

Abstract
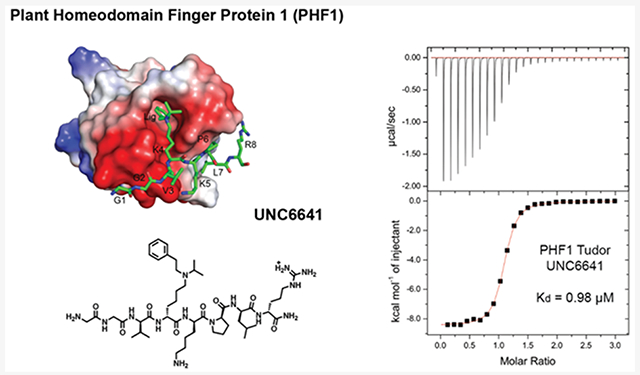
Plant homeodomain finger protein 1 (PHF1) is an accessory component of the gene silencing complex polycomb repressive complex 2 and recognizes the active chromatin mark, trimethylated lysine 36 of histone H3 (H3K36me3). In addition to its role in transcriptional regulation, PHF1 has been implicated as a driver of endometrial stromal sarcoma and fibromyxoid tumors. We report the discovery and characterization of UNC6641, a peptidomimetic antagonist of the PHF1 Tudor domain which was optimized through in silico modeling and incorporation of non-natural amino acids. UNC6641 binds the PHF1 Tudor domain with a Kd value of 0.96 ± 0.03 μM while also binding the related protein PHF19 with similar potency. A crystal structure of PHF1 in complex with UNC6641, along with NMR and site-directed mutagenesis data, provided insight into the binding mechanism and requirements for binding. Additionally, UNC6641 enabled the development of a high-throughput assay to identify small molecule binders of PHF1.
Graphical Abstract
INTRODUCTION
Chromatin regulation is a dynamic process requiring oscillation between transcriptionally silent heterochromatin and transcriptionally active euchromatin which is tightly controlled by a network of epigenetic factors including DNA methylation, chromatin remodelers, histone variants, and histone post-translational modifications (PTMs). Regulation of this network is essential for proper gene transcription, while dysregulation of these factors has been linked to a variety of diseases from cancer to neurological disorders.1-4 Histone PTMs make up an extensive language known as the “histone code” which helps to dictate chromatin structure and function.5,6 Examples of histone PTMs include acetylation, methylation, phosphorylation, and ubiquitylation, which are most commonly found on the flexible tails of histones H2A, H2B, H3, and H4. These marks can act both individually and in combination, enabling a diverse and complex network of downstream signals. Molecularly, these modifications typically function in one of two ways to influence chromatin accessibility: (1) by directly influencing histone—DNA interactions or (2) by recruiting specific effector proteins.
Lysine mono-, di-, and trimethylation (Kme1/2/3) have been well characterized as essential and ubiquitous PTMs.7,8 Kme1/2/3 are selectively recognized by a class of proteins known as methyl-lysine “readers”, which engage the methylammonium group largely through cation—π interactions with an aromatic cage.9-11 The selectivity of this molecular recognition event for a specific methylation state is generally based on the size of the cage and number of aromatic residues in the cage, while selectivity for a certain lysine residue is often dictated by the surrounding histone sequence. Previous work has shown that the selectivity and potency of Kme readers for their cognate peptide substrates can be tuned by modifying the electronics of the aromatic cage residues.12-14
The systematic design of peptidomimetic ligands based on histone substrates can provide valuable insight into the molecular details of the binding event and an excellent starting point for ligand discovery.15-19 Our lab has demonstrated the utility of such peptidomimetic ligands in a variety of applications, ranging from cellular target inhibition15,17,18 and activation16 to assay development.20,21
In this paper, we focus on the Tudor domain of plant homeodomain finger protein 1 (PHF1), which functions as a reader for the active chromatin mark, trimethylated lysine 36 of histone H3 (H3K36me3).22-27 PHF1 and its closely related homologue, PHF19, are the only annotated readers of H3K36me3 with affinities in the micromolar range. This recognition event has been shown to facilitate gene repression via the recruitment of the epigenetic silencing complex, polycomb repressive complex 2 (PRC2), to a subset of target genes.23,28-31 Specifically, PHF1 and PHF19 levels increase dramatically during differentiation of mouse embryonic stem cells (mESCs),32 and following differentiation, PHF1 and PHF19 facilitate silencing of stem cell genes by recruiting PRC2 which deposits the repressive mark, trimethylated lysine 27 on histone H3 (H3K27me3), to active genes previously marked by H3K36me3.26,33,34 Additionally, the dysregulation of PHF1 has been implicated as a hallmark of disease. For example, aberrant fusions of PHF1 have been linked to endometrial stromal sarcoma and ossifying fibromyxoid tumors.35-38
Despite the extensive validation of PHF1’s significance in normal and malignant biology, no high-quality ligands have been reported to date. Development of a potent and selective chemical probe for PHF1 will be essential to further validate the role of PHF1 in gene regulation and disease and assess its suitability for therapeutic intervention. As the downstream effects of altering protein expression via genetic methods can become convoluted,39 especially for proteins that regulate transcription, small molecule antagonism of the Tudor domain of PHF1 could produce a functional phenotype that would directly elucidate the role of the Tudor domain in normal and disease states.
Using the endogenous binding partner of PHF1, H3K36me3, as a starting point, we sought to design potent and selective peptidomimetic ligands for PHF1. Through rational mutations and incorporation of synthetic Kme3 mimetics, we discovered UNC6641, which contains an aromatic lysine mimetic and binds the PHF1 Tudor domain with a Kd value of 0.96 ± 0.03 μM. X-ray crystallography revealed that the (isopropyl)phenethyl lysine mimetic of UNC6641 both strengthened existing interactions and created new contacts with the aromatic cage residues of PHF1. Most notably, the phenethyl group of UNC6641 demonstrated face-to-face and edge-to-face π—π stacking interactions with all four aromatic cage residues, likely contributing to its increased affinity for the Tudor domain. This appears to be the first example of a peptidomimetic ligand for a Kme reader domain that utilizes an aromatic substituent on the lysine amino group to better engage the aromatic cage. Through careful characterization of the molecular recognition of UNC6641 by the PHF1 Tudor domain, we have paved the way for future assay development, small molecule discovery, and investigation of the biological role of PHF1.
RESULTS AND DISCUSSION
Structure-Based Ligand Design.
We began the development of PHF1 ligands by determining the minimal H3K36me3 sequence required to maintain binding to the Tudor domain. H3K36me3 binds PHF1 in a surface-groove binding mode,22 making key interactions with a relatively exposed portion of the protein (Figure S1, Supporting Information). Previous literature has reported binding affinities for PHF1 of 4, 14, and 16 μM using H3K36me3 peptides spanning 15, 14, and 10 residues, respectively.23,27,40 However, smaller peptides are more attractive starting points for ligand development, as they are more easily synthesized, enable concise and tractable structure—activity relationship (SAR) exploration, and demonstrate improved cell permeability. In order to decrease the peptide size while maintaining affinity, we synthesized a series of H3K36me3 variants based on the eight residues reported to make direct contacts with the PHF1 surface, H3(33—40), and known peptide SAR.22,23,31,41 We assessed binding affinities for these peptides using isothermal titration calorimetry (ITC), the results of which are outlined in Table 1 (Figure S2). Our 8-mer peptide spanning H3(33—40) gave a Kd value of 22.3 ± 1.9 μM, a slighter weaker affinity than the previously reported 10-mer peptide H3(31—40) (Kd of 16 ± 4 μM).27
Table 1.
Binding of H3K36me3 Variants to PHF1 Tudora
| Histone Residues | Peptide Sequence | ITC Kd (μM)a |
|---|---|---|
| H3(33-40) | GGVKme3KPHR | 22.3 ± 1.9 |
| H3(33-39) | GGVKme3KPH | 52.0 ± 6.1 |
| H3(33-39)b | VGVKme3KPH | 48.0 ± 0.2 |
| H3(33-39)b | VGVKme3KPL | 16.3 ± 3.0 |
| H3(33-40)b | GGVKme3KPLR | 18.7 ± 4.1 |
Kd values were determined by two independent ITC experiments (mean ± SD).
Peptide sequence differs from natural histone H3 sequence at residues denoted in blue.
In order to further assess the minimal binding sequence required, we next truncated the C-terminal R40 residue. This residue is solvent-exposed in the reported X-ray crystal structure of H3K36me3 bound to the PHF1 Tudor domain (PDB ID: 4HCZ),22 leading us to believe that it did not make stable contacts with the PHF1 surface. However, truncation of R40 resulted in a modest 2-fold decrease in affinity, likely due to loss of transient hydrogen bonds formed with the PHF1 surface. In an attempt to recoup this loss in affinity, we incorporated a G33V mutation into the H3(33–39) peptide. This mutation was previously reported to increase the affinity of H3K36me3 for the PHF1 Tudor domain, as shown by Western blot.40 However, we observed the same 2-fold loss in affinity resulting from the R40 truncation and no significant effect from the G33V mutation. Gratifyingly, we regained affinity by mutation of H39 to a leucine residue (H39L), which we hypothesized would better interact with the hydrophobic patch of PHF1 made up of L38, L45, L46, and L48.22 Incorporation of the H39L mutant into the initial 8-mer peptide spanning H3(33–40) afforded a small boost in potency for a final Kd value of 18.7 ± 4.1 μM.
Prior work has demonstrated the utility of non-natural Kme mimics in improving the binding affinity and selectivity of histone peptides and peptidomimetics for their target reader proteins.15,17,18,42-44 In addition, replacement of the quaternary amine in Kme3 with a secondary or tertiary amine is attractive in that it is likely to improve passive cell permeability by eliminating the permanent positive charge. While removal of the trimethylammonium can weaken the characteristic cation—π interactions with the aromatic cage residues, this can sometimes be overcome via incorporation of additional hydrophobic interactions. In the context of the GGVKme3KPLR peptide sequence, we substituted Kme3 with synthetic analogues that take advantage of the hydrophobic nature of the aromatic cage, as well as the potential for hydrogen bonding and π—π interactions (Table 2, Figure S2). Encouragingly, we observed that replacing Kme3 with the corresponding (ethyl)isopropyl-lysine residue (UNC7643, Kd = 10.1 ± 1.2 μM) was well tolerated and moderately beneficial. The addition of a hydrogen bond acceptor in the (ethyl)ethylfuran-lysine (UNC6640, Kd = 17.9 ± 4.6 μM) did not further improve potency from the trimethylated peptide. The most significant improvement came with the incorporation of the aromatic (isopropyl)phenethyl-lysine residue in UNC6641, which resulted in a 20-fold increase in affinity versus the parent peptide to give a Kd value of 0.96 ± 0.03 μM.
Table 2.
Binding of Peptidomimetic Ligands with Non-Natural Kme Mimetics to PHF1 Tudor
| ITC | ||||
|---|---|---|---|---|
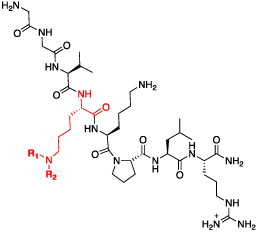 |
R1 | R2 | Kd (μM)a | Compound ID |
| 10.1 ± 1.2 | UNC7643 | |||
| 17.9 ± 4.6 | UNC6640 | |||
| 0.96 ± 0.03 | UNC6641 | |||
| TR-FRET | ||||
| R1 | R2 | IC50 (μM)b | Compound ID | |
| 1.6 ± 0.5 | UNC6641 | |||
| 8 ± 3 | UNC7259 | |||
| 19 ± 4 | UNC7253 | |||
| 21 ± 1 | UNC7258 | |||
Kd values were determined by two independent ITC experiments (mean ± SD).
IC50 values were determined by three technical replicates (mean ± SD).
