

Abstract
Designing covalent inhibitors is increasingly important, although it remains challenging. Here, we present covalentizer, a computational pipeline for identifying irreversible inhibitors based on structures of targets with non-covalent binders. Through covalent docking of tailored focused libraries, we identify candidates that can bind covalently to a nearby cysteine while preserving the interactions of the original molecule. We found ∼11,000 cysteines proximal to a ligand across 8,386 complexes in the PDB. Of these, the protocol identified 1,553 structures with covalent predictions. In a prospective evaluation, five out of nine predicted covalent kinase inhibitors showed half-maximal inhibitory concentration (IC50) values between 155 nM and 4.5 μM. Application against an existing SARS-CoV Mpro reversible inhibitor led to an acrylamide inhibitor series with low micromolar IC50 values against SARS-CoV-2 Mpro. The docking was validated by 12 co-crystal structures. Together these examples hint at the vast number of covalent inhibitors accessible through our protocol.
Keywords: irreversible inhibitors, covalent inhibitors, covalent docking, computer-aided drug discovery, DOCKovalent, COVID-19, SARS-CoV-2, Mpro
Graphical abstract
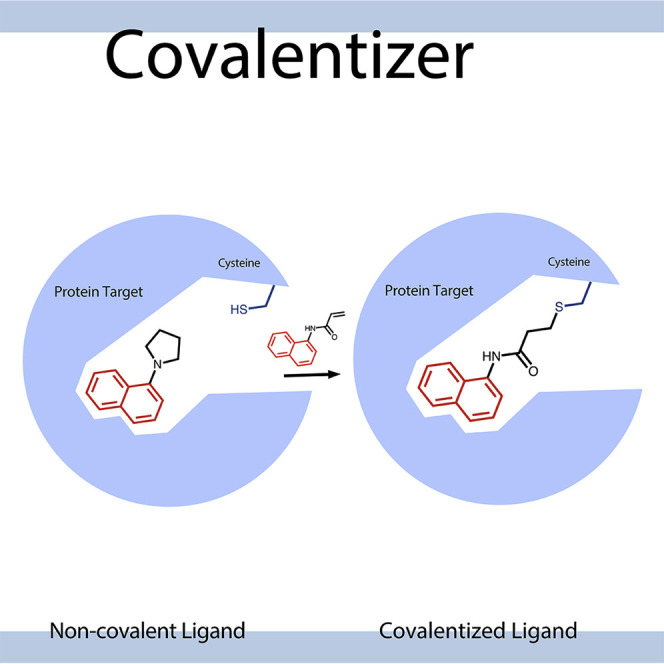
Designing covalent inhibitors is a challenging task. Covalentizer is a computational pipeline for identifying irreversible inhibitors based on known non-covalent binders. Within the PDB, Zaidman and Gehrtz et al. identified >1,553 structures with covalent predictions, leading to the discovery of covalent kinase inhibitors and an inhibitor of SARS-CoV-2 Mpro.
Introduction
Covalent irreversible inhibitors have become increasingly popular over the last decade as chemical probes and drugs. Most often these inhibitors target a cysteine residue to form the covalent bond. Several rationally designed irreversible inhibitors targeting cysteines were approved by the FDA in recent years, with notable examples, such as ibrutinib (Burger and Buggy, 2013), afatinib (Sequist et al., 2013), and osimertinib (Oxnard et al., 2016). Irreversible inhibitors offer a variety of advantages over non-covalent ones. These include: (1) prolonged residence time (Bradshaw et al., 2015), (2) an ability to compete with high-affinity natural substrates (Lonsdale and Ward, 2018; Michalczyk et al., 2008; Yun et al., 2008),(3) their improved selectivity when targeting non-conserved cysteine residues (Cohen et al., 2005; Ghosh et al., 2019), (4) targeting shallow binding sites (Sutanto et al., 2020), and (5) covalent binding can enable targeting of especially challenging targets such as the G12C oncogenic K-Ras mutation (Canon et al., 2019; Nnadi et al., 2018; Zeng et al., 2017).
Historically, most covalent inhibitors were designed by the addition of an electrophile to an already known reversible inhibitor that suitably binds next to a cysteine residue (Angst et al., 2020; Dubiella et al., 2015; Hagel et al., 2015; Vazquez-Rodriguez and Wright, 2019; Ward et al., 2015; Weisner et al., 2015). More recently, covalent inhibitors are also being discovered by empirical screening of covalent fragment libraries (Backus et al., 2016; Craven et al., 2018; Johansson et al., 2019; Kathman et al., 2014, 2015; Parker et al., 2017; Resnick et al., 2019) and by covalent virtual screening (Bensinger et al., 2019; Chowdhury et al., 2019; London et al., 2014; Nnadi et al., 2018; Rachman et al., 2019; Scarpino et al., 2018; Shraga et al., 2019; Toledo Warshaviak et al., 2014). While covalent fragment and virtual screening can potentially discover new scaffolds, the binding affinity of primary hits may be relatively low, and often require laborious medicinal chemistry to reach suitable potency.
Covalent derivatization of an already known reversible binder, can endow the compound with the added benefits of irreversible binding mentioned above, while retaining at least part of the binding energy from the reversible recognition, and possibly even have improved potency. Still, this approach is far from trivial. Three crucial questions have to be answered: (1) Which electrophilic moiety to use? (2) What is the optimal vector on the scaffold to attach through? (3) What linker, if any, would optimize the placement of the electrophile with respect to the binding mode of the scaffold and the position of the target residue? There are numerous possible answers for these questions. Furthermore, the covalentized (derivative containing the electrophile) version of the non-covalent inhibitor should be synthetically accessible. Therefore, tools that would be able to address this design problem algorithmically, would significantly simplify covalent inhibitor design and has the potential to discover many potent covalent binders for a large variety of targets.
Computational approaches to address this challenge are scarce. DUckCov (Rachman et al., 2019), a covalent virtual screening method, begins with non-covalent docking of a library of covalent compounds, while using pharmacophoric constraints for hydrogen bonds, as well as for the covalent warhead. This is followed by covalent docking of the ligands with the strongest non-covalent affinities. CovaDOTS (Hoffer et al., 2019) uses a set of synthetic schemes and available building blocks to create covalent analogs of existing non-covalent ligands, but was only assessed retrospectively. Cov_FB3D (Wei et al., 2020) constructs de novo covalent ligands and was retrospectively assessed on recapitulation on known covalent inhibitors.
Here, we present a computational pipeline to identify potential existing non-covalent binders for covalentization (creation of a covalent analog). Given a complex structure or model of a ligand in the vicinity of a cysteine residue, we elaborate the ligand or its substructures with various electrophiles. This ad hoc library of covalent analogs is covalently docked to the target protein and the original (non-covalent) structure is used as a filter to identify high-confidence covalent candidates. We applied this protocol—covalentizer—to the entire PDB to identify thousands of potential candidates amenable for irreversible inhibition, and made both the protocol and the database of pre-computed candidates publicly available to the community (https://covalentizer.weizmann.ac.il). We have prospectively synthesized and tested several predictions of various covalent kinase inhibitors proposed by the protocol and succeeded in five out of nine designs with half-maximal inhibitory concentration (IC50) values of between 155 nM and 4.2 μM.
In early February 2020, the COVID-19 pandemic started to spread globally (Wu et al., 2020; Zhu et al., 2020). We turned to the pre-compiled database of covalentizer results to look for possible candidate inhibitors for SARS-CoV-2 proteins. The search found a reversible small-molecule inhibitor designed against the main protease of the SARS-CoV virus (PDB: 3V3M; Jacobs et al., 2013), which has 96% sequence identity to the main protease of SARS-CoV-2, with a promising covalent prediction. We synthesized the prediction and validated irreversible binding to the SARS-CoV-2 main protease (Mpro). We further optimized the non-covalent affinity of the compound, resulting in improved analogs. Co-crystal structures confirmed the computational model. This example highlights the strength of our method—the design was already available, and enabled very rapid development. The database suggests that hundreds more such examples await testing.
