

Abstract
There is widespread interest in facile methods for generating potent neutralizing antibodies, nanobodies, and other affinity proteins against SARS-CoV-2 and related viruses to address current and future pandemics. While isolating antibodies from animals and humans are proven approaches, these methods are limited to the affinities, specificities, and functional activities of antibodies generated by the immune system. Here we report a surprisingly simple directed evolution method for generating nanobodies with high affinities and neutralization activities against SARS-CoV-2. We demonstrate that complementarity-determining region swapping between low-affinity lead nanobodies, which we discovered unintentionally but find is simple to implement systematically, results in matured nanobodies with unusually large increases in affinity. Importantly, the matured nanobodies potently neutralize both SARS-CoV-2 pseudovirus and live virus, and possess drug-like biophysical properties. We expect that our methods will improve in vitro nanobody discovery and accelerate the generation of potent neutralizing nanobodies against diverse coronaviruses.
Keywords: COVID-19, neutralization, affinity, maturation, nanobody, yeast, receptor-binding domain, RBD, ACE2, spike
Graphical abstract
Zupancic et al. report potent neutralizing nanobodies against SARS-CoV-2. They demonstrate an approach that involves swapping the complementarity-determining regions of low-affinity clones to generate matured nanobodies with large increases in affinity and neutralization activity.
Introduction
The COVID-19 pandemic has resulted in widespread interest in developing antibodies and other affinity reagents that recognize the SARS-CoV-2 virus with high affinity and specificity for diagnostic and therapeutic applications. Most antibody generation efforts against SARS-CoV-2 have involved either immunizing animals (Alsoussi et al., 2020; Hanke et al., 2020; Hansen et al., 2020) with the spike (S1) protein (or receptor-binding domain [RBD] thereof) or isolating antigen-specific antibodies from humans after infection (Hansen et al., 2020; Shi et al., 2020; Wu et al., 2020). These approaches have yielded diverse types of antibodies for sensitive virus detection and potent inhibition of viral infection, including multiple antibodies now being used as therapeutics in humans (Baum et al., 2020a; Chen et al., 2020; Hansen et al., 2020).
Despite the many strengths of in vivo antibody generation methods, they possess limitations relative to in vitro antibody generation methods, including those that use antibody display technologies such as phage and yeast surface display. The most important limitation is that in vivo methods lack the ability to robustly control antigen presentation to the immune system (Boder et al., 2000; Bradbury et al., 2011; Foote and Eisen, 2000; Tiller and Tessier, 2015). This, in turn, limits the ability to use such methods to select antibodies with predefined affinities, specificities, and functional activities that are optimal for different applications. Even antibodies generated in vivo are commonly affinity matured using in vitro display methods to achieve ultra-high affinities and/or cross-species reactivities (Jackson et al., 1995).
We have evaluated the potential of directed evolution methods for selecting high-affinity nanobodies against the SARS-CoV-2 spike protein from a non-immune library (McMahon et al., 2018). In particular, we tested if nanobody variants could be identified that would possess similar or superior affinities and neutralizing activities relative to a potent SARS-CoV-2 neutralizing nanobody (Ty1) generated via immunization (Hanke et al., 2020) and a potent neutralizing SARS-CoV-2 human antibody isolated after infection (CB6; Shi et al., 2020). Herein, we report an unexpected finding that high-affinity nanobodies can be isolated from non-immune libraries by complementarity-determining region (CDR) swapping between low-affinity lead clones without additional mutagenesis. We demonstrate that this surprising finding, which was initially discovered by accident due to inadvertent recombination of two low-affinity lead clones, can be easily employed in a systematic manner during initial library sorting to identify high-affinity nanobodies without the need for subsequent affinity maturation.
Results
In vitro discovery and affinity maturation of potent neutralizing nanobodies
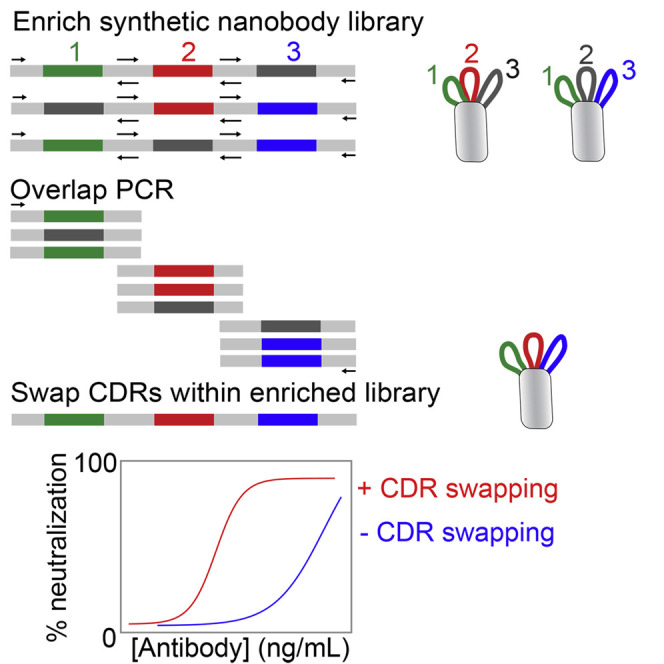
A synthetic nanobody library was first systematically sorted to isolate nanobodies that bind to the RBD of the SARS-CoV-2 spike protein S1 subunit (residues V16-R685; GenBank: QHD43416; Figure 1 ). This library has been previously reported for use in isolating nanobodies that bind to a diverse range of antigens (McMahon et al., 2018). For use in this study, the library was transferred to a yeast surface display system in which the nanobody N-terminus is linked to Aga2. We found that this Aga2 display system increased the percentage of yeast cells within the library that display nanobodies on the cell surface compared with a glycophosphatidylinositol anchor display system (Figure S1). The library was first sorted against a soluble biotinylated SARS-CoV-2 RBD via magnetic-activated cell sorting (MACS) to enrich the library and reduce the diversity to a level that could be feasibly processed by fluorescence-activated cell sorting (FACS). The enriched library was then sorted by FACS five times against RBD-Fc, biotinylated RBD, or spike protein trimer. We found that the use of a bivalent antigen, RBD-Fc, was necessary for the first three rounds of FACS in order to distinguish a clear binding population within the library. Biotinylated RBD or spike protein trimer was used in later rounds of sorting after greater enrichment of a binding population was achieved.
Figure 1.

Summary of the discovery and affinity maturation of nanobodies against the spike protein of SARS-CoV-2
A synthetic nanobody library displayed on yeast was screened against the receptor-binding domain (RBD), spike (S1) protein, and spike protein trimer of SARS-CoV-2 by MACS and FACS. Two lead clones (KA1 and KC3) were identified and affinity matured using error-prone PCR. The sublibraries were screened against the S1 protein by FACS to isolate nanobody variants (KA1.ep1, KC3.ep3, and KC3.ep5) with superior binding activity relative to a potent neutralizing nanobody generated via immunization (Ty1).
Next, unique nanobody sequences that were enriched by library sorting were identified via Sanger sequencing, expressed on the yeast surface, and tested for binding to biotinylated S1 protein (50 nM). Two lead clones were selected from a sort against the RBD (KA1) and spike protein trimer (KC3) for further examination and affinity maturation. A third lead clone was also observed in sequencing from the spike protein trimer sort (KC1), which possessed more modest affinity compared with the two selected lead clones. The lead clones have the same frameworks because the library consists of nanobody variants with a common framework and diversity only in their CDRs (McMahon et al., 2018). The two selected lead nanobodies have similar sequences for CDR1 with only one residue difference between them (Figures 2 and S2). However, the sequences of CDR2 differ by five residues, and the sequences of CDR3 differ by eight residues and display minor differences in length (13 residues for KA1 and 11 residues for KC3).
Figure 2.

Affinity-matured nanobodies possess a combination of CDRs from the two lead clones
(A) Affinity maturation of lead nanobodies KA1 and KC3 via error-prone PCR results in nanobody variants that possess one CDR from each lead nanobody (CDR2 [red] from KA1 and CDR3 [blue] from KC3) in addition to one CDR (CDR1 [green]) that differs by only a single mutation.
(B) Nanobody sequences (Kabat numbering) for the three affinity-matured variants. Residues that are different from KA1.ep1 are indicated with an amino acid letter.
KA1 and KC3 were then affinity matured using error-prone PCR at a low mutational frequency of approximately 1.2–1.5 mutations per nanobody on average (Figure 1). Four rounds of FACS were performed to select for clones with improved affinity for the S1 protein. The concentration of S1 antigen was decreased in each subsequent round until the library was enriched for superior binding relative to that of a leading SARS-CoV-2 nanobody (Ty1) with potent neutralization activity that was generated via immunization (Hanke et al., 2020). In the final round of sorting, the enriched nanobodies displayed clear binding at 100 pM S1, and the observed binding was stronger than for Ty1. Only cells that bound at levels higher than that observed for Ty1 were collected. Clones were isolated from these terminal sorts for analysis and Sanger sequencing. Unexpectedly, all of the nine unique clones contained CDR2 from KA1 and CDR3 from KC3 (Figures 2 and S2 and data not shown), including one clone (KA1.ep1) without any additional mutations in the CDRs or framework regions. KC3.ep3 and KC3.ep5 both contained a few (two or three) additional mutations in their CDRs and frameworks resulting from error-prone PCR (Figures 2 and S1). Herein, we refer to the replacement of one or more CDRs in a given nanobody (e.g., CDR2 in nanobody 1) with one or more CDRs from another nanobody (e.g., CDR2 from nanobody 2) as CDR swapping.
CDR-swapped nanobodies display large increases in neutralization activity and affinity
Both lead and affinity-matured clones were next analyzed for their ability to neutralize SARS-CoV-2 infection as nanobody-Fc fusion proteins using a lentivirus-based SARS-CoV-2 pseudovirus assay (Figure 3 A). The nanobody-Fc fusion proteins were produced in HEK293-6E cells and isolated at high purity via Protein A chromatography (Figure S3). Their neutralization activity was directly compared with that of two previously published neutralizers, namely a nanobody (Ty1; Hanke et al., 2020) and an antibody (CB6; Shi et al., 2020). Strikingly, the three affinity-matured clones demonstrated dramatic (>300-fold) improvement in neutralization activity relative to their parental nanobodies (Figure 3A). Moreover, the affinity-matured variants displayed complete neutralization at concentrations lower than for Ty1 and CB6. KA1.ep1 (half-maximal inhibitory concentration [IC50] of 4.8 ± 2.6 ng/mL), KC3.ep3 (IC50 of 1.9 ± 1.2 ng/mL), and KC3.ep5 (IC50 of 2.3 ± 1.3 ng/mL) displayed the lowest IC50 values, with KC3.ep3 being the most potent neutralizer (Figure 3A). The IC50 values determined for Ty1 (IC50 of 16 ± 7 ng/mL) and CB6 (IC50 of 23 ± 6 ng/mL) were higher and comparable to those previously reported for these antibodies (Hanke et al., 2020; Shi et al., 2020).
