

Abstract
Genomic imbalance caused by changing the dosage of individual chromosomes (aneuploidy) has a more detrimental effect than varying the dosage of complete sets of chromosomes (ploidy). We examined the impact of both increased and decreased dosage of 15 distal and 1 interstitial chromosomal regions via RNA-seq of maize (Zea mays) mature leaf tissue to reveal new aspects of genomic imbalance. The results indicate that significant changes in gene expression in aneuploids occur both on the varied chromosome (cis) and the remainder of the genome (trans), with a wider spread of modulation compared with the whole-ploidy series of haploid to tetraploid. In general, cis genes in aneuploids range from a gene-dosage effect to dosage compensation, whereas for trans genes the most common effect is an inverse correlation in that expression is modulated toward the opposite direction of the varied chromosomal dosage, although positive modulations also occur. Furthermore, this analysis revealed the existence of increased and decreased effects in which the expression of many genes under genome imbalance are modulated toward the same direction regardless of increased or decreased chromosomal dosage, which is predicted from kinetic considerations of multicomponent molecular interactions. The findings provide novel insights into understanding mechanistic aspects of gene regulation.
Genomic imbalance caused by the addition or subtraction of chromosomal segments leads to modulations of gene expression inversely or directly related to chromosomal dosage but also to nonlinear responses.
Introduction
It has been known for nearly a century that changing the dosage of individual chromosomes or chromosomal segments (aneuploidy) has more phenotypic effects than changing the dosage of the whole set of chromosomes in a genome (ploidy) (Blakeslee et al., 1920; Blakeslee, 1921, 1934; Sinnott and Blakeslee, 1922; Bridges, 1925). This phenomenon became known as genetic imbalance and was postulated to result from a dosage effect of the varied genes. This concept has been studied molecularly in many organisms such as Drosophila and maize (Zea mays) (Grell, 1962; O’Brien and Gethmann, 1973; Birchler, 1979, 1981; Birchler and Newton, 1981; Rabinow et al., 1991). Early studies illustrated that varying the dosage of a gene would produce a directly proportional amount of gene product, known as a gene-dosage effect (Grell, 1962; Carlson, 1972; O’Brien and Gethmann, 1973). In other cases, however, genes on the varied chromosome produced a nearly equivalent amount of gene product between aneuploids and diploids, a phenomenon referred to as dosage compensation (Birchler, 1979, 1981; Birchler and Newton, 1981). Further, by investigating the modulation of gene expression from the unvaried portion of the genome, an inverse correlation between the dosage of the varied chromosome and the amount of gene product encoded elsewhere in the genome was observed (Birchler, 1979; Birchler and Newton, 1981; Rabinow et al., 1991; Guo and Birchler, 1994) with positive modulations being found to a lesser degree. The basis of dosage compensation was determined in several studies to result from a gene-dosage effect being cancelled by an inverse effect produced together by the varied segment of the genome (Birchler, 1981; Birchler and Newton, 1981; Birchler et al., 1990). In contrast, relative changes in gene expression in a whole-genome ploidy series were not as prominent (Birchler and Newton, 1981; Guo et al., 1996; Yao et al., 2011; Robinson et al., 2018).
The genes responsible for these dosage effects were sought in Drosophila and could be reduced to the action of single genes (Rabinow et al., 1991; Birchler et al., 2001; Xie and Birchler, 2012). When the molecular functions were identified, they included transcription factors (TFs), signal transduction components, and chromatin proteins (Birchler et al., 2001). Similar conclusions were reached in other taxa and the unifying aspect was that they were typically members of macromolecular complexes and multicomponent interactions including with DNA (Seidman and Seidman, 2002; Veitia, 2002; Papp et al., 2003; Kondrashov and Koonin, 2004; Veitia et al., 2013; Defoort et al., 2019; Shi et al., 2020). Furthermore, copy-number variants of these components are frequently associated with human diseases (Ionita-Laza et al., 2009; Makino and McLysaght, 2010). These findings led to the gene balance hypothesis, which states that varying the stoichiometry of members of multicomponent interactions will affect their kinetics, mode of assembly and function of the whole, thus causing negative fitness consequences (Birchler et al., 2005; Birchler and Veitia, 2007, 2010, 2012). This hypothesis is in parallel with the phenotypic observation of aneuploids versus the whole-genome ploidy series (Blakeslee, 1921). When there is a partial rather than a whole-genomic change that alters the stoichiometry of certain subunits relative to others, the function of the whole multiunit complex would be altered.
A case of autosomal dosage compensation was examined in Drosophila with regard to the alcohol dehydrogenase gene (Birchler et al., 1990). When the segmental trisomy was broken into smaller segments, the structural gene showed a dosage effect and an adjacent small region, included in the larger trisomy, had an inverse effect on the studied gene (Birchler et al., 1990). When a phenotypic promoter–reporter construct was tested, the inverse effect was shown to operate on the 5′-regulatory sequences. In a different system, when an individual gene that produced an inverse dosage effect on the target white eye color gene was tested with promoter deletions, the deletion alleles were found not to respond (Rabinow et al., 1991). These examples show that promoter regions are necessary and sufficient to confer the regulatory inverse effect on a target gene.
The gene balance hypothesis is also supported by evolutionary genomics. Across eukaryotes, certain classes of genes are maintained as duplicates for a longer period of evolutionary time after whole-genome duplication (WGD), typically those composing macromolecular interactions including TFs and signal transduction genes (Simillion et al., 2002; Blanc and Wolfe, 2004; Maere et al., 2005; Aury et al., 2006; Freeling and Thomas, 2006; Thomas et al., 2006; Freeling et al., 2008; Jiao et al., 2011; Tasdighian et al., 2017; Defoort et al., 2019; Du et al., 2020; Shi et al., 2020). Deletion of one member of a balanced duplicated pair was postulated to have a detrimental effect on fitness and thus would be selected against (Birchler et al., 2005; Freeling and Thomas, 2006). In contrast, there is an underrepresentation of genes encoding subunits of multiunit complexes retained after small-scale duplications (Maere et al., 2005; Freeling et al., 2008; Coate et al., 2016; Tasdighian et al., 2017). In this case, these duplications would mimic an aneuploid effect, which upsets the genome balance, resulting in detrimental defects that would be selected against. Despite the distinct fates for whole genome and small-scale duplication events, both scenarios indicate that genomic balance, which is found experimentally, operates over evolutionary time. Collectively, the fact that TFs and signaling components are dosage sensitive, together with these extensive evolutionary genomics data, illustrates that regulatory processes lie at the basis of genomic balance.
In addition, the concept of balance is further supported by the observation that quantitative traits could be affected by multiple loci that exhibit dosage effects (Tanksley, 1993). TFs and signal transduction genes were found to be major contributors in the identification of several quantitative trait loci (Frary et al., 2000; Cong et al., 2002, 2008; Liu et al., 2002; Xu et al., 2016; Huang et al., 2018). The parallels between quantitative genetics and the effect of multiple aneuploidies were noted in that any one trait can be affected by multiple aneuploidies and by multiple quantitative trait loci (Guo and Birchler, 1994; Birchler and Veitia, 2012).
Because TFs and signaling components operate in cascades affecting targets, which include other TFs, varying a portion of the genome would be predicted to have global impacts across the genome (Birchler et al., 2001). Indeed, large aneuploidies in Drosophila produce such extensive modulations (Sun et al., 2013a, 2013b). Further, in a study of global gene expression modulations using mRNA-sequencing (mRNA-seq), a set of all five trisomies and a whole-genome ploidy series of diploids, triploids, and tetraploids was examined in Arabidopsis (Hou et al., 2018). A greater spread of modulation was observed in the trisomies than the ploidy series. In general, expression of genes on the varied chromosome ranged from dosage compensation to a gene-dosage effect, whereas genes from the remainder of the genome ranged from no effect to an inverse effect with some positive effects observed. Furthermore, genome-wide DNA methylation analysis indicates genomic imbalance is generally unrelated to DNA methylation although it can be modulated by trisomy. Studies of yeast disomies (Torres et al., 2007) and mouse trisomic cells (Williams et al., 2008) were originally interpreted as showing mainly cis gene-dosage effects with minimal trans effects because of the normalization of expression for the varied chromosome to the remainder of the genome. However, a re-analysis of those data revealed prevalent trans-acting modulations, primarily of an inverse nature (Hou et al., 2018), consistent with historical observations tracing back 100 years in which every trisomy of Datura modulated pigment levels (Blakeslee, 1921). Recent studies of a dosage series of the human sex chromosomes demonstrate global modulations of a subset of genes across the genome, also with a predominance of inverse effects (Raznahan et al., 2018; Zhang et al., 2020).
The aforementioned previous studies examined the global genomic impacts on gene expression in genotypes with additional chromosomes out of necessity because reductions in chromosome number to only one copy are either lethal or impossible to construct genetically in the various organisms. Among diploid genetic model organisms, maize is the most tolerant to aneuploidy as evidenced by the fact that it is possible to recover monosomies and trisomies for all chromosomes (Carlson, 1988). Also, it is possible to produce tetrasomies for most of the genome as well as haploid disomies together with a range of ploidies. This fact is likely the case because of a history of WGD (Rhoades, 1951; Gaut and Doebley, 1997; Schnable et al., 2009) but nevertheless this allows one to address several questions about genomic balance that are not readily investigated in other species. Principal among these questions is how global modulations compare between monosomy and trisomy. Such comparisons provide valuable new data for developing theoretical underpinnings for the nature of genomic balance, the kinetics of gene expression, and the molecular basis of aneuploid syndromes with implications across eukaryotes.
In this study, 15 segmentally distal and one interstitial segmental aneuploid lines, created by translocations between the normal chromosomes and the supernumerary B chromosome (B–A translocations) (Roman, 1948; Beckett, 1978, 1991; Carlson, 1988) containing various copies of chromosomal segments, were studied in concert with a whole-genome ploidy series of haploid, diploid, triploid, and tetraploid. The aneuploids in total cover approximately 74.5% of the genes in the maize W22 genome. The various genotypes were assayed via mRNA-seq of maize leaf tissue to determine the common and unique trends of how variation of different partial genomes or whole-genome doses affects gene expression on a global scale. By examining the corresponding monosomies and trisomies for large and small regions comprising a substantial portion of the genome, the results provide new insight into the impact of genetic imbalance on quantitative gene expression and illustrate its pervasive nature. The study revealed not only linear effects of chromosomal dosage on gene expression but also extensive nonlinear responses that would be predicted from the kinetics of models of gene regulation.
Results
Aneuploidy causes modulation of gene expression both in cis and in trans
We analyzed plants from a collection of 15 maize B–A translocation lines containing various copies of chromosomal segments, with one to three copies for 1S (short arm of chromosome 1), 1L (long arm of chromosome 1), 3S, 3L, 4S, 4L, 5L, 6L, 7L, 8L, 9S, and 9L, 2–4 copies for 6S and one to four copies for 5S and 10L, whose genotypes were screened and verified by fluorescence in situ hybridization (FISH). These lines are referred to as distal aneuploids as their segmental aneuploidy does not include the respective centromeres. In addition, interstitial segmental trisomies and tetrasomies with breakpoints spanning the centromere of chromosome 4, proximal duplication 4 (Dp4) (Zheng and Carlson, 1997), produced by overlapping reciprocal B–A translocations were examined and compared with their diploid siblings. By way of comparison, a collection of haploids, triploids, tetraploids, and their corresponding diploid controls was also analyzed. The crosses and grouping information of these plants are described in Material and methods and listed in Supplemental Table S1. For convenience, monosomies, diploids, trisomies, and tetrasomies in each aneuploid were referred to as 1D, 2D, 3D, and 4D (D designates dosage of the chromosomal segment); whereas haploids, diploids, triploids, and tetraploids in the ploidy series were named 1X, 2X, 3X, and 4X, respectively.
mRNA-seq was performed on the leaf tissue of the abovementioned materials to examine the effect of aneuploidy and polyploidy on gene expression. Principal component analysis (PCA) was performed using normalized read counts to determine the similarity of gene expression levels among biological replicates (Supplemental Figure S1). To identify possible outliers, the mean and standard deviation (sd) of the principal components (PCs) among biological replicates were calculated and compared. For all genotypes, values of PC1 and PC2 for each replicate were within two SDs from the mean, indicating none could be considered as an outlier.
