

Abstract
Ca2+/calmodulin-dependent protein kinase II (CaMKII) is a ubiquitous serine threonine kinase with established roles in physiological and pathophysiological vascular remodeling. Based on our previous study demonstrating that CaMKIIδ promotes thrombin-induced endothelial permeability and recent reports that CaMKII may contribute to inflammatory remodeling in the heart, we investigated CaMKIIδ-dependent regulation of endothelial function downstream of an interleukin-6 (IL-6)/JAK/STAT3 signaling axis. Upon treatment with IL-6 and its soluble receptor (sIL-6R), CaMKIIδ expression is significantly induced in HUVEC. Using pharmacological inhibitors of JAK and siRNA targeting STAT3, we demonstrated that activation of STAT3 is sufficient to induce CaMKIIδ expression. Under these conditions, rather than promoting IL-6-induced permeability, we found that CaMKIIδ promotes endothelial cell migration as measured by live cell imaging of scratch wound closure and single cell motility analysis. In a similar manner, endothelial cell proliferation was attenuated upon knockdown of CaMKIIδ as determined by growth curves, cell cycle analysis, and capacitance of cell-covered electrodes as measured by ECIS. Using inducible endothelial-specific STAT3 knockout mice, we demonstrate that STAT3 signaling promotes developmental angiogenesis in the neonatal mouse retina assessed at postnatal day 6. CaMKIIδ expression in retinal endothelium was attenuated in these animals as measured by qPCR. STAT3’s effects on angiogenesis were phenocopied by the endothelial specific knockout of CaMKIIδ, with significantly reduced vascular outgrowth and number of junctions in the developing P6 retina. For the first time, we demonstrate that transcriptional regulation of CaMKIIδ by STAT3 promotes endothelial motility, proliferation, and in vivo angiogenesis.
Keywords: Angiogenesis, CaMKII, STAT3 signaling, Cell motility, Endothelial biology
Introduction
Ca2+/calmodulin–dependent protein kinase II (CaMKII) is a multimeric protein of about 600 kDa that is temporally and spatially regulated by intracellular calcium flux and has over 30 discrete splice variants between its four main subtypes (1). Its subcellular function has been well studied in the brain, heart, and vascular smooth muscle (VSM) (2–4), but is relatively unexplored in the endothelium. Our lab has shown that the CaMKIIδ2 splice variant, containing a unique 21-aa C-terminal tail, regulates VSM cell migration and proliferation in the context of post-injury vascular remodeling (5–9). In the endothelium, we have shown that CaMKIIδ6, a tailless splice variant of CaMKIIδ, is the predominant isoform and regulates thrombin-dependent increase in vascular permeability in a RhoA-dependent mechanism (10). Additionally, a recent publication has identified unique roles for CaMKIIδ and CaMKIIγ in pathological neoangiogenesis following vessel ablation in a mouse model of retinopathy of prematurity (ROP) (11). Despite its functional significance in multiple organ and cell systems, little is known about mechanisms regulating CaMKII expression.
Signal transducer and activator of transcription factor 3 (STAT3) is a key transcription factor that has been demonstrated to have conserved roles in embryonic development and homeostatic tissue function (12,13). It is largely accepted that its main function is modification of inflammation and innate immunity (14). Activation of STAT3 by the proinflammatory cytokine interleukin-6 (IL-6) in the endothelium has been demonstrated to play a key role in acute inflammation by increasing vascular permeability to allow for immune cell trafficking and pathogen clearance (15). Both STAT3 and IL-6 have been suggested to play critical roles in regulating tumor, cardiac, and AAA-related angiogenesis. The implied mechanism by which activation of STAT3 regulates these processes is by upregulation of proangiogenic factors such as VEGF-A, bFGF, and MMP’s (16–20). The primary downstream target of activated STAT3 signaling is suppressor of cytokine signaling 3 (SOCS3), which primarily functions in a negative feedback mechanism as an inhibitor of STAT3 activation. It has been reported that SOCS3 also functions as a negative modulator of pathological angiogenesis in the context of retinal vascularization and melanoma tumor growth and vessel density (21). Other downstream targets of active STAT3 signaling that directly contribute to angiogenic processes have not been thoroughly investigated.
Blood vessel development is critical for providing oxygen and nutrients to hypoxic parenchyma. This process is known as angiogenesis, an essential developmental process for organ formation and organism growth. However, dysregulation of angiogenesis has been demonstrated to play key roles in multiple pathologies such as rheumatoid arthritis, psoriasis, multiple forms of retinopathy, cancer metastasis, infertility, heart disease, and stroke (22). Angiogenesis is regulated by a diverse array of signaling pathways involving endothelial cell motility, proliferation, cell fate determination, tube formation, and recruitment of mural cells (23,24).
In this study, we report that CaMKIIδ expression is significantly induced following treatment with IL-6 according to RNAseq, qPCR, and immunoblot and is sensitive to JAK inhibition. We also demonstrate that CaMKIIδ is transcriptionally regulated by STAT3 but does not contribute to IL-6 induced vascular permeability in vitro. Instead, we demonstrate that STAT3-dependent expression of CaMKIIδ promotes endothelial cell motility and proliferation in vitro as well as in vivo developmental angiogenesis.
Materials and Methods
Cell Culture.
Human umbilical vein endothelial cells (HUVEC) were isolated in-house as previously reported (15). Briefly, umbilical cords for scheduled Cesarian sections were washed with 70% ethanol and PBS before 30-minute incubation in 0.2% collagenase. Cells were then collected in phenol red-free EBM media (Lonza: catalog no. CC-3129) and EGM-2 SingleQuots (Lonza: catalog no. CC-4176). HUVEC cell lines expressing non-targeting and shRNA against CaMKIIδ were described in a previous publication from our lab (10). All cells used for experiments from passage number 4–8. Unless specified seeding densities, HUVEC are seeded at full confluence: 1X105 cells/cm2. Cell culture and all seeded experiments were maintained with Endothelial Cell Basal Medium 2 (PromoCell: catalog no. C-22211) with SupplementPack (PromoCell: catalog no. C-39211).
Coating anti-Rat IgG Dyna Beads.
Dyna M-450 sheep anti-rat IgG magnetic beads (Invitrogen, catalog no. 11035) are washed 3x with bead wash buffer (0.1% BSA (Krackeler Scientific, catalog no. A3059) in PBS) before incubation with 2 μL of rat anti-mouse CD31 antibody (BD Pharmingen, catalog no. 550274) per 20 μL of beads for 2h at room temperature. Beads were then washed 3x with bead wash buffer before resuspension in final desired volume and stored at 4°C for up to 1 week.
Endothelial Cell Retina Prep.
Postnatal day 6 (P6) mice were sacrificed by decapitation and the eyes were elucidated. Eyes were processed (remove sclera, choroid, lens, and iris) in an ice-cold Petri dish. Retina beds were then digested in prewarmed mix of 5 mg collagenase II (Worthington, catalog no. LS004176), 1% BSA, and 100 U DNase (Zymo kit, lister in RNA isolation section) in 15 mL HBSS. The solution was then incubated for 40 minutes at 37°C before trituration with an 18-gauge needle. Following addition 500 μL FBS, solutions were centrifuged at 300g for 5 minutes at 4° C. Pellets were then resuspended in 5% BSA in HBSS and incubated with 20 μL/mL of anti-CD31-coated beads with agitation for 3 hours at 4°C. Tubes were then placed on a magnetic separator and washed 4x with 1% BSA in HBSS. Beads were then resuspended in Trizol reagent (Thermo Fisher Scientific, catalog no. 10296010) and proceeded into the RNA isolation protocol.
Antibodies and Reagents.
Recombinant human IL-6 (catalog no. 206-IL-050) and sIL-6R (catalog no. 227-SR-025/CF) were purchased from R&D Systems. Pharmalogocial JAK inhibitor ruxolitinib was purchased from Selleck Chem (catalog no. S1378) and transcriptional inhibitor Actinomycin D was purchased from Sigma-Aldrich (catalog no. A9415). Polyclonal antibodies to pan-CaMKII,the δ2 isoform, the δ6 isoform, and the γ isoforms were previously described (10). Antibodies against STAT3 (catalog no. 8768S) and pY705-STAT3 (catalog no. 94994) were purchased from Cell Signaling Technologies. Anti-CD31 antibody (catalog no. BD550274) was purchased from BD Biosciences, anti-β-actin (catalog no. A1978) was purchased from Sigma-Aldrich, and DAPI stain (catalog no. D3571) was purchased from Invitrogen. For western blots, anti-mouse IgG HRP conjugate (catalog no. W402B) was purchased from Promega and ECL Rabbit IgG, HRP-Linked Whole Antibody, Donkey (catalog no. NA934–1ML) was purchased from GE Healthcare. For Immunofluorescence, Alexa Fluor 647 goat anti-rat IgG (catalog no. A21247) was purchased from Life Technologies. For validation of CAMK2A and CAMK2B primers, total human brain RNA was purchased from Takara Bio (catalog no. 636530).
siRNA.
Small interfering RNA (siRNA) against CAMK2D was obtain from Dharmacon as a SmartPool On Target Plus (catalog No. L-004042–00-0005). Additionally, siRNA against STAT3 was designed as previously described (15). Target siRNAs were complexed with Lipofectamine siRNA iMAX (catalog no. 13778075) and used to transfect HUVEC. Non-targeting sequence (NTS) siRNA (catalog no. D-001810–10-05) from Dharmacon was used to transfect HUVEC as a negative control.
RNA Isolation and Primers.
HUVEC RNA isolation is performed using Trizol (Lifetech: catalog no. 15596018) and the Direct-zol RNA Miniprep w/ Zymo-Spin IIC Columns (Zymo Research: catalog no. R2052). RNA concentrations were quantified with NanoDrop 1000 Spectrophotometer from Thermo Scientific. Reverse transcription of 500 ng RNA into cDNA was performed using QuantiTect Rev. Transcription Kit (Qiagen: catalog no. 205313).
Quantitative PCR.
Quantitative PCR was performed with SsoAdvanced Universal SYBR green Super Mix (BioRad Laboratories: catalog no. 1725272) and CFX Connect Real-Time System from BioRad (catalog no. 1855200). Primers for qPCR were ordered from Integrated DNA Technologies, sequences can be found in Table 1.
Table 1. Oligonucleotides.
