

Abstract
Abstract
The accumulation of massive data in the plethora of Cheminformatics databases has made the role of big data and artificial intelligence (AI) indispensable in drug design. This has necessitated the development of newer algorithms and architectures to mine these databases and fulfil the specific needs of various drug discovery processes such as virtual drug screening, de novo molecule design and discovery in this big data era. The development of deep learning neural networks and their variants with the corresponding increase in chemical data has resulted in a paradigm shift in information mining pertaining to the chemical space. The present review summarizes the role of big data and AI techniques currently being implemented to satisfy the ever-increasing research demands in drug discovery pipelines.
Graphic abstract
Keywords: Artificial intelligence, Big data, Drug discovery, Machine learning, Deep learning, Autoencoders
Introduction
The advancements in technologies coupled with reducing instrumentation cost have resulted in increased data generation in both quantity and diversity, leading to numerous data resources [1]. Big data comprises this collection of data of enormous volume and complexity. The drastic increment of data has resulted in this data’s availability across varied platforms, in public and commercial resources [2]. The resulting data-centric environment has mandated the acquisition, integration and analysis of big data to decipher complex medical and scientific problems. This gigantic complex data mining to uncover the underlying meaningful hidden patterns is equally significant and is referred to as big data analytics [3]. In the modern era, the emergence of big data has revolutionized the process and strategies to tackle drug development [4]. It has also facilitated and accelerated the translation of basic research discoveries into clinical practice and transformed the process of conventional drug discovery to a data-driven approach [4–6]. The availability of data-rich resources has encouraged the exploitation of artificial intelligence (AI) that mimics human intelligence to solve multifaceted challenges in the drug discovery process, from design and identification of novel drug molecules, drug repurposing, testing and clinical trial to personalized medicine [7–10]. Thus, AI applications related to big data analytics in the pharmaceutical space are witnessing a constant interest in making the multipronged approach of the multifaceted drug development process more promising and less time-consuming. However, some hurdles still need to be overcome despite numerous advancements, leaving sufficient room for further data-driven AI-led innovations [11].
The evolution of big data and artificial intelligence has reformed the strategies adopted to shorten the drug development process. The artificial intelligence approach has enabled the development of drug candidates in a more structured and economical manner and within a considerably shorter time period. The computational resources and algorithms in the drug discovery process utilize existing data to provide better analytics and assessment, from identifying a drug candidate to the pharmaceutical industry’s manufacturing process [11–13]. Hence, prior to the synthesis and experimental evaluation of the drug molecule, the AI-driven analysis facilitates identifying and screening the drug candidates against the desired disease effectively and efficiently.
Presently, AI is a rapidly evolving field that involves various domains, such as reasoning, knowledge representation, and machine learning (ML). Machine learning has been widely implemented for numerous drug discovery applications pertaining to large data sets. It uses various algorithms and techniques to recognize templates and patterns within the given data set [14]. Its primary application in drug designing is to identify and exploit the relationship between the chemical structure and their biological activities, referred to as the structure–activity relationship (SAR). The advent of massive sequencing approaches like next-generation sequencing (NGS) has resulted in the exponential growth of sequences, thus identifying potential fruitful putative novel drug targets [15]. Machine learning (ML) approaches have contributed significantly to drug target prediction from the available large-scale data sources. ML methods have been classified under two broad subcategories, supervised learning and unsupervised learning methods. The prominent algorithms in drug discovery applications are random forest (RF), support vector machine (SVM), gradient boosted machine with trees (GBM), elastic net regulation (EN), deep learning (DL), and deep neural network (DNN) [16, 17]. The continuous increment in data and limitations within the ML approaches has led to the emergence of deep learning (DL) methodology, a subfield of machine learning that uses the power of artificial neural network (ANN) [7]. The quantitative structure–activity relationship (QSAR) methods widely used in drug design are regression models used to predict the biological activity of the chemical compounds. Increasingly, ANN methods are now being frequently utilized in the pharmaceutical space for drug designing by parameterizing the QSAR model nonlinearly. The basic concept of ANN is to mimic the functioning of electrical impulses generated by neurons in the human brain. This is achieved by computing units referred to as ‘perceptrons’ which are interconnected like the neurons in the brain and possess self-learning capabilities [18]. The artificial perceptrons in ANN constitute a set of nodes required for data input and output to solve biological problems. It is commonly used in drug discovery to resolve the complexity of screening compounds and to estimate the pharmacokinetics and pharmacodynamics parameters [19]. Other types of ANN include multilayer perceptron networks (MLP), recurrent neural networks (RNNs), convolutional neural network (CNNs) and autoencoders, which use either supervised or unsupervised learning methods [20]. The advancement of ANN, called deep neural network (DNN), is now gaining attention for its successful application in drug discovery-related areas such as, to generate novel molecules, predicting the biological activity as well as the absorption, distribution, metabolism, excretion and toxicity (ADMET) properties of the drug candidate molecules. Like the ML approach, deep learning method was found to be effective in building the QSAR/QSAP models [21].
In this review, the emphasis is on the role of big data and artificial intelligence in the area of drug design. It attempts to provide a current conceptual framework and “state-of-the-art” snapshot of this domain. Several ML architectures, including the supervised and unsupervised methods and their application in small molecule drug discovery, have also been emphasized. Various other articles available in the public domain have focused either on the role of machine learning [14] or deep learning [22–24] methods, while some have discussed the big data resources in the drug discovery [10, 11, 23, 25]. However, there is currently no single review paper that has covered all these aspects of drug design, from the big data resources to an overview and explanation of the development of the implemented algorithms. This review attempts to fill these lacunae and presents in a nutshell how these algorithms were developed and implemented to uplift the drug discovery process in the modern AI era. Thus, this review comprises an insight into the deployment of big data resources in the modern ‘big data’ era by engaging advanced AI algorithms and providing an integrated, synthesized summary of the current state of knowledge regarding machine learning and big data in drug discovery.
Advent of AI in drug design
Drug discovery is a complex and lengthy venture which requires a multidisciplinary approach. A drug molecule to reach the market passes through multiple defined stages, wherein each step has its challenges, timeline and cost. Despite numerous advancements in the understanding of biological systems, identifying a novel drug molecule for therapeutic purposes still remains largely a lengthy, costly and complicated process [26]. The human genome project (HGP) has facilitated several advancements in drug development, including precision medicine and target identification for a disease. Compared to the traditional approach, both in vitro and in silico methods have a greater propensity to lower drug discovery costs. These computational approaches in the early stages of drug development also minimize the time span to distinguish a drug candidate with suitable therapeutic effects by excluding compounds exhibiting complex side effects. The modern drug discovery pipelines integrate hierarchical steps that engage various phases such as target identification, target validation, screening of lead candidates against the desired target, optimization of identified hits to increase the affinity, selectivity, metabolic stability, and oral bioavailability. Once a lead molecule is recognized and evaluated, it undergoes preclinical and clinical trials. Finally, the identified molecule that complies with all these investigations moves forward for approval as a drug.
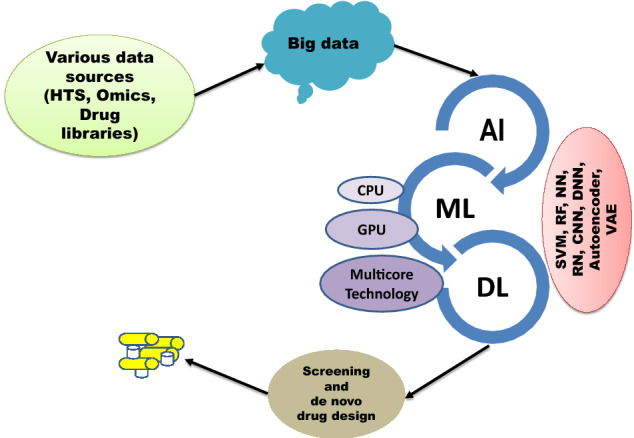
The advancements over time in computational chemistry and high throughput screening (HTS) strategies have fast-tracked the prompt screening of millions of compounds against the specific identified drug targets. These techniques produce a large quantity of biological data accumulated in the databases and public repositories. The generation of massive data due to the advancement in technology for drug and drug candidates has shifted the modern drug discovery approaches towards the big data era. Previously, big data analytics was widely used in information technology, but nowadays, with the available large-scale data, it has been frequently implemented in all the engineering and science domains, including drug discovery. Data mining of this complex and heterogeneous data across many resources is highly crucial. This has resulted in big data-related novel computational tools and algorithms for its curation and management and put forth challenges and opportunities for the research communities [27]. Moreover, advancements in high computing facilities, together with the emergence of artificial intelligence (AI) and machine learning (ML) algorithms play a prominent part in computer-aided drug design technology to screen and mine the lead-like molecules against the desired target more efficaciously with reduced cost and time (Fig. 1) [19].
Fig. 1.

Growth of machine learning with the subsequent increase in big data and computation power; KB—Kilobyte, MB—Megabyte, CPU—Central processing unit, GPU—Graphics processing unit, HTS—High throughput sequencing
Currently, there exist several opportunities to apply both AI and ML associated with big data in drug discovery applications, such as protein folding prediction, protein–protein interaction, virtual screening, QSAR, de novo drug designing and drug repurposing. Several approaches like high throughput virtual screening (HTVS), molecular docking, pharmacophore modelling, QSAR and molecular dynamics simulation are widely used for drug discovery [28]. Computer-based drug discovery implements virtual screening (VS) as the primary method to filter out novel small molecules from large compound libraries against the desired target for therapeutic effect in the early phase of drug discovery [29]. It also helps to determine the novel scaffolds for further optimization of the hit molecules. Computer-based drug discovery can be broadly classified into structure-based drug discovery (SBDD) and ligand-based drug discovery (LBDD). In structure-based drug discovery, the target structure is used to identify a potent drug molecule against a particular disease, whereas the ligand-based drug discovery is an effective method based on the structural knowledge of chemical scaffolds to design compounds with improved biological activity. The pharmacophore modelling method is used in both the structure-based and ligand-based drug discovery approach, while molecular docking, and molecular dynamics (MD) simulation studies are extensively used in structure-based drug discovery. In contrast, scaffold hopping and QSAR are the widely used methods for ligand-based drug discovery. [19, 30].
Similar to computer-based drug design, virtual screening methods also fall under two broad categories depending on the available structural information: structure-based virtual screening (SBVS) and ligand-based virtual screening (LBVS). Structure-based virtual screening (SBVS) explores the interaction between the ligand molecule and binding site residues. In contrast, the ligand-based virtual screening (LBVS) method uses the chemical similarity approach to identify a drug molecule. However, both are an integral part of the drug design method and have their merits and demerits. Structure-based virtual screening is widely used when the structure of the target is known, and it exploits the information gleaned from protein–ligand interaction during the docking study through the scoring function analysis to identify the potent drug molecules against the desired target. Whereas, the ligand-based virtual screening method is not generally based on the availability of the target structure but on the chemical similarity approach to identify the drug candidate and hence may be biased towards the reference scaffold. The exponential increase in structural and protein–ligand binding data has necessitated engaging AI methods to deduce these interactions to enable further development of SBVS. ML-based methods such as support vector machine (SVM), random forest (RF) and boosting help us to establish the nonlinear dependence of molecular interactions between the ligand and target [31]. Loss of relevant information during feature extraction in ML can be solved through the deep learning (DL)-based approach. Deep learning methods permit automatic generation of higher level hierarchical abstractions from big data that can be used as features, thus reducing the dependency for feature generation in ML. Another type of DL, the convolutional neural network (CNN), has been notably adapted for virtual screening as it implements feature extraction based on small sections of the input image referred to as receptive fields. DeepVS is a deep learning-based programme that utilizes CNN methodology for screening compounds against the desired target [32]. PTPD another tool based on CNN, has been developed for designing peptide-based molecules [33].