As ITC is low throughput, we capitalized on our discovery of a more potent PHF1 ligand to develop a time-resolved fluorescence resonance energy transfer (TR-FRET) assay using a biotinylated analogue of UNC6641 as a bait ligand. Biotin-UNC6641 binds to the PHF1 Tudor domain about equipotently to UNC6641 (Kd ~ 3 μM, Figure S2). TR-FRET assays have been used previously to screen Kme reader domains in a robust fashion, but they typically require a bait ligand that binds in the low micromolar range.21 This had not previously been feasible due to the low affinity of the endogenous histone substrate for the PHF1 Tudor domain. Using this assay, we were able to screen a larger panel of peptidomimetic ligands based on UNC6641, some of which are shown in Table 2 (Figure S3). It should be noted that the IC50 of UNC6641 as determined by TR-FRET closely agrees with the dissociation constant measured by ITC (Kd = 0.96 ± 0.03 μM, IC50 = 1.6 ± 0.5 μM). We observed that increasing or decreasing the distance between the K36 amine and phenyl ring by a single methylene as in UNC7259 and UNC7253 resulted in a 5- and 12-fold loss in potency, respectively. This suggests that the phenyl substituent makes a fairly specific interaction(s) with the Tudor domain and the increase in potency is not solely the result of an increase in hydrophobicity and/or steric bulk. Additionally, removing the potential for π—π interactions by swapping the phenyl group for a cyclohexyl ring in UNC7258 also decreased the potency 13-fold relative to UNC6641. Taken together, these data suggest that the potent binding of UNC6641 to PHF1 Tudor is unique and specific, depending both on the aromaticity and the position of the phenethyl substituent.
Confirmation of the UNC6641 Binding Mode by NMR and Crystallography.
Preliminary analysis of the binding mode of UNC6641 was performed by docking UNC6641 into the PHF1 Tudor domain (PDB ID: 4HCZ) using Schrödinger Glide docking (Figure S4). The docking model suggested that the (isopropyl)phenethyl-lysine of UNC6641 filled the aromatic cage more efficiently than the Kme3 substituent of the natural histone peptide (Figure S4a). In addition, the model offered a possible explanation for the increased affinity of the peptide for the PHF1 Tudor domain. Introduction of the phenethyl substituent appeared to provide additional π interactions with the aromatic cage through π—π stacking of the phenyl group of UNC6641 and PHF1 Y47 and W41 (Figure S4b).
To further validate this model, we investigated the binding of UNC6641 to the PHF1 Tudor domain by NMR. Following expression and purification of the uniformly 15N-labeled PHF1 Tudor domain (14—87), 1H,15N heteronuclear single quantum coherence (HSQC) spectra of the protein were collected while UNC6641 was added stepwise. Substantial chemical shift changes in the intermediate to slow exchange regime on the NMR time scale were observed upon addition of UNC6641, indicating direct and tight binding (Figure 1a). Specific differences in chemical shifts for backbone amides of the PHF1 Tudor domain in the apo state and the UNC6641 bound state (at a molar protein to UNC6641 ratio of 1:4) are shown in the histogram in Figure 1b. The Tudor domain residues most perturbed upon UNC6641 binding, including W41, L46, Y47, G49, F65, and E66, were then mapped onto the surface of the PHF1 Tudor domain (Figure 1c). As expected, these residues included and clustered around the aromatic cage residues of W41, Y47, F65, and F71 (Figure 1d) and showed a similar CSP pattern to the titration of H3K36me3 reported previously22 (Figure S5), further supporting that UNC6641 binds in the H3K36me3 binding site.
Figure 1.

Characterization of the interaction between the PHF1 Tudor domain and UNC6641 by NMR. (a) Overlay of 1H,15N HSQC spectra of the 15N-labeled PHF1 Tudor domain collected before and after the addition of UNC6641. Spectra are color coded according to the protein–peptide molar ratio. (b) Histogram showing chemical shift perturbations of backbone amides of the PHF1 Tudor domain at a 1:4 molar ratio of protein to UNC6641. The dashed line indicates an averaged chemical shift change plus one standard deviation. (c) The surface representation of the PHF1 Tudor domain (PDB ID: 4HCZ). Residues that exhibit substantial changes in part b are colored purple. (d) The PHF1 Tudor domain (PDB ID: 4HCZ) is depicted as a gray ribbon with the aromatic cage residues W41, Y47, F65, and F71 shown as pink sticks and labeled.
To gain further insight into the molecular basis of the PHF1 Tudor—UNC6641 interaction, we co-crystallized the Tudor domain with UNC6641 and obtained a crystal structure of the complex at 1.85 Å resolution (Figure 2). The X-ray diffraction and structure refinement statistics are summarized in Supplementary Table 1. UNC6641 is bound in an elongated groove of the Tudor domain (Figure 2a). The electron-density map of the UNC6641 peptide chain from G1 to R8 and the phenethyl and isopropyl lysine substituents can be unambiguously traced (Figure 2b).
Figure 2.

Molecular mechanism for the association of the PHF1 Tudor domain with UNC6641 (PDB ID: 7LKY). (a) The electrostatic surface potential of the PHF1 Tudor domain. Bound UNC6641 is colored green, and the phenyl substituent of K4 is indicated as Lig. (b) The 2Fo-Fc electron-density map of UNC6641 contoured at 1 σ shown as a gray mesh. (c) Structure of the PHF1 Tudor domain in complex with UNC6641. The PHF1 Tudor domain is shown as a gray ribbon, UNC6641 is shown as green sticks, and the protein residues interacting with UNC6641 are shown as gray sticks (except for W41, Y47, F65, and F71, which are colored yellow). The hydrogen bonds are indicated by yellow dashed lines. (d) Zoomed-in view of the UNC6641 binding site. (e) Overlay of the structures of the of PHF1 Tudor domain in complex with the H3K36me3 peptide (purple sticks) (PDB ID: 4HCZ) and UNC6641 (green sticks).
The structure belongs to the P1 space group with eight molecules of the PHF1 Tudor domain (each bound to UNC6641) present in one asymmetric unit. In the complex, the Tudor domain adopts a five-stranded β-barrel fold and contains the aromatic cage, which is comprised of W41, Y47, F65, and F71 (Figure 2c). The loops between β3 and β4 strands form an acidic groove, which is occupied by the N-terminal portion of UNC6641 (G1-K5), whereas L38, L45, L46, and L48 of the β3 strand form a hydrophobic patch where the C-terminal part of UNC6641 (P6-R8) is bound. The protein—ligand complex is further stabilized by several hydrogen bonds (Figure 2d). Specifically, the N-terminal amino group of UNC6641 is restrained by a hydrogen bond with the side chain carboxyl group of D68. The side chain amino group of K5 of UNC6641 is hydrogen bonded to the carboxyl group of E66, and the backbone amino group of L7 of UNC6641 forms a hydrogen bond with the backbone carbonyl of L46. These important hydrogen bonding interactions are in agreement with the large resonance perturbations observed for these residues in NMR titration experiments (Figure 1b).
Additionally, the pyrrolidine ring of P6 of UNC6641 lies parallel to the hydroxyphenyl moiety of PHF1 Y47, interacting with it through C—H…π contacts. The hydrophobic contacts involving the backbone of G2 and the side chains of K4, L7, and R8 of UNC6641 and D67, Y47, L38, and L45 of the PHF1 Tudor domain further contribute to the interaction (Figure S6, Ligplot).
The K4 side chain of UNC6641 contains two substituents, a phenethyl group and an isopropyl group. In the complex, the phenethyl group of UNC6641 inserts deeply into the aromatic cage of the Tudor domain and participates in hydrophobic interactions with W41, Y47, F65, F71, and A39 of the protein. The aromatic ring of the phenethyl group is sandwiched between the aromatic side chains of F65 and W41 of the Tudor aromatic cage residues. Specifically, the benzene ring is engaged with the phenyl group of F65 through a π—π interaction (the distance between these aromatic moieties is ~3.4 Å). Furthermore, the benzene ring is positioned almost perpendicular to the phenyl ring of F71 and to the hydroxyphenyl group of Y47, suggesting productive T-shaped edge-to-face interactions. The strength of these π-interactions seems to depend upon the precise placement of the benzene ring as dictated by the ethyl linker. As seen with UNC7259 and UNC7253, increasing or decreasing the linker length by a single methylene resulted in a modest decrease in potency. Finally, the two methyl groups of the isopropyl lysine substituent of UNC6641 interact via hydrophobic interactions with the side chains of W41, Y47, and F71 of the Tudor domain, likely stabilizing the binding of the phenethyl substituent (Figure 2c,d).
Structural overlay of the PHF1 Tudor domain in complex with the H3K36me3 peptide and UNC6641 shows that the majority of residues within the peptide chains superimpose well, indicating similar binding modes of these ligands (Figure 2e). However, several important differences are apparent. In the complex with the H3K36me3 peptide, G33 of the peptide does not interact with D68 of the Tudor domain, whereas G1 of UNC6641 forms a hydrogen bond with D68. Further, H39 in the H3K36me3 peptide is replaced with a leucine in UNC6641, which augments the association of UNC6641 with the hydrophobic patch. Importantly, the phenethyl substituent of UNC6641 is involved in π stacking and other productive interactions with the aromatic cage residues of the Tudor domain, which likely accounts for the tighter binding of UNC6641 than that of the H3K36me3 peptide (Figure 2e).
Biochemical Analysis of the UNC6641 Binding Mode.
To tease apart the key π-stacking interactions between the Kme mimetic of UNC6641 and the PHF1 Tudor aromatic cage residues, we performed alanine scanning mutagenesis of residues W41, Y47, and F65. We used UNC7643 as a control compound, as it maintains the (ethyl)isopropyl lysine but lacks the characteristic phenyl group. UNC7643 binds the wildtype PHF1 Tudor domain about 10-fold less potently than UNC6641. Previous literature has shown that single-residue mutations in the aromatic cage can significantly decrease the binding affinity of H3K36me3 for PHF1 due to disruption of the essential cation—π interactions.23,27 However, with the added affinity from π—π stacking, we hypothesized that UNC6641 but not UNC7643 may maintain binding to these aromatic cage residue mutants, although likely to a lesser degree than wildtype. UNC7643 failed to bind any PHF1 aromatic cage mutants, while UNC6641 retained moderate binding to PHF1 Tudor W41A and F65A (Table 3, Figure S7). Interestingly, UNC6641 showed no measurable binding to the Y47A mutant, suggesting a preferential dependence on the interactions with this residue.
Table 3.
Mutation of Aromatic Cage Residues to Assess the Key Molecular Interactions between UNC6641 and the PHF1 Tudor Domain
| UNC7643 | ||
|---|---|---|
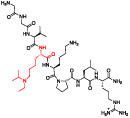 |
Mutation | ITC Kd(μM) |
| WT | 10.1 ± 1.7 | |
| W41A | >100 | |
| Y47A | >100 | |
| F65A | >100 | |
| UNC6641 | ||
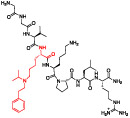 |
Mutation | ITC Kd (μM) |
| WT | 0.96 ± 0.03 | |
| W41A | 4.9 ± 0.6 | |
| Y47A | >100 | |
| F65A | 13.7 ± 1.3 | |
Kd values were determined by two independent ITC experiments (mean ± SD).
Selectivity of UNC6641.