Results
The covalentizer pipeline
For a given complex structure with a non-covalent ligand in the vicinity of a target cysteine residue, the pipeline (Figure 1 ) comprises four consecutive steps: fragmentation, electrophile diversification, covalent docking, and root-mean-square deviation (RMSD) filtering.
Figure 1.

An overview of the covalentizer computational protocol
The protocol comprises four consecutive steps.
(A) Fragmentation: the molecule is broken and divided into fragments (red arrows) using synthetically accessible bonds (Lewell et al., 1998). Murcko scaffolds (Bemis and Murcko, 1996) of the fragments (blue arrows) are also added to the list of fragments.
(B) Electrophilic diversification: for each substructure, a library of potential electrophilic analogs is generated, a few hundred compounds in size. We used four kinds of nitrogen-based electrophiles ranging in reactivity: vinyl sulfones, chloroacetamides, acrylamides, and propynamides. We also considered various linkers between the fragment and the electrophile.
(C) Docking: the target structure is then docked against its appropriate analog library using all available cysteine rotamers. Finally, RMSD calculation: for each docked compound, an RMSD is calculated between the MCS (maximal common substructure) of the reversible compound and the covalent analog found by covalentizer. We show examples of predictions with increasing RMSDs, for binders of (1) nitrate reductase from Ulva prolifera (PDB: 5YLY; You et al., 2018), (2) human mineralocorticoid receptor (PDB: 5HCV; Lotesta et al., 2016), and (3) human progesterone receptor (PDB: 1A28; Williams and Sigler, 1998).
Fragmentation
In this step, the ligand is broken down and divided into two parts via synthetically accessible bonds (Lewell et al., 1998). Doing this recursively, results in a list of substructures (Figure 1A). For each substructure, we augment the list with its corresponding Murcko scaffold (Bemis and Murcko, 1996), which is the naked ring system, without any decoration, to allow more exit vectors from which the electrophile can be added next. The motivation for this fragmentation step is 3-fold. First, as mentioned, fragmenting the molecule exposes new vectors on which to install the electrophile (see Figure 1C, example 2). Second, the additional constraint of forming the covalent bond might cause a slight shift to the molecule's binding mode from the original crystal structure. Such a shift may propagate and cause a steric clash between the protein and a ligand moiety distal to the electrophile. Since adding the covalent bond is expected to increase the overall potency, we “sacrifice” parts of the molecule to enable the addition of an electrophile. The final ranking of candidate covalent analogs relies on covalent docking, which is sensitive to sub-Å shifts. Hence, occasionally, a truncated version of the ligand will dock well, while the full ligand will not. Thus, including the substructures and their scaffolds maximizes the number of candidates. Since covalent docking accuracy was shown to deteriorate with ligand size and number of rotatable bonds (London et al., 2014), we filter the final list of substructures, to those with 8–25 heavy atoms and up to 5 rotatable bonds. We should note that, after successful docking of a fragment, additional parts of the reversible scaffold that do not interfere with electrophile installation can be added to the design.
Electrophile diversification
For each substructure or scaffold, we generate a library of potential electrophilic analogs, typically resulting in a few hundred analogs (Figure 1B). We consider four kinds of electrophiles ranging in reactivity—vinyl sulfonamides, chloroacetamides, acrylamides, and propynamides—that can all be installed in one step onto a free amine. We add these electrophiles to the substructures using simple connection rules, which, however, do not guarantee synthetic accessibility (see the STAR Methods for more details). We also consider various linkers between the fragment and the electrophile. In our application below, we considered either a methylene linker or various di-amine linkers (Figure S1).
Covalent docking
The structure of the complex is prepared for docking, using all available cysteine rotamers. We use DOCKovalent (London et al., 2014) to dock the electrophile library we described above against the protein (after removing the crystallographic ligand).
RMSD filtering
Compounds that are able to form a covalent bond with the target cysteine while still maintaining the same binding mode of the original reversible ligand are likely candidates for covalent analogs. To assess this, we evaluate the RMSD between each docking prediction and the crystallographic ligand. Due to the fact that RMSD is calculated between two sets of matching atoms, and the reversible ligand is different from the irreversible one, it was calculated based on the maximal common substructure (MCS) between the two molecules. Figure 1C exemplifies predictions with varying RMSDs. For a PDB-wide application of the pipeline, we focused on covalent analogs with a docking position of <1.5 Å RMSD from the crystallographic ligand.
Covalent kinase inhibitors benchmark
To benchmark the pipeline, we wanted to test whether it is able to find known covalent inhibitors, given only their reversible part as input. To achieve this, we used the kinase subset of a recently published covalent docking benchmark (Scarpino et al., 2018). This set included 35 kinase covalent inhibitor complex structures with acrylamides, chloroacetamides, or vinyl sulfonamides (after excluding seven inhibitors with uncommon electrophiles). To form the input for covalentizer, we removed the electrophiles while leaving only a free amine. For substituted acrylamides we removed β-substitutions as well. In parallel, we covalently docked the original non-fragmented covalent inhibitors to compare the performance of covalentizer with that of regular covalent docking.
Out of the 35 structures, the pipeline identified the crystallographic covalent inhibitor in 14 (40%) of the cases, with a threshold of 1.5 Å MCS-RMSD (Figure 2 ; Data S1A). In the rest of the cases no accurate docking poses were identified. When docking the original covalent molecules only 3 (8.6%) of them show an RMSD of less than 1.5 Å from the crystallographic pose. These results suggest that covalentizer is able to rediscover a substantial portion of known covalent inhibitors, but at the same time emphasize the fact that the success of our protocol is dependent on the performance of the docking program, which is limited.
Figure 2.

Covalentizer successfully recapitulates known covalent kinase inhibitors
Examples of covalent kinase inhibitors (green) for which covalentizer was able to find a substructure match (magenta) under the 1.5 Å threshold.
(A) ERK2, PDB: 4ZZO (Ward et al., 2015).
(B) EphB3, PDB: 5L6P (Kung et al., 2016).
(C) EGFR (T790M), PDB: 4I24 (Gajiwala et al., 2013).
(D) JAK3, PDB: 5TOZ (Telliez et al., 2016). The electrophiles span acrylamides (A, D), a substituted acrylamide (C) and chloroacetamide (B).
Covalentizing the PDB
Encouraged by the results in recapitulating known covalent kinase inhibitors we aimed to apply our protocol to the entire PDB. We started from the set of all the protein-small molecule X-ray structures (<3.0 Å resolution) that contained a small molecule with a molecular weight greater than 300 Da, and no DNA/RNA chains. We did not apply a maximum molecular weight cutoff, since the fragmentation process will address larger ligands. As of the date of the search (July 4, 2019) this resulted in 44,990 structures. We filtered these to structures in which a ligand has one of its atoms within 6 Å of the sulfur atom of a free cysteine residue. Disulfides or covalently modified cysteines were excluded. After applying this filter, we ended up with 8,386 such structures, and ∼11,000 cysteines.
These structures, which constitute the target space for our protocol, contain substantial redundancy. Clustering them with a threshold of 90% sequence identity, results in 2,227 representatives; 38% of the structures are of human proteins and the rest span many other organisms, including rodents, bacteria, and yeast. They also span seven different enzyme classes, with the most prevalent being transferases (41.4%). A total of 928 structures (11.1% of the entire dataset) are kinases. These ∼8,400 proteins contain 3,673 different ligands, each binding next to a cysteine (Data S1B). The ligand that is most abundant in this database is flavin-adenine dinucleotide occurring in 504 structures, whereas 3,058 ligands (83% of the compounds) occur only in a single structure. The most common ligands were nucleotide or nucleotide-like molecules.
