

Abstract
Ketogenic diets (KDs) alter brain metabolism. Multiple mechanisms may account for their effects, and different brain regions may variably respond. Here, we considered how a KD affects brain neuron and astrocyte transcription. We placed male C57Bl6/N mice on either a 3-month KD or chow diet, generated enriched neuron and astrocyte fractions, and used RNA-Seq to assess transcription. Neurons from KD-treated mice generally showed transcriptional pathway activation while their astrocytes showed a mix of transcriptional pathway suppression and activation. The KD especially affected pathways implicated in mitochondrial and endoplasmic reticulum function, insulin signaling, and inflammation. An unbiased analysis of KD-associated expression changes strongly implicated transcriptional pathways altered in AD, which prompted us to explore in more detail the potential molecular relevance of a KD to AD. Our results indicate a KD differently affects neurons and astrocytes, and provide unbiased evidence that KD-induced brain effects are potentially relevant to neurodegenerative diseases such as AD.
Keywords: Ketogenic diet, Alzheimer’s disease, RNA-Seq, mitochondria, neurons, astrocytes
Graphical Abstract
We proposed different brain cell types would respond differently to a ketogenic diet (KD). Here, we focused on neuron and astrocyte transcription responses. Neurons from KD-treated mice generally showed transcriptional pathway activation while astrocytes showed a mix of transcriptional suppression and activation. The KD especially affected pathways implicated in mitochondrial and endoplasmic reticulum function, insulin signaling, and inflammation. An unbiased analysis of KD-associated expression changes strongly implicated pathways altered in Alzheimer’s disease. Our results indicate a KD uniquely affects neurons and astrocytes and show a KD alters the brain in ways that establish its relevance to specific neurodegenerative diseases.
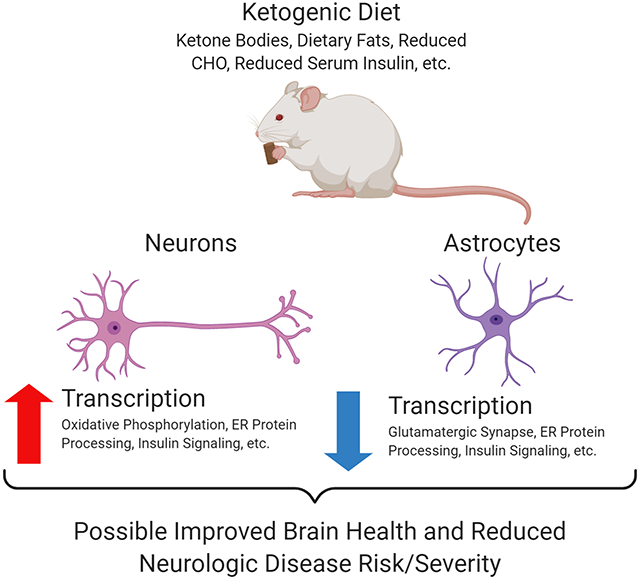
Introduction
The ketone bodies D-β-hydroxybutyrate and acetoacetate supply and support brain energy-generating carbon fluxes (Koppel & Swerdlow 2018). In starvation states they supplement and spare brain glucose catabolism. By activating liver ketone body production ketogenic diets (KDs) increase circulating levels of D-β-hydroxybutyrate and acetoacetate, which freely cross the blood brain barrier and access neurons and astrocytes via monocarboxylate transporters (MCTs) 2 and 1 respectively. (Owen et al. 1967; Vijay & Morris 2014). In ketolytic tissues such as neurons ketone bodies are converted to acetyl-CoA, which accesses the mitochondrial TCA cycle and generates reducing equivalents that enter the electron transport chain (Fukao et al. 1997).
KDs reduce seizure frequency in persons with epilepsy (Kossoff et al. 2009), which suggests manipulating brain energy metabolism in some settings confers neurologic benefits. Different mechanisms may contribute as this intervention can support bioenergetics, enhance ROS scavenging, modulate neurotransmitters, induce post-translational protein modifications, increase neurotrophin signaling, and activate signaling receptors (Marosi et al. 2016; Rahman et al. 2014; Kimura et al. 2011; Shimazu et al. 2013; Xie et al. 2016; Rardin et al. 2013; Sullivan et al. 2004; Erecinska et al. 1996). Some advocate a potential role for KDs in treating neurodegenerative diseases that feature defective brain bioenergetics, including Alzheimer’s disease (AD), in which mitochondrial dysfunction and glucose hypometabolism can precede cognitive decline (Taylor et al. 2017; Taylor et al. 2019; Cunnane et al. 2016a; Cunnane et al. 2016b; Swerdlow et al. 1989).
Some of the above-noted mechanisms may not depend on the presence of ketone bodies, although overall ketone bodies presumably play an important role. A KD may also uniquely affect distinct parts of the brain. Here, we considered whether neurons and astrocytes could respond differently to a KD. To address this, we placed C57Bl6/N male mice on either a 3-month KD or chow diet and generated enriched neuron and astrocyte fractions. We then used RNA-Seq KEGG pathway analysis, gene set enrichment analysis (GSEA), Ingenuity Pathway Analysis (IPA), and an AD-pertinent, hypothesis-based approach to analyze KD-induced transcriptome changes.
Materials and Methods
KD Intervention in C57Bl6/N Mice
The University of Kansas Medical Center Institutional Animal Care and Use Committee approved all experimental animal protocols and procedures (Animal Care and Use Protocol #2020-2558). This study was not pre-registered. This study was exploratory and was designed for a 90-day dietary intervention period. Power calculations were performed using the PROPER method to estimate an appropriate number of mice (Wu et al. 2014). For power calculations we assumed a sampling of approximately 20,000 genes, primary comparison of diet effect within cell-type (comparison of 2 groups), and 5% of genes being differentially expressed with a magnitude of effect defined from a normal distribution with standard deviation equal to 1 on a log2 scale. From this we determined that groups of 8, 10, and 12 would provide marginal power of 0.72, 0.75, and 0.77 respectively with a nominal false discovery rate of 0.1.
Sixteen -week old C57Bl6/N male mice (Charles River Laboratories, RRID: MGI: 5656552) were acclimated for one week before obtaining baseline weight, blood glucose, and blood ketone levels. Mice were group-housed to a maximum of 5 littermates per cage in Touch Slim Line IVC cages (Techniplast). Mice were separated on an individual basis upon recommendation of the institutional veterinary staff for excessive fighting and fight wounds. Singly housed mice were provided additional domed plastic housing for environmental enrichment per institutional standard operating procedures. Blood was obtained by facial vein phlebotomy and metabolites measured using a Precision Xtra meter (Abbott Diabetes Care, Inc. cat no. #9881465) with blood glucose (Abbott cat no. #9972865) and ketone test strips (Abbott cat no. #7074565). Mice were distributed into dietary treatment groups via simple randomization stratified by weight to obtain equivalent mean initial body weights prior to initiating the KD.
Mice were maintained on either a standard chow (LabDiet cat no. #5053) or ketogenic diet (Bioserve cat no. #F3666) for 90 days with ad libitum access to food and water. Mouse weight and blood parameters were monitored periodically throughout the dietary intervention. Food intake was measured by weighing food prior to and following a 24-hour feeding cycle for three days with a mean intake value per mouse (g) being determined and multiplied by the associated diet kcal/g to determine the daily consumed kcal.
We began the study by utilizing ten mice for protocol development. For the dietary intervention initial cohorts of 15 mice per group were generated, to facilitate a final comparison of 12 mice per group at study termination. In total, 40 mice contributed to the present study.
Cell Type Enrichment and RNA Isolation
Figure S1 illustrates the overall experimental timeline and cell enrichment procedure. At the end of the 90-day intervention mice were sacrificed for brain cell separation and RNA isolation. Euthanasia began in the morning with sample isolation completed by early afternoon. Mice that completed the diet interventions were euthanized by rapid decapitation, using an inspected and certified mouse guillotine and brains were removed from the skull and placed in digestion buffer within 30 seconds of decapitation. For these mice anesthesia and CO2 asphyxiation were not utilized to avoid disruptions to brain metabolism prior to euthanasia (Mena et al. 2010; Richerson 1995; Dean et al. 2003). Animals that were euthanized prior to study completion for humane reasons received primary CO2 asphyxiation in home cages followed by secondary decapitation.
