly decaffeination protocols using genetic engineering of Coffea.304 Recent efforts in microbial engineering for de novo production of xanthine alkaloids have also garnered moderate success, with benchmark titers of 0.27 mg/L and 21 mg/L in S. cerevisiae and E. coli respectively.92,89 In both studies, low levels of endogenous xanthosine represented a key hurdle that was approached using two different methods. McKeague et al. devised a xanthine 109 salvage pathway in yeast, using xanthine phosphoribosyltransferase (XPT) to revert flux towards 116 (Fig. 32C). A combination of genomic integration and low copy expression of the biosynthetic and salvage pathways using strong constitutive promoters provided maximum caffeine titers of 0.031 mg/L following 6 days of culture. In the same study, a key observation was made that xanthine could be accepted by caffeine synthase, which enabled construction of a theophylline production strain. Bench scale fermentations of customized strains permitted improved production titers of caffeine 4 (0.27 mg/L), theophylline 108 (0.06 mg/L), and 3-methylxanthine 117 (3.71 mg/L).
In E. coli, a xanthosine-to-caffeine conversion pathway was leveraged, taking advantage of background xanthine methylation activity exhibited by the CsTCS1 (Fig. 32D). Li et al. employed plasmid-based expression using inducible promoters to enhance xanthine and SAM biosynthesis.89 Following bioprospecting, codon optimization, and media optimization, a 4-day shake flask culture enabled production of caffeine at 21 mg/L. Despite these efforts, microbes lack the optimized flavor profiles and titers of caffeine plant products. In each of these studies, however, accumulation of monomethylated xanthines was observed, indicating the potential for metabolic engineers to produce valuable pathway intermediates of low natural abundance.
3.3. Nicotine
The pyridine alkaloids (PyAs) are comprised of the highly-addictive stimulant nicotine 5, along with the structurally related anabasine 118 and nornicotine 119 (Fig. 33 and 34). Nicotine 5 is produced by numerous members of the Solanaceae (nightshade) family of flowering plants, and like the xanthine alkaloids, pyridine alkaloids are bitter antifeedants. In fact, the nicotine scaffold served as inspiration for the controversial neonicotinoid insecticides, the use of which has been linked to honey-bee health and colony collapse disorder.305,306 Most of the nightshades, including potatoes, tomatoes, and eggplant, produce PyAs in trace amounts (~0.00001 percent dry weight);307 selective breeding has been used to generate tobacco cultivars containing up to 3.0 percent dry weight nicotine.308 Discovered by the native people of Mesoamerica and South America, tobacco was traditionally used in spiritual ceremonies as well as for its medicinal properties, owing to its analgesic effects when smoked.128 Binding of nicotine to nicotinic acetylcholine receptors results in activation of the mesolimbic pathway and subsequent release of dopamine; at higher concentrations, the activity shifts from stimulant to sedative via dampening of neural activity.309 Recent evidence suggests that cotinine, a metabolic degradation product of 5, is responsible for at least some of tobacco’s psychoactive effects.310
Fig. 33.

Nicotiana tabacum leaves contain 2 to 8 percent dry weight nicotine.
Fig. 34. Summary of the nicotine biosynthetic pathway, including known and proposed enzymatic steps.
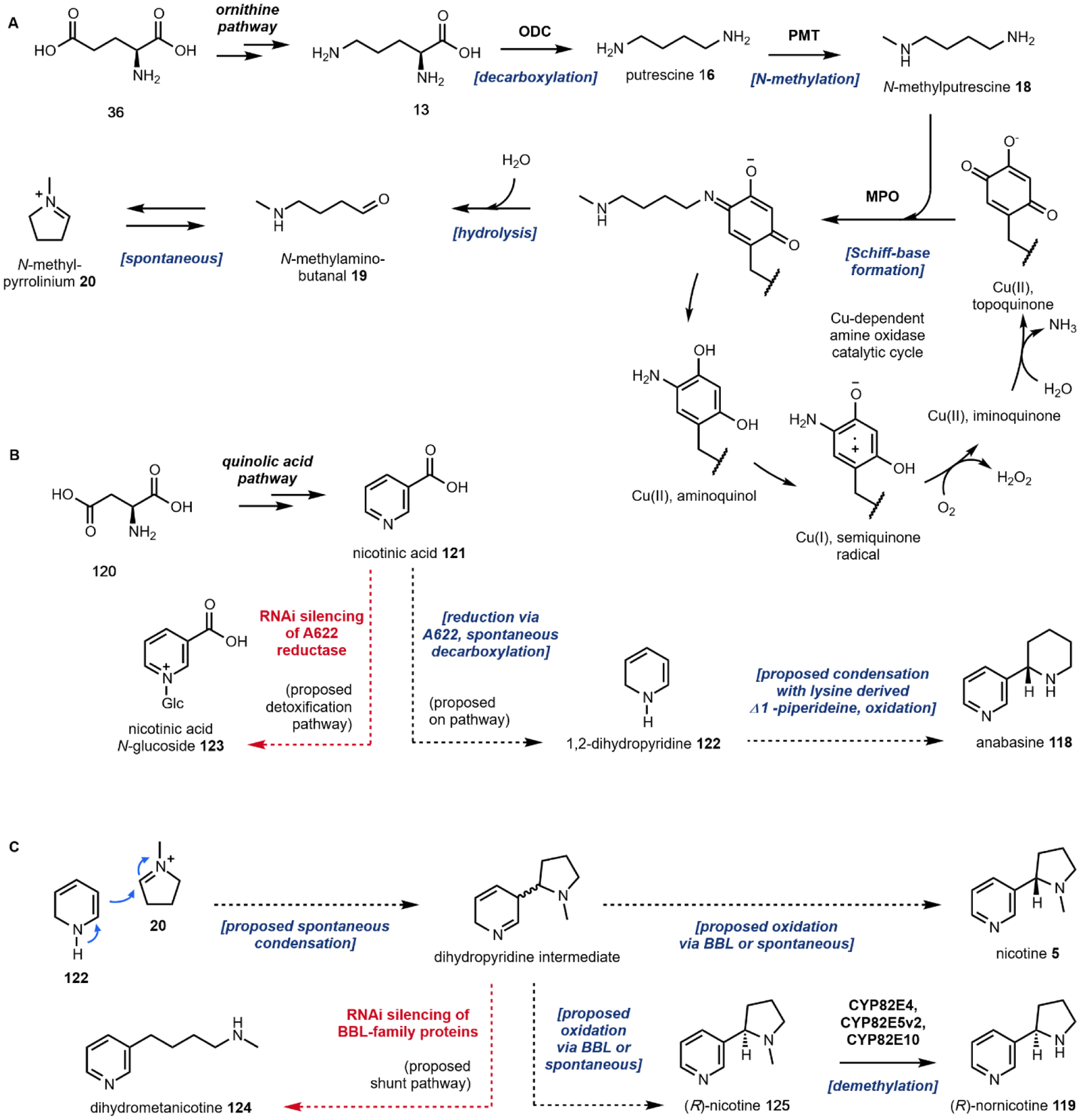
(A) N-methylpyrrolinium formation via the polyamine pathway. (B) Proposed reduction of nicotinic acid via A622. (C) Proposed oxidation of condensation products via BBL towards nicotine, nornicotine.
3.3.1. Biosynthesis of pyridine alkaloids
Initially isolated as tobacco’s active constituent in 1828, nicotine was structurally characterized in 1893 and synthesized by Pictet in 1904.311 Efforts to isolate nicotine and oxidation products by Weidel also led to the discovery of an aspartatic acid 120-derived nicotinic acid (niacin, 121), precursor to the universal cellular redox currency nicotinamide adenine dinucleotide cofactors.312 Investigations into the biosynthesis commenced rapidly in the late 1950s with the discovery that metabolic precursors could be incorporated into nicotine via sterile root cultures. Tandem feeding studies by Byerrum et al. indicated that 2-14C-labeled l-glutamic acid313 36 and l-ornithine314 13 were incorporated into two different positions of nicotine 5, sparking interest in a proposed “symmetrical intermediate.” Leete was the first to propose the diamine putrescine 16, derived via l-ornithine decarboxylation, as a pyridine alkaloid precursor.315 Later studies confirmed incorporation of putrescine 16,316 N-methylputrescine 18,317 and N-methylpyrrolinium 20.318 Tamaki and coworkers subsequently identified putrescine N-methyltransferase319 and N-methylputrescine oxidase320 activity in tobacco roots, confirming the pathway to 20 shown in Fig. 34.
Genes in tobacco encoding the responsible enzymes were later identified by comparison to a low-nicotine mutant and cloned into E. coli for functional characterization.321 l-Glutamic acid 36 derived l-ornithine 13 is first decarboxylated by an ornithine decarboxylate (ODC), which is subsequently methylated by putrescine N-methyltransferase (PMT). Sequence analysis indicates that PMTs are closely related to spermidine synthases (SPDSs), which utilize decarboxylated SAM as the coenzyme to transfer an aminopropyl group onto putrescine. PMTs likely evolved via gene duplication and neofunctionalization, as mutational studies indicated that changing only a few amino acids resulted in the generation of PMT activity in SPDS proteins.322 Next, oxidative deamination of N-methyl putrescine 18 to N-methylaminobutanal 19 is carried out by N-methylputrescine oxidase (NMO), which was functionally verified using E. coli expression.323 NMO belongs to the superfamily of copper-containing amine oxidases, which employ a covalently bound topaquinone (TPQ) cofactor generated via autooxidation of a conserved tyrosine residue. (Fig. 34A) The Cu-TPQ complex enables radical-based oxidative deamination consisting of two steps. Following formation of a Schiff base between the TPQ and substrate amine, proton abstraction and hydrolysis result in aldehyde release. Then, reduced TPQ is reoxidized with molecular oxygen through two sequential single electron transfers via a Cu(I)-semiquinone radical intermediate, releasing H2O2 and NH4+. A homodimer, NMO exhibits 7-fold greater catalytic efficiency towards 18 compared to 16, achieved via a substantial decrease in KM. The NMO product N-methylaminobutanal 19 spontaneously cyclizes to give the Schiff base N-methylpyrrolinium cation, 20.
Surprisingly, the identity of the final stereospecific enzyme in the pathway, nicotine synthase, has yet to be elucidated. Little progress has been made since Frieson and Leete demonstrated formation of nicotine from N-methylpyrrolinium cation 20 and [2-3H]-labeled-121 using crude extracts.324 The loss of the C-6 hydrogen suggested a hydride mediated formation of the 3,6-dihydronicotinic acid, which would readily decarboxylate to give the 1,2-dihydropyridine 122 (Fig. 34B). Hashimoto and coworkers utilized RNAi knockdown of the A622 gene in tobacco (belonging to the PIP family of NADPH-dependent reductases), which resulted in the decrease in the formation of nicotine and accumulation of nicotinic acid N-glucoside 123, a presumed detoxification product.325 While additional studies have confirmed the involvement of A622 in nicotine biosynthesis, more biochemical evidence is needed to ascertain its catalytic function.
Following reduction of aspartic acid 120 derived niacin 121, a proposed nucleophilic attack of N-methylpyrrolinium 20 by 122 is believed to occur either spontaneously or enzymatically in a Mannich-like reaction, providing the dihydropyridine precursor to the final product nicotine 5. Subsequent RNAi targeting of a vacuolar berberine bridge enzyme-like (BBL) protein resulted in accumulation of a new nicotine-related metabolite, dihydrometanicotine 124 (Fig. 34C).326 Direct conversion of the ring-open 124 by the BBL protein would explain the enantiomeric purity of (S)-nicotine 5 (whereas the (R)-enantiomer only accounts for just 0.2% of the total nicotine). However, in experiments using recombinant BBL protein as well as crude tobacco cell extracts, oxidative conversion of 124 was not observed. Enantioselective demethylation of (R)-nicotine 125 by several P450s (CYP82E4, CYP82E5v2, and CYP82E10) has been postulated to explain how tobacco maintains the (S)-nicotine 5 and (R)-nornicotine 119 pools. Further studies are required in order to definitively establish the identity and mechanism of nicotine synthase, as well as the predicted anabasine 118 synthase.
3.3.2. Heterologous production of pyridine alkaloids
Complete reconstitution of the nicotine pathway in a heterologous host has not been possible due to missing steps in the biosynthesis. However, identification of genes involved in nicotine precursor formation have been used in genome mining and N-methylpyrrolinium platform engineering. Complete pathway elucidation will enable the use of such chassis strains for the production of rare or unnatural pyridine alkaloids. Now an undisputed model organism and biotechnological chassis, Nicotiana plants have been extensively engineered for applications ranging from production of biopharmaceuticals to heterologous natural product biosynthesis.327,328 Synthetic biology tools analogous to those developed for microbial engineering have been extended to N. tabacum and N. benthamiana; the future of such Nicotiana “plant biofactories” has been recently reviewed.329 Identification and silencing of the nicotine N-demethylase using RNAi led to suppression of P450-mediated conversion of (R)-nicotine 125 to the carcinogenic nornicotine 119.330 Recently, the CRISPR-mediated simultaneous knockout of the BBL-family protein and five additional isoforms was shown to reduce the amount of nicotine produced in tobacco by >99%, providing additional evidence for the involvement of this BBL protein in nicotine formation.331 Optimizing the alkaloid profiles of “nonfood” industrial crops such as tobacco expands the capabilities of an additional chassis for industrial chemical manufacture.
3.4. Cocaine
Tropane alkaloids encompass the third class of nitrogen-containing stimulants discussed, and are defined by their characteristic 8-methyl-8-azabicyclo(3.2.1)octane (“tropane”) ring system. While just a few tropane alkaloids are known to be psychoactive, this family of molecules includes the well-known illicit stimulant cocaine 6. Named for Erythroxylum coca (endogenous to South America, Mexico, Indonesia, and the West Indies), cocaine use can be traced to indigenous populations in the Andes. As early as 1000 BC, the leaves of coca were chewed for religious and medicinal purposes. At present, Columbia is the largest producer of cocaine; hundreds of metric tons are extracted and exported annually.332 The product may be refined and distributed as the cocaine hydrochloride salt, or neutralized and distributed as the inhalable free base, a subtle difference which has resulted in extreme sentencing discrepancies.333 Despite its current Schedule II status in the United States, a myriad of medicinal uses for cocaine have been described, and in 2020 a cocaine hydrochloride formulation was approved for use as a topical anesthetic. The principal mechanism of action is to block monoamine transporters, resulting in the accumulation of dopamine 17 in the synaptic cleft and strong sympathomimetic effect.334 In addition to cocaine, the tropane alkaloids include the hallucinogenic scopolamine 126 as well as catuabines and calystegines.
3.4.1. Biosynthesis of tropane alkaloids
Tropane structure and biosynthesis has been a topic of intense investigation for over a century. Structural confirmation of the tropane core in cocaine accompanied Willstätter’s lengthy synthesis, which warranted a 1915 Nobel Prize in Chemistry.335 Two years later, Robinson published a one-pot tropinone formation by the addition of succinaldehyde to an acetone-dicarboxylic equivalent, which is widely regarded as the first biomimetic synthesis.13 This elegant method stimulated Robinson’s 1955 proposal of an analogous biosynthesis involving condensation of a pyrrolidine ring with an “acetone equivalent.” Indeed, work by Leete confirmed incorporation of l-ornithine 13 into tropanes,336 establishing a pathway identical to the N-methylpyrrolinium 20 formation described in nicotine biosynthesis. Feeding of labeled acetate and advanced intermediates hinted that condensation of 20 with malonate units occurs via a polyketide synthase (PKS).337 Subsequent advancements in molecular biology and genomics have rapidly facilitated the near complete pathway elucidation and engineering of tropane alkaloids.
Using differential transcriptomics of plant tissues, Bedewitz et al. identified a type III PKS (AbPYKS) expressed in the roots of Atropa belladonna, confirming its involvement in tropinone biosynthesis using virus-induced gene silencing.338 Recombinant expression of AbPYKS in E. coli indicated direct use of the N-methylpyrrolinium cation 20 as a starter substrate prior to incorporation of two malonyl-CoA 105 units, forming 4-(1-methyl-2-pyrrolidinyl)-3-oxobutanoic acid 106 (Fig. 36). Subsequently, Huang and coworkers proposed an alternative route to 106 following additional crystallographic and mechanistic studies.339 In the absence of 20, AbPYKS was shown to produce 3-oxo-glutaric acid 21; this compound undergoes non-enzymatic condensation with 20 via an intermolecular Mannich reaction, the kinetics of which were unaffected by the presence of AbPYKS.339 The resulting racemic 128 is thought to be the divergence point between the tropinone 129 pathway (leading to scopolamine – resolved, Fig. 37) and methylecognone 130 pathway (leading to cocaine – unresolved, Fig. 38).
Fig. 36.
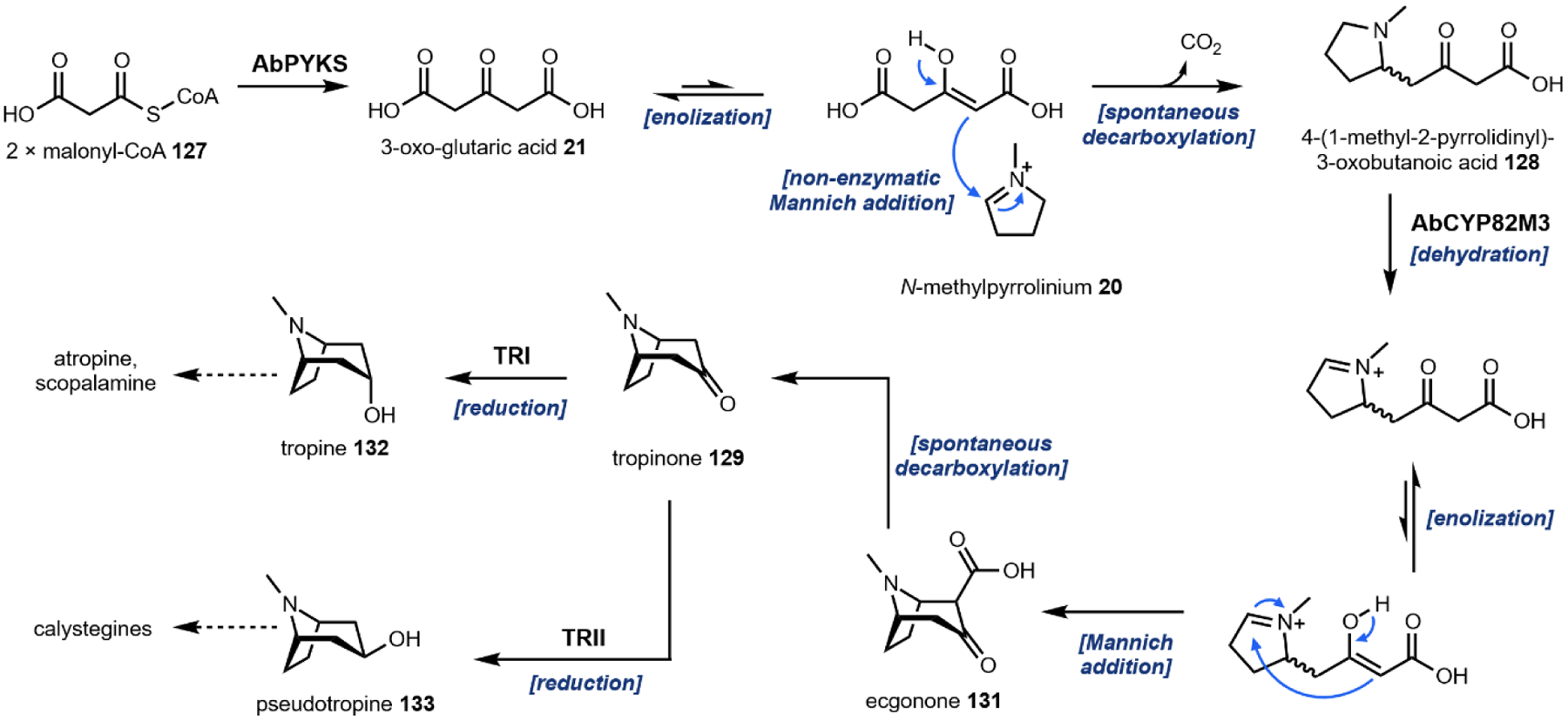
Formation of tropine, pseudotropine.
Fig. 37.
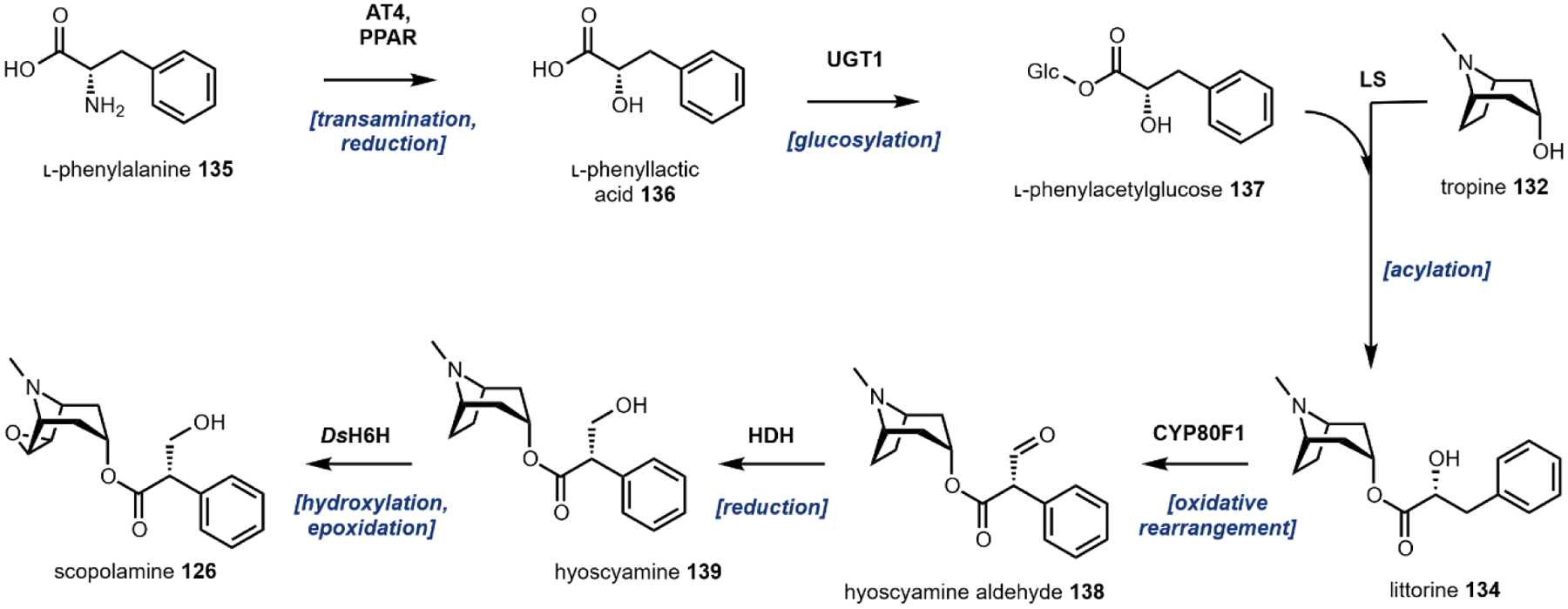
Scopolamine biosynthesis from phenylalanine and tropine.
Fig. 38. Cocaine biosynthesis.
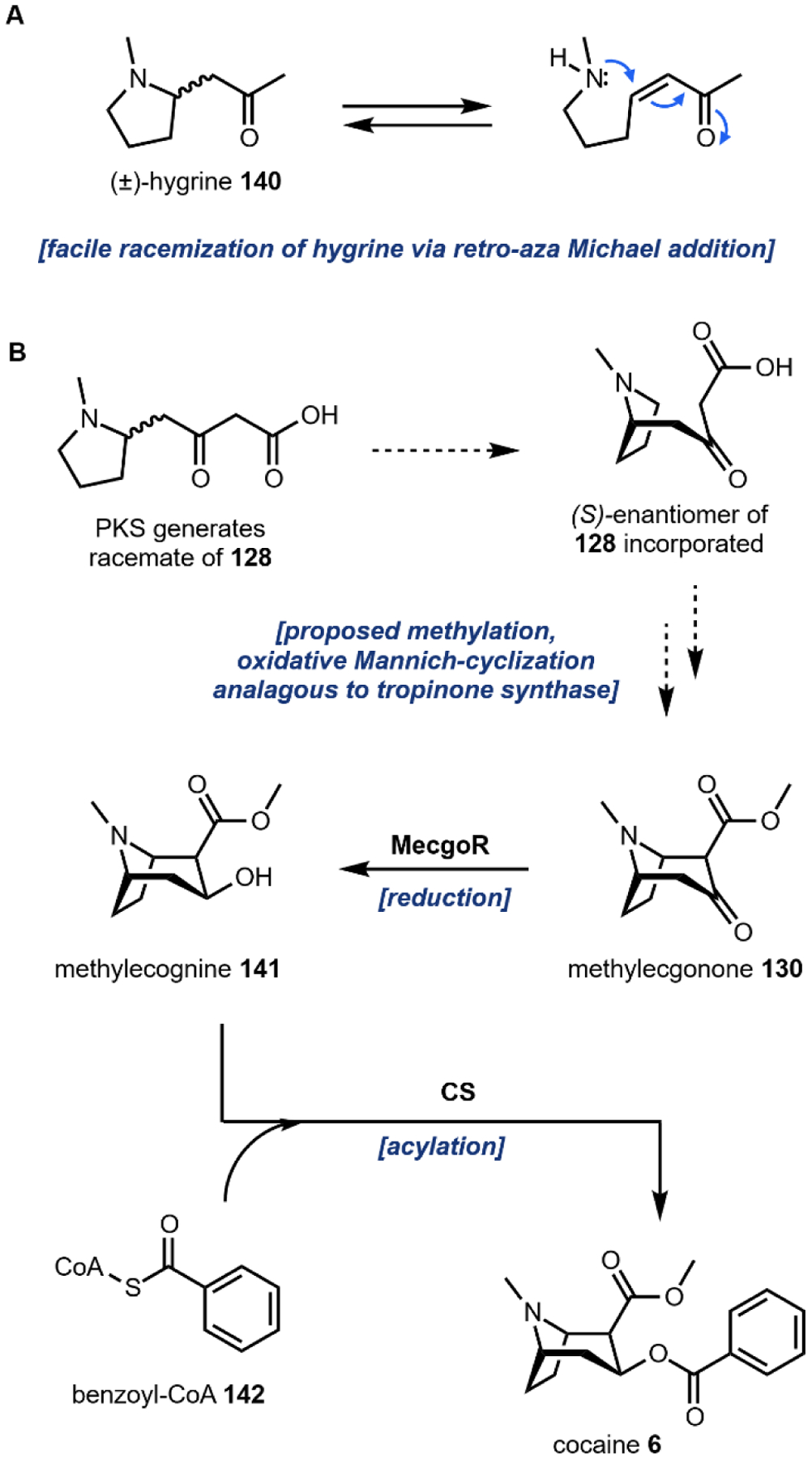
(A) Racemization of the cocaine pathway intermediate decarboxylation product hygrine. (B) Proposed biosynthesis of methylecognone and subsequent formation of cocaine.
