

Abstract
Development of macromolecules provides applicable platforms for the delivery of therapeutics. In this general overview, we focus on the design principles of synthetic polymers, with disulfide bonds located in either the polymer backbone or side chains. We also discuss the role of disulfide bonds, as well as the remaining questions to better understand their applications in therapeutic delivery systems.
Graphical Abstract
Introduction
Disulfide bonds are formed by the oxidization of two cysteine residues in proteins, often to enhance their structural stability.[1-3] In eukaryotic cells, the formation of such covalent bonds occurs in oxidizing environments such as in endoplasmic reticulum (ER) and mitochondrial intermembrane space (IMS).[4] These disulfide bonds are specifically important for the folding and assembly of secreted proteins and cell surface proteins.[5, 6] Inspired by the existence of disulfide bonds in biomacromolecules (i.e. proteins), installing disulfide bonds in synthetic macromolecules (i.e. polymers) presents a straightforward method to covalently connect polymer chains, forming crosslinked structures and therefore increasing the stability of such structure.[7, 8]
Disulfide-containing polymers have facilitated the development of therapeutic delivery because of the improvement of material stability. Therapeutic cargos such as small-molecule drugs, nucleic acids, and proteins, are susceptible to hydrolysis and enzymatic degradation within biological fluids.[9] To ensure the stability of these cargos, synthetic delivery vehicles have been taken into consideration. Along with small-molecule lipids[10, 11] and inorganic nanomaterials,[12-16] macromolecules have been widely engineered to transport therapeutics into cells.[17-21] Specifically, regarding the disulfide bonds, building such cross-linkages intramolecularly or intermolecularly among polymeric vehicles results in a covalent “cage” for therapeutic cargos.[22] Meanwhile, conjugating therapeutic cargos with polymeric vehicles with disulfide bond is also an option for enhanced stability, improving the therapeutic efficacy of cargos.[23]
Incorporation of disulfide bonds in delivery systems also allows a controlled release mechanism for therapeutic cargos.[24-28] The basis of such mechanism originates from glutathione (GSH), an abundant intracellular antioxidant (1-10 mM) that is involved in the cellular reduction-oxidation (redox) processes, while GSH concentration is only at 2-20 μM in extracellular fluids.[29, 30] The reduced form of glutathione (GSH) is a cysteine-containing tripeptide. The disulfide bonds in delivery vehicles undergo thiol-disulfide exchange reactions in the presence of GSH, eventually leading to the cleavage of disulfide bonds within the delivery vehicles. Based on the structural design of polymers, disulfide-containing macromolecules can be broadly classified by the location of the disulfide bonds, in either the polymeric backbone or the side chains. Herein, we will briefly overview the development of disulfide-containing macromolecules for therapeutic delivery applications, highlighting the structural design principles for disulfide-containing macromolecules.
Disulfide bond in polymeric backbone
The primary advantage of disulfide bonds in the polymeric backbone is to generate biodegradable (or bioreducible) polymers. Comparing to the solely carbon-based polymeric backbones, such disulfide moieties can be cleaved in the presence of GSH, triggering the degradation of polymer chains. Nonetheless, an efficient polymeric delivery system requires not only the existence of disulfide bonds, but also the availability of functional groups on polymers to enable their interaction with the therapeutic cargos. Moreover, after the complexation between polymeric vehicles and cargo materials, the cellular internalization of such complexes is an essential step to determine the performance of the delivery system.
One straightforward design to obtain disulfides in the polymeric backbone is to copolymerize disulfide-containing bis(acrylamide) with amines via Michael addition, forming poly(amidoamine)s with disulfide bonds in their backbone.[31, 32] Considering the variety of amine derivatives and bis(acrylamide) derivatives, such copolymerization enables a palette of structural variations within poly(amidoamine)s, creating libraries of polymer analogs to evaluate their therapeutic delivery performance. In an example, a series of disulfide-containing poly(amidoamine)s was synthesized by the Michael addition between N,N’-cystamine-bisacrylamide and multiple primary amines (Figure 1).[33] In the presence of 2.5 mM dithiothreitol (DTT) to mimic the intracellular reductive environment, the disulfide-containing polymers were rapidly degraded. These poly(amidoamines) were shown to efficiently condense DNA and formed positively charged complexes via electrostatic interaction. The polyplexes delivered DNA into monkey kidney cells, demonstrating successful in vitro gene transfection. Similar structural designs have also been achieved using peptide coupling[34] and click reaction,[35] to introduce disulfide bonds in the polymer backbone.