Although we focused our initial studies on the antagonism of PHF1, we were curious to evaluate the binding of UNC6641 to other closely related Kme reader domains. PHF1 belongs to the polycomb-like (PCL) family of proteins which also consists of plant homeodomain finger protein 19 (PHF19) and metal response element binding transcription factor 2 (MTF2). While PHF1 and PHF19 each contain an aromatic cage composed of four key aromatic residues, MTF2 contains a serine in place of F71/Y75 which is thought to be responsible for “closing” the aromatic cage (Figure 3a). In many cases, PHF1 and PHF19 have been shown to play redundant roles in normal biology distinct from that of MTF2,32 suggesting that a dual ligand for PHF1 and PHF19 may be a valuable tool to investigate PCL biology. We were pleased to see that UNC6641 binds the PHF19 Tudor domain with a Kd value of 2.1 ± 0.2 μM by ITC, only 2-fold weaker than PHF1 (Figure S2). In contrast, UNC6641 binds MTF2 with a Kd value of 18 ± 2 μM, ~10- and 20-fold weaker than PHF19 and PHF1, respectively (Figure S8). We next conducted a sequence alignment within the Tudor family, and those domains most closely related to the PCL proteins are shown in Figure 3a, all of which share 3 of the 4 aromatic cage residues with the PCL proteins (for the full Tudor domain phylogenetic tree, see Figure S9). However, none of these Tudor domains including those of 53BP1, PHF20, and PHF20L1 demonstrated measurable binding to UNC6641 by ITC (Figure 3b, Figure S8). Furthermore, preliminary screening of representative members of other Kme reader families with UNC6641 by ITC and/or TR-FRET showed >20-fold selectivity for PHF1/19 (Figure S10). Overall, this suggests that both the sequence and unique Kme mimetic of UNC6641 confer selectivity over related proteins.
Figure 3.

Selectivity of UNC6641 across closely related Tudor domains. (a) Sequence alignment of Tudor domains most closely related to PHF1. Aromatic cage residues are in orange. * = Fully conserved. (b) Binding affinities of UNC6641 for closely related Tudor domains. Kd values were determined by two independent ITC experiments (mean ± SD).
CONCLUSION
PHF1 and its homologue, PHF19, are valuable accessory components of the ubiquitous repressive complex, PRC2, through their roles as readers of H3K36me3. A recent uptick in literature investigating these proteins revealed key roles for PHF1 and PHF19 in malignant biology and encouraged our pursuit of ligands that could be used in the development of a chemical probe.32,33,35,45-47 In this paper, we describe the discovery and characterization of UNC6641, which binds the Tudor domains of PHF1 and PHF19 with Kd’s of 0.96 ± 0.03 and 2.1 ± 0.2 μM, respectively.
UNC6641 is an 8-mer peptidomimetic ligand that contains a unique (isopropyl)phenethyl-lysine residue in place of K36me3. To the best of our knowledge, this is the first example of an aromatic substituent on a lysine residue facilitating enhanced interactions with a Kme reader domain. Co-crystallization of UNC6641 with the PHF1 Tudor domain revealed key interactions that contribute to the potent binding of this ligand. Namely, the distinctive phenethyl group of UNC6641 is sandwiched between the PHF1 aromatic cage residues W41 and F65, engaging in face-to-face π—π stacking interactions with these residues. Additionally, UNC6641 engages in T-shaped edge-to-face π stacking interactions with the remaining cage residues Y47 and F71. Combined with the additional hydrogen bonds and hydrophobic interactions created from our rational sequence mutations, we were able to determine structure—activity relationships that resulted in a clear gain in affinity relative to the endogenous histone peptide. Additionally, we used site-directed mutagenesis of the aromatic cage residues to gauge which ligand interactions are most critical for binding, which revealed that the phenethyl group of UNC6641 makes critical contacts with PHF1 Y47. UNC6641 showed selectivity for PHF1/19 over the closely related protein MTF2, likely due to the dependence on π interactions with all four aromatic cage residues. Although MTF2 has high sequence similarity to PHF1/19, it lacks the final aromatic cage residue that is present in PHF1/19.
While UNC6641 demonstrates the ligandability of PHF1 and shows promising potency and selectivity, peptidomimetic ligands are often limited in their utility by low cell permeability. However, the discovery and characterization of UNC6641 allowed us to develop and optimize a robust TR-FRET displacement assay for the PHF1 Tudor domain, which had previously not been feasible due to the low affinity of the endogenous histone substrate. Importantly, this assay can facilitate the screening of larger compound libraries toward the discovery of novel PHF1 Tudor domain antagonists. Additional screening efforts will shed light on the likelihood of uncovering potent small molecule chemical probes that bind PHF1. Recent reports have also demonstrated that improved potency and selectivity can be achieved for Tudor domain containing proteins such as Spindlin1 through the development of bidentate ligands which target multiple consecutive reader domains.48,49 While simultaneously targeting the Tudor and adjacent PHD domain (PHD1) of PHF1 may be another strategy to achieve potent antagonism, data supporting the reader activity of PHF1 PHD1 remains largely elusive.50
A potent and cell permeable small molecule chemical probe for PHF1 would be a valuable tool for researchers invested in studying the role of the PCL proteins as well as PRC2 regulation. Additionally, a PHF1 chemical probe has the potential to catalyze new drug discovery efforts toward the treatment of endometrial stromal sarcomas and fibroxymoid tumors, as these diseases depend heavily on aberrant PHF1 fusion proteins.
EXPERIMENTAL SECTION
Synthesis of Peptides and Peptidomimetics.
General Procedures.
Reverse phase column chromatography was performed with a Teledyne ISCO CombiFlashRf 200 using C18 RediSepRf Gold columns with the UV detector set to 220 and 254 nm. The mobile phases used are indicated for each compound. Preparative HPLC was performed using an Agilent Prep 1200 series with the UV detector set to 220 and 254 nm. Samples were injected onto a Phenomenex Luna 250 × 30 mm2, 5 μm, C18 column at 25 °C. Mobile phases of A (H2O + 0.1% TFA) and B (CH3CN + 1% H2O) were used with a flow rate of 40 mL/min. A general gradient of 0—25 min increasing from 10 to 100% B, followed by a 100% B flush for another 5 min, was used. Small variations in this purification method were made as needed to achieve ideal separation for each compound. Analytical LC-MS data for all compounds were acquired using an Agilent 1260 Infinity II Series system with the UV detector set to 220 and 254 nm. Samples were injected (5 μL) onto an Agilent Eclipse Plus 4.6 × 3 × 50 mm3, 1.8 mm, C18 column at 25 °C. Mobile phases A (H2O + 0.1% acetic acid) and B (CH3CN + 0.1% acetic acid) were used with a linear gradient from 10 to 100% B in 5.0 min, followed by a flush at 100% B for another 2 min at a flow rate of 1.0 mL/min. Mass spectra (MS) data were acquired in positive ion mode using an Agilent 6110 single quadrupole mass spectrometer with an electrospray ionization (ESI) source or matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI TOF/TOF) using an AB Sciex 5800. Nuclear magnetic resonance (NMR) spectra were recorded on a Varian Mercury spectrometer at 400 MHz for proton (1H) and carbon (13C) NMR; chemical shifts are reported in ppm (δ) relative to residual protons in deuterated solvent peaks, and coupling constants are reported in Hz. All compounds reported had >95% purity, as determined by LC-MS.
Solid-Phase Peptide Synthesis (SPPS).
Peptide compounds listed in Table 1 were synthesized by SPPS on Fmoc Rink amide resin on polystyrene beads (0.81 mmol/g loading, 1 equiv, Anaspec). The resin was first swelled in DCM for 5 min followed by equilibration in DMF for another 5 min. The resin was then Fmoc deprotected in a solution of 2.5% 1,8-diazabicycloundec-7-ene (DBU) and 2.5% pyrrolidine in DMF for 10 min. Then, the resin was filtered and washed twice with DMF, methanol, DMF, and DCM (6 mL each) before adding the first amino acid for coupling. Fmoc-protected amino acids (3 equiv) except Fmoc-Arg(Pmc)-OH were preactivated with HBTU (3 equiv), HOAt (3 equiv), and DIPEA (10 equiv) for 5 min with periodic swirling in 5 mL of DMF and 3 mL of DCM. For Fmoc-Arg(Pmc)-OH coupling, the longer preincubation was omitted to prevent formation of the intramolecular cyclization byproduct. The solution was then added to the resin and left on a shaker at room temperature for 1 h. The resin was filtered and washed twice with DCM, DMF, methanol, and DMF again (6 mL each). The N-terminal Fmoc protecting group was then removed in a solution of 2.5% DBU and 2.5% pyrrolidine in DMF for 10 min. The resin was filtered and washed twice with DMF, methanol, DMF, and DCM (6 mL each) before adding the next amino acid for coupling. Following installation of the final residue, the resin was rinsed six times with DCM. Cleavage cocktail (95% trifluoroacetic acid, 2.5% triisopropylsilane, and 2.5% water) was added to the resin, the mixture was left on the shaker for 2 h, and the filtrate was collected. The resin was rinsed twice with DCM, and filtrates were pooled and concentrated under a vacuum. The crude product was then submitted to preparative HPLC for purification as described in the General Procedures section. The solvent was removed in vacuo, dissolved in water, and lyophilized to yield the desired products.
(S)-6-(((S)-6-Amino-1-((S)-2-(((S)-1-(((S)-1-amino-5-((amino-(iminio)methyl)amino)-1-oxopentan-2-yl)amino)-3-(1H-imidazol-4-yl)-1-oxopropan-2-yl)carbamoyl)pyrrolidin-1-yl)-1-oxohexan-2-yl)amino)-5-((S)-2-(2-(2-aminoacetamido)acetamido)-3-methylbutanamido)-N,N,N-trimethyl-6-oxohexan-1-aminium (H3 Peptide: GGVKme3KPHR).
White powder; yield 18%. 1H NMR (MeOD-d4, 400 MHz): δ 8.81 (d, J = 1.4 Hz, 1H), 7.43 (d, J = 1.4 Hz, 1H), 4.74–4.70 (m, 1H), 4.64 (dd, J = 8.6, 5.1 Hz, 1H), 4.44–4.38 (m, 2H), 4.36 (dd, J = 8.7, 5.2 Hz, 1H), 4.16 (d, J = 7.3 Hz, 1H), 4.05–3.93 (m, 2H), 3.84–3.77 (m, 1H), 3.76 (s, 2H), 3.68–3.62 (m, 1H), 3.21 (dt, J = 9.1, 6.6 Hz, 3H), 3.13 (s, 9H), 2.95 (t, J = 7.4 Hz, 2H), 2.28–2.16 (m, 1H), 2.12–1.73 (m, 12H), 1.68 (dt, J = 13.3, 7.1 Hz, 6H), 1.55–1.33 (m, 5H), 0.98 (dd, J = 6.8, 1.4 Hz, 6H); MS (ESI): m/z calcd for [C41H76N16O8]3+/2 460.80, found 460.30.
(S)-6-(((S)-6-Amino-1-((S)-2-(((S)-1-amino-3-(1H-imidazol-4-yl)-1-oxopropan-2-yl)carbamoyl)pyrrolidin-1-yl)-1-oxohexan-2-yl)-amino)-5-((S)-2-(2-(2-aminoacetamido)acetamido)-3-methylbutanamido)-N,N,N-trimethyl-6-oxohexan-1-aminium (H3 Peptide: GGVKme3KPH).
White powder; yield 10%. 1H NMR (MeOD-d4, 400 MHz): δ 8.82 (d, J = 1.4 Hz, 1H), 7.41 (d, J = 1.3 Hz, 1H), 4.65 (td, J = 9.1, 8.5, 5.3 Hz, 2H), 4.39 (ddd, J = 11.3, 8.8, 5.5 Hz, 2H), 4.15 (d, J = 7.2 Hz, 1H), 4.05–3.91 (m, 2H), 3.86–3.78 (m, 1H), 3.76 (s, 2H), 3.73–3.63 (m, 1H), 3.13 (s, 9H), 2.95 (t, J = 7.4 Hz, 2H), 2.22 (ddd, J = 15.1, 7.4, 4.2 Hz, 1H), 2.14–1.74 (m, 10H), 1.69 (ddt, J = 14.0, 10.9, 4.8 Hz, 3H), 1.54–1.40 (m, 4H), 1.38–1.31 (m, 3H), 0.97 (d, J = 6.5 Hz, 6H); MS (ESI): m/z calcd for [C35H63N12O7]2+ 764.59, found 764.30.
