

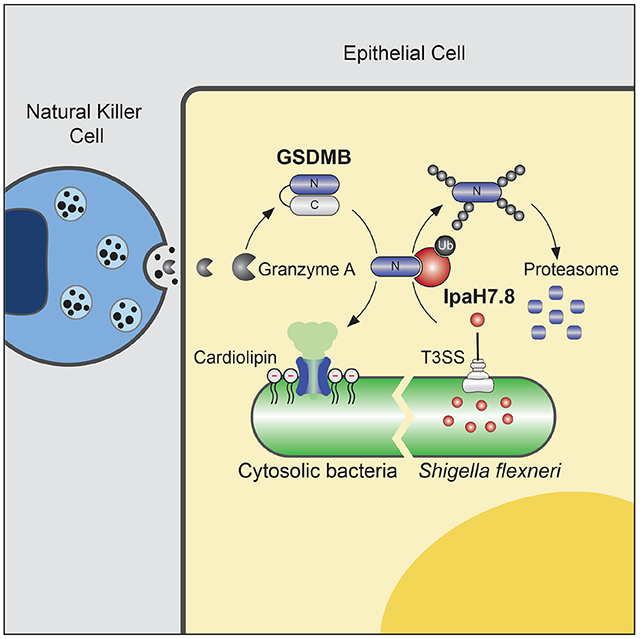
SUMMARY
Gasdermin B (GSDMB) belongs to a large family of pore forming cytolysins that execute inflammatory cell death programs. While genetic studies have linked GSDMB polymorphisms to human disease, its function in the immunological response to pathogens remains poorly understood. Here, we report a dynamic host-pathogen conflict between GSDMB and the IpaH7.8 effector protein secreted by enteroinvasive Shigella flexneri. We show that IpaH7.8 ubiquitinates and targets GSDMB for 26S proteasome destruction. This virulence strategy protects Shigella from the bacteriocidic activity of Natural Killer cells by suppressing Granzyme-A mediated activation of GSDMB. In contrast to the canonical function of most Gasdermin-family members, GSDMB does not inhibit Shigella by lysing host cells. Rather, it exhibits direct microbiocidal activity through recognition of phospholipids found on Gram-negative bacterial membranes. These findings place GSDMB as a central executioner of intracellular bacterial killing and reveals a mechanism employed by pathogens to counteract this host defense system.
Graphical Abstract
In Brief
Repurposing a cell death mechanism for antibacterial defense, human immune cells combat infection by punching holes in bacterial membranes. A human pathogen has evolved a way to fight back.
INTRODUCTION
The Gasdermin-family of mammalian pore forming cytolysins execute pyroptotic lytic cell death characterized by release of proinflammatory molecules (Evavold et al., 2018; He et al., 2015; Kayagaki et al., 2015; Shi et al., 2015a). Recent structural and biochemical studies have revealed a detailed picture of Gasdermin activation by cellular proteases and lytic pore formation through plasma membrane oligomerization (Aglietti et al., 2016; Ding et al., 2016; Kuang et al., 2017; Liu et al., 2016; Ruan et al., 2018; Sborgi et al., 2016). In general, these proteins are composed of an N-terminal cytotoxic domain (GSDM-N), an interdomain linker, and a C-terminal autoinhibitory domain (GSDM-C). Proteolytic cleavage of the linker region releases the N-terminus from autoinhibition. This event results in GSDM-N membrane insertion, oligomerization, and pore formation. While the majority of Gasdermin-family members execute pyroptotic cell death in response to inflammatory stimulation, it has been suggested that they can also target prokaryotic membranes (Liu et al., 2016). It is unclear however if bacteria are indeed physiologically relevant targets of Gasdermin-mediated immunity.
Humans encode six Gasdermin genes. GSDMB is unique among these family members as it is the only GSDM gene that is not found in rodents (Tamura et al., 2007). This observation suggests that GSDMB is not a general component of the mammalian immune system, but rather, has evolved a specialized function within humans and other mammals. Consistent with this idea, Genome Wide Association Studies (GWAS) link polymorphisms in GSDMB (17q12-q21 locus) to an increased risk for asthma, type I diabetes, primary biliary cirrhosis, and Crohn’s disease (Spycher et al., 2012; Tulah et al., 2013; Yu et al., 2011). While these analyses indicate that GSDMB plays an important function in the human immune system, its propensity to form lytic pores in cells is controversial. Several reports have indicated that the GSDMB-N subunit fails to form pores when expressed in cultured cells (Chen et al., 2019; Shi et al., 2015b). Consistent with this in vitro observation, mice expressing a human GSDMB transgene develop an asthma-like phenotype characterized by airway responsiveness and remodeling in the absence of lung inflammation (Das et al., 2016). Conversely, GSDMB was shown to be a key executioner of inflammatory lysis of cancer cells targeted by innate lymphocytes (Zhou et al., 2020). It is unclear whether GSDMB oligomerizes in the plasma membrane of cancer cells or if cancer cell death is a consequence of downstream Gasdermin-family member activation as observed in other cellular contexts (Zhang et al., 2021; Zhou et al., 2020). Nevertheless, these findings reinforce the idea that GSDMB exhibits a non-canonical function.
Shigella spp. are a human enteroinvasive pathogen that infects an estimated 165 million people, resulting in more than 200,000 deaths annually (Khalil et al., 2018; Kotloff et al., 1999). The pathogenic lifecycle of S. flexneri is fueled by its multiple interactions with macrophages and colonic epithelial cells. For example, S. flexneri traverses the mucosal barrier of the colon, where it is engulfed by tissue-resident macrophages. These phagocytes undergo Inflammasome/Caspase-1/GSDMD-mediated cell death, a key immunological event that alerts immune cells to the location of infection (Sansonetti et al., 2000; Watson et al., 2019; Zychlinsky et al., 1992). Interestingly, inflammasome activation may also enhance virulence as macrophage lysis results in S. flexneri escape into the extracellular milieu and subsequent reinvasion of the colonic epithelium. Once established in the epithelial cell layer, S. flexneri replicates to high levels which increases tissue transmission and disease severity. While lytic cell death of macrophages by GSDMD is a critical event in the S. flexneri lifecycle, the Gasdermin family members are also expressed in a wide variety of other tissues and cell types (Broz et al., 2020). This observation raises the question: how do intracellular pathogens suppress the lytic activity of Gasdermin-proteins in host cells that function as reservoirs for bacterial replication?
Virulence associated with S. flexneri infection requires expression of the Mxi-Spa Type III secretion system (T3SS) and delivery of over 30 effector proteins into the host cell (Schroeder and Hilbi, 2008). Among these effector proteins are the Invasion plasmid antigen H genes (ipaH) that encode bacterial E3-ubiquitin ligases (Ashida et al., 2007; Rohde et al., 2007). While these effector proteins are known to hijack the host E1/E2 ubiquitin conjugation machinery to post-translationally modify target substrates, the pathogenic function of most IpaH genes remains poorly understood. Using an unbiased screen for bacterial effector protein substrates, we found that the S. flexneri Type 3 Secretion System (T3SS) effector protein IpaH7.8 targets human GSDMB for ubiquitin-mediated proteolysis. IpaH7.8 did not interact with or ubiquitinate GSDMD or any other Gasdermin-family members. Rather, IpaH7.8 secretion prevented NK cells from clearing S. flexneri from epithelial cells by suppressing GZMA-activated GSDMB. Unexpectedly, GSDMB activation did not induce lytic cell death of epithelial cells, but instead caused direct lysis of bacteria. We describe how alternative lipid binding by GSDMB promotes bacterial recognition and lysis while limiting damage to epithelial cells that function as a critical immunological barrier to microbial infection.
RESULTS
Identification of GSDMB as a cellular substrate of IpaH7.8
A technique termed Ubiquitin Activated Interaction Traps (UBAIT) was adapted for identifying host substrates of orphan bacterial E3-ubiquitin ligases (O'Connor et al., 2015). As diagramed in Figure 1A, the human ubiquitin gene was cloned at the 3' end of each ipaH effector to generate a carboxy-terminal fusion between IpaH protein and ubiquitin (termed IpaHUBAIT). In theory, the protein chimera can perform each of the essential steps required for substrate ubiquitination: (1) activation of C-terminal Ub through E2-mediated transfer to the IpaH catalytic cysteine, (2) substrate recognition through the N-terminal Leucine Rich Repeat (LRR) domain of IpaH and (3) thiol-mediated ligation between the C-terminal Ub in IpaHUBAIT and a host substrate Lysine. Successful ubiquitination would result in an iso-peptide bond formed between IpaHUBAIT and its substrate, which can be purified using affinity tags introduced into each of the IpaHUBAIT constructs (Figure 1A).
Figure 1. Characterization of GSDMB ubiquitination by IpaH7.8.

(A) Diagram showing UBAIT reaction. GST, Glutathione S Transferase; LRR, Leucine Rich Repeat; E3, E3 ligase-enzyme domain
(B-D) UBAIT reaction between the IpaH proteins and the substrates indicated. Western blots (W.B.) of the GSDMs and Coomassie stain gels of protein inputs are shown.
(E) Co-immunoprecipitation between FLAG-tagged GSDM family members and GFP-tagged IpaH7.8C357S.
(F) Glutathione pulldown experiments between GST-tagged proteins and His-IpaH2.5 or His-IpaH7.8.
(G) Size exclusion chromatography traces of the proteins indicated.
(H-J) In vitro ubiquitination assays with IpaH7.8 and the Gasdermin family members indicated. GSDM modification was assessed using anti-GST (H and J) or anti-GSDMB (I). Ubiquitin was detected using anti-ubiquitin (H) and IpaH7.8 was detected by Coomassie stain (H and I) or anti-His (J).
(B-J) Results shown are representative of at least three independent biological repeats.
See also Figure S1.
To test the efficiency of the UBAIT technology, ubiquitin on IpaH2.5UBAIT and IpaH9.8UBAIT was charged with human E1 (UbE1) and E2 (UbcH5b) enzymes in vitro and then incubated with Caco-2 epithelial cell lysates. Consistent with our previous findings (de Jong et al., 2016), IpaH2.5UBAIT reacted with the linear ubiquitination chain assembly complex (LUBAC) subunit HOIP as well as several other components of the TNF receptor signaling complex including BIRC2 (Baculoviral IAP repeat containing protein 2), TRAF2 (TNR Receptor associated factor 2), and DBLOH (Diablo IAP-Binding mitochondrial protein) (Figure S1A-C). In addition, IpaH9.8UBAIT captured GBP1, 2 and 4 from lysates prepared from interferon gamma (IFNγ) stimulated Caco-2 cells (Figure S1A and B) (Li et al., 2017; Piro et al., 2017; Wandel et al., 2017). The capture of known IpaH2.5 and IpaH9.8 substrates demonstrated that UBAIT is a powerful technique for identifying substrates of bacterial E3-ubiquitin ligases.
Out of seven IpaHUBAIT proteins produced in vitro, IpaH7.8UBAIT uniquely reacted with the Gasdermin family members GSDMB and GSDMD in Caco-2 lysates (Figure S1A and B). The UBAIT reaction was confirmed using purified proteins. IpaH7.8UBAIT formed an SDS-resistant covalent bond with GSDMB that relied on the catalytic cysteine 357 residue of IpaH7.8 and diglycine ligation between fused ubiquitin and the GSDMB protein (Figure 1B). Interestingly, while IpaH7.8UBAIT formed a stable complex with GSDMB, it reacted poorly with GSDMD under these conditions (Figure 1C). In addition, among other IpaHUBAIT proteins examined, only IpaH7.8UBAIT reacted with GSDMB (Figure 1D).
IpaH7.8 selectively binds and ubiquitinates GSDMB
The N-terminal LRR domain of IpaH effectors is required for substrate interactions, whereas the C-terminal domain harbors the ligase activity required for ubiquitin transfer and chain elongation (Keszei et al., 2014; Miao et al., 1999; Rohde et al., 2007; Singer et al., 2008; Zhu et al., 2008). To assess whether IpaH7.8 binds Gasdermin family members, the catalytic cysteine (C) residue 357 was mutated to serine (S) as a method to prevent substrate turnover and stabilize the interaction between IpaH7.8 and substrate (Pierce et al., 2009). Co-immunoprecipitation studies revealed that IpaH7.8C357S bound to GSDMB in cells but not to other Gasdermin family members (Figure 1E). In addition, IpaH7.8 and GSDMB formed a 1:1 stoichiometric complex in solution, whereas IpaH2.5 did not bind GSDMB in this assay (Figure 1F and 1G). Interestingly, IpaH7.8 did not recognize the C-terminal autoinhibitory domain of GSDMB (GSDMB-C), suggesting that the N-terminal pore forming subunit (GSDMB-N) contributes to the binding specificity (Figure 1F, lane 6). Consistent with this interpretation, IpaH7.8 did not bind recombinant GSDMD, which shares less than 30% sequence identity to the N-terminus of GSDMB (Figure S1D).
To determine if IpaH7.8 ubiquitinates GSDMB as the UBAIT reaction and binding studies indicate, recombinant IpaH7.8 and GSDMB were incubated with E1 (UbE1), E2 (UbcH5b), ubiquitin, and ATP in vitro. IpaH7.8, but not the IpaH7.8C357S mutant, transferred free ubiquitin to GSDMB in a dose dependent manner (Figure 1H). Mass spectrometry analysis revealed two high-confidence ubiquitin modified residues in the N- (K166) and C- (K308) terminal domains of GSDMB (Figure S1E). However, mutations of these residues to arginine (K166/308R) did not prevent GSDMB ubiquitination suggesting that mulitple lysine residues are modified by IpaH7.8 (Figure S1F). Consistent with this interpretation, GSDMB migrated at a high molecular weight indicative of multi-site ubiquitination when a ubiquitin variant incapable of serving as an acceptor for polyubiquitin chain formation (ΔK ubiquitin) was used in the ligation reaction (Figure 1I). In addition, deletion of the C-terminal glycine (ΔG ubiquitin) required for lysine modification of substrates eliminated IpaH7.8 ubiquitination of GSDMB (Figure 1I). We also found that IpaH7.8 catalyzed K48 ubiquitin chains similar to other IpaH family members (K48 ubiquitin, Figure 1I) (Rohde et al., 2007; Zhu et al., 2008). In contrast, ubiquitin chain formation on GSDMB was not observed in reactions supplemented with the K48R ubiquitin mutant (Figure 1I). Lastly, in agreement with the substrate specificity observed in the Co-IP studies, IpaH7.8 ubiquitinated GSDMB but not other Gasdermin family members (Figure 1J). We conclude that GSDMB is a highly specific substrate of IpaH7.8 and that K48 ubiquitin modification of several lysine residues may allow S. flexneri to alter GSDMBs immunological function.