Bedewitz et al. also hypothesized that a P450 may be responsible for the cyclization of nascent 128 via amine oxidation. Pathway reconstitution of candidate P450s identified via transcriptomics indicated that AbCYP82M3 encodes a tropinone synthase (TS), which was directly confirmed by conversion of 128 to 129 using yeast microsomes.338 The proposed mechanism involves hydroxylation and dehydration of the pyrrolidinyl to generate the pyrrolinium intermediate. Oxidation of 128 sets up the intramolecular Mannich cyclization to produce ecgonone 131, establishing the tropane skeleton; subsequent nonenzymatic decarboxylation produces 129. As discussed in Section 1.2.2, iminium formation and intramolecular Mannich-cyclization is a common cascade observed in the biogenesis of diverse plant alkaloid scaffolds.340 Two different tropinone reductases (TPI and TPII) were identified in Datura stramonium of high sequence identity (64% identity), each performing stereospecific reduction of 129 to either tropine 132 (TPI) or pseudotropine 133 (TPII), the precursor to the calystegines.341
The phenylacetate unit required for littorine 134 biosynthesis is derived from phenylalanine 135, which is transaminated by an aromatic amino acid aminotransferase (AT4)342 and reduced by a phenylpyruvic acid reductase (PPAR)343 to provide phenyllactic acid 136. This compound is subsequently glucosylated by phenyllactate UDP‐glycosyltransferase (UGT1).344 The resulting phenylacetylglucose 137 is then used by littorine synthase (LS) to acylate 132, forming littorine.344 The longstanding mystery around rearrangement of littorine was solved in 2006, wherein 134 was converted into hyoscamine aldehyde 138 by CYP80F1 via a benzylic carbocation intermediate.345,346 A recently identified hyoscyamine dehydrogenase (HDH) then reduces 138 to hyoscamine 139 followed by epoxidation catalyzed by an α-ketoglutarate-dependent hydroxylase/dioxygenase (DsH6H) to complete the biosynthetic pathway to scopolamine 126.73
The majority of the pathway towards cocaine 6 has been established, with the exception of the enzymes responsible for production of the precursor methylecognone 130. Evidence suggests a sequence analogous to tropinone 129 formation starting from a PKS product. During tropinone 129 biogenesis, the spontaneous decarboxylation following cyclization permits the use of either stereoisomer of 128. The retention of the carboxymethyl in the methylecognone 130 scaffold, however, necessitates incorporation of the (S)-enantiomer. The decarboxylation product of 128, hygrine 140, is known to racemize rapidly at physiological conditions. The proposed mechanism involves a retro-aza-Michael addition (Fig. 38A). Stereospecific incorporation of (S)-128 into cocaine may involve selective methylation and cyclization, facilitated by spontaneous or enzyme catalyzed stereoinversion of (R)-128. A proposed methylation of (S)-128 followed by a P450-mediated Mannich-cyclization by an enzyme homologous to tropinone synthase would yield the confirmed on pathway metabolite methylecognone 130. Product methylation is believed to take place before cyclization, otherwise rapid decarboxylation of the putative β-keto acid would occur. This hypothesis is supported by a feeding study in which a low but observable amount of the methyl ester of 128 painted on coca leaves was incorporated into cocaine.347 Following cyclization, methylecognine 141 is formed via methylecognine reductase (MecgoR).348 MecgoR belongs to the aldo-keto reductase family of enzymes, indicating tropine ester formation evolved independently in E. coca and A. belladonna. The final enzyme, cocaine synthase, is a BAHD acyltransferase which condenses methylecognine with activated benzoyl-CoA 142.349
3.4.2. Heterologous production of tropane alkaloids
Extensive engineering efforts by Srinivasan and Smolke allowed for the first reported de novo production of hyoscyamine 139 (10.3 μg/L) and scopolamine 126 (0.87 μg/L) in yeast (Fig. 35).73 This synthetic biology achievement builds upon previous works to reconstitute segments of the tropane alkaloid biosynthetic pathway in E. coli and yeast.108,350,351 The fully integrated yeast strain contains 26 additional genes from yeast, E. coli and five different plants along with disruption of 8 native yeast genes for a total of 34 chromosomal modifications (Fig. 39). The authors organized the biosynthetic pathway with five modules, each comprised of a distinct pathway segment.
Fig. 35. Erythroxylum coca leaves contain ~0.7 percent dry weight cocaine.
Image on left courtesy of Danna Lizeth Guevara Prieto via CC-4.0.
Fig. 39.
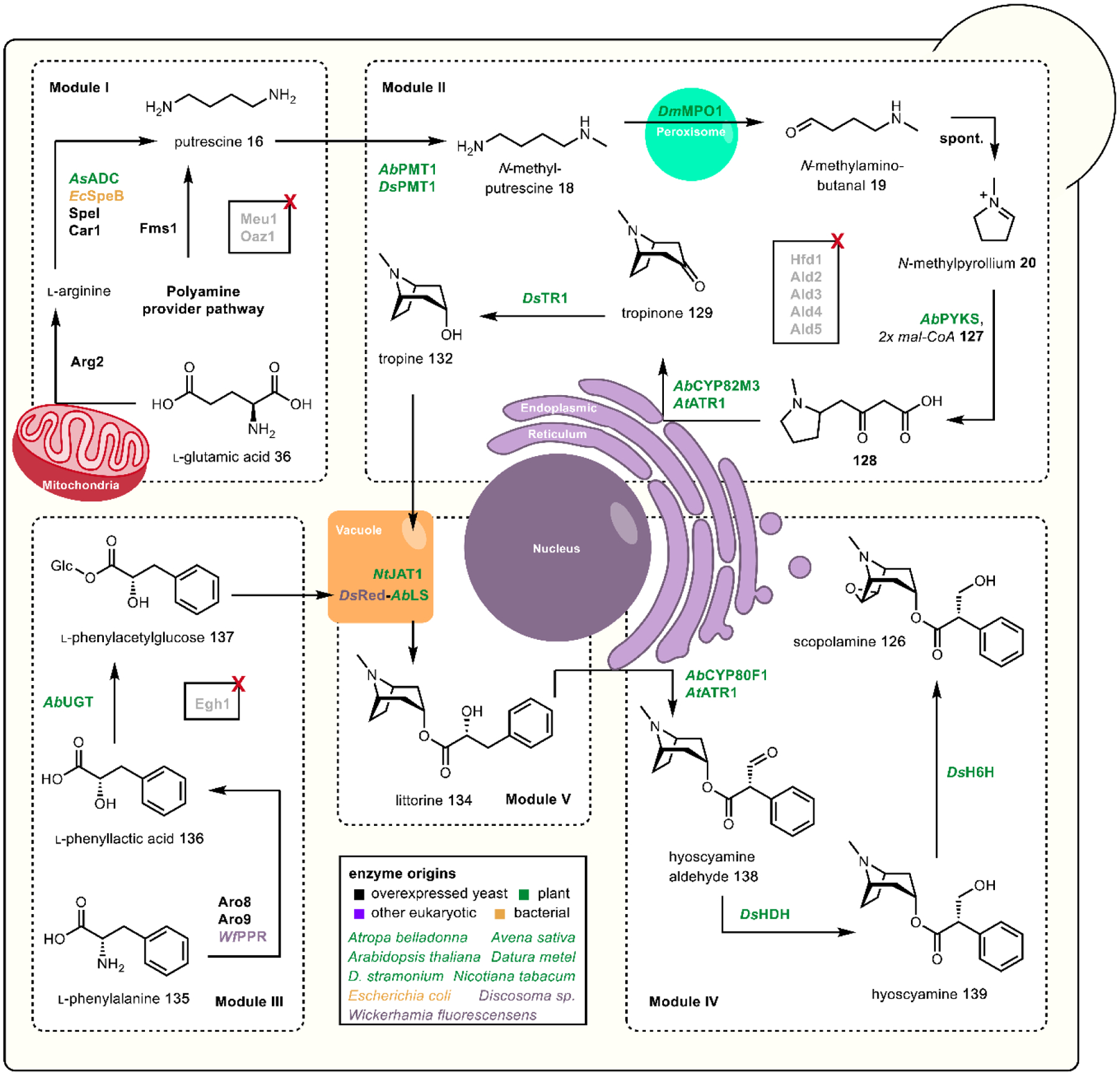
Production of tropane alkaloids in yeast.73
Module I is dedicated to putrescine 16 production and contains heterologous plant (AsADC) and bacterial (SpeB) putrescine pathway genes as well as additional copies of native yeast putrescine biosynthesis genes (Arg2, Fms1, Car1, Spe1) to maximize putrescine 16 accumulation. The authors also disrupted two yeast genes MEU1 and OAZ1 involved in off-pathway polyamine formation that reduce putrescine 16 accumulation. Module II then contains the genes encoding for the enzymes required to transform putrescine 16 into tropine 132 along with disruptions of five endogenous aldehyde dehydrogenases (Ald2–5 and Hfd1) that were previously determined to decrease N-methylaminobutanal 19 titers.108 These two modules were a part of the platform strain from previous work by Srinivasan et al. that were leveraged to produce the non-canonical tropane alkaloid, cinnamoyltropine, from the acyl donor cinnamoyl-CoA.351 This acyl donor is also used in the biogenesis of the polyketide-derived kavalactones, which are the anxiolytic sedatives found in the kava plant, Piper methysticum.98
The next module, Module III, contains the genes required for biotransformation of phenylalanine 135 into the acyl donor, phenylacetyl glucose 137. The pathway intermediate phenyllactic acid 136 is likely produced non-specifically by action of an endogenous yeast lactate dehydrogenase. However, the authors determined that expression of a phenylpyruvic acid reductase from the fungus Wickerhamia fluorescens increased phenyllactic acid 136 titers by nearly 80-fold. Yeast glucosidase Egh1 was disrupted to prevent hydrolysis of the heterologous glucoside, phenylacetyl glucose 137.
Module IV contains the genes encoding for enzymes to transform the TA scaffold into medicinal alkaloids, hyoscyamine 139 and scopolamine 126, including a newly identified hyoscyamine dehydrogenase (HDH) that was discovered by manually screening 12 putative HDHs genome mined from available A. belladonna transcriptome datasets. The final module, Module V, contains genes that encode for the vacuole transporter NtJAT1 and an engineered, chimeric AbLS to form littorine 134 in the yeast vacuole. Initial expression of AbLS resulted in growth defects and no activity in vivo which the authors attributed to difficulties in post-translational processing stemming from differences in glycosylation pattern recognition and transport factors between yeast and plants. Srinivasan et al. determined AbLS may be stalled in the secretion pathway upstream of the trans-Golgi network based on an N-terminal signal peptide and designed a chimera with DsRed linked to the N-terminus of AbLS to mask said signal peptide. This modification allowed the chimeric AbLS to be properly sorted to the yeast vacuole and restored activity in vivo. To allow ample supply of tropine 132 and phenyllactic acid glucoside 137 into the vacuole to access the sorted AbLS, several vacuole transporters from plants were tested and expression of the transporter NtJAT1 resulted in the highest titers of the AbLS product, littorine 134.
Like heterologous production of iboga alkaloids, a yeast-based platform for medicinal tropane alkaloids demonstrates the potential of a more sustainable and reliable pipeline for production. While titers on the microgram scale do not make this platform competitive to current processes for obtaining tropane alkaloids, further host engineering combined with fermentation optimization could result in an economically viable strain. In recent years, sub-cellular localization has been a popular method to greatly improve product titers and cellular fitness via forming enzymatic cascades, accessing rich chemical environments, and sequestering toxic intermediates.105,352,353 Here, the recapitulation of the endogenous plant vacuole sorting and intermediate transport system in yeast is an innovative approach to sub-cellular localization that is a promising strategy that may benefit other yeast systems heterologously expressing complex, spatially-organized plant pathways.
3. Cannabinoids
Cannabis indica, C. sativa (Fig. 40), and C. ruderalis are traditional plants that have been used as medicine, recreationally, and as a fiber for all of recorded history.354 This plant treats epilepsy,355 inflammatory bowel disorder,356 fibromyalgia357 and holds promise in treating cancer,358 psychiatric disorders,359 multiple sclerosis,360 basal ganglia disorders,361 and others.362 Despite this lengthy history and myriad of medicinal applications, the usage of this plant has remained controversial.363 For example, in the 1930s, Harry Anslinger demonized usage of Cannabis for his own political benefit and reshaped the American consensus on the plant.363 Because of this, today, previous colloquialisms such as ‘marijuana’ are currently being depopularized, as the term was used to purposely force negative associations with the Latino community. This is reestablishing the scientific name and “pot,” “weed,” or “bud” as common names.
Fig. 40.
The Cannabis sativa plant typically contains 5–16% tetrahydrocannabinol (7).364
Arguably, the Cannabis plant is unparalleled in morphological and biochemical diversity as well as its gamut of bioactive compounds – producing more than 80 biologically active compounds.354,365 Cannabinoids covered in this section are psychoactive Cannabis plant-based hybrid meroterpenoid natural products, containing terpene and polyketide fragments. These structures all contain a 5-pentylbenzene-1,3-diol (olivetol) substituted with a dimethyl octadiene chain (geranyl) (Fig. 41). The geranyl diene arm undergoes various intramolecular reactions with the polyketide core to form classic cannabinoid tetrahydrobenzochromene, vinyl biphenyl, and related chromene scaffolds.
Fig. 41.
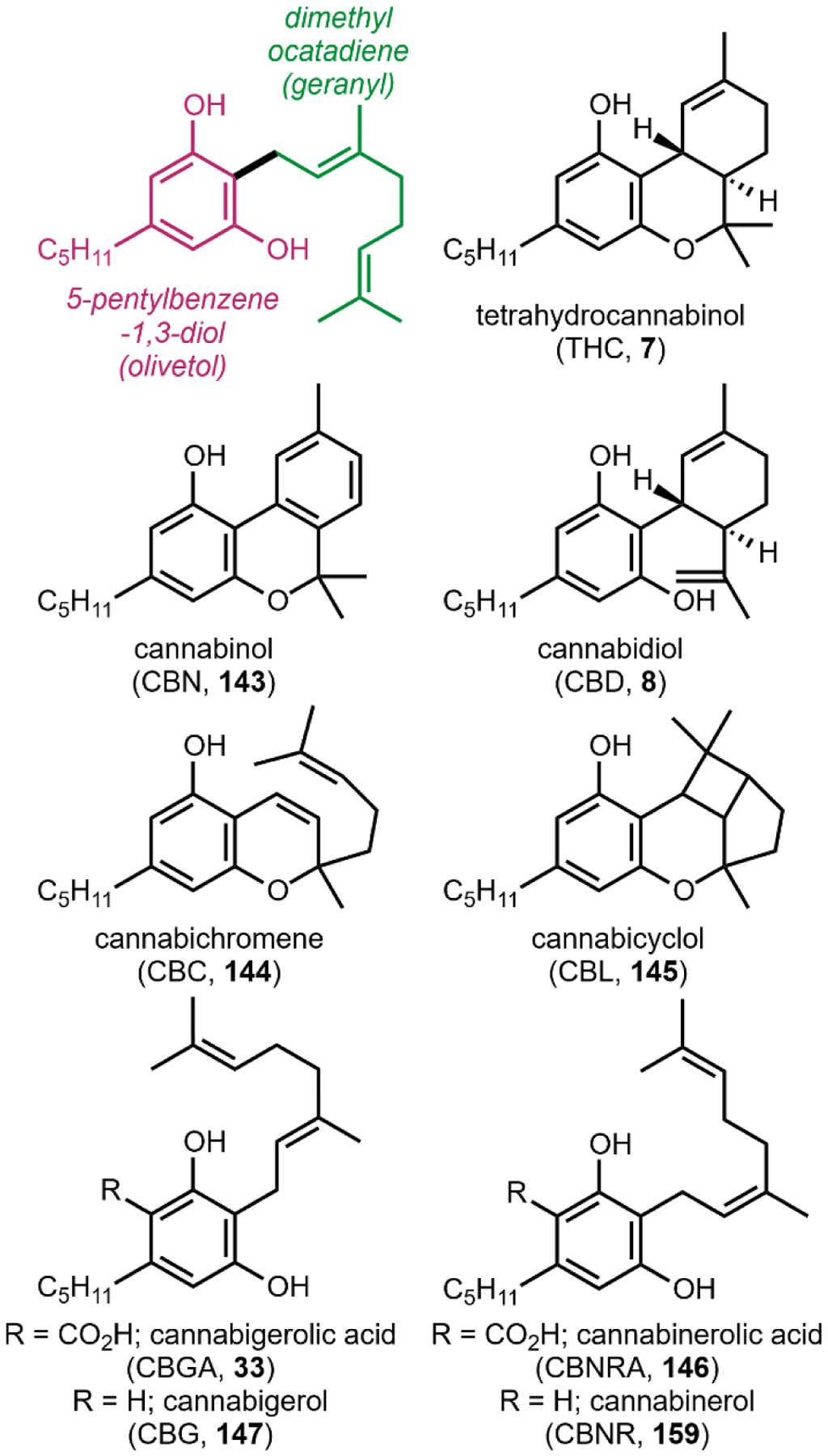
Structural motifs and examples of isolated natural products from the Cannabis plant.
Tetrahydrocannabinol (THC, 7) is the major psychoactive cannabinoid and is metabolized to cannabinol (CBN, 143) (Fig. 41).366 7 and 143 define the classic cannabinoid benzochromene skeleton. Typically, Cannabis plants contain, depending on variety, between 5–16% 7 content, but in some cases can be as high as 18% with a theoretical maximum of 54%.354
Cannabidiol (CBD, 8) has a vinyl biphenyl skeleton and is a non-euphoric compound that is produced in a 1:1 ratio with 7 in C. indica.367 Fascinatingly, 8 is reported to act against adverse effects of 7.368 Some reports refer to 8 as non-psychoactive as it is not intoxicating nor euphoric, but this compound does alter brain behavior369–371 and therefore referred to herein as psychoactive. The pharmacology of 8 is complex as it interacts with many types of receptors and enzymes.372–374
Cannabichromene (CBC, 144) is a natural chromene racemate that functions through non-cannabinoid receptor mechanisms activating the ankyrin transient receptor potential channels 1 (TRPA1).375 144 has also been reported to modulate 7 activity.376 Cannabicyclol (CBL, 145) is the photochemical formal [2+2] cycloadduct of 144 – this process is nonenzymatic and dependent on storing the plant material in light.377 Formation of 145 likely occurs during medicinal and recreational smoking of the Cannabis plant. However, there is no published pharmacological data on 145.
Natural products 7, 8, and 143–145 all derive from cannabigerolic acid (CBGA) 33 and cannabinerolic acid (CBNA) 146. This pathway was originally hypothesized in 1964 upon isolation of the decarboxylated product cannabigerol (CBG, 147).378 Until recently, 147 has not been studied, and like 8, was believed to be non-psychoactive despite modulating multiple receptors.369,379
Previously, the cannabinoid natural product scaffolds were thought to be exclusively produced by the Cannabis plant. With an increase in interest in such compounds and modern characterization techniques, related scaffolds have been discovered in rhododendrons (Rhododendron dauricum), liverworts (Radula perrottetii), indigo bushes (Amphora fruticosa), and even fungi (Cylindrocarpon olidum) (Fig. 42).380–383 The key difference in many of these scaffolds is the alkyl chain substituent changing from an alkyl to an aralkyl (148, 149) or β-aralkyl substituent (150). In other cases, this alkyl chain is truncated (151). Intriguingly, some of these compounds were shown to exhibit similar bioactivities to classical cannabinoids.381
Fig. 42. Exemplary structurally related cannabinoid-like natural products isolated from other plant and fungal sources (italics).
Structural deviations highlighted in red. Amorfrutin 2 (B) (148) is a 148 derivative,380 (–)-cis-perrottetinene (149) is a 7 derivative,381 machaeridiol (150) is a 8 derivative,382 and 6-chloro-cannabiorchichromene (151) is a 144 derivative.383
4.1. Cannabinoid Receptors
Isolation of 7, 8, and 143 and then chemical synthesis of 7 facilitated the discovery and characterization of the G protein-coupled receptors (GPCR) named the cannabinoid receptor type 1 and 2 (CB1 and CB2).384–386 CB1 and CB2 cooperatively function with heterotrimeric G protein alpha subunits (Gi/o) to inhibit adenylyl cyclase activity and activate mitogen-activated protein kinase (MAPK).387 The CB1 and CB2 receptors are involved in achieving homeostasis after exposure to physical or mental stimuli, and therefore are attractive as therapeutic targets to treat various pathologies.388 The CB1 receptor is primarily found in the central nervous system at the terminals of central and peripheral neurons. The location of the CB1 correlates receptor activation with effects on motor function, cognition and memory, and analgesia. The CB2 is found in the immune system cells and affect immune cell migration. These GPCR receptors share 44% sequence homology overall, and 68% homology between transmembrane domains.385 The functional equivalence of CB1 and CB2 is evident in cannabinoids half maximal inhibitory concentration (IC50), where typically these small molecules inhibit the receptors at near similar concentrations (Fig. 43). Synthetic analogues and some natural cannabinoids have been found to selectively potentiate the cannabinoid receptors (Fig. 43).
Fig. 43.
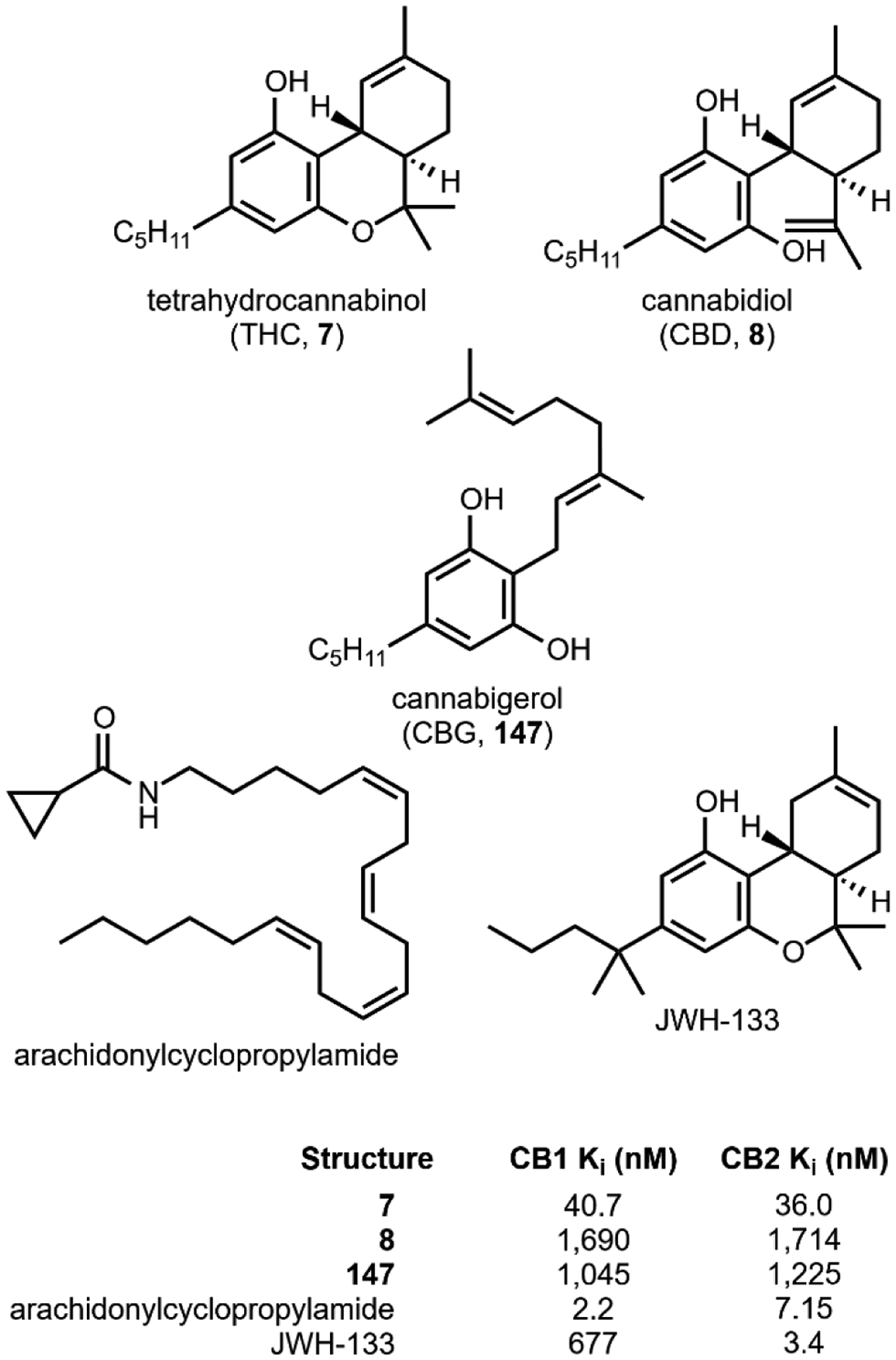
CB1 and CB2 activity for 7, 8, 147, the natural endocannabinoid arachidonylcyclopropylamide, and synthetic analogue JWH-133.
Over the years, the structure-activity relationships between cannabinoids and receptors have been established. A key discovery was that molecule potency is proportional to the C3 chain length.389 Cannabinoid analogues have been isolated and synthesized with C3 alkyl chain lengths ranging from 1–7 carbons; the CB1 and CB2 inhibitory activities of propyl and heptyl-substituted analogs are highlighted in Fig. 44. The propyl-substituted THC and CBD derivatives, tetrahydrocannabivarin (THCV, 152) and cannabidivarin (CBDV, 153), have weaker inhibitory activities as the alkyl chain cannot adequately fill the hydrophobic channel of CB1 and CB2.390–392 This means that THCV and CBDV have a more subtle or even no psychoactive effect, giving these molecules other therapeutic potentials.393,394 Recently, tetrahydrocannabiphorol (THCP, 154) and cannabidiphorol (CBDP, 155) were isolated from C. sativa L. that feature a C3 heptyl-substituent and are currently the most potent natural CB1 and CB2 modulators.395
Fig. 44.
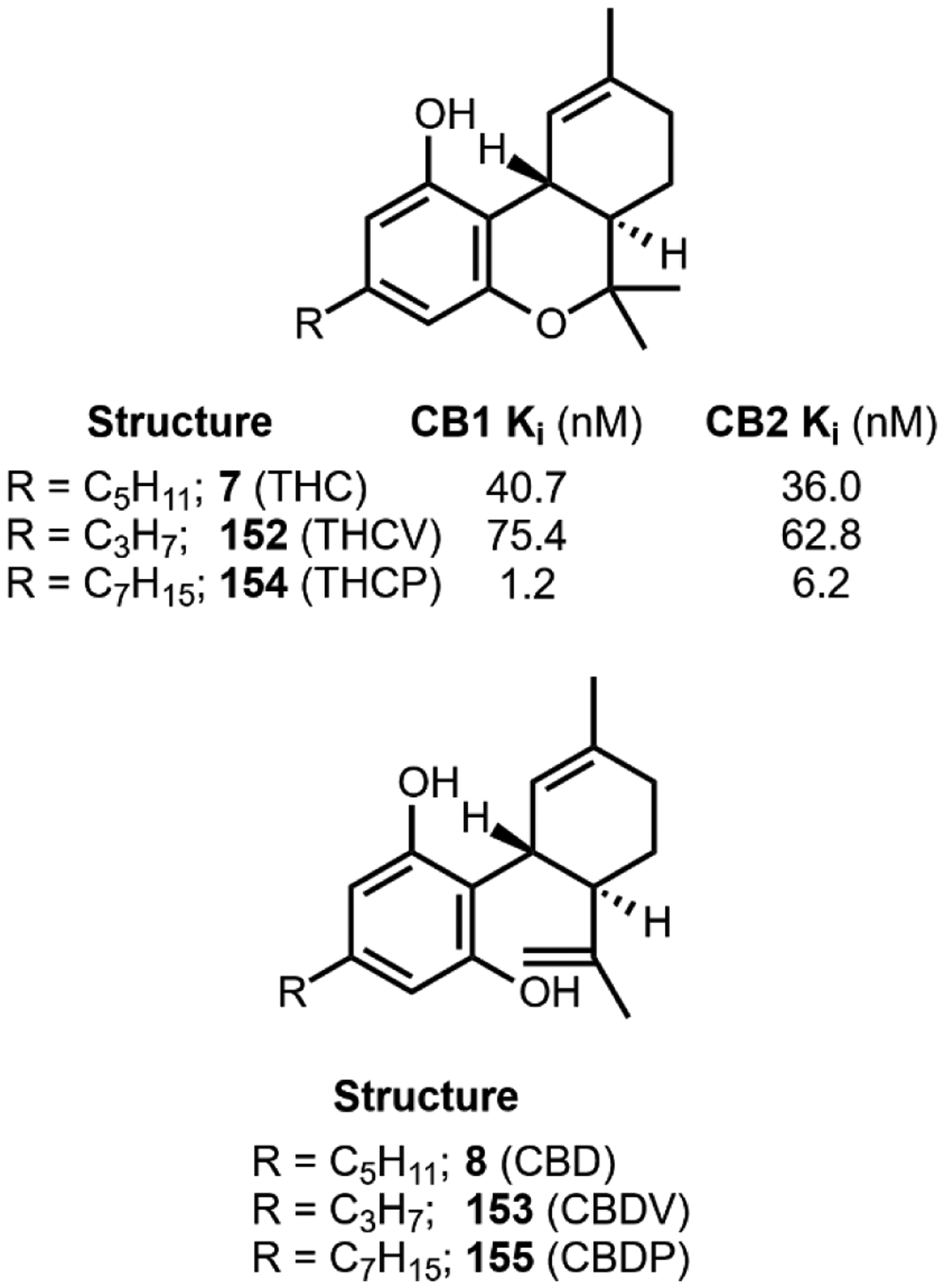
CB1 and CB2 activity of THC (7) with varying C3 alkyl chain lengths, propyl (varin, 152) and heptyl (phorol, 154). CBD alkyl chain length derivatives also shown for clarity.
Shortly after the discovery of CB1 and CB2 as targets of cannabinoids, Mechoulam et al. discovered the entourage effect.396,397 When biologically inactive compounds are administered with THC (7), these ‘entourage’ compounds modulate the observed psychoactivity. This effect is observed in vivo with fatty acid amides, terpenes, cannabinoids, and other compounds.396,398 Before naming this effect, other researchers have noted similar properties, for example a Cannabis extract produced a psychoactive effect two to four fold of pure 7.399 The entourage effect could explain why consumers of Cannabis might prefer to smoke or vaporize the plant material versus taking single, purified compounds.
