

Abstract
Single-cell profiling methods have had a profound impact on the understanding of cellular heterogeneity. While genomes and transcriptomes can be explored at the single-cell level, single-cell profiling of proteomes is not yet established. Here we describe new single-molecule protein sequencing and identification technologies alongside innovations in mass spectrometry that will eventually enable broad sequence coverage in single-cell profiling. These technologies will in turn facilitate biological discovery and open new avenues for ultrasensitive disease diagnostics.
The emergence of next-generation sequencing and single-molecule DNA sequencing technologies has revolutionized genomics and, consequently, has profoundly altered precision medicine diagnostics. Proteomics awaits similar transformative waves of protein sequencing techniques that will allow for the examination of proteins at the single-cell and ultimately single-molecule level, even with low-abundance proteins. The proteome is not a direct reflection of the transcriptome, and the way that RNA abundance relates to protein abundance varies from transcript to transcript. Further, the post-translationally modified proteome is inaccessible from the transcriptome. Therefore, whole-proteome sequencing and profiling of the vast repertoire of cell types is expected to fundamentally enhance understanding of all living systems. This necessitates analysis of the proteome with ultra-high resolution, complementing today’s single-cell RNA sequencing studies.
DNA sequencing technologies are routinely used for whole-genome and whole-transcriptome profiling with extensive read depths and high sequence coverage. In the absence of an amplification method similar to those available with DNA, conventional bottom–up mass spectrometry (MS)-based proteomics assays fall short of providing the same breadth of view for proteins (Box 1). Analysis of complex protein mixtures is particularly challenging because the more than 20,000 genes in the human genome1 are translated into a diversity of proteoforms that may include millions of variants as a result of post-translational modifications, alternative splicing and germline variants2. In cancer, for example, the proteoform landscape can be aberrant with many new protein variants resulting from non-canonical splicing, mutations, fusions and post-translational modifications. Characterization of such proteoforms is likely to benefit from improvements in current protein sequencing techniques and the emergence of new methods.
Box 1 |. Mass spectrometry-based global proteomics.
The last decade saw the maturation of MS use in global proteomics. The typical proteomics workflow is ‘bottom–up’ in nature and involves digesting a protein sample using a protease and characterizing the resulting peptides by MS114. Two types of measurements are typically made in succession: (1) MS1 spectra survey the masses of a set of peptides present in the mass spectrometer at a given moment and (2) MS2 spectra probe the structures of peptide ion species identified in the MS1 survey by isolating, fragmenting and measuring the fragment masses of one or a few of them. Peptides identified from the MS2 spectra are then mapped back to proteins to infer overall protein abundance.
Current mass spectrometers have drawbacks in terms of their dynamic range, the read length (peptide length) of ‘sequenced’ peptides and biases in detectability arising from the ionization mechanism, transmission and the mass analyzer used. Consequently, although ‘top–down’ proteomics methods capable of analyzing intact proteins exist115, most state-of-the-art proteomics approaches characterize the proteome with high numbers of proteins but on average characterize proteins with low sequence coverage and low sequencing depth. Different sample preparation strategies, instruments and elution profiles can improve the numbers and average sequence coverage of the proteins identified in an experiment. Summarizing the best single-sample run from 47 experiments (a summary of over 1,000 distinct samples) in ProteomicsDB116 revealed that, even with complex sample preparation, the mean sequence coverage (the average percentage of amino acids covered in an identified protein) for a single sample reaches just 33%. The resulting challenge in proteoform inference is demonstrated in studies evaluating the sensitivity of detection for various cancer aberrations in proteomics datasets. For example, in a study of over 30 sample process replicates, only about 10% of germline and somatic single-nucleotide variants detected at both the DNA and RNA level were detectable as peptides, and an even smaller proportion of peptides corresponding to novel splice junctions were detected that had been observed with RNA sequencing117.
Sequence coverage in global proteomics studies.
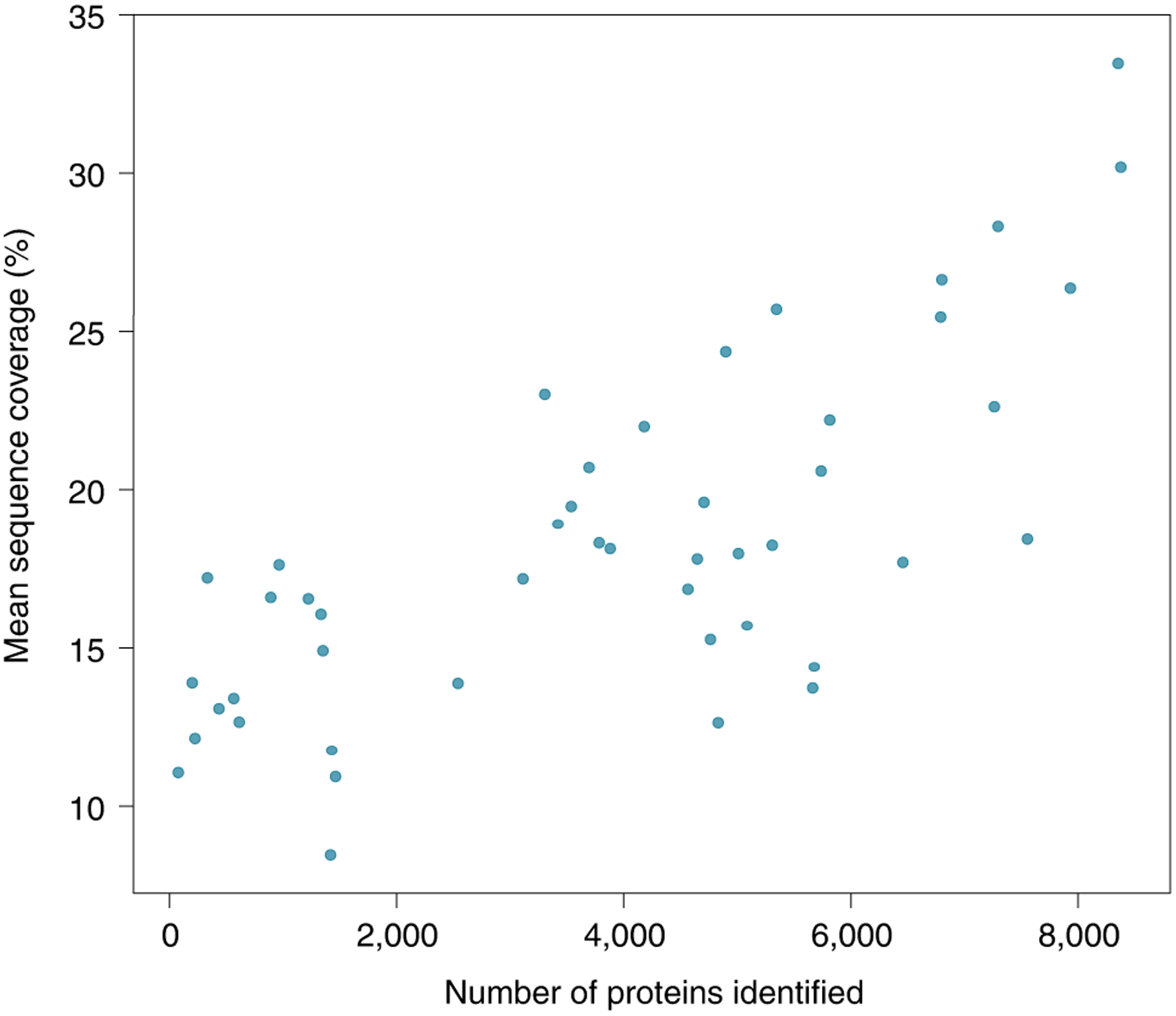
MS-based global proteomics studies identify and quantify proteins with variable sequence coverage. The single best run from the 47 publications present in ProteomicsDB shows how sample-specific protein sequence coverage improves with sample preparation methods. Sequence coverage generally decreases with sample complexity and increases with time (cost) dedicated to studying the sample.
MS remains a staple of protein identification and continues to develop toward single-cell methods (Box 2). In addition, a diverse range of protein sequencing and identification techniques have emerged that aim to increase the sensitivity of proteomics to the single-molecule level. Many of these techniques rely on fluorescence and nanopores for single-molecule sensing as an alternative means to sequence or identify proteins (Fig. 1). The landscape of emerging proteomics technologies is already vast, with different approaches at various stages of development, some of which have already secured industry investment3,4, an important step toward broad dissemination to the research community. Other technologies have shown great promise and gained popularity among the single-molecule biophysics community, while others are available as proofs of concept at just one or a few laboratories.
Box 2 |. Mass spectrometry-based single-cell proteomics.
The dream of extending MS-based proteomics to the single-cell level has eluded researchers for decades. Even as the sensitivity of MS instrumentation has improved to provide single-cell-compatible detection limits, in practice, samples comprising at least thousands of cells have been required to obtain an in-depth proteome profile. Two recent advances have made single-cell proteomics a reality. Miniaturized sample processing workflows such as nanodroplet processing in one pot for trace samples (nanoPOTS)118 have dramatically increased the efficiency of single-cell sample preparation. NanoPOTS utilizes a robotic nanopipettor to interface with a microfabricated nanowell plate. The reduced surface contact and increased protein concentrations within the nanoliter-sized droplets dramatically enhance digestion kinetics and increase sample recovery for single cells and other trace samples. Concurrently, multiplexed strategies (for example, single-cell proteomics by mass spectrometry, SCoPE-MS)119 have been developed in which proteins from single cells are labeled with unique isobaric tags and several cells are analyzed together in the presence of a larger carrier sample. The single cells and carrier provide a combined MS signal for each protein, and unique reporter ions released upon fragmentation enable protein quantification for each cell. While nanoPOTS and SCoPE-MS originally enabled quantification of hundreds of proteins119,120, the combination of these two techniques, as well as advances in miniaturized liquid chromatography and gas-phase separation, now enables more than 1,000 proteins to be quantified from single mammalian cells121.
Fig. 1 |. The emerging landscape of single-molecule protein sequencing and fingerprinting technologies.