(S)-6-(((S)-6-Amino-1-((S)-2-(((S)-1-amino-3-(1H-imidazol-4-yl)-1-oxopropan-2-yl)carbamoyl)pyrrolidin-1-yl)-1-oxohexan-2-yl)-amino)-5-((S)-2-(2-((S)-2-amino-3-methylbutanamido)acetamido)-3-methylbutanamido)-N,N,N-trimethyl-6-oxohexan-1-aminium (H3 Peptide: VGVKme3KPH).
White powder; yield 12%. 1H NMR (MeOD-d4, 400 MHz): δ 8.83 (d, J = 1.4 Hz, 1H), 7.41 (d, J = 1.3 Hz, 1H), 4.65 (ddd, J = 8.6, 5.4, 2.9 Hz, 2H), 4.39 (ddd, J = 11.7, 8.7, 5.6 Hz, 2H), 4.18 (d, J = 7.0 Hz, 1H), 4.06–3.94 (m, 2H), 3.84 (dt, J = 10.0, 6.4 Hz, 1H), 3.72–3.64 (m, 2H), 3.17 (d, J = 7.7 Hz, 1H), 3.14 (s, 1H), 3.13 (s, 9H), 2.95 (t, J = 7.4 Hz, 2H), 2.30–1.60 (m, 16H), 1.47 (dt, J = 21.3, 7.5 Hz, 4H), 1.07 (dd, J = 6.9, 1.4 Hz, 6H), 0.98 (dd, J = 6.8, 2.5 Hz, 6H); MS (ESI): m/z calcd for [C38H69N12O7]2+ 806.54, found 806.40.
(S)-6-(((S)-6-Amino-1-((S)-2-(((S)-1-amino-4-methyl-1-oxopen-tan-2-yl)carbamoyl)pyrrolidin-1-yl)-1-oxohexan-2-yl)amino)-5-((S)-2-(2-((S)-2-amino-3-methylbutanamido)acetamido)-3-methylbutanamido)-N,N,N-trimethyl-6-oxohexan-1-aminium (H3 Peptide: VGVKme3KPL).
White powder; yield 50%. 1H NMR (MeOD-d4, 400 MHz): δ 4.64–4.60 (m, 1H), 4.46–4.31 (m, 3H), 4.17 (dd, J = 7.2, 4.8 Hz, 1H), 4.07–3.92 (m, 2H), 3.80 (dd, J = 11.1, 5.2 Hz, 1H), 3.73–3.62 (m, 2H), 3.13 (s, 9H), 2.95 (t, J = 7.3 Hz, 2H), 2.21 (dt, J = 13.3, 6.6 Hz, 2H), 2.13–1.96 (m, 4H), 1.94–1.36 (m, 17H), 1.07 (d, J = 6.9 Hz, 6H), 0.99–0.92 (m, 12H); MS (ESI): m/z calcd for [C38H73N10O7]2+/2 391.29, found 391.30.
(S)-6-(((S)-6-Amino-1-((S)-2-(((S)-1-(((S)-1-amino-5-((amino-(iminio)methyl)amino)-1-oxopentan-2-yl)amino)-4-methyl-1-oxo-pentan-2-yl)carbamoyl)pyrrolidin-1-yl)-1-oxohexan-2-yl)amino)-5-((S)-2-(2-(2-aminoacetamido)acetamido)-3-methylbutanamido)-N,N,N-trimethyl-6-oxohexan-1-aminium (H3 Peptide: GGVKme3KPLR).
White powder; yield 44%. 1H NMR (MeOD-d4, 400 MHz): 4.62 (dd, J = 8.3, 5.4 Hz, 1H), 4.45 (dd, J = 8.4, 4.3 Hz, 1H), 4.42–4.30 (m, 3H), 4.16 (d, J = 7.3 Hz, 1H), 4.05–3.91 (m, 2H), 3.84–3.79 (m, 1H), 3.76 (s, 2H), 3.68–3.59 (m, 1H), 3.21 (q, J = 7.6 Hz, 2H), 3.13 (s, 9H), 2.94 (t, J = 7.4 Hz, 2H), 2.27–2.18 (m, 1H), 2.04 (ddt, J = 25.1, 14.3, 7.4 Hz, 4H), 1.91–1.57 (m, 16H), 1.46 (ddd, J = 24.7, 11.6, 7.4 Hz, 4H), 0.95 (dd, J = 17.1, 6.5 Hz, 12H); MS (ESI): m/z calcd for [C41H80N14O8]+ 895.63, found 895.50.
Mixed Synthesis of Peptidomimetic Compounds.
Peptidomimetic compounds were synthesized using an integrated approach of solid- and solution-phase synthesis. Kme3 mimetics were synthesized in solution as Fmoc-protected amino acids using reductive aminations as described below and added to the corresponding peptide by SPPS.
Protocol A.
Fmoc-L-lysine (1 equiv) was dissolved in ethanol and stirred at rt. Aldehyde and/or ketone (8 equiv) was added to the cloudy solution, and the mixture was left to stir for 10 min followed by addition of acetic acid (3 equiv). Sodium cyanoborohydride (5 equiv) was then added to the reaction. The mixture was stirred at rt for 2 h. The crude mixture was concentrated in vacuo and purified by reverse phase flash chromatography (H2O + 0.1% trifluoroacetic acid and CH3CN) to yield the final product as a TFA salt. The product was redissolved in water and lyophilized to a hygroscopic, white powder.
Protocol B.
Fmoc-L-lysine (1 equiv) was dissolved in ethanol and stirred at rt. Ketone (8 equiv) was added to the cloudy solution, and the mixture was left to stir for 10 min followed by addition of acetic acid (3 equiv). Sodium cyanoborohydride (5 equiv) was then added to the reaction. The mixture was stirred at 60 °C for 16 h. Aldehyde (8 equiv) and an additional 5 equiv of sodium cyanoborohydride were added to the reaction. The reaction mixture was again stirred at 60 °C for 16 h. The crude mixture was concentrated in vacuo and purified by reverse phase flash chromatography (H2O + 0.1% trifluoroacetic acid and CH3CN) to yield the final product as a TFA salt. The product was redissolved in water and lyophilized to a hygroscopic, white powder.
N2-(((9H-Fluoren-9-yl)methoxy)carbonyl)-N6-ethyl-N6-isopropyl-L-lysine (1). Protocol A.
White powder; yield 50%. 1H NMR (MeOD-d4, 400 MHz): δ 7.76 (d, J = 7.5 Hz, 2H), 7.64 (t, J = 8.2 Hz, 2H), 7.36 (t, J = 7.4 Hz, 2H), 7.28 (td, J = 7.5, 1.2 Hz, 2H), 4.39–4.26 (m, 2H), 4.21–4.12 (m, 2H), 3.62 (p, J = 6.7 Hz, 1H), 3.21–2.88 (m, 4H), 1.90 (dtd, J = 13.5, 8.1, 7.7, 4.9 Hz, 1H), 1.73 (qd, J = 9.4, 5.8 Hz, 3H), 1.51–1.40 (m, 2H), 1.28 (q, J = 6.6, 6.2 Hz, 9H); MS (ESI): m/z calcd for [C26H34N2O4]+ 439.25, found 439.20.
Amino(((S)-5-amino-4-((S)-2-((S)-1-((2S,5S,8S)-14-amino-2-(4-aminobutyl)-5-(4-(ethyl(isopropyl)amino)butyl)-8-isopropyl-4,7,10,13-tetraoxo-3,6,9,12-tetraazatetradecanoyl)pyrrolidine-2-carboxamido)-4-methylpentanamido)-5-oxopentyl)amino)methaniminium (UNC7643).
White powder; yield 40%.1H NMR (MeOD-d4, 400 MHz): δ 4.61 (dd, J = 8.3, 5.3 Hz, 1H), 4.45 (dd, J = 8.2, 4.1 Hz, 1H), 4.39–4.29 (m, 3H), 4.17 (d, J = 7.2 Hz, 1H), 3.99 (s, 2H), 3.83–3.77 (m, 1H), 3.76 (s, 2H), 3.69 (td, J = 15.3, 14.3, 6.5 Hz, 2H), 3.28–3.11 (m, 5H), 3.06 (dd, J = 16.0, 7.6 Hz, 1H), 2.94 (t, J = 7.4 Hz, 2H), 2.27–2.17 (m, 1H), 2.16–1.94 (m, 4H), 1.90–1.79 (m, 3H), 1.77–1.60 (m, 12H), 1.50 (p, J = 8.2 Hz, 4H), 1.37–1.32 (m, 9H), 0.99–0.92 (m, 12H); MS (ESI): m/z calcd for [C43H83N14O8]2+ 924.65, found 924.50.
N2-(((9H-Fluoren-9-yl)methoxy)carbonyl)-N6-ethyl-N6-(1-(furan-2-yl)ethyl)-L-lysine (2). Protocol B.
White powder; yield 40%. 1H NMR (MeOD-d4, 400 MHz): δ 7.80 (d, J = 7.6 Hz, 2H), 7.66 (q, J = 12.8, 10.9 Hz, 3H), 7.40 (t, J = 7.5 Hz, 2H), 7.31 (t, J = 7.3 Hz, 2H), 6.71 (s, 1H), 6.50 (d, J = 15.2 Hz, 1H), 4.44–4.31 (m, 2H), 4.20 (dt, J = 24.1, 5.7 Hz, 2H), 3.02 (dd, J = 97.5, 53.8 Hz, 4H), 1.90 (dd, J = 13.9, 6.3 Hz, 1H), 1.70 (t, J = 7.5 Hz, 7H), 1.43 (s, 2H), 1.31 (t, J = 6.7 Hz, 3H); MS (ESI): m/z calcd for [C29H34N2O5]+ 491.25, found 491.25.
Amino(((4S)-5-amino-4-((2S)-2-((2S)-1-((2S,5S,8S)-14-amino-2-(4-aminobutyl)-5-(4-(ethyl(1-(furan-2-yl)ethyl)amino)butyl)-8-isopropyl-4,7,10,13-tetraoxo-3,6,9,12-tetraazatetradecanoyl)pyrrolidine-2-carboxamido)-4-methylpentanamido)-5-oxopentyl)amino)methaniminium (UNC6640).
White powder; yield 25%. 1H NMR (MeOD-d4, 400 MHz): δ 7.68 (dt, J = 1.9, 0.9 Hz, 1H), 6.74 (d, J = 3.4 Hz, 1H), 6.54 (dd, J = 3.4, 1.9 Hz, 1H), 4.61 (dd, J = 8.2, 5.4 Hz, 1H), 4.45 (dd, J = 8.3, 4.0 Hz, 1H), 4.39–4.30 (m, 3H), 4.18 (d, J = 7.1 Hz, 1H), 3.99 (s, 2H), 3.80 (dt, J = 10.0, 6.5 Hz, 1H), 3.76 (s, 2H), 3.68–3.60 (m, 1H), 3.22 (tt, J = 14.3, 6.9 Hz, 4H), 3.14–3.05 (m, 1H), 2.94 (t, J = 7.4 Hz, 2H), 2.22 (dt, J = 15.4, 9.1 Hz, 1H), 2.12–1.95 (m, 4H), 1.94–1.57 (m, 19H), 1.55–1.38 (m, 4H), 1.33 (d, J = 8.2 Hz, 4H), 0.99–0.91 (m, 12H); MS (ESI): m/z calcd for [C46H83N14O9]2+ 976.64, found 976.50.
N2-(((9H-Fluoren-9-yl)methoxy)carbonyl)-N6-isopropyl-N6-phenethyl-L-lysine (3). Protocol B.