The digestion buffer contained 2 parts Neurobasal medium (Gibco cat no. #21103-049) to 1-part Accutase (Gibco cat no. #A11105-01) supplemented with DNAse I (Sigma cat no. #DN25) to a final concentration of 0.3 mg/mL. Brains were incubated in digestion buffer at 37°C for 20 minutes. Brains were supplemented with an additional 10 mL of neurobasal medium and then triturated via 10 passages through 10 mL serological pipettes, followed by 10 passages through 5 mL serological pipettes, and a final 20 passages through 1,000 μL pipette tips in a 50 mL conical tube. We removed large debris by letting it settle to the bottom of the tube. The medium layer was collected and passed through a 70 μm cell strainer (Fisher cat no. #22-363-549) pre-hydrated with 5 mL neurobasal medium. The pass-through was collected and strained through a 40 μm cell strainer (Fisher cat no. #22-363-547) pre-hydrated with 5 mL neurobasal medium.
The final pass-through was collected and centrifuged at 300 rcf for 10 minutes at 4°C to pellet cells. The supernatant was discarded. Cells were resuspended in 1.8 mL of ice-cold cell stain buffer composed of low-fluorescence Hibernate A (BrainBits, LLC), 0.5% bovine serum albumin (Boston Bioproducts cat no. #P-753), and SuperaseIn RNAse inhibitor (ThermoFisher cat no. #AM2696) and sterile filtered. 200 μL of myelin removal beads (Miltenyi Biotec cat no. #130-096-433) were added and cells were incubated for 15 minutes at 4°C on a rocking shaker. 10 mL of fresh ice-cold cell stain buffer were added per sample and cells were again centrifuged. The supernatant was discarded, and the cell pellet resuspended in 4 mL of cell stain buffer. The cell suspension was evenly distributed by 1 mL volumes into clean, autoclaved microcentrifuge tubes. Tubes were placed into a magnetic tube rack (ThermoFisher cat no. #12321D) at 4°C for 10 minutes. The unbound supernatant was carefully removed through pipetting and transferred to a new, clean 15 mL conical tube. Collections were centrifuged under prior conditions and the supernatant discarded. Cell pellets were resuspended in 720 μL of cell stain buffer and evenly distributed into two clean microcentrifuge tubes for cell-type specific enrichment protocols.
For neuron enrichment, 80 μL of biotinylated antibody cocktail (Miltenyi Biotec cat no. #130-115-389) targeting non-neuronal cell types was added to the cell suspension per manufacturer instructions. Samples were incubated at 4°C for 10 minutes on a rotating tube rack (Fisher cat no. #05-450-127). Samples were centrifuged at 300 g for 10 minutes at 4°C and the supernatant carefully removed. Pellets were resuspended in 360 μL of cell stain buffer and 80 μL of anti-biotin magnetic beads added. Cells were again incubated at 4°C for 10 minutes on a rotating tube rack. Samples were diluted with 1 mL of ice-cold cell stain buffer and placed in the magnetic stand for 10 minutes at 4°C. Unbound supernatant containing neuronal cells was collected so as not to disturb the sedimented layer along the magnet containing glia and endothelium and transferred to a clean microcentrifuge tube. Collections were centrifuged as described above and the supernatants discarded. Cell pellets were dissolved in 1 mL of TRI reagent (ThermoFisher cat no. #15596018) for ten minutes before proceeding to RNA isolation. RNA was isolated via phenol-chloroform extraction with TRI reagent, with RNA purity and content measured by A260/280 ratio.
For astrocyte enrichment, 80 μL of biotinylated antibody targeting glutamate aspartate transporter 1 (GLAST) (Miltenyi Biotec cat no. #130-095-826, RRID: AB_2733472) was added to the cell suspension per manufacturer instructions. Samples were incubated at 4°C for 10 minutes on a rotating tube rack. Samples were centrifuged at 300 g for 10 minutes at 4°C and the supernatant was removed. Pellets were resuspended in 360 μL of cell stain buffer and 80 μL of anti-biotin magnetic beads were added. Cells were next incubated at 4°C for 15 minutes on a rotating tube rack. Samples were centrifuged as described previously and the supernatant removed. Pellets were resuspended in 1 mL of ice-cold cell stain buffer and placed in the magnetic stand for 10 minutes at 4°C. Unbound supernatant containing non-astrocytic cells was removed so as not to disturb the sedimented layer along the magnet containing astrocytes. The sedimented magnetized cells were washed in 1 mL of cell stain buffer and centrifuged to a pellet. The supernatant was removed, and pellets were dissolved in 1 mL of TRI reagent and allowed to stand for ten minutes before proceeding to RNA isolation. RNA was isolated via phenol-chloroform extraction with TRI reagent, with RNA purity and content measured by A260/280 ratio. The procedure time from sacrifice to TRI reagent was just under 3 hours.
Fluorescence Activated Cell Sorting (FACS) Cell Enrichment Analysis
To facilitate a FACS analysis of cell enrichment, we stained an initial set of cell suspensions with fluorescent antibodies directed to different brain cell populations. At different stages of the cell enrichment protocols, cell counts were ascertained and adjusted to 1 x 106 cells/mL with cell stain buffer and aliquoted into clean microcentrifuge tubes for the quantitative determination of negative, single channel positive, and combined positive labeling. The stains/antibodies were as follows: GLAST-PE antibody for astrocytes (Miltenyi Biotec cat no. #130-118-344, RRID: AB_2733722), CD11b-PE/Cy7 antibody (Miltenyi Biotec cat no. #101216) for microglia, O4-APC (Miltenyi Biotec cat no. #130-099-211, RRID: AB_2751644) for oligodendrocyte precursors, and DAPI (1 μg/mL) for live/dead discrimination. Live/dead discrimination was also performed using Ghost Dye 450 (TONBO Biosciences cat no. #13-0863-T100). Verification of nucleated events was performed using Vybrant DyeCycle Green stain (ThermoFisher cat no. #V35004). All antibodies were added at 1 μL per 1 X 106 cells.
Staining proceeded for 30 minutes on ice in the dark. Samples were then centrifuged as before and washed 3x in cell stain buffer. Following the final spin cells were resuspended in 500 μL and analyzed by flow cytometry. Samples were processed and analyzed by FACS on a BD FACSAria II cytometer (BD Biosciences, San Jose, CA RRID: SCR_018091). Cells were initially gated by forward scatter complexity (FSC) and side scatter complexity (SSC) to identify debris and multicellular events. Events that were DAPI negative but positive in more than one color channel were considered multicellular events or non-specific antibody labeling. Astrocytic events were considered as singly positive for GLAST-PE and neuronal events were considered as those negative for all glial markers.
Generation of Total Stranded RNA Library and Performance of RNA-Seq
Stranded Total RNA-Seq was performed using an Illumina NovaSeq 6000 Sequencing System (RRID: SCR_016387). Total RNA (input range: 315 ng – 1417 ng) was used to initiate the Stranded Total RNA-Seq library preparation protocol. The total RNA fraction underwent ribosomal reduction, size fragmentation (6, 4, 3 or 2 minutes based on %DV200 calculation), reverse transcription into cDNA, and ligation with the appropriate indexed adaptors using the TruSeq Stranded Total RNA HT Sample Preparation Kit (Illumina cat no. #RS-122-2203). Following Agilent Bioanalyzer QC of the library preparation and library quantification using the Roche Lightcycler96 with FastStart Essential DNA Green Master (Roche cat no. #06402712001), the RNA-Seq libraries were adjusted to a 2 nM concentration and pooled for multiplexed sequencing on a NovaSeq 6000. The onboard clonal clustering procedure was automated during the NovaSeq 6000 sequencing run. The 100-cycle paired end sequencing was performed using the NovaSeq 6000 S1 Reagent Kit - 200 cycle (Illumina cat no. #20012864). Following collection, sequence data were converted from .bcl file format to fastQ files and de-multiplexed into individual sequences for further downstream analyses.
RNA-Seq Data Quality Assessment and Preprocessing
To assess the quality of the RNA-Seq data, we used the FastQC tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). The QC report suggested high sequence duplication level in the samples; a high duplication level may result in lower counts per sample and will subsequently reduce the power to detect differentially expressed genes. RNA-Seq by Expectation Maximization (RSEM, RRID: SCR_013027) was then used to map sequences to the mouse mm10 genome assembly (Li & Dewey 2011). The bowtie2 (Bowtie, RRID: SCR_005476) was selected as the aligner within RSEM (Langmead & Salzberg 2012). RSEM produced low alignment rates as the unique alignment rates were observed to be <10 % for most samples.
