

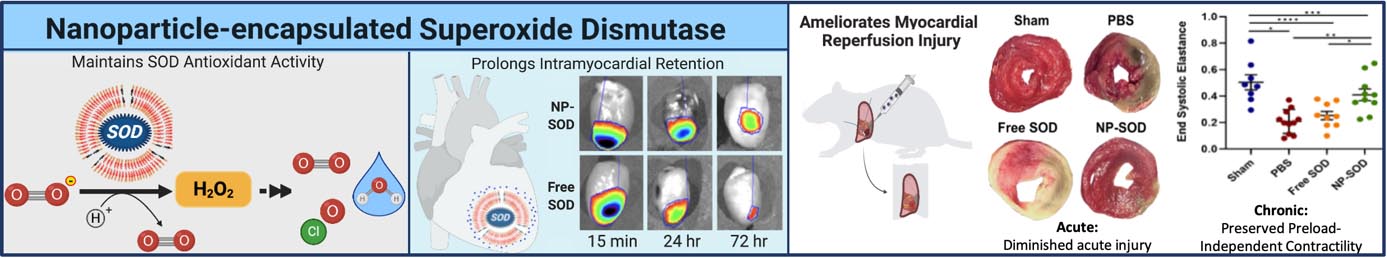
Abstract
Early revascularization is critical to reduce morbidity after myocardial infarction, although reperfusion incites additional oxidative injury. Superoxide dismutase (SOD) is an antioxidant that scavenges reactive oxygen species (ROS) but has low endogenous expression and rapid myocardial washout when administered exogenously. This study utilizes a novel nanoparticle carrier to improve exogeneous SOD retention while preserving enzyme function. Its role is assessed in preserving cardiac function after myocardial ischemia-reperfusion (I/R) injury. Here, nanoparticle-encapsulated SOD (NP-SOD) exhibits similar enzyme activity as free SOD, measured by ferricytochrome-c assay. In an in vitro I/R model, free and NP-SOD reduce active ROS, preserve mitochondrial integrity and improve cell viability compared to controls. In a rat in vivo I/R injury model, NP-encapsulation of fluorescent-tagged SOD improves intramyocardial retention after direct injection. Intramyocardial NP-SOD administration in vivo improves left ventricular contractility at 3-hours post-reperfusion by echocardiography and 4-weeks by echocardiography and invasive pressure-volume catheter analysis. These findings suggest that NP-SOD mitigates ROS damage in cardiac I/R injury in vitro and maximizes retention in vivo. NP-SOD further attenuates acute injury and protects against myocyte loss and chronic adverse ventricular remodeling, demonstrating potential for translating NP-SOD as a therapy to mitigate myocardial I/R injury.
Keywords: ischemia-reperfusion injury, superoxide dismutase, nanoparticle, semipermeable, reactive oxygen species
Graphical Abstract
Encapsulating superoxide dismutase, a potent but transitory antioxidant, within a semipermeable nanoparticle construct protects from proteolytic cleavage while maintaining its antioxidant activity. In an in vivo model of myocardial ischemia/reperfusion injury, direct intramyocardial administration of this antioxidant-nanoparticle construct protects myocardium against damage from reactive oxygen species. This preserves ventricular structure and function in both acute and chronic settings.
1. Introduction
Ischemic heart disease remains the leading cause of morbidity and mortality worldwide, with over seven million people experiencing a myocardial infarction (MI) annually.[1,2] Untreated, mortality associated with MI exceeds 30%.[3] Current treatment paradigms prioritize urgent revascularization through percutaneous coronary intervention (PCI), which are critical in limiting ischemic injury and preserving cardiac function. Reperfusion, however, generates additional cellular injury due to a buildup of reactive oxygen species (ROS) related to an abrupt change from a hypoxic to a hyperoxic milieu.[4]
ROS accumulate both intracellularly within the cardiomyocytes and the vascular endothelium, as well as extracellularly as the immune system undergoes oxidative burst.[4–6] Intracellularly, ROS disrupt Ca2+ equilibrium, impair paracrine signaling, and result in cell death.[7] This occurs through depletion of the sarcoplasmic reticulum and increased Ca2+ ion flux between depolarized mitochondria and the cytosol, opening the mitochondrial permeability transition pore (mPTP) and dissipating the chemiosmotic gradient required for ATP production.[8–12] Increased cytosolic Ca2+ and depleted oxidative phosphorylation have also been linked to decrements in contractility and viability in cardiomyocytes secondary to ischemia-reperfusion (I/R) injury.[13,14]
Innate protective mechanisms exist to attenuate I/R injury. One important early ROS scavenging enzyme is superoxide dismutase (SOD), which functions inside the mitochondria and cytosol and catalyzes the conversion of free radical superoxide (O2•−) into hydrogen peroxide (H2O2) in a pathway ultimately yielding oxygen and water. Given its potential to act early in mitigating oxidative damage, SOD is an attractive potential therapeutic target in reducing I/R injury. Recombinant SOD has been shown to reduce free radical accumulation in isolated rabbit myocardium[15,16] and mitigate I/R injury in cardiomyocytes,[17] while in vivo I/R injury was attenuated in transgenic mice overexpressing SOD.[18]
Despite its potent antioxidant properties, use of exogeneous SOD has yielded inconsistent results.[15,19,20] This has been attributed to the enzyme’s intrinsic properties, namely, a short half-life, rapid tissue washout, and limited membrane permeability.[21–23]
Coupling antioxidant enzymes to carrier vehicles has been increasingly used to enhance enzyme bioavailability. Nanoparticle carriers have been shown to protect enzymes against proteolysis in neuronal I/R injury and other models of oxidative stress,[24–29] similar to larger microparticles and polyethylene-glycol (PEG)-based delivery modalities.[30,31] This study utilized a novel NP-encapsulated SOD (NP-SOD) which, unlike many other carriers, allows for delivery of unmodified enzyme, protection from proteolysis, and access to ROS via a highly porous membrane. Previously shown to be beneficial in a model of neurologic injury,[24] this antioxidant-nanoparticle construct was adapted for use in cardiac I/R injury. The goal of this study was to evaluate the efficacy of NP-SOD in reducing myocardial I/R injury through enhanced enzyme stability and bioavailability.
2. Results
2.1. SOD-loaded porous polymersomes retain SOD while maintaining enzyme activity
NP-SOD was developed by containing SOD within polymersomes composed of 75 mol% Poly(ethylene glycol) (900)-polybutadiene (1800) copolymer (denoted PEG-PBD) / 25 mol% poly(ethylene glycol) (450)-poly(propylene oxide) (1400) copolymer (denoted PEG-PPO), Figure 1A. The SOD encapsulation efficiency within these NPs was ~ 20.17%. Dynamic light scattering (DLS) showed that NP-SOD had a mean diameter of 116 nm (Figure 1B). Transmission electron microscopy (TEM) further confirmed the formation of nanoparticles, Supplementary Figure S1. As shown in Figure 1C, SOD displayed high catalytic activity following nanoparticle encapsulation, indicating that superoxide radicals can access the encapsulated SOD through the porous membrane of PEG-PBD/PEG-PPO polymersomes. The activity of SOD following disruption of the polymersome with Triton X-100 was similar to the activity of NP-SOD without Triton X-100 treatment. In contrast, when SOD was encapsulated in nonporous polymersomes made from 100 mol% PEG-PBD, SOD activity was only detected after disrupting the nonporous polymersomes with Triton X-100 treatment. These results indicate that SOD-encapsulated PEG-PBD/PEG-PPO polymersomes provide a permeable membrane that allows free superoxide radicals to pass into the aqueous interior and interact with the encapsulated antioxidant enzyme SOD. In addition, DLS measurements showed that there were no significant changes in the hydrodynamic diameter when NP-SOD were incubated in phosphate buffered saline (PBS) for 1 week (Figure 1D) and in serum for 24 hours (Figure 1E).
Figure 1. Porous nanoparticle construct retains SOD while maintaining activity.