Plotting ratio distributions of experimental to control read counts provides a broad view of trends of gene expression, especially for those that are subtly modulated with experimental and control values close to each other. Normalized counts of biological replicates were averaged and ratios of individual genes of each experimental condition to the control were plotted as a histogram. The distributions for the aneuploids were partitioned into genes that are present on the varied chromosome (cis) versus those that are in the remainder of the genome (trans) according to the breakpoint in each line listed in Supplemental Table S2. Location of the breakpoints in aneuploids was determined by analyzing DNA-sequencing (DNA-seq) data generated from aneuploids as described in Materials and methods. The mean, median, and sd for each distribution were computed (Supplemental Data Set 1). Also, various statistical tests were performed, including determinations of deviation of the distributions from normal (Supplemental Data Set 2), or comparisons of distributions for differences with Kolmogorov–Smirnov (K–S) tests of significance (Supplemental Data Set 3), or comparisons of variances across distributions using Bartlett’s test (Supplemental Data Set 4). Scatter plots were used to illustrate the fold change and significance of differential gene expression (DGE), as a cross-validation to the ratio distributions (Supplemental Figure S2).
The aneuploids show a wide range of distinct effects both on the cis and trans chromosomes compared with their diploid control (Figures 1–3). In general, cis genes in aneuploids range from dosage compensation to a gene-dosage effect. For each monosomy (1D/2D), the cis effect shows a peak grouping between a gene-dosage effect (0.5) and dosage compensation (1.0), but the range of effects extends above and below these levels (Figure 1). Some monosomies exhibit greater extents of cis modulation than others. The major cis peak for 3S and 5L monosomies is modulated slightly below a gene-dosage effect, while the greatest cis peaks for 1S, 1L, 4S, and 5S monosomies are closer to a gene-dosage effect than dosage compensation. There is a spread of ratios extending below and above toward compensation for most arms. Trisomies (3D/2D) display cis effects with similar patterns but a greater trend toward dosage compensation compared with monosomies (1D/2D). Cis peaks of each trisomy lie between a gene-dosage effect (1.5) and dosage compensation (1.0), with those for 1S, 4S, 4L, 5S, 6S, and 6L showing a close approximation to a generalized dosage effect while those for 3S and 5L are closer to dosage compensation. The major peaks for others are either intermediate or closer to a gene-dosage effect (Figure 2). The spread of effects generally spans between a dosage effect and compensation. However, for some regions, there are minor peaks that coincide with an inverse reduction in cis, even below the diploid control, notably for 1S, 1L, 3S, 4S, 5L, 6L, 7L, 8L, 9S, 9L, and 10L. These peaks represent a subset of genes that apparently have multiple independent inverse modulations, a situation also observed in Drosophila (Sun et al., 2013b). As for tetrasomies (4D/2D), the cis peak for 10L shows a closer approximation to a gene-dosage effect (2.0) than dosage compensation (1.0), whereas those for 5S and 6S are intermediate (Figure 3).
Figure 1.

Ratio distributions of gene expression in each monosomy compared with diploids (1D/2D). Genes were partitioned into those encoded on the varied chromosome (cis) versus those encoded on the remainder of the genome that were not varied in dosage (trans). Normalized read counts for each gene were averaged across biological replicates and were then used for the generation of ratios comparing each experimental group to the control. Cis distributions were painted in orange, while trans distributions were painted in blue. Gene ratios for each pair of comparison were plotted on the x-axis with a bin width of 0.05. The y-axis notes the number of genes per bin (frequency). A ratio of 0.5 represents a gene-dosage effect in cis, whereas 1.0 represents dosage compensation. A ratio of 2.0 represents the inverse ratio of gene expression in trans, whereas 1.0 represents no change and 0.5 represents a positive modulation. These ratio values are demarcated with labeled vertical lines in red (0.5), black (1.0) and purple (2.0).
Figure 2.

Ratio distributions of gene expression in each trisomy compared with diploids (3D/2D). Analysis was conducted as described in Figure 1. A ratio of 1.5 represents a gene-dosage effect in cis, whereas 1.0 represents dosage compensation. A ratio of 0.67 represents the inverse ratio of gene expression in trans, whereas 1.0 represents no change and 1.5 represents a positive modulation. These ratio values are demarcated with labeled vertical lines in pink (0.67), black (1.0), and green (1.5).
Figure 3.

Ratio distributions of gene expression in each tetrasomy compared with diploids (4D/2D) or trisomies (4D/3D). Analysis was conducted as described in Figure 1. Ratios of 2.0 (4D/2D) and 1.33 (4D/3D) represent a gene-dosage effect in cis, whereas 1.0 represents dosage compensation. Ratios of 0.5 (4D/2D) and 0.75 (4D/3D) represent the inverse ratio of gene expression in trans, whereas 1.0 represents no change. Ratios of 2.0 (4D/2D) and 1.33 (4D/3D) represent a positive modulation in trans. These ratio values are demarcated with labeled vertical lines in red (0.5), orange (0.75), black (1.0), blue (1.33), and purple (2.0).
There is a wide spread of modulation for trans genes, but the most common effect for trans genes is an inverse effect in that expression decreases with increasing doses of chromosomal dosage or increases with decreased doses. Monosomic 1S, 1L, 4S, 5S, 8L, 9S, and 9L (1D/2D) display a trend of increased generalized gene expression whereas trisomic 1L, 3S, 3L, 5S, 5L, 6S, 8L, 9L, and 10L (3D/2D) experience decreased gene expression, both toward the inverse level (Figures 1 and 2). Similar trends are also observed in tetrasomic 5S, 6S, and 10L (4D/2D) (Figure 3). A few aneuploids, such as monosomic 3S, 5L, and 10L, as well as trisomic 4S, exhibit a positive relationship in that expression slightly increases with increasing chromosomal dosage (Figures 1 and 2). Scatter plots of fold change and DGE complement the results of ratio distributions (Supplemental Figure S2). Quantitative PCR (qPCR) of specific genes for relative expression levels in 5S aneuploids was conducted and the ratios agree with those generated in the ratio distributions (Supplemental Figure S3). A Pearson correlation coefficient (R) between ratios computed from qPCR and mRNA-seq equals 0.930 (P-value = 2.29e-08).
To examine whether the cis and trans effects in aneuploids are progressive, with greater chromosomal dosage producing greater effects, we further plotted the ratio distribution of tetrasomies to trisomies (4D/3D) (Figure 3). The cis peak of 10L is near 1.33, in proportion to the chromosome dosage, while that of 5S and 6S is intermediate between 1.0 and 1.33. Scatter plots of DGE in comparing tetrasomies to trisomies further demonstrate the progressive cis effect on gene expression is statistically significant (Figure 4 and Supplemental Figure S2C). Together with the analysis on 1L tetrasomies and trisomies in a previous study (Johnson et al., 2020), a progressive cis effect positively occurring with chromosomal dosage is a common scenario. In the trans comparison, ratio distributions of tetrasomies are subtly different compared to trisomies, as the peak is distributed near ratio 1.0. However, the fact that many genes are significantly differentially expressed in the 4D/3D trans comparison demonstrates a progressive effect also exists in trans, although expression of many genes remain unchanged (Figure 4 and Supplemental Figure S2C). The companion paper (Yang et al., 2021) examined haploid disomies, which also have greater imbalance than trisomy, with the finding of generalized greater modulations in trans.
Figure 4.

Number of cis genes and TFs in each aneuploidy line as well as the number of upregulated and downregulated DEGs in each comparison. Up, upregulated DEGs; down, downregulated DEGs; total, all DEGs in each comparison. The blue color is scaled to the number (no.)/proportion (%) of cis genes while the orange color to the number of cis TFs. The red color is scaled to the number of total DEGs. DEGs were computed by significance (q value <0.05), as in each scatter plot in Supplemental Figure S2.
Of the B–A translocation lines analyzed, the percentage of the genome for cis genes in each line range over 10-fold from 0.5% to 7.9% of all genes in W22 (Figure 4). The number or proportion of cis genes is not related to the extent of modulation in both ratio distributions and scatter plots (Figures 1–3; Supplemental Figure S2). In addition, no relationship is observed between the number of cis genes or the cis TFs to the number of DEGs in each comparison (Figure 4). However, a general trend emerges when examining the global trend of each comparison. Medians of cis and trans distributions in each comparison are highly positively correlated across different cis regions for both monosomy and trisomy (Figure 5, A and B), indicating cis and trans peaks in each comparison were shifting in the same direction. In other words, when gene expression approaches closer to the inverse level in trans, cis distributions proceed more toward dosage compensation. There is a positive correlation between the proportion of up-regulated cis DEGs and that of the upregulated trans DEGs in the 14 monosomic comparisons (1D/2D). This relationship is also the case in the comparison between the proportion of downregulated cis DEGs and that of the downregulated trans DEGs in trisomies (3D/2D) (Figure 5, C and D). These observations indicate the inversely affected trans genes are similarly affected as the cis genes that trend toward dosage compensation, suggesting both groups are affected by a related mechanism.
Figure 5.

Relationship between expression of cis and trans genes in each distal aneuploidy comparison. R, Pearson correlation coefficient; p, P-values for Pearson correlation. Each data point represents one comparison out of the 14 distal aneuploids, with 6S excluded due to a lack of monosomy. A and B, Correlation between the median of cis and trans distributions for monosomies (1D/2D) and trisomies (3D/2D). Medians of cis and trans genes in each comparison were plotted on the x- and y-axes, respectively, computed as in Supplemental Data Set 1. C, Correlation between up-regulated cis and trans DEGs in monosomies (1D/2D). D, Correlation between down-regulated cis and trans DEGs in trisomies (3D/2D). DEGs were computed as described in Supplemental Figure S2.
Ratio distributions permit an evaluation of general trends, including subtle modulations holistically that are difficult to detect statistically in experiments that are restricted in scope due to practical considerations. However, to examine how gene expression is modulated at the individual gene level, we compared the direction of gene regulation of DEGs in monosomies versus trisomies in the 14 distal aneuploidy lines (Supplemental Table S3). There are 0.78%– 44.10% of cis genes that are differentially expressed in both monosomies and trisomies compared with diploids, the plurality of which is downregulated in monosomies and upregulated in trisomies. This observation indicates that a gene-dosage effect is the plurality trend for individual cis gene expression upon changes of chromosome dosage in the range of aneuploid sizes examined. An analysis of trans genes eliminates the variable of gene copy number that is present with cis genes. The percentage of trans genes being differentially expressed in both monosomies and trisomies ranges between 0.30% and 33.07%. Although the ratio distribution analysis, which detects more subtle modulations, found that the inverse effect was the most prominent global response to aneuploidy, the DGE analysis of individual genes revealed that many trans DEGs are modulated toward the same direction regardless of whether there is increased or decreased chromosomal dosage relative to diploids. This type of response has been noted on the individual gene level (Guo and Birchler, 1994), but this effect has not been previously addressed globally. Percentages of DEGs being upregulated or downregulated in both types of aneuploids outnumbered DEGs modulated toward opposite directions in 11 out of 14 lines. These gene expression patterns are therefore referred to as an increased effect (both upregulated) and a decreased effect (both downregulated), respectively, in the following paragraphs.
Gene Ontology (GO) term enrichment analysis regarding biological process of DEGs exhibiting different expression patterns was performed using PANTHER (Thomas et al., 2003; Mi et al., 2013) (Supplemental Data Set 5). Under- or over-representation of GO terms shared across different distal aneuploids was summarized by analyzing DEGs being modulated toward opposite directions in monosomies versus trisomies (inverse or positive effect). None of these GO terms are shared between any 2 distal aneuploidy lines out of 14 (Supplemental Data Set 5). Similarly, DEGs that showed an increased or a decreased effect in both types of aneuploids were analyzed and GO terms being under- or over-represented are shared among no more than 5 distal aneuploidy lines out of 14 (Supplemental Data Set 5). Thus, DEGs exhibiting different expression patterns are modulated in a manner specific to each dosage series rather than a uniform reaction to genomic imbalance.
Ploidy shows a lesser spread of modulation than aneuploidy
Ratio distributions comparing ploidies of haploid, triploid, and tetraploid with the corresponding diploid control show much less spread with a sharp peak near ratio 1.0 compared with aneuploidy (Figure 6A). In each case, the peak is slightly modulated above or below 1.0 with an inverse relationship to ploidy. Statistical tests were performed to compare distributions between aneuploidy and ploidy with the same number of varied individual chromosomes or sets of chromosomes, namely, trans distributions of monosomies (1D/2D) compared with haploids (1X/2X), trisomies (3D/2D) compared with triploids (3X/2X) and tetrasomies (4D/2D) compared with tetraploids (4X/2X). K–S tests for different distributions and Bartlett’s tests for variations both showed highly significant differences (Supplemental Data Sets 3 and 4), suggesting a greater level of disruption of global gene expression in aneuploidy than in ploidy. This observation is further validated by tests for DGE as shown in the scatter plots, with fewer DEGs found in each ploidy comparison than most aneuploidy comparisons (Figure 6; Supplemental Table S4). The number of DEGs in each comparison varies, with 1,749 DEGs in haploids, 144 DEGs in triploids, and 855 DEGs in tetraploids compared with diploids. However, only a small fraction of genes is differentially expressed in more than one comparison. The W22 1X/2X and 4X/2X comparisons shared more overlapping DEGs, likely because of their same genetic background in contrast to 3X/2X, which originated from inbred line Mo17 (Figure 6C).