Raw gene counts were then normalized according to library size and genes with low transcription, defined as less than 1 count per million (CpM) in at least 2 out of 27 samples, were filtered out. After the filtering of low expressed genes, a total of ~23,000 genes were retained for downstream statistical analyses.
Statistical Analysis
Comparisons of physiologic parameters and dietary intake were assessed via unpaired two-tailed Welch’s T-test using GraphPad Prism 9.0.0 software (RRID: SCR_002798). The Bioconductor package “edgeR” (RRID: SCR_012802), a software package for examining differential expression of replicated count data, was used for pair-wise comparisons of gene expression between mice given a 3-month KD and mice given a chow diet (Robinson et al. 2010). Given the count nature characteristic of RNA-seq data, “edgeR” implements novel statistical methods based on the negative binomial distribution as a model for count variability, including empirical Bayes methods, exact tests, and generalized linear models. Given the assumed negative binomial distribution for the models we fit using “edgeR”, no formal test for normality was conducted on our data. When appropriate, the presence of outliers was assessed using the ROUT method with q = 1% in GraphPad Prism 9.0.0 software (RRID: SCR_002798). No outliers were identified by this method. The edgeR differential gene expression analysis was performed separately for astrocytes and neuron samples. Given the large number genes, and subsequently, the large number of tests being implemented, multiple testing adjustment was carried out by computing the Benjamini-Hochberg false discovery rate (FDR).
Pathways Analysis
KEGG (RRID: SCR_012773) pathway analysis and visualization were performed using the R Bioconductor packages “gage” (RRID: SCR_017067) and “pathview” (RRID: SCR_002732) (Luo et al. 2009; Luo & Brouwer 2013). To identify biological pathways enriched with differentially expressed genes, we performed a gene set enrichment analysis (GSEA, RRID: SCR_003199) (Subramanian et al. 2005). Datasets were analyzed in reference to the h.all.v.7.0.symbols.gmt gene set database on the chip platform Mouse_Gene_Symbol_Remapping_MSigDB of the Molecular Signature Database (MSigDB, RRID: SCR_016863) (Liberzon et al. 2011). Network activation analysis amongst the top 2,000 differentially expressed genes was also performed by Ingenuity Pathway Analysis (Qiagen Inc., RRID: SCR_008653). Differentially expressed genes were defined as genes whose transcripts differed between groups by a minimum 2-fold increase or decrease when compared to the chow diet groups. Analysis was performed in reference to the Ingenuity Knowledge Base (RRID: SCR_008117) including direct and indirect interactions with filters applied for mouse and central nervous system.
Blinding
Investigators involved in mouse husbandry were aware of the diet intervention status through the brain cell separation and RNA quality assessment stages. They were uninvolved in further sample management or statistical analyses following the submission of RNA samples to core facilities for cDNA library generation, quality control, and subsequent pathway analysis. Genomics Core and Biostatistics Core personnel were blinded to intervention status and remained so through the completion of all quality control steps and pathway analyses.
Figure Generation
Figures within this publication were generated using GraphPad Prism v 9.0.0, Microsoft PowerPoint, and BioRender (SCR_018361).
Results
Validation of Cell Population Enrichment
We initially analyzed pooled brains from 3 mice to validate our cell population enrichment procedures. Following the myelin removal step, we used FSC and SSC profiling to identify potential cells, and DAPI, which labels the nuclei of cells that lack intact plasma membranes, to assess basic cell integrity. Within the potential cell population DAPI negative events constituted 92.86 ± 1.66% of the total events (Fig. 1A&B). To further determine what percentage of DAPI negative events represented live cells and not subcellular debris we used Dye Cycle Green to label nucleated cells, and Ghost Dye eFluor 450 to label dead cells. Dye Cycle Green labeling indicated 85.45 ± 3.00% of the DAPI negative events came from nucleated cells rather than from subcellular debris. Ghost Dye labeling indicated 68.03 ± 2.69% of the events derived from viable cells.
Figure 1. FACS enrichment analysis.

These data derive from three pooled brains (n = 3). Representative images of individual sorts are shown here. A) For this myelin-depleted, unenriched cell population, red events falling outside the predicted cell FSC and SSC boundaries are considered non-cells. Events falling within the predicted cell FSC and SSC boundaries represent either whole cells or subcellular debris. Within the gated area, purple indicates DAPI negative events, and black indicates DAPI positive events. Most events in the gated region are DAPI negative. B) We used FACS to further quantify the events in the gated area from (A). This enabled a quantitative assessment of what percentage of the events were DAPI negative. C) Brain cell samples not subjected to cell enrichment protocols demonstrated large numbers of events in all quadrants/staining combinations. Multi-positive events are debris/nonspecific binding. GLAST served as the astrocyte marker, CD11b as the microglial marker, and O4 as the oligodendrocyte (precursor) marker. D) Following the neuron enrichment procedure, the neuron fraction produced cells that mostly sorted into the 3rd quadrant (negative for all markers), while the neuron-negative fraction generated during the neuron-enrichment procedure showed frequent glial antibody-labeled events. E) Following the astrocyte enrichment procedure, the proportion of singly positive GLAST labeled events (quadrant 4) increased, while the astrocyte negative fraction generated during the glial-enrichment procedure showed a reduced number of GLAST-positive events.
Applying our neuronal enrichment protocol to the brains of these 3 mice increased the percentage of glial marker-free events from 37.93 ± 6.88% in an unenriched cell fraction to 95.85 ± 2.10% in the final neuron-enriched fraction (Fig. 1C&D). Applying our astrocyte enrichment protocol increased the percentage of singly positive GLAST-PE labeled events from 38.23 ± 15.23% in an unenriched cell fraction to 56.58 ± 23.83% in the enriched astrocyte fraction, with an associated reduction within the non-astrocyte fraction to 8.34 ± 8.31% (Fig. 1C&E).
Diet Macronutrient Profiles and Physiologic Effects
We placed 15 C57Bl6/N mice into each of the two diet groups, with the goal of obtaining 12 samples per group for analysis. One mouse in the KD group was euthanized with cancer, and five mice (3 chow, 2 KD) were euthanized due to fight wounds. Twelve mice in each group, therefore, completed the 3-month diet intervention.
For mice on the KD calories derived mostly from fat, and for mice on the chow diet calories derived mostly from carbohydrate and protein (Fig 2A). Mice on the KD (n=12), but not the chow diet (n=12), increased their BHB level, from 0.29 ± 0.10 mM to a final value of 0.82 ± 0.19 mM (p< 0.001) (Fig 2B). Blood glucose levels progressively fell in the KD mice, from 149.13 ± 26.68 mg/dL to 75.23 ± 35.37 mg/dL (p=0.011) (Fig 2C). The weight of the KD mice fell initially from 29.10 ± 3.06 g to a low of 26.40 ± 5.36 g, before rebounding to a final value of 28.68 ± 6.67 g. Chow-fed mice, on the other hand, steadily increased their weight from 31.16 ± 4.07 g at the start of the intervention to 38.49 ± 4.80 g at the end (Fig 2D). KD mice consumed significantly less food by weight (chow 5.29 ± 0.89 g/day; KD 2.34 ± 0.43 g/day; mean ± SD, p < 0.001), but the daily amount of kcal consumed was equivalent (chow 18.03 ± 3.02 kcal/day, KD 16.96 ± 3.09 kcal/day; mean ± SD, p=0.36) (Fig. 2E).
Figure 2. Diet composition and physiologic effects.

Twelve mice in each diet group completed the three-month intervention and contributed to the data shown here (n = 12, 12 for chow diet and keto diet respectively). A) Chow diet kcals were 62.14% carbohydrate, 24.65% protein, and 13.21% fat. Ketogenic diet kcals were 93.37% fat, 4.70% protein, and 1.80% carbohydrate. B) The KD induced ketosis. C) The KD progressively reduced blood glucose. D) Mice on the KD did not gain weight. E) KD mice consumed less food by mass. F) Both diets showed equivalent Kcal intake. Error bars represent standard error of the mean. *p< 0.05, **p<0.01, ***p<0.001, ****p< 0.0001. ns=not significant.