Figure 1.

(a) Schematic illustration of DNA complexation using poly(amidoamine)s and subsequent triggered release. i, formation of DNA-polymer complexes that are stable in the extracellular environment. ii, intracellular reduction of the disulfide linkages in the polymer within the complexes. iii, DNA release from the complexes. (b) Synthesis of disulfide-containing poly(amido amine)s. Adapted with permission.[33] Copyright 2007, American Chemical Society.
Another method to insert disulfide moieties into polymer chain is to react nucleophiles with cyclic thioimidate or thiolactone,[36, 37] generating a thiol group in the product readily for subsequent polymerization. For example, primary amines in polymers such as polyethylenimine (PEI) and polylysine can be thiolated by 2-iminothiolane, converting the amine functional group to a thiol terminal.[38] In one report, low molecular weight branched PEI was reacted with 2-iminothiolane, forming a thiol group to be further oxidized as disulfide bonds, and simultaneously presenting a amidine residue on the resultant polymer.[39] Such disulfide-containing cationic polymers were demonstrated to be a suitable delivery vehicle for plasmid DNA. In a recent example, our group reported the combination of thiolactone ring-opening and disulfide-thiol exchange reactions, developing a cascaded step-growth polymerization methodology to prepare disulfide-containing polyamides (Figure 2).[40] The polymerization initiates with the amino group in pyridyl disulfide ethylamine as a nucleophile to open the thiolactone ring, eventually forming amide side-chains with a disulfide-containing backbone. Note that by using homocysteine thiolactone derivatives, a variety of functional groups have been successfully incorporated into the polymer side chains, adding the stimuli-sensitive features of such polymers beyond DTT-based redox stimulus.
Figure 2.
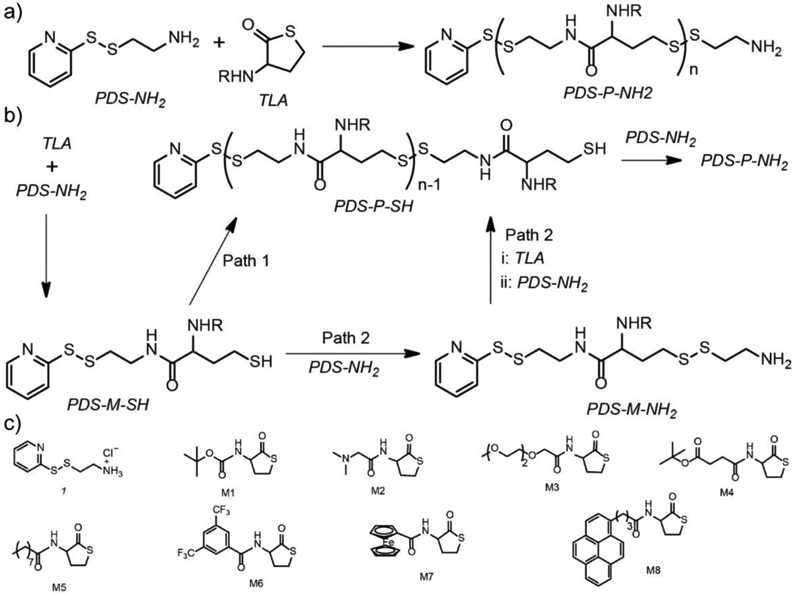
(a) Step growth polymerization for disulfide-containing polyamide with the combination between thiolactone ring opening and thiol-disulfide exchange reactions. (b) Proposed mechanism of the polymerization. (c) Structural variations of monomers. Adapted with permission.[40] Copyright 2019, American Chemical Society.