GSDMB is targeted for 26S proteasomal degradation by Shigella IpaH7.8
The majority of IpaH family members target substrates for 26S proteasome destruction (Rohde et al., 2007; Singer et al., 2008; Zhu et al., 2008). In line with this conserved activity, we were unable to detect GSDMB expression in cells that were co-transfected with IpaH7.8 (Figure 2A, S2A). In contrast, IpaH7.8 had no effect on the expression levels of HOIP or GBP1, the host targets of IpaH1.4/2.5 and IpaH9.8, respectively (Figure 2A, S2A). No other IpaH family members reduced GSDMB protein levels under these conditions (Figure 2A, S2A). To determine if IpaH7.8 exhibits target specificity for GSDMB, we co-transfected IpaH7.8 with GSDMB, GSDMC, and GSDMD. Consistent with the selectivity observed in binding and ubiquitination assays, only GSDMB protein levels were depleted by IpaH7.8 (Figure 2B). Catalytically inactive IpaH7.8C357S failed to reduce GSDMB expression, indicating that loss of the protein was due to site directed ubiquitination (Figure 2B). Indeed, application of the 26S-proteasome inhibitor MG132 prevented loss of GSDMB in IpaH7.8 expressing cells, confirming that IpaH7.8 targets GSDMB for ubiquitin-mediated proteolysis (Figure 2C, S2B).
Figure 2. GSDMB is targeted for 26S proteasome destruction by IpaH7.8.

(A-C) Cellular degradation assays showing host protein abundance in cells expressing the IpaH proteins indicated. Cells in (C) were treated with DMSO or MG132.
(D) GSDMB ubiquitination in non-infected (NI), WT, or ΔipaH7.8 (Δ7.8) S. flexneri (MPI=20; 2.5hrs) infected cells as indicated. Cells were treated with MG132 to prevent GSDMB proteosome degradation.
(E) Cellular degradation assay showing GSDMB protein levels in cells either non-infected (NI) or infected with the indicated S. flexneri strains (MOI=300; 4 hrs). GSDMB protein was probed as in (A-C) and quantified by densitometry and reported as the ratio of GSDMB to actin found in S. flexneri infected cells compared to NI cells. Data are represented as mean ± SD (n=3).
(F) Immunofluorescence microscopy of GSDMB expressing cells infected with the S. flexneri strains (MOI=100; 2.5hrs) indicated.
(G) HCC1954 cells were infected with indicated S. flexneri strains (MOI=300; 4 hrs). Endogenous GSDMB protein levels were probed with anti-GSDMB antibody and S. flexneri levels were quantified by determining CFU/well. Data are represented as mean ± SD (n=4).
(A-G) Results shown are representative of at least three independent biological repeats. Statistical analyses can be found in Method Details.
See also Figure S2.
Shigella IpaH7.8 targets GSDMB during infection
Next, we performed Shigella infection studies to determine if GSDMB is a physiologically relevant target of bacterially produced and secreted IpaH7.8. Ubiquitinated GSDMB was detected in cells infected with S. flexneri but not in cells infected with S. flexneri ΔipaH7.8 (Figure 2D, note that MG132 proteasome inhibitor was used to prevent GSDMB degradation in this assay). Consistent with this result, GSDMB protein was significantly reduced by S. flexneri infection compared to non-infected (NI) controls (Figure 2E and S2C). Genetic studies revealed that the Mxi-SPA T3SS (Allaoui et al., 1993) was necessary for GSDMB degradation and that IpaH7.8 was the essential effector required for this reaction (Figure 2E, S2C). Fluorescence microscopy confirmed that GSDMB was eliminated in host cells that were infected with S. flexneri but not S. flexneri ΔipaH7.8 (Figure 2F). Lastly, S. flexneri eliminated endogenous GSDMB from the human epithelial cell line HCC1954 in an IpaH7.8 dependent manner (Figure 2G). These data support the conclusion that IpaH7.8 eliminates GSDMB as a virulence strategy of S. flexneri.
Comparison between GSDMD and GSDMB pore formation in vitro
GWAS have linked GSDMB polymorphisms to human pathology, yet its role in host immunity is still poorly understood. Unlike its related family members including GSDMD, GSDMB is not activated by inflammatory Caspases (Figure S3A-D) and does not induce inflammation in a murine model of asthma (Chao et al., 2017; Das et al., 2016). However, the specificity by which GSDMB was targeted by IpaH7.8 suggested that it may play a central role in human defense against pathogens. To then interrogate the membrane binding properties of GSDMB, we developed an in vitro activation system to directly compare the well characterized pore forming activity of GSDMD to the currently uncharacterized activity of GSDMB. A Tobacco Etch Virus protease (TEVp) cleavage site was introduced into the linker region of both GSDMB and GSDMD (denoted GSDMBTEV and GSDMDTEV) (Figure S3E and S3F). Incubation with TEVp resulted in similar levels of GSDMBTEV and GSDMDTEV activation (Figure S3E and S3F).
Previous studies have shown that activated GSDMD binds anionic phospholipids enriched on the inner leaflet of the mammalian cell plasma membrane (Aglietti et al., 2016; Ding et al., 2016; Evavold et al., 2018; Liu et al., 2016). As anticipated, the N-terminal domain of GSDMD (GSDMD-N) bound to liposomes consisting of 20 mol% Phosphoinositide (PI), PI(4,5)P2 and sulfatide (Figure 3A, lanes 4, 8, and 12; and S3G). To our surprise, neither full-length nor the N-terminal pore forming subunit of GSDMB (GSDMB-N) substantially interacted with membranes composed of these lipids (Figure 3A lanes 2, 6, and 10; and S3G). Because GSDMs may require multiple lipid species to insert into the plasma membrane, we generated liposomes from bovine liver extracts composed of 42% PC, 26% PE, 9% PI, 1% Lyso-PI, 5% cholesterol, and 17% neutral lipids. GSDMD-N interacted with this membrane preparation whereas neither full length nor the GSDMB-N subunit bound to these liposomes (Figure 3B). These data indicate that GSDMD and GSDMB exhibit distinct membrane binding properties and that the major lipid species found in mammalian cell membranes do not support GSDMB pore formation.
Figure 3. GSDMB-N forms functional oligomeric pores in E. coli derived membranes.

(A-C) Liposome binding assays with TEVp cleaved GSDMBTEV or GSDMDTEV. Proteins in the supernatant (S) or liposome pellet (P) fractions were detected after centrifugation. Ratio of the GSDM-N subunit found in the S and P fraction was determined by densitometry.
(D-E) Scanning electron microscopy (SEM) of GSDMB pores embedded in E. coli liposomes (D) or purified from liposomes by deoxycholate extraction (E).
(F) Graph showing measurement of the diameter of GSDMB-N oligomers from (D) (n=231) or from (E) (n=261). The average pore size was 13.5nm and 15.6nm, respectively and the size comparison between GSDMB and GSDMA3 (18nm) is shown.
(G-H) Diagram (G) and graph (H) of liposome dye release assay used to measure GSDMB pores in E. coli and liver liposomes as indicated.
(A-H) Results shown are representative of at least three independent biological repeats.
See also Figure S3.
GSDMB-N forms lytic pores in bacterial-mimetic membranes
The inability of GSDMB to interact with liposomes that approximate the mammalian cell plasma membrane suggested an alternative target. Because S. flexneri has evolved a sophisticated mechanism to eliminate GSDMB, we tested whether a bacterial lipid might serve as a binding substrate. Remarkably, over 90% of the GSDMB N-terminal subunit interacted with unilamellar liposomes generated from E. coli polar extracts (Figure 3C, lane 2). Neither full length nor the C-terminus of GSDMB bound to this lipid bilayer. In addition, GSDMB did not bind to liver liposomes in a side-by-side comparison (Figure 3B, lanes 2 and 4), suggesting that the GSDMB N-terminus may insert and form pores in bacterial-mimetic membranes. Indeed, negative stain electron microscopy (EM) revealed that activated GSDMB formed oligomers in artificial E. coli membrane bilayers, but not in bilayers composed of mammalian lipids (Figure 3D and S3H). Consistent with GSDMB functioning as a pore forming protein, the detergent extracted oligomers of GSDMB were comparable in size to the GSDMA3 pore (~18 nm) (Figure 3E, 3F, and S3I) (Ruan et al., 2018). In addition, activated GSDMBTEV induced carboxyfluorescein dye release from E. coli liposomes but not from liposomes generated from liver extracts (Figure 3G and 3H).
The GSDMB-N targets cardiolipin and Lipid A for binding and pore formation
The inner membrane of Gram-negative bacteria is primarily composed of phosphatidylethanolamine (PE), phosphatidylglycerol (PG), cardiolipin (CL), and glycolipids (GLs) (shown in Figure 4A) while the outer leaflet of the outer membrane is composed of Di[3-deoxy-D-manno-octulosonyl]-lipid A (KLA) (Bond and Sansom, 2004; Bos and Tommassen, 2004). To define the mechanism of bacterial membrane recognition by GSDMB, each of these lipids were mixed with phosphatidylcholine (PC) carrier lipid in a 20:80 molar ratio (Figure 4A and 4B). PE containing liposomes did not interact with GSDMB, whereas PG pelleted approximately 22% of the total GSDMB N-terminus (Figure 4B). In contrast to this moderate interaction, over 70% of the GSDMB-N subunit sedimented with liposomes containing 20 mol% CL, a product of PE and PG condensation and an integral component of the Gram-negative bacterial inner membrane (Figure 4B). CL also supported GSDMB membrane oligomerization and pore formation as assessed by negative stain EM and dye release assays, respectively (Figure 4C and 4D). Lastly, we found that the lipid component of LPS (20 mol% KLA) bound GSDMB-N, albeit at a lower relatively affinity than that observed for cardiolipin (Figure 4B).
Figure 4. GSDMB-N suppresses bacterial viability through cardiolipin, phosphatidyl-glycerol, and lipid A interactions.

(A) Chemical structures of bacterial membrane phospholipids.
(B and E) Liposome binding assays as in Figure 3A using liposomes of the lipid composition indicated. The percent of GSDMB-N found in the pellet fraction was quantified (mean ± SD; n=4).
(C) Liposomes generated from the lipids indicated were treated with control or TEVp activated GSDMB. Liposomes were imaged with 2% uranyl acetate negative staining and visualized by scanning electron microscopy (SEM).
(D and F) Dye release assays as described in Figure 3G. PC liposomes (D) or liver liposomes (F) were supplemented with 20 mol% cardiolipin (CL) as indicated.
(G) Dilution series of S. flexneri in TBS treated with TEVp and GSDMBTEV as indicated. After incubation at 37°C, reactions were diluted in 10 fold increments, and plated on LB agar.
(H and I) Quantification of recovery of exponential or stationary phase S. flexneri (H) and indicated exponential phase bacterial species (I) after in vitro incubation with activated GSDMB-N. Colony forming units (CFU) recovered after each treatment were calculated as the CFUs recovered from treated bacteria compared to CFUs recovered from TBS treated bacteria. Data are represented as mean ± SD (n=3 or 4).
(A-I) Results shown are representative of at least three independent biological repeats (B-G). Statistical analyses (B,E, H, and I) can be found in Method Details.
See also Figure S4.
These studies indicate that CL, and to a lesser degree PG and KLA, function as binding substrates for activated GSDMB. They also suggest that GSDMB discriminates between distinct chemical structures of lipids rather than simply binding negatively charged membrane surfaces. In theory, such a mechanism would allow GSDMB to distinguish between the bacterial inner membrane and mammalian cell plasma membrane despite both surfaces being negatively charged. Consistent with this notion, addition of 20mol% CL to liver polar extracts supported GSDMB membrane binding and pore formation (Figure 4E and 4F). The ability of GSDMB to interact with a lipid bilayer that mimics the plasma membrane only when reconstituted with cardiolipin, but not with other negatively charged lipids, suggests that membrane binding specificity of GSDMB has evolved through a "lock and key" mechanism.
GSDMB targets actively dividing bacteria for lytic destruction
CL, PG, and KLA are essential components of Gram-negative bacterial membranes. To determine if GSDMB selectively targets bacteria we assessed viability of S. flexneri exposed to GSDMB in vitro. Co-incubation of GSDMBTEV and TEVp inhibited recovery of S. flexneri by over 50 fold (Figure 4G and 4H). The morphology of bacteria treated with activated GSDMB was strikingly different compared to bacteria treated with inactive GSDMB as assessed by Scanning Electron Microscopy (SEM) (Figure S4A). Less than 1 μM of GSDMB-N was sufficient to inhibit S. flexneri in vitro (Figure S4B). In addition, the killing activity was greatest when GSDMB was incubated with S. flexneri in exponential growth phase (Figure 4H). These data indicate that actively dividing bacteria are more vulnerable to the pore forming activity of GSDMB, possibly because the septum between dividing cells is enriched with CL (Mileykovskaya and Dowhan, 2009). Neither the full-length nor the C-terminal domain of GSDMB inhibited S. flexneri, confirming GSDMB N-terminus is the bactericidal subunit (Figure S4B).
We then examined the ability of GSDMB to target a panel of Gram-negative and Gram-positive bacteria. The viability of E. coli DH5α, Enterohemorrhagic E. coli (EHEC) O157:H7, Citrobacter rodentium, Enterobacter cloacae, and Salmonella enterica serovar Typhimurium was significantly reduced by activated GSDMB (Figure 4I). In contrast, Gram-positive bacteria (Listeria monocytogenes, Staphylococcus aureus, and Lactococcus lactis) were unaffected (Figure 4I). Interestingly, Y. pseudotuberculosis viability was also unaffected by GSDMB activation (Figure 4I). These data suggest that alteration in bacterial membrane structure or composition may dictate target specificity by GSDMB (Hitchen et al., 2002; Rebeil et al., 2004).
NK cells activate GSDMB in response to intracellular pathogen infection
GSDMB is cleaved and activated by Granzyme A (GZMA) (Zhou et al., 2020), a serine proteinase that is expressed by tissue resident innate lymphoid cells (ILC) and delivered into bacterial or viral infected target cells (Chowdhury and Lieberman, 2008; Gasteiger et al., 2015; Panda and Colonna, 2019; Walch et al., 2014). Consistent with this report, GSDMB that was cleaved by GZMA formed pores in bacterial-mimetic liposomes and mammalian liposomes reconstituted with CL (Figure S5A-C). Additionally, GZMA activated GSDMB inhibited S. flexneri viability in vitro (Figure S5D). These studies suggest that GZMA may activate GSDMB in response to bacterial infection in vivo.