RNA Sample Preparation and Quality Assessments
Of the 48 neuron and astrocyte samples generated from these 24 mice, 32 samples (8 per group) were selected for cDNA library generation based upon %DV200 quality ≥ 30% (Hester et al. 2016; Illumina 2015). Post-library generation we found two samples did not meet QC criteria. These samples were excluded from further processing and analysis, leaving 7 chow neuron, 7 KD neuron, 8 chow astrocyte, and 8 KD astrocyte samples.
After obtaining gene counts from RSEM, the correlation between samples was calculated to assess mislabeling. The correlation analysis revealed three samples were potentially mislabeled or otherwise significantly distinct from other samples within their group. These three samples were excluded prior to any subsequent statistical analyses. This resulted in a final count of 7 chow neuron, 7 KD neuron, 7 chow astrocyte, and 6 KD astrocyte samples.
For the 14 neuron and 13 astrocyte samples subjected to RNA-Seq, DV200 assessments showed equivalent RNA quality across the separation protocols and dietary interventions. DV200 mean percentages were as follows: chow neurons 42.69 ± 16.76%, KD neurons 45.81 ± 17.95%, chow astrocytes 47.25 ± 14.31%, and KD astrocytes 49.72 ± 15.36% (p=0.9586, two-way ANOVA) (Fig. 3A). Neuron RNA yields exceeded astrocyte RNA yields, but the diets did not affect RNA amount (Fig 3B). The mean RNA yields were as follows: chow neurons 2,964 ± 2,497 ng, chow astrocytes 1,160 ± 432 ng, KD neurons 2,857 ± 1,592 ng, and KD astrocytes 1,004 ± 301 ng (p=0.005 for cell type by two-way ANOVA).
Figure 3. Cell-type enrichment of the transcriptome libraries.

Seven mice contributed Chow neuron and Chow astrocyte data (n = 7, 7). Seven mice contributed KD neuron data, and six mice contributed KD astrocyte data (n = 7, 6). A) RNA quality did not vary by diet or enrichment protocol. B) Astrocytes generated less RNA than neurons. C) Astrocyte-enriched libraries demonstrated higher CpMs of the astrocyte-specific genes Gfap, Slc1a3, Aldh1l1, and S100b than neuron-enriched libraries. D) Neuron and astrocyte-enriched samples demonstrated expression of the neuron specific genes Rbfox3, Syp, Nefm, and Nefh, although in astrocytes the CpM levels for three of these genes were clearly lower than their CpM levels of the astrocyte-specific genes. E) Ratioing the aggregate CpMs of neuron-specific genes to astrocyte-specific genes within individual samples demonstrated a general enrichment for genes in accordance with their cell enrichment protocol. Thirteen of 14 neuron samples had higher ratios than those observed in astrocyte samples. F) Intrasample transcriptome correlation analysis indicated high degrees of correlation between samples that underwent the same enrichment protocol regardless of dietary intervention. Yellow indicates a perfect correlation of 1.00, and blue indicates no correlation of 0.00. Samples are identified by mouse number and enrichment fraction (“N” and “A” correspond to neuron and astrocyte respectively). Not every mouse contributed to both experimental groups. Bars indicate means ± standard deviation. **** indicates p-value < 0.0001.
To verify cell population enrichment in these samples, we quantified select cell-type specific transcripts. RNA levels for genes primarily expressed in astrocytes were higher in the astrocyte fractions (Fig. 3C). This is not surprising given FACS data shown in Fig. 1 predict our neuron fractions should contain very little astrocyte contamination. Relative to levels in neurons, astrocyte Gfap expression was 6.1, Slc1a3 was 5.7, Aldh1l1 was 5.3, and S100b was 8.4 times higher. RNA levels for genes primarily expressed by neurons were also higher in the astrocyte fractions (Fig. 3D). This too is not surprising given FACS data shown in Fig. 1 predict our astrocyte fractions should contain some neuron contamination, and Fig. 3B data that show neurons contain more RNA than astrocytes. Relative to neuron levels, astrocyte Rbfox3 was 2.0, Syp was 2.2, Nefm was 1.7, and Nefh was 4.1 times higher (Fig. 3D).
Given the high raw neuronal gene count in astrocyte samples, we ratioed each sample’s summated neuronal gene counts to its summated astrocyte gene counts. Compared to the astrocyte samples, the neuron:astrocyte gene expression ratio was higher in all but one of the neuron samples (neurons 0.9926 ± 0.302, astrocytes 0.292 ± 0.102; mean ± SD, p < 0.0001) (Fig. 3E). The single deviant sample did not meet statistical significance in an outlier analysis and was identified while considering the expression levels of defined genes. For these reasons we did not exclude this sample. For the analyzed genes, expression changes within and between KD and chow-treated mice strongly correlated most strongly by cell type (Fig. 3F).
To determine whether either protocol generated robust contamination by other cell types, we measured RNA levels of microglia, oligodendrocyte, and endothelial cell genes. The expression levels for most of these genes was quite low (Figure S2).
Changes in Individual Gene Expression
We compared gene expression levels for the KD vs. chow diet neuron and KD vs. chow diet astrocyte samples. Tables S1 and S2 shows the 30 genes with the smallest uncorrected p-values from each cell population. After correcting for multiple comparisons, no genes were identified as differentially expressed.
KEGG Molecular Pathway Analysis
KEGG analysis found that in neurons, the KD activated 96 molecular pathways and suppressed 0 molecular pathways. In astrocytes, the KD activated 5 molecular pathways and suppressed 67 molecular pathways. The different cell types showed pathway overlap, as 58 of the 96 activated neuron pathways were among the 67 suppressed astrocyte pathways (Fig. 4A; Table S3).
Figure 4. KEGG pathway analysis.

A) Venn diagram indicating the number of neuron and astrocyte pathway modules altered by the KD, as well as pathways that overlapped between neurons and astrocytes (n = 7, 7, 7, 6). B) The insulin signaling pathway was upregulated in neurons from KD mice. For each gene transcript displayed, individual mice fed a KD are represented by vertical color bars placed over the gene name to indicate relative expression compared to the mean of the chow cohort for the same gene. Blue reflects a decrease in relative expression, red represents increased relative expression, and grey indicates no change. The color scheme ranges from −1 to +1 indicating a scaled color spectrum. C) The insulin signaling pathway was downregulated in astrocytes from KD mice. In (B) and (C), differences appear clustered around JNK, PI3K, Akt, Ras, Raf, ERK1/2, and PKA. PEPCK deviates from the overall neuron and astrocyte expression shifts, as its expression appears suppressed in neurons and increased in astrocytes.
Our KD-treated mice consistently showed reduced blood glucose levels, which could potentially impact insulin signaling. The unbiased KEGG molecular pathway analysis did reveal the insulin signaling pathway changed in both neurons and astrocytes, but in different directions. Neurons tended to show increased, and astrocytes decreased, insulin pathway gene expression (Fig. 4B&C). Multiple molecular signaling pathways implicated by KEGG analysis seemed to be driven by underlying shifts in JNK, PI3K, Ras/Raf, ERK1/2, and PKA expression. We also observed increased neuron expression of oxidative phosphorylation-related genes and an activation of genes relevant to protein trafficking (Table S3).
GSEA of Molecular Pathways
We used the Molecular Signatures Database (MSigDB) to perform a GSEA analysis of all isolated transcripts. As we saw in the KEGG pathway analysis, the KD favored activation of neuron transcription pathways and the top results by normalized enrichment score (NES) again identified oxidative phosphorylation, insulin-related, and protein trafficking modules. Only one module, the one for “coagulation,” was significantly downregulated in neurons from KD mice (Fig 5A). Astrocyte responses again showed a combination of KD upregulated and downregulated expression. The top activated pathways in KD mouse astrocytes were primarily inflammation-associated signaling modules. Downregulated pathways in KD mouse astrocytes included Myc targets, mTORC1 signaling, and DNA repair modules (Fig. 5B).
Figure 5. GSEA.

Negative enrichment scores, shown in blue, indicate significant pathway downregulation for KD samples versus chow samples. Positive enrichment scores, shown in red, indicate significant pathway upregulation for KD samples versus chow samples. A) For neurons (n = 7, 7). B) For astrocytes (n = 7, 6).