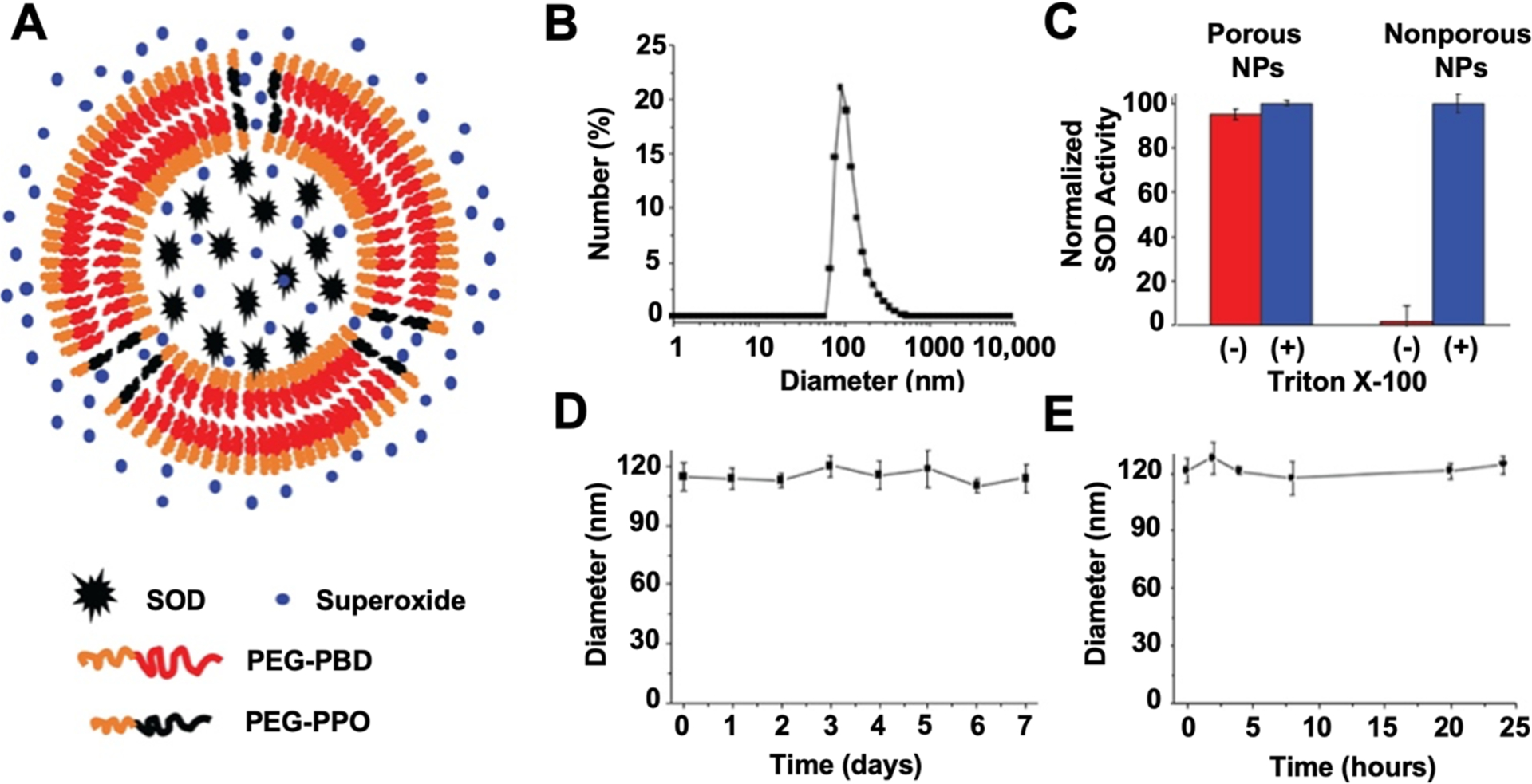
A) SOD remains encapsulated within a porous nanoparticle, composed of PEG-PBD and PEG-PPO, while free superoxide can diffuse through the pores. B) Diameter of NP-SOD as measured by DLS. C) SOD activity was tested before and after the treatment of porous and non-porous nanoparticles, with encapsulated SOD, with Triton X-100. Triton X-100 is used to disrupt the nanoparticle membranes and release the encapsulated SOD. Non-porous NP-SOD were composed entirely of PEG-PBD. D) Stability of NP-SOD in PBS for 1 week. E) Stability of NP-SOD in serum for 24 hours.
2.2. SOD provides protection against cellular oxidative stress in vitro
The therapeutic effect of SOD in an in vitro model of I/R was assessed using the general oxidative stress fluorescence indicator dye H2DCFDA. ROS cleavage of H2DCFDA was diminished and fluorescent intensity reduced in free and NP-SOD treated cells compared to those treated with empty NP or PBS, Figure 2A/B. Cellular redox state was further assessed via mitochondrial membrane permeability. As mPTP opening is an early and lethal event in the cellular mechanisms behind ROS damage,[9,13] preservation of membrane polarization indicates a more favorable redox state. JC-1 aggregate/monomer intensity ratios in untreated, uninjured cells was 2.48; NP-SOD (2.47) preserved mitochondrial membrane potential compared to free SOD (1.56, p=0.003), empty NP (0.96, p<0.001) and PBS (0.66, p<0.001; Figure 2C/D). Cytotoxicity of NP-SOD, free SOD and empty NP were additionally analyzed by measuring cardiomyoblast lactate dehydrogenase (LDH) release while cell viability was measured through 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. At 24 hours, NP-SOD, free SOD and empty NP-treated cells demonstrated comparable, minimal cytotoxicity and cell viability to PBS-treated cells, Supplementary Figure S2.
Figure 2. SOD minimizes oxidative injury in H9C2 cells following simulation of I/R injury.
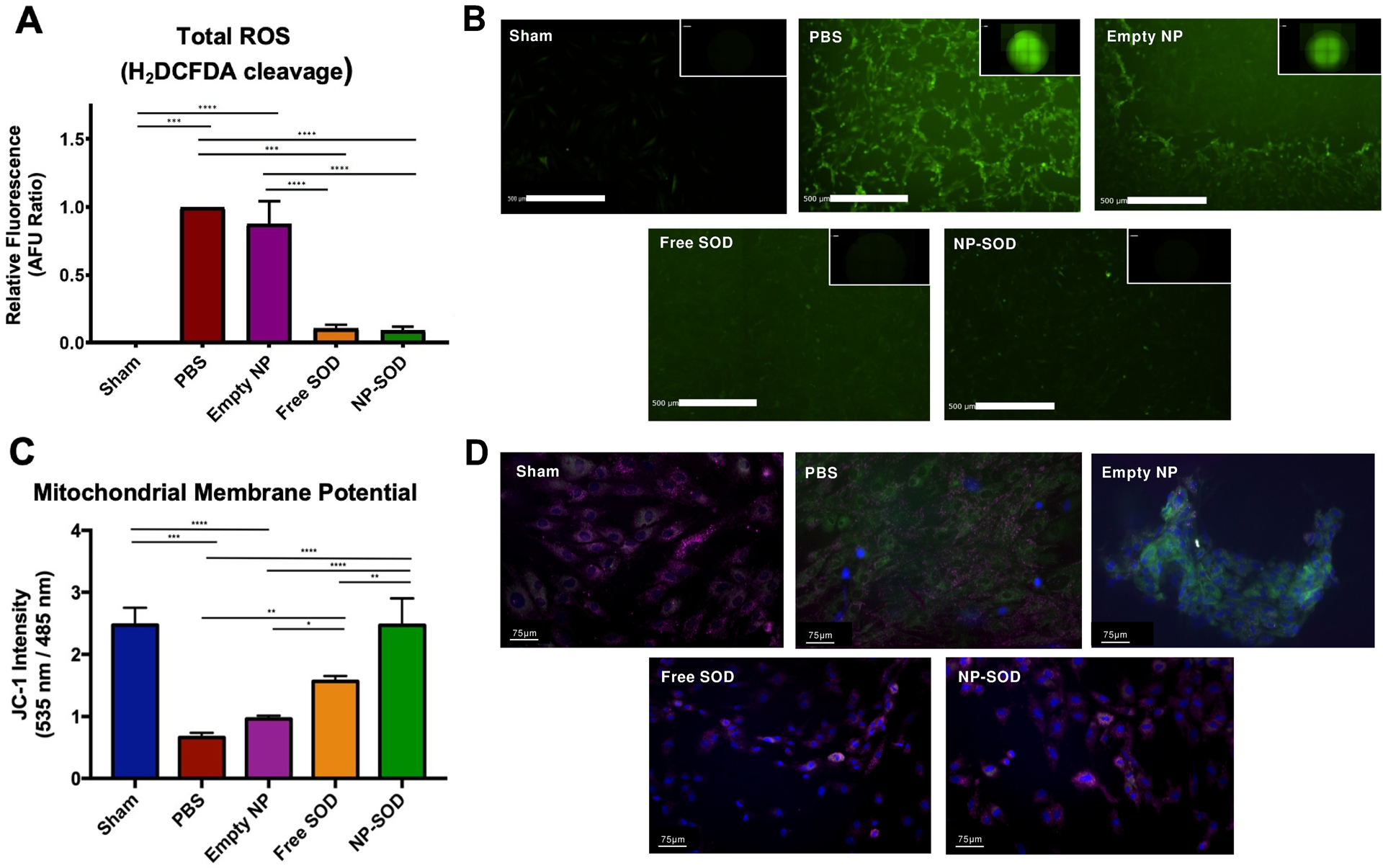
H9C2 cells were subjected to 1 hour of ischemia (0.5% O2) prior to administration of treatment groups and were returned to normoxic conditions (21% O2). A-B) Presence of ROS free radicals at 3 hours post I/R was quantified by fluorescence intensity (Ex:485 nm) produced by cleavage of H2DCFDA. C-D) H9C2 cells were seeded in multi-well chamber slides and subjected to either normoxic or I/R conditions before staining with JC-1. Samples were then mounted with DAPI and imaged on a Leica fluorescent microscope at 405/485/535 nm at 10, 40, and 100X magnifications. Image intensities of each excitation wavelength were quantified utilizing an automated ImageJ algorithm and set as a ratio of JC-1 aggregates (pink: 535 nm) to JC-1 monomers (green: 485nm). Significance represented as follows: *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
2.3. Nanoparticle encapsulation improves intramyocardial SOD retention in vivo
An important theorized distinction between antioxidant enzyme-nanoparticle therapy and antioxidant enzyme alone lies in the former’s ability for enhanced retention within the myocardium. Using an in vivo rat I/R injury model, fluorescent-tagged SOD was more effectively retained within the myocardium at longer timepoints when NP-encapsulated rather than freely administered (Figure 3). At 24 hours after reperfusion NP-SOD retention was 65.06% of baseline compared to 44.79% in freely administered SOD (p=0.003); at 72 hours, retention was 63.25% that of baseline in NP-SOD vs. 30.46% in free SOD (p<0.001).
Figure 3. NP-SOD increases enzyme retention in the myocardium following I/R injury.