Primers and oligonucleotides used in these experiments where specific; listed in 5’ to 3’ orientation.
| Oligonucleotide | Forward | Reverse |
|---|---|---|
| siRNA SmartPool | ||
| siCaMKIIδ 1 | GCUAGAAUCUGCCGUCUUU | NA |
| siCaMKIIδ 2 | AAACCAAUCCACACUAUUA | NA |
| siCaMKIIδ 3 | GCGACUUCAUGAUAAGCAUA | NA |
| siCaMKIIδ 4 | UCACCUAAAUGGCAUAGUU | NA |
| siSTAT3 | GAGAUUGACCAGCAGUAUA | NA |
| qPCR Primers | ||
| CAMK2A | CAGTTCCAGCGTTCAGTTAATG | TTCGTGTAGGACTCAAAATCTCC |
| CAMK2B | CTCTGACATCCTGAACTCTGTG | CCGTGGTCTTAATGATCTCCTG |
| CAMK2D | GGCACACCTGGATATCTTTCTC | AGTCTGTGTTGGTCTTCATCC |
| CAMK2G | ATCCCATCTGTAGCGTTGTG | ATCCTCACGACCATGCTTG |
| GAPDH | GTCTCCTCTGACTTCAACAGCG | ACCACCCTGTTGCTGTAGCCAA |
| mCamk2a | ACCCTCTACTTTCTCTCCTCC | ACTTTGGTGTCTTCGTCCTC |
| mCamk2b | TCAAGCCCCAGACAAACAG | ATTCCTTCCCGTCCACTG |
| mCamk2d | GACGAGTATCAGCTCTTTGAGG | GTTTCTGATGGTCCCTAGCAG |
| mCamk2g | CGACTACCAGCTTTTCGAGG | GCCTCTCGTTCTAGTTTCTGATG |
| mSocs3 | GGACCAAGAACCTACGCATCCA | CACCAGCTTGAGTACACAGTCG |
| mCdh5 | GAACGAGGACAGCAACTTCACC | GTTAGCGTGCTGGTTCCAGTCA |
| mCd31 | CCAAAGCCAGTAGCATCATGGTC | GGAT GGT GAAGTT GGCTACAGG |
| mVwf | CGATGGACTCACAGGAGCAAGT | AACAGACGATGGTGGACTCAGC |
| mCdh1 | GGTCATCAGTGTGCTCACCTCT | GCTGTTGTGCTCAAGCCTTCAC |
| mActa2 | TGCTGACAGAGGCACCACTGAA | CAGTTGTAC GT C CAGAGGCATAG |
| mGAPDH | GTAGTTGAGGTCAATGAAGGG | TCGTCTCATAGACAAGATGGT |
Electrophoresis and Immunoblotting.
Cells were grown in 6 or 12-well dishes at 37°C and 5% CO2 before treatment with 200 ng/mL IL-6 + 100 ng/mL sIL-6R and incubation for 24 hours. The media was then aspirated from each well and replaced with 200 μL of lysis buffer (Nonidet P-40 lysis buffer at 4 °C (50 mm Tris (pH 7.4), 50 mm NaF, 0.1 mm NaVO4, 0.5% Nonidet P-40, 0.1 mM phenylmethylsulfonyl fluoride, and 0.2 U/ml aprotinin). Lysates were then boiled and centrifuged at 14,000 rpm. A total of 20 μL of lysate was loaded into each well of standard 10% acrylamide SDS –PAGE gels and run at 200V for approximately 45 minutes. The gels were also run with 5 μL prestained protein ladder (Invitrogen: catalog no. 10748010). Proteins are transferred onto nitrocellulose membranes (Fisher Scientific: catalog no. WP4HY00010). Membranes were blocked for an hour with 5% nonfat dry milk in Tris-buffered saline containing 0.2% Tween 20 (TBST) (Krackeler Scientific: catalog no. P9416). After washing three times with TBST, membranes are placed into 5% milk with primary antibody diluted to a specified concentration (1:1,000 unless otherwise specified) either overnight at 4°C or 1 hour at room temperature. Secondary HRP-conjugated rabbit or mouse antibodies were then incubated at room temperature for 1 hour. Membranes were then developed with SuperSignal West Pico Chemiluminescent kit (Thermo Scientific: catalog no. PI34580) and imaged with ChemiDoc MP Imaging System (BioRad: catalog no. 12003154).
Measurement of Transendothelial Electrical Resistance (TEER).
Endothelial monolayer permeability was detected using an electrical cell-substrate impedance sensor (ECIS) from Applied BioPhysics. HUVEC were seeded at confluence on 8 well, 10-electrode (8W10E+) cultureware that had been pre-coated with 0.1% gelatin. At 1V and 4,000 Hz AC, resistance of the endothelial monolayer was calculated and monitored in real time for 24 hours following treatment (IL-6+sIL-6R, as above). Data is represented as a function of resistance over time. All experiments were performed in full growth media as listed in Cell Culture.
Migration Assays.
HUVEC were seeded with NTS or siRNA against CaMKIIδ at confluence on pre-gelatinized 6 or 12-well dishes in full growth media. Using a P20 pipette tip and a ruler for guidance, a linear scratch wound is administered to the endothelial monolayer 24 hours following IL-6 + sIL-6R treatment. Using an inverted microscope, phase images were collected at time zero to mark the initial wound area. Every six hours, phase images were taken of the wound. Calculation of remaining wound area relative to the original wound size with ImageJ allows for comparison of migratory rates in wounded HUVEC monolayers under varying experimental conditions. HUVEC cell lines expressing a non-targeting shRNA or shRNA against CAMK2D were seeded at 10,000 cells in a 6-well dish for analysis of individual cell migration. Using a stagetop incubator (LiveCell Pathology Devices Inc.) and a Leica DMI6000 B, we tracked individual cells for 24 hours, acquiring TIFF images every 15 minutes. These images were then stacked in ImageJ and run through the Manual Cell Tracker and ChemoTaxis plugins, which calculates directionality, migration velocity, and total Euclidean distance.
Proliferation Assays.
HUVEC are seeded in 6-well dishes at 20,000 cells per well with NTS or siRNA against CaMKIIδ in full growth media. Cells were treated 24 hours later with AdLacZ or AdY640F STAT3 and then allowed to proliferate for three additional days. On the fourth day, cells were lifted with 1X trypsin with EDTA (Invitrogen: catalog no. 23400120) and counted using Moxi Z Mini automated cell counter (Orflo: product code MXZ001). Additionally, cells were seeded at 5,000 cells per well in 8-well microscopy slides (ibidi: catalog no. 80826) and treated in the same manor. They were then fixed with 4% PFA (Fisher Scientific: catalog no. AAJ19943K2) for 30 minutes at room temperature and permeabilized with 0.1% Triton X-100 (Sigma-Aldrich) in PBS (PBST). HUVEC were then treated with DAPI at 1:500 for one hour and imaged using Cytation5 plate reader and imager. We counted cells and plotted per binned integral DAPI content for cell cycle analysis. Additionally, we utilized the ECIS 96W1E+ to measure impedance of freshly seeded sub-confluent monolayers as readout of proliferation. Cells were seeded at 5,000 cells per well with NTS or siRNA against CaMKIIδ. Impedance and capacitance were tracked over the first 48 hours following seeding and the rates of increase and decrease, respectively were compared between NTS and siCaMKIIδ-transfected cells.
Mice Strains.
The C57J/B6 mice used in these experiments are of the following genotypes:
B6; Camk2δtm1Eno/ Tg (Cdh5-cre/ERT2)1Rha/J
B6; Stat3tm1Xyfu/ Tg (Cdh5-cre/ERT2)1Rha
Induction of these animals with tamoxifen resulted in endothelial-specific deletion of CaMKIIδ and endothelial-specific loss of function of STAT3 in vivo.
Neonatal Retina Angiogenesis.
We utilized the in vivo retina angiogenesis model used in this study as previously described (25). We administered 100 μg in 10 μL of tamoxifen via oral gavage at postnatal days 1, 2, and 3. At postnatal day 6, we collected eyes and removed the sclera, choroid, lens, and iris under a dissecting microscope. We then fixed the retinal beds with overnight incubation with 4% PFA before labelled the vasculature with AlexaFluor488-conjugated Isolectin B4 at a concentration of 1:3,000 at room temperature for two hours. Retinas were then mounted on glass coverslips with ProLong™ Diamond Antifade Mountant and imaged with the Cytation5 plate reader and imager.
RNAseq.
IL-6-induced gene expression differences observed through RNA-Sequencing (N. Martino, A. Adam, unpublished), which has been submitted to Gene Expression Omnibus (GEO): submission number: GSE163649. Briefly, confluent HUVEC grown on 0.1% gelatin pre-coated 6-well plates were pre-treated for 30 min with 2 μM Ruxolitinib or 0.1% DMSO control and then treated with IL-6+R or PBS for 3 hours in triplicate. RNA was isolated with Trizol following manufacturer’s instructions. RNA enrichment and next-generation sequencing was performed by Genewiz.
Statistics.
All graphs and statistical analyses were done in GraphPad Prism version 6.0e. All values are expressed as Mean ± SEM. Statistical tests include Two-way ANOVA with Neuman-Keuls post-hoc analysis as well as Student’s t-test. In all comparisons, statistical significance was determined by p ≤ 0.05.
Results
Determining basal CaMKII isoform expression in human endothelium –
A prior study from our lab has demonstrated that CaMKIIδ is the dominant isoform expressed in HUVEC, BAEC, and HDMEC (10). However, it has also been reported that the endothelium has a CaMKII expression profile more like the brain: with high expression of CaMKIIα and CaMKIIβ (26). To confirm the basal expression profile of CaMKII isoforms in the endothelium, RNA from confluent HUVECs was harvested and qPCR analysis was performed with primers designed to each of the four subtypes (α, β, δ, γ). Under these conditions, CaMKIIα and β were nearly undetectable while CaMKIIδ was shown to be the dominant isoform with modest expression of CaMKIIγ (Fig. 1A). Total human brain RNA (Takara) was used as a positive control to validate CaMKIIα and β primers (Supplemental Fig. 1A). We next utilized custom CaMKII antibodies to detect different splice isoforms of CaMKIIδ and CaMKIIγ. Earlier, we reported that the CaMKIIδ2 isoform, containing a unique 21 amino acid tail on its C-terminus, is a critical modulator of VSM motility and proliferation (5). When compared to rVSM lysates, HUVEC express significantly less CaMKIIδ2, as detected by antibodies raised against the 21-amino acid C-terminal tail. Consistent with a previous publication from our lab (10), HUVEC predominantly express the “tailless” CaMKIIδ6 isoform. An antibody selective for CaMKIIγ revealed expression of a 58 kDa band in cultured HUVEC, consistent with the CaMKIIγB isoform. (Fig. 1B). These data are foundational for the proceeding study and identify CaMKIIδ6 and CaMKIIγB as the predominant isoforms expressed in human endothelium.
Figure 1. Determining basal CaMKII isoform expression in human and murine endothelium.