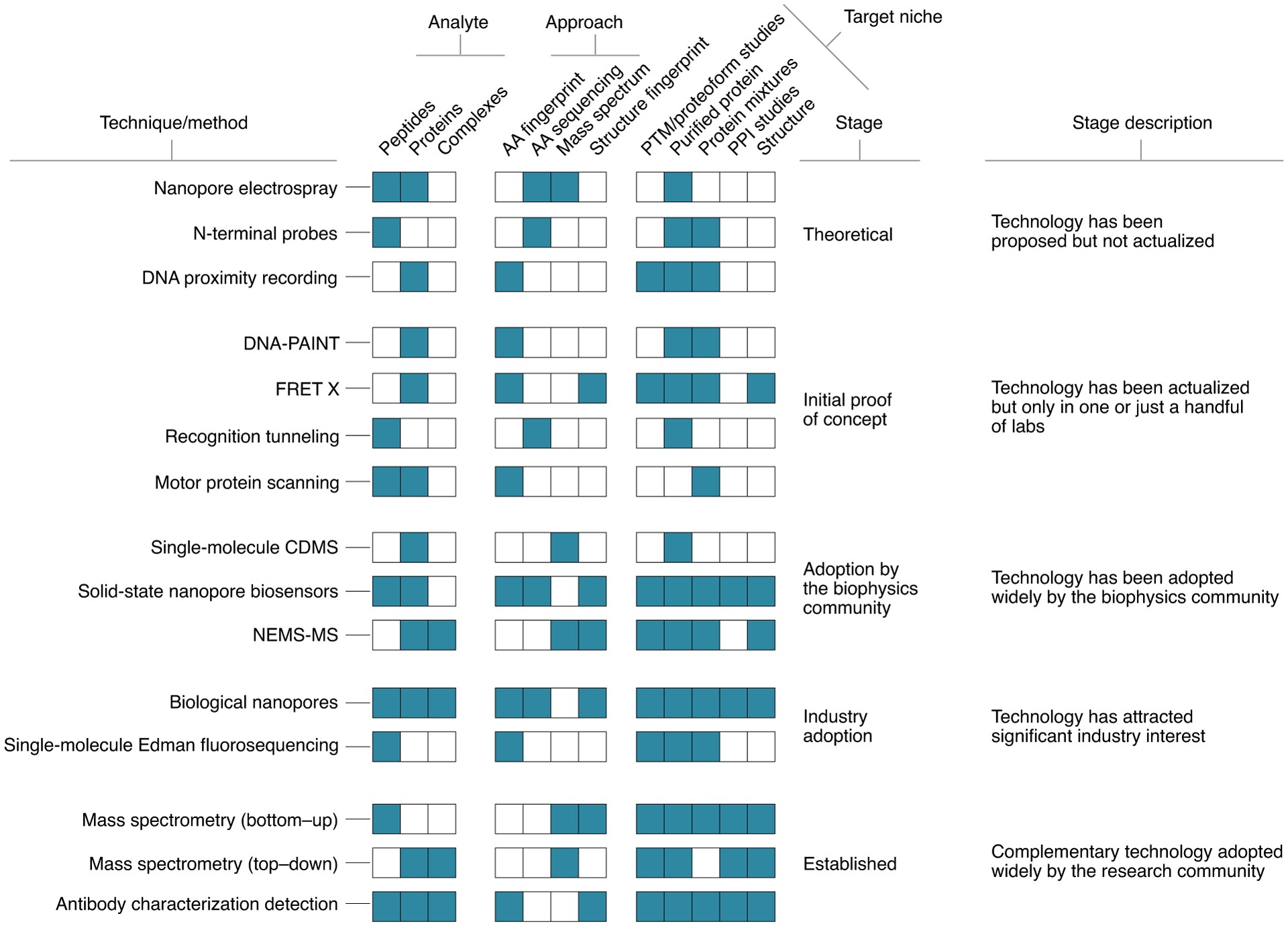
The new technologies address a range of analytes, methods of protein identification and target niches. Various techniques, particularly those involving complex readout signals, are suitable for characterizing short peptide sequences, while others are primed to characterize full-length proteins or larger complexes. The method of protein identification may fingerprint certain classes of amino acids (AA fingerprint) or reveal each amino acid down to its physiochemical class or better (AA sequencing). Technologies might characterize proteins by their mass or the mass of their fragments (mass spectrum). Other methods aim to characterize the properties of folded proteins (structure fingerprint). PTM, post-translational modification; PPI, protein–protein interaction; NEMS-MS, nanoelectromechanical systems MS.
Here we describe prominent emerging protein sequencing and fingerprinting techniques in the context of mature methods such as MS-based proteomics, discuss challenges for their real-world application and assess their transformative potential.
A renaissance of classical techniques
Edman degradation, MS and enzyme-linked immunosorbent assay (ELISA) have been broadly used for protein/peptide sequencing and identification for several decades; therefore, it is no surprise that further enhancements of these classical technologies are being sought. The biophysics community has been developing methods to increase the throughput5 and sensitivity6 of single-molecule ELISA, Edman degradation, single-particle MS, neutral-particle nanomechanical MS and single-particle electrospray. Even established tools commonly used in materials science, such as electric tunneling and direct current measurements, can be repurposed for protein sequencing.
Massively parallel Edman degradation.
Edman degradation7 was the first method to determine the amino acid sequence of a purified peptide. The method entails chemical modification of the N-terminal amino acid, cleavage of this amino acid from the peptide and determination of the identity of the cleaved labeled amino acid using high-performance liquid chromatography. Until recently, conducting sequencing of this sort in a massively parallel fashion was not feasible because the method requires highly purified peptides. However, recent multiplex strategies that use peptide arrays and either sequence chemically labeled peptides (‘fluorosequencing’) or successively detect the N-terminal amino acid are making breakthroughs.
Fluorosequencing combines Edman chemistry, single-molecule microscopy and stable synthetic fluorophore chemistry (Fig. 2a). Proteins are digested to shorter peptides and immobilized on a glass surface using the C terminus8. Millions of individual fluorescently labeled peptides can be visualized in parallel, and changing fluorescence intensities are monitored as N-terminal amino acids are sequentially removed through multiple rounds of Edman degradation. The resulting fluorescence signatures serve to uniquely identify individual peptides8. This method allows for millions of distinct peptide molecules to be sequenced in parallel, identified and digitally quantified on a zeptomole scale9. Specific amino acids are covalently labeled with spectrally distinguishable fluorophores, and the peptide fingerprint comes from measuring the decrease in fluorescence of peptides following Edman degradation9. Much as in MS, the partial sequence is mapped back to a reference proteome within a probabilistic framework.
Fig. 2 |. The renaissance of classic techniques.
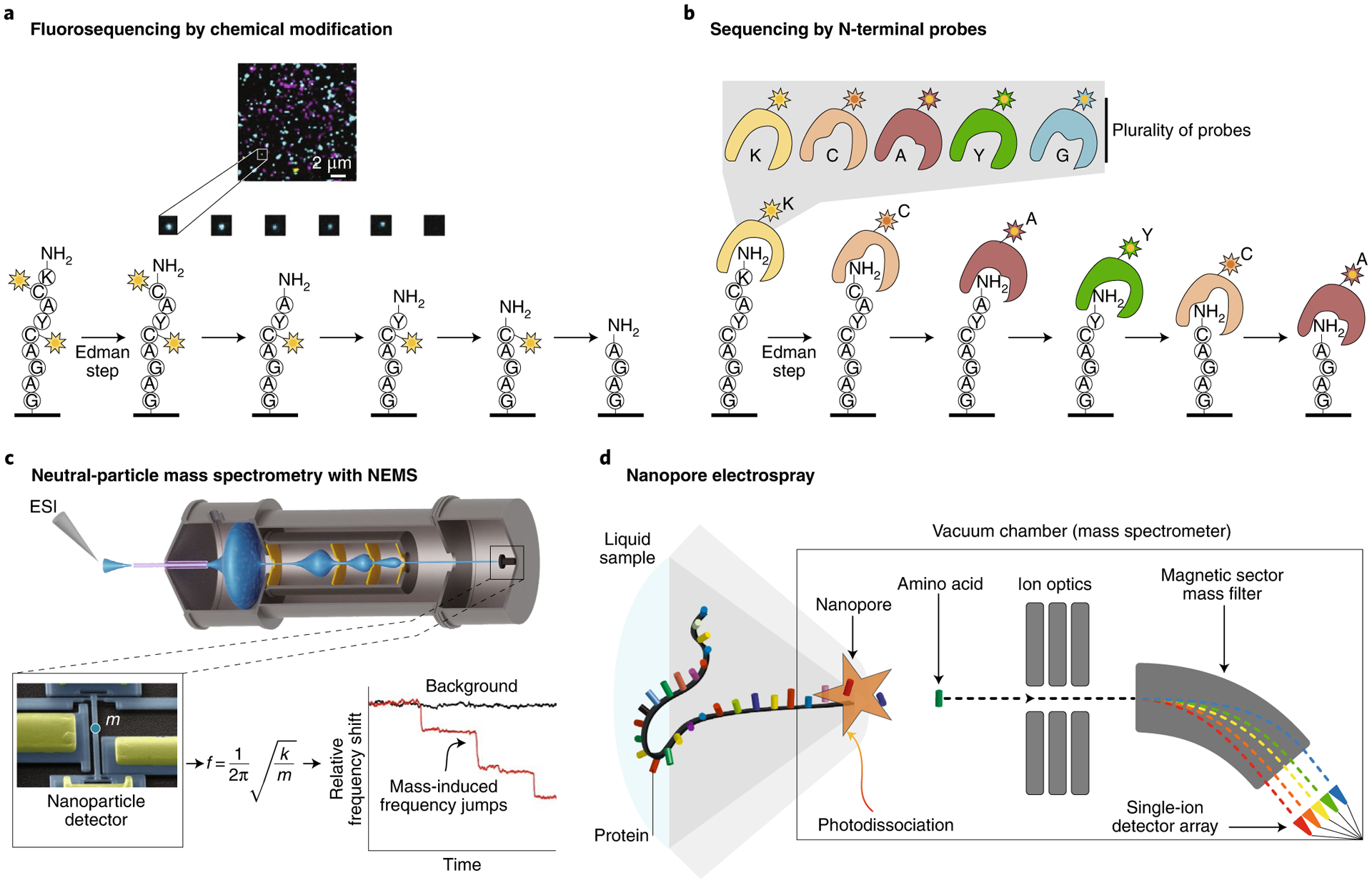
a,b, High-throughput fluorosequencing by Edman degradation featuring amino acid-specific chemical modification of peptides with fluorophores (a) and N-terminal amino acid recognition using a plurality of probes (b). c, Neutral-particle MS is a promising technique to characterize proteoforms. Currently, the technology can be used to characterize large megadalton-scale complexes using silicon-based nanosensors. Graphene nanosensors and further developments may push the technology toward smaller and smaller proteins and potentially lead to increased sequence coverage in global proteomics. ESI, electrospray ionization. d, Nanopore electrospray is a marriage of nanopores, classical electrospray and single-particle detection techniques to sequence single proteins by measuring amino acids one at a time. Panel a adapted with permission from ref.9, Springer Nature.
The technology is not without challenges, as the reagents used for Edman degradation chemistry lead to increased rates of fluorescent dye destruction, which in turn limits the read length. These reagents include slightly basic structures such as pyridine, strong acids such as trifluoroacetic acid and the electrophile phenyl isothiocyanate. Furthermore, the reliance on chemical labeling leads to partial sequencing of the peptide, with the unidentified remainder inferred by comparison to a reference proteome. In addition, inefficient labeling can lead to errors that must be modeled into the reference proteome comparison, spurring the development of new protocols to increase yields10. Exciting new proposals could add the dimension of protonation-based sequencing. The pKa of the N-terminal amino acid could be used for identification by observing and interpreting the protonation–deprotonation signal of the peptide at fixed pH through the Edman degradation process11. Much like fluorosequencing, the signal observed would be for the whole peptide and the decay pattern would be interpreted to derive a pKa for each N-terminal amino acid.
Several natural proteins and RNA molecules recognize specific amino acids either as free amino acids or as a part of a polypeptide chain12. These proteins and nucleic acids provide different solutions for N-terminal amino acid recognition. Each N-terminal amino acid binder (NAAB) probe selectively identifies a specific N-terminal amino acid or an N-terminal amino acid derivative. With each cycle, another amino acid is revealed in the sequence of the peptide. However, further directed evolution and engineering of NAAB probes is required to meet the stringent affinity, selectivity and stability requirements for error-free sequencing applications. In addition, such probes would need to discriminate among all amino acids, including the same amino acid in alternative positions in the peptide sequence. Probes that bind a class of N-terminal amino acids (for example, short aliphatic residues) could also be useful but would introduce ambiguity in the sequencing process. Different probes could also be designed to recognize short N-terminal k-mers, which would increase the number of probes needed but reduce the ambiguity in the resulting sequencing information. To circumvent this limitation, it may be possible to sequence the N-terminal amino acid by selective recognition using a plurality of probes in each cycle of Edman degradation13,14 (Fig. 2b).
Single-molecule mass spectrometry.
MS is a century-old method that measures the mass-to-charge (m/z) ratio of ions, in particular, charged peptides/proteins and their assemblies. Single-ion detection has been possible since the 1990s, for example, in Fourier-transform ion cyclotron resonance instruments15. Charge detection MS (CDMS) is a single-ion method where the charge assignment of each individual ion is determined directly, enabling conversion of the mass-to-charge ratio into the neutral mass domain. This approach has focused on the analysis of large biomolecular complexes, especially viruses in the range of 1–100 MDa16. While previously CDMS was limited to specialized instrumentation, the past year has seen breakthroughs built on early work producing mass spectra of single ions in Orbitrap mass analyzers17–19. Today, these mass analyzers can be used to directly derive the charge states of single proteins and even their fragment ions20. Orbitrap instruments are particularly useful because the readout of individual ions can be multiplexed by 100- to 1,000-fold in Orbitrap-based CDMS20. Individual ion MS has already shown resolution of mixtures with approximately 1,000 proteoforms that provided no data using standard MS20,21. This has greatly expanded the top–down approach to confirm DNA-inferred sequences of whole proteins, including localization of their post-translational modifications20–22. Without extensive alteration, Orbitrap mass analyzers can therefore measure tens of thousands of proteins in a matter of minutes. With these rapidly evolving technologies, charting the full human proteoform atlas has already begun23, making strides toward a comprehensive human proteoform project. However, ionization is a critical requirement for MS of proteins and peptides, and not all peptides are efficiently ionized and transmitted through the mass spectrometer. This might restrict some of the proteoform mapping efforts, providing a niche for the other technologies in Fig. 1.