4.2. Biosynthesis of cannabinoids
Cannabinoid biosynthesis begins with the Claisen and aldol condensations of malonyl- and hexanoyl-CoA (127 and 156) – which is produced by the acyl activating enzyme (AAE1)400 – to form the polyketide olivetolic acid 32 (Fig. 40). Taura et al. discovered a type III polyketide synthase (tetraketide synthase, TKS) and proposed its function in cannabinoid biosynthesis, but at the time were unable to produce olivetolic acid in vitro.401 Later, Page et al. showed that this TKS enzyme cooperatively functions with olivetolic acid cyclase (OAC) to form 32 – this marked the first example of type III PKS and polyketide cyclase acting in concert to form cyclic polyketides in planta.402 The biosynthesis starts with incorporation of the hexanoyl unit as the starter unit for TKS, followed by three rounds of decarboxylative chain extension with malonyl-CoA to form a tetraketide. OAC then catalyzes the Claisen-like cyclization to form the dihydroxybenzene ring, followed by hydrolytic release of 32.
The next step in cannabinoid biosynthesis is the electrophilic addition of a terpene unit to C6 of 32 (also see Fig. 4D). The aromatic prenyltransferase (APT) enzyme403 selectively prenylates C6 of 32 to form either CBGA (33) or cannabinerolic acid (CBNRA, 157).404 These molecules 33 and 157 are (E) and (Z) isomers and are derived from geranyl or neryl pyrophosphate (82 or 158), respectively. The activity of APT is dependent on the carboxylic acid of 32404 as the reaction does not occur with a decarboxylated olivetol substrate. This indicates that the decarboxylation to form cannabigerol 147 and cannabinerol 159 occurs after prenylation. Despite this C2 substituent requirement, APT is actually quite promiscuous and able to accommodate varying alkyl-chains at the neighboring C3 position.75
The diverse psychoactive cannabinoid skeletons all diverge from 33 and 157 as shown in Fig. 41. The aptly named tetrahydrocannabinolic acid synthase (THCAS),405 cannabidiolic acid synthase (CBDAS),406 and cannabichromenic acid synthase (CBCAS)407 catalyze the oxidative cyclizations to form tetrahydrocannabinolic acid (160), cannabidiolic acid (161), and cannabichromenic acid (162) respectively. These compounds can transform nonenzymatically to further generate structural diversity, either at elevated temperatures or in sunlight. Subsequent modifications can lead to the decarboxylated 7, 8 and 144, aromatized 143 and cannabinodiol (CBND, 163), and further cyclized products 145, cannabielsoin (CBE, 164), and isotetrahydrocannabinol (ITHC, 165). There are other known, further functionalized cannabinoid skeletons, but all of them – including 145, 164, and 165 – are proposed to form nonenzymatically due to environmental stimuli. However, many of these molecules have no published pharmacological data. Despite our chemical interest in these structures, we do not speculate on the potential herein.
Biochemical characterization and crystal structures of the cyclization enzymes have revealed the likely mechanism through which 33 is modified into the more advanced cannabinoids.407–409 These transformations are catalyzed by FAD-dependent berberine bridge enzymes (BBEs). Data indicates that upon binding of 33, oxidation likely occurs by an active site tyrosine-484 deprotonating the resorcinol C5 proton followed by FAD-catalyzed dehydrogenation of the exocyclic methylene to form a key quinone methide intermediate 166 (Fig. 47A). Interestingly, enzyme activity likely requires the C2 carboxylic acid to be present in the substrate 33, as cyclization of the decarboxylated 147 has not been observed..408 The carboxylic acid is likely an electronic requirement for reactivity with FAD, as when the acid is protonated the pKa of the C5 phenolic hydrogen will decrease and when deprotonated the electron density at C6 will increase. In either protonation state, the carboxylic acid makes the FAD-catalyzed dehydrogenation more facile.
Fig. 47. Key proposed step in biosynthesis of cannabis natural products converting cannabigerolic or cannabinerolic acid to THCA (160).
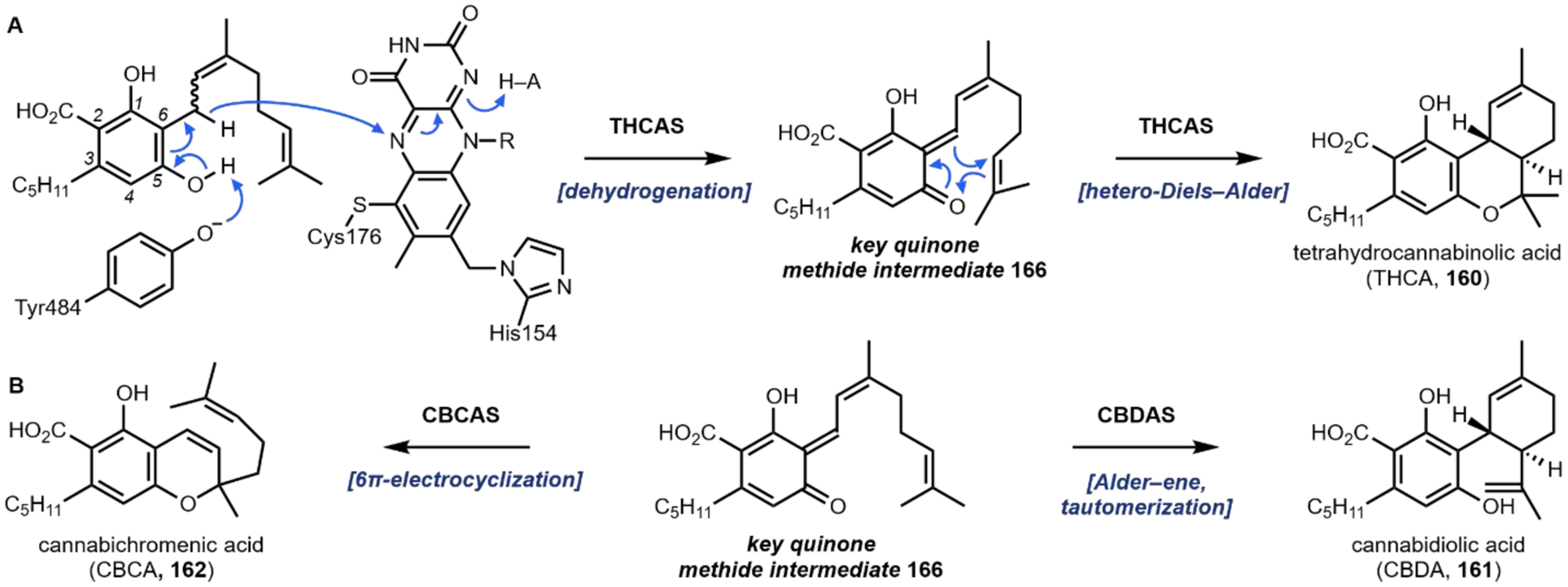
(A) Enzymatic dehydrogenation reaction leads to a reactive quinone methide intermediate 166 that can undergo various pericyclic reactions to yield all cannabis scaffolds. Flavin adenine dinucleotide (FAD), R = C16H26N5O13P2. (B) Related enzymatic transformations by CBCAS and CBDAS form CBCA (162) and CBDA (161)
From this key quinone methide intermediate 166, all three cannabinoid scaffolds (160, 161, and 162) can be formed by hetero-Diels–Alder, Alder-ene, or electrocyclization reactions, respectively (Fig. 47A, B). This proposed mechanism indicates that these enzymes THCAS, CBDAS, and CBCAS can be considered as multifunctional pericyclases – enzymes that catalyze pericyclic reactions.410 Very recently, the plant BBE MaDa that shares 45% identity with THCAS has been characterized to catalyze the Diels–Alder reaction.411 Our laboratory has also shown enzymes groups that share >70% homology catalyze stereoselective dehydrations and concomitant pericyclic reactions – either hetero-Diels–Alder or Alder-ene.412 These findings point us back to the THCAS, CBDAS, and CBCAS enzymes and led us to ask: are these reactions pericyclic? Another aspect of this transformation that warrants further investigation is the 33 substrate Δ8,9-alkene configuration. 33 is in the (E) configuration, but the products of THCAS, CBDAS, and CBCAS are all in the (Z) configuration. Authors have shown that THCAS can convert either cannabigerolic acid (33) or cannabinerolic acid (157) into 160.407 This implies that the enzyme facilitates isomerization upon quinone methide formation and before cyclization, but there is no evidence for the mechanism of isomerization. Further research needs to be conducted in order to fully understand the mechanism in which the psychoactive cannabinoid skeletons are forged.
4.3. Heterologous production of cannabinoids
Keasling and coworkers realized heterologous production of 160 and 161 in Saccharomyces cerevisiae from galactose (Fig. 48).75 In order to produce cannabinoids in yeast, it was crucial to optimize the flux of geranyl pyrophosphate (82) and hexanoyl-CoA (156) by introducing an upregulated mevalonate pathway, a mutant (F96W, N127W) of the endogenous farnesyl pyrophosphate synthase (ERG20), and incorporation of an acyl activating enzyme from Cannabis sativa to form hexanoyl-CoA (156). The use of the mutant ERG20 is to attenuate the conversion of GPP to FPP, as discussed in Section 2.8 in strictosidine biosynthesis. Despite efforts to incorporate APT and catalyze the electrophilic prenylation to form 33, no activity could be observed when expressed in yeast. The authors searched Cannabis transcriptomes for enzymes that share homology with the well-functioning soluble aromatic prenyl transferase, NphB (vide infra), of Streptomyces sp. and discovered the enzyme CsPT4 – which not only efficiently catalyzes the reaction, but is clustered with other prenyltransferases in Cannabis. Incorporation of all genes above led to a 1.4 mg·L–1 titer of 33. To functionally reconstitute the final oxidative cyclization by THCAS or CBDAS in yeast, the N-terminal domain of THCAS and CBDAS were replaced with a vacuolar localization tag. In total, integrating all genes into a single strain and culturing with galactose yielded titers of 8.0 mg·L–1 160 or 4.2 μg·L–1 161.
Fig. 48.
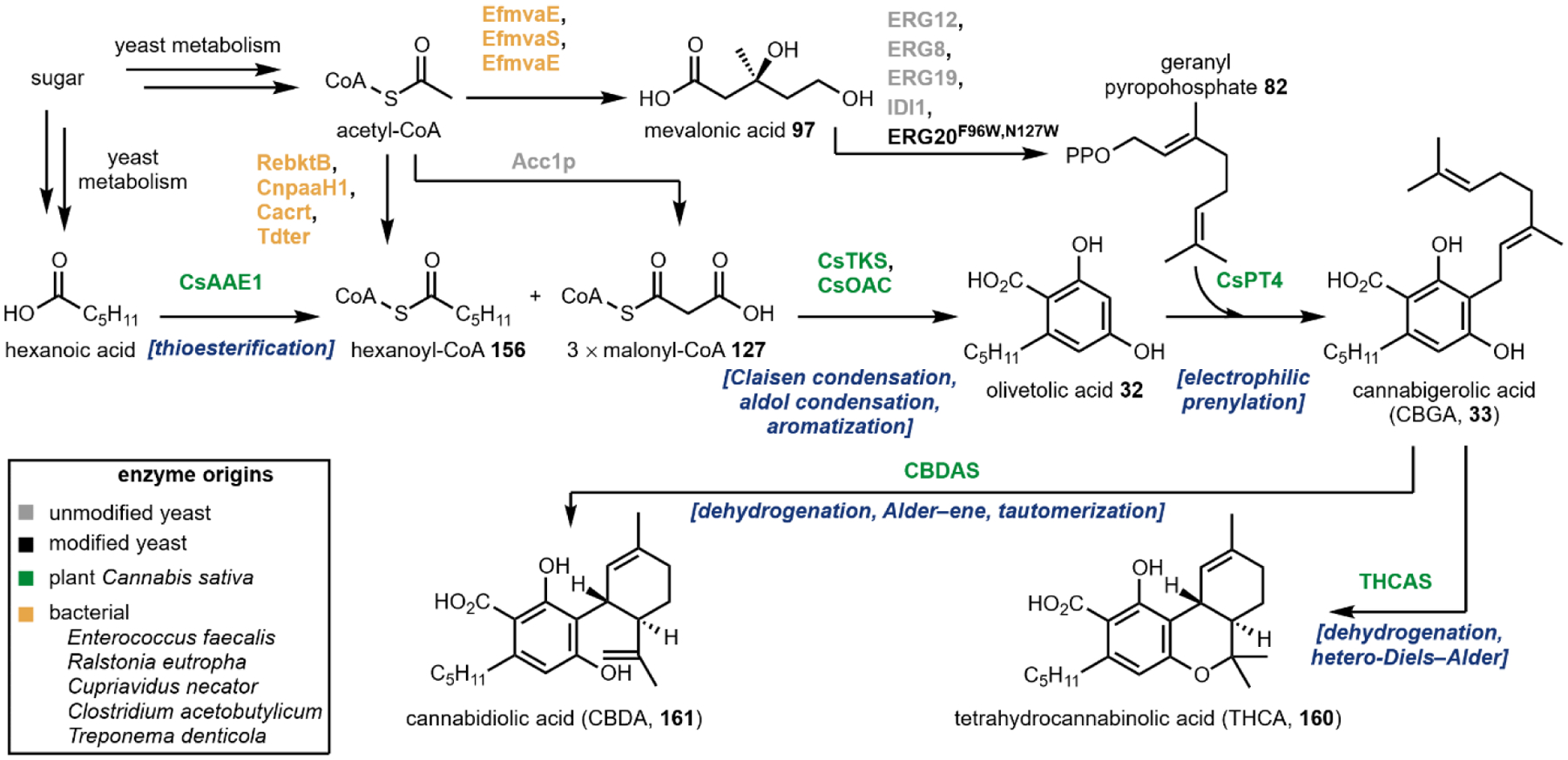
Heterologous production of tetrahydrocannabinolic acid (160) and cannabidiolic acid (161).
Due to the substrate promiscuity of OAC, Keasling et al. also used this platform to produce cannabinoid C3 alkyl chain derivatives. Starting from various fatty acids, 32, 33 and 160 could be produced with a propyl, butyl, pentenyl, 3-methylpentyl, hexyl, and hexynyl C3 substituents. This heterologous expression showcases the feasibility of complete cannabinoid and cannabinoid derivative production in yeast. Improvements to this method for microbial cannabinoid production methods are currently being pursued by different synthetic biology startup companies.
Cell-free platforms for cannabinoid production have also garnered much interest and success. As geranyl pyrophosphate (82) levels are challenging to optimize in cells, cell-free methods circumvent inherent issues of forming prenylated natural products in large quantities. The Bowie laboratory has successfully used cell-free platforms to produce CBGA (33) with a 1.25 g·L–1 titer53 and, most recently, using a far simpler and more cost-effective system were able to realize a 0.48 ± 0.12 g·L–1 titer.102
Perhaps a more important discovery than this titer improvement was the implementation and engineering promiscuous bacterial prenyltransferases to catalyze the electrophilic addition of a geranyl pyrophosphate to 32 and 32 derivatives.53,413,414 This strategy avoids the native integral membrane bound Cannabis prenyltransferase that is intrinsically difficult to work with both in vivo and in cell-free systems.403 Previous work by Kuzuyama and coworkers showed that the enzyme NphB could prenylate a wide variety of substrates including olivetol to form 148.413,415,416 Wild type NphB prenylates olivetolic acid nonspecifically generating a mixture of the desired cannabigerolic acid 33 and undesired O-prenylated product with a very low kcat (0.002 min–1). Bowie and coworkers expanded on this work by using Rosetta to computationally redesign NphB.53 The endpoint was a soluble, easy-to-work-with enzyme – named M23 – that was highly selective for the desired electrophilic prenylation of C6 to form 33 and exhibited 1,000-fold increase in kcat from the wild-type enzyme. A variant (M31) was also designed to function with divarinic acid 167 (the C3 propyl derivative of olivetolic acid 32). Now, NphB and its variants can be expressed heterologously or used in cell-free systems to produce 33 and derivatives thereof.
Cell-free systems for divarinic and olivetolic acid (32 and 163) production are becoming fairly effective in producing large titers. Recently, Bowie and coworkers used a six-enzyme system to produce 32 and 163 (Fig. 49A) as well as further develop their platform for geranyl pyrophosphate production (Fig. 49B). These methods are generalizable and applicable to many molecules. The authors build off of a previously discovered route417,418 in order to minimize the number of expensive adenosine triphosphate (ATP) and coenzyme A (CoA) molecules required for cell-free synthesis. As shown Fig. 49A, first acetic acid is phosphorylated by AckA and then thioesterified to form acetyl-CoA. The CoA group of acetyl-CoA is then transferred to malonic acid to form malonyl-CoA 127 which continues on the canonical pathway to form olivetolic acid 32. The authors used ThiM to phosphorylate isoprenol and then a subsequent phosphorylation by IPK.417,418 Typically ThiM, a hydroxyethylthiazole kinase, phosphorylates 2-hydroxyethyl thiazoles. Here they have used this enzyme to catalyze the same reaction on a simpler acyclic starting material. The following steps to form geranyl pyrophosphate were reported previously101 using typical isopentyl-diphosphate delta-isomerase (IDI) and a modified farnesyl pyrophosphate synthase (FPPS) enzyme that generates the C10 dimethylallyl derived geranyl pyrophosphate. Ultimately, this strategy is a highly modular method to make various malonyl-CoA products and useful for making high-titers of geranylated natural products.
Fig 49.
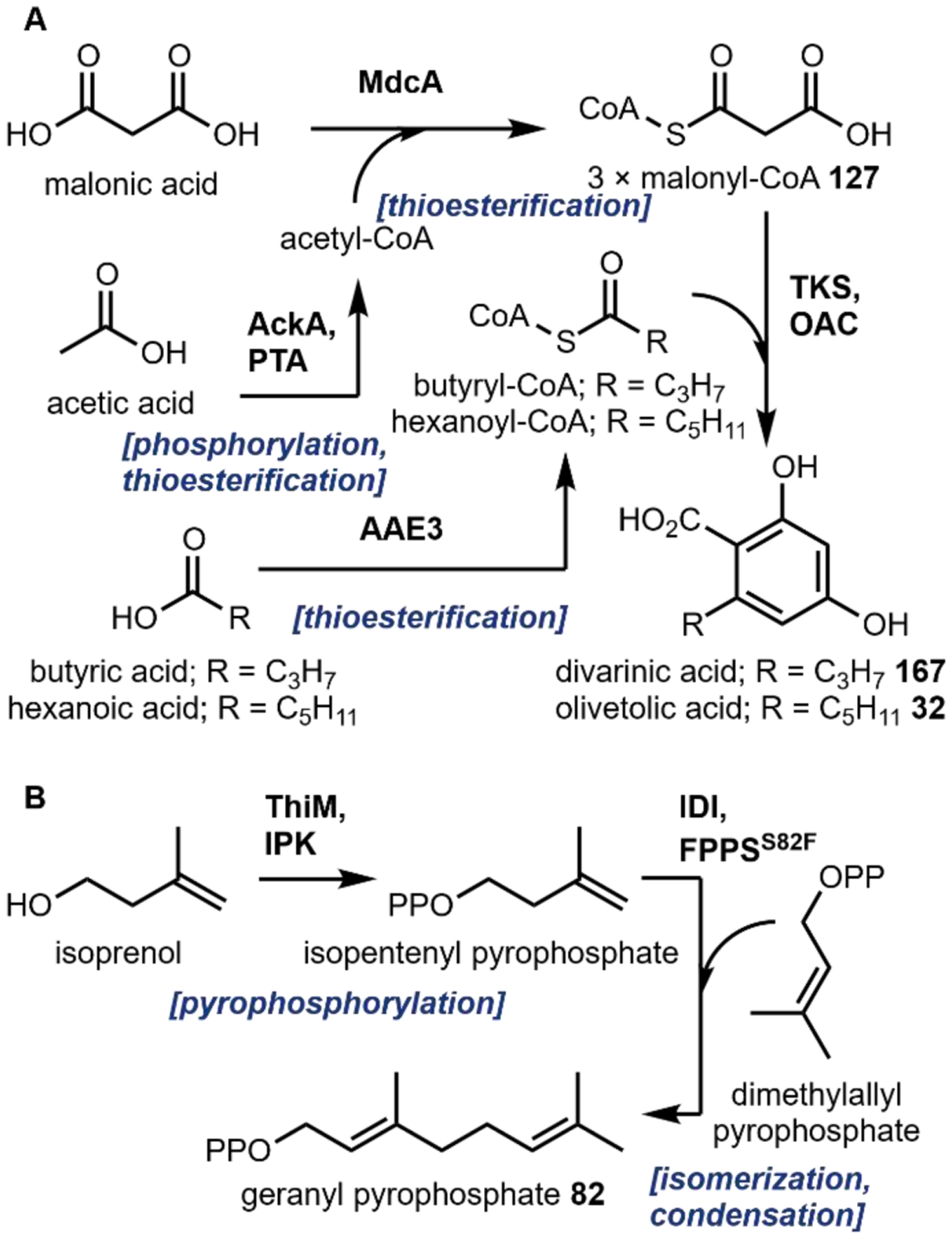
Cell-free system for improved olivetolic acid, divarinic acid, and geranyl pyrophosphate production.
5. Opioids
Western medicine was born from the poppy plant Papaver somniferum. Opium has been scraped from the P. somniferum bulb and used both recreationally and medicinally for all of written history.419,420 For example, in the 16th century, people used the botanical tincture laudanum,421 which is mixture of ambergris, musk, alcohol and opium, for the promise of good health. In the 1800s, morphine (9), the major component of opium, was isolated and sold as the first single-molecule drug (Fig. 50).422 This began the contemporary 150-year medicinal tradition of prescribing single-molecule drugs versus botanical tinctures.
Fig. 50. Image of bulbs and bloom of the poppy plant, Papaver somniferum.
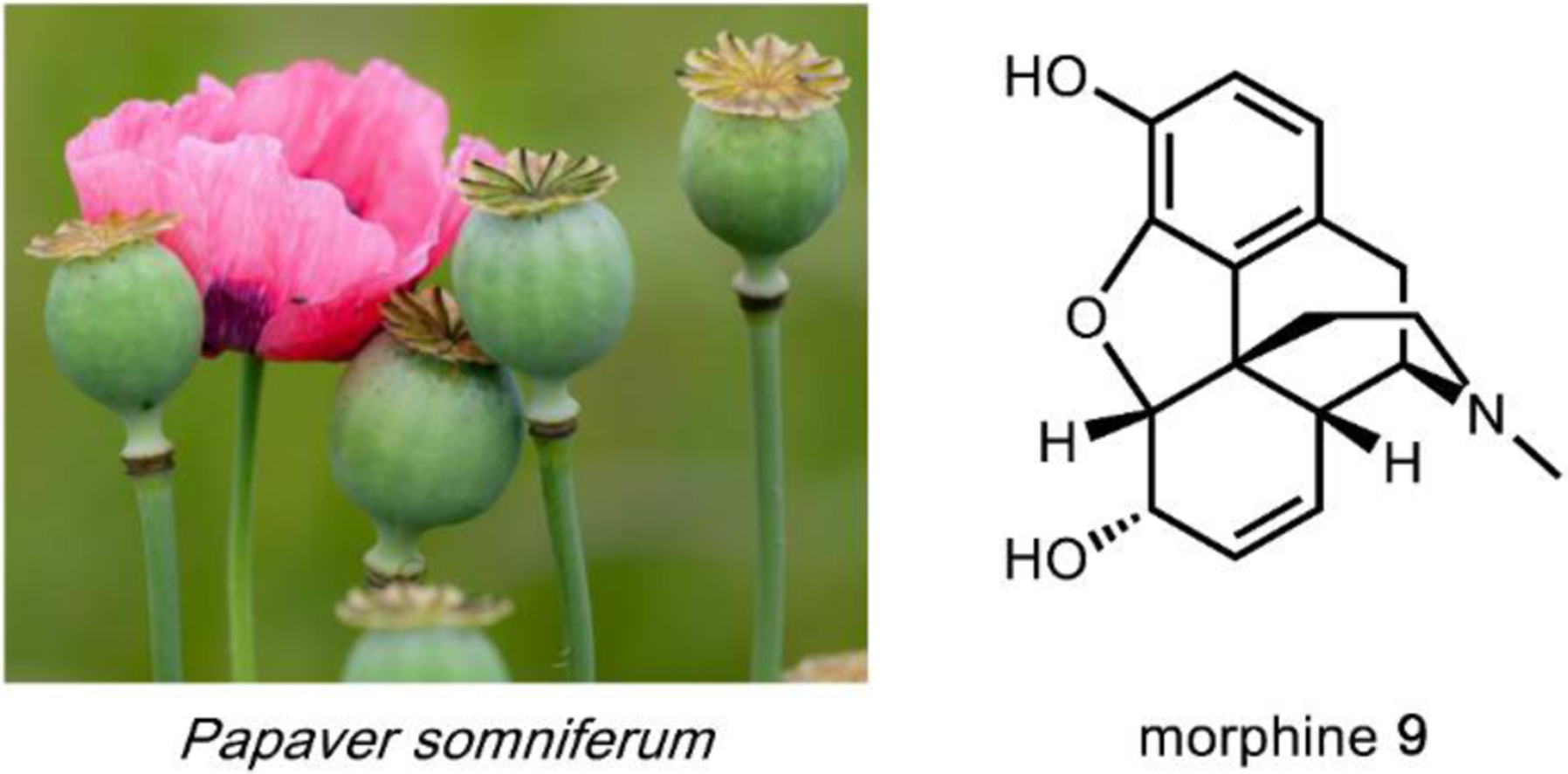
On average, poppy bulbs contain 16% by weight morphine 9.423
5.1. Opioid receptors
There are four class A GPCR opioid receptors, μ, ∂, K, and N.424 The μ opioid receptor (MOP) is named for binding morphine. The ∂ opioid receptor (DOP) is named for being expressed in the vas deferens. The K opioid receptor (KOP) is named for binding the synthetic ligand ketocyclazocine. The N opioid receptor (NOP) is named after the endogenous mammalian peptide nociceptin. These names are not truly informative and have been subject to debate. Opioid receptors are distributed throughout the central nervous system and partially in the vas deferens, joints, and immune system. MOP, DOP, and KOP are sometimes referred to as classical opioid receptors. Whereas NOP is ‘less’ classical; NOP was discovered later and now is considered an opioid receptor as well. As the ligand was not originally known for NOP, the receptor is sometimes still referred to as an opioid-like receptor or an orphan receptor. Other opioid-like receptors have been identified based on binding, however these are not bona fide opioid receptors. The σ receptor (named for binding SKF10047) binds opioids as well as other drugs of abuse like phenylcyclidine. Other receptors that do exhibit related pharmacology to MOP, DOP, KOP, or NOP have been identified but are not fully characterized; for example, the ζ receptor is an opioid growth factor receptor, and the ƛ receptor and ε binding site have been proposed in β-endorphin binding.
The structure function relationship of opioid receptors is relatively well understood.425–427 MOP, DOP, KOP, and NOP share ~ 60% homology with highly conserved fingerprints of class A GPCRs and a homologous binding cavity. The small molecule ligands like morphine bind to the conserved receptor residues in the homologous binding cavities.
5.2. Opium alkaloids
The morphinan alkaloids are the most colloquial family of opium alkaloids as morphine (9) is the flagship molecule of the family. This family features a tetracyclic phenanthrene fused piperidine core, a so-called morphinan scaffold (Fig. 51). In early 1800s, Friedrich Sertürner isolated 9 (Fig. 51) from Papaver somniferum.428 In the following years, Pierre-Jean Robiquet isolated the O-methylated morphine derivative, codeine 168 (Fig. 51).429 These discoveries led chemists to develop related compounds, i.e. heroin 169, that were more potent, safer (minimizes hypoventilation), and touted as “free from abuse liability.”419 This claim marked the first falsification of opioids being safe to use without risk of addiction, which were most recently repeated by the Sacklers at Purdue Pharma. The morphinan alkaloids are potent analgesics that have been used for thousands of years as opium mixtures and now as isolated pure compounds.424 Natural products oripavine (170) and thebaine (171) do not exhibit safe pharmacology, but are highly useful morphinan alkaloids as synthetic building blocks for derivatization.
Fig. 51.
