

Abstract
Small cell lung cancer (SCLC) is a particular subtype of lung cancer with high mortality. Recent advances in understanding SCLC genomics and breakthroughs of immunotherapy have substantially expanded existing knowledge and treatment modalities. However, challenges associated with SCLC remain enigmatic and elusive. Most of the conventional drug discovery approaches targeting altered signaling pathways in SCLC end up in the ‘grave-yard of drug discovery’, which mandates exploring novel approaches beyond inhibiting cell signaling pathways. Epigenetic modifications have long been documented as the key contributors to the tumorigenesis of almost all types of cancer, including SCLC. The last decade witnessed an exponential increase in our understanding of epigenetic modifications for SCLC. The present review highlights the central role of epigenetic regulations in acquiring neoplastic phenotype, metastasis, aggressiveness, resistance to chemotherapy, and immunotherapeutic approaches of SCLC. Different types of epigenetic modifications (DNA/histone methylation or acetylation) that can serve as predictive biomarkers for prognostication, treatment stratification, neuroendocrine lineage determination, and development of potential SCLC therapies are also discussed. We also review the utility of epigenetic targets/epidrugs in combination with first-line chemotherapy and immunotherapy that are currently under investigation in preclinical and clinical studies. Altogether, the information presents the inclusive landscape of SCLC epigenetics and epidrugs that will help translate the knowledge of epigenetics to improve SCLC outcomes.
Keywords: Epigenetic modifications, histone acetylation, DNA methylation, small cell lung cancer, neuroendocrine carcinoma
1. Introduction
Small cell lung cancer (SCLC) remains a highly metastatic neuroendocrine lung cancer subtype with high lethality [1]. SCLC accounts for 13–15% of total lung cancer cases, with more than 30,000 new cases annually in the United States [2]. Depending on the extent of spread at diagnosis, SCLC is divided into limited-stage SCLC (~30%) or extensive-stage SCLC (~70%). Despite excellent initial response to conventional chemotherapy (platinum/etoposide), the overall 5-year survival rate of SCLC is bleak (nearly 5–6%) [3–5]. Due to high metastasis, late diagnosis, and recalcitrant behavior of this cancer, a limited number of therapies are available that improve the outcomes in SCLC [6, 7]. Although smoking is the foremost risk factor in most SCLC cases, other risk factors are also associated like exposure to radon, asbestos, and other polycyclic aromatic hydrocarbon pollutants [8].
High mutational rate and genomic instability are additional interesting features of SCLC. This is possibly due to the frequent loss of function mutations in TP53 and retinoblastoma 1 (RB1), which are the primary tumor suppressors which maintain or genomic integrity [9, 10]. These genetic accumulations lead to different epigenetic alterations that ultimately result in the aberrant regulation of key DNA repair/housekeeping genes, oncogenes, and tumor suppressor genes [1, 11]. Overall, the probability of occurrence of these pathological events is mainly dependent on the phenotypic variability and individual exposome. Therefore, the likelihood of occurrence for all cancers is a composite of different risk factors, including lifestyle-related, environmental, phenotypic variabilities, and individual exposomes [12]. In isolation, the frequency of high-penetrance genetic mutations is low and accounts for a small percentage of all cancer cases [13, 14]. Several Genome-wide association studies (GWAS) have discovered the existence of common heritable components of cancers and suggested the spread of these components across the common germline variants [15, 16]. Nevertheless, in a small fraction, these genetic heterogeneity studies revealed the role of these variants in conferring risk of progressive increase in lung cancer susceptibility. Still, as a whole, it fails to explain the basic phenomenon of SCLC [17]. In short, it is difficult to explain the enigma of SCLC only in terms of genetic factors, as ‘fate of every cell is not always written in the genes’ [17]. These observations suggest that the remainder of SCLC heritability might eventually be explained through extensive association studies and could be effectuated at a broader scale through omics-based analyses, such as epigenomics [18]. Epigenetic variabilities, including histone modifications, DNA methylation, and expression of non-coding RNAs directly contribute to cancer development or progression. Thus epigenomics has a great potential to divulge more substantial effects and define immediate risk for SCLC [19–21]. In addition to genetics, epigenetic studies guide us to understand the pathogenesis of disease and enhance our knowledge of SCLC.
Tumor development in the lungs (including SCLC) is proceeds through a multistage, step by step, journey that sequentially accumulates genetic and epigenetics deformities in the lung or respiratory epithelium [1, 20, 22–24]. Genetic changes like somatic mutations and change in copy numbers are the established mechanisms for oncogenesis or cancer induction, but in lung cancer, especially SCLC, epigenetic modifications appear to be more prominent than somatic genetic aberrations [10, 13, 25, 26]. SCLC exhibits multiple epigenetic abnormalities, and different studies have defined the crucial role of epigenetic disruptions in disease progression and characterization [27]. In SCLC, epigenetic abnormalities contribute to the acquisition of cancerous phenotype, aggressiveness, and resistance to treatment [28–31], suggesting their pivotal role, and introduces new possibilities for identifying novel therapeutic targets and developing effective epigenetic therapies for SCLC [32]. Thus, the detailed understanding of these epigenetic modifiers of SCLC may explain the role of different molecules, or mechanisms responsible for the development and progression of SCLC. Moreover, the continued expansion of our understanding of various epigenetic events involved with different types of lung cancer expands the potential battery of diagnostic and prognostic biomarkers available to clinicians. It introduces new avenues for the discovery of novel therapeutic targets [32, 33]. This review’s primary objective is to summarize the common epigenetic events in SCLC (Figure 1) and potential translational applications of these events for the management of this disease.
Figure 1:
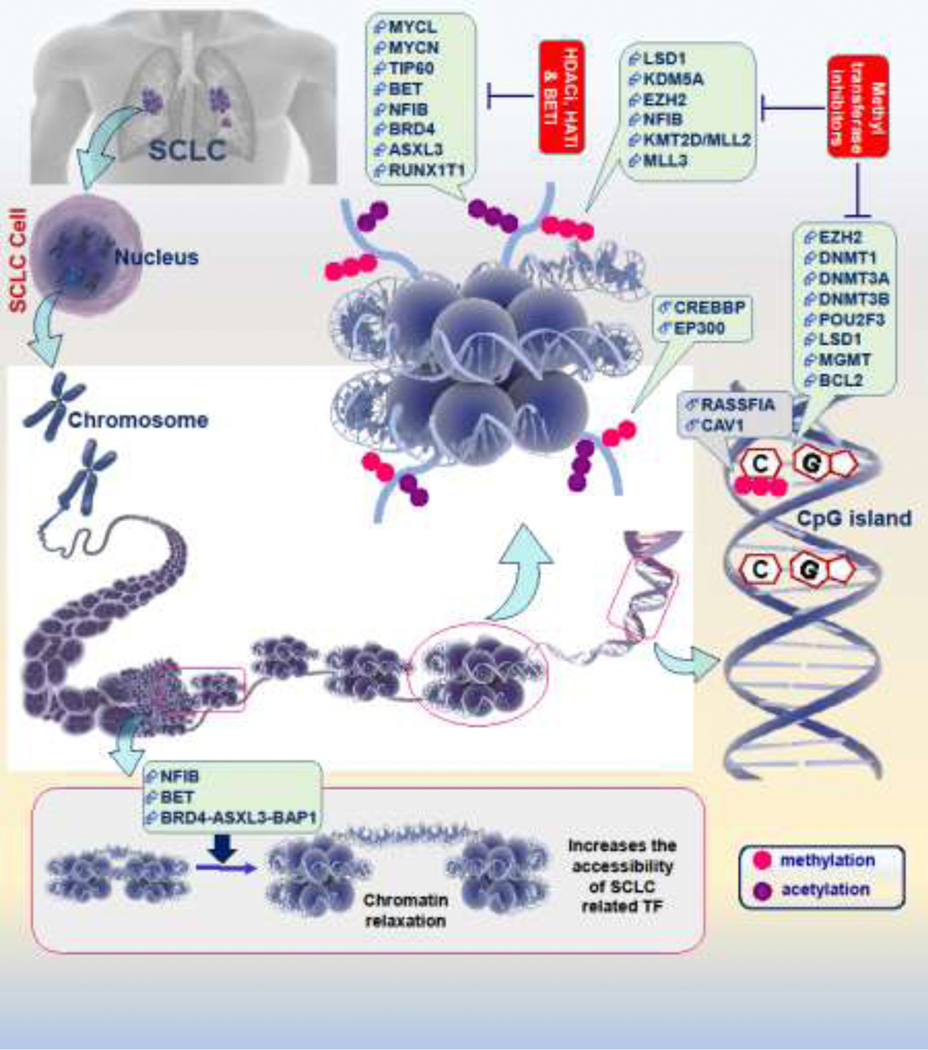
Epigenetic landscape of SCLC genome is regulated at the DNA & histone level by multiple regulators. The prime features of this regulation involve modifications of basic chromatin unit ‘nucleosome’ an octamer of histone that works as a spool for DNA wrapping. The key post-translational modifications of histones mainly involves acetylation and methylation of lysine residues that controls chromatin transformations and regulates the expression of target genes (other modifications like sumoylation, phosphorylation, and ubiquitination, are not shown here). A number of chromatin modifier proteins are involved performing this task including readers, writers, and erasers (like HDACS, HATs, and methyltransferases). SCLC-specific key epigenetic genes that regulate the histone/DNA related epigenetic modifications have been reviewed and summarized in the illustration. Similar to writer, readers, and erasers, the overexpression of other factors like NFIB induces the chromatin relaxation (as shown in the lower side panel) and increases the accessibility of transcription factors to the DNA that ultimately induces the expression of SCLC related genes. Epigenetic drugs (HDACi: histone deacetylase inhibitors, HATi: histone acetyltransferase inhibitors, BETi: bromodomain and extra-terminal domain protein inhibitors, and inhibitors of methyltransferases) targets these chromatin modifiers.
In addition to the aforesaid importance of epigenetics, it is an emerging paradigm for understanding the details of SCLC and opens an active area of clinical investigations. In this review, we have highlighted the ‘central role’ of epigenetics in the development and progression of SCLC. The clinical/translational aspects and run-down of the potential drug targets along with the implications of epigenetic drugs for the perturbation of SCLC pathogenesis have also been summarized here.
2. Epigenetic predictors in SCLC
Over the past two decades, the misprogramming of epigenetic regulations has been considered as a core component of cancer initiation and progression mechanisms [34–36]. Some of the observations, like the genome of stem cells, specifically marked or transiently silenced by protein members of the polycomb group, becomes hypermethylated and silenced completely during cancer development, this has been a key step for understanding the consequences of epigenetic misprogramming [34, 37].
Epigenetic traits did not confer the usual sequence of nitrogenous bases as the case of genetic code but can be inherited mitotically and meiotically (or transgenerationally). Instead, epigenetics is a collective dynamic process that regulates gene expression and fine-tunes various molecular pathways. It is a process that provides the genomic plasticity, affords the cellular identity at specific time points in early development, and during adulthood that helps in the functioning and maintenance of tissues and thus can be considered as genome ‘editor’[38]. Despite the recent advances in the pathobiology of SCLC, the heterogeneity of the disease restricts the generalization of patient outcomes [39]. The epigenetic changes in SCLC carcinogenesis mainly include the irregular status of DNA methylation (hyper- and hypomethylation) and histone modifications like acetylation/methylations (Figure 1) [32, 40, 41].
2.1. Altered DNA methylation in SCLC
It is a universal fact that cancer cells have altered DNA methylation patterns compared to normal cells, and the same is true for SCLC with some specific and unique DNA methylation predictors [32, 41]. In addition to TP53 and RB1, the genome studies identified other somatic alterations in SCLC cells, including inactivation of PTEN tumor suppressor, amplification of MYC family members (MYC, MYCL1, and MYCN), mutations in KMT2A (MLL), KMT2D (MLL2), EP300, CREBBP, histone-modifying proteins encoding genes, NOTCH family genes, FHIT and CDKN2A genes [1, 10, 11, 32, 41–43]. Other genomic alterations noticed in SCLC specimens include somatic changes in the TP73 gene, overexpression of CCND1, mutations in EPHA7, SLIT2, and focal amplification of FGFR1 [36, 43]. Smoking-associated signatures or epigenetic changes have also been reported in SCLC tumors [44]. Specific alterations in the methylation patterns of SCLC related genes/targets is outlined in Figure 1.
2.1.1. Methylation of tumor suppressor and oncogenes
The carcinogenic journey of SCLC starts with the inactivation of two major tumor suppressor genes RB1 and TP53, which is a hallmark of SCLC and despite some reported mutations, the main cause of inactivation for these tumor suppressor genes is promotor hypermethylation [1, 10]. During the course of tumor development, promoter methylation of tumor suppressors along with the hypermethylation of other genes are responsible for SCLC aggressiveness [45]. Often promoter methylation of tumor suppressor genes is coupled with the deletion or mutation events, suggesting different events of inactivation for each allele [46, 47]. The possible reason behind this is the haploinsufficiency for the dominant suppressor gene loci, and the inactivation of a single allele is insufficient for clonal selection as one normal allele is sufficient for the production of the protein. Contrary to this, there are reports which suggest that the inactivation of both copies of the allele may not be necessary to adversely impact the cell behavior that leads to carcinogenesis. In such cases, the promoter methylation of even a single allele leads to clonal selection [18, 48].
Promoter hyper/hypomethylation or, more specifically, C5 methylation of cytosine within CpG doublet is the most frequent and stable event in epigenomics that can be altered both by inherent and non-inherent reasons [49, 50]. CpG islands are predominantly located within the promoter regions, and in most cancer tissues; CpG hypermethylation is noticed with respect to a global hypomethylation background. These conditions are the hallmarks of epigenetic modulations and are generally found associated with aberrant gene expression profiles witnessed through different cancer types [24].
Some of the tumor suppressor genes hypermethylated in SCLC are also frequently hypermethylated in other types of tumors like TP53, but some are SCLC specific, like the case of somatic mutations. Promoter methylation is the most studied and crucial symbol of epigenetic modification that is frequently observed in the genes involved in cell regulatory functions, including proliferation, cell cycle regulation, DNA repair, apoptosis, cellular adhesions and motility [51]. For the initiation of carcinogenesis, DNA hypo- or hypermethylation follows three different molecular mechanisms, (a) microsatellite instability that leads to increase the mutational load in cancer-specific genes through the stimulation of retrotransposons or repetitive elements (SINE/LINE, Long interspersed element), (b) transcriptional and translational activation of oncogenes, (c) irregularities in the expression of imprinted genes or loss of imprinting (LOI) [52–54]. Interestingly, the global hypomethylation of DNA is a characteristic of malignant transformation; on the other hand, the local or promoter hypermethylation of tumor suppressor (TS) genes leads to the development of SCLC [32, 53, 55–59]. This means that hypo- or hypermethylation are two different epigenetic modifications, which can be considered inclusive events compared globally, or exclusive events for a specific DNA sequence. In the case of TS genes, two genes RASSF1A and caveolin-1 (CAV1) were downregulated through promoter hypermethylation in over 60% of studied SCLC cell lines and tumor tissues [59–64]. RASSF1A regulates apoptosis and cell cycle, whereas CAV1 is known to regulate different pathways in SCLC, including Hh signaling, autophagy, MAPK signaling, and cell growth through hormone-dependent pathways [63, 65–67]. The consistent inactivation of RASSF1A and CAV1 through hypermethylation suggests the role of epigenetics in deregulated apoptosis, cell-cycle, and autophagy of SCLC cells.
The most crucial oncogene related to methylation in SCLC is EZH2, a chromatin modifier, and the main enzymatic component of polycomb repressive complex (PRC) found overexpressed in the majority of SCLC cases, and the critical regulator of extensive promoter hypermethylation. Specifically, it is a component of histone methyltransferase complex, and its overexpression is coupled with the trimethylation of histone H3 at lysine 27 that fashions aberrant DNA methylation in the majority of the cancers, including SCLC [45, 68, 69]. The expression of EZH2 is regulated by elongation factor E2F, and due to loss of function mutations or copy number loss of RB1, E2F is highly activated in SCLC as the repressor for E2F is encoded by RB1. EZH2 is situated downstream of the pRB- E2F pathway, one of the key regulatory pathways of SCLC progression [57, 70]. EZH2 also plays an essential role in cigarette smoke-induced epigenetic changes. Chronic cigarette smoking induces steady changes in the methylation patterns of chromatin through EZH2 [25]. It has been shown that overexpression of EZH2 downregulates the transforming growth factor-β (TGF-β)-SMAD pathway through methylation, which further upregulates Achaete-scute complex homologue 1 (ASCL1) and promotes SCLC progression [71].
The aberrant DNA methylation by EZH2 activates DNA methyltransferase 1 (DNMT1), induces epithelial-to-mesenchymal transition, decreases the anchorage dependency for growth, and activates different oncogenic signaling cascades with the silencing of TS genes by hypermethylation [25, 72–75]. Apart from this, EZH2 mediated epigenetic modifications (histone modifications) leads to the upregulation of TWIST1 and suppression of SLFN11 in SCLC that confers chemoresistance to cisplatin and etoposide [76]. All these studies suggest that EZH2 is an important initiator of SCLC and underscore the importance of EZH2 or EZH2 mediated epigenetic alterations in the initiation, progression, and development of SCLC. EZH2 also regulates histone modifications in SCLC, as further discussed in the section of histone modifications. The next important differentially methylated and commonly studied oncogene in SCLC is BCL2 [57]. In normal lung epithelial cells, this gene is silenced through methylation, but it shows minimal methylation and differential expression in different stages of SCLC [57, 77]. It encodes cell survival protein (Bcl-2) and thus helps in SCLC cell survival.
2.1.2. Methylation of SCLC or neuroendocrine (NE) fate determinants
There are clues that the DNA methylation landscape of SCLC defines the differentiation fate of NE cells. The ontological analysis of genes has demonstrated the enrichment of transcription factors (TF) that are regulated by methylation and involved in the differentiation of neuronal lineage. The tumor-specific methylation sites show the enrichment of motifs that possess the binding sites for different NE-fate determining TFs, including NEUROD1, ZNF423, REST, and HANDI [78, 79]. Based on the DNA methylation patterns, these studies provide two possible mechanisms for the transition of tumor progenitor cells towards SCLC. One mechanism suggests the loss of cell fate-determining TF by promoter methylation, and the other includes the functional inactivation of respective binding sites of TF through DNA methylation [78, 80]. These are the major epigenetic aberrations during the differentiation of NE cells that promote the differentiation of tumor progenitor cells’ to SCLC phenotype. Another important observation associated with the DNA methylation that leads to the development of an aggressive SCLC phenotype of lung cancer is the enrichment of epithelial adult stem cell (ASC) transcriptional signatures [81].
The integral part of ASC signatures is DNA methyltransferase, and in SCLC, the expression of DNA methyltransferases (DNMT1, DNMT3A, and DNMT3B) is very high and further enriches with the progression of tumors towards neuroendocrine features (Figure 2). These findings uncovered an epigenomic profile for SCLC and established an important link between DNA methylation status, NE fate, and aggressiveness of the disease (as shown in Figure 2) [81, 82]. POU2F3, a master regulator of transcription, is used to define the lineage of SCLC and is a marker of the tuft cell-like lineage [79, 83]. The expression of POU2F3 is associated with SCLC variants that lack neuroendocrine (NE) markers and are chemosensitive. The expression of POU2F3 is epigenetically downregulated by promoter hypermethylation suggesting the critical role of DNA methylation for the development and characterization of SCLC lineages (Figure 2) [41, 83].
Figure 2:

Epigenetic modifiers describing different SCLC subtypes. Key SCLC studies described in the text help to define the SCLC subtypes specific epigenetically regulated genes (as aligned on the circumference of each subtype). The proposed SCLC nomenclature was followed by Rudin et al., [41]. ASCL1, achaete-scute homologue 1; NE, neuroendocrine; NeuroD1, neurogenic differentiation factor 1; POU2F3, POU class 2 homeobox 3; YAP1, yes-associated protein 1.
Integrative analyses of the transcriptional and epigenetic landscapes of NE cells in SCLC suggested some of the hyper-accessible (LHX1, LHX2, LHX3, and ISL1 as LHX family TF motifs) and hypo-accessible (OCT2 and OCT6 as OCT family TF motifs, and EHF and ELF5 as ETS family TF motifs) regions of chromatin compared to normal lung epithelial cells. The hyper-accessible regions correspond to the SCLC related TFs or proneural TF including NEUROD1, NEUROG2, ASCL1, OLIG2, and NKX family TF (NKX2.1, NKX2.2, NKX2.5 and NKX6.1) [84]. Neurogenic differentiation 1 (NeuroD1) and ASCL1 are the lineage-specific markers of SCLC, and are critical TFs for the initiation and survival of SCLC cells (Figure 2) [85–88]. These TFs act negatively to the NOTCH-signaling, which plays an indispensable role in the differentiation of NE cells that further control cell proliferation, migration, EMT, and chemoresistance [88]. Lysine demethylase 1 (LSD1) is highly overexpressed in SCLC cell lines or tumors, and small molecule inhibition studies of LSD1 suggested that the DNA hypomethylation status is directly correlated with the efficacy of LSD1 inhibitors [89]. These studies identify a DNA hypomethylation signature that can be used as a biomarker for the activity of, or the stratification of SCLC patients. Enrichment of SMAD2 binding motifs in the differentially methylated sites suggest the involvement of TGF beta pathway in the epigenetic regulation of SCLC and as a mechanism of LSD1 inhibition.
2.1.3. Methylation of DNA repair pathway genes
The DNA repair pathway is one of the frequently affected corridors in SCLC. Studies suggest that in most of the SCLC cases, the genes of DNA repair pathway are regulated by methylation (promoter methylation or gene methylation). Some of the important signatures in this category involve O6-methylguanine DNA methyltransferase (MGMT) gene, that encodes for an enzyme regulating DNA repair by removing the alkyl groups from guanine (from O6 position) [90, 91]. Hypermethylation of MGMT promoter was observed in ~90% of SCLC cell lines, whereas the MGMT gene was found methylated in nearly 20–30% of SCLC tissues (Figure 1) [92–94].
Another member in this category is a tumor suppressor gene named fragile histidine triad (FHIT) that regulates p53-independent cell-cycle regulation, apoptosis, and confers protection against chemically induced lung cancer [32, 41, 67, 95]. The promoter methylation and subsequent inactivation or downregulation of FHIT was noticed in a majority of SCLC tissue samples or cell lines [1, 41, 96].