Ligand-based virtual screening depends on the data set of ligands which are further classified into the active and inactive set for classification and regression purposes to predict the activity of the compounds. Based on the physicochemical analysis and spatial similarities between the active ligands, it identifies and predicts other ligand molecules with higher bioactivities. This method predicts the active ligand when the target structure is missing or structural accuracy is low for the known targets. Like structure-based drug design, the adoption of machine learning methods in ligand-based drug designing leads to an improved rate of predicted hits by minimizing the rate of false hit prediction [34]. With the ever-increasing data size and number of active compounds in the chemical space, the development of ML algorithms has become indispensable to handle the big data sets without compromising speed and accuracy. The limitations in addressing the large data set were overcome by the emergence of deep learning (DL) methods that could efficiently manage large data sets [35]. Deep learning is a sub-branch of machine learning. It emphasizes on the neural network with multiple layers of the perceptron, which help in learning data with multiple layers of abstraction that are beneficial for supervised and unsupervised learning [36]. Recent progress in computational power to comprehend big data and convert it for reusable knowledge gain has further boosted AI in the drug design process [37]. The popular deep learning-based libraries such as Tensorflow and PyTorch are widely used to screen big data for drug discovery applications.
Big data resources in drug design
The large-scale data exists in diverse forms and data types which can be raw or processed, standardized or unstandardized. The extraction of meaningful information from this heterogeneous data is a challenging task. The drug discovery process relies on data from several disciplines such as clinical data, bioassay, pharmacological and structural biology. These data generated from distinct domains and sources encompass a divergent array of large data sets where artificial intelligence plays a significant role in solving the complexity present in the data [38]. The continuous incrementation in big data requires greater computational resources and advanced computational algorithms to analyse the resulting complex data. The demand for enhanced computational power has resulted in a paradigm shift from personal computers to high-performance computing, cloud computing, and graphical processing units (GPUs) to analyse big data [3]. The accumulated big data utilized for drug discovery can be classified into various categories or databases such as a collection of chemical compounds (e.g. PubChem, ChEMBL), drug/drug-like compounds (e.g. Drugbank, e-Drug3D), collection of drug targets, including the genomic and proteomic data (e.g. Binding DB, Supertarget), databases containing the collection of assay screening, metabolism and efficacy studies (e.g. HMDB, TTD) (Table 1). Over the years, several data-sharing projects have been initiated parallel to the development of high throughput screening (HTS) techniques [39].
Table 1.
Data sources used in drug discovery
| S. no. | Database | Url link | Data type | Data size (as on 15 March 2021) | References |
|---|---|---|---|---|---|
| Chemical collections | |||||
| 1 | ChEMBL | https://www.ebi.ac.uk/chembl/ | Chemical database containing bioactive and drug-like molecules | 1,800,000 compounds | [49] |
| 2 | PubChem | https://pubchem.ncbi.nlm.nih.gov/ | Chemical database containing chemicals and their activity against biological targets | 110 Million Compounds, 271 Million Substance, 297 Million Bioactivities | [50] |
| 3 | Small molecule pathway database (SMPDB) | https://smpdb.ca/ | Database containing small molecule pathway of humans | 48,690 pathways | [51] |
| 4 | ZINC | http://zinc.docking.org/ | Database containing curated chemical compounds | > 750 Million compounds | [52] |
| 5 | Human metabolome database (HMDB) | https://hmdb.ca/ |
Database containing 1) Chemical data, 2) clinical data, and 3) molecular biology/biochemistry data |
114,304 metabolite entries | [53] |
| 6 | Binding database (BindingDB) | https://www.bindingdb.org/bind/index.jsp | Database containing small molecules binding affinity data and protein targets | 2,240,573 binding data for 8503 protein targets, 971,073 small molecules | [54] |
| Drug/Drug-like compound | |||||
| 1 | DrugBank | https://go.drugbank.com/ | Database containing information about drug and drug targets | 13,441 drugs | [55] |
| 2 | Drugs@FDA database | https://www.accessdata.fda.gov/scripts/cder/daf/ | Database containing FDA approved drug molecules | 1600 FDA approved drugs | [56] |
| 3 | Drug central | https://drugcentral.org/ | Database containing active chemical entities, pharmaceutical product and drug mode of action | 4642 drugs, 110,577 pharmaceutical products | [57] |
| Drug Targets | |||||
| 1 | Supertarget | https://bioinformatics.charite.de/supertarget/ | Database containing drug target information | 3,32,828 drug target interaction | [58] |
| 2 | Ligand Depot | http://ligand-depot.rutgers.edu/ | Database containing information of chemical and structural information about small molecules | 30,480 ligand entry | [59] |
| 3 | BioGRID | https://thebiogrid.org/ | Database information about protein, genetic and chemical interactions | 2,015,809 protein and genetic interactions, 29,093 chemical interactions and 1,017,123 post-translational modifications | [60] |
| Protein–Protein Interaction | |||||
| 1 | Database of interacting proteins (DIP) | https://dip.doe-mbi.ucla.edu/dip/Main.cgi | Database containing information of protein–protein interaction | 40,678 interaction information | [61] |
| 2 | Therapeutic target database (TTD) | http://db.idrblab.net/ttd/ | Database containing information of known therapeutic protein and nucleic acid targets | 2458 protein target, 5059 patented drugs | [62] |
| 3 | Potential drug target database (PDTD) | http://crdd.osdd.net/pmics.php | Database containing information about drug target with known 3d structure | 1100 entries covering > 800 known potential drug targets | [63] |
| Efficacy and assay screening | |||||
| 1 | BioCyc | https://biocyc.org/ | Database containing information of organism specific Pathway/ Genome Databases | 18,030 Pathway/Genome information | [64] |
| 2 | BRENDA | https://www.brenda-enzymes.org/ | Database containing information of enzyme function data | 84,000 Enzyme data | [65] |
| 3 | Reactome | https://reactome.org/ | A curated database containing information of biological pathways, including the metabolic, protein trafficking and signalling pathways | > 9600 proteins, 9800 reactions and 2000 pathways for humans | [66] |
| 4 | KEGG | https://www.genome.jp/kegg/ | Database containing information of genomic, chemical and functional information | 18,778 chemical compound metabolite, 7062 Genome information, 1312 Network information | [67] |
| Drug toxicity prediction | |||||
| 1 | Comparative toxicogenomics database | http://ctdbase.org/ | Database containing information about environmental exposures to human health | 2.7 million manually curated chemical gene, chemical phenotype, chemical disease, gene-disease and chemical exposure interactions | [68] |
| 2 | TOXNE | https://www.nlm.nih.gov/toxnet/index.html | Database containing hazardous substance data | 5800 chemical substance | [69] |
| 3 | DrugMatrix | https://ntp.niehs.nih.gov/data/drugmatrix/ | Database of toxicogenomic reference resources | 600 drug molecules and 10,000 genes | [70] |
Big data is required at different stages of the drug discovery process. The initial step in the drug discovery process involves the screening of gigantic libraries containing chemical compounds to wean out probable lead drug candidates. The chemical compound library space is enormous and comprises both virtual, designed, and synthesized compounds with descriptions of their properties and distribution sourced across both public and subscribed databases. Thus, these data sources are massive and provide a range of multidimensional data for drug discovery and development, including the chemical structure, chemical assay, target structure, clinical data. The quantity and mass of these data resources are expanding exponentially with time, unlocking avenues to exploit artificial intelligence and machine learning for rapid and effective drug discovery solutions.
Feature/descriptor representation
Most machine learning algorithms cannot use the protein sequence information or molecular structure information directly from the databases. The protein sequences and molecular structures need to be transformed through mathematical equations before they can be handled by machine learning algorithms. The protein sequence-based features like physicochemical properties, amino acid composition, dipeptide composition, pseudo-amino acid composition (captures long range sequence correlation) and amino acid distribution, exploit numerical techniques to convert these variable length protein sequences into fixed length feature vectors for input to machine learning algorithms. Similarly, numerical features consisting of 1D (molecular weight etc.), 2D (molecular fingerprints etc.) and 3D (volume etc.) descriptors are calculated for the molecules to make them suitable for machine learning-based analytics (Table 2). Simplified molecular-input line-entry system (SMILES) and strings are some of the commonly utilized molecular representations or notations. With an increase in the dimensionality of the descriptor class, information content about the descriptors is also expanding. Several software resources like Open Babel [40], PaDEL [41], Dragon [42], MOE [43], PeptiDesCalculator [44], AlvaDes [45], QuBiLS-MAS [46] are currently available which can calculate a wide set of different descriptors (OD/1D/2D/3D) from the SMILES format or 2D structure of the chemical compounds.
Table 2.
Different classes of descriptors with their examples
| S. no. | Descriptor class | Property of particular class of descriptors |
|---|---|---|
| 1 | 0D or count descriptors | Atom counts, bond counts, molecular weight |
| 2 | 1D or fingerprints | Molecular weight |
| 3 | 2D or topological descriptors | Atom and bond count, connectivity between atoms, Pharmacophore features, adjacency and distance matrix, molecular fingerprint |
| 4 | 3D or geometrical descriptors | Potential energy, surface area, volume and shape, conformational charge |
Artificial intelligence methods and their role in drug discovery
Artificial intelligence (AI) can explore and sort through available data, recognize and learn patterns from the input unstructured/structured data to extract gainful insights from the input data. AI can be classified into different categories such as reasoning and problem solving, representation of knowledge, planning and social intelligence, perception, machine learning, robotics and natural language processing (NLP) [47]. General intelligence remains amongst the long-term goals of AI. The various tools exploited in AI include statistical methods, computational intelligence, optimization, logic, methods based on probability and related methods to solve problems of interdisciplinary areas such as, computer science, mathematics, psychology, linguistics, drug discovery, and neuroscience. Speech recognition technology has also been empowered by the use of AI to automate transcription service. In speech recognition, AI enables us to convert the voice message into text and aids individual recognition based on their voice command.
On the other hand, NLP enables us to understand the natural human language and categorize it into different subsets such as classification, machine translational, and text generation based on their utility. The popular examples of NLP currently widely accepted are virtual assistants like Google assist, Siri and Alexa [48]. Machine learning (ML) and deep learning (DL) are the subsets of AI technology and are extensively used for prediction and classification purposes. ML algorithms recognize patterns from the data set for further classification [14]. DL, a subfield of machine learning, deploys artificial neural networks (ANNs) for different tasks. Adopting AI for solving data-intensive processes has opened up newer possibilities in the drug design space [7]. AI has, thus, revolutionized and accelerated rational drug designing from machine learning and finally to deep learning in the present big data era.
Artificial intelligence methods: advantages and pitfalls
Machine learning
Machine learning methods can be defined as a set of algorithms that do not require human intervention and explicit instructions for learning [71]. Big data has opened immense opportunities for machine learning methods to be developed specifically to handle the four V’s: Volume, Variety, Velocity and Veracity, and mine interesting patterns [72]. Big data’s sheer size or volume presents several challenges for traditional machine learning algorithms, such as processing time and memory requirement [73]. The second ‘V’, variety, comprises different forms/structures of data that can be unstructured, semi-structured, or structured. Velocity refers to the speed/ frequency with which the incoming data needs to be processed. Veracity concerns the trustworthiness/ reliability of the data. Machine learning algorithms are generally employed for classification and regression tasks. In the former case, the objective is to discriminate between two or more classes (binary and multiclass classification problems). In contrast, the problem of regression involves predicting a real-valued quantity or variable [74]. The typical steps for implementing machine learning-based prediction methods consist of data preprocessing, model learning, and evaluation. The data preprocessing steps comprise preparing the data suitable for the various machine learning algorithms, such as discretization and standardization. The model learning phase constitutes the actual implementation of the machine learning algorithms. The final phase involves performance evaluation methods and metrics to assess the numerous trained machine learning models (Fig. 2).
Fig. 2.