White, sticky solid; yield 42%. 1H NMR (MeOD-d4, 400 MHz): δ 7.68 (dt, J = 1.9, 0.9 Hz, 1H), 6.74 (d, J = 3.4 Hz, 1H), 6.54 (dd, J = 3.4, 1.9 Hz, 1H), 4.61 (dd, J = 8.2, 5.4 Hz, 1H), 4.45 (dd, J = 8.3, 4.0 Hz, 1H), 4.39–4.30 (m, 3H), 4.18 (d, J = 7.1 Hz, 1H), 3.99 (s, 2H), 3.80 (dt, J = 10.0, 6.5 Hz, 1H), 3.76 (s, 2H), 3.68–3.60 (m, 1H), 3.22 (tt, J = 14.3, 6.9 Hz, 4H), 3.14–3.05 (m, 1H), 2.94 (t, J = 7.4 Hz, 2H), 2.22 (dt, J = 15.4, 9.1 Hz, 1H), 2.12–1.95 (m, 4H), 1.94–1.57 (m, 19H), 1.55–1.38 (m, 4H), 1.33 (d, J = 8.2 Hz, 4H), 0.99–0.91 (m, 12H); MS (ESI): m/z calcd for [C32H38N2O4]+ 515.28, found 515.30.
Amino(((S)-5-amino-4-((S)-2-((S)-1-((2S,5S,8S)-14-amino-2-(4-aminobutyl)-8-isopropyl-5-(4-(isopropyl(phenethyl)amino)butyl)-4,7,10,13-tetraoxo-3,6,9,12-tetraazatetradecanoyl)pyrrolidine-2-carboxamido)-4-methylpentanamido)-5-oxopentyl)amino)methaniminium (UNC6641).
White powder; yield 21%. 1H NMR (MeOD-d4, 400 MHz): δ 7.38–7.25 (m, 5H), 4.61 (dd, J = 8.4, 5.2 Hz, 1H), 4.45 (dd, J = 8.3, 4.2 Hz, 1H), 4.39–4.29 (m, 3H), 4.17 (d, J = 7.2 Hz, 1H), 3.98 (d, J = 5.1 Hz, 2H), 3.83–3.74 (m, 4H), 3.65 (dd, J = 10.8, 4.0 Hz, 1H), 3.38 (dt, J = 17.9, 8.3 Hz, 1H), 3.27–3.12 (m, 4H), 3.06 (dq, J = 12.2, 6.5, 5.7 Hz, 2H), 2.94 (t, J = 7.4 Hz, 2H), 2.23 (ddd, J = 12.7, 8.2, 3.8 Hz, 1H), 2.15–1.93 (m, 4H), 1.93–1.55 (m, 16H), 1.51 (q, J = 7.8 Hz, 4H), 1.37 (dd, J = 6.7, 2.0 Hz, 6H), 1.02–0.88 (m, 12H); MS (ESI): m/z calcd for [C49H87N14O8]2+/2 500.32, found 500.40.
N2-(((9H-Fluoren-9-yl)methoxy)carbonyl)-N6-isopropyl-N6-(3-phenylpropyl)-L-lysine (4). Protocol B.
White, sticky solid; yield 58%. 1H NMR (MeOD-d4, 400 MHz): δ 7.77 (d, J = 7.5 Hz, 2H), 7.65 (dd, J = 10.4, 7.5 Hz, 2H), 7.37 (t, J = 7.4 Hz, 2H), 7.30 (dd, J = 7.5, 1.2 Hz, 2H), 7.28–7.25 (m, 2H), 7.24–7.19 (m, 3H), 4.38 (dd, J = 10.5, 6.9 Hz, 1H), 4.30 (dd, J = 10.4, 7.1 Hz, 1H), 4.22–4.15 (m, 2H), 3.61 (p, J = 7.5, 7.0 Hz, 1H), 3.12–2.94 (m, 4H), 2.70–2.65 (m, 2H), 1.99 (s, 2H), 1.79–1.57 (m, 4H), 1.45 (dt, J = 15.0, 6.3 Hz, 2H), 1.26–1.23 (m, 6H); MS (ESI): m/z calcd for [C33H40N2O4]+ 529.30, found 529.30.
Amino(((S)-5-amino-4-((S)-2-((S)-1-((2S,5S,8S)-14-amino-2-(4-aminobutyl)-8-isopropyl-5-(4-(isopropyl(3-phenylpropyl)amino)butyl)-4,7,10,13-tetraoxo-3,6,9,12-tetraazatetradecanoyl)pyrrolidine-2-carboxamido)-4-methylpentanamido)-5-oxopentyl)amino)methaniminium (UNC7259).
White powder; yield 21%. 1H NMR (MeOD-d4, 400 MHz): δ 7.35–7.17 (m, 5H), 4.62 (dd, J = 8.5, 5.2 Hz, 1H), 4.48–4.42 (m, 1H), 4.35 (tt, J = 9.4, 5.2 Hz, 3H), 4.17 (d, J = 7.2 Hz, 1H), 4.00 (d, J = 3.0 Hz, 1H), 3.81–3.76 (m, 3H), 3.21 (p, J = 6.9 Hz, 3H), 3.16–3.05 (m, 4H), 2.96 (t, J = 7.3 Hz, 2H), 2.74 (t, J = 7.6 Hz, 2H), 2.25 (d, J = 7.5 Hz, 1H), 2.16–1.94 (m, 5H), 1.92–1.59 (m, 17H), 1.53–1.40 (m, 4H), 1.32 (dd, J = 6.6, 1.6 Hz, 6H), 1.29 (q, J = 2.3 Hz, 1H), 0.99–0.92 (m, 12H); MS (ESI): m/z calcd for [C50H89N14O8]+ 1013.70, found 1013.50.
N2-(((9H-Fluoren-9-yl)methoxy)carbonyl)-N6-benzyl-N6-isopropyl-L-lysine (5). Protocol B.
White, sticky solid; yield 53%. 1H NMR (MeOD-d4, 400 MHz): δ 7.46 (s, 6H), 7.29 (d, J = 3.9 Hz, 7H), 5.31–5.26 (m, 1H), 5.17 (dd, J = 12.1, 1.9 Hz, 1H), 4.36 (dd, J = 13.1, 1.9 Hz, 1H), 4.11 (dd, J = 13.3, 10.3 Hz, 1H), 3.89 (dd, J = 13.7, 3.9 Hz, 2H), 3.59 (dd, J = 13.1, 6.6 Hz, 3H), 1.79–1.66 (m, 2H), 1.52 (d, J = 12.6 Hz, 1H), 1.40 (d, J = 6.6 Hz, 3H), 1.38–1.29 (m, 5H), 1.25–1.16 (m, 1H); MS (ESI): m/z calcd for [C31H36N2O4]+ 501.27, found 501.30.
Amino(((S)-5-amino-4-((S)-2-((S)-1-((2S,5S,8S)-14-amino-2-(4-aminobutyl)-5-(4-(benzyl(isopropyl)amino)butyl)-8-isopropyl-4,7,10,13-tetraoxo-3,6,9,12-tetraazatetradecanoyl)pyrrolidine-2-carboxamido)-4-methylpentanamido)-5-oxopentyl)amino)methaniminium (UNC7253).
White powder; yield 12%. 1H NMR (MeOD-d4, 400 MHz): δ 7.57–7.45 (m, 5H), 4.60 (dd, J = 8.5, 5.2 Hz, 1H), 4.48–4.23 (m, 6H), 4.16 (d, J = 6.9 Hz, 1H), 3.98 (s, 6H), 3.69 (dt, J = 13.8, 6.8 Hz, 1H), 3.27–3.12 (m, 3H), 3.10–2.98 (m, 1H), 2.94 (t, J = 7.4 Hz, 2H), 2.22 (dt, J = 7.6, 3.3 Hz, 1H), 2.14–1.54 (m, 19H), 1.50 (q, J = 7.8 Hz, 2H), 1.39 (td, J = 19.3, 6.3 Hz, 8H), 0.94 (dd, J = 16.8, 6.3 Hz, 12H); MS (MALDI): m/z calcd for [C48H85N14O8]+ 985.67, found 985.62.
N2-(((9H-Fluoren-9-yl)methoxy)carbonyl)-N6-(2-cyclohexylethyl)-N6-isopropyl-L-lysine (6). Protocol B.
White powder; yield 62%. 1H NMR (MeOD-d4, 400 MHz): δ 7.76 (t, J = 6.6 Hz, 2H), 7.68–7.60 (m, 2H), 7.36 (t, J = 7.6 Hz, 2H), 7.28 (t, J = 7.5 Hz, 2H), 4.31 (dtd, J = 17.6, 10.4, 9.0, 4.1 Hz, 2H), 4.19 (q, J = 7.5, 7.0 Hz, 2H), 3.59 (dt, J = 13.1, 6.8 Hz, 1H), 3.01 (s, 4H), 2.13 (d, J = 6.7 Hz, 1H), 1.98–1.86 (m, 1H), 1.86–1.32 (m, 19H), 1.23–1.08 (m, 4H); MS (ESI): m/z calcd for [C32H44N2O4]+ 521.33, found 521.35.
Amino(((S)-5-amino-4-((S)-2-((S)-1-((2S,5S,8S)-14-amino-2-(4-aminobutyl)-5-(4-((2-cyclohexylethyl)(isopropyl)amino)butyl)-8-isopropyl-4,7,10,13-tetraoxo-3,6,9,12-tetraazatetradecanoyl)pyrrolidine-2-carboxamido)-4-methylpentanamido)-5-oxopentyl)amino)methaniminium (UNC7258).
White powder; yield 10%. 1H NMR (MeOD-d4, 400 MHz): δ 4.48–4.44 (m, 3H), 4.35 (ddd, J = 14.8, 9.0, 5.6 Hz, 4H), 4.18 (d, J = 7.2 Hz, 1H), 4.01 (d, J = 1.9 Hz, 1H), 3.80 (d, J = 5.9 Hz, 3H), 3.50–3.46 (m, 1H), 3.22 (q, J = 6.8 Hz, 2H), 3.18–3.03 (m, 4H), 2.97 (q, J = 7.3 Hz, 2H), 2.03–1.95 (m, 2H), 1.86 (s, 2H), 1.73 (d, J = 21.8 Hz, 14H), 1.69–1.61 (m, 7H), 1.51 (s, 4H), 1.35 (d, J = 6.5 Hz, 6H), 1.33–1.21 (m, 6H), 1.05 (d, J = 11.7 Hz, 2H), 1.00–0.92 (m, 12H); MS (MALDI): m/z calcd for [C49H93N14O8]+ 1005.73, found 1005.65.
Synthesis of Biotin-UNC6641.
UNC6641 was first coupled on-bead to propargylamine to provide a clickable handle for attachment of azido-PEG5-biotin. The peptide was then capped with an additional glycine residue using SPPS as described. Following cleavage from the resin, the alkynyl peptide was coupled to PEG5-biotin using click chemistry.
Amino(((S)-5-amino-4-((S)-2-((S)-1-((2S,5S,8S)-2-(4-aminobutyl)-18-glycyl-8-isopropyl-5-(4-(isopropyl(phenethyl)amino)butyl)-4,7,10,13,16-pentaoxo-3,6,9,12,15,18-hexaazahenicos-20-ynoyl)-pyrrolidine-2-carboxamido)-4-methylpentanamido)-5-oxopentyl)-amino)methaniminium (alkynyl-UNC6641).
To UNC6641 (0.215 g, 0.215 mmol, 1 equiv) on resin was added 1 M bromoacetic acid in DMF (2.5 mL, 12 equiv) and 1 M diisopropylmethanediimine in DMF (2.5 mL, 12 equiv). The mixture was stirred on the shaker at rt for 30 min after which the solution was expelled and the resin was washed with DMF 7× (5 mL each). One M propargylamine (5 mL, 27 equiv) was then added to the resin and stirred on the shaker at rt for 2 h. Again, the solution was expelled and the beads were washed with DMF 7× (5 mL each). The terminal glycine was added and the final peptide cleaved and purified as described in the Solid-Phase Peptide Synthesis (SPPS) section to yield a white solid (20 mg, 15%). MS (ESI): m/z calcd for [C56H95N16O10]+ 1152.74, found 1152.60.