We generated transgenic mice expressing human GSDMB (note that mice do not have a GSDMB homolog) (Tamura et al., 2007), and confirmed broad tissue expression of the transgene in vivo (Figure 5A and S5E) (Chu et al., 2016; Schwenk et al., 1995). Because Shigella does not naturally infect mucosal tissues of mice, we examined GSDMB activation in context of the murine tropic pathogen Salmonella enterica serovar Typhimurium (STm). Previous studies have shown that STm establishes a replicative niche in intestinal epithelial cells and is transmitted to systemic tissues (Carter and Collins, 1974). We found that GSDMB was readily activated by STm in the caecum of orally infected mice (Figure 5B). Surprisingly however, STm had no effect on GSDMB activation levels in the liver or spleen despite robust colonization of the pathogen in these tissues (Figure 5B). We conclude that despite the ubiquitous expression of GZMA in the three tissues examined, GSDMB is selectively activated in mucosal tissues in response to bacterial infection (Figure 5B). It is important to note that transgenic expression of GSDMB had little effect on STm replication in the intestinal tract or at systemic sites in this conventional model of infection (Figure 5C). It is plausible that STm is protected from GSDMB activation by its replication vacuole or by unknown virulence mechanisms in these conventionally raised mice. Alternative models in which cytosolic STm contributes significantly to murine disease will be needed to determine the precise impact of GSDMB activation on this pathogen (Kaiser et al., 2012; Santos et al., 2001).
Figure 5. GSDMB is activated and inhibits S. flexneri intracellular growth in response to NK cells and GZMA.

(A) Diagram of the human GSDMB transgene and insertion site at the Rosa26 locus.
(B) GSDMB expression in tissues of mice infected with 1X109 CFU of WT STm by oral gavage. Western blots are representative multiple animals tested.
(C) Graphs showing the STm burden in tissues of GSDMB− (n=17) or GSDMB+ (n=12) mice. Data are represented as CFU/g tissue (log10) from two independent cohorts.
(D) Schematic for S. flexneri infection (MOI=20) of GSDMB expressing HEK293T cells followed by NK cell co-culture and quantitative analysis of bacterial recovery.
(E and F) Western blots (E) showing GZMA delivery (middle WB) and GSDMB activation (upper WB) in HEK293 cells infected with the S. flexneri strains indicated and incubated with NK-92 MI cells (or mock control). Graph (F) showing the S. flexneri CFUs recovered from WT (GSDMB−) or GSDMB+ cells incubated with NK cells. Data are represented as mean ± SD (n=3).
(C and F) Statistical analyses can be found in Method Details.
See also Figure S5.
Natural Killer (NK) cells comprise a subset of tissue resident ILCs that deliver GZMA to infected target cells (Gasteiger et al., 2015; Panda and Colonna, 2019; Walch et al., 2014). We therefore sought to determine if NK cells activate GSDMB as a mechanism of host protection against a susceptible cytosolic pathogen. GSDMB expressing cells were pre-infected with WT S. flexneri or S. flexneri ΔipaH7.8 and the remaining extracellular bacteria were eliminated using gentamicin. IL-2 activated NK cells were then co-cultured with either uninfected or infected cells (Figure 5D). Interestingly, NK cells failed to induce cleavage of GSDMB in uninfected target cells (Figure 5E, lanes 1 and 4). In addition, the N-terminal subunit of GSDMB was not detected in cells infected with WT S. flexneri (Figure 5E, lane 5). In contrast, a 28kDa fragment corresponding in size to the GSDMB N-terminal pore-forming subunit was detected in S. flexneri Δipah7.8 infected cells when co-cultured with NK cells (Figure 5E, lane 6). Thus, GSDMB activation is a direct response to NK cell recognition of pathogen infected cells, and this activation is neutralized by IpaH7.8 in cells infected with WT S. flexneri.
Next, we sought to determine if GSDMB activation by NK cells inhibits intracellular viability of a disarmed pathogen (Figure 4H). Following the procedure described in Figure 5D, we found that NK cells potently inhibited S. flexneri ΔipaH7.8 viability in GSDMB-positive cells (Figure 5F, bar 4). By comparison, NK cells had little effect on intracellular WT S. flexneri, indicating that IpaH7.8 protects the pathogen from activated GSDMB through ubiquitin-mediated proteolysis (Figure 5F, bar 2). In control experiments, NK cells did not suppress either WT or S. flexneri ΔipaH7.8 viability in GSDMB-negative cells (Figure 5F, bars 1 and 3). Collectively, these data suggest that NK cell activation of GSDMB is a host response to infection and a means for killing cytosolic bacteria.
The GZMA-GSDMB immune axis eliminates S. flexneri independent of host cell death.
A central question raised by these studies is whether GZMA-dependent activation of GSDMB eliminates pathogens by inducing host cell death or by forming pores in bacterial membranes. To address this question, we reconstituted GZMA delivery to infected cells using purified components. GSDMB-positive HEK293T cells were infected with S. flexneri, incubated with gentamicin, and then transfected with recombinant GZMA (Figure 6A). As early as two hours post GZMA transfection, GSDMB activation was detected in S. flexneri Δipah7.8, but not WT S. flexneri infected cells (Figure 6B, lanes 4 and 8). Consistent with this timing of GSDMB activation, S. flexneri ΔipaH7.8 growth and viability were markedly reduced by GZMA transfection whereas WT S. flexneri was protected (Figure 6C, S6A). The anti-bacterial effect of GZMA was caused by GSDMB activation since GZMA transfection had no effect on S. flexneri ΔipaH7.8 in GSDMB-negative cells (Figure S6A). To then assess the role of host cell death in this process, we monitored cellular release of lactate dehydrogenase (LDH) as a marker of plasma membrane disruption. S. flexneri infection caused naive (GSDMB-negative) HEK293T cells to release lactate dehydrogenase (LDH), a sign that plasma membrane integrity was disrupted by the pathogen (Figure S6B). However, the membrane damage caused by S. flexneri was not further exacerbated by GSDMB activation (Figure 6D and S6B). These studies suggest that GSDMB can limit intracellular bacterial viability and replication by a mechanism independent of host cell death. In agreement with this conclusion, microinjection of Caspase-1 into GSDMD expressing cells induced cytosolic leakage (Figure S6C), whereas delivery of GZMA had no discernable effect on the membrane integrity of GSDMB expressing cells at time points similar to those used to assess S. flexneri infection (Figure S6D). Together, these findings support the non-canonical function of GSDMB as a bacterial targeting cytolysin.
Figure 6. GSDMB is activated by GZMA and inhibits S. flexneri intracellular growth.

(A) Schematic for S. flexneri infection (MOI=20) of GSDMB expressing HEK293T cells followed by GZMA protein transfection and quantitative analysis of bacterial recovery.
(B-D) Western blots (B) showing GZMA delivery (middle WB) and GSDMB activation (upper WB) in HEK293 cells infected with the S. flexneri strains indicated and transfected with GZMA over time. Graph (C) showing S. flexneri WT (dark blue bars) or ΔipaH7.8 (light blue bars) CFUs recovered form GSDMB+ cells transfected with GZMA for the indicated times. Data are represented as mean ± SD (n=3). Graph (D) showing the levels of Lactose Dehydrogenase (LDH) in the media collected from experiments in B and C. Data are represented as mean ± SD of the CFU/well (C) and Percent LDH Release (D) (n=3).
(E-F) Diagram of the S. flexneri life cycle and the sites of GSDMD and GSDMB activity in the gut mucosa. The role of GZMA-mediated GSDMB activation and pore formation in S. flexneri is shown (inset). T3SS delivery of IpaH7.8 induced ubiquitin mediated degradation of GSDMB as a bacterial defense strategy against pore formation.
(C and D) Statistical analyses can be found in Method Details.
See also Figure S6.
DISCUSSION
In this study, we show that human GSDMB functions as an anti-bacterial cytolysin that forms pores in bacterial-mimetic membranes in vitro and restricts the growth of intracellular cytosolic bacteria in response to NK cell mediated activation. The physiological relevance of this anti-bacterial pathway for host immunity is underscored by the specificity in which a food-borne pathogen activates GSDMB in the intestinal tract of mice and by the finding that S. flexneri has evolved a strategy to counteract this host defense mechanism. Specifically, the bacterial E3 ubiquitin ligase IpaH7.8 marks GSDMB for 26S proteasomal destruction. We further show that IpaH7.8 protects S. flexneri from the bactericidal activity of NK cells via GSDMB elimination. Together, these studies shed light on the molecular arms race between S. flexneri and a mechanism of innate immunity that controls intracellular replication of cytosolically localized pathogens (Figure 6E and F).
Pore forming proteins are an ancient form of immunity that have been co-opted by mammals to fight both intracellular and extracellular pathogens (Liu and Lieberman, 2020). For example, cytolytic proteins released from the granules of lymphocytes can exhibit direct anti-bacterial activity (Dotiwala et al., 2017; Stenger et al., 1998; Walch et al., 2014). Prior to our work, Gasdermin-family members had been implicated in growth restriction of non-pathogenic E. coli (Liu et al., 2016). However, it has been unclear if direct targeting of bacteria by GSDMs is a physiologically relevant mechanism of mammalian immunity, or simply results from non-specific interactions between GSDM proteins and anionic phospholipids that are found in both prokaryotic and eukaryotic membranes. Here, we demonstrate that GSDMB indeed limits the replication of S. flexneri in context of the host cell. The specificity by which GSDMB targets bacteria is generated by its unique lipid binding profile. Previous studies suggested that full length GSDMB bound to phospholipids immobilized to a nitrocellulose support (Chao et al., 2017). However, we found that these interactions are not sufficient to support GSDMB pore formation in a membrane bilayer (Figure 3A). Rather, GSDMB binds and forms pores in membranes enriched in cardiolipin and other bacterial lipids including phosphatidylglycerol and lipid A (Figure 4B-D, S5B). These results indicate that GSDMB can discriminate between the chemical properties of lipids found in cellular membranes. Consistent with this idea, GSDMB was unable to insert into a plasma-membrane mimetic bilayer generated from a complex lipid mixture without addition of cardiolipin (Figure 4E, 4F, S5C). Moreover, activation of GSDMB by NK cells or GZMA delivery potently inhibited S. flexneri through a mechanism independent of host cell death. These studies provide compelling evidence that bacteria are a physiologically relevant target of the activated GSDMB protein.
To date, the molecular function of GSDMB in normal physiology and human disease has been enigmatic. Recent studies indicate that lytic cell death induced by GSDMB is a key event in the immune response to cancer (Zhou et al., 2020). However, this mechanism is difficult to reconcile with studies showing that the GSDMB N-terminus does not cause mammalian cell lysis through a mechanism similar to other Gasdermin family members (Chen et al., 2019; Shi et al., 2015b). The work presented here supports an alternative mechanism of membrane recognition by GSDMB that may help explain these discrepancies. We found that GSDMB-N does not interact with phospholipids commonly found in the mammalian cell plasma membrane. It is therefore intriguing to speculate that GSDMB induces cell death pathways by recognizing the mis-localization of lipids that could occur in diseased tissues or infected cells. For example, cardiolipin is a constituent of the inner mitochondrial membrane. Surface exposure of cardiolipin could trigger GSDMB pore formation and a mitochondrial-dependent cell death pathway (Chu et al., 2013). Alternatively, cardiolipin, or its precursor PG may mis-localize to the plasma membrane in transformed cancer cells where they function as GSDMB substrates. Determining the breadth of lipids that drives GSDMB pore formation and its cellular location could shed light on the diverse role of this pore forming cytolysin in autoinflammation, microbial infection, and cancer.
While this study has elucidated a unique function of GSDMB in host immunity, it also sheds light on the role of IpaH7.8 in S. flexneri pathogenesis. Upon crossing the epithelial cell barrier of the gut, S. flexneri infects tissue resident macrophage. This event results in macrophage lysis and re-infection of colonic epithelial cells that serve as a reservoir for pathogen replication (Figure 6E). IpaH7.8 appears to play two distinct roles in the S. flexneri lifecycle. First, IpaH7.8 activates macrophage cell death by ubiquitinating and degrading negative regulatory components of the NLRP1/NLRP3 inflammasomes (Sandstrom et al., 2019; Suzuki et al., 2014; Suzuki et al., 2018). While NLRP1 inflammasome activation has been proposed to enhance immunity to microbial infection, lysis of macrophage also promotes the dissemination of S. flexneri from immune cells to the colonic epithelium (Schroeder and Hilbi, 2008; Zychlinsky et al., 1992; Zychlinsky et al., 1996). Thus, inflammasome activation appears to benefit both the host and the pathogen. Our data are consistent with these studies as IpaH7.8 does not ubiquitinate the primary executioner of inflammasome mediated cell death GSDMD. Second, IpaH7.8 potently suppresses NK cell mediated killing of S. flexneri by ubiquitinating and targeting GSDMB for proteolytic destruction (Figure 6F). Importantly, GSDMB is primarily expressed in barrier epithelial cells (Carl-McGrath et al., 2008). Thus, IpaH7.8 supports replication of S. flexneri in its predominant cellular niche by protecting the pathogen from GSDMB pore formation. It is currently unclear how IpaH7.8 distinguishes between GSDMB and other Gasdermin family members. Elucidating this mechanism may explain how S. flexneri navigates the complex cellular environment of the gut mucosa by controlling multiple arms of the innate immune system.
While Shigella encodes a Type III effector protein that evolved to inhibit the GZMA/GSDMB innate immune signaling axis in humans, other bacterial pathogens may exhibit alternative mechanisms of protection. For example, bacteria that reside in a phagocytic vacuole, such as STm, would likely have minimal exposure to activated GSDMB in the cytosol. However, it remains to be determined if GSDMB targets the small but biologically significant proportion of STm found in the cytosol of epithelial cells (Knodler et al., 2014; Malik-Kale et al., 2012). We also found that unlike other Gram-negative bacteria, Yersinia was unaffected by GSDMB activation in vitro. It will be informative to determine if the cell surface lipid structure of this pathogen is refractory to GSDMB recognition, or if it secretes a virulence factor as a mechanism of protection similar to other Gasdermin activation pathways (Orning et al., 2018; Sarhan et al., 2018). Future studies will be needed to define the breadth of bacterial species targeted by GSDMB and to determine if human pathogens have indeed evolved mechanisms to counteract this arm of the innate immune system. If so, then activation of the GZMA/GSDMB signaling axis may be a critical factor in determining human disease outcomes to infection by numerous bacterial pathogens.
Limitations of Study
We have yet to determine if NK cells eliminate additional Gram-negative bacteria through GSDMB activation or if other pathogens have evolved virulence strategies to counteract this host defense mechanism. In addition, the current studies on S. flexneri are limited to cell-based assays since this pathogen does not naturally infect genetically tractable mammals and because GSDMB is a human gene not found in mice. Newly developed mouse models of S. flexneri infection (Mitchell et al., 2020) combined with transgenic expression of GSDMB will be needed to define this mechanism of S. flexneri virulence in vivo. Similarly, determining the cell types of the gut mucosa targeted by NK cells in this model of GSDMB function requires further investigation.
STAR METHODS
RESOURCE AVAILABILITY
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Neal M. Alto (neal.alto@utsouthwestern.edu). Plasmids, primers, recombinant protein, experimental strains, and any other research reagents generated by the authors will be distributed upon request to other research investigators under a Material Transfer Agreement.