After running the aforementioned algorithm against the ∼8,400 structures that passed our filtering (Figure 3 A), 1,553 structures produced at least one candidate below the 1.5 Å RMSD cutoff. These structures represent roughly 380 proteins (representative set at 95% sequence identity). A total of 1,051 structures are of human proteins, 338 are structures of kinases. Eighty of the structures had produced a covalent analog prediction that was docked <0.5 Å from the original ligand, representing very high-confidence candidates (Figure 3B; Data S1C). The distribution of selected electrophiles is almost uniform (Figure 3C). All of the predictions are made available through a public website (https://covalentizer.weizmann.ac.il), which is automatically updated weekly with new PDB entries.
Figure 3.
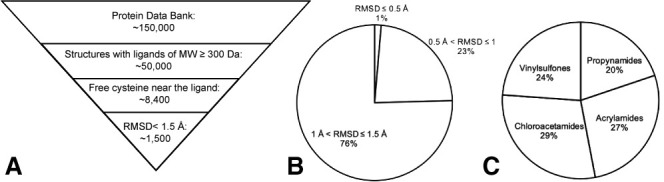
PDB-wide application of covalentizer identifies candidate irreversible inhibitors for more than 1,500 structures
(A) We filtered the PDB for structures that had only protein chains (no DNA/RNA), and contained a small molecule of at least 300 Da. This threshold was set to ensure some minimal initial fit/binding affinity to the target, as well as to filter out non-ligand small molecules, such as crystallization reagents. We used a PyMOL-based script to filter only the structures in which at least one ligand atom is <6 Å away from the sulfur atom of a cysteine residue. This cysteine also has to be free (no disulfide or other covalent modifications). After running the covalentizer protocol and filtering only for results with <1.5 Å RMSD of the MCS between the reversible ligand and the covalent analog generated by covalentizer, there were 1,553 structures for which at least one such prediction was obtained.
(B) The top 1% of results have an RMSD under 0.5 Å; 23% are between 0.5 and 1 Å, and 76% are between 1 and 1.5 Å.
(C) The distribution of the four electrophiles used is balanced, with 29% chloroacetamides, 27% acrylamides, 24% vinylsulfones, and 20% propynamides.
Exploring additional linkers
As mentioned above, the entire database was processed using direct attachments of the electrophiles to atoms of the substructures, as well as with a methylene linker. The use of longer and more diverse linkers for the addition of an electrophile would allow the targeting of cysteines further from the ligand, thus increasing the available target space, as well as diversifying the introduced chemistry. To investigate this further, we searched the covalent inhibitor discovery literature (Caldwell et al., 2019; Engel et al., 2015; Liang et al., 2017; Shi et al., 2019) for the most common di-amine linkers used in the last decade, which led to the selection of 9 aromatic linkers and 17 aliphatic linkers (Figure S1). Since including all of these linkers increases the computational demands of the pipeline, we restricted its application to the subset of liganded kinase structures in the PDB. Since these linkers can enable ligands to reach further cysteines at extended distances, the search criteria was extended to a distance of up to 10 Å from the ligand (instead of 6 Å, previously).
The final subset includes 1,880 PDB structures that contain a Cys residue of up to 10 Å away from one of 1,398 various ligands. The size of the custom-made libraries of electrophilic analogs for a particular reversible ligand, containing these linkers, now extends to a few thousand compounds. Overall, we generated in silico over 3 million electrophilic compounds with di-amine linkers for the kinase subset. The results show candidates of <1.5 Å MCS-RMSD between the original reversible ligand and the electrophilic candidate for 411 protein structures. A total of 186 of these structures (45%) was not found in the previous run, showing the potential of using these more sophisticated linkers to reach farther cysteines and to covalentize more ligands. Out of these 186, some target new cysteines that are further than 6 Å from the ligand, while others are due to better docking of the fragments to the same cysteine made possible by the new linkers.
Covalent inhibitors found for various kinases
Kinase inhibitors comprise 22% of the covalentizer results. We selected a subset of these for prospective validation. We chose the candidates based on three features: (1) low RMSD relative to the parent reversible ligand; (2) the addition of the electrophile is not predicted to interfere with the kinase hinge binding region; and (3) ease of synthesis. This required manual inspection of pre-selected low RMSD results. Overall, we made and tested nine compounds (Figure 4 ) targeting five different kinases. In some cases, addition of the electrophile required removal of large parts of the parent reversible ligand (Figure S2). The compounds were each tested in a kinase activity assay against the target kinase in the structure from which the covalentizer result was derived. The assay was performed at ATP concentration equal to the Km of the kinase in question, with a 2 h pre-incubation of the inhibitor at 25°C.
Figure 4.

Prospective prediction identifies irreversible kinase inhibitors
(A) Chemical structures and in vitro kinase activity assay IC50 values for nine prospective covalentizer predictions. See Figure S2 for the parent compounds, pose predictions, and RMSD values.
(B) Dose-response curves for each of the nine compounds. Each compound was tested against its corresponding target kinase. For compound 1, n = 3; for compounds 2, 4, 5, and 9, n = 2; error bars represent standard deviation.
(C) Deconvoluted mass spectra obtained by intact protein LC/MS of recombinant ERK2 (10 μM) incubated with 100 μM of 1 or 2 for 3 h at room temperature, shows full irreversible binding by both compounds.
Four of the nine compounds did not show inhibition under the assay conditions (IC50 > 10 μM). Three compounds targeting ERK2 showed IC50 values of 2.9–4.52 μM. The remaining covalentizer hits included a 2.01 μM inhibitor (3) of FGFR4 derived from the non-selective kinase inhibitor ponatinib, and a 155 nM inhibitor (4) of GSK3β.
We focused on two of the ERK2 inhibitors (1 and 2) for further characterization. We assessed their irreversible binding to ERK2 by intact protein mass spectrometry (10 μM ERK2, 100 μM compound, 3 h incubation at room temperature). The expected protein-compound adducts were detected (100% and 85% labeling, respectively; peak-to-peak Δm 265–270 Da for both compounds; Figure 4B) with no additional adducts derived from multiple reactions, highlighting the moderate reactivity of the designed α-chloroacetamides. Even at much more stringent conditions of 1:1 molar ratio and 1 h at room temperature we still see 25% and 33% labeling with 1 and 2, respectively (Figure S3A). To validate that the labeling site corresponds to the covalentizer results we performed liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis (Figure S3C; Data S2) which identified the computationally targeted Cys166 as the irreversible binding site. We also synthesized non-covalent acetylated versions of 1 and 2 (1-Ac and 2-Ac; Figure S3B). While 2-Ac lost all activity, 1-Ac actually retained similar potency to 1 (IC50 = 2.79 μM; Figure S3B).
A covalent SARS-CoV-2 main protease inhibitor
When the COVID-19 pandemic started spreading around the globe, we searched the database for relevant entries. At the time there were no structures of SARS-CoV-2 proteins. However, we found a candidate for the SARS-CoV Mpro protease, based on a reversible inhibitor ML188 (IC50 = 4.8 ± 0.8 μM, racemate; 1.5 ± 0.3 μM, (R)-enantiomer) of the SARS-CoV main protease (PDB: 3V3M; Jacobs et al., 2013; Figure 5 A). Upon the release of the first structure of the new SARS-CoV-2 Mpro protease (PDB: 6LU7; Jin et al., 2020) we noticed that the active site is nearly identical to that of SARS-CoV. The entire protein is highly conserved with 96% sequence identity. This made us confident that the reversible ligand would have had similar binding mode when bound to the SARS-CoV-2 Mpro. Later indeed, a new crystal structure of ML188 bound to SARS-CoV-2 Mpro (Lockbaum et al., 2021) validated this assumption.