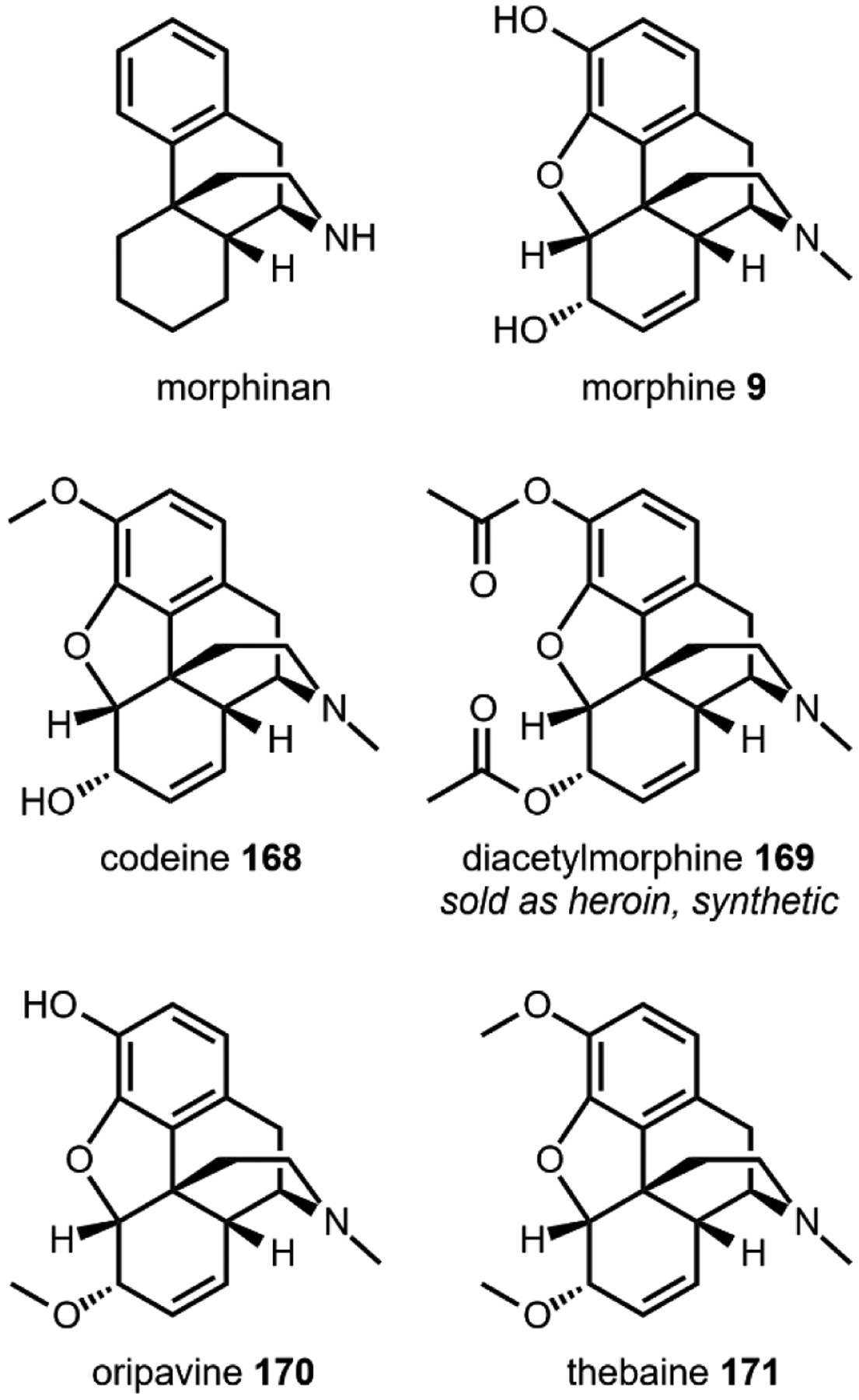
Structures of natural morphinan opioids and synthetic compound diacetylmorphine (heroin, 169).
The reticulines are key early pathway intermediates that many families of opioid scaffolds diverge from. These structures are simple benzylisoquinolines (Fig. 52). (S)-reticuline (172) can epimerize to (R)-reticuline (28) and continue to morphinan biosynthesis or directly undergo a C–C coupling reaction and lead to the phthalide isoquinolines and protoberberines families of opioids. (S)-norcoclaurine (27) is formed from dopamine 17 and 4-hydroxyphenylacetaldehyde 26 by a Pictet-Spenglerase and marks the first dedicated step in opioid biosynthesis (also see Fig. 3). Though these compounds are not known to be psychoactive, they are the building blocks to form many psychoactive natural products. Of note, the oxidation of the isoquinoline to form papaverine (173) alters the pharmacological properties as 173 is an approved antispasmodic drug.
Fig. 52.
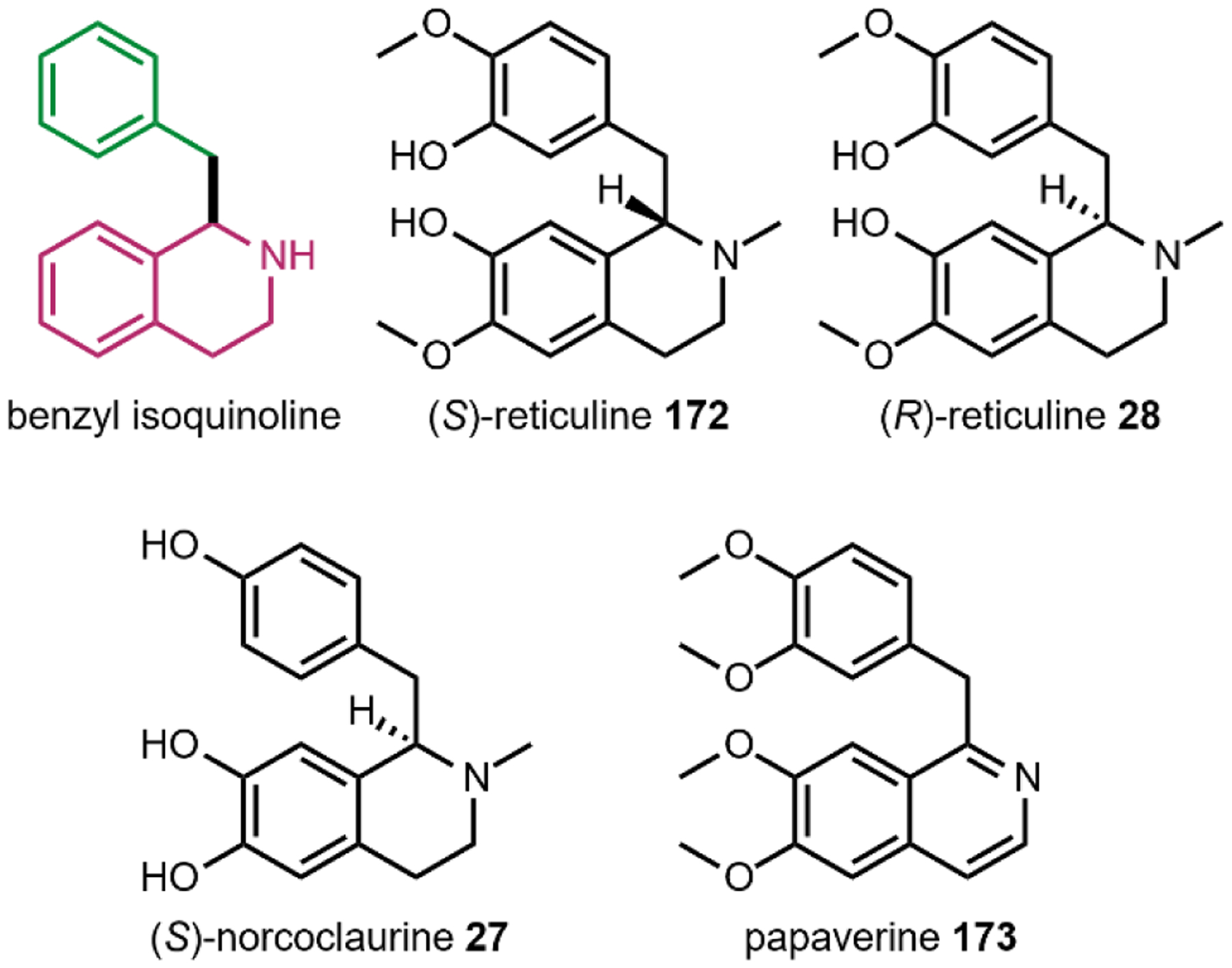
Structures of simple benzyl isoquinolines that play key roles in opioid biosynthesis (172, 28, 27) and as antispasmodic drugs (173).
The phthalide isoquinolines class of opioids encompass two general structures in which the isoquinoline is either intact or open as a dimethyl amino sidechain, as exemplified by noscapine (174) and narceine (175). 174 and 175 are non-narcotic, antitussives with minor hypnotic, euphoric, and analgesic properties. 174 has a lengthy history as a pharmaceutical with its isolation in 1817 and first use as an anti-malarial drug until 1930.430 Now, many are rediscovering 174 as an anti-cancer drug candidate.431,432 Such compounds are produced by many species of the Papveraceae poppy plant.
Aporphine opioids are C–C phenol coupled benzylisoquinolines that feature a functionalized aporphine structure (Fig. 54). These natural products have a range of activity from anticonvulsant (corytuberine, 176) to antinociceptive ((S)-glaucine, 177). The unnatural (R)-isomer of glaucine 178 is known to be a potent hallucinogen that modulates the 5-HT2A receptor and is sometimes used recreationally. Such compounds can be isolated from a variety of Papaveraceae species such as Glaucium flavum and Corydalis yanhusuo.
Fig. 54.
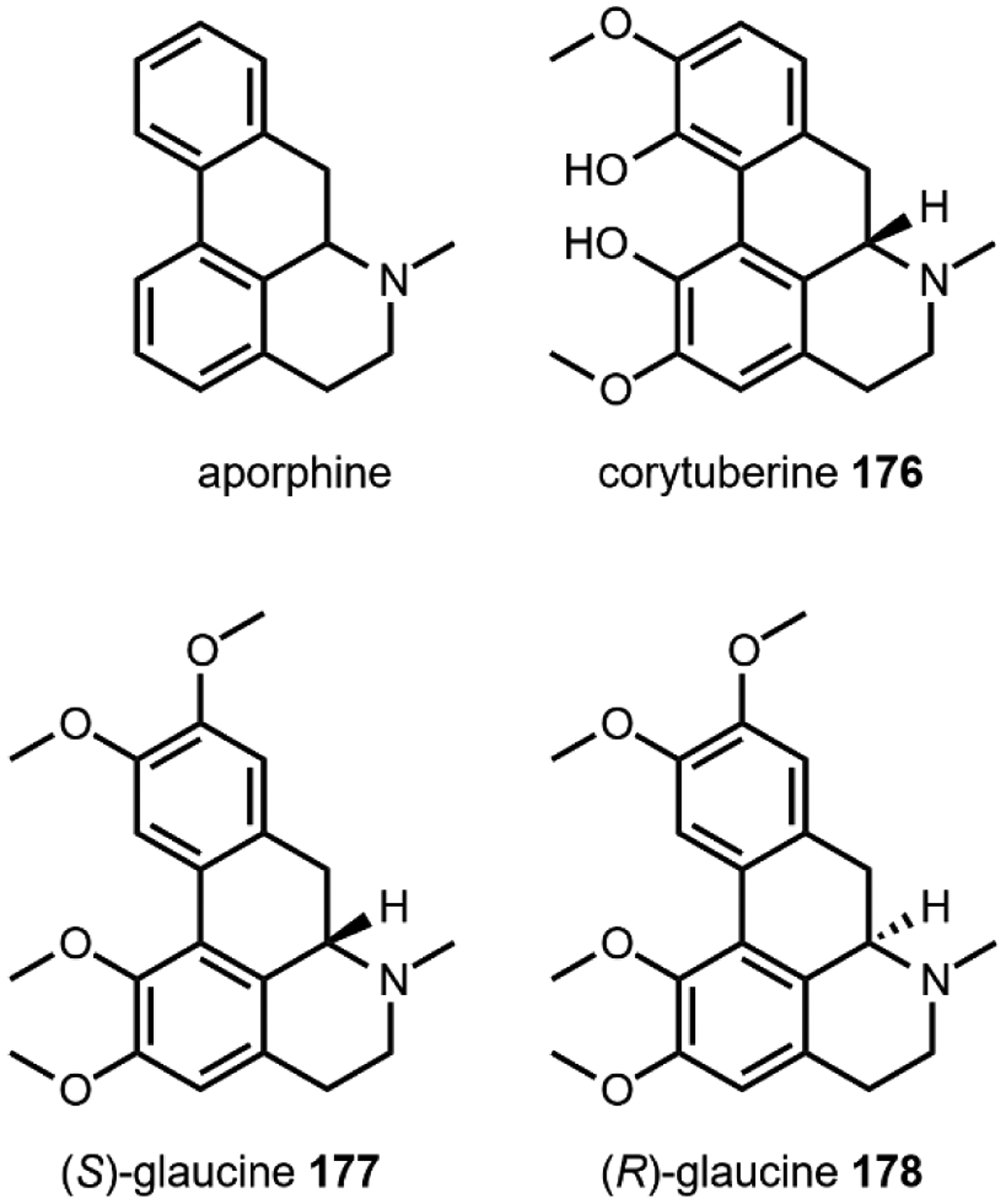
Aporphine opioids corytuberine (176), natural (S)-glaucine (177) and unnatural (R)-glaucine (178).
Lastly, the berberine opioids are pentacycles with a dibenzoquinizolium core. These molecules are quaternary ammonium salts. Berberine (179) is a traditional natural yellow dye and sanguinarine (180) is an escharotic toxin that also causes epidemic dropsy. The berberine opioids have been isolated from Papaver somniferum and Macleaya cordata.
5.2.1. Biosynthesis of opium alkaloids
Biosynthesis of morphinan opioids requires more than 10 enzymatic steps starting from dopamine 17 and 4-hydroxyphenylacetaldehyde 26. The elucidation of this route has taken more than 30 years of research and is condensed into a single figure, Fig. 56. The first dedicated step in opioid biosynthesis is a Pictet-Spengler reaction catalyzed by norcoclaurine synthase (NCS) to forge the tetrahydroisoquinoline core of (S)-norcoclaurine (27) – which is a simple example of the benzylisoquinoline alkaloid natural product family.433,434 There has been many mechanistic studies of this enzyme that are not discussed herein (also see Fig. 3).43,435,436
Fig. 56.
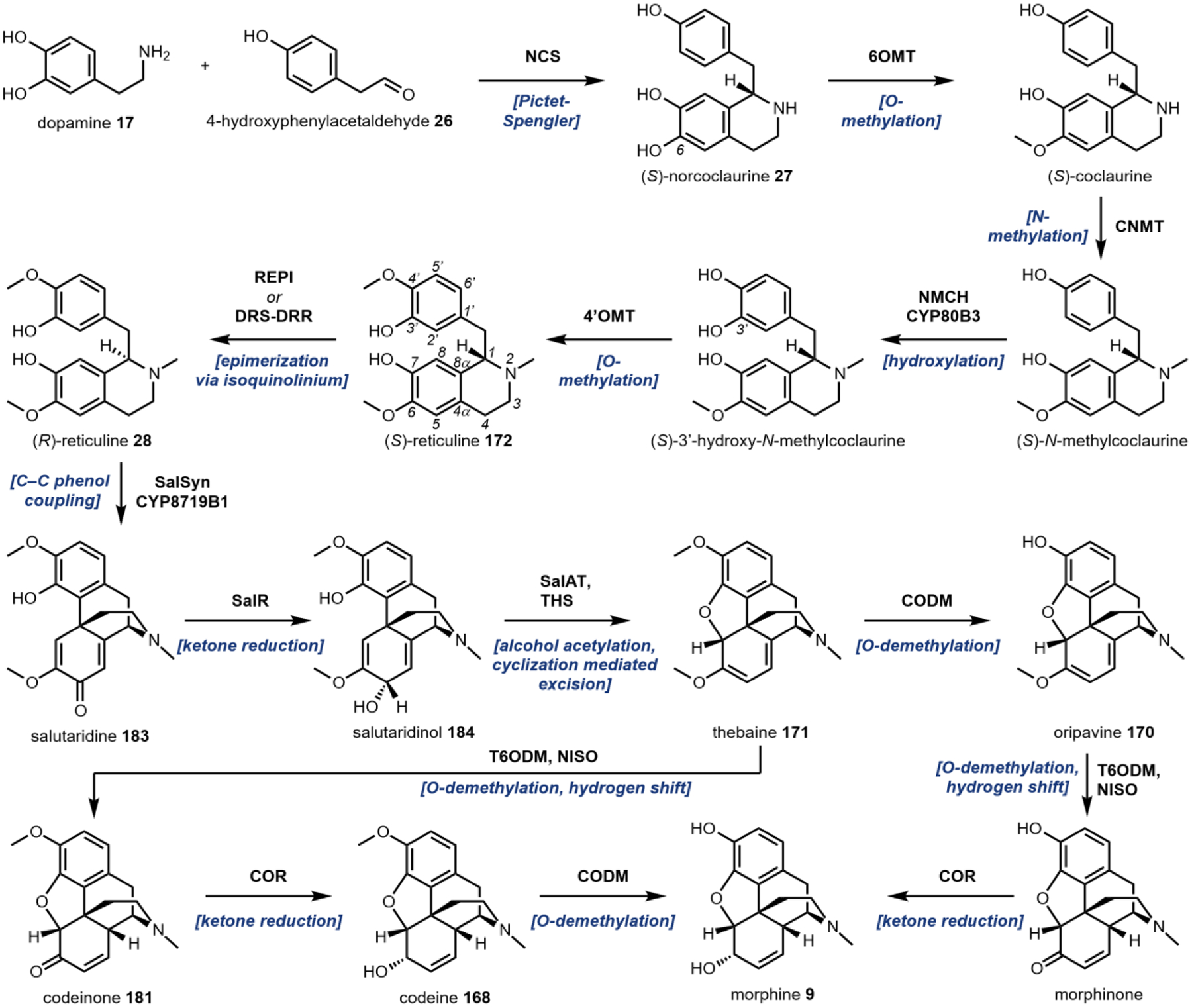
Biosynthesis of the morphine opioids from dopamine 17 and 4-hydroxyphenylacetaldehyde 26.
(S)-reticuline (172) is formed from 27 by two hydroxylations, an N-methylation, and an O-methylation by the enzymes norcoclaurine 6-O-methyltransferase (6OMT), coclaurine N-methyltransferase (CNMT), N-methylcoclaurine 3’-hydroxylase (NMCH CYP80B1), and 3’-hydroxy-N-methylcoclaurine 4’-O-methyltransferase (4’OMT), respectively.437–440 These steps can occur in a variety of orders with similar efficiencies and are drawn above in a typical order in Fig. 56. 172 is a key branch point in opioid biosynthesis from which many benzylisoquinoline scaffolds can form. Recently, the epimerase enzymes STORR reticuline epimerase (REPI) and 1,2-dehydroreticuline synthase-1,2-dehydroreticuline reductase (DRS-DRR) were discovered to convert (S)- to (R)-reticuline by dehydrogenation at C1 to form an iminium cation which is hydrated from the opposing face.441,442 In order to discover these genes, laboratories turned to RNA interface mediated silencing of the codeinone reductase (COR) gene. Silencing COR, which operates several steps downstream from the epimerization of reticuline, results in accumulation of (S)-reticuline versus the substrate codeinone 181. This could occur due to off-target co-silencing of related oxidoreductases that catalyze the epimerization of (S) to (R) reticuline. Using this strategy, a fusion protein REPI and DRS-DRR was identified that was able to catalyze the crucial epimerization reaction.
In order to discover the enzyme without access to the opium poppy, the Smolke lab searched the 1000 Plants Project and PhytoMetaSyn databases for COR-like enzymes in Papaver species. This revealed several genes that encoded for a two-domain enzyme with P450 82Y1-like and COR-like domains. From these two domains it was reasonable to hypothesize that the (S)-reticuline 172 could be oxidized to an isoquinilonium (P450) and then reduced to (R)-reticuline 28 by the COR domain. One of the gene candidates from P. somniferum, named DRS-DRR was cultured with P450 reductases, CNMT, 6OMT and 1 mM norlaudanosoline 182, a desmethoxy derivative of reticuline, for 72 hours, and >50% conversion to (R)-reticuline 28 was observed.
(R)-reticuline (28) is the substrate for the salutaridine synthase (SalSyn) CYP8719B1-catalyzed oxidative phenol coupling reaction that forms a carbon-carbon bond between C2’ and C4ɑ yielding salutaridine (183).58,443 This reaction is proposed to occur by the iron oxo heme compound I abstracting the hydrogen from the C3’ hydroxyl of 28 to generate compound II, which then abstracts the remaining phenol hydrogen to facilitate the cyclization (also see Fig. 5B).58 The direct di-keto product readily enolizes to form 183.
Salutaridine (183) is converted to thebaine (171) in three enzymatic steps. First, salutaridine reductase (SalR) reduces the quinone ketone to form salutaridinol 184 which is then acylated by the acyl transferase enzyme salutaridinol 7-O-acetyltransferase (SalAT) and was believed to slowly cyclizes nonenzymatically to form thebaine (171).444–446 Recently, thebaine synthase (THS) was discovered and isolated in Papaver somniferum opium poppy latex and found to accelerate this cyclization to form the morphine skeleton of 171.447
The final four enzymatic steps in morphine (9) biosynthesis are two O-demethylations, an isomerization and ketone reduction that are catalyzed by codeine O-demethylase (CODM), thebaine 6-O-demthylase (T6ODM), neopinone isomerase (NISO), and codeinone reductase (COR), respectively.74,448,449 There are two established paths that differ in the first O-demethylation, which can lead to either oripavine 170 or codeinone 181. In 2018, the crystal structure for T6ODM was solved, but the mechanism for the O-demethylation is still unknown.450 Further O-demethylation and isomerization (a formal 1,5-hydrogen shift) produces morphinone 185 which is, finally, reduced to form morphine 9.
In 2012, a 10-gene cluster responsible for noscapine (174) biosynthesis was discovered.451 Noscapine (174) biosynthesis diverges from 9 biosynthesis after (S)-reticuline (172) formation (Fig. 57). First, 172 is transformed to (S)-scoulerine 186 by a berberine bridge enzyme (BBE).452,453 This BBE catalyzes an FAD-dependent dehydrogenation of the N-methyl group to form a methylene isoquinolinium. This reactive intermediate then undergoes C–C bond formation between the methylene and C2’ that is facilitated by glutamate sidechain hydrogen bonding to the C3’ phenolic hydrogen. Mechanistic studies have proposed that complete proton transfer is not required,452 but the C3’ hydroxyl – which increases the nucleophilicity of C2’ – is required for catalysis.454 Multiple alkaloid classes derive from 186; for example, the protoberbines, benzophenanthridines, protopines, and the phthalideisoquinolines.
Fig. 57.
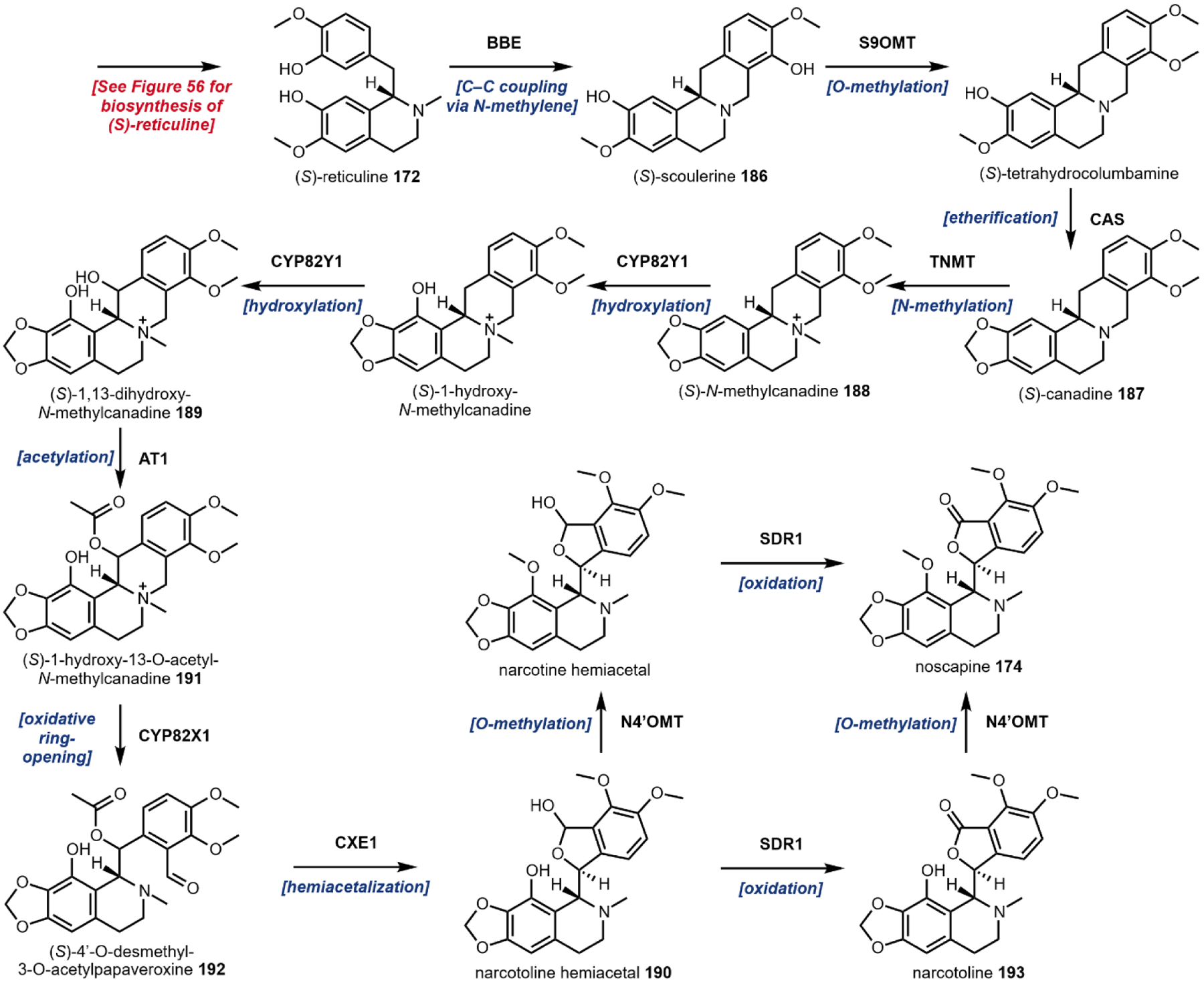
Noscapine biosynthesis from (S)-reticuline.
(S)-canadine 187, an antioxidant, is formed by subsequent O-methylation and etherification of (S)-scoulerine 186 by the scoulerine 9-O-methyltransferase (S9OMT) and canadine synthase (CAS) enzymes, respectively.455,456 187 undergoes N-methylation by tetrahydroprotoberberine N-methyltransferase (TNMT) to form the isoquinolinium core of (S)-N-methylcanadine 188 that can undergo dihydroxylation by CYP82Y1 to form (S)-1,13-dihydroxy-N-methylcanadine 189.457–459
The noscapine core is formed by the oxidative ring opening and cyclization to yield narcotoline hemiacetal 190. These transformations begin with acetylation of (S)-1,13-dihydroxy-N-methylcanadine (189) to form (S)-1-hydroxy-13-O-acetyl-N-canadine 191 by the acetyltransferase AT1 (see Fig. 4B).460 This enzymatically-installed acetyl group is essential for CYP82X1 hydroxylation activity and has been proposed to function as a protecting group to alleviate from precocious hemiacetalization.460 Following acetylation, a CYP82X1 installs a hydroxyl ortho to the nitrogen that facilitates a spontaneous oxidative ring opening to form (S)-4’-O-desmethyl-3-O-acetylpapaveroxine 192.
(S)-4’-O-desmethyl-3-O-acetylpapaveroxine (192) undergoes three final enzymatic transformations to form noscapine (174): hemiacetalization, oxidation and O-methylation. The enzyme CXE1 catalyzes the hemiacetalization to form the phthalideisoquinoline core of narcotoline hemiacetal (190) which is then oxidized to the lactone (narcotoline, 193) by the enzyme SDR1.460,461 Lastly, noscapine 170 is formed by the O-methylation by N4’OMT.455 Of note, these last two steps can occur in either order; N4’OMT O-methylation can preclude SDR1 lactonization.
5.2.2. Heterologous production of opium alkaloids
There have been many efforts in heterologous production of opioids.109,462–467 These pathways, at the time, were the longest biosynthetic pathways reconstituted in yeast.466 However, almost all studies stopped at (S)-reticuline 172 or begin at highly functionalized opioids, like thebaine 171. This had to do with the fact that the crucial epimerase that forms (R)-reticuline 28 was not characterized until 2015. At this time, Smolke’s laboratory had already realized heterologous production of thebaine 171 and hydrocodone 194 in yeast (Fig. 58).77 To complete biosynthetic reconstitution, the laboratory had to overcome two main challenges: (1) discover an enzyme that racemizes (S)-reticuline 172 to (R)-reticuline 28; and (2) engineer the aryl coupling P450 SalSyn to be fully functional when expressed in yeast. A further challenge was implicit in the task; simply expressing >20 genes and obtaining high efficiency with each enzymatic transformation. In spite of these challenges, Galanie et al. engineered a fully integrated yeast strain that produced 6.4 ± 0.3 μg/L of thebaine 171 and with additional downstream enzymes, ~0.3 μg/L of hydrocodone 194 in a culmination of decades of research.78,109
Fig. 58.