Thiol-disulfide exchange reaction alone has also been shown to prepare polymers with repeating disulfide moieties in the backbone. During the polymerization, a nucleophilic thiol initiator opens a strained disulfide-based monomer such as lipoic acid derivatives, forming a disulfide bond between the initiator and the monomer, as well as liberating a thiol on the same molecule to react with another strained disulfide monomer.[41, 42] The polymerization propagation can be terminated by an iodoacetamide, with the possibility to introduce additional drugs or probes by varying the structure of the chain terminus. Likewise, the choice of thiol initiator provides another variable to functionalize the polymer chain end.[42, 43] For example, such polymerization has been used to prepare polymers with disulfide bond in the backbone of each repeating unit, allowing a rapid degradation of polymers in the presence of glutathione (Figure 3).[44] Meanwhile, a guanidinium cation is affiliated within the repeating unit to equip the polymer with cell-penetrating capability. Interestingly, the cellular uptake of these guanidinium cation-decorated poly(disulfide)s did not improve by structural modifications on the polymer side chains. Moreover, replacing lipoic acid with asparagusic acid monomers lead to significant loss in cellular uptake of the resultant polymers. The polymerization system has also been further applied to facilitate intracellular delivery of small-molecule drugs and proteins.[45, 46]
Figure 3.
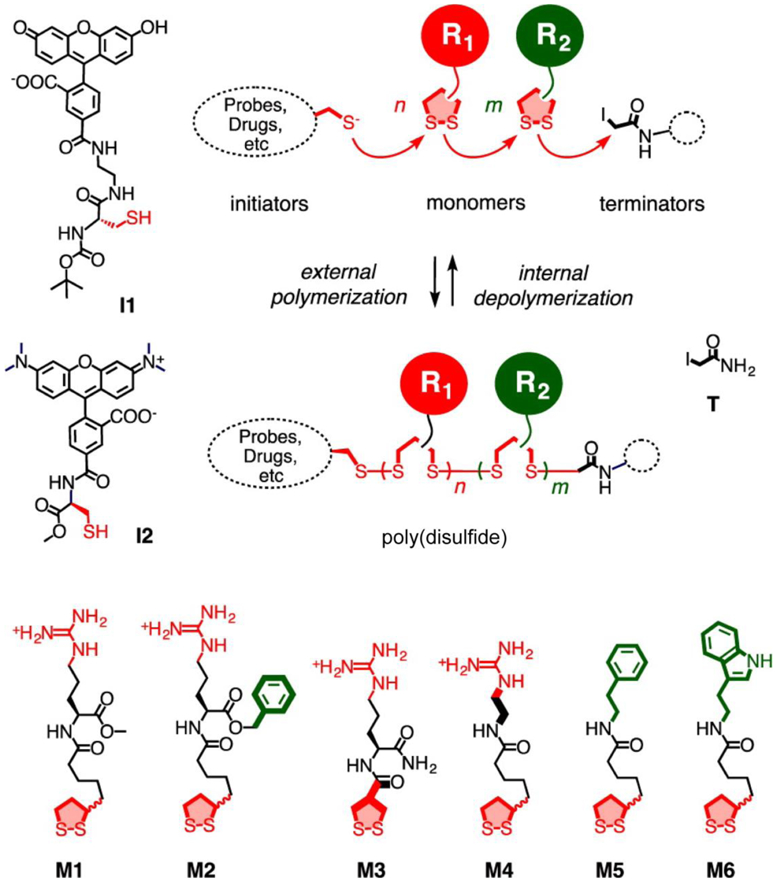
Synthesis of poly(disulfide) based on thiol-disulfide exchange reactions. I1 and I2, initiators. T, terminator. M1~M6, monomers. Adapted with permission.[44] Copyright 2014, American Chemical Society.