2.1.4. Methylation of metastasis or EMT related genes
Cell adhesion and metastasis-related genes that maintain the normal architecture of tissues and inhibit SCLC progression are the genes impacted significantly by methylation patterns. These include cadherin genes, especially cadherin 1 (CDH1 or E-cadherin), cadherin 13 (CDH13 or H-cadherin), tissue inhibitor of metalloproteinase 3 (TIMP3), MMPs, and snail [91]. Hypermethylation of CDH1 promoter is a very common phenomenon in SCLC [64, 97]. Different genes or pathways are responsible for this, like snail2 and NFIB, they epigenetically downregulate the expression of CDH1 [98, 99]. CDH13 is the other member of the cadherin family whose expression is regulated by methylation, and is associated with SCLC metastasis [91]. In other lung cancer subtypes, methylation of CDH13 is associated with cisplatin resistance [100]. DNA polymerase β (Pol β), a critical enzyme for DNA base excision repair and genomic stability/maintenance, modulates the methylation of CDH13. It works as a demethylase and decreases the promoter methylation of CDH13 [101]. The downregulation of CDH13 results in enhanced migration and angiogenesis in SCLC. Recently, the analysis of circulating cell-free DNA from patients with early-stage lung cancer also confirmed extensive hypermethylation of CDH13, marking the potential of these epigenetic modifications towards the establishment of early-stage lung cancer diagnosis [102].
2.2. Histone modifications in SCLC
Histones are highly basic proteins (rich in arginine and lysine) in the nuclei that act as spools to pack the DNA into nucleosomes [103, 104]. Thus, any alteration in the wrapping of genetic material ultimately harms the wrapped object (DNA), which suggests that if there is any aberration in histones, it eventually alters the expression pattern of the related gene [35, 105]. Thus, histone modifications turn out to be a critical component of epigenetic regulations. Following synthesis, nucleosomal histones go through massive post-translational modifications that involve acetylation, methylation, phosphorylation, ubiquitylation, sumoylation, and succinylation (Figure 1) [106–108]. These modifications are preserved by restricting the activities of histone acetyltransferases (HATs) and histone deacetylases (HDACs) [109].
The N-terminal tails of nucleosomal histones (H2A, H2B, H3 & H4) comprise of a 20–30 amino acid long unstructured region dominated by lysine residues available easily for different types of covalent modifications [110, 111]. The histone modifications and aforementioned methylation anomalies play a mutual aid role in modifying the chromatin conformation and modulate gene expression. In different types of cancer, including SCLC, the CpG hypermethylation of TS genes (TSG) or methylation dynamics is associated with a particular type of histone modification like deacetylation of histone H3/H4, trimethylation of histone H3 (H3K9/H3K27), and loss of lysine 4 methylation in histone H3 (H3K4) [112–115].
2.2.1. Histone modification in MLL2 family
The trimethylation and acetylation status of H2 and H3 differs significantly in NSCLC and SCLC, helping to identify the subpopulations along with the differential prognosis [116]. This marks the importance of histone-associated epigenetic changes in lung cancer. In the case of SCLC, the modifications or mutations in chromatin remodeling enzymes have been identified, suggesting the importance of these chromatin keepers in the development or progression of the disease. Similar to the changes in the methylation status of TSG or oncogenes, the corresponding change in the methylation status of nearby histone plays a role in SCLC development. Supporting this, the demethylation of histone H4 leads to the growth of NE tumors and correspondingly increases the proliferation [117]. Nearly 8% of SCLC tumors and 17% SCLC cell lines harbor several types of mutations (mainly truncating) in histone methyltransferase encoding gene named lysine methyltransferase 2D gene (KMT2D), also known as MLL2 [11]. KMT2D is an important transcriptional enhancer regulator and reported for the methylation (monomethylation and dimethylation) of histone H3 lysine 4 (Figure 1) [118, 119]. The monomethylation of H3K4 is considered as a chromatin marker linked with transcriptional enhancers [120, 121]. In SCLC, mutations in KMT2D reduces H3K4 monomethylation locally and dimethylation globally, and thus suggesting that the methylation status of H3K4 is directly correlated with SCLC.
The major nonsense mutations in KMT2D histone methyltransferase are G4779X, S2590X, and Q809fs, whereas homozygous point mutations have also been reported in exon 51 that leads to I5430M substitution [11]. This mutation was mapped within the conserved SET domain, critical for the methyltransferase activity of KMT2D. These mutations lead to the loss of protein expression and correspondingly decrease the methyltransferase activity. Truncating mutations are also noticed in polybromo 1 gene (PBRM1), an important chromatin remodeling gene [11]. PBRM1 encodes a bromodomain-containing protein BAF180, which is an important element of the PBAF SWI/SNF complex (a chromatin remodeling complex) [122]. Although no detailed studies are available for PBRM1 in SCLC, this gene is located at 3p21, a frequently deleted or mutated locus in SCLC [61, 123]. The studies on PBRM1 or SWI/SNF complex in other cancers suggested that the components of this complex found mutated in more than 20% of all studied cancers, and mutations in PBRM1 altered the chromatin accessibilities to different transcription factors. PBRM1 is also responsible for T-cell mediated immune response, chemoresistance and regulates MYC, an important oncogene for different cancers [124–126]. Studies suggested that loss of PBAF altered chromatin structure in a way making it easily accessible for response elements of IFN-γ in interferon-stimulated gene (ISG) promoter regions and thus increasing the expression of ISGs. Under physiological conditions, PBAF might cooperate with EZH2 (chromatin modifier) and reduces the chromatin accessibility to IFN-γ elements [125, 127]. Thus, these chromatin modifications can potentially determine T-cell response and immune escape properties of cancer cells, including SCLC.
2.2.2. Histone modifications of CREBBP-EP300 axis
The other important regulator of SCLC in the category of the transcriptional enhancer is CREB binding protein gene (CREBBP), which encodes an acetyltransferase, and is one of the most frequently mutated gene in SCLC [128–131]. The inactivation of CREBBP accelerates SCLC in the autochthonous mouse model. Expression analyses revealed that the inactivation of CREBBP results in the decreased expression of cell adhesions and tight junctions proteins including CDH1 across neuroendocrine cells (typical SCLC cell) [128]. The loss of CEBBP reduces the acetylation of histones (Figure 1). These studies supported the implication of deacetylase inhibitors (like pracinostat), as the treatment of pracinostat increases the acetylation of histones and expression of CDH1. A subset of the Rb1/Trp53/Crebbp deficient SCLC mice showed remarkable sensitivity and response to pracinostat [128]. The outcomes of genome-wide loss-of-function screens and gene analysis established CREBBP as a predictive biomarker for volasertib (an inhibitor of polo-like kinase 1) [132]. Interestingly, these studies showed that the effect of PLK1 inhibitors (like volasertib) and deacetylase inhibitors (like pracinostat) depend on the mutational status of CREBBP or acetylation of histones, and further established the importance of histone acetylation for developing SCLC therapies targeting epigenetic alterations. The other partner of CREBBP is the E1A binding protein p300 gene (EP300), which is mutated or inactivated in conjunction with CREBBP. Ep300 also has an intrinsic HAT activity and acts as a critical regulator of different biological functions like cell growth, embryonic development, and homeostasis by modulating chromatin remodeling and accessibility to other transcription factors [133, 134].
The missense mutations in the HAT encoding domains of CREBBP and EP300 signify the functioning of these TSG, and mutual selectiveness between CREBBP/EP300 mutations affecting HAT domains also suggesting dominant-negative behavior on wild type proteins/functional paralogues [1]. In SCLC, the significance of mutant CREBBP-EP300 has been studied using Rb1/Trp53-mutant GEMMs and precancerous cells of SCLC. The CRISPR-mediated mutational studies of HAT domains suggested the tumor-suppressive function of HAT containing transcriptional enhancers or co-factors [11, 134]. CREBBP-EP300 acetylates H3K27 to facilitate the transcription of target genes in concert with MLL3/4-UTX demethylase complex and antagonize the PRC2-mediated histone methylation that generally downregulates the expression of different genes, especially TSG [135–138]. In the context of SCLC, the altered CREBBP/EP300 activities modify the global acetylation of H3K27 and most likely affects the expression of TSG through methylation. Further identification of the genes targeted by CREBBP-EP300 will augment our understanding of SCLC tumorigenesis.
At a lower frequency, gene inactivating mutations are noticed in KAT6B, chromodomain helicase DNA binding protein 7 gene (CHD7), histone demethylase UTX gene, and chromatin remodeling factors like ARID1A and ARID1B [1, 11, 139]. Interestingly, KAT6B also encodes an acetyltransferase that acetylates histone H3 at lysine 23 (substrate similar to CREBBP/EP300) and acts as a tumor suppressor in SCLC [139]. KAT6B undergoes homozygous deletion in SCLC, and this genomic loss confers sensitivity to Irinotecan (a chemotherapeutic drug) [139]. In relation to the previous studies, the KAT6B deletion established that MYST family of HATs supported the ATM-mediated DNA-damage response and in KAT6B deficient SCLC tumors irinotecan induced damage cannot be repaired efficiently, and thus become a contributing factor towards the sensitivity of SCLC [139, 140]. The phosphorylation of histone H2AX also decreases upon irinotecan treatment. This also suggests developing targeted therapies for SCLC based on histone modifications or deletion/inactivation of HATs.
2.2.3. Histone modifications in LSD1–ASCL1 and NOTCH axis
KDM5A is the central component of NOTCH-RBP-J repressor complex and dynamically erases the methylation of histone H3 lysine 4 and increases the expression of ASCL1 [141]. ASCL1 holds immense potential in SCLC as it regulates different pathways of SCLC. An interesting study recently reported the role of a histone demethylase KDM5A/RBP2 in the regulation of NOTCH signaling that sustains the hallmark capability of NE differentiation and SCLC progression [29]. The results of this study highlighted that KDM5A promotes proliferation and NE differentiation phenotype of SCLC by supporting the expression of NE transcription factor ASCL1. The KDM5A endures the levels of ASCL1and NE phenotype by repressing NOTCH2 and NOTCH related genes. KDM5A decreases the trimethylation of H3K4 and increases the expression of ASCL1 [29]. SCLC cells with high ASCL1 levels have low expression of NOTCH receptors and target genes (Figure 2) [1]. Another recent study demonstrated the role of ASCL1 in Wnt11 pathways that regulate cell proliferation, NE differentiation, and epithelial-to-mesenchymal transition (EMT) in SCLC [142]. ASCL1 directly regulates the expression of Wnt11 through enhancer region acetylation of lysine H3K27. Wnt11 is specifically upregulated in SCLC, and in the presence of ASCL1, it modulates the expression of E-cadherin, snail, and NE markers in a context-dependent manner [142].
The indirect inhibition of ASCL1 by inhibiting the LSD1 (lysine-specific demethylase) has also been reported recently that establishes a novel LSD1–NOTCH–ASCL1 therapeutic axis in SCLC. [131]. In most of the gene expression frameworks, LSD1 demethylates lysine of histone H3 (H3K4me1/2); however, SCLC exhibits different histone modifications. LSD1 inhibition in SCLC activates NOTCH1 expression and decreases ASCL1, and these changes are associated with the acetylation status of histone (H3K27Ac) [131, 143]. Iadademstat (a covalent inhibitor of LSD1, also known as ORY-1001) treatment was found to increase H3K27Ac at NOTCH1 around LSD1 binding site both in SCLC cells and PDX models, suggesting that LSD1 modulates the acetylation of H3K27Ac [131]. It means that the consequences of LSD1 inhibition are directly associated with epigenetic changes in SCLC, resulting in NOTCH activation and ASCL1 downregulation [131, 143]. NOTCH further inhibited the differentiation or NE lineage of SCLC through ASCL1 downregulation, which is a crucial factor for the survival of SCLC cells [1, 144].
Two independent drug development studies also found that inhibition of LSD1 using GSK2879552/ORY-1001 and T-3775440 (small molecules inhibitors for LSD1) exerted anticancer effects in SCLC cells that were mechanistically dependent on the hypomethylation status of histones or other genes including ZEB1, IGFBP2, SNAIL [89, 145]. These inhibitors also decrease the expression of NE fate associated genes in SCLC like ASCL1, whose expression is associated with the methylation and acetylation status of histones (Figure 1 & 2). Although the detailed mechanism of LSD1 mediated SCLC development and progression remains elusive, however these studies have established the role of epigenetic modifications pertaining to LSD1, NOTCH, and ASCL1, and their potential as targets for developing unique therapeutic approaches for SCLC.
2.2.4. Histone modifications in the members of polycomb repressive complex
Recently, BRD4-ASXL3-BAP1 axis which modulates chromatin enhancers, has emerged as another epigenetic determinant specifically associated with SCLC-A subtype [146, 147]. BRCA1-associated protein 1 (BAP1) is a major histone deubiquitinase of polycomb repressive complex that modifies histone H2A at lysine 119. BAP1 also regulates the recruitment of several other epigenetic complexes like lysine methyltransferase KMT2C and other members of COMPASS family [148, 149]. Additional sex combs-like protein 3 (ASXL3) works as a linking channel between BAP1 and bromodomain-containing protein 4 (BRD4) in SCLC-A subtype where ASCL1 expression is very high. ASXL3 possesses a BRD4 binding motif that physically interacts with the extra-terminal domain of BRD4 and regulates the chromatin occupancy. Genetic deletion of ASXL3 globally decreases the acetylation of H3K27 and gene expression of BRD4 target genes in SCLC. BET specific pharmacological inhibitors or degraders decrease the cell proliferation of specific SCLC-subtypes where high expression of ASXL3 is noticed [146].
In another interesting study, Shukla et al., [147] generated induced pluripotent stem cells (iPSC) from human small airway/lung epithelium and demonstrated the epigenetic role of ASXL3 in pluripotency and stemness of SCLC. ASXL3 is highly overexpressed in lung–iPSC, cell lines, and clinical samples of SCLC. Silencing of ASXL3 inhibited clonogenicity, proliferation, and teratoma induction by lung-iPSC, and in-vivo tumorigenesis of SCLC cells. Epigenetic analysis of lung-iPSC established a decrease in the trimethylation of H3K27 (repressive histone mark) without a corresponding increase in the trimethylation of H3K4 (histone activation mark) in a number of genes such as MAGE-A1, MAGE-A3, and ESO-1. They also demonstrated the hypomethylation of NANOG and OCT4 promoter regions with decreased H3K27 and increased H3K4 trimethylation in lung-iPSC [147]. These observations suggest that the epigenetic modifier ASXL3 maintains the pluripotency of lung-iPSC and also acts as an oncogene in SCLC. In these lung-iPSCs, the expression of EZH2 (the main component of PRC2 complex) is also very high, and the physical interactions of AXSL proteins with EZH2 and BAP1 is also reported [146, 147, 150]. Interestingly, germane to SCLC, ASXL3 is also known to interact with KDM1A/LSD1, which also co-localizes with NANOG and OCT4, and together with PRC2, maintains the chromatin of lineage-specific undifferentiated stem cells [147, 151].
2.2.5. Histone modifications impacting MYC family
MYC family, including MYCL and MYCN, is a family of proto-oncogenes with structural homology, but functional diversity stands out as an essential driver for SCLC and constitutes a novel therapeutic axis [1, 10, 44]. Apart from the epigenetic abnormalities of HATs, MYC family members’ have been implicated in SCLC and the functional role of MYC family members in SCLC has been extensively discussed in multiple studies [28, 43, 44, 152, 153]. Different genetic and epigenetic alterations have been reported in MYC family members that exacerbate SCLC. MYC members are the important helix-loop-helix (HLH) type leucine zipper TFs that bind to E-box elements and trigger the expression of target genes in cooperation with other HLH protein MAX. MYC genes encode highly conserved domains that help to recruit transcriptional machinery for functional regulation [43, 154].
There is an interesting correlation between the differential expressions of MYC family members (MYC, MYCN, and MYCL) and subtypes of SCLC; for example, two substantially different transcripts of MYCL have been noticed in SCLC; similarly inactivation of MYCL or MYCN in mouse SCLC cell lines reduces tumorigenic properties while inactivation of MYC does not [43, 152, 155]. Thus, there are mechanistic differences among family members of MYC on how they impact SCLC or each member have different impact on SCLC progression [43]. MYC regulates the expression of several genes that contribute to SCLC development through different genetic and epigenetic regulations like the recruitment of basal TFs, RNA polymerases, chromatin remodeling enzymes, and histone acetylases [156–159]. MYC is a TF, that binds to specific sites of DNA and promotes the acetylation of H3 and H4 histones [158]. MYC is found associated with TIP60/KAT5, which is a crucial acetyltransferase. MYC-induced acetylation of H3 and H4 histones increases the expression of MYC target genes [158]. TIP60 also plays a regulatory role in cell proliferation and apoptosis [160] and the association of MYC with TIP60 further suggest MYC driven epigenetic regulation of these cancer-associated pathways. Other epigenetic regulators through which MYC causes epigenetic alterations in SCLC are bromodomain and extra-terminal (BET) family proteins [161, 162]. BET proteins are the critical epigenetic writers who interacted with different chromatin modifiers, including HATs and HDACs [163]. These proteins bind explicitly to acetylated-lysine residues of histones and change the chromatin accessibility and expression pattern of target genes [164].
Genetic and pharmacological inhibition of BET or MYC genes decreases the growth and proliferation of SCLC cells [161, 162]. Similarly, a combination of DNA-demethylation agents depletes MYC and increases the efficacy of HDAC inhibitors in lung cancer [165]. Recent reports suggested the diverse implications of MYCN and MYCL in SCLC metabolism and drug resistance [166, 167]. MYC-driven SCLC cells and tumors mainly depend on arginine-mediated metabolic pathways like polyamine biosynthesis and mTOR [166]. In chemoresistant SCLC cells or tumors, MYCN associated synthetic vulnerabilities has been identified, and it was found that pharmacological inhibition of USP7 (a deubiquitinase) resensitizes the SCLC PDX to chemotherapy [167]. These studies did not provide the direct role of epigenetic regulation/histone modifications in presently reported SCLC mechanisms; however, there are reports in other cancer types that prove the role of these epigenetic regulations or histone modifications in the regulation of similar pathways [168–170]. Thus, these reports also provided a link between MYC related epigenetic alterations concerning SCLC metabolism and drug resistance. Further, these studies imply the utility of these epigenetic modifiers in relation to MYC for SCLC therapies.
2.2.6. Histone modifications in metastatic determinants of SCLC: NFIB
Metastasis is a characteristic feature and the leading cause of SCLC related deaths. Different mechanisms have been proposed for SCLC metastasis of which amplification of MYC and nuclear factor I B (NFIB) are the two significant determinants [171–173]. Both molecules serve as TFs and extensively regulate chromatin modifications and are themselves regulated by epigenetic changes [174, 175]. Genome-wide analyses of pure SCLC population from GEMM suggested the potential role of NFIB in chromatin accessibility and histone modifications. The amplification of NFIB causes chromatin relaxation, increases the accessibility of intergenic regions, and enriches the TF binding sites (Figure 1) [171, 174, 176]. These chromatin modifications lead to the global reprogramming of pro-metastatic genes and enhance the metastatic potential of SCLC. Approximately 5–15% of invasive primary SCLC tumor cells have high NFIB, whereas almost all metastatic SCLC cells exhibit NFIB amplification [171]. The importance of NFIB in histone modifications is also revealed by its associated partners, like a recent report showed that it forms a complex with histone deacetylase 3 (HDAC3) [134, 177]. HDAC3 is a critical deacetylase responsible for the development of neural stem and progenitor cells [177].
The NFIB mediated chromatin relaxation/accessibility enriches a unique histone modification signature by regulating the trimethylation of histone at two different sites, H3K4me3 (active) and H3K27me3 (repressive) [178, 179]. These histone modifications play a regulatory role in cell fate decisions, differentiation, and gene expression [178]. The second important direct epigenetic target of NFIB is EZH2 that mainly altered the trimethylation of H3K27, and regulates the expression of different oncogenes [180]. Interestingly, NFIB also plays a role in lineage fate determination and stem cell maintenance [174, 181]. It is proposed that NFIB not only acts as a metastatic gate for SCLC, it can also serve as a biomarker for SCLC classification due to its involvement in cell fate determination (Figure 2). In addition, these studies put forward many questions for future research that will identify other epigenetic partners of NFIB for the identification of additional therapeutic targets.
A recent study reported RUNX1 partner transcriptional co-repressor 1 (RUNX1T1) as an epigenetic modifier in SCLC [182]. RUNX1T1 encodes a putative zinc finger TF and recruits various corepressors to facilitate transcriptional repression [183]. Overexpression of RUNX1T1 decreases the expression of CDKN1A (p21) and enhances the transcriptional activity of E2F TF (a commonly altered molecule in SCLC). RUNX1T1 interacts with the promoter region of CDKN1A and decreases the acetylation of histone 3. HDAC inhibitor trichostatin-A restored the expression of CDKN1A while the knockdown of RUNX1T1 increased the global acetylation of histone and CDKN1A promoter [182]. Thus, these studies have established the role of histone-modifying proteins/enzymes in SCLC progression and development, provide opportunities to interrogate these targets further. The aforementioned exciting findings related to the expression, functioning, and epigenetic modifications concerning MYC, NFIB, USP7, ASXL3, LSD1, PRC2, EZH2, BAP1, BRD4, ASCL1, and NOTCH have provided novel insight into SCLC carcinogenesis. The clinical phenotypes established by these epigenetic landscapes further underscore the utility of epigenetic modulators as novel therapeutic targets for the development of SCLC therapies (Figure 1 & 2, Table 1).