Workflow of machine learning (ML) process in drug discovery
Big data also presents a challenge in evaluating the imbalanced distribution of available data [75, 76]. The dataset is imbalanced when the instances for a particular class overwhelms the instances of other class/classes with its sheer number [77, 78]. When the dataset is imbalanced, the accuracy of the learned model tends to shift towards the majority class compared to the minority class resulting in majority class classifiers [79, 80]. The models trained on imbalanced datasets are biased towards predicting the majority class over the minority class (which is often the class of interest). To diminish the effects of imbalanced datasets, generally, two types of approaches are undertaken: (i) changes at the algorithm level to make them suitable for handling the imbalanced datasets, (ii) Resampling methods: which are non-algorithm specific and consist of different types of sampling methods. Random undersampling concerns balancing the majority and minority class instances and involves the random removal of a percentage of majority class instances. Since this engages random deletion of instances, it can lead to bias and drop of information triggering loss of unique instances. To mitigate the shortcomings of random undersampling, K-means clustering based sampling and Kennard stone sampling is exploited [80]. Other variants of under sampling in practice include cluster centroid-based, K-nearest neighbour-based, etc. [81]. Another approach to handle imbalanced datasets is oversampling. Random oversampling may result in sample redundancy as there is ample chance that similar instances are replicated during balancing. Random oversampling is just the reverse of random undersampling, where a fixed proportion of minority class samples are randomly replicated. The outcome is duplication of samples culminating in redundant information. These methods are significant for clinical research in drug sampling and drug epidemiology. SMOTE [82] and its variants, such as borderline-SMOTE, SVM-SMOTE [83], present an effective way of balancing without much bias [84]. K-means, along with SMOTE, further reduces the bias. SMOTE is a nearest neighbour-based method that uses a predefined number of neighbouring minority samples to interpolate a new synthetic minority sample [77].
Deep learning
The rise of deep learning neural networks (DLNN) has revolutionized the analysis of big data. DLNNs have greatly benefitted from using ReLu activation function to avoid the vanishing gradient problems, which have plagued the shallow neural networks since their inception. They consist of an input layer, an output layer and more than two hidden layers in their architecture [85]. As the number of hidden layers increases, the network’s capability to extract more and more features enhances. Hence, the complexity of the features to be extracted is directly proportional to the number of hidden layers. The successful training of DLNNs usually requires vast amount of data as the number of parameters is quite large (e.g. every weight associated with each connection between the neurons in the network can be considered as a parameter). It has been observed that using a small amount of data for training results in suboptimal trained networks. Apart from parameters learned during the training process, some hyperparameters are also to be considered for optimal training of a DLNN [86]. Hyperparameters are crucial as they decide how the network is trained and significantly impact the model’s performance. These are also referred to as the ‘tuning parameters’ as some of them are iteratively fine-tuned using an appropriate algorithm. In DLNN, the number of layers, number of neurons per layer, activation function are some of the common hyperparameters [87]. Optimal hyperparameter setting changes with each dataset, as they are tuned for the individual datasets. When DLNNs are trained with a stochastic gradient descent algorithm, the network weights are updated depending on the learning rate (a hyperparameter). A large learning rate results in faster training of the model but may result in suboptimal solutions, while a smaller learning rate results in slow training of the network. A suitable learning rate results in the best approximate solution depending on the predefined number of training epochs. For obtaining the optimal set of hyperparameters, random search along a grid is often used. Overfitting occurs when the learning algorithm learns the minute details of the dataset instead of generalization. The accuracy of DLNNs can be improved by employing regularization parameters, such as L1 (lasso regression) and L2 (ridge regression) regression models, which help to avoid overfitting. Regularization imposes higher penalty on complex models as compared to simpler models but not at the cost of reduction in predictive performance. L1 regularization adds the absolute value of the coefficient and results in shrinking the less significant feature’s coefficients to zero and facilitates feature selection since features with zero coefficients can be removed from the model.
Loss function with L1 regularization can be given by Eq. (1)
| 1 |
where y = true value; = predicted value; λ = parameter governing the magnitude of penalty applicable to the model; n = number of features; βi = model coefficient.
In contrast, L2 regularization utilizes the square magnitude of the feature’s coefficients and results in shrinking coefficients evenly. It prevents overfitting of data and is especially useful in cases where collinear features are present.
Loss function with L2 regularization can be given by Eq. (2)
| 2 |
where y = true value; = predicted value; λ = parameter governing the magnitude of penalty applicable to the model; n = number of features; βi = model coefficient.
Dropout has also proved to be an important technique in reducing the effect of overfitting [88]. Dropout involves the random deletion of a specified percentage of neurons and their connections in different deep network layers. This results in making the network more robust to memorization and increases generalization (Fig. 3).
Fig. 3.

a Deep learning neural network (DLNN) without dropout b Deep learning neural network (DLNN) with dropout
Deep learning variants
Generative adversarial networks (GANs) are combination of two competing neural networks; a generative network and a discriminator network. The purpose of the discriminator network is to classify and distinguish the real data from the fake data. The generative network produces the fake data by using feedback from the discriminator which is trained on real labelled data (i.e. consisting of class information). The iterative procedure of optimizing fake data to resemble the real data by the generative network and its discrimination by the discriminative network continues until local Nash equilibrium is attained, at which there is no further reduction in the cost of both generator and the discriminator [89]. Many novel applications of GANs in cheminformatics and computer-aided drug design have emerged recently [90]. The modification of GANs such as conditional GAN [91] and Wasserstein GAN [92] have proved to be very useful in various tasks such as novel molecule design (Fig. 4) [93, 94] and for optimization of molecules with desired properties [95, 96].
Fig. 4.

De novo chemical design using generative adversarial networks (GANs)
Convolutional neural networks (CNN) (such as VGGNet, VGG19) [97] are variants of DLNNs, which are mainly used for computer vision and image classification. CNNs consist of three components—convolution layer, pooling layer and the fully connected layer. The convolution layer is involved with recognizing the colour and edges of an image and results in the generation of activation maps. The pooling layer reduces the spatial dimension of the activation maps, and the fully connected network executes the image classification. A different variant of CNNs such as Inception [98] and ResNet are considered state of the art in computer vision/ image classification [99]. High-accuracy CNN models have been implemented for the diagnosis of diseases such as cancer [100]. Recently, CNNs are being trained to mine protein–ligand interactions [101, 102], text mining [103] and toxicity prediction of compounds from their graphic images [104]. Recurrent neural networks (RNN) [105] can model sequential information. Long short-term memory (LSTM) units are primarily used for constructing the RNNs [106]. They can also be used for generative purposes [35, 107]. The concept of multitask learning [108, 109] involves training a learning algorithm on similar tasks rather than on a single task, proving to be very effective in cheminformatics, such as toxicity prediction [110, 111].
Hybrid approaches like LSTM-GAN (long short-term memory–generative adversarial network), DCGAN (deep convolutional generative adversarial network), gcWGAN (guided conditional Wasserstein GAN), which are constructed using different deep learning paradigms, have been successfully used in de novo protein design [112, 113].
One major drawback of DLNNs is that they are like black boxes and do not interpret the decision-making/classification process. Recently, to mitigate the black box assumption, VIP (Variable Importance) charts and SHAP (SHapely Additive exPlanations) plots were introduced, which have lessened the black-box nature of DLNNs to some extent. SHAP is based on game theory and has been mainly adopted to deduce the importance of an individual feature and its distribution over the target variable [114, 115]. Platforms like H2O (https://www.h2o.ai/), TensorFlow [116], Keras (https://keras.io/) are being implemented to train DLNNs with big data. Traditional visualization methods may not be optimal for these enormous datasets, whereas newer methods such as t-sne can be exploited readily [117].
Autoencoder
Autoencoders are unsupervised neural networks that are trained to reproduce the input (reconstructed input) at its output nodes (Fig. 5). In between the hidden layers, the autoencoders transform the input into hierarchical higher-order representations (Fig. 6). As different attributes/features of the data present a different facet, therefore, it is not known in prior as to which feature /attribute will result in better training for a machine learning algorithm [118]. These higher-order representations can be used as features/attributes in the training of learning algorithms. Autoencoders are mainly used for dimensionality reduction and anomaly detection [119]. In relation to drug discovery, they have been mainly practised for dimensionality reduction of features for drug target interaction prediction [120], initialization of model parameters [121] and assessing drug similarities [122].
Fig. 5.
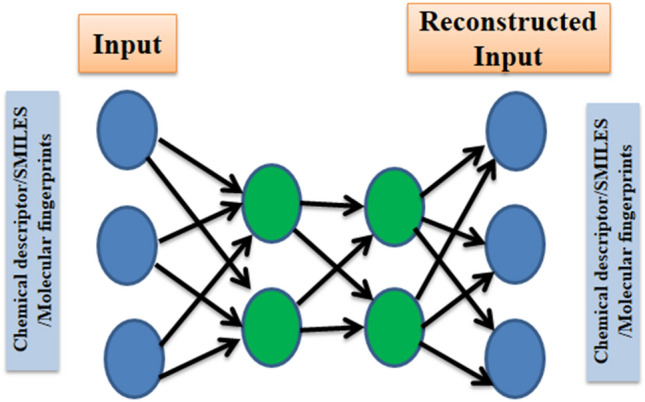
Representation of an autoencoder. The green circles represent the hidden layer
Fig. 6.
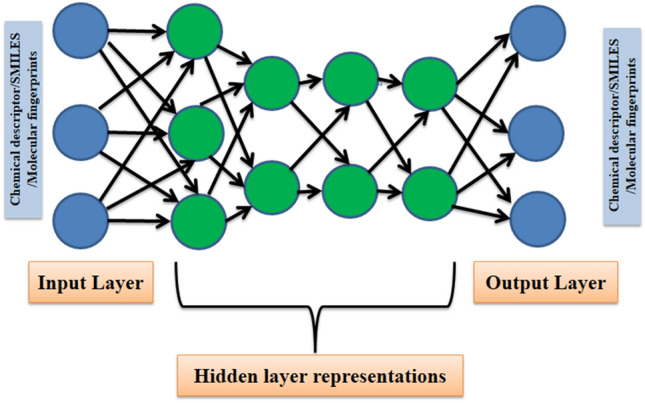
A deep autoencoder with hidden layers. The hierarchical representations from the hidden layers can be used as features in the training of learning algorithms
Ensemble learning
It is also possible to apply several different classifiers together for constructing the final classification and regression tasks. The ensemble learning approach [123] utilizes many different base classifiers in the initial phase and their decision fusion in the final stage. This provides a critical advantage as each base classifier’s deficiency can be possibly compensated by other different base classifiers. Stacking, StackingC, and Voted ensemble classifiers are most commonly used to construct ensemble classification systems [124]. In stacking, the first step involves training different base classifiers, and the second step consists of combining the outputs of the base classifiers using a metaclassifier (Fig. 7). Voted ensemble classifiers can be constructed by using the more popular majority voting scheme where the class is predicted based on the votes by different base classifiers. It can also be implemented using the average vote rule, maximum and minimum probability rule, and product probability rule [125]. The concept of ensemble learning has also been implemented for regression problems where instead of a discreet class, a real numbered value (target variable) is being predicted [126]. Two prominent ensemble learning algorithms are Bagging and AdaBoost employed for QSAR modelling.
Fig. 7.

Schematic representation of stacking ensemble approach
Deep belief networks
Deep belief networks (DBN) are generative graphical deep learning networks [13] that consist of restricted Boltzmann machines or autoencoders and are characterized by the absence of connections between units present in the same layer. They can be employed to train both in a supervised and unsupervised manner [127]. They have found definitive applications in virtual screening [128], multilabel classification of multi-target drugs [129] and in the classification of small molecules into drugs and non-drugs [130].
Performance evaluation metrics
The machine learning algorithms have to be assessed critically for their performance. The performance evaluation metrics such as accuracy, sensitivity, specificity, G-means are commonly used, which are calculated from the various quadrants of a confusion matrix (TP: true positives, TN: true negatives, FP: false positives, FN: false negatives) [131]. The evaluation of regression models is mainly assessed by determining the mean absolute error, mean squared error, and root-mean-squared error. The various evaluation parameters for measuring the performance of machine learning algorithms include:
Accuracy: This is the total number of all correct predictions out of the total number of samples as shown in Eq. (3).
| 3 |
Sensitivity: It is the percentage of the correctly predicted positive class represented by Eq. (4).
| 4 |
Specificity: It is the percentage of the correctly predicted negative class (Eq. (5)).
| 5 |
G-means: It is a very useful metric to gauge the machine learning model’s performance in class imbalance scenarios, as shown in Eq. (6).
| 6 |
F-score: It is also known as the F1 score and is defined as the harmonic mean of precision and recall, as given in Eq. (7).
| 7 |
Cohen’s Kappa (K): It is a quantitative measure of the reliability of two classifiers that classify the same thing and quantify the agreement between the classification outcomes (Eq. (8)). A score of 0 means there is an agreement due to chance alone, a score of 1 means complete agreement and a score below 0 means less agreement than expected due to chance alone
| 8 |
where P0: observed agreement; Pe: the expected probability of chance agreement.