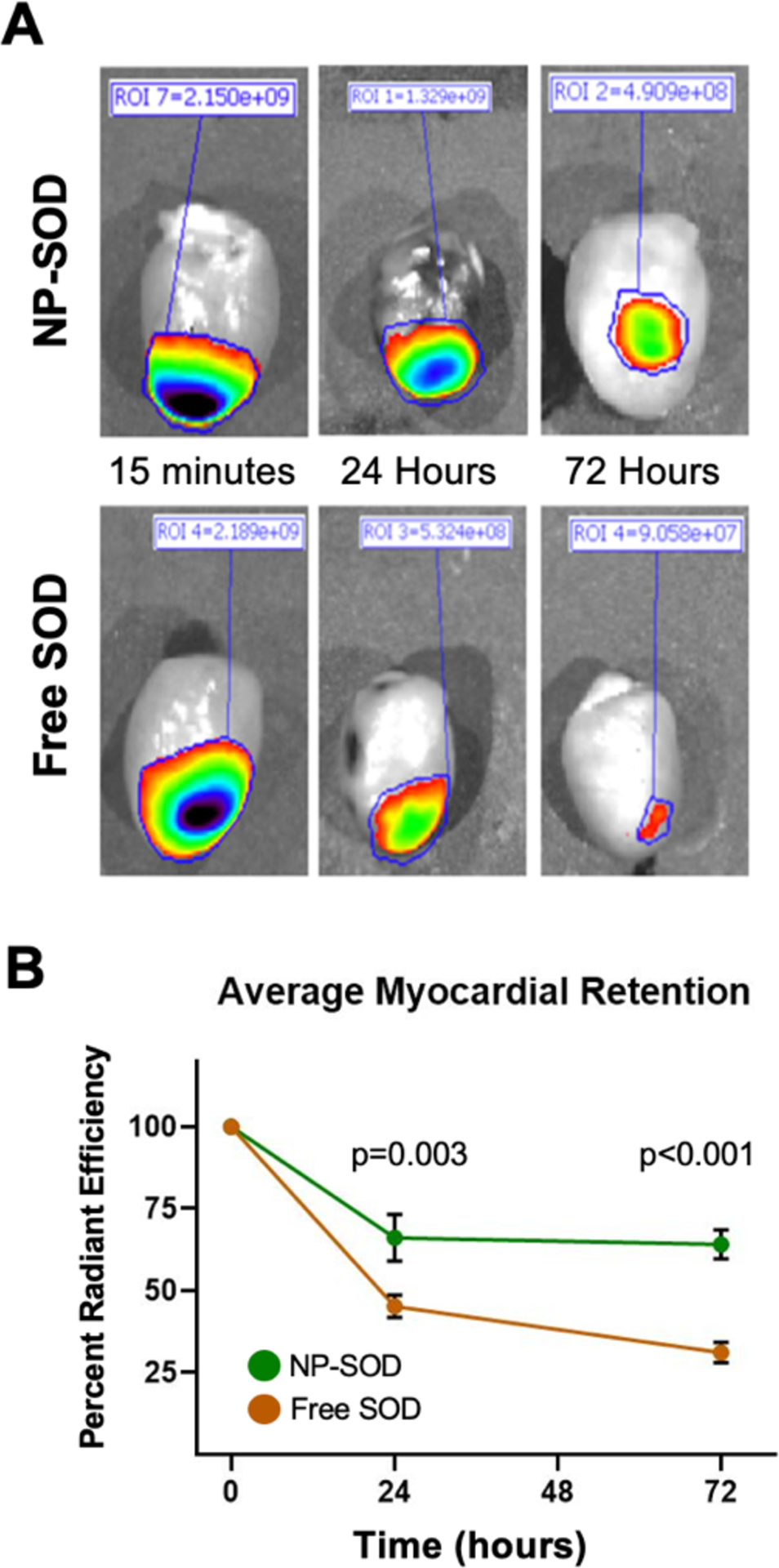
A-B) Free SOD and NP-SOD were tagged with an IRDye 800CW dye and injected following 1 hour of myocardial ischemia. Hearts were explanted at 0.25, 24, and 72 hours post injection and imaged using the Spectrum In Vivo Imaging System (IVIS) imager (ex/em: 760/800nm). Excitation of IRDye 800CW fluorescence was demarcated in regions of interest (ROIs) and magnification of signal was quantified using Perkin Elmer software and normalized to myocardial area. N=4 animals per group per timepoint.
2.4. NP-SOD reduces I/R injury in vivo
2.4.1. NP-SOD protects against acute oxidative injury following I/R in rats
Exogenous SOD administration was assessed in vivo during the acute phase of myocardial I/R injury. At 3 hours, 7 NP-SOD, 5 free SOD, 6 PBS and 4 sham animals were assessed. NP-SOD administration decreased post-reperfusion myocardial ischemia histologically (Figure 4A). Mean area at risk (AAR) of mid-papillary cross sections in sham animals was 0%, while NP-SOD (AAR 12.1%) was lower than both PBS (33.0%, p=0.044) and free SOD (53.7%, p<0.001), Figure 4B. To explain this protective effect, lipid peroxidation was measured via free malondialdehyde (MDA) concentration in border-zone tissue lysates. MDA concentration was reduced in NP-SOD treated rats compared to those receiving PBS, (32.95 vs. 40.74 μM, p=0.039), while lipid peroxidation in NP-SOD was comparable to free SOD (32.23 μM, p=0.961), Figure 4C. Functional degree of acute myocardial injury was then measured by echocardiography. Comparing sham (n=4), NP-SOD (n=7), PBS (n=6) and free SOD (n=5), left ventricular (LV) function was preserved in the NP-SOD group (ejection fraction [EF]=50.6 +/− 2.0%) compared to PBS (33.2 +/− 2.5%, p<0.001) and free SOD (34.8 +/− 1.5%, p<0.001), Figure 4D. There was no significant difference between PBS and free SOD-treated animals (p=0.949).
Figure 4. NP-SOD administration minimizes acute oxidative injury following I/R in vivo.
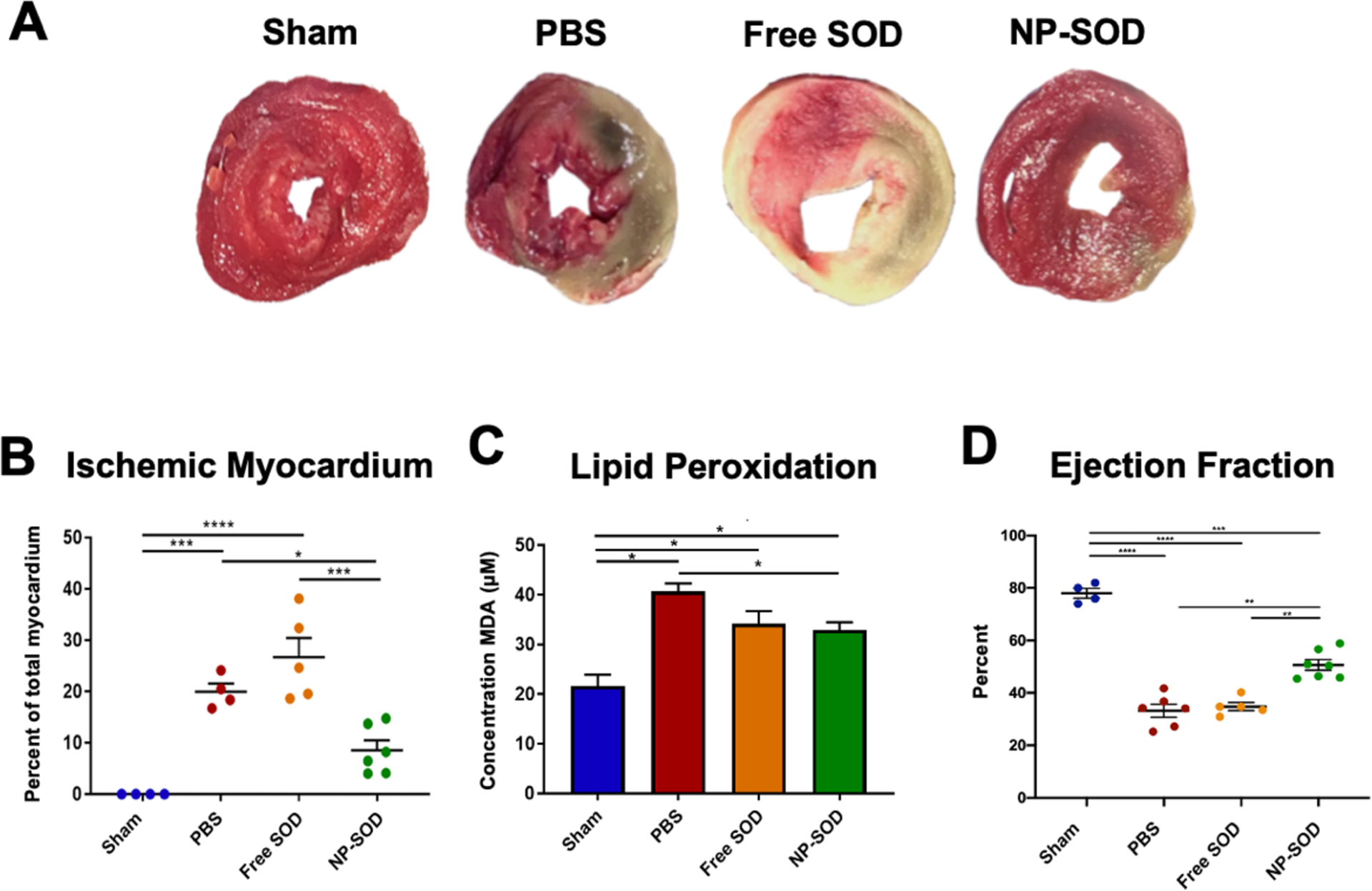
A-B) Explanted hearts were flushed with PBS and cut into 2mm sections along the short axis. Apical, mid, and basilar sections were incubated in 1% 2,3,5-Triphenyltetrazolium chloride (TTC) for 20 minutes at 37°C before fixation in 4% PFA. Sections were imaged and the area of ischemic tissue across all apical and middle regions was quantified as a portion of the whole myocardium demonstrating the myocardial area at risk. C) LV myocardium was lysed and precipitated with Thiobarbituric Acid (TBA). MDA concentration was determined by colorimetric detection of TBA-MDA adduct formation as a measure of ROS induced lipid peroxidation. D) At 3-hours post I/R, echocardiography was performed across short and long axes. End-systolic and end-diastolic volumes were obtained in order to derive LV ejection fraction. Treatment groups assessed were as follows: Sham (n=4), PBS (n=6), free SOD (n=5), NP-SOD (n=7). Significance represented as follows: *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
2.4.2. NP-SOD attenuates chronic adverse ventricular remodeling
Treatment effects were additionally analyzed in the chronic setting. Sham (n=9), NP-SOD (n=13), PBS (n=12) or free SOD (n=10) treated rats were assessed 28 days post I/R injury via terminal hemodynamic measurements with subsequent heart explant. Comparing extent of adverse LV remodeling via fibrosis, there was a stepwise decrement in LV scar area from PBS (10.4 +/− 1.7%, n=7) to free SOD (6.9 +/− 1.1%, n=5), while NP-SOD demonstrated the least scar burden (4.6 +/− 0.6%, n=8; NP-SOD to PBS p=0.007), Figure 5A/5B. Assessing myocardial remodeling at a microscopic level revealed even greater NP-SOD therapeutic efficacy, as fibrotic scar replacement encompassed 1.4 +/− 0.2% of LV myocardium in NP-SOD animals (n=5) compared to 9.2 +/− 1.7% in PBS (n=5, p<0.001) and 6.9 +/− 0.6% in free SOD animals (n=4, p=0.006), Figure 5C/5D. These results were further corroborated by Hematoxylin & Eosin (H&E) staining of hearts subjected to I/R injury which demonstrated greater myocyte density and decreased fibrosis in NP-SOD treated hearts compared to free SOD and PBS, Supplementary Figure S3.