Amino(((S)-5-amino-4-((S)-2-((S)-1-((2S,5S,8S)-20-amino-2-(4-aminobutyl)-8-isopropyl-5-(4-(isopropyl(phenethyl)amino)butyl)-4,7,10,13,16,19-hexaoxo-18-((1-(19-oxo-23-((3aR,4R,6aS)-2-oxo-hexahydro-1H-thieno[3,4-d]imidazol-4-yl)-3,6,9,12,15-pentaoxa-18-azatricosyl)-1H-1,2,3-triazol-5-yl)methyl)-3,6,9,12,15,18-hexaazaicosanoyl)pyrrolidine-2-carboxamido)-4-methylpentanamido)-5-oxopentyl)amino)methaniminium (Biotin-UNC6641).
Alkynyl-UNC6641 (0.014 g, 0.012 mmol, 1 equiv) was dissolved in DMF (0.2 mL). Aqueous solutions of 0.6 M copper(II) sulfate (0.2 mL, 10 equiv) and 0.12 M sodium ascorbate (1 mL, 10 equiv) were added followed by N-(17-azido-3,6,9,12,15-pentaoxaheptadecyl)-5-((3aS,4S,6aR)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)-pentanamide. The mixture was stirred at rt for 2 h. The solution was then filtered and concentrated in vacuo, followed by purification via preparative HPLC as described in the General Procedures section. The solvent was removed in vacuo, redissolved in water, and lyophilized to yield the desired products as a white solid (0.018 g, 89%). 1H NMR (MeOD-d4, 400 MHz): δ 7.38–7.25 (m, 5H), 4.71 (d, J = 5.0 Hz, 2H), 4.64–4.53 (m, 3H), 4.47 (ddd, J = 16.4, 8.1, 4.2 Hz, 2H), 4.39–4.29 (m, 4H), 4.21 (d, J = 21.9 Hz, 3H), 4.12 (dd, J = 7.4, 2.5 Hz, 1H), 3.98 (s, 3H), 3.92–3.85 (m, 4H), 3.79 (p, J = 6.6 Hz, 2H), 3.66–3.58 (m, 18H), 3.53 (t, J = 5.5 Hz, 2H), 3.35 (d, J = 1.8 Hz, 4H), 3.21 (q, J = 7.2 Hz, 4H), 3.07 (dt, J = 11.4, 7.0 Hz, 2H), 2.93 (dt, J = 10.2, 6.1 Hz, 3H), 2.70 (d, J = 12.8 Hz, 1H), 2.22 (t, J = 7.3 Hz, 3H), 2.16–1.91 (m, 4H), 1.91–1.61 (m, 18H), 1.60–1.42 (m, 8H), 1.38 (d, J = 6.7 Hz, 6H), 0.99–0.91 (m, 12H); MS (ESI): m/z calcd for [C78H135N22O17S]2+ 1685.91, found 1685.60.
Protein Expression and Purification.
Unlabeled Protein Expression Constructs.
The Tudor domains of PHF1 (residues 25–87 of NP_077084) and PHF19 (residues 49–115 of NP_001273769) were expressed with N-terminal His tags in pET28 expression vectors. PHF1 Tudor domain point mutants were generated using a QuickChange II Site-Directed Mutagenesis kit (Agilent, Santa Clara, CA) and the following primer sets:
| W41A | 5′-agcagcccatcagtcgctctggccagcacatc-3′ |
| 5′-gatgtgctggccagagcgactgatgggctgct-3′ | |
| Y47A | 5′-ctttttgatggtacccaaggctagcagcccatcagtccat-3′ |
| 5′-atggactgatgggctgctagccttgggtaccatcaaaaag-3 | |
| F65A | 5′-aactgcgaatcatcctcagcctggaccagacacacctc-3′ |
| 5′-gaggtgtgtctggtccaggctgaggatgattcgcagtt-3′ |
The Tudor domain of 53BP1 (residues 1484–1603 of NP_005648) was expressed with an N-terminal His tag in a pET15 expression vector. The Tudor domains of MTF2 (residues 1–160 of NP_031384), PHF20 (residues 58–148 of NP_057520), and PHF20L1 (residues 1–150 of NP_057102) were expressed with N-terminal GST tags in pGEX expression vectors.
Unlabeled Protein Expression and Purification.
All expression constructs were transformed into Rosetta2 BL21(DE3)pLysS competent cells (Novagen, EMD Chemicals, San Diego, CA). Protein expression was induced by growing cells at 37 °C with shaking until the OD600 reached ~0.6–0.8 at which time the temperature was lowered to 18 °C and expression was induced by adding 0.5 mM IPTG and continuing shaking overnight. Cells were harvested by centrifugation, and pellets were stored at −80 °C.
His-tagged proteins were purified by resuspending thawed cell pellets in 30 mL of lysis buffer (50 mM sodium phosphate pH 7.2, 50 mM NaCl, 30 mM imidazole, 1× EDTA free protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN)) per liter of culture. Cells were lysed on ice by sonication with a Branson Digital 450 Sonifier (Branson Ultrasonics, Danbury, CT) at 40% amplitude for 12 cycles with each cycle consisting of a 20 s pulse followed by a 40 s rest. The cell lysate was clarified by centrifugation and loaded onto a HisTrap FF column (GE Healthcare, Piscataway, NJ) that had been pre-equilibrated with 10 column volumes of binding buffer (50 mM sodium phosphate pH 7.2, 500 mM NaCl, 30 mM imidazole) using an AKTA FPLC (GE Healthcare, Piscataway, NJ). The column was washed with 15 column volumes of binding buffer, and protein was eluted in a linear gradient to 100% elution buffer (50 mM sodium phosphate pH 7.2, 500 mM NaCl, 500 mM imidazole) over 20 column volumes. Peak fractions containing the desired protein were pooled and concentrated to 2 mL in Amicon Ultra-15 concentrators with a 3000 molecular weight cutoff (Merck Millipore, Carrigtwohill Co. Cork IRL). Concentrated protein was loaded onto a HiLoad 26/60 Superdex 75 prep grade column (GE Healthcare, Piscataway, NJ) that had been pre-equilibrated with 1.2 column volumes of sizing buffer (25 mM Tris pH 7.5, 250 mM NaCl, 2 mM DTT, 5% glycerol) using an ATKA Purifier (GE Healthcare, Piscataway, NJ). Protein was eluted isocratically in sizing buffer over 1.3 column volumes at a flow rate of 2 mL/min collecting 3 mL fractions. Peak fractions were analyzed for purity by SDS-PAGE, and those containing pure protein were pooled and concentrated using Amicon Ultra-15 concentrators with a 3000 molecular weight cutoff (Merck Millipore, Carrigtwohill Co. Cork IRL).
GST-tagged proteins were purified by resuspending thawed cell pellets in 30 mL of lysis buffer (1× PBS, 5 mM DTT, 1× EDTA free protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN)) per liter of culture. Cells were lysed on ice by sonication as described for His-tagged proteins. Clarified cell lysate was loaded onto a GSTrap FF column (GE Healthcare, Piscataway, NJ) that had been preequilibrated with 10 column volumes of binding buffer (1× PBS, 5 mM DTT) using a AKTA FPLC (GE Healthcare, Piscataway, NJ). The column was washed with 10 column volumes of binding buffer, and protein was eluted in 100% elution buffer (50 mM Tris pH 7.5,150 mM NaCl, 10 mM reduced glutathione) over 10 column volumes. Peak fractions containing the desired protein were pooled and concentrated to 2 mL in Amicon Ultra-15 concentrators with a 10,000 molecular weight cutoff (Merck Millipore, Carrigtwohill Co. Cork IRL). Concentrated protein was loaded onto a HiLoad 26/60 Superdex 200 prep grade column (GE Healthcare, Piscataway, NJ) that had been pre-equilibrated with 1.2 column volumes of sizing buffer (25 mM Tris pH 7.5, 250 mM NaCl, 2 mM DTT, 5% glycerol) using an ATKA FPLC (GE Healthcare, Piscataway, NJ). Protein was eluted isocratically in sizing buffer over 1.3 column volumes at a flow rate of 2 mL/min collecting 3 mL fractions. Peak fractions were analyzed for purity by SDS-PAGE, and those containing pure protein were pooled and concentrated using Amicon Ultra-15 concentrators with a 10,000 molecular weight cutoff (Merck Millipore, Carrigtwohill Co. Cork IRL).
GST Tag Removal.
The N-terminal GST tag was removed from MTF2, PHF20, and PHF20L1 by thrombin cleavage according to the manufacturer’s recommendations (Novagen, EMD Chemicals, San Diego, CA). Briefly, purified protein was incubated with biotinylated thrombin at a final concentration of 1 unit of thrombin/mg of tagged protein for 16 h at 4 °C. The cleavage reaction was then passed over a GSTrap FF column to remove protein that still retained the tag and free GST. Tag free proteins were further purified by collecting and concentrating the column flow through and running over a HiLoad 26/60 Superdex 75 prep grade column as described above. Peak fractions containing the desired protein were pooled and concentrated using Amico Ultra-15 concentrators with a 3000 molecular weight cutoff (Merck Millipore, Carrigtwohill Co. Cork IRL). All proteins were dialyzed into a buffer containing 25 mM Tris–HCl pH 7.5, 150 mM NaCl, and 2 mM β-mercaptoethanol prior to use for ITC.
15N-Labeled Protein Expression and Purification.
The PHF1 Tudor domain constructs comprising residues 14–87 and 28–87 were expressed and purified as described previously.10 Briefly, the proteins were expressed in Escherichia coli BL21 (DE3) RIL cells grown in LB media or M9 minimal media supplemented with 15NH4Cl. Expression was induced by 0.2 mM IPTG with shaking at 16 °C for 20 h. Cells were harvested and lysed by sonication. The GST-tagged PHF1 Tudor proteins were purified on glutathione Sepharose 4B beads (GE Healthcare) in 20 mM Tris (pH 7.5) buffer supplemented with 200 mM NaCl, 5 mM DTT, and 1 mM phenylmethanesulfonyl fluoride. The GST tag was removed overnight at 4 °C with PreScission protease. Proteins were further purified by size exclusion chromatography.
Isothermal Titration Calorimetry (ITC) Experiments.
All ITC measurements were recorded at 25 °C with an Auto-iTC200 isothermal titration calorimeter (MicroCal Inc., USA). All protein and compound stock samples were stored in ITC buffer (25 mM Tris–HCl pH 7.5, 150 mM NaCl, and 2 mM β-mercaptoethanol) and then diluted to achieve the desired concentrations. Typically, 100 μM protein and 1 mM compound were used; variations in these concentrations always maintained a 10:1 compound to protein ratio for all ITC experiments. The concentration of the protein stock solution was established using the Edelhoch method, whereas compound stock solutions were prepared based on mass. The experimental protocol included a single 0.2 mL compound injection into a 200 mL cell filled with protein, followed by 26 subsequent 1.5 mL injections of compound. Injections were performed with a spacing of 180 s and a reference power of 8 cal/s. The initial data point was routinely deleted. The titration data was analyzed using Origin 7 Software (MicroCal Inc., USA) by the nonlinear least-squares method, fitting the heats of binding as a function of the compound to protein ratio to a one-site binding model. At these concentrations, a reasonable curve could be fit up to Kd ~ 100 μM. Subsequently, curves too weak to be fit were reported as >100 μM.
General Time-Resolved Fluorescence Resonance Energy Transfer (TR-FRET) Assay Protocol.
The general TR-FRET protocol for Kme readers was followed as described previously.21 Assays were run using white, low-volume, flat-bottom, nonbinding, 384-well microplates (Greiner, 784904) containing a total assay volume of 10 μL per well. The assay buffer was composed of 20 mM Tris (pH 7.5), 150 mM NaCl, 0.05% Tween 20, and 2 mM DTT. LANCE Europium (Eu)-W1024 Streptavidin conjugate (2 nM) and LANCE Ultra ULight-anti6x-His antibody (10 nM) were used as donor and acceptor fluorophores associated with the tracer ligand and protein, respectively. Final assay concentrations of 60 nM 6× histidine tagged PHF1 Tudor protein (residues 28–87, N-terminal tag) and 90 nM biotin-UNC6641 tracer ligand were used for final compound testing. For PHF19, final assay concentrations of 5 nM 6× histidine tagged PHF19 Tudor protein (residues 49–115, N-terminal tag) and 50 nM of biotin-UNC6641 tracer ligand were used. Assay performance was evaluated using the Z′ factor calculation at varying DMSO concentrations up to 3%. Low signals were obtained using 100 μM UNC6641 to obtain complete inhibition, and high signals were obtained without compound (DMSO only). The Z′ factors for PHF1 and PHF19 were consistent at each DMSO concentration, revealing a DMSO tolerance of up to 3% for both proteins (PHF1 Z′ at 1% = 0.81, at 3% = 0.75; PHF19 Z′ at 1% = 0.90, at 3% = 0.90).