Mean absolute error (MAE): It is defined as the absolute difference between the actual target value and the value predicted by the trained model. It is represented as Eq. (9).
| 9 |
where n: number of samples; Y: actual target value; : predicted target value.
Mean squared error (MSE): It is defined as the average of the squared differences between the actual target values and the predicted target values (Eq. (10)).
| 10 |
where n: number of samples; Y: actual target value; : predicted target value.
Root-mean-squared error (RMSE): It is defined as the square root of the average of the squared differences between the actual target values and the predicted target values. RMSE is used in cases where large errors are to be penalized as represented by Eq. (11).
| 11 |
where n: number of samples; Y: actual target value; : predicted target value.
Applications in drug discovery
Computer resources are essential for effective AI execution. Thus, the rise of high-performance computing clusters, development in graphics processing unit (GPU) power, cloud-based sources and accumulation of massive chemical informatics data have further augmented the evolution of artificial intelligence (AI) technology [132]. This technology has turned the drug discovery paradigm uphill and completely transformed the pharmaceutical space work culture. AI capitalizes on the predictive hypothesis from the available large data sets compared to the traditional trial and error approach for drug discovery [1]. Currently, R&D sectors of renowned pharmaceutical companies such as Pfizer, GlaxoSmithKline, Novartis, Merck, Sanofi, Genentech and Takeda are adopting machine learning and artificial intelligence to manage the enormous generated data to deliver cost-effective solutions. It is proposed that the market of AI-based drug discovery will reach $1.43bn dollars in 2024, with an annual enhancement of 40.8%. The increase in the number of cross-industry collaborations and partnerships to control the escalating drug discovery costs are major factors responsible for the rise of the AI market in drug discovery and development [24, 133].
The drug discovery approach encompasses various steps from target identification to the clinical phase. The recent breakthrough in AI technology and its incorporation has benefitted the various phases of drug discovery and the pharmaceutical industry. This technology provides innovative solutions in all aspects of the multifaceted drug discovery process such as, in the identification of drug targets, screening of lead compounds from data libraries, drug repurposing, predicting the toxicity of compounds, predicting bioactivity of compounds, de novo design and in automation of compound synthesis [134–136]. The different areas where AI has significantly contributed to the various stages of drug design are shown in Fig. 8.
Fig. 8.
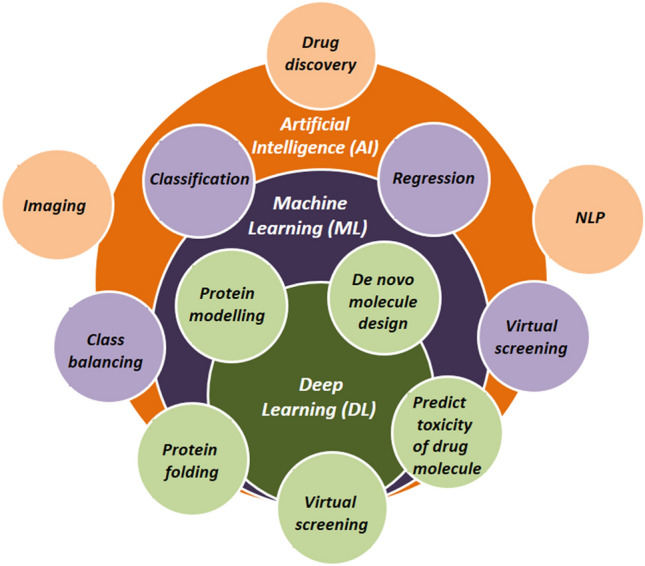
Role of AI technology in different phases of drug discovery
In structure-based drug discovery, a target is essential for the successful design of a drug molecule. Homology modelling and de novo protein design are the traditional methods for structure modelling. The emergence of AI technology has contributed enormously in predicting the 3D structure of the protein as well as in determining the effect of a compound on the designed target. Recurrent neural network (RNN) and deep neural network algorithms are widely exploited in target modelling studies. Alpha fold, an AI tool that relies on DNN, is widely used to predict the 3D structure from its primary sequence [137]. The feature extraction potential of deep learning makes it a promising method to predict the secondary structure, backbone torsional angle and residue contacts in protein. Thus, protein folding study can be determined from its sequences with the help of AI methods [138, 139]. DN-fold is another deep learning network method widely used for protein folding and can efficiently predict the structural fold of the protein [140]. With the growth of protein sequence data, AI methods also significantly contribute in predicting the protein–protein interaction studies by using the DNN called DeepPPI, which outperforms (prediction accuracy 80.82%) the traditional ML-based approach (prediction accuracy 65.80%), as the latter approach is faced with the problem of manual feature extraction [141].
Apart from protein modelling, AI has a role in drug screening, where it reduces the time to identify a drug-like compound. ML algorithms such as nearest neighbour classifiers, RF, extreme learning machines, SVMs, and DNNs are used for the drug molecule’s virtual screening and synthetic feasibility. ML-based drug screening has been successfully applied to identify drug-like molecules against various diseases such as cancer and neurogenerative disorders [142–144]. The incorporation of AI has opened up newer avenues and transformed the drug discovery process. AI and ML implementation has guided the exploration of low molecular weight compounds for their therapeutic potential. Zhavoronkov et al. performed a deep learning analysis to discover novel inhibitors of an enzyme, DDR1 kinase [145]. McCloskey et al. employed ML models like Graph CNN and RF to identify novel small drug-like molecules against three different proteins [146]. Small molecules were predicted against rheumatoid arthritis using an integrated approach of ML and DL [147]. Another study performed using an AI-based method identified the hepatotoxic ingredient from Chinese traditional medicine [148]. Predictive models have been developed for screening liver toxicity induced due to drugs using ML algorithms [110]. This technology has contributed to the current pandemic scenario to recognize drug-like molecules against the different SARS-CoV-2 targets. Numerous studies have been performed to identify potent lead molecules against the novel coronavirus using traditional medicine. Xu et al. used ML and molecular modelling to identify the inhibitors against 3CL proteinase [149]. The deep learning approach has also assisted in the identification of potential drug targets for SARS-CoV-2 [150]. Studies have also encompassed drug repurposing approaches against targets of novel coronavirus using AI methods [151, 152]. DL-based platform DeepDTA has been deployed on marketed antiviral drugs to predict possible therapeutic agents against COVID-19 [153].
Pharmaceutical industries, namely Bayer, Roche, and Pfizer, have collaborated with the IT companies to develop an AI-enabled platform for therapeutics discovery in areas such as immune-oncology and cardiovascular diseases [154]. Apart from drug screening, AI has considerably improved the scoring functions of docking methods to evaluate drug molecule binding affinity towards the target. ML-based approaches such as RF and SVM aided the development of scoring functions by effectively extracting the geometric, chemical and physical force field features. Due to the advancement of deep learning methods in image processing, CNN has been incorporated successfully to extract features from the protein–ligand image and predict protein–ligand binding affinity [155, 156]. DeepVS, a deep learning-based software used for molecular docking studies, is extensively employed over traditional docking programmes based on its scoring functions [32].
After identifying the hit or lead molecules in the drug discovery pipeline, a series of tests and evaluation studies are executed to assess the physicochemical and toxicity properties of the candidate drug molecule. Thus, early identification and weaning of drug candidates with poor physical and chemical properties reduce the failure rate during the drug discovery process [157]. The AI-based methods aid the execution of this process in a time-efficient manner from a large dataset to effectively predict the physicochemical properties of the compounds [158, 159]. Both ML and deep learning-based algorithms are employed in this process. Various tools based on CNN, deep neural network, RF are available, namely TargeTox [160], DeepTox [110], DeepNeuralnetQSAR [161], eToxPred [162], DeepDTA [163], GraphDTA [164], and DeepAffinity [165], which can afford the prediction of the toxicity and physicochemical properties of the compounds from the large compound libraries.
AI-based methods are comparatively more effective and widely used nowadays in de novo drug design and compound synthesis automation [166]. Established automated techniques such as solid phase are currently used to synthesize several compounds, including peptides and oligonucleotides. This method suffered from the lack of standardized digital automation to control the chemical reaction due to the absence of a suitable universal programming language. Thus, with the advancement of AI methodology, the deep learning approach has been incorporated to generate new chemical entities with its powerful learning capabilities. Deep neural network (DNN), reinforcement learning (RL), variational autoencoder (VAE) and multilayer perceptron (MLP) are currently adopted for de novo drug design and automation process [167, 168]. Chemputer is a recently developed platform that gives a detailed recipe for molecule synthesis and is exercised in compound synthesis automation. Three pharmaceutical compounds, diphenhydramine hydrochloride, rufinamide, and sildenafil, have been successfully automated through this method [169, 170]. The purity and yield of the synthesized compounds were comparable with or better than the manual synthesis. Thus, AI has moved forth in the pharmaceutical industry to automate and up scale the bench chemistry with an edge over the safety, efficacy and accessibility of the identified complex molecules.
AI has also contributed immensely to the various steps involved in clinical trial research. It can be deployed for remote surveillance to access real-time data with increased efficacy. AI can assist in decision-making for patient recruitment from a defined cohort, replanning patient treatment regime through patient response monitoring to a drug, determining patient dropout rate and the final efficacy of the drug [171]. BioXcel Therapeutics (https://www.bioxceltherapeutics.com/) have successfully identified BXCL701, a candidate molecule using AI technology that is effective against schizophrenia and bipolar disorder. BXCL701 is also currently in different phases of clinical trials against pancreatic cancer, for which it has obtained FDA approval [172]. Thus, conventional drug discovery concepts combined with advanced computational approaches provide an excellent platform for research and development to enhance the drug discovery and development process.
Available AI computational tools for drug design
The power of computer software in the area of drug design is evident from the initial stages of drug discovery. The advancement in software and its availability opens new opportunities for their application in research and learning processes. Open-source software has gained popularity due to its easy availability and accessibility. Many researchers also share their programmes on Github and other platforms to accelerate and permit widespread use of the drug discovery process through these AI resource (Table 3). Several open-source deep learning frameworks are also available for users, such as TensorFlow, Pytorch, Keras, scikit learn, MXNet, Gluon, Swift, and Chainer ONNX. These frameworks require high-performance computing resources across various platforms, including CPUs, GPUs, and tensor processing units (TPUs) [173]. The inbuilt libraries are based on the deep learning framework and are applicable in multiple areas of science and technology, including health care. TensorFlow, Pytorch, Keras, and Scikit-learn based on python-based libraries are widely used in drug discovery where large datasets are present. TensorFlow (TF) is a framework from Google that can be utilized to develop models to predict the molecular activity of the compound dataset using the deep learning approach. Keras is an advancement over TensorFlow and is user-friendly and easy to debug. Pytorch is also an open-source project used to define and train models to gain insight into the complex link between the drugs and accelerate the drug discovery process [174]. Scikit-learn presents an open-source, user-friendly platform for classification, regression, and dimensionality reduction purposes. Some softwares are also available such as Weka, which is extensively utilized for machine learning-based applications in drug discovery, classification and clustering purposes.
Table 3.