Disulfide bond in polymer side chains
Embedding disulfide units in polymer backbone offers an excellent degradability of polymer chains in reducing conditions. However, the disulfide bonds in polymer backbone have limited possibilities for subsequent functionalization over the polymer. From this perspective, inserting disulfide bonds in polymer side chains creates the opportunities for convenient chemical modifications through thiol-disulfide exchange reactions. Specifically, pyridyl disulfide (PDS) derivatives have been employed to construct polymers with disulfide units in the side chains.[47-51] PDS derivatives efficiently react with thiols and release a stable and unreactive byproduct, 2-pyridinethione. As the byproduct does not present a free thiol in the tautomer form, it cannot be involved in further thiol-disulfide exchange reaction with the reactant thiol. As a result, PDS moieties on the polymer side chain allows the incorporation of a wide range of functionality into the polymer.[50]
One advantage of presenting PDS-based side chains on polymers is the capability of generating self-crosslinked polymeric assemblies. For this purpose, a random copolymer was synthesized with oligo(ethylene glycol) and PDS units as side-chain functionalities (Figure 4).[52] Polymeric micelles that formed by such copolymers in water were reacted with DTT, covalently connecting two PDS units together and forming polymeric nanogels that contains disulfide cross-linkages. The disulfide cross-linkages allowed non-covalent encapsulation of hydrophobic guest molecules into the nanogels.[53] These encapsulated guest molecules were shown to release in response to GSH. By varying the crosslinking degree within the nanogels, the GSH-triggered guest release kinetics can be fine-tuned. The polymeric nanogels were used as carriers for a hydrophobic anti-cancer drug (doxorubicin) and successfully delivered the drug into breast cancer cells.[52] Moreover, the PDS-containing polymers allow the decoration of thiolated active targeting ligands or cell penetrating peptides on the polymeric nanogels, presenting a functionalizable scaffold to modulate the cellular uptake of polymeric delivery vehicles.[54-56]
Figure 4.

(a) Structure of the polymer with pyridyl disulfide (PDS)-based side chains and crosslinked nanogels: i, partial cleavage of PDS groups by dithiothreitol (DTT); ii, nanogel formation by inter/intrachain disulfide cross-linking. (b) Schematic illustration of the biodegradable nanogel formation. Adapted with permission.[52] Copyright 2010, American Chemical Society.
Through thiol-disulfide exchange reactions, the functionalizable feature of PDS-containing polymers can be utilized to modify them based on the available reactive group of therapeutic cargos, covalently connecting polymers with cargos for delivery applications. For example, the reduced cysteine residues on caspase were reacted with the PDS side chains of an amphiphilic copolymer, resulting in a polymer-protein conjugate that can reversibly control the corresponding protein activity.[57] Comparing to reduced thiol from cysteine residues, amine groups from lysine residues are broadly available on protein surface. Considering the nucleophilicity of amines, a p-nitrophenylcarbonate (NPC) moiety was installed on PDS-containing polymers through disulfide exchange reaction (Figure 5).[58] The reaction between protein surface amine and the NPC moiety within the polymer resulted in a polymer-protein conjugate that connected by a carbamate. During the formation of the conjugate, protein acted as a template to form the polymeric network. Note that a redox-sensitive disulfide moiety is placed at the β-position relative to the carbamate oxygen. Reductive cleavage of the disulfide bond will cause the cleavage of carbamate and release the original amine, eventually allowing the protein to be tracelessly liberated from the polymer. The disulfide-containing polymeric platform was shown to successfully deliver proteins with varied molecular weight (MW) and pI, including lysozyme, RNase A and cytochrome C (MW within 12~15 kDa and pI > 8.5), as well as green fluorescent protein (GFP, MW ~27 kDa, pI 5.5).
Figure 5.

Schematic illustration for the conjugation between the p-nitrophenylcarbonate moieties of polymers and the surface lysine residues of proteins, followed by the traceless release of protein in a reducing environment. Adapted with permission.[58] Copyright 2017, American Chemical Society.