Figure 5.

Computational prediction and experimental validation of an irreversible SARS-CoV-2 Mpro inhibitor
(A) The covalentizer prediction of 10 (magenta) overlaid on the non-covalent compound it is based on (ML188 (Jacobs et al., 2013); green; PDB: 3V3M). The protocol suggested to substitute the furanyl moiety of ML188 with an acrylamide to bind the catalytic cysteine. The RMSD between the covalent fragment and the original reversible inhibitor is 0.65 Å.
(B) The crystal structure of 12, one of the covalent analogs of 10 (PDB: 5RH5; cyan) overlaid on ML188 (green).
(C) Overlay of all the 12 crystal structures of compound 10 analogs, all exhibiting the same predicted binding mode. PDB: 5RGT, 5RH5, 5RH6, 5RH7, 5RH9, 5RL0, 5RL1, 5RL2, 5RL3, 5RL4, 5RL5, and 7NW2. For individual structures see Data S3F.
(D) The chemical structures of ML188 and 10.
(E) Chemical structure of Ugi compounds exploring the S3 pocket, with the R group that is shown in the crystal structure in (B).
(F) Deconvoluted mass spectra obtained by intact protein LC/MS of recombinant SARS-CoV-2 Mpro 2 μM incubated with 2–200 μM 10 for 1.5 h at room temperature. We should note we did not detect multiple labeling events by this compound.
(G) Further analogs of 10 with their associated biochemical potencies.
(H) The dose-response curves for the seven compounds shown in (G) (n = 2, error bars represent standard deviation).
We re-synthesized and tested racemic ML188 against SARS-CoV-2 Mpro, which showed an IC50 of 3.14 μM (Figure S4A), similar to what has been reported for SARS-CoV. ML188 was synthesized using the Ugi four-component reaction (4-CR), and the covalent prediction was easily accessible by replacing one reactant (2-furoic acid to acrylic acid) to give 10, synthesized and isolated as the racemate (Figure 5D). We initially assessed irreversible binding of 10 toward recombinant SARS-CoV-2 Mpro using intact protein mass spectrometry (2 μM protein, 1.5 h incubation with electrophile at 25°C; Figure 5F). The expected adduct was detected with 19% labeling at 2 μM compound, and up to 88% labeling at 200 μM compound (Figure 5F).
Despite the irreversible binding, this initial compound did not show strong inhibition in a fluorescence-based enzymatic assay (IC50 > 99 μM, 13% inhibition at 20 μM; 15 min pre-incubation; Figure 5H). However, it was a promising starting point for additional optimization. Due to the modular nature of the Ugi 4-CR procedure, it was possible to synthesize and test large libraries of analogs by systematically varying each reactant to target different pockets. We designed those libraries based on computational modeling of in-silico-generated Ugi products, as well as an exhaustive screen of commercially available isocyanides (Data S3). A few of the early combinatorial synthetic results, which had low biochemical potency (comparable with our starting Ugi compound), allowed for crystallographic analysis in the presence of Mpro. In these cases, the expected binding mode was recapitulated experimentally and showed low deviation from the non-covalent starting point (Figures 5B and 5C; Data S3F), thus proving the covalentizer prediction to be correct. In all crystal structures, the electrophile formed the expected covalent bond with the catalytic cysteine residue.
To optimize 10, we have made and tested close to 140 analogs (Data S3; Figures 5 and S5), exploring all three components of the Ugi reaction while keeping the acrylamide fixed. We explored a variety of replacements for the initial p-tert-butylphenyl motif protruding into the S2 pocket (Figure 5), most of them did not result in improved potency (Figure S5C). Similarly, independent optimization of binding to the S1 pocket only led to the identification of one beneficial change (23, IC50 = 65.58 μM), with a meta chloro-substitution of the pyridine (Figure S5A). Other substituents (-Br, -OMe, -OEt, -CF2CH3) led to inactive compounds.
Beyond further optimization of the S1 and S2 pocket binding it was clear that extension of the ligand toward the S3 and S4 pockets should prove fruitful. For example, a reversible covalent α-ketoamide inhibitor (Zhang et al., 2020) (biochemical IC50 = 0.67 ± 0.18 μM) probes the S3/4 region with an additional hydrogen bond to the backbone of Glu166. In a large-scale fragment screen, numerous fragments were able to bind in these pockets (Douangamath et al., 2020). In this case, we exhaustively synthesized analogs of 10, using 34 available isocyanides. Starting from 10, simple alkyl chain extension resulted in compounds with improved potency (Figure 5D). Namely compound 11, harboring a phenethylamide motif, was particularly potent with an IC50 of 2.95 μM (Figure 5G) and Kinact/Ki of 18.4 M−1 s−1 (Figure S4C). After chiral chromatography (see Methods S1) of the molecule, one enantiomer showed slightly better inhibition (IC50 = 2.86 μM), while the other enantiomer was inactive (IC50 = 86.32 μM; Figure S4D). A co-crystal structure of 11 complexed with Mpro showed that the fluorophenyl binds through hydrophobic interactions with Met165 and Gln189, explaining its improved potency (Figure 5C; Data S3F).
It appears that relief of steric strain around the amide nitrogen also plays a part, since change to a methyl amide (in 12, relative to 10) also resulted in increased potency. Opposed to our initial assumption of independent optimization of S1–3 pocket binding, the combination of beneficial structural motifs in a third generation of Ugi products led to inhibitors with diminished potency compared with 11 (see Figure S5B; Data S3). One explanation for this behavior is the high plasticity of Mpro, leading to induced fit effects.
Removal of the furanyl in ML188 and replacement with an electrophile in 10 initially led to a loss in potency, which in this case was overcome by optimization of the non-covalent affinity in the S3 region to give compound 11. Re-installing the furanyl ring in combination with the S3-optimized phenethylamide motif led to compound 17 with a similar IC50 (2.72 μM; Figure S4B), suggesting that the marked improvement of this side chain is particular to the covalently bound conformation.
In conclusion, we successfully executed a mode of action change toward irreversible targeting of the catalytic cysteine residue in Mpro, which may have improved activity in cells as well as long-term strategic benefits to safeguard against viral evolution.
Discussion
Designing new covalent inhibitors is challenging. Here, we leveraged the subset of protein targets for which a structure of a known binder is available, to computationally enumerate and evaluate exhaustive sets of covalent derivatives. Automating the protocol allowed us to apply it to the entire PDB and assess the applicability of this approach. Prospective testing against six real-world targets demonstrated that irreversible ligands can be reached with little synthesis, and structures validated the binding-pose prediction.
A main advantage of our work is the wide exploration of X-ray structures, which produced an extensive list of candidates waiting to be explored. This allowed us to quickly find a promising lead series against SARS-CoV-2 Mpro. This prediction, which was based on a historic non-covalent SARS-CoV Mpro inhibitor (Jacobs et al., 2013), was pre-calculated and ready for synthesis at a moment's notice. We have made thousands of such predictions available through a public website (https://covalentizer.weizmann.ac.il) that updates weekly with the release of new structures to the PDB. It also allows covalentizing of user uploaded structures. The covalentizer was also integrated into PDBe-KB (Protein Data Bank in Europe-Knowledge Base) (PDBe-KB consortium, 2020), a community-driven resource collating functional annotations and predictions for structure data in the PDB archive. PDBe-KB allows understanding covalentizer data in the context of results from a diverse group of related bioinformatics resources and research teams. We believe these would enable wide application and experimental testing of new covalent inhibitors.
Despite the success of our protocol, several caveats remain. First is the fact that currently the protocol does not take into account the synthetic feasibility of the proposed designs. When selecting candidates for prospective evaluation, we found that some of the molecules required complicated synthesis. Incorporating into our pipeline a strategy, such as DOTS (Hoffer et al., 2018, 2019), other retrosynthesis algorithms (Delépine et al., 2018; Law et al., 2009; Watson et al., 2019) or even the use of synthetic feasibility scores (Ertl and Schuffenhauer, 2009; Huang et al., 2011; Podolyan et al., 2010), can improve the quality of proposed candidates in the future.