Figure 6.

Ratio distributions and scatter plots of gene expression in each whole-ploidy series compared with diploids, and tetraploids compared with triploids. A, Ratio distributions of each ploidy compared with diploids and tetraploids to triploids. Analysis of each ploidy to diploids was conducted as described in Figure 1. Ratios of 4X/3X were generated by dividing the ratio of 4X/2X by 3X/2X as described in “Materials and methods” section. B, Scatter plots of each ploidy. The x-axis represents the log-fold change with base 2 of each experimental genotype to the control, whereas the y-axis notes the mean of normalized counts of each experimental genotype and the control. Data points with an q value (or adjusted P-value in 4X/3X) <0.05 and a corresponding logFC >0 were depicted in magenta, while points with an q value < 0.05 and a corresponding logFC <0 were depicted in green. A and B, A ratio of 1.0 represents no change. Ratios of 0.5 (1X/2X), 1.33 (4X/3X), 1.5 (3X/2X), and 2.0 (4X/2X) represent a gene-dosage effect, whereas ratios of 2.0 (1X/2X), 0.75 (4X/3X), 0.67 (3X/2X), and 0.5 (4X/2X) represent the inverse ratio of gene expression. These ratio values are demarcated with labeled vertical lines in red (0.5), pink (0.67), orange (0.75), black (1.0), blue (1.33), green (1.5), and purple (2.0). C, Venn diagram of DEGs in each ploidy compared with diploids. DEGs were determined as in (B). Numbers in the brackets denote the total number of DEGs in each comparison. D, Relative (Rel.) transcriptome size of the whole-ploidy series. Error bar, sd across ratios of the eight genes being assayed as described in Material and methods.
We further investigated the ratio distribution comparing tetraploids to triploids (4X/3X) by computing the ratios of ratios in the 4X/2X comparison to those in the 3X/2X comparison (Figure 6A). The sharp peak lies slightly below 1.0, indicating a slightly linear relationship between the inverse trend of gene modulation and the degree of ploidy. A Student’s t test was performed to detect significantly different ratios in the 4X/3X comparison. The fact that the ratios of only 12 genes are significantly different shows the increased inverse trend of gene modulation from triploids to tetraploids is detectable on the global landscape, more so than on an individual gene basis (Supplemental Table S4), as is generally the case in this study.
Epidermal cell area and transcriptome size are correlated with chromosome number in polyploids
It is generally accepted that there is a linear relation between cell size, number of chromosomes, and transcriptome size in Arabidopsis with cell size and transcriptome size showing the strongest correlation (Miller et al., 2012; Tsukaya, 2013; Robinson et al., 2018). The center of ratio distributions of the ploidy series compared with diploids is near 1.0, suggesting that larger cell size and correlated changes in gene expression per cell are observed with increased number of chromosomal sets. This hypothesis is further confirmed by measurement of cell size of each ploidy using epidermal cell imprints (Figure 7). An increase in the number of chromosome sets (1X, 2X, and 4X) results in significantly greater cell area with a strong linear relationship (R ∼ 1.0; P = 0.038). For aneuploidy, dosage series of chromosome arms 5S and 6S were used for demonstration. A slight numerical increase of cell area is observed with the increase of the dosage of 5S; however, none of the 5S aneuploids show a significant change of cell area compared to their diploid siblings (Figure 7A). In addition, no change was observed when examining the cell area of plants carrying two to five copies of 6S (Figure 7B). Therefore, it is unlikely that a measurable shift in transcriptome size occurs in the 5S and 6S aneuploid series.
Figure 7.
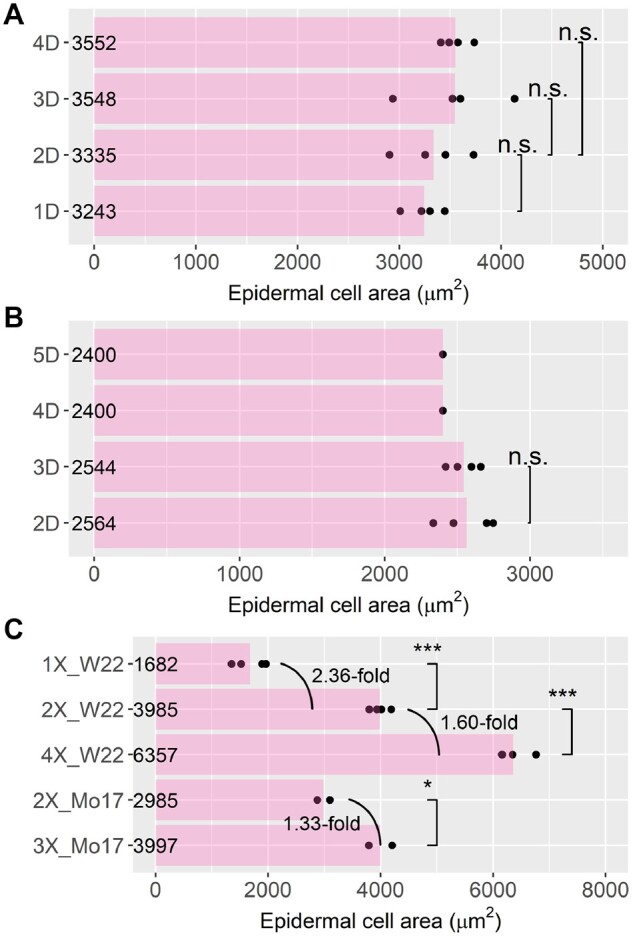
Epidermal cell area of aneuploids and polyploids. Asterisks indicate significant differences (Student’s t test: *P <0.05; ***P <0.001; n.s., P ≥0.05). Epidermal cell area of 5S aneuploids (A), 6S aneuploids (B), and the ploidy series (C). Each point represents data for one biological replicate.
To further understand how the transcriptome size varies with increased number of chromosome sets, we measured the transcriptome size of the ploidy series relative to the diploid control. An overall estimate of relative transcriptome size was computed by dividing the per cell expression ratios (droplet digital PCR, ddPCR) by per transcriptome expression ratios (RNA-Seq) (Coate and Doyle, 2010) (Supplemental Figure S4) as described in the companion paper (Yang et al., 2021). The transcriptome sizes of haploids and triploids relative to diploids are proportional to the ratios of their corresponding chromosome sets, while the relative transcriptome size of tetraploids to diploids is >2.0 (Figure 6D).
Genes in different functional groups exhibit diverse responses to aneuploidy and ploidy
Genes were partitioned into several functional groups to determine whether there were any specific responses to aneuploidy and ploidy. Genes in each functional group were assembled from resources of various types including databases, tools predicting subcellular localizations, and tools predicting GO terms, as described in a previous study (Johnson et al., 2020) (Supplemental Data Set 6). Ratio distributions were plotted for each functional group while scatter plots for significance of DGE were performed as a complement.
TFs and signal transduction genes in cis showed similar distributions compared with all genes (Supplemental Figures S5 and S6). However, in some aneuploids (e.g. monosomic 1S), cis distributions of those genes show closer approximation to a gene-dosage effect than those of all genes, but the difference is not statistically significant (Supplemental Data Set 3). TFs of many aneuploids distribute differently compared to all genes in trans, validated by K–S tests (Supplemental Data Set 3). Significant changes of distributions are observed in both monosomic and trisomic 3S, 4S, 5L, 8L, and 10L, whereas varied copy number of 1S, 1L, 3L, 5S, 6S, 7L, and 9L only leads to significant variation of distributions in either monosomy or trisomy. In addition, statistical determinations by K–S tests demonstrate that signal transduction genes in most aneuploidy comparisons have similar trans distributions compared with all genes, although a few trisomies distribute differently (Supplemental Figure S6 and Supplemental Data Set 3). In contrast, both TFs and signal transduction genes from the ploidy series distribute similarly compared with all genes (Supplemental Figure S7), statistically verified by K–S tests (Supplemental Data Set 3).
Distributions of genes encoding structural components of the ribosome exhibit the most significant changes in contrast to those of all genes among all functional groups investigated (Supplemental Figure S8). Ribosomal genes in cis display increased or decreased expression compared with all genes in aneuploidy; however, most comparisons are not statistically significant due to the limited gene numbers (Supplemental Data Set 3). A generalized trend toward dosage compensation was observed for cis ribosomal genes in trisomies and tetrasomies rather than monosomies. Ribosomal genes in trans show a narrower spread of modulation than all genes, with P values in Bartlett’s tests for variances approaching zero (Supplemental Data Set 4). Ribosomal genes are modulated to various degrees as the greatest peaks in their distributions are shifted to diverse positions compared with all genes. Trans peaks of 3L, 4S, 5L, and 10L aneuploids are modulated toward the same direction but with different extents, regardless of whether the plants have increased or decreased chromosomal dosage, with those of all 5L and 10L aneuploids approaching 0.5. In contrast, peaks of trans distributions of 4L, 6L, 8L, and 9L aneuploids are shifted toward the direction of a positive effect, while those of 1S, 5S, 7L, and 9S move toward the inverse level. In addition, trans distributions of ribosomal genes show a significant nonlinear relationship to chromosomal dosage, given that peaks of 4D/3D comparisons deviate from 1.0 (Supplemental Figure S8C). Tetrasomic 5S and 6S are more inversely modulated than the respective trisomies whereas tetrasomic 10L is less downregulated than trisomic 10L. Significance of the different distributions of the above-mentioned comparisons was validated by K–S tests (Supplemental Data Set 3).
In further comparisons, trans genes encoding the subunits of the proteasome distribute significantly differently than all genes in most aneuploids (Supplemental Figure S9 and Supplemental Data Set 3), exhibiting a narrow range of modulation with significantly less variation (Supplemental Data Set 4). Proteasomal trans genes in a few monosomies and trisomies display various extents of increased expression. In general, a greater degree of upregulation is observed in monosomies than in trisomies, with modulations in monosomic 1S, 1L, 4S, and 5S being most significant. Ribosomal and proteasomal genes in the ploidy series also distribute statistically differently with a narrower spread than all genes (Supplemental Figure S7 and Supplemental Data Sets 3 and 4); their distributions are shifted slightly upward or downward compared with all genes. In addition, many fewer ribosomal and proteasomal genes are significantly differentially expressed in ploidy comparisons than in aneuploidy comparisons (Supplemental Table S4). Similar distributions between stress-related genes and all genes in aneuploidy and polyploidy conditions indicate that a varied number of individual chromosomes or chromosome sets did not induce extensive stress-related responses in these normal growth conditions (Supplemental Figure S10).
Plant cells contain two semiautonomous organelles, the chloroplast and the mitochondrion. They each contain a small genome that relies largely on nuclear factors for maintenance and expression (Newton et al., 2009). We investigated the distributions of nuclear genes whose products are subsequently transported to the chloroplast or mitochondrion. Cis distributions of genes encoding chloroplast-targeted proteins (hereafter referred to as nuclear chloroplast genes) in a few aneuploids are significantly different (Supplemental Data Set 3). The sharp cis peaks of some monosomies and trisomies are slightly shifted to the left or to the right compared with those of all genes (Supplemental Figure S11). Nuclear chloroplast genes in trans distribute differently compared with all genes in all aneuploids, with a second peak or the greatest peak observed near the ratio of 0.5 (1D/2D) or 0.67 (3D/2D) in distributions of some aneuploids such as 3S, 5L, and 10L (Supplemental Data Set 3). A trend of decreased expression of nuclear chloroplast genes is observed regardless of the increase or decrease of chromosomal dosage, indicating a disruption of expression of them that is conditioned by genome imbalance. Cis distributions of genes encoding mitochondrially targeted proteins (hereafter referred to as nuclear mitochondrial genes) are similar to all genes, while trans distributions in some aneuploids display a significant difference (Supplemental Figure S12). Nuclear mitochondrial genes do not show as much differential modulations in comparison to those for the chloroplast in trans, although small peaks are observed in some aneuploids near the level of a positive trans dosage effect. Nuclear chloroplast and mitochondrial RNAs in the ploidy series distribute statistically differently compared with all genes (Supplemental Figure S7 and Supplemental Data Set 3); however, the difference is minor considering the ploidy series shows a similar trend of gene expression modulation with similar values of mean and median and contains fewer number of DEGs overall compared with aneuploids (Supplemental Data Set 1 and Supplemental Table S4).
Peroxisomes are highly dynamic organelles and are involved in a wide range of plant processes (Hu et al., 2012). Very different from the chloroplast and mitochondrion, they are surrounded by only a single membrane. Peroxisomes do not contain DNA or ribosomes and therefore are devoid of organelle-synthesized components. Distributions of peroxisomal-targeted genes in both aneuploids and polyploids are very similar to all genes (Supplemental Figures S7, S13, and Supplemental Data Set 3).