Figure 3.

Affinity-matured nanobodies potently neutralize SARS-CoV-2 pseudovirus and live virus
(A) Neutralization results for nanobodies as bivalent Fc-fusion proteins (KA1, KC3, KA1.ep1, KC3.ep3, KC3.ep5, and Ty1) and an antibody (CB6) for inhibiting pseudovirus infectivity in a luciferase-based, HEK293T reporter cell line. Pseudovirus particles were preincubated with antibodies and added to reporter cells, and luciferase signal was measured after 48 h.
(B) Neutralization results for nanobodies as bivalent Fc-fusion proteins (KC3.ep3, Ty1) and antibodies (CB6) for inhibiting live virus infection of VeroE6 cells. Nanobody and antibody dilutions were tested in eight replicate wells each. After cells were incubated with virus and nanobodies or antibody for 3 days, the cells were examined microscopically for visible cytopathic effect. Wells with any degree of visible, virus-induced cytopathic effect were scored as positive for infection. In (A), the data are averages of four or five repeats, and the error bars are standard deviations. In (B), the data are averages of two to four repeats, and the error bars are standard deviations.
Based on the promising results from the pseudovirus assay, we further examined the neutralization activity of KC3.ep3 and Ty1 (as Fc-fusion proteins) and CB6 (immunoglobulin G [IgG]) against live SARS-CoV-2 virus (Figure 3B). Neutralization of the live virus was determined by observing the cytopathic effect of the virus on VeroE6 cells. Measurement of neutralization activity using the cytopathic effect has been previously observed to result in higher IC50 values than those reported using other detection methods for both live virus and pseudovirus assays (Shi et al., 2020). Nevertheless, our best affinity-matured variant (KC3.ep3) was significantly more potent at inhibiting the live virus (IC50 of 39 ± 4 ng/mL) than Ty1 (IC50 of 211 ± 42 ng/mL) and CB6 (IC50 of 655 ± 287 ng/mL). These results are consistent with the pseudovirus assay and demonstrate the potent neutralization activity of our CDR-swapped nanobody.
We also characterized the affinities for our matured nanobodies relative to their parental nanobodies in the monovalent and bivalent formats (Figure 4 ). Monovalent affinities of the nanobodies for the SARS-CoV-2 RBD were examined using yeast surface display (Figure 4A). The different monovalent nanobodies were expressed on the yeast surface (as Aga2-nanobody fusions) at similar levels (Figure S4). Notably, the monovalent affinities of the affinity-matured nanobodies (KD of 5.1–5.5 nM) were superior to those of leading nanobodies generated in vivo (KD of 8–19 nM for Ty1 and VHH-72), and much higher than those of the parental nanobodies (KD >100 nM). We also observed similar differences for the apparent affinities (half-maximal effective concentration [EC50]) of bivalent nanobodies (Figure 4B). These experiments were performed using nanobodies formatted as soluble Fc-fusion proteins and RBD immobilized on magnetic Dynabeads. The affinity-matured nanobodies displayed stronger binding (EC50 of 34–48 pM) than the previously reported nanobodies (EC50 of 55–69 pM for Ty1 and VHH-72) and antibodies (EC50 of 69–78 pM for CB6 and CR3022), and much stronger binding (>20-fold improved affinity) than the parental nanobodies (>1,000 pM for KA1 and KC3). In summary, the affinity-matured nanobodies demonstrate improved monovalent and bivalent affinities compared with leading nanobodies and antibodies generated in vivo, which is consistent with their superior neutralization activities.
Figure 4.

Potent neutralizing nanobodies display high monovalent and bivalent affinities for the SARS-CoV-2 receptor-binding domain
(A) Monovalent binding of nanobodies displayed on the surface of yeast to biotinylated SARS-CoV-2 receptor-binding domain.
(B) Bivalent binding of nanobodies (Fc-fusion proteins) and antibodies (IgGs) to biotinylated SARS-CoV-2 receptor-binding domain immobilized on magnetic beads. The results are averages from three independent experiments, and the error bars are standard deviations.
KC3.ep3 recognizes an epitope in the receptor-binding domain common to other potent neutralizers
The RBDs from the SARS-CoV-2 and SARS-CoV viruses share >70% sequence similarity (Tian et al., 2020). It has been observed that some antibodies, including VHH-72, CR3022, and S309, bind to the RBDs of both viruses (Pinto et al., 2020; Tian et al., 2020; Wrapp et al., 2020). Therefore, we evaluated the affinity of monovalent KC3.ep3 for the RBDs of SARS-CoV and SARS-CoV-2 to compare its specificity relative to VHH-72 (Figure S5). Both KC3.ep3 and VHH-72 demonstrated strong binding to the SARS-CoV-2 RBD, but KC3.ep3 did not show detectable binding to the RBD of SARS-CoV, indicating that KC3.ep3 recognizes an epitope that is unique in SARS-CoV-2 RBD, while VHH-72 strongly recognizes the SARS-CoV RBD (KD 1.6 ± 0.6 nM), indicating that VHH-72 and KC3.ep3 recognize distinct RBD epitopes.
We further probed the epitope of KC3.ep3 by examining its competition for binding to the RBD of SARS-CoV-2 with the ACE2 receptor, previously reported nanobodies (Ty1 and VHH-72), and antibodies (CB6 with class 1 epitope [Shi et al., 2020], C119 with class 2 epitope [Barnes et al., 2020], S309 with class 3 epitope [Pinto et al., 2020], and CR3022 with class 4 RBD epitope [Yuan et al., 2020]) that recognize distinct epitopes on the SARS-CoV-2 RBD (Figure 5 ). Soluble biotinylated RBD (5 nM) was preincubated with soluble ACE2 receptor or bivalent antibodies (nanobody-Fc fusions or IgGs) at a range of antibody concentrations (0.05–500 nM), and then these receptor-antigen or antibody-antigen complexes were incubated with yeast-surface-displayed monovalent KC3.ep3.
Figure 5.

Affinity-matured nanobody recognizes an epitope in the receptor-binding domain that overlaps with epitopes recognized by ACE2 and other potent SARS-CoV-2 neutralizing nanobodies and antibodies
Bivalent nanobodies (KC3.ep3, VHH-72, and Ty1), antibodies (S309, CR3022, CB6, and C119) and ACE2 were preincubated with biotinylated receptor-binding domain of SARS-CoV-2 (5 nM) over a range of nanobody, antibody, and ACE2 concentrations, and then the mixtures were added to yeast cells displaying monovalent KC3.ep3. The percentage bound receptor-binding domain is reported relative to the amount bound in the absence of preblocking. The results are averages from three independent repeats, and the error bars are standard deviations.
Notably, the binding of monovalent KC3.ep3 to RBD was inhibited by preincubation of ACE2 with RBD, suggesting that the KC3.ep3 and ACE2 binding sites on RBD overlap (Figure 5). This is consistent with the ability of KC3.ep3 to potently neutralize the virus. Moreover, the binding of monovalent KC3.ep3 to RBD was strongly inhibited by preincubation of RBD with bivalent KC3.ep3, as expected. KC3.ep3 binding to RBD was also inhibited by bivalent Ty1, CB6, or C119, although not as strongly as for bivalent KC3.ep3. Thus, the epitope of KC3.ep3 appears to overlap with that of Ty1, CB6 (class 1 epitope), and C119 (class 2 epitope).
Conversely, monovalent KC3.ep3 binding was weakly impacted or even enhanced when the RBD was preincubated with bivalent VHH-72, S309 (class 3 epitope), and CR3022 (class 4 epitope; Figure 5), revealing that the epitope of KC3.ep3 does not overlap with these antibodies. The fact that KC3.ep3 and VHH-72 do not compete for binding is in agreement with our finding that KC3.ep3 recognizes an epitope in the SARS-CoV-2 RBD that is absent from the SARS-CoV RBD, while VHH-72 recognizes an epitope that is present in both RBDs (Figure S5). Likewise, CR3022 has been previously demonstrated to cross-react with both viruses (Tian et al., 2020), and most residues in its class 4 epitope are conserved between the two viruses (Yuan et al., 2020), indicating that its epitope would also be expected to be distinct from that of KC3.ep3. Likewise, S309 has been observed to cross-react with SARS-CoV, and the absence of competition between KC3.ep3 and S309 also agrees with our observation that KC3.ep3 competes with ACE2 and previous observations that S309 does not compete with ACE2 (Pinto et al., 2020).
Neutralizing nanobodies display drug-like biophysical properties
For use in therapeutic and diagnostic applications, nanobodies need to be easily produced and possess favorable biophysical and biomanufacturing properties, including high stability, high solubility, low levels of aggregation, low non-specific binding, and high expression levels. Therefore, we first quantified the expression yields of nanobodies in this study via transient transfection of HEK293 cells. It has been previously shown that this expression system can be used to express nanobody-Fc fusion proteins at yields ranging from ∼20 to 140 mg/L (Zhang et al., 2009). We observed similar high purification yields for the affinity-matured nanobodies obtained via directed evolution (∼27–110 mg/L), which were also comparable to the purification yields for the nanobodies obtained via immunization (Ty1 and VHH-72; 52–85 mg/L). The purity (SDS-PAGE) and homogeneity (size-exclusion chromatography) of the affinity-matured nanobodies were also excellent (e.g., >95% monomer), and similar to those of the nanobodies generated via immunization (Figures S3 and S6).