Another point for improvement is the relatively weak potency of our prospective designs in comparison with their parent compounds (Figure S2). One likely explanation for these lower affinities is the removal of non-covalent affinity elements, which are not sufficiently compensated by the gains from covalent bond formation. For example, in compound 2 (derived from PDB: 4QTA) more than 350 Da of the original compound (Chaikuad et al., 2014) is removed (Figure S2), resulting in three orders of magnitude loss in potency. However, the remaining covalent fragment, despite its very small size, still shows substantial inhibition of ERK2. Another example is compound 1, its parent compound (PDB: 4QP9) has an IC50 of 71 nM (Burdick et al., 2015); however, the propyl-pyrazole group we have omitted to accommodate the electrophile (Figure S2) improved the parent reversible binder by more than 150-fold. Even omitting only the propyl moiety from the parent compound already decreased its potency by ∼5-fold. A third example is compound 8, which did not show inhibition, while its parent compound (PDB: 5IHA) has an IC50 of 9 nM (Touré et al., 2016). Although keeping the hinge binding motif, we omitted groups from both ends, one due to clash with the added electrophile, and the other due to synthetic difficulty. While the same fragment was not tested by the authors of the parent compound paper, they did test a matched pair, showing that a single N to C substitution in the pyridine ring that we removed caused a decrease of 50-fold in potency, underscoring pyridine's importance.
The acetyl versions of 1 and 2 we have made and tested corroborate this hypothesis. Both non-covalent compounds showed marked reduction in potency compared with the parent compound. 2-Ac became inactive and 1-Ac lost more than 40-fold potency, suggesting that the decrease in potency compared with the parent compound originated from the trimming of the propyl-pyrazole group rather than from the covalentization process itself. Finally, the loss of a hydrogen bond between the Mpro backbone NH of Gly143 and the furanyl oxygen of ML188 (PDB: 3V3M), due to the replacement with the acrylamide, decreased potency, under our assay conditions of 15 min pre-incubation, by more than 30-fold. These results suggest that a careful examination of the binding energy contribution is required for the parts that are omitted to accommodate the new electrophile.
However, as we saw for both ERK2 (1 versus 5) and Mpro (10 versus 11), improving reversible recognition is able to improve potency, and even to surpass the parent compound in the case of 11. For this series we believe that further structure-based optimization of binding to the S3 pocket, including H bonding to Glu166, can pave the way for sub-micromolar Ugi-type covalent Mpro inhibitors. Thus, in many cases where irreversible binding is needed, to avoid resistance, for instance, or improve selectivity, our protocol can provide a promising starting point for optimization.
Another possible explanation for the relatively low affinity of the irreversible binders are slight inaccuracies in the covalent warhead positioning, which results in sub-optimal covalent bond formation. Perhaps due to the fact that the docking program does not take into account the actual formation of the covalent bond, and ignores, for instance, the transition state energy of the rate-determining step of the covalent bond formation, but rather evaluates the binding energy of the adduct. The performance of covalentizer is limited by the performance of the docking program that it uses. Having said that, the fragmentation technique does allow to find solutions that might not have been found using the full molecules (Data S1A). An obvious way to improve covalentizer is to improve the covalent docking program, or to replace it with a better performing one. Several programs have been reported retrospectively to perform better than DOCKovalent (Scarpino et al., 2018) and thus may be used to replace it in the protocol. We should also note that, while some successful covalent virtual screening prospective results are reported, these were typically a result of testing several predictions per target, whereas here we tested a very limited number of predictions per protein.
For the above reasons, we suspect that only a subset of the results that passed the 1.5 Å threshold are worth pursuing, depending on the position on which the electrophile is installed and whether the addition is mutually exclusive with an important part of the reversible recognition element of the original molecule. For example, while selecting kinase inhibitors, we only considered predictions for which the electrophile can be added without removing the hinge binding region, which is known to be crucial for binding (Xing et al., 2015). Another important aspect in selecting candidates for testing is the complementation of the docked fragment to include as much as possible of the reversible scaffold that is not mutually exclusive with the electrophile. The importance of this is demonstrated by the cases of 5 and 1, as well as in the case of 10, for which only a fragment of the molecule docked well but we complemented it to the full compound. In the future such considerations may also be automated.
It is interesting to note the similar activity of a methylene-chloroacetamide (1), compared with its acrylamide analog (6). Such electrophile replacements can be very useful in rational design of irreversible inhibitors, especially if they prove to work across various scaffolds. Geometrically, the additional methylene before the chloroacetamide makes the distance from the ring to the thiol similar to that of the acrylamide (Figure S2). In terms of reactivity, however, the acrylamide, conjugated to the azaindole is activated (Flanagan et al., 2014) and thus is likely closer in reactivity to the chloroacetamide. Indeed, a methylene linker would be the minimal linker element required to insulate against π-conjugation, allowing easier prediction of intrinsic reactivity. No-linker designs connected to extended π-systems, such as heteroarenes, often exhibit a range of intrinsic reactivities (Flanagan et al., 2014; Resnick et al., 2019), which remain challenging to predict computationally (Lonsdale et al., 2017; Palazzesi et al., 2019, 2020) and thus require careful evaluation.
Many additional designs remain to be discovered beyond the ones we made available through the covalentizer server. New electrophiles and linkers, which will enable new geometric trajectories between the cysteine and the molecule, can considerably expand the design space. We tested this idea computationally using a library of linkers curated from the literature (Figure S1), on a subset of kinases from our database, showing an increase in the number of structures that can be covalentized. Recent covalent “warheads,” including reversible covalent warheads, such as cyanoacrylamides (Bandyopadhyay and Gao, 2016), and chlorofluoroacetamides (Shindo et al., 2019), became available, both for cysteine residues (Backus, 2019), but also for other amino acids (Martín-Gago and Olsen, 2019; Ray and Murkin, 2019; Shannon and Weerapana, 2015). These can be incorporated with little effort into the covalentizer pipeline. Since cysteine is one of the least-abundant natural amino acids, additional covalent chemistries will substantially expand the number of ligands that can be potentially addressed.
In summary, we show that, using covalent docking, we were able to make irreversible analogs of ligands for which a complex structure is available. We made our discoveries public in the form of a database of the results we obtained by running our protocol on the entire PDB, which is automatically updated weekly with newly released entries, as well as a web-tool for applying the protocol on new targets given by users. Using the protocol, we discovered several covalent kinase inhibitors and optimized a potent covalent COVID-19 protease inhibitor, with a low-cost, modular, and fast synthesis. We hope our results will encourage researchers to apply covalent inhibitors for a wide range of targets.
Significance
Covalent inhibitors have increased in popularity over the past decade, owing to several advantages over non-covalent ligands. However, their discovery and rational design is still challenging. By developing the covalentizer we show that many non-covalent small-molecule binders for which a structure is available, with a variety of proteins, have the potential to be transformed into covalent binders. We prospectively validated our protocol against various kinases, as well as in a rapid drug discovery campaign against the SARS-CoV-2 main protease. These results point to many other potential targets that may be targeted by a covalent analog of a known ligand, potentially shortening the time and effort in finding future covalent inhibitors. Our web server https://covalentizer.weizmann.ac.il/ allows users with little to no computational background to use our predictions for covalent analogs, as well as using the covalentizer on their own models. We hope that our results and server will facilitate a broad use of covalent inhibitors.