DATA AND CODE AVAILABILITY
This study did not generate/analyze unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
E. coli DH5α chemically competent cells were used for molecular cloning. E. coli BL21 chemically competent cells were used for protein expression. E. coli were grown in LB medium cultured at 37°C. For cellular infections, Shigella flexneri was grown overnight at 30°C in LB and then subcultured 1:50 in fresh LB at 37°C the day of the infection to OD600 0.6-0.8. For mouse infection, Salmonella enterica serovar Typhimurium (strain SL1344, Strepr) was grown overnight at 37°C and then subcultured the next day in fresh LB at 37°C to an OD600 0.6. For in vitro bacterial killing assays, bacteria were cultured in BHI at 37°C overnight and then subcultured another 3 hours the next day at 37°C. HEK293T cells (human; sex: female, kidney epithelial) were grown in Dulbecco's Modified Eagle Medium (DMEM) high glucose, supplemented with 10% Fetal Bovine Serum, 1% Non-Essential Amino Acids (NEAA). H1299 (human; sex: male, lung epithelial) HCC1954 (human; sex: female, breast epithelial) cells were grown in RPMI 1640 with L-glutamine 10% Fetal Bovine Serum, and 1% NEAA. NK-92 MI cells (human; sex: male, peripheral blood Natural Killer) were grown in Alpha Minimal Essential medium (αMEM) without ribonucleotides and deoxyribonucleosides, 20% Fetal Bovine Serum, 2mM L-glutamine, 1.5g/L sodium bicarbonate, 0.2mM myo-inositol, 0.02mM Folic Acid, 10mM HEPES, and 0.1mM 2-mercaptoethanol. All cell lines were grown in at 37°C under 5% CO2. HEK293T and NK-92 MI cells were obtained from ATCC. H1299 and HCC1954 cells were obtained from the Minna Lab at UTSW. Mice were female 6-8 weeks old. To obtain experimental mice C57BL6 GSDMB+/+ female mice were crossed with male CMV-Cre+/− mice. CMV-Cre is X-linked and thus all female pups will be GSDMB+/− CMV-Cre+/−. These female mice were housed as littermates in 5 mice/cage and used in infection experiments. After treatment, Western blots were performed to determine GSDMB expression. Since CMV-Cre is X-chromosome linked GSDMB expression was detectable in half of all mice (GSDMB+ group) and silenced in the other half (GSDMB-group).
METHOD DETAILS
Purification of recombinant proteins.
To obtain full length untagged GSDMB and IpaH7.8, E. coli BL21 cells were transformed with the pET28b-6xHis-MBP-His-TEV-GSDMB vector or pProEx-HTa-IpaH7.8 vector and were grown in LB medium supplemented with kanamycin (pET28b) or ampicillin (pProEx-HTa). After cultures reached OD600 of 0.7, protein expression was induced with 0.5 mM IPTG overnight at 18°C. Cells were lysed using an Emulsiflex C5 (Avestin) in buffer containing 20mM HEPES, 150mM NaCl, 1mM TCEP and 25mM Imidazole at pH7.5. The fusion proteins were affinity purified using Ni-NTA agarose (Qiagen). His-MBP-His-GSDMB or His-IpaH7.8 were eluted in buffer containing 500mM Imidazole and dialyzed into 20mM HEPES, 150mM NaCl, 1mM TCEP (pH7.5). The His-MBP-His- or His-tags were removed by overnight digestion with TEV protease at room temperature. TEV protease was removed from the cleavage reactions with Ni-NTA agarose and the untagged proteins were further purified using a Superdex highload 200 16/600 gel filtration chromatography column (GE Healthcare life sciences).
To obtain C-terminal 5xHis-tagged GSDMBTEV, E. coli BL21 carrying pET21a-GSDMBTEV were grown in LB media supplemented with ampicillin. After the cultures reached OD600 0.7, protein expression was induced with 0.5mM IPTG at 17°C overnight. Cells were lysed using an Emulsflex C5 (Avestin) in buffer containing 20mM HEPES, 150mM NaCl, 1mM TCEP, and 25mM Imidazole (HBSi) with the final pH at 7.5. The fusion protein was affinity purified using TALON Metal Affinity Resin (Takara Biosciences) and eluted in HEPES buffer containing 500mM Imidazole. The protein was concentrated using a 10,000 MWCO Amicon 50 spin concentrator and further purified using a Superdex highload 200 16/600 gel filtration column (GE Healthcare Life Sciences).
To obtain untagged GSDMDTEV, the GSDMD coding region including a TEV cleavage site in the interdomain linker, was cloned into pET28b-MBP. The HIS-MBP-HIS-3C-GSDMDTEV protein was purified from E. coli BL21 using TALON Metal Affinity Resin (Takara Biosciences) in HBSi. GST-3C protease was added to the HIS-MBP-HIS-3C-GSDMDTEV bound to TALON Resin and resin suspension was agitated overnight at 4°C. The resin suspension was run over a chromatography column (BioRad) packed with GST-Agarose (GE) to remove the TALON resin and the GST-3C protease. The flow-through was then concentrated in an 30,000 MWCO Amicon 50 spin filter and purified over a Superdex highload 200 16/600 gel filtration column (GE Healthcare Life Sciences).
To obtain GST-tagged GSDMA, GSDMB, GSDMD and GSDME, E. coli BL21 were transformed with the respective Gasdermin constructs cloned in pGEX4T-1 and grown in LB media supplemented with ampicillin. After the cultures reached OD600 0.7, protein expression was induced with 0.5mM IPTG at 17°C overnight. Cells were lysed using an Emulsiflex C5 (Avestin) in buffer containing 20mM HEPES, 150mM NaCl, and 1mM TCEP pH 7.5. The fusion proteins were affinity purified using glutathione sepharose beads (GE Healthcare Life Sciences). GST-GSDMs were eluted in buffer containing 50mM reduced glutathione at pH8.0 and dialyzed overnight at 4°C into HEPES buffer without glutathione.
To obtain recombinant GST-IpaH-3xFLAG-Ub proteins for UBAIT experiments, full length IpaH2.5, IpaH4.5, IpaH7.8, and IpaH9.8 were cloned into pGEX6P-1 together with a 3xFLAG peptide followed by the coding sequence for ubiquitin using Gibson Cloning (NEB). Constructs were sequence confirmed and then transformed into E. coli BL21 and purified using the same protocol as other GST tagged proteins.
To obtain recombinant GZMA protein, HEK293T cells were plated at a density of 4x106 cells per 15cm dish. The next day cells were transfected with pHL SEC GZMA plasmid using Calcium-Phosphate transfection (Au - Dotiwala et al., 2015). Three days later GZMA was purified from the media using TALON Resin (Takara). Resin was washed with 20 volumes of 50mM TRIS pH 8.0, 150mM NaCl, and then eluted in the same buffer with 250mM Imidazole. Elution was concentrated to 6ml and buffer exchanged into 4L of 50mM TRIS pH 8.0, 50mM NaCl, and 4mM CaCl2 for 4 hours at room temperature. Enterokinase was then added to the dialysis cassette with GZMA and incubated at room temperature overnight in fresh buffer. The next day the dialysis cassette was placed in 4L 50mM bisTRIS pH 5.8, 150mM NaCl and buffer exchanged for 4 hours at room temperature. GZMA protein was then loaded onto a Capto-S column (GE/Cytiva) and eluted on a linear gradient using 50mM bisTRIS pH 5.8, 1M NaCl. Fractions were collected, pooled, and activity was tested by monitoring cleavage of GSDMB.
UBAIT Substrate Capture Assays.
For UBAIT assays to capture unknown substrates from cell lysates, CaCo-2 cells were grown to confluency in 3 dishes (15 cm). Cells were washed in cold PBS and lysed in 300μl Lysis Buffer (1x Ubiquitination buffer (Boston Biochem), 1% Triton X-100, 1x protease inhibitor (Sigma), 1mM DTT). Then the lysate was centrifuged at 13,000g for 10 mins at 4°C. The supernatant (~460μl) was moved into a new tube and 25μg of the GST-IpaH-3xFLAG-Ub construct was added and rotated 1h at 4°C. Then His-UbE1 (100nM), His-UbcH5b (2000nM), ATP (1mM), MgCl (1mM) and 1x Ubiquitination buffer (Boston Biochem) were added to a final volume of 500μl. The reaction was left at 30°C for 10 minutes and then 300μl TBS buffer (25mM Tris-HCl pH7.4, 150mM NaCl, 1mM DTT) and 30μl of washed GST beads were added and rotated for 2h at 4°C. Beads were washed 4 times in TBS-T (TBS + 0.5% Triton-X100) and 2 times in TBS and then eluted in 150μl reduced glutathione (pH8.0). Beads were centrifuged and the supernatant taken to a new tube. 0.25% SDS (final concentration) and 5mM DTT (final concentration) were added and the solution was boiled 5 min. at 95°C. 1.2ml TBS was added followed by 20μl M2-FLAG beads (Sigma) and tube was rotated for 2h at 4°C. Then beads were washed 4 times in TBS-T followed by 2 times in TBS. Finally, 35μl of hot (95°C) SDS-Loading buffer was added and the beads were boiled an additional 5 mins at 95°C. Samples were electrophoresed on a precast SDS-PAGE gradient gel (Biorad) and Coomassie stained. To identify captured proteins, a lane of gel above the unmodified IpaH-UBAIT band was excised and proteins were digested in-gel with trypsin and run on a Q Exactive MS platform at the Proteomics Core Facility at University of Texas Southwestern Medical Center.
For assays involving capture of purified proteins, 25μg GST-IpaH-3xFLAG-Ub constructs were combined with 25μg recombinant GST-HOIP (de Jong et al., 2016), GST-GSDMB or GST-GSDMD in a similar Ub reaction as shown above (but without addition of cell lysate). In this assay 35μl of final sample in SDS-LB was split in 2 equal volumes and run on 2 SDS-PAGE gels: one for Coomassie and one for Western blotting using anti-HOIP (R&D Systems), anti-GSDMB (Covance) or anti-GSDMD (Cell Signaling) antibodies.
Protein binding assays.
30μg GST, GST- GSDMBFL, GST-GSDMBCT or GST-GSDMDFL was incubated for 1h with 30μl GST agarose with end-over-end mixing at 4°C in 750μl 20mM HEPES, 150mM NaCl, 1mM TCEP (pH7.5). Protein-bound agarose was washed 4 times with 750μl binding buffer. Then, 30μg His-IpaH2.5 or His-IpaH7.8 was added to the protein-bound agarose in 750μl binding buffer and incubated for 1h at 4°C end-over-end. Protein-bound agarose was washed 4 times with 750μl binding buffer. Proteins were eluted by resuspending the GST agarose in 35μl 2x Loading buffer with BME and boiling for 10 min. Eluted proteins were separated by SDS-PAGE and stained with Coomassie brilliant blue. Inputs were generated by loading 10μg of each protein for SDS-PAGE.
For size exclusion chromatography (SEC) based binding assays, untagged full length GSDMB and IpaH7.8 were first run individually over a S200 10/300 GL increase column. Then, GSDMB and IpaH7.8 were mixed at equimolar concentrations and incubated for 1h at 4°C end-over-end, and then run over the S200 10/300 GL increase column.
Mass Spectrometry.
To determine which lysines in GSDMB are ubiquitinated by IpaH7.8, a ubiquitination reaction (30μl) was performed with His-IpaH7.8WT or His-IpaH7.8C357S. After 5h reactions were run on an SDS-PAGE gel (BioRad), Coomassie-stained bands were excised and proteins were digested in-gel with trypsin and run on a Q Exactive MS platform at the Proteomics Core Facility at University of Texas Southwestern Medical Center.
In vitro ubiquitination reactions.
In vitro ubiquitination reactions were performed in 50mM HEPES (pH7.5), 150mM NaCl, 20mM MgCl2, and 10mM ATP (Stieglitz et al., 2012). Components were mixed as indicated at the following concentrations 1μM UbE1 (E1), 5μM UbcH5b (E2), 500nM-10μM IpaH7.8WT or IpaH7.8C357S, 50μM Ubiquitin (WT, K48R, K48 only, ΔK, or 1-75aa (ΔG )), and 5μM GST-tagged GSDMA, GSDMB, GSDMD, GSDME, or un-tagged GSDMB. Reactions were incubated for 2-5h at 30°C, and then stopped with 2x Loading buffer containing β-Mercaptoethanol (BME) and boiling for 10 min. Samples were processed for Western Blotting and blotted with anti-GST, anti-His, anti-GSDMB, or anti-Ubiquitin antibodies.
Immunoprecipitation assays.
FLAG-GSDMA, FLAG-GSDMB, FLAG-GSDMD, and FLAG-GSDME were expressed in combination with GFP-IpaH7.8C357S in 293T cells. After 24h, cells were collected and lysed in 25mM Tris-HCl (pH7.4), 150mM NaCl, 1% Triton, 1x protease inhibitor (Sigma Aldrich). Lysate was added to 15μl FLAG-M2 agarose beads (Sigma Aldrich) and incubated for 1h at 4°C with end-over-end mixing. Beads were washed 10 times in lysis buffer and 50μl 2x SDS loading buffer (BioRad) was added.
For detection of GSDMB ubiquitination during Shigella flexneri infection, 300,000 GSDMB-FLAG HEK293T cells were infected with WT or ΔipaH7.8 at an MOI of 50 for 1.5 hours then cells were washed with DMEM/20mM HEPES, then replaced with media containing 50ug/mL gentamicin for 1 hour. Cells were then was two times with 2x cold PBS and lysed in 50mM TRIS (pH8.0) 150mM NaCl, 1% Triton, 1x protease inhibitors, and benzonase. Lysate was added to 40ul FLAG-M2 agarose beads (Sigma Aldrich) and incubated for 2 hours at 4°C with end-over-end mixing. Beads were washed six times with lysis buffer and eluted by boiling in 44μl 3x SDS loading buffer (BioRad).
Cellular degradation assays.
This assay was performed as described previously (de Jong et al., 2016). Briefly all non-redundant Shigella flexneri IpaH genes were cloned into pEGFP-C2 and substrates HOIP, GSDMA GSDMB, GSDMC, GSDMD, GSDME or GBP1 were cloned into pCMV-FLAG-6b. For assays, 150ng of pEGFP-C2 plasmid was co-transfected together with 350ng pCMV-FLAG-6b plasmid into HEK293T cells seeded in a 24 well plate at 1x105 cells per well. After 40h, cells were lysed in PBS + 1% Triton-X100, added to an equal volume of 2x SDS Loading buffer (Biorad) and processed for Western blotting. For MG132 assays, cells were transfected as above and 16h after transfection 10μM MG132 or equal volume of vehicle (DMSO) was added to wells. After 24h samples were processed as described above.
Production of anti-GSDMB antibodies in rabbits.
GSDMB antibody used in Figure 1C and 1D was raised against the C-terminus of GSDMB (GSDMB-CT). GSDMB-CT was first cloned into pET28-MBP and purified from E. coli BL21 using Ni-NTA beads. His-MBP was then cleaved from the protein using TEV protease and removed by running the sample on a His-trap column. GSDMB antibody was produced from inoculation of 2 rabbits and purified from serum using an affinity column loaded with GSDMB-CT (Covance). The resulting anti-GSDMB antibody was tested in our lab for activity by Western blotting. The antibody specifically detects purified GST tagged, His-MBP-tagged and FLAG-tagged GSDMB full length proteins.