The stability of the affinity-matured nanobodies was also examined (Figure 6 A). It is a concern that mutations accumulated by nanobodies, and antibodies in general, during affinity maturation have an increased risk for reducing stability (Julian et al., 2017; Rabia et al., 2018; Shehata et al., 2019). This is even more concerning for CDR-swapped variants with CDRs of different lengths, as observed for the CDR3-swapped variant KA1.ep1, because these changes could have an impact on the local structure of the nanobody. Therefore, we analyzed the folding stability (melting temperature, T m) of the lead nanobody clones and their affinity-matured variants relative to previously reported SARS-CoV-2 nanobodies and monoclonal antibodies (mAbs). The lead nanobodies displayed high stabilities, as both KA1 and KC3 displayed melting temperatures >68°C (KA1 T m of 71.3°C ± 0.4°C and KC3 T m of 68.3°C ± 1.0°C). Notably, the affinity-matured variants displayed similar stabilities (KA1.ep1 T m of 69.5°C ± 0.8°C, KC3.ep3 T m of 69.6°C ± 0.8°C, and KC3.ep5 of T m 68.6°C ± 0.6°C), suggesting that the affinity-enhancing CDR swaps maintained high stability. Moreover, the stabilities of the affinity-matured nanobodies were similar to those for nanobodies generated via immunization (Ty1 T m of 69.3°C ± 1.0°C and VHH-72 T m of 69.1°C ± 0.6°C). As expected, the stability of the mAbs (CB6 and CR3022) was higher (T m > 77°C) because of their stabilizing light chains and constant regions (CH1 and CL).
Figure 6.

Affinity-matured nanobodies display high stability and specificity
(A) Melting temperatures of bivalent nanobodies and antibodies evaluated via differential scanning fluorimetry.
(B) Non-specific binding of bivalent nanobodies and antibodies (immobilized on magnetic beads) was evaluated via incubation with biotinylated soluble membrane proteins from CHO cells and detection of non-specific binding via flow cytometry. Control antibodies with high (emibetuzumab) and low (elotuzumab) non-specific binding were also evaluated for comparison. The two control antibodies are not identical to the actual drugs, as they have the variable regions of the actual drugs and a common IgG1 framework. In (A) and (B), the results are averages from three independent repeats, and the error bars are standard deviations.
Finally, the specificity (non-specific binding) of the affinity-matured variants was examined by testing their ability to bind complex mixtures of soluble membrane proteins obtained from HEK293 cells (Figure 6B) (Xu et al., 2013). It has been previously reported that approved antibody drugs typically display lower levels of non-specific binding, including to soluble membrane proteins, than antibodies that either failed in clinical development or are still in development (Jain et al., 2017). Further, low antibody specificity has also been shown to correlate with poor pharmacokinetic properties (Hötzel et al., 2012). Notably, the lead nanobodies and affinity-matured variants in this work displayed extremely low binding to soluble membrane proteins. The observed levels of non-specific binding were comparable to those of other SARS-CoV-2 nanobodies and antibodies generated in vivo and similar to a clinical-stage mAb with low levels of non-specific binding (elotuzumab) and much lower than a clinical-stage antibody with high levels of non-specific binding (emibetuzumab). These results collectively demonstrate that the potent neutralizing nanobodies reported in this work have drug-like biophysical properties that are similar to those of SARS-CoV-2 nanobodies and antibodies generated by the immune system.
Systematic CDR-swapping mutagenesis for identifying high-affinity nanobodies without additional affinity maturation
Given that unintentional CDR swapping between low-affinity lead nanobodies led to unexpectedly large increases in affinity, we next asked whether the introduction of intentional CDR swapping during the initial library sorting process would enable identification of high-affinity nanobodies without the need for lead clone evaluation and affinity maturation. Therefore, we isolated the enriched library of nanobody plasmids prior to the terminal sort of our original sorting efforts (after five rounds of sorting against RBD and related reagents; Figure 1), shuffled their three CDRs via standard PCR methods (see STAR Methods for details), and sorted the CDR-swapped library two additional times against RBD.
Encouragingly, Sanger sequencing revealed that this simple mutagenesis method is able to identify multiple known or promising high-affinity nanobodies (Figure S7). These include a high-affinity nanobody that we discovered in our initial two-step library sorting and affinity-maturation process (KA1.ep1) as well as nanobodies not observed previously (K7.13 and K7.19). The most common nanobody identified was KA1.ep1, which is logical because this high-affinity nanobody is a CDR-swapped version of KA1 and KC3 without any additional mutations. We also identified a nanobody (K7.19) that was a variant of KA1.ep1 with one mutation. Notably, we also identified a nanobody (K7.13) with a unique CDR3, which is particularly interesting because we originally identified a low-affinity lead clone (KC1) with the same CDR3 but with different CDR1 and CDR2 loops. Our initial observations of CDR swapping in KA1.ep1, KC3.ep3, and KC3.ep5 resulted from a combination of CDRs from KA1 and KC3. Similarly, K7.13 contains CDR1 and CDR2 from KA1 and CDR3 from KC1 (Figure S7).
Finally, we evaluated the monovalent affinities of the nanobodies generated by CDR-swapping mutagenesis (Figure 7 ). Notably, the nanobodies identified from intentional CDR-swapping mutagenesis displayed single-digit monovalent binding affinities (KD 3–4 nM) that were similar to that of KA1.ep1. These binding affinities were much stronger than those of the lead clones identified in our original sorting efforts (KA1, KC1, and KC3) despite the fact that the CDRs of the high-affinity clones are identical to or closely related to those of the low-affinity lead clones (Figure S7). In summary, these results indicate that CDR swapping between common framework nanobodies has great potential to enable the facile isolation of high-affinity nanobodies, and this mutagenesis strategy can be easily incorporated during the initial sorting process to avoid the need for lead clone evaluation and subsequent affinity maturation.
Figure 7.

Nanobodies with nanomolar monovalent affinities can be generated via CDR-swapping mutagenesis without the need for lead clone identification and subsequent affinity maturation
Nanobodies were selected from a non-immune library with (KA1.ep1, K7.13, K7.19) or without (KC3, KA1, KC1) CDR-swapping mutagenesis, and the selected clones were evaluated in terms of their monovalent binding affinities for the SARS-CoV-2 receptor-binding domain. The results are averages from two independent experiments, and the error bars are standard deviations.
Discussion
We have demonstrated that common-framework, non-immune nanobody libraries can be used in a surprisingly simple manner to generate high-affinity nanobodies without the need for lead clone identification or additional mutagenesis in the framework or CDRs. Some previous reports of antibodies against unrelated targets have optimized individual CDRs separately, and then combined the optimized CDRs to further increase affinity (Steidl et al., 2008; Yang et al., 1995). However, these studies are for antibodies that already have relatively high affinity and, thus, address a simpler challenge of affinity maturation and result in much lower synergistic improvements in affinity after CDR swapping than we observed in our studies. In contrast, our work addresses a more challenging problem of how to identify high-affinity clones without the need to first identify lead clones with modest affinity and select individual clones for affinity maturation. We expect that this approach of combining multiple low-affinity clones via CDR and/or framework swapping holds great potential for rapidly generating nanobodies and, more generally, antibodies with high affinity with much less effort than is typically required. Despite this discovery being unintentional, we demonstrate that it is simple to perform CDR swapping using standard PCR methods, and it could be used as a facile method for identifying high-affinity clones, even without additional mutagenesis.
Compared with CDR swapping, we observed much smaller improvements in affinity from additional CDR and framework mutations (KC3.ep3 and KC3.ep5) due to error-prone PCR. Nevertheless, the average number of mutations incorporated by our error-prone PCR method (1.2–1.5 mutations per nanobody) was low in comparison to the changes incorporated though CDR swapping. A higher error rate could have the potential to introduce a greater number of beneficial mutations in combination with or following CDR swapping.
The epitopes of our neutralizing nanobodies relative to previously reported neutralizing nanobodies and antibodies also deserve further consideration. The epitope is of particular interest for identifying pairs of nanobodies or antibodies that bind to different sites and can therefore aid in the prevention of infection by SARS-CoV-2 escape mutants (Baum et al., 2020b; Greaney et al., 2020; Weisblum et al., 2020). Like KC3.ep3, the epitopes of several other potent neutralizing antibodies in the RBD have also been reported to overlap with the ACE2 binding site (Barnes et al., 2020). It is notable that KC3.ep3 competes with a class 1 antibody (CB6), as CB6 binds the RBD only in the “up” conformation and competes with ACE2. CB6 has been shown to sterically hinder ACE2 binding to RBD, and the epitope recognized by CB6 overlaps with the region bound by ACE2 (Shi et al., 2020). Further, class 1 antibodies have been observed to have short (<15 residues) HCDR3 loops (Barnes et al., 2020). KC3.ep3 similarly has a CDR3 that consists of only 11 residues.
The fact that KC3.ep3 also competes with a class 2 antibody (C119) is notable because C119 binds to the RBD in both the “up” and “down” conformations and also competes with ACE2. Consistent with its competition with class 1 and class 2 antibodies, the binding of KC3.ep3 was also reduced when the RBD was preincubated with ACE2 (Figure 5). Thus, KC3.ep3 appears to recognize an epitope that overlaps with the ACE2 binding site and is common to potent neutralizing antibodies that also block ACE2 binding. KC3.ep3 competes similarly with a potent neutralizing nanobody, Ty1. Like class 2 antibodies, Ty1 has previously been demonstrated to reduce the ability of RBD to bind ACE2, and structural analysis indicates that Ty1 sterically hinders this binding when the RBD is in both the up and the down conformations (Hanke et al., 2020). Moreover, preincubation of RBD with ACE2 did not reduce the ability of KC3.ep3 to bind RBD as strongly as preincubation with the class 1 and class 2 antibodies, indicating that the KC3.ep3 epitope does not completely overlap with the ACE2 binding site.
However, KC3.ep3 does not compete for binding with antibodies that cross-react with both SARS-CoV and SARS-CoV-2, namely VHH-72, CR3022, and S309. CR3022 has been reported to be a class 4 antibody, indicating that it binds the RBD in the up conformation but does not compete with ACE2 (Barnes et al., 2020). Structural analysis has further shown that CR3022 binds to the SARS-CoV-2 RBD but does not compete for binding with ACE2, and CR3022 weakly neutralizes the SARS-CoV-2 virus (Yuan et al., 2020). Interestingly, the epitopes for CR3022 and VHH-72 overlap, but due to different angles of binding, VHH-72 indirectly hinders RBD binding to ACE2, while CR3022 does not (Wrapp et al., 2020). As both VHH-72 and CR3022 cross-react with the SARS-CoV RBD, while KC3.ep3 does not, it would be expected that KC3.ep3 would not recognize an overlapping epitope. Therefore, the reported epitopes are consistent with the lack of competition observed between KC3.ep3 and both CR3022 and VHH-72.