For higher-molecular-weight species, the ionization of proteins and complexes yields a mixture of macro ions with variable charge states, resulting in a net reduction of sensitivity as the signal distributes over multiple peaks in the mass-to-charge dimension. Moreover, charge state distributions may overlap above a certain mass or in the case of mixtures, creating challenges in species identification. Since their inception24, nanomechanical mass sensors have made tremendous progress toward protein characterization25. Such devices, which take the shape of cantilevers or beams with lateral dimensions in the range of hundreds of nanometers, can detect individual particles accreting onto their active surface through changes in vibration frequency. Importantly, as the inertial mass of a particle is determined directly from the frequency change, these devices are insensitive to charge states26. This realization prompted the development of new MS instrument designs devoid of ion guides, which no longer depend on electromagnetic fields to collect and transmit analytes (Fig. 2c). Such a nanomechanical resonator-based MS system has recently been shown to have the ability to characterize large protein assemblies such as individual viral capsids above 100 MDa in size27. Outside of proteomics, a resolution of 1 Da has been demonstrated with carbon nanotubes28. Moreover, recent reports suggest the possibility of determining other physical parameters such as the stiffness or shape of the analyte by monitoring multiple vibrational modes29,30. These previously inaccessible metrics may open new avenues to discriminate peptides, proteins and their complexes. Nonetheless, one of the challenges of the nanoresonator mass spectrometer lies in devising efficient ways to bring individual proteins onto the resonator’s active surface for mass sensing.
Ionization is commonly achieved by electrospray ionization of a solution containing the compound(s) of interest. The use of ever-smaller electrospray ion source apertures has led to substantial improvements in the sensitivity of MS31,32. Mass spectrometers with a nanopore ion source have been developed for the purpose of sequencing single proteins33 (Fig. 2d). A nanopore electrospray can potentially deliver individual amino acid ions directly into a high-vacuum gas phase, where the ions can be efficiently detected by their mass-to-charge ratios. This opens a path to sequencing peptides one amino acid at a time. The concept makes use of nanopores to guide a protein into a linear configuration so that its monomers can be delivered into the mass spectrometer sequentially34. Individual amino acids must be cleaved from the protein molecule as it transits the nanopore, which could potentially be accomplished with photodissociation35 or chemical digestion methods. The 100-MHz bandwidth of the channeltron single-ion detectors used in this setup is also sufficient to resolve the arrival order of the ions. The high mass resolution makes this technique promising for identifying post-translational modifications, which change the masses of particular amino acids by predictable amounts. One challenge on the path for this technology will be achieving high throughput, which might require a strategy for parallelizing mass analysis.
Tunneling conductance measurements.
The appearance of the scanning tunneling microscope in the 1980s introduced a new way to analyze molecules. Small organic molecules can be transiently trapped between two metal electrodes with sub-nanometer separation, with the tunneling currents between the electrodes reporting on the molecular signature of the analyte. Recently, several technical advances have been made to move toward single-molecule amino acid and protein analysis. Extracting insightful information from electron tunneling is complicated by the noise resulting from water and contaminants reaching the electrode surfaces. To overcome this problem, recognition tunneling has been developed in which the electrodes are covalently modified with adaptor molecules that form transient but well-defined links to the target molecule36. The rapidly fluctuating tunnel current signals are processed using machine learning algorithms, which makes it possible to distinguish individual amino acids and small peptides37. Moreover, smaller electrode gaps have been introduced to obtain distinct signals from different amino acids and post-translational modifications38. Further development of the technology will depend on a reliable source of tunnel junctions with a defined gap to replace the cumbersome scanning tunneling microscopy, but it is clear that both the sequence and post-translational modifications of small peptides can be determined37. Currently, tunneling conductance is a proof-of-concept technology for fully sequencing short peptides that could one day be used for the analysis of protein digests and expanded to analysis of post-translational modifications (Fig. 1).
Recently, it was discovered that electrical charges can be transmitted through a protein if the electrodes are bridged by a protein via formation of chemical bonds or ligand binding39. Specifically, changes in protein conformation upon nucleotide addition could be followed in real time from the direct currents passing through a DNA polymerase40. Although the observation was preliminary, the electronic signatures were distinctive when the polymerase was associated with different DNA sequences, enabling a new approach to label-free single-molecule DNA sequencing. A similar approach could potentially be used for protein sequencing with enzymes such as proteases or glycopeptidases that process substrates sequentially.
DNA nanotechnologies for protein sequencing
DNA nanotechnologies, in which a large number of sequences with prescribed pairing interactions and dynamic properties can be custom designed, have facilitated developments in fields ranging from synthetic biology to diagnostics and drug delivery41. For example, programmable transient binding between short DNA strands is central to the super-resolution technique of DNA-based point accumulation for imaging in nanoscale topography (DNA-PAINT)42–44 (Box 3). Here we describe the application of DNA-PAINT and DNA-based local and global pairwise distance measurement methods for single-molecule protein detection and identification.
Box 3 |. DNA-PAINT.
DNA-PAINT relies on the transient binding of dye-labeled DNA strands (imagers) to their complementary target sequence (docking site) attached to a molecule of interest. The transient binding of imager strands is detected as ‘blinking’ in an intensity versus time trace. DNA-PAINT has a few unique advantages. First, the blinking kinetics (on and off rates) can be tuned over a wide range, by altering the length and sequence of the imager strands or buffer conditions, making the method compatible with different sample conditions. Second, repetitive binding with different imager strands makes the target ‘non-bleachable’, allowing for the collection of a large number of high-quality and high-precision blinking events and for high-sensitivity imaging on single-molecule targets with discrete molecular resolution (<5 nm). Finally, in combination with orthogonal sequence labels, DNA-PAINT can be multiplexed by imaging with up to dozens of molecular species (Exchange-PAINT).
Fingerprinting via DNA-PAINT.
DNA-PAINT uses repetitive binding between designed docking and imager DNA strands to allow for imaging with molecular-level resolution (Box 3). This method provides a promising way to fingerprint proteins on the level of single molecules. A simple approach to characterize proteins could involve amino acid counting using quantitative DNA-PAINT (qPAINT)44. In this technique, the total blinking rate of a region of interest is measured, which linearly reflects the number of molecular targets in the region. It has been proposed that high-efficiency DNA labeling of specific amino acids (Fig. 3a) followed by qPAINT could lead to single-molecule protein fingerprinting of intact proteins (Fig. 3b)45.
Fig. 3 |. DNA-facilitated protein sequencing.
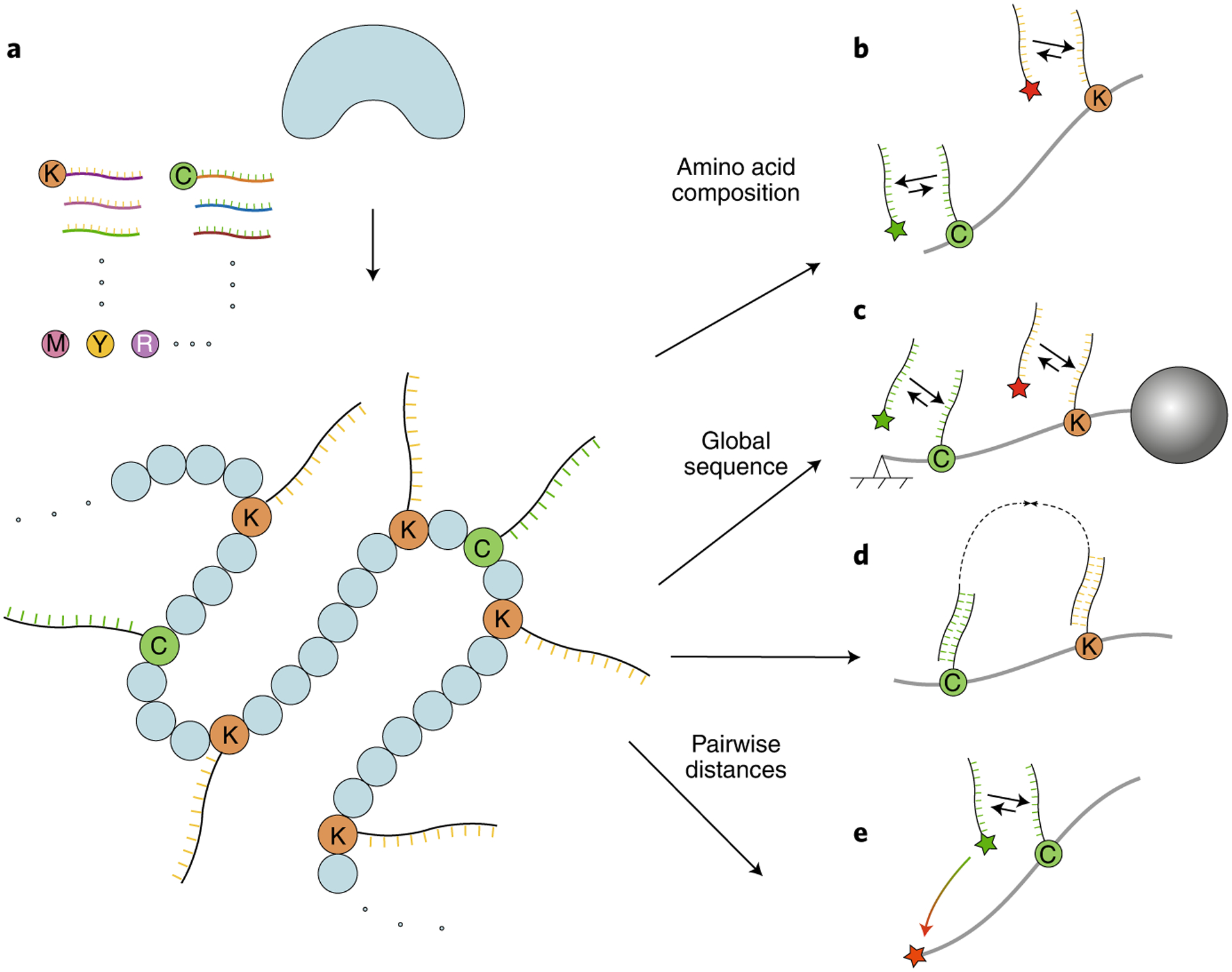
a, Schematic of specific amino acid labeling on a denatured protein with DNA strands. Each DNA strand contains a barcode for the specific amino acid and (optionally) a UMI. b–e, Various readout strategies of DNA-labeled samples for protein identification. b, Protein kinetic fingerprinting using qPAINT. c, Protein linear barcoding using molecular-resolution DNA-PAINT. d, DNA proximity recording. e, Protein structural fingerprinting using FRET-X.
The recent development of DNA-PAINT has allowed discrete molecular imaging (DMI) of individual molecular targets with spatial resolution below 5 nm43. Therefore, protein identification by fingerprinting of amino acids along an extended protein backbone is a possibility. DMI was achieved by combining a systematic analysis and optimization of the DNA-PAINT super-resolution workflow and a high-accuracy (<1 nm) drift correction method. To effectively unfold and extend the protein backbone, N- and C-terminal-specific modifications should be used to attach surface and microbead anchors. The protein can then be subjected to mechanical or electromagnetic extension force (Fig. 3c). Proposals to combine protein extension methods with high-resolution DMI45 indicate that, with lysine labeling alone and 5-nm effective imaging resolution, more than 50% of the human proteome could be uniquely identified, even with up to 20% amino acid imaging error. Labeling lysine and cysteine would allow coverage of the proteome to increase to more than 75%.
Protein fingerprinting using DNA-PAINT single-molecule imaging combines the ultra-high imaging resolution and quantitative capacity of this technique and the inherent throughput of wide imaging-based methods. qPAINT can produce signals linearly (with <5% deviation), based on the amino acid composition of a particular protein. The proposed methods will be particularly useful for global proteomics analysis of complex protein mixtures and post-translational modification patterns as well as combinatorial analysis of PTM patterns at the single-molecule level.