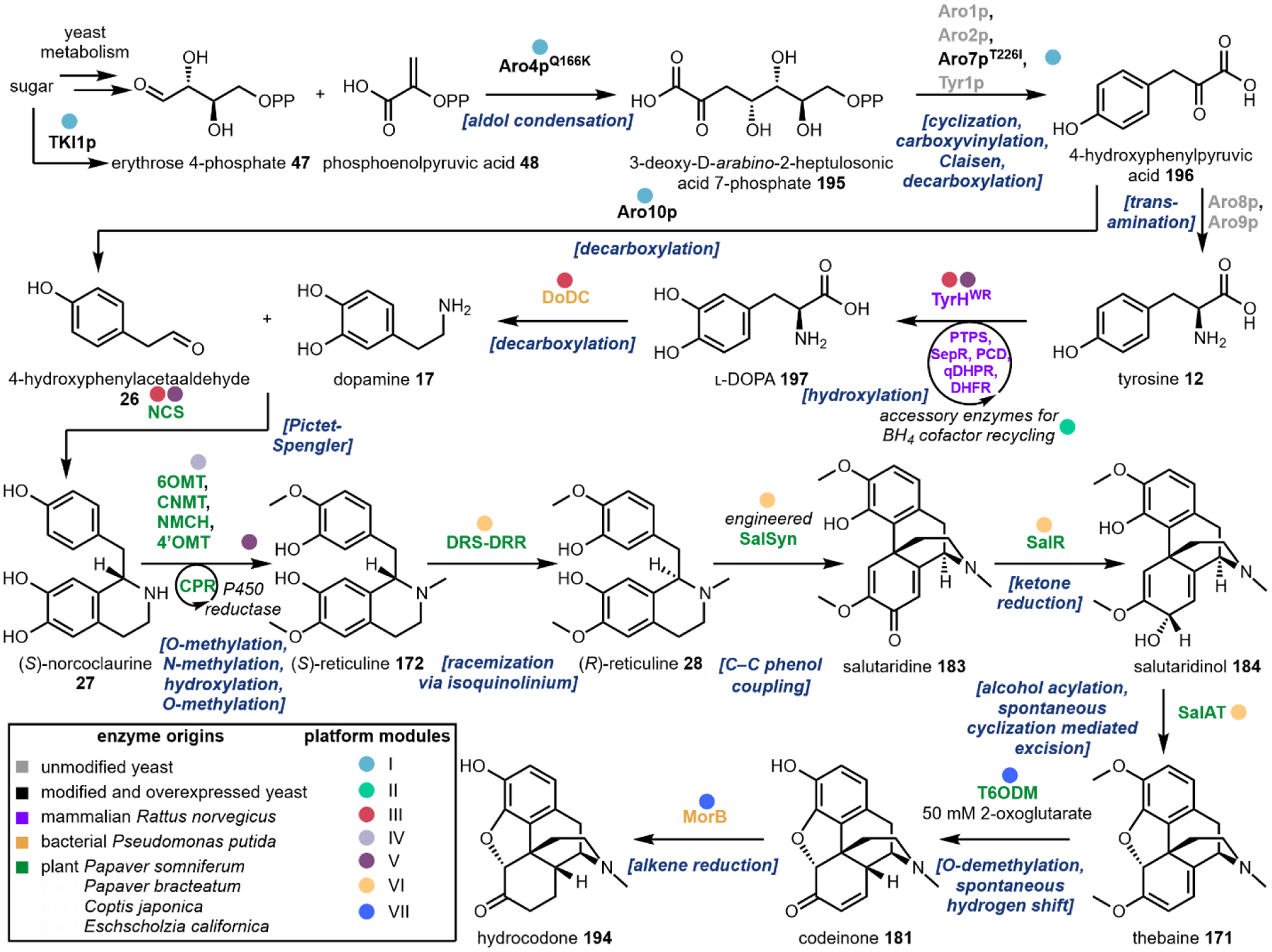
Heterologous production of thebaine and hydrocodone from sugar in yeast.
The engineered strain contained 19 heterologously expressed mammalian, bacterial, and plant enzymes, two modified yeast enzymes, two overexpressed native yeast enzymes and one inactivated enzyme for a total of 24 chromosomal modifications. These modifications were split between seven modules for both pathway and chromosomal organization.
Module I consists of overexpression of two modified shikimate pathway enzymes and two native yeast genes. The Q166K point mutation in Aro4p, which catalyzes the aldol condensation of erythrose 4-phosphate 47 and phosphoenolpyruvic acid 48 to form 3-deoxy-d-arabino-2-heptulosonic acid 7-phosphate 195, renders the enzyme feedback inhibition resistant. Similarly, the T226I mutation in Aro7p, which is one of the enzymes involved in the biotransformation of 195 into 4-hydroxyphenolpyruvic acid 196, makes the enzyme feedback resistant. Overexpression of Aro10p and Tkl1 resulted in shifting metabolic flux towards the pathway.
The next module (II) focuses on producing and recycling the mammalian redox cofactor, tetrahydrobiopterin (BH4). This cofactor is essential for the selective C3 hydroxylation of l-tyrosine 12 to form l-DOPA 71 catalyzed by mammalian tyrosine hydroxylase (TyrH) and is not native to yeast. 6-pyruvoyl-tetrahydropterin (PTPS) and sepiapterin reductase (SepR) are used to produce BH4 from dihydroneopterin, a yeast metabolite. Quinonoid dihydropteridine reductase (QDHPR) and pterin carbinolamine dehydratase (PCD) are then used to recycle BH4 back to its active form.
Module III uses bacterial, plant, and mammalian enzymes to catalyze formation of the first BIA scaffold. Dihydrofolate reductase (DHFR) is another BH4 salvage enzyme that works with TyrHWT, a mutant that is more inhibition resistant. Following hydroxylation, l-DOPA 71 undergoes decarboxylation catalyzed by DOPA decarboxylase (DoDC) to form dopamine 17 followed by a Pictet-Spengler reaction between 4-hydroxyphenylacetaldehyde 26 and 17 by norcoclaurine synthase (NCS) to form (S)-norcoclaurine 27.
The remaining modules consists of the biosynthetic pathway enzymes towards thebaine 171 and hydrocodone 194 and the discovered enzyme for (S)-reticuline epimerization. The native P450 enzyme SalSyn had low activity when initially expressed in yeast. This was hypothesized to be due to incorrect translocation of nascent SalSyn to the endoplasmic reticulum (ER) lumen as opposed to correct anchoring to the outer ER membrane based on nonnative N-glycosylation patterns. Mistranslocation could stem from a poorly recognized N-terminus and thus the authors replaced the N-terminus portion of SalSyn with that from a homologous, non-glycosylated P450,, Cheilanthifoline synthase, that shares 61% identity and exhibits high activity in yeast.468 The engineered chimeric SalSyn enzyme exhibited nearly 6-fold improvement in conversion of (R)-reticuline 28 to salutaridine 183 compared to the wild type enzyme.
After establishing ~6 μg/L thebaine 171 production with their platform, the authors sought to introduce downstream enzymes towards hydrocodone 194 production. Upon coexpression of two more enzymes, MorB and T6ODM and supplementation with 50 mM oxoglutarate, the strain produced 0.3 μg·L–1 194. The Smolke lab previously used MorB, an NADH-dependent morphinone reductase from a bacteria Pseudomonas putida M10 that was originally discovered in an opium poppy processing factory, for production of natural and semi-synthetic opioids.465,469 Expression of such a long pathway required careful codon-optimization of multiple enzymes and led to proof-of-concept titers that highlight the potential of chassis species for pharmaceutical production.
In 2018, the Smolke lab modified this pathway to produce noscapine 174.470 The new work branches at (S)-reticuline 172, using the BBE to produce (S)-scoulerine 186. Therein, more than 30 enzymes were heterologously expressed, including five plant P450s which are notoriously difficult to express in yeast. To overcome challenges in P450 activity and other pathway bottlenecks, the authors (i) deleted the first 24 amino acids of NCS corresponding to an N-terminal signal vacuole translocation peptide to avoid detrimental sorting of the nascent peptide,471 (ii) codon optimized the TyrH R37E, R38E, W166Y (TyrHWR), (iii) incorporated an NADPH regenerating system, (iv) and lastly, optimized media and fermentation conditions which led to the largest gain (~300-fold) in production. Overall, the combined strategies resulted in a noscapine 174 titer of 2.21 mg/L–1 in 72 h. Finally, Li et al. demonstrated the versatility of their yeast platform by generating halogenated BIA derivatives through feeding modified l-tyrosines.
5.3. Kratom
In addition to the opium alkaloids, more than 50 kratom alkaloids have been isolated from the Mitragyna speciosa plant, several of which exhibit opioid-like properties.472 Native to Southeast Asia, kratom (Mitragyna speciosa) has been used in traditional Thai medicine for centuries. The use in the United States has increased rapidly since early 2000s, both recreationally and to relieve chronic pain or opioid withdrawal symptoms. Compared to conventional opium alkaloids, kratom alkaloids exhibit “unique binding and functional profiles” suggesting that plant extracts may be effective alternative to the benzylisoquinoline-based pain treatments.473 However, similar to opium alkaloids, repeated use of kratom may lead to addiction, and the FDA has not approved kratom for any medical use; as a result, the DEA lists kratom as a Drug of Concern. The first reported and most abundant kratom alkaloid is mitragynine 10, comprising up to 66% of the alkaloid content in Thai cultivars.474 The less abundant 7-hydroxymitragynine 197 and its rearrangement product mitragynine pseudoindoxyl 198 are potent partial agonists of human μ-opioid receptors at nanomolar concentrations.27,475
5.3.1. Biosynthesis of mitragynine
Kratom alkaloids belong to the MIA family, and are presumed to be derived from the universal MIA precursor strictosidine. The 12-step pathway leading to the formation of strictosidine 25 from primary the primary metabolites l-tryptophan 11 and isopentenyl pyrophosphate 98 has been elucidated in C. roseus and is discussed in Section 2.8. While the remaining biosynthetic steps leading to the formation of mitragynine are currently unknown, we have proposed the pathway shown in Fig. 60 based on a number of biochemical observations. It is known that following deglucosylation of 25 by strictosidine-O-β-glucosidase (SGD) and subsequent rearrangement, a reductase converts the reactive aglycone 87 isomer into a more stable pathway intermediate.476 Examples from literature include tetrahydroalstonine synthase, geissoschizine synthase, and vitrosamine synthase, which are all NADPH-dependent reductases.242,477,478 Moreover, O’Connor and coworkers recently identified a dihydrocorynantheine aldehyde synthase (CpDCS) from Cinchona pubescens involved in quinine biosynthesis.479CpDCS performs iterative reduction of geissoschizine 87 to provide a demethylcorynantheidine 200 isomer. The authors identified an orthologue in Mitragyna speciosa named MsDCS, postulating that following deglycosylation of 25, two successive reductions of the conjugated iminium 87 would provide the stable demethylcorynantheidine 199. Reduction of conjugated iminiums has also been demonstrated in the formation of other late stage MIAs including tabersonine and catharanthine.236,237 Additionally, production of 199 has been reported in Uncaria rhynchophylla, which like kratom belongs to the family Rubiaceae.480 Methylation of the putative 199 would provide corynantheidine 200, which has been isolated from Mitragyna and differs from mitragynine by one methoxy group.472 Following aromatic hydroxylation and methylation, mitragynine 10 is likely further hydroxylated to 7-hydroxymitragynine 197. The P450-mediated conversion of 10 to 197 has been demonstrated in both mouse and human liver preparations.238 A semi-pinacol rearrangement to provide the mitragynine pseudoindoxyl 198 may occur either spontaneously or enzymatically481 as has been described in analogous transformations by FAD-dependent oxidases in fungal alkaloid pathways.482,483 Identification of the M. speciosa biosynthetic enzymes will provide biocatalytic tools necessary for heterologous production of kratom alkaloids in existing seco-iridoid producing yeast platforms.76
Fig. 60.
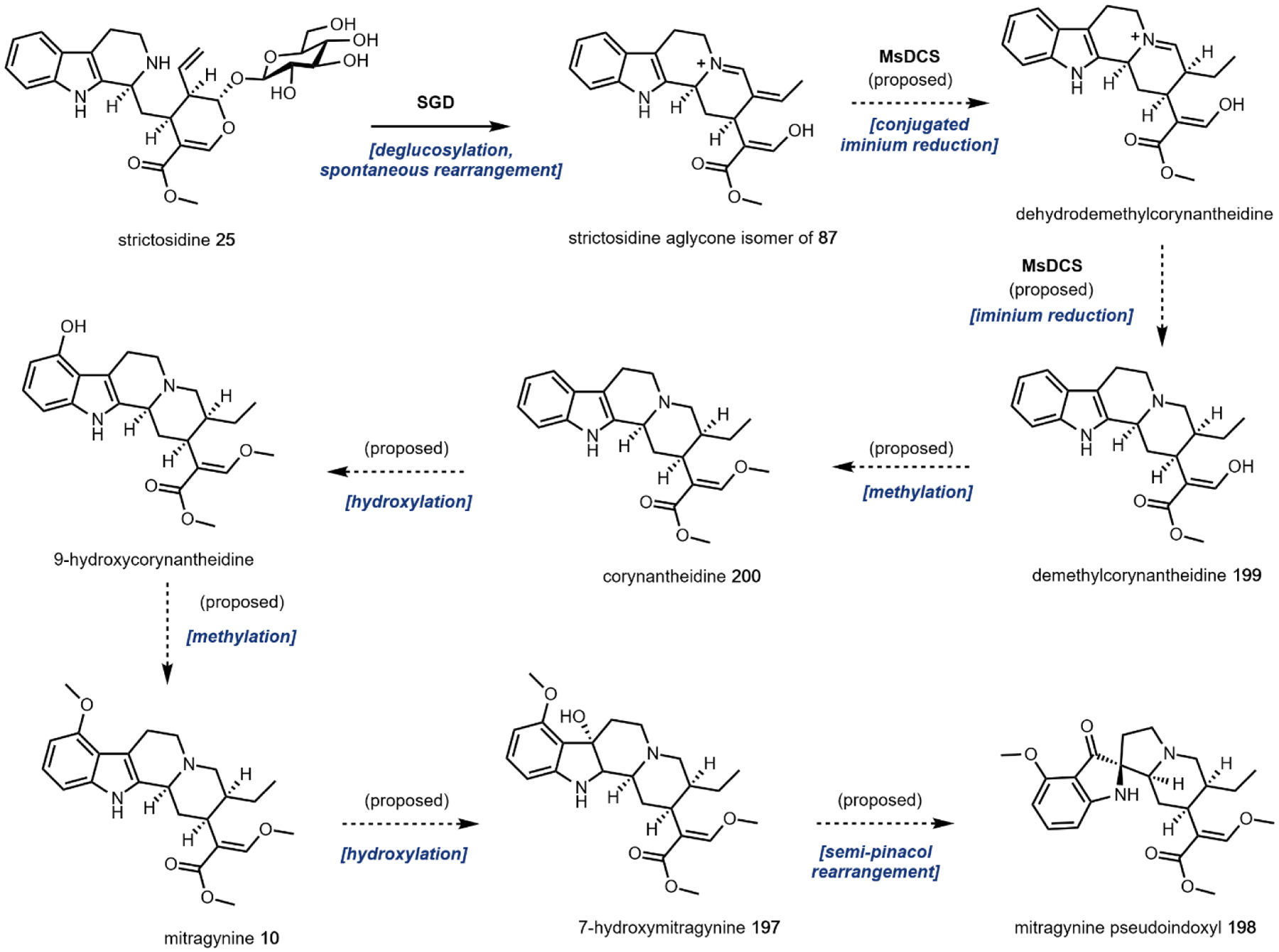
Proposed biosynthetic route to kratom alkaloids.
6. Conclusions and perspective
The natural products described in this review run the gamut of metabolic origin, psychoactive effect, and biological source. While most of the compounds discussed have been isolated from plants, we have highlighted several well-known psychoactive natural products produced by fungi and one of animal origin. Given the immense structural diversity exhibited by such molecules, the wide array of psychoactivities is not surprising. We have noted that the majority of the compounds originate from amino acid metabolism, however prominent examples of compound biogenesis via terpenoid and polyketide metabolism have been provided. Moreover, we have featured a number of remarkable enzymatic transformations that not only provide inspiration for biomimetic syntheses, but have been directly used in chemoenzymatic applications; these include completely stereoselective nucleophilic additions, tightly controlled scaffold rearrangements, and regioselective group transfer reactions on deprotected substrates. We have also chosen to outline biosynthetic pathways ranging from fully elucidated to almost entirely incomplete. Ongoing efforts towards total pathway elucidation are necessitated by the multitude of synthetic biology applications that benefit from a complete set of biosynthetic information. Given the rapid expansion of molecular biology techniques and prominent early successes in pathway refactoring, such synthetic biology technologies will very likely play a role in 21st century biomanufacturing. Major questions around the cost, ethics, and legality of synthetic-biology-based production of psychoactive substances must be answered in the very near future.
At present, the natural products covered in this review are either regulated or unregulated, however this legal binary is currently being traversed by Cannabis products. Popular culture and the media have sensationalized cannabinoid research. Despite the wide interest in cannabinoids, the research is still highly controversial and difficult to fund. This is rapidly changing as the World realizes there is ‘money in cannabinoids.’ Western medicine prefers pure, single molecule therapeutics to botanical extracts. In 1996, California legalized botanical cannabis for medicinal use.484 This goes against the western medicine doctrine and begs the question: as pharmacology, chemistry, and biochemistry all advance, will western medicine move towards curated complex mixtures of small molecules that emulate botanical tinctures? Perhaps, the cannabinoids will represent a case study that other scheduled substances will follow. Evidentiary developments indicate that Cannabis components such as 8 modulate adverse effects of 7, a phenomenon commonly described as the entourage effect.396,397 However the majority of cannabinoid compounds do not have published pharmacological data. And, the molecules that are well-studied are still controlled substances. For example, 8 is a non-euphoric compound with a safe pharmacokinetic profile, yet it is a controlled substance. As Di Marzo and coworkers say, “This anomaly makes clear that, despite considerable scientific evidence, talks about legalization, and the many industrial and medical uses of the plant, stigma around cannabis still hinders the conclusive assessment of the therapeutic potential of the plant’s most abundant components. Further education is needed to reduce the negative impact of these factors on research.”485
Undoubtedly, this paradigm extends beyond cannabinoids, as immense untapped therapeutic potential exists in regards to the other compounds described. As Western medicine has long cannibalized indigenous discoveries, however, we must prioritize the rights of practitioners of traditional medicine as we unpack the potential applications of these natural products. Ironically, the same reductionist vision of single molecule therapeutics has resulted in a persistent rejection of holistic approaches to medicine. However, the tides are changing, and practitioners of science are more readily acknowledging the limitations of reductionist frameworks. In the same way that we have reduced the extraordinary complexity of metabolic networks into linear biosynthetic pathways, reductionism should be used to complement holism. From this perspective, medicine, culture, and technology can all be beneficiaries of a comprehensive understanding of Nature’s biosynthetic routes to psychoactive natural products.
Fig. 31.

Coffea arabica (the dominant coffee cultivar) contains ~1.2 percent dry weight caffeine.
Fig. 45.
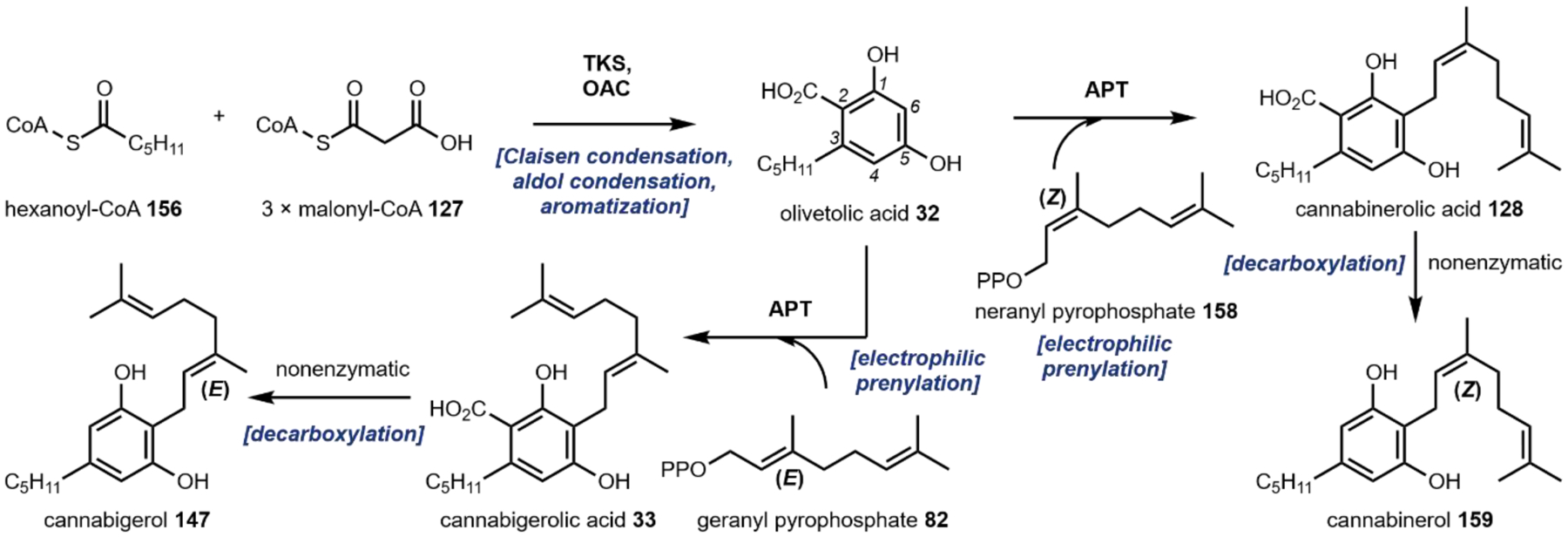
Biosynthesis of cannabigerol (147) and cannabinerol (159) from hexanoyl-CoA 156 and malonyl-CoA 127.
Fig. 46.
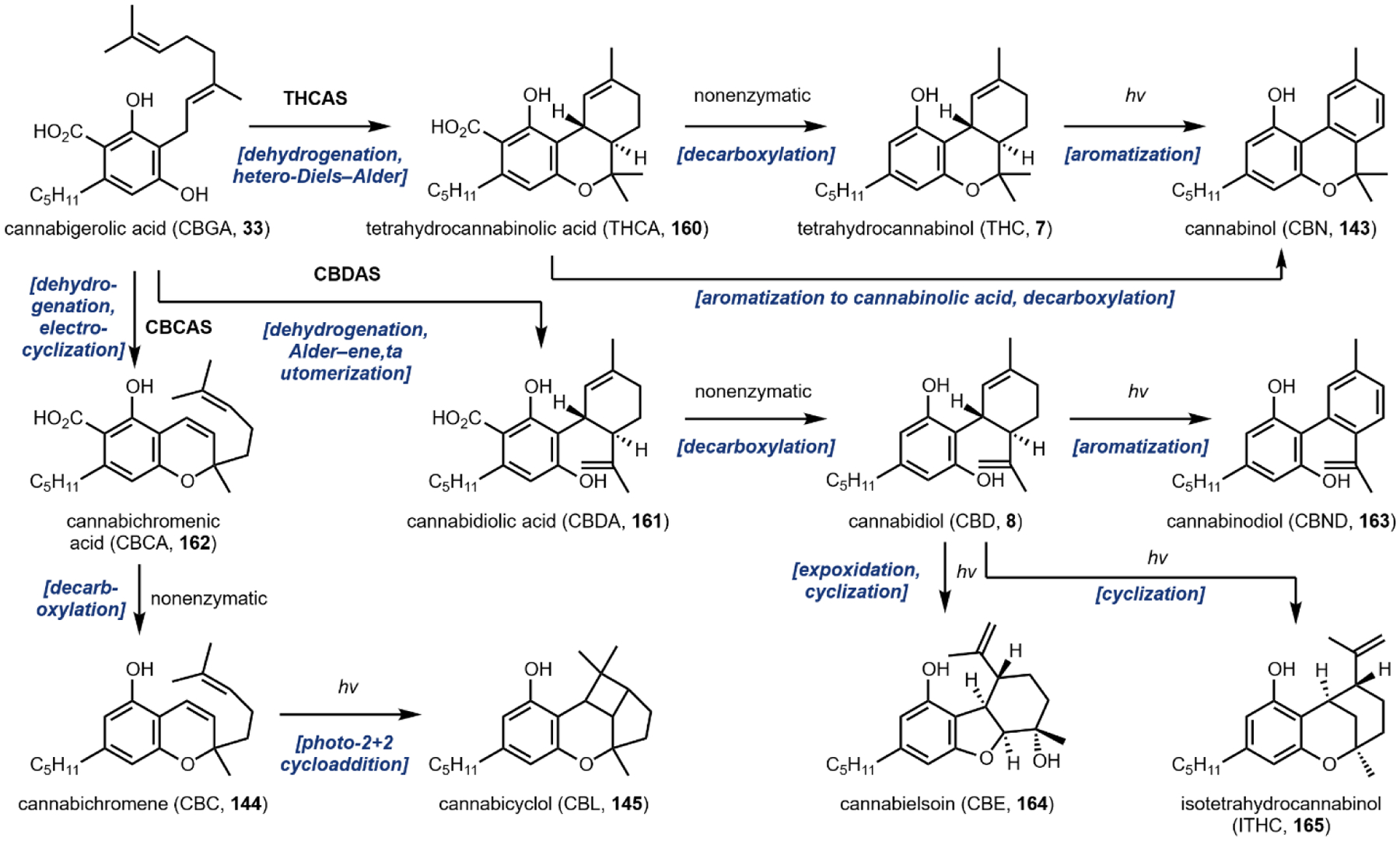
Biosynthesis of tetrahydrocannabinol (7), cannabidiol (8), cannabichromene (144), and further nonenzymatic derivatized products.
Fig. 53.
Phthalide isoquinoline opioid natural products, noscapine (174) and narceine (175).
Fig. 55.
Examples of berberine opioids, berberine (179) and sanguinarine (180).
Fig. 59. Mitragyna speciosa cultivars may contain up to one percent dry weight mitragynine.
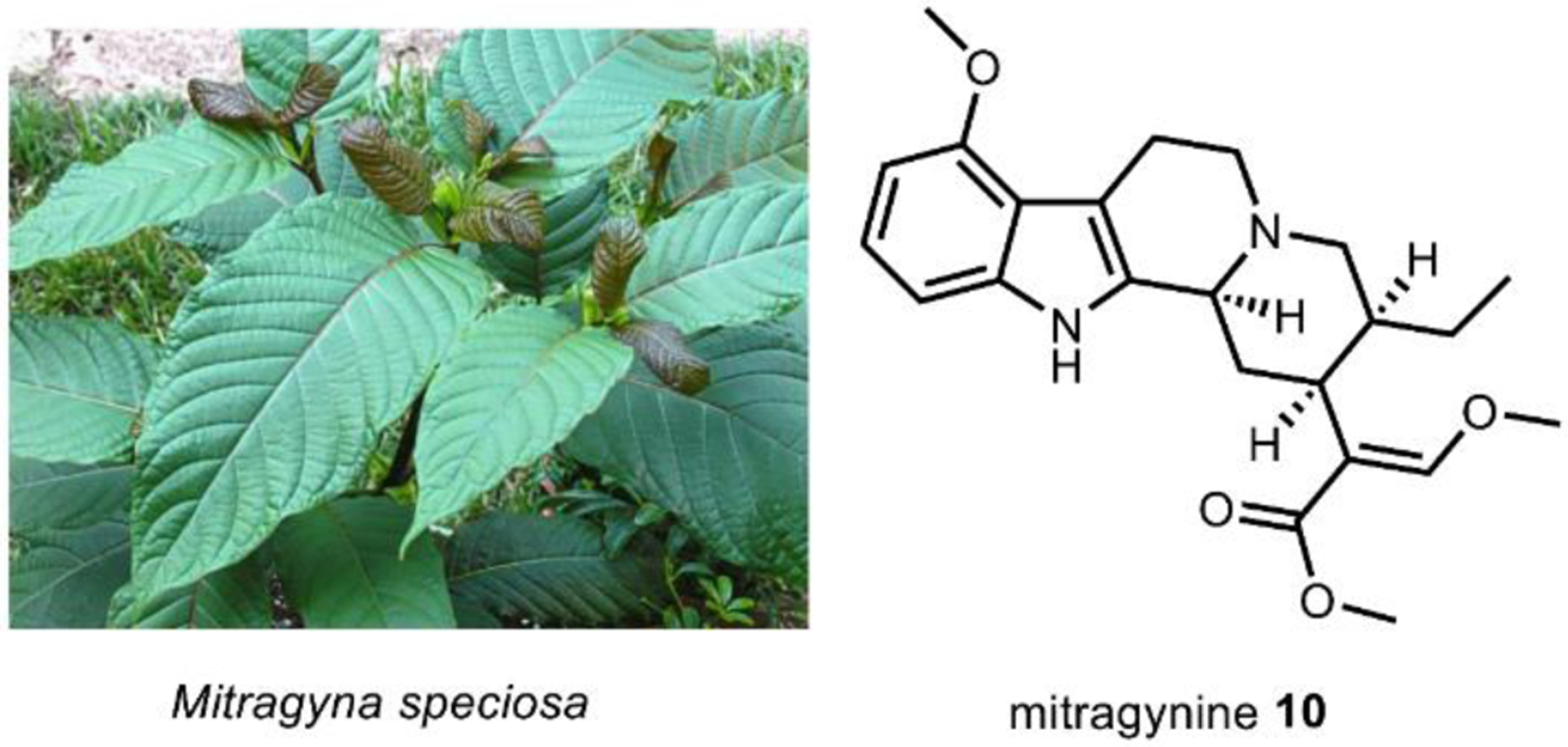
Image on left courtesy of Thor Porre via CC-3.0.