Apart from the disulfide moiety, the pyridine moiety within PDS provides another site for the functionalization of PDS-containing polymers. In a recent report, N-methylation reaction was achieved on the PDS side chains of a random copolymer, generating a positively charged copolymer (Figure 6).[59] These cationic polymers were subsequently complexed with RNA through electrostatic interaction. The methylated PDS moieties on the polymer side chain feature several advantages for RNA delivery systems. After forming the RNA-polymer polyelectrolyte, varied amount of DTT can be introduced to achieve tunable disulfide cross-linkage percentage within the RNA-polymer complex. Most importantly, the DTT-crosslinking process simultaneously removes the cationic moieties from the complex.[59, 60] As positive surface charge of materials at high dosage generally cause severe cytotoxicity, the design therefore significantly improves the biocompatibility of the polymer-based RNA delivery system. Moreover, when partially crosslinking the RNA-polymer complex, the remaining methylated PDS moieties are reactive sites to post-functionalize the complex, allowing the incorporation of thiolated small-molecule targeting ligands or biomacromolecules for future material design (Figure 6).[61] Overall, such a disulfide-containing platform has been shown to formulate complexes with nucleic acids at different lengths, from small interfering RNA (siRNA, 20~25 bp) [60, 61] to long double-stranded DNA (~900 bp).[61]
Figure 6.

Formulation process of the RNA–Polymer complex that contains disulfide bonds. Adapted with permission.[61] Copyright 2019, American Chemical Society.
Role of disulfide bond during cellular uptake
The general purpose of involving disulfide bonds in delivery vehicles is to take advantage of the overall reducing environment of the cytosol, aiming for the intracellular release of the therapeutic cargoes upon the cleavage of disulfide. The GSH-induced intracellular cleavage of disulfide bonds can be evaluated by comparing the therapeutic effect of the delivery system at different intracellular GSH levels. For example, supplementing glutathione monoester (GSH-OEt) increases the intracellular GSH concentration, as esterases cleave GSH-OEt into GSH.[62] Buthionine sulfoximine-treatment decreases the intracellular GSH concentration by inhibiting gamma-glutamylcysteine synthetase.[63] With disulfide bonds embedded in the polymeric delivery vehicles, enhanced intracellular delivery efficiency for small-molecule drugs has been observed comparing to the polymer analog without disulfide bonds.[64] Such enhanced therapeutic efficacy is presumably due to the cleavage of the disulfide bonds in the reducing cellular environment, resulting in a faster release rate of the cargos from the delivery vehicle.[65]
Since the majority of delivery vehicles undergo the endocytic process that starts with the endosomes,[66, 67] it is important to understand the redox environment within endosomes. Previous results have revealed that endosomes and lysosomes have comparable oxidizing environment to endoplasmic reticulum.[68] These results indicate that disulfide bonds in exogenic materials may remain stable in endosomes and lysosomes, and can be cleaved in the cytosolic reducing environment.[69, 70] However, the endosomal escape process of disulfide-containing macromolecules is not well understood yet.[44] In fact, the overall cellular uptake process of disulfide-containing polymers remain to be deeply understood,[71] including but not limited to the participation of cell surface thiols,[72] protein disulfide isomerase (PDI),[73, 74] and gamma-interferon-inducible lysosomal thiol reductase (GILT).[75, 76]
Conclusions and outlook
Disulfide-containing macromolecules inherently possess outstanding degradability in reducing environments, enhancing their biocompatibility and providing controlled bond cleavage mechanisms for biological applications. Meanwhile, versatile design principles are available to display disulfide bonds at different sites of polymers, and chemical modifications can be conveniently tailored on the polymers to accommodate the therapeutic cargos. These features make disulfide-containing macromolecules attractive scaffolds for therapeutic delivery applications. However, thorough investigations are still required on the cellular uptake process of disulfide-containing macromolecules, specifically regarding the role of disulfide bond. Understanding these processes will fundamentally benefit the materials design of macromolecules, eventually moving the field forward to tackle future challenges in therapeutic delivery.
Acknowledgement
We thank the support from the NIGMS of the National Institutes of Health (GM-128181) and the U. S. Army Research Office (W911NF-15-1-0568).
Footnotes
The authors declare no competing interest.