Table 1:
Common epigenetic predictors in SCLC.
| Gene | Epigenetic Mark | Outcomes | Reference |
|---|---|---|---|
| EZH2 | DNA methylation Histone methylation (H3K27) | -Smoke induced epigenetic changes -Modulates TGF-β-SMAD pathway -Upregulates ASCL1 -Activates DNMT1 -Upregulates TWIST1 -Suppression of SLFN11 -Apoptosis -Chemoresistance |
[25, 32, 57, 70-76] |
| DNMT1 | DNA methylation | -NE fate determination (ASCL1 high) | [81, 82] |
| DNMT3A/B | DNA methylation | -NE fate determination (NEUROD1 high) | [81, 82] |
| POU2F3 | Promoter hypermethylation | -NE fate determination (POU2F3 high) | [41, 79, 83] |
| MGMT | Promoter hypermethylation | -DNA repair pathway | [92-94] |
| FHIT | Promoter hypermethylation | -p53 independent cell-cycle regulation -Apoptosis |
[1, 41, 96] |
| BCL2 | Promoter hypermethylation | -Cell survival | [57, 77] |
| RASSFIA | Promoter hypermethylation | -Dysregulated cell cycle -Apoptosis |
[59-61, 64] |
| CAV1 | Promoter hypermethylation | -Autophagy -Hedgehog and MAPK signaling -Cell growth |
[62, 63, 65-67] |
| CDH1 | Promoter hypermethylation | -High metastasis/EMT | [64, 91, 97] |
| CDH13 | Promoter hypermethylation | -High metastasis/EMT | [91] |
| ASCL1 | Histone acetylation (H3K27) | -NE differentiation -Regulates Wnt11 pathway -Modulates EMT (CDH1, Snail) |
[142] |
| LSD1 | Histone demethylation (H3K4, H3K9) Histone acetylation (H3K27) | -Modulates NOTCH-ASCL1 axis -NE differentiation -Modulates EMT (ZEB1, Snail) -Chemoresistance -Cell growth, migration, and invasion |
[1, 32, 89, 131, 143-145] |
| KDM5A | Histone demethylation (H3K4) | -NE differentiation -Supports ASCL1 expression -Regulates NOTCH signaling |
[29, 141] |
| NFIB | Histone acetylation Histone methylation (H3K4, H3K27) | -Chromatin remodeling/relaxation -Chromatin accessibility -EMT (downregulate CDH1) -Metastasis -Modulates HDAC3, EZH2 -Lineage fate determination -Stem cell maintenance |
[134, 171-174, 176-181] |
| KMT2D/MLL2 | Histone methylation (H3K4) | -Cell proliferation -Growth of NE tumors -Regulates transcriptional enhancers |
[118-121] |
| CREBBP | Histone acetylation (H3K27) | -Decreases the expression of cell adhesions/tight junction genes (like CDH1) in NE cells | [1, 128-131] |
| EP300 | Histone acetylation (H3K27) | -Cell growth -Chromatin accessibility |
[1, 133, 134] |
| KAT6B | Histone acetylation (H3K23) | -DNA damage response | [139, 140] |
| MYC (L/N) | Histone acetylation (H3 and H4) | -Chromatin remodeling -Chemoresistance -Metabolism -Recruitment of transcription factors -Recruitment histone acetylases -Regulates TIP60/KAT5, BET proteins -Cell proliferation, apoptosis -Metastasis |
[1, 10, 43, 44, 152, 155, 161, 162, 166, 167] |
| BET | Histone acetylation | -Chromatin accessibility -Interacts with HATs and HDACs -Cell growth and proliferation |
[163, 164] |
| BRD4 | -Chromatin accessibility | [146, 147] | |
| ASXL3 | Histone acetylation (H3K27) Histone methylation (H3K4, H3K27) | -Chromatin accessibility -Expression of BRD4 target genes -Pluripotency and stemness -Cell proliferation -Interacts with LSD1 |
[146, 147, 150, 151] |
| BAP1 | Histone deubiquitination (H2AK119) Histone methylation | -Chromatin accessibility -Regulate KMT2C or CAMPASS members |
[146–149] |
3. Epigenetic targets and drugs in SCLC
Target identification and drug development are the two indispensable components of any drug-discovery program. The drug targets are generally protein molecules or enzymes playing an essential role in catalysis for the completion of important biological reactions. The druggable epigenetic regulators are mainly divided into three categories; writers, readers, and erasers. The protein molecules that ‘mark’ DNA or histone by incorporating chemical modifications or groups are known as ‘writers’ such as methyltransferases, HATs, and kinases. ‘Readers’ are the molecules that read these epigenetic modifications and alter the molecular mechanism accordingly consist of chromodomain and bromodomain proteins, whereas ‘erasers’ as the name suggested, wipe out the incorporated epigenetic changes include histone lysine demethylases and HDACs [184]. In the context of epigenetics, the process of epigenetic modifications (methylation, acetylation, sumoylation, and phosphorylation) is regulated by a number of important molecules such as protein methyltransferases, histone deacetylases (HDACs), and protein kinases. These epigenetic modifications and epigenetic regulators contribute to therapy resistance and disease aggressiveness. Recent reports showed a significant correlation between DNA methylation marks and sensitivity to chemo- or radiotherapy in SCLC [32, 45]. Downregulation of three prime repair exonuclease 1 (TREX1, a gene that encodes an exonuclease or STING antagonist) through promoter hypermethylation is associated with the sensitivity to Aurora kinase inhibitors (AZD-1152, SCH-1473759, SNS-314, and TAK-901), CDK inhibitor (R-547), Vertex ATR inhibitor (Cpd 45), and the spindle disruptor vinorelbine. Multiple epigenetic mechanisms like 3′UTR methylation of CEP350, EPAS1, and MLPH were associated with sensitivity to Aurora kinase inhibitors. Methylation of EPAS1, LSD1/KDM1A was associated with the response of PLK-1, BCL-2, and KSP inhibitors. Promoter hypermethylation of SLFN11 decreases the protein expression and contributes to resistance to DNA damaging agents. Similarly, the 5′ UTR of EZH2 and methylation of YAP1 have been shown to be associated with the response of Aurora kinase and mTOR inhibitors, respectively [32].
Promoter hypermethylation increases hTERT expression and promotes overexpression of EZH2 that finally alters methylation of H3K27 and contributes to the development of radiotherapy resistance in SCLC [45]. Epigenetic regulations of NE differentiation-related genes (ASCL1, NEUROD1, NEUROG2, OLIG2, and NKX homeodomain TFs) and RASSF1 also contribute towards the drug sensitivity of SCLC [32, 84, 185]. Overall, epigenetic regulators play the diverse roles in modulating therapy response and hold promise as targets for drug development for SCLC. For the targeting of SCLC, two main classes of drug targets are deacetylates (like HDACs) and acetyl-/methyl transferases (like LSD-1) that regulate the development and progression of SCLC. The utility of these epigenetic machinery components as drug targets and the targeting properties of their inhibitors is discussed in the following sections.
3.1. Inhibitors of LSD-1/KDM1A for SCLC
LSD1 is a flavin-containing protein that functions as a histone demethylase and corepressor of transcription [186]. LSD1 is the widely studied member of the LSD family and is overexpressed in multiple cancers, including SCLC subtypes. It erases the mono- and dimethylation signatures from histone 3 lysine 4 (H3K4), resulting in transcriptional repression or H3K9 that leads to activation. Several chemically synthesized and natural inhibitors have been reported for LSD1 that have shown efficacy in various cancers and are currently under clinical trial [187–189]. Interestingly, the inhibition of LSD1 provides a novel epigenetic therapeutic approach for the management of SCLC [89, 131]. Augert et al., have shown that the ORY-1001 (a selective LSD1 inhibitor) mediated LSD1 inhibition results in the activation of NOTCH pathway and downregulates the expression of ASCL1 that decreases the tumorigenesis of SCLC [131]. The activation of NOTCH, along with the inhibition of ASCL1, holds therapeutic promise in SCLC as NOTCHhigh-ASCL1low also suppresses the differentiation of NE lineage. The ORY-1001 inhibitor exhibited efficacy in chemoresistance patient-derived xenografts (PDX) models of SCLC.
Mohammad et al., reported another selective, orally bioavailable, and highly potent LSD1 inhibitor (GSK2879552) that shows anticancer properties in SCLC cell lines and tumor models [89]. The efficacy of GSK2879552 was dependent on the methylation status of DNA, and a DNA hypomethylation signature that predicts the sensitivity of SCLC to the inhibitors of LSD1 was identified [89, 190]. The cohesive analysis of epigenetic modifications associated with LSD1 inhibition also suggests changes in the expression of NE genes associated with the cell fate determination or SCLC stem cells [89, 190]. GSK2879552 completed phase I clinical trial for SCLC (Trial number: NCT02034123) that was terminated early, while another phase I clinical trial (Trial number: NCT02913443) was completed recently. In the latter, the potential of orally available LSD1 inhibitor (RG6016/ RO7051790) for relapsed and extensive stage SCLC was evaluated [191]. As a subset of SCLC shows the sensitivity of LSD1 inhibitors, these studies demonstrate the utility of LSD1 mediated epigenetic signatures to stratify the SCLC patients for the selection of therapy.
The three-dimensional structure of human LSD1 was solved in 2006 (PDB ID: 2HKO, PDB ID: 2IW5) [192, 193], and has served as an important platform for the development of specific small molecule inhibitors with wide implications. Structural features of LSD1 show that it contains an N-terminal SWIRM domain and amine oxidase domain at C-terminal, and it forms a unique complex with REST corepressor 1 (CoREST) (Figure 3). SANT2, a C-terminal domain of CoREST, acts as a stimulator for LSD1 demethylase activity on nucleosomes or histones. LSD1 alone is sufficient to demethylate histone (histone tail), but for the demethylation of a bulky nucleosome, it requires SANT2 of CoREST [193]. Structural arrangements suggested that CoREST SANT2 is situated ~100Å away from the LSD1 active site (Figure 3). These observations provide a critical rationale for designing selective inhibitors for LSD1-mediated epigenetic modifications, for inhibiting the demethylation of either histone tails or intact nucleosome. The detailed characterization of LSD1 associated epigenetic changes in SCLC will further establish the signatures that can be targeted using small molecule inhibitors. Overall, the inhibition studies of LSD1 in SCLC suggest its potential for the development of epidrugs or epigenetic therapies for SCLC. Some of the reported inhibitors of LSD1 are summarized in Table 2.
Figure 3:
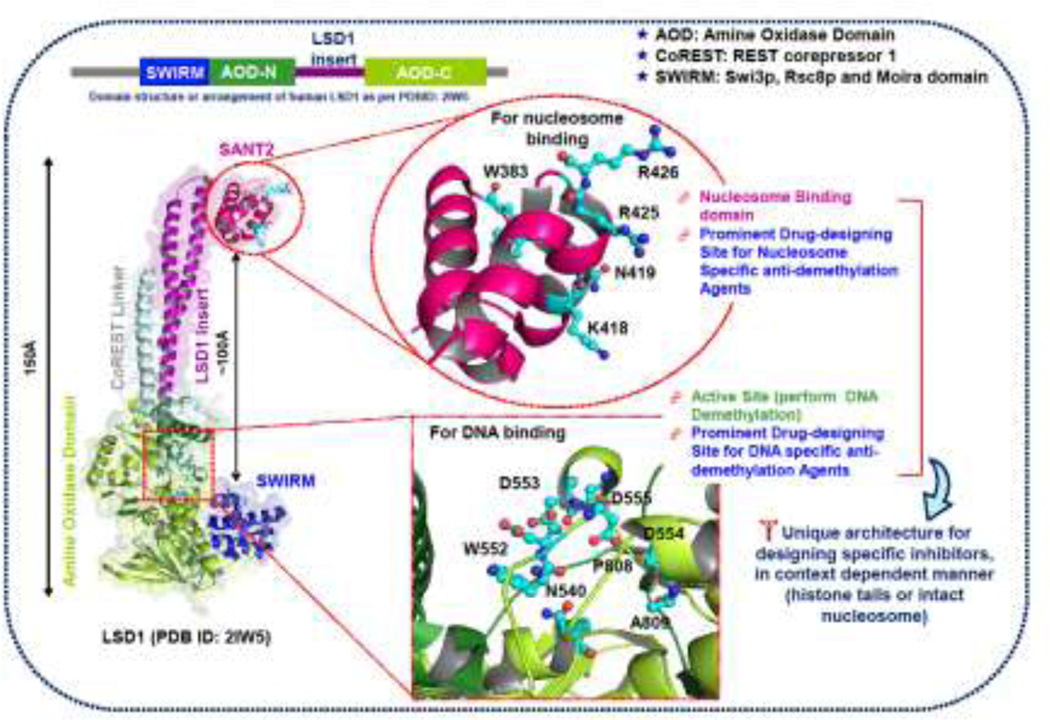
Three-dimensional structure of LSD1-CoREST complex showing the unique architecture of two different binding/active sites for the development of specific inhibitors of LSD1 with implications in SCLC. The same color scheme was used to represent linear domain organization (upper part) and subsequent domains in the three-dimensional cartoon model. The structural model was generated using PyMOL, and structure coordinates were taken from Protein Data Bank (PDB ID: 2IW5) [193].
Table 2:
Overview of epigenetic drugs/inhibitors along with the structures implicated in SCLC.
| Drug/Inhibitor | Structure | Target | Outcomes | Reference |
|---|---|---|---|---|
| Iadademstat (GSK2879552/ORY-1001) |
|
LSD1 | -Activates NOTCH -Decreases ASCL1 |
[89, 131] |
| T-3775440 |
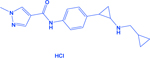
|
LSD1 | -Acts on LSD1-INSM1 interactions -Decreases ASCL1 |
[145, 265] |
| Vorinostat |
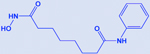
|
HDAC | -Increases acetylation of histone H3 -Enhances the efficacy of cisplatin & BCL2 inhibitors |
[195, 202, 203] |
| Belinostat |
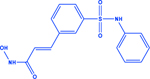
|
HDAC | -Increases lysine acetylation -Effective in NE subtypes |
[195, 201] |
| Romidepsin |
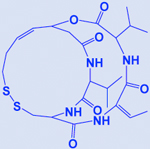
|
HDAC | -Increases acetylation of histone H3 & H4 | [195, 266] |
| Panobinostat/LBH589 |
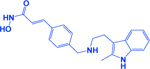
|
HDAC | -Increases efficacy of DNMT inhibitors -Sustains stable diseases |
[204, 267] |
| Trichostatin A |
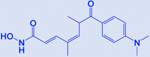
|
HDAC | -Effective in EGFR mutated distinct subtype -Increases acetylation of histone H4 |
[200, 268] |
| Pracinostat |
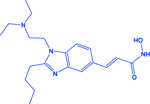
|
HDAC | -Increases acetylation of histone 3 (H3K27ac) -Increases CDH1 expression -Effective in CREBBP deleted SCLC tumors |
[128] |
| Ricolinostat |
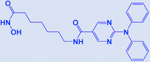
|
HDAC | -Enhances efficacy of BET inhibitors -Activates NK cell mediated innate immunity |
[207] |
| MGCD0103/Mocetinostat |
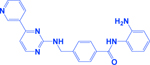
|
HDAC | -Increases efficacy of topoisomerase inhibitors -Increases tumor antigen presentation -Enhances check-point inhibitor therapy |
[205, 252] |
| Valproate/valproic acid |
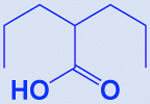
|
HDAC | -Augments cisplatin and etoposide activity (standard first-line chemotherapy) | [206] |
| 5-AzaC |
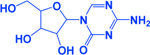
|
-Modulates death receptor & caspase-8 expression | [253, 269] | |
| JQ1 |
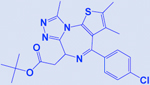
|
BET | -Enhances efficacy of HDAC inhibitors -Activates NK cell mediated innate immunity |
[207] |
| EPZ011989 |
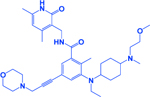
|
EZH2 | -Decreases methylation of histone H3 (H3K27me3) -Increases SLFN11 expression -Prevents emergence of chemoresistance -Augments chemoresponse |
[76, 270] |
| Tazemetostat/ EPZ-6438 |
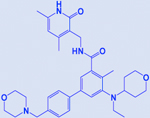
|
EZH2 | [76] |
3.2. HDAC/HAT inhibitors in SCLC
Acetylation is a post-translational modification that neutralizes the positive charge of lysine residues, reducing the electrostatic interactions with negatively charged DNA molecules, which finally help in chromatin relaxation and facilitate the gene expression [194, 195]. The overexpression of HDACs erases the acetylation marks of chromatin and divert the gene expression patterns towards cancerous phenotype. HDAC overexpression is closely associated with neuroendocrine tumors, and the other exciting features associated with HDACs are occasional somatic mutations, whereas loss of function mutations are often in HATs [11, 196–198]. Histone acetylation is an important epigenetic hallmark that modulates the expression of different genes responsible for the onset, progression, and aggressiveness of SCLC (as discussed in the previous section). The two main components that determine the acetylation status of histone is HDACs (erases the acetyl groups) and HATs (writer molecule for lysine acetylation in histone tails). The HDAC family consists of four subclasses (I, IIa, IIb, and IV and includes eleven HDACs [195, 199]. The binding of HDAC inhibitors (HDACi) to the specific HDAC inhibits the deacetylation of lysine (H4K5ac, H4K8ac, H3K9ac, H4K16ac, H3K18ac, H3K23ac, and H3K56ac) residues and maintains the global acetylation of chromatin. CREBBP and EP300 have widely investigated HATs affected by the loss of function mutations and play a significant role in the pathogenesis of SCLC [11, 128, 132, 198].
Presently, four HDACi are FDA approved, including vorinostat, belinostat, panobinostat, romidepsin, while a fifth one chidamide got regulatory approval in China [195]. The observed role of HDACs in the development of NE tumors led the foundation for the evaluation of HDACi in SCLC therapies. HDACi have diverse implications in SCLC as these inhibitors are reported to show efficacies in different SCLC models and subtypes, like trichostatin A (a pan-HDAC inhibitor) is highly effective in EGFR-mutated SCLC subtypes [200]. Trichostatin A treatment has been shown to sensitize the EGFR-mutated SCLC to conventional chemotherapy (cisplatin and etoposide) [200].
Jia et al., reported that the loss of CREBBP promotes the development of SCLC and enhances the efficacy of HDACi in SCLC [128]. CREBBP works as an acetylase for multiple histones, including H3K27. In SCLC, CREBBP loss epigenetically suppresses CDH1 expression and exacerbates the aggressiveness of the disease. Jia et al., used pracinostat (HDACi currently being in clinical trials for different cancers) and observed that it exhibits efficacy in SCLC models (cell lines, mouse models, and PDX) independent of CREBBP status, but the responses were higher in CREBBP deleted SCLC cases [128]. Loss of CREBBP and EP300 is an established feature for SCLC [1, 11, 128], so the other recognizable outcome of this study associated with HDACi, is the novel direction for the combination of HDACs and HATs inhibitors for SCLC or utilization of CREBBP loss to stratify the SCLC patients for HDACi based epigenetic therapies.
In a completed phase I clinical trial (NCT00926640) in SCLC patients, belinostat in combination with cisplatin and etoposide has shown promising results for efficacy and exhibited potential to move towards a phase II trial [201]. The combination of vorinostat (another FDA approved HDACi) with cisplatin augmented the antitumorigenic effects in SCLC [202]. Similarly, another report has shown that the combination of vorinostat and BCl2 inhibitor (Navitoclax/ABT-263) sensitizes the navitoclax resistant SCLC cell lines and suggested that HDACi could be used as an alternative strategy for resistant SCLC tumors [203]. The results of a phase II clinical trial also established the potential of panobinostat (or LBH589, FDA-approved pan-HDACi) where it shows that as a monotherapy it induces the shrinkage of SCLC tumors [204]. Though this trial was discontinued, but initial results suggested the evaluation of panobinostat or other HDACi in combinational regimens with standard chemotherapeutic agents [204].
Preclinical studies of HDACi (MGCD0103 and vorinostat) and topoisomerase inhibitors combination also showed superior cytotoxicity in SCLC cell lines and make a case for the future clinical evaluation of this combination [205]. Hubaux et al., provided the potential of valproate (HDACi) in combination with cisplatin and etoposide for augmented anticancer activity as a first-line chemotherapy of SCLC [206]. A recent study identified HDAC6 as a synthetic lethal drug target with JQ1 (an FDA approved BET inhibitor) and showed the efficacy of combination treatment in SCLC-xenograft with ricolinostat (or ACY-1215, a HDAC6 inhibitor) and JQ1 in reducing tumor growth. This combination (JQ1 and ricolinostat) was demonstrated to induce the innate immunity response in SCLC and suggested a novel treatment strategy for SCLC [207]. Other important inhibitors of epigenetic targets in SCLC like BET, CREBBP, BCL2, and EZH2 are summarized in Table 2.
4. Epigenetics and SCLC Immunotherapy
The past decade has witnessed the emergence of immunotherapy as a leading approach for cancer treatment and has revolutionized the management of various cancers, including SCLC [208–210]. Early in 2014, FDA approval of immune-checkpoint inhibition through ipilimumab (an anti-cytotoxic T lymphocyte antigen 4 or CTLA-4) introduced a unique clinical standard in cancer therapy of advanced-stage melanoma [211–213]. This cancer immunotherapy landscape further improved with the approval of two more antibodies against programmed cell death 1 (PD1) named nivolumab and pembrolizumab [214–221]. Since then, the inhibitors targeting PD-1 and CTLA-4 not only proved useful to improve the outcomes of melanoma, also found promising in lung cancer, renal cell carcinoma, Hodgkin’s lymphoma, and other malignancies [217, 219, 221].
The experts predicted that SCLC might respond favorably to immunotherapy, due to their high tumor mutational burden (TMB) to generate neoantigens that could be targeted by immunotherapy [222]. The relatively high TMB of SCLC is perceived due to the heavy smoking exposures, deficits in DNA repair pathways, and other genomic instabilities of SCLC [1, 11, 223]. The studies related to TMB established it as a predictive biomarker for immunotherapy response in NSCLC, which shows that combination of nivolumab and ipilimumab is more effective and beneficial than chemotherapy in patients where TMB is >10 mutations/megabase compared to the patients with TMB <10 mutations/megabase [224]. Immunotherapy has immense potential, but due to the restricted expression of receptor antigen, most of the patients did not respond or develop resistance [225–228]. Various immunotherapeutic approaches evaluated in SCLC failed to meet the desired end-point. Maintenance pembrolizumab after first-line chemotherapy evaluated in phase II trial did not improve progression-free survival (PFS) and overall survival (OS) [229]. Similarly, the maintenance ipilimumab and nivolumab (CheckMate 451) failed to meet the criteria of OS survival in a phase III trial [230, 231]. Another report announced the failure of CheckMate 331 (a phase III study, NCT02481830) to meet its primary endpoint of OS, comparing the topotecan and nivolumab in relapsed SCLC [232, 233].