DNA proximity recording.
An alternative method for DNA-based protein identification attaches DNA probes to specific amino acids on a protein and uses enzymatic DNA amplification between nearby probes to generate DNA ‘records’ that vary in length and abundance according to pairwise distances within a protein46, as exemplified by autocycling proximity recording (APR)47 (Fig. 3d). The distribution of the lengths of these molecular records is then analyzed to decode the pairwise distance between two DNA tags. It is possible to use unique molecular identifier (UMI) barcoding and repetitive enzymatic recording, such that each lysine and cysteine residue can be studied and used to construct a pairwise distance map, allowing for single-molecule protein identification48,49. DNA proximity recording takes advantage of high-throughput next-generation DNA sequencing methods for efficient protein fingerprinting analysis and will be useful for the analysis of both purified proteins and complex protein mixtures.
Protein fingerprinting using FRET.
A different approach that allows for global pairwise distance measurements combines DNA technology with single-molecule Förster resonance energy transfer (FRET)50. The current state of the art for single-molecule FRET analysis allows only one or two FRET pairs to be probed at a time51, and new high-resolution FRET using transient binding between DNA tags allows for one FRET pair to be probed at a time while many probes are collectively present on a single protein50. Similarly to the approaches described above, specific amino acids (for example, lysine, cysteine, etc.) required for fingerprinting have to be labeled with a set of different DNA docking strands. Furthermore, a fixed position on the protein (either the N or C terminus) is labeled with the acceptor fluorophore. Only a single FRET pair forms at a time using DNA strands that are complementary to only a single docking strand. Measurements are then repeated to probe the remaining docking strands and thus the amino acids. The output of this approach is a FRET histogram containing information on the position (referred to as FRET fingerprint) of each detected amino acid relative to one of the reference points. This information is compared to a database consisting of predicted FRET fingerprints, allowing for identification of the protein species (Fig. 3e). The proposed high-resolution FRET approach (named FRET using DNA eXchange, or FRET X) benefits from the immobilization of protein molecules, allowing users to probe each protein multiple times to obtain fingerprints with high resolution. FRET X is a particularly promising tool for targeted proteomics or proteoform analysis as it is able to distinguish small structural changes.
Biological and solid-state nanopores
Since its first demonstration as a single-biomolecule sensor52, nanopore sensing has dramatically advanced, ultimately achieving the goal of single-molecule DNA sequencing53. Many of the nanopore sequencing applications thus far have materialized using an ultra-small device54 that features vast arrays of biological nanopores, each coupled to its own current amplifier, allowing readout of hundreds of DNA strands simultaneously. Owing primarily to the long read lengths and portability capabilities of this technology, nanopore-based DNA and direct RNA sequencing have become key players in the sequencing field. Nanopore sensing involves drawing biomolecules through the nanopore in a single-file manner. During their passage, the analytes partially block the flow of the ionic current through the pore, leading to time-dependent and sequence-specific electrical signals. Over the past two decades, a variety of synthetic nanopore biosensors have shown substantial progress and are currently used in diverse applications beyond sequencing, including the detection of epigenetic variations and ultra-sensitive detection of mRNA expression55, among many others.
Just like gel electrophoresis, nanopores may serve as a generic tool to analyze biomolecules. Therefore, as nanopore-based DNA sequencing continues to advance, this technique is poised to extend to proteins, metabolites and other analytes. But despite the remarkable advances in DNA and RNA sequencing, nanopore-based protein sensing is still in its infancy, facing challenges unique to proteins and proteomics. In particular, proteins span a large range of sizes and have a stable three-dimensional folded structure. In contrast to nucleic acids, the backbones of peptides are not naturally charged, complicating the possibility of single-file electrokinetic threading into nanopores. In addition, proteins are composed of combinations of 20 different amino acids instead of 4 nucleobases, further complicating the task of relating the ionic current signals to the amino acid sequence.
While substantial progress in nanopore-based protein sensing has already been made, the development of full-protein sequencers and single-protein identification based on nanopores remains a topic of intense focus. Here we elaborate on three of the principal directions in this field (Fig. 4): (1) single-file threading and direct sensing of the sequence of a polypeptide’s amino acids, analogous to the nanopore DNA sequencing principle—in this approach, translocation of either full-length proteins or shorter polypeptide digests of proteins may be targeted; (2) protein identification methods based on sensing unique fingerprints in linearized proteins, without de novo amino acid sequencing; and (3) identification of folded proteins on the basis of specific patterns in their current blockade while in the nanopore. In the following sections, we provide short overviews of the current state of these approaches and refer to additional methods.
Fig. 4 |. Three strategies of nanopore-based protein sequencing and sensing.
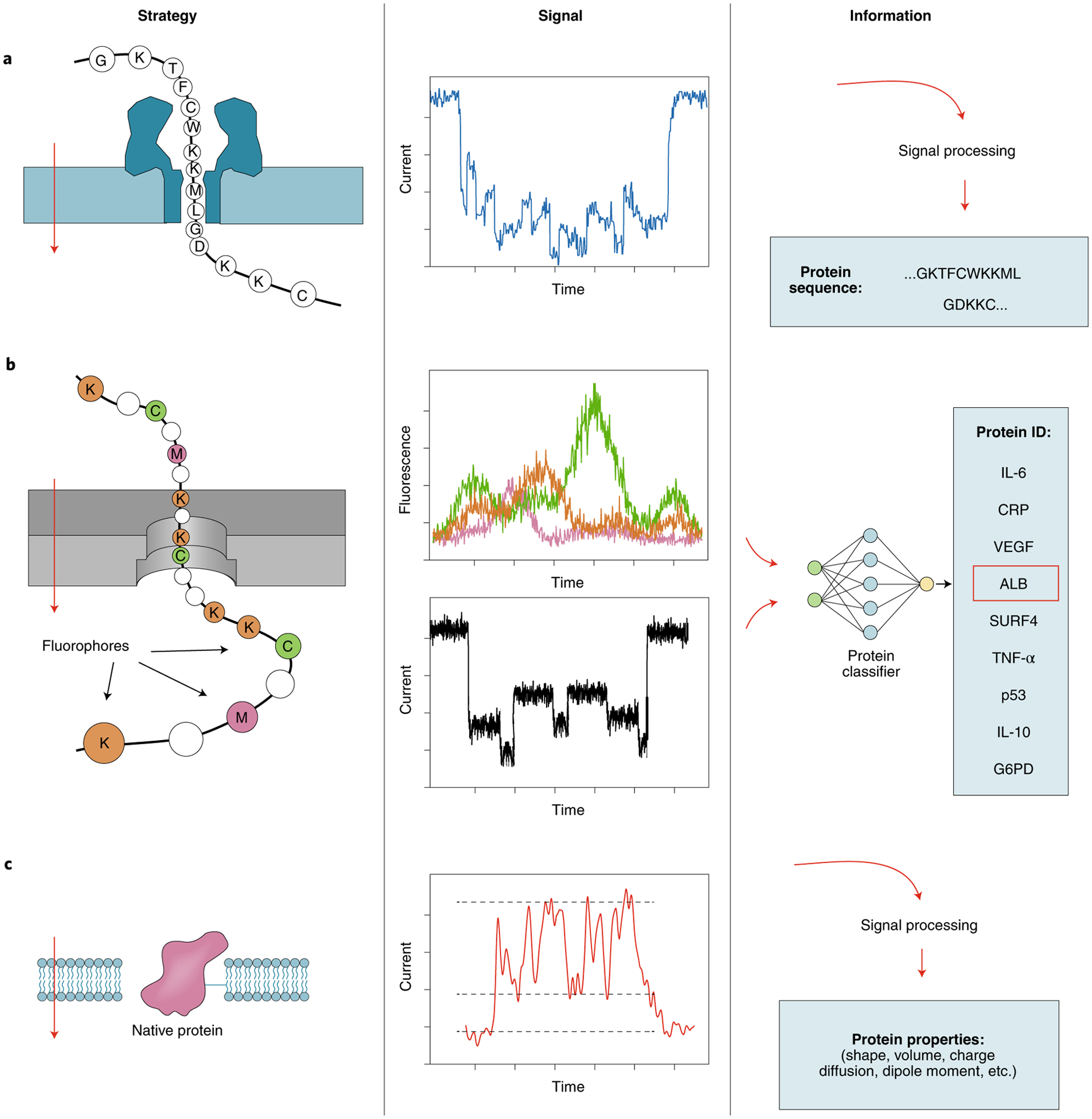
In all cases, a voltage bias is applied across an insulating membrane (left panels) and the analytes translocate through the nanopore from top to bottom (red arrows). a, Reading unlabeled proteins or peptides using a biological nanopore. b, Identification of whole proteins or peptides by fingerprinting with deep learning algorithms. Residue-specific fluorescent labels (for example, at lysine, cysteine and methionine) can be used to fingerprint proteins and peptides alongside electrical current sensing. c, Identification of folded proteins using lipid tethering. Other possible tethers include DNA carriers, DNA origami anchors and plasmonic trapping.
Reading the amino acid sequence of linearized peptides.
In this proposed approach, a single protein or peptide is linearized and threaded through a nanopore and the resulting ionic current is interpreted to yield an amino acid sequence (Fig. 4a). All-atom molecular dynamics simulations using the α-hemolysin pores have demonstrated a global correlation between the volume of an amino acid and the current blockade in homopolymers56. Computationally efficient predictions using coarse-grained models have also performed well in comparison to all-atom molecular dynamics simulations for both solid-state and biological pores57.
Discrimination among peptides differing by one amino acid (alanine to glutamate substitution) has been demonstrated using engineered fragaceatoxin C (FraC) nanopores58. Moreover, single-amino acid differences within short polyarginine peptides were resolved with superb resolution, using the aerolysin protein pore in its wild-type conformation59. Combining molecular dynamics simulations and single-channel experiments, Cao et al. rationally introduced specific point mutations in aerolysin to fine-tune the charge and diameter of the pore, which enhanced its sensitivity and selectivity as showcased experimentally using DNA and peptides60. Notably, protein pore sensors were used for the analysis of bodily fluids (blood, sweat, etc.), indicating a substantial potential for applications in diagnostics61. As an alternative to nanopore sequencing of intact polypeptide chains, smaller digested fragments can also be analyzed, allowing for detection of minute differences in amino acid composition62. Even post-translational modifications can be detected, including individual phosphorylation and glycosylation modifications, using the FraC protein pore63.
An essential step in the development of nanopore-based DNA sequencing came with the application of an enzymatic stepping motor (for example, a helicase) that facilitated nucleotide-by-nucleotide progression of the DNA through the nanopore. A similar system is being pursued for single-molecule protein sequencing: molecular motors of the type II secretion system (SecY)64 and the AAA family (ClpX)65 are known to unfold and pull protein substrates through pores in an ATP-dependent manner. Nivala et al.66,67 used ClpXP (or ClpX alone) to unfold and translocate a multidomain fusion protein through the α-hemolysin pore using energy derived from ATP hydrolysis. In this approach, the motor is at the exit of the nanopore and the step size of translocation is therefore dependent on stable structural motifs that resist translocation, rather than being controlled by the enzyme. This approach is currently being expanded by several groups who conjugated ClpXP covalently to α-hemolysin at the entrance of the nanopore to form a combination sensor and substrate delivery machine. The Maglia laboratory genetically introduced a nanopore directly into an archaeal proteasome and found that assisted transport across the nanopore was not influenced by the unfolding of the protein. These nanoscale constructs would also allow a ‘chop-and-drop’ approach in which single proteins are recognized by their pattern of peptide fragments as they are sequentially cleaved by the peptidase above the nanopore68. Knyazev et al. introduced a protein-secreting ATPase as an additional natural choice for a potential peptide-translocating motor69,70. Other proteins have the potential to control protein translocation through nanopores, beyond secretases and unfoldases, including chaperones (Hsp70), via processes resembling protein translocation into the mitochondrial matrix71. Recently, Rodriguez-Larrea’s group has discussed how protein refolding at the entry and exit compartments can oppose and promote protein translocation, respectively72,73, and the use of deep learning networks to analyze raw ionic current signals for accurate classification of single point mutations in a translocating protein74. In addition, Cardozo et al. built a library of approximately 20 proteins that are orthogonally barcoded with an intrinsic peptide sequence and successfully read them with nanopore sensors75.