Figure 5. Chronic ventricular remodeling following I/R injury is attenuated by NP-SOD treatment.
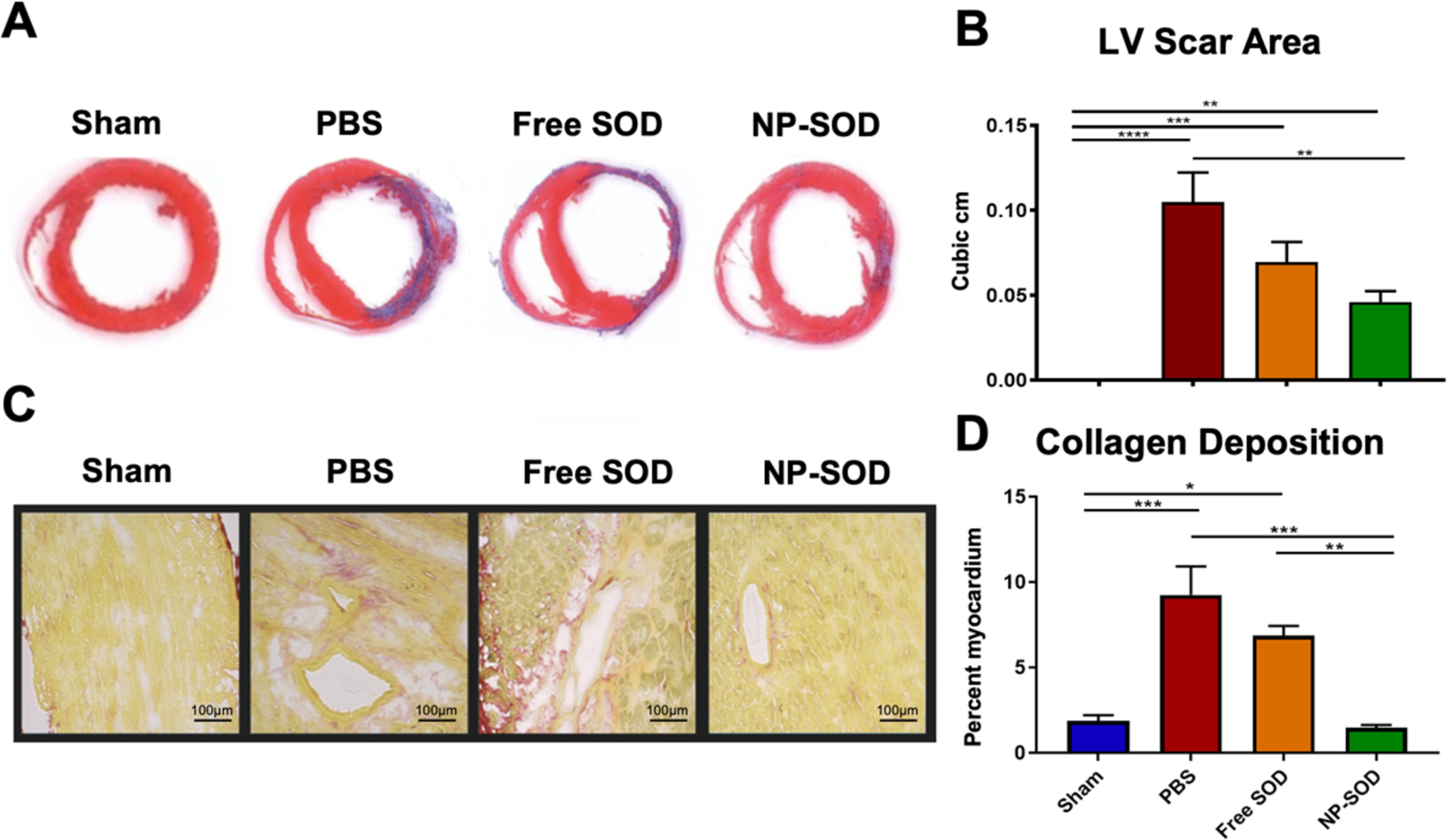
Hearts were explanted at 28-days post I/R, washed, sectioned and mounted at 10 μM in OCT. Sections including mid-papillary muscles in the LV were selected as representative regions affected by previous I/R injury. Masson’s Trichrome and Picrosirius red stains were performed on sections before bright field imaging and automated ImageJ quantification of area containing blue fibrotic scar (A) with quantitative analysis (B) and red type I collagen (C) with quantitative analysis (D) for the respective stains. Treatment groups assessed were as follows: Sham (n=9), PBS (n=12), free SOD (n=10), NP-SOD (n=13). Significance represented as follows: *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
Hemodynamic function at 28 days was assessed through echocardiography and invasive intra-ventricular pressure-volume loop recordings. The primary hemodynamic outcome, end systolic pressure-volume relation (ESPVR), assesses for preload-independent contractility; this was significantly higher in the NP-SOD group, Figure 6. ESPVR of sham animals was 0.50 +/− 0.05 (n=8) and 0.41 +/− 0.043 in NP-SOD animals (n=10, p=0.376). ESPVR in the NP-SOD group was 49.1% greater than PBS (n=11, p=0.003) and 38.1% greater than free SOD (n=9, p=0.042). Other preload-dependent markers of LV function similarly showed NP-SOD superiority. Mean EF, measured by echocardiography, was 64.8% in NP-SOD animals compared to 45.1% in the PBS group (p<0.001) and 50.2% in those receiving free SOD (p<0.001). Other pressure-volume catheter measured parameters were heart rate, cardiac output, stroke volume and stroke work. Heart rate was similar across treatment groups, while stroke volume demonstrated superiority in NP-SOD compared to both PBS and free SOD groups.
Figure 6. Cardiac function is preserved in animals treated with NP-SOD at 28 days following I/R injury.