Thus, the above-mentioned upshots mandate the development of innovative combinatorial approaches that will improve the efficacies or potentiate the response of immunotherapies [234, 235]. Combinations of epigenetic approaches/chemotherapies with immune-checkpoint inhibition holds promise and are currently being tested in clinical trials for different cancers [23, 236–238]. The rationale for the development of epigenetic combinations for immunotherapy of cancer is the key role of various epigenetic regulations in immune cell functioning, related immune responses and direct correlation of patient response with TMB [236, 237, 239, 240]. Satisfying the criteria of SCLC stratification on the basis of TMB and the implications of combinatorial immunotherapy has proved a fruitful approach compared to unselected cohorts. The results of CheckMate 032, in SCLC patients stratified on the basis of ‘low’; ‘medium’; or ‘high’ TMB, and receiving nivolumab with ipilimumab or nivolumab alone showed higher overall response rate (ORR) to nivolumab or ipilimumab/nivolumab combination in high TMB cohort [241–243]. The results of CheckMate 032 accelerated the FDA approval of nivolumab as a third-line treatment [244].
Initially, these studies provided a rationale for the combination of immunotherapy with chemotherapy, as the chemotherapy mediated damage releases tumor-specific antigens and increases the T-cell activation [245]. IMpower 133, a phase III trial, evaluated atezolizumab in combination with first-line chemotherapy and demonstrated a gain in PFS and OS compared to standard chemotherapy [246]. Based on the success of the trial, FDA approved atezolizumab in combination with first-line chemotherapy. On the other hand, a phase III trial of ipilimumab with first-line chemotherapy has failed to add any benefit for using immunotherapy [247]. The recent CASPIAN trial (a phase III clinical trial, NCT03043872), evaluated durvalumab alone or with tremelimumab in combination with platinum-etoposide showed the improvement in the OS of extensive-stage SCLC patients [248]. These outcomes of immunotherapy combinations speculated that the genetic factors alone are not responsible for differential outcomes of trials, there could be several factors within the tumor microenvironment that play a crucial role in enhancing the efficacy of immunotherapies.
Preliminary research in different cancers, including lung cancer showed that epigenetic modification plays a prominent role in the modulation of these factors [249, 250]. Several studies have tested combinations of epigenetic drugs like histone deacetylases inhibitors, hypomethylation agents to induce the changes within the tumor microenvironment and increases the applicability and efficacy of checkpoint inhibitors [251, 252]. Recent trials of immune-epigenetic therapies combining CC-486 (oral formulation of azacitidine) and vorinostat (a histone deacetylase inhibitors/HDACi) have shown enhanced overall response rate (ORR) among the patients not responding previously to immunotherapy alone [253, 254]. Another study performed on NSCLC patients showed that the epigenetic milieu of the tumors identifies/stratifies the patients most likely to benefit from the pembrolizumab and nivolumab [237]. Specifically, methylation status of FOXP1 served as a predictive biomarker for the clinical benefits of anti-PD-1 therapies. The combination of immunotherapy and epigenetic drugs in SCLC needs escalation as no significant reports are available pertaining to these combinations in SCLC.
A recent study established the role of MHC class-I antigen and PRC2 in immunotherapy resistance of SCLC [255]. PRC2 mediated transcriptional silencing of MHC-I antigen presentation in SCLC cells decreases the T-cell mediated immune response. MHC-I low SCLC cells harbor H3K4 trimethylation (activating) and H3K27 trimethylation (repressive) histone modifications and show a conserved mechanism through which SCLC tumors achieve high PRC2 activity and execute immune evasion [255, 256]. There are studies in different cancers that show that the epigenetic regulators such as MYC family, EZH2, NOTCH, PTEN, LSD-1, and BET members plays an important role in determining the response of immunotherapies [131, 165, 256–263]. There are some promising combinations that hold investigative potential in SCLC. For example, LSD-1, MYC, EZH2 plays an inclusive role in immune modulation, drug resistance as well as overall development or differentiation of SCLC lineages [32, 76, 165, 167, 257–261]. These observations make a strong case for the combined evaluation of established epigenetic therapies of these targets with immunotherapies. The future implementation of these epigenetic-immunotherapies in SCLC will depend on the observed efficacies from clinical trials, however the outcomes of trials from other cancers give hope to see the development of these novel approaches involving emerging immunotherapeutic modalities combined with next-generation epigenetic drugs.
5. Concluding remarks and future prospective
SCLC remains a recalcitrant tumor with high lethality. There are significant obstacles with SCLC translational research. The first one is the scarcity of tumor tissues as surgical resections are not very common, unlike NSCLC. Despite the importance of drug resistance, the repeated biopsies of the recurrent disease are also rare. Secondly, is the observed disappointments of clinical trials though several putative targets are available that yield promising preclinical results. Further, there is limited success of immunotherapy due to the restricted expression of immune targets. However, the outcomes of genome-wide association studies combined with the “genomics”, “transcriptomics”, and “epigenomics” have defined the relevance of different epigenetic marks that regulate the biology of SCLC and have provided a roadmap for designing therapeutic modalities targeting epigenetic vulnerabilities.
The studies in the last decade prove the importance of epigenetic modifications in various aspects of SCLC, including NE differentiation, lineage specificity, drug resistance, and metastasis. These studies place SCLC research beyond genomics and establish the importance of epigenetics in the etiology of SCLC. The so far collected data of SCLC epigenetics has left us with a number of questions and set the stage for epigenetics research on multiple fronts. For example; LSD1 inhibition may be implemented as a potential therapeutic approach in SCLC patients, but the identification of specific biomarkers for the activity will be needed because LSD1 expression in SCLC is lineage-specific. Some studies suggest that ASCL1 expression could be an indicator of response to LSD1 inhibitors, but it is likely that other regulators of NOTCH pathway could help to predict the response of LSD1 inhibition, as its expression is directly correlated with NOTCH signaling. Also, the heterogeneity studies of SCLC tumors show that they are composed of NE and non-NE populations that are regulated through endogenous NOTCH and other pathways [176, 264]. So, it will be interesting to study the impact of epigenetic modification on the ratios of NE and non-NE cells induced by LSD1 inhibition. These outcomes will help to carefully design the combination therapies (chemotherapies/immunotherapies) because LSD1 expression is related to the immune response as well as drug resistance.
While expression of NFIB is associated with metastasis of SCLC [171, 176], and NFIB increases chromatin accessibility, the exact mechanism that show NFIB drives metastasis, and its interplay with the key epigenetic modifiers or target genes remains elusive. Elucidation of the mechanisms and identification of associated epigenetic regulators will indeed offer additional epigenetic therapeutic targets. The dynamicity of DNA methylation and other epigenetic changes, and their contribution to the chemoresistance of SCLC is also an important avenue for future investigations. The other undermined branch of SCLC epigenetics is the elucidation of the functional role of different microRNAs, long non-coding RNAs, and circular RNAs that can further help to identify novel diagnostic or prognostic biomarkers and therapeutic strategies.
Enhanced understanding of ‘what and how’ epigenetic modifications contribute to the SCLC will open the epigenetic doors for translating the observations into clinical relevance and augment our capabilities to manage SCLC accordingly. Further, the refinement of molecular mechanisms associated with epigenetic regulations, including the remarkable progress in the last decade, will surely help the researchers and clinicians to remove the tag of ‘recalcitrant disease’ from SCLC.
Acknowledgments
We thank our colleagues for their valuable suggestions, critical reading, and valuable comments on this review. Our apology to colleagues for not citing their work in this review owing to space limitations. Also, we thank Ar. Abdul Qadir for help with the illustrations. SCLC studies in our laboratory are supported by the National Institutes of Health (NIH) grants R01CA218545 to MWN and VA Merit Review I01 BX004676 to AKG. The work of MJ and SKB are supported by NIH R01CA247471 and R01CA195586. The work of RS is supported by the National Cancer Institute of the NIH under award number P30CA033572, 1U54CA209978, R01CA247471, and R01CA218545. Interpretations, opinions, conclusions and recommendations presented in this manuscript are those of the authors and does not necessarily represent the official views of the National Institutes of Health and other funding agencies.
Funding Source
SCLC studies in our laboratory are supported by the National Institutes of Health grants R01CA218545 to MWN and VA Merit Review I01 BX004676 to AKG.
The work of MJ and SKB are supported by NIH R01CA247471 and R01CA195586. The work of RS is supported by the National Cancer Institute of the NIH under award number P30CA033572, 1U54CA209978–01A1, R01CA247471 and R01CA218545.
Interpretations, opinions, conclusions and recommendations presented in this manuscript are those of the author and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- SCLC
Small cell lung cancer
- TS
Tumor suppressor
- TF
Transcription factor
- NE
Neuroendocrine
- non-NE
Non-neuroendocrine
- PRC
polycomb repressive complex
- PRC2
polycomb repressive complex 2
- ASC
Adult stem cell
- hTERT
human telomerase reverse transcriptase
- ASCL1
Achaete-scute complex homologue 1
- MGMT
O6-methylguanine DNA methyltransferase
- HATs
fragile histidine triad (FHIT), histone acetyltransferases
- HATi
histone acetyltransferase inhibitors
- HDACs
histone deacetylases
- HDACi
histone deacetylase inhibitors
- CREBBP
CREB binding protein gene
- EP300
E1A binding protein p300 gene
- CHD7
chromodomain helicase DNA binding protein 7 gene
- ISG
interferon-stimulated gene
- KMT2D
lysine methyltransferase 2D gene
- PBRM1
polybromo 1 gene
- CHD7
chromodomain helicase DNA binding protein 7 gene
- KDM5A
Lysine (K)-Specific Demethylase 5A
- RUNX1T1
RUNX1 partner transcriptional co-repressor 1
- TREX1
three prime repair exonuclease 1
- BET
bromodomain and extra-terminal
- BAP1
BRCA1-associated protein 1
- ASXL3
additional sex combs-like protein 3
- BRD4
bromodomain-containing protein 4
- iPSC
induced pluripotent stem cells
- ORR
overall response rate
- PFS
progression free survival
- OS
overall survival
- PDX
patient-derived xenografts
- CoREST
REST corepressor 1
Footnotes
Conflict of interest statement
SKB is co-founder of Sanguine Diagnostics and Therapeutics, Inc. AKG has served as a consultant to AstraZeneca and Genentech. He is on the advisory board for AstraZeneca, Blueprint Medicines, G1 Therapeutics and Cardinal Health. He has received research support from Takeda Pharmaceuticals and Oncoceutics. Other authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, Leenders F, Lu X, Fernandez-Cuesta L, Bosco G, Muller C, Dahmen I, Jahchan NS, Park KS, Yang D, Karnezis AN, Vaka D, Torres A, Wang MS, Korbel JO, Menon R, Chun SM, Kim D, Wilkerson M, Hayes N, Engelmann D, Putzer B, Bos M, Michels S, Vlasic I, Seidel D, Pinther B, Schaub P, Becker C, Altmuller J, Yokota J, Kohno T, Iwakawa R, Tsuta K, Noguchi M, Muley T, Hoffmann H, Schnabel PA, Petersen I, Chen Y, Soltermann A, Tischler V, Choi CM, Kim YH, Massion PP, Zou Y, Jovanovic D, Kontic M, Wright GM, Russell PA, Solomon B, Koch I, Lindner M, Muscarella LA, la Torre A, Field JK, Jakopovic M, Knezevic J, Castanos-Velez E, Roz L, Pastorino U, Brustugun OT, Lund-Iversen M, Thunnissen E, Kohler J, Schuler M, Botling J, Sandelin M, Sanchez-Cespedes M, Salvesen HB, Achter V, Lang U, Bogus M, Schneider PM, Zander T, Ansen S, Hallek M, Wolf J, Vingron M, Yatabe Y, Travis WD, Nurnberg P, Reinhardt C, Perner S, Heukamp L, Buttner R, Haas SA, Brambilla E, Peifer M, Sage J, Thomas RK, Comprehensive genomic profiles of small cell lung cancer, Nature 524(7563) (2015) 47–53. 10.1038/nature14664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Siegel RL, Miller KD, Jemal A, Cancer statistics, 2019, CA Cancer J Clin 69(1) (2019)7–34. 10.3322/caac.21551 [DOI] [PubMed] [Google Scholar]
- [3].Siegel RL, Miller KD, Jemal A, Cancer statistics, 2020, CA Cancer J Clin 70(1) (2020) 7–30. 10.3322/caac.21590 [DOI] [PubMed] [Google Scholar]
- [4].van Meerbeeck JP, Ball D, Small-cell lung cancer: local therapy for a systemic disease?, Lancet 385(9962) (2015) 9–10. 10.1016/S0140-6736(14)61252-6 [DOI] [PubMed] [Google Scholar]
- [5].van Meerbeeck JP, Fennell DA, De Ruysscher DK, Small-cell lung cancer, Lancet 378(9804) (2011) 1741–55. 10.1016/S0140-6736(11)60165-7 [DOI] [PubMed] [Google Scholar]
- [6].Li L, Ng SR, Colon CI, Drapkin BJ, Hsu PP, Li Z, Nabel CS, Lewis CA, Romero R, Mercer KL, Bhutkar A, Phat S, Myers DT, Muzumdar MD, Westcott PMK, Beytagh MC, Farago AF, Vander Heiden MG, Dyson NJ, Jacks T, Identification of DHODH as a therapeutic target in small cell lung cancer, Sci Transl Med 11(517) (2019). 10.1126/scitranslmed.aaw7852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Byers LA, Rudin CM, Small cell lung cancer: where do we go from here?, Cancer 121(5) (2015) 664–72. 10.1002/cncr.29098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dela Cruz CS, Tanoue LT, Matthay RA, Lung cancer: epidemiology, etiology, and prevention, Clin Chest Med 32(4) (2011) 605–44. 10.1016/j.ccm.2011.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, Bergbower EA, Guan Y, Shin J, Guillory J, Rivers CS, Foo CK, Bhatt D, Stinson J, Gnad F, Haverty PM, Gentleman R, Chaudhuri S, Janakiraman V, Jaiswal BS, Parikh C, Yuan W, Zhang Z, Koeppen H, Wu TD, Stern HM, Yauch RL, Huffman KE, Paskulin DD, Illei PB, Varella-Garcia M, Gazdar AF, de Sauvage FJ, Bourgon R, Minna JD, Brock MV, Seshagiri S, Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer, Nat Genet 44(10) (2012) 1111–6. 10.1038/ng.2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, Plenker D, Leenders F, Sun R, Zander T, Menon R, Koker M, Dahmen I, Muller C, Di Cerbo V, Schildhaus HU, Altmuller J, Baessmann I, Becker C, de Wilde B, Vandesompele J, Bohm D, Ansen S, Gabler F, Wilkening I, Heynck S, Heuckmann JM, Lu X, Carter SL, Cibulskis K, Banerji S, Getz G, Park KS, Rauh D, Grutter C, Fischer M, Pasqualucci L, Wright G, Wainer Z, Russell P, Petersen I, Chen Y, Stoelben E, Ludwig C, Schnabel P, Hoffmann H, Muley T, Brockmann M, Engel-Riedel W, Muscarella LA, Fazio VM, Groen H, Timens W, Sietsma H, Thunnissen E, Smit E, Heideman DA, Snijders PJ, Cappuzzo F, Ligorio C, Damiani S, Field J, Solberg S, Brustugun OT, Lund-Iversen M, Sanger J, Clement JH, Soltermann A, Moch H, Weder W, Solomon B, Soria JC, Validire P, Besse B, Brambilla E, Brambilla C, Lantuejoul S, Lorimier P, Schneider PM, Hallek M, Pao W, Meyerson M, Sage J, Shendure J, Schneider R, Buttner R, Wolf J, Nurnberg P, Perner S, Heukamp LC, Brindle PK, Haas S, Thomas RK, Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer, Nat Genet 44(10) (2012) 1104–10. 10.1038/ng.2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Augert A, Zhang Q, Bates B, Cui M, Wang X, Wildey G, Dowlati A, MacPherson D, Small Cell Lung Cancer Exhibits Frequent Inactivating Mutations in the Histone Methyltransferase KMT2D/MLL2: CALGB 151111 (Alliance), J Thorac Oncol 12(4) (2017) 704–713. 10.1016/j.jtho.2016.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wu S, Powers S, Zhu W, Hannun YA, Substantial contribution of extrinsic risk factors to cancer development, Nature 529(7584) (2016) 43–7. 10.1038/nature16166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Langevin SM, Kratzke RA, Kelsey KT, Epigenetics of lung cancer, Transl Res 165(1) (2015) 74–90. 10.1016/j.trsl.2014.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lu Y, Ek WE, Whiteman D, Vaughan TL, Spurdle AB, Easton DF, Pharoah PD, Thompson DJ, Dunning AM, Hayward NK, Chenevix-Trench G, M. Q, A. Investigators, S. Anecs, S. Ukops, B. Consortium, S. Macgregor, Most common ‘sporadic’ cancers have a significant germline genetic component, Hum Mol Genet 23(22) (2014) 6112–8. 10.1093/hmg/ddu312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Widschwendter M, Jones A, Evans I, Reisel D, Dillner J, Sundstrom K, Steyerberg EW, Vergouwe Y, Wegwarth O, Rebitschek FG, Siebert U, Sroczynski G, de Beaufort ID, Bolt I, Cibula D, Zikan M, Bjorge L, Colombo N, Harbeck N, Dudbridge F, Tasse AM, Knoppers BM, Joly Y, Teschendorff AE, Pashayan N, Consortium F, Epigenome-based cancer risk prediction: rationale, opportunities and challenges, Nat Rev Clin Oncol 15(5) (2018) 292–309. 10.1038/nrclinonc.2018.30 [DOI] [PubMed] [Google Scholar]
- [16].Yurgelun MB, Chenevix-Trench G, Lippman SM, Translating Germline Cancer Risk into Precision Prevention, Cell 168(4) (2017) 566–570. 10.1016/j.cell.2017.01.031 [DOI] [PubMed] [Google Scholar]
- [17].Langevin SM, Kelsey KT, The fate is not always written in the genes: epigenomics in epidemiologic studies, Environ Mol Mutagen 54(7) (2013) 533–41. 10.1002/em.21762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bond DR, Uddipto K, Enjeti AK, Lee HJ, Single-cell epigenomics in cancer: charting a course to clinical impact, Epigenomics 12(13) (2020) 1139–1151. 10.2217/epi-2020-0046 [DOI] [PubMed] [Google Scholar]
- [19].Koch A, Joosten SC, Feng Z, de Ruijter TC, Draht MX, Melotte V, Smits KM, Veeck J, Herman JG, Van Neste L, Van Criekinge W, De Meyer T, van Engeland M, Analysis of DNA methylation in cancer: location revisited, Nat Rev Clin Oncol 15(7) (2018) 459–466. 10.1038/s41571-018-0004-4 [DOI] [PubMed] [Google Scholar]
- [20].Duruisseaux M, Esteller M, Lung cancer epigenetics: From knowledge to applications, Semin Cancer Biol 51 (2018) 116–128. 10.1016/j.semcancer.2017.09.005 [DOI] [PubMed] [Google Scholar]
- [21].Shi YX, Sheng DQ, Cheng L, Song XY, Current Landscape of Epigenetics in Lung Cancer: Focus on the Mechanism and Application, J Oncol 2019 (2019) 8107318. 10.1155/2019/8107318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Fouad YA, Aanei C, Revisiting the hallmarks of cancer, Am J Cancer Res 7(5) (2017) 1016–1036. [PMC free article] [PubMed] [Google Scholar]