Medians of trans ratios in each ratio distribution were plotted to illustrate the overall trend of trans gene modulation in each functional group (Figure 8). The finding that the medians of ratios of all genes and many functional groups distribute over or under a ratio of 1.0 (no change) further supports the conclusion that the predominant effect in trans is an inverse modulation. This result also demonstrates that the structural components of the ribosome are modulated to a greater extent compared with other classes.
Figure 8.
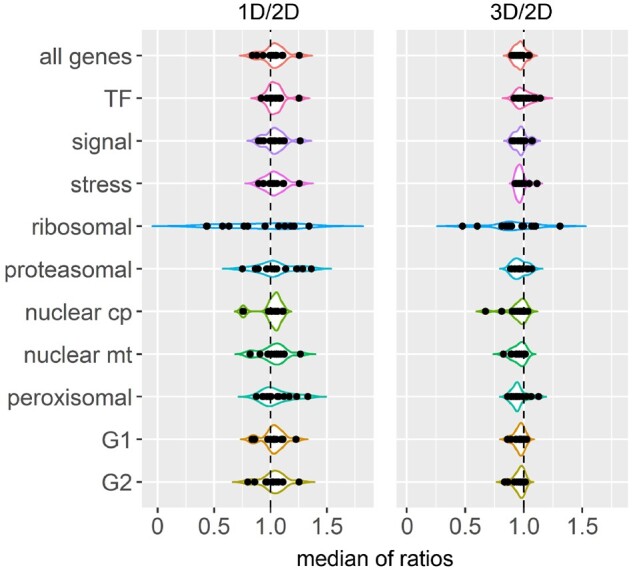
Median of trans ratios of each functional class and maize subgenome in each distal aneuploid line. X-axis refers to median of trans ratios computed as in Supplemental Data Set 1, whereas the y axis denotes functional classes or maize subgenomes (G1 and G2). Each data point represents the median of one distal aneuploid in the corresponding group. Medians of ratios in the 1D/2D comparison are shown on the left while those in the 3D/2D comparison are presented on the right. The black dashed line represents no change of gene expression in trans (ratio 1.0). Cp, chloroplast; mt, mitochondrial.
An interstitial proximal aneuploid exhibits more dosage compensation than distal aneuploids
Apart from the 15 distal aneuploids analyzed above, we assayed an interstitial aneuploid line, Dp4, which varies the centromeric region of chromosome 4 (Zheng and Carlson, 1997). Breakpoints of Dp4 were determined by DNA-seq as described in the “Materials and methods” section (Supplemental Figure S14). Cis distributions of genes in trisomic and tetrasomic Dp4 approach the level of dosage compensation (1.0) whereas trans genes exhibit reduced expression toward the inverse level (Figure 9A). The extent of these modulations toward dosage compensation and inverse levels is more extensive in Dp4 than in most distal aneuploids, especially in trisomic Dp4 (Figures 1–3 and 9). Such cis effects in Dp4 are further supported by the scatter plots of significance (Figure 9B). However, there are fewer trans DEGs in Dp4 than in many other terminal aneuploid lines, likely due to a high variance across biological replicates (Figure 4). Data analyzing expression levels of genes around the cis region of Dp4 in the diploid controls indicate that the greater percentage of dosage compensated genes is not due to a generalized lower expression of genes in this heterochromatic centromeric region (Supplemental Figure S14B).
Figure 9.

Ratio distributions and scatter plots of differential gene expression of the Dp4 dosage series. Distributions (A) were generated as described in Figure 1, whereas scatter plots (B) were generated as described in Figure 6 with genes partitioned into cis and trans.
Genes in different functional groups for Dp4 show a similar trend of modulation compared with the 15 distal aneuploids. TFs, signal transduction genes, structural components of the ribosome and the proteasome, as well as nuclear chloroplast genes distribute differently, whereas stress-related genes, nuclear mitochondrial, and peroxisomal genes show similar distributions compared with all genes, statistically verified by K–S tests (Supplemental Figure S15 and Supplemental Data Set 3). However, the expression modulation of functional groups in Dp4 still exhibits some differences in contrast to other aneuploids in that less modulation is observed in trans distributions of ribosomal and nuclear chloroplast genes (Supplemental Figures S8, S11, and S15). The largest peaks of these distributions are closer to no change (1.0) compared with those of all genes.
When comparing tetrasomic Dp4 to trisomic Dp4, the cis peak is located around the dosage effect (1.33), while the trans peak is slightly below 1.0 (Figure 9A). Thus, a progressive cis effect positively correlated with chromosome dosage is also observed in Dp4 as in distal aneuploids. However, another aspect that is different between Dp4 and distal aneuploids is that trans distributions in the 4D/3D comparisons of signaling and stress-related genes shift toward the inverse level (0.75), while ribosomal, proteasomal, and chloroplast-targeted genes distribute in approximation to the dosage-effect level (1.33). This distinction might be due to the specific regulatory gene content of Dp4 compared to other regions as opposed to this region being primarily heterochromatic. Peaks of the remaining groups for Dp4 are centered around 1.0 as in distal aneuploids (Supplemental Figure S15). On the contrary, deviation of trans peaks in the 4D/3D comparisons in distal aneuploids is only observed for the functional group of ribosomal genes (Supplemental Figure S8). This phenomenon is likely due to a greater spread of modulation in tetrasomic Dp4 in comparison to trisomic Dp4, so that there is a nonlinear relationship between gene expression and chromosomal dosage. Indeed, we observe a higher degree of dosage compensation in trisomic Dp4 than in tetrasomic Dp4 (Figure 9A).
Maize subgenomes respond similarly to genomic imbalance
Many species that have experienced ancient polyploidy show fractionation bias in gene loss and retention between duplicate genomic regions (Thomas et al., 2006; Woodhouse et al., 2010; Garsmeur et al., 2014; Edger et al., 2017, 2019). Maize contains two subgenomes, maize1 (G1) and maize2 (G2), that are differentiated by ongoing fractionation. G1 has experienced less gene loss and its homologs are expressed to a higher level in duplicate gene pairs compared with those from G2 (Schnable et al., 2011). Such fractionation bias could be caused by a selection against loss of the genes with greater expression for a duplicate gene pair, which could have greater negative fitness consequences. Thus, it is of interest to determine whether aneuploidy or polyploidy conditions would result in different modulations of gene expression between G1 and G2. Gene lists of high-confidence retained homologs were obtained as reported (Schnable et al., 2011) (Supplemental Data Set 6). Ratio distributions and statistical determinations indicate a similar expression pattern between G1 and G2 genes in both aneuploids and polyploids (Supplemental Figure S16 and Supplemental Data Set 3). Thus, despite different expression levels, genes in G1 and G2 are modulated as target genes in a similar way under the impact of aneuploidy and polyploidy. Thus, while stoichiometry appears to impact the retention or loss of genes in G1 and G2, per se, with regard to target genes the two genomes are indistinguishable.
Discussion
In this study, a comprehensive gene expression study was performed on 16 aneuploid lines with varied dosage of multiple chromosomal segments with widely varying genic content, in concert with a whole-genome ploidy series including haploids, diploids, triploids, and tetraploids. In general, aneuploids show a wider spread of modulation compared with the whole-ploidy series. Cis genes generally range from a gene-dosage effect to dosage compensation, whereas the most common effect for trans genes is an inverse correlation in that expression is modulated toward the opposite direction of altered chromosomal dosage, although both positive and negative modulations are observed. Genes in different functional classes exhibit diverse responses to aneuploidy, with TFs, signaling genes, ribosomal, and proteasomal components, as well as nuclear chloroplast and mitochondrial genes behaving differently compared with stress-related genes and peroxisomal-targeted genes.
Because only added chromosomal copies were examined in most previous studies of aneuploidy in a variety of organisms, the ability to generate monosomies in maize makes it an excellent model for studying how individual genes respond to changes both above and below the diploid level. When comparing hyperploids with hypoploids sharing the same varied region, a linear relationship between dosage changes and the direction of modulation is observed for many cis DEGs and for a fraction of trans DEGs. However, a nonlinear relationship was also found between chromosomal dosage and the global trend of gene modulation as depicted in ratio distributions depending on the region examined, and between chromosomal dosage and differential expression patterns on a per gene basis. For instance, ratio distributions of trans genes in 10L monosomies are shifted toward the same direction as in trisomies. Also, a large proportion of trans DEGs on the individual gene level is modulated in the same direction regardless of increased or decreased chromosomal dosage. Therefore, apart from the gene-dosage effect, dosage compensation, and inverse-dosage effect, which were regarded as the most common responses to hyperploid dosage changes, this study, with the ability to assay monosomies routinely, shows that genome imbalance can also impact gene expression in additional ways. Genomic imbalance triggered by aneuploidy can result in an increased or decreased effect in both hypoploids and hyperploids compared with normal diploids. A few GO terms are over- or under-represented in common in those DEGs showing different expression patterns (e.g inverse, positive, increased or decreased effects). However, these GO terms are shared among only a few aneuploidy lines, indicating genome balance affects gene expression through different pathways, largely dependent on the specificity of genes or regions being modulated.
Theoretic implications for gene regulatory processes
Although there is now extensive evidence from genetics and evolutionary genomics that stoichiometry plays an important role in quantitative gene expression, little is known about the molecular parameters. Gene regulation is mediated by macromolecular interactions but virtually nothing is known about their assembly except some theoretical modeling (Veitia et al., 2008, 2013; Bray and Lay, 1997). In the case of aneuploid effects, a simplistic model can view the varied chromosomal region as contributing one component relative to the rest of the genome. Although this is clearly an oversimplification, it provides a means to begin to make predictions for future experimentation in this realm.
The nonlinear relationships of the DEGs in the cases of increased or decreased effects could be explained by the nonlinearity in macromolecular assembly of their regulatory machinery. The kinetics of assembly of bridge molecules in relation to other subunits to form multicomponent interactions are positively correlated between their relative amounts and the number of functional complexes at low concentration of the individual varied subunit (Bray and Lay, 1997; Veitia et al., 2008; Birchler et al., 2016). When the ratio of all subunits approaches the same ratio as in the molecular complex, the quantity of functional assemblies reaches the peak before a negative relationship results from further increase in the concentration of the bridge subunit relative to the others (Bray and Lay, 1997; Veitia et al., 2008, 2013; Birchler et al., 2016). In genetic terms, overexpression of bridge components leads to a dominant negative effect. If the dosage balance is approximated in diploids, the number of functional regulatory complexes would certainly drop in both hyperploids and hypoploids if a bridge subunit concentration is altered by gene dosage, thus, impacting gene expression of their targets toward the same direction in the two types of aneuploids. Note that the modulations in these cases still fall more or less within the limits of the dosage imbalance and are not reflective of strongly induced or repressed expression, which would be expected to exceed these limits. This phenomenon likely contributes to the observation that quantitative expression variation across lines is not useful in developing gene regulatory networks but instead needs to rely on presence/absence variation (Zhou et al., 2020). In contrast, if the concentration of a peripheral subunit of the multicomponent regulatory interactions increases, the number of functional complexes would increase until a plateau is reached when the concentration of the peripheral subunit exceeds that of the bridge molecule (Birchler et al., 2016). Of course, the rate of synthesis and degradation of the gene products contributing to a macromolecular complex would also influence the ultimate functional concentration.
The common and unique trends of gene expression in aneuploidy
The cis and trans distributions from the various genomic regions studied shift in concert toward the same direction. In other words, there is a relationship between the inversely regulated trans genes and the cis genes that trend toward dosage compensation, which suggests a common molecular basis. Nevertheless, it is of note that there is an overall greater extent of gene modulation toward the inverse level in hyperploids compared with hypoploids, although hypoploidy triggered more trans DEGs than hyperploidy, likely due to the former causing a wider spread of trans distributions. That hyperploidy exhibits a greater trend of inverse effects than hypoploidy fits the kinetic model of regulatory multicomponent interactions mentioned above. Indeed, in the companion study (Yang et al., 2021) of haploid disomies, which have greater hyperploid imbalance, inverse effects are by far the most prevalent.
Fifteen distal aneuploids were examined in this study, which in total cover almost three quarters of the genes in the W22 genome. As noted above, for each comparison, when gene expression approaches closer to the inverse level in trans, cis distributions proceed more toward dosage compensation. However, the uniqueness of modulation of each aneuploid line should be noted. The extent of increased or decreased expression and the width of spread of modulation depend on the region being altered. The variation of these two features is not related to the number of cis genes nor cis TFs in the respective region. Also, the fraction of DEGs being modulated toward the same versus different directions in monosomies and trisomies is quite varied across aneuploids. Furthermore, DEGs from different aneuploid lines with the same expression patterns are mostly over- or under-represented in different GO terms. Thus, it is likely that the varied copy number of specific genes in each chromosomal segment rather than the size of the region per se has more effect on global gene expression. Also, the fact that every region tested modulated gene expression extensively in trans, including those in the lower part of the size range, suggests that the effects of larger regions are the conglomerate of many and illustrates the extensive nature of the genomic balance network.