The fact that KC3.ep3 also does not compete with a class 3 antibody (S309) also deserves further consideration. Class 3 antibodies can bind to the RBD in both the up and the down conformations but do not compete with ACE2. Interestingly, the binding of KC3.ep3 was enhanced when RBD was incubated with S309 (Figure 5). Given that KC3.ep3 competes with ACE2, its epitope would be expected to be distinct from that of a class 3 antibody, such as S309. Enhancement of neutralization activity has previously been reported for antibody cocktails composed of S309 and an antibody targeting a distinct epitope (Pinto et al., 2020). A pair of class 1 and class 3 antibodies (Barnes et al., 2020), REGEN10933 and REGN10967, are currently being used in combination for SARS-CoV-2 treatment (Baum et al, 2020a, 2020b). This analysis of the epitope of KC3.ep3 indicates that it is likely to compete for binding to the SARS-CoV-2 RBD with class 1 and class 2 antibodies, but the observed enhancement of KC3.ep3 binding in the presence of S309 suggests that the combination of KC3.ep3 with a class 3 antibody could be beneficial in terms of affinity and potentially neutralization activity as well.
The identification of neutralizing nanobodies and antibodies that target SARS-CoV-2 represents a rapidly evolving area of research. As such, consideration should also be given to the affinity and neutralization activities of the nanobodies that we report in the context of previously reported nanobodies and antibodies in addition to the targeted epitope. In terms of nanobody affinity and neutralization activity, a broad range of these properties has been reported in the literature. Several recent studies have reported nanobodies that bind to the SARS-CoV-2 virus (Chi et al., 2020; Hanke et al., 2020; Huo et al., 2020; Schoof et al., 2020; Wrapp et al., 2020; Xiang et al., 2020). Multiple studies have reported nanobodies isolated from naive and synthetic libraries using various in vitro panning and sorting strategies (Chi et al., 2020; Custódio et al., 2020; Huo et al., 2020; Schoof et al., 2020). Similar to the strategy that we have reported here, some of these studies have incorporated affinity maturation in order to further improve the properties of the isolated lead candidates (Huo et al., 2020; Schoof et al., 2020). Direct comparison between various studies and the results reported here are complicated by the use of different experimental methodologies and nanobody constructs (e.g., monovalent versus bivalent). Nevertheless, the nanobodies that we report appear to compare favorably in terms of affinity and neutralization activity to those reported previously. Similar to several nanobodies previously isolated using in vitro methods (Chi et al., 2020; Custódio et al., 2020; Huo et al., 2020), the nanobodies we report here demonstrate nanomolar monovalent affinities (Figure 4A). Direct comparison to nanobodies and antibodies isolated in three previous studies also indicates that the nanobodies reported here have comparable or improved affinities (Figure 4) and neutralization activities (Figure 3) relative to multiple leading nanobodies isolated from immunization (VHH-72 [Wrapp et al., 2020] and Ty1 [Hanke et al., 2020]) and an antibody isolated from infected humans (CB6 [Shi et al., 2020]). While recent studies also demonstrate that extensive, large-scale efforts can identify nanobodies with higher affinities and increased neutralization activities (Xiang et al., 2020), it would be simple to further affinity mature our nanobodies to achieve even higher affinities. More generally, our findings demonstrate the power of directed evolution methods to rapidly generate high-affinity nanobodies with epitopes that overlap with leading neutralizing nanobodies and antibodies and which result in highly potent neutralization activities.
Significance
The COVID-19 pandemic currently represents a global health crisis for which therapeutic options are limited. Neutralizing antibodies, nanobodies, and other affinity proteins are urgently needed as therapeutics and diagnostic reagents for analysis of SARS-CoV-2 and related viruses. Here we have reported a surprisingly simple yet powerful approach for generating nanobodies that bind to the receptor-binding domain of SARS-CoV-2 with high affinity and which potently neutralize both the pseudovirus and the live virus. We demonstrate that a synthetic library may be used to isolate potent neutralizing nanobodies and that CDR swapping between multiple clones can be used to rapidly increase both the affinity and the neutralization activity of isolated nanobodies. We demonstrate that CDR-swapping mutagenesis can be systematically incorporated into library sorting through a simple PCR step. Further, we demonstrate that the isolated nanobodies are highly specific and display drug-like biophysical properties, including high purification yields, stabilities, and specificities. We expect that the nanobodies described here could be applied to several applications within the rapidly developing research field surrounding this and related viruses. The modularity of single-domain antibody fragments enables their incorporation into multivalent scaffolds to inhibit virus infection with dramatic increases in potency (Schoof et al., 2020; Xiang et al., 2020). Further, the nanobodies reported here have the potential to be combined with nanobodies that target distinct epitopes, in either multi-epitopic constructs or cocktails with other nanobodies or antibodies, to further increase their activities to yield ultra-potent neutralizing agents.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Emibetuzumab | Jain et al., 2017 | N/A |
| Elotuzumab | Jain et al., 2017 | N/A |
| Mouse anti-Myc-Tag antibody (9B11) | Cell Signaling Technology | Cat#2276S; RRID: AB_331783 |
| Chicken anti-His-Tag antibody | Invitrogen | Cat#PA1-9531; RRID: AB_1069887 |
| Goat anti-mouse IgG AlexaFluor 488 | Invitrogen | Cat#A11001; RRID: AB_2534069 |
| Donkey anti-chicken IgY F(ab’)2 AlexaFluor 647 | Jackson ImmunoResearch | Cat#703-606-155; RRID: AB_2340380 |
| Goat anti-human IgG AlexaFluor 647 | Jackson ImmunoResearch | Cat#109-605-098; RRID: AB_2337889 |
| Goat anti-human Fc F(ab’)2 AlexaFluor 488 | Invitrogen | Cat#H10120; RRID: AB_2536548 |
| Bacterial and virus strains | ||
| DH5α | Julian et al., 2017 | N/A |
| SARS-Related Coronavirus 2, Isolate USA-WA1/2020 | BEI | Cat#NR-52281 |
| Chemicals, peptides, and recombinant proteins | ||
| SARS-CoV-2 (COVID-19) S protein RBD, Fc Tag (RBD-Fc) | Acro Biosystems | Cat#SPD-C5255 |
| Biotinylated SARS-CoV-2 (COVID-19) S protein RBD, His, Avitag (bRBD) | Acro Biosystems | Cat#SPD-C82E9 |
| Biotinylated SARS-CoV-2 (COVID-19) S1 protein, His, Avitag | Acro Biosystems | Cat#S1N-C82E8 |
| SARS-CoV S protein RBD, His Tag | Acro Biosystems | Cat#SPD-S52H6 |
| SARS-CoV-2 S protein (R683A, R685A), His Tag, active trimer | Acro Biosystems | Cat#SPN-C52H8 |
| Recombinant Human Angiotensin-converting Enzyme 2 (ACE2) | RayBiotech | Cat#230-30165 |
| Taq DNA Polymerase with Standard Taq Buffer | New England Biolabs | Cat#M0273L |
| 8-Oxo-2'-deoxyguanosine-5'-Triphosphate | TriLink Biotechnologies | Cat#N-2034 |
| 2’-Deoxy-P-nucleoside-5'-Triphosphate | TriLink Biotechnologies | Cat#N-2037 |
| Q5 High-Fidelity DNA Polymerase | New England Biolabs | Cat#M0491L |
| NheI-HF | New England Biolabs | Cat#R3131L |
| HindIII-HF | New England Biolabs | Cat#R3104L |
| Quick CIP calf intestinal alkaline phosphatase | New England Biolabs | Cat#M0525L |
| T4 ligase | New England Biolabs | Cat#M0202L |
| F17 media | Thermo Fisher Scientific | Cat#A1383502 |
| Glutamine | Invitrogen | Cat#25030081 |
| Kolliphor | Fisher Scientific | Cat#NC0917244 |
| G418 | Thermo Fisher Scientific | Cat#10131035 |
| Yeastolate | BD Biosciences | Cat#292804 |
| Luciferase substrate | Promega ONE-Glo | Cat#E6110 |
| Streptavidin AlexaFluor 647 | Invitrogen | Cat#S32357 |
| Protein Thermal Shift Dye | Applied Biosystems | Cat#4461146 |
| cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail | Sigma Aldrich | Cat#4693159001 |
| cOmplete, EDTA-free Protease Inhibitor Cocktail | Sigma Aldrich | Cat#11873580001 |
| 1% n-dodecyl-b-D-maltopyranoside | Sigma Aldrich | Cat#D4641 |
| Sulfo-NHS-LC-biotin | Thermo Fisher Scientific | Cat#21335 |
| Critical commercial assays | ||
| DC Protein Assay Reagents Package | BioRad | Cat#5000116 |
| Deposited data | ||
| KA1 | This paper | GenBank: MZ133804 |
| KC3 | This paper | GenBank: MZ133805 |
| KC1 | This paper | GenBank: MZ133806 |
| KA1.ep1 | This paper | GenBank: MZ133807 |
| KC3.ep3 | This paper | GenBank: MZ133808 |
| KC3.ep5 | This paper | GenBank: MZ133809 |
| K7.13 | This paper | GenBank: MZ133810 |
| K7.19 | This paper | GenBank: MZ133811 |
| Ty1 | Hanke et al., 2020 | PDB: 6ZXN |
| VHH-72 | Wrapp et al., 2020 | PDB: 6WAQ |
| CB6 | Shi et al., 2020 | PDB: 7C01 |
| C119 |
Barnes et al., 2020 Provided by laboratories of Pamela Bjorkman (California Institute of Technology) and Michel Nussenzweig (The Rockefeller University) |
PDB: 7K8W |
| S309 |
Pinto et al., 2020 Provided by laboratories of Pamela (California Institute of Technology) Bjorkman and Michel Nussenzweig (The Rockefeller University) |
PDB: 6WPS |
| CR3022 | Yuan et al. | PDB: 6W41 |
| Experimental models: Cell lines | ||
| Human: HEK293-6E | National Research Council (NRC) of Canada | N/A |
| Human: Lenti-X 293T | Takara | Cat#632180 |
| Human: 293T-ACE2 | BEI | Cat#NR-52511 |
| Cercopithecus aethiops: VeroE6 | ATCC | Cat#CRL1586 |
| Hamster: CHO | Gibco | Cat#A29133 |
| Yeast: EBY100 | Julian et al. 2017 | N/A |
| Recombinant DNA | ||
| HDM-Hgpm2 plasmid encoding HIV Gag-Pol under CMV promoter | BEI | Cat#NR-52517 |
| HDM-tat1b plasmid encoding HIV Tat under CMV promoter | BEI | Cat#NR-52518 |
| pRC14 CMV-Rev1b plasmid encoding HIV Rev | BEI | Cat#NR-52519 |
| pHAGE-CMV-Luc2-IRES-ZsGreen-W lentiviral transfer plasmid encoding co-expression of luciferase and ZsGreen | BEI | Cat#NR-52516 |
| pCMV3 SARS-CoV2 S Untagged Delta 19AA C-term plasmid encoding the SARS-CoV-2 spike (S) protein with a 19-amino acid deletion at the C-terminus | Provided by the laboratory of Marilia Cascalho (University of Michigan) | N/A |
| Software and algorithms | ||
| Python version 3.7 | Python Software Foundation | https://www.python.org |
| Code for curve fitting | This paper | https://GitHub.com/mduncans/Data_Fitting |
| Others | ||
| Synthetic nanobody library |
McMahon et al., 2018 Provided by the laboratory of Andrew Kruse (Harvard Medical School) |
N/A |
| Streptavidin MicroBeads | Miltenyi Biotec | Cat#130-048-101 |
| Zymoprep Yeast Plasmid Miniprep II | Zymo Research | Cat#D2004 |
| Streptavidin Dynabeads | Invitrogen | Cat#11047 |
| Protein A magnetic beads | Invitrogen | Cat#88846 |
| QIAquick Gel Extraction Kit | Qiagen | Cat#28704 |
| QIAquick PCR Purification Kit | Qiagen | Cat#28104 |
| QIAprep Spin Miniprep Kit | Qiagen | Cat#27106 |
| Protein A Agarose | Thermo Fisher Scientific | Cat#20333 |
| Centrifuge columns | Thermo Fisher Scientific | Cat#89898 |
| Zeba Spin Desalting Columns | Thermo Fisher Scientific | Cat#89894; Cat#89892; Cat#89890 |
| Invitrogen NuPAGE 10% Bis-Tris, 1.0 mm, Midi Protein GEL (SDS-PAGE) | Thermo Fisher Scientific | Cat#WG1203BOX |
| Lenti-X Concentrator | Takara | Cat#631232 |
| Superdex 200 Increase 10/300 GL column | GE | Cat#28990944 |
| 96-well plates | VWR | Cat#650261 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Peter Tessier (ptessier@umich.edu).