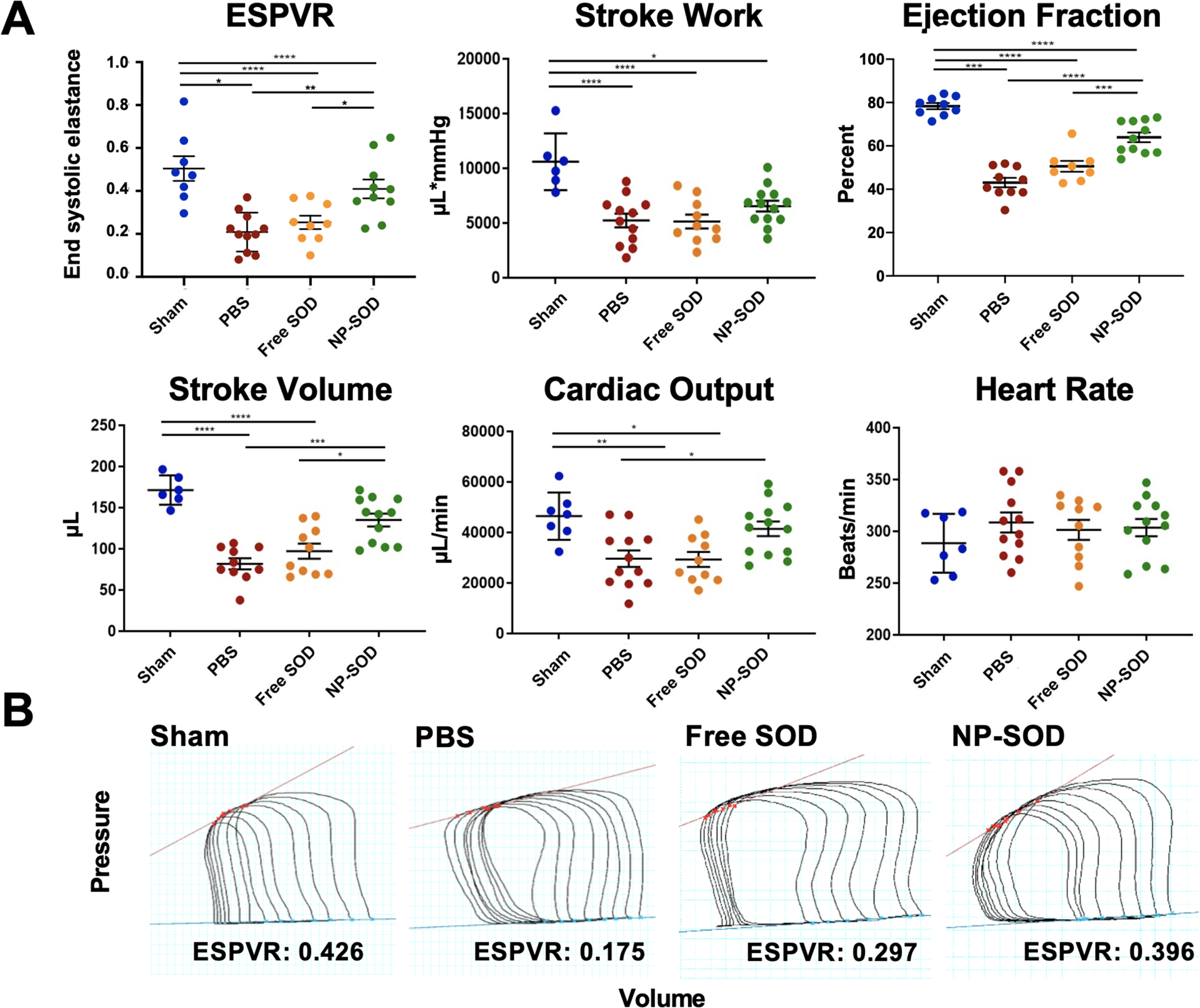
A) Hemodynamic analysis was performed using transthoracic echocardiography and intraventricular pressure-volume loop displacement. Clinical measures for preload-independent contractility (ESPVR) and other preload-dependent volumetric parameters were derived using Millar Pressure-Volume systems and LabChart analysis software. B) Representative PV loops during occlusion of the inferior vena cava (IVC) as are utilized in deriving ESPVR values. Y-axis represents LV pressure and X-axis represents volume. Treatment groups assessed were as follows: Sham (n=8), PBS (n=11), free SOD (n=9), NP-SOD (n=10). Significance represented as follows: *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
2.5. Discussion
This manuscript has described the creation and use of a novel nanoparticle construct to encapsulate SOD and allow for more efficient, sustained antioxidant function in the setting of myocardial I/R injury. This was done by first characterizing the unique permeable, biocompatible nature of the nanoparticle itself. The NP-SOD construct was further shown to maintain therapeutic efficacy in an in vitro I/R setting, in which NP-SOD and free SOD were both able to efficiently scavenge ROS and maintain mitochondrial integrity without causing additional cytotoxicity or cell death. Translating to an in vivo myocardial I/R setting, SOD retention within the myocardium was improved with NP-encapsulated; this correlated with NP-SOD protecting against acute injury as well as chronic ventricular remodeling. SOD has long been considered an attractive therapy in reducing oxidative damage but has failed to translate into pre-clinical success, and this construct provides promise into restoring its translational potential.
To date, treatment failure of SOD has been attributed to its intrinsic characteristics such as short half-life (4–8 minutes),[21] tissue washout,[32] and rapid proteolysis.[33] Attempts to address these shortcomings to-date have successfully enlisted the use of delivery vehicles by harboring SOD within liposomal[34] or polymersome[24] constructs or enmeshing it within hydrogels.[35–37] Despite these advances, prior studies have observed limitations in delivery vehicles’ ability to balance the need for stability and preservation of SOD with the accessibility and permeability required to treat affected tissue. For example, in order for SOD-loaded liposomes or polymersomes to be used as an efficient antioxidant, they should allow ROS (e.g. O2•−) to pass into the aqueous interior and interact with encapsulated SOD. Unfortunately, most liposomes or polymersomes have a low membrane permeability. The novelty of this study’s therapy is in the utilization of nanoparticles that enable stable, yet efficient delivery of SOD. By doping diblock copolymer PEG-PPO into PEG-PBD polymersomes, the construct is able maintain the to harbor and retain the antioxidant enzyme within the nanoparticle while small molecules, such as free superoxide radicals, are able to pass through the permeable membrane of polymersomes.
As oxidative injury leads to both acute myocardial dysfunction as well as chronic LV dysfunction and adverse remodeling, the therapeutic efficacy of NP-SOD was assessed in both the acute and chronic stages using a I/R model. Acute injury three hours after I/R was measured via quantifying oxidative damage by ROS induced lipid peroxidation. NP-SOD-treated animals were able to scavenge free radical species more efficiently than those treated with PBS as measured by levels of the reactive aldehyde MDA. When comparing free SOD to PBS, however, no statistically significant treatment effect was observed. These subtleties became more evident as injury was analyzed from a macroscopic view. Histologic quantification of myocardial AAR not only demonstrated significantly greater myocardial preservation following administration of NP-SOD compared to PBS, it also showed that free SOD administration alone was, at best, equivalent to placebo injection. Ultimately, when assessing clinical utility, organ function is the most critical assessment of treatment effect; this study demonstrated a 17% improvement in EF in NP-SOD treated rats compared to the PBS group, while free SOD alone did not improve LV function over PBS.
In addition to attenuation of acute injury, NP-SOD also improved enzyme retention and bioavailability, likely owing to the protective quality of the polymersome construct against enzymatic degradation. Similar benefits have been observed in previous studies assessing carrier-based delivery systems.[22,38,39] As up to 50% of total permanent infarct in myocardial I/R is due to reperfusion itself, prolonged antioxidant availability and function using NP-SOD treatment are significant. This study demonstrated a reduction in collagen deposition, less fibrosis and preservation of functional myocardium in NP-SOD compared to saline alone at both cellular and whole-organ levels, while free SOD failed to replicate these treatment benefits. Importantly, these findings translated to significant improvement in chronic LV function in NP-SOD treated animals over those treated with saline or unencapsulated SOD.
Several studies have shown that ischemic preconditioning may be a reliable and effective method to reduce I/R injury;[40–42] however, its clinical utility is limited as the vast majority of PCI therapy for myocardial infarction occurs in the emergent, unplanned setting. Similarly, while direct intramyocardial administration is more invasive than catheter-based or peripherally delivered therapy, its use mitigates potential treatment variability related to targeting and absorption and raises the potential for undesired systemic consequences. Future studies in enhancing peripheral delivery may follow recent promising advancements using nanocarriers in cancer models, [43–45] or may rely upon alternative modalities such as bispecific antibody binding. [46] Until these modalities show the potential for clinical translatability, however, this study’s treatment construct and delivery model provides the most direct and straightforward assessment of antioxidant-nanoparticle based therapy in an in vivo model of cardiac I/R injury.
This study has several limitations. One stems from the rapidity in which superoxide and other ROS are degraded, making direct in vivo quantification of ROS injury a challenge. In vitro data was relied upon to ensure that the enzyme of interest was functioning in an antioxidative capacity; this mirrored, although could not replicate, an in vivo model. Additionally, significant morbidity and mortality in I/R injury is caused by fatal arrhythmias,[47] as ROS generation itself impairs local electrophysiology by modifying proteins central to excitation-contraction coupling and altering myofilament sensitivity.[48] While this study assessed acute injury from mechanistic, histologic, and functional perspectives, it did not quantify injury at an electrophysiologic level. Procedural mortality was low, however, at 10% with two mortalities in the 28-day cohort (4.4%) occurring post-operatively. One death occurred after PBS injection and one after NP-SOD and may have been due to a number of different factors including fatal arrhythmia. Injection itself may further cause injury via disruption of already compromised microvasculature, which may represent a potential area requiring alternative therapy delivery modalities moving forward with preclinical translation.
3. Conclusion
This study provides a promising foundation for further development and assessment of a nanoparticle construct which delivers stabilized, bioavailable, exogeneous SOD. Described in this study is a therapeutic model which demonstrates the ability to utilize the potent antioxidative properties of SOD while preserving enzyme integrity within a semipermeable, amphiphilic diblock nanoparticle. The transience of SOD has been a long-standing deterrent to its exogeneous administration as a potential cardio-protectant in myocardial I/R injury. With this NP-SOD construct, the intent was to stabilize SOD function within the myocardium for extended durations. The findings observed here, particularly NP-SOD’s ability to preserve LV native structure and function in both acute and chronic settings, warrant further investigation into the molecular, metabolomic and immunologic responses of the myocardium to NP-SOD treatment. Ongoing analysis will inform future therapeutic strategy in achieving clinical translatability.
4. Experimental Section/Methods
4.1. Animal Use
All experiments conformed to the National Institute of Health Guide for Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania. Rattus norvegicus (Wistar) rats were obtained from Charles River Laboratories, Inc (Boston, MA).