References
- [1].Betz SF, Protein Sci. 1993, 2, 1551–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Trivedi MV, Laurence JS, Siahaan TJ, Curr. Protein Peptide Sci 2009, 10, 614–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dombkowski AA, Sultana KZ, Craig DB, FEBS Lett. 2014, 588, 206–212. [DOI] [PubMed] [Google Scholar]
- [4].Riemer J, Bulleid N, Herrmann JM, Science 2009, 324, 1284. [DOI] [PubMed] [Google Scholar]
- [5].Wedemeyer WJ, Welker E, Narayan M, Scheraga HA, Biochemistry 2000, 39, 4207–4216. [DOI] [PubMed] [Google Scholar]
- [6].Bechtel TJ, Weerapana E, Proteomics 2017, 17, 1600391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Appel EA, del Barrio J, Loh XJ, Scherman OA, Chem. Soc. Rev 2012, 41, 6195–6214. [DOI] [PubMed] [Google Scholar]
- [8].Oh JK, Drumright R, Siegwart DJ, Matyjaszewski K, Prog. Polym. Sci 2008, 33, 448–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Stern R, Biochem. Biophys. Res. Commun 1970, 41, 608–614. [DOI] [PubMed] [Google Scholar]
- [10].Immordino ML, Dosio F, Cattel L, Int. J. Nanomed 2006, 1, 297–315. [PMC free article] [PubMed] [Google Scholar]
- [11].Allen TM, Cullis PR, Adv. Drug Del. Rev. 2013, 65, 36–48. [DOI] [PubMed] [Google Scholar]
- [12].Xu ZP, Zeng QH, Lu GQ, Yu AB, Chem. Eng. Sci 2006, 61, 1027–1040. [Google Scholar]
- [13].Sun C, Lee JSH, Zhang M, Adv. Drug Del. Rev 2008, 60, 1252–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Davis ME, Chen Z, Shin DM, Nat. Rev. Drug Discov 2008, 7, 771–782. [DOI] [PubMed] [Google Scholar]
- [15].Qi L, Gao X, Expert Opin. Drug Deliv 2008, 5, 263–267. [DOI] [PubMed] [Google Scholar]
- [16].Petros RA, DeSimone JM, Nat. Rev. Drug Discov 2010, 9, 615–627. [DOI] [PubMed] [Google Scholar]
- [17].Uhrich KE, Cannizzaro SM, Langer RS, Shakesheff KM, Chem. Rev 1999, 99, 3181–3198. [DOI] [PubMed] [Google Scholar]
- [18].Duncan R, Nat. Rev. Drug Discov 2003, 2, 347–360. [DOI] [PubMed] [Google Scholar]
- [19].Pack DW, Hoffman AS, Pun S, Stayton PS, Nat. Rev. Drug Discov 2005, 4, 581–593. [DOI] [PubMed] [Google Scholar]
- [20].Liechty WB, Kryscio DR, Slaughter BV, Peppas NA, Annu. Rev. Chem. Biomol. Eng. 2010, 1, 149–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lv J, Fan Q, Wang H, Cheng Y, Biomaterials 2019, 218, 119358. [DOI] [PubMed] [Google Scholar]
- [22].Kloxin CJ, Bowman CN, Chem. Soc. Rev. 2013, 42, 7161–7173. [DOI] [PubMed] [Google Scholar]
- [23].Meng F, Hennink WE, Zhong Z, Biomaterials 2009, 30, 2180–2198. [DOI] [PubMed] [Google Scholar]
- [24].Mura S, Nicolas J, Couvreur P, Nat. Mater. 2013, 12, 991. [DOI] [PubMed] [Google Scholar]
- [25].Huo M, Yuan J, Tao L, Wei Y, Polym. Chem. 2014, 5, 1519–1528. [Google Scholar]
- [26].Phillips DJ, Gibson MI, Antioxid. Redox Signal 2014, 21, 786–803. [DOI] [PubMed] [Google Scholar]
- [27].Lee MH, Yang Z, Lim CW, Lee YH, Dongbang S, Kang C, Kim JS, Chem. Rev. 2013, 113, 5071–5109. [DOI] [PubMed] [Google Scholar]
- [28].Lee MH, Sessler JL, Kim JS, Acc. Chem. Res. 2015, 48, 2935–2946. [DOI] [PubMed] [Google Scholar]