AI computational tools for drug design
| S. no. | Tools | Algorithm used | Url | References |
|---|---|---|---|---|
| 1 | AlphaFold | Predicts tertiary structure of a protein using deep neural network | https://deepmind.com/blog/alphafold | [175] |
| 2 | Chemputer | Give detailed recipe for compound synthesis | https://zenodo.org/record/1481731 | [170] |
| 3 | Conv_qsar_fast | Predict molecular properties based CNN method | https://github.com/connorcoley/conv_qsar_fast | [176] |
| 4 | Chemical VAE | Automated chemical design using variational autoencoder (VAE) | https://github.com/aspuru-guzik-group/chemical_vae | [177] |
| 5 | DeepChem | An open-source Python library uses a deep learning algorithm for compound identification | https://github.com/deepchem/deepchem | [23] |
| 6 | DeepTox | Predict the toxicity of chemical compounds using deep learning algorithm | www.bioinf.jku.at/research/DeepTox | [110] |
| 7 | DeepNeuralNetQSAR | Predict molecular activity using multilevel deep neural network (DNN) | https://github.com/Merck/DeepNeuralNet-QSAR | [161] |
| 8 | DeltaVina | Predict small molecule binding affinity with drug with a combination of random forest (RF) and AutoDock scoring function | https://github.com/chengwang88/deltavina | [178] |
| 9 | Hit Dexter | Predict frequent hitter by using machine learning (ML) algorithm | http://hitdexter2.zbh.uni-hamburg.de | [179] |
| 10 | InnerOuterRNN | Predicts the physical, chemical and biological properties using inner- and outer recursive neural networks | https://github.com/Chemoinformatics/InnerOuterRNN | [180] |
| 11 | JunctionTree VAE | De novo molecule design using junction tree variational autoencoder (VAE) | https://github.com/wengong-jin/icml18-jtnn | [181] |
| 12 | Neural graph fingerprint | Predict the property of novel compounds using CNN | https://github.com/HIPS/neural-fingerprint | [182] |
| 13 | NNScore | Predict the affinity of protein–ligand interaction using neural network-based scoring function | http://www.nbcr.net/software/nnscore | [183] |
| 14 | ORGANIC | De novo design of organic molecule and polymer using ML algorithm | https://github.com/aspuru-guzik-group/ORGANIC | [184] |
| 15 | Open Drug Discovery Toolkit (ODDT’s) | Chemoinformatics pipeline using random forest score (RF)-Score and NNScore | https://github.com/oddt/oddt | [185] |
| 16 | PotentialNet | Predict binding affinity using graph convolutional neural network (CNN) | https://pubs.acs.org/doi/full/10.1021/acscentsci.8b00507 | [186] |
| 17 | PPB2 | Predict the target of query molecule using nearest neighbour and machine learning algorithm | https://ppb2.gdb.tools/ | [187] |
| 18 | QML | Python toolkit for quantum machine learning | https://www.qmlcode.org/ | [188] |
| 19 | REINVENT | De novo design of molecule using RNN (recurrent neural network) and RL (reinforcement learning) | https://github.com/MarcusOlivecrona/REINVENT | [189] |
Apart from the freely available resources, some companies, namely, Janssen, AstraZeneca, Novartis, Sanofi, are currently exploring the potential of AI technology in the healthcare sector. They have collaborated with the software and data science companies namely IBM Watson, Microsoft, PointR data, Numerate, BenevolentAI, Atomwise which provide them support and a cloud-based/server to implement AI according to their requirement for research purposes in drug discovery against various diseases (Table 4).
Table 4.
Collaborations of AI organization with pharmaceutical companies
| S. no. | Company | Role of AI | Collaboration with the pharmaceutical company | Platform developed/Clinical trial candidates |
|---|---|---|---|---|
| 1 | Numerate | A platform for AI-based drug design against oncology and gastroenterology | Takeda | Drug candidate S48168 in Phase 1 clinical trial against Ryanodine receptor 2 |
| 2 | Numerate | A platform for AI-based drug design against oncology and gastroenterology | Servier | Drug development for oncology, gastroenterology and central nervous system disorders |
| 3 | Atomwise | A platform for AI-based structure modelling | Lilly | Drug candidate BBT-401 in Phase 2 clinical trial |
| 4 | Atomwise | A platform for AI-based structure modelling | Bridge Biotherapeutics | Expansion of Pellino Inhibitor Pipeline; BBT-401 in Phase-2a clinical trial |
| 5 | Benevolent AI | AI-enabled Judgement Augmented Cognition System (JACS) to develop novel clinical candidate against neurodegenerative diseases | Janssen | New range of drug molecules to be developed through this collaboration |
| 6 | Benevolent AI | AI enable platforms to develop novel clinical candidate against chronic kidney diseases | AstraZeneca | Drug candidate Placebo in Phase 2b clinical trial as a drug candidate for chronic kidney disease |
| 7 | Exscientia | A platform for AI-based drug discovery and lead optimization | Sanofi | Research in obsessive–compulsive disorder, Drug candidate DSP-1181 in Phase I clinical trial. Developed Centaur Chemist™ platform for AI-based drug discovery |
| 8 | IBM Watson Health | Provide a platform for clinical and health-related data research | Pfizer | Fast-tracking drug discovery research in immuno-oncology |
| 9 | IBM Watson Health | Provide a platform for clinical and health-related data research | Novartis | Real-time monitoring of patients to improve breast cancer patient outcome |
| 10 | Microsoft | A platform for image processing and cell and gene-based therapeutics | Novartis | Establishing an AI Innovation lab to transform the drug discovery process and its commercialization |
| 11 | Owkin | Provide a platform for a clinical trial based on ML technology | Roche | Developed Owkin’s Studio platform using AI technology |
| 12 | Sensyne health | A platform for clinical AI technology | Bayer | Developed Sensyne Health’s proprietary clinical AI technology platform |
| 13 | XtalPi | A platform for Target identification and validation based on QM and ML algorithm | Pfizer | Prediction and Optimization of crystalline forms of drug candidates for early drug screening |
| 14 | BioXcel therapeutics | A platform for the drug discovery application using AI technology | Pfizer | Drug candidate BXCL501-in Phase 3 clinical trial Drug candidate BXCL701-in Phase 2 clinical trial |
Challenges and future perspectives
The advent of faster and lower-cost technology coupled with development in computing power has accelerated the pace for data generation leading to several enormous compound data sources. This mandated implementing numerous artificial intelligence and machine learning approaches at various drug discovery stages to mine pharmaceutical knowledge from large-scale ‘big’ data. The knowledge gleaned from applying these AI algorithms in big data has provided a stimulus to design and discover novel molecules and their further optimization. This technique has helped push forward the drug discovery process by automating and customizing the process and affirming big data’s significance. The impact of artificial intelligence is gaining steadily in the academic sector and the pharmaceutical companies concomitantly with a surge of startups and AI-based R&D companies. Compared to the traditional high throughput screening methodology, an AI-based computational pipeline can screen virtual compound libraries rapidly to identify preclinical candidates. Besides drug screening, AI tools can be witnessed in different stages of the drug discovery cycle, such as predicting the physical properties, bioactivity, toxicity of the molecules, ADME properties, protein structure prediction and patient recruitment and surveillance.
Apart from the varied application of AI-based technology, some limitations and challenges still need to be overcome. The triumph of AI-based technology relies on the ease and frequency of data availability to the users. The multiple ‘V’ features of big data such as volume, velocity, variety, and volatility require improved data curation and management and user-friendly web portals. Thus, reliable and high-quality curated data is essential to glean insightful information. Though AI technology is slowly revolutionizing the drug discovery process through accelerated drug design methods and lower failure rates, the lack of adequate curated data and data accessibility can prove to be a hurdle. Other rate limitation steps include difficulty in the constant and expeditious updation of the available software as per the format of generated data and recently developed algorithm. Additionally, skilled personnel for the full-fledged operation of AI-based applications in drug discovery are not readily available. Despite the advances and popularity of machine learning approaches, some aspects still remains to be extensively explored, such as predicting conformational changes in protein and the binding affinity between the drug molecule and the target. Since deep learning requires massive data, this technique is limited only by the data extent and quality. Thus, rapid transfer of learning technology development can be a better approach to solving this problem. Although these advanced approaches displayed high prediction accuracy and performance, deep learning still works as a “black box” approach and its mechanism to solve the problem remains unclear. Moreover, though AI technology and gigantic data sources have contributed enormously to speeding up the drug design pipeline, experiments still need to be conducted before the drugs can be approved. Regardless of the limitations, AI has changed the landscape of drug discovery, and with its surging demand, it will soon become an essential, integral tool in the search for novel drugs and their targets and the pharmaceutical sector in the not too distant future.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Dash S, Shakyawar SK, Sharma M, Kaushik S. Big data in healthcare: management, analysis and future prospects. J Big Data. 2019;6:54. doi: 10.1186/s40537-019-0217-0. [DOI] [Google Scholar]
- 2.De Mauro A, Greco M, Grimaldi M. A formal definition of Big Data based on its essential features. Libr Rev. 2016;65:122–135. doi: 10.1108/LR-06-2015-0061. [DOI] [Google Scholar]
- 3.Sivarajah U, Kamal MM, Irani Z, Weerakkody V. Critical analysis of Big Data challenges and analytical methods. J Bus Res. 2017;70:263–286. doi: 10.1016/j.jbusres.2016.08.001. [DOI] [Google Scholar]
- 4.Alemayehu D, Berger ML. Big Data: transforming drug development and health policy decision making. Heal Serv Outcomes Res Methodol. 2016;16:92–102. doi: 10.1007/s10742-016-0144-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim RS, Goossens N, Hoshida Y. Use of big data in drug development for precision medicine. Expert Rev Precis Med drug Dev. 2016;1:245–253. doi: 10.1080/23808993.2016.1174062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qian T, Zhu S, Hoshida Y. Use of big data in drug development for precision medicine: an update. Expert Rev Precis Med drug Dev. 2019;4:189–200. doi: 10.1080/23808993.2019.1617632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Najafabadi MM, Villanustre F, Khoshgoftaar TM, et al. Deep learning applications and challenges in big data analytics. J Big Data. 2015 doi: 10.1186/s40537-014-0007-7. [DOI] [Google Scholar]
- 8.Schneider P, Walters WP, Plowright AT, et al. Rethinking drug design in the artificial intelligence era. Nat Rev Drug Discov. 2020;19:353–364. doi: 10.1038/s41573-019-0050-3. [DOI] [PubMed] [Google Scholar]
- 9.Kuang Z, Bao Y, Thomson J, et al. A machine-learning-based drug repurposing approach using baseline regularization. Methods Mol Biol. 2019;1903:255–267. doi: 10.1007/978-1-4939-8955-3_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang L, Ding J, Pan L, et al. Artificial intelligence facilitates drug design in the big data era. Chemom Intell Lab Syst. 2019 doi: 10.1016/j.chemolab.2019.103850. [DOI] [Google Scholar]