IPA of Gene Expression
We performed an IPA analysis of the 2000 genes with the greatest diet-related differential expression. This again revealed a KD-related increase in neuron pathways that could relate to insulin signaling, including those that involve mTOR, ERK/MAPK, PI3K/Akt, and SAPK/JNK, as well as oxidative phosphorylation-related gene expression (Fig. 6A). KD astrocytes increased their expression of genes that mediate glucuronidation; implicated glucuronidation-dependent pathways included ones for nicotine, serotonin, melatonin, and thyroid hormone degradation. Other activated glial responses included several inflammation-related pathways (Fig. 6B).
Figure 6. IPA.

Orange bars indicate upregulation of KD samples versus chow samples, and blue bars indicate downregulation of KD samples versus chow samples. Degree of saturation of color bars indicates relative strength of prediction scores. White bars indicate no predicted activation or inactivation. Grey bars indicate pathways where not enough information exists to predict activation status. A) For neurons (n = 7, 7). B) For astrocytes (n = 7, 6).
KEGG Pathology Analysis
KEGG analysis found that in neurons, the KD activated 30 pathology-associated modules and suppressed 0 pathology-associated modules. In astrocytes, the KD activated 1 pathology-associated module and suppressed 14 pathology-associated modules. The different cell types showed module overlap, as all the suppressed astrocyte pathology-associated modules were activated in neurons (Fig. 7A; Table S4).
Figure 7. KEGG pathology and disease analysis.

A) Venn diagram indicating the number of neuron and astrocyte pathology and disease modules altered by the KD, as well as modules that overlapped between neurons and astrocytes (n = 7, 7, 7, 6). B) The AD module was altered in neurons from KD mice. For each gene transcript displayed, individual mice fed a KD are represented by vertical color bars placed over the gene name to indicate relative expression compared to the mean of the chow cohort for the same gene. Blue reflects a decrease in relative expression, red represents increased relative expression, and grey indicates no change. The color scheme ranges from −1 to +1 indicating a log2 fold change. C) KD-induced changes in neuron oxidative phosphorylation gene expression helped implicate the AD pathway. The protocol we used did not capture mtDNA-encoded genes, so changes in mtDNA-encoded RNAs are unknown. In most samples, oxidative phosphorylation subunits showed some degree of increased expression.
For the 30 neuron KD-activated pathology-associated transcriptional modules 11 were associated with cancer, 5 with neurodegenerative diseases, 5 with infection, 3 with metabolic disorders, 4 with addiction, and 2 with cardiovascular disease. Within astrocytes, the only upregulated pathology module was one that associates with the response to S. aureus infection. Of the remaining suppressed modules, 7 were associated with cancer, 3 with addiction, 2 with metabolic disorders, 1 with infection, and 1 with cardiovascular disease.
In this unbiased transcriptomic analysis of KD brain effects, AD was the disease/pathology module with the strongest q-value. The curated KEGG AD pathway revealed enhanced transcription of mitochondrial oxidative phosphorylation genes contributed to this finding (Fig. 7B&C). Enhanced transcription of amyloid precursor protein (APP), apolipoprotein E (APOE), the APOE receptor LRP1, tau, the tau kinase GSK3β, and multiple proteins involved in modulating intracellular calcium also contributed.
IPA of Disease States
IPA associated 59 disease states with the observed KD-altered neuron transcription patterns. Seventeen of the 59 significant z-scores were in the negative (range −0.124 to −3.442), and 7 of the 17 predicted reduced risk (z-score range −2.919 to −3.442) (Table S5). The 7 reduced risk modules included neurodegeneration of the central nervous system, hypoplasia of the brain, degeneration of the central nervous system, neurodegeneration of the brain, congenital malformation of the brain, degeneration of the brain, and neurodegeneration. Only two modules were found to have a positive z-score, although they were not predicted to be activated by the software analysis. These two modules were damage of the hippocampus (z-score +1.406) and damage of the cerebral cortex (z-score +1.085).
IPA associated 9 disease states with the observed KD-altered astrocyte transcription patterns. Of these, only the “brain lesion” module had an associated z-score, of +1.745, although the software did not predict its activation (Table S5).
Hypothesis-Driven Evaluation of AD-Associated Transcriptional Changes
As the KEGG analysis specifically revealed a KD alters the expression of genes in a pattern that links it to AD, we looked for KD-induced expression changes by mean fold change of genes previously tied to AD through linkage, genome wide association, and molecular studies (Karch & Goate 2015; Kunkle et al. 2019). Neurons from KD mice showed a frequent although not exclusive pattern of enhanced expression of genes associated with APP metabolism, cholesterol metabolism, endocytosis, and cytoskeleton/axon development, and a muted expression pattern of genes associated with the inflammatory mediator Ms4a superfamily (Fig. 8A). When considered from a more direct perspective of genes that encode APP-processing proteins or proteins believed to regulate APP levels, levels of its derivatives, or related molecules such as Notch neurons from KD frequently (but not exclusively) showed increased expression. This included components of the α, β, and γ -secretases (Fig. 8B). Expression of the tau (MAPT) gene, along with tau kinase and phosphatase genes (Fig. 8C), frequently (but not exclusively) showed increased expression. Relative to changes seen in neurons, astrocyte response patterns often shifted in the opposite direction.
Figure 8. Hypothesis-driven heat maps of AD-relevant genes show a KD frequently affects neurons and astrocytes differently.

Each row corresponds to a single gene. Neuron responses to the KD are shown on the left, and astrocyte responses are shown on the right. The color for each cell indicates the relative mean fold change of the KD samples compared to the normalized mean of the chow-fed samples (n = 7, 7, 7, 6). Red indicates increased expression, blue decreased expression, and white no change in expression. Saturation of color indicates the magnitude of the associated change. A) Relative mRNA expression of genes or locus markers demonstrated through genome wide association or other genetic studies to influence AD risk. B) Relative mRNA expression of genes that encode proteins that interact with APP or influence APP or beta amyloid processing, handling, or expression. C) Relative mRNA expression of MAPT and genes implicated in tau biology, including recognized tau kinases and phosphatases.
Discussion
In this study, a KD increased the gene expression of multiple neuron molecular pathways while simultaneously suppressing their astrocyte expression. The observed patterns of change robustly overlapped with those identified in some neurodegenerative diseases that feature altered brain bioenergetics, including AD.
Empirical experience from the epilepsy field reveals KDs affect brain function, but the precise mechanisms and mediators remain unresolved (Koppel & Swerdlow 2018; Li et al. 2013; McNally & Hartman 2012). Molecular studies intended to address these knowledge gaps often consider the brain as a single compartment. This perhaps limits their ability to resolve such questions, especially given emerging data that indicate neurons and astrocytes are not bioenergetically equivalent (Pellerin & Magistretti 2004; Guzman & Blazquez 2001). Our data support the view that energy metabolism in neurons and astrocytes fundamentally differs and suggest activities from one cell type may complement the activities of the other. Regardless, it seems obvious that if the expression of a gene increases in one cell type and decreases in another, mixing those cell types together would limit the ability to detect intervention-induced changes.
Instead of sampling individual neurons and astrocytes, we attempted to pool as many neurons and astrocytes as possible within separate, enriched fractions. While this approach sacrifices levels of purity achievable through laser capture, it should reduce the impact of sampling variation. We appeared to generate high levels of neuron enrichment, with less robust astrocyte enrichment. Despite this, the fact that we saw inverse relationships argues the levels of enrichment achieved were adequate to reveal cell-specific pathway changes.
A KD certainly changes systemic insulin signaling endpoints and our data clearly reveal this extends to the brain. The overall view, though, is complex as the expression of genes that would favor insulin signaling increases in neurons while their expression in astrocytes decreases. Given the emerging perspective that astrocytes provide energy-rich carbon molecules to neurons (Guzman & Blazquez 2001; Pellerin & Magistretti 2004), this could indicate neurons sensitize themselves for glucose utilization, or at least increase their sensitivity to insulin (Bouzier-Sore et al. 2003; Liu et al. 2017; Stobart & Anderson 2013). On the other hand, under KD conditions astrocytes appear to spare glucose utilization, at least by neurons, and do not increase their own sensitivity to insulin.