4.2. Nanoparticle construct
4.2.1. Materials
PEG-PBD and PEG-PPO were purchased from Polymer Source (Dorval, Quebec, Canada). IRDye 800CW NHS Ester was obtained from LI-COR, Inc. Calbiochem (EMD Millipore, Billerica, MA) provided Cu,Zn-Superoxide dismutase (MW 32500) from bovine erythrocytes. All other chemicals were used as received. All buffer solutions were prepared with deionized water.
4.2.2. Synthesis of fluorescent labeled SOD
IRDye 800CW-labeled SOD was prepared for retention assay and was synthesized utilizing a molar ratio of 2:1 of IRDye 800CW NHS Ester : SOD. For preparation, 1 mL 9.5 mg mL−1 SOD (in 1 M sterile PBS) was mixed with 58.5 μL 10 mM IRDye 800CW NHS Ester (in anhydrous DMSO). After shaking at room temperature for 2 hours, unconjugated IRDye 800CW NHS Ester was removed by centrifugal filter devices (Amicon Ultra-4, 3000 MWCO, Millipore Corp.). The purified IRDye 800CW-labeled SOD was stored in darkness at 4°C.
4.2.3. Preparation of SOD-encapsulated porous nanoparticles
Nanometer-sized porous polymersomes were prepared using the film hydration technique.[22] A 75 mol % PEG-PBD/25 mol % PEG-PPO mixture was prepared in chloroform in a glass vial using a total of 20 mg of PEG-PBD. The chloroform solvent was removed using a direct stream of nitrogen prior to vacuum desiccation overnight. After the formation of a dried film, 1 mL of 9.5 mg/mL SOD or IRDye 800CW-SOD in 1 M PBS (pH 7.4) was added to the dried polymer film. Samples were subjected to 5 freeze−thaw−vortex cycles in liquid nitrogen and warm H2O (55°C), followed by extrusion 21 times through a 200 nm Nuclepore polycarbonate filter using a stainless-steel extruder (Avanti Polar Lipids). Non-entrapped SOD was removed via size exclusion chromatography using Sepharose CL-4B (Sigma-Aldrich). The sample was further purified and concentrated by centrifugal filter devices (Amicon Ultra-4, 100,000 MWCO, Millipore Corp.).
4.2.4. SOD activity measurement
SOD activity was measured using the ferricytochrome c assay.[49] Hypoxanthine (HX) and xanthine oxidase (XO) were used as a source of superoxide anion, while cytochrome c indicated scavenging of superoxide radical competing with SOD. Working solutions contained 50 mM phosphate buffer (pH 7.8), 0.1 EDTA, 50 μM HX, 20 μM cytochrome c and nanoparticle samples (before and after polymersome dissolution with Triton X-100). The reaction was initiated by the addition of XO (0.2 U/ml final concentration) and the absorbance was monitored at 550 nm using Synergy H1 hybrid multi-mode microplate reader (BioTek). One unit of SOD activity was defined as the amount of the enzyme which inhibited the rate of cytochrome c reduction by 50%. Differences in SOD activity before and after Triton X-100 treatment were determined using separate Student’s t-tests for each formulation. The SOD encapsulation efficiency was calculated utilizing the following equation:
4.2.5. Assessing nanoparticle morphology
The morphology of the NPs was imaged on a Tecnai-12 electron microscope. A drop of the samples were placed on a carbon coated 200-mesh copper grids for 2–3 minutes, then washed with water. The grids were stained with 2% phosphotungstic acid and analyzed at an acceleration voltage of 120 kV.
4.2.6. Instrumentation
DLS measurements were performed on a Zetasizer Nano from Malvern Instruments. The scattering angle was held constant at 90°. Fluorescence spectra measurements were done on a SPEX FluoroMax-3 spectrofluorometer (Horiba Jobin Yvon).
4.3. Cell culture and in vitro analysis
4.3.1. In vitro I/R model
Embryonic Rattus norvegicus cell line, H9C2 cardiomyoblasts (ATCC® CRL-1446™) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS). Cells were maintained in a humidified incubator at 37°C (21% O2 / 5% CO2) and utilized for experiments from passages 2–8. Ischemia-reperfusion injury was modeled by exposure to hypoxia (0.5% O2 / 5% CO2 / 95% N2 / 37°C) for 60 mins before treatment administration and return to normoxia (21% O2 / 5% CO2 / 74% N2 / 37°C). For in vitro experiments, 10 U of free SOD or NP-SOD dissolved in cell culture grade distilled water was administered for every 25,000 cells.
4.3.2. ROS quantification in vitro
H9C2 cells were seeded in black-wall, clear bottom 96-well plates at a density of 25,000 cells per well and allowed to adhere overnight. Ischemia was induced for one hour before administration of either PBS, empty NP, free SOD or NP-SOD and reperfusion in normoxic conditions. A 20 mM stock solution of H2DCFDA was dissolved in anhydrous DMSO and stored at −20°C for up to one week. A final concentration of 20 μM H2DCFDA was dissolved immediately prior to use in sterile PBS and added to each well for a 20 min incubation at room temperature in the dark. The H2DCFDA solution was aspirated and replaced with sterile PBS before reading the fluorescence intensity on the Nexcelom Celigo Cell Image Cytometer (Ex/Em: 485 nm/535 nm).
4.3.3. JC-1 mitochondrial membrane staining:
In multi-well chamber slides, H9C2 cells were seeded at a density of 50,000 cells per chamber and incubated in growth media overnight. Cells were then placed into either hypoxic or normoxic conditions for 1 hour before administration of PBS, empty NP, free SOD or NP-SOD. Slides were then returned to normoxia for 3 hours of reperfusion and stained with 10 μg/mL JC-1 working solution (Abcam) for 10 min in the dark at room temperature. Slides were then washed with PBS and mounted with Vectashield DAPI conjugated mounting medium and imaged on a Leica Fluorescence microscope. The intensity of JC-1 aggregate (Ex/Em: 535 nm/595 nm) and monomer (Ex/Em: 485 nm/535 nm) intensities were then quantified with ImageJ.
4.3.4. Cellular cytotoxicity and viability
H9C2 cells in cell culture medium were seeded at a density of 10,000 cells per well before incubation with 20 μL of the specified treatment at 21% O2 / 5% CO2. To assess cytotoxicity, samples of culture media from each treatment group were harvested in a time-dependent fashion and diluted 1:100 in LDH Storage Buffer. Samples and standards were incubated in equivolume LDH Detection Reagent and Reductase for 60 minutes according to supplier instructions before luminescence was recorded (Promega LDH-Glo). Viability was assessed after incubating treated cells with 10 μL of 12 mM Vybrant MTT (Invitrogen) for 4 hours at 37°C. Culture media was then removed after 24 hours and formazin crystals were dissolved using DMSO. Absorbance at 540 nm was measured.
4.4. In vivo analysis
4.4.1. In vivo I/R model
A rat model of myocardial I/R injury was used to study the efficacy of NP-SOD in vivo. General anesthesia was induced with 5% isoflurane, after which rats were endotracheally intubated with a 16-gauge angiocatheter and mechanically ventilated. Confirmation of anesthesia to a surgical plane was confirmed by absence of palpebral and pedal reflex. Isoflurane was maintained at 1–3% intraoperatively. Subcutaneous injections of 0.05 mg/kg buprenorphine were administered for analgesia. After positioning in right lateral decubitus position, a left sided thoracotomy was performed via the fourth interspace and the pericardium was opened. The left anterior descending artery was then exposed and suture ligated 1 mm below the left atrial appendage with a 7–0 polypropylene suture, creating an anterolateral infarction encompassing 30–40% of the LV. Ischemia was maintained for 60 minutes as described previously.[50] At 60 minutes, five separate 20 μL intramyocardial injections of selected treatment (100 μL total) were administered circumferentially along the ‘border zone’ of perfused and non-perfused tissue. The ligation was then relieved immediately after intramyocardial injection with treatment modalities, allowing for reperfusion. The chest was then closed in three layers, analgesia administered through 1mg/kg intercostal bupivacaine and 2 mg/kg subcutaneous meloxicam injections and the animal recovered. When applicable, an additional group of sham surgery subjects underwent thoracotomy alone without ligation or injection.
4.4.2. Confirmation of I/R injury by plasma Troponin I concentration
Whole blood was collected before ligation and 3 hours post reperfusion in heparinized tubes and centrifuged at 2500 × g for 15 minutes at 4°C. Plasma was separated out and stored at −80°C. Troponin I ELISA (Abcam) was then conducted with samples prepared according to instructions for quantification of plasma troponin I. Samples with elevation of plasma troponin I above 20,000 pg/mL were confirmed to have myocardial I/R injury.
4.4.3. In vivo SOD retention
Using the in vivo I/R model described above, intramyocardial enzyme retention was assessed using fluorescent-labeled SOD. IRDye 800CW fluorophore (10 mM) was conjugated to SOD [500 U/mL] and injected into the myocardium as either free or nanoparticle-encapsulated enzyme. Hearts were explanted in a time-course fashion at 15-minutes, 24- and 72-hours post injection (n=4 per group per timepoint). Imaging was performed with near-IR IVIS microscopy (Spectrum, Ex/Em 760 nm/800 nm), and retention quantified by proportional radiant efficiency [p/s/cm2/sr]/[μW/cm2] of the region of interest (ROI) to that of baseline after subtracting background signal.
4.4.4. Assessing in vivo therapeutic efficacy
Subjects were divided into four treatment groups to assess for therapeutic efficacy. Again using the in vivo I/R injury model, animals received intramyocardial injections of 1) free SOD [500 U/mL], 2) NP-SOD [500 U/mL], 3) PBS or 4) sham surgery alone. Analyses were performed at 3 hours to measure the extent of acute injury and 28 days to assess chronic ventricular remodeling.