- 11.Zhao L, Ciallella HL, Aleksunes LM, Zhu H. Advancing computer-aided drug discovery (CADD) by big data and data-driven machine learning modeling. Drug Discov Today. 2020;25:1624–1638. doi: 10.1016/j.drudis.2020.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Réda C, Kaufmann E, Delahaye-Duriez A. Machine learning applications in drug development. Comput Struct Biotechnol J. 2020;18:241–252. doi: 10.1016/j.csbj.2019.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang B, Pan Z, Yin K, Khateeb A. Recent advances of deep learning in bioinformatics and computational biology. Front Genet. 2019 doi: 10.3389/fgene.2019.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vamathevan J, Clark D, Czodrowski P, et al. Applications of machine learning in drug discovery and development. Nat Rev Drug Discov. 2019;18:463–477. doi: 10.1038/s41573-019-0024-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qin D. Next-generation sequencing and its clinical application. Cancer Biol Med. 2019;16:4–10. doi: 10.20892/j.issn.2095-3941.2018.0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagarajan N, Yapp EKY, Le NQK, et al. Application of computational biology and artificial intelligence technologies in cancer precision drug discovery. Biomed Res Int. 2019 doi: 10.1155/2019/8427042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang X, Wang Y, Byrne R, et al. Concepts of artificial intelligence for computer-assisted drug discovery. Chem Rev. 2019;119:10520–10594. doi: 10.1021/acs.chemrev.8b00728. [DOI] [PubMed] [Google Scholar]
- 18.Abiodun OI, Jantan A, Omolara AE, et al. State-of-the-art in artificial neural network applications: a survey. Heliyon. 2018 doi: 10.1016/j.heliyon.2018.e00938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sliwoski G, Kothiwale S, Meiler J, Lowe EW., Jr Computational methods in drug discovery. Pharmacol Rev. 2013;66:334–395. doi: 10.1124/pr.112.007336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abiodun OI, Jantan A, Omolara AE, et al. Comprehensive review of artificial neural network applications to pattern recognition. IEEE Access. 2019;7:158820–158846. doi: 10.1109/ACCESS.2019.2945545. [DOI] [Google Scholar]
- 21.Jing Y, Bian Y, Hu Z, et al. Deep learning for drug design: an artificial intelligence paradigm for drug discovery in the Big Data era. AAPS J. 2018 doi: 10.1208/s12248-018-0210-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang L, Tan J, Han D, Zhu H. From machine learning to deep learning: progress in machine intelligence for rational drug discovery. Drug Discov Today. 2017;22:1680–1685. doi: 10.1016/j.drudis.2017.08.010. [DOI] [PubMed] [Google Scholar]
- 23.Zhu H. Big Data and artificial intelligence modeling for drug discovery. Ann Rev Pharmacol Toxicol. 2020;60:573–589. doi: 10.1146/annurev-pharmtox-010919-023324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paul D, Sanap G, Shenoy S, et al. Artificial intelligence in drug discovery and development. Drug Discov Today. 2021;26:80–93. doi: 10.1016/j.drudis.2020.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chan HCS, Shan H, Dahoun T, et al. Advancing drug discovery via artificial intelligence. Trends Pharmacol Sci. 2019 doi: 10.1016/j.tips.2019.07.013. [DOI] [PubMed] [Google Scholar]
- 26.Mohs RC, Greig NH. Drug discovery and development: role of basic biological research. Alzheimer’s Dement Transl Res Clin Interv. 2017;3:651–657. doi: 10.1016/j.trci.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roy A, McDonald PR, Sittampalam S, Chaguturu R. Open access high throughput drug discovery in the public domain: a Mount Everest in the making. Curr Pharm Biotechnol. 2010;11:764–778. doi: 10.2174/138920110792927757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vatansever S, Schlessinger A, Wacker D, et al. Artificial intelligence and machine learning-aided drug discovery in central nervous system diseases: State-of-the-arts and future directions. Med Res Rev. 2020 doi: 10.1002/med.21764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meng X-Y, Zhang H-X, Mezei M, Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des. 2011;7:146–157. doi: 10.2174/157340911795677602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hughes JP, Rees S, Kalindjian SB, Philpott KL. Principles of early drug discovery. Br J Pharmacol. 2011;162:1239–1249. doi: 10.1111/j.1476-5381.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schapire R. The boosting approach to machine learning: an overview. Nonlin Estim Classif Lect Notes Stat. 2002 doi: 10.1007/978-0-387-21579-2_9. [DOI] [Google Scholar]
- 32.Pereira JC, Caffarena ER, dos Santos CN. Boosting docking-based virtual screening with deep learning. J Chem Inf Model. 2016;56:2495–2506. doi: 10.1021/acs.jcim.6b00355. [DOI] [PubMed] [Google Scholar]
- 33.Wu C, Gao R, Zhang Y, De Marinis Y. PTPD: predicting therapeutic peptides by deep learning and word2vec. BMC Bioinform. 2019 doi: 10.1186/s12859-019-3006-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lionta E, Spyrou G, Vassilatis DK, Cournia Z. Structure-based virtual screening for drug discovery: principles, applications and recent advances. Curr Top Med Chem. 2014;14:1923–1938. doi: 10.2174/1568026614666140929124445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kell DB, Samanta S, Swainston N. Deep learning and generative methods in cheminformatics and chemical biology: navigating small molecule space intelligently. Biochem J. 2020;477:4559–4580. doi: 10.1042/bcj20200781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lundervold AS, Lundervold A. An overview of deep learning in medical imaging focusing on MRI. Z Med Phys. 2019;29:102–127. doi: 10.1016/j.zemedi.2018.11.002. [DOI] [PubMed] [Google Scholar]
- 37.Jiménez-Luna J, Grisoni F, Schneider G. Drug discovery with explainable artificial intelligence. Nat Mach Intell. 2020;2:573–584. doi: 10.1038/s42256-020-00236-4. [DOI] [Google Scholar]
- 38.de Souza A, Bittker JA, Lahr DL, et al. An overview of the challenges in designing, integrating, and delivering BARD: a public chemical-biology resource and query portal for multiple organizations, locations, and disciplines. J Biomol Screen. 2014;19:614–627. doi: 10.1177/1087057113517139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pereira DA, Williams JA. Origin and evolution of high throughput screening. Br J Pharmacol. 2007;152:53–61. doi: 10.1038/sj.bjp.0707373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Boyle NM, Banck M, James CA, et al. Open babel: an open chemical toolbox. J Cheminform. 2011 doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yap CW. PaDEL-descriptor: an open source software to calculate molecular descriptors and fingerprints. J Comput Chem. 2011;32:1466–1474. doi: 10.1002/jcc.21707. [DOI] [PubMed] [Google Scholar]
- 42.Moriwaki H, Tian Y-S, Kawashita N, Takagi T. Mordred: a molecular descriptor calculator. J Cheminform. 2018 doi: 10.1186/s13321-018-0258-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vilar S, Cozza G, Moro S. Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Curr Top Med Chem. 2008;8:1555–1572. doi: 10.2174/156802608786786624. [DOI] [PubMed] [Google Scholar]
- 44.Barigye SJ, Gómez-Ganau S, Serrano-Candelas E, Gozalbes R. PeptiDesCalculator: software for computation of peptide descriptors. Definition, implementation and case studies for 9 bioactivity endpoints. Proteins Struct Funct Bioinforma. 2021;89:174–184. doi: 10.1002/prot.26003. [DOI] [PubMed] [Google Scholar]
- 45.Mauri A (2020) alvaDesc: a tool to calculate and analyze molecular descriptors and fingerprints
- 46.Valdés-Martiní JR, Marrero-Ponce Y, García-Jacas CR, et al. QuBiLS-MAS, open source multi-platform software for atom- and bond-based topological (2D) and chiral (2.5D) algebraic molecular descriptors computations. J Cheminform. 2017 doi: 10.1186/s13321-017-0211-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tecuci G. Artificial intelligence. WIREs Comput Stat. 2012;4:168–180. doi: 10.1002/wics.200. [DOI] [Google Scholar]
- 48.Hoy MB. Alexa, Siri, Cortana, and more: an introduction to voice assistants. Med Ref Serv Q. 2018;37:81–88. doi: 10.1080/02763869.2018.1404391. [DOI] [PubMed] [Google Scholar]
- 49.Mendez D, Gaulton A, Bento AP, et al. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res. 2019;47:D930–D940. doi: 10.1093/nar/gky1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim S, Chen J, Cheng T, et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 2021;49:D1388–D1395. doi: 10.1093/nar/gkaa971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frolkis A, Knox C, Lim E, et al. SMPDB: the small molecule pathway database. Nucleic Acids Res. 2010;38:D480–D487. doi: 10.1093/nar/gkp1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Irwin JJ, Shoichet BK. ZINC–a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wishart DS, Tzur D, Knox C, et al. HMDB: the human metabolome database. Nucleic Acids Res. 2007;35:D521–D526. doi: 10.1093/nar/gkl923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gilson MK, Liu T, Baitaluk M, et al. BindingDB in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016;44:D1045–D1053. doi: 10.1093/nar/gkv1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wishart DS, Knox C, Guo AC, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668–D672. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sahdeo S, Tomilov A, Komachi K, et al. High-throughput screening of FDA-approved drugs using oxygen biosensor plates reveals secondary mitofunctional effects. Mitochondrion. 2014;17:116–125. doi: 10.1016/j.mito.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ursu O, Holmes J, Knockel J, et al. DrugCentral: online drug compendium. Nucleic Acids Res. 2017;45:D932–D939. doi: 10.1093/nar/gkw993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hecker N, Ahmed J, von Eichborn J, et al. SuperTarget goes quantitative: update on drug-target interactions. Nucleic Acids Res. 2012;40:D1113–D1117. doi: 10.1093/nar/gkr912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Feng Z, Chen L, Maddula H, et al. Ligand Depot: a data warehouse for ligands bound to macromolecules. Bioinformatics. 2004;20:2153–2155. doi: 10.1093/bioinformatics/bth214. [DOI] [PubMed] [Google Scholar]
- 60.Stark C, Breitkreutz B-J, Reguly T, et al. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006;34:D535–D539. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xenarios I, Rice DW, Salwinski L, et al. DIP: the database of interacting proteins. Nucleic Acids Res. 2000;28:289–291. doi: 10.1093/nar/28.1.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen X, Ji ZL, Chen YZ. TTD: therapeutic target database. Nucleic Acids Res. 2002;30:412–415. doi: 10.1093/nar/30.1.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gao Z, Li H, Zhang H, et al. PDTD: a web-accessible protein database for drug target identification. BMC Bioinformatics. 2008 doi: 10.1186/1471-2105-9-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Caspi R, Billington R, Fulcher CA, et al (2019) BioCyc: a genomic and metabolic web portal with multiple omics analytical tools. FASEB J 33:473.2–473.2 10.1096/fasebj.2019.33.1_supplement.473.2
- 65.Schomburg I, Chang A, Schomburg D. BRENDA, enzyme data and metabolic information. Nucleic Acids Res. 2002;30:47–49. doi: 10.1093/nar/30.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Croft D, O’Kelly G, Wu G, et al. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res. 2011;39:D691–D697. doi: 10.1093/nar/gkq1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mattingly CJ, Rosenstein MC, Davis AP, et al. The comparative toxicogenomics database: a cross-species resource for building chemical-gene interaction networks. Toxicol Sci. 2006;92:587–595. doi: 10.1093/toxsci/kfl008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fonger GC, Stroup D, Thomas PL, Wexler P. Toxnet: a computerized collection of toxicological and environmental health information. Toxicol Ind Health. 2000;16:4–6. doi: 10.1177/074823370001600101. [DOI] [PubMed] [Google Scholar]
- 70.Ganter B, Snyder RD, Halbert DN, Lee MD. Toxicogenomics in drug discovery and development: mechanistic analysis of compound/class-dependent effects using the DrugMatrix® database. Pharmacogenomics. 2006;7:1025–1044. doi: 10.2217/14622416.7.7.1025. [DOI] [PubMed] [Google Scholar]
- 71.Koza JR, Bennett FH, Andre D, Keane MA. Automated design of both the topology and sizing of analog electrical circuits using genetic programming. In: Gero JS, Sudweeks F, editors. Artificial Intelligence in Design ‘96. Netherlands, Dordrecht: Springer; 1996. pp. 151–170. [Google Scholar]
- 72.Kitchin R, McArdle G. What makes Big Data, Big Data? Exploring the ontological characteristics of 26 datasets. Big Data Soc. 2016 doi: 10.1177/2053951716631130. [DOI] [Google Scholar]
- 73.Bhadani A, Jothimani D (2017) Big Data: challenges, opportunities and realities. CoRR abs/1705.0