Peptides were tested in a high-throughput manner using 384-well assay-ready plates in standard plate format: columns 1 and 2 were used for low-signal controls (100% inhibition with UNC6641), columns 23 and 24 were used for high-signal controls (DMSO only), and columns 3–22 were used for 16-point, 3-fold serial dilutions of each compound. First, controls were added to a mother plate where columns 1 and 2 were filled with 10 mM stock of UNC6641 in DMSO and columns 23 and 24 were filled with DMSO. Test compounds were dispensed across the mother plate at 100× concentration in columns 3–22 using a TECAN Freedom EVO liquid handling workstation. Using a TTP Labtech Mosquito HTS liquid handling instrument, assay-ready plates were prepared by stamping 100 nL of control compound into columns 1 and 2, 10 nL of compounds from the mother plate into columns 3–22, and 25 nL of DMSO into columns 23 and 24. Protein, biotinylated tracer ligand, and the TR-FRET reagents were added together and gently mixed by pipetting and rocking. Ten μL was then added to each well of an assay-ready plate using a Multidrop Combi (Thermo Fisher, Waltham, MA). Percent inhibition was calculated on a scale of 0% (i.e., activity with DMSO vehicle only) to 100% (100 μM UNC6641) from the full column controls on each plate.
Molecular Docking.
Molecular docking was performed using Maestro version 12.6.149 from Schrödinger Suite 2020-4 (Schrödinger Inc.). PHF1 Tudor (PDB ID: 4HCZ) was prepared for docking using Prime tools followed by OPLS3e force field minimization after deletion of water molecules. Ligands were uploaded as SDF structures and prepared for docking using LigPrep, which ionizes and standardizes molecules at pH 7.0 ± 2.0. For peptides, stereoisomer generation was limited to 5. The Glide docking grid was created using the H3K36me3 peptide from the original structure. Ligands were docked using standard precision peptide Glide docking with the backbone atoms constrained by the H3K36me3 peptide from the original structure. Ring-to-ring distances were measured from centroid to centroid, and ring-to-atom distances were measured from centroid to atom.
HSQC NMR Spectroscopy.
NMR experiments were carried out at 298 K on a Varian INOVA 600 spectrometer as described.51 The NMR sample contained 0.2 mM uniformly 15N-labeled PHF1 Tudor domain (residue 14–87) in 20 mM Tris (pH 6.8) buffer supplemented with 150 mM NaCl, 5 mM DTT, and 10% D2O. Binding was characterized by monitoring chemical shift changes in the proteins induced by UNC6641. Spectra were processed using NMRPipe.52
Crystallization, Data Collection, and Structure Determination.
The PHF1 Tudor domain (residue 28–87) was concentrated to 10 mg/mL in a buffer containing 20 mM HEPES (pH 7.0), 200 mM NaCl, and 1 mM TCEP. Protein was incubated with UNC6641 at a 1:2 molar ratio for 1 h before crystallization. The preliminary plate-cluster crystals were obtained using the sitting drop vapor diffusion method by mixing 1 μL of the protein–peptide solution with 1 μL of reservoir solution containing 0.01 M ZnCl2, 0.1 M MES pH 6.0, and 20% PEG 6000. The strike seeding method was applied in order to obtain single well diffracting crystals. The crystals were cryoprotected with addition of 20% glycerol before being flash-frozen in liquid nitrogen. X-ray diffraction data were collected at the Advanced Light Source beamline 4.2.2 administrated by the Molecular Biology Consortium. The data set was indexed, integrated, and scaled by the XDS package.53 The substituents (the phenethyl and isopropyl groups) on K4 of UNC6641 were generated using the Phenix eLBOW program.54 The complex structure was determined by the Phaser-MR program in Phenix using the PHF1 Tudor domain-H3K36me3 peptide complex structure (PDB ID: 4HCZ)10 as the search model. The initial models were built with Coot and refined using Phenix.55,56 The data collection and structure refinement statistics are summarized in Supplementary Table 1.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institute on Drug Abuse, US NIH (grant R61DA047023-01), to L.I.J., the National Institute of General Medical Sciences, US NIH (grant R01GM100919), to S.V.F., and US NIH (grants CA252707 and HL151334) to T.G.K. The authors thank Jay Nix and Hardin John for help with data collection and processing, Justin Rectenwald for assistance with development of the TR-FRET assay, and Bryce Hart, Devan Shell, and Jarod Waybright for reviewing the primary data supporting this manuscript.
ABBREVIATIONS USED
- PTM
post-translational modification
- Kme
methyl-lysine
- PHF1
plant homeodomain finger protein 1
- PRC2
polycomb repressive complex 2
- H3K36me3
histone H3 lysine 36 trimethyl
- ITC
isothermal titration calorimetry
- TR-FRET
time-resolved fluorescence resonance transfer
- PCL
polycomblike
- PHF19
plant homeodomain finger protein 19
- MTF2
metal response element binding transcription factor 2
- CSP
chemical shift perturbation
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c00430.
Supporting figures and tables (PDF)
Molecular formula strings (CSV)
Accession Codes
Protein Data Bank ID 7LKY (UNC6641). Authors will release the atomic coordinates and experimental data upon article publication.
The authors declare no competing financial interest.
Contributor Information
Isabelle A. Engelberg, Center for Integrative Chemical Biology and Drug Discovery, Division of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States
Jiuyang Liu, Department of Pharmacology, University of Colorado School of Medicine, Aurora, Colorado 80045, United States.
Jacqueline L. Norris-Drouin, Center for Integrative Chemical Biology and Drug Discovery, Division of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States
Stephanie H. Cholensky, Center for Integrative Chemical Biology and Drug Discovery, Division of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States
Samantha A. Ottavi, Center for Integrative Chemical Biology and Drug Discovery, Division of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States
Stephen V. Frye, Center for Integrative Chemical Biology and Drug Discovery, Division of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Tatiana G. Kutateladze, Department of Pharmacology, University of Colorado School of Medicine, Aurora, Colorado 80045, United States.
Lindsey I. James, Center for Integrative Chemical Biology and Drug Discovery, Division of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
REFERENCES
- (1).Macdonald IA; Hathaway NA Epigenetic Roots of Immunologic Disease and New Methods for Examining Chromatin Regulatory Pathways. Immunol. Cell Biol 2015, 93 (3), 261–270. [DOI] [PubMed] [Google Scholar]
- (2).Mirabella AC; Foster BM; Bartke T Chromatin Deregulation in Disease. Chromosoma 2016, 125 (1), 75–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Valencia AM; Kadoch C Chromatin Regulatory Mechanisms and Therapeutic Opportunities in Cancer. Nat. Cell Biol 2019, 21 (2), 152–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Bird A The Methyl-CpG-Binding Protein MeCP2 and Neurological Disease. Biochem. Soc. Trans 2008, 36 (4), 575–583. [DOI] [PubMed] [Google Scholar]
- (5).Strahl BD; Allis CD The Language of Covalent Histone Modifications. Nature 2000, 403, 41–45. [DOI] [PubMed] [Google Scholar]
- (6).Jenuwein T; Allis CD Translating the Histone Code. Science 2001, 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- (7).Musselman CA; Khorasanizadeh S; Kutateladze TG Towards Understanding Methyllysine Readout. Biochim. Biophys. Acta, Gene Regul. Mech 2014, 1839, 686–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Beaver JE; Waters ML Molecular Recognition of Lys and Arg Methylation. ACS Chem. Biol 2016, 643–653. [DOI] [PubMed] [Google Scholar]
- (9).Taverna SD; Li H; Ruthenburg AJ; Allis CD; Patel DJ How Chromatin-Binding Modules Interpret Histone Modifications: Lessons from Professional Pocket Pickers. Nat. Struct. Mol. Biol 2007, 14, 1025–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Musselman CA; Lalonde M-E; Côté J; Kutateladze TG Perceiving the Epigenetic Landscape through Histone Readers. Nat. Struct. Mol. Biol 2012, 19 (12), 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Andrews FH; Strahl BD; Kutateladze TG Insights into Newly Discovered Marks and Readers of Epigenetic Information. Nat. Chem. Biol 2016, 12, 662–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Baril SA; Koenig AL; Krone MW; Albanese KI; He CQ; Lee GY; Houk KN; Waters ML; Brustad EM Investigation of Trimethyllysine Binding by the HP1 Chromodomain via Unnatural Amino Acid Mutagenesis. J. Am. Chem. Soc 2017, 139 (48), 17253–17256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lee Y-J; Schmidt MJ; Tharp JM; Weber A; Koenig AL; Zheng H; Gao J; Waters ML; Summerer D; Liu WR Genetically Encoded Fluorophenylalanines Enable Insights into the Recognition of Lysine Trimethylation by an Epigenetic Reader. Chem. Commun. (Cambridge, U. K.) 2016, 52 (85), 12606–12609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Milosevich N; Hof F Chemical Inhibitors of Epigenetic Methyllysine Reader Proteins. Biochemistry 2016, 55 (11), 1570–1583. [DOI] [PubMed] [Google Scholar]