Fingerprinting linearized proteins.
Accurate quantification of different protein species in the proteome with single-molecule resolution would in itself be an achievement of great importance. This can be realized through single-molecule fingerprinting, that is, through the identification of individual protein molecules on the basis of prior knowledge of their amino acid sequences or specific signal patterns, recognized by machine learning8,76,77 (Fig. 4b). To this end, several nanopore approaches have been pursued: Restrepo-Pérez et al.78 established a fingerprinting approach using six chemical tags, which were placed on a dipolar peptide79. Additionally, Wang et al. reported the ability to distinguish individual lysine and cysteine residues in short polypeptides through specific coupling to fluorescent tags while using a solid-state nanopore with low fluorescence background80. In all these approaches, separating the proteins by mass before single-molecule sensing may have greatly facilitated the identification of proteins in complex samples containing many different proteins81.
Nanopore protein fingerprinting can make extensive use of advanced deep learning artificial intelligence (AI) strategies to identify patterns in noisy signals. Ohayon et al. recently showed computationally that more than 95% of the proteins in the human proteome can be identified with high confidence when labeling three amino acids (lysine, cysteine and methionine) and threading them linearly through a solid-state nanopore77. These simulations predict that even partial labeling of proteins would be sufficient to achieve a high degree of accurate whole-proteome identification, owing to the ability of AI functions to correctly recognize partial protein patterns. This identification method involves the incorporation of sub-wavelength light localization in the proximity of the nanopore using plasmonic nanostructures82. The work in this field benefits from recent advances in nanofabrication and nanopatterning technologies allowing for the formation of complex metallic nanostructures to localize fluorescence through plasmon resonance83.
Characterization and identification of folded proteins.
Thus far, nanopores have been successfully used to detect specific sets of folded proteins and protein oligomers84 (Fig. 4c) such as large globular proteins, various cytokines and even low-molecular-weight proteins such as ubiquitin. Holding proteins in their folded state inside the nanopore for sufficiently long periods of time is a key requirement. Early studies have shown that globular proteins in the molecular weight range of roughly 5 to 50 kDa can only be detected for a few tens of microseconds or less85, which is too short for characterization. Several approaches to overcome this challenge have been devised. A lipid bilayer coating of a solid-state nanopore can be used to tether the proteins for extended periods of time86. Lipid-tethered proteins86 and, more recently, freely diffusing proteins (using a higher-bandwidth sensing system)87 have been characterized with respect to their size, shape, charge, dipole and rotational diffusion coefficient88. Various strategies are being pursued to ‘trap’ proteins in a nanopore. One such strategy is to use plasmonics to hold a protein in a nanopore for seconds or even minutes89,90. More recently, single proteins have been held at the nanopore’s most sensitive region for minutes to hours using the nanopore electro-osmotic trap (NEOtrap), which exploits strong electro-osmotic water flows created in situ by a charged, permeable object, such as a DNA origami structure91. Another approach for slowing down the translocation of proteins involves the use of nanopores smaller than those in earlier studies to increase the hydrodynamic drag, thus resulting in longer translocation dwell times that are easier to measure92,93. In addition, high-bandwidth measurements can resolve differential size and conformational flexibility between and within folded proteins92–96. Biological nanopores with a diameter of 5.5 or 10 nm97 can also be used to measure folded proteins, including protein conformations98 and post-translational modifications99 such as ubiquitination. Lastly, Aramesh et al.100 used a combination of atomic-force microscopy and nanopore technology to carry out the first steps of nanopore sensing directly inside cells. Altogether, the detection, identification and sequencing of proteins using single-nanopore approaches has become a highly active, thriving research field, with great potential to revolutionize proteomics, medical diagnostics and also the fundamental biosciences.
Chemistry for next-generation proteomics technologies
Single-molecule protein fingerprinting has underlined the need for innovative approaches to attach various functional groups to peptides, such as fluorescent moieties. A high degree of chemical specificity is required to avoid downstream misidentification of amino acids, which could lead to sequencing errors. Chemists are making headway on a suite of selective and high-yield methods for labeling specific amino acid side chains, amino acid termini and post-translational modifications with minimal cross-reactivity (Box 4).
Box 4 |. Chemistry concepts in protein sequencing.
Labeling efficiency and stability.
The challenges in labeling efficiency and stability are well characterized in fluorosequencing, which uses harsh conditions (including neat trifluoroacetic acid) that can lead to reversal of maleimide labeling of cysteine residues. To circumvent this reversal, fluorosequencing instead uses iodoacetamide chemistry, which generates a more stable bond. Another point of complexity is that full conversion is dictated by the solvent accessibility of the targeted amino acid side chains, which can influence labeling efficiency. However, modeling suggests that labeling efficiencies and stabilities substantially below 100% can be compensated for computationally, at least to some degree, during the reference database matching process8.
Labeling side chains.
The most widely accessible labels are those that target lysine using NHS esters and cysteine using maleimide and iodoacetamide reactive groups. Additionally, the phenol ring of tyrosine can be labeled using benzyl diazo groups122; however, the attachment of fluorescent molecules generally requires a two-step labeling procedure owing to the cross-reactivity with fluorescent molecules. Another robust bioconjugation method to selectively target tyrosine side chains is an ene-like reaction with cyclic diazodicaboxamides in aqueous buffer123. Carboxylic acids have also been labeled on peptides, but, owing to the similar reactivities of aspartate, glutamate and the C terminus, this has primarily been used on synthetic peptides. The method makes use of a standard technique (EDC coupling) for binding amines covalently to carboxylic acids, forming an amide bond. In a recently reported promising bioconjugation approach, light-activated 2,5-disubstituted tetrazoles have been shown to be able to convert glutamate and aspartate residues with high yield124. Finally, tryptophan can be labeled at the C2 position using sulfenyl chlorides. However, this comes with the limitations that the reaction is extremely water sensitive and the reactive group must be made in situ101. There are also promising new methods that allow for chemical modification of other amino acids. Methionine, for example, can either be elegantly labeled with hypervalent iodine reagents125 or by the use of urea-derived oxaziridines126,127. Recently, a histidine-selective conjugation methodology was reported where thiophosphorodichloridates selectively form a covalent bond with the histidine residues in proteins128.
C-terminal labeling.
Labeling of the C terminus is a challenge in that the C terminus must be differentiated from aspartate and glutamate, which carry the same functionality. A photoredox reaction on the C terminus of peptides and proteins entailing decarboxylation of the C-terminal carboxylic acid followed by an alkylation step with a Michael acceptor has recently been reported129. Because of their higher oxidation potential, the carboxylates of internal amino acid chains are less prone to this modification, making the method highly site selective. This technique has been applied for a variety of peptide substrates as well as for C-terminus-specific alkylation of human insulin chain A.
N-terminal labeling.
Several methods exist for modifying the N terminus130. Classic approaches such as reductive amination with aldehydes or acylation with NHS esters, which rely on pH control to increase selectivity, are not sufficiently specific. Other strategies involve the side chain of the N-terminal amino acid. Native chemical ligation131 or condensation reactions with aldehydes132 could be used to label N-terminal cysteine, serine, threonine or tryptophan residues. Furthermore, oxidizing N-terminal serine or threonine residues to their corresponding aldehydes allows oxime conjugation with hydrazides or hydroxylamines133. A more general methodology has also emerged where the N-terminal amine condenses with 2-pyridinecarboxaldehyde, forming an imine structure that further reacts via cyclization with the nearby amide nitrogen of the second amino acid to form a stable imidazolidinone product134. This reaction has recently been shown to be useful for single-molecule peptide sequencing as a method for the immobilization of peptides onto a solid-phase resin, multiple chemical derivatization steps without purification and subsequent traceless release before fluorosequencing10.
Post-translational modifications.
As an example of elimination replacement chemistries, phospho-serine and phospho-threonine residues can be labeled by β-elimination followed by Michael addition (BEMA). In MS-based phosphoproteomics, this is used to introduce an additional trypsin cleavage site at the phosphorylated amino acid135, while at the single-molecule level it can be used to site specifically attach a fluorescent label. Such an approach has been established for the Edman degradation described above9.
Protein glycosylation can be complex, featuring many different types of monomeric units bound in possibly branching polymer structures. Full structural characterization often requires derivatization and is done on glycans that are released from the protein. Therefore, schemes for understanding site-specific and simple glycosylation events should be the current focus. N-glycan-anchoring asparagine residues can be converted to aspartate by glycan removal with PNGase F for practically all protein sequencing approaches, reducing complexity in the detection of this modification. Another possibility to introduce site-selective labels is the incorporation of azide-tagged glycans, achieved by adding modified carbohydrates to the cell medium136. In other detection schemes, the location of a modification could also be inferred using glycan-specific reporter molecules such as lectins, engineered proteins or aptamers137.
Chemistry for protein sequencing.
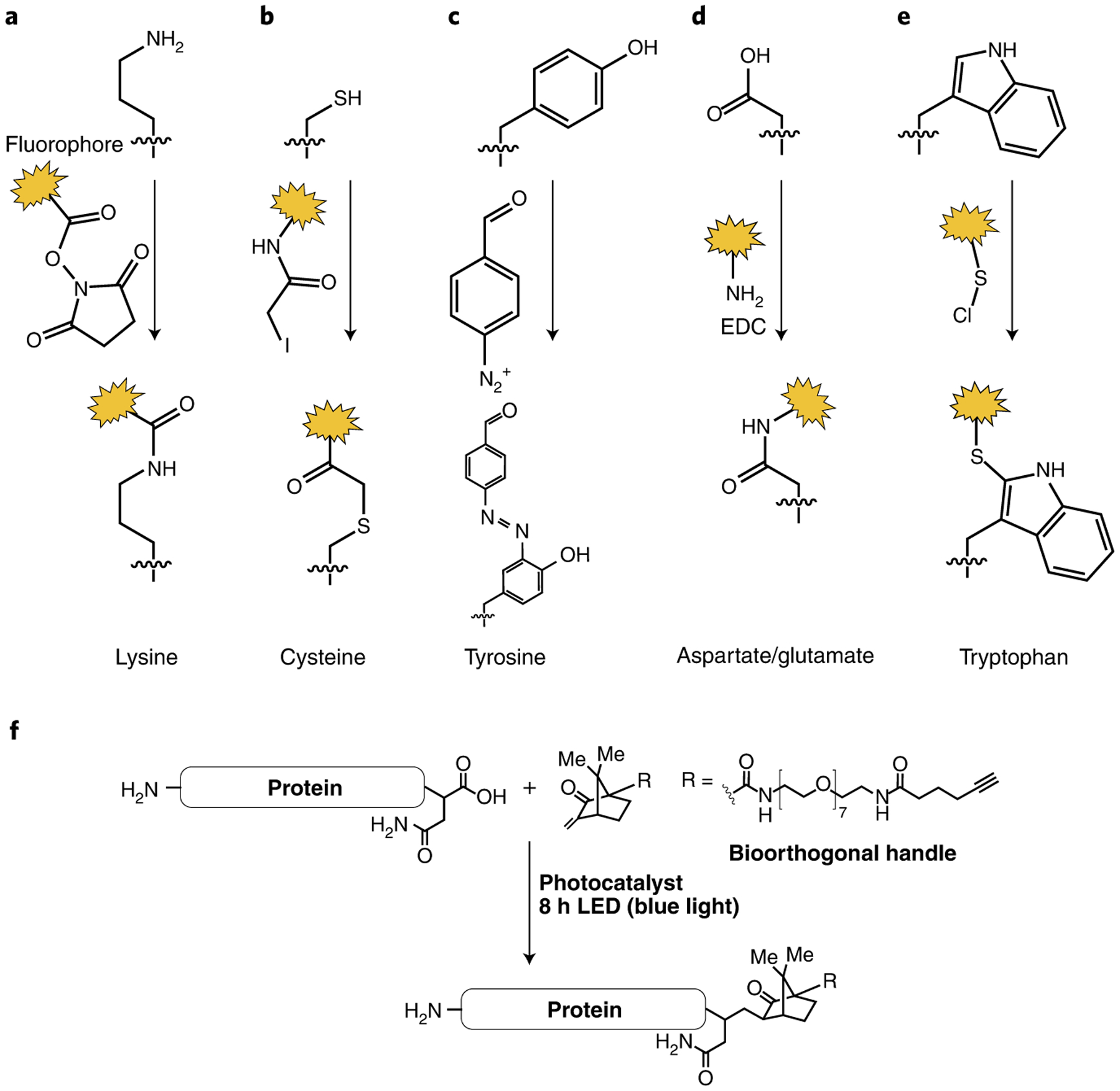
a, Lysine labeling with NHS esters. b, Cysteine labeling with iodoacetamide reactive groups. c, Strategies for labeling the phenol ring of tyrosine. d, Aspartate/glutamate labeling. e, Tryptophan labeling with sulfenyl chlorides. f, C-terminal derivatization through monoalkylation of the insulin A chain (yield 41%).