The three pathway analysis programs we used found that in neurons, a KD favors the expression of oxidative phosphorylation-related genes. Astrocytes showed neither an increase nor decrease in oxidative phosphorylation-related genes. This could reflect fundamental differences between neuron and astrocyte mitochondria. Neuron mitochondria may emphasize ATP production through oxidative phosphorylation, and astrocyte mitochondria may emphasize the strategic production of carbon intermediates that go on to support molecule biosynthesis or neuron respiration (Pellerin & Magistretti 2004; Guzman & Blazquez 2001). An increase in neuron hypoxia-related gene expression, detected by GSEA, suggests a KD causes neurons to increase their oxygen consumption, which is potentially consistent with the possibility that an increased expression of oxidative phosphorylation-related genes does in fact enhance neuron oxidative phosphorylation.
In most cases, findings from the different analytical platforms nicely complement each other. This typically featured obvious confirmations, but also more obscure ones. For example, GSEA showed a KD activated the xenobiotic metabolism module. This is consistent with the KEGG analysis finding that a KD alters molecular pathways that are relevant to drug addiction.
The unbiased KEGG pathology analysis found a KD altered molecular pathways modified in neurodegenerative diseases that feature bioenergetic dysfunction, with the three smallest q-values implicating AD, Parkinson’s disease, and Huntington’s disease. Interest in using KDs to treat the bioenergetic lesions observed in some of these diseases is not new (Swerdlow et al. 1989), and recent studies suggest a KD could potentially benefit persons with AD or the mild cognitive impairment syndrome that frequently precedes its diagnosis (Taylor et al. 2017; Krikorian et al. 2012; Brandt et al. 2019; Neth et al. 2020). In AD oxidative phosphorylation gene expression declines (Brooks et al. 2007; Wang et al. 2020; Liang et al. 2008). Our data suggest that in neurons, a KD may counteract that change. Neurons from AD brains also show evidence of insulin resistance (Steen et al. 2005), and our data suggest a KD may additionally counteract that change.
Other KD-induced neuron molecular pathways with potential relevance to AD include those that involve protein processing, endosomes, and neurotrophin signaling (Joshi & Wang 2015; Nixon 2005; Xu et al. 2018; Ginsberg et al. 2019; Marcelli et al. 2018). Our astrocyte data indicate a KD can increase glial inflammation-related activity. The functional consequences of any of these observed changes are unclear; such interpretations must await perspective from clinical trials.
Our hypothesis-driven analysis of AD-relevant genes provided additional insight into KD-AD molecular pathway associations and tended to confirm or complement those of the unbiased analyses. Reminiscent of the KEGG analysis, we saw a relative increase in endocytosis-related gene expression. Converse to the increase in astrocyte inflammation-related pathway expression revealed by KEGG, GSEA, and IPA, multiple genes relating to inflammation-related Ms4a activity were downregulated in neurons. With respect to genes implicated in the biology of APP and tau, we continued to observe expression changes whose directions differed between neurons and astrocytes. These changes once again tended to feature upregulation in neurons and downregulation in astrocytes. Because it is unclear whether observed changes promote a compensation, reverse a compensation, alleviate a pathological event, or enhance a pathological event, interpretations of how these shifts impact AD clinical outcomes should await perspective from clinical trials.
One mouse in the KD group developed a large soft-tissue mass and was euthanized prior to study completion. As it is unusual for young mice to develop soft tumor masses, it is worth considering the relationship of the KD to this anomalous event. KDs are high in fatty acids, which have been shown to drive tumorigenesis in a number of tissue types, including glioma (Lin et al. 2017; Lewis et al. 2015; Mashimo et al. 2014; Griffiths et al. 2013; Peck et al. 2016). Although our study focused on brain rather than cancer biology, our KEGG analyses notably revealed effects on cancer-associated pathways including those for choline metabolism, microRNAs, central carbon metabolism, and proteoglycan biology with inverse effects observed between neurons and astrocytes. Other implicated cancer-relevant pathways include insulin/IGF-1 signaling, PI3K-Akt-mTOR, DNA Repair, Myc Targets, MAPK signaling, and Ras signaling. Overall, our data suggest studies to evaluate how a KD influences tumor incidence and progression are warranted.
Following corrections for multiple hypothesis testing we did not observe the significantly altered expression of any individual genes. Some nominally identified genes, however, share functional similarities. Three of the top ten implicated neuron genes, Rnf115 (ring finger protein 115/Rabring7), Ubac2, and Rnf14 participate in ubiquitin signaling. Rnf115 just barely missed significance with a false discovery rate of 0.16. Rnf115 and Rnf14 both function as E3 ubiquitin ligases and are thought to play a role in membrane receptor internalization, ubiquitination, and trafficking content between the endosome and lysosome (Smith et al. 2013).
In addition to our use of enriched as opposed to pure cell fractions, our study features several technical and conceptual limitations. Our RNA quality was limited. We cannot rule out a disproportionate loss of transcripts with short poly-A tails, which could lead to a possible 3′-UTR bias in our transcriptome dataset. Our data also did not inform the status of mitochondrial DNA (mtDNA) expression. In assessing the effects of a KD, we only considered RNA-Seq-detected transcriptional changes and did not pursue orthogonal validation. RNA data may not reflect levels or activation states of proteins, so our data merely implicate biological processes without determining how their changes affect functional outcomes. We enriched for cell type irrespective of brain region, and it is possible that just as neurons differ from astrocytes, neurons or astrocytes from one region may fundamentally differ from other neurons or astrocytes from a different region. From a translational perspective, our KD featured a higher proportion of fat calories and a lower proportion of carbohydrate calories than most human KDs can achieve.
Our study used only male mice. As such, we were unable to identify sex-specific responses. Sex reportedly influences various KD endpoints including degree of ketosis, extent of weight and fat reduction, GLP-1 and PYY serum levels, and γ-glutamyl transferase changes (Lyngstad et al. 2019; D'Abbondanza et al. 2020). Although our unbiased analysis specifically identified AD as a disease of interest, our experiments did not extend our neuron-astrocyte directed analysis to AD transgenic mice. Doing so could potentially inform a number of studies that find ketone-based interventions variably affect histology and behavioral phenotypes in these models (Van der Auwera et al. 2005; Yao et al. 2011; Aso et al. 2013; Beckett et al. 2013; Brownlow et al. 2013; Kashiwaya et al. 2013). Regardless, as we did not include in our study measurements of protein levels or post translational protein modifications, and as our study did not include an AD transgenic mouse model, we cannot say how a KD intervention might affect neuron versus glia gene expression, AD-relevant protein levels, or AD-relevant post-translational modifications in such models. We cannot comment on any correlations between any parameter we measured with mouse cognition or brain electrophysiology.
Our study also does not establish the extent to which different KD-induced physiologic changes contributed to the observed transcriptional changes. Even considering just direct ketone body-mediated mechanisms becomes complex. Cells evolved intricate protein and protein-based networks to monitor and respond to bioenergetic states, and bioenergetic changes impact this infrastructure through post-translational protein modification and by altering gene expression (Selfridge et al. 2015). D-β-hydroxybutyrate itself can inhibit histone deacetylases, (Rardin et al. 2013; Shimazu et al. 2013), serve as a substrate for lysine β-hydroxybutyrylation (Xie et al. 2016), and bind G-protein coupled receptors (Rahman et al. 2014; Kimura et al. 2011; Miyamoto et al. 2019). In addition to causing ketonemia, the KD mice experienced relative hypoglycemia, elevated fat intake, and avoided a longitudinal weight gain. KDs should also lower insulin levels, which could profoundly impact molecular pathways.
At a minimum, our study provides insight that can inform the design of future studies intended to address how KDs or ketone bodies affect the brain. Although our study does not address whether a KD may benefit persons with AD or any other disease, it does though support the view that KDs can affect brain molecular biology in ways that impact neurodegenerative diseases that feature bioenergetic dysfunction, including AD.