Materials availability
All constructs used in this study are available upon reasonable request from the lead contact. All constructs were sequence-verified via Sanger sequencing.
Data and code availability
The data generated in this study are available upon reasonable request from the lead contact. The sequences of antibodies developed in this study have been deposited in GenBank under accession numbers MZ133804-MZ133811. Accession numbers for each antibody can also be found in the Key resources table. Code for curve fitting has been deposited in GitHub and can be accessed at https://GitHub.com/mduncans/Data_Fitting.
Experimental model and subject details
HEK 293 cells are an immortalized human female cell line. VeroE6 cells are a Cerocopithecus aethiops (grivet) female cell line. CHO cells are a hamster female cell line. EBY100 are a yeast (Saccharomyces cerevisae) cell line (MATa).
Cell lines
All cell culture work was performed using standard aseptic technique. Cell culture and experiments were performed in biosafety level 2 or 3 facilities as appropriate for the involved materials, and appropriate safety procedures for the respective biosafety levels were observed. HEK 293-6E cells are a human embryonic kidney cell line licensed from the National Research Council of Canada for the transient expression of protein. These cells were maintained in F17 media supplemented with Glutamine, Kolliphor, and G418, and cultured at 37°C with 5% CO2 with agitation (250 RPM). Cells were passaged or transfected after reaching a density of approximately 1.5-2 million cells per mL. Transfection was performed as described in the Method details.
Lenti-X 293T cells (Takara, 632180) are a human embryonic kidney cell line modified for the facile expression of viral protein. Lenti-X 293T cells were cultured in RPMI media supplemented with 10% Fetal Bovine Serum (FBS) and 1% penicillin/streptomycin (P/S) at 37°C with 5% CO2. Production of pseudovirus particles using Lenti-X 293 cells is described in the Method details. HEK-293T-hACE2 cells (BEI, NR52511) are a human embryonic kidney cell line which constitutively expresses human angiotensin-converting enzyme 2 (ACE2). HEK-293T-hACE2 cells were cultured in DMEM media supplemented with 10% FBS and 1% P/S at 37°C with 5% CO2. Pseudovirus neutralization assays performed with HEK-293T-hACE2 cells are described in the Methods details. VeroE6 cells (ATCC, CRL1586) are a Cerocopithecus aethiops (grivet) kidney cell line. VeroE6 cells were cultured in DMEM supplemented with 10% FBS and 1% P/S at 37°C with 5% CO2. Live virus neutralization assays performed with VeroE6 cells are described in the Method details.
EBY100 is a yeast cell line used for yeast surface display of antibody fragments. EBY100 cells were first cultured in YPD media (20 g/L dextrose, 20 g/L peptone, and 10 g/L yeast extract) supplemented with antibiotics (100 μg/mL ampicillin, 100 μg/mL kanamycin, and 100x dilution of P/S). After transformation of plasmid encoding nanobody, EBY100 cells were grown in SDCAA (20 g/L dextrose, 6.7 g/L yeast nitrogen base without amino acids, 5 g/L casamino acids, 16.75 g/L sodium citrate, and 4 g/L citric acid) supplemented with antibiotics (100 μg/mL ampicillin, 100 μg/mL kanamycin, and 100x dilution of P/S) with agitation (225 RPM) at 30°C for approximately 16-24 h and induced in SDGCAA (8.56 g/L sodium phosphate dibasic dihydrate, 6.76 g/L sodium phosphate monobasic monohydrate, 6.7 g/L yeast nitrogen base without amino acids, 5 g/L casamino acids, 20 g/L galactose, and 2 g/L dextrose) supplemented with antibiotics (100 μg/mL ampicillin, 100 μg/mL kanamycin, and 100x dilution of P/S) with agitation (225 RPM) at either 20°C for approximately 36-40 h or 30°C for approximately 16-24 h.
Method details
Lead nanobody isolation and maturation
The original nanobody library (McMahon et al., 2018) was cloned into an Aga2-based yeast surface display plasmid (Julian et al., 2019). The nanobodies were expressed on the yeast surface as C-terminal fusion proteins to Aga2 (Aga2-nanobody). In the first round, MACS was performed against biotinylated RBD (bRBD) of SARS-CoV-2 (Acro, SPD-C82E9). 109 cells were incubated with 300 nM biotinylated RBD in PBS supplemented with 1 g/L BSA (PBSB) and 1% milk at room temperature for 3 h. Post incubation, the cells were washed once and incubated with streptavidin microbeads (Miltenyi, 130-048-141) with gentle rocking for 30 min at 4°C. Following incubation, the cells were washed once with ice-cold PBSB and passed through a MACS column under magnetic field to isolate cells bound to beads. The captured beads were washed once with ice-cold PBSB while employing the magnetic field. After washing, the beads were eluted into low pH SDCAA (20 g/L of dextrose, 6.7 g/L of yeast nitrogen base without amino acids, 5 g/L of casamino acids, 16.75 g/L of sodium citrate and 4 g/L of citric acid) liquid media and grown at 30°C for 2 d. All subsequent sorting was performed by FACS. In rounds 2, 3 and 4, a selection was performed against RBD-Fc (Acro, SPD-C5255; 100 nM for rounds 2 and 3 and 50 nM for round 4 respectively). In round 5, a selection against bRBD was performed at 100 nM followed by selection against 100 nM bRBD, 100 nM bS1 (S1 domain of SARS-CoV-2; Acro, S1N-C82E8) or 50 nM S protein trimer (Acro, SPN-C52H8).
Three lead nanobodies (KA1, KC3 and KC1) from the initial discovery campaign were isolated with modest affinities. Two of these clones (KA1 and KC3) were affinity matured by first preparing error-prone PCR libraries as previously described (Chao et al., 2006). Briefly, the DNA region encoding only KA1 or KC3 was amplified using Taq DNA Polymerase with Standard Taq Buffer (New England Biolabs, M0273L) in the presence of non-natural nucleotides, 8-Oxo-2'-deoxyguanosine-5'-Triphosphate (TriLink Biotechnologies, N-2034-1) and 2’-Deoxy-P-nucleoside-5'-Triphosphate (TriLink Biotechnologies, N-2037). Ten PCR cycles were used to amplify the DNA, and nanobody DNA was gel purified in a 1% agarose gel. To increase the number of mutations, DNA was amplified using Q5 High-Fidelity DNA Polymerase (New England Biolabs, M0491L), gel purified, and a second error-prone PCR with Taq DNA polymerase (New England Biolabs, M0320L) was performed under identical conditions. DNA encoding the region of plasmid surrounding KA1 or KC3 was added by overlap PCR. Total insert DNA was then amplified, and DNA was transformed into EBY100 as previously described (Benatuil et al., 2010). Four rounds of FACS selections were performed for each library, and the antigen concentration was progressively reduced, including 50 nM biotinylated S1 in round 1, 10 nM biotinylated S1 in round 2, 2 nM biotinylated S1 in round 3, and 100 pM biotinylated S1 in round 4.
CDR-swapping mutagenesis and clone evaluation
CDR-swapping mutagenesis was intentionally introduced after sort 5 of the initial discovery campaign. DNA was isolated from yeast cells that were collected after the fifth sort of the initial synthetic library. DNA segments of the nanobody gene comprising CDR1 (framework 1 to framework 2), CDR2 (framework 2 to framework 3) and CDR3 (framework 3 to framework 4) were PCR amplified to facilitate overlap PCR. The DNA encoding each CDR was then mixed at an equal mass ratio, and overlap PCR was used to reassemble and amplify DNA encoding the entire nanobody. The CDR-swapped nanobody DNA library was inserted into the yeast display plasmid by homologous recombination. The transformation efficiency for this CDR-swapped nanobody library was ∼5 x 107. Next, two rounds of sorting were performed by FACS using biotinylated RBD (100 nM in sort 1 and 10 nM in sort 2). Yeast cells collected from the terminal sort were miniprepped and Sanger sequenced.