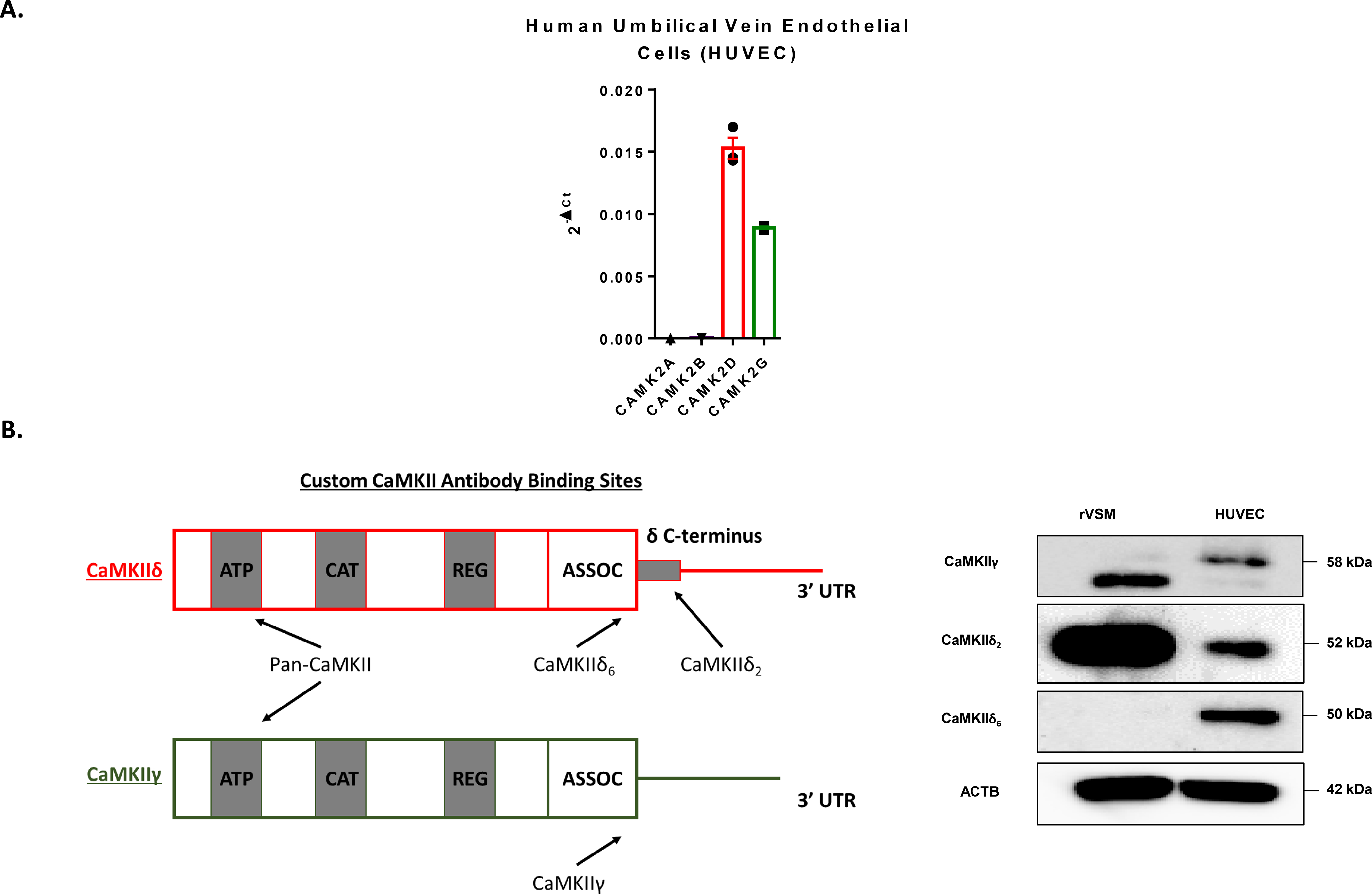
A. qPCR analysis of CaMKII subtypes CAMK2A, CAMK2B, CAMK2D, and CAMK2G in cultured HUVEC normalized to GAPDH. n = 3. B. Diagram of custom CaMKII antibody binding sites on CaMKIIδ and γ recognizing unique splice isoforms. Immunoblot for the variable domain of γ and the C-terminal tail of CaMKIIδ reveals differential isoform expression in endothelial cells when compared to rat vascular smooth muscle cells (rVSM). n = 3.
CaMKIIδ is upregulated by Interleukin-6 (IL-6) –
RNAseq analysis of HUVEC stimulated with IL-6 revealed that CaMKIIδ is significantly upregulated as compared to other CaMKII subtypes after 3 hours (Fig. 2A). Subsequent qPCR analysis is consistent with the findings of the RNAseq experiment: CaMKIIδ is significantly upregulated about 3 to 4-fold following 3 hours of IL-6 treatment relative to PBS control. We found that levels of CaMKIIγ were not significantly changed following treatment with IL-6 (Fig. 2B). To ensure that this increase in RNA message is due to nascent transcription, we pretreated HUVECs with transcriptional inhibitor Actinomycin D. Pretreatment with Actinomycin D significantly attenuated the IL-6-dependent increase in CaMKIIδ transcription, suggesting that this increase is, in fact, due to active induction of transcription (Fig. 2C). Since we have demonstrated that CaMKIIδ6 is the dominant isoform expressed in the endothelium, we further tested whether the nascent upregulation of CaMKIIδ by IL-6 treatment was also δ6 or whether alternative splicing was taking place. Utilizing custom antibody against the variable C-terminal tail of the CaMKIIδ subtype, we were able to validate with immunoblot that, following IL-6 treatment, there is no change in splicing of either CaMKIIδ or CaMKIIγ (Fig. 2D). Together, these data suggest that CaMKIIδ is upregulated following treatment with IL-6, which has been demonstrated to regulate transcription in a JAK/STAT3-dependent manner (15,27).
Figure 2. CaMKIIδ is upregulated by Interleukin-6 (IL-6).
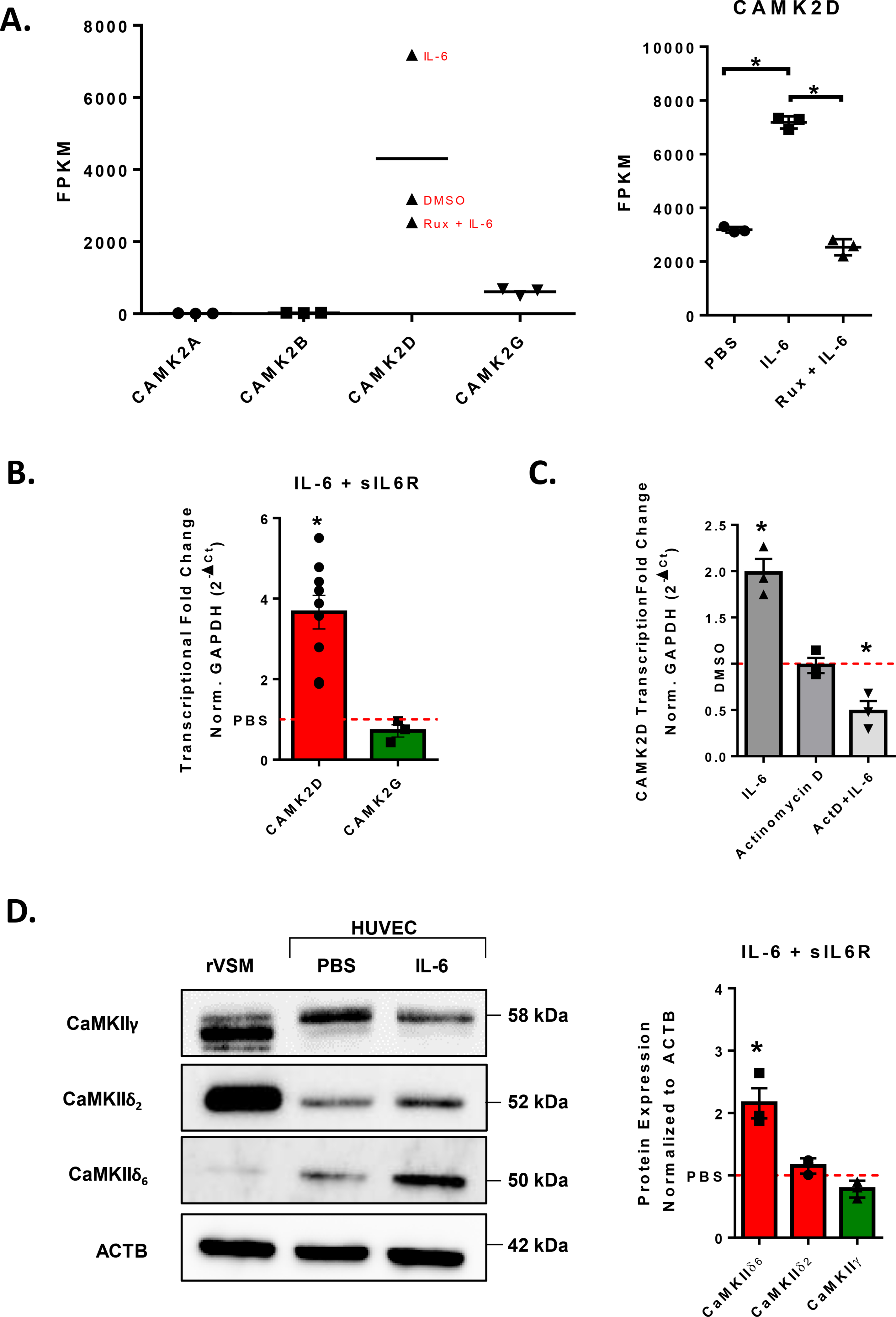
A. RNAseq data of CAMK2A, CAMK2B, CAMK2D, AND CAMK2G following treatment with DMSO, 200 ng/mL IL-6 and 100 ng/mL sIL6r for three hours, or IL-6/sIL-6r with 30-minute pretreatment with 2 μM ruxolitinib. Statistical analyses performed by Genewiz. B. qPCR analysis of CAMK2D and CAMK2G following treatment with 200 ng/mL IL-6 and 100 ng/mL sIL6r for three hours, normalized to GAPDH, n = 4. Student’s t-test, (*) p <0.05 C. Confluent HUVEC monolayers were pretreated with Actinomycin D (10 μg/ mL) for 30 minutes before treatment with IL-6/sIL-6r for 3 hours. qPCR analysis of CAMK2D normalized to GAPDH, n = 3. Two-Way ANOVA, (*) p < 0.05. D. Custom CaMKII antibody immunoblot to CaMKIIγ, CaMKIIδ2, and CaMKIIδ6 normalized to ACTB. Confluent HUVEC treated with IL-6/sIL-6r for 24h. n = 3. Student’s t-test, (*) p <0.05.
CaMKIIδ is upregulated in a JAK-dependent manner –
As described above, RNAseq analysis of HUVEC stimulated with IL-6 for 3 hours revealed that expression of CaMKIIδ is significantly upregulated. Importantly, this upregulation was attenuated by pretreatment with JAK inhibitor ruxolitinib (Fig. 2A). Following 30-minute pretreatment with ruxolitinib, the IL-6-dependent upregulation of CaMKIIδ was attenuated at both the message and protein level when analyzed using qPCR and immunoblot, respectively (Fig. 3). These results demonstrated that IL-6-dependent upregulation of CaMKIIδ expression is JAK-dependent.
Figure 3. CaMKIIδ is upregulated in a JAK-dependent manner.