- [23].Topper MJ, Vaz M, Marrone KA, Brahmer JR, Baylin SB, The emerging role of epigenetic therapeutics in immuno-oncology, Nat Rev Clin Oncol 17(2) (2020) 75–90. 10.1038/s41571-019-0266-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Flavahan WA, Gaskell E, Bernstein BE, Epigenetic plasticity and the hallmarks of cancer, Science 357(6348) (2017). 10.1126/science.aal2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Vaz M, Hwang SY, Kagiampakis I, Phallen J, Patil A, O’Hagan HM, Murphy L, Zahnow CA, Gabrielson E, Velculescu VE, Easwaran HP, Baylin SB, Chronic Cigarette Smoke-Induced Epigenomic Changes Precede Sensitization of Bronchial Epithelial Cells to Single-Step Transformation by KRAS Mutations, Cancer Cell 32(3) (2017) 360–376 e6. 10.1016/j.ccell.2017.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hua X, Zhao W, Pesatori AC, Consonni D, Caporaso NE, Zhang T, Zhu B, Wang M, Jones K, Hicks B, Song L, Sampson J, Wedge DC, Shi J, Landi MT, Genetic and epigenetic intratumor heterogeneity impacts prognosis of lung adenocarcinoma, Nat Commun 11(1) (2020) 2459. 10.1038/s41467-020-16295-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rudin CM, Poirier JT, Small-cell lung cancer in 2016: Shining light on novel targets and therapies, Nat Rev Clin Oncol 14(2) (2017) 75–76. 10.1038/nrclinonc.2016.203 [DOI] [PubMed] [Google Scholar]
- [28].Ireland AS, Micinski AM, Kastner DW, Guo B, Wait SJ, Spainhower KB, Conley CC, Chen OS, Guthrie MR, Soltero D, Qiao Y, Huang X, Tarapcsak S, Devarakonda S, Chalishazar MD, Gertz J, Moser JC, Marth G, Puri S, Witt BL, Spike BT, Oliver TG, MYC Drives Temporal Evolution of Small Cell Lung Cancer Subtypes by Reprogramming Neuroendocrine Fate, Cancer Cell 38(1) (2020) 60–78 e12. 10.1016/j.ccell.2020.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Oser MG, Sabet AH, Gao W, Chakraborty AA, Schinzel AC, Jennings RB, Fonseca R, Bonal DM, Booker MA, Flaifel A, Novak JS, Christensen CL, Zhang H, Herbert ZT, Tolstorukov MY, Buss EJ, Wong KK, Bronson RT, Nguyen QD, Signoretti S, Kaelin WG Jr., The KDM5A/RBP2 histone demethylase represses NOTCH signaling to sustain neuroendocrine differentiation and promote small cell lung cancer tumorigenesis, Genes Dev 33(23–24) (2019) 1718–1738. 10.1101/gad.328336.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Pillai RN, Owonikoko TK, Small cell lung cancer: therapies and targets, Semin Oncol 41(1) (2014) 133–42. 10.1053/j.seminoncol.2013.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Teicher BA, Targets in small cell lung cancer, Biochem Pharmacol 87(2) (2014) 211–9. 10.1016/j.bcp.2013.09.014 [DOI] [PubMed] [Google Scholar]
- [32].Krushkal J, Silvers T, Reinhold WC, Sonkin D, Vural S, Connelly J, Varma S, Meltzer PS, Kunkel M, Rapisarda A, Evans D, Pommier Y, Teicher BA, Epigenome-wide DNA methylation analysis of small cell lung cancer cell lines suggests potential chemotherapy targets, Clin Epigenetics 12(1) (2020) 93. 10.1186/s13148-020-00876-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sui JSY, Martin P, Gray SG, Pre-clinical models of small cell lung cancer and the validation of therapeutic targets, Expert Opin Ther Targets 24(3) (2020) 187–204. 10.1080/14728222.2020.1732353 [DOI] [PubMed] [Google Scholar]
- [34].Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, Mohammad HP, Chen W, Daniel VC, Yu W, Berman DM, Jenuwein T, Pruitt K, Sharkis SJ, Watkins DN, Herman JG, Baylin SB, A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing, Nat Genet 39(2) (2007) 237–42. 10.1038/ng1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Weichenhan D, Lipka DB, Lutsik P, Goyal A, Plass C, Epigenomic technologies for precision oncology, Semin Cancer Biol (2020). 10.1016/j.semcancer.2020.08.004 [DOI] [PubMed] [Google Scholar]
- [36].Robinson NJ, Parker KA, Schiemann WP, Epigenetic plasticity in metastatic dormancy: mechanisms and therapeutic implications, Ann Transl Med 8(14) (2020) 903. 10.21037/atm.2020.02.177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, Eden E, Yakhini Z, Ben-Shushan E, Reubinoff BE, Bergman Y, Simon I, Cedar H, Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer, Nat Genet 39(2) (2007) 232–6. 10.1038/ng1950 [DOI] [PubMed] [Google Scholar]
- [38].Bergman Y, Cedar H, DNA methylation dynamics in health and disease, Nat Struct Mol Biol 20(3) (2013) 274–81. 10.1038/nsmb.2518 [DOI] [PubMed] [Google Scholar]
- [39].Zhang W, Girard L, Zhang YA, Haruki T, Papari-Zareei M, Stastny V, Ghayee HK, Pacak K, Oliver TG, Minna JD, Gazdar AF, Small cell lung cancer tumors and preclinical models display heterogeneity of neuroendocrine phenotypes, Transl Lung Cancer Res 7(1) (2018) 32–49. 10.21037/tlcr.2018.02.02 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bonder MJ, Luijk R, Zhernakova DV, Moed M, Deelen P, Vermaat M, van Iterson M, van Dijk F, van Galen M, Bot J, Slieker RC, Jhamai PM, Verbiest M, Suchiman HE, Verkerk M, van der Breggen R, van Rooij J, Lakenberg N, Arindrarto W, Kielbasa SM, Jonkers I, van ‘t Hof P, Nooren I, Beekman M, Deelen J, van Heemst D, Zhernakova A, Tigchelaar EF, Swertz MA, Hofman A, Uitterlinden AG, Pool R, van Dongen J, Hottenga JJ, Stehouwer CD, van der Kallen CJ, Schalkwijk CG, van den Berg LH, van Zwet EW, Mei H, Li Y, Lemire M, Hudson TJ, Consortium B, Slagboom PE, Wijmenga C, Veldink JH, van Greevenbroek MM, van Duijn CM, Boomsma DI, Isaacs A, Jansen R, van Meurs JB, t Hoen PA, Franke L, Heijmans BT, Disease variants alter transcription factor levels and methylation of their binding sites, Nat Genet 49(1) (2017) 131–138. 10.1038/ng.3721 [DOI] [PubMed] [Google Scholar]
- [41].Rudin CM, Poirier JT, Byers LA, Dive C, Dowlati A, George J, Heymach JV, Johnson JE, Lehman JM, MacPherson D, Massion PP, Minna JD, Oliver TG, Quaranta V, Sage J, Thomas RK, Vakoc CR, Gazdar AF, Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data, Nat Rev Cancer 19(5) (2019) 289–297. 10.1038/s41568-019-0133-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Thomas A, Pommier Y, Small cell lung cancer: Time to revisit DNA-damaging chemotherapy, Sci Transl Med 8(346) (2016) 346fs12. 10.1126/scitranslmed.aaf6282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bragelmann J, Bohm S, Guthrie MR, Mollaoglu G, Oliver TG, Sos ML, Family matters: How MYC family oncogenes impact small cell lung cancer, Cell Cycle 16(16) (2017) 1489–1498. 10.1080/15384101.2017.1339849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Mollaoglu G, Guthrie MR, Bohm S, Bragelmann J, Can I, Ballieu PM, Marx A, George J, Heinen C, Chalishazar MD, Cheng H, Ireland AS, Denning KE, Mukhopadhyay A, Vahrenkamp JM, Berrett KC, Mosbruger TL, Wang J, Kohan JL, Salama ME, Witt BL, Peifer M, Thomas RK, Gertz J, Johnson JE, Gazdar AF, Wechsler-Reya RJ, Sos ML, Oliver TG, MYC Drives Progression of Small Cell Lung Cancer to a Variant Neuroendocrine Subtype with Vulnerability to Aurora Kinase Inhibition, Cancer Cell 31(2) (2017) 270–285. 10.1016/j.ccell.2016.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Zhai G, Li J, Zheng J, An P, Chen X, Wang X, Li C, hTERT promoter methylation promotes small cell lung cancer progression and radiotherapy resistance, J Radiat Res 61(5) (2020) 674–683. 10.1093/jrr/rraa052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Licchesi JD, Westra WH, Hooker CM, Herman JG, Promoter hypermethylation of hallmark cancer genes in atypical adenomatous hyperplasia of the lung, Clin Cancer Res 14(9) (2008) 2570–8. 10.1158/1078-0432.CCR-07-2033 [DOI] [PubMed] [Google Scholar]
- [47].Berger AH, Knudson AG, Pandolfi PP, A continuum model for tumour suppression, Nature 476(7359) (2011) 163–9. 10.1038/nature10275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lareau CA, Ludwig LS, Muus C, Gohil SH, Zhao T, Chiang Z, Pelka K, Verboon JM, Luo W, Christian E, Rosebrock D, Getz G, Boland GM, Chen F, Buenrostro JD, Hacohen N, Wu CJ, Aryee MJ, Regev A, Sankaran VG, Massively parallel single-cell mitochondrial DNA genotyping and chromatin profiling, Nat Biotechnol (2020). 10.1038/s41587-020-0645-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Safi-Stibler S, Gabory A, Epigenetics and the Developmental Origins of Health and Disease: Parental environment signalling to the epigenome, critical time windows and sculpting the adult phenotype, Semin Cell Dev Biol 97 (2020) 172–180. 10.1016/j.semcdb.2019.09.008 [DOI] [PubMed] [Google Scholar]
- [50].Cavalli G, Heard E, Advances in epigenetics link genetics to the environment and disease, Nature 571(7766) (2019) 489–499. 10.1038/s41586-019-1411-0 [DOI] [PubMed] [Google Scholar]
- [51].Han X, Chen S, Flynn E, Wu S, Wintner D, Shen Y, Distinct epigenomic patterns are associated with haploinsufficiency and predict risk genes of developmental disorders, Nat Commun 9(1) (2018) 2138. 10.1038/s41467-018-04552-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ehrlich M, DNA hypomethylation in cancer cells, Epigenomics 1(2) (2009) 239–59. 10.2217/epi.09.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Herman JG, Baylin SB, Gene silencing in cancer in association with promoter hypermethylation, N Engl J Med 349(21) (2003) 2042–54. 10.1056/NEJMra023075 [DOI] [PubMed] [Google Scholar]
- [54].Jelinic P, Shaw P, Loss of imprinting and cancer, J Pathol 211(3) (2007) 261–8. 10.1002/path.2116 [DOI] [PubMed] [Google Scholar]
- [55].Friso S, Udali S, Guarini P, Pellegrini C, Pattini P, Moruzzi S, Girelli D, Pizzolo F, Martinelli N, Corrocher R, Olivieri O, Choi SW, Global DNA hypomethylation in peripheral blood mononuclear cells as a biomarker of cancer risk, Cancer Epidemiol Biomarkers Prev 22(3) (2013) 348–55. 10.1158/1055-9965.EPI-12-0859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Smith LT, Lin M, Brena RM, Lang JC, Schuller DE, Otterson GA, Morrison CD, Smiraglia DJ, Plass C, Epigenetic regulation of the tumor suppressor gene TCF21 on 6q23-q24 in lung and head and neck cancer, Proc Natl Acad Sci U S A 103(4) (2006) 982–7. 10.1073/pnas.0510171102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Poirier JT, Gardner EE, Connis N, Moreira AL, de Stanchina E, Hann CL, Rudin CM, DNA methylation in small cell lung cancer defines distinct disease subtypes and correlates with high expression of EZH2, Oncogene 34(48) (2015) 5869–78. 10.1038/onc.2015.38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Lerman MI, Minna JD, The 630-kb lung cancer homozygous deletion region on human chromosome 3p21.3: identification and evaluation of the resident candidate tumor suppressor genes. The International Lung Cancer Chromosome 3p21.3 Tumor Suppressor Gene Consortium, Cancer Res 60(21) (2000) 6116–33. [PubMed] [Google Scholar]
- [59].Burbee DG, Forgacs E, Zochbauer-Muller S, Shivakumar L, Fong K, Gao B, Randle D, Kondo M, Virmani A, Bader S, Sekido Y, Latif F, Milchgrub S, Toyooka S, Gazdar AF, Lerman MI, Zabarovsky E, White M, Minna JD, Epigenetic inactivation of RASSF1A in lung and breast cancers and malignant phenotype suppression, J Natl Cancer Inst 93(9) (2001) 691–9. 10.1093/jnci/93.9.691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Nunes SP, Diniz F, Moreira-Barbosa C, Constancio V, Silva AV, Oliveira J, Soares M, Paulino S, Cunha AL, Rodrigues J, Antunes L, Henrique R, Jeronimo C, Subtyping Lung Cancer Using DNA Methylation in Liquid Biopsies, J Clin Med 8(9) (2019). 10.3390/jcm8091500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Du M, Thompson J, Fisher H, Zhang P, Huang CC, Wang L, Genomic alterations of plasma cell-free DNAs in small cell lung cancer and their clinical relevance, Lung Cancer 120 (2018) 113–121. 10.1016/j.lungcan.2018.04.008 [DOI] [PubMed] [Google Scholar]
- [62].Sunaga N, Miyajima K, Suzuki M, Sato M, White MA, Ramirez RD, Shay JW, Gazdar AF, Minna JD, Different roles for caveolin-1 in the development of non-small cell lung cancer versus small cell lung cancer, Cancer Res 64(12) (2004) 4277–85. 10.1158/0008-5472.CAN-03-3941 [DOI] [PubMed] [Google Scholar]
- [63].Wu J, Di D, Zhao C, Pan Q, Liu Y, Zhang X, Zhao X, Chen H, Clinical Significance of Gli-1 And Caveolin-1 Expression in the Human Small Cell Lung Cancer, Asian Pac J Cancer Prev 19(2) (2018) 401–406. 10.22034/APJCP.2018.19.2.401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Roncarati R, Lupini L, Miotto E, Saccenti E, Mascetti S, Morandi L, Bassi C, Rasio D, Callegari E, Conti V, Rinaldi R, Lanza G, Gafa R, Papi A, Frassoldati A, Sabbioni S, Ravenna F, Casoni GL, Negrini M, Molecular testing on bronchial washings for the diagnosis and predictive assessment of lung cancer, Mol Oncol 14(9) (2020) 2163–2175. 10.1002/1878-0261.12713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Sun MZ, Guan Z, Liu S, Zhou X, Wang N, Shao S, Lin D, Caveolin-1 interferes cell growth of lung cancer NCI-H446 cell through the interactions with phospho-ERK1/2, estrogen receptor and progestin receptor, Biomed Pharmacother 66(4) (2012) 242–8. 10.1016/j.biopha.2011.11.003 [DOI] [PubMed] [Google Scholar]
- [66].Yang X, Xiong H, Guan ZZ, Okai I, Ye D, Song Y, Li X, Wang L, Liu L, Du S, Lin D, Shao S, Higher expression of Caveolin-1 inhibits human small cell lung cancer (SCLC) apoptosis in vitro, Cancer Invest 30(6) (2012) 453–62. 10.3109/07357907.2012.675384 [DOI] [PubMed] [Google Scholar]
- [67].Gazdar AF, Bunn PA, Minna JD, Small-cell lung cancer: what we know, what we need to know and the path forward, Nat Rev Cancer 17(12) (2017) 725–737. 10.1038/nrc.2017.87 [DOI] [PubMed] [Google Scholar]
- [68].Chang CJ, Hung MC, The role of EZH2 in tumour progression, Br J Cancer 106(2) (2012) 243–7. 10.1038/bjc.2011.551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Jiao L, Shubbar M, Yang X, Zhang Q, Chen S, Wu Q, Chen Z, Rizo J, Liu X, A partially disordered region connects gene repression and activation functions of EZH2, Proc Natl Acad Sci U S A 117(29) (2020) 16992–17002. 10.1073/pnas.1914866117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K, EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer, EMBO J 22(20) (2003) 5323–35. 10.1093/emboj/cdg542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Murai F, Koinuma D, Shinozaki-Ushiku A, Fukayama M, Miyaozono K, Ehata S, EZH2 promotes progression of small cell lung cancer by suppressing the TGF-beta-Smad-ASCL1 pathway, Cell Discov 1 (2015) 15026. 10.1038/celldisc.2015.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Saito M, Saito K, Shiraishi K, Maeda D, Suzuki H, Minamiya Y, Kono K, Kohno T, Goto A, Identification of candidate responders for anti-PD-L1/PD-1 immunotherapy, Rova-T therapy, or EZH2 inhibitory therapy in small-cell lung cancer, Mol Clin Oncol 8(2) (2018) 310–314. 10.3892/mco.2017.1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Pezzuto F, Fortarezza F, Lunardi F, Calabrese F, Are there any theranostic biomarkers in small cell lung carcinoma?, J Thorac Dis 11(Suppl 1) (2019) S102–S112. 10.21037/jtd.2018.12.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Lu L, Luo F, Liu Y, Liu X, Shi L, Lu X, Liu Q, Posttranscriptional silencing of the lncRNA MALAT1 by miR-217 inhibits the epithelial-mesenchymal transition via enhancer of zeste homolog 2 in the malignant transformation of HBE cells induced by cigarette smoke extract, Toxicol Appl Pharmacol 289(2) (2015) 276–85. 10.1016/j.taap.2015.09.016 [DOI] [PubMed] [Google Scholar]
- [75].Anzalone G, Arcoleo G, Bucchieri F, Montalbano AM, Marchese R, Albano GD, Di Sano C, Moscato M, Gagliardo R, Ricciardolo FLM, Profita M, Cigarette smoke affects the onco-suppressor DAB2IP expression in bronchial epithelial cells of COPD patients, Sci Rep 9(1) (2019) 15682. 10.1038/s41598-019-52179-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Gardner EE, Lok BH, Schneeberger VE, Desmeules P, Miles LA, Arnold PK, Ni A, Khodos I, de Stanchina E, Nguyen T, Sage J, Campbell JE, Ribich S, Rekhtman N, Dowlati A, Massion PP, Rudin CM, Poirier JT, Chemosensitive Relapse in Small Cell Lung Cancer Proceeds through an EZH2-SLFN11 Axis, Cancer Cell 31(2) (2017) 286–299. 10.1016/j.ccell.2017.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Gardner EE, Connis N, Poirier JT, Cope L, Dobromilskaya I, Gallia GL, Rudin CM, Hann CL, Rapamycin rescues ABT-737 efficacy in small cell lung cancer, Cancer Res 74(10) (2014) 2846–56. 10.1158/0008-5472.CAN-13-3460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Kalari S, Jung M, Kernstine KH, Takahashi T, Pfeifer GP, The DNA methylation landscape of small cell lung cancer suggests a differentiation defect of neuroendocrine cells, Oncogene 32(30) (2013) 3559–68. 10.1038/onc.2012.362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Pozo K, Minna JD, Johnson JE, Identifying a missing lineage driver in a subset of lung neuroendocrine tumors, Genes Dev 32(13–14) (2018) 865–867. 10.1101/gad.316943.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Miyoshi T, Umemura S, Matsumura Y, Mimaki S, Tada S, Makinoshima H, Ishii G, Udagawa H, Matsumoto S, Yoh K, Niho S, Ohmatsu H, Aokage K, Hishida T, Yoshida J, Nagai K, Goto K, Tsuboi M, Tsuchihara K, Genomic Profiling of Large-Cell Neuroendocrine Carcinoma of the Lung, Clin Cancer Res 23(3) (2017) 757–765. 10.1158/1078-0432.CCR-16-0355 [DOI] [PubMed] [Google Scholar]
- [81].Smith BA, Balanis NG, Nanjundiah A, Sheu KM, Tsai BL, Zhang Q, Park JW, Thompson M, Huang J, Witte ON, Graeber TG, A Human Adult Stem Cell Signature Marks Aggressive Variants across Epithelial Cancers, Cell Rep 24(12) (2018) 3353–3366 e5. 10.1016/j.celrep.2018.08.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Atala A, Re: A Human Adult Stem Cell Signature Marks Aggressive Variants across Epithelial Cancers, J Urol 201(3) (2019) 446. 10.1097/01.JU.0000553705.00347.f3 [DOI] [PubMed] [Google Scholar]
- [83].Huang YH, Klingbeil O, He XY, Wu XS, Arun G, Lu B, Somerville TDD, Milazzo JP, Wilkinson JE, Demerdash OE, Spector DL, Egeblad M, Shi J, Vakoc CR, POU2F3 is a master regulator of a tuft cell-like variant of small cell lung cancer, Genes Dev 32(13–14) (2018) 915–928. 10.1101/gad.314815.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Park JW, Lee JK, Sheu KM, Wang L, Balanis NG, Nguyen K, Smith BA, Cheng C, Tsai BL, Cheng D, Huang J, Kurdistani SK, Graeber TG, Witte ON, Reprogramming normal human epithelial tissues to a common, lethal neuroendocrine cancer lineage, Science 362(6410) (2018) 91–95. 10.1126/science.aat5749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Ito T, Kudoh S, Ichimura T, Fujino K, Hassan WA, Udaka N, Small cell lung cancer, an epithelial to mesenchymal transition (EMT)-like cancer: significance of inactive Notch signaling and expression of achaete-scute complex homologue 1, Hum Cell 30(1) (2017) 1–10. 10.1007/s13577-016-0149-3 [DOI] [PubMed] [Google Scholar]
- [86].Jiang T, Collins BJ, Jin N, Watkins DN, Brock MV, Matsui W, Nelkin BD, Ball DW, Achaete-scute complex homologue 1 regulates tumor-initiating capacity in human small cell lung cancer, Cancer Res 69(3) (2009) 845–54. 10.1158/0008-5472.CAN-08-2762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Osborne JK, Larsen JE, Shields MD, Gonzales JX, Shames DS, Sato M, Kulkarni A, Wistuba II, Girard L, Minna JD, Cobb MH, NeuroD1 regulates survival and migration of neuroendocrine lung carcinomas via signaling molecules TrkB and NCAM, Proc Natl Acad Sci U S A 110(16) (2013) 6524–9. 10.1073/pnas.1303932110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Kudoh S, Tenjin Y, Kameyama H, Ichimura T, Yamada T, Matsuo A, Kudo N, Sato Y, Ito T, Significance of achaete-scute complex homologue 1 (ASCL1) in pulmonary neuroendocrine carcinomas; RNA sequence analyses using small cell lung cancer cells and Ascl1-induced pulmonary neuroendocrine carcinoma cells, Histochem Cell Biol 153(6) (2020) 443–456. 10.1007/s00418-020-01863-z [DOI] [PubMed] [Google Scholar]