Supplementary Material
Acknowledgements
This study was supported by the University of Kansas Alzheimer’s Disease Center (P30 AG035982), a Ruth L. Kirchstein National Research Service Award (NIA F30 AG058397), the KUMC Neurological & Rehabilitation Sciences Training Program (NICHD T32 HD057850), the Stop Alzheimer’s Now Foundation, the Snyder Family Foundation, a Paul G. Roofe Award from the KU Institute for Neurologic Discoveries, and a Mabel A. Woodyard Memorial Fellowship from the KU Institute for Neurologic Discoveries. The KU Medical Center Flow Cytometry Core Laboratory is sponsored in part by NIH/NIGMS P20 GM103326, the Kansas Intellectual and Developmental Disabilities Research Center (NIH U54 HD 090216), the Molecular Regulation of Cell Development and Differentiation COBRE (P20 GM122731-03), and an NIH S10 High-End Instrumentation Grant (NIH S10OD021743) to the University of Kansas Medical Center, Kansas City, KS 66160. The Ingenuity Pathways Analysis (IPA) software and the Compute nodes and storage at the HPC at KU-L used in this publication were supported by the Biostatistics and Informatics Shared Resource, funded by the National Cancer Institute Cancer Center Support Grant P30CA168524; the Kansas IDeA Network of Biomedical Research Excellence Bioinformatics Core, supported in part by the National Institute of General Medical Science award P20GM103418; and the Kansas Institute for Precision Medicine COBRE, supported by the National Institute of General Medical Science award P20GM130423.
Abbreviations:
- AD
Alzheimer’s disease
- Akt
protein kinase B
- Aldh1l1
aldehyde dehydrogenase 1 family member L1
- ApoE
apolipoprotein E
- APP
amyloid precursor protein
- ATP
adenosine triphosphate
- βOHB
β-hydroxybutyrate
- Clu
clusterin
- CNS
central nervous system
- CpM
count per million
- DV200%
percentage of RNA fragments greater than 200 bp
- ERK 1/2
extracellular signal-regulated kinases 1/2
- ETC
electron transport chain
- FACS
fluorescence assisted cell sorting
- FSC
forward scatter
- Gfap
glial fibrillary acidic protein
- GLAST
glutamate aspartate transporter 1
- GSEA
gene set enrichment analysis
- IPA
ingenuity pathway analysis
- JNK
Jun amino-terminal kinase
- KD
ketogenic diet
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- PI3K
phosphoinositide triphosphate kinase
- PKA
protein kinase A
- MCT
monocarboxylate transporter
- mtDNA
mitochondrial DNA
- mTOR
mammalian target of rapamycin
- Nefh
neurofilament heavy
- Nefm
neurofilament medium
- NeuN
neurofilament
- Rbfox3
NeuN
- Rnf115
ring finger protein 115
- Rnf14
ring finger protein 14
- RRID
research resource identifier (see scicrunch.org)
- S100b
S100-beta
- SAPK
stress-activated protein kinase
- Slc1a3
solute carrier protein 1a3 (GLAST)
- SSC
side scatter Syp synaptophysin
- Ubac2
ubiquitin associated domain containing 2
Footnotes
--Human subjects --
Involves human subjects:
If yes: Informed consent & ethics approval achieved:
=> if yes, please ensure that the info "Informed consent was achieved for all subjects, and the experiments were approved by the local ethics committee." is included in the Methods.
ARRIVE guidelines have been followed:
Yes
=> if it is a Review or Editorial, skip complete sentence => if No, include a statement in the "Conflict of interest disclosure" section: "ARRIVE guidelines were not followed for the following reason:"
(edit phrasing to form a complete sentence as necessary).
=> if Yes, insert in the "Conflict of interest disclosure" section:
"All experiments were conducted in compliance with the ARRIVE guidelines." unless it is a Review or Editorial
The authors declare no conflict of interest.
REFERENCES
- Aso E, Semakova J, Joda L, Semak V, Halbaut L, Calpena A, Escolano C, Perales JC and Ferrer I (2013) Triheptanoin supplementation to ketogenic diet curbs cognitive impairment in APP/PS1 mice used as a model of familial Alzheimer's disease. Curr Alzheimer Res 10, 290–297. [DOI] [PubMed] [Google Scholar]
- Beckett TL, Studzinski CM, Keller JN, Paul Murphy M and Niedowicz DM (2013) A ketogenic diet improves motor performance but does not affect β-amyloid levels in a mouse model of Alzheimer's disease. Brain Res 1505, 61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzier-Sore AK, Serres S, Canioni P and Merle M (2003) Lactate involvement in neuron-glia metabolic interaction: (13)C-NMR spectroscopy contribution. Biochimie 85, 841–848. [DOI] [PubMed] [Google Scholar]
- Brandt J, Buchholz A, Henry-Barron B, Vizthum D, Avramopoulos D and Cervenka MC (2019) Preliminary Report on the Feasibility and Efficacy of the Modified Atkins Diet for Treatment of Mild Cognitive Impairment and Early Alzheimer’s Disease. Journal of Alzheimer’s disease : JAD 68, 969–981. [DOI] [PubMed] [Google Scholar]
- Brooks WM, Lynch PJ, Ingle CC, Hatton A, Emson PC, Faull RL and Starkey MP (2007) Gene expression profiles of metabolic enzyme transcripts in Alzheimer's disease. Brain Res 1127, 127–135. [DOI] [PubMed] [Google Scholar]
- Brownlow ML, Benner L, D'Agostino D, Gordon MN and Morgan D (2013) Ketogenic diet improves motor performance but not cognition in two mouse models of Alzheimer's pathology. PLoS One 8, e75713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunnane SC, Courchesne-Loyer A, St-Pierre V, Vandenberghe C, Pierotti T, Fortier M, Croteau E and Castellano CA (2016a) Can ketones compensate for deteriorating brain glucose uptake during aging? Implications for the risk and treatment of Alzheimer's disease. Annals of the New York Academy of Sciences 1367, 12–20. [DOI] [PubMed] [Google Scholar]
- Cunnane SC, Courchesne-Loyer A, Vandenberghe C et al. (2016b) Can Ketones Help Rescue Brain Fuel Supply in Later Life? Implications for Cognitive Health during Aging and the Treatment of Alzheimer's Disease. Frontiers in molecular neuroscience 9, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Abbondanza M, Ministrini S, Pucci G, Nulli Migliola E, Martorelli EE, Gandolfo V, Siepi D, Lupattelli G and Vaudo G (2020) Very Low-Carbohydrate Ketogenic Diet for the Treatment of Severe Obesity and Associated Non-Alcoholic Fatty Liver Disease: The Role of Sex Differences. Nutrients 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean JB, Mulkey DK, Garcia AJ 3rd, Putnam RW and Henderson RA 3rd (2003) Neuronal sensitivity to hyperoxia, hypercapnia, and inert gases at hyperbaric pressures. J Appl Physiol (1985) 95, 883–909. [DOI] [PubMed] [Google Scholar]
- Erecinska M, Nelson D, Daikhin Y and Yudkoff M (1996) Regulation of GABA level in rat brain synaptosomes: fluxes through enzymes of the GABA shunt and effects of glutamate, calcium, and ketone bodies. J Neurochem 67, 2325–2334. [DOI] [PubMed] [Google Scholar]
- Fukao T, Song XQ, Mitchell GA, Yamaguchi S, Sukegawa K, Orii T and Kondo N (1997) Enzymes of ketone body utilization in human tissues: protein and messenger RNA levels of succinyl-coenzyme A (CoA):3-ketoacid CoA transferase and mitochondrial and cytosolic acetoacetyl-CoA thiolases. Pediatric research 42, 498–502. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Malek-Ahmadi MH, Alldred MJ et al. (2019) Selective decline of neurotrophin and neurotrophin receptor genes within CA1 pyramidal neurons and hippocampus proper: Correlation with cognitive performance and neuropathology in mild cognitive impairment and Alzheimer's disease. Hippocampus 29, 422–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths B, Lewis CA, Bensaad K et al. (2013) Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer Metab 1, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman M and Blazquez C (2001) Is there an astrocyte-neuron ketone body shuttle? Trends in endocrinology and metabolism: TEM 12, 169–173. [DOI] [PubMed] [Google Scholar]
- Hester SD, Bhat V, Chorley BN, Carswell G, Jones W, Wehmas LC and Wood CE (2016) Editor's Highlight: Dose-Response Analysis of RNA-Seq Profiles in Archival Formalin-Fixed Paraffin-Embedded Samples. Toxicological sciences : an official journal of the Society of Toxicology 154, 202–213. [DOI] [PubMed] [Google Scholar]
- Illumina (2015) Technical Note: Expression Analysis of FFPE Samples.