Nanobody-Fc expression and purification
Yeast cells from the terminal rounds of sorting were mini-prepped (Zymo Research, D2004) and plasmids were recovered. Nanobody genes were amplified by performing PCR on yeast mini-prepped DNA with forward and reverse primers containing NheI and HindIII restriction sites, respectively. The PCR products were purified via a 1% agarose gel and extracted with DNA purification kit (Qiagen, 28704). The nanobody genes were then digested with NheI-HF (New England Biolabs, R3131L) and HindIII-HF (New England Biolabs, R3104L), as instructed by the manufacturer’s protocol, followed by purification (Qiagen, 28104). Nanobody-Fc expression plasmid was digested with NheI-HF and HindIII-HF, as instructed by the manufacturer’s protocol, followed by treatment with calf intestinal alkaline phosphatase (New England Biolabs, M0525L). The digested vector was purified by 1% agarose gel electrophoresis and followed by DNA extraction. The digested vector and inserts were ligated with T4 ligase (New England Biolabs, M0202L) followed by transformation into competent DH5α cells. Transformed cells were plated on LB plates supplemented with ampicillin (100 ug/mL) overnight at 37°C. Individual colonies were picked and grown in LB media (with ampicillin) overnight followed by mini-prepping (Qiagen, 27106). Plasmids from colonies were sequenced using Sanger sequencing.
HEK 293-6E cells (National Research Council of Canada) were grown, maintained and passaged at a density of 1.5-2 million cells per mL in F17 media (Thermo Fisher Scientific, A1383502) supplemented with Glutamine (Invitrogen, 25030081), Kolliphor (Fisher Scientific, NC0917244) and G418 (Thermo Fisher Scientific, 10131035). Nanobody-Fc plasmid (15 μg) was mixed with PEI (45 μg) at room temperature with F17 media (without supplements) for 10-15 min and added to cells at a density of 1.5-1.8 million cells per mL. Cells were fed with 20% w/v Yeastolate (BD Biosciences, 292804) 24-48 h post transfection and were grown for an additional 2-4 d at 37°C. Post expression, media was harvested by centrifuging cells at 4000 xg for 40 min. Media was collected, transferred to new tubes and 0.5-1 mL Protein A bead (Thermo Fisher Scientific, 20333) slurry was added followed by gently rocking at 4°C overnight. Protein A beads were collected from media with filter columns (Thermo Fisher Scientific, 89898) under vacuum followed by washing with 50-100 mL of PBS. Protein was eluted from Protein A beads using 0.1 M glycine buffer (pH 3.0) followed by 1x buffer exchange into 20 mM acetate (pH 5.0) using Zeba desalting columns (Thermo Fisher Scientific, 89894). Proteins were then filtered using 0.2 μm filters, aliquoted and stored at -80°C. Nanobody concentrations were evaluated by measuring absorbance at 280 nm and purity was evaluated by SDS-PAGE (Thermo Fisher Scientific, WG1203BOX).
Pseudovirus neutralization analysis
SARS-CoV-2 pseudovirus neutralization assay was adapted from a previous report (Crawford et al., 2020). To prepare SARS-CoV-2 pseudovirus particles, Lenti-X 293T cells (Takara, 632180) were seeded at 5x105 cells per well in 6-well plates in RPMI media supplemented with 10% Fetal Bovine Serum (FBS), 1% penicillin/streptomycin (P/S) and cultured at 37°C with 5% CO2. Upon reaching a target confluency of 50-70%, cells were transfected with third generation lentivirus 5 plasmid system (0.22, 0.22, 0.22, 1, or 0.34 μg respectively): HDM-Hgpm2 plasmid (BEI catalog number NR-52517) encoding HIV Gag-Pol under CMV promoter, HDM-tat1b plasmid (BEI catalog number NR-52518) encoding HIV Tat under CMV promoter, pRC-CMV-Rev1b plasmid (BEI catalog number NR-52519) encoding HIV Rev, pHAGE-CMV-Luc2-IRES-ZsGreen-W (BEI catalog number NR-52516) lentiviral transfer plasmid encoding co-expression of luciferase and ZsGreen, pCMV3 SARS-CoV2 S Untagged Delta 19AA C-term plasmid encoding the SARS-CoV-2 spike (S) protein with a 19-amino acid deletion at the C-terminus.
At 24 h post-transfection, cell media was changed to fresh RPMI with 10% FBS and 1% P/S. At 72 h post-transfection, cell supernatant was collected and passed through a 0.45 μm filter to remove cellular debris. SARS-CoV-2 pseudovirus was then concentrated via Lenti-X Concentrator (Takara, 631232) without ultracentrifugation. Briefly, Lenti-X Concentrator was added to cell culture supernatant at a volume ratio of 1:3 and incubated overnight at 4°C. The mixture was then centrifuged at 1500 xg for 45 min. Supernatant was discarded, and the pseudovirus pellet was resuspended in Opti-MEM media in a volume of 50 μL Opti-MEM per well of virus harvest.
To determine virus titer, 293T-ACE2 cells (BEI resources catalog NR-52511) were seeded at 8,000 cells per well in a 96-well plate in DMEM with 10% FBS and 1% P/S and cultured at 37°C with 5% CO2. At 24 h post-seeding, cells were infected with varying dilutions of virus, diluted in DMEM media in the presence of 5 μg/mL polybrene, 10% FBS, and 1% P/S. At 48 h post-infection, the percentage of ZsGreen-expressing cells was determined via flow cytometry using a Bio-Rad ZE5 cell analyzer and further corroborated via fluorescence microscopy. Tissue culture infectious units (TCIU) per mL of virus was then calculated.
For neutralization assays, 293T-ACE2 cells were seeded at 8,000 cells per well in white bottom 96-well plates (Corning, 3917) in DMEM (10% FBS and 1% P/S) and cultured at 37°C with 5% CO2. At 24 h post-seeding, 293T-ACE2 cells were infected with 350 TCIU SARS-CoV-2 pseudovirus per well in the presence of antibody treatments. Briefly, 4-fold serial dilutions of antibody were prepared, mixed with SARS-CoV-2 pseudovirus, and incubated for 1 h at 37°C. Following this incubation, 293T-ACE2 cells were treated with SARS-CoV-2 pseudovirus-antibody mixtures in the presence of 5 μg/mL polybrene. At 48 h post-infection, neutralization activity was determined via bioluminescence detection using a microplate reader. Briefly, 96-well plates were equilibrated to room temperature for 15 min. Media volume in each well was then reduced to 80 μL via micropipette. Luciferase substrate (80 μL; Promega ONE-Glo, E6110) was added to each well, the plate was incubated at room temperature for 10 min, and bioluminescence was detected using Molecular Devices SpectraMax microplate reader with 500 millisecond integration/well.
Live virus neutralization analysis
For antibody neutralization assays, 96-well plates were seeded with VeroE6 (ATCC CRL1586) cells at 10,000 cells per well and incubated at 37°C and 5% CO2 for 24 h. Antibodies were diluted in DMEM with 2% FBS in 96-well plates at a 2x final concentration in a volume of 50 μL. Cell culture plates and antibody dilution plates were then transferred to a BSL3 facility. 50 μL of diluted SARS-CoV-2 (2000 pfu/mL or 100 pfu/well) was added to each well containing 50 μL of diluted antibodies. The antibody-virus mixtures were incubated at 37°C for 1 h. Growth media was then aspirated from cell culture plates and replaced with 100 μL of the virus-antibody solution. Final media composition for neutralization assays was DMEM media supplemented with 2% FBS and 1% P/S. Antibody dilutions were tested in eight replicate wells each. Plates were incubated at 37°C and 5% CO2 for 3 d and examined microscopically for visible cytopathic effect (CPE). Wells with any degree of visible, virus-induced CPE were scored as positive. All antibody neutralization screening experiments were conducted following standard operating procedures of an approved Biosafety Level 3 Facility.
Nanobody affinity and specificity analysis
The monovalent affinities for the nanobodies were evaluated in yeast surface display format. The nanobodies were expressed on the yeast surface as C-terminus fusion proteins (Aga2-nanobody). For affinity measurements, 105 yeast cells per sample that express each nanobody were washed twice with PBSB and incubated with mouse anti-Myc antibody (1000x dilution) and biotinylated RBD over a range of concentrations in 1% milk at room temperature for 3 h. Post incubation, the cells were centrifuged at 2500 xg for 5 min followed by washing once with ice-cold PBSB. Next, the cells were then incubated with goat anti-mouse IgG AF488 (200x dilution; Invitrogen, A11001) and streptavidin AF647 (1000x dilution; Invitrogen, S32357) on ice for 4 min. Post-secondary antibodies incubation, cells were centrifuged and washed once with ice-cold PBSB, re-suspended in PBSB and evaluated on Bio-Rad ZE5 analyzer.
For specificity analysis, the binding was evaluated for antibodies KC3.ep3 and VHH-72 to receptor-binding domain (RBD) of SARS-CoV (Acro, SPD-S52H6) and SARS-CoV-2 virus. The antigen binding was performed in similar way as described above. Post antigen binding, the cells were washed once with ice-cold PBSB and incubated with mouse anti-Myc (1000x dilution) and chicken anti-His (1000x dilution; Invitrogen, PA1-9531) antibodies on ice for 20 min. Post primary incubation, the cells were washed with ice-cold PBSB and incubated with goat anti-mouse IgG AF488 (200x dilution) and donkey anti-chicken IgY F(ab’)2 fragment AF647 (500x dilution; Jackson ImmunoResearch, 703-606-155) antibodies on ice for 4 min. Post-secondary incubation, the cells were washed once with ice-cold PBSB and evaluated by flow cytometry.
For affinity measurements of soluble antibodies in the bivalent format, biotinylated RBD was first immobilized on streptavidin Dynabeads (Invitrogen, 11047). Antigen loading was 0.1 μg protein for 107 beads in a final volume of 400 μL. Beads were washed twice with PBSB and blocked with 10% milk in PBSB by end-over-end mixing at room temperature for 1 h. Post blocking, the beads were washed once with PBSB and incubated with varying concentrations of antibodies (105 beads per sample) in PBSB with 1% milk at room temperature for 3 h. After antibody incubation, the beads were centrifuged and washed once with ice-cold PBSB followed by incubation with goat anti-human IgG AF647 (Jackson ImmunoResearch, 109-605-098) on ice for 4 min. Post labeling, the beads were washed once with ice-cold PBSB and evaluated by flow cytometry.