- [89].Mohammad HP, Smitheman KN, Kamat CD, Soong D, Federowicz KE, Van Aller GS, Schneck JL, Carson JD, Liu Y, Butticello M, Bonnette WG, Gorman SA, Degenhardt Y, Bai Y, McCabe MT, Pappalardi MB, Kasparec J, Tian X, McNulty KC, Rouse M, McDevitt P, Ho T, Crouthamel M, Hart TK, Concha NO, McHugh CF, Miller WH, Dhanak D, Tummino PJ, Carpenter CL, Johnson NW, Hann CL, Kruger RG, A DNA Hypomethylation Signature Predicts Antitumor Activity of LSD1 Inhibitors in SCLC, Cancer Cell 28(1) (2015) 57–69. 10.1016/j.ccell.2015.06.002 [DOI] [PubMed] [Google Scholar]
- [90].Pietanza MC, Kadota K, Huberman K, Sima CS, Fiore JJ, Sumner DK, Travis WD, Heguy A, Ginsberg MS, Holodny AI, Chan TA, Rizvi NA, Azzoli CG, Riely GJ, Kris MG, Krug LM, Phase II trial of temozolomide in patients with relapsed sensitive or refractory small cell lung cancer, with assessment of methylguanine-DNA methyltransferase as a potential biomarker, Clin Cancer Res 18(4) (2012) 1138–45. 10.1158/1078-0432.CCR-11-2059 [DOI] [PubMed] [Google Scholar]
- [91].Toyooka S, Toyooka KO, Maruyama R, Virmani AK, Girard L, Miyajima K, Harada K, Ariyoshi Y, Takahashi T, Sugio K, Brambilla E, Gilcrease M, Minna JD, Gazdar AF, DNA methylation profiles of lung tumors, Mol Cancer Ther 1(1) (2001) 61–7. [PubMed] [Google Scholar]
- [92].Hiddinga BI, Pauwels P, Janssens A, van Meerbeeck JP, O(6)-Methylguanine-DNA methyltransferase (MGMT): A drugable target in lung cancer?, Lung Cancer 107 (2017) 91–99. 10.1016/j.lungcan.2016.07.014 [DOI] [PubMed] [Google Scholar]
- [93].Pietanza MC, Waqar SN, Krug LM, Dowlati A, Hann CL, Chiappori A, Owonikoko TK, Woo KM, Cardnell RJ, Fujimoto J, Long L, Diao L, Wang J, Bensman Y, Hurtado B, de Groot P, Sulman EP, Wistuba II, Chen A, Fleisher M, Heymach JV, Kris MG, Rudin CM, Byers LA, Randomized, Double-Blind, Phase II Study of Temozolomide in Combination With Either Veliparib or Placebo in Patients With Relapsed-Sensitive or Refractory Small-Cell Lung Cancer, J Clin Oncol 36(23) (2018) 2386–2394. 10.1200/JCO.2018.77.7672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Lu H, Qin J, Xu H, Han N, Xie F, Mao W, O(6)-methyl-guanine-DNA methyltransferase methylation and IDH1/2 mutation in small cell lung cancer, Exp Ther Med 14(1) (2017) 398–402. 10.3892/etm.2017.4476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Zandi R, Xu K, Poulsen HS, Roth JA, Ji L, The effect of adenovirus-mediated gene expression of FHIT in small cell lung cancer cells, Cancer Invest 29(10) (2011) 683–91. 10.3109/07357907.2011.626475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Zochbauer-Muller S, Fong KM, Maitra A, Lam S, Geradts J, Ashfaq R, Virmani AK, Milchgrub S, Gazdar AF, Minna JD, 5’ CpG island methylation of the FHIT gene is correlated with loss of gene expression in lung and breast cancer, Cancer Res 61(9) (2001) 3581–5. [PubMed] [Google Scholar]
- [97].Krishnamurthy K, Mishra TK, Saxena A, Daga MK, Khurana N, Masroor M, Jamatia E, Evaluating NISCH and CDH1 Promoter Hypermethylation in Nonsmokers, Cancer Free Smokers and Lung Cancer Patients: A Case Control Study, Indian J Clin Biochem 34(4) (2019) 458–464. 10.1007/s12291-018-0767-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Semenova EA, Kwon MC, Monkhorst K, Song JY, Bhaskaran R, Krijgsman O, Kuilman T, Peters D, Buikhuisen WA, Smit EF, Pritchard C, Cozijnsen M, van der Vliet J, Zevenhoven J, Lambooij JP, Proost N, van Montfort E, Velds A, Huijbers IJ, Berns A, Transcription Factor NFIB Is a Driver of Small Cell Lung Cancer Progression in Mice and Marks Metastatic Disease in Patients, Cell Rep 16(3) (2016) 631–43. 10.1016/j.celrep.2016.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Hu Y, Zheng Y, Dai M, Wu J, Yu B, Zhang H, Kong W, Wu H, Yu X, Snail2 induced E-cadherin suppression and metastasis in lung carcinoma facilitated by G9a and HDACs, Cell Adh Migr 13(1) (2019) 285–292. 10.1080/19336918.2019.1638689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Wang Y, Zhang L, Yang J, Li B, Wang J, CDH13 promoter methylation regulates cisplatin resistance of non-small cell lung cancer cells, Oncol Lett 16(5) (2018) 5715–5722. 10.3892/ol.2018.9325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Wang M, Long K, Li E, Li L, Li B, Ci S, He L, Pan F, Hu Z, Guo Z, DNA polymerase beta modulates cancer progression via enhancing CDH13 expression by promoter demethylation, Oncogene 39(33) (2020) 5507–5519. 10.1038/s41388-020-1386-1 [DOI] [PubMed] [Google Scholar]
- [102].Yang Z, Qi W, Sun L, Zhou H, Zhou B, Hu Y, DNA methylation analysis of selected genes for the detection of early-stage lung cancer using circulating cell-free DNA, Adv Clin Exp Med 28(3) (2019) 355–360. 10.17219/acem/84935 [DOI] [PubMed] [Google Scholar]
- [103].Littau VC, Burdick CJ, Allfrey VG, Mirsky SA, The role of histones in the maintenance of chromatin structure, Proc Natl Acad Sci U S A 54(4) (1965) 1204–12. 10.1073/pnas.54.4.1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Loppin B, Berger F, Histone Variants: The Nexus of Developmental Decisions and Epigenetic Memory, Annu Rev Genet (2020). 10.1146/annurev-genet-022620-100039 [DOI] [PubMed] [Google Scholar]
- [105].Martire S, Banaszynski LA, The roles of histone variants in fine-tuning chromatin organization and function, Nat Rev Mol Cell Biol 21(9) (2020) 522–541. 10.1038/s41580-020-0262-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Jing Y, Ding D, Tian G, Kwan KCJ, Liu Z, Ishibashi T, Li XD, Semisynthesis of site-specifically succinylated histone reveals that succinylation regulates nucleosome unwrapping rate and DNA accessibility, Nucleic Acids Res 48(17) (2020) 9538–9549. 10.1093/nar/gkaa663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Tan C, Takada S, Nucleosome allostery in pioneer transcription factor binding, Proc Natl Acad Sci U S A 117(34) (2020) 20586–20596. 10.1073/pnas.2005500117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Qin N, Li Y, Wang C, Zhu M, Dai J, Hong T, Albanes D, Lam S, Tardon A, Chen C, Goodman G, Bojesen SE, Landi MT, Johansson M, Risch A, Wichmann HE, Bickeboller H, Rennert G, Arnold S, Brennan P, Field JK, Shete S, Le Marchand L, Melander O, Brunnstrom H, Liu G, Hung RJ, Andrew A, Kiemeney LA, Zienolddiny S, Grankvist K, Johansson M, Caporaso N, Woll P, Lazarus P, Schabath MB, Aldrich MC, Stevens VL, Jin G, Christiani DC, Hu Z, Amos CI, Ma H, Shen H, Comprehensive functional annotation of susceptibility variants identifies genetic heterogeneity between lung adenocarcinoma and squamous cell carcinoma, Front Med (2020). 10.1007/s11684-020-0779-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Xia C, Tao Y, Li M, Che T, Qu J, Protein acetylation and deacetylation: An important regulatory modification in gene transcription (Review), Exp Ther Med 20(4) (2020) 2923–2940. 10.3892/etm.2020.9073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Cedar H, Bergman Y, Linking DNA methylation and histone modification: patterns and paradigms, Nat Rev Genet 10(5) (2009) 295–304. 10.1038/nrg2540 [DOI] [PubMed] [Google Scholar]
- [111].Dong C, Nakagawa R, Oyama K, Yamamoto Y, Zhang W, Dong A, Li Y, Yoshimura Y, Kamiya H, Nakayama JI, Ueda J, Min J, Structural basis for histone variant H3tK27me3 recognition by PHF1 and PHF19, Elife 9 (2020). 10.7554/eLife.58675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Brzezianska E, Dutkowska A, Antczak A, The significance of epigenetic alterations in lung carcinogenesis, Mol Biol Rep 40(1) (2013) 309–25. 10.1007/s11033-012-2063-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Kondo Y, Epigenetic cross-talk between DNA methylation and histone modifications in human cancers, Yonsei Med J 50(4) (2009) 455–63. 10.3349/ymj.2009.50.4.455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Neganova ME, Klochkov SG, Aleksandrova YR, Aliev G, Histone modifications in epigenetic regulation of cancer: Perspectives and achieved progress, Semin Cancer Biol (2020). 10.1016/j.semcancer.2020.07.015 [DOI] [PubMed] [Google Scholar]
- [115].Dunican DS, Mjoseng HK, Duthie L, Flyamer IM, Bickmore WA, Meehan RR, Bivalent promoter hypermethylation in cancer is linked to the H327me3/H3K4me3 ratio in embryonic stem cells, BMC Biol 18(1) (2020) 25. 10.1186/s12915-020-0752-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Barlesi F, Giaccone G, Gallegos-Ruiz MI, Loundou A, Span SW, Lefesvre P, Kruyt FA, Rodriguez JA, Global histone modifications predict prognosis of resected non small-cell lung cancer, J Clin Oncol 25(28) (2007) 4358–64. 10.1200/JCO.2007.11.2599 [DOI] [PubMed] [Google Scholar]
- [117].Li F, Ye B, Hong L, Xu H, Fishbein MC, Epigenetic modifications of histone h4 in lung neuroendocrine tumors, Appl Immunohistochem Mol Morphol 19(5) (2011) 389–94. 10.1097/PAI.0b013e3182108e2e [DOI] [PubMed] [Google Scholar]
- [118].Lee JE, Wang C, Xu S, Cho YW, Wang L, Feng X, Baldridge A, Sartorelli V, Zhuang L, Peng W, Ge K, H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation, Elife 2 (2013) e01503. 10.7554/eLife.01503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Jang Y, Wang C, Zhuang L, Liu C, Ge K, H3K4 Methyltransferase Activity Is Required for MLL4 Protein Stability, J Mol Biol 429(13) (2017) 2046–2054. 10.1016/j.jmb.2016.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Gorkin DU, Barozzi I, Zhao Y, Zhang Y, Huang H, Lee AY, Li B, Chiou J, Wildberg A, Ding B, Zhang B, Wang M, Strattan JS, Davidson JM, Qiu Y, Afzal V, Akiyama JA, Plajzer-Frick I, Novak CS, Kato M, Garvin TH, Pham QT, Harrington AN, Mannion BJ, Lee EA, Fukuda-Yuzawa Y, He Y, Preissl S, Chee S, Han JY, Williams BA, Trout D, Amrhein H, Yang H, Cherry JM, Wang W, Gaulton K, Ecker JR, Shen Y, Dickel DE, Visel A, Pennacchio LA, Ren B, An atlas of dynamic chromatin landscapes in mouse fetal development, Nature 583(7818) (2020) 744–751. 10.1038/s41586-020-2093-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, Ching KA, Antosiewicz-Bourget JE, Liu H, Zhang X, Green RD, Lobanenkov VV, Stewart R, Thomson JA, Crawford GE, Kellis M, Ren B, Histone modifications at human enhancers reflect global cell-type-specific gene expression, Nature 459(7243) (2009) 108–12. 10.1038/nature07829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].He S, Wu Z, Tian Y, Yu Z, Yu J, Wang X, Li J, Liu B, Xu Y, Structure of nucleosome-bound human BAF complex, Science 367(6480) (2020) 875–881. 10.1126/science.aaz9761 [DOI] [PubMed] [Google Scholar]
- [123].Horikawa I, Barrett JC, cDNA cloning of the human polybromo-1 gene on chromosome 3p21, DNA Seq 13(4) (2002) 211–5. 10.1080/1042517021000021590 [DOI] [PubMed] [Google Scholar]
- [124].Loo CS, Gatchalian J, Liang Y, Leblanc M, Xie M, Ho J, Venkatraghavan B, Hargreaves DC, Zheng Y, A Genome-wide CRISPR Screen Reveals a Role for the Non-canonical Nucleosome-Remodeling BAF Complex in Foxp3 Expression and Regulatory T Cell Function, Immunity 53(1) (2020) 143–157 e8. 10.1016/j.immuni.2020.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma AM, Tsoucas D, Qiu X, Lim K, Rao P, Long HW, Yuan GC, Doench J, Brown M, Liu XS, Wucherpfennig KW, A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing, Science 359(6377) (2018) 770–775. 10.1126/science.aao1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Shi H, Tao T, Abraham BJ, Durbin AD, Zimmerman MW, Kadoch C, Look AT, ARID1A loss in neuroblastoma promotes the adrenergic-to-mesenchymal transition by regulating enhancer-mediated gene expression, Sci Adv 6(29) (2020) eaaz3440. 10.1126/sciadv.aaz3440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Ghorani E, Quezada SA, Chromatin regulation and immune escape, Science 359(6377) (2018) 745–746. 10.1126/science.aat0383 [DOI] [PubMed] [Google Scholar]
- [128].Jia D, Augert A, Kim DW, Eastwood E, Wu N, Ibrahim AH, Kim KB, Dunn CT, Pillai SPS, Gazdar AF, Bolouri H, Park KS, MacPherson D, Crebbp Loss Drives Small Cell Lung Cancer and Increases Sensitivity to HDAC Inhibition, Cancer Discov 8(11) (2018) 1422–1437. 10.1158/2159-8290.CD-18-0385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Mohan S, Foy V, Ayub M, Leong HS, Schofield P, Sahoo S, Descamps T, Kilerci B, Smith NK, Carter M, Priest L, Zhou C, Carr TH, Miller C, Faivre-Finn C, Blackhall F, Rothwell DG, Dive C, Brady G, Profiling of Circulating Free DNA Using Targeted and Genome-wide Sequencing in Patients with SCLC, J Thorac Oncol 15(2) (2020) 216–230. 10.1016/j.jtho.2019.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Hu J, Wang Y, Zhang Y, Yu Y, Chen H, Liu K, Yao M, Wang K, Gu W, Shou T, Comprehensive genomic profiling of small cell lung cancer in Chinese patients and the implications for therapeutic potential, Cancer Med 8(9) (2019) 4338–4347. 10.1002/cam4.2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Augert A, Eastwood E, Ibrahim AH, Wu N, Grunblatt E, Basom R, Liggitt D, Eaton KD, Martins R, Poirier JT, Rudin CM, Milletti F, Cheng WY, Mack F, MacPherson D, Targeting NOTCH activation in small cell lung cancer through LSD1 inhibition, Sci Signal 12(567) (2019). 10.1126/scisignal.aau2922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Carazo F, Bertolo C, Castilla C, Cendoya X, Campuzano L, Serrano D, Gimeno M, Planes FJ, Pio R, Montuenga LM, Rubio A, DrugSniper, a Tool to Exploit Loss-Of-Function Screens, Identifies CREBBP as a Predictive Biomarker of VOLASERTIB in Small Cell Lung Carcinoma (SCLC), Cancers (Basel) 12(7) (2020). 10.3390/cancers12071824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Francetic T, Le May M, Hamed M, Mach H, Meyers D, Cole PA, Chen J, Li Q, Regulation of Myf5 Early Enhancer by Histone Acetyltransferase p300 during Stem Cell Differentiation, Mol Biol 1 (2012). 10.4172/2168-9547.1000103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Kim DW, Kim KC, Kim KB, Dunn CT, Park KS, Transcriptional deregulation underlying the pathogenesis of small cell lung cancer, Transl Lung Cancer Res 7(1) (2018) 4–20. 10.21037/tlcr.2017.10.07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Kasper LH, Qu C, Obenauer JC, McGoldrick DJ, Brindle PK, Genome-wide and single-cell analyses reveal a context dependent relationship between CBP recruitment and gene expression, Nucleic Acids Res 42(18) (2014) 11363–82. 10.1093/nar/gku827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Wang XQ, Dostie J, Reciprocal regulation of chromatin state and architecture by HOTAIRM1 contributes to temporal collinear HOXA gene activation, Nucleic Acids Res 45(3) (2017) 1091–1104. 10.1093/nar/gkw966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Lavarone E, Barbieri CM, Pasini D, Dissecting the role of H3K27 acetylation and methylation in PRC2 mediated control of cellular identity, Nat Commun 10(1) (2019) 1679. 10.1038/s41467-019-09624-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Jambhekar A, Dhall A, Shi Y, Roles and regulation of histone methylation in animal development, Nat Rev Mol Cell Biol 20(10) (2019) 625–641. 10.1038/s41580-019-0151-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Simo-Riudalbas L, Perez-Salvia M, Setien F, Villanueva A, Moutinho C, Martinez-Cardus A, Moran S, Berdasco M, Gomez A, Vidal E, Soler M, Heyn H, Vaquero A, de la Torre C, Barcelo-Batllori S, Vidal A, Roz L, Pastorino U, Szakszon K, Borck G, Moura CS, Carneiro F, Zondervan I, Savola S, Iwakawa R, Kohno T, Yokota J, Esteller M, KAT6B Is a Tumor Suppressor Histone H3 Lysine 23 Acetyltransferase Undergoing Genomic Loss in Small Cell Lung Cancer, Cancer Res 75(18) (2015) 3936–45. 10.1158/0008-5472.CAN-14-3702 [DOI] [PubMed] [Google Scholar]
- [140].Vonlaufen N, Naguleswaran A, Coppens I, Sullivan WJ Jr., MYST family lysine acetyltransferase facilitates ataxia telangiectasia mutated (ATM) kinase-mediated DNA damage response in Toxoplasma gondii, J Biol Chem 285(15) (2010) 11154–61. 10.1074/jbc.M109.066134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Liefke R, Oswald F, Alvarado C, Ferres-Marco D, Mittler G, Rodriguez P, Dominguez M, Borggrefe T, Histone demethylase KDM5A is an integral part of the core Notch-RBP-J repressor complex, Genes Dev 24(6) (2010) 590–601. 10.1101/gad.563210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Tenjin Y, Kudoh S, Kubota S, Yamada T, Matsuo A, Sato Y, Ichimura T, Kohrogi H, Sashida G, Sakagami T, Ito T, Ascl1-induced Wnt11 regulates neuroendocrine differentiation, cell proliferation, and E-cadherin expression in small-cell lung cancer and Wnt11 regulates small-cell lung cancer biology, Lab Invest 99(11) (2019) 1622–1635. 10.1038/s41374-019-0277-y [DOI] [PubMed] [Google Scholar]
- [143].Kim J, Sage J, Taking SCLC on a Bad LSD(1) Trip One NOTCH Further, Trends Mol Med 25(4) (2019) 261–264. 10.1016/j.molmed.2019.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Ouadah Y, Rojas ER, Riordan DP, Capostagno S, Kuo CS, Krasnow MA, Rare Pulmonary Neuroendocrine Cells Are Stem Cells Regulated by Rb, p53, and Notch, Cell 179(2) (2019) 403–416 e23. 10.1016/j.cell.2019.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Takagi S, Ishikawa Y, Mizutani A, Iwasaki S, Matsumoto S, Kamada Y, Nomura T, Nakamura K, LSD1 Inhibitor T-3775440 Inhibits SCLC Cell Proliferation by Disrupting LSD1 Interactions with SNAG Domain Proteins INSM1 and GFI1B, Cancer Res 77(17) (2017) 4652–4662. 10.1158/0008-5472.CAN-16-3502 [DOI] [PubMed] [Google Scholar]
- [146].Szczepanski AP, Zhao Z, Sosnowski T, Goo YA, Bartom ET, Wang L, ASXL3 bridges BRD4 to BAP1 complex and governs enhancer activity in small cell lung cancer, Genome Med 1 2(1) (2020) 63. 10.1186/s13073-020-00760-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Shukla V, Rao M, Zhang H, Beers J, Wangsa D, Wangsa D, Buishand FO, Wang Y, Yu Z, Stevenson HS, Reardon ES, McLoughlin KC, Kaufman AS, Payabyab EC, Hong JA, Zhang M, Davis S, Edelman D, Chen G, Miettinen MM, Restifo NP, Ried T, Meltzer PA, Schrump DS, ASXL3 Is a Novel Pluripotency Factor in Human Respiratory Epithelial Cells and a Potential Therapeutic Target in Small Cell Lung Cancer, Cancer Res 77(22) (2017) 6267–6281. 10.1158/0008-5472.CAN-17-0570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Wang L, Zhao Z, Ozark PA, Fantini D, Marshall SA, Rendleman EJ, Cozzolino KA, Louis N, He X, Morgan MA, Takahashi YH, Collings CK, Smith ER, Ntziachristos P, Savas JN, Zou L, Hashizume R, Meeks JJ, Shilatifard A, Resetting the epigenetic balance of Polycomb and COMPASS function at enhancers for cancer therapy, Nat Med 24(6) (2018) 758–769. 10.1038/s41591-018-0034-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Sze CC, Ozark PA, Cao K, Ugarenko M, Das S, Wang L, Marshall SA, Rendleman EJ, Ryan CA, Zha D, Douillet D, Chen FX, Shilatifard A, Coordinated regulation of cellular identity-associated H3K4me3 breadth by the COMPASS family, Sci Adv 6(26) (2020) eaaz4764. 10.1126/sciadv.aaz4764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Katoh M, Functional proteomics of the epigenetic regulators ASXL1, ASXL2 and ASXL3: a convergence of proteomics and epigenetics for translational medicine, Expert Rev Proteomics 12(3) (2015) 317–28. 10.1586/14789450.2015.1033409 [DOI] [PubMed] [Google Scholar]
- [151].Adamo A, Sese B, Boue S, Castano J, Paramonov I, Barrero MJ, Izpisua Belmonte JC, LSD1 regulates the balance between self-renewal and differentiation in human embryonic stem cells, Nat Cell Biol 13(6) (2011) 652–9. 10.1038/ncb2246 [DOI] [PubMed] [Google Scholar]