Labeling stability and efficiency are paramount to the success of sequencing technologies but are also a challenge. First, modification of most or all individual residues of one amino acid type is desired for explicit identification of a peptide sequence, which requires selective and highly efficient reactions. Second, error-free sequence prediction requires multiple chemical labels, but the stability of the chemical labels has been an issue in some sequencing techniques. Such issues have been best characterized for fluorosequencing (Box 4).
For many of the sequencing techniques, amino acids must be labeled with a chemical tag to allow for differentiation between them. While it is theoretically possible to obtain broad coverage of the proteome with labeling for a minimal set of amino acids, specific identification of peptides and broader sequence coverage require a larger suite of labels. Overall, there are 12 distinct side chain types in peptides, ranging from those for highly reactive amino acids such as lysine and cysteine to functional groups that are more challenging to modify, such as amides (glutamine and asparagine) and alkanes (alanine, glycine, isoleucine, leucine, proline and valine). There are a large number of methods to label amino acids; however, some chemistries do not provide sufficiently stable bonds for some single-molecule sequencing approaches. Thus far, labeling for only eight amino acids (lysine, cysteine, glutamate, aspartate, tyrosine, tryptophan, histidine and arginine) has been shown to be stable, selective and reactive enough for the single-molecule fluorosequencing approach9,101. Research is ongoing to test a wide variety of other labeling conditions to cover all of the proteinogenic amino acids (Box 4).
Chemical modification of protein termini is highly desired for several sequencing techniques such as the fluorosequencing, nanopore and DNA-PAINT approaches where end labeling or ligation is required (Figs. 2–4). The terminus provides an attachment point for surface immobilization and can offer a simple way to remove excess chemical reagents during procedures that require multiple labeling steps. Two terminus-specific methods have shown great promise for single-molecule sequencing, C-terminal labeling using decarboxylative alkylation and modification of the N terminus with 2-pyridinecarboxaldehyde (Box 4).
The long-term goal of characterizing proteoforms requires methods to detect and differentiate post-translational modifications. Such modifications can be recognized by MS through the mass shifts they cause on a protein, peptide and their fragments102,103, and databases of the expected mass shifts such as Unimod are used to support identification104. However, these databases show that there can be substantial overlap between post-translational modifications of the same or similar mass, suggesting that orthogonal methods are needed. Single-molecule protein sequencing methods rely on either site-specific labeling or elimination and replacement chemistries (Box 4).
Discussion: a spectrum of opportunities
An emerging landscape of single-molecule protein sequencing and fingerprinting technologies is unfolding with the promise of resolving the full proteome of single cells with single-protein resolution, opening up unprecedented opportunities in basic science and in medical diagnostics. For example, resolving the cellular and spatial heterogeneity in tissue proteomes with integration of other layers of the central dogma could open new research avenues from embryonic development to cancer research. Diagnostics could benefit from the ultimate single-molecule resolution by resolving very low amounts of protein in bodily samples. The detection of rare proteins with copy numbers as low as one or a few may uncover new molecular regulatory networks within cells. Some of the emerging technologies described here are still at early proof-of-concept stages in development, whereas others, including sequencing by Edman degradation and nanopore sequencing technologies, have already attracted industry funding. Additional single-molecule approaches are also promoted by commercial entities but are outside the scope of this Perspective.
A real-world application of a technology that is not MS or antibody based for whole-proteome characterization is yet to be achieved. Meanwhile, MS will continue to improve in its capacity to support single-ion detection22 and ultimately single-cell proteomics105. Similarly, antibody-based methods such as immunoassays that rely on specific antigen–antibody interactions have served as the standard methods for protein identification and quantification for the last few decades. Specifically, antibody-based methods have enabled multiplexed protein analysis with improvements of several orders of magnitude in sensitivity over conventional immunoassays. A notable example is the Single Molecule Array technology (Simoa)106 by Quanterix, a digital immunoassay based on single-molecule counting used for the analysis of minute biological samples with up to sub-femtomolar sensitivity107. The coronavirus disease 2019 (COVID-19) pandemic has accelerated the development of high-throughput serological tests of clinical samples using Simoa108 based on ultra-small blood samples. These sensitive antibody-based methods will continue to have a main role in molecular diagnostics, in parallel with other single-molecule techniques that will permit comprehensive proteoform inference or differentiation.
The emerging landscape of alternative protein sequencing and fingerprinting technologies in Fig. 1 could one day help to sequence human proteoforms in a more complete way. High-throughput Edman degradation could pair with bottom–up MS strategies to alleviate current limitations on sequence coverage (Box 1). These bottom–up methods could benefit from nanopore sequencing and DNA fluorescence-based methods that aim for long-read sequencing and structural fingerprinting of whole proteins. Integration of both existing and emerging technologies promises to iteratively reveal an atlas of full-length proteoforms, which could itself assist these up-and-coming technologies to infer what cannot be directly measured in terms of protein primary sequence and structure.
An additional far-reaching goal for single-molecule proteomics lies in the analysis of protein–protein interactions. A map covering a wide range of proteoforms and their interactions is an unmet milestone needed to uncover protein networks in normal tissues and in disease. Bottom–up MS-based approaches, such as cross-linking109,110 and affinity purification, are implemented to identify physical111 and proximal112 interactions. However, these techniques present either biochemical or sample processing yield limitations, as a result of challenges such as over-representation of intra-protein cross-linking, loss of protein–protein interactions upon solubilization and limitations inherent to MS analysis, hindering single-cell interactome analysis. Currently, single-molecule analysis of protein–protein interactions has not reached mainstream proteomics, which is even more true for single-cell interactomics. Achieving these goals would be of great interest in accurately defining, for example, protein organization within highly dynamic membraneless organelles113, such as in resolving protein condensates and spatial and temporal organization at a single-organelle or single-cell scale, and would provide an unprecedented resolution for the organization of protein–protein interactions.
Challenges for next-generation protein sequencing.
Two grand challenges await technological innovations and need to be addressed to enable the high-throughput sequencing of complex protein mixtures. First, there is no method to amplify the copy number of proteins similar to the methods used for nucleic acids. New techniques focus on characterizing individual proteins. The aim is to sequence proteomes starting from a low number of cells or minute samples that often contain just a few or single copies of specific proteins. This presents a second problem: a single eukaryotic cell contains billions of proteins. While the presented methods may enable single-molecule protein identification, they must reach an extremely high sensing throughput to profile all proteins in the cell and permit whole-cell analysis on a reasonable timescale. These two seemingly contractionary requirements (single-protein molecule sensitivity and extremely high throughput) present one of the main challenges to the field, and striking an optimal balance between them will be key for all the technologies discussed. Of the orthogonal methods presented, nanopores, fluorosequencing and protein linear barcoding using DNA-PAINT, to name a few, stand a chance to eventually measure billions of proteins within a few hours.
Emerging technologies will be evaluated in terms of their sensitivity, proteome coverage (fraction of whole proteins in the sample covered), sequence coverage (average fraction of protein sequences covered), peptide read length (mean number of amino acids in a single read), accuracy (error in calling an amino acid or in identifying a whole protein), cost and throughput. In this regard, additional research and validation will be required to demonstrate the benefits of these orthogonal technologies. The formation of a dedicated global academic/scientific community in single-protein sequencing may catalyze further development and implementation of these technologies for more widespread use. Multidisciplinary meetings that bring together experts in chemistry, physics, engineering, computer sciences and other relevant areas of expertise (for example, pathologists and clinicians) with a clear vision of the most relevant problems and unmet needs will need to be embraced.
Acknowledgements
We thank all the presenting delegates of the 2019 Single-Molecule Protein Sequencing conference (Jerusalem). We thank the PL-Grid and CI-TASK Infrastructure, Poland, for providing their hardware and software resources. S.S. acknowledges Postdoc Mobility fellowship no. P400PB 180889 from the Swiss National Science Foundation. E.M.M. and E.V.A. acknowledge funding from the NIH (R35 GM122480 and R01 DK110520 to E.M.M.), Welch Foundation (F1515 to E.M.M. and F-0046 to E.V.A.), Army Research Office grant W911NF-12-1-0390 and Erisyon. E.M.M. and E.V.A. are co-founders and shareholders of Erisyon. R.T.K. acknowledges funding from NIGMS (R01 GM138931). P.Y. acknowledges funding from an NIH Director’s New Innovator Award (1DP2OD007292), an NIH Transformative Research Award (1R01EB018659), an NIH Pioneer Award (1DP1GM133052), and the Molecular Robotics Initiative fund at the Wyss Institute for Biologically Inspired Engineering. M.D. acknowledges funding from a Systems Biology Department Fellowship from Harvard Medical School and a Technology Development Fellowship from Wyss Institute for Biologically Inspired Engineering. C.C. acknowledges the Peter and Traudl Engelhorn Foundation. C.D. acknowledges the ERC Advanced Grant Looping DNA (no. 883684) and the NWO programs NanoFront and Basyc. E.M.M., E.V.A. and C.J.H. are co-inventors on patents relevant to this work. S.O. acknowledges the support of the Azrieli fellowship foundation. N.L.K. acknowledges funding from the Paul G. Allen Frontiers Program (11715), the NIH HuBMAP program (UH3 CA246635) and NIGMS (P41 GM108569). J.P.M. and Z.K. acknowledge internal funding from NIST and are co-inventors on patents relevant to this work. M. Wanunu acknowledges funding from the NIH (HG009186). K.S. and A.A. acknowledge funding from the NSF (PHY-1430124). C.J., C.D. and R.E. acknowledge funding from NWO-I (SMPS). C.J. acknowledges funding from HFSP (RGP0026/2019). A.P. acknowledges Bekker fellowship no. PPN/BEK/2018/1/00296 from the Polish National Agency for Academic Exchange. C.M. and S.H. acknowledge funding from the European Research Council (ERC ‘Enlightened’, GA 616251) and the CEA Transverse Program ‘Instrumentation and Detection’ (PTC-ID VIRIONEMS). Support from the Proteomics French Infrastructure (PROFI) is also gratefully acknowledged. G.D. acknowledges funding from FNR (C17/BM/11642138). M.M. acknowledges funding from the Adolphe Merkle Foundation, the Michael J. Fox Foundation for Parkinson’s Research (grant 17924) and the Swiss National Science Foundation (grant no. 200021-169304). A.M. acknowledges funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement no. 833399-ERC NanoProt-ID and ISF award 3485/19. M.C. acknowledges computational resources from CINECA (NATWE project) and the Swiss National Super-Computing Centre (CSCS), under projects sm11 and s865. E.C. acknowledges funding from I-Site Lille, Région Hauts-de-France, and the European Union’s Horizon 2020 Marie Skłodowska-Curie no. 843052. The study was supported by the project ‘International Centre for Cancer Vaccine Science’ that is carried out within the International Agendas Programme of the Foundation for Polish Science co-financed by the European Union under the European Regional Development Fund. D.G. thanks Genome Canada and Genome British Columbia for financial support for Genomics Technology Platforms (GTP) funding for operations and technology development (264PRO). We thank V. Globyte for critical reading.