- [29].Pastore A, Federici G, Bertini E, Piemonte F, Clin. Chim. Acta 2003, 333, 19–39. [DOI] [PubMed] [Google Scholar]
- [30].Cheng R, Feng F, Meng F, Deng C, Feijen J, Zhong Z, J. Controlled Release 2011, 152, 2–12. [DOI] [PubMed] [Google Scholar]
- [31].Emilitri E, Ranucci E, Ferruti P, J. Polym. Sci., Part A: Polym. Chem 2005, 43, 1404–1416. [Google Scholar]
- [32].Chen J, Wu C, Oupický D, Biomacromolecules 2009, 10, 2921–2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lin C, Zhong Z, Lok MC, Jiang X, Hennink WE, Feijen J, Engbersen JFJ, Bioconjugate Chem. 2007, 18, 138–145. [DOI] [PubMed] [Google Scholar]
- [34].Zeng H, Little HC, Tiambeng TN, Williams GA, Guan Z, J. Am. Chem. Soc 2013, 135, 4962–4965. [DOI] [PubMed] [Google Scholar]
- [35].Liu J, Jiang X, Xu L, Wang X, Hennink WE, Zhuo R, Bioconjugate Chem. 2010, 21, 1827–1835. [DOI] [PubMed] [Google Scholar]
- [36].King TP, Li Y, Kochoumian L, Biochemistry 1978, 17, 1499–1506. [DOI] [PubMed] [Google Scholar]
- [37].Espeel P, Goethals F, Du Prez FE, J. Am. Chem. Soc 2011, 133, 1678–1681. [DOI] [PubMed] [Google Scholar]
- [38].Miyata K, Kakizawa Y, Nishiyama N, Harada A, Yamasaki Y, Koyama H, Kataoka K, J. Am. Chem. Soc 2004, 126, 2355–2361. [DOI] [PubMed] [Google Scholar]
- [39].Kang HC, Kang H-J, Bae YH, Biomaterials 2011, 32, 1193–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhuang J, Zhao B, Thayumanavan S, ACS Macro Lett. 2019, 8, 245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bang E-K, Lista M, Sforazzini G, Sakai N, Matile S, Chem. Sci 2012, 3, 1752–1763. [Google Scholar]
- [42].Bang E-K, Gasparini G, Molinard G, Roux A, Sakai N, Matile S, J. Am. Chem. Soc 2013, 135, 2088–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bang E-K, Ward S, Gasparini G, Sakai N, Matile S, Polym. Chem 2014, 5, 2433–2441. [Google Scholar]
- [44].Gasparini G, Bang E-K, Molinard G, Tulumello DV, Ward S, Kelley SO, Roux A, Sakai N, Matile S, J. Am. Chem. Soc 2014, 136, 6069–6074. [DOI] [PubMed] [Google Scholar]
- [45].Fu J, Yu C, Li L, Yao SQ, J. Am. Chem. Soc 2015, 137, 12153–12160. [DOI] [PubMed] [Google Scholar]
- [46].Qian L, Fu J, Yuan P, Du S, Huang W, Li L, Yao SQ, Angew. Chem. Int. Ed 2018, 57, 1532–1536. [DOI] [PubMed] [Google Scholar]
- [47].Sui B, Cheng C, Xu P, Adv. Ther 2019, 1900062. [Google Scholar]
- [48].Wang L, Kristensen J, Ruffner DE, Bioconjugate Chem. 1998, 9, 749–757. [DOI] [PubMed] [Google Scholar]
- [49].Bulmus V, Woodward M, Lin L, Murthy N, Stayton P, Hoffman A, J. Controlled Release 2003, 93, 105–120. [DOI] [PubMed] [Google Scholar]
- [50].Ghosh S, Basu S, Thayumanavan S, Macromolecules 2006, 39, 5595–5597. [Google Scholar]
- [51].Ryu J-H, Roy R, Ventura J, Thayumanavan S, Langmuir 2010, 26, 7086–7092. [DOI] [PubMed] [Google Scholar]
- [52].Ryu J-H, Chacko RT, Jiwpanich S, Bickerton S, Babu RP, Thayumanavan S, J. Am. Chem. Soc 2010, 132, 17227–17235. [DOI] [PubMed] [Google Scholar]