Construction of a Shigella ΔipaH7.8 strain.
A Shigella flexneri M90T ΔipaH7.8 strain was constructed by replacement of ipaH7.8 coding region with a kanamycin resistance cassette by homologous recombination followed by flipase catalyzed removal of the cassette (Datsenko and Wanner, 2000). The deletion was confirmed by sequencing.
Construction of GSDMB with TEV cleavage site (GSDMBTEV).
Amino acid residues 240-246 in the loop between the N- and C-terminal domains of GSDMB were replaced with a TEV protease cleavage sequence (ENLYFQS) and cloned into the bacterial expression vector pET21 (for His-tag purification from E. coli BL21), or into pCMVFLAG-6b (for expression in mammalian cells).
Caspase cleavage assay.
10μg of untagged recombinant GSDMB and His-MBP-GSDMD were incubated with 1 unit of recombinant active human Caspases 1-10 in 1x reaction buffer supplied from the manufacturer Biovision. Reactions were incubated for 3 hours at 30°C, stopped with addition of 2x Loading buffer, separated by SDS-PAGE and stained with Coomassie.
Liposome preparation.
All lipids and lipid extracts were purchased from Avanti Polar Lipids. To prepare unilamelar liposomes from E. coli and bovine liver lipids, polar extracts were hydrated at 5mg/ml with 20mM HEPES, 150mM NaCl (pH7.5) and then vortexed continuously for 5min at room temperature. To prepare unilamellar liposomes of defined lipid composition, Phosphatidylcholine (PC) was mixed with either phosphoinositide (PI), phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2), sulfatide, phosphatidylethanolamine (PE), phosphatidylglycerol (PG), cardiolipin (CL), or Di[3-deoxy-D-manno-octulosonyl]-lipid A (Kdo2-Lipid A, KLA) at an 80% to 20% molar ratio. Lipids were hydrated at 5mM with 20mM HEPES, 150mM NaCl (pH7.5) and then vortexed continuously for 5min at room temperature. Liposomes were generated by extrusion of the hydrated lipids through a 100nm polycarbonate filter (Whatman) 31 times using a Mini-Extruder device from Avanti Polar Lipids Inc. The CL complemented bovine liver liposomes were made by dissolving 20mg of bovine polar liver extract in chloroform. Then 5mg of CL dissolved in chloroform was added, and vortexed continuously for 5 minutes. The lipids, dissolved in chloroform, were placed on a heat block set to 50°C and evaporated under a stream of nitrogen. The lipid film was then hydrated in HEPES buffer and extruded as before.
Liposome Binding Assay.
Liposome binding assays were adapted from (Ding et al., 2016). GSDMBTEV/GSDMDTEV (5μM) and TEV protease (4μM) were incubated along with the indicated liposomes (500μM lipids) at 4°C overnight in a total volume of 80μl. After overnight incubation, samples were centrifuged in a Beckman Optima Max-TL Ultracentrifuge at 4°C for 20min. at 40,000 rpm to pellet liposomes. The supernatant (S1) fraction was collected to examine proteins not bound to the liposomes. The pellet (P) fraction was washed once in 500μl 20mM HEPES, 150mM NaCl by re-centrifugation and brought to equal volume as the supernatant. The S and P fractions were analyzed by SDS-PAGE followed by Coomassie blue staining.
Scanning Electron Microscopy of Liposomes, GSDMB pores, and S. flexneri.
For imaging GSDMB pores in liposomes, GSDMBTEV (5μM) and TEV (4μM) were incubated together with the indicated liposomes (500μM lipids) at 4°C for 7h in a total volume of 40μl. Samples were then diluted 1:1 with HEPES buffer and 10μl of the sample was transferred to carbon 400 mesh support films (Figure 3D, 3E, 4C, S3H). For imaging purified GSDMB pores, GSDMBTEV was cleaved with TEVp in the presence of E. coli polar liposomes overnight at 4°C. Reactions were then pelleted at 40,000 rpm for 20min at 4°C. The supernatant (S1) was removed and 4x loading buffer was added. The pellet fraction containing GSDMB-N were either resuspended in HEPES buffer or buffer containing 50mM deoxycholate and incubated at room temperature for 1 hour. The reactions were then centrifuged at 17,000g for 30 minutes. The supernatant (S2) was collected to examine solubilized GSDMB-N. The pellet fraction was then resuspended in 1x loading buffer. The S1, S2, and P fractions were analyzed by SDS-PAGE followed by Coomassie blue staining (Figure S3E). The soluble GSDMB-N S2 fraction was diluted 1:1 with 50mM deoxycholate containing buffer and 10μl of sample was transferred to carbon 400 mesh support films. Samples were stained with 2% uranyl acetate for visualizing membrane bound GSDMB-N and 1% uranyl formate for visualizing soluble GSDMB-N (adapted from (Liu et al., 2019)) (Figure 3E). For imaging S. flexneri, GSDMB-N treated bacteria were fixed with 2% glutaraldehyde, stained with 2% uranyl acetate, and transferred to Formvar/carbon- 400 mesh support films. All samples were imaged on a Tecnai G2 Spirit Biotwin (FEI) at 120kV. All Images were obtained using a Gatan 2k x 2k CCD camera. GSDMB-N diameter of embedded and solubilized pores were measured by importing the raw EM files into Image J and using measure function.
Liposome Dye Release Assay.
Liposome dye release assays were adapted from (Peng et al., 2019) and (Ding et al., 2016). 5,6-carboxyfluorescein encapsulated liposomes were prepared by resuspending E. coli, liver polar, liver polar + CL, 100% PC, or 80% PC/20% CL in buffer containing 20mM HEPES 150mM NaCl and 100mM 5(6)-carboxyfluorescein (pH7.5) (Peng et al., 2019). The lipid suspension was extruded 31 times through a 100nm polycarbonate membrane. Liposomes containing carboxyfluorescein dye were diluted to 300μM in HEPES buffer and the Ft0 was measured using excitation at 480nm and emission at 517nm for 5 cycles or approximately 2.5min to obtain a baseline reading. GSDMBTEV or TEVp cleaved GSDMBTEV were added to reactions at 1.0μM in a final volume of 100μl and dye release was monitored over 45min. After 45min. 10μl 1% Triton X-100 was added to each well and fluorescence measured for an additional 2.5min. Percent dye release was calculated with the baseline set at 0% and after Triton X-100 addition set at 100%. Conditions for GSDMB cleaved by GZMA were the same as in TEVp cleaved GSDMBTEV reaction.
LDH release assay.
LDH release assays were performed following manufacturer’s protocol (Promega).
Construction of GSDMB transgenic mouse.
To test GSDMB function in a mouse model we cooperated with the UT Southwestern Medical Center Transgenic Core to generate a mouse GSDMB-1 (isoform 1) knock-in strain. Human GSDMB was 3xFLAG tagged at the C-terminus and inserted into the mouse Rosa26 locus using CRISPR/Cas9 according to methods published previously (Chu et al., 2016). The plasmid pRP26-GSDMB-FLAG was constructed and was used as template to repair CRISPR/Cas9 mediated breaks in the Rosa26 locus leading to the insertion of GSDMB-3xFLAG coding region into the mouse Rosa26 locus. The pRP26-GSDMB-FLAG construct was injected into WT C57BL/6 (Charles River) embryos, which were implanted into pseudo pregnant female C57BL/6 mice. This resulted in the birth of a total of 60 pups and screening these pups by PCR identified one female mouse positive for GSDMB and Neo and with correct Rosa26 integration (R26F4/SAR PCR). This female founder was bred with WT C57BL/6 (Charles River) male mouse resulting in 8 pups. PCR analysis on these mice resulted in the identification of 4 GSDMB−/+ mice. These mice were intercrossed to obtain homozygous GSDMB+/+ female mice used for further breeding with CMV-Cre (Schwenk et al., 1995) male mice.
Mouse infection experiments.
All experiments conducted were in accordance with the policies of the Institutional Animal Care and Use Committee at UT Southwestern Medical Center. For mouse infection, Salmonella enterica serovar Typhimurium (strain SL1344, Strepr) was grown overnight at 37°C and then subcultured the next day in fresh LB at 37°C to an OD600 0.6. GSDMB+/− CMV-Cre+/− mice that were 6-8 weeks old were orally infected by gavage with 1x109 STm . After 4 days, mice were euthanized and spleen, liver, and ceacum were removed, weighed, washed, and homogenized in PBS. Extracellular bacteria were eliminated from the caecum by incubation with gentamicin (100μg/ml, 30 minutes) prior to tissue homogenization. For CFU counts, homogenates were 10-fold serial diluted and plated on LB agar plates + Streptomycin. For Western Blotting, homogenates were centrifuged at 5000g at 4°C and supernatants were added to equal volumes of SDS-LB (Biorad). Antibodies used to detect GSDMBFLAG expression were anti-FLAG (Sigma) or anti-GSDMB (Abcam).
In vitro bacterial killing assay.
Bacteria were grown overnight in BHI and diluted next day 1:50 in BHI (for log phase sample) or kept undiluted (for stationary sample) and grown another 3h at 37°C. Then 1ml of the culture was centrifuged at 4000g for 2min., washed 1x in TBS and then resuspended in TBS at 1x109 cells/ml (volume depending on OD 600 measurement). For the killing assay 15μl reactions were set up in TBS (50mM Tris pH8.0 150mM NaCl) and included TBS alone, 0.2μg/μl TEV alone, 64μM GSDMBTEV alone or 0.2μg/μl TEV + 64μM GSDMBTEV. Bacteria (5x106 cells in 5μl) were immediately added to the reaction and incubated 2h at 37°C. Then serial dilutions were plated on BHI agar to determine recovered CFU. For GZMA mediated activation of GSDMB, 20μl reactions were set up in TBS and included TBS alone, 1μM GZMA alone, 1μM GSDMB alone, or 1μM GZMA + 3.8μM GSDMB. Shigella flexneri (5x106 cells in 5 μl) were immediately added to the reaction and incubated 2h at 37°C. Then serial dilutions were plated on LB agar to determine recovered CFU.
Generation of GSDMB and GSDMD overexpressing cell lines.
GSDMB-FLAG was cloned into pTRIP-IRES-RFP (Schoggins et al., 2011) and pSRBL(Richardson et al., 2018) and lentivirus was produced as described previously (Perelman et al., 2016). HEK293T (pTRIP) or H1299 (pSCRBL) cells were then transduced with GSDMB-FLAG pTRIP-IRES-RFP or GSDMB-FLAG pSCRBL lentivirus via spin inoculation for 1 hour and incubation for an addition 6 hours at 37°C. Cells were then incubated at 37°C for two days. GSDMB-FLAG HEK293T cells were then sorted for RFP expression. Cells were allowed to recover and expand prior to determining GSDMB-FLAG expression by western blot. For GSDMB-FLAG H1299 cells, cells plated on 15cm dishes and were selected in media containing 6μg/mL blasticidin for eight days. Colonies were picked from the plate and expanded. GSDMB-FLAG expression in H1299 cells was determined via western blotting. GSDMD was cloned into pSCRBL lentivirus was produced and H1299 cells were transduced, selected for using the method described above.
Shigella infection of human cell lines.
HCC1954 and HEK293T cells were plated at 1.5x105 and 2x105 cells per well, respectively, the day before infection with Shigella flexneri. 3ml LB overnight cultures of Shigella flexneri M90T WT, ΔipaH7.8, and Δmxid were started on the day cells were plated and grown at 30°C. The next day cultures were diluted 1:50 in LB and grown for 2.5h at 37°C. OD600 of the cultures was checked and 1ml of the cultures were centrifuged at 4000g for 2min., washed in PBS, and then resuspended in DMEM/20mM HEPES at 1x109 cells/ml and incubated at 37°C for 15min. Cells were infected at an MOI of 300 and spun for 15min. at 1000g. Cells were placed back in the incubator for 1.5h. Then cells were washed with PBS, media containing 50μg/ml gentamicin was added and cells were placed back in incubator for an additional 2.5h. Cells for CFU assay were lysed in 200μl PBS 1% Triton X-100 and serial dilutions were made and plated on LB agar plates. Cells for western blot were lysed in 75μl 2x Loading buffer with BME. Lysates were separated by SDS-PAGE, transferred to nitrocellulose membranes and blotted with anti-GSDMB and anti-actin antibodies.
For the S. flexneri infection time-course, GSDMB-FLAG HEK293T cells were plated at 3.0x104 cells per well, the day before infection. Shigella flexneri cultures were prepared same as above. GSDMB-FLAG cells were infected at an MOI of 20 for, spun at 800g for 10 minutes and incubated at 37°C for 1.5 hours. Then cells were washed with DMEM/20mM HEPES once and media containing 50μg/ml gentamicin was added and cells were placed back in incubated for 0-4 hours. Cells for western blotting were lysed in 25μl lysis buffer (50mM TRIS, 150mM NaCl, 1% Triton X-100, 2% sodium deoxycholate at pH 8.0) containing fresh protease inhibitors and benzonase. SDS was added to the lysate at a final concentration of 1% and loading buffer with was added prior to boiling. GSDMB protein levels were determined by blotting with anti-GSDMB and anti-actin antibodies. Cells for CFU assay were lysed in 200μl PBS 1% Triton X-100 and serial dilutions were made and plated on LB agar plates.
Immunofluorescence staining and image processing.
H1299 cells (WT or stably expressing GSDMB-FLAG) were plated at 50,000 cells/well on glass coverslips in a 24-well plate. Cells were infected with Shigella (MOI = 100) using a standard infection protocol (previously described). Cells were incubated with bacteria for 1.5 hrs, washed once with complete media, incubated with media containing 50 μg/ml gentamicin for 1 hr, washed again and incubated an additional 2hrs in media containing 50 μg/ml gentamicin. Cells were then washed twice with PBS and fixed with 4% PFA (in PBS) for 10 min. at room temperature. After fixation, cells were washed 3 times with PBS, permeabilized in 0.5% TX-100 + 1% BSA (in PBS) for 5 min., washed twice with PBS and blocked in 1% BSA (in PBS) for 30 min. Cells were incubated with rabbit anti-GSDMB primary antibody (1:1000, abcam 215729) 1 h at RT, washed 3 times in PBS and then incubated with goat anti-rabbit TX Red (1:200, Invitrogen T2767) for 1 h. After antibody incubation, cells were washed 3x with PBS, once with water and coverslips were mounted on slides with Prolong Glass mounting media (Invitrogen P36980) and imaged on an Observer.Z1 inverted microscope (Zeiss).
Microinjection of cells.