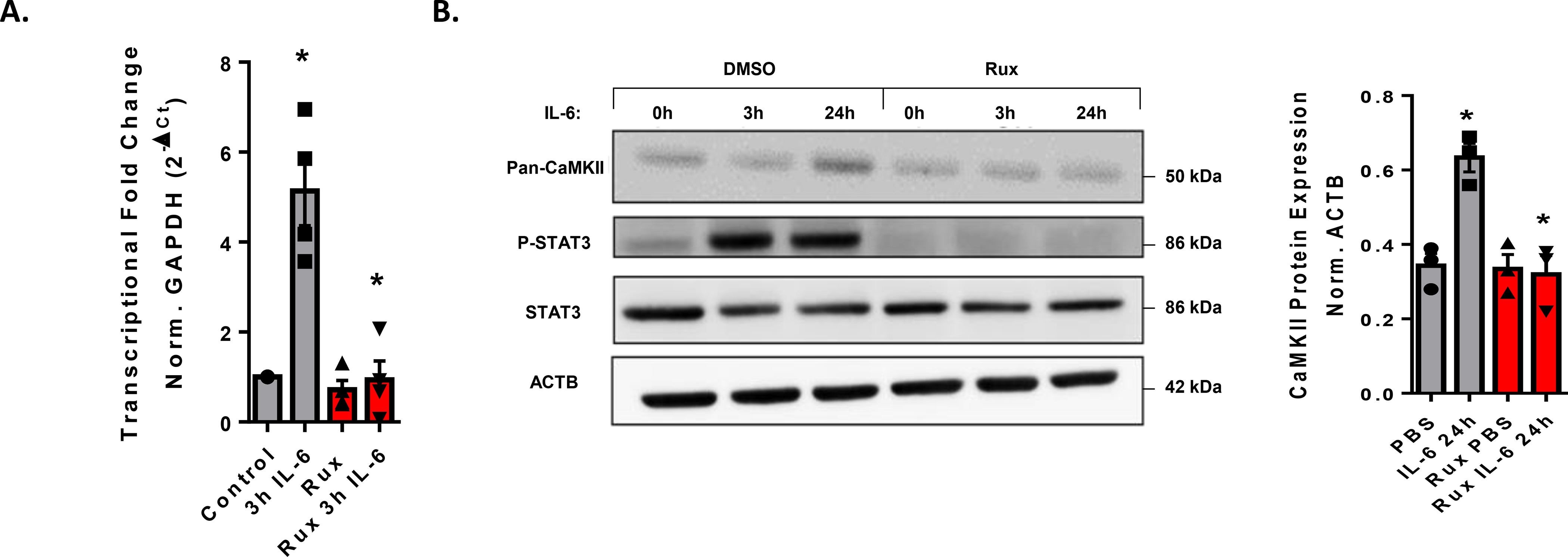
A. qPCR analysis of CaMKIIδ expression after 3h IL-6/sIL-6r treatment following a 30-minute treatment with 2 μM ruxolitinib. n = 4, Two-way ANOVA, p < 0.05 B. Immunoblot of confluent HUVEC treated with or without 2 μM ruxolitinib and 3, 24h IL-6/sIL-6r treatment. Normalization of pan-CaMKII expression relative to the β-actin expression is shown. n = 3, Two-way ANOVA, (*) p < 0.05.
STAT3 promotes CaMKIIδ expression in vitro –
There are several pathways shown to be dependent on JAK-activation including ERK1/2 and PI3K, but the primary downstream signaling axis is through STAT3 (15). To test whether CaMKIIδ expression is promoted by STAT3 activation, transfected HUVEC with siRNA against STAT3 and compared transcriptional and translational changes to its nontargeting sequence (NTS) control following treatment with IL-6. Knocking down STAT3 with siRNA attenuated the IL-6-dependent increase in CaMKIIδ message and protein according to qPCR and immunoblot, respectively. Immunoblotting for total STAT3 confirmed nearly 100% knockdown using this method (Fig. 4A, B). To test if STAT3 activation is sufficient to drive CaMKIIδ expression, we treated confluent HUVEC with LacZ-expressing adenovirus or adenovirus expressing a reported constitutively active (CA) STAT3 mutant, Y640F (28). To confirm STAT3 activation in Y640F-transfected HUVEC, we used qPCR analysis to detect transcript levels of primary downstream target of STAT3 signaling, SOCS3 (29). In these analyses, SOCS3 expression was upregulated approximately 50-fold in Y640F transfected samples and served as a positive control for STAT3 activity. Relative to LacZ-expressing adenovirus controls, the Y640F-treated HUVEC displayed 3 to 4-fold upregulation of CaMKIIδ as detected by qPCR analysis (Fig. 4C). A significant increase in CaMKIIδ6 and a modest increase in CaMKIIδ2 were also observed via immunoblot following treatment with Y640F adenovirus, which is consistent with the upregulation observed after treatment with IL-6 for 24h (Fig. 4D). We also observed little change in message or protein expression of CaMKIIγ in Y640F-treated HUVEC. To interrogate whether CaMKIIδ contains STAT3 binding sites in its promoter or enhancer regions, we utilized the 2020 update of ReMap’s array of 2764 new human ChIP-seq datasets (30). This analysis enabled us to identify STAT3 binding within conserved enhancer and promoter regions of the CAMK2D gene on chromosome 4, as labelled in green (Fig. 4E). Together, these data demonstrate that activate STAT3 is both necessary and sufficient to drive CaMKIIδ expression in vitro.
Figure 4. STAT3 activation promotes CaMKIIδ expression in vitro.
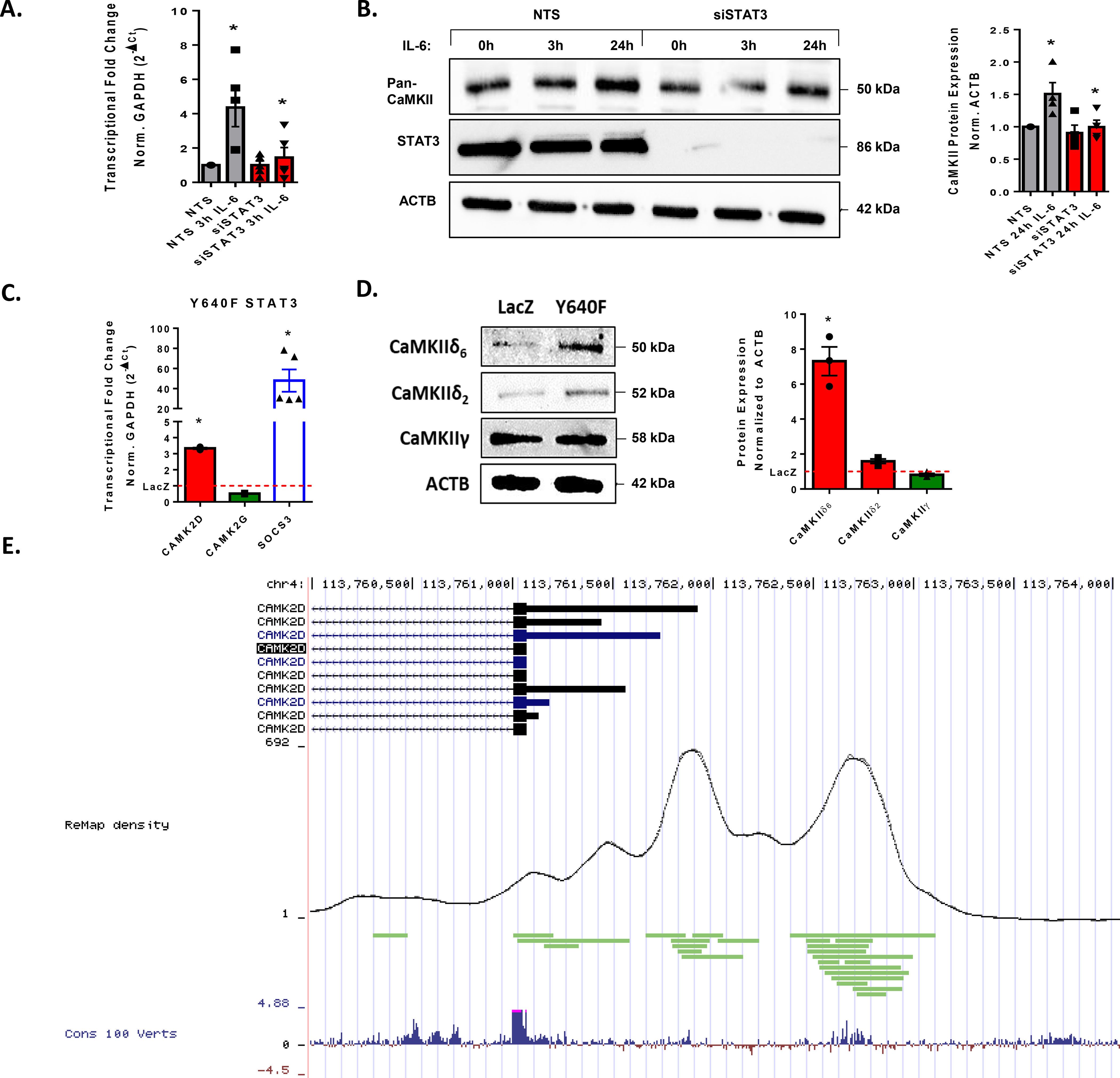
A. qPCR analysis of HUVEC seeded with NTS siRNA or siCaMKIIδ treated with IL-6/sIL-6r for three hours. n = 4, Two-way ANOVA, (*) p < 0.05. B. Immunoblot for pan-CaMKII and STAT3α in HUVEC seeded with NTS or siCaMKIIδ and treated for 3, 24h IL-6/sIL-6r. Normalized to ACTB, n = 4, Two-way ANOVA, (*) p < 0.05. C. qPCR analysis of CAMK2D AND CAMK2G in confluent HUVEC treated overnight with LacZ-expressing adenovirus or Y640F-STAT3 expressing adenovirus normalized to GAPDH. SOCS as a positive control. n = 3, Student’s t-test, (*) p < 0.05. D. Immunoblot for CaMKIIδ6, CaMKIIδ2, and CaMKIIγ in HUVEC transfected with AdLacZ or AdY640F, normalized to ACTB. n = 3, Student’s t-test, (*) p<0.05. E. ReMap2020 analysis of human ChIP data displaying STAT3 binding sites (green) within the promoter and enhancer region of the CAMK2D gene. These sites are compared with density of total binding sites as represented in black by the ReMap density and visualized as conserved domains among vertebrates in blue.
CaMKIIδ does not contribute to the IL-6/JAK/STAT3-dependent increase in vascular permeability –
IL-6 has an accepted role of increasing vascular permeability in the context of acute inflammation. Data from our lab has shown that CaMKIIδ6 promotes thrombin-dependent increases in vascular permeability in a RhoA-dependent manner (10). Therefore, we interrogated CaMKIIδ’s contribution to IL-6-induced increase in endothelial permeability utilizing electrical cell-substrate impedance sensing (ECIS) to measure trans-endothelial electrical resistance (TEER). In confluent HUVEC monolayers, we overexpressed: the wild-type CaMKIIδ6, a kinase-dead form of rodent CaMKIIδ with a point mutation within the catalytic domain (K43A), and a constitutively active form of the rodent CaMKIIδ with a point mutation at the autophosphorylation site (T287D), allowing for Ca2+-independent activity. Treatment of HUVECs with IL-6 causes an approximate 40% decrease in resistance that is sustained for more than 24 hours (15). None of these constructs had any effect on either the basal integrity of the barrier or the IL-6 dependent decrease in barrier function. HUVEC transfected with siRNA against CaMKIIδ, had a statistically significant, yet modest effect on junctional integrity (Supplemental Fig. 2). These data suggest that CaMKIIδ does not play a significant role in IL-6-induced increase in vascular permeability.