4.4.5. Measuring LV hemodynamic function
LV function was measured 3 hours post reperfusion. They were re-induced with 5% isoflurane. After endotracheal intubation and mechanical ventilation, rats were placed in supine position and isoflurane decreased to 1–3% to maintain adequate sedation. EF was then measured by transthoracic echocardiography (Philips SONOS 5500). Ventricular function was assessed in the parasternal short-axis view at the ventricular base, mid-papillary region and apex, as well as in parasternal long-axis.
LV function was assessed 28 days after the initial operation in the same manner. These data are represented as the average of individual mid-papillary and apical EF measurements, taken in triplicate. Following echocardiography recordings, the right neck was dissected, and the right common carotid artery exposed. A 2-Fr pressure-conductance catheter (Millar, Inc., Houston, TX) was then passed in retrograde fashion into the LV through an arteriotomy in the right common carotid artery. Primary hemodynamic measurements assessed preload-independent contractility through ESPVR. This was obtained by measuring the slope of end-systole during occlusion of the IVC.[51] Additional hemodynamic parameters measured were EF, cardiac output, stroke volume, stroke work, and heart rate.
4.4.6. Histologic quantification
Following terminal hemodynamic measures, hearts were explanted, washed with sterile PBS and prepared for further analysis. Hearts assessed 3 hours post-surgery were filled at −25°C for 5 minutes and sectioned into 2 mm slices. Alternating 2 mm sections were stained with TTC for 20 minutes at 37°C and fixed with 4% paraformaldehyde. Sections were photographed and the injured area at risk was assessed by quantifying the relative area of white (unstained, non-perfused) to red (stained, perfused) myocardium using ImageJ. Remaining sections were flash frozen for future analysis.
For animals assessed 28 days post-surgery, hearts were filled and frozen in OCT at −80°C. Utilizing a Leica CM3050S Cryostat, 10 mm sections were obtained and mounted for histological analysis. Hearts were stained with Masson’s Trichrome Stain (Sigma Aldrich) and the area of gross left ventricular scar represented as Aniline Blue positive myocardial fibrosis was quantified using ImageJ. Picrosirius Red (Abcam) was administered to frozen sections for 1 hour and dehydrated as previously described[52] before quantification of LV area containing collagen deposits using ImageJ. Additional 10μM cryosections were fixed in 4% paraformaldehyde and stained for one minute in both Mayer’s Hematoxylin and Eosin (H&E) before dehydration with ethanol and xylene. Slides were mounted and imaged in bright field across the length of the left ventricle and septum.
4.4.7. Lipid peroxidation quantification
MDA is an end product of ROS induced lipid peroxidation.[53] Frozen myocardial tissue was lysed in MDA extraction buffer with butylated hydroxytoluene and precipitated with TBA according to instructions (Abcam). Samples and standards were heated at 95°C for 1 hour. Samples were then transferred to a 96 well plate and absorbance was recorded at OD 532 nm in the BioTek Gen5 Synergy 2 plate reader.
4.5. Statistical analysis
Treatment across groups was randomly generated and subjects identified with random identifiers. Investigators were blinded to treatment during both data acquisition and analysis. Unless otherwise specified, all analyses are represented as mean +/− standard error of the mean (SEM). Comparisons across groups utilized one-way analysis of variance (ANOVA), while individual comparisons between groups were performed using Tukey’s honestly significant difference test. P-values of <0.05 were considered significant. Data analysis was performed using GraphPad Prism 9.0 (GraphPad Software, Inc., San Diego, CA).Error bars on all plots represent mean +/− SEM.
Supplementary Material
Acknowledgments
This work was supported in part by the National Institutes of Health R01NS100892 (Z.C.), R01HL135090 (PA), and the PENN ITMAT-CT3N Pilot Project (ZC). PJA was supported by National Institutes of Health institutional training grant T32GM008562.
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest.
Contributor Information
Lijun Luo, Department of Bioengineering, University of Pennsylvania, 210 South 33rd Street, 240 Skirkanich Hall, Philadelphia, PA 19104, USA.
Mark R. Helmers, Division of Cardiovascular Surgery, Department of Surgery, University of Pennsylvania, 3400 Spruce Street, Philadelphia, PA 19104, USA
Bonirath Chhay, Department of Bioengineering, University of Pennsylvania, 210 South 33rd Street, 240 Skirkanich Hall, Philadelphia, PA 19104, USA.
Pavan Atluri, Division of Cardiovascular Surgery, Department of Surgery, University of Pennsylvania, 3400 Spruce Street, Philadelphia, PA 19104, USA.
References
- [1].Roth GA, Johnson C, Abajobir A, Abd-Allah F, Abera SF, Gebre A, Ahmed M, Aksut B, Alam T, Alam K, Alla F, Alvis-Guzman N, Amrock S, Ansari H, Arnlov J, et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J Am Coll Cardiol. 2017;70(1):1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Khan MA, Hashim MJ, Mustafa H, Baniyas MY, Suwaidi SKBMA, AlKatheeri R, Alblooshi FMK, Almatrooshi MEAH, Alzaabi MEH, Darmaki RSA, Lootah SNAH. Global Epidemiology of Ischemic Heart Disease: Results from the Global Burden of Disease Study. Cureus. 2020;12(7):e9349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Law MR, Watt HC, Wald NJ. The underlying risk of death after myocardial infarction in the absence of treatment. Arch Intern Med. 2002;162(21):2405–2410. [DOI] [PubMed] [Google Scholar]
- [4].Braunersreuther V, Jaquet V. Reactive Oxygen Species in Myocardial Reperfusion Injury: From Physiopathology to Therapeutic Approaches. Curr Pharm Biotechnol. 2011;13(1):97–114. [DOI] [PubMed] [Google Scholar]
- [5].Yang Q, He GW, Underwood MJ, Yu CM. Cellular and molecular mechanisms of endothelial ischemia/reperfusion injury: Perspectives and implications for postischemic myocardial protection. Am J Transl Res. 2016;8(2):765–777. [PMC free article] [PubMed] [Google Scholar]
- [6].Cadenas S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radic Biol Med. 2018;117:76–89. [DOI] [PubMed] [Google Scholar]
- [7].Görlach A, Bertram K, Hudecova S, Krizanova O. Calcium and ROS: A mutual interplay. Redox Biol. 2015;6:260–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Scherer NM, Deamer DW. Oxidative stress impairs the function of sarcoplasmic reticulum by oxidation of sulfhydryl groups in the Ca2+-ATPase. Arch Biochem Biophys. 1986;246(2):589–601. [DOI] [PubMed] [Google Scholar]
- [9].Batandier C, Leverve X, Fontaine E. Opening of the mitochondrial permeability transition pore induces reactive oxygen species production at the level of the respiratory chain complex I. J Biol Chem, 2004;279(17),17197–17204. [DOI] [PubMed] [Google Scholar]
- [10].Jacobson J, Duchen MR. Mitochondrial oxidative stress and cell death in astrocytes—requirement for stored Ca2+ and sustained opening of the permeability transition pore. J Cell Sci. 2002;115(6),1175–1188. [DOI] [PubMed] [Google Scholar]
- [11].Odagiri K, Katoh H, Kawashima H, Tanaka T, Ohtani H, Saotome M, Urushida T, Satoh H, Hayashi H. Local control of mitochondrial membrane potential, permeability transition pore and reactive oxygen species by calcium and calmodulin in rat ventricular myocytes. J Mol Cell Cardiol. 2009;46(6), 989–997. [DOI] [PubMed] [Google Scholar]
- [12].Pérez MJ, Quintanilla RA. Development or disease: duality of the mitochondrial permeability transition pore. Dev Biol. 2017;426(1), 1–7. [DOI] [PubMed] [Google Scholar]
- [13].Piper HM. Energy deficiency, calcium overload or oxidative stress: possible causes of irreversible ischemic myocardial injury. Klin Wochenschr. 1989;67(9), 465–476. [DOI] [PubMed] [Google Scholar]
- [14].Arslan F, Lai RC, Smeets MB, Akeroyd L, Andre C, Aguor ENE, Timmers L, van Rijen HV, Doevendans PA, Pasterkamp G, Lim SK, de Kleijn DP. Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res. 2013;10(3), 301–312. [DOI] [PubMed] [Google Scholar]
- [15].Grill HP, Zweier JL, Kuppusamy P, Weisfeldt ML, Flaherty JT. Direct Measurement of Myocardial Free Radical Generation in an In Vivo Model: effects of Postischemic Reperfusion and Treatment With Human Recombinant Superoxide Dismutase. J Am Coll Cardiol. 1992;20:1604–11. [DOI] [PubMed] [Google Scholar]
- [16].Omar BA, McCord JM. Interstitial equilibration of superoxide dismutase correlates with its protective effect in the isolated rabbit heart. J Mol Cell Cardiol. 1991;23(2):149–159. [DOI] [PubMed] [Google Scholar]
- [17].Liu J, Hou J, Xia ZY, Zeng W, Wang X, Li R, Ke C, Xu J, Lei S, Xia Z. Recombinant PTD-Cu/Zn SOD attenuates hypoxia-reoxygenation injury in cardiomyocytes. Free Radic Res. 2013;47(5):386–93. [DOI] [PubMed] [Google Scholar]