H1299 cells (WT or stably expressing GSDMB-Flag or GSDMD) plated at 150,000 cells/well in 60 mm culture dishes 2 days before microinjection. Just prior to microinjection, culture media was replaced with media supplemented with 20 mM HEPES. Cells were located using an Axio Observer.A1 microscope (Zeiss) and microinjected using pulled glass pipettes (Sutter Instruments P-97) and an Eppendorf FemtoJet and InjectMan NI 2 according to manufacturer’s instructions. Cells were injected with a solution containing 2 mg/ml purified GZMA (WT and GSDMB cells) or 0.125 U/μl Caspase 1 (Biovision 1081-25; WT and GSDMD cells) along with 1 mg/ml of either Texas Red Dextran 10,000 MW (Invitrogen D1863 ) or Cascade Blue Dextran 10,000 MW (Invitrogen D1976) in PBS. Live microinjected cells were identified by Texas Red fluorescence and the same field of live cells was imaged at 0h, 2h and 4 h after microinjection using an Eclipse Ti-S microscope (Nikon). Cells were then washed 3 times with PBS, fixed in 4% paraformaldehyde (in PBS) for 10 min., washed 3 times in PBS and coverslips were mounted onto glass slides using VectaShield Plus Antifade Mounting Medium (VectaShield H1900). Microinjected cells were identified by Cascade blue labeling and imaged on an Observer.Z1 inverted microscope (Zeiss).
Measurements of intracellular bacteria and host cell death.
WT and GSDMB-FLAG expressing HEK293 cells were plated in a 48 well dish coated in poly-lysine at a density of 3x104 cells per well the day before infection with Shigella flexneri. 3ml LB overnight cultures of Shigella flexneri M90T WT and Δipah7.8 were started on the day cells were plated and grown overnight at 30°C. The next day cultures were diluted 1:50 in fresh LB and grown for an additional 2.5h at 37°C. OD600 of the cultures was checked and 1ml of the cultures was centrifuged at 4000g for 2 min., washed with PBS, and then resuspended in DMEM/20mM HEPES resulting in 1x109 cfu/ml and incubated at 37°C for 15min. Cells were infected at an MOI of 20 and spun for 10min at 800g. Cells were placed back in the incubator for 1.5h. After 1.5h cells were washed with DMEM/20mM HEPES, media with 50μg/ml gentamycin was added and cells were placed back in the incubator for 1h. After incubation, cells were washed with DMEM/20mM HEPES and then transfected with 15μg GZMA protein or mock transfected using the Pierce Protein Transfection Reagent according to the manufacturers protocol (Thermofisher). After 4h of transfection, 50μl of media was aliquoted in quadruplicate to a 96-well plate. The Cytotox 96 non-radioactive Cytotoxicity Assay (Promega) was used to measure LDH release from cells according to the manufacturer’s protocol. For western blot, cells were lysed in the well with 25μl lysis buffer (25mM Tris pH8.0, 150mM NaCl, 1% Triton X-100, 2% Sodium Deoxycholate with fresh protease inhibitors (Millipore-Sigma) and benzonase (Novagen). Lysate was transferred to a separate tube and 2.5μL 10% SDS, and 4x Loading buffer (Bio Rad) were added. Lysates were boiled at 98°C for 10min. and 16μl of lysate per sample was run on 12.5% acrylamide SDS-PAGE, transferred to nitrocellulose membranes, and blotted with antibodies against GSDMB, GZMA, and actin. For CFU, cells were lysed in 200μl 1% Triton X-100 in PBS and serial dilutions were made and plated on LB agar. CFU recovered was calculated as the percentage of colonies recovered compared to mock transfected control per Shigella flexneri genotype.
For the intracellular Shigella flexneri killing time-course, the protocol was the same as above except samples for western blot, LDH release, and CFU analysis were taken and processed at 0, 1, 2, and 4 hours post transfection with GZMA.
For NK-92 MI cell mediated intracellular bacteria killing, HEK293 GSDMB-FLAG and 293 WT cells were infected under the same conditions as with GZMA transfection. After 1h in media containing gentamicin, cells were washed 1 time with NK cell media, then NK-92 MI cells were added to 293 cells at an effector target ratio of 10:1 in 200μl media or as a control 200μl media was added alone. The plate was then briefly spun at 100g to enhance effector and target cell proximity, then incubated at 37°C for 5h. Cells were then washed 3 times in PBS and processed for western blot and CFU counts as before.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification of western blot densitometry for GSDMB was performed by determining pixel density using Adobe photoshop. Quantification of Coomassie stained gel bands of GSDMB/D-N subunits was performed using Image Lab software from BioRad. The following statistical tests were performed. Figure 2E: Statistical significance was determined by One-way ANOVA (p=0.0011), all samples compared to NI (control) using Dunnett’s multiple comparisons post hoc test. Figure 2G: Statistical significance was determined by unpaired Students T-Test. NS= Not significant. Figure 4 B and E: Statistical significance was determined using a One-way ANOVA) and Dunnett’s multiple comparisons post hoc test comparing GSDMB-N pelleting in the indicated liposomes compared to those generated from 100% PC (B) or Liver polar extract (E). Figure 4 H and I: Statistical significance for each bacterial phase was determined using One-way ANOVA (exponential growth phase p<0.0001; stationary growth phase p<0.0001) followed by Dunnett’s multiple comparisons test with all samples compared to TBS control (H). Students unpaired T-Tests for each bacterial species (I). Figure 5C: statistical significance was determined using unpaired two-tailed Students T-tests. Because the data were non-normally distributed, a non-parametric Mann-Whitney Test (rank test) was also applied and the results agreed with those presented (caecum p=0.6526; liver p=0.3245; and spleen p=0.2273). Figure 5F: Statistical significance was determined using a Two-way ANOVA (interaction p=0.0034; Shigella genotype p=0.0024; cell type and treatment p=0.0016) followed by Sidak’s multiple comparisons post hoc test with samples compared to GSDMB negative cells for each Shigella genotype. Figure 6C and D: Statistical significance was determined using Two-way ANOVAs WT (interaction p=0.464; time p<0.0001; treatment p=0.3097) Δipah7.8 (interaction p<0.0001; time p<0.0001; treatment p<0.0001) for (C) and WT (interaction p=0.7089; time p<0.0001; treatment p=0.075) Δipah7.8 (interaction p=0.80; time p<0.0001; treatment p=0.15) for (D). Post hoc comparisons were made using Sidak’s multiple comparisons test with treated (+GZMA) samples compared to untreated (−GZMA) samples at each timepoint within each Shigella flexneri genotype. NS=Not significant. GraphPad Prism 8 was used for all statistical analysis.
Supplementary Material
Figure S1 ∣ IpaHUBAIT capture of substrates, Data Supporting Figure 1.
(A) Eluates from UBAIT experiments were electrophoresed on SDS-PAGE gels and proteins were visualized using Coomassie blue stain. A gel slice was excised above the free IpaH-UBAIT bands from the lanes where Caco-2 lysate was included in the UBAIT reactions, and proteins were trypsin digested in gel and peptides were identified by tandem mass spectrometry (MS/MS). IpaH2 is NCBI protein SF5M90T_1825, IpaH3 is SF5M90T_1355, IpaH4 is SF5M90T_1966 and IpaH5 is SF5M90T_2665.
(B) Table with top 10 (based on number of spectral counts) hits from UBAIT screens determined by tandem mass spectrometry (MS/MS) identification of proteins in eluate fractions. Results from IpaHUBAIT proteins were compared and commonly found hits were eliminated.
(C) UBAIT reaction between the IpaHUBAIT proteins indicated and recombinant HOIP. Western blot using anti-HOIP antibody shown. Coomassie stain SDS PAGE gels show protein inputs.
(D) GST pulldown assay with full length GST-GSDMD incubated with His-IpaH2.5 or His-IpaH7.8. Protein inputs are shown (right).
(E) Tandem mass spectrometry (MS/MS) analysis of GSDMB sample treated with IpaH7.8 revealed IpaH7.8 mediated ubiquitin modification on K166 on the N-terminal domain and K308 on the C-terminal domain. High confidence assignment of ubiquitination on K166 and K308 was based on sequence coverage from Y and B ions and mis-cleavage by trypsin due to GlyGly modification on the lysine residues. In addition, these lysines were not ubiquitinated in IpaH7.8C357S treated samples (data not shown).
(F) In vitro ubiquitination of GST-GSDMB WT and K166/308R using increasing concentrations of IpaH7.8 WT. Controls with buffer alone are indicated (first lane) and IpaH7.8 C/S designates reactions containing catalytically inactive IpaH7.8. Anti-ubiquitin antibody was used to detect ubiquitinated protein and coomassie stain SDS PAGE gels show protein inputs.
(A,B) Representative data shown are from two independent experiments.
(C,D,F) Representative data shown are from three independent experiments.
Figure S2 ∣ S. flexneri infection induces degradation of GSDMB, Data Supporting Figure 2.
(A) Graph of GSDMB/actin densitometry ratio from three independent experiments corresponding to Figure 2A. Graph shows mean ± SD. Statistical significance was determined by One-way ANOVA (p=.0033) followed by Dunnett’s multiple comparisons post hoc test with empty vector as the control (A).
(B) Graph of GSDMB/actin densitometry ratio from three independent experiments corresponding to Figure 2C. Graph shows mean ± SD. Statistical significance was determined with Two-way ANOVA (interaction p=0.19; vector p=0.052; treatment p=0.0145). Post Hoc testing was performed using Sidak’s test comparing to MG132 treated samples.
(C) Time-course of GSDMB degradation in S. flexneri infected GSDMB-FLAG HEK293T cells. Anti-GSDMB and anti-actin antibodies used to detect host proteins and is a representative of three independent experiments. The ratio of GSDMB/Actin densitometry is show below each lane. S. flexneri levels were quantified by determining CFU/well of infected cells and graph shows mean ± SD. Statistical significance was determined with Two-way ANOVA (interaction p=0.98; time p=0.0002; shigella genotype p=0.714). Post Hoc testing was performed using Sidak’s test and comparing Shigella genotypes within each timepoint. NS=Not Significant.
Figure S3 ∣ GSDMB-N does not form pores in liver liposomes, Data Supporting Figure 3.
(A) Model of the GSDMB protein showing the predicted molecular weight (kDa) of the N- and C- functional domains and the caspase cleavage site found at residue 91. The predicted product of caspase cleavage is shown (lower diagram).
(B) Model of the HIS-MBP tagged GSDMD protein showing predicted molecular weight (kDa) of the N- and C- functional domains and the caspase cleavage site found at residue 275. The predicted product of caspase cleavage is shown (lower diagram).
(C) Coomassie stained gel showing GSDMB cleavage by the indicated caspases. Caspases 3, 6, and 7 cleaved GSDMB into fragments that confirm previously published data showing that cleavage site after D91 in the GSDMB N-terminus producing p10 and p36 fragments (Chao et al., 2017).
(D) Coomassie stained gel showing HIS-MBP-GSDMD cleavage by the indicated caspases.
(E and F) Schematic showing amino acids in GSDMB (E) and GSDMD (F) that were replaced with the TEV cleavage sequence ENLYFQS. Coomassie stained gel showing cleavage of GSDMBTEV (E) and GSDMDTEV (F) by TEVp in vitro.
(G) Graph showing quantification GSDMB/D-N binding to liposomes shown in Figure 3A. The percent GSDMB/D-N bound to liposomes were quantified from four independent biological replicates and the mean is plotted ± SD Statistical significance was determined using a Two-way ANOVA (interaction p<0.00001; liposomes p<0.0001; GSDM protein p<0.0001) with Sidak’s multiple comparisons post hoc test comparing pelleting of GSDMB-N and GSDMD-N. NS=Not significant.
(H) Liposomes generated from Liver lipids and treated with the proteins indicated were incubated with 2% uranyl acetate negative staining and visualized by scanning electron microscopy (SEM).
(I) Coomassie stained gel showing detergent extraction of GSDMB-N from E. coli liposomes. The pellet (P) fraction from liposome pulldown assays was resuspended in the indicated detergents and then centrifuged. The S2 fraction contained solubilized GSDMB-N when extracted with deoxycholate. S2 fraction material was used for SEM imaging (Figure 3E) and quantification (Figure 3F).
(C-F),(H) and (I) Results shown are representative of three independent biological repeats.
Figure S4 ∣ Activated GSDMB perturbs the S. flexneri cell membrane, Data Supporting Figure 4.
(A) Represented SEM images of S. flexneri treated as indicated. Boxes show magnification of the bacteria surface showing the rough exterior of untreated bacteria and an altered "smooth" exterior of GSDMB treated bacteria reminiscent of the inner-membrane. Images shown are representatives of three independent biological repeats.
(B) Dose curve of activated GSDMB used to determine the minimum concentration for inhibition of exponential growing S. flexneri. The indicated concentrations of GSDMBTEV, HIS-MBP-TEV-GSDMB in which the TEV site is placed in-between the MBP-tag and GSDMB (there is no TEV protease site in the interdomain linker), or the GSDMB-C were incubated with exponentially growing S. flexneri and TEVp was added where indicated. Viability of bacteria was calculated as in Figure 4 H and I. Input protein levels and GSDMB activation in this assay was assessed by Coomassie stained SDS-PAGE gel (bottom). Graph shows the mean ± SD of the percent CFU recovered calculated from three independent biological experiments. Statistical significance was determined using an unpaired Two-Tailed Students T-Test with the indicated comparison. The Coomassie gel is a representative from three independent biological repeats.
Figure S5 ∣ GSDMB protein detected in tissues from GSDMB+/+ transgenic mouse, Data Supporting Figure 5.
(A) Model of the GSDMB isoform 1 (Uniprot: Q8TAX-1; 411 aa) showing the predicted molecular weight (kDa) of the N- and C- functional domains and the putative GZMA cleavage site and products. Coomassie stained gel showing GSDMB cleavage by GZMA and is a representative from three independent biological repeats.
(B and C) Carboxyfluorescein dye release assay using E. coli liposomes (B) and liver liposomes supplemented with 20mol% cardiolipin (CL) (C) with liver liposomes as a control for both and incubated with GZMA activated GSDMB as indicated. Graphs are presented as described in Figure 4F and are representatives of three independent experiments.
(D) Quantification of recovery of S. flexneri after in vitro incubation with the indicated proteins. Colony forming units (CFU) recovered after each treatment were calculated as the CFUs recovered from treated bacteria compared to CFUs recovered from TBS treated bacteria. Graph shows the mean ± SD of the percent CFU recovered calculated from 3 independent biological experiments. Statistical significance was determined using a One-way ANOVA (p=0.0003) followed by Dunnett’s multiple comparisons test with all samples compared to TBS control.
(E) Tissue distribution of GSDMB expression in transgenic mice as determined by anti-FLAG, and anti-actin Western blots.
Figure S6 ∣ GZMA microinjection into GSDMB+ cells does not induced membrane leakage, Data Supporting Figure 6.