Consortia
Covid-Moonshot Consortium contributors: Aarif Shaikh, Aaron Morris, Adam P. Smalley, Ailsa Powell, Alessandro Contini, Alex Dias, Alice Douangamath, Aline M. Nakamura, Alpha Lee, Amir Ben-Shmuel, Anastassia L. Kantsadi, Andre S. Godoy, Andrew Jajack, Andrew Thompson, Anna Carbery, Annette von Delft, Anthony Aimon, Anthony Tumber, Arathy Jose, Assa Sittner, Austin Clyde, Benjamin Perry, Boaz Politi, Boris Kovar, Bruce A. Lefker, C. David Owen, Carina Gileadi, Charles J. Eyermann, Charlie Weatherall, Charline Giroud, Chris Schofield, Claire Strain-Damerell, Conor Francis Wild, Daniel Guest, Daniel Zaidman, Daren Fearon, David L. Dotson, Demetri Moustakas, Dominic Rufa, Ed J. Griffen, Efrat Resnick, Einat B. Vitner, Elad Bar-David, Elizabeth M. MacLean, Emma Cattermole, Eric Jnoff, Finny S. Varghese, Frank von Delft, Gabriela D. Noske, Galit Cohen, Garrett M. Morris, Gijs J. Overheul, Glaucius Oliva, Gregory R. Bowman, Hadas Tamir, Hadeer Zidane, Hagit Achdout, Haim Barr, Haim Levy, Halina Mikolajek, Hannah Bruce Macdonald, Holly Foster, Ioannis Vakonakis, Itai Glinert, J. L. Kiappes, Jag Heer, Jakir Pinjari, James Bennett, Jason Cole, Jenny C. Taylor, Jin Pan, Jiye Shi, John D. Chodera, John Spencer, Jose Brandao Neto, Joseph E. Coffland, Juliane Brun, Khriesto Shurrush, Lauren Reid, Leonardo J. Solmesky, Lisa Cox, Lizbé Koekemoer, Lori Ferrins, Louise Dunnett, Maneesh Pingle, Marian Gorichko, Mariana Vaschetto, Mark Calmiano, Martin Walsh, Matteo P. Ferla, Matthew C. Robinson, Matthew F.D. Hurley, Matthew Wittmann, Melissa L. Boby, Melody Jane Morwitzer, Michelle Hill, Mihaela D. Smilova, Mike Fairhead, Milan Cvitkovic, Nathan David Wright, Nicole Zitzmann, Nir London, Nir Paran, Noam Erez, Oleg Fedorov, Oleg Michurin, Pascal Miesen, Paul Gehrtz, Peter K. Eastman, Peter W. Kenny, Petra Lukacik, Rachael E. Tennant, Rachael Skyner, Rafaela S. Fernandes, Ralph P. Robinson, Rambabu N. Reddi, Reut Puni, Richard Foster, Robert Glen, Ronald P. van Rij, Ronen Gabizon, Ruby Pai, Sam Horrell, Sarma BVNBS, Sharon Melamed, Shay Weiss, Shirly Duberstein, St Patrick Reid, Storm-Hassell-Hart, Susana Tomásio, Tatiana Matviiuk, Tika R. Malla, Tim Dudgeon, Tobias John, Tobias Krojer, Tomer Israely, Tyler Gorrie-Stone, Victor L. Rangel, Victor O. Gawriljuk, Vincent A. Voelz, Vishwanath Swamy, Vladas Oleinikovas, Vladimír Pšenák, Walter Ward, Warren Thompson, Warren Thompson, Willam McCorkindale, William G. Glass, Yfat Yahalom-Ronen.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| E. Coli BL21(DE3)-R3-pRARE | Walsh Lab | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| pGEX-6P-1-Mpro plasmid | Walsh Lab | N/A |
| Deposited Data | ||
| SARS-CoV-2 Mpro with LON-WEI-b8d98729-23 (Mpro-x2540) | This paper | PDB: 5RGT |
| SARS-CoV-2 Mpro with LON-WEI-b8d98729-43 (Mpro-x2694) | This paper | PDB: 5RH5 |
| SARS-CoV-2 Mpro with LON-WEI-b8d98729-18 (Mpro-x2703) | This paper | PDB: 5RH6 |
| SARS-CoV-2 Mpro with LON-WEI-b8d98729-20 (Mpro-x2705) | This paper | PDB: 5RH7 |
| SARS-CoV-2 Mpro with LON-WEI-b8d98729-8 (Mpro-x2776) | This paper | PDB: 5RH9 |
| SARS-CoV-2 Mpro with LON-WEI-adc59df6-2 (Mpro-x3110) | This paper | PDB: 5RL0 |
| SARS-CoV-2 Mpro with LON-WEI-adc59df6-27 (Mpro-x3113) | This paper | PDB: 5RL1 |
| SARS-CoV-2 Mpro with LON-WEI-adc59df6-26 (Mpro-x3115) | This paper | PDB: 5RL2 |
| SARS-CoV-2 Mpro with LON-WEI-adc59df6-39 (Mpro-x3117) | This paper | PDB: 5RL3 |
| SARS-CoV-2 Mpro with LON-WEI-adc59df6-3 (Mpro-x3124) | This paper | PDB: 5RL4 |
| SARS-CoV-2 Mpro with LON-WEI-adc59df6-30 (Mpro-x3359) | This paper | PDB: 5RL5 |
| SARS-CoV-2 Mpro with LON-WEI-adc59df6-47 (Mpro-P0047) | This paper | PDB: 7NW2 |
| SARS-CoV Mpro with ML188 | (Jacobs et al., 2013) | PDB: 3V3M |
| Nitrate reductase from Ulva prolifera | (You et al., 2018) | PDB: 5YLY |
| Human mineralocorticoid receptor | (Lotesta et al., 2016) | PDB: 5HCV |
| Human progesterone receptor | (Williams and Sigler 1998) | PDB: 1A28 |
| ERK2 | (Ward et al., 2015) | PDB: 4ZZO |
| EphB3 | (Kung et al., 2016) | PDB: 5L6P |
| EGFR (T790M) | (Gajiwala et al., 2013) | PDB: 4I24 |
| JAK3 | (Telliez et al., 2016) | PDB: 5TOZ |
| Software and Algorithms | ||
| Covalentizer | London Lab |
https://covalentizer.weizmann.ac.il/ https://github.com/LondonLab/Covalentizer |
| PyMOL | Schrödinger | https://pymol.org/ |
| Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| DOCKovalent 3.7 | (London et al., 2014) | http://dock.compbio.ucsf.edu/DOCK3.7/ |
| Illustrator | Adobe | http://adobe.com/ |
| OpenBabel | OpenBabel | http://openbabel.org/wiki/Main_Page |
| RDKit version 2018.09.3 | RDKit | RDKit.org |
| Marvin 17.21.0 | ChemAxon | https://www.chemaxon.com |
| Rosetta modeling suite | RosettaCommons | https://www.rosettacommons.org/ |
| OpenLynx | Waters | https://www.waters.com/ |
| Other Items | ||
| Assay-ready plates (384 wells, low volume) | Greiner | 784900 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Nir London (nir.london@weizmann.ac.il)
Materials availability
Small molecules synthesized in this study can be custom synthesized following the synthetic procedures in the supplemental information. This study did not generate other new unique reagents.
Data and code availability
The code generated in this study is available at GitHub: https://covalentizer.weizmann.ac.il/
https://github.com/LondonLab/Covalentizer.
The Covalentizer database and webserver is available online: http://covalentizer.weizmann.ac.il/
Experimental model and subject details
Microbe strains
The pGEX-6P-1-Mpro plasmid was transformed into a competent E. coli expression cell line based on a multi-phage resistant BL21(DE3) derivative, containing the pRARE2 plasmid for improved expression of proteins that utilise rare E. coli tRNA genes (BL21(DE3)-R3-pRARE). Multiple transformant colonies were used to inoculate a starter culture supplemented with 100 μg/ml Carbenicillin. The culture was then grown to log phase for ∼8 h. Ten millilitres of the starter culture was used to inoculate one litre of auto induction medium supplemented with 10 ml of glycerol and 100 μg/ml carbenicillin. The cultures were grown at 37°C, 200 rpm for 5 h then switched to 18°C, 200 rpm for 10 h.