Distinct effect of interstitial aneuploidy on gene expression
Apart from the distal aneuploids, we analyzed the expression of an interstitial aneuploid, Dp4, surrounding the centromeric region of chromosome 4 that carries extra copies of over 600 genes. Dp4 generally shows similar cis and trans modulations compared with distal aneuploids. However, a larger proportion of cis genes in Dp4 is dosage compensated than in distal aneuploids as the major peaks of cis distributions are close to 1.0 in trisomic and tetrasomic Dp4 compared with diploids. We considered the possibility that this phenomenon might be due to the specific location of cis regions in Dp4 as it spans the heterochromatic regions of chromosome 4. Repeats and heterochromatin are usually highly enriched around centromeric regions, possibly resulting in epigenetic repression of gene expression. However, reduced levels of gene expression in the cis region of Dp4 were not observed in our data, suggesting extensive dosage compensation is not due to epigenetic repression. The cis distributions of trisomic Dp4 display a more pronounced trend of dosage compensation than tetrasomic Dp4, suggesting that the increased dosage in the latter might trigger a shift in a responsible regulatory network. Although the degree of reduced trans expression of all genes in Dp4 is proportional to its chromosomal dosage from two to four copies, that of several functional classes deviates from the linear relationship in the 4D/3D comparison. Trisomic Dp4 also shows a higher degree of dosage compensation in cis and inverse effects in trans than most distal trisomies. Although this example of aneuploidy of centromeric heterochromatin is distinct from the distal aneuploidies studied, it is a single case. Additional heterochromatic regions will need to be examined to determine if this difference is generalizable.
Considerations of cell and transcriptome size
First identified in Datura, the relationship between cell size and the level of ploidy has been repeatedly found in studies of many organisms (Sinnott and Blakeslee, 1922; Dobzhansky, 1929; Steinitz-Sears, 1963; Yao et al., 2011; Miller et al., 2012; Tsukaya, 2013; Robinson et al., 2018). This phenomenon was also observed in this study in that cell size increases with ascending ploidy from haploids to tetraploids. There is a greater amount of total RNA per cell with an increased level of ploidy (Loven et al., 2012; Robinson et al., 2018; Song et al., 2020). Therefore, cell size is strongly related to transcriptome size or the amount of total RNA. The normalization method used in this study does not reflect the expression level per cell. As a result, ratios of the whole-genome ploidy series distributing around 1.0 indicates the effect of an increased amount of total RNA is canceled by the effect of increased cell size. Thus, the differences found reflect the relative changes of gene expression in a polyploid series, which are minor.
Our measurement of genome-normalized expression level using ddPCR further supports the minor changes of gene expression in the whole-ploidy series (Supplemental Figure S4B). The epidermal cell size of haploid, triploid, and tetraploid is 0.43-, 1.33-, and 1.60-fold relative to that of diploid; whereas the relative transcriptome size is 0.53-, 1.48-, and 3.70-fold in relation to that of diploids (Figures 6D and 7C). This relationship is generally consistent with a greater transcriptome size with increasing ploidy with a larger step for the tetraploid with this genotype.
In contrast to polyploidy, a generalized relationship between cell size and varied copy of individual chromosomes or chromosomal segments has been rarely observed in Datura, Drosophila, or Arabidopsis (Sinnott and Blakeslee, 1922; Dobzhansky, 1929; Sinnott et al., 1934; Steinitz-Sears, 1963; Lo et al., 2014). Our data showing that varied copies of aneuploid segments in the 5S and 6S series did not significantly impact the cell size is consistent with these previous findings. The insignificant variation in cell size indicates that the trends of expression modulation in aneuploids are not, or at least minimally, due to changes in transcriptome size. However, in the companion study of haploid disomies, a few regions of the genome inversely affect the transcriptome size in this tissue (Yang et al., 2021).
Regarding the question of whether the rRNA transcriptome, the major constituent of total RNA, is altered in aneuploids, we used TB-6Sa as a control. TB-6Sa is a B–A translocation line that carries part of the nucleolar organizer region (NOR) and a very short portion of 6S (Lin, 1955; Beckett, 1978), with a cis region covering approximately 200 genes (Supplemental Figure S14D). Historically, maize plants carrying multiple copies of the B6 chromosome with the NOR were examined and a linear relationship between the total amount of RNA being present within the nucleolus and the number of the B6 chromosomes was observed (Lin, 1955). Also, the amount of RNA in the nucleolus in triploids was increased to the same level as in trisomic maize plants carrying an extra chromosome 6. However, in the present study, only a few genes showing an expression approaching the inverse level are observed in trisomic and tetrasomic 6S with most gene expression being similar. Considering that the rRNA is the major component of the total RNA, if the varied copy number of the NOR was able to alter the total amount of rRNA independently of mRNA in the total RNA extractions, there would be an apparent generalized shift of almost all gene expression to the inverse level, which was not found. Furthermore, if the amount of rRNA would be altered, the relative total transcriptome size of hyperploid 6S would be significantly increased, leading to increased cell size, which was also not found (Figure 7). There is no evidence in the literature that changing the dosage of the NOR affects cell size despite the increased RNA output within the nucleolus (Lin, 1955; Phillips et al., 1971). Collectively, these results indicate there is an increase in the amount of rRNA in the nucleolus with increasing dosage of the NOR, but this increase is not reflected in the steady state level of rRNA in the ribosomes of the cells. In another study, the ratio of rRNA per DNA was measured in a large number of maize monosomies and trisomies (TB-6Sa was not used in that study), yet no evidence of altered amount of rRNA was found (Guo and Birchler, 1994). In Drosophila, the amount of rRNA per DNA was also reported to be unchanged (Birchler et al., 1989; 1990). Considering in Drosophila there is one NOR on each X and Y chromosome and that three copies of the NOR did not affect the amount of rRNA isolated in the total RNA extractions, it is likely that the amount of rRNA in total RNA extractions directly reflects the amount of rRNA present in the ribosomes (Birchler et al., 1989). This assumption is supported by our analyses on the dosage series of 6S. All these results indicate that the transcribed amounts of rRNA are not reflected in the total RNA extractions in aneuploids.
Implications for quantitative characteristics
Phenotypically, monosomies and trisomies are both detrimental compared with balanced diploids (Lee et al 1996a, 1996b; Sheridan and Auger, 2008; Brunelle and Sheridan, 2014). An extensive genomic scan of induced copy number variants in poplar illustrates this fact on a global fine-scale (Bastiannse et al., 2019). Specific examples of how the subunit stoichiometry of signaling or TF protein interactions can affect plant morphology have been described (Chen et al., 2010; Stahl et al., 2013; Clark et al., 2020); our results suggest a related behavior is a widespread aspect of regulatory mechanisms. Although a relationship between global gene expression during development and the phenotype of whole organisms is complex, influenced by the environment, and not understood, it has been noted that, as a general rule, there are potentially limiting expressions in both monosomies and trisomies (Birchler and Newton, 1981). Monosomies have reduced expression from cis dosage effects and trans positive modulations. Trisomies have limiting expression from the predominant inverse effects operating in this aneuploid type. Tetrasomies have reductions that can be greater in magnitude in general. Indeed, extensive evidence from evolutionary genomics, cited above, indicates that changing the stoichiometry of regulatory machineries on the individual gene level can affect fitness and selection against altered balance. Our results indicate that there is a rheostat of target gene expression illustrating that there is an astronomical number of potential subtle quantitative phenotypes that can occur for any one trait, assuming variation. Individual genes can have an impact resulting from different stoichiometries among the components of regulatory macromolecular interactions. Coupled with the potential spectrum of quantitative effects of dissected 5′ regulatory regions of target genes (Rodriquez-Leal et al., 2017), the number of possible phenotypes is compounded further. Taken together, the results illustrate the need to incorporate balance principles into the field of quantitative genetics.
Quantitative and theoretical population genetics modeling tends to classify allelic alternatives as plus or minus. The results of the dosage analyses indicate that any one level of quantitative regulatory expression could have a positive or negative influence on target gene expression that is also influenced by the level of expression of interactors in the background and therefore is not necessarily predictable from individual to individual. Thus, to some degree population and quantitative genetics theory needs to be revisited to incorporate the concept of stoichiometry among regulatory genes that produce a gradient of target gene expression.
The impact of B chromosomes on gene expression in aneuploids
Maize with stable B–A translocations was used to study the effect of distal aneuploidy on global gene expression. Except for the 10L and 5S diploids, which contain the B chromosome, other diploid lines in the control groups are composed of 20 normal A chromosomes (Supplemental Table S1). In some comparisons, plants in the experimental group (distal aneuploidy) have part or the equivalent of a whole B chromosome or more, whereas plants in the control group (diploid) do not contain any B chromosome. Although the maize B chromosome is a nonessential chromosome that is dispensable for the normal growth and reproduction of the plant (Randolph, 1941), it has been reported to impact the expression of over a hundred genes transcribed from the A chromosome (Huang et al., 2016). Results of the DGE analysis of haploids with one or two copies of the B chromosome compared with haploids without any B chromosome in the companion study further support this conclusion (Yang et al., 2021). Thus, the possibility that the B chromosome could impact gene expression in aneuploids created by B–A translocations should not be overlooked. However, the B chromosome is not the major contributor to global gene modulation in the distal aneuploids assayed in this study. In the 5S and 10L comparisons, plants in both the control and experimental groups contain all of the parts of the B chromosome, yet similar patterns of gene modulation were observed as in other distal aneuploids. Furthermore, in the companion study, ratio distributions of haploids with one or two copies of the B chromosome compared with normal haploids center around 1.0 and are significantly different from the A chromosome disomy groups, suggesting the effects observed in A chromosome disomies are trivially impacted by the B chromosome portion, if at all (Yang et al., 2021).
Concluding remarks
In general, the results of this study illustrate that there is an overall trend for greater gene expression modulation in aneuploids than in a ploidy series, which provides insight into the century-old observation that changing the dosage of individual chromosomes has more phenotypic effects than changing the whole set. Unlike what was generally accepted in the field that altered expression induced by aneuploidy mainly occurs in the cis region, this study demonstrates that aneuploidy leads to global gene modulation both in cis and in trans. Furthermore, monosomies were examined in this study along with the respective hyperploidies with genomic imbalance assayed both globally and on a per gene basis. Apart from the predominant dosage compensation and gene-dosage effects in cis and the inverse effect in trans, a range of effects such as increased and decreased effects appear in some cases. The coexistence of linear and nonlinear relationships between chromosomal dosage and the direction of genomic modulation further supports the kinetic model of regulatory genomic balance. Despite similarities, gene expression trends in each aneuploid line showed unique features, as expected if specific genes in the respective varied regions trigger global cascading modulations. The prevalence of the inverse and positive modulations and their potential interrelationship is counterintuitive to current views of gene regulation but these genetic results indicate the need to incorporate these principles into mechanistic models of gene regulation.
Materials and methods
Plant material and morphological analysis
To obtain maize (Zea mays) plants with one to four doses of chromosomal arms, maize plants with B–A translocations of 15 distinct chromosome arms converged to inbred line W22 were karyotyped for numbers of normal and translocated chromosomes using FISH (Supplemental Table S1). In general, for maize plants with one to three doses of chromosome arms, their male parents were hyperploid for the translocated BA chromosome, while their female parents were testers. For obtaining plants with four doses, maize plants hyperploid for the BA chromosome were selfed, which resulted in the spectrum of one to four doses of the translocated arm. For Dp4, plants heterozygous for the proximal overlap of B–A translocations for chromosome 4 were self-pollinated to produce a segregating progeny with two, three, and four doses of the region.
FISH was performed for counting chromosome numbers in the progeny using a fluorescence microscope (Olympus BX61). Chromosomes were stained with 4′,6′-diamidino-2-phenylindole (DAPI) (Thermo Fisher) and then probed with oligo probes including the CentC centromere repeat and the microsatellite TAG repeat, in addition to a nick-translated B chromosome-specific centromere repeat probe (Albert et al., 2019). Afterwards, plants with desired karyotypes were grown in the Sears greenhouse at the University of Missouri (16-h light, 25°C day/20°C night).
Four biological replicates were obtained for each karyotype for each distal aneuploid line. Monosomies (1D), trisomies (3D), and tetrasomies (4D) were compared to diploids (2D) as the control. Except for the 10L and 5S diploid, which contained one BA and one AB chromosome, other diploid lines were composed of 20 normal A chromosomes (Supplemental Table S1). Diploid controls used in each comparison were siblings to the corresponding experimental group as both groups segregated from the same cross, with the exception of the controls used in the comparison regarding chromosome arms 5L and 3S, which were the same diploids used for the 1S comparison. However, little variance would be caused by this substitution, given that these diploid controls had the same genotype and were grown and sequenced simultaneously as the 5L and 3S aneuploids. In addition, four biological replicates were gathered for trisomic and diploid Dp4, whereas only three were obtained for tetrasomic Dp4.