CaMKIIδ promotes in vitro endothelial cell motility –
Angiogenesis is an integrated multicellular process coordinating endothelial cell motility and proliferation (31). To test whether STAT3-dependent CaMKIIδ expression promotes endothelial cell migration, we performed a scratch wound assay on confluent HUVEC monolayers transfected with either NTS or siRNA against CaMKIIδ and treated with IL-6 or PBS 24h before wounding. These data showed that IL-6 treatment increased the rate of wound closure 6 hours post-injury and silencing of CaMKIIδ dramatically attenuated the rate of wound closure (Fig. 5A). We tracked leading edge endothelial cells using time lapse live-cell imaging over the course of 24 hours following wounding. Treatment with IL-6 led to an increase in migration velocity, total distance migrated, and Euclidean distance. Silencing of CaMKIIδ with siRNA attenuated both basal and IL-6-induced motility (Fig. 5B). Treatment with Y640F STAT3 (CA STAT3) adenovirus also contributed to an increased rate of wound closure six hours post-injury that was attenuated by silencing of CaMKIIδ (Fig. 5C). This data suggests that wound closure is promoted by activation of STAT3 signaling and STAT3-dependent expression of CaMKIIδ is critical for endothelial cell motility in this context.
Figure 5. CaMKIIδ promotes in vitro endothelial cell motility.
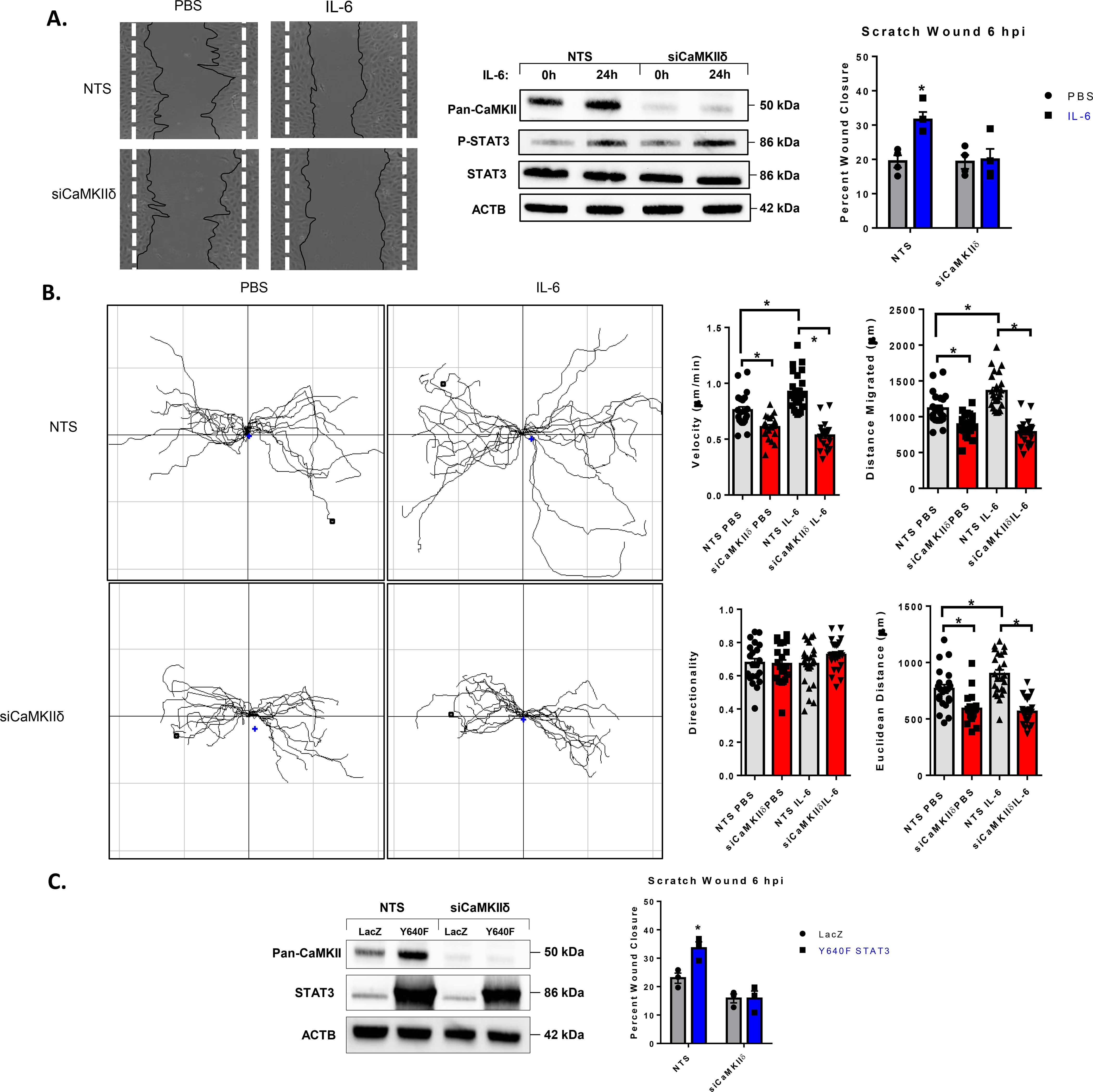
A. HUVEC seeded with NTS siRNA or siCaMKIIδ were grown to confluence, treated with IL-6/sIL-6r for 24h, and wounded with a P20 pipette tip. Original wound borders are marked by dashed white lines. Six hours post-injury, secondary 4x brightfield images were taken, wound edges are labelled by black tracings. Percent wound closure is determined by normalizing the remaining wound area to the initial wound area at time zero. Immunoblot of pan-CaMKII and P-STAT3 (Y705) were utilized to ensure silencing of CaMKIIδ and activation of STAT3. n =4, Two-way ANOVA, (*) p < 0.05. B. Time-lapse live imaging (20x) of leading-edge cells in both NTS and siCaMKIIδ-treated HUVEC following IL-6/sIL-6r treatment. Data acquired from manual tracking of cells was input into ImageJ Chemotaxis Tool plugin for velocity, total distance, Euclidean distance, and directionality. 22 PBS treated cells and 24 IL-6 treated cells total, n = 3, Two-way ANOVA, (*) p <0.05. C. The same scratch wound assay utilized in A. with AdY640F transfection in place of IL-6/sIL-6r treatment. n = 3, Two-way ANOVA, (*) p< 0.05.
CaMKIIδ regulates endothelial cell migration velocity and directionality.
To further establish the effect of STAT3/CaMKIIδ regulation on endothelial cell motility, we analyzed the effects of CaMKIIδ knockdown on individual cells from HUVEC cell lines expressing either a control shRNA (shN) or one targeted to CaMKIIδ. After sparse seeding, shN and shCaMKIIδ cells were placed in a stage-top incubator and tracked for 24h following treatment with either PBS or IL-6. CaMKIIδ knockdown attenuated both migration velocity as well as total Euclidean distance when compared to shN-expressing controls (Supplemental Fig. 3). However, in this experiment, IL-6 treatment did not have a significant effect on velocity, total distance, or Euclidean distance. One interpretation of this data may be that the primary effect of IL-6 on the endothelium is junctional integrity and is permissive to endothelial motility in the context of a wounded monolayer (Fig. 6).
Figure 6. CaMKIIδ promotes in vitro endothelial cell proliferation.
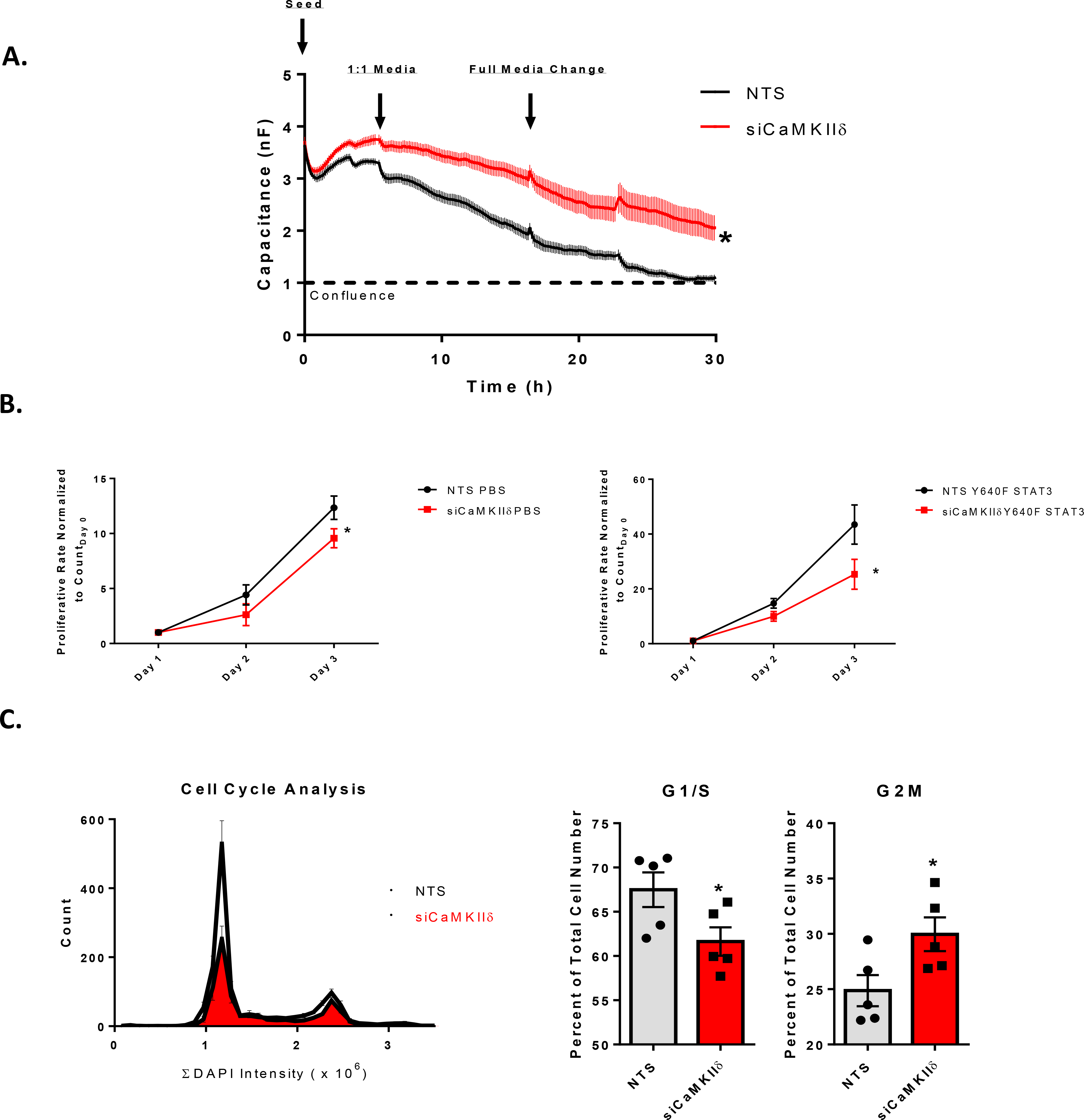
A. ECIS-based analysis of endothelial cell proliferation. 96W1E cultureware seeded with NTS or siCaMKIIδ-transfected HUVEC were seeded, supplemented with a 1:1 ratio of full growth media 6h later, and media changed the following morning. Measurement of capacitance for the first 30 hours reflects the rate of endothelial growth and coverage of the electrode. n = 3, Two-way ANOVA with repeated measures, (*) p < 0.05. B. Growth curve assay of HUVEC seeded with NTS or siCaMKIIδ. HUVEC were seeded at 10,000 cells in a 6-well plate and fixed, stained for DAPI, and counted (4x imaging) at one, two-, and three-days post-seeding. Cells were transfected with either AdLacZ or AdY640F on the first day after seeding. n = 3, Two-way ANOVA, (*) p <0.05. C. Cell cycle analysis performed with the Cytation5 plate reader and imager. Ibidi 8-well imaging slides were seeded at 10% confluence and allowed to grow for two days before fixation and staining for DAPI. DAPI+ nuclei were counted with the cellular analysis tool within the Cytation5 software, and integral DAPI content was computed and plotted and compared to number of cells within the field containing the same DAPI intensity. G1/S and G2M populations were extrapolated from this dataset and normalized to total cell number, giving relative populations within each group. n = 3, Two-way ANOVA, (*) p< 0.05.