- Joshi G and Wang Y (2015) Golgi defects enhance APP amyloidogenic processing in Alzheimer's disease. Bioessays 37, 240–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch CM and Goate AM (2015) Alzheimer's disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry 77, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwaya Y, Bergman C, Lee JH et al. (2013) A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer's disease. Neurobiol Aging 34, 1530–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura I, Inoue D, Maeda T, Hara T, Ichimura A, Miyauchi S, Kobayashi M, Hirasawa A and Tsujimoto G (2011) Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). Proc Natl Acad Sci U S A 108, 8030–8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppel SJ and Swerdlow RH (2018) Neuroketotherapeutics: A modern review of a century-old therapy. Neurochem Int 117, 114–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kossoff EH, Zupec-Kania BA and Rho JM (2009) Ketogenic diets: an update for child neurologists. J Child Neurol 24, 979–988. [DOI] [PubMed] [Google Scholar]
- Krikorian R, Shidler MD, Dangelo K, Couch SC, Benoit SC and Clegg DJ (2012) Dietary ketosis enhances memory in mild cognitive impairment. Neurobiology of aging 33, 425 e419–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkle BW, Grenier-Boley B, Sims R et al. (2019) Genetic meta-analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nature genetics 51, 414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B and Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis CA, Brault C, Peck B et al. (2015) SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene 34, 5128–5140. [DOI] [PubMed] [Google Scholar]
- Li B and Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HF, Zou Y and Ding G (2013) Therapeutic Success of the Ketogenic Diet as a Treatment Option for Epilepsy: a Meta-analysis. Iran J Pediatr 23, 613–620. [PMC free article] [PubMed] [Google Scholar]
- Liang WS, Reiman EM, Valla J et al. (2008) Alzheimer's disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proceedings of the National Academy of Sciences of the United States of America 105, 4441–4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P and Mesirov JP (2011) Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Patel S, Affleck VS, Wilson I, Turnbull DM, Joshi AR, Maxwell R and Stoll EA (2017) Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro Oncol 19, 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, MacKenzie KR, Putluri N, Maletic-Savatic M and Bellen HJ (2017) The Glia-Neuron Lactate Shuttle and Elevated ROS Promote Lipid Synthesis in Neurons and Lipid Droplet Accumulation in Glia via APOE/D. Cell Metab 26, 719–737.e716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W and Brouwer C (2013) Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 29, 1830–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Friedman MS, Shedden K, Hankenson KD and Woolf PJ (2009) GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics 10, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyngstad A, Nymo S, Coutinho SR, Rehfeld JF, Truby H, Kulseng B and Martins C (2019) Investigating the effect of sex and ketosis on weight-loss-induced changes in appetite. Am J Clin Nutr 109, 1511–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelli S, Corbo M, Iannuzzi F, Negri L, Blandini F, Nistico R and Feligioni M (2018) The Involvement of Post-Translational Modifications in Alzheimer's Disease. Curr Alzheimer Res 15, 313–335. [DOI] [PubMed] [Google Scholar]
- Marosi K, Kim SW, Moehl K, Scheibye-Knudsen M, Cheng A, Cutler R, Camandola S and Mattson MP (2016) 3-Hydroxybutyrate regulates energy metabolism and induces BDNF expression in cerebral cortical neurons. J Neurochem 139, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashimo T, Pichumani K, Vemireddy V et al. (2014) Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 159, 1603–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally MA and Hartman AL (2012) Ketone bodies in epilepsy. J Neurochem 121, 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mena M, Perucho J, Rubio I and de Yébenes JG (2010) Studies in animal models of the effects of anesthetics on behavior, biochemistry, and neuronal cell death. Journal of Alzheimer's disease : JAD 22 Suppl 3, 43–48. [DOI] [PubMed] [Google Scholar]
- Miyamoto J, Ohue-Kitano R, Mukouyama H et al. (2019) Ketone body receptor GPR43 regulates lipid metabolism under ketogenic conditions. Proc Natl Acad Sci U S A 116, 23813–23821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neth BJ, Mintz A, Whitlow C et al. (2020) Modified ketogenic diet is associated with improved cerebrospinal fluid biomarker profile, cerebral perfusion, and cerebral ketone body uptake in older adults at risk for Alzheimer's disease: a pilot study. Neurobiology of aging 86, 54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA (2005) Endosome function and dysfunction in Alzheimer's disease and other neurodegenerative diseases. Neurobiol Aging 26, 373–382. [DOI] [PubMed] [Google Scholar]
- Owen OE, Morgan AP, Kemp HG, Sullivan JM, Herrera MG and Cahill GF Jr. (1967) Brain metabolism during fasting. J Clin Invest 46, 1589–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peck B, Schug ZT, Zhang Q et al. (2016) Inhibition of fatty acid desaturation is detrimental to cancer cell survival in metabolically compromised environments. Cancer Metab 4, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L and Magistretti PJ (2004) Neuroenergetics: calling upon astrocytes to satisfy hungry neurons. Neuroscientist 10, 53–62. [DOI] [PubMed] [Google Scholar]
- Rahman M, Muhammad S, Khan MA et al. (2014) The beta-hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages. Nat Commun 5, 3944. [DOI] [PubMed] [Google Scholar]
- Rardin MJ, Newman JC, Held JM et al. (2013) Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc Natl Acad Sci U S A 110, 6601–6606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richerson GB (1995) Response to CO2 of neurons in the rostral ventral medulla in vitro. J Neurophysiol 73, 933–944. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ and Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selfridge JE, Wilkins HM, E, L. et al. (2015) Effect of one month duration ketogenic and non-ketogenic high fat diets on mouse brain bioenergetic infrastructure. Journal of bioenergetics and biomembranes 47, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazu T, Hirschey MD, Newman J et al. (2013) Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 339, 211–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CJ, Berry DM and McGlade CJ (2013) The E3 ubiquitin ligases RNF126 and Rabring7 regulate endosomal sorting of the epidermal growth factor receptor. Journal of Cell Science 126, 1366. [DOI] [PubMed] [Google Scholar]
- Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR and de la Monte SM (2005) Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease--is this type 3 diabetes? Journal of Alzheimer's disease : JAD 7, 63–80. [DOI] [PubMed] [Google Scholar]
- Stobart JL and Anderson CM (2013) Multifunctional role of astrocytes as gatekeepers of neuronal energy supply. Front Cell Neurosci 7, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PG, Rippy NA, Dorenbos K, Concepcion RC, Agarwal AK and Rho JM (2004) The ketogenic diet increases mitochondrial uncoupling protein levels and activity. Annals of neurology 55, 576–580. [DOI] [PubMed] [Google Scholar]
- Swerdlow R, Marcus DM, Landman J, Harooni M and Freedman ML (1989) Brain glucose and ketone body metabolism in patients with Alzheimer's disease. Clin Res 37, 461A. [Google Scholar]
- Taylor MK, Sullivan DK, Mahnken JD, Burns JM and Swerdlow RH (2017) Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer's disease. Alzheimers Dement (N Y) 4, 28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MK, Swerdlow RH and Sullivan DK (2019) Dietary Neuroketotherapeutics for Alzheimer's Disease: An Evidence Update and the Potential Role for Diet Quality. Nutrients 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera I, Wera S, Van Leuven F and Henderson ST (2005) A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer's disease. Nutr Metab (Lond) 2, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijay N and Morris ME (2014) Role of Monocarboxylate Transporters in Drug Delivery to the Brain. Current pharmaceutical design 20, 1487–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Zhao F, Ma X, Perry G and Zhu X (2020) Mitochondria dysfunction in the pathogenesis of Alzheimer's disease: recent advances. Mol Neurodegener 15, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Wang C and Wu Z (2014) PROPER: comprehensive power evaluation for differential expression using RNA-seq. Bioinformatics 31, 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Zhang D, Chung D et al. (2016) Metabolic Regulation of Gene Expression by Histone Lysine beta-Hydroxybutyrylation. Mol Cell 62, 194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Fang F, Ding J and Wu C (2018) Dysregulation of Rab5-mediated endocytic pathways in Alzheimer's disease. Traffic 19, 253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Chen S, Mao Z, Cadenas E and Brinton RD (2011) 2-Deoxy-D-glucose treatment induces ketogenesis, sustains mitochondrial function, and reduces pathology in female mouse model of Alzheimer's disease. PLoS One 6, e21788. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.