Method details
Programs and libraries
RDKit was used for 2D molecular handling, conformation generation and RMSD calculation. RDKit: Open-source cheminformatics; version 2018.09.3; RDKit.org. Marvin was used in the process of preparing the molecules for docking, Marvin 17.21.0, ChemAxon (https://www.chemaxon.com). OpenBabel (http:// openbabel.org/wiki/Main_Page) was used to switch between molecular file formats. DOCKovalent (London et al., 2014) was used for virtual covalent docking. The Covalentizer code is available at https://github.com/LondonLab/Covalentizer.
Curating target structures from the PDB
Using pymol scripts (The PyMOL Molecular Graphics System, Version 2.0.4 Schrödinger, LLC), we filtered only the structures that have a ligand in which one of its atoms is within 6 Å cysteine residue. We further filtered the list to include only cysteines with a free thiol group (defined as a sulfur atom that is only connected to the residue’s Cβ). By doing this, we discarded any disulfides, as well as cysteines that are already covalently attached to a ligand. We further removed any ligands which had more than one copy per chain in the structure, and ligands on which processing of the ligand’s SMILES failed.
Enumerating substructures for covalentization
Fragmentation and scaffold extraction was done using RDKit’s implementation of the Recap algorithm (Lewell et al., 1998) and the Murcko Scaffold (Bemis and Murcko, 1996) functionality respectively.
Covalentizing a substructure
For each substructure or scaffold, we generated a library of potential electrophilic analogs using SMARTS based reactions. The reaction rules were: 1. Adding an electrophile (including the nitrogen) to any non-substituted aromatic carbon, as well as all aliphatic carbons with one or two bonded atoms, excluding carbons which are already connected to nitrogen. 2. Adding an electrophile to a free amine, either primary or secondary. In this case the nitrogen is completed with the rest of the electrophile. The first rule will usually require more complicated synthesis, whereas the second rule, will allow to use the same ligand as a starting material for a nucleophilic substitution of the acyl form of the electrophile with the free amine.
Docking and RMSD calculation
RDKit and Marvin were used to create 250 conformations for each electrophilic analog. Covalent docking was done using DOCKovalent – a virtual screening program. We docked the appropriate analog library for each target, while saving 10 structures for each analog to increase the number of final candidates. When docking the larger linker based libraries we only used the top scoring structure for each analog, due to the large number of structures for analysis. Alternative rotamers for the cysteine residue were generated with pymol based scripts. DOCKovalent does not allow for protein flexibility, however, sampling the cysteine’s rotamers before docking does introduce some flexibility. Water molecules as well as any co-crystallised ligand were not taken into account during docking. We used RDKit to filter only for results with a MCS that has an RMSD of less than 1.5 Å to the original ligand. For this we used the FindMCS function with both FindMCSringMatchesRingOnly and completeRingsOnly attributes set to True.
Hardware and run time
Running Covalentizer on all suitable structures from the PDB was done using a PBS scheduler, on a computer cluster with 528 cores. The overall runtime was several days. Running Covalentizer on a single structure on a similar sized cluster takes only a few hours, and many structures can run in parallel.
Computational optimisation of the Mpro inhibitor
We used the RDKit reaction functionalities, as well as OpenBabel (http://openbabel.org/wiki/Main_Page) to prepare virtual libraries of analogs of compound 10. The Ugi reaction has three reactants: amine, isocyanide, aldehyde and carboxylic acid. The carboxylic acid is set constant to acrylic acid, since we didn’t want to change the electrophilic component. In the virtual libraries, we left it as the reversible furan moiety for convenience in modeling. We thus created three such libraries, each one by replacing one of the three other Ugi reactants with commercially available building blocks. Using RDKit, we generated up to 100 constrained conformations of each molecule, by fixing the conformation of three components as in the crystal structure, and changing only the conformation of the variable part. We then used the Rosetta modeling suite in order to choose the best conformation for each compound, when bound to the protease. For each molecule, we then defined this set of constrained conformations as an extra residue for Rosetta, and used Rosetta Packer (Kuhlman and Baker, 2000) to choose the best conformation, while allowing side-chain flexibility. Eventually, we chose analogs only for the amine and the isocyanide components, as the aldehyde component was highly optimised already. We chose 9 isocyanide replacements and 14 amine replacements (one of them was not based on docking). Most combinations of these components were made by Enamine and tested as part of the Covid-Moonshot effort (Chodera et al., 2020; Douangamath et al., 2020).
Intact protein LC/MS
The LC/MS runs were performed on a Waters ACQUITY UPLC class H instrument, in positive ion mode using electrospray ionization. UPLC separation used a C4 column (300 Å, 1.7 μm, 21 mm × 100 mm). The column was held at 40°C and the autosampler at 10°C. The mass data were collected on a Waters SQD2 detector with an m/z range of 2−3071.98. The desolvation temperature was 500°C with a flow rate of 1000 L/h. The voltages used were 0.69 kV for the capillary and 46 V for the cone. Raw data was processed using openLYNX and deconvoluted using MaxEnt.
2 μM recombinant Mpro was incubated with 2 - 200 μM for 90 minutes in 50 mM Tris pH 8 300 mM NaCl in room temperature. The column was held at 40°C and the autosampler at 10°C. Mobile solution A was 0.1% formic acid in water, and mobile phase B was 0.1% formic acid in acetonitrile. The run flow was 0.4 mL/min with gradient 20% B for 4 min, increasing linearly to 60% B for 2 min, holding at 60% B for 0.5 min, changing to 0% B in 0.5 min, and holding at 0% for 1 min. The mass data were collected at a range of 1000−2000 m/z. Raw data was processed deconvoluted using a 20000 - 60000 kDa window, 1 Da/channel resolution.
10 μM recombinant ERK2 was incubated with 100 μM for 3 hours in 50 mM Tris pH 8 300 mM NaCl in room temperature. 2 μl sample was diluted with 22 μl buffer + 6 μl 2.4% formic acid, and 10 μl were injected with the following gradient at 0.4 ml/min: 20% acetonitrile + 0.1% formic acid for 2 minutes, followed by a rise to 80% acetonitrile over 2.5 minutes, 0.5 minutes at 80% acetonitrile, fall back to 20% acetonitrile over 0.2 minutes, and 0.8 minutes at 20% acetonitrile. The mass data were collected at a range of 750-1750 m/z. Raw data was processed deconvoluted using a 37000-51000 kDa window, 1 Da/channel resolution.
LC/MS/MS
50 μl of 10 μM Recombinant ERK2 was incubated in Tris 50 mM pH = 8, 0.3 M NaCl with 100 μM compounds or DMSO. The compounds were then removed by methanol-chloroform precipitation of the protein. The dry pellet was dissolved in 40 μl of 50 mM Tris pH = 8 + 5% SDS and heated to 95°C for 6 minutes. The concentration of the protein was estimated using BCA assay (using purified ERK2 heated in 50 mM Tris pH = 8 + 5% SDS as the standard). 50 pmol of each sample (∼2 μg) were diluted to 20 μl with Tris 50 mM pH = 8 + 5% SDS, reduced with DTT (1 μl of 0.1 M in 5% SDS/Tris 50 mM pH = 8, 45 minutes 65°C), cooled to room temperature, then alkylated with 1 μl of 0.2 M iodoacetamide in water (30 minutes room temperature in the dark). The protein was then isolated and trypisinized on s-traps (Protifi) according to the manufacturer's instructions. Triplicates were prepared for each molecule.