- 74.Larose DT, Larose CD (2015) Data mining and predictive analytics, Wiley Publishing
- 75.Fernández A, del Río S, Chawla NV, Herrera F. An insight into imbalanced Big Data classification: outcomes and challenges. Complex Intell Syst. 2017;3:105–120. doi: 10.1007/s40747-017-0037-9. [DOI] [Google Scholar]
- 76.Hasanin T, Khoshgoftaar TM, Leevy JL, Bauder RA. Severely imbalanced Big Data challenges: investigating data sampling approaches. J Big Data. 2019 doi: 10.1186/s40537-019-0274-4. [DOI] [Google Scholar]
- 77.Nath A, Subbiah K. Maximizing lipocalin prediction through balanced and diversified training set and decision fusion. Comput Biol Chem. 2015;59:101–110. doi: 10.1016/j.compbiolchem.2015.09.011. [DOI] [PubMed] [Google Scholar]
- 78.Nath A, Karthikeyan S. Enhanced prediction and characterization of CDK inhibitors using optimal class distribution. Interdiscip Sci Comput Life Sci. 2017;9:292–303. doi: 10.1007/s12539-016-0151-1. [DOI] [PubMed] [Google Scholar]
- 79.Wei Q, Dunbrack RL., Jr The role of balanced training and testing data sets for binary classifiers in bioinformatics. PLoS ONE. 2013 doi: 10.1371/journal.pone.0067863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nath A, Subbiah K. The role of pertinently diversified and balanced training as well as testing data sets in achieving the true performance of classifiers in predicting the antifreeze proteins. Neurocomputing. 2018;272:294–305. doi: 10.1016/j.neucom.2017.07.004. [DOI] [Google Scholar]
- 81.Barigye SJ, García de la Vega JM, Castillo-Garit JA. Undersampling: case studies of flaviviral inhibitory activities. J Comput Aided Mol Des. 2019;33:997–1008. doi: 10.1007/s10822-019-00255-3. [DOI] [PubMed] [Google Scholar]
- 82.Chawla NV, Bowyer KW, Hall LO, Kegelmeyer WP. SMOTE: synthetic minority over-sampling technique. J Artif Int Res. 2002;16:321–357. [Google Scholar]
- 83.Wang Q, Luo Z, Huang J, et al. A novel ensemble method for imbalanced data learning: bagging of extrapolation-SMOTE SVM. Comput Intell Neurosci. 2017 doi: 10.1155/2017/1827016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gulowaty B, Ksieniewicz P, Yin H, et al. SMOTE algorithm variations in balancing data streams. Cham: Springer International Publishing; 2019. pp. 305–312. [Google Scholar]
- 85.Zemouri R, Omri N, Fnaiech F, et al. A new growing pruning deep learning neural network algorithm (GP-DLNN) Neural Comput Appl. 2020 doi: 10.1007/s00521-019-04196-8. [DOI] [Google Scholar]
- 86.Nath A, Sahu GK. Exploiting ensemble learning to improve prediction of phospholipidosis inducing potential. J Theor Biol. 2019;479:37–47. doi: 10.1016/j.jtbi.2019.07.009. [DOI] [PubMed] [Google Scholar]
- 87.Nath A, Karthikeyan S. Enhanced prediction of recombination hotspots using input features extracted by class specific autoencoders. J Theor Biol. 2018;444:73–82. doi: 10.1016/j.jtbi.2018.02.016. [DOI] [PubMed] [Google Scholar]
- 88.Srivastava N, Hinton GE, Krizhevsky A, et al. Dropout: a simple way to prevent neural networks from overfitting. J Mach Learn Res. 2014;15:1929–1958. [Google Scholar]
- 89.Fedus W, Rosca M, Lakshminarayanan B, et al (2017) Many paths to equilibrium: GANs do not need to decrease aDivergence at every step
- 90.Lin E, Lin C-H, Lane H-Y. Relevant applications of generative adversarial networks in drug design and discovery: molecular de novo design, dimensionality reduction, and de novo peptide and protein design. Molecules. 2020;25:3250. doi: 10.3390/molecules25143250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ge Q, Huang X, Fang S, et al. Conditional generative dversarial networks for individualized treatment effect estimation and treatment selection. Front Genet. 2020 doi: 10.3389/fgene.2020.585804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mirza M, Osindero S (2014) Conditional generative adversarial nets. ArXiv abs/1411.1
- 93.Kadurin A, Aliper A, Kazennov A, et al (2017) The cornucopia of meaningful leads: Applying deep adversarial autoencoders for new molecule development in oncology. Oncotarget 8:10883–10890. 10.18632/oncotarget.14073 [DOI] [PMC free article] [PubMed]
- 94.Kadurin A, Nikolenko S, Khrabrov K, et al. druGAN: an advanced generative adversarial autoencoder model for de novo generation of new molecules with desired molecular properties in silico. Mol Pharm. 2017;14:3098–3104. doi: 10.1021/acs.molpharmaceut.7b00346. [DOI] [PubMed] [Google Scholar]
- 95.Maziarka Ł, Pocha A, Kaczmarczyk J, et al. Mol-CycleGAN: a generative model for molecular optimization. J Cheminform. 2020 doi: 10.1186/s13321-019-0404-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Prykhodko O, Johansson SV, Kotsias P-C, et al. A de novo molecular generation method using latent vector based generative adversarial network. J Cheminform. 2019 doi: 10.1186/s13321-019-0397-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Simonyan K, Zisserman A (2015) Very deep convolutional networks for large-scale image recognition. CoRR abs/1409.1
- 98.Szegedy C, Vanhoucke V, Ioffe S, et al. Rethinking the inception architecture for computer vision. IEEE Conf Comput Vis Pattern Recognit. 2016;2016:2818–2826. [Google Scholar]
- 99.He K, Zhang X, Ren S, Sun J. Deep residual learning for image recognition. IEEE Conf Comput Vis Pattern Recognit. 2016;2016:770–778. [Google Scholar]
- 100.Chougrad H, Zouaki H, Alheyane O. Deep convolutional neural networks for breast cancer screening. Comput Methods Programs Biomed. 2018;157:19–30. doi: 10.1016/j.cmpb.2018.01.011. [DOI] [PubMed] [Google Scholar]
- 101.Ragoza M, Hochuli J, Idrobo E, et al. Protein-ligand scoring with convolutional neural networks. J Chem Inf Model. 2017;57:942–957. doi: 10.1021/acs.jcim.6b00740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Stepniewska-Dziubinska MM, Zielenkiewicz P, Siedlecki P. Development and evaluation of a deep learning model for protein–ligand binding affinity prediction. Bioinformatics. 2018;34:3666–3674. doi: 10.1093/bioinformatics/bty374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhao Z, Yang Z, Luo L, et al. Drug drug interaction extraction from biomedical literature using syntax convolutional neural network. Bioinformatics. 2016;32:3444–3453. doi: 10.1093/bioinformatics/btw486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fernandez M, Ban F, Woo G, et al. Toxic colors: the use of deep learning for predicting toxicity of compounds merely from their graphic images. J Chem Inf Model. 2018;58:1533–1543. doi: 10.1021/acs.jcim.8b00338. [DOI] [PubMed] [Google Scholar]
- 105.Dvornek N, Li X, Zhuang J, Duncan J. Jointly discriminative and generative recurrent neural networks for learning from fMRI. Mach Learn Med imaging MLMI. 2019;11861:382–390. doi: 10.1007/978-3-030-32692-0_44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hochreiter S, Schmidhuber J. Long short-term memory. Neural Comput. 1997;9:1735–1780. doi: 10.1162/neco.1997.9.8.1735. [DOI] [PubMed] [Google Scholar]
- 107.Li X, Xu Y, Yao H, Lin K. Chemical space exploration based on recurrent neural networks: applications in discovering kinase inhibitors. J Cheminform. 2020;12:42. doi: 10.1186/s13321-020-00446-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Caruana R. Multitask learning. Mach Learn. 1997;28:41–75. doi: 10.1023/A:1007379606734. [DOI] [Google Scholar]
- 109.Caruana R (1993) Multitask learning: a knowledge-based source of inductive bias. In: Proc. Tenth Int. Conf. Int. Conf. Mach. Learn, pp 41–48
- 110.Mayr A, Klambauer G, Unterthiner T, Hochreiter S. DeepTox: toxicity prediction using deep learning. Front Environ Sci. 2016 doi: 10.3389/fenvs.2015.00080. [DOI] [Google Scholar]
- 111.Jain S, Siramshetty VB, Alves VM, et al. Large-scale modeling of multispecies acute toxicity end points using consensus of multitask deep learning methods. J Chem Inf Model. 2021;61:653–663. doi: 10.1021/acs.jcim.0c01164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sabban S, Markovsky M (2020) RamaNet: computational <em>de novo</em> helical protein backbone design using a long short-term memory generative neural network. bioRxiv 671552 10.1101/671552
- 113.Karimi M, Zhu S, Cao Y, Shen Y. De novo protein design for novel folds using guided conditional wasserstein generative adversarial networks. J Chem Inf Model. 2020;60:5667–5681. doi: 10.1021/acs.jcim.0c00593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lundberg S, Lee S-I (2017) A unified approach to interpreting model predictions. In: NIPS
- 115.Nohara Y, Matsumoto K, Soejima H, Nakashima N (2019) Explanation of machine learning models using improved shapley additive explanation. In: Proc. 10th ACM Int. Conf. Bioinformatics, Comput. Biol. Heal. Informatics, p 546
- 116.Abadi M, Agarwal A, Barham P et al (2015) TensorFlow : large-scale machine learning on heterogeneous distributed systems
- 117.van der Maaten L, Hinton G. Visualizing data using t-SNE. J Mach Learn Res. 2008;9:2579–2605. [Google Scholar]
- 118.P Baldi (2011) Autoencoders, unsupervised learning and deep architectures. In: Proc. 2011 Int Conf Unsupervised Transf Learn Work, vol 27, pp 37–50
- 119.Vincent P, Larochelle H, Bengio Y, Manzagol P-A (2008) Extracting and composing robust features with denoising autoencoders. In: Proc. 25th Int. Conf. Mach. Learn, 1096–1103
- 120.Peng J, Li J, Shang X. A learning-based method for drug-target interaction prediction based on feature representation learning and deep neural network. BMC Bioinform. 2020 doi: 10.1186/s12859-020-03677-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hu Q, Feng M, Lai L, Pei J. Prediction of drug-likeness using deep autoencoder neural networks. Front Genet. 2018;9:585. doi: 10.3389/fgene.2018.00585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ma T, Xiao C, Zhou J, Wang F (2018) Drug similarity integration through attentive multi-view graph auto-encoders. In: Proc. 27th Int. Jt. Conf. Artif. Intell, pp 3477–3483
- 123.Polikar R. Ensemble based systems in decision making. IEEE Circuits Syst Mag. 2006;6:21–45. doi: 10.1109/MCAS.2006.1688199. [DOI] [Google Scholar]
- 124.Seewald A (2002) How to make stacking better and faster while also taking care of an unknown weakness
- 125.Kuncheva LI. A theoretical study on six classifier fusion strategies. IEEE Trans Pattern Anal Mach Intell. 2002;24:281–286. doi: 10.1109/34.982906. [DOI] [Google Scholar]
- 126.Moreira J, Soares C, Jorge A, Sousa J. Ensemble approaches for regression a survey. ACM Comput Surv. 2012;45(1):10–40. doi: 10.1145/2379776.2379786. [DOI] [Google Scholar]
- 127.Wang D, Shang Y. Modeling physiological data with deep belief networks. Int J Inf Educ Technol. 2013;3:505–511. doi: 10.7763/IJIET.2013.V3.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fitriawan A, Wasito I, Syafiandini AF et al (2016) Deep belief networks using hybrid fingerprint feature for virtual screening of drug design. In: 2016 international conference on computer, control, informatics systems (ICACSIS), pp 417–420
- 129.Fitriawan A, Wasito I, Syafiandini AF et al (2016) Multi-label classification using deep belief networks for virtual screening of multi-target drug. In: 2016 international conference on computer, control, informatics and its applications (IC3INA), pp 102–107
- 130.Hooshmand SA, Jamalkandi SA, Alavi SM, Masoudi-Nejad A. Distinguishing drug/non-drug-like small molecules in drug discovery using deep belief network. Mol Divers. 2020 doi: 10.1007/s11030-020-10065-7. [DOI] [PubMed] [Google Scholar]
- 131.Bal M, Amasyali MF, Sever H, et al. Performance Evaluation of the Machine Learning Algorithms Used in Inference Mechanism of a Medical Decision Support System. Sci World J. 2014 doi: 10.1155/2014/137896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Grelck C, Niewiadomska-Szynkiewicz E, Aldinucci M, et al. Why high-performance modelling and simulation for big data applications matters BT—High-performance modelling and simulation for big data applications: selected results of the COST action IC1406 cHiPSet. In: González-Vélez H, et al., editors. Kołodziej J. Cham: Springer International Publishing; 2019. pp. 1–35. [Google Scholar]
- 133.Lake F (2019) Artificial intelligence in drug discovery: what is new, and what is next? Futur Drug Discov 1:FDD19 10.4155/fdd-2019-0025