- [152].Kim DW, Wu N, Kim YC, Cheng PF, Basom R, Kim D, Dunn CT, Lee AY, Kim K, Lee CS, Singh A, Gazdar AF, Harris CR, Eisenman RN, Park KS, MacPherson D, Genetic requirement for Mycl and efficacy of RNA Pol I inhibition in mouse models of small cell lung cancer, Genes Dev 30(11) (2016) 1289–99. 10.1101/gad.279307.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Kim KB, Dunn CT, Park KS, Recent progress in mapping the emerging landscape of the small-cell lung cancer genome, Exp Mol Med 51(12) (2019) 1–13. 10.1038/s12276-019-0349-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].DePinho RA, Hatton KS, Tesfaye A, Yancopoulos GD, Alt FW, The human myc gene family: structure and activity of L-myc and an L-myc pseudogene, Genes Dev 1(10) (1987) 1311–26. 10.1101/gad.1.10.1311 [DOI] [PubMed] [Google Scholar]
- [155].Kaye F, Battey J, Nau M, Brooks B, Seifter E, De Greve J, Birrer M, Sausville E, Minna J, Structure and expression of the human L-myc gene reveal a complex pattern of alternative mRNA processing, Mol Cell Biol 8(1) (1988) 186–95. 10.1128/mcb.8.1.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Vervoorts J, Luscher-Firzlaff JM, Rottmann S, Lilischkis R, Walsemann G, Dohmann K, Austen M, Luscher B, Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP, EMBO Rep 4(5) (2003) 484–90. 10.1038/sj.embor.embor821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Cheng SW, Davies KP, Yung E, Beltran RJ, Yu J, Kalpana GV, c-MYC interacts with INI1/hSNF5 and requires the SWI/SNF complex for transactivation function, Nat Genet 22(1) (1999) 102–5. 10.1038/8811 [DOI] [PubMed] [Google Scholar]
- [158].Frank SR, Parisi T, Taubert S, Fernandez P, Fuchs M, Chan HM, Livingston DM, Amati B, MYC recruits the TIP60 histone acetyltransferase complex to chromatin, EMBO Rep 4(6) (2003) 575–80. 10.1038/sj.embor.embor861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Thomas LR, Wang Q, Grieb BC, Phan J, Foshage AM, Sun Q, Olejniczak ET, Clark T, Dey S, Lorey S, Alicie B, Howard GC, Cawthon B, Ess KC, Eischen CM, Zhao Z, Fesik SW, Tansey WP, Interaction with WDR5 promotes target gene recognition and tumorigenesis by MYC, Mol Cell 58(3) (2015) 440–52. 10.1016/j.molcel.2015.02.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Liang Z, Yu Q, Ji H, Tian D, Tip60-siRNA regulates ABCE1 acetylation to suppress lung cancer growth via activation of the apoptotic signaling pathway, Exp Ther Med 17(4) (2019) 3195–3202. 10.3892/etm.2019.7302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE, Selective inhibition of BET bromodomains, Nature 468(7327) (2010) 1067–73. 10.1038/nature09504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Jahchan NS, Lim JS, Bola B, Morris K, Seitz G, Tran KQ, Xu L, Trapani F, Morrow CJ, Cristea S, Coles GL, Yang D, Vaka D, Kareta MS, George J, Mazur PK, Nguyen T, Anderson WC, Dylla SJ, Blackhall F, Peifer M, Dive C, Sage J, Identification and Targeting of Long-Term Tumor-Propagating Cells in Small Cell Lung Cancer, Cell Rep 16(3) (2016) 644–56. 10.1016/j.celrep.2016.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Morgado-Pascual JL, Rayego-Mateos S, Tejedor L, Suarez-Alvarez B, Ruiz-Ortega M, Bromodomain and Extraterminal Proteins as Novel Epigenetic Targets for Renal Diseases, Front Pharmacol 10 (2019) 1315. 10.3389/fphar.2019.01315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Jung M, Gelato KA, Fernandez-Montalvan A, Siegel S, Haendler B, Targeting BET bromodomains for cancer treatment, Epigenomics 7(3) (2015) 487–501. 10.2217/epi.14.91 [DOI] [PubMed] [Google Scholar]
- [165].Topper MJ, Vaz M, Chiappinelli KB, DeStefano Shields CE, Niknafs N, Yen RC, Wenzel A, Hicks J, Ballew M, Stone M, Tran PT, Zahnow CA, Hellmann MD, Anagnostou V, Strissel PL, Strick R, Velculescu VE, Baylin SB, Epigenetic Therapy Ties MYC Depletion to Reversing Immune Evasion and Treating Lung Cancer, Cell 171(6) (2017) 1284–1300 e21. 10.1016/j.cell.2017.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Chalishazar MD, Wait SJ, Huang F, Ireland AS, Mukhopadhyay A, Lee Y, Schuman SS, Guthrie MR, Berrett KC, Vahrenkamp JM, Hu Z, Kudla M, Modzelewska K, Wang G, Ingolia NT, Gertz J, Lum DH, Cosulich SC, Bomalaski JS, DeBerardinis RJ, Oliver TG, MYC-Driven Small-Cell Lung Cancer is Metabolically Distinct and Vulnerable to Arginine Depletion, Clin Cancer Res 25(16) (2019) 5107–5121. 10.1158/1078-0432.CCR-18-4140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Grunblatt E, Wu N, Zhang H, Liu X, Norton JP, Ohol Y, Leger P, Hiatt JB, Eastwood EC, Thomas R, Ibrahim AH, Jia D, Basom R, Eaton KD, Martins R, Houghton AM, MacPherson D, MYCN drives chemoresistance in small cell lung cancer while USP7 inhibition can restore chemosensitivity, Genes Dev 34(17–18) (2020) 1210–1226. 10.1101/gad.340133.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Vadla R, Haldar D, Mammalian target of rapamycin complex 2 (mTORC2) controls glycolytic gene expression by regulating Histone H3 Lysine 56 acetylation, Cell Cycle 17(1) (2018) 110–123. 10.1080/15384101.2017.1404207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Shi L, Chen X, Zang A, Li T, Hu Y, Ma S, Lu M, Yin H, Wang H, Zhang X, Zhang B, Leng Q, Yang J, Xiao H, TSC1/mTOR-controlled metabolic-epigenetic cross talk underpins DC control of CD8+ T-cell homeostasis, PLoS Biol 17(8) (2019) e3000420. 10.1371/journal.pbio.3000420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Seo JH, Agarwal E, Bryant KG, Caino MC, Kim ET, Kossenkov AV, Tang HY, Languino LR, Gabrilovich DI, Cohen AR, Speicher DW, Altieri DC, Syntaphilin Ubiquitination Regulates Mitochondrial Dynamics and Tumor Cell Movements, Cancer Res 78(15) (2018) 4215–4228. 10.1158/0008-5472.CAN-18-0595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Denny SK, Yang D, Chuang CH, Brady JJ, Lim JS, Gruner BM, Chiou SH, Schep AN, Baral J, Hamard C, Antoine M, Wislez M, Kong CS, Connolly AJ, Park KS, Sage J, Greenleaf WJ, Winslow MM, Nfib Promotes Metastasis through a Widespread Increase in Chromatin Accessibility, Cell 166(2) (2016) 328–342. 10.1016/j.cell.2016.05.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Bottger F, Semenova EA, Song JY, Ferone G, van der Vliet J, Cozijnsen M, Bhaskaran R, Bombardelli L, Piersma SR, Pham TV, Jimenez CR, Berns A, Tumor Heterogeneity Underlies Differential Cisplatin Sensitivity in Mouse Models of Small-Cell Lung Cancer, Cell Rep 27(11) (2019) 3345–3358 e4. 10.1016/j.celrep.2019.05.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Dooley AL, Winslow MM, Chiang DY, Banerji S, Stransky N, Dayton TL, Snyder EL, Senna S, Whittaker CA, Bronson RT, Crowley D, Barretina J, Garraway L, Meyerson M, Jacks T, Nuclear factor I/B is an oncogene in small cell lung cancer, Genes Dev 25(14) (2011) 1470–5. 10.1101/gad.2046711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Adam RC, Yang H, Ge Y, Infarinato NR, Gur-Cohen S, Miao Y, Wang P, Zhao Y, Lu CP, Kim JE, Ko JY, Paik SS, Gronostajski RM, Kim J, Krueger JG, Zheng D, Fuchs E, NFI transcription factors provide chromatin access to maintain stem cell identity while preventing unintended lineage fate choices, Nat Cell Biol 22(6) (2020) 640–650. 10.1038/s41556-020-0513-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Nepon-Sixt BS, Bryant VL, Alexandrow MG, Myc-driven chromatin accessibility regulates Cdc45 assembly into CMG helicases, Commun Biol 2 (2019) 110. 10.1038/s42003-019-0353-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Yang D, Denny SK, Greenside PG, Chaikovsky AC, Brady JJ, Ouadah Y, Granja JM, Jahchan NS, Lim JS, Kwok S, Kong CS, Berghoff AS, Schmitt A, Reinhardt HC, Park KS, Preusser M, Kundaje A, Greenleaf WJ, Sage J, Winslow MM, Intertumoral Heterogeneity in SCLC Is Influenced by the Cell Type of Origin, Cancer Discov 8(10) (2018) 1316–1331. 10.1158/2159-8290.CD-17-0987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Li L, Jin J, Yang XJ, Histone Deacetylase 3 Governs Perinatal Cerebral Development via Neural Stem and Progenitor Cells, iScience 20 (2019) 148–167. 10.1016/j.isci.2019.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Liu X, Wang C, Liu W, Li J, Li C, Kou X, Chen J, Zhao Y, Gao H, Wang H, Zhang Y, Gao Y, Gao S, Distinct features of H3K4me3 and H3K27me3 chromatin domains in pre-implantation embryos, Nature 537(7621) (2016) 558–562. 10.1038/nature19362 [DOI] [PubMed] [Google Scholar]
- [179].Xie H, Zhang W, Zhang M, Akhtar T, Li Y, Yi W, Sun X, Zuo Z, Wei M, Fang X, Yao Z, Dong K, Zhong S, Liu Q, Shen Y, Wu Q, Wang X, Zhao H, Bao J, Qu K, Xue T, Chromatin accessibility analysis reveals regulatory dynamics of developing human retina and hiPSC-derived retinal organoids, Sci Adv 6(6) (2020) eaay5247. 10.1126/sciadv.aay5247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Fane ME, Chhabra Y, Hollingsworth DEJ, Simmons JL, Spoerri L, Oh TG, Chauhan J, Chin T, Harris L, Harvey TJ, Muscat GEO, Goding CR, Sturm RA, Haass NK, Boyle GM, Piper M, Smith AG, NFIB Mediates BRN2 Driven Melanoma Cell Migration and Invasion Through Regulation of EZH2 and MITF, EBioMedicine 16 (2017) 63–75. 10.1016/j.ebiom.2017.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Minna JD, Johnson JE, Opening a Chromatin Gate to Metastasis, Cell 166(2) (2016) 275–276. 10.1016/j.cell.2016.06.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].He T, Wildey G, McColl K, Savadelis A, Spainhower K, McColl C, Kresak A, Tan AC, Yang M, Abbas A, Dowlati A, Identification of RUNX1T1 as a potential epigenetic modifier in small cell lung cancer, Mol Oncol (2020). 10.1002/1878-0261.12829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Calabi F, Cilli V, CBFA2T1, a gene rearranged in human leukemia, is a member of a multigene family, Genomics 52(3) (1998) 332–41. 10.1006/geno.1998.5429 [DOI] [PubMed] [Google Scholar]
- [184].Biswas S, Rao CM, Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy, Eur J Pharmacol 837 (2018) 8–24. 10.1016/j.ejphar.2018.08.021 [DOI] [PubMed] [Google Scholar]
- [185].Zhang T, Li Y, Zhang H, Wang X, Liu X, Li L, The Role of RASSF1 Methylation in Lung Carcinoma, Adv Exp Med Biol 1255 (2020) 99–108. 10.1007/978-981-15-4494-1_8 [DOI] [PubMed] [Google Scholar]
- [186].Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y, Histone demethylation mediated by the nuclear amine oxidase homolog LSD1, Cell 119(7) (2004) 941–53. 10.1016/j.cell.2004.12.012 [DOI] [PubMed] [Google Scholar]
- [187].Fang Y, Yang C, Yu Z, Li X, Mu Q, Liao G, Yu B, Natural products as LSD1 inhibitors for cancer therapy, Acta Pharm Sin B (2020). 10.1016/j.apsb.2020.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Yang GJ, Lei PM, Wong SY, Ma DL, Leung CH, Pharmacological Inhibition of LSD1 for Cancer Treatment, Molecules 23(12) (2018). 10.3390/molecules23123194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Fang Y, Liao G, Yu B, LSD1/KDM1A inhibitors in clinical trials: advances and prospects, J Hematol Oncol 12(1) (2019) 129. 10.1186/s13045-019-0811-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Stewart CA, Byers LA, Altering the Course of Small Cell Lung Cancer: Targeting Cancer Stem Cells via LSD1 Inhibition, Cancer Cell 28(1) (2015) 4–6. 10.1016/j.ccell.2015.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Milletti F, Cheng W-Y, Maes T, Lunardi S, DeMario M, Pierceall WE, Mack F, Neuroendocrine gene transcript expression is associated with efficacy to lysine-specific demethylase-1 inhibitor RG6016 in small cell lung cancer-derived cell lines, Cancer Research, , p. 76:4708. 10.1158/1538-7445.AM2016-4708 [DOI] [Google Scholar]
- [192].Chen Y, Yang Y, Wang F, Wan K, Yamane K, Zhang Y, Lei M, Crystal structure of human histone lysine-specific demethylase 1 (LSD1), Proc Natl Acad Sci U S A 103(38) (2006) 13956–61. 10.1073/pnas.0606381103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Yang M, Gocke CB, Luo X, Borek D, Tomchick DR, Machius M, Otwinowski Z, Yu H, Structural basis for CoREST-dependent demethylation of nucleosomes by the human LSD1 histone demethylase, Mol Cell 23(3) (2006) 377–87. 10.1016/j.molcel.2006.07.012 [DOI] [PubMed] [Google Scholar]
- [194].Drazic A, Myklebust LM, Ree R, Arnesen T, The world of protein acetylation, Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics 1864(10) (2016) 1372–1401. 10.1016/j.bbapap.2016.06.007 [DOI] [PubMed] [Google Scholar]
- [195].Bates SE, Epigenetic Therapies for Cancer, N Engl J Med 383(7) (2020) 650–663. 10.1056/NEJMra1805035 [DOI] [PubMed] [Google Scholar]
- [196].Alvarez MJ, Subramaniam PS, Tang LH, Grunn A, Aburi M, Rieckhof G, Komissarova EV, Hagan EA, Bodei L, Clemons PA, A precision oncology approach to the pharmacological targeting of mechanistic dependencies in neuroendocrine tumors, Nature genetics 50(7) (2018) 979–989. 10.1038/s41588-018-0138-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Klieser E, Urbas R, Stättner S, Primavesi F, Jäger T, Dinnewitzer A, Mayr C, Kiesslich T, Holzmann K, Di Fazio P, Comprehensive immunohistochemical analysis of histone deacetylases in pancreatic neuroendocrine tumors: HDAC5 as a predictor of poor clinical outcome, Human pathology 65 (2017) 41–52. 10.1016/j.humpath.2017.02.009 [DOI] [PubMed] [Google Scholar]
- [198].Attar N, Kurdistani SK, Exploitation of EP300 and CREBBP lysine acetyltransferases by cancer, Cold Spring Harbor perspectives in medicine 7(3) (2017) a026534. 10.1101/cshperspect.a026534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Lin SQ, Li X, Epigenetic Therapies and Potential Drugs for Treating Human Cancer, Curr Drug Targets 21(11) (2020) 1068–1083. 10.2174/1389450121666200325093104 [DOI] [PubMed] [Google Scholar]
- [200].Lin CA, Yu SL, Chen HY, Chen HW, Lin SU, Chang CC, Yu CJ, Yang PC, Ho CC, EGFR-Mutant SCLC Exhibits Heterogeneous Phenotypes and Resistance to Common Antineoplastic Drugs, J Thorac Oncol 14(3) (2019) 513–526. 10.1016/j.jtho.2018.11.021 [DOI] [PubMed] [Google Scholar]
- [201].Balasubramaniam S, Redon CE, Peer CJ, Bryla C, Lee MJ, Trepel JB, Tomita Y, Rajan A, Giaccone G, Bonner WM, Figg WD, Fojo T, Piekarz RL, Bates SE, Phase I trial of belinostat with cisplatin and etoposide in advanced solid tumors, with a focus on neuroendocrine and small cell cancers of the lung, Anticancer Drugs 29(5) (2018) 457–465. 10.1097/CAD.0000000000000596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Pan CH, Chang YF, Lee MS, Wen BC, Ko JC, Liang SK, Liang MC, Vorinostat enhances the cisplatin-mediated anticancer effects in small cell lung cancer cells, BMC Cancer 16(1) (2016) 857. 10.1186/s12885-016-2888-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Nakajima W, Sharma K, Hicks MA, Le N, Brown R, Krystal GW, Harada H, Combination with vorinostat overcomes ABT-263 (navitoclax) resistance of small cell lung cancer, Cancer Biol Ther 17(1) (2016) 27–35. 10.1080/15384047.2015.1108485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].de Marinis F, Atmaca A, Tiseo M, Giuffreda L, Rossi A, Gebbia V, D’Antonio C, Dal Zotto L, Al-Batran SE, Marsoni S, Wolf M, A phase II study of the histone deacetylase inhibitor panobinostat (LBH589) in pretreated patients with small-cell lung cancer, J Thorac Oncol 8(8) (2013) 1091–4. 10.1097/JTO.0b013e318293d88c [DOI] [PubMed] [Google Scholar]
- [205].Gray J, Cubitt CL, Zhang S, Chiappori A, Combination of HDAC and topoisomerase inhibitors in small cell lung cancer, Cancer Biol Ther 13(8) (2012) 614–22. 10.4161/cbt.19848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Hubaux R, Vandermeers F, Crisanti MC, Kapoor V, Burny A, Mascaux C, Albelda SM, Willems L, Preclinical evidence for a beneficial impact of valproate on the response of small cell lung cancer to first-line chemotherapy, Eur J Cancer 46(9) (2010) 1724–34. 10.1016/j.ejca.2010.03.021 [DOI] [PubMed] [Google Scholar]
- [207].Liu Y, Li Y, Liu S, Adeegbe DO, Christensen CL, Quinn MM, Dries R, Han S, Buczkowski K, Wang X, Chen T, Gao P, Zhang H, Li F, Hammerman PS, Bradner JE, Quayle SN, Wong KK, NK Cells Mediate Synergistic Antitumor Effects of Combined Inhibition of HDAC6 and BET in a SCLC Preclinical Model, Cancer Res 78(13) (2018) 3709–3717. 10.1158/0008-5472.CAN-18-0161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Goetze TO, Immunotherapy: a new era in small-cell lung cancer, Lancet 394(10212) (2019) 1884–1885. 10.1016/S0140-6736(19)32235-4 [DOI] [PubMed] [Google Scholar]
- [209].Esposito G, Palumbo G, Carillio G, Manzo A, Montanino A, Sforza V, Costanzo R, Sandomenico C, La Manna C, Martucci N, La Rocca A, De Luca G, Piccirillo MC, De Cecio R, Botti G, Totaro G, Muto P, Picone C, Normanno N, Morabito A, Immunotherapy in Small Cell Lung Cancer, Cancers (Basel) 12(9) (2020). 10.3390/cancers12092522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Subbiah S, Nam A, Garg N, Behal A, Kulkarni P, Salgia R, Small Cell Lung Cancer from Traditional to Innovative Therapeutics: Building a Comprehensive Network to Optimize Clinical and Translational Research, J Clin Med 9(8) (2020). 10.3390/jcm9082433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ, Improved survival with ipilimumab in patients with metastatic melanoma, N Engl J Med 363(8) (2010) 711–23. 10.1056/NEJMoa1003466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ, Davidson N, Richards J, Maio M, Hauschild A, Miller WH Jr., Gascon P, Lotem M, Harmankaya K, Ibrahim R, Francis S, Chen TT, Humphrey R, Hoos A, Wolchok JD, Ipilimumab plus dacarbazine for previously untreated metastatic melanoma, N Engl J Med 364(26) (2011) 2517–26. 10.1056/NEJMoa1104621 [DOI] [PubMed] [Google Scholar]
- [213].Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, Patt D, Chen TT, Berman DM, Wolchok JD, Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma, J Clin Oncol 33(17) (2015) 1889–94. 10.1200/JCO.2014.56.2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M, Safety, activity, and immune correlates of anti-PD-1 antibody in cancer, N Engl J Med 366(26) (2012) 2443–54. 10.1056/NEJMoa1200690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, Dronca R, Gangadhar TC, Patnaik A, Zarour H, Joshua AM, Gergich K, Elassaiss-Schaap J, Algazi A, Mateus C, Boasberg P, Tumeh PC, Chmielowski B, Ebbinghaus SW, Li XN, Kang SP, Ribas A, Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma, N Engl J Med 369(2) (2013) 134–44. 10.1056/NEJMoa1305133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Motzer RJ, Rini BI, McDermott DF, Redman BG, Kuzel TM, Harrison MR, Vaishampayan UN, Drabkin HA, George S, Logan TF, Margolin KA, Plimack ER, Lambert AM, Waxman IM, Hammers HJ, Nivolumab for Metastatic Renal Cell Carcinoma: Results of a Randomized Phase II Trial, J Clin Oncol 33(13) (2015) 1430–7. 10.1200/JCO.2014.59.0703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry D, Freeman GJ, Rodig SJ, Chapuy B, Ligon AH, Zhu L, Grosso JF, Kim SY, Timmerman JM, Shipp MA, Armand P, PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma, N Engl J Med 372(4) (2015) 311–9. 10.1056/NEJMoa1411087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Reck M, Taylor F, Penrod JR, DeRosa M, Morrissey L, Dastani H, Orsini L, Gralla RJ, Impact of Nivolumab versus Docetaxel on Health-Related Quality of Life and Symptoms in Patients with Advanced Squamous Non-Small Cell Lung Cancer: Results from the CheckMate 017 Study, J Thorac Oncol 13(2) (2018) 194–204. 10.1016/j.jtho.2017.10.029 [DOI] [PubMed] [Google Scholar]