(A and B) GSDMB-FLAG and WT HEK293T cells were infected with the S. flexneri strains indicated at an MOI of 20 prior to transfection with GZMA protein and incubation for four hours. (A) Western blots showing activation of GSDMB using anti GSDMB, GZMA, and actin antibodies, and are representatives from three independent biological repeats. Graph showing the recovered CFUs of the S. flexneri strains indicated from either naive (GSDMB-; light green bars) or GSDMB expressing (GSDMB+; dark green bars) HEK293T cells transfected with GZMA protein. Data shown in is percent CFU recovered from cells treated with GZMA compared to mock treatment. (B) Graph showing the levels of Lactose Dehydrogenase (LDH) in the media collected from GSDMB− or GSDMB+ HEK293T cells infected with the S. flexneri strains indicated and then transfected with GZMA. Percent LDH release was calculated by dividing experimental values by a max LDH release value determined for each experiment by lysing a well of untreated cells with lysis buffer provided with Cytotox 96 kit from Promega. Graphs show the mean ± SD of the percent CFU recovered (A) and Percent LDH Release (B) from three independent biological experiments. Statistical significance for (A) was determined using Two-way ANOVA (interaction p=0.0034; shigella genotype p=0.0024; cell type p=0.0016) followed by Sidak’s multiple comparisons test with samples compared to GSDMB negative cells for each Shigella genotype. Statistical significance for (B) was determined using Two-Way ANOVAs for NI (interaction p=0.3536; treatment p=.52; cell type p=.15), WT (interaction p=0.11; treatment p=0.27; cell type p=0.073), Δipah7.8 (interaction p=0.94; treatment p=0.86; cell type p=0.53) followed by Tukey’s multiple comparisons post hoc tests NS=Not significant.
(C) GSDMD expressing and WT H1299 cells were microinjected with active recombinant caspase 1, cell permeability was monitored overtime by visualizing the Texas Red conjugated 10kDa dextran.
(D) GSDMB-FLAG and WT H1299 cell were microinjected with GZMA protein, cell permeability was monitored overtime by visualizing the Texas Red conjugated 10kDa dextran.
(C and D) Images shown are representatives of three independent biological repeats.
Table S1. Primers used in Study, Related to Key Resources Table
Highlights.
The Type 3 effector IpaH7.8 targets GSDMB for proteasome destruction.
GSDMB forms pores in bacterial-derived membranes.
Lymphocyte activation of GSDMB protects epithelial cells from cytosolic pathogens.
Shigella flexneri prevents GSDMB-mediated lysis by secreting IpaH7.8.
ACKNOWLEDGEMENTS
We would like to thank Dr. Kim Orth, Dr. Michael Reese, Dr. Vanessa Sperandio, Dr. Zixu Liu, Benjamin Kocsis and members of the Alto laboratory for helpful discussions. We thank the UT Southwestern Medical Center Proteomics Core and Electron Microscopy Core Facilities for assistance with aspects of experimental design and execution. This research was supported by grants from the National Institutes of Health (grant no. AI083359), The Welch Foundation (grant no. I-1704), the Burroughs Welcome Fund (grant no. 1011019) and the Howard Hughes Medical Institute and Simons Foundation Faculty Scholars Program (grant no. 55108499).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We worked to ensure diversity in experimental samples through the selection of cell lines. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
REFERENCES
- Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, Ciferri C, Dixit VM, and Dueber EC (2016). GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A 113, 7858–7863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allaoui A, Sansonetti PJ, and Parsot C (1993). MxiD, an outer membrane protein necessary for the secretion of the Shigella flexneri lpa invasins. Mol Microbiol 7, 59–68. [DOI] [PubMed] [Google Scholar]
- Ashida H, Toyotome T, Nagai T, and Sasakawa C (2007). Shigella chromosomal IpaH proteins are secreted via the type III secretion system and act as effectors. Mol Microbiol 63, 680–693. [DOI] [PubMed] [Google Scholar]
- Au - Dotiwala F, Au - Fellay I, Au - Filgueira L, Au - Martinvalet D, Au - Lieberman J, and Au - Walch M (2015). A High Yield and Cost-efficient Expression System of Human Granzymes in Mammalian Cells. JoVE, e52911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond PJ, and Sansom MS (2004). The simulation approach to bacterial outer membrane proteins. Mol Membr Biol 21, 151–161. [DOI] [PubMed] [Google Scholar]
- Bos MP, and Tommassen J (2004). Biogenesis of the Gram-negative bacterial outer membrane. Curr Opin Microbiol 7, 610–616. [DOI] [PubMed] [Google Scholar]
- Broz P, Pelegrin P, and Shao F (2020). The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol 20, 143–157. [DOI] [PubMed] [Google Scholar]
- Carl-McGrath S, Schneider-Stock R, Ebert M, and Rocken C (2008). Differential expression and localisation of gasdermin-like (GSDML), a novel member of the cancer-associated GSDMDC protein family, in neoplastic and non-neoplastic gastric, hepatic, and colon tissues. Pathology 40, 13–24. [DOI] [PubMed] [Google Scholar]
- Carter PB, and Collins FM (1974). The route of enteric infection in normal mice. J Exp Med 139, 1189–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao KL, Kulakova L, and Herzberg O (2017). Gene polymorphism linked to increased asthma and IBD risk alters gasdermin-B structure, a sulfatide and phosphoinositide binding protein. Proc Natl Acad Sci U S A 114, E1128–E1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Shi P, Wang Y, Zou D, Wu X, Wang D, Hu Q, Zou Y, Huang Z, Ren J, et al. (2019). GSDMB promotes non-canonical pyroptosis by enhancing caspase-4 activity. J Mol Cell Biol 11, 496–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury D, and Lieberman J (2008). Death by a thousand cuts: granzyme pathways of programmed cell death. Annu Rev Immunol 26, 389–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, Tyurin VA, Yanamala N, Shrivastava IH, Mohammadyani D, et al. (2013). Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol 15, 1197–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu VT, Weber T, Graf R, Sommermann T, Petsch K, Sack U, Volchkov P, Rajewsky K, and Kuhn R (2016). Efficient generation of Rosa26 knock-in mice using CRISPR/Cas9 in C57BL/6 zygotes. BMC Biotechnol 16, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Miller M, Beppu AK, Mueller J, McGeough MD, Vuong C, Karta MR, Rosenthal P, Chouiali F, Doherty TA, et al. (2016). GSDMB induces an asthma phenotype characterized by increased airway responsiveness and remodeling without lung inflammation. Proceedings of the National Academy of Sciences of the United States of America 113, 13132–13137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, and Wanner BL (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proceedings of the National Academy of Sciences of the United States of America 97, 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong MF, Liu Z, Chen D, and Alto NM (2016). Shigella flexneri suppresses NF-kappaB activation by inhibiting linear ubiquitin chain ligation. Nat Microbiol 1, 16084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, and Shao F (2016). Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116. [DOI] [PubMed] [Google Scholar]
- Dotiwala F, Sen Santara S, Binker-Cosen AA, Li B, Chandrasekaran S, and Lieberman J (2017). Granzyme B Disrupts Central Metabolism and Protein Synthesis in Bacteria to Promote an Immune Cell Death Program. Cell 171, 1125–1137 e1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evavold CL, Ruan J, Tan Y, Xia S, Wu H, and Kagan JC (2018). The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 48, 35–44 e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger G, Fan X, Dikiy S, Lee SY, and Rudensky AY (2015). Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science 350, 981–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, and Han J (2015). Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res 25, 1285–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchen PG, Prior JL, Oyston PC, Panico M, Wren BW, Titball RW, Morris HR, and Dell A (2002). Structural characterization of lipo-oligosaccharide (LOS) from Yersinia pestis: regulation of LOS structure by the PhoPQ system. Mol Microbiol 44, 1637–1650. [DOI] [PubMed] [Google Scholar]
- Kaiser P, Diard M, Stecher B, and Hardt WD (2012). The streptomycin mouse model for Salmonella diarrhea: functional analysis of the microbiota, the pathogen's virulence factors, and the host's mucosal immune response. Immunol Rev 245, 56–83. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671. [DOI] [PubMed] [Google Scholar]
- Keszei AF, Tang X, McCormick C, Zeqiraj E, Rohde JR, Tyers M, and Sicheri F (2014). Structure of an SspH1-PKN1 complex reveals the basis for host substrate recognition and mechanism of activation for a bacterial E3 ubiquitin ligase. Mol Cell Biol 34, 362–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil IA, Troeger C, Blacker BF, Rao PC, Brown A, Atherly DE, Brewer TG, Engmann CM, Houpt ER, Kang G, et al. (2018). Morbidity and mortality due to shigella and enterotoxigenic Escherichia coli diarrhoea: the Global Burden of Disease Study 1990-2016. Lancet Infect Dis 18, 1229–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knodler LA, Crowley SM, Sham HP, Yang H, Wrande M, Ma C, Ernst RK, Steele-Mortimer O, Celli J, and Vallance BA (2014). Noncanonical inflammasome activation of caspase-4/caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe 16, 249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotloff KL, Winickoff JP, Ivanoff B, Clemens JD, Swerdlow DL, Sansonetti PJ, Adak GK, and Levine MM (1999). Global burden of Shigella infections: implications for vaccine development and implementation of control strategies. Bull World Health Organ 77, 651–666. [PMC free article] [PubMed] [Google Scholar]
- Kuang S, Zheng J, Yang H, Li S, Duan S, Shen Y, Ji C, Gan J, Xu XW, and Li J (2017). Structure insight of GSDMD reveals the basis of GSDMD autoinhibition in cell pyroptosis. Proc Natl Acad Sci U S A 114, 10642–10647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Jiang W, Yu Q, Liu W, Zhou P, Li J, Xu J, Xu B, Wang F, and Shao F (2017). Ubiquitination and degradation of GBPs by a Shigella effector to suppress host defence. Nature 551, 378–383. [DOI] [PubMed] [Google Scholar]
- Liu J, Wu H, Huang C, Lei D, Zhang M, Xie W, Li J, and Ren G (2019). Optimized Negative-Staining Protocol for Lipid-Protein Interactions Investigated by Electron Microscopy. Methods Mol Biol 2003, 163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, and Lieberman J (2020). Knocking 'em Dead: Pore-Forming Proteins in Immune Defense. Annu Rev Immunol 38, 455–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, and Lieberman J (2016). Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik-Kale P, Winfree S, and Steele-Mortimer O (2012). The bimodal lifestyle of intracellular Salmonella in epithelial cells: replication in the cytosol obscures defects in vacuolar replication. PLoS One 7, e38732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao EA, Scherer CA, Tsolis RM, Kingsley RA, Adams LG, Baumler AJ, and Miller SI (1999). Salmonella typhimurium leucine-rich repeat proteins are targeted to the SPI1 and SPI2 type III secretion systems. Mol Microbiol 34, 850–864. [DOI] [PubMed] [Google Scholar]
- Mileykovskaya E, and Dowhan W (2009). Cardiolipin membrane domains in prokaryotes and eukaryotes. Biochim Biophys Acta 1788, 2084–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell PS, Roncaioli JL, Turcotte EA, Goers L, Chavez RA, Lee AY, Lesser CF, Rauch I, and Vance RE (2020). NAIP–NLRC4-deficient mice are susceptible to shigellosis. bioRxiv, 2020.2005.2016.099929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor HF, Lyon N, Leung JW, Agarwal P, Swaim CD, Miller KM, and Huibregtse JM (2015). Ubiquitin-Activated Interaction Traps (UBAITs) identify E3 ligase binding partners. EMBO Rep 16, 1699–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, Brooks A, Xia S, Wu H, Kelliher MA, et al. (2018). Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362, 1064–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda SK, and Colonna M (2019). Innate Lymphoid Cells in Mucosal Immunity. Front Immunol 10, 861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W, de Souza Santos M, Li Y, Tomchick DR, and Orth K (2019). High-resolution cryo-EM structures of the E. coli hemolysin ClyA oligomers. PLoS One 14, e0213423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perelman SS, Abrams ME, Eitson JL, Chen D, Jimenez A, Mettlen M, Schoggins JW, and Alto NM (2016). Cell-Based Screen Identifies Human Interferon-Stimulated Regulators of Listeria monocytogenes Infection. PLoS Pathog 12, e1006102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce NW, Kleiger G, Shan SO, and Deshaies RJ (2009). Detection of sequential polyubiquitylation on a millisecond timescale. Nature 462, 615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piro AS, Hernandez D, Luoma S, Feeley EM, Finethy R, Yirga A, Frickel EM, Lesser CF, and Coers J (2017). Detection of Cytosolic Shigella flexneri via a C-Terminal Triple-Arginine Motif of GBP1 Inhibits Actin-Based Motility. mBio 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebeil R, Ernst RK, Gowen BB, Miller SI, and Hinnebusch BJ (2004). Variation in lipid A structure in the pathogenic yersiniae. Mol Microbiol 52, 1363–1373. [DOI] [PubMed] [Google Scholar]
- Richardson RB, Ohlson MB, Eitson JL, Kumar A, McDougal MB, Boys IN, Mar KB, De La Cruz-Rivera PC, Douglas C, Konopka G, et al. (2018). A CRISPR screen identifies IFI6 as an ER-resident interferon effector that blocks flavivirus replication. Nat Microbiol 3, 1214–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde JR, Breitkreutz A, Chenal A, Sansonetti PJ, and Parsot C (2007). Type III secretion effectors of the IpaH family are E3 ubiquitin ligases. Cell Host Microbe 1, 77–83. [DOI] [PubMed] [Google Scholar]
- Ruan J, Xia S, Liu X, Lieberman J, and Wu H (2018). Cryo-EM structure of the gasdermin A3 membrane pore. Nature 557, 62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandstrom A, Mitchell PS, Goers L, Mu EW, Lesser CF, and Vance RE (2019). Functional degradation: A mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansonetti PJ, Phalipon A, Arondel J, Thirumalai K, Banerjee S, Akira S, Takeda K, and Zychlinsky A (2000). Caspase-1 activation of IL-1beta and IL-18 are essential for Shigella flexneri-induced inflammation. Immunity 12, 581–590. [DOI] [PubMed] [Google Scholar]
- Santos RL, Zhang S, Tsolis RM, Kingsley RA, Adams LG, and Baumler AJ (2001). Animal models of Salmonella infections: enteritis versus typhoid fever. Microbes Infect 3, 1335–1344. [DOI] [PubMed] [Google Scholar]
- Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, Rongvaux A, Bunnell SC, Shao F, Green DR, et al. (2018). Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc Natl Acad Sci U S A 115, E10888–E10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sborgi L, Ruhl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, Farady CJ, Muller DJ, Broz P, and Hiller S (2016). GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J 35, 1766–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, and Rice CM (2011). A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder GN, and Hilbi H (2008). Molecular pathogenesis of Shigella spp.: controlling host cell signaling, invasion, and death by type III secretion. Clin Microbiol Rev 21, 134–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenk F, Baron U, and Rajewsky K (1995). A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res 23, 5080–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, and Shao F (2015a). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. [DOI] [PubMed] [Google Scholar]
- Shi P, Tang A, Xian L, Hou S, Zou D, Lv Y, Huang Z, Wang Q, Song A, Lin Z, et al. (2015b). Loss of conserved Gsdma3 self-regulation causes autophagy and cell death. Biochem J 468, 325–336. [DOI] [PubMed] [Google Scholar]
- Singer AU, Rohde JR, Lam R, Skarina T, Kagan O, Dileo R, Chirgadze NY, Cuff ME, Joachimiak A, Tyers M, et al. (2008). Structure of the Shigella T3SS effector IpaH defines a new class of E3 ubiquitin ligases. Nat Struct Mol Biol 15, 1293–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spycher BD, Henderson J, Granell R, Evans DM, Smith GD, Timpson NJ, and Sterne JA (2012). Genome-wide prediction of childhood asthma and related phenotypes in a longitudinal birth cohort. J Allergy Clin Immunol 130, 503–509 e507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenger S, Hanson DA, Teitelbaum R, Dewan P, Niazi KR, Froelich CJ, Ganz T, Thoma-Uszynski S, Melian A, Bogdan C, et al. (1998). An antimicrobial activity of cytolytic T cells mediated by granulysin. Science 282, 121–125. [DOI] [PubMed] [Google Scholar]
- Stieglitz B, Morris-Davies AC, Koliopoulos MG, Christodoulou E, and Rittinger K (2012). LUBAC synthesizes linear ubiquitin chains via a thioester intermediate. EMBO Rep 13, 840–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Mimuro H, Kim M, Ogawa M, Ashida H, Toyotome T, Franchi L, Suzuki M, Sanada T, Suzuki T, et al. (2014). Shigella IpaH7.8 E3 ubiquitin ligase targets glomulin and activates inflammasomes to demolish macrophages. Proc Natl Acad Sci U S A 111, E4254–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Suzuki T, Mimuro H, Mizushima T, and Sasakawa C (2018). Shigella hijacks the glomulin-cIAPs-inflammasome axis to promote inflammation. EMBO Rep 19, 89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura M, Tanaka S, Fujii T, Aoki A, Komiyama H, Ezawa K, Sumiyama K, Sagai T, and Shiroishi T (2007). Members of a novel gene family, Gsdm, are expressed exclusively in the epithelium of the skin and gastrointestinal tract in a highly tissue-specific manner. Genomics 89, 618–629. [DOI] [PubMed] [Google Scholar]
- Tulah AS, Holloway JW, and Sayers I (2013). Defining the contribution of SNPs identified in asthma GWAS to clinical variables in asthmatic children. BMC Med Genet 14, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walch M, Dotiwala F, Mulik S, Thiery J, Kirchhausen T, Clayberger C, Krensky AM, Martinvalet D, and Lieberman J (2014). Cytotoxic cells kill intracellular bacteria through granulysin-mediated delivery of granzymes. Cell 157, 1309–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wandel MP, Pathe C, Werner EI, Ellison CJ, Boyle KB, von der Malsburg A, Rohde J, and Randow F (2017). GBPs Inhibit Motility of Shigella flexneri but Are Targeted for Degradation by the Bacterial Ubiquitin Ligase IpaH9.8. Cell Host Microbe 22, 507–518 e505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JL, Sanchez-Garrido J, Goddard PJ, Torraca V, Mostowy S, Shenoy AR, and Clements A (2019). Shigella sonnei O-Antigen Inhibits Internalization, Vacuole Escape, and Inflammasome Activation. mBio 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Kang MJ, Kim BJ, Kwon JW, Song YH, Choi WA, Shin YJ, and Hong SJ (2011). Polymorphisms in GSDMA and GSDMB are associated with asthma susceptibility, atopy and BHR. Pediatr Pulmonol 46, 701–708. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Zhang Y, and Lieberman J (2021). Lighting a Fire: Can We Harness Pyroptosis to Ignite Antitumor Immunity? Cancer Immunol Res 9, 2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, Wang Y, Li D, Liu W, Zhang Y, et al. (2020). Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Li H, Hu L, Wang J, Zhou Y, Pang Z, Liu L, and Shao F (2008). Structure of a Shigella effector reveals a new class of ubiquitin ligases. Nat Struct Mol Biol 15, 1302–1308. [DOI] [PubMed] [Google Scholar]
- Zychlinsky A, Prevost MC, and Sansonetti PJ (1992). Shigella flexneri induces apoptosis in infected macrophages. Nature 358, 167–169. [DOI] [PubMed] [Google Scholar]
- Zychlinsky A, Thirumalai K, Arondel J, Cantey JR, Aliprantis AO, and Sansonetti PJ (1996). In vivo apoptosis in Shigella flexneri infections. Infect Immun 64, 5357–5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 ∣ IpaHUBAIT capture of substrates, Data Supporting Figure 1.