- (15).Stuckey JI; Dickson BM; Cheng N; Liu Y; Norris JL; Cholensky SH; Tempel W; Qin S; Huber KG; Sagum C; Black K; Li F; Huang XP; Roth BL; Baughman BM; Senisterra G; Pattenden SG; Vedadi M; Brown PJ; Bedford MT; Min J; Arrowsmith CH; James LI; Frye SV A Cellular Chemical Probe Targeting the Chromodomains of Polycomb Repressive Complex 1. Nat. Chem. Biol 2016, 12 (3), 180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Stuckey JI; Simpson C; Norris-Drouin JL; Cholensky SH; Lee J; Pasca R; Cheng N; Dickson BM; Pearce KH; Frye SV; James LI Structure-Activity Relationships and Kinetic Studies of Peptidic Antagonists of CBX Chromodomains. J. Med. Chem 2016, 59 (19), 8913–8923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lamb KN; Bsteh D; Dishman SN; Moussa HF; Fan H; Stuckey JI; Norris JL; Cholensky SH; Li D; Wang J; Sagum C; Stanton BZ; Bedford MT; Pearce KH; Kenakin TP; Kireev DB; Wang GG; James LI; Bell O; Frye SV Discovery and Characterization of a Cellular Potent Positive Allosteric Modulator of the Polycomb Repressive Complex 1 Chromodomain, CBX7. Cell Chem. Biol 2019, 26 (10), 1365–1379.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Barnash KD; The J; Norris-Drouin JL; Cholensky SH; Worley BM; Li F; Stuckey JI; Brown PJ; Vedadi M; Arrowsmith CH; Frye SV; James LI Discovery of Peptidomimetic Ligands of EED as Allosteric Inhibitors of PRC2. ACS Comb. Sci 2017, 19 (3), 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Suh JLJL; Barnash KDKD; Abramyan TMTM; Li F; The J; Engelberg IA; Vedadi M; Brown PJPJ; Kireev DBDB; Arrowsmith CHCH; James LILI; Frye SVSV Discovery of Selective Activators of PRC2Mutant EED-I363M. Sci. Rep 2019, 9 (1), 6524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Barnash KD; Lamb KN; Stuckey JI; Norris JL; Cholensky SH; Kireev DB; Frye SV; James LI Chromodomain Ligand Optimization via Target-Class Directed Combinatorial Repurposing. ACS Chem. Biol 2016, 11 (9), 2475–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Rectenwald JM; Hardy PB; Norris-Drouin JL; Cholensky SH; James LI; Frye SV; Pearce KH A General TR-FRET Assay Platform for High-Throughput Screening and Characterizing Inhibitors of Methyl-Lysine Reader Proteins. SLAS Discovery Adv. Life Sci. R&D 2019, 24 (6), 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Musselman CA; Avvakumov N; Watanabe R; Abraham CG; Lalonde M-EE; Hong Z; Allen C; Roy S; Nuñez JK; Nickoloff J; Kulesza CA; Yasui A; Côté J; Kutateladze TG Molecular Basis for H3K36me3 Recognition by the Tudor Domain of PHF1. Nat. Struct. Mol. Biol 2012, 19 (12), 1266–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Cai L; Rothbart SB; Lu R; Xu B; Chen W-YY; Tripathy A; Rockowitz S; Zheng D; Patel DJ; Allis CD; Strahl BD; Song J; Wang GG An H3K36 Methylation-Engaging Tudor Motif of Polycomb-like Proteins Mediates PRC2 Complex Targeting. Mol. Cell 2013, 49 (3), 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Qin S; Guo Y; Xu C; Bian C; Fu M; Gong S; Min J Tudor Domains of the PRC2 Components PHF1 and PHF19 Selectively Bind to Histone H3K36me3. Biochem. Biophys. Res. Commun 2013, 430 (2), 547–553. [DOI] [PubMed] [Google Scholar]
- (25).Ballaré C; Lange M; Lapinaite A; Martin GM; Morey L; Pascual G; Liefke R; Simon B; Shi Y; Gozani O; Carlomagno T; Benitah SA; Di Croce L Phf19 Links Methylated Lys36 of Histone H3 to Regulation of Polycomb Activity. Nat. Struct. Mol. Biol 2012, 19 (12), 1257–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Brien GL; Gambero G; O’Connell DJ; Jerman E; Turner SA; Egan CM; Dunne EJ; Jurgens MC; Wynne K; Piao L; Lohan AJ; Ferguson N; Shi X; Sinha KM; Loftus BJ; Cagney G; Bracken AP Polycomb PHF19 Binds H3K36me3 and Recruits PRC2 and Demethylase NO66 to Embryonic Stem Cell Genes during Differentiation. Nat. Struct. Mol. Biol 2012, 19 (12), 1273–1281. [DOI] [PubMed] [Google Scholar]
- (27).Gatchalian J; Kingsley MC; Moslet SD; Rosas Ospina RD; Kutateladze TG An Aromatic Cage Is Required but Not Sufficient for Binding of Tudor Domains of the Polycomblike Protein Family to H3K36me3. Epigenetics 2015, 10 (6), 467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Nekrasov M; Klymenko T; Fraterman S; Papp B; Oktaba K; Köcher T; Cohen A; Stunnenberg HG; Wilm M; Müller J Pcl-PRC2 Is Needed to Generate High Levels of H3-K27 Trimethylation at Polycomb Target Genes. EMBO J. 2007, 26 (18), 4078–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Sarma K; Margueron R; Ivanov A; Pirrotta V; Reinberg D Ezh2 Requires PHF1 To Efficiently Catalyze H3 Lysine 27 Trimethylation In Vivo. Mol. Cell. Biol 2008, 28 (8), 2718–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Choi J; Bachmann AL; Tauscher K; Benda C; Fierz B; Müller J DNA Binding by PHF1 Prolongs PRC2 Residence Time on Chromatin and Thereby Promotes H3K27 Methylation. Nat. Struct. Mol. Biol 2017, 24 (12), 1039–1047. [DOI] [PubMed] [Google Scholar]
- (31).Li H; Liefke R; Jiang J; Kurland JV; Tian W; Deng P; Zhang W; He Q; Patel DJ; Bulyk ML; Shi Y; Wang Z Polycomb-like Proteins Link the PRC2 Complex to CpG Islands. Nature 2017, 549 (7671), 287–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).van Mierlo G; Veenstra GJC; Vermeulen M; Marks H The Complexity of PRC2 Subcomplexes. Trends Cell Biol. 2019, 29 (8), 660–671. [DOI] [PubMed] [Google Scholar]
- (33).Healy E; Mucha M; Glancy E; Fitzpatrick DJ; Conway E; Neikes HK; Monger C; Van Mierlo G; Baltissen MP; Koseki Y; Vermeulen M; Koseki H; Bracken AP PRC2.1 and PRC2.2 Synergize to Coordinate H3K27 Trimethylation. Mol. Cell 2019, 76 (3), 437–452.e6. [DOI] [PubMed] [Google Scholar]
- (34).Perino M; van Mierlo G; Wardle SMT; Marks H; Veenstra GJC Two Distinct Functional Axes of Positive Feedback-Enforced PRC2 Recruitment in Mouse Embryonic Stem Cells. 2019, bioRxiv 669960. biorxiv.org e-Print archive. https://www.biorxiv.org/content/10.1101/669960v1. [Google Scholar]
- (35).Micci F; Brunetti M; Dal Cin P; Nucci MR; Gorunova L; Heim S; Panagopoulos I Fusion of the Genes BRD8 and PHF1 in Endometrial Stromal Sarcoma. Genes, Chromosomes Cancer 2017, 56 (12), 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Panagopoulos I; Micci F; Thorsen J; Gorunova L; Eibak AM; Bjerkehagen B; Davidson B; Heim S Novel Fusion of MYST/Esa1-Associated Factor 6 and PHF1 in Endometrial Stromal Sarcoma. PLoS One 2012, 7 (6), e39354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Gebre-Medhin S; Nord KH; Möller E; Mandahl N; Magnusson L; Nilsson J; Jo VY; Vult Von Steyern F; Brosjö O; Larsson O; Domanski HA; Sciot R; Debiec-Rychter M; Fletcher CDM; Mertens F Recurrent Rearrangement of the PHF1 Gene in Ossifying Fibromyxoid Tumors. Am. J. Pathol 2012, 181 (3), 1069–1077. [DOI] [PubMed] [Google Scholar]
- (38).Endo M; Kohashi K; Yamamoto H; Ishii T; Yoshida T; Matsunobu T; Iwamoto Y; Oda Y Ossifying Fibromyxoid Tumor Presenting EP400-PHF1 Fusion Gene. Hum. Pathol 2013, 44 (11), 2603–2608. [DOI] [PubMed] [Google Scholar]
- (39).Weiss WA; Taylor SS; Shokat KM Recognizing and Exploiting Differences between RNAi and Small-Molecule Inhibitors. Nat. Chem. Biol 2007, 3, 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Kycia I; Kudithipudi S; Tamas R; Kungulovski G; Dhayalan A; Jeltsch A The Tudor Domain of the PHD Finger Protein 1 Is a Dual Reader of Lysine Trimethylation at Lysine 36 of Histone H3 and Lysine 27 of Histone Variant H3t. J. Mol. Biol 2014, 426 (8), 1651–1660. [DOI] [PubMed] [Google Scholar]
- (41).Dong C; Nakagawa R; Oyama K; Yamamoto Y; Zhang W; Dong A; Li Y; Yoshimura Y; Kamiya H; Nakayama JI; Ueda J; Min J Structural Basis for Histone Variant H3tk27me3 Recognition by Phf1 and Phf19. eLife 2020, 9, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Simhadri C; Daze KD; Douglas SF; Quon TTH; Dev A; Gignac MC; Peng F; Heller M; Boulanger MJ; Wulff JE; Hof F Chromodomain Antagonists That Target the Polycomb-Group Methyllysine Reader Protein Chromobox Homolog 7 (CBX7). J. Med. Chem 2014, 57 (7), 2874–2883. [DOI] [PubMed] [Google Scholar]
- (43).Milosevich N; Gignac MC; McFarlane J; Simhadri C; Horvath S; Daze KD; Croft CS; Dheri A; Quon TTH; Douglas SF; Wulff JE; Paci I; Hof F Selective Inhibition of CBX6: A Methyllysine Reader Protein in the Polycomb Family. ACS Med. Chem. Lett 2016, 7 (2), 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Wang S; Denton KE; Hobbs KF; Weaver T; McFarlane JMB; Connelly KE; Gignac MC; Milosevich N; Hof F; Paci I; Musselman CA; Dykhuizen EC; Krusemark CJ Optimization of Ligands Using Focused DNA-Encoded Libraries to Develop a Selective, Cell-Permeable CBX8 Chromodomain Inhibitor. ACS Chem. Biol 2020, 15 (1), 112–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Højfeldt JW; Hedehus L; Laugesen A; Tatar T; Wiehle L; Helin K Non-Core Subunits of the PRC2 Complex Are Collectively Required for Its Target-Site Specificity. Mol. Cell 2019, 76 (3), 423–436.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Brunetti M; Gorunova L; Davidson B; Heim S; Panagopoulos I; Micci F Identification of an EPC2-PHF1 Fusion Transcript in Low-Grade Endometrial Stromal Sarcoma. Oncotarget 2018, 9 (27), 19203–19208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Hoang L; Chiang S; Lee C-H Endometrial Stromal Sarcomas and Related Neoplasms: New Developments and Diagnostic Considerations. Pathology 2018, 50 (2), 162–177. [DOI] [PubMed] [Google Scholar]
- (48).Bae N; Viviano M; Su X; Lv J; Cheng D; Sagum C; Castellano S; Bai X; Johnson C; Khalil MI; Shen J; Chen K; Li H; Sbardella G; Bedford MT Developing Spindlin1 Small-Molecule Inhibitors by Using Protein Microarrays. Nat. Chem. Biol 2017, 13 (7), 750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Fagan V; Johansson C; Gileadi C; Monteiro O; Dunford JE; Nibhani R; Philpott M; Malzahn J; Wells G; Faram R; Cribbs AP; Halidi N; Li F; Chau I; Greschik H; Velupillai S; Allali-Hassani A; Bennett J; Christott T; Giroud C; Lewis AM; Huber KVM; Athanasou N; Bountra C; Jung M; Schüle R; Vedadi M; Arrowsmith C; Xiong Y; Jin J; Fedorov O; Farnie G; Brennan PE; Oppermann U A Chemical Probe for Tudor Domain Protein Spindlin1 to Investigate Chromatin Function. J. Med. Chem 2019, 62 (20), 9008–9025. [DOI] [PubMed] [Google Scholar]
- (50).Liu R; Gao J; Yang Y; Qiu R; Zheng Y; Huang W; Zeng Y; Hou Y; Wang S; Leng S; Feng D; Yu W; Sun G; Shi H; Teng X; Wang Y PHD Finger Protein 1 (PHF1) Is a Novel Reader for Histone H4R3 Symmetric Dimethylation and Coordinates with PRMT5-WDR77/CRL4B Complex to Promote Tumorigenesis. Nucleic Acids Res. 2018, 46 (13), 6608–6626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Gatchalian J; Wang X; Ikebe J; Cox KL; Tencer AH; Zhang Y; Burge NL; Di L; Gibson MD; Musselman CA; Poirier MG; Kono H; Hayes JJ; Kutateladze TG Accessibility of the Histone H3 Tail in the Nucleosome for Binding of Paired Readers. Nat. Commun 2017, 8 (1), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Delaglio F; Grzesiek S; Vuister GW; Zhu G; Pfeifer J; Bax A NMRPipe: A Multidimensional Spectral Processing System Based on UNIX Pipes. J. Biomol. NMR 1995, 6 (3), 277–293. [DOI] [PubMed] [Google Scholar]
- (53).Kabsch W Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66 (2), 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Moriarty NW; Grosse-Kunstleve RW; Adams PD Electronic Ligand Builder and Optimization Workbench (ELBOW): A Tool for Ligand Coordinate and Restraint Generation. Acta Crystallogr., Sect. D: Biol. Crystallogr 2009, 65 (10), 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and Development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66 (4), 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Adams PD; Afonine PV; Bunkóczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: A Comprehensive Python-Based System for Macro-molecular Structure Solution. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66 (2), 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.