Footnotes
Competing interests
S.H. and C.M. are co-inventors on the patent application EP14158255. E.M.M. and E.V.A. are co-inventors on patent 9625469. D.S. is sponsored by Oxford Nanopore for his work on nanotip MS. E.M.M. and E.V.A. are co-founders and shareholders of Erisyon. B.K. and M. Wilhelm are founders and shareholders of OmicScouts and MSAID. They have no operational role in either company. M.D. and P.Y. are co-inventors on US patent 10006917. P.Y. is an inventor on US patent 10697974 and provisional patent and patent applications on various aspects of DNA nanotechnology–based protein sequencing methods described in this article. P.Y. is a co-founder, director and consultant of Ultivue Inc. and Spear Bio Inc. All remaining authors declare no competing interests. Some authors may be bound by confidentiality agreements that prevent them from disclosing their competing interests in this work; the corresponding authors are not aware of such cases.
References
- 1.Breuza L et al. The UniProtKB guide to the human proteome. Database 2016, bav120 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith LM et al. Proteoform: a single term describing protein complexity. Nat. Methods 10, 186–187 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seattle Times Business Staff. Seattle biotech startup Nautilus to get $350 million, stock listing in blank-check deal. The Seattle Times https://www.seattletimes.com/business/seattle-biotech-startup-nautilus-to-get-350-million-stock-listing-in-blank-check-deal/ (8 February 2021).
- 4.Reuters Staff. Protein sequencing firm Quantum-Si to go public via $1.46 billion SPAC merger. Reuters https://www.reuters.com/article/us-quantum-si-m-a-highcape-capital-idUSKBN2AI1HT (18 February 2021).
- 5.Cohen L & Walt DR Single-molecule arrays for protein and nucleic acid analysis. Annu. Rev. Anal. Chem 10, 345–363 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Aggarwal V & Ha T Single-molecule fluorescence microscopy of native macromolecular complexes. Curr. Opin. Struct. Biol 41, 225–232 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Edman P A method for the determination of the amino acid sequence in peptides. Arch. Biochem 22, 475–476 (1949). [PubMed] [Google Scholar]
- 8.Swaminathan J, Boulgakov AA & Marcotte EM A theoretical justification for single molecule peptide sequencing. PLoS Comput. Biol 11, 1076–1082 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swaminathan J et al. Highly parallel single-molecule identification of proteins in zeptomole-scale mixtures. Nat. Biotechnol 36, 1076–1082 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Howard CJ et al. Solid-phase peptide capture and release for bulk and single-molecule proteomics. ACS Chem. Biol 15, 1401–1407 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miclotte G, Martens K & Fostier J Computational assessment of the feasibility of protonation-based protein sequencing. PLoS ONE 15, e0238625 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tullman J, Marino JP & Kelman Z Leveraging nature’s biomolecular designs in next-generation protein sequencing reagent development. Appl. Microbiol. Biotechnol 104, 7261–7271 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Rodriques SG, Marblestone AH & Boyden ES A theoretical analysis of single molecule protein sequencing via weak binding spectra. PLoS ONE 14, e0212868 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tullman J, Callahan N, Ellington B, Kelman Z & Marino JP Engineering ClpS for selective and enhanced N-terminal amino acid binding. Appl. Microbiol. Biotechnol 103, 2621–2633 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Smith RD, Cheng X, Brace JE, Hofstadler SA & Anderson GA Trapping, detection and reaction of very large single molecular ions by mass spectrometry. Nature 369, 137–139 (1994). [Google Scholar]
- 16.Keifer DZ & Jarrold MF Single-molecule mass spectrometry. Mass Spectrom. Rev 36, 715–733 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Rose RJ, Damoc E, Denisov E, Makarov A & Heck AJ High-sensitivity Orbitrap mass analysis of intact macromolecular assemblies. Nat. Methods 9, 1084–1086 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Makarov A & Denisov E Dynamics of ions of intact proteins in the Orbitrap mass analyzer. J. Am. Soc. Mass Spectr 20, 1486–1495 (2009). [DOI] [PubMed] [Google Scholar]
- 19.Kafader JO et al. Measurement of individual ions sharply increases the resolution of Orbitrap mass spectra of proteins. Anal. Chem 91, 2776–2783 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Kafader JO et al. Multiplexed mass spectrometry of individual ions improves measurement of proteoforms and their complexes. Nat. Methods 17, 391–394 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wörner TP et al. Resolving heterogeneous macromolecular assemblies by Orbitrap-based single-particle charge detection mass spectrometry. Nat. Methods 17, 395–398 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Kafader JO et al. Individual ion mass spectrometry enhances the sensitivity and sequence coverage of top down mass spectrometry. J. Proteome Res 19, 1346–1350 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith L et al. The human proteoform project: a plan to define the human proteome. Preprint at Preprints 10.20944/preprints202010.0368.v1 (2020). [DOI]
- 24.Ekinci KL, Huang XMH & Roukes ML Ultrasensitive nanoelectromechanical mass detection. Appl. Phys. Lett 84, 4469–4471 (2004). [Google Scholar]
- 25.Hanay MS et al. Single-protein nanomechanical mass spectrometry in real time. Nat. Nanotechnol 7, 602–608 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sage E et al. Neutral particle mass spectrometry with nanomechanical systems. Nat. Commun 6, 6482 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dominguez-Medina S et al. Neutral mass spectrometry of virus capsids above 100 megadaltons with nanomechanical resonators. Science 362, 918–922 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Chaste J et al. A nanomechanical mass sensor with yoctogram resolution. Nat. Nanotechnol 7, 301–304 (2012). [DOI] [PubMed] [Google Scholar]
- 29.Hanay MS et al. Inertial imaging with nanomechanical systems. Nat. Nanotechnol 10, 339–344 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malvar O et al. Mass and stiffness spectrometry of nanoparticles and whole intact bacteria by multimode nanomechanical resonators. Nat. Commun 7, 13452 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilm M & Mann M Analytical properties of the nanoelectrospray ion source. Anal. Chem 68, 1–8 (1996). [DOI] [PubMed] [Google Scholar]
- 32.El-Faramawy A, Siu KM & Thomson BA Efficiency of nano-electrospray ionization. J. Am. Soc. Mass Spectr 16, 1702–1707 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Bush J et al. The nanopore mass spectrometer. Rev. Sci. Instrum 88, 113307 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maulbetsch W, Wiener B, Poole W, Bush J & Stein D Preserving the sequence of a biopolymer’s monomers as they enter an electrospray mass spectrometer. Phys. Rev. Appl 6, 054006 (2016). [Google Scholar]
- 35.Brodbelt JS Photodissociation mass spectrometry: new tools for characterization of biological molecules. Chem. Soc. Rev 43, 2757–2783 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang S et al. Tunnelling readout of hydrogen-bonding-based recognition. Nat. Nanotechnol 4, 297–301 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao Y et al. Single-molecule spectroscopy of amino acids and peptides by recognition tunnelling. Nat. Nanotechnol 9, 466–473 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohshiro T et al. Detection of post-translational modifications in single peptides using electron tunnelling currents. Nat. Nanotechnol 9, 835–840 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Zhang B et al. Observation of giant conductance fluctuations in a protein. Nano Futures 1, 035002 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang B et al. Engineering an enzyme for direct electrical monitoring of activity. ACS Nano 14, 1360–1368 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seeman NC & Sleiman HF DNA nanotechnology. Nat. Rev. Mater 3, 17068 (2017). [Google Scholar]
- 42.Schnitzbauer J, Strauss MT, Schlichthaerle T, Schueder F & Jungmann R Super-resolution microscopy with DNA-PAINT. Nat. Protoc 12, 1198–1228 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Dai M, Jungmann R & Yin P Optical imaging of individual biomolecules in densely packed clusters. Nat. Nanotechnol 11, 798–807 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jungmann R et al. Quantitative super-resolution imaging with qPAINT. Nat. Methods 13, 439–442 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dai M & Yin P Methods and compositions relating to super-resolution imaging and modification. US patent 10006917 (2018).
- 46.Woo S & Yin P Methods and compositions for protein identification. US patent 10697974 (2020).
- 47.Schaus TE, Woo S, Xuan F, Chen X & Yin P A DNA nanoscope via auto-cycling proximity recording. Nat. Commun 8, 696 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kishi JY, Schaus TE, Gopalkrishnan N, Xuan F & Yin P Programmable autonomous synthesis of single-stranded DNA. Nat. Chem 10, 155–164 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gopalkrishnan N, Punthambaker S, Schaus TE, Church GM & Yin P A DNA nanoscope that identifies and precisely localizes over a hundred unique molecular features with nanometer accuracy. Preprint at bioRxiv 10.1101/2020.08.27.271072 (2020). [DOI]
- 50.Filius M, Kim SH, Severins I & Joo C High-resolution single-molecule FRET via DNA eXchange (FRET X). Nano Lett. 21, 3295–3301 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lerner E et al. Toward dynamic structural biology: two decades of single-molecule Förster resonance energy transfer. Science 359, eaan1133 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kasianowicz JJ, Brandin E, Branton D & Deamer DW Characterization of individual polynucleotide molecules using a membrane channel. Proc. Natl Acad. Sci. USA 93, 13770–13773 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Deamer D, Akeson M & Branton D Three decades of nanopore sequencing. Nat. Biotechnol 34, 518–524 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Loman NJ & Watson M Successful test launch for nanopore sequencing. Nat. Methods 12, 303–304 (2015). [DOI] [PubMed] [Google Scholar]
- 55.Rozevsky Y et al. Quantification of mRNA expression using single-molecule nanopore sensing. ACS Nano 14, 13964–13974 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Di Muccio G, Rossini AE, Di Marino D, Zollo G & Chinappi M Insights into protein sequencing with an α-hemolysin nanopore by atomistic simulations. Sci. Rep 9, 6440 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wilson J, Sarthak K, Si W, Gao L & Aksimentiev A Rapid and accurate determination of nanopore ionic current using a steric exclusion model. ACS Sens. 4, 634–644 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang G, Voet A & Maglia G FraC nanopores with adjustable diameter identify the mass of opposite-charge peptides with 44 dalton resolution. Nat. Commun 10, 835 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Piguet F et al. Identification of single amino acid differences in uniformly charged homopolymeric peptides with aerolysin nanopore. Nat. Commun 9, 966 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cao C et al. Single-molecule sensing of peptides and nucleic acids by engineered aerolysin nanopores. Nat. Commun 10, 4918 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Galenkamp NS, Soskine M, Hermans J, Wloka C & Maglia G Direct electrical quantification of glucose and asparagine from bodily fluids using nanopores. Nat. Commun 9, 4085 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ouldali H et al. Electrical recognition of the twenty proteinogenic amino acids using an aerolysin nanopore. Nat. Biotechnol 38, 176–181 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Restrepo-Pérez L, Wong CH, Maglia G, Dekker C & Joo C Label-free detection of post-translational modifications with a nanopore. Nano Lett. 19, 7957–7964 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Korotkov KV, Sandkvist M & Hol WG The type II secretion system: biogenesis, molecular architecture and mechanism. Nat. Rev. Microbiol 10, 336–351 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Olivares AO, Baker TA & Sauer RT Mechanical protein unfolding and degradation. Annu. Rev. Physiol 80, 413–429 (2018). [DOI] [PubMed] [Google Scholar]
- 66.Nivala J, Marks DB & Akeson M Unfoldase-mediated protein translocation through an α-hemolysin nanopore. Nat. Biotechnol 31, 247–250 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nivala J, Mulroney L, Li G, Schreiber J & Akeson M Discrimination among protein variants using an unfoldase-coupled nanopore. ACS Nano 8, 12365–12375 (2014). [DOI] [PubMed] [Google Scholar]
- 68.Zhang S et al. Bottom–up fabrication of a multi-component nanopore sensor that unfolds, processes and recognizes single proteins. Preprint at bioRxiv 10.1101/2020.12.04.411884 (2020). [DOI]