8. Acknowledgements
Related work in the Tang lab is supported by NIH 1R01AT010001. C.S. Jamieson is grateful for additional funding from the Saul Winstein fellowship, the Foote fellowship, and a UCLA Dissertation Year Fellowship. J. Misa is supported by NIGMS-funded predoctoral fellowship T32 GM136614. The authors wish to acknowledge the indigenous peoples whose immense knowledge of the natural world has facilitated the study of psychoactive natural products. We recognize that many of the scientific discoveries described in this review would not be possible were it not for the expert observations of indigenous people of the Sierra Mazateca, indigenous groups in the Amazon basin, indigenous populations in the Andes, indigenous Bwiti practitioners in Gabon, and countless others to whom we pay our respects.
Biographies

Cooper S. Jamieson was born in New York, NY and raised in San Luis Obispo, CA. In 2016, he received a B.A. in Chemistry and a B.A. in Art from Lewis & Clark College in Portland, OR. He moved to far-West Marfa, Texas and worked in art conservation at the Chinati Foundation. Now, Cooper has returned to academia and is in Los Angeles, CA finishing his Ph.D. under the direction of Prof. K. N. Houk and Prof. Yi Tang at UCLA on pericyclases and pericyclic reactions in nature.

Joshua Misa was born and raised in Riverside, CA. He received his B.S. in Chemical Engineering with an emphasis in Biochemical Engineering from the University of California, Riverside (UCR) in 2018, graduating magna cum laude. At UCR he worked in Prof. Ian Wheeldon’s lab on the development of CRISPR tools for engineering non-conventional yeasts as a Chancellor’s Research Fellow. Joshua is currently a Ph.D. candidate working under Prof. Yi Tang on developing yeast-based platforms for production of plant natural products and new, biosynthetic analogues.

Yi Tang received his undergraduate degree in Chemical Engineering and Material Science from Penn State University. He received his Ph.D. in Chemical Engineering from California Institute of Technology in 2002. After NIH postdoctoral training in Chemical Biology at Stanford University, he started his independent career at the University of California Los Angeles in 2004. He is currently Professor in the Department of Chemical and Biomolecular Engineering at UCLA, and holds joint appointments in the Department of Chemistry and Biochemistry; and Department of Bioengineering. His lab is interested in natural product biosynthesis, biocatalysis and protein engineering.

John M. Billingsley grew up in Cambria, NY, and attended the University at Buffalo pursuing a B.S. in Chemical Engineering. There, John developed a budding interest in natural product biosynthesis during an internship in Prof. Andrew Gulick’s lab at the Hauptmann-Woodward Medical Research Institute. John received a Ph.D. in Chemical Engineering from UCLA in 2019 under the guidance of Yi Tang, where they investigated and engineered the biosynthesis of plant and fungal alkaloids. John currently lives in West Hollywood, is a Visiting Scientist at UCLA and Principal Scientist at Invizyne Technologies in Monrovia, CA.
Footnotes
Conflicts of interest
The authors declare the following competing financial interest(s):
John Billingsley is an employee of Invizyne, Technologies (Monrovia, CA, USA), a company seeking to commercialize synthetic biochemistry.
9 References
- 1.Merlin MD, Econ. Bot, 2003, 57, 295–323. [Google Scholar]
- 2.Guerra-Doce E, Time Mind, 2015, 8, 91–112. [Google Scholar]
- 3.Buenz EJ, Verpoorte R and Bauer BA, Annu. Rev. Pharmacol. Toxicol, 2018, 58, 509–530. [DOI] [PubMed] [Google Scholar]
- 4.Efferth T, Banerjee M, Paul NW, Abdelfatah S, Arend J, Elhassan G, Hamdoun S, Hamm R, Hong C, Kadioglu O, Naß J, Ochwangi D, Ooko E, Ozenver N, Saeed MEM, Schneider M, Seo EJ, Wu CF, Yan G, Zeino M, Zhao Q, Abu-Darwish MS, Andersch K, Alexie G, Bessarab D, Bhakta-Guha D, Bolzani V, Dapat E, Donenko FV, Efferth M, Greten HJ, Gunatilaka L, Hussein AA, Karadeniz A, Khalid HE, Kuete V, Lee IS, Liu L, Midiwo J, Mora R, Nakagawa H, Ngassapa O, Noysang C, Omosa LK, Roland FH, Shahat AA, Saab A, Saeed EM, Shan L and Titinchi SJJ, Phytomedicine, 2016, 23, 166–173. [DOI] [PubMed] [Google Scholar]
- 5.George JR, Michaels TI, Sevelius J and Williams MT, J. Psychedelic Stud, 2019, 4, 4–15. [Google Scholar]
- 6.Gootenberg P, Am. Anthropol, 2005, 107, 152–153. [Google Scholar]
- 7.Kean S, Science (80-.), 2019, 364, 16–20. [DOI] [PubMed] [Google Scholar]
- 8.Provine DM, Annu. Rev. Law Soc. Sci, 2011, 7, 41–60. [Google Scholar]
- 9.Sullivan RJ and Hagen EH, Addiction, 2002, 97, 389–400. [DOI] [PubMed] [Google Scholar]
- 10.Martini A, J. Wine Res, 1993, 4, 165–176. [Google Scholar]
- 11.Brito GSL, Cad. Saude Publica, 1994, 10, 403–405. [Google Scholar]
- 12.Dias DA, Urban S and Roessner U, Metabolites, 2012, 2, 303–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson R, J. Chem. Soc. Trans, 1917, 111, 762–768. [Google Scholar]
- 14.Kornfeld EC, Fornefeld EJ, Bruce Kline G, Mann MJ, Morrison DE, Jones RG and Woodward RB, J. Am. Chem. Soc, 1956, 78, 3087–3114. [Google Scholar]
- 15.Walsh CT and Tang Y, Natural Product Biosynthesis: Chemical and Enzymatic Logic, Royal Society of Chemistry, 2017. [Google Scholar]
- 16.Pluskal T and Weng JK, Chem. Soc. Rev, 2018, 47, 1592–1637. [DOI] [PubMed] [Google Scholar]
- 17.Valenstein ES, Brain Cogn., 2002, 49, 73–95. [DOI] [PubMed] [Google Scholar]
- 18.Murnane KS, in Progress in Brain Research, 2018, vol. 242, pp. 25–67. [DOI] [PubMed] [Google Scholar]
- 19.Nadim F and Bucher D, Curr. Opin. Neurobiol, 2014, 29, 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bracco L and Kearsey J, Trends Biotechnol., 2003, 21, 346–353. [DOI] [PubMed] [Google Scholar]
- 21.Contet C, Kieffer BL and Befort K, Curr. Opin. Neurobiol, 2004, 14, 370–378. [DOI] [PubMed] [Google Scholar]
- 22.Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg SA, Ernsberger P and Rothman RB, Proc. Natl. Acad. Sci. U. S. A, 2002, 99, 11934–11939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nutt D, PLOS Biol., 2015, 13, e1002047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carhart-Harris RL, Bolstridge M, Day CMJ, Rucker J, Watts R, Erritzoe DE, Kaelen M, Giribaldi B, Bloomfield M, Pilling S, Rickard JA, Forbes B, Feilding A, Taylor D, Curran HV and Nutt DJ, Psychopharmacology (Berl)., 2018, 235, 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carhart-Harris RL, Leech R, Williams TM, Erritzoe D, Abbasi N, Bargiotas T, Hobden P, Sharp DJ, Evans J, Feilding A, Wise RG and Nutt DJ, Br. J. Psychiatry, 2012, 200, 238–244. [DOI] [PubMed] [Google Scholar]
- 26.Cameron LP, Tombari RJ, Lu J, Pell AJ, Hurley ZQ, Ehinger Y, Vargas MV, McCarroll MN, Taylor JC, Myers-Turnbull D, Liu T, Yaghoobi B, Laskowski LJ, Anderson EI, Zhang G, Viswanathan J, Brown BM, Tjia M, Dunlap LE, Rabow ZT, Fiehn O, Wulff H, McCorvy JD, Lein PJ, Kokel D, Ron D, Peters J, Zuo Y and Olson DE, Nature, 2021, 589, 474–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salim V, Yu F, Altarejos J and De Luca V, Plant J., 2013, 76, 754–765. [DOI] [PubMed] [Google Scholar]
- 28.Barone JJ and Roberts HR, Food Chem. Toxicol, 1996, 34, 119–129. [DOI] [PubMed] [Google Scholar]
- 29.Fiore MC, Smith SS, Jorenby DE and Baker TB, JAMA J. Am. Med. Assoc, 1994, 271, 1940–1947. [PubMed] [Google Scholar]
- 30.Grinspoon L and Bakalar JB, J. Ethnopharmacol, 1981, 3, 149–159. [DOI] [PubMed] [Google Scholar]
- 31.Amin MR and Ali DW, in Advances in Experimental Medicine and Biology, 2019, vol. 1162, pp. 151–165. [DOI] [PubMed] [Google Scholar]
- 32.Rosenblum A, Marsch LA, Joseph H and Portenoy RK, Exp. Clin. Psychopharmacol, 2008, 16, 405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iijima Y, Gang DR, Fridman E, Lewinsohn E and Pichersky E, Plant Physiol., 2004, 134, 370–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang M-C, Zou Y, Watanabe K, Walsh CT and Tang Y, Chem. Rev, 2017, 117, 5226–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walsh CT and Moore BS, Angew. Chemie Int. Ed, 2019, 58, 6846–6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin C-I, McCarty RM and Liu H, Angew. Chemie Int. Ed, 2017, 56, 3446–3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walsh CT and Tang Y, The Chemical Biology of Human Vitamins, Royal Society of Chemistry, 2018. [Google Scholar]
- 38.Blicke FF, Org. React, 2011, 303–341.
- 39.Leete E, Planta Med, 1990, 56, 339–352. [DOI] [PubMed] [Google Scholar]
- 40.Kohnen-Johannsen K and Kayser O, Molecules, 2019, 24, 796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Allen JRF and Holmstedt BR, Phytochemistry, 1980, 19, 1573–1582. [Google Scholar]
- 42.Maresh JJ, Giddings L-A, Friedrich A, Loris EA, Panjikar S, Trout BL, Stöckigt J, Peters B and O’Connor SE, J. Am. Chem. Soc, 2008, 130, 710–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ilari A, Franceschini S, Bonamore A, Arenghi F, Botta B, Macone A, Pasquo A, Bellucci L and Boffi A, J. Biol. Chem, 2009, 284, 897–904. [DOI] [PubMed] [Google Scholar]
- 44.Walsh CT, Tu BP and Tang Y, Chem. Rev, 2018, 118, 1460–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miettinen K, Dong L, Navrot N, Schneider T, Burlat V, Pollier J, Woittiez L, Van Der Krol S, Lugan R, Ilc T, Verpoorte R, Oksman-Caldentey KM, Martinoia E, Bouwmeester H, Goossens A, Memelink J and Werck-Reichhart D, Nat. Commun, 2014, 5, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Metzger U, Schall C, Zocher G, Unsöld I, Stec E, Li S-M, Heide L and Stehle T, Proc. Natl. Acad. Sci, 2009, 106, 14309 LP–14314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tanner ME, Nat. Prod. Rep, 2015, 32, 88–101. [DOI] [PubMed] [Google Scholar]
- 48.Grundmann A and Li SM, Microbiology, 2005, 151, 2199–2207. [DOI] [PubMed] [Google Scholar]
- 49.Kato H, Yoshida T, Tokue T, Nojiri Y, Hirota H, Ohta T, Williams RM and Tsukamoto S, Angew. Chemie - Int. Ed, 2007, 46, 2254–2256. [DOI] [PubMed] [Google Scholar]
- 50.Li SM, Nat. Prod. Rep, 2010, 27, 57–78. [DOI] [PubMed] [Google Scholar]
- 51.Walsh CT, ACS Chem. Biol, 2014, 9, 2718–2728. [DOI] [PubMed] [Google Scholar]
- 52.Lin HC, Chiou G, Chooi YH, McMahon TC, Xu W, Garg NK and Tang Y, Angew. Chemie - Int. Ed, 2015, 54, 3004–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Valliere MA, Korman TP, Woodall NB, Khitrov GA, Taylor RE, Baker D and Bowie JU, Nat. Commun, 2019, 10, 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Walsh CT and Wencewicz TA, Nat. Prod. Rep, 2013, 30, 175–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yamamoto H, Katano N, Ooi A and Inoue K, Phytochemistry, 2000, 53, 7–12. [DOI] [PubMed] [Google Scholar]
- 56.Irmler S, Schröder G, St-Pierre B, Crouch NP, Hotze M, Schmidt J, Strack D, Matern U and Schröder J, Plant J., 2000, 24, 797–804. [DOI] [PubMed] [Google Scholar]
- 57.Kishimoto S, Sato M, Tsunematsu Y and Watanabe K, Mol., 2016, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gesell A, Rolf M, Ziegler J, Chávez MLD, Huang FC and Kutchan TM, J. Biol. Chem, 2009, 284, 24432–24442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shipkowski KA, Betz JM, Birnbaum LS, Bucher JR, Coates PM, Hopp DC, MacKay D, Oketch-Rabah H, Walker NJ, Welch C and Rider CV, Food Chem. Toxicol, 2018, 118, 963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hollingsworth RG, Armstrong JW and Campbell E, Ann. Appl. Biol, 2003, 142, 91–97. [Google Scholar]
- 61.Leonard E, Runguphan W, O’Connor S and Prather KJ, Nat. Chem. Biol, 2009, 5, 292–300. [DOI] [PubMed] [Google Scholar]
- 62.Cravens A, Payne J and Smolke CD, Nat. Commun, 2019, 10, 2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA and Smith HO, Nat. Methods, 2009, 6, 343–345. [DOI] [PubMed] [Google Scholar]
- 64.Gresham D, Dunham MJ and Botstein D, Nat. Rev. Genet, 2008, 9, 291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Urnov FD, Rebar EJ, Holmes MC, Zhang HS and Gregory PD, Nat. Rev. Genet, 2010, 11, 636–646. [DOI] [PubMed] [Google Scholar]
- 66.Kosuri S and Church GM, Nat. Methods, 2014, 11, 499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang Z, Gerstein M and Snyder M, Nat. Rev. Genet, 2009, 10, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hsu PD, Lander ES and Zhang F, Cell, 2014, 157, 1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Khalil AS and Collins JJ, Nat. Rev. Genet, 2010, 11, 367–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Osbourn A, Plant Physiol, 2010, 154, 531–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Field B, Fiston-Lavier AS, Kemen A, Geisler K, Quesneville H and Osbourn AE, Proc. Natl. Acad. Sci. U. S. A, 2011, 108, 16116–16121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu Z, Cheema J, Vigouroux M, Hill L, Reed J, Paajanen P, Yant L and Osbourn A, Nat. Commun, 2020, 11, 5354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Srinivasan P and Smolke CD, Nature, 2020, 585, 614–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dastmalchi M, Chen X, Hagel JM, Chang L, Chen R, Ramasamy S, Yeaman S and Facchini PJ, Nat. Chem. Biol, 2019, 15, 384–390. [DOI] [PubMed] [Google Scholar]
- 75.Luo X, Reiter MA, d’Espaux L, Wong J, Denby CM, Lechner A, Zhang Y, Grzybowski AT, Harth S, Lin W, Lee H, Yu C, Shin J, Deng K, Benites VT, Wang G, Baidoo EEK, Chen Y, Dev I, Petzold CJ and Keasling JD, Nature, 2019, 567, 123–126. [DOI] [PubMed] [Google Scholar]
- 76.Brown S, Clastre M, Courdavault V and O’Connor SE, Proc. Natl. Acad. Sci. U. S. A, 2015, 112, 3205–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Galanie S, Thodey K, Trenchard IJ, Interrante MF and Smolke CD, Science (80-.), 2015, 349, 1095–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Deloache WC, Russ ZN, Narcross L, Gonzales AM, Martin VJJ and Dueber JE, Nat. Chem. Biol, 2015, 11, 465–471. [DOI] [PubMed] [Google Scholar]
- 79.Packer MS and Liu DR, Nat. Rev. Genet, 2015, 16, 379–394. [DOI] [PubMed] [Google Scholar]
- 80.Liu N, Santala S and Stephanopoulos G, Curr. Opin. Biotechnol, 2020, 62, 15–21. [DOI] [PubMed] [Google Scholar]
- 81.Adams AM, Kaplan NA, Wei Z, Brinton JD, Monnier CS, Enacopol AL, Ramelot TA and Jones JA, Metab. Eng, 2019, 56, 111–119. [DOI] [PubMed] [Google Scholar]
- 82.Billingsley JM, DeNicola AB, Barber JS, Tang MC, Horecka J, Chu A, Garg NK and Tang Y, Metab. Eng, 2017, 44, 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Copeland WB, Bartley BA, Chandran D, Galdzicki M, Kim KH, Sleight SC, Maranas CD and Sauro HM, Metab. Eng, 2012, 14, 270–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Finnigan W, Hepworth LJ, Flitsch SL and Turner NJ, Nat. Catal, 2021, 4, 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sheppard MJ, Kunjapur AM, Wenck SJ and Prather KLJ, Nat. Commun, 2014, 5, 5031. [DOI] [PubMed] [Google Scholar]
- 86.Bachmann BO, Nat. Chem. Biol, 2010, 6, 390–393. [DOI] [PubMed] [Google Scholar]
- 87.Casini A, Chang FY, Eluere R, King AM, Young EM, Dudley QM, Karim A, Pratt K, Bristol C, Forget A, Ghodasara A, Warden-Rothman R, Gan R, Cristofaro A, Borujeni AE, Ryu MH, Li J, Kwon YC, Wang H, Tatsis E, Rodriguez-Lopez C, O’Connor S, Medema MH, Fischbach MA, Jewett MC, Voigt C and Gordon DB, J. Am. Chem. Soc, 2018, 140, 4302–4316. [DOI] [PubMed] [Google Scholar]
- 88.Carbonell P, Le Feuvre R, Takano E and Scrutton NS, Synth. Biol, 2020, 5, ysaa020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li M, Sun Y, Pan SA, Deng WW, Yu O and Zhang Z, RSC Adv., 2017, 7, 56382–56389. [Google Scholar]
- 90.Nakagawa A, Matsumura E, Koyanagi T, Katayama T, Kawano N, Yoshimatsu K, Yamamoto K, Kumagai H, Sato F and Minami H, Nat. Commun, 2016, 7, 10390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Milne N, Thomsen P, Mølgaard Knudsen N, Rubaszka P, Kristensen M and Borodina I, Metab. Eng, 2020, 60, 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.McKeague M, Wang YH, Cravens A, Win MN and Smolke CD, Metab. Eng, 2016, 38, 191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Skellam E, Trends Biotechnol., 2019, 37, 416–427. [DOI] [PubMed] [Google Scholar]
- 94.Hoefgen S, Lin J, Fricke J, Stroe MC, Mattern DJ, Kufs JE, Hortschansky P, Brakhage AA, Hoffmeister D and Valiante V, Metab. Eng, 2018, 48, 44–51. [DOI] [PubMed] [Google Scholar]
- 95.Ohashi M, Liu F, Hai Y, Chen M, Tang M, Yang Z, Sato M, Watanabe K, Houk KN and Tang Y, Nature, 2017, 549, 502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Reed J, Stephenson MJ, Miettinen K, Brouwer B, Leveau A, Brett P, Goss RJM, Goossens A, O’Connell MA and Osbourn A, Metab. Eng, 2017, 42, 185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lau W and Sattely ES, Science (80-.), 2015, 349, 1224–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pluskal T, Torrens-Spence MP, Fallon TR, De Abreu A, Shi CH and Weng JK, Nat. Plants, 2019, 5, 867–878. [DOI] [PubMed] [Google Scholar]
- 99.Nett RS, Lau W and Sattely ES, Nature, 2020, 584, 148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Opgenorth PH, Korman TP and Bowie JU, Nat. Chem. Biol, 2016, 12, 393–395. [DOI] [PubMed] [Google Scholar]
- 101.Korman TP, Opgenorth PH and Bowie JU, Nat. Commun, 2017, 8, 15526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Valliere MA, Korman TP, Arbing MA and Bowie JU, Nat. Chem. Biol, 2020, 16, 1427–1433. [DOI] [PubMed] [Google Scholar]
- 103.Roze LV, Chanda A and Linz JE, Fungal Genet. Biol, 2011, 48, 35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dueber JE, Wu GC, Malmirchegini GR, Moon TS, Petzold CJ, Ullal AV, Prather KLJ and Keasling JD, Nat. Biotechnol, 2009, 27, 753–759. [DOI] [PubMed] [Google Scholar]
- 105.Hammer SK and Avalos JL, Nat. Chem. Biol, 2017, 13, 823–832. [DOI] [PubMed] [Google Scholar]
- 106.Renault H, Bassard JE, Hamberger B and Werck-Reichhart D, Curr. Opin. Plant Biol, 2014, 19, 27–34. [DOI] [PubMed] [Google Scholar]
- 107.Leonard E and Koffas MAG, Appl. Environ. Microbiol, 2007, 73, 7246–7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ping Y, Li X, Xu B, Wei W, Wei W, Kai G, Zhou Z and Xiao Y, ACS Synth. Biol, 2019, 8, 257–263. [DOI] [PubMed] [Google Scholar]
- 109.Hawkins KM and Smolke CD, Nat. Chem. Biol, 2008, 4, 564–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bowie JU, Sherkhanov S, Korman TP, Valliere MA, Opgenorth PH and Liu H, Trends Biotechnol., 2020, 38, 766–778. [DOI] [PubMed] [Google Scholar]
- 111.Clomburg JM, Crumbley AM and Gonzalez R, Science (80-.), 2017, 355, aag0804. [DOI] [PubMed] [Google Scholar]
- 112.Kumar V, Bhalla A and Rathore AS, Biotechnol. Prog, 2014, 30, 86–99. [DOI] [PubMed] [Google Scholar]
- 113.Gilman J, Walls L, Bandiera L and Menolascina F, ACS Synth. Biol, 2021, 10, 1–18. [DOI] [PubMed] [Google Scholar]
- 114.Xia P-F, Ling H, Foo JL and Chang MW, Biotechnol. Adv, 2019, 37, 107393. [DOI] [PubMed] [Google Scholar]
- 115.Win MN, Klein JS and Smolke CD, Nucleic Acids Res., 2006, 34, 5670–5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Portnoy VA, Bezdan D and Zengler K, Curr. Opin. Biotechnol, 2011, 22, 590–594. [DOI] [PubMed] [Google Scholar]
- 117.Nichols DE, Pharmacol. Rev, 2016, 68, 264–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.O’Neill-Dee C, Spiller HA, Casavant MJ, Kistamgari S, Chounthirath T, Smith GA, O’Neill-Dee C, Spiller HA, Casavant MJ, Kistamgari S, Chounthirath T and Smith GA, Clin. Toxicol, 2020, 58, 813–820. [DOI] [PubMed] [Google Scholar]
- 119.Tylš F, Páleníček T and Horáček J, Eur. Neuropsychopharmacol, 2014, 24, 342–356. [DOI] [PubMed] [Google Scholar]
- 120.Halberstadt AL, Behav. Brain Res, 2015, 277, 99–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nichols DE and Nichols CD, Chem. Rev, 2008, 108, 1614–1641. [DOI] [PubMed] [Google Scholar]
- 122.Beliveau V, Ganz M, Feng L, Ozenne B, Højgaard L, Fisher PM, Svarer C, Greve DN and Knudsen GM, J. Neurosci, 2017, 37, 120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.López-Giménez JF and González-Maeso J, in Current Topics in Behavioral Neurosciences, Springer Verlag, 2017, vol. 36, pp. 45–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nichols DE, Wiley Interdiscip. Rev. Membr. Transp. Signal, 2012, 1, 559–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wacker D, Wang S, McCorvy JD, Betz RM, Venkatakrishnan AJ, Levit A, Lansu K, Schools ZL, Che T, Nichols DE, Shoichet BK, Dror RO and Roth BL, Cell, 2017, 168, 377–389.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Carbonaro TM and Gatch MB, Brain Res. Bull, 2016, 126, 74–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Miller MJ, Albarracin-Jordan J, Moore C and Capriles JM, Proc. Natl. Acad. Sci. U. S. A, 2019, 166, 11207–11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Carod-Artal FJ, Neurol. (English Ed, 2015, 30, 42–49. [DOI] [PubMed] [Google Scholar]
- 129.Weil AT and Davis W, J. Ethnopharmacol, 1994, 41, 1–8. [DOI] [PubMed] [Google Scholar]
- 130.Most A, Bufo alvarius: the psychedelic toad of the Sonoran Desert, Venom Press, 1984. [Google Scholar]
- 131.de Lima OG, Arq. do Inst. Pesqui. Agronômicas, 1946, 4, 45–80. [Google Scholar]
- 132.Szára S, Experientia, 1956, 12, 441–442. [DOI] [PubMed] [Google Scholar]
- 133.Barker SA, Front. Neurosci, 2018, 12, 536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Szabo A, Kovacs A, Riba J, Djurovic S, Rajnavolgyi E and Frecska E, Front. Neurosci, 2016, 10, 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cameron LP, Benson CJ, Dunlap LE and Olson DE, ACS Chem. Neurosci, 2018, 9, 1582–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ly C, Greb AC, Cameron LP, Wong JM, Barragan EV, Wilson PC, Burbach KF, Soltanzadeh Zarandi S, Sood A, Paddy MR, Duim WC, Dennis MY, McAllister AK, Ori-McKenney KM, Gray JA and Olson DE, Cell Rep., 2018, 23, 3170–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Djura P, Stierle DB, Sullivan B, Faulkner DJ, Arnold E and Clardy J, J. Org. Chem, 1980, 45, 1435–1441. [Google Scholar]
- 138.Longeon A, Copp BR, Quévrain E, Roué M, Kientz B, Cresteil T, Petek S, Debitus C and Bourguet-Kondracki ML, Mar. Drugs, 2011, 9, 879–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kochanowska AJ, Rao KV, Childress S, El-Alfy A, Matsumoto RR, Kelly M, Stewart GS, Sufka KJ and Hamann MT, J. Nat. Prod, 2008, 71, 186–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.LOVENBERG W, WEISSBACH H and UDENFRIEND S, J. Biol. Chem, 1962, 237, 89–93. [PubMed] [Google Scholar]
- 141.Axelrod J, Science (80-.), 1961, 134, 343–343. [DOI] [PubMed] [Google Scholar]
- 142.Thompson MA and Weinshilboum RM, J. Biol. Chem, 1998, 273, 34502–34510. [DOI] [PubMed] [Google Scholar]
- 143.Hofmann A, Heim R, Brack A, Kobel H, Frey A, Ott H, Petrzilka T and Troxler F, Helv. Chim. Acta, 1959, 42, 1557–1572. [Google Scholar]
- 144.Nichols DE, J. Antibiot. (Tokyo), 2020, 73, 679–686. [DOI] [PubMed] [Google Scholar]
- 145.Hasler F, Bourquin D, Brenneisen R and Vollenweider FX, J. Pharm. Biomed. Anal, 2002, 30, 331–339. [DOI] [PubMed] [Google Scholar]
- 146.Hasler F, Grimberg U, Benz MA, Huber T and Vollenweider FX, Psychopharmacology (Berl)., 2004, 172, 145–156. [DOI] [PubMed] [Google Scholar]
- 147.Mahapatra A and Gupta R, Ther. Adv. Psychopharmacol, 2017, 7, 54–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Grob CS, Danforth AL, Chopra GS, Hagerty M, McKay CR, Halberstad AL and Greer GR, Arch. Gen. Psychiatry, 2011, 68, 71–78. [DOI] [PubMed] [Google Scholar]
- 149.Johnson MW, Garcia-Romeu A, Cosimano MP and Griffiths RR, J. Psychopharmacol, 2014, 28, 983–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Agurell S and Nilsson JL, Acta Chem. Scand, 1968, 22, 1210–1218. [DOI] [PubMed] [Google Scholar]
- 151.Fricke J, Blei F and Hoffmeister D, Angew. Chemie - Int. Ed, 2017, 56, 12352–12355. [DOI] [PubMed] [Google Scholar]
- 152.Wakimoto T, Egami Y, Nakashima Y, Wakimoto Y, Mori T, Awakawa T, Ito T, Kenmoku H, Asakawa Y, Piel J and Abe I, Nat. Chem. Biol, 2014, 10, 648–655. [DOI] [PubMed] [Google Scholar]
- 153.Zhu S, Hegemann JD, Fage CD, Zimmermann M, Xie X, Linne U and Marahiel MA, J. Biol. Chem, 2016, 291, 13662–13678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Reynolds HT, Vijayakumar V, Gluck-Thaler E, Korotkin HB, Matheny PB and Slot JC, Evol. Lett, 2018, 2, 88–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Awan AR, Winter JM, Turner D, Shaw WM, Suz LM, Bradshaw AJ, Ellis T and Dentinger BTM, bioRxiv, 2018, 374199. [Google Scholar]
- 156.Paddon CJ, Westfall PJ, Pitera DJ, Benjamin K, Fisher K, McPhee D, Leavell MD, Tai A, Main A, Eng D, Polichuk DR, Teoh KH, Reed DW, Treynor T, Lenihan J, Jiang H, Fleck M, Bajad S, Dang G, Dengrove D, Diola D, Dorin G, Ellens KW, Fickes S, Galazzo J, Gaucher SP, Geistlinger T, Henry R, Hepp M, Horning T, Iqbal T, Kizer L, Lieu B, Melis D, Moss N, Regentin R, Secrest S, Tsuruta H, Vazquez R, Westblade LF, Xu L, Yu M, Zhang Y, Zhao L, Lievense J, Covello PS, Keasling JD, Reiling KK, Renninger NS and Newman JD, Nature, 2013, 496, 528–532. [DOI] [PubMed] [Google Scholar]
- 157.Osamu S, Katsumata Y and Masakazu O, Biochem. Pharmacol, 1981, 30, 1353–1358. [PubMed] [Google Scholar]
- 158.McKenna D and Riba J, in Current Topics in Behavioral Neurosciences, Springer Verlag, 2018, vol. 36, pp. 283–311. [DOI] [PubMed] [Google Scholar]
- 159.Estrella-Parra EA, Almanza-Pérez JC and Alarcón-Aguilar FJ, Nat. Products Bioprospect, 2019, 9, 251–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ayala Flores F and Lewis WH, Econ. Bot, 1978, 32, 154–156. [Google Scholar]
- 161.Morales-García JA, De La Fuente Revenga M, Alonso-Gil S, Rodríguez-Franco MI, Feilding A, Perez-Castillo A and Riba J, Sci. Rep, 2017, 7, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Callaway JC, Brito GS and Neves ES, J. Psychoactive Drugs, 2005, 37, 145–150. [DOI] [PubMed] [Google Scholar]
- 163.Brito-da-costa AM, Dias-da-silva D, Gomes NGM, Dinis-oliveira RJ and Madureira-carvalho Á, Pharmaceuticals, 2020, 13, 1–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Blei F, Dörner S, Fricke J, Baldeweg F, Trottmann F, Komor A, Meyer F, Hertweck C and Hoffmeister D, Chem. - A Eur. J, 2020, 26, 729–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Stolle K and Gröger D, Arch. Pharm. (Weinheim), 1968, 301, 561–571. [DOI] [PubMed] [Google Scholar]
- 166.Nettleship L and Slaytor M, Phytochemistry, 1974, 13, 735–742. [Google Scholar]
- 167.Dewick PM, Medicinal Natural Products: A Biosynthetic Approach: Third Edition, Second., 2009.