- 134.Ramesh AN, Kambhampati C, Monson JRT, Drew PJ. Artificial intelligence in medicine. Ann R Coll Surg Engl. 2004;86:334–338. doi: 10.1308/147870804290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kalyane D, Sanap G, Paul D et al (2020) Chapter 3—Artificial intelligence in the pharmaceutical sector: current scene and future prospect. In: Tekade RKBT-TF of PPD and R (ed) Advances in pharmaceutical product development and research, Academic Press, pp 73–107
- 136.Mak K-K, Pichika MR. Artificial intelligence in drug development: present status and future prospects. Drug Discov Today. 2019;24:773–780. doi: 10.1016/j.drudis.2018.11.014. [DOI] [PubMed] [Google Scholar]
- 137.Zhong F, Xing J, Li X, et al. Artificial intelligence in drug design. Sci China Life Sci. 2018;61:1191–1204. doi: 10.1007/s11427-018-9342-2. [DOI] [PubMed] [Google Scholar]
- 138.Qian N, Sejnowski TJ. Predicting the secondary structure of globular proteins using neural network models. J Mol Biol. 1988;202:865–884. doi: 10.1016/0022-2836(88)90564-5. [DOI] [PubMed] [Google Scholar]
- 139.Spencer M, Eickholt J, Cheng J. A deep learning network approach to ab initio protein secondary structure prediction. IEEE/ACM Trans Comput Biol Bioinforma. 2015;12:103–112. doi: 10.1109/TCBB.2014.2343960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jo T, Hou J, Eickholt J, Cheng J. Improving protein fold recognition by deep learning networks. Sci Rep. 2015 doi: 10.1038/srep17573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Du X, Sun S, Hu C, et al. DeepPPI: boosting prediction of protein-protein interactions with deep neural networks. J Chem Inf Model. 2017;57:1499–1510. doi: 10.1021/acs.jcim.7b00028. [DOI] [PubMed] [Google Scholar]
- 142.Rifaioglu AS, Atas H, Martin MJ, et al. Recent applications of deep learning and machine intelligence on in silico drug discovery: methods, tools and databases. Brief Bioinform. 2019;20:1878–1912. doi: 10.1093/bib/bby061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Carpenter KA, Huang X. Machine learning-based virtual screening and its applications to Alzheimer’s drug discovery: a review. Curr Pharm Des. 2018;24:3347–3358. doi: 10.2174/1381612824666180607124038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Jiang D, Lei T, Wang Z, et al. ADMET evaluation in drug discovery. 20. Prediction of breast cancer resistance protein inhibition through machine learning. J Cheminform. 2020 doi: 10.1186/s13321-020-00421-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhavoronkov A, Ivanenkov YA, Aliper A, et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat Biotechnol. 2019;37:1038–1040. doi: 10.1038/s41587-019-0224-x. [DOI] [PubMed] [Google Scholar]
- 146.McCloskey K, Sigel EA, Kearnes S, et al. Machine learning on DNA-encoded libraries: a new paradigm for hit finding. J Med Chem. 2020;63:8857–8866. doi: 10.1021/acs.jmedchem.0c00452. [DOI] [PubMed] [Google Scholar]
- 147.Xing G, Liang L, Deng C, et al. Activity prediction of small molecule inhibitors for antirheumatoid arthritis targets based on artificial intelligence. ACS Comb Sci. 2020;22:873–886. doi: 10.1021/acscombsci.0c00169. [DOI] [PubMed] [Google Scholar]
- 148.He S, Zhang X, Lu S, et al. A computational toxicology approach to screen the hepatotoxic ingredients in traditional Chinese medicines polygonum multiflorum thunb as a case study. Biomol. 2019;9:577. doi: 10.3390/biom9100577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Xu Z, Yang L, Zhang X, et al. Discovery of potential flavonoid inhibitors against COVID-19 3CL proteinase based on virtual screening strategy. Front Mol Biosci. 2020;7:247. doi: 10.3389/fmolb.2020.556481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hooshmand SA, Zarei Ghobadi M, Hooshmand SE, et al. A multimodal deep learning-based drug repurposing approach for treatment of COVID-19. Mol Divers. 2020 doi: 10.1007/s11030-020-10144-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zhou Y, Hou Y, Shen J, et al. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020 doi: 10.1038/s41421-020-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Tripathi MK, Sharma S, Singh TP et al (2021) Computational intelligence in drug repurposing for COVID-19 BT—Computational intelligence methods in COVID-19: surveillance, prevention, prediction and diagnosis. In: Raza K (ed), Springer Singapore, Singapore, pp 273–294
- 153.Beck BR, Shin B, Choi Y, et al. Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Comput Struct Biotechnol J. 2020;18:784–790. doi: 10.1016/j.csbj.2020.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Schuhmacher A, Gassmann O, McCracken N, Hinder M. Open innovation and external sources of innovation. An opportunity to fuel the R&D pipeline and enhance decision making? J Transl Med. 2018 doi: 10.1186/s12967-018-1499-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zhao J, Cao Y, Zhang L. Exploring the computational methods for protein-ligand binding site prediction. Comput Struct Biotechnol J. 2020;18:417–426. doi: 10.1016/j.csbj.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wang X, Zhao Y, Pourpanah F. Recent advances in deep learning. Int J Mach Learn Cybern. 2020;11:747–750. doi: 10.1007/s13042-020-01096-5. [DOI] [Google Scholar]
- 157.Kiriiri GK, Njogu PM, Mwangi AN. Exploring different approaches to improve the success of drug discovery and development projects: a review. Futur J Pharm Sci. 2020;6:27. doi: 10.1186/s43094-020-00047-9. [DOI] [Google Scholar]
- 158.Lo Y-C, Rensi SE, Torng W, Altman RB. Machine learning in chemoinformatics and drug discovery. Drug Discov Today. 2018;23:1538–1546. doi: 10.1016/j.drudis.2018.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ahuja AS. The impact of artificial intelligence in medicine on the future role of the physician. PeerJ. 2019;7:e7702–e7702. doi: 10.7717/peerj.7702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lysenko A, Sharma A, Boroevich KA, Tsunoda T. An integrative machine learning approach for prediction of toxicity-related drug safety. Life Sci alliance. 2018;1:e201800098–e201800098. doi: 10.26508/lsa.201800098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ghasemi F, Mehridehnavi A, Fassihi A, Pérez-Sánchez H. Deep neural network in QSAR studies using deep belief network. Appl Soft Comput. 2018;62:251–258. doi: 10.1016/j.asoc.2017.09.040. [DOI] [Google Scholar]
- 162.Pu L, Naderi M, Liu T, et al. eToxPred: a machine learning-based approach to estimate the toxicity of drug candidates. BMC Pharmacol Toxicol. 2019;20:2. doi: 10.1186/s40360-018-0282-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Öztürk H, Özgür A, Ozkirimli E. DeepDTA: deep drug–target binding affinity prediction. Bioinformatics. 2018;34:i821–i829. doi: 10.1093/bioinformatics/bty593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Nguyen T, Le H, Quinn TP et al (2020) GraphDTA: Predicting drug–target binding affinity with graph neural networks. bioRxiv 684662 10.1101/684662 [DOI] [PubMed]
- 165.Karimi M, Wu D, Wang Z, Shen Y. DeepAffinity: interpretable deep learning of compound–protein affinity through unified recurrent and convolutional neural networks. Bioinformatics. 2019;35:3329–3338. doi: 10.1093/bioinformatics/btz111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Mouchlis VD, Afantitis A, Serra A, et al. Advances in de novo drug design: from conventional to machine learning methods. Int J Mol Sci. 2021 doi: 10.3390/ijms22041676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chan HCS, Shan H, Dahoun T, et al. Advancing drug discovery via artificial intelligence. Trends Pharmacol Sci. 2019;40:592–604. doi: 10.1016/j.tips.2019.06.004. [DOI] [PubMed] [Google Scholar]
- 168.Mäde V, Els-Heindl S, Beck-Sickinger AG. Automated solid-phase peptide synthesis to obtain therapeutic peptides. Beilstein J Org Chem. 2014;10:1197–1212. doi: 10.3762/bjoc.10.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hardwick T, Ahmed N. Digitising chemical synthesis in automated and robotic flow. Chem Sci. 2020;11:11973–11988. doi: 10.1039/D0SC04250A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Steiner S, Wolf J, Glatzel S, et al. Organic synthesis in a modular robotic system driven by a chemical programming language. Science. 2019 doi: 10.1126/science.aav2211. [DOI] [PubMed] [Google Scholar]
- 171.Harrer S, Shah P, Antony B, Hu J. Artificial intelligence for clinical trial design. Trends Pharmacol Sci. 2019;40:577–591. doi: 10.1016/j.tips.2019.05.005. [DOI] [PubMed] [Google Scholar]
- 172.Aggarwal RR, Costin D, O’Neill VJ, et al. Phase 1b study of BXCL701, a novel small molecule inhibitor of dipeptidyl peptidases (DPP), combined with pembrolizumab (pembro), in men with metastatic castration-resistant prostate cancer (mCRPC) J Clin Oncol. 2020;38:e17581–e17581. doi: 10.1200/JCO.2020.38.15_suppl.e17581. [DOI] [Google Scholar]
- 173.Nguyen G, Dlugolinsky S, Bobák M, et al. Machine learning and deep learning frameworks and libraries for large-scale data mining: a survey. Artif Intell Rev. 2019;52:77–124. doi: 10.1007/s10462-018-09679-z. [DOI] [Google Scholar]
- 174.Paszke A, Gross S, Massa F et al (2019) PyTorch: An imperative style, high-performance deep learning library. In: Wallach H, Larochelle H, Beygelzimer A et al (eds) Advances in neural information processing systems, Curran Associates, Inc.
- 175.Senior AW, Evans R, Jumper J, et al. Improved protein structure prediction using potentials from deep learning. Nature. 2020;577:706–710. doi: 10.1038/s41586-019-1923-7. [DOI] [PubMed] [Google Scholar]
- 176.Coley CW, Barzilay R, Green WH, et al. Convolutional embedding of attributed molecular graphs for physical property prediction. J Chem Inf Model. 2017;57:1757–1772. doi: 10.1021/acs.jcim.6b00601. [DOI] [PubMed] [Google Scholar]
- 177.Gómez-Bombarelli R, Wei JN, Duvenaud D, et al. Automatic Chemical design using a data-driven continuous representation of molecules. ACS Cent Sci. 2018;4:268–276. doi: 10.1021/acscentsci.7b00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wang C, Zhang Y. Improving scoring-docking-screening powers of protein–ligand scoring functions using random forest. J Comput Chem. 2017;38:169–177. doi: 10.1002/jcc.24667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Stork C, Chen Y, Šícho M, Kirchmair J. Hit dexter 2.0: machine-learning models for the prediction of frequent hitters. J Chem Inf Model. 2019;59:1030–1043. doi: 10.1021/acs.jcim.8b00677. [DOI] [PubMed] [Google Scholar]
- 180.Urban G, Subrahmanya N, Baldi P. Inner and outer recursive neural networks for chemoinformatics applications. J Chem Inf Model. 2018;58:207–211. doi: 10.1021/acs.jcim.7b00384. [DOI] [PubMed] [Google Scholar]
- 181.Jin W, Barzilay R, Jaakkola T (2018) Junction tree variational autoencoder for molecular graph generation. In: Dy J, Krause A (ed) proceedings of the 35th international conference on machine learning. PMLR, Stockholmsmässan, Stockholm Sweden, pp 2323–2332
- 182.Duvenaud DK, Maclaurin D, Iparraguirre J et al (2015) Convolutional networks on graphs for learning molecular fingerprints. In: Cortes C, Lawrence N, Lee D et al (eds) Advances in neural information processing systems, Curran Associates, Inc
- 183.Durrant JD, McCammon JA. NNScore 2.0: a neural-network receptor-ligand scoring function. J Chem Inf Model. 2011;51:2897–2903. doi: 10.1021/ci2003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Sanchez-Lengeling B, Outeiral C, Guimaraes GL, Aspuru-Guzik A (2017) Optimizing distributions over molecular space. An objective-reinforced generative adversarial network for inverse-design chemistry (ORGANIC)
- 185.Wójcikowski M, Zielenkiewicz P, Siedlecki P. Open Drug Discovery toolkit (ODDT): a new open-source player in the drug discovery field. J Cheminform. 2015 doi: 10.1186/s13321-015-0078-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Feinberg EN, Sur D, Wu Z, et al. PotentialNet for molecular property prediction. ACS Cent Sci. 2018;4:1520–1530. doi: 10.1021/acscentsci.8b00507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Awale M, Reymond J-L. Polypharmacology browser PPB2: target prediction combining nearest neighbors with machine learning. J Chem Inf Model. 2019;59:10–17. doi: 10.1021/acs.jcim.8b00524. [DOI] [PubMed] [Google Scholar]
- 188.Cho A. No room for error. Science. 2020;369:130–133. doi: 10.1126/science.369.6500.130. [DOI] [PubMed] [Google Scholar]
- 189.Blaschke T, Arús-Pous J, Chen H, et al. REINVENT 2.0: an AI tool for de novo drug design. J Chem Inf Model. 2020;60:5918–5922. doi: 10.1021/acs.jcim.0c00915. [DOI] [PubMed] [Google Scholar]