Nanobody competition analysis
To evaluate the epitope of KC3.ep3, competitive binding analysis was performed with other SARS-CoV-2 antibodies and nanobodies. Biotinylated RBD (5 nM) was first pre-incubated with soluble nanobodies/antibodies in the bivalent format or ACE2 (RayBiotech, 230-30165) over a range of concentrations (0.05, 0.5, 5, 50 and 500 nM) for 2 h at room temperature with mild agitation. Next, the antibody-antigen complexes were incubated with yeast cells expressing monovalent KC3.ep3, along with anti-Myc antibody (1000x dilution), in PBSB with 1% milk at room temperature for 3 h. Post incubation, cells were washed once with ice-cold PBSB and incubated with streptavidin AF647 (1000x dilution) and goat anti-mouse IgG AF488 (200x dilution) on ice for 4 min. Following secondary incubation, cells were washed once with ice-cold PBSB and analyzed by flow cytometry.
Nanobody biophysical characterization
Melting temperature analysis
The melting temperatures of the proteins in this work were determined using differential scanning fluorimetry. Proteins were prepared at 0.12 mg/mL and mixed with Protein Thermal Shift Dye (Applied Biosystems, 4461146) at a volume ratio of 7:1 protein:dye to reach a final concentration of 1x dye. The protein-dye mixture was added to individual wells of a clear 384-well plate. Background signals were determined from 2-3 wells of 1x PBS mixed with dye. Samples were submitted to the University of Michigan Advanced Genomics core for analysis. Samples were centrifuged in the 384-well plate at 1000-2000 rpm for 1 min. The plates were then inserted into an ABI Prism 7900HT Sequence Detection System (Applied Biosystems), and thermal cycle conditions were set to examine increasing temperatures between 25-98°C over a period of 45 min. Background signals were subtracted from samples, and melting temperatures were determined from the temperatures at which the maximum signals (first derivative equals zero) were observed.
Analytical size-exclusion chromatography
The purity of the proteins after the Protein A purification was evaluated using size-exclusion chromatography with a Shimadzu Prominence HPLC System outfitted with a LC-20AT pump, SIL-20AC autosampler and FRC-10A fraction collector. Proteins in 20 mM acetate (pH 5) were buffer exchanged into PBS (pH 7.4). For analytical SEC, 100 μL of protein sample (diluted to 0.1 mg/mL) was loaded onto the column (Superdex 200 Increase 10/300 GL column; GE, 28990944) and analyzed at 0.75 mL/min using a PBS running buffer supplemented with 200 mM arginine (pH 7.4). Absorbance was monitored at 220 and 280 nm, and the 280 nm signal was primarily used for analysis. The percentage of protein monomer was evaluated by analyzing the area under the peak between 8 and 22 min (exclusion volume to solvent elution times). Proteins with less than 90% monomer were further purified via size-exclusion chromatography. Protein fractions were collected, buffer exchanged into PBS (pH 7.4), filtered, aliquoted and stored at -80°C.
Polyspecificity analysis
The polyspecificity reagent (PSR) was prepared as previously (Xu et al., 2013). CHO cells (109, Gibco, A29133) were pelleted, the cell pellets were washed separately with PBSB and Buffer B (50 mM HEPES, 0.15 M NaCl, 2 mM CaCl2, 5 mM KCl, 5 mM MgCl2, 10% Glycerol, pH 7.2), and then pelleted again. The pellets were resuspended in 5 mL of Buffer B supplemented with a protease inhibitor (Sigma Aldrich, 4693159001). Next, the resuspended cells were homogenized for 90 s (three cycles of 30 s) followed by sonication for 90 s (three cycles of 30 s). The cell suspension was then spun down at 40,000 xg for 1 h and the supernatant was discarded.
The pellet, comprising the enriched membrane fraction, was resuspended in Buffer B with a Dounce homogenizer for 30 strokes. The protein concentration was determined using a detergent compatible protein assay kit (BioRad, 5000116). The enriched membrane fraction was diluted to a theoretical concentration of 1 mg/mL in solubilization buffer (pH 7.2), the latter of which contained 50 mM HEPES, 0.15 M NaCl, 2 mM CaCl2, 5 mM KCl, 5 mM MgCl2, 1% n-dodecyl-b-D-maltopyranoside (Sigma Aldrich, D4641), and a protease inhibitor (Sigma Aldrich, 11873580001). The solution was then mixed overnight (end-over-end) at 4°C. The soluble membrane protein fraction was centrifuged at 40,000 xg for 1 h and the supernatant was collected. The final concentration of supernatant was ∼0.8-0.9 mg/mL.
Sulfo-NHS-LC-biotin (Thermo Fisher Scientific, PI21335) was dissolved in distilled water at ∼11.5 mg/mL. Stock solution of Sulfo-NHS-LC-biotin (150 mL) and the PSR reagent (4.5 mL at 0.8-0.9 mg/mL) were mixed via end-over-end mixing at room temperature (45 min). The reaction was quenched (10 mL of 1.5 M hydroxylamine at pH 7.2), and biotinylated PSR was aliquoted and stored at -80°C.
Protein A magnetic beads (Invitrogen, 88846) were washed three times with PBSB and incubated with antibodies at a range of concentrations in 96-well plates (VWR, 650261) overnight at 4°C. The antibodies were purified either via one-step (Protein A chromatography) or two-step (Protein A and size-exclusion chromatography) purification methods. Protein immobilization concentrations ranged from 0.03x to 10x of saturation of reported bead binding capacity for IgGs. Protein concentrations were normalized by molarity to maintain the same Fc concentration and bead saturation. Next, the protein-coated beads were washed by centrifuging the 96-well plates at 2500 xg for 4 min and washed twice with PBSB. Afterward, the beads were resuspended with a 10x diluted solution of biotinylated PSR and incubated on ice for 20 min. Beads were washed once with PBSB and incubated with 1000x diluted solution of streptavidin AF-647 (Invitrogen, S32357) and 1000x diluted solution of goat anti-human Fc F(ab’)2 AF-488 (Invitrogen, H10120) on ice (4 min). Beads were washed once, resuspended in PBSB, and analyzed via flow cytometry. The antibody binding steps were performed in PBSB, and three independent repeats were performed. The control antibodies used were the variable regions of elotuzumab (specific control) and emibetuzumab (polyspecific control) grafted onto a common IgG1 framework, which results in differences in the evaluated antibodies and the actual clinical-stage drugs. The control antibodies were two-step purified (Protein A and SEC) and were used to normalize results from all replicates between 0 and 1.
Quantification and statistical analysis
The number replicates performed for each experiment can be found in the figure caption. The average and standard deviation of the IC50, KD, and EC50 values are given in the figures. Curve fitting was performed in Python. The average and standard deviation of melting temperatures are given in the Results.
Acknowledgments
We thank Andrew Kruse for providing the nanobody library used in this work. We also thank Michel Nussenzweig, Pamela Bjorkman, Christopher Barnes, and Anna Gazumyan for providing class 2 and 3 SARS-CoV-2 antibodies. Moreover, we thank Adam Lauring for providing reagents and advice related to the pseudovirus neutralization assay. We acknowledge BEI Resources (NIAID, NIH) for providing the SARS-Related Coronavirus 2, Wuhan-Hu-1 Spike-Pseudotyped Lentiviral Kit (NR-5294) and SARS-Related Coronavirus 2, Isolate USA-WA1/2020 (NR-52281; originally deposited by the Centers for Disease Control and Prevention). We also thank members of the Tessier lab for their helpful suggestions. This work was supported by the National Institutes of Health (RF1AG059723, R01AG050598, and R35GM136300 to P.M.T., U54GM103297 and R21AI152865 to A.W.T., and R01 AI 151588 01 to M.C.), National Science Foundation (CBET 1159943, 1605266, and 1813963 to P.M.T., Graduate Research Fellowship to M.D.S.), Biointerfaces Institute (to P.M.T.), Albert M. Mattocks Chair (to P.M.T), MICHR Education PTSP 2020 (U069943 to M.G.M.B), COVID-19: CVC Impact Research Ignitor Grant Award (to M.C.), University of Michigan MICHR Accelerating Synergy Award (to M.C.), and University of Michigan Institutional Funds (to T.M.L).
Author contributions
J.M.Z., A.A.D., J.S.S., and P.M.T. designed the research. J.M.Z. and A.A.D. modified and constructed the mutant nanobody libraries and performed the antibody sorting. J.M.Z., A.A.D., J.S.S., G.P., E.M.K., and A.A.K. produced and/or performed the characterization of antibodies. T.M.L., M.C., and M.G.M.B. planned and constructed reagents for the pseudovirus assay. M.D.S. performed antibody sequence and bioinformatics analysis. J.M.Z., A.A.D., J.S.S., A.A.K., A.W.T., and P.M.T. analyzed the data. J.M.Z., A.A.D., and P.M.T. wrote the paper with input from the co-authors.
Declaration of interests
The authors declare no competing interests.
Published: June 24, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.chembiol.2021.05.019.