CaMKIIδ promotes in vitro endothelial cell proliferation –
In order to test whether CaMKII is necessary for endothelial cell proliferation, we seeded 5,000 cells per well into 96W1E+ cultureware and transfected them with NTS of siRNA against CaMKIIδ. We measured the capacitance of gold electrodes as seeded HUVEC proliferated to reach confluence and found that the NTS treated cells reach confluence (capacitance = 1 nF) at approximately 26 hours post-seeding while the siCaMKIIδ cells were significantly delayed (Fig. 6A). This result may reflect either a deficit in endothelial cell proliferation or attachment and spreading. To test the role of CaMKIIδ in promoting proliferation more directly, we seeded 10,000 cells in each well of a 6-well dish with either NTS or siCaMKIIδ siRNA. We tracked these cells over the course of three additional days following transfection with either LacZ or Y640F STAT3-expressing adenovirus on the first day. Growth curve plots demonstrated that CaMKIIδ knockdown attenuates endothelial cell proliferation in both LacZ and Y640F transfected HUVEC (Fig. 6B). Additionally, using the Cytation5 plate reader and imager, we were able to quantify the integral DAPI content in either NTS of siCaMKIIδ-transfected HUVEC and found that silencing CaMKIIδ led to a decrease in cells found in the G1/S phase and a larger relative percentage of cells were in the G2M phase of the cell cycle (Fig. 6C). Taken together, these data suggest that CaMKIIδ promotes endothelial cell proliferation in vitro.
STAT3-dependent expression of CaMKIIδ promotes angiogenesis in vivo-
To test if STAT3 regulates CaMKIIδ expression in vivo to promote angiogenesis, we utilized the neonatal mouse retina as a model (Fig. 7A). We utilized both STAT3 f/f Cdh5 Cre and CaMKIIδf/f Cdh5 Cre for endothelial-specific knockout of STAT3 and CaMKIIδ. We extracted retina endothelial cells using anti-CD31-conjugated magnetic beads to isolate RNA and protein. Endothelial enrichment was validated by detection of endothelial marker gene expression Cdh5, Cd31, and Vwf and low expression of epithelial marker Cdh1 and mesenchymal marker Acta2. Consistent with the expression profiles in cultured HUVEC, we found that CaMKIIδ6 is the dominant isoform in the murine retinal endothelium based on qPCR and western blot analysis (Fig. 7B). In inducible STAT3 knockout animals, we found that CaMKIIδ transcript levels were significantly decreased relative to Cre (–) littermate controls (Fig. 7C). Additionally, STAT3 iEKO animals exhibited a delay in vascular outgrowth when visualized with AlexaFluor 488-conjugated Isolectin B4. The retinas were quantified by vessel area normalized to total retinal bed area and number of junctions were computed by Angiotool2.0. The STAT3 iEKO mice were phenocopied by the CaMKIIδ iEKO mice, presenting with an even more dramatic defect in vascular outgrowth and fewer junctions (Fig. 7D). Collectively, these data demonstrate that CaMKIIδ is the dominant isoform in murine endothelium, STAT3 is activated in the developing retina endothelium and promotes angiogenesis, STAT3 regulates CaMKIIδ expression in vivo, and CaMKIIδ is a critical promoter of developmental angiogenesis.
Figure 7. STAT3-dependent expression of CaMKIIδ promotes angiogenesis in vivo.
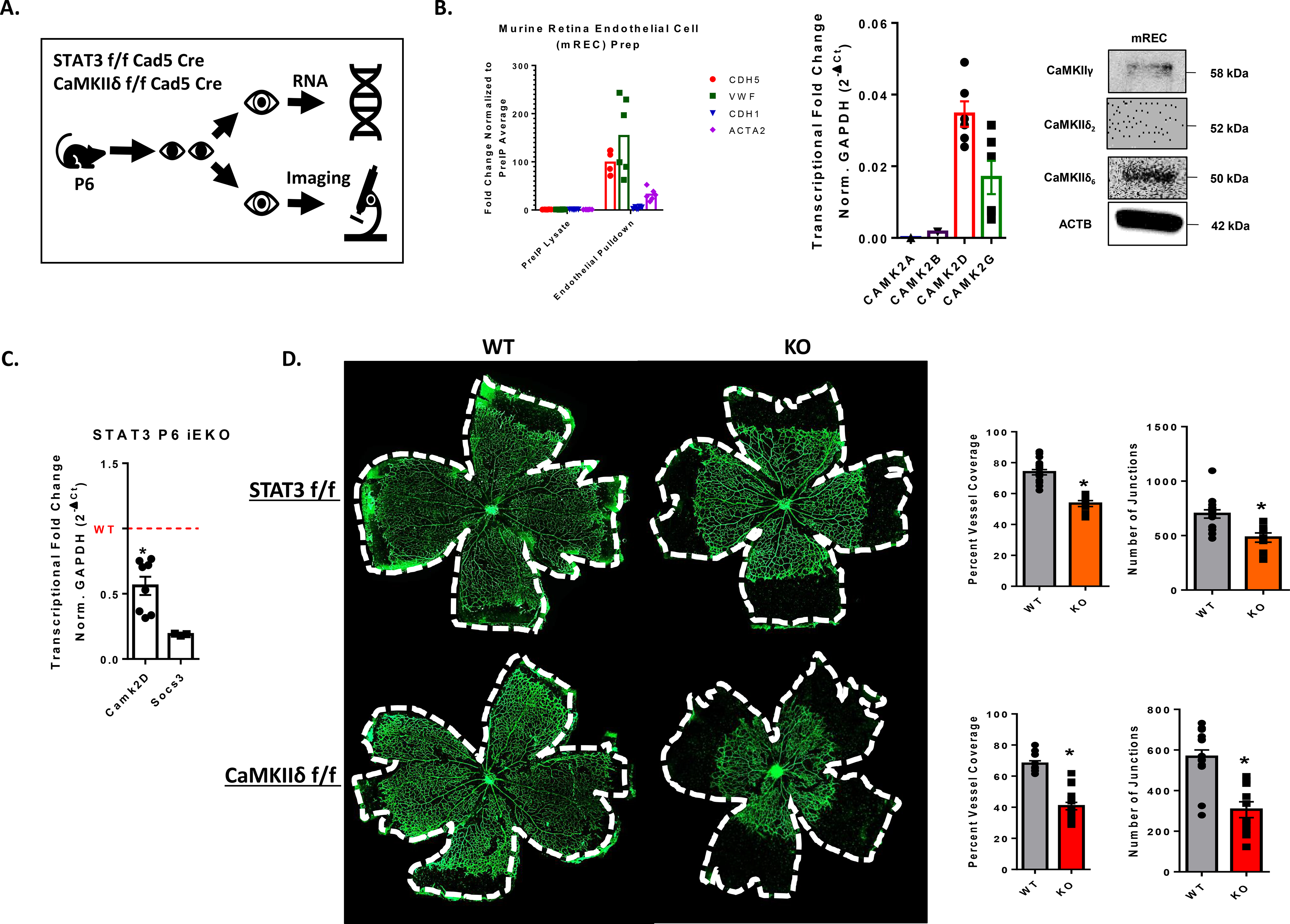
A. Schematic for strategy for measuring in vivo angiogenesis. Pups were oral gavaged with10 μL of 100 mg/mL tamoxifen at postnatal day 1, 2, and 3. At postnatal day 6, mice eyes were either collected and fixed for whole mount imaging or digested with collagenase solution and incubated with anti-CD31 beads for RNA isolation. B. qPCR analysis of endothelial marker genes (Cdh5, Vwf), epithelial marker Cdh1, and pericyte/VSM marker Acta2 expression in RNA isolated from antiCD31-pulldown lysates and non-bound fraction lysates extracted from murine retinas at postnatal day 6 (P6). qPCR analysis of Camk2a, Camk2b, Camk2d, and Camk2g expression in isolated murine retinal endothelial cells (mREC) normalized to GAPDH, n = 3. Western blot analysis of antiCD31-pulldown lysates with custom CaMKII antibodies targeted to CaMKIIδ2, CaMKIIδ6, and CaMKIIγ isoforms. n = 3. C. RNA from CD31-pulldown enriched retinal endothelial cells was used for qPCR analysis to detect relative amounts of CAMK2D in our inducible STAT3 loss of function animals. Data normalized to GAPDH transcript levels and Socs3 was utilized as a positive control for loss of STAT3 function. n = 3, Student’s t-test, (*) p< 0.05. D. Whole mount retinas from P6 endothelial-specific STAT3 loss of function mice and CaMKIIδ knockout mice. Retina vessels lableled with AlexaFluor488-IsolectinB4 and imaged using 4x tiling with the Cytation5 plate reader and imager. Percent Vessel coverage was calculated by normalizing the area covered by vessels to the area of the total retinal bed. Number of junctions was quantified using Angiotool2.0 software. n = 3 litters each, Student’s t-test, (*) p<0.05.
Discussion
In the present study we support that CaMKIIδ is the dominant isoform in human endothelium and identify for the first time that the tailless CaMKIIδ6 is also the main isoform in murine retina endothelial cells. Additionally, we identify novel induction of CaMKIIδ expression by the IL-6/STAT3 regulatory axis both in vitro and in vivo. STAT3 has been reported to regulate vascular permeability, wound healing, and angiogenesis (15,32,33). However, the specific downstream effectors of these processes other than VEGF-A, bFGF, and MMP’s (16,18) in the endothelium are yet to be identified. Based on our data, which demonstrates that CaMKIIδ is upregulated in a JAK/STAT3-dependent manner in vitro and in vivo and is required for endothelial cell migration, proliferation, and in vivo retina angiogenesis, we propose that CaMKIIδ belongs in that STAT3-dependent proangiogenic gene set.