- [219].Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, Waterhouse D, Ready N, Gainor J, Aren Frontera O, Havel L, Steins M, Garassino MC, Aerts JG, Domine M, Paz-Ares L, Reck M, Baudelet C, Harbison CT, Lestini B, Spigel DR, Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer, N Engl J Med 373(2) (2015) 123–35. 10.1056/NEJMoa1504627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Forde PM, Chaft JE, Smith KN, Anagnostou V, Cottrell TR, Hellmann MD, Zahurak M, Yang SC, Jones DR, Broderick S, Battafarano RJ, Velez MJ, Rekhtman N, Olah Z, Naidoo J, Marrone KA, Verde F, Guo H, Zhang J, Caushi JX, Chan HY, Sidhom JW, Scharpf RB, White J, Gabrielson E, Wang H, Rosner GL, Rusch V, Wolchok JD, Merghoub T, Taube JM, Velculescu VE, Topalian SL, Brahmer JR, Pardoll DM, Neoadjuvant PD-1 Blockade in Resectable Lung Cancer, N Engl J Med 378(21) (2018) 1976–1986. 10.1056/NEJMoa1716078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Hellmann MD, Ciuleanu TE, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, Minenza E, Linardou H, Burgers S, Salman P, Borghaei H, Ramalingam SS, Brahmer J, Reck M, O’Byrne KJ, Geese WJ, Green G, Chang H, Szustakowski J, Bhagavatheeswaran P, Healey D, Fu Y, Nathan F, Paz-Ares L, Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden, N Engl J Med 378(22) (2018) 2093–2104. 10.1056/NEJMoa1801946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Yarchoan M, Hopkins A, Jaffee EM, Tumor Mutational Burden and Response Rate to PD-1 Inhibition, N Engl J Med 377(25) (2017) 2500–2501. 10.1056/NEJMc1713444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, Schrock A, Campbell B, Shlien A, Chmielecki J, Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden, Genome medicine 9(1) (2017) 34. 10.1186/s13073-017-0424-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Hellmann MD, Ciuleanu TE, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, Minenza E, Linardou H, Burgers S, Salman P, Borghaei H, Ramalingam SS, Brahmer J, Reck M, O’Byrne KJ, Geese WJ, Green G, Chang H, Szustakowski J, Bhagavatheeswaran P, Healey D, Fu Y, Nathan F, Paz-Ares L, Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden, New England Journal of Medicine 378(22) (2018) 2093–2104. 10.1056/NEJMoa1801946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, Schrock AB, Hartmaier RJ, Trabucco SE, Gay L, Ali SM, Elvin JA, Singal G, Ross JS, Fabrizio D, Szabo PM, Chang H, Sasson A, Srinivasan S, Kirov S, Szustakowski J, Vitazka P, Edwards R, Bufill JA, Sharma N, Ou SI, Peled N, Spigel DR, Rizvi H, Aguilar EJ, Carter BW, Erasmus J, Halpenny DF, Plodkowski AJ, Long NM, Nishino M, Denning WL, Galan-Cobo A, Hamdi H, Hirz T, Tong P, Wang J, Rodriguez-Canales J, Villalobos PA, Parra ER, Kalhor N, Sholl LM, Sauter JL, Jungbluth AA, Mino-Kenudson M, Azimi R, Elamin YY, Zhang J, Leonardi GC, Jiang F, Wong KK, Lee JJ, Wistuba V.A. Papadimitrakopoulou II, Miller VA, Frampton GM, Wolchok JD, Shaw AT, Janne PA, Stephens PJ, Rudin CM, Geese WJ, Albacker LA, Heymach JV, STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma, Cancer Discov 8(7) (2018) 822–835. 10.1158/2159-8290.CD-18-0099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Auvray M, Auclin E, Barthelemy P, Bono P, Kellokumpu-Lehtinen P, Gross-Goupil M, De Velasco G, Powles T, Mouillet G, Vano YA, Gravis G, Mourey L, Priou F, Rolland F, Escudier B, Albiges L, Second-line targeted therapies after nivolumab-ipilimumab failure in metastatic renal cell carcinoma, Eur J Cancer 108 (2019) 33–40. 10.1016/j.ejca.2018.11.031 [DOI] [PubMed] [Google Scholar]
- [227].Tokaca N, Wotherspoon A, Nicholson AG, Fotiadis N, Thompson L, Popat S, Lack of response to nivolumab in a patient with EGFR-mutant non-small cell lung cancer adenocarcinoma sub-type transformed to small cell lung cancer, Lung Cancer 111 (2017) 65–68. 10.1016/j.lungcan.2017.07.012 [DOI] [PubMed] [Google Scholar]
- [228].Hellmann MD, Callahan MK, Awad MM, Calvo E, Ascierto PA, Atmaca A, Rizvi NA, Hirsch FR, Selvaggi G, Szustakowski JD, Sasson A, Golhar R, Vitazka P, Chang H, Geese WJ, Antonia SJ, Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination with Ipilimumab in Small-Cell Lung Cancer, Cancer Cell 33(5) (2018) 853–861 e4. 10.1016/j.ccell.2018.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Gadgeel SM, Pennell NA, Fidler MJ, Halmos B, Bonomi P, Stevenson J, Schneider B, Sukari A, Ventimiglia J, Chen W, Galasso C, Wozniak A, Boerner J, Kalemkerian GP, Phase II Study of Maintenance Pembrolizumab in Patients with Extensive-Stage Small Cell Lung Cancer (SCLC), J Thorac Oncol 13(9) (2018) 1393–1399. 10.1016/j.jtho.2018.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Squibb B-M, Bristol-Myers Squibb announces CheckMate-451 study did not meet primary endpoint of overall survival with Opdivo plus Yervoy Vs. placebo as a maintenance therapy in patients with extensive-stage small cell lung cancer after completion of firstline. New York, NY: Bristol-Myers Squibb; 2018 [November 26], 2019. https://news.bms.com/news/corporate-financial/2018/Bristol-Myers-Squibb-Announces-CheckMate−−451-Study-Did-Not-Meet-Primary-Endpoint-of-Overall-Survival-with-Opdivo-Plus-Yervoy-Vs-Placebo-as-A-Maintenance-Therapy-in-Patients-with-Extensive-Stage-Small-Cell-Lung-Cancer-After-Completion-of-First-Line/default.aspx [Google Scholar]
- [231].Ready N, Owonikoko TK, Postmus PE, Reck M, Peters S, Pieters A, Selvaggi G, Fairchild JP, Govindan R, CheckMate 451: A randomized, double-blind, phase III trial of nivolumab (nivo), nivo plus ipilimumab (ipi), or placebo as maintenance therapy in patients (pts) with extensive-stage disease small cell lung cancer (ED-SCLC) after first-line platinum-based doublet chemotherapy (PT-DC), Journal of Clinical Oncology 34(15_suppl) (2016) TPS8579-TPS8579. 10.1200/JCO.2016.34.15_suppl.TPS8579 [DOI] [Google Scholar]
- [232].Squibb B-M, Bristol-Myers Squibb announces Phase 3 CheckMate-331 study does not meet primary endpoint of overall survival with Opdivo versus chemotherapy in patients with previously treated relapsed small cell lung cance r. New York, NY: Bristol-Myers Squibb; 2018 [October 12], 2019. https://news.bms.com/news/corporate-financial/2018/Bristol-Myers-Squibb-Announces-Phase-3-CheckMate−−331-Study-Does-Not-Meet-Primary-Endpoint-of-Overall-Survival-with-Opdivo-Versus-Chemotherapy-in-Patients-with-Previously-Treated-Relapsed-Small-Cell-Lung-Cancer/default.aspx [Google Scholar]
- [233].Horn L, Reck M, Gettinger SN, Spigel DR, Antonia SJ, Rupnow BA, Pieters A, Selvaggi G, Fairchild JP, Peters S, CheckMate 331: An open-label, randomized phase III trial of nivolumab versus chemotherapy in patients (pts) with relapsed small cell lung cancer (SCLC) after first-line platinum-based chemotherapy (PT-DC), Journal of Clinical Oncology 34(15_suppl) (2016) TPS8578-TPS8578. 10.1200/JCO.2016.34.15_suppl.TPS8578 [DOI] [Google Scholar]
- [234].Weng X, Huang X, Li H, Lin S, Rao X, Guo X, Huang P, First-Line Treatment With Atezolizumab Plus Nab-Paclitaxel for Advanced Triple-Negative Breast Cancer: A Cost-Effectiveness Analysis, Am J Clin Oncol 43(5) (2020) 340–348. 10.1097/COC.0000000000000671 [DOI] [PubMed] [Google Scholar]
- [235].Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Dieras V, Hegg R, Im SA, Shaw Wright G, Henschel V, Molinero L, Chui SY, Funke R, Husain A, Winer EP, Loi S, Emens LA, Investigators IMT, Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer, N Engl J Med 379(22) (2018) 2108–2121. 10.1056/NEJMoa1809615 [DOI] [PubMed] [Google Scholar]
- [236].Sundar R, Huang KK, Qamra A, Kim KM, Kim ST, Kang WK, Tan ALK, Lee J, Tan P, Epigenomic promoter alterations predict for benefit from immune checkpoint inhibition in metastatic gastric cancer, Ann Oncol 30(3) (2019) 424–430. 10.1093/annonc/mdy550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Duruisseaux M, Martinez-Cardus A, Calleja-Cervantes ME, Moran S, Castro de Moura M, Davalos V, Pineyro D, Sanchez-Cespedes M, Girard N, Brevet M, Giroux-Leprieur E, Dumenil C, Pradotto M, Bironzo P, Capelletto E, Novello S, Cortot A, Copin MC, Karachaliou N, Gonzalez-Cao M, Peralta S, Montuenga LM, Gil-Bazo I, Baraibar I, Lozano MD, Varela M, Ruffinelli JC, Palmero R, Nadal E, Moran T, Perez L, Ramos I, Xiao Q, Fernandez AF, Fraga MF, Gut M, Gut I, Teixido C, Vilarino N, Prat A, Reguart N, Benito A, Garrido P, Barragan I, Emile JF, Rosell R, Brambilla E, Esteller M, Epigenetic prediction of response to anti-PD-1 treatment in non-small-cell lung cancer: a multicentre, retrospective analysis, Lancet Respir Med 6(10) (2018) 771–781. 10.1016/S2213-2600(18)30284-4 [DOI] [PubMed] [Google Scholar]
- [238].Morel D, Jeffery D, Aspeslagh S, Almouzni G, Postel-Vinay S, Combining epigenetic drugs with other therapies for solid tumours - past lessons and future promise, Nat Rev Clin Oncol 17(2) (2020) 91–107. 10.1038/s41571-019-0267-4 [DOI] [PubMed] [Google Scholar]
- [239].Ricciuti B, Kravets S, Dahlberg SE, Umeton R, Albayrak A, Subegdjo SJ, Johnson BE, Nishino M, Sholl LM, Awad MM, Use of targeted next generation sequencing to characterize tumor mutational burden and efficacy of immune checkpoint inhibition in small cell lung cancer, J Immunother Cancer 7(1) (2019) 87. 10.1186/s40425-019-0572-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Sabari JK, Leonardi GC, Shu CA, Umeton R, Montecalvo J, Ni A, Chen R, Dienstag J, Mrad C, Bergagnini I, Lai WV, Offin M, Arbour KC, Plodkowski AJ, Halpenny DF, Paik PK, Li BT, Riely GJ, Kris MG, Rudin CM, Sholl LM, Nishino M, Hellmann MD, Rekhtman N, Awad MM, Drilon A, PD-L1 expression, tumor mutational burden, and response to immunotherapy in patients with MET exon 14 altered lung cancers, Ann Oncol 29(10) (2018) 2085–2091. 10.1093/annonc/mdy334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Hellmann MD, Callahan MK, Awad MM, Calvo E, Ascierto PA, Atmaca A, Rizvi NA, Hirsch FR, Selvaggi G, Szustakowski JD, Sasson A, Golhar R, Vitazka P, Chang H, Geese WJ, Antonia SJ, Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination with Ipilimumab in Small-Cell Lung Cancer, Cancer Cell 35(2) (2019) 329. 10.1016/j.ccell.2019.01.011 [DOI] [PubMed] [Google Scholar]
- [242].Antonia SJ, López-Martin JA, Bendell J, Ott PA, Taylor M, Eder JP, Jäger D, Pietanza MC, Le DT, de Braud F, Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial, The Lancet Oncology 17(7) (2016) 883–895. 10.1016/S1470-2045(16)30098-5 [DOI] [PubMed] [Google Scholar]
- [243].Ready N, Farago AF, de Braud F, Atmaca A, Hellmann MD, Schneider JG, Spigel DR, Moreno V, Chau I, Hann CL, Eder JP, Steele NL, Pieters A, Fairchild J, Antonia SJ, Third-Line Nivolumab Monotherapy in Recurrent SCLC: CheckMate 032, J Thorac Oncol 14(2) (2019) 237–244. 10.1016/j.jtho.2018.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].U. FDA, FDA grants nivolumab accelerated approval for third-line treatment of metastatic small cell lung cancer. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-nivolumab-accelerated-approval-third-line-treatment-metastatic-small-cell-lung-cancer
- [245].Mak DWS, Li S, Minchom A, Challenging the recalcitrant disease-developing molecularly driven treatments for small cell lung cancer, Eur J Cancer 119 (2019) 132–150. 10.1016/j.ejca.2019.04.037 [DOI] [PubMed] [Google Scholar]
- [246].Horn L, Mansfield AS, Szczesna A, Havel L, Krzakowski M, Hochmair MJ, Huemer F, Losonczy G, Johnson ML, Nishio M, Reck M, Mok T, Lam S, Shames DS, Liu J, Ding B, Lopez-Chavez A, Kabbinavar F, Lin W, Sandler A, Liu SV, I.M.S. Group, First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer, N Engl J Med 379(23) (2018) 2220–2229. 10.1056/NEJMoa1809064 [DOI] [PubMed] [Google Scholar]
- [247].Reck M, Luft A, Szczesna A, Havel L, Kim S-W, Akerley W, Pietanza MC, Wu Y.-l., Zielinski C, Thomas M, Phase III randomized trial of ipilimumab plus etoposide and platinum versus placebo plus etoposide and platinum in extensive-stage small-cell lung cancer, Journal of Clinical Oncology 34(31) (2016) 3740–3748. 10.1200/JCO.2016.67.6601 [DOI] [PubMed] [Google Scholar]
- [248].Paz-Ares L, Dvorkin M, Chen Y, Reinmuth N, Hotta K, Trukhin D, Statsenko G, Hochmair MJ, Özgüroğlu M, Ji JH, Durvalumab plus platinum–etoposide versus platinum– etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): a randomised, controlled, open-label, phase 3 trial, The Lancet 394(10212) (2019) 1929–1939. 10.1016/S0140-6736(19)32222-6 [DOI] [PubMed] [Google Scholar]
- [249].Cao J, Yan Q, Cancer Epigenetics, Tumor Immunity, and Immunotherapy, Trends Cancer 6(7) (2020) 580–592. 10.1016/j.trecan.2020.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Kommalapati A, Tanvetyanon T, Epigenetic modulation of immunotherapy cofactors to enhance tumor response in lung cancer, Hum Vaccin Immunother (2020) 1–4. 10.1080/21645515.2020.1764273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Zheng H, Zhao W, Yan C, Watson CC, Massengill M, Xie M, Massengill C, Noyes DR, Martinez GV, Afzal R, Chen Z, Ren X, Antonia SJ, Haura EB, Ruffell B, Beg AA, HDAC Inhibitors Enhance T-Cell Chemokine Expression and Augment Response to PD-1 Immunotherapy in Lung Adenocarcinoma, Clin Cancer Res 22(16) (2016) 4119–32. 10.1158/1078-0432.CCR-15-2584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Briere D, Sudhakar N, Woods DM, Hallin J, Engstrom LD, Aranda R, Chiang H, Sodre AL, Olson P, Weber JS, Christensen JG, The class I/IV HDAC inhibitor mocetinostat increases tumor antigen presentation, decreases immune suppressive cell types and augments checkpoint inhibitor therapy, Cancer Immunol Immunother 67(3) (2018) 381–392. 10.1007/s00262-017-2091-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Levy BP, Giaccone G, Besse B, Felip E, Garassino MC, Domine Gomez M, Garrido P, Piperdi B, Ponce-Aix S, Menezes D, MacBeth KJ, Risueno A, Slepetis R, Wu X, Fandi A, Paz-Ares L, Randomised phase 2 study of pembrolizumab plus CC-486 versus pembrolizumab plus placebo in patients with previously treated advanced non-small cell lung cancer, Eur J Cancer 108 (2019) 120–128. 10.1016/j.ejca.2018.11.028 [DOI] [PubMed] [Google Scholar]
- [254].Gray JE, Saltos A, Tanvetyanon T, Haura EB, Creelan B, Antonia SJ, Shafique M, Zheng H, Dai W, Saller JJ, Chen Z, Tchekmedyian N, Goas K, Thapa R, Boyle TA, Chen DT, Beg AA, Phase I/Ib Study of Pembrolizumab Plus Vorinostat in Advanced/Metastatic Non-Small Cell Lung Cancer, Clin Cancer Res 25(22) (2019) 6623–6632. 10.1158/1078-0432.CCR-19-1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Burr ML, Sparbier CE, Chan KL, Chan YC, Kersbergen A, Lam EYN, Azidis-Yates E, Vassiliadis D, Bell CC, Gilan O, Jackson S, Tan L, Wong SQ, Hollizeck S, Michalak EM, Siddle HV, McCabe MT, Prinjha RK, Guerra GR, Solomon BJ, Sandhu S, Dawson SJ, Beavis PA, Tothill RW, Cullinane C, Lehner PJ, Sutherland KD, Dawson MA, An Evolutionarily Conserved Function of Polycomb Silences the MHC Class I Antigen Presentation Pathway and Enables Immune Evasion in Cancer, Cancer Cell 36(4) (2019) 385–401 e8. 10.1016/j.ccell.2019.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [256].Villanueva L, Alvarez-Errico D, Esteller M, The Contribution of Epigenetics to Cancer Immunotherapy, Trends Immunol 41(8) (2020) 676–691. 10.1016/j.it.2020.06.002 [DOI] [PubMed] [Google Scholar]
- [257].Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, Gouw AM, Baylot V, Gutgemann I, Eilers M, Felsher DW, MYC regulates the antitumor immune response through CD47 and PD-L1, Science 352(6282) (2016) 227–31. 10.1126/science.aac9935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, Li Y, Chen H, Yang H, Hsu PH, Van Allen EM, Freeman GJ, De Carvalho DD, He HH, Sharpe AH, Shi Y, LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade, Cell 174(3) (2018) 549–563 e19. 10.1016/j.cell.2018.05.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [259].Qin Y, Vasilatos SN, Chen L, Wu H, Cao Z, Fu Y, Huang M, Vlad AM, Lu B, Oesterreich S, Davidson NE, Huang Y, Inhibition of histone lysine-specific demethylase 1 elicits breast tumor immunity and enhances antitumor efficacy of immune checkpoint blockade, Oncogene 38(3) (2019) 390–405. 10.1038/s41388-018-0451-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].Adeegbe DO, Liu S, Hattersley MM, Bowden M, Zhou CW, Li S, Vlahos R, Grondine M, Dolgalev I, Ivanova EV, Quinn MM, Gao P, Hammerman PS, Bradner JE, Diehl JA, Rustgi AK, Bass AJ, Tsirigos A, Freeman GJ, Chen H, Wong KK, BET Bromodomain Inhibition Cooperates with PD-1 Blockade to Facilitate Antitumor Response in Kras-Mutant Non-Small Cell Lung Cancer, Cancer Immunol Res 6(10) (2018) 1234–1245. 10.1158/2326-6066.CIR-18-0077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [261].Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, Mischel PS, Stokoe D, Pieper RO, Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma, Nat Med 13(1) (2007) 84–8. 10.1038/nm1517 [DOI] [PubMed] [Google Scholar]
- [262].Melaiu O, Mina M, Chierici M, Boldrini R, Jurman G, Romania P, D’Alicandro V, Benedetti MC, Castellano A, Liu T, PD-L1 is a therapeutic target of the bromodomain inhibitor JQ1 and, combined with HLA class I, a promising prognostic biomarker in neuroblastoma, Clinical Cancer Research 23(15) (2017) 4462–4472. 10.1158/1078-0432.CCR-16-2601 [DOI] [PubMed] [Google Scholar]
- [263].Kim EY, Kim A, Kim SK, Chang YS, MYC expression correlates with PD-L1 expression in non-small cell lung cancer, Lung Cancer 110 (2017) 63–67. 10.1016/j.lungcan.2017.06.006 [DOI] [PubMed] [Google Scholar]
- [264].Lim JS, Ibaseta A, Fischer MM, Cancilla B, O’Young G, Cristea S, Luca VC, Yang D, Jahchan NS, Hamard C, Antoine M, Wislez M, Kong C, Cain J, Liu YW, Kapoun AM, Garcia KC, Hoey T, Murriel CL, Sage J, Intratumoural heterogeneity generated by Notch signalling promotes small-cell lung cancer, Nature 545(7654) (2017) 360–364. 10.1038/nature22323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [265].Ishikawa Y, Gamo K, Yabuki M, Takagi S, Toyoshima K, Nakayama K, Nakayama A, Morimoto M, Miyashita H, Dairiki R, Hikichi Y, Tomita N, Tomita D, Imamura S, Iwatani M, Kamada Y, Matsumoto S, Hara R, Nomura T, Tsuchida K, Nakamura K, A Novel LSD1 Inhibitor T-3775440 Disrupts GFI1B-Containing Complex Leading to Transdifferentiation and Impaired Growth of AML Cells, Mol Cancer Ther 16(2) (2017) 273–284. 10.1158/1535-7163.MCT-16-0471 [DOI] [PubMed] [Google Scholar]
- [266].Otterson GA, Hodgson L, Pang H, Vokes EE, Cancer B. Leukemia Group, Phase II study of the histone deacetylase inhibitor Romidepsin in relapsed small cell lung cancer (Cancer and Leukemia Group B 30304), J Thorac Oncol 5(10) (2010) 1644–8. 10.1097/JTO.0b013e3181ec1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [267].Luszczek W, Cheriyath V, Mekhail TM, Borden EC, Combinations of DNA methyltransferase and histone deacetylase inhibitors induce DNA damage in small cell lung cancer cells: correlation of resistance with IFN-stimulated gene expression, Mol Cancer Ther 9(8) (2010) 2309–21. 10.1158/1535-7163.MCT-10-0309 [DOI] [PubMed] [Google Scholar]
- [268].Platta CS, Greenblatt DY, Kunnimalaiyaan M, Chen H, The HDAC inhibitor trichostatin A inhibits growth of small cell lung cancer cells, J Surg Res 142(2) (2007) 219–26. 10.1016/j.jss.2006.12.555 [DOI] [PubMed] [Google Scholar]
- [269].Hopkins-Donaldson S, Ziegler A, Kurtz S, Bigosch C, Kandioler D, Ludwig C, Zangemeister-Wittke U, Stahel R, Silencing of death receptor and caspase-8 expression in small cell lung carcinoma cell lines and tumors by DNA methylation, Cell Death & Differentiation 10(3) (2003) 356–364. 10.1038/sj.cdd.4401157 [DOI] [PubMed] [Google Scholar]
- [270].Campbell JE, Kuntz KW, Knutson SK, Warholic NM, Keilhack H, Wigle TJ, Raimondi A, Klaus CR, Rioux N, Yokoi A, Kawano S, Minoshima Y, Choi HW, Porter Scott M, Waters NJ, Smith JJ, Chesworth R, Moyer MP, Copeland RA, EPZ011989, A Potent, Orally-Available EZH2 Inhibitor with Robust in Vivo Activity, ACS Med Chem Lett 6(5) (2015) 491–5. 10.1021/acsmedchemlett.5b00037 [DOI] [PMC free article] [PubMed] [Google Scholar]