- [18].Wang P, Chen H, Qin H, Sankarapandi S, Becher MW, Wong PC, Zweier JL. Overexpression of human copper, zinc-superoxide dismutase (SOD1) prevents postischemic injury. Proc Natl Acad Sci U S A. 1998;95(8):4556–4560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tanaka M, Stoler RC, FitzHarris GP, Jennings RB, Reimer KA. Evidence against the “early protection-delayed death” hypothesis of superoxide dismutase therapy in experimental myocardial infarction. Polyethylene glycol-superoxide dismutase plus catalase does not limit myocardial infarct size in dogs. Circ Res. 1990;67(3):636–644. [DOI] [PubMed] [Google Scholar]
- [20].Flaherty JT, Pitt B, Gruber JW, Heuser RR, Rothbaum DA, Burwell LR, George BS, Kereiakes DJ, Deitchman D, Gustafson N. Recombinant human superoxide dismutase (h-SOD) fails to improve recovery of ventricular function in patients undergoing coronary angioplasty for acute myocardial infarction. Circulation. 1994;89(5):1982–1991. [DOI] [PubMed] [Google Scholar]
- [21].Kabu S, Gao Y, Kwon BK, Labhasetwar V. Drug delivery, cell-based therapies, and tissue engineering approaches for spinal cord injury. J Control Release. 2015;219:141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hood E, Simone E, Wattamwar P, Dziubla T, Muzykantov V. Nanocarriers for vascular delivery of antioxidants. Nanomedicine. 2011;6(7):1257–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Salvador A, Sousa J, Pinto RE. Hydroperoxyl, superoxide and pH gradients in the mitochondrial matrix: A theoretical assessment. Free Radic Biol Med. 2001;31(10):1208–1215. [DOI] [PubMed] [Google Scholar]
- [24].Kartha S, Yan L, Weisshaar CL, Ita ME, Shuvaev VV, Muzykantov VR, Tsourkas A, Winkelstein BA, Cheng Z. Superoxide Dismutase-Loaded Porous Polymersomes as Highly Efficient Antioxidants for Treating Neuropathic Pain. Adv Healthc Mater. 2017;6(17):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Reddy MK, Wu L, Kou W, Ghorpade A, Labhasetwar V. Superoxide dismutase-loaded PLGA nanoparticles protect cultured human neurons under oxidative stress. Appl Biochem Biotechnol. 2008;151(2–3):565–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Rajkovic O, Gourmel C, d’Arcy R, Wong R, Rajkovic I, Tirelli N, Pinteaux E. Reactive Oxygen Species-Responsive Nanoparticles for the Treatment of Ischemic Stroke. Adv Ther (Weinh). 2019;2(7):1900038 [Google Scholar]
- [27].Zhou X, Lv J, Li G, Qian T, Jiang H, Xu J, Cheng Y, Hong J. Rescue the retina after the ischemic injury by polymer-mediated intracellular superoxide dismutase delivery. Biomaterials. 2021;268:120600. [DOI] [PubMed] [Google Scholar]
- [28].Ni D, Wei H, Chen W, Bao Q, Rosenkrans ZT, Barnhart TE, Ferreira CA, Wang Y, Yao H, Sun T, Jiang D, Li S, Cao T, Liu Z, Engle JW, et al. Ceria Nanoparticles Meet Hepatic Ischemia-Reperfusion Injury: The Perfect Imperfection. Adv Mater. 2019;31(40):e1902956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Han SJ, Williams RM, D’Agati V, Jaimes EA, Heller DA, Lee HT. Selective nanoparticle-mediated targeting of renal tubular Toll-like receptor 9 attenuates ischemic acute kidney injury. Kidney Int. 2020;98(1):76–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chi LG, Tamura Y, Hoff PT, Macha M, Gallagher KP, Schork MA, Lucchesi BR. Effect of superoxide dismutase on myocardial infarct size in the canine heart after 6 hours of regional ischemia and reperfusion: a demonstration of myocardial salvage. Circ Res. 1989;64(4):665–675. [DOI] [PubMed] [Google Scholar]
- [31].Seshadri G, Sy JC, Brown M, Dikalov S, Yang SC, Murthy N, Davis ME. The delivery of superoxide dismutase encapsulated in polyketal microparticles to rat myocardium and protection from myocardial ischemia-reperfusion injury. Biomaterials. 2010;31(6):1372–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Uraizee A, Reimer KA, Murry CE, Jennings RB. Failure of superoxide dismutase to limit size of myocardial infarction after 40 minutes of ischemia and 4 days of reperfusion in dogs. Circulation. 1987;75(6):1237–1248. [DOI] [PubMed] [Google Scholar]
- [33].Salo DC, Pacifici RE, Lin SW, Giulivi C, Davies KJ. Superoxide dismutase undergoes proteolysis and fragmentation following oxidative modification and inactivation. J Biol Chem. 1990;265(20):11919–11927. [PubMed] [Google Scholar]
- [34].Luisa Corvo M, Jorge JC, van’t Hof R, Cruz ME, Crommelin DJ, Storm G. Superoxide dismutase entrapped in long-circulating liposomes: formulation design and therapeutic activity in rat adjuvant arthritis. Biochim Biophys Acta. 2002;1564(1):227–236. [DOI] [PubMed] [Google Scholar]
- [35].Li Z, Wang F, Roy S, Sen CK, Guan J. Injectable, highly flexible, and thermosensitive hydrogels capable of delivering superoxide dismutase. Biomacromolecules. 2009;10(12):3306–3316. [DOI] [PubMed] [Google Scholar]
- [36].Bae S, Park M, Kang C, Dilmen S, Kang TH, Kang DG, Ke Q, Lee SU, Lee D, Kang PM. Hydrogen Peroxide-Responsive Nanoparticle Reduces Myocardial Ischemia/Reperfusion Injury. J Am Heart Assoc. 2016;5(11):e003697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Nagaoka K, Matoba T T, Mao Y, Nakano Y, Ikeda G, Egusa S, Tokutome M, Nagahama R, Nakano K, Sunagawa K, Egashira K. A New Therapeutic Modality for Acute Myocardial Infarction: Nanoparticle-Mediated Delivery of Pitavastatin Induces Cardioprotection from Ischemia-Reperfusion Injury via Activation of PI3K/Akt Pathway and Anti-Inflammation in a Rat Model. PLoS One. 2015;10(7):e0132451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Youan BB. Microencapsulation of superoxide dismutase into biodegradable microparticles by spray-drying. Drug Deliv. 2004;11(3):209–214. [DOI] [PubMed] [Google Scholar]
- [39].Su S, Kang PM. Systemic Review of Biodegradable Nanomaterials in Nanomedicine. Nanomaterials (Basel). 2020;10(4):656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Dong S, Cao Y, Li H, Tian J, Yi C, Sang W. Impact of ischemic preconditioning on ischemia-reperfusion injury of the rat sciatic nerve. Int J Clin Exp Med. 2015;8(9):16245–16251. [PMC free article] [PubMed] [Google Scholar]
- [41].Donato M, Evelson P, Gelpi RJ. Protecting the heart from ischemia/reperfusion injury: an update on remote ischemic preconditioning and postconditioning. Curr Opin Cardiol. 2017;32(6):784–790. [DOI] [PubMed] [Google Scholar]
- [42].Loukogeorgakis SP, Panagiotidou AT, Broadhead MW, Donald A, Deanfield JE, MacAllister RJ. Remote ischemic preconditioning provides early and late protection against endothelial ischemia-reperfusion injury in humans: role of the autonomic nervous system. J Am Coll Cardiol. 2005;46(3):450–456. [DOI] [PubMed] [Google Scholar]
- [43].Bölükbas DA, Datz S, Meyer-Schwickerath C, Morrone C, Doryab A, Gößl D, Vreka M, Yang L, Arygo C, van Rijt SH, Lindner M, Eickelberg O, Stoeger T, Schmid O, Lindstedt S, et al. Organ-Restricted Vascular Delivery of Nanoparticles for Lung Cancer Therapy. Adv Ther (Weinh). 2020;3(7):2000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hossen S, Hossain MK, Basher MK, Mia MNH, Rahman MT, Uddin MJ. Smart nanocarrier-based drug delivery systems for cancer therapy and toxicity studies: A review. J Adv Res. 2019;15:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rosenblum D, Joshi N, Tao W, Karp JM, Peer D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat Commun. 2018;12;9(1):1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Huang K, Li Z, Su T, Shen D, Hu S, Cheng K. Bispecific Antibody Therapy for Effective Cardiac Repair through Redirection of Endogenous Stem Cells. Adv Ther (Weinh). 2019;2(10):1900009. [Google Scholar]
- [47].Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357(11):1121–1135. [DOI] [PubMed] [Google Scholar]
- [48].Takimoto E, Kass DA. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension. 2007;49:241–248. [DOI] [PubMed] [Google Scholar]
- [49].McCord JM, Fridovich I. Superoxide dismutase an enzymic function for erythrocuprein (hemocuprein). J Biol Chem. 1969;244(22): 6049–6055. [PubMed] [Google Scholar]
- [50].He B, Xiao J, Ren AJ, Zhang YF, Zhang H, Chen M, Xie B, Gao XG, Wang YW. Role of miR-1 and miR-133a in myocardial ischemic postconditioning. J Biomed Sci. 2011;18(1):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Pacher P, Nagayama T, Mukhopadhyay P, Bátkai S, Kass DA. Measurement of cardiac function using pressure-volume conductance catheter technique in mice and rats. Nat Protoc. 2008;3(9):1422–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lattouf R, Younes R, Lutomski D, Naaman N, Godeau G, Senni K, Changotade S. Picrosirius Red Staining: A Useful Tool to Appraise Collagen Networks in Normal and Pathological Tissues. J Histochem Cytochem. 2014;62(10):751–758. [DOI] [PubMed] [Google Scholar]
- [53].Tsikas D. Assessment of lipid peroxidation by measuring malondialdehyde (MDA) and relatives in biological samples: Analytical and biological challenges. Anal Biochem. 2017;524:13–30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.