For the polyploid series, we compared four W22 haploids to four W22 diploids (see Yang et al., 2021 for generation of haploids), four W22 tetraploids to four W22 diploids (Kato and Birchler, 2006), and two Mo17 triploids to three of their diploid siblings (Yao et al., 2013). The numbers of chromosomes in these samples were determined by counting DAPI-stained chromosomes with a fluorescence microscope (Olympus BX61). A 30.48 cm of leaf tissue was collected from the fifth and sixth leaves from the bottom of each plant 45 days after germination during late mornings to minimize circadian differences in expression. Only the leaf blade was kept, and the midrib was discarded. The tissue was frozen in liquid nitrogen and then stored at –80°C for further analysis.
Epidermal cell size measurement
For the 5S dosage series and the whole-ploidy series, 45-day-old plants were used to obtain the epidermal cell imprints for cell area measurements as described previously (Ferris and Taylor, 1994; Ferris et al., 2002; Yao et al., 2011). Imprints were taken from the abaxial surface of the second leaf from the bottom of each plant and were examined by a brightfield microscope (Olympus BX61). For each plant, four imprints were taken. We measured the area of 25 epidermal cells from each imprint using Fiji (Schindelin et al., 2012), calculated the mean value for the area of 100 cells for each plant, and performed Student’s t test of means for statistical significance. Data were collected from the same plants from which RNA was extracted for mRNA-seq. For the 6S dosage series, seeds were obtained from a selfed plant derived from R-scm2 W22 with multiple B6Sa chromosomes. Plants were grown in the field in July 2020 in Columbia, Missouri and the cell imprints were taken 43 days after planting. Genotypes of these plants were confirmed by FISH assays. For each plant, 10 imprints were taken and 15 cells from each imprint were measured.
Transcriptome size measurement
The sizes of the transcriptome of the haploid, triploid, and tetraploid relative to diploid were measured as described (Yang et al., 2021), according to the approach previously developed (Coate and Doyle, 2010). In brief, RNA and gDNA were coextracted using Dr P Isolation Kit (BioChain) from the same plants used for RNA-seq. ddPCR was performed to measure the RNA/gDNA ratio using primers specifically designed to amply either the cDNA or gDNA so that a genome-normalized expression level of each gene of interest was obtained. Details of the ddPCR and the corresponding primers being used were described (Yang et al., 2021). The per genome expression ratio (experiment/control) equals the ratio of genome-normalized expression level of the experimental group to that of the control. Subsequently, the per cell expression ratio was computed by multiplying the per genome expression ratio by the ratio of chromosome sets. For example, the per cell ratio (1X/2X) equals its per genome ratio times 0.5 (3X/2X × 1.5 and 4X/2X × 2.0, respectively). To estimate the relative transcriptome size of haploid, triploid, and tetraploid in comparison to diploids, we then divided the per cell expression ratios obtained from the ddPCR assay by the per transcriptome expression ratios derived from the RNA-Seq. The mean of eight independent estimates of the transcriptome size of each polyploid relative to diploid transcriptome was taken as the overall estimate of relative transcriptome size.
RNA isolation and RNA-seq library construction
Total RNA was extracted from maize leaves using mirVana miRNA Isolation Kit (Thermo Fisher Scientific). Total RNA was quantified by a Qubit fluorometer (Invitrogen) using the Qubit RNA HS RNA assay kit (Thermo Fisher Scientific). The RNA integrity was checked using the Fragment Analyzer automated electrophoresis system (Agilent). A 5 µg of total RNA was spiked with the External RNA Controls Consortium (ERCC) RNA Spike-In Mix (Thermo Fisher Scientific) according to the manufacturer’s protocol. Spiked total RNA was depleted for rRNA with Ribo-Zero rRNA Removal Kit (Illumina) and used for library construction with TruSeq Stranded mRNA Library Preparation Kit (Illumina). The rRNA was removed from total RNA by hybridization with the probes instead of poly(A) RNA enrichment. All samples were sequenced in seven independent mRNA-seq experiments on the NextSeq500 platform using 75-bp single-end sequencing. Libraries from each mRNA-seq experiment were mixed and then aliquoted to each flow cell (NextSeq High Output Flow Cell-SE75) to reduce the technical error between experimental and control groups in each comparison. Sequencing was performed at the DNA core (University of Missouri). Read mapping statistics of each mRNA-seq experiment are listed in Supplemental Data Set 7. Each experiment generated 41–78 million raw read counts on average.
mRNA-seq data processing
Adaptors at the 3′ end of the reads were trimmed using cutadapt version 1.16 (Martin, 2011). Low-quality reads were removed using the FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/, fastq_quality_filter -Q33 -q20 -p80). Subsequently, mRNA reads were aligned to ERCC sequences using Bowtie 2 with default parameters (Langmead and Salzberg, 2012). A table of ERCC counts together with known relative concentrations was used as input and output for an R script to generate a linear regression model. R-squared values were extracted from the model to evaluate the quality of the sequenced library (Supplemental Data Set 7). Subsequently, the remaining non-ERCC reads were aligned to maize chloroplast and mitochondrial genomes using TopHat2 with default parameters so that the organellar transcripts are excluded from further study (Clifton et al., 2004; Kim et al., 2013; Bosacchi et al., 2015). The remaining reads were then mapped to the maize reference genome W22v2 along with the maize chloroplast and mitochondrial genomes using TopHat2 with default parameters (Clifton et al., 2004; Bosacchi et al., 2015; Springer et al., 2018). Less than 1% of organellar reads were retained. A 9–24 million uniquely mapped nonorganellar read counts were obtained for each experiment on average (Supplemental Data Set 7). Normalized read counts were generated by Cuffdiff (raw-mapped-norm) for each comparison (e.g. aneuploids with different dosage for chromosome 5S were normalized against each other) (Trapnell et al., 2013). The lowly-expressed genes (mean of the control group + mean of the experimental group ≤ 1) were excluded from further study. PCA plots were generated in R using normalized read counts (Supplemental Figure S1).
DNA isolation, library construction, and breakpoint determination
DNA was extracted using DNeasy Plant Mini Kit (QIAGEN). Libraries were prepared according to TruSeq DNA PCR-Free Library Prep (Illumina). DNA samples isolated from one tetrasomic Dp4 and its diploid sibling were sequenced on a NextSeq High Output Flow Cell - SE75 (Illumina) at the DNA core of University of Missouri. DNA samples extracted from a trisomic 10L18, 6Sa, and their respective diploid sibling were subjected to the same procedure. Adaptor trimming and low-quality read removal were performed the same as in the mRNA-seq data process. Then the remaining DNA reads were aligned to the maize reference genome W22v2 along with the maize chloroplast and mitochondrial genome using Bowtie 2 with default parameters (Langmead and Salzberg, 2012). Uniquely mapped read counts were extracted and then subjected to Reads Per Kilobase per Million mapped reads (RPKM) normalization. Subsequently, ratios of normalized counts of tetrasomic Dp4 to diploid were generated on a per gene basis, which was used for determination of Dp4 breakpoints later (Supplemental Figure S14A). Ratios of normalized counts of trisomic 10L or 6S to the diploid were computed to determine the breakpoint of TB-10L18 and TB-6Sa (Supplemental Figure S14). Breakpoints of other distal aneuploids were determined as described in the companion study (Yang et al., 2021).
Ratio distribution and scatter plots
Ratio distribution plots were generated as described in previous studies (Hou et al., 2018; Shi et al., 2020). In brief, means of normalized counts of biological replicates of each experimental genotype (e.g. 3D of chromosome 5S) and the control group (e.g. 2D of chromosome 5S) were computed. The ratio was generated by dividing the mean of treatment counts by the mean of control counts and is visualized as a histogram. For scatter plots, corrected P value (q value) and log fold change with base 2 (log2fold_change, or logFC) produced by Cuffdiff were used. The logFC between treatment and control was plotted on the x-axis, while the mean of normalized counts of the treatment and control group was plotted on the y-axis. Data points with a q value <0.05 and a corresponding logFC >0 were depicted in magenta, while points with an q value <0.05 and a corresponding logFC <0 were depicted in green. Otherwise, they were designated in black.
All ratio distributions and scatter plots were generated using the method above except for the comparison between triploids and tetraploids (4X/3X), considering that this comparison involves two sets of controls with different backgrounds. For plotting ratio distributions, normalized read counts for each tetraploid were divided by the mean of their diploid control (4X/2X ratio), while normalized read counts for each triploid were divided by the mean of their diploid control (3X/2X ratio). Then the 4X/2X ratio was used as the experimental value whereas the 3X/2X ratio as the control in the generation of novel ratios used for the ratio distribution. For scatter plots, instead of DGE, we performed a Student’s t test in comparing ratios of 4X/2X to 3X/2X, which tests for the significance of whether the mean log2 ratio of 4X/2X differs from the mean log2 ratio of 3X/2X. q values were calculated by adjusting P values by the Benjamini–Hochberg algorithm for computing false discovery rates. Genes with adjusted P <0.05 were defined as DEGs. The log2 ratio on the x-axis denotes the log ratio of 4X/2X to 3X/2X with base 2, while the mean of normalized counts of 4X, 3X, and their diploid controls was plotted on the y-axis.
Quantitative PCR
A 1 µg total RNA was spiked in 2 µL 1:100 diluted ERCC RNA Spike-In Mix (Thermo Fisher Scientific). cDNA was synthesized using the SuperScript IV Reverse Transcriptase (Thermo Fisher Scientific) with gene specific primers. RT-qPCR was performed with a StepOnePlus Real-Time PCR System (Thermo Fisher Scientific) and the PowerUp SYBR Green Master Mix (Thermo Fisher Scientific). A 2-ng cDNA was used as the template for RT-qPCR. Three technical replicates were carried out for each sample. Four biological replicates were used for each genotype. RNA accumulation levels of selected genes were measured in relation to levels of ERCC-00111 as a control. Primers are listed in Supplemental Table S5.
GO term enrichment analysis
GO term enrichment analysis was performed using PANTHER online tools (Thomas et al., 2003; Mi et al., 2013). Genes of interest were tested for over- or under-representation using Fisher's exact test against all maize genes expressed in each condition (lowly expressed genes were excluded as described in section “mRNA-seq data processing”). Only significant terms (Bonferroni-corrected P <0.05) were used for further study.
Statistical analysis
Statistical tests were performed using R with extreme values (ratio >6 or < 1/6) excluded. Mean, median, and sd of each comparison were computed (Supplemental Data Set 1). Normality assumption was checked by the Lilliefors test (Supplemental Data Set 2). In addition, similarity between two ratio distributions was also validated by the K–S test (Supplemental Data Set 3). We also used the Bartlett’s test to examine if variances are equal across different groups (Supplemental Data Set 4).
Accession numbers
All sequencing data were deposited at the Gene Expression Omnibus repository under the accession number GSE149186.
Supplemental data
The following materials are available in the online version of this article.
Supplemental Figure S1. PCA plots of aneuploids and polyploids.
Supplemental Figure S2. Scatter plots of significant differential expression for each gene in aneuploids.
Supplemental Figure S3. Ratios of selected genes generated from mRNA-seq and qPCR in 5S aneuploids.
Supplemental Figure S4. Expression ratios generated from ddPCR and mRNA-seq in the whole-ploidy series comparisons.
Supplemental Figure S5. Ratio distributions and scatter plots of differential gene expression for the functional class of TFs in aneuploids.
Supplemental Figure S6. Ratio distributions and scatter plots of differential gene expression for the functional class of signal transduction genes in aneuploids.
Supplemental Figure S7. Ratio distributions and scatter plots of differential gene expression for various functional groups in the ploidy series.
Supplemental Figure S8. Ratio distributions and scatter plots of differential gene expression for the functional class of structural components of the ribosome in aneuploids.
Supplemental Figure S9. Ratio distributions and scatter plots of differential gene expression for the functional class of structural components of the proteasome in aneuploids.
Supplemental Figure S10. Ratio distributions and scatter plots of differential gene expression for the functional class of stress-related genes in aneuploids.
Supplemental Figure S11. Ratio distributions and scatter plots of differential gene expression for the functional class of nuclear chloroplast genes in aneuploids.
Supplemental Figure S12. Ratio distributions and scatter plots of differential gene expression for the functional class of nuclear mitochondrial genes in aneuploids.
Supplemental Figure S13. Ratio distributions and scatter plots of differential gene expression for the functional class of peroxisomal-targeted genes in aneuploids.
Supplemental Figure S14. Ratios of normalized DNA read counts and RNA read counts of Dp4, TB-6Sa and TB-10L18.
Supplemental Figure S15. Ratio distributions and scatter plots of differential gene expression for different functional classes in Dp4.
Supplemental Figure S16. Ratio distributions of high-confidence retained homologs from maize1 (G1) and maize2 (G2) subgenomes.