RNAseq data from cultured HUVEC identified that CaMKIIδ expression was transcriptionally responsive to 3 hours of IL-6 treatment and sensitive to JAK inhibition. qPCR data was consistent with the RNAseq data: transcript levels increasing by about 3-fold following IL-6 treatment. We then asked whether the CaMKIIδ6 splice variant remained the dominant isoform as reported (10) following IL-6 treatment. Immunoblot with antibodies against the alternatively spliced C-terminal tail of the enzyme revealed that, indeed, CaMKIIδ6 is the dominant isoform in the human endothelium and is the isoform upregulated by IL-6 treatment. CaMKIIγ, which is also expressed in the endothelium, was not changed following treatment with IL-6 at the message or protein level. Additionally, we gained novel insight into the CaMKII isoform distribution in the murine retina, where CaMKIIδ6 and CaMKIIγB are also the predominantly expressed splice isoforms. We focused on comparing relative abundance of CaMKIIδ2 and CaMKIIδ6 expression in HUVEC and mREC (Fig. 1) because of our experience studying them as the predominant functional isoforms in VSM and endothelial cells, respectively (5,10).
Our data demonstrated that CaMKIIδ expression was upregulated in an IL-6/JAK/STAT3-dependent manner using RNAseq, qPCR, and immunoblot. The predominant functional role for this pathway in the endothelium is to increase vascular permeability and has been demonstrated to lead to a 40% decrease in barrier function (15). Despite the accepted role for activated STAT3 signaling and data from our lab demonstrating that CaMKIIδ is required for thrombin-induced permeability, we found that neither overexpression nor silencing of CaMKIIδ had a significant effect on monolayer permeability. CaMKII is a multifunctional kinase: very rarely is it the lone mediator of any one cellular process and its function is highly context specific. There are likely many other mediators associated with both the thrombin and IL-6-dependent signaling pathways that govern unique functional roles for CaMKIIδ. It is possible that CaMKIIδ has a role in thrombin-induced permeability as a result of intracellular Ca2+ flux (34), whereas IL-6 binding to its receptor is not known to directly elicit a Ca2+ response. Instead of directly activating the enzyme, overall levels of CaMKIIδ are increased following IL-6 stimulation. Our plan is to continue investigating whether increasing the abundance of CaMKIIδ with activated STAT3 signaling potentiates the endothelium to a secondary Ca2+-dependent agonist like VEGF-A or thrombin. It has been reported by our lab that CaMKIIδ and γ have seemingly antagonistic roles in the context of VSM remodeling in vivo (8,9,35). Additionally, a recent publication proposes that CaMKIIδ is preferentially activated by VEGF and bFGF while CaMKIIγ is more responsive to IGF-1 (11). We may therefore investigate whether increasing CaMKIIδ with IL-6 treatment sensitizes the endothelium to VEGF and bFGF-dependent signaling and if IGF-1 signaling is dampened by increasing the ratio of CaMKIIδ to CaMKIIγ. This data may be critical for our understanding integration of inflammatory and proangiogenic signaling pathways.
In the context of an in vitro scratch wound, we found that silencing of CaMKIIδ significantly decreased the rate of wound closure and the velocity of leading-edge cell migration. The same is true in the supplemental single cell analysis: where velocity and total Euclidean distance are significantly decreased in shCaMKIIδ-expressing cells. What differs, however, is the primary effect of IL-6 treatment on endothelial cell motility. In the single-cell assay, we found that IL-6 treatment did not have a significant effect on migration velocity, total distance or Euclidean distance, unlike in the scratch wound assay where pretreatment with IL-6 promoted wound closure. We postulate that this discrepancy exists due to the nature of the assays themselves, as IL-6 has a key accepted role of regulating endothelial junctional integrity (15) and intercellular junction dynamics do not affect the outcome of single-cell analysis. Collectively, from these data we suggest that IL-6/STAT3 regulates endothelial cell motility partly through upregulation of CaMKIIδ and partly through its effects on intercellular junction integrity.
Our data has also demonstrated that CaMKIIδ regulates endothelial cell proliferation. When seeded at 5,000 cells HUVEC per well, cells transfected with siCaMKIIδ reached confluence at half the rate of NTS-treated cells. Cell counting and cell cycle analysis also confirmed these findings. We found that there was no significant change in proliferation following treatment with IL-6 in the same proliferation assay (data not shown). We still observed an independent effect on HUVEC proliferation by silencing CaMKIIδ with siRNA. Based on these data we concluded that CaMKIIδ is required in part for basal growth-media stimulated endothelial cell proliferation and STAT3-dependent CaMKIIδ expression promotes proliferation, but IL-6 itself had no significant effect. We found a larger percentage of cells that were stuck in the G2M phase of the cell cycle in siCaMKIIδ cells. Our lab has observed this distribution in VSM, where a higher number of multinucleated cells was also observed. This suggests that CaMKIIδ regulates a key cytokinetic element such as a nuclear phosphatase or a cytoskeletal element; potentially actomyosin. CaMKII has also been suggested to play key roles in regulating apoptosis and necroptosis in the context of ischemia/reperfusion injury in the heart (36,37). Therefore, we plan to investigate whether the observed deficit in cell number observed with silencing of CaMKIIδ is due in part to downstream effects on endothelial cell death.
Our data demonstrates that activation of STAT3 induces CaMKIIδ expression in the endothelium and functionally promotes cell motility and proliferation. However, the downstream effector by which STAT3-dependent CaMKIIδ functions is still an active area of investigation by our lab. Given our lab’s experience studying CaMKIIδ’s role in promoting VSM motility and proliferation, one candidate signaling molecule is ERK1/2 (2,7). However, in our only other study on CaMKIIδ in the endothelium, we found that CaMKIIδ promotes thrombin-induced permeability in an ERK1/2-independent manner (10). Additionally, while collecting data for this study we have found that constitutively active STAT3 promotes ERK1/2 phosphorylation (T202/Y204) in a CaMKIIδ-independent manner (data not shown). We therefore will continue investigating the downstream signaling events following STAT3-dependent CaMKII expression. Some other potential effectors downstream of CaMKIIδ have been suggested in a previous study investigating a role of CaMKII in pathological angiogenesis including JNK, Src, FAK, and β-catenin (11). Additionally, we acknowledge that angiogenesis is a coordinated process of both cytoskeletal rearrangement and transcriptional changes regulating cell identity and differentiation. While CaMKIIδ has been demonstrated to regulate transcription through phosphorylation and regulation of HDAC4/5 subcellular localization (38), CaMKIIδ-dependent gene expression in the context of angiogenesis has not been investigated and will be a central focus of our work going forward.
Finally, we identified a key role for the STAT3/CaMKIIδ regulatory axis in the in vivo model of retina angiogenesis. STAT3 iEKO animals exhibit delayed vascular outgrowth at postnatal day 6 and decreased expression of CaMKIIδ according to qPCR of RNA enriched from antiCD31-enrichment of retinal endothelial cells. Additionally, CaMKIIδ iEKO animals display delayed vascular outgrowth and decreased number of junctions. These data demonstrate that STAT3 activation promotes CaMKIIδ expression in vivo, and the STAT3/CaMKIIδ regulatory axis regulates in vivo angiogenesis. This study utilized IL-6 as an activator of STAT3 in order to study STAT3-dependent CaMKIIδ expression and its functional role in the endothelium. A clear role for IL-6 specifically was not well elucidated. IL-6 has been demonstrated to be a critical molecular in retinal inflammation and pathological angiogenesis (19,39,40), not necessarily canonical processes of development such as the ones studied here. We therefore propose that investigating IL-6’s role in stimulating this pathway would be more relevant to an inflammatory angiogenesis-related disease model such as retinopathy of prematurity, tumor metastasis, or abdominal aortic aneurysm (AAA).
We also plan on investigating the differential roles of the CaMKIIδ and CaMKIIγ isoforms in regulating angiogenesis. Our lab has demonstrated that CaMKIIδ and γ have reciprocal functions in the context of vascular remodeling following injury: where CaMKIIδ promotes migration and proliferation of synthetic phenotype VSM cells which is negatively regulated by CaMKIIγ (2,8,35). In the endothelium, CaMKIIδ and γ have been demonstrated to have varying sensitivities to growth factors, as mentioned above. Additionally, using an oxygen-induced retinopathy model, it has been demonstrated that global CaMKIIδ knockout animals display decreased preretinal neovascularization and reparative angiogenesis, where global CaMKIIγ knockout animals also display decreased preretinal neovascularization but show increased reparative angiogenesis in ischemic regions (11). So far, our lab has collected substantial preliminary data supporting these studies. Using our inducible endothelial-specific knockout animals, we plan on investigating the roles of each isoform at multiple stages of development and using high-throughput proteomic and transcriptomic approaches to interrogate the unique functional roles of the CaMKIIδ and CaMKIIγ isoforms in the endothelium during angiogenesis.
In summary, these data have identified novel transcriptional regulation of CaMKIIδ in the endothelium by activation of the JAK/STAT3 signaling axis. Additionally, these data suggest that CaMKIIδ’s role in regulating vascular permeability may be agonist-specific, since previously published data from our lab has established a role for CaMKIIδ in thrombin-induced permeability but no significant effect was observed on IL-6-induced permeability with overexpression or silencing of the enzyme. This study also demonstrates novel STAT3-dependent regulation of CaMKIIδ in vivo, which is critical for regulating developmental angiogenesis. Not only does this elucidate a key angiogenic effector downstream of STAT3, but it also suggests that CaMKIIδ is a suitable therapeutic target for diseases of hypervascularity. One such disease is abdominal aortic aneurysm (AAA), a progressive vascular disease that is common in elderly males and is responsible for around 5,000 deaths in the United States annually (41). An underappreciated contributor to AAA progress is neovascularization of the medial wall, where the hypoxic environment of the aneurysm promotes angiogenesis, exacerbating immune cell recruitment and matrix destruction. (20). It has also been demonstrated that proinflammatory signaling pathways such as IL-6/JAK/STAT3 contribute positively to AAA rupture (42,43). Thus, we will be testing our hypothesis that STAT3/CaMKIIδ promotes medial neovascularization and progression of AAA in a subsequent study.
Supplementary Material
Acknowledgements:
The authors would like to thank Nina Martino for her insight and expertise in HUVEC cell culture and in vitro protocols for IL-6-dependent signaling. We would also like to thank Dr. Qingfen Li for her guidance in the murine retina angiogenesis assay.
Nonstandard Abbreviations:
- CaMKII
Ca2+/Calmodulin-dependent protein kinase II
- IL-6
interleukin-6
- JAK
janus kinase
- STAT3
signal transducer and activator of transcription 3
- SOCS3
suppressor of cytokine signaling 3
- ECIS
electrical cell-substrate impedance sensing
- HUVEC
human umbilical vein endothelial cells
- mREC
murine retina endothelial cells
- NTS
nontargeting sequence
- Cre
Cre recombinase
- iEKO
inducible endothelial-specific knockout
- P6
postnatal day 6
Footnotes
Conflict of Interest: The listed authors have no conflict of interest.