- 168.Nichols DE, Pharmacol. Ther, 2004, 101, 131–181. [DOI] [PubMed] [Google Scholar]
- 169.Lorenz N, Haarmann T, Pažoutová S, Jung M and Tudzynski P, Phytochemistry, 2009, 70, 1822–1832. [DOI] [PubMed] [Google Scholar]
- 170.Wallwey C and Li SM, Nat. Prod. Rep, 2011, 28, 496–510. [DOI] [PubMed] [Google Scholar]
- 171.van Dongen PWJ and de Groot ANJA, Eur. J. Obstet. Gynecol. Reprod. Biol, 1995, 60, 109–116. [DOI] [PubMed] [Google Scholar]
- 172.Jakubczyk D, Cheng JZ and O’Connor SE, Nat. Prod. Rep, 2014, 31, 1328–1338. [DOI] [PubMed] [Google Scholar]
- 173.Gerhards N, Neubauer L, Tudzynski P and Li SM, Toxins (Basel)., 2014, 6, 3281–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sharma N, Sharma V, Manikyam H and Krishna A, European J. Med. Plants, 2016, 14, 1–17. [Google Scholar]
- 175.Gröcer D and Floss HG, Alkaloids Chem. Biol, 1998, 50, 171–218. [Google Scholar]
- 176.Lee SL, Floss HG and Heinstein P, Arch. Biochem. Biophys, 1976, 177, 84–94. [DOI] [PubMed] [Google Scholar]
- 177.Yin WB, Grundmann A, Cheng J and Li SM, J. Biol. Chem, 2009, 284, 100–109. [DOI] [PubMed] [Google Scholar]
- 178.Winkelblech J and Li SM, ChemBioChem, 2014, 15, 1030–1039. [DOI] [PubMed] [Google Scholar]
- 179.Schultz AW, Lewis CA, Luzung MR, Baran PS and Moore BS, J. Nat. Prod, 2010, 73, 373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Yu X, Liu Y, Xie X, Zheng XD and Li SM, J. Biol. Chem, 2012, 287, 1371–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Fan A, Chen H, Wu R, Xu H and Li SM, Appl. Microbiol. Biotechnol, 2014, 98, 10119–10129. [DOI] [PubMed] [Google Scholar]
- 182.Li SM, Appl. Microbiol. Biotechnol, 2009, 84, 631–639. [DOI] [PubMed] [Google Scholar]
- 183.Mori T, J. Nat. Med, 2020, 74, 501–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Tudzynski P, Hölter K, Correia T, Arntz C, Grammel N and Keller U, Mol. Gen. Genet, 1999, 261, 133–141. [DOI] [PubMed] [Google Scholar]
- 185.Haarmann T, Machado C, Lübbe Y, Correia T, Schardl CL, Panaccione DG and Tudzynski P, Phytochemistry, 2005, 66, 1312–1320. [DOI] [PubMed] [Google Scholar]
- 186.Rigbers O and Li SM, J. Biol. Chem, 2008, 283, 26859–26868. [DOI] [PubMed] [Google Scholar]
- 187.Goetz KE, Coyle CM, Cheng JZ, O’Connor SE and Panaccione DG, Curr. Genet, 2011, 57, 201–211. [DOI] [PubMed] [Google Scholar]
- 188.Lorenz N, Olšovská J, Šulc M and Tudzynski P, Appl. Environ. Microbiol, 2010, 76, 1822–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ryan KL, Moore CT and Panaccione DG, Toxins (Basel)., 2013, 5, 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Nielsen CA, Folly C, Hatsch A, Molt, Schröder H, O’Connor SE and Naesby, Microb. Cell Fact, 2014, 13, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Chen KL, Lai CY, Pham MT, Chein RJ, Tang Y and Lin HC, Org. Lett, 2020, 22, 3302–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Yao Y, An C, Evans D, Liu W, Wang W, Wei G, Ding N, Houk KN and Gao SS, J. Am. Chem. Soc, 2019, 141, 17517–17521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Iijima Y, Gang DR, Fridman E, Lewinsohn E and Pichersky E, Plant Physiol., 2004, 134, 370–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Wallwey C, Heddergott C, Xie X, Brakhage AA and Li SM, Microbiol. (United Kingdom), 2012, 158, 1634–1644. [DOI] [PubMed] [Google Scholar]
- 195.Coyle CM, Cheng JZ, O’Connor SE and Panaccione DG, Appl. Environ. Microbiol, 2010, 76, 3898–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Cheng JZ, Coyle CM, Panaccione DG and O’Connor SE, J. Am. Chem. Soc, 2010, 132, 12835–12837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Jakubczyk D, Caputi L, Hatsch A, Nielsen CAF, Diefenbacher M, Klein J, Molt A, Schröder H, Cheng JZ, Naesby M and O’Connor SE, Angew. Chemie, 2015, 127, 5206–5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Matuschek M, Wallwey C, Xie X and Li SM, Org. Biomol. Chem, 2011, 9, 4328–4335. [DOI] [PubMed] [Google Scholar]
- 199.Maier W, Schumann B and Gröger D, J. Basic Microbiol, 1988, 28, 83–93. [Google Scholar]
- 200.Haarmann T, Ortel I, Tudzynski P and Keller U, ChemBioChem, 2006, 7, 645–652. [DOI] [PubMed] [Google Scholar]
- 201.Robinson SL and Panaccione DG, Appl. Environ. Microbiol, 2014, 80, 6465–6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Schardl CL, Panaccione DG and Tudzynski P, Alkaloids Chem. Biol, 2006, 63, 45–86. [DOI] [PubMed] [Google Scholar]
- 203.Schwab JM and Henderson BS, Chem. Rev, 1990, 90, 1203–1245. [Google Scholar]
- 204.Chen J-JJ, Han M-YY, Gong T, Yang J-LL and Zhu P, Recent progress in ergot alkaloid research, Royal Society of Chemistry, 2017, vol. 7. [Google Scholar]
- 205.Bruhn JG, Lindgren JE, Holmstedt B and Adovasio JM, Science (80-.), 1978, 199, 1437–1438. [DOI] [PubMed] [Google Scholar]
- 206.Braun U and Kalbhen D, Pharmacology, 1973, 9, 312–316. [DOI] [PubMed] [Google Scholar]
- 207.Stein U, Greyer H and Hentschel H, Forensic Sci. Int, 2001, 118, 87–90. [DOI] [PubMed] [Google Scholar]
- 208.Shulgin AT, Nature, 1964, 201, 1120–1121. [DOI] [PubMed] [Google Scholar]
- 209.Shulgin AT and Shulgin A, Pihkal : a chemical love story, Transform Press, Berkeley, CA, 1991. [Google Scholar]
- 210.Shulgin AT and Shulgin A, Tihkal : the continuation, Transform Press, Berkeley, CA, 1997. [Google Scholar]
- 211.AXELROD J and TOMCHICK R, J. Biol. Chem, 1958, 233, 702–705. [PubMed] [Google Scholar]
- 212.Kopin IJ, in Catecholamines, Springer Berlin Heidelberg, Berlin, Heidelberg, 1972, pp. 270–282. [Google Scholar]
- 213.Benington F and Morin RD, Experientia, 1968, 24, 33–34. [DOI] [PubMed] [Google Scholar]
- 214.Friedhoff AJ, Schweitzer JW and Miller J, Nature, 1972, 237, 454–455. [DOI] [PubMed] [Google Scholar]
- 215.Agurell S, Lundström J and Sandberg F, Tetrahedron Lett., 1967, 8, 2433–2435. [DOI] [PubMed] [Google Scholar]
- 216.Kapadia GJ, Vaishnav YN and Fayez MBE, J. Pharm. Sci, 1969, 58, 1157–1159. [DOI] [PubMed] [Google Scholar]
- 217.Lundström J and Agurell S, Tetrahedron Lett., 1969, 10, 3371–3374. [DOI] [PubMed] [Google Scholar]
- 218.Lundström J, Acta Pharm. Suec, 1970, 7, 651–666. [PubMed] [Google Scholar]
- 219.Lundström J, Acta Chem. Scand, 1971, 25, 3489–3499. [DOI] [PubMed] [Google Scholar]
- 220.Ibarra-Laclette E, Zamudio-Hernández F, Pérez-Torres CA, Albert VA, Ramírez-Chávez E, Molina-Torres J, Fernández-Cortes A, Calderón-Vázquez C, Olivares-Romero JL, Herrera-Estrella A and Herrera-Estrella L, BMC Genomics, 2015, 16, 657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Lundström J, Acta Chem. Scand, 1971, 25, 3489–3499. [DOI] [PubMed] [Google Scholar]
- 222.Krogsgaard‐Larsen P, Hjeds H, Curtis DR, Lodge D and Johnston GAR, J. Neurochem, 1979, 32, 1717–1724. [DOI] [PubMed] [Google Scholar]
- 223.Chandra D, Halonen LM, Linden AM, Procaccini C, Hellsten K, Homanics GE and Korpi ER, Neuropsychopharmacology, 2010, 35, 999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Winn P, Tarbuck A and Dunnett SB, Neuroscience, 1984, 12, 225–240. [DOI] [PubMed] [Google Scholar]
- 225.Tsujikawa K, Kuwayama K, Miyaguchi H, Kanamori T, Iwata Y, Inoue H, Yoshida T and Kishi T, J. Chromatogr. B Anal. Technol. Biomed. Life Sci, 2007, 852, 430–435. [DOI] [PubMed] [Google Scholar]
- 226.Stebelska K, Ther. Drug Monit, 2013, 35, 420–442. [DOI] [PubMed] [Google Scholar]
- 227.Takemoto T, Nakajima T, T., and Yokobe, J. Pharm. Soc. Japan, 1964, 84, 1232–1233. [PubMed] [Google Scholar]
- 228.Obermaier S and Müller M, Angew. Chemie - Int. Ed, 2020, 59, 12432–12435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Alper KR, in Alkaloids: Chemistry and Biology, Academic Press, 2001, vol. 56, pp. 1–38. [DOI] [PubMed] [Google Scholar]
- 230.O’Connor SE and Maresh JJ, Nat. Prod. Rep, 2006, 23, 532. [DOI] [PubMed] [Google Scholar]
- 231.Pan Q, Mustafa NR, Tang K, Choi YH and Verpoorte R, Phytochem. Rev, 2016, 15, 221–250. [Google Scholar]
- 232.Chierrito TP, Aguiar AC, De Andrade IM, Ceravolo IP, Gonçalves RA, De Oliveira AJ and Krettli AU, Malar. J, 2014, 13, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.MačiIulaitis R, Kontrimavičiute V, Bressolle FMM and Briedis V, Hum. Exp. Toxicol, 2008, 27, 181–194. [DOI] [PubMed] [Google Scholar]
- 234.Li S, Long J, Ma Z, Xu Z, Li J and Zhang Z, Curr. Med. Res. Opin, 2004, 20, 409–415. [DOI] [PubMed] [Google Scholar]
- 235.Alper KR, Lotsof HS and Kaplan CD, J. Ethnopharmacol, 2008, 115, 9–24. [DOI] [PubMed] [Google Scholar]
- 236.Qu Y, Easson MEAM, Simionescu R, Hajicek J, Thamm AMK, Salim V and De Luca V, Proc. Natl. Acad. Sci. U. S. A, 2018, 115, 3180–3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Caputi L, Franke J, Farrow SC, Chung K, Payne RME, Nguyen TD, Dang TTT, Teto Carqueijeiro IS, Koudounas K, De Bernonville TD, Ameyaw B, Jones DM, Curcino Vieira IJ, Courdavault V and O’Connor SE, Science (80-.), 2018, 360, 1235–1239. [DOI] [PubMed] [Google Scholar]
- 238.Salim V, Yu F, Altarejos J and De Luca V, Plant J., 2013, 76, 754–765. [DOI] [PubMed] [Google Scholar]
- 239.Asada K, Salim V, Masada-Atsumi S, Edmunds E, Nagatoshi M, Terasaka K, Mizukami H and De Luca V, Plant Cell, 2013, 25, 4123–4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Qu Y, Easson MLAE, Froese J, Simionescu R, Hudlicky T and DeLuca V, Proc. Natl. Acad. Sci. U. S. A, 2015, 112, 6224–6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Qu Y, Thamm AMK, Czerwinski M, Masada S, Kim KH, Jones G, Liang P and De Luca V, Planta, 2018, 247, 625–634. [DOI] [PubMed] [Google Scholar]
- 242.Tatsis EC, Carqueijeiro I, Dugé De Bernonville T, Franke J, Dang TTT, Oudin A, Lanoue A, Lafontaine F, Stavrinides AK, Clastre M, Courdavault V and O’Connor SE, Nat. Commun, 2017, 8, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Lichman BR, Godden GT, Hamilton JP, Palmer L, Kamileen MO, Zhao D, Vaillancourt B, Wood JC, Sun M, Kinser TJ, Henry LK, Rodriguez-Lopez C, Dudareva N, Soltis DE, Soltis PS, Robin Buell C and O’Connor SE, Sci. Adv, 2020, 6, eaba0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Farrow SC, Kamileen MO, Caputi L, Bussey K, Mundy JEA, McAtee RC, Stephenson CRJ and O’Connor SE, J. Am. Chem. Soc, 2019, 141, 12979–12983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Croteau R, Chem. Rev, 1987, 87, 929–954. [Google Scholar]
- 246.Bouwmeester HJ, Gershenzon J, Konings MCJM and Croteau R, Plant Physiol., 1998, 117, 901–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Collu G, Unver N, Peltenburg-Looman AMG, Van der Heijden R, Verpoorte R and Memelink J, FEBS Lett., 2001, 508, 215–220. [DOI] [PubMed] [Google Scholar]
- 248.Geu-Flores F, Sherden NH, Glenn WS, O’connor SE, Courdavault V, Burlat V, Nims E, Wu C and Cui Y, Nature, 2012, 492, 138–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Lichman BR, Kamileen MO, Titchiner GR, Saalbach G, Stevenson CEM, Lawson DM and O’Connor SE, Nat. Chem. Biol, 2019, 15, 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Murata J, Roepke J, Gordon H and De Luca V, Plant Cell, 2008, 20, 524–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Guirimand G, Guihur A, Ginis O, Poutrain P, Héricourt F, Oudin A, Lanoue A, St-Pierre B, Burlat V and Courdavault V, FEBS J., 2011, 278, 749–763. [DOI] [PubMed] [Google Scholar]
- 252.Courdavault V, Papon N, Clastre M, Giglioli-Guivarc’h N, St-Pierre B and Burlat V, Curr. Opin. Plant Biol, 2014, 19, 43–50. [DOI] [PubMed] [Google Scholar]
- 253.Larsen B, Fuller VL, Pollier J, Van Moerkercke A, Schweizer F, Payne R, Colinas M, O’Connor SE, Goossens A and Halkier BA, Plant Cell Physiol., 2017, 58, 1507–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Bracher D and Kutchan TM, Arch. Biochem. Biophys, 1992, 294, 717–723. [DOI] [PubMed] [Google Scholar]
- 255.Battersby AR, in Alkaloids, 2007, vol. 1, pp. 31–47. [Google Scholar]
- 256.McCoy E and O’Connor SE, J. Am. Chem. Soc, 2006, 128, 14276–14277. [DOI] [PubMed] [Google Scholar]
- 257.Bernhardt P, McCoy E and O’Connor SE, Chem. Biol, 2007, 14, 888–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Runguphan W, Qu X and O’Connor SE, Nature, 2010, 468, 461–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Payne RME, Xu D, Foureau E, Teto Carqueijeiro MIS, Oudin A, De Bernonville TD, Novak V, Burow M, Olsen CE, Jones DM, Tatsis EC, Pendle A, Halkier BA, Geu-Flores F, Courdavault V, Nour-Eldin HH and O’Connor SE, Nat. Plants, 2017, 3, 16208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Guirimand G, Courdavault V, Lanoue A, Mahroug S, Guihur A, Blanc N, Giglioli-Guivarc’h N, St-Pierre B and Burlat V, BMC Plant Biol., 2010, 10, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Konno K, Hirayama C, Yasui H and Nakamura M, Proc. Natl. Acad. Sci. U. S. A, 1999, 96, 9159–9164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Luijendijk TJC, Stevens LH and Verpoorte R, Plant Physiol. Biochem, 1998, 36, 419–425. [Google Scholar]
- 263.Geerlings A, Martinez-Lozano Ibañez M, Memelink J, Van Der Heijden R and Verpoorte R, J. Biol. Chem, 2000, 275, 3051–3056. [DOI] [PubMed] [Google Scholar]
- 264.Gerasimenko I, Sheludko Y, Ma X and Stöckigt J, Eur. J. Biochem, 2002, 269, 2204–2213. [DOI] [PubMed] [Google Scholar]
- 265.Farrow SC, Kamileen MO, Meades J, Ameyaw B, Xiao Y and O’Connor SE, J. Biol. Chem, 2018, 293, 13821–13833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Caputi L, Franke J, Bussey K, Farrow SC, Vieira IJC, Stevenson CEM, Lawson DM and O’Connor SE, Nat. Chem. Biol, 2020, 16, 383–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Zhan ZJ, Yu Q, Wang ZL and Shan WG, Bioorganic Med. Chem. Lett, 2010, 20, 6185–6187. [DOI] [PubMed] [Google Scholar]
- 268.Steyer D, Erny C, Claudel P, Riveill G, Karst F and Legras JL, Food Microbiol., 2013, 33, 228–234. [DOI] [PubMed] [Google Scholar]
- 269.Ortega A, Blount JF and Manchand PS, J. Chem. Soc. Perkin Trans 1, 1982, 2505. [Google Scholar]
- 270.Valdes LJ, Butler WM, Hatfield GM, Paul AG and Koreeda M, J. Org. Chem, 1984, 49, 4716–4720. [Google Scholar]
- 271.Siebert DJ, J. Ethnopharmacol, 1994, 43, 53–56. [DOI] [PubMed] [Google Scholar]
- 272.Prisinzano TE, Life Sci., 2005, 78, 527–531. [DOI] [PubMed] [Google Scholar]
- 273.Coffeen U and Pellicer F, J. Pain Res, 2019, Volume 12, 1069–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Kivell BM, Ewald AWM and Prisinzano TE, in Advances in Pharmacology, 2014, vol. 69, pp. 481–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Yan F, Mosier PD, Westkaemper RB, Stewart J, Zjawiony JK, Vortherms TA, Sheffler DJ and Roth BL, Biochemistry, 2005, 44, 8643–8651. [DOI] [PubMed] [Google Scholar]
- 276.Gupta A, Gomes I, Bobeck EN, Fakira AK, Massaro NP, Sharma I, Cavé A, Hamm HE, Parello J, Devi LA and Snyder SH, Proc. Natl. Acad. Sci. U. S. A, 2016, 113, 6041–6046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Siebert DJ, Ann. Bot, 2004, 93, 763–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Kutrzeba L, Dayan FE, Howell J, Feng J, Giner JL and Zjawiony JK, Phytochemistry, 2007, 68, 1872–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Chen X, Berim A, Dayan FE and Gang DR, J. Exp. Bot, 2017, 68, 1109–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Pelot KA, Mitchell R, Kwon M, Hagelthorn DM, Wardman JF, Chiang A, Bohlmann J, Ro DK and Zerbe P, Plant J., 2017, 89, 885–897. [DOI] [PubMed] [Google Scholar]
- 281.Nunn N and Qian N, J. Econ. Perspect, 2010, 24, 163–188. [Google Scholar]
- 282.DeVido JJ, in Absolute Addiction Psychiatry Review, 2020, pp. 185–203. [Google Scholar]
- 283.Ashihara H and Suzuki T, Front. Biosci, 2004, 9, 1864–1876. [DOI] [PubMed] [Google Scholar]
- 284.Nathanson JA, Science (80-.), 1984, 226, 184–187. [DOI] [PubMed] [Google Scholar]
- 285.Chou CH and Waller GR, J. Chem. Ecol, 1980, 6, 643–654. [Google Scholar]
- 286.Wright GA, Baker DD, Palmer MJ, Stabler D, Mustard JA, Power EF, Borland AM and Stevenson PC, Science (80-.), 2013, 339, 1202–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Carpenter B and Lebon G, Front. Pharmacol, 2017, 8, 898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Waldvogel SR, Angew. Chemie Int. Ed, 2003, 42, 604–605. [DOI] [PubMed] [Google Scholar]
- 289.Fischer E, Berichte der Dtsch. Chem. Gesellschaft, 1898, 31, 3266–3277. [Google Scholar]
- 290.Anderson L and Gibbs M, J. Biol. Chem, 1962, 237, 1941–1944. [PubMed] [Google Scholar]
- 291.Negishi O, Ozawa T and Imagawa H, Agric. Biol. Chem, 1985, 49, 887–890. [Google Scholar]
- 292.Negishi O, Ozawa T and Imagawa H, Agric. Biol. Chem, 1988, 52, 169–175. [Google Scholar]
- 293.Kato M, Mizuno K, Fujimura T, Iwama M, Irie M, Crozier A and Ashihara H, Plant Physiol., 1999, 120, 579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Kato M, Mizuno K, Crozier A, Fujimura T and Ashihara H, Nature, 2000, 406, 956–957. [DOI] [PubMed] [Google Scholar]
- 295.Mizuno K, Kato M, Irino F, Yoneyama N, Fujimura T and Ashihara H, FEBS Lett., 2003, 547, 56–60. [DOI] [PubMed] [Google Scholar]
- 296.Ashihara H, Monteiro AM, Gillies FM and Crozier A, Plant Physiol., 1996, 111, 747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Negishi O, Ozawa T and Imagawa H, Biosci. Biotechnol. Biochem, 1992, 56, 499–503. [DOI] [PubMed] [Google Scholar]
- 298.Mizuno K, Okuda A, Kato M, Yoneyama N, Tanaka H, Ashihara H and Fujimura T, FEBS Lett., 2003, 534, 75–81. [DOI] [PubMed] [Google Scholar]
- 299.Ogawa M, Herai Y, Koizumi N, Kusano T and Sano H, J. Biol. Chem, 2001, 276, 8213–8218. [DOI] [PubMed] [Google Scholar]
- 300.Huang R, O’Donnell AJ, Barboline JJ and Barkman TJ, Proc. Natl. Acad. Sci. U. S. A, 2016, 113, 10613–10618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Koshiishi C, Kato A, Yama S, Crozier A and Ashihara H, FEBS Lett., 2001, 499, 50–54. [DOI] [PubMed] [Google Scholar]
- 302.Waldhauser SSM and Baumann TW, Phytochemistry, 1996, 42, 985–996. [Google Scholar]
- 303.Kato A, Crozier A and Ashihara H, Phytochemistry, 1998, 48, 777–779. [Google Scholar]
- 304.Ogita S, Uefuji H, Yamaguchi Y, Nozomu K and Sano H, Nature, 2003, 423, 823. [DOI] [PubMed] [Google Scholar]
- 305.Lu C, Warchol KM and Callahan RA, Bull. Insectology, 2014, 67, 125–130. [Google Scholar]
- 306.Tsvetkov N, Samson-Robert O, Sood K, Patel HS, Malena DA, Gajiwala PH, Maciukiewicz P, Fournier V and Zayed A, Science (80-.), 2017, 356, 1395–1397. [DOI] [PubMed] [Google Scholar]
- 307.Siegmund B, Leitner E and Pfannhauser W, J. Agric. Food Chem, 1999, 47, 3113–3120. [DOI] [PubMed] [Google Scholar]
- 308.Lewis RS, Drake-Stowe KE, Heim C, Steede T, Smith W and Dewey RE, Front. Plant Sci, 2020, 11, 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Benowitz NL, Annu. Rev. Pharmacol. Toxicol, 2009, 49, 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Moran VE, Front. Pharmacol, 2012, 3, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Pictet A and Rotschy A, Berichte der Dtsch. Chem. Gesellschaft, 1904, 37, 1225–1235. [Google Scholar]
- 312.Weidel H, Justus Liebigs Ann. Chem, 1873, 165, 328–349. [Google Scholar]
- 313.Cheng JZ, Coyle CM, Panaccione DG and O’Connor SE, J. Am. Chem. Soc, 2010, 132, 12835–12837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Lamberts BL, Dewey LJ and Byerrum RU, BBA - Biochim. Biophys. Acta, 1959, 33, 22–26. [DOI] [PubMed] [Google Scholar]
- 315.Leete E, in Alkaloids: chemical and biological perspectives, The Society, 1983, vol. 1, pp. 85–151. [Google Scholar]
- 316.Walton NJ, Robins RJ and Rhodes MJC, Plant Sci., 1988, 54, 125–131. [Google Scholar]
- 317.Leete E and McDonell JA, J. Am. Chem. Soc, 1981, 103, 658–662. [Google Scholar]
- 318.Mizusaki S, Kisaki T and Tamaki E, Plant Physiol., 1968, 43, 93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Mizusaki S, Tanabe Y, Noguchi M and Tamaki E, Plant Cell Physiol., 1971, 12, 633–640. [Google Scholar]
- 320.Mizusaki S, Tanabe Y, Noguchi M and Tamaki E, Phytochemistry, 1972, 11, 2757–2762. [Google Scholar]
- 321.Hibi N, Higashiguchi S, Hashimoto T and Yamada Y, Plant Cell, 1994, 6, 723–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Biastoff S, Reinhardt N, Reva V, Brandt W and Dräger B, FEBS Lett., 2009, 583, 3367–3374. [DOI] [PubMed] [Google Scholar]
- 323.Moriyama S, Tanaka H, Uwataki M, Muguruma M and Ohta K, J. Biosci. Bioeng, 2003, 96, 324–331. [DOI] [PubMed] [Google Scholar]
- 324.Brent Friesen J and Leete E, Tetrahedron Lett., 1990, 31, 6295–6298. [Google Scholar]
- 325.Kajikawa M, Hirai N and Hashimoto T, Plant Mol. Biol, 2009, 69, 287–298. [DOI] [PubMed] [Google Scholar]
- 326.Kajikawa M, Shoji T, Kato A and Hashimoto T, Plant Physiol., 2011, 155, 2010–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Goulet M-C, Gaudreau L, Gagné M, Maltais A-M, Laliberté A-C, Éthier G, Bechtold N, Martel M, D’Aoust M-A, Gosselin A, Pepin S and Michaud D, Front. Plant Sci, 2019, 10, 735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Romanowski S and Eustáquio AS, Curr. Opin. Chem. Biol, 2020, 58, 137–145. [DOI] [PubMed] [Google Scholar]
- 329.Molina-Hidalgo FJ, Vazquez-Vilar M, D’Andrea L, Demurtas OC, Fraser P, Giuliano G, Bock R, Orzáez D and Goossens A, Trends Biotechnol.,, DOI: 10.1016/j.tibtech.2020.11.012. [DOI] [PubMed] [Google Scholar]
- 330.Lewis RS, Jack AM, Morris JW, Robert VJM, Gavilano LB, Siminszky B, Bush LP, Hayes AJ and Dewey RE, Plant Biotechnol. J, 2008, 6, 346–354. [DOI] [PubMed] [Google Scholar]
- 331.Schachtsiek J and Stehle F, Plant Biotechnol. J, 2019, 17, 2228–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.United Nations, World Drug Report 2018, 2018.