- 69.Sachelaru I et al. YidC and SecYEG form a heterotetrameric protein translocation channel. Sci. Rep 7, 101 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Knyazev DG, Kuttner R, Zimmermann M, Sobakinskaya E & Pohl P Driving forces of translocation through bacterial translocon SecYEG. J. Membr. Biol 251, 329–343 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Backes S & Herrmann JM Protein translocation into the intermembrane space and matrix of mitochondria: mechanisms and driving forces. Front. Mol. Biosci 4, 83 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Feng J et al. Transmembrane protein rotaxanes reveal kinetic traps in the refolding of translocated substrates. Commun. Biol 3, 159 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rosen CB, Bayley H & Rodriguez-Larrea D Free-energy landscapes of membrane co-translocational protein unfolding. Commun. Biol 3, 160 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rodriguez-Larrea D Single-aminoacid discrimination in proteins with homogeneous nanopore sensors and neural networks. Biosens. Bioelectron 180, 113108 (2021). [DOI] [PubMed] [Google Scholar]
- 75.Cardozo N et al. Multiplexed direct detection of barcoded protein reporters on a nanopore array. Preprint at bioRxiv 10.1101/837542 (2019). [DOI] [PMC free article] [PubMed]
- 76.Yao Y, Docter M, Van Ginkel J, de Ridder D & Joo C Single-molecule protein sequencing through fingerprinting: computational assessment. Phys. Biol 12, 055003 (2015). [DOI] [PubMed] [Google Scholar]
- 77.Ohayon S, Girsault A, Nasser M, Shen-Orr S & Meller A Simulation of single-protein nanopore sensing shows feasibility for whole-proteome identification. PLoS Comput. Biol 15, e1007067 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Restrepo-Pérez L et al. Resolving chemical modifications to a single amino acid within a peptide using a biological nanopore. ACS Nano 13, 13668–13676 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Asandei A et al. Placement of oppositely charged aminoacids at a polypeptide termini determines the voltage-controlled braking of polymer transport through nanometer-scale pores. Sci. Rep 5, 10419 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang R et al. Single-molecule discrimination of labeled DNAs and polypeptides using photoluminescent-free TiO2 nanopores. ACS Nano 12, 11648–11656 (2018). [DOI] [PubMed] [Google Scholar]
- 81.Zrehen A, Ohayon S, Huttner D & Meller A On-chip protein separation with single-molecule resolution. Sci. Rep 10, 15313 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Assad ON et al. Light-enhancing plasmonic-nanopore biosensor for superior single-molecule detection. Adv. Mater 29, 1605442 (2017). [DOI] [PubMed] [Google Scholar]
- 83.Spitzberg JD, Zrehen A, van Kooten XF & Meller A Plasmonic-nanopore biosensors for superior single-molecule detection. Adv. Mater 31, 1900422 (2019). [DOI] [PubMed] [Google Scholar]
- 84.Houghtaling J, List J & Mayer M Nanopore-based, rapid characterization of individual amyloid particles in solution: concepts, challenges, and prospects. Small 14, 1802412 (2018). [DOI] [PubMed] [Google Scholar]
- 85.Plesa C et al. Fast translocation of proteins through solid state nanopores. Nano Lett. 13, 658–663 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yusko EC et al. Controlling protein translocation through nanopores with bio-inspired fluid walls. Nat. Nanotechnol 6, 253–260 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Houghtaling J et al. Estimation of shape, volume, and dipole moment of individual proteins freely transiting a synthetic nanopore. ACS Nano 13, 5231–5242 (2019). [DOI] [PubMed] [Google Scholar]
- 88.Yusko EC et al. Real-time shape approximation and fingerprinting of single proteins using a nanopore. Nat. Nanotechnol 12, 360–367 (2017). [DOI] [PubMed] [Google Scholar]
- 89.Pang Y & Gordon R Optical trapping of a single protein. Nano Lett. 12, 402–406 (2012). [DOI] [PubMed] [Google Scholar]
- 90.Verschueren D, Shi X & Dekker C Nano-optical tweezing of single proteins in plasmonic nanopores. Small Methods 3, 1800465 (2019). [Google Scholar]
- 91.Schmid S, Stömmer P, Dietz H & Dekker C Nanopore electro-osmotic trap for the label-free study of single proteins and their conformations. Preprint at bioRxiv 10.1101/2021.03.09.434634 (2021). [DOI] [PubMed]
- 92.Larkin J, Henley RY, Muthukumar M, Rosenstein JK & Wanunu M High-bandwidth protein analysis using solid-state nanopores. Biophys. J 106, 696–704 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nir I, Huttner D & Meller A Direct sensing and discrimination among ubiquitin and ubiquitin chains using solid-state nanopores. Biophys. J 108, 2340–2349 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Waduge P et al. Nanopore-based measurements of protein size, fluctuations, and conformational changes. ACS Nano 11, 5706–5716 (2017). [DOI] [PubMed] [Google Scholar]
- 95.Varongchayakul N, Hersey JS, Squires A, Meller A & Grinstaff MW A solid-state hard microfluidic-nanopore biosensor with multilayer fluidics and on-chip bioassay/purification chamber. Adv. Funct. Mater 28, 1804182 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hu R et al. Differential enzyme flexibility probed using solid-state nanopores. ACS Nano 12, 4494–4502 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Huang G et al. Electro-osmotic vortices promote the capture of folded proteins by PlyAB nanopores. Nano Lett. 20, 3819–3827 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Soskine M, Biesemans A & Maglia G Single-molecule analyte recognition with ClyA nanopores equipped with internal protein adaptors. J. Am. Chem. Soc 137, 5793–5797 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wloka C et al. Label-free and real-time detection of protein ubiquitination with a biological nanopore. ACS Nano 11, 4387–4394 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Aramesh M et al. Localized detection of ions and biomolecules with a force-controlled scanning nanopore microscope. Nat. Nanotechnol 14, 791–798 (2019). [DOI] [PubMed] [Google Scholar]
- 101.Hernandez ET, Swaminathan J, Marcotte EM & Anslyn EV Solution-phase and solid-phase sequential, selective modification of side chains in KDYWEC and KDYWE as models for usage in single-molecule protein sequencing. New J. Chem 41, 462–469 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D & Nesvizhskii AI MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat. Methods 14, 513–520 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhong J et al. Proteoform characterization based on top–down mass spectrometry. Brief. Bioinform 22, 1729–1750 (2021). [DOI] [PubMed] [Google Scholar]
- 104.Creasy DM & Cottrell JS Unimod: protein modifications for mass spectrometry. Proteomics 4, 1534–1536 (2004). [DOI] [PubMed] [Google Scholar]
- 105.Marx V A dream of single-cell proteomics. Nat. Methods 16, 809–812 (2019). [DOI] [PubMed] [Google Scholar]
- 106.Rissin DM et al. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat. Biotechnol 28, 595–599 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wu C, Garden PM & Walt DR Ultrasensitive detection of attomolar protein concentrations by dropcast single molecule assays. J. Am. Chem. Soc 142, 12314–12323 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Norman M et al. Ultrasensitive high-resolution profiling of early seroconversion in patients with COVID-19. Nat. Biomed. Eng 4, 1180–1187 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liu F, Rijkers DT, Post H & Heck AJ Proteome-wide profiling of protein assemblies by cross-linking mass spectrometry. Nat. Methods 12, 1179–1184 (2015). [DOI] [PubMed] [Google Scholar]
- 110.Iacobucci C, Götze M & Sinz A Cross-linking/mass spectrometry to get a closer view on protein interaction networks. Curr. Opin. Biotechnol 63, 48–53 (2020). [DOI] [PubMed] [Google Scholar]
- 111.Dunham WH, Mullin M & Gingras A-C Affinity-purification coupled to mass spectrometry: basic principles and strategies. Proteomics 12, 1576–1590 (2012). [DOI] [PubMed] [Google Scholar]
- 112.Gentzel M, Pardo M, Subramaniam S, Stewart AF & Choudhary JS Proteomic navigation using proximity-labeling. Methods 164, 67–72 (2019). [DOI] [PubMed] [Google Scholar]
- 113.Zhao YG & Zhang H Phase separation in membrane biology: the interplay between membrane-bound organelles and membraneless condensates. Dev. Cell 55, 30–44 (2020). [DOI] [PubMed] [Google Scholar]
- 114.Steen H & Mann M The ABC’s (and XYZ’s) of peptide sequencing. Nat. Rev. Mol. Cell Biol 5, 699–711 (2004). [DOI] [PubMed] [Google Scholar]
- 115.Toby TK, Fornelli L & Kelleher NL Progress in top–down proteomics and the analysis of proteoforms. Annu. Rev. Anal. Chem 9, 499–519 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Samaras P et al. ProteomicsDB: a multi-omics and multi-organism resource for life science research. Nucleic Acids Res. 48, D1153–D1163 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ruggles KV et al. An analysis of the sensitivity of proteogenomic mapping of somatic mutations and novel splicing events in cancer. Mol. Cell. Proteomics 15, 1060–1071 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhu Y et al. Nanodroplet processing platform for deep and quantitative proteome profiling of 10–100 mammalian cells. Nat. Commun 9, 882 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Budnik B, Levy E, Harmange G & Slavov N SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 19, 161 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhu Y et al. Proteomic analysis of single mammalian cells enabled by microfluidic nanodroplet sample preparation and ultrasensitive NanoLC–MS. Angew. Chem. Int. Ed 57, 12370–12374 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kelly RT Single-cell proteomics: progress and prospects. Mol. Cell. Proteomics 19, 1739–1748 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gavrilyuk J, Ban H, Nagano M, Hakamata W & Barbas CF Formylbenzene diazonium hexafluorophosphate reagent for tyrosine-selective modification of proteins and the introduction of a bioorthogonal aldehyde. Bioconjugate Chem. 23, 2321–2328 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ban H, Gavrilyuk J & Barbas CF III Tyrosine bioconjugation through aqueous ene-type reactions: a click-like reaction for tyrosine. J. Am. Chem. Soc 132, 1523–1525 (2010). [DOI] [PubMed] [Google Scholar]
- 124.Bach K, Beerkens BL, Zanon PR & Hacker SM Light-activatable, 2,5-disubstituted tetrazoles for the proteome-wide profiling of aspartates and glutamates in living bacteria. ACS Cent. Sci 6, 546–554 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Taylor MT, Nelson JE, Suero MG & Gaunt MJ A protein functionalization platform based on selective reactions at methionine residues. Nature 562, 563–568 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lin S et al. Redox-based reagents for chemoselective methionine bioconjugation. Science 355, 597–602 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Christian AH et al. A physical organic approach to tuning reagents for selective and stable methionine bioconjugation. J. Am. Chem. Soc 141, 12657–12662 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jia S, He D & Chang CJ Bioinspired thiophosphorodichloridate reagents for chemoselective histidine bioconjugation. J. Am. Chem. Soc 141, 7294–7301 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bloom S et al. Decarboxylative alkylation for site-selective bioconjugation of native proteins via oxidation potentials. Nat. Chem 10, 205–211 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rosen CB & Francis MB Targeting the N terminus for site-selective protein modification. Nat. Chem. Biol 13, 697–705 (2017). [DOI] [PubMed] [Google Scholar]
- 131.Busch GK et al. Specific N-terminal protein labelling: use of FMDV 3C pro protease and native chemical ligation. Chem. Commun 29, 3369–3371 (2008). [DOI] [PubMed] [Google Scholar]
- 132.Bandyopadhyay A, Cambray S & Gao J Fast and selective labeling of N-terminal cysteines at neutral pH via thiazolidino boronate formation. Chem. Sci 7, 4589–4593 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Agten SM, Dawson PE & Hackeng TM Oxime conjugation in protein chemistry: from carbonyl incorporation to nucleophilic catalysis. J. Pept. Sci 22, 271–279 (2016). [DOI] [PubMed] [Google Scholar]
- 134.MacDonald JI, Munch HK, Moore T & Francis MB One-step site-specific modification of native proteins with 2-pyridinecarboxyaldehydes. Nat. Chem. Biol 11, 326–331 (2015). [DOI] [PubMed] [Google Scholar]
- 135.Matheron L et al. Improving the selectivity of the phosphoric acid β-elimination on a biotinylated phosphopeptide. J. Am. Soc. Mass Spectr 23, 1981–1990 (2012). [DOI] [PubMed] [Google Scholar]
- 136.Du J et al. Metabolic glycoengineering: sialic acid and beyond. Glycobiology 19, 1382–1401 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tommasone S et al. The challenges of glycan recognition with natural and artificial receptors. Chem. Soc. Rev 48, 5488–5505 (2019). [DOI] [PubMed] [Google Scholar]