Supplemental Table S1. Karyotype, number of biological replicates being sequenced and parental lines of distal aneuploid maize.
Supplemental Table S2. Number of cis genes and position of translocation breakpoints in each aneuploid line.
Supplemental Table S3. Percentage of DEGs with different expression patterns upon changes of chromosomal dosage.
Supplemental Table S4. The number of DEGs in each functional group in aneuploids and polyploids.
Supplemental Table S5. Sequence for primer sets used in qPCR.
Supplemental Data Set 1. Mean, median, and standard deviation (sd) for each ratio distribution.
Supplemental Data Set 2. Normality test for each ratio distribution.
Supplemental Data Set 3. Distribution comparisons with K–S tests of significance.
Supplemental Data Set 4. Comparisons of distribution variances with Bartlett’s test.
Supplemental Data Set 5. GO term enrichment analysis regarding biological process on DEGs with inverse, positive, increased or decreased effects in monosomies and trisomies compared with diploids.
Supplemental Data Set 6. Gene lists of diverse functional groups and maize subgenomes used in this study.
Supplemental Data Set 7. Read mapping statistics and group information of each mRNA-seq experiment.
Funding
Funding from National Science Foundation (grants IOS-1545780, IOS-1444514, NSF 1615789, and NSF 1853556) is acknowledged.
Conflict of interest statement. We declare no conflicts of interest.
Supplementary Material
Contributor Information
Xiaowen Shi, Division of Biological Sciences, University of Missouri, Columbia, Missouri 65211, USA.
Hua Yang, Division of Biological Sciences, University of Missouri, Columbia, Missouri 65211, USA.
Chen Chen, Department of Electrical Engineering and Computer Science, University of Missouri, Columbia, Missouri 65211, USA.
Jie Hou, Department of Electrical Engineering and Computer Science, University of Missouri, Columbia, Missouri 65211, USA.
Katherine M Hanson, Division of Biological Sciences, University of Missouri, Columbia, Missouri 65211, USA.
Patrice S Albert, Division of Biological Sciences, University of Missouri, Columbia, Missouri 65211, USA.
Tieming Ji, Department of Statistics, University of Missouri, Columbia, Missouri 65211, USA.
Jianlin Cheng, Department of Electrical Engineering and Computer Science, University of Missouri, Columbia, Missouri 65211, USA.
James A Birchler, Division of Biological Sciences, University of Missouri, Columbia, Missouri 65211, USA.
J.B., J.C., X.S., and H.Y. designed the research. X.S., H.Y., C.C., J.H., K.M., and P.A. performed research. C.C., J.H., T.J., X.S., H.Y., and J.C. contributed analytical/computational tools. X.S., H.Y., C.C., J.H., T.J., and J.B. analyzed data. X.S., H.Y., and J.B. wrote the article.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://academic.oup.com/plcell) is: James A. Birchler (BirchlerJ@missouri.edu).
References
- Albert PS, Zhang T, Semrau K, Rouillard J-M, Kao Y-H, Wang C-JR, Danilova TV, Jiang J, Birchler JA (2019) Whole-chromosome paints in maize reveal rearrangements, nuclear domains, and chromosomal relationships. Proc Natl Acad Sci U S A 116: 1679–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aury JM, et al. (2006) Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia. Nature 444: 171–178 [DOI] [PubMed] [Google Scholar]
- Bastiannse H, Zinkgraf M, Canning C, Tsai H, Lieberman M, Comai L, Henry I, Groover A (2019) A comprehensive genomic scan reveals gene dosage balance impacts on quantitative traits in Populus trees. Proc Natl Acad Sci U S A 116: 13690–13699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckett JB (1978) B-A translocations in maize I. Use in locating genes by chromosome arms. J Hered 69: 27–36 [Google Scholar]
- Beckett JB (1991) Cytogenetic, genetic and plant breeding applications of B–A translocations in maize, Chromosome engineering in plants: genetics, breeding, evolution. Part A. In Developments in Plant Genetics and Breeding. Elsevier, Amsterdam, Netherlands, pp 493–529 [Google Scholar]
- Birchler JA (1979) A study of enzyme activities in a dosage series of the long arm of chromosome one in maize. Genetics 92: 1211–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler JA (1981) The genetic basis of dosage compensation of alcohol dehydrogenase-1 in maize. Genetics 97: 625–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler JA, Bhadra U, Bhadra MP, Auger DL (2001) Dosage-dependent gene regulation in multicellular eukaryotes: implications for dosage compensation, aneuploid syndromes, and quantitative traits. Dev Biol 234: 275–288 [DOI] [PubMed] [Google Scholar]
- Birchler JA, Hiebert JC, Krietzman M (1989) Gene expression in adult metafemales of Drosophila melanogaster. Genetics 122: 869–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler JA, Hiebert JC, Paigen K (1990) Analysis of autosomal dosage compensation involving the alcohol dehydrogenase locus in Drosophila melanogaster. Genetics 124: 677–686 [PMC free article] [PubMed] [Google Scholar]
- Birchler JA, Johnson AF, Veitia RA (2016) Kinetics genetics: incorporating the concept of genomic balance into an understanding of quantitative traits. Plant Sci 245: 128–134 [DOI] [PubMed] [Google Scholar]
- Birchler JA, Newton KJ (1981) Modulation of protein levels in chromosomal dosage series of maize: the biochemical basis of aneuploid syndromes. Genetics 99: 247–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler JA, Riddle NC, Auger DL, Veitia RA (2005) Dosage balance in gene regulation: biological implications. Trends Genet 21: 219–226 [DOI] [PubMed] [Google Scholar]
- Birchler JA, Veitia RA (2012) Gene balance hypothesis: connecting issues of dosage sensitivity across biological disciplines. Proc Natl Acad Sci U S A 109: 14746–14753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler JA, Veitia RA (2007) The gene balance hypothesis: from classical genetics to modern genomics. Plant Cell 19: 395–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler JA, Veitia RA (2010) The gene balance hypothesis: implications for gene regulation, quantitative traits and evolution. New Phytol 186: 54–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakeslee AF (1934) New Jimson Weeds from Old Chromosomes. J Hered 25: 81–108 [Google Scholar]
- Blakeslee AF (1921) Types of mutations and their possible significance in evolution. Am Nat 55: 254–267 [Google Scholar]
- Blakeslee AF, Belling J, Farnham ME (1920) Chromosomal duplication and Mendelian phenomena in Datura mutants. Science 52: 388–390 [DOI] [PubMed] [Google Scholar]
- Blanc G, Wolfe KH (2004) Functional divergence of duplicated genes formed by polyploidy during Arabidopsis evolution. Plant Cell 16: 1679–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosacchi M, Gurdon C, Maliga P (2015) Plastid genotyping reveals the uniformity of cytoplasmic male sterile-T maize cytoplasms Plant Physiol 169: 2129–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray D, Lay S (1997) Computer-based analysis of the binding steps in protein complex formation. Proc Natl Acad Sci U S A 94: 13493–13498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges CB (1925) Sex in relation to chromosomes and genes. Am Nat 59: 127–137 [Google Scholar]
- Brunelle DC, Sheridan WF (2014) The effect of varying chromosome arm dosage on maize plant morphogenesis. Genetics 198: 171–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson PS (1972) Locating genetic loci with aneuploids. Mol Gen Genet 114: 273–280 [DOI] [PubMed] [Google Scholar]
- Carlson WR (1988) B chromosomes as a model system for nondisjunction. In Vig BK, Sandberg AA, eds, Aneuploidy: Induction and Test Systems (Part B). Alan R. Liss Inc., NY, pp 199–207 [Google Scholar]
- Carlson WR (1988) The cytogenetics of corn. Corn Corn Improvement 18: 259–343. [Google Scholar]
- Chen Y-F, Gao Y, Kerris RJ III, Wang W, Binder BM, Schaller GE (2010) Ethylene receptors function as components of high molecular-mass protein complexes in Arabidopsis. PLoS One 5: e8640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark NM, Fisher AP, Berckmans B, Van den Broek L, Nelson EC, Nguyen TT, Bustillo-Avendano E, Zebell SG, Moreno-Risueno MA, Simon R, et al. (2020) Protein complex stoichiometry and expression dynamics of transcription factors modulate stem cell division. Proc Natl Acad Sci U S A 117: 15332–15342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifton SW, Minx P, Fauron CM, Gibson M, Allen JO, Sun H, Thompson M, Barbazuk WB, Kanuganti S, Tayloe C, et al. (2004) Sequence and comparative analysis of the maize NB mitochondrial genome. Plant Physiol 136: 3486–3503 [DOI] [PMC free article] [PubMed]
- Cong B, Barrero LS, Tanksley SD (2008) Regulatory change in YABBY-like transcription factor led to evolution of extreme fruit size during tomato domestication. Nat Genet 40: 800–804 [DOI] [PubMed] [Google Scholar]
- Cong B, Liu J, Tanksley SD (2002) Natural alleles at a tomato fruit size quantitative trait locus differ by heterochronic regulatory mutations. Proc Natl Acad Sci U S A 99: 13606–13611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coate JE, Doyle JJ (2010) Quantifying whole transcriptome size, a prerequisite for understanding transcriptome evolution across species: an example from a plant allopolyploid. Genome Biol Evol 2:534–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coate JE, Song MJ, Bombarely A, Doyle JJ (2016) Expression-level support for gene dosage sensitivity in three Glycine subgenus Glycine polyploids and their diploid progenitors. New Phytol 212: 1083–1093 [DOI] [PubMed] [Google Scholar]
- Defoort J, Van de Peer Y, Carretero-Paulet L (2019) The evolution of gene duplicates in angiosperms and the impact of protein-protein interactions and the mechanism of duplication. Genome Biol Evol 11: 2292−2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobzhansky T (1929) The influence of the quantity and quality of chromosomal material on the size of the cells in Drosophila melanogaster. Wilhelm Roux Arch Entwickl Mech Org 115: 363–379 [DOI] [PubMed] [Google Scholar]
- Du K, et al. (2020) The sterlet sturgeon genome sequence and the mechanism of segmental rediploidization. Nat Ecol Evol 4: 841–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edger PP, Smith R, McKain MR, Cooley AM, Vallejo-Marin M, Yuan Y, Bewick AJ, Ji L, Platts AE, Bowman MJ, et al. (2017) Subgenome dominance in an interspecific hybrid, synthetic allopolyploid, and a 140-year-old naturally established neo-allopolyploid monkeyflower. Plant Cell 29: 2150–2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edger PP, Poorten TJ, VanBuren R, Hardigan MA, Colle M, McKain MR, Smith RD, Teresi SJ, Nelson ADL, Wai CM, et al. (2019) Origin and evolution of the octoploid strawberry genome. Nat Genet 51: 541–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris R, Long L, Bunn SM, Robinson KM, Bradshaw HD, Rae AM, Taylor G (2002) Leaf stomatal and epidermal cell development: identification of putative quantitative trait loci in relation to elevated carbon dioxide concentration in poplar. Tree Physiol 22: 633–640 [DOI] [PubMed] [Google Scholar]
- Ferris R, Taylor G (1994) Stomatal characteristics of four native herbs following exposure to elevated CO2. Ann Bot 73: 447–453 [Google Scholar]
- Frary A, Nesbitt TC, Grandillo S, Knaap E, Cong B, Liu J, Meller J, Elber R, Alpert KB, Tanksley SD (2000) fw2.2: a quantitative trait locus key to the evolution of tomato fruit size. Science 289: 85–88 [DOI] [PubMed] [Google Scholar]
- Freeling M, Lyons E, Pedersen B, Alam M, Ming R, Lisch D (2008) Many or most genes in Arabidopsis transposed after the origin of the order Brassicales. Genome Res 18: 1924–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeling M, Thomas BC (2006) Gene-balanced duplications, like tetraploidy, provide predictable drive to increase morphological complexity. Genome Res 16: 805–814 [DOI] [PubMed] [Google Scholar]
- Garsmeur O, Schable JC, Almeida A, Jourda C, D’Hont A, Freeling M (2014) Two evolutionarily distinct classes of paleopolyploidy. Mol Biol Evol 31: 448–454 [DOI] [PubMed] [Google Scholar]
- Gaut BS, Doebley JF (1997) DNA sequence evidence for the segmental allotetraploid origin of maize. Proc Natl Acad Sci U S A 94(13): 6809–6814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grell EH (1962) The dose effect of ma-l+ and ry+ on xanthine dehydrogenase activity in Drosophila melanogaster. Zeitschrift für Vererbungslehre 93: 371–377 [Google Scholar]
- Guo M, Birchler JA (1994) Trans-acting dosage effects on the expression of model gene systems in maize aneuploids. Science 266: 1999–2002 [DOI] [PubMed] [Google Scholar]