- [53].Jiwpanich S, Ryu J-H, Bickerton S, Thayumanavan S, J. Am. Chem. Soc 2010, 132, 10683–10685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ryu J-H, Jiwpanich S, Chacko R, Bickerton S, Thayumanavan S, J. Am. Chem. Soc 2010, 132, 8246–8247. [DOI] [PubMed] [Google Scholar]
- [55].Ryu J-H, Bickerton S, Zhuang J, Thayumanavan S, Biomacromolecules 2012, 13, 1515–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].González-Toro DC, Ryu J-H, Chacko RT, Zhuang J, Thayumanavan S, J. Am. Chem. Soc 2012, 134, 6964–6967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ventura J, Eron SJ, González-Toro DC, Raghupathi K, Wang F, Hardy JA, Thayumanavan S, Biomacromolecules 2015, 16, 3161–3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Dutta K, Hu D, Zhao B, Ribbe AE, Zhuang J, Thayumanavan S, J. Am. Chem. Soc 2017, 139, 5676–5679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Jiang Z, Cui W, Prasad P, Touve MA, Gianneschi NC, Mager J, Thayumanavan S, Biomacromolecules 2019, 20, 435–442. [DOI] [PubMed] [Google Scholar]
- [60].Dutta K, Bochicchio D, Ribbe AE, Alfandari D, Mager J, Pavan GM, Thayumanavan S, ACS Appl. Mater. Interfaces 2019, 11, 24971–24983. [DOI] [PubMed] [Google Scholar]
- [61].Jiang Z, Cui W, Mager J, Thayumanavan S, Ind. Eng. Chem. Res 2019, 58, 6982–6991. [Google Scholar]
- [62].Levy EJ, Anderson ME, Meister A, Proc. Natl. Acad. Sci. U.S.A 1993, 90, 9171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Griffith OW, J. Biol. Chem 1982, 257, 13704–13712. [PubMed] [Google Scholar]
- [64].Zhu Y, Zhang J, Meng F, Deng C, Cheng R, Feijen J, Zhong Z, J. Controlled Release 2016, 233, 29–38. [DOI] [PubMed] [Google Scholar]
- [65].Sun H, Guo B, Cheng R, Meng F, Liu H, Zhong Z, Biomaterials 2009, 30, 6358–6366. [DOI] [PubMed] [Google Scholar]
- [66].Conner SD, Schmid SL, Nature 2003, 422, 37–44. [DOI] [PubMed] [Google Scholar]
- [67].Sahay G, Alakhova DY, Kabanov AV, J. Controlled Release 2010, 145, 182–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Austin CD, Wen X, Gazzard L, Nelson C, Scheller RH, Scales SJ, Proc. Natl. Acad. Sci. U.S.A 2005, 102, 17987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Brülisauer L, Gauthier MA, Leroux J-C, J. Controlled Release 2014, 195, 147–154. [DOI] [PubMed] [Google Scholar]
- [70].Wang Z, Luo M, Mao C, Wei Q, Zhao T, Li Y, Huang G, Gao J, Angew. Chem. Int. Ed 2017, 56, 1319–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Jiang Z, He H, Liu H, Thayumanavan S, Biomacromolecules 2019, 20, 4407–4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Torres AG, Gait MJ, Trends Biotechnol. 2012, 30, 185–190. [DOI] [PubMed] [Google Scholar]
- [73].Sullivan DC, Huminiecki L, Moore JW, Boyle JJ, Poulsom R, Creamer D, Barker J, Bicknell R, J. Biol. Chem 2003, 278, 47079–47088. [DOI] [PubMed] [Google Scholar]
- [74].Wilkinson B, Gilbert HF, Biochim. Biophys. Acta 2004, 1699, 35–44. [DOI] [PubMed] [Google Scholar]
- [75].Phan UT, Arunachalam B, Cresswell P, J. Biol. Chem 2000, 275, 25907–25914. [DOI] [PubMed] [Google Scholar]
- [76].Hastings KT, Cresswell P, Antioxid. Redox Signal 2011, 15, 657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]