References
- 1.Bayer KU, and Schulman H (2001) Regulation of signal transduction by protein targeting: the case for CaMKII. Biochem Biophys Res Commun 289, 917–923 [DOI] [PubMed] [Google Scholar]
- 2.Singer HA (2012) Ca2+/calmodulin-dependent protein kinase II function in vascular remodelling. J Physiol 590, 1349–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Erickson JR (2014) Mechanisms of CaMKII Activation in the Heart. Front Pharmacol 5, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saneyoshi T, Matsuno H, Suzuki A, Murakoshi H, Hedrick NG, Agnello E, O’Connell R, Stratton MM, Yasuda R, and Hayashi Y (2019) Reciprocal Activation within a Kinase-Effector Complex Underlying Persistence of Structural LTP. Neuron 102, 1199–1210 e1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mercure MZ, Ginnan R, and Singer HA (2008) CaM kinase II delta2-dependent regulation of vascular smooth muscle cell polarization and migration. Am J Physiol Cell Physiol 294, C1465–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saddouk FZ, Ginnan R, and Singer HA (2017) Ca(2+)/Calmodulin-Dependent Protein Kinase II in Vascular Smooth Muscle. Adv Pharmacol 78, 171–202 [DOI] [PubMed] [Google Scholar]
- 7.Ginnan R, and Singer HA (2002) CaM kinase II-dependent activation of tyrosine kinases and ERK1/2 in vascular smooth muscle. Am J Physiol Cell Physiol 282, C754–761 [DOI] [PubMed] [Google Scholar]
- 8.House SJ, Ginnan RG, Armstrong SE, and Singer HA (2007) Calcium/calmodulin-dependent protein kinase II-delta isoform regulation of vascular smooth muscle cell proliferation. Am J Physiol Cell Physiol 292, C2276–2287 [DOI] [PubMed] [Google Scholar]
- 9.House SJ, and Singer HA (2008) CaMKII-delta isoform regulation of neointima formation after vascular injury. Arterioscler Thromb Vasc Biol 28, 441–447 [DOI] [PubMed] [Google Scholar]
- 10.Wang Z, Ginnan R, Abdullaev IF, Trebak M, Vincent PA, and Singer HA (2010) Calcium/Calmodulin-dependent protein kinase II delta 6 (CaMKIIdelta6) and RhoA involvement in thrombin-induced endothelial barrier dysfunction. J Biol Chem 285, 21303–21312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ashraf S, Bell S, O’Leary C, Canning P, Micu I, Fernandez JA, O’Hare M, Barabas P, McCauley H, Brazil DP, Stitt AW, McGeown JG, and Curtis TM (2019) CAMKII as a therapeutic target for growth factor-induced retinal and choroidal neovascularization. JCI Insight 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, Kishimoto T, and Akira S (1997) Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci U S A 94, 3801–3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takeda K, and Akira S (2000) STAT family of transcription factors in cytokine-mediated biological responses. Cytokine Growth Factor Rev 11, 199–207 [DOI] [PubMed] [Google Scholar]
- 14.Hirano T (2010) Interleukin 6 in autoimmune and inflammatory diseases: a personal memoir. Proc Jpn Acad Ser B Phys Biol Sci 86, 717–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alsaffar H, Martino N, Garrett JP, and Adam AP (2018) Interleukin-6 promotes a sustained loss of endothelial barrier function via Janus kinase-mediated STAT3 phosphorylation and de novo protein synthesis. Am J Physiol Cell Physiol 314, C589–C602 [DOI] [PubMed] [Google Scholar]
- 16.Chen Z, and Han ZC (2008) STAT3: a critical transcription activator in angiogenesis. Med Res Rev 28, 185–200 [DOI] [PubMed] [Google Scholar]
- 17.Gao P, Niu N, Wei T, Tozawa H, Chen X, Zhang C, Zhang J, Wada Y, Kapron CM, and Liu J (2017) The roles of signal transducer and activator of transcription factor 3 in tumor angiogenesis. Oncotarget 8, 69139–69161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei D, Le X, Zheng L, Wang L, Frey JA, Gao AC, Peng Z, Huang S, Xiong HQ, Abbruzzese JL, and Xie K (2003) Stat3 activation regulates the expression of vascular endothelial growth factor and human pancreatic cancer angiogenesis and metastasis. Oncogene 22, 319–329 [DOI] [PubMed] [Google Scholar]
- 19.Kayakabe K, Kuroiwa T, Sakurai N, Ikeuchi H, Kadiombo AT, Sakairi T, Matsumoto T, Maeshima A, Hiromura K, and Nojima Y (2012) Interleukin-6 promotes destabilized angiogenesis by modulating angiopoietin expression in rheumatoid arthritis. Rheumatology (Oxford) 51, 1571–1579 [DOI] [PubMed] [Google Scholar]
- 20.Kaneko H, Anzai T, Takahashi T, Kohno T, Shimoda M, Sasaki A, Shimizu H, Nagai T, Maekawa Y, Yoshimura K, Aoki H, Yoshikawa T, Okada Y, Yozu R, Ogawa S, and Fukuda K (2011) Role of vascular endothelial growth factor-A in development of abdominal aortic aneurysm. Cardiovasc Res 91, 358–367 [DOI] [PubMed] [Google Scholar]
- 21.Stahl A, Joyal JS, Chen J, Sapieha P, Juan AM, Hatton CJ, Pei DT, Hurst CG, Seaward MR, Krah NM, Dennison RJ, Greene ER, Boscolo E, Panigrahy D, and Smith LE (2012) SOCS3 is an endogenous inhibitor of pathologic angiogenesis. Blood 120, 2925–2929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chung AS, and Ferrara N (2011) Developmental and pathological angiogenesis. Annu Rev Cell Dev Biol 27, 563–584 [DOI] [PubMed] [Google Scholar]
- 23.Geudens I, and Gerhardt H (2011) Coordinating cell behaviour during blood vessel formation. Development 138, 4569–4583 [DOI] [PubMed] [Google Scholar]
- 24.Hamik A, Wang B, and Jain MK (2006) Transcriptional regulators of angiogenesis. Arterioscler Thromb Vasc Biol 26, 1936–1947 [DOI] [PubMed] [Google Scholar]
- 25.Fantin A, and Ruhrberg C (2015) The Embryonic Mouse Hindbrain and Postnatal Retina as In Vivo Models to Study Angiogenesis. Methods Mol Biol 1332, 177–188 [DOI] [PubMed] [Google Scholar]
- 26.Toussaint F, Charbel C, Blanchette A, and Ledoux J (2015) CaMKII regulates intracellular Ca(2)(+) dynamics in native endothelial cells. Cell Calcium 58, 275–285 [DOI] [PubMed] [Google Scholar]
- 27.Hou T, Tieu BC, Ray S, Recinos Iii A, Cui R, Tilton RG, and Brasier AR (2008) Roles of IL-6-gp130 Signaling in Vascular Inflammation. Curr Cardiol Rev 4, 179–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walker S, Wang C, Walradt T, Hong BS, Tanner JR, Levinsohn JL, Goh G, Subtil A, Lessin SR, Heymann WR, Vonderheid EC, King BA, Lifton RP, and Choi J (2016) Identification of a gain-of-function STAT3 mutation (p.Y640F) in lymphocytic variant hypereosinophilic syndrome. Blood 127, 948–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Babon JJ, Varghese LN, and Nicola NA (2014) Inhibition of IL-6 family cytokines by SOCS3. Semin Immunol 26, 13–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheneby J, Menetrier Z, Mestdagh M, Rosnet T, Douida A, Rhalloussi W, Bergon A, Lopez F, and Ballester B (2020) ReMap 2020: a database of regulatory regions from an integrative analysis of Human and Arabidopsis DNA-binding sequencing experiments. Nucleic Acids Res 48, D180–D188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Folkman J (2006) Angiogenesis. Annu Rev Med 57, 1–18 [DOI] [PubMed] [Google Scholar]
- 32.Gallucci RM, Simeonova PP, Matheson JM, Kommineni C, Guriel JL, Sugawara T, and Luster MI (2000) Impaired cutaneous wound healing in interleukin-6-deficient and immunosuppressed mice. FASEB J 14, 2525–2531 [DOI] [PubMed] [Google Scholar]
- 33.Gopinathan G, Milagre C, Pearce OM, Reynolds LE, Hodivala-Dilke K, Leinster DA, Zhong H, Hollingsworth RE, Thompson R, Whiteford JR, and Balkwill F (2015) Interleukin-6 Stimulates Defective Angiogenesis. Cancer Res 75, 3098–3107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Minami T, Sugiyama A, Wu SQ, Abid R, Kodama T, and Aird WC (2004) Thrombin and phenotypic modulation of the endothelium. Arterioscler Thromb Vasc Biol 24, 41–53 [DOI] [PubMed] [Google Scholar]
- 35.Saddouk FZ, Sun LY, Liu YF, Jiang M, Singer DV, Backs J, Van Riper D, Ginnan R, Schwarz JJ, and Singer HA (2016) Ca2+/calmodulin-dependent protein kinase II-gamma (CaMKIIgamma) negatively regulates vascular smooth muscle cell proliferation and vascular remodeling. FASEB J 30, 1051–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vila-Petroff M, Salas MA, Said M, Valverde CA, Sapia L, Portiansky E, Hajjar RJ, Kranias EG, Mundina-Weilenmann C, and Mattiazzi A (2007) CaMKII inhibition protects against necrosis and apoptosis in irreversible ischemia-reperfusion injury. Cardiovasc Res 73, 689–698 [DOI] [PubMed] [Google Scholar]
- 37.Salas MA, Valverde CA, Sanchez G, Said M, Rodriguez JS, Portiansky EL, Kaetzel MA, Dedman JR, Donoso P, Kranias EG, and Mattiazzi A (2010) The signalling pathway of CaMKII-mediated apoptosis and necrosis in the ischemia/reperfusion injury. J Mol Cell Cardiol 48, 1298–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ginnan R, Sun LY, Schwarz JJ, and Singer HA (2012) MEF2 is regulated by CaMKIIdelta2 and a HDAC4-HDAC5 heterodimer in vascular smooth muscle cells. Biochem J 444, 105–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tzeng HE, Tsai CH, Chang ZL, Su CM, Wang SW, Hwang WL, and Tang CH (2013) Interleukin-6 induces vascular endothelial growth factor expression and promotes angiogenesis through apoptosis signal-regulating kinase 1 in human osteosarcoma. Biochem Pharmacol 85, 531–540 [DOI] [PubMed] [Google Scholar]
- 40.Rojas M, Zhang W, Lee DL, Romero MJ, Nguyen DT, Al-Shabrawey M, Tsai NT, Liou GI, Brands MW, Caldwell RW, and Caldwell RB (2010) Role of IL-6 in angiotensin II-induced retinal vascular inflammation. Invest Ophthalmol Vis Sci 51, 1709–1718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aggarwal S, Qamar A, Sharma V, and Sharma A (2011) Abdominal aortic aneurysm: A comprehensive review. Exp Clin Cardiol 16, 11–15 [PMC free article] [PubMed] [Google Scholar]
- 42.Kokje VBC, Gabel G, Koole D, Northoff BH, Holdt LM, Hamming JF, and Lindeman JHN (2016) IL-6: A Janus-like factor in abdominal aortic aneurysm disease. Atherosclerosis 251, 139–146 [DOI] [PubMed] [Google Scholar]
- 43.Xiao J, Wei Z, Chen X, Chen W, Zhang H, Yang C, Shang Y, and Liu J (2020) Experimental abdominal aortic aneurysm growth is inhibited by blocking the JAK2/STAT3 pathway. Int J Cardiol 312, 100–106 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.