- Guo M, Davis D, Birchler JA (1996) Dosage effects on gene expression in a maize ploidy series. Genetics 142: 1349–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J, et al. (2018) Global impacts of chromosomal imbalance on gene expression in Arabidopsis and other taxa. Proc Natl Acad Sci U S A 115: E11321–E11330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Baker A, Bartel B, Linka N, Mullen RT, Reumann S, Zolman BK (2012) Plant peroxisomes: biogenesis and function. Plant Cell 24: 2279–2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Du Y, Zhao X, Jin W (2016) B chromosome contains active genes and impacts the transcription of A chromosomes in maize (Zea mays L.). BMC Plant Biol 16: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Tang L, Yu Y, Dairymple J, Lippman ZB, Xu C (2018) Control of flowering and inflorescence architecture in tomato by synergistic interactions between ALOG transcription factors. J Genet Genomics 45: 557–560 [DOI] [PubMed] [Google Scholar]
- Ionita-Laza I, Rogers AJ, Lange C, Raby BA, Lee C (2009) Genetic association analysis of copy-number variation (CNV) in human disease pathogenesis. Genomics 93: 22–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Wickett NJ, Ayyampalayam S, Chanderbali AS, Landherr L, Ralph PE, Tomsho LP, Hu Y, Liang H, Soltis PS, et al. (2011). Ancestral polyploidy in seed plants and angiosperms. Nature 473: 97-100 [DOI] [PubMed] [Google Scholar]
- Johnson AF, Hou J, Yang H, Shi X, Chen C, Islam MS, Ji T, Cheng J, Birchler JA (2020) Magnitude of modulation of gene expression in aneuploid maize depends on the extent of genomic imbalance. J Genet Genom 47: 93–103 [DOI] [PubMed] [Google Scholar]
- Kato A, Birchler JA (2006) Induction of tetraploid derivatives of maize inbred lines by nitrous oxide gas treatment. J Hered 97: 39–44 [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions Genome Biol 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov FA, Koonin EV (2004) A common framework for understanding the origin of genetic dominance and evolutionary fates of gene duplications. Trends Genet 20: 287–290 [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EA, Darrah LL, Coe EH (1996a) Genetic variation in dosage effects in maize aneuploids. Genome 39: 711–721 [DOI] [PubMed] [Google Scholar]
- Lee EA, Darrah LL, Coe EH (1996b) Dosage effects on morphological and quantitative traits in maize aneuploids. Genome 39: 898–908 [DOI] [PubMed] [Google Scholar]
- Lin M (1955) Chromosomal control of nuclear composition in maize. Chromosoma 7: 340–370 [DOI] [PubMed] [Google Scholar]
- Liu J, Van Eck J, Cong B, Tanksley SD (2002) A new class of regulatory genes underlying the cause of pear-shaped tomato fruit. Proc Natl Acad Sci U S A 99: 13302–13306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo K-L, Wang L-C, Chen I-J, Liu Y-C, Chung M-C, Lo W-S (2014) Transcriptional consequence and impaired gametogenesis with high-grade aneuploidy in Arabidopsis thaliana. PLoS One 9: e114617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loven J, Orlando DA, Sigova AA, Lin CY, Rahl PB, Burge CB, Levens DL, Lee TI, Young RA (2012) Revisiting global gene expression analysis. Cell 151: 476–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maere S, Bodt SD, Raes J, Casneuf T, Montagu MV, Kuiper M, de Peer YV (2005) Modeling gene and genome duplications in eukaryotes. Proc Natl Acad Sci U S A 102: 5454–5459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino T, McLysaght A (2010) Ohnologs in the human genome are dosage balanced and frequently associated with disease. Proc Natl Acad Sci U S A 107: 9270–9274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17: 10–12 [Google Scholar]
- Mi H, Muruganujan A, Thomas PD (2013) PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res 41: D377–D386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M, Zhang C, Chen ZJ (2012) Ploidy and hybridity effects on growth vigor and gene expression in Arabidopsis thaliana hybrids and their parents. G3 (Bethesda) 2: 505–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton KJ, Stern DB, Gabay-Laughnan S (2009) Mitochondria and chloroplasts. In Bennetzen J.L. and Hake S., eds, Handbook of Maize: Genetics and Genomics. Springer New York, New York, NY, pp 481–503 [Google Scholar]
- O’Brien SJ, Gethmann RC (1973) Segmental aneuploidy as a probe for structural genes in Drosophila: mitochondrial membrane enzymes. Genetics 75: 155–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp B, Pál C, Hurst LD (2003) Dosage sensitivity and the evolution of gene families in yeast. Nature 424: 194–197 [DOI] [PubMed] [Google Scholar]
- Phillips RL, Kleese RA, Wang SS (1971) The nucleolar organizer region of maize (Zea mays L.): Chromosomal site for DNA complementary to ribosomal RNA. Chromosoma 36: 79–88 [Google Scholar]
- Rabinow L, Nguyen-Huynh AT, Birchler JA (1991) A trans-acting regulatory gene that inversely affects the expression of the white, brown and scarlet loci in Drosophila. Genetics 129: 463–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randolph LF (1941) Genetic characteristics of the B chromosomes in maize. Genetics 26: 608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raznahan A , Parikshak NN, Chandran V, Blumenthal JD, Clasen LS, Alexander-Block F, Zinn AR, Wangsa D, Wise J, Murphy DGM, et al. (2018) Sex-chromosome dosage effects on gene expression in humans. Proc Natl Acad Sci U S A 115: 7398–7403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoades MM (1951) Duplicate genes in maize. Am Nat 821: 105–110 [Google Scholar]
- Robinson DO, Coate JE, Singh A, Hong L, Bush M, Doyle JJ, Roeder AHK (2018) Ploidy and size at multiple scales in the Arabidopsis sepal. Plant Cell 30: 2308–2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriquez-Leal D, Lemmon ZH, Man J, Bartlett ME, Lippman ZB (2017) Engineering quantitative trait variation for crop improvement by genome editing. Cell 171: 470–480 [DOI] [PubMed] [Google Scholar]
- Roman H. (1948) Directed fertilization in maize. Proc Natl Acad Sci U S A 34: 36–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnable JC, Springer NM, Freeling M (2011) Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss. Proc Natl Acad Sci U S A 108: 4069–4074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, et al. (2009) The B73 maize genome: complexity, diversity, and dynamics. Science 326: 1112-1115 [DOI] [PubMed] [Google Scholar]
- Seidman JG, Seidman C (2002) Transcription factor haploinsufficiency: when half a loaf is not enough. J Clin Invest 109: 451–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan WF, Auger DL (2008) Chromosome segmental dosage analysis of maize morphogenesis using B-A-A translocations. Genetics 180: 755–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi T, Rahmani RS, Gugger PF, Wang M, Li H, Zhang Y, Li Z, Wang Q, Yves VP, Marchal L, et al. (2020) Distinct expression and methylation patterns for genes with different fates following a single whole-genome duplication in flowering plants. Mol Biol Evol 37: 2394-2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Chen C, Yang H, Hou J, Ji T, Cheng J, Veitia RA and Birchler JA (2020) The gene balance hypothesis: epigenetics and dosage effects in plants. Methods in Molecular Biology (Clifton, NJ) 2093: 161-171 [DOI] [PubMed] [Google Scholar]
- Simillion C, Vandepoele K, Van Montagu MCE, Zabeau M, Van de Peer Y (2002) The hidden duplication past of Arabidopsis thaliana. Proc Natl Acad Sci U S A 99: 13627–13632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnott EW, Blakeslee AF (1922) Structural changes associated with factor mutations and with chromosome mutations in Datura. Proc Natl Acad Sci U S A 8: 17–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnott EW, Houghtaling H, Blakeslee AF (1934) The Comparative Anatomy of Extra-chromosomal Types in Datura stramonium. Carnegie Institution of Washington, Washington, DC [Google Scholar]
- Song MJ, Potter B, Doyle JJ, Coate JE (2020) Gene balance predicts transcriptional responses immediately following ploidy change in Arabidopsis thaliana. Plant Cell 32: 1434–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer NM, Anderson SN, Andorf CM, Ahern KR, Bai F, Barad O, Barbazuk WB, Bass HW, Baruch K, Ben-Zvi G, et al. (2018) The maize W22 genome provides a foundation for functional genomics and transposon biology. Nat Genet 50: 1282–1288 [DOI] [PubMed] [Google Scholar]
- Stahl Y, Grabowski S, Bleckmann A, Kuhnemuth R, Weldtkamp-Peters S, Pinto KG, Kirschner GK, Schmid JB, Wink RH, Hulsewede A, et al. (2013) Moderation of Arabidopsis root stemness by CLAVATA1 and ARABIDOPSIS CRINKLY4 receptor kinase complexes. Current Biology 23: 362–371 [DOI] [PubMed] [Google Scholar]
- Steinitz-Sears LM (1963) Chromosome studies in Arabidopsis thaliana. Genetics 48: 483–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Johnson AF, Donohue RC, Li J, Cheng J, Birchler JA (2013a) Dosage compensation and inverse effects in triple X metafemales of Drosophila. Proc Natl Acad Sci U S A 110: 7383–7388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Johnson AF, Li J, Lambdin AS, Cheng J, Birchler JA (2013b) Differential effect of aneuploidy on the X chromosome and genes with sex-biased expression in Drosophila. Proc Natl Acad Sci U S A 110: 16514–16519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanksley SD (1993) Mapping polygenes. Annu Rev Genet 27: 205–233 [DOI] [PubMed] [Google Scholar]
- Tasdighian S, Van Bel M, Li Z, Van de Peer Y, Carretero-Paulet L, Maere S (2017) Reciprocally retained genes in the angiosperm lineage show the hallmarks of dosage balance sensitivity. Plant Cell 29: 2766–2785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas BC, Pedersen B, Freeling M (2006) Following tetraploidy in an Arabidopsis ancestor, genes were removed preferentially from one homeolog leaving clusters enriched in dose-sensitive genes. Genome Res 16: 934–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A (2003) PANTHER: a library of protein families and subfamilies indexed by function. Genome Res 13: 2129–2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ and Amon A (2007) Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317: 916-924 [DOI] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L (2013) Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 31: 46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukaya H (2013) Does ploidy level directly control cell size? Counterevidence from Arabidopsis genetics. PLoS One 8: e83729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veitia RA (2002) Exploring the etiology of haploinsufficiency. BioEssays 24:175–184 [DOI] [PubMed]
- Veitia RA, Bottani S, Birchler JA (2008) Cellular reactions to gene dosage imbalance: genomic, transcriptomic and proteomic effects. Trends Genet 24: 390–397 [DOI] [PubMed] [Google Scholar]
- Veitia RA, Bottani S, Birchler JA (2013) Gene dosage effects: nonlinearities, genetic interactions, and dosage compensation. Trends Genet 29: 385–393 [DOI] [PubMed] [Google Scholar]
- Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE and Amon A (2008) Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science 322: 703-709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhouse MR, Schnable JC, Pedersen BS, Lyons E, Lisch D, Subramaniam S, Freeling M (2010) Following tetraploidy in maize, a short deletion mechanism removed genes preferentially from one of the two homologs. PLoS Biol 8: e1000409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Birchler JA (2012) Identification of Inverse Regulator-a (Inr-a) as synonymous with pre-mRNA cleavage complex II protein (Pcf11) in Drosophila. G3 2: 701–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Park SJ, Van Eck J, Lippman ZB (2016) Control of inflorescence architecture in tomato by BTB/POZ transcriptional regulators. Genes Dev 30: 2048–2061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Shi X, Chen C, Hou J, Ji T, Cheng J, Birchler JA (2021) Predominantly inverse modulation of gene expression in genomically unbalanced disomic haploid maize. Plant Cell 33: 1016–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H, Dogra Gray A, Auger DL, Birchler JA (2013) Genomic dosage effects on heterosis in triploid maize. Proc Natl Acad Sci U S A 110: 2665–2669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H, Kato A, Mooney B, Birchler JA (2011) Phenotypic and gene expression analyses of a ploidy series of maize inbred Oh43. Plant Mol Biol 75: 237–251 [DOI] [PubMed] [Google Scholar]
- Zhang X, Hong D, Ma S, Ward T, Ho M, Pattni R, Duren Z, Stankov A, Shrestha SB, Hallmayer J, et al. (2020) Integrated functional genomic analyses of Klinefelter and Turner syndromes reveal global network effects of altered X chromosome dosage. Proc Natl Acad Sci U S A 117: 4864-4873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng YZ, Carlson WR (1997) Further construction of proximal duplication stocks. Maize Newsletter 71: 37–39 [Google Scholar]
- Zhou P, Li Z, Magnusson E, Gomez Cano FA, Crisp PA, Noshay JM, Grotewold E, Hirsch CN, Briggs SP, Springer NM (2020) Meta gene regulatory networks in maize highlight functionally relevant regulatory interactions. Plant Cell 32: 1377–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.