(A) Eluates from UBAIT experiments were electrophoresed on SDS-PAGE gels and proteins were visualized using Coomassie blue stain. A gel slice was excised above the free IpaH-UBAIT bands from the lanes where Caco-2 lysate was included in the UBAIT reactions, and proteins were trypsin digested in gel and peptides were identified by tandem mass spectrometry (MS/MS). IpaH2 is NCBI protein SF5M90T_1825, IpaH3 is SF5M90T_1355, IpaH4 is SF5M90T_1966 and IpaH5 is SF5M90T_2665.
(B) Table with top 10 (based on number of spectral counts) hits from UBAIT screens determined by tandem mass spectrometry (MS/MS) identification of proteins in eluate fractions. Results from IpaHUBAIT proteins were compared and commonly found hits were eliminated.
(C) UBAIT reaction between the IpaHUBAIT proteins indicated and recombinant HOIP. Western blot using anti-HOIP antibody shown. Coomassie stain SDS PAGE gels show protein inputs.
(D) GST pulldown assay with full length GST-GSDMD incubated with His-IpaH2.5 or His-IpaH7.8. Protein inputs are shown (right).
(E) Tandem mass spectrometry (MS/MS) analysis of GSDMB sample treated with IpaH7.8 revealed IpaH7.8 mediated ubiquitin modification on K166 on the N-terminal domain and K308 on the C-terminal domain. High confidence assignment of ubiquitination on K166 and K308 was based on sequence coverage from Y and B ions and mis-cleavage by trypsin due to GlyGly modification on the lysine residues. In addition, these lysines were not ubiquitinated in IpaH7.8C357S treated samples (data not shown).
(F) In vitro ubiquitination of GST-GSDMB WT and K166/308R using increasing concentrations of IpaH7.8 WT. Controls with buffer alone are indicated (first lane) and IpaH7.8 C/S designates reactions containing catalytically inactive IpaH7.8. Anti-ubiquitin antibody was used to detect ubiquitinated protein and coomassie stain SDS PAGE gels show protein inputs.
(A,B) Representative data shown are from two independent experiments.
(C,D,F) Representative data shown are from three independent experiments.
Figure S2 ∣ S. flexneri infection induces degradation of GSDMB, Data Supporting Figure 2.
(A) Graph of GSDMB/actin densitometry ratio from three independent experiments corresponding to Figure 2A. Graph shows mean ± SD. Statistical significance was determined by One-way ANOVA (p=.0033) followed by Dunnett’s multiple comparisons post hoc test with empty vector as the control (A).
(B) Graph of GSDMB/actin densitometry ratio from three independent experiments corresponding to Figure 2C. Graph shows mean ± SD. Statistical significance was determined with Two-way ANOVA (interaction p=0.19; vector p=0.052; treatment p=0.0145). Post Hoc testing was performed using Sidak’s test comparing to MG132 treated samples.
(C) Time-course of GSDMB degradation in S. flexneri infected GSDMB-FLAG HEK293T cells. Anti-GSDMB and anti-actin antibodies used to detect host proteins and is a representative of three independent experiments. The ratio of GSDMB/Actin densitometry is show below each lane. S. flexneri levels were quantified by determining CFU/well of infected cells and graph shows mean ± SD. Statistical significance was determined with Two-way ANOVA (interaction p=0.98; time p=0.0002; shigella genotype p=0.714). Post Hoc testing was performed using Sidak’s test and comparing Shigella genotypes within each timepoint. NS=Not Significant.
Figure S3 ∣ GSDMB-N does not form pores in liver liposomes, Data Supporting Figure 3.
(A) Model of the GSDMB protein showing the predicted molecular weight (kDa) of the N- and C- functional domains and the caspase cleavage site found at residue 91. The predicted product of caspase cleavage is shown (lower diagram).
(B) Model of the HIS-MBP tagged GSDMD protein showing predicted molecular weight (kDa) of the N- and C- functional domains and the caspase cleavage site found at residue 275. The predicted product of caspase cleavage is shown (lower diagram).
(C) Coomassie stained gel showing GSDMB cleavage by the indicated caspases. Caspases 3, 6, and 7 cleaved GSDMB into fragments that confirm previously published data showing that cleavage site after D91 in the GSDMB N-terminus producing p10 and p36 fragments (Chao et al., 2017).
(D) Coomassie stained gel showing HIS-MBP-GSDMD cleavage by the indicated caspases.
(E and F) Schematic showing amino acids in GSDMB (E) and GSDMD (F) that were replaced with the TEV cleavage sequence ENLYFQS. Coomassie stained gel showing cleavage of GSDMBTEV (E) and GSDMDTEV (F) by TEVp in vitro.
(G) Graph showing quantification GSDMB/D-N binding to liposomes shown in Figure 3A. The percent GSDMB/D-N bound to liposomes were quantified from four independent biological replicates and the mean is plotted ± SD Statistical significance was determined using a Two-way ANOVA (interaction p<0.00001; liposomes p<0.0001; GSDM protein p<0.0001) with Sidak’s multiple comparisons post hoc test comparing pelleting of GSDMB-N and GSDMD-N. NS=Not significant.
(H) Liposomes generated from Liver lipids and treated with the proteins indicated were incubated with 2% uranyl acetate negative staining and visualized by scanning electron microscopy (SEM).
(I) Coomassie stained gel showing detergent extraction of GSDMB-N from E. coli liposomes. The pellet (P) fraction from liposome pulldown assays was resuspended in the indicated detergents and then centrifuged. The S2 fraction contained solubilized GSDMB-N when extracted with deoxycholate. S2 fraction material was used for SEM imaging (Figure 3E) and quantification (Figure 3F).
(C-F),(H) and (I) Results shown are representative of three independent biological repeats.
Figure S4 ∣ Activated GSDMB perturbs the S. flexneri cell membrane, Data Supporting Figure 4.
(A) Represented SEM images of S. flexneri treated as indicated. Boxes show magnification of the bacteria surface showing the rough exterior of untreated bacteria and an altered "smooth" exterior of GSDMB treated bacteria reminiscent of the inner-membrane. Images shown are representatives of three independent biological repeats.
(B) Dose curve of activated GSDMB used to determine the minimum concentration for inhibition of exponential growing S. flexneri. The indicated concentrations of GSDMBTEV, HIS-MBP-TEV-GSDMB in which the TEV site is placed in-between the MBP-tag and GSDMB (there is no TEV protease site in the interdomain linker), or the GSDMB-C were incubated with exponentially growing S. flexneri and TEVp was added where indicated. Viability of bacteria was calculated as in Figure 4 H and I. Input protein levels and GSDMB activation in this assay was assessed by Coomassie stained SDS-PAGE gel (bottom). Graph shows the mean ± SD of the percent CFU recovered calculated from three independent biological experiments. Statistical significance was determined using an unpaired Two-Tailed Students T-Test with the indicated comparison. The Coomassie gel is a representative from three independent biological repeats.
Figure S5 ∣ GSDMB protein detected in tissues from GSDMB+/+ transgenic mouse, Data Supporting Figure 5.
(A) Model of the GSDMB isoform 1 (Uniprot: Q8TAX-1; 411 aa) showing the predicted molecular weight (kDa) of the N- and C- functional domains and the putative GZMA cleavage site and products. Coomassie stained gel showing GSDMB cleavage by GZMA and is a representative from three independent biological repeats.
(B and C) Carboxyfluorescein dye release assay using E. coli liposomes (B) and liver liposomes supplemented with 20mol% cardiolipin (CL) (C) with liver liposomes as a control for both and incubated with GZMA activated GSDMB as indicated. Graphs are presented as described in Figure 4F and are representatives of three independent experiments.
(D) Quantification of recovery of S. flexneri after in vitro incubation with the indicated proteins. Colony forming units (CFU) recovered after each treatment were calculated as the CFUs recovered from treated bacteria compared to CFUs recovered from TBS treated bacteria. Graph shows the mean ± SD of the percent CFU recovered calculated from 3 independent biological experiments. Statistical significance was determined using a One-way ANOVA (p=0.0003) followed by Dunnett’s multiple comparisons test with all samples compared to TBS control.
(E) Tissue distribution of GSDMB expression in transgenic mice as determined by anti-FLAG, and anti-actin Western blots.
Figure S6 ∣ GZMA microinjection into GSDMB+ cells does not induced membrane leakage, Data Supporting Figure 6.
(A and B) GSDMB-FLAG and WT HEK293T cells were infected with the S. flexneri strains indicated at an MOI of 20 prior to transfection with GZMA protein and incubation for four hours. (A) Western blots showing activation of GSDMB using anti GSDMB, GZMA, and actin antibodies, and are representatives from three independent biological repeats. Graph showing the recovered CFUs of the S. flexneri strains indicated from either naive (GSDMB-; light green bars) or GSDMB expressing (GSDMB+; dark green bars) HEK293T cells transfected with GZMA protein. Data shown in is percent CFU recovered from cells treated with GZMA compared to mock treatment. (B) Graph showing the levels of Lactose Dehydrogenase (LDH) in the media collected from GSDMB− or GSDMB+ HEK293T cells infected with the S. flexneri strains indicated and then transfected with GZMA. Percent LDH release was calculated by dividing experimental values by a max LDH release value determined for each experiment by lysing a well of untreated cells with lysis buffer provided with Cytotox 96 kit from Promega. Graphs show the mean ± SD of the percent CFU recovered (A) and Percent LDH Release (B) from three independent biological experiments. Statistical significance for (A) was determined using Two-way ANOVA (interaction p=0.0034; shigella genotype p=0.0024; cell type p=0.0016) followed by Sidak’s multiple comparisons test with samples compared to GSDMB negative cells for each Shigella genotype. Statistical significance for (B) was determined using Two-Way ANOVAs for NI (interaction p=0.3536; treatment p=.52; cell type p=.15), WT (interaction p=0.11; treatment p=0.27; cell type p=0.073), Δipah7.8 (interaction p=0.94; treatment p=0.86; cell type p=0.53) followed by Tukey’s multiple comparisons post hoc tests NS=Not significant.
(C) GSDMD expressing and WT H1299 cells were microinjected with active recombinant caspase 1, cell permeability was monitored overtime by visualizing the Texas Red conjugated 10kDa dextran.
(D) GSDMB-FLAG and WT H1299 cell were microinjected with GZMA protein, cell permeability was monitored overtime by visualizing the Texas Red conjugated 10kDa dextran.
(C and D) Images shown are representatives of three independent biological repeats.
Table S1. Primers used in Study, Related to Key Resources Table
Data Availability Statement
This study did not generate/analyze unique datasets or code.