Supplemental information
References
- Alsoussi W.B., Turner J.S., Case J.B., Zhao H., Schmitz A.J., Zhou J.Q., Rita E., Lei T., Rizk A.A., Mcintire K.M., et al. A potently neutralizing antibody protects mice against SARS-CoV-2 infection. J. Immunol. 2020;205:915–922. doi: 10.4049/jimmunol.2000583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes C.O., Jette C.A., Abernathy M.E., Dam K.M.A., Esswein S.R., Gristick H.B., Malyutin A.G., Sharaf N.G., Huey-Tubman K.E., Lee Y.E., et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature. 2020;588:682–687. doi: 10.1038/s41586-020-2852-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum A., Ajithdoss D., Copin R., Zhou A., Lanza K., Negron N., Ni M., Wei Y., Mohammadi K., Musser B., et al. REGN-COV2 antibodies prevent and treat SARS-CoV-2 infection in rhesus macaques and hamsters. Science. 2020;370:1110–1115. doi: 10.1126/science.abe2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum A., Fulton B.O., Wloga E., Copin R., Pascal K.E., Russo V., Giordano S., Lanza K., Negron N., Ni M., et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science. 2020;369:1014–1018. doi: 10.1126/science.abd0831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benatuil L., Perez J.M., Belk J., Hsieh C.-M. An improved yeast transformation method for the generation of very large human antibody libraries. Protein Eng. Des. Sel. 2010;23:155–159. doi: 10.1093/protein/gzq002. [DOI] [PubMed] [Google Scholar]
- Boder E.T., Midelfort K.S., Wittrup K.D. Directed evolution of antibody fragments with monovalent femtomolar antigen-binding affinity. Proc. Natl. Acad. Sci. U S A. 2000;97:10701–10705. doi: 10.1073/pnas.170297297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury A.R.M., Sidhu S., Dübel S., McCafferty J. Beyond natural antibodies: the power of in vitro display technologies. Nat. Biotechnol. 2011;29:245–254. doi: 10.1038/nbt.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao G., Lau W.L., Hackel B.J., Sazinsky S.L., Lippow S.M., Wittrup K.D. Isolating and engineering human antibodies using yeast surface display. Nat. Protoc. 2006;1:755–768. doi: 10.1038/nprot.2006.94. [DOI] [PubMed] [Google Scholar]
- Chen P., Nirula A., Heller B., Gottlieb R.L., Boscia J., Morris J., Huhn G., Cardona J., Mocherla B., Stosor V., et al. SARS-CoV-2 neutralizing antibody LY-CoV555 in outpatients with covid-19. N. Engl. J. Med. 2020;384:229–237. doi: 10.1056/NEJMoa2029849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi X., Liu X., Wang C., Zhang X., Li X., Hou J., Ren L., Jin Q., Wang J., Yang W. Humanized single domain antibodies neutralize SARS-CoV-2 by targeting the spike receptor binding domain. Nat. Commun. 2020;11:4528. doi: 10.1038/s41467-020-18387-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford K.H.D., Eguia R., Dingens A.S., Loes A.N., Malone K.D., Wolf C.R., Chu H.Y., Tortorici M.A., Veesler D., Murphy M., et al. Protocol and reagents for pseudotyping lentiviral particles with SARS-CoV-2 spike protein for neutralization assays. Viruses. 2020;12:13–15. doi: 10.3390/v12050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Custódio T.F., Das H., Sheward D.J., Hanke L., Pazicky S., Pieprzyk J., Sorgenfrei M., Schroer M.A., Gruzinov A.Y., Jeffries C.M., et al. Selection, biophysical and structural analysis of synthetic nanobodies that effectively neutralize SARS-CoV-2. Nat. Commun. 2020;11:5588. doi: 10.1038/s41467-020-19204-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foote J., Eisen H.N. Breaking the affinity ceiling for antibodies and T cell receptors. Proc. Natl. Acad. Sci. U S A. 2000;97:10679–10681. doi: 10.1073/pnas.97.20.10679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaney A.J., Starr T.N., Gilchuk P., Zost S.J., Binshtein E., Loes A.N., Hilton S.K., Huddleston J., Eguia R., Crawford K.H.D., et al. Complete mapping of mutations to the SARS-CoV-2 spike receptor-binding domain that escape antibody recognition. Cell Host Microbe. 2020;29:1–14. doi: 10.1016/j.chom.2020.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke L., Vidakovics Perez L., Sheward D.J., Das H., Schulte T., Moliner-Morro A., Corcoran M., Achour A., Karlsson Hedestam G.B., Hällberg B.M., et al. An alpaca nanobody neutralizes SARS-CoV-2 by blocking receptor interaction. Nat. Commun. 2020;11:1–9. doi: 10.1038/s41467-020-18174-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen J., Baum A., Pascal K.E., Russo V., Giordano S., Wloga E., Fulton B.O., Yan Y., Koon K., Patel K., et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science. 2020;369:1010–1014. doi: 10.1126/science.abd0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hötzel I., Theil F.P., Bernstein L.J., Prabhu S., Deng R., Quintana L., Lutman J., Sibia R., Chan P., Bumbaca D., et al. A strategy for risk mitigation of antibodies with fast clearance. mAbs. 2012;4:753–760. doi: 10.4161/mabs.22189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo J., Le Bas A., Ruza R.R., Duyvesteyn H.M.E., Mikolajek H., Malinauskas T., Tan T.K., Rijal P., Dumoux M., Ward P.N., et al. Neutralizing nanobodies bind SARS-CoV-2 spike RBD and block interaction with ACE2. Nat. Struct. Mol. Biol. 2020;27:846–854. doi: 10.1038/s41594-020-0469-6. [DOI] [PubMed] [Google Scholar]
- Jackson J.R., Sathe G., Rosenberg M., Sweet R. In vitro antibody maturation. Improvement of a high affinity, neutralizing antibody against IL-1 beta. J. Immunol. 1995;154:3310–3319. [PubMed] [Google Scholar]
- Jain T., Sun T., Durand S., Hall A., Houston N.R., Nett J.H., Sharkey B., Bobrowicz B., Caffry I., Yu Y., et al. Biophysical properties of the clinical-stage antibody landscape. Proc. Natl. Acad. Sci. U S A. 2017;114:944–949. doi: 10.1073/pnas.1616408114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julian M.C., Li L., Garde S., Wilen R., Tessier P.M. Efficient affinity maturation of antibody variable domains requires co-selection of compensatory mutations to maintain thermodynamic stability. Sci. Rep. 2017;7:1–13. doi: 10.1038/srep45259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julian M.C., Rabia L.A., Desai A.A., Arsiwala A., Gerson J.E., Paulson H.L., Kane R.S., Tessier P.M. Nature-inspired design and evolution of anti-amyloid antibodies. J. Biol. Chem. 2019;294:8438–8451. doi: 10.1074/jbc.RA118.004731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon C., Baier A.S., Pascolutti R., Wegrecki M., Zheng S., Ong J.X., Erlandson S.C., Hilger D., Rasmussen S.G.F., Ring A.M., et al. Yeast surface display platform for rapid discovery of conformationally selective nanobodies. Nat. Struct. Mol. Biol. 2018;25:289–296. doi: 10.1038/s41594-018-0028-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D., Park Y.J., Beltramello M., Walls A.C., Tortorici M.A., Bianchi S., Jaconi S., Culap K., Zatta F., De Marco A., et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature. 2020;583:290–295. doi: 10.1038/s41586-020-2349-y. [DOI] [PubMed] [Google Scholar]
- Rabia L.A., Desai A.A., Jhajj H.S., Tessier P.M. Understanding and overcoming trade-offs between antibody affinity, specificity, stability and solubility. Biochem. Eng. J. 2018;137:365–374. doi: 10.1016/j.bej.2018.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoof M., Faust B., Saunders R.A., Sangwan S., Rezelj V., Hoppe N., Boone M., Billesbølle C.B., Puchades C., Azumaya C.M., et al. An ultrapotent synthetic nanobody neutralizes SARS-CoV-2 by stabilizing inactive Spike. Science. 2020;3255:1473–1479. doi: 10.1126/science.abe3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shehata L., Maurer D.P., Wec A.Z., Lilov A., Champney E., Sun T., Archambault K., Burnina I., Lynaugh H., Zhi X., et al. Affinity maturation enhances antibody specificity but compromises conformational stability. Cell Rep. 2019;28:3300–3308.e4. doi: 10.1016/j.celrep.2019.08.056. [DOI] [PubMed] [Google Scholar]
- Shi R., Shan C., Duan X., Chen Z., Liu P., Song J., Song T., Bi X., Han C., Wu L., et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature. 2020;584:120–124. doi: 10.1038/s41586-020-2381-y. [DOI] [PubMed] [Google Scholar]
- Steidl S., Ratsch O., Brocks B., Dürr M., Thomassen-Wolf E. In vitro affinity maturation of human GM-CSF antibodies by targeted CDR-diversification. Mol. Immunol. 2008;46:135–144. doi: 10.1016/j.molimm.2008.07.013. [DOI] [PubMed] [Google Scholar]
- Tian X., Li C., Huang A., Xia S., Lu S., Shi Z., Lu L., Jiang S., Yang Z., Wu Y., et al. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg. Microbes Infect. 2020;9:382–385. doi: 10.1080/22221751.2020.1729069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiller K.E., Tessier P.M. Advances in antibody design. Annu. Rev. Biomed. Eng. 2015;17:191–216. doi: 10.1146/annurev-bioeng-071114-040733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisblum Y., Schmidt F., Zhang F., DaSilva J., Poston D., Lorenzi J.C.C., Muecksch F., Rutkowska M., Hoffmann H.H., Michailidis E., et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. eLife. 2020;9:e61312. doi: 10.7554/eLife.61312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrapp D., De Vlieger D., Corbett K.S., Torres G.M., Wang N., Van Breedam W., Roose K., van Schie L., Hoffmann M., Pöhlmann S., et al. Structural basis for potent neutralization of betacoronaviruses by single-domain camelid antibodies. Cell. 2020;181:1004–1015.e15. doi: 10.1016/j.cell.2020.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., Wang F., Shen C., Peng W., Li D., Zhao C., Li Z., Li S., Bi Y., Yang Y., et al. A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2. Science. 2020;368:1274–1278. doi: 10.1126/science.abc2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y., Nambulli S., Xiao Z., Liu H., Sang Z., Duprex W.P., Schneidman-Duhovny D., Zhang C., Shi Y. Versatile and multivalent nanobodies efficiently neutralize SARS-CoV-2. Science. 2020;370:1479–1484. doi: 10.1126/science.abe4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Roach W., Sun T., Jain T., Prinz B., Yu T.-Y., Torrey J., Thomas J., Bobrowicz P., Vasquez M., et al. Addressing polyspecificity of antibodies selected from an in vitro yeast presentation system: a FACS-based, high-throughput selection and analytical tool. Protein Eng. Des. Sel. 2013;26:663–670. doi: 10.1093/protein/gzt047. [DOI] [PubMed] [Google Scholar]
- Yang W.P., Green K., Pinz-Sweeney S., Briones A.T., Burton D.R., Barbas C.F. CDR walking mutagenesis for the affinity maturation of a potent human anti-HIV-1 antibody into the picomolar range. J. Mol. Biol. 1995;254:392–403. doi: 10.1006/jmbi.1995.0626. [DOI] [PubMed] [Google Scholar]
- Yuan M., Wu N.C., Zhu X., Lee C.C.D., So R.T.Y., Lv H., Mok C.K.P., Wilson I.A. A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science. 2020;368:630–633. doi: 10.1126/science.abb7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Liu X., Bell A., To R., Baral T.N., Azizi A., Li J., Cass B., Durocher Y. Transient expression and purification of chimeric heavy chain antibodies. Protein Expr. Purif. 2009;65:77–82. doi: 10.1016/j.pep.2008.10.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available upon reasonable request from the lead contact. The sequences of antibodies developed in this study have been deposited in GenBank under accession numbers MZ133804-MZ133811. Accession numbers for each antibody can also be found in the Key resources table. Code for curve fitting has been deposited in GitHub and can be accessed at https://GitHub.com/mduncans/Data_Fitting.