- 333.Walker LS and Mezuk B, BMC Int. Health Hum. Rights, 2018, 18, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Nestler E, Sci. Pract. Perspect, 2005, 3, 4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Humphrey AJ and O’Hagan D, Nat. Prod. Rep, 2001, 18, 494–502. [DOI] [PubMed] [Google Scholar]
- 336.Leete E, Marion L and Spenser ID, Nature, 1954, 174, 650–651. [DOI] [PubMed] [Google Scholar]
- 337.Duran-Patron R, O’Hagan D, Hamilton JTG and Wong CW, Phytochemistry, 2000, 53, 777–784. [DOI] [PubMed] [Google Scholar]
- 338.Bedewitz MA, Jones AD, D’Auria JC and Barry CS, Nat. Commun, 2018, 9, 5281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Huang J-P, Fang C, Ma X, Wang L, Yang J, Luo J, Yan Y, Zhang Y and Huang S-X, Nat. Commun, 2019, 10, 4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Lichman BR, Nat. Prod. Rep, 2021, 38, 103–129. [DOI] [PubMed] [Google Scholar]
- 341.Dräger B, Phytochemistry, 2006, 67, 327–337. [DOI] [PubMed] [Google Scholar]
- 342.Gonzales-Vigil E, Góngora-Castillo E, Uebler JB, Gonzales-Vigil E, Wiegert-Rininger KE, Childs KL, Hamilton JP, Vaillancourt B, Yeo YS, Chappell J, Della DP, Daniel Jones A, Robin Buell C and Barry CS, Plant Cell, 2014, 26, 3745–3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Nichols DE and Nichols CD, Chem. Rev, 2008, 108, 1614–1641. [DOI] [PubMed] [Google Scholar]
- 344.Qiu F, Zeng J, Wang J, Huang JP, Zhou W, Yang C, Lan X, Chen M, Huang SX, Kai G and Liao Z, New Phytol., 2020, 225, 1906–1914. [DOI] [PubMed] [Google Scholar]
- 345.Li R, Reed DW, Liu E, Nowak J, Pelcher LE, Page JE and Covello PS, Chem. Biol, 2006, 13, 513–520. [DOI] [PubMed] [Google Scholar]
- 346.Nasomjai P, Reed DW, Tozer DJ, Peach MJG, Slawin AMZ, Covello PS and O’Hagan D, ChemBioChem, 2009, 10, 2382–2393. [DOI] [PubMed] [Google Scholar]
- 347.Leete E, Bjorklund JA, Couladis MM and Kim SH, J. Am. Chem. Soc, 1991, 113, 9286–9292. [Google Scholar]
- 348.Jirschitzka J, Schmidt GW, Reichelt M, Schneider B, Gershenzon J and D’Auria JC, Proc. Natl. Acad. Sci. U. S. A, 2012, 109, 10304–10309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Schmidt GW, Jirschitzka J, Porta T, Reichelt M, Luck K, Torre JCP, Dolke F, Varesio E, Hopfgartner G, Gershenzon J and D’auria JC, Plant Physiol., 2015, 167, 89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Ping Y, Li X, You W, Li G, Yang M, Wei W, Zhou Z and Xiao Y, ACS Synth. Biol, 2019, 8, 1257–1262. [DOI] [PubMed] [Google Scholar]
- 351.Srinivasan P and Smolke CD, Nat. Commun, 2019, 10, 3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Wheeldon I, Minteer SD, Banta S, Barton SC, Atanassov P and Sigman M, Nat. Chem, 2016, 8, 299–309. [DOI] [PubMed] [Google Scholar]
- 353.DeLoache WC, Russ ZN and Dueber JE, Nat. Commun, 2016, 7, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Russo EB, Chem. Biodivers, 2007, 4, 1614–1648. [DOI] [PubMed] [Google Scholar]
- 355.Press CA, Knupp KG and Chapman KE, Epilepsy Behav., 2015, 45, 49–52. [DOI] [PubMed] [Google Scholar]
- 356.Lal S, Prasad N, Ryan M, Tangri S, Silverberg MS, Gordon A and Steinhart H, Eur. J. Gastroenterol. Hepatol, 2011, 23, 891–896. [DOI] [PubMed] [Google Scholar]
- 357.Fiz J, Durán M, Capellà D, Carbonell J and Farré M, PLoS One, 2011, 6, e18440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Abrams DI and Guzman M, Clin. Pharmacol. Ther, 2015, 97, 575–586. [DOI] [PubMed] [Google Scholar]
- 359.Sarris J, Sinclair J, Karamacoska D, Davidson M and Firth J, BMC Psychiatry, 2020, 20, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Corey-Bloom J, Wolfson T, Gamst A, Jin S, Marcotte TD, Bentley H and Gouaux B, Can. Med. Assoc. J, 2012, 184, 1143 LP–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Müller-Vahl KR, Kolbe H, Schneider U and Emrich HM, Complement. Med. Res, 1999, 6(suppl 3), 23–27. [DOI] [PubMed] [Google Scholar]
- 362.Wei D, Dinh D, Lee D, Li D, Anguren A, Moreno-Sanz G, Gall CM and Piomelli D, Cannabis Cannabinoid Res., 2016, 1, 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Solomon R, Cannabis Cannabinoid Res., 2020, 5, 2–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Russo EB, Chem. Biodivers, 2007, 4, 1614–1648. [DOI] [PubMed] [Google Scholar]
- 365.Sharpless NE, White Oak, MD, 2019.
- 366.Gaoni Y and Mechoulam R, J. Am. Chem. Soc, 1971, 93, 217–224. [DOI] [PubMed] [Google Scholar]
- 367.Mechoulam R and Shvo Y, Tetrahedron, 1963, 19, 2073–2078. [DOI] [PubMed] [Google Scholar]
- 368.Niesink RJM and van Laar MW, Front. Psychiatry, 2013, 4, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.De Petrocellis L, Ligresti A, Moriello AS, Allarà M, Bisogno T, Petrosino S, Stott CG and Di Marzo V, Br. J. Pharmacol, 2011, 163, 1479–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Laprairie RB, Bagher AM, Kelly MEM and Denovan-Wright EM, Br. J. Pharmacol, 2015, 172, 4790–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Thomas A, Baillie GL, Phillips AM, Razdan RK, Ross RA and Pertwee RG, Br. J. Pharmacol, 2007, 150, 613–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Rock EM, Bolognini D, Limebeer CL, Cascio MG, Anavi-Goffer S, Fletcher PJ, Mechoulam R, Pertwee RG and Parker LA, Br. J. Pharmacol, 2012, 165, 2620–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Russo EB, Burnett A, Hall B and Parker KK, Neurochem. Res, 2005, 30, 1037–1043. [DOI] [PubMed] [Google Scholar]
- 374.Yamaori S, Kushihara M, Yamamoto I and Watanabe K, Biochem. Pharmacol, 2010, 79, 1691–1698. [DOI] [PubMed] [Google Scholar]
- 375.De Petrocellis L, Vellani V, Schiano-Moriello A, Marini P, Magherini PC, Orlando P and Di Marzo V, J. Pharmacol. Exp. Ther, 2008, 325, 1007 LP–1015. [DOI] [PubMed] [Google Scholar]
- 376.DeLong GT, Wolf CE, Poklis A and Lichtman AH, Drug Alcohol Depend., 2010, 112, 126–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Hanuš LO, Meyer SM, Muñoz E, Taglialatela-Scafati O and Appendino G, Nat. Prod. Rep, 2016, 33, 1357–1392. [DOI] [PubMed] [Google Scholar]
- 378.Proc. Chem. Soc, 1964, 73. [Google Scholar]
- 379.Cascio MG, Gauson LA, Stevenson LA, Ross RA and Pertwee RG, Br. J. Pharmacol, 2010, 159, 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Fuhr L, Rousseau M, Plauth A, Schroeder FC and Sauer S, J. Nat. Prod, 2015, 78, 1160–1164. [DOI] [PubMed] [Google Scholar]
- 381.Chicca A, Schafroth MA, Reynoso-Moreno I, Erni R, Petrucci V, Carreira EM and Gertsch J, Sci. Adv, 2018, 4, eaat2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Muhammad I, Li X-C, Jacob MR, Tekwani BL, Dunbar DC and Ferreira D, J. Nat. Prod, 2003, 66, 804–809. [DOI] [PubMed] [Google Scholar]
- 383.Quaghebeur K, Coosemans J, Toppet S and Compernolle F, Phytochemistry, 1994, 37, 159–161. [DOI] [PubMed] [Google Scholar]
- 384.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC and Bonner TI, Nature, 1990, 346, 561–564. [DOI] [PubMed] [Google Scholar]
- 385.Munro S, Thomas KL and Abu-Shaar M, Nature, 1993, 365, 61–65. [DOI] [PubMed] [Google Scholar]
- 386.Mechoulam R, Lander N, Varkony TH, Kimmel I, Becker O, Ben-Zvi Z, Edery H and Porath G, J. Med. Chem, 1980, 23, 1068–1072. [DOI] [PubMed] [Google Scholar]
- 387.Howlett AC, Prostaglandins Other Lipid Mediat., 2002, 68–69, 619–631. [DOI] [PubMed] [Google Scholar]
- 388.Pertwee RG, Howlett AC, Abood ME, Alexander SPH, Di Marzo V, Elphick MR, Greasley PJ, Hansen HS, Kunos G, Mackie K, Mechoulam R and Ross RA, Pharmacol. Rev, 2010, 62, 588–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Bow EW and Rimoldi JM, Perspect. Medicin. Chem, 2016, 8, 17–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Vollner L, Bieniek D and Korte F, Tetrahedron Lett., 1969, 10, 145–147. [DOI] [PubMed] [Google Scholar]
- 391.Merkus FWHM, Nature, 1971, 232, 579–580. [DOI] [PubMed] [Google Scholar]
- 392.Linciano P, Citti C, Luongo L, Belardo C, Maione S, Vandelli MA, Forni F, Gigli G, Laganà A, Montone CM and Cannazza G, J. Nat. Prod, 2020, 83, 88–98. [DOI] [PubMed] [Google Scholar]
- 393.Cascio MG, Zamberletti E, Marini P, Parolaro D and Pertwee RG, Br. J. Pharmacol, 2015, 172, 1305–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Rock EM, Sticht MA, Duncan M, Stott C and Parker LA, Br. J. Pharmacol, 2013, 170, 671–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Citti C, Linciano P, Russo F, Luongo L, Iannotta M, Maione S, Laganà A, Capriotti AL, Forni F, Vandelli MA, Gigli G and Cannazza G, Sci. Rep, 2019, 9, 20335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Ben-Shabat S, Fride E, Sheskin T, Tamiri T, Rhee M-H, Vogel Z, Bisogno T, De Petrocellis L, Di Marzo V and Mechoulam R, Eur. J. Pharmacol, 1998, 353, 23–31. [DOI] [PubMed] [Google Scholar]
- 397.Mechoulam R and Ben-Shabat S, Nat. Prod. Rep, 1999, 16, 131–143. [DOI] [PubMed] [Google Scholar]
- 398.Russo EB, Br. J. Pharmacol, 2011, 163, 1344–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Carlini EA, Karniol IG, Renault PF and Schuster CR, Br. J. Pharmacol, 1974, 50, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Stout JM, Boubakir Z, Ambrose SJ, Purves RW and Page JE, Plant J, 2012, 71, 353–365. [DOI] [PubMed] [Google Scholar]
- 401.Taura F, Tanaka S, Taguchi C, Fukamizu T, Tanaka H, Shoyama Y and Morimoto S, FEBS Lett., 2009, 583, 2061–2066. [DOI] [PubMed] [Google Scholar]
- 402.Gagne SJ, Stout JM, Liu E, Boubakir Z, Clark SM and Page JE, Proc. Natl. Acad. Sci. U. S. A, 2012, 109, 12811–12816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.US9765308B2, 2009.
- 404.Fellermeier M and Zenk MH, FEBS Lett., 1998, 427, 283–285. [DOI] [PubMed] [Google Scholar]
- 405.Sirikantaramas S, Morimoto S, Shoyama Y, Ishikawa Y, Wada Y, Shoyama Y and Taura F, J. Biol. Chem, 2004, 279, 39767–39774. [DOI] [PubMed] [Google Scholar]
- 406.Taura F, Sirikantaramas S, Shoyama Y, Yoshikai K, Shoyama Y and Morimoto S, FEBS Lett., 2007, 581, 2929–2934. [DOI] [PubMed] [Google Scholar]
- 407.Morimoto S, Komatsu K, Taura F and Shoyama Y, Phytochemistry, 1998, 49, 1525–1529. [DOI] [PubMed] [Google Scholar]
- 408.Shoyama Y, Tamada T, Kurihara K, Takeuchi A, Taura F, Arai S, Blaber M, Shoyama Y, Morimoto S and Kuroki R, J. Mol. Biol, 2012, 423, 96–105. [DOI] [PubMed] [Google Scholar]
- 409.Zirpel B, Kayser O and Stehle F, J. Biotechnol, 2018, 284, 17–26. [DOI] [PubMed] [Google Scholar]
- 410.Jamieson CS, Ohashi M, Liu F, Tang Y and Houk KN, Nat. Prod. Rep, 2019, 36, 698–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Gao L, Su C, Du X, Wang R, Chen S, Zhou Y, Liu C, Liu X, Tian R, Zhang L, Xie K, Chen S, Guo Q, Guo L, Hano Y, Shimazaki M, Minami A, Oikawa H, Huang N, Houk KN, Huang L, Dai J and Lei X, Nat. Chem, 2020, 12, 620–628. [DOI] [PubMed] [Google Scholar]
- 412.Ohashi M, Jamieson CS, Cai Y, Tan D, Kanayama D, Tang M-C, Anthony SM, V Chari J, Barber JS, Picazo E, Kakule TB, Cao S, Garg NK, Zhou J, Houk KN and Tang Y, Nature, 2020, 586, 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Kuzuyama T, Noel JP and Richard SB, Nature, 2005, 435, 983–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Zirpel B, Degenhardt F, Martin C, Kayser O and Stehle F, J. Biotechnol, 2017, 259, 204–212. [DOI] [PubMed] [Google Scholar]
- 415.Kumano T, Richard SB, Noel JP, Nishiyama M and Kuzuyama T, Bioorg. Med. Chem, 2008, 16, 8117–8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Kumano T, Tomita T, Nishiyama M and Kuzuyama T, J. Biol. Chem, 2010, 285, 39663–39671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Chatzivasileiou AO, Ward V, Edgar SM and Stephanopoulos G, Proc. Natl. Acad. Sci, 2019, 116, 506 LP–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Lund S, Hall R and Williams GJ, ACS Synth. Biol, 2019, 8, 232–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Brownstein MJ, Proc. Natl. Acad. Sci. U. S. A, 1993, 90, 5391–5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Singh A, Menéndez-Perdomo IM and Facchini PJ, Phytochem. Rev, 2019, 18, 1457–1482. [Google Scholar]
- 421.Sigerist HE and Paracelsus, Paracelsus in the light of four hundred years, Columbia University Press, 1941. [Google Scholar]
- 422.Jurna I, Schmerz, 2003, 17, 280–283. [DOI] [PubMed] [Google Scholar]
- 423.Krenn L, Glantschnig S and Sorgner U, Chromatographia, 1998, 47, 21–24. [Google Scholar]
- 424.Waldhoer M, Bartlett SE and Whistler JL, Annu. Rev. Biochem, 2004, 73, 953–990. [DOI] [PubMed] [Google Scholar]
- 425.Quock RM, Burkey TH, Varga E, Hosohata Y, Hosohata K, Cowell SM, Slate CA, Ehlert FJ, Roeske WR and Yamamura HI, Pharmacol. Rev, 1999, 51, 503 LP–532. [PubMed] [Google Scholar]
- 426.Minami M and Satoh M, Neurosci. Res, 1995, 23, 121–145. [DOI] [PubMed] [Google Scholar]
- 427.Pogozheva ID, Lomize AL and Mosberg HI, Biophys. J, 1998, 75, 612–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Schmitz R, Pharm. Hist, 1985, 27, 61–74. [PubMed] [Google Scholar]
- 429.Robiquet P-J, Ann. der Chemie, 1833, 5, 82–111. [Google Scholar]
- 430.Rida PCG, Livecche D, Ogden A, Zhou J and Aneja R, Med. Res. Rev, 2015, 35, 1072–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Ye K, Ke Y, Keshava N, Shanks J, Kapp JA, Tekmal RR, Petros J and Joshi HC, Proc. Natl. Acad. Sci, 1998, 95, 1601 LP–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Ke Y, Ye K, Grossniklaus HE, Archer DR, Joshi HC and Kapp JA, Cancer Immunol. Immunother, 2000, 49, 217–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Samanani N and Facchini PJ, J. Biol. Chem, 2002, 277, 33878–33883. [DOI] [PubMed] [Google Scholar]
- 434.Liscombe DK, MacLeod BP, Loukanina N, Nandi OI and Facchini PJ, Phytochemistry, 2005, 66, 2501–2520. [DOI] [PubMed] [Google Scholar]
- 435.Lichman BR, Gershater MC, Lamming ED, Pesnot T, Sula A, Keep NH, Hailes HC and Ward JM, FEBS J, 2015, 282, 1137–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Lichman BR, Sula A, Pesnot T, Hailes HC, Ward JM and Keep NH, Biochemistry, 2017, 56, 5274–5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Pauli HH and Kutchan TM, Plant J., 1998, 13, 793–801. [DOI] [PubMed] [Google Scholar]
- 438.Facchini PJ and Park SU, Phytochemistry, 2003, 64, 177–186. [DOI] [PubMed] [Google Scholar]
- 439.Gurkok T, Ozhuner E, Parmaksiz I, Özcan S, Turktas M, İpek A, Demirtas I, Okay S and Unver T, Front. Plant Sci, 2016, 7, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Bennett MR, Thompson ML, Shepherd SA, Dunstan MS, Herbert AJ, Smith DRM, Cronin VA, Menon BRK, Levy C and Micklefield J, Angew. Chemie - Int. Ed, 2018, 57, 10600–10604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Farrow SC, Hagel JM, Beaudoin GAW, Burns DC and Facchini PJ, Nat. Chem. Biol, 2015, 11, 728–732. [DOI] [PubMed] [Google Scholar]
- 442.Winzer T, Kern M, King AJ, Larson TR, Teodor RI, Donninger SL, Li Y, Dowle AA, Cartwright J, Bates R, Ashford D, Thomas J, Walker C, Bowser TA and Graham IA, Science (80-.), 2015, 349, 309–312. [DOI] [PubMed] [Google Scholar]
- 443.Wijekoon CP and Facchini PJ, Plant J., 2012, 69, 1052–1063. [DOI] [PubMed] [Google Scholar]
- 444.Lenz R and Zenk MH, J. Biol. Chem, 1995, 270, 31091–31096. [DOI] [PubMed] [Google Scholar]
- 445.Grothe T, Lenz R and Kutchan TM, J. Biol. Chem, 2001, 276, 30717–30723. [DOI] [PubMed] [Google Scholar]
- 446.Ziegler J, Brandt W, Geißler R and Facchini PJ, J. Biol. Chem, 2009, 284, 26758–26767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Chen X, Hagel JM, Chang L, Tucker JE, Shiigi SA, Yelpaala Y, Chen HY, Estrada R, Colbeck J, Enquist-Newman M, Ibáñez AB, Cottarel G, Vidanes GM and Facchini PJ, Nat. Chem. Biol, 2018, 14, 738–743. [DOI] [PubMed] [Google Scholar]
- 448.Farrow SC and Facchini PJ, J. Biol. Chem, 2013, 288, 28997–29012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Unterlinner B, Lenz R and Kutchan TM, Plant J., 1999, 18, 465–475. [DOI] [PubMed] [Google Scholar]
- 450.Kluza A, Niedzialkowska E, Kurpiewska K, Wojdyla Z, Quesne M, Kot E, Porebski PJ and Borowski T, J. Struct. Biol, 2018, 202, 229–235. [DOI] [PubMed] [Google Scholar]
- 451.Winzer T, Gazda V, He Z, Kaminski F, Kern M, Larson TR, Li Y, Meade F, Teodor R, Vaistij FE, Walker C, Bowser TA and Graham IA, Science (80-.), 2012, 336, 1704–1708. [DOI] [PubMed] [Google Scholar]
- 452.Kutchan TM and Dittrich H, J. Biol. Chem, 1995, 270, 24475–24481. [DOI] [PubMed] [Google Scholar]
- 453.Facchini PJ, Penzes C, Johnson AG and Bull D, Plant Physiol, 1996, 112, 1669–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Gaweska HM, Roberts KM and Fitzpatrick PF, Biochemistry, 2012, 51, 7342–7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Dang TTT and Facchini PJ, Plant Physiol., 2012, 159, 618–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Dang TTT and Facchini PJ, FEBS Lett., 2014, 588, 198–204. [DOI] [PubMed] [Google Scholar]
- 457.Liscombe DK and Facchini PJ, J. Biol. Chem, 2007, 282, 14741–14751. [DOI] [PubMed] [Google Scholar]
- 458.Lang DE, Morris JS, Rowley M, Torres MA, Maksimovich VA, Facchini PJ and Ng KKS, J. Biol. Chem, 2019, 294, 14482–14898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Dang TTT and Facchini PJ, J. Biol. Chem, 2014, 289, 2013–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Dang TTT, Chen X and Facchini PJ, Nat. Chem. Biol, 2015, 11, 104–106. [DOI] [PubMed] [Google Scholar]
- 461.Chen X and Facchini PJ, Plant J., 2014, 77, 173–184. [DOI] [PubMed] [Google Scholar]
- 462.Nakagawa A, Minami H, Kim JS, Koyanagi T, Katayama T, Sato F and Kumagai H, Nat. Commun, 2011, 2, 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Nakagawa A, Minami H, Kim JS, Koyanagi T, Katayama T, Sato F and Kumagai H, Bioeng. Bugs, 2012, 3, 49–53. [DOI] [PubMed] [Google Scholar]
- 464.Kim JS, Nakagawa A, Yamazaki Y, Matsumura E, Koyanagi T, Minami H, Katayama T, Sato F and Kumagai H, Biosci. Biotechnol. Biochem, 2013, 77, 2166–2168. [DOI] [PubMed] [Google Scholar]
- 465.Thodey K, Galanie S and Smolke CD, Nat. Chem. Biol, 2014, 10, 837–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Fossati E, Ekins A, Narcross L, Zhu Y, Falgueyret JP, Beaudoin GAW, Facchini PJ and Martin VJJ, Nat. Commun, 2014, 5, 3283. [DOI] [PubMed] [Google Scholar]
- 467.Pyne ME, Kevvai K, Grewal PS, Narcross L, Choi B, Bourgeois L, Dueber JE and Martin VJJ, Nat. Commun, 2020, 11, 3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Trenchard IJ and Smolke CD, Metab. Eng, 2015, 30, 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.French CE and Bruce NC, Biochem. J, 1994, 301 (Pt 1, 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Li Y, Li S, Thodey K, Trenchard I, Cravens A and Smolke CD, Proc. Natl. Acad. Sci. U. S. A, 2018, 115, E3922–E3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Li J, Lee E-J, Chang L and Facchini PJ, Sci. Rep, 2016, 6, 39256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Flores-Bocanegra L, Raja HA, Graf TN, Augustinović M, Wallace ED, Hematian S, Kellogg JJ, Todd DA, Cech NB and Oberlies NH, J. Nat. Prod, 2020, 83, 2165–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Todd DA, Kellogg JJ, Wallace ED, Khin M, Flores-Bocanegra L, Tanna RS, McIntosh S, Raja HA, Graf TN, Hemby SE, Paine MF, Oberlies NH and Cech NB, Sci. Rep, 2020, 10, 19158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Takayama H, Chem. Pharm. Bull, 2004, 52, 916–928. [DOI] [PubMed] [Google Scholar]
- 475.Yamamoto LT, Horie S, Takayama H, Aimi N, Sakai SI, Yano S, Shan J, Pang PKT, Ponglux D and Watanabe K, Gen. Pharmacol, 1999, 33, 73–81. [DOI] [PubMed] [Google Scholar]
- 476.Stavrinides A, Tatsis EC, Caputi L, Foureau E, Stevenson CEM, Lawson DM, Courdavault V and O’Connor SE, Nat. Commun, 2016, 7, 12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Stavrinides A, Tatsis EC, Foureau E, Caputi L, Kellner F, Courdavault V and O’Connor SE, Chem. Biol, 2015, 22, 336–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Stavrinides AK, Tatsis EC, Dang TT, Caputi L, Stevenson CEM, Lawson DM, Schneider B and O’Connor SE, ChemBioChem, 2018, 19, 940–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Trenti F, Yamamoto K, Hong B, Paetz C, Nakamura Y and O’Connor SE, Org. Lett, 2021, 23, 1793–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Kong F, Ma Q, Huang S, Yang S, Fu L, Zhou L, Dai H, Yu Z and Zhao Y, Nat. Prod. Res, 2017, 31, 1403–1408. [DOI] [PubMed] [Google Scholar]
- 481.Kamble SH, León F, King TI, Berthold EC, Lopera-Londonõ C, Siva Rama Raju K, Hampson AJ, Sharma A, Avery BA, McMahon LR and McCurdy CR, ACS Pharmacol. Transl. Sci, 2020, 3, 1063–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Tsunematsu Y, Ishikawa N, Wakana D, Goda Y, Noguchi H, Moriya H, Hotta K and Watanabe K, Nat. Chem. Biol, 2013, 9, 818–825. [DOI] [PubMed] [Google Scholar]
- 483.Ye Y, Du L, Zhang X, Newmister SA, McCauley M, Alegre-Requena JV, Zhang W, Mu S, Minami A, Fraley AE, Adrover-Castellano ML, Carney NA, Shende VV, Qi F, Oikawa H, Kato H, Tsukamoto S, Paton RS, Williams RM, Sherman DH and Li S, Nat. Catal, 2020, 3, 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Bonn-Miller MO, ElSohly MA, Loflin MJE, Chandra S and Vandrey R, Int. Rev. Psychiatry, 2018, 30, 277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Cristino L, Bisogno T and Di Marzo V, Nat. Rev. Neurol, 2020, 16, 9–29. [DOI] [PubMed] [Google Scholar]