LC/MS/MS running
ULC/MS grade solvents were used for all chromatographic steps. Each sample was loaded using split-less nano-Ultra Performance Liquid Chromatography (10 kpsi nanoAcquity; Waters, Milford, MA, USA). The mobile phase was: A) H2O + 0.1% formic acid and B) acetonitrile + 0.1% formic acid. Desalting of the samples was performed online using a reversed-phase Symmetry C18 trapping column (180 μm internal diameter, 20 mm length, 5 μm particle size; Waters). The peptides were then separated using a T3 HSS nano-column (75 μm internal diameter, 250 mm length, 1.8 μm particle size; Waters) at 0.35 μL/min. Peptides were eluted from the column into the mass spectrometer using the following gradient: 4% to 30%B in 155 min, 35% to 90%B in 5 min, maintained at 90% for 5 min and then back to initial conditions. The nanoUPLC was coupled online through a nanoESI emitter (10 μm tip; New Objective; Woburn, MA, USA) to a quadrupole orbitrap mass spectrometer (Q Exactive HFX, Thermo Scientific) using a FlexIon nanospray apparatus (Proxeon). Data was acquired in data dependent acquisition (DDA) mode, using a Top10 method. MS1 resolution was set to 120,000 (at 200 m/z), mass range of 375-1650 m/z, AGC of 3e6 and maximum injection time was set to 60 msec. MS2 resolution was set to 15,000, quadrupole isolation 1.7 m/z, AGC of 1e5, dynamic exclusion of 45 sec and maximum injection time of 60 msec.
LC/MS/MS analysis
Analysis was done using MaxQuant 1.6.3.4. The sequence of ERK2 was used for the analysis. The digestion enzyme was set to Trypsin/P with a maximum number of missed cleavages of 2. Carbamidomethyl and the modification by the molecules were included as variable modifications on cysteine. The “Re-quantify” option was enabled. Contaminants were included. Peptides were searched with a minimum peptide length of 7 and a maximum peptide mass of 4,500 Da. “Second peptides” were enabled and “Dependent peptides” were disabled. The option “Match between runs” was enabled with a Match time window of 0.7 min and an alignment window of 20 min. An FDR of 0.01 was used for Protein FDR, PSM FDR and XPSM FDR. The triplicate measured for each compound (or for DMSO-treated protein) was analyzed separately. Following MaxQuant analysis, only fully cleaved peptides were quantified and cysteine-containing peptides that were not modified by either iodoacetamide or compound were ignored. The intensity for each peptide was calculated as the average of the three triplicates. If the intensity was zero for one of the replicates, the average of the two others was used, and if two replicates showed zero intensity the peptide was ignored. The intensities for the non-cysteine containing peptides were averaged for each data set and used to normalize the intensity of cysteine containing peptides.
Biochemical assays
While covalent inhibitors display time-dependent inhibition, determination of IC50’s at a specific incubation time allows their evaluation. For kinase inhibition we determined all IC50’s at 2h pre-incubation. For Mpro inhibition we determined all IC50’s at 15 min. pre-incubation.
Kinase activity assays
Biochemical Kinase inhibition assays for compounds 1-9, 1-Ac and 2-Ac, were carried out at Nanosyn, Santa Clara. Test compounds were diluted in 100% DMSO using 3-fold dilution steps. Final compound concentration in assay ranged from 10 μM to 0.0565 nM. Compounds were tested in a single well for each dilution, and the final concentration of DMSO in all assays was kept at 1%. Reference compound, Staurosporine, was tested in an identical manner. Compounds were preincubated in 25°C for 2 hours before the measurements, and the kinase reactions were then performed for an additional 3 hours. For ERK2, the kinase concentration was 0.25-0.35 nM, the ATP concentration was 25 μM. For MELK, the kinase concentration was 0.06 nM, the ATP concentration was 30 μM. For VEGFR2, the kinase concentration was 0.25 nM, the ATP concentration was 80 μM. For GSK3B, the kinase concentration was 0.09 nM, the ATP concentration was 10 μM. For FGFR4, the kinase concentration was 0.17 nM, the ATP concentration was 250 μM.
Biochemical Mpro inhibition assay
All UGI Compounds and in particular compounds 10-40 were seeded into assay-ready plates (Greiner 384 low volume 784900) using an Echo 555 acoustic dispenser, and DMSO was back-filled for a uniform concentration in assay plates (maximum 1%). Reagents for Mpro assay were dispensed into the assay plate in 10 μl volumes for a final of 20 μl. Final reaction concentrations were 20 mM HEPES pH=7.3, 1mM TCEP, 50 mM NaCl, 0.01% Tween-20, 10% glycerol, 5 nM Mpro, 375 nM fluorogenic peptide substrate ([5-FAM]-AVLQSGFR-[Lys(Dabcyl)]-K-amide). Mpro was pre-incubated for 15 minutes at room temperature with compound before addition of substrate. Protease reaction was measured continuously in a BMG Pherastar FS with a 480/520 ex/em filter set. Data was mapped and normalized in Genedata Screener.
MproCrystallography Mpro protein was expressed and purified as discussed previously (Douangamath et al., 2020). The pGEX-6P-1-Mpro plasmid was transformed into a competent E. coli expression cell line based on a multi-phage resistant BL21(DE3) derivative, containing the pRARE2 plasmid. Transformant colonies were used to inoculate a starter culture supplemented with 100 μg/ml Carbenicillin. The culture was then grown to log phase for ∼8 h. Ten millilitres of the starter culture was used to inoculate one litre of auto induction medium supplemented with 10 ml of glycerol and 100 μg/ml carbenicillin. The cultures were grown at 37 °C, 200 rpm for 5 h then switched to 18 °C, 200 rpm for 10 h. The cells were harvested by centrifugation and stored at −80 °C. For purification, cells were resuspended in 50 mM Tris pH 8, 300 mM NaCl, 10 mM Imidazole, 0.03 μg/ml Benzonase. The cells were disrupted on a high-pressure homogeniser (3 passes, 30 kpsi, 4 °C). The lysate was clarified by centrifugation at 50,000 × g. The supernatant was then applied to a Nickel-NTA gravity column and washed and eluted with 50 mM Tris pH 8, 300 mM NaCl, and 25–500 mM imidazole pH 8. N-terminal His-tagged HRV 3 C Protease was then added to the eluted protein at 1:10 w/w ratio. The mixture was then dialysed overnight at 4 °C against 50 mM Tris pH 8, 300 mM NaCl, 1 mM TCEP. The following day, the HRV 3 C protease and other impurities were removed from the cleaved target protein by reverse Nickel-NTA. The relevant fractions were concentrated and applied to an S200 16/60 gel filtration column equilibrated in 20 mM Hepes pH 7.5, 50 mM NaCl buffer. The protein was concentrated to 30 mg/ml using a 10 kDa MWCO centrifugal filter device.
Apo Mpro crystals were grown using the sitting drop vapour diffusion method at 20°C by adding 150 nl of protein (5 mg/ml in 20 mM Hepes pH 7.5, 50 mM NaCl) to 300 nl of crystallisation solution (11% PEG 4K, 6% DMSO, 0.1M MES pH 6.7) and 50 nl of seed stock prepared from initial crystal hits. 55 nl of a 100 mM compound stock solution in DMSO was added directly to the crystallisation drops using an ECHO liquid handler (final concentration 10% DMSO) and drops were incubated for approximately 1 hour prior to mounting and flash freezing in liquid nitrogen. Data were collected at Diamond Light Source on beamline I04-1 at 100K and processed using XDS (Kabsch, 2010) and either xia2 (Winter et al., 2013), autoPROC (Vonrhein et al., 2011) or DIALS (Winter et al., 2018). Further analysis was performed with XChemExplorer (Krojer et al., 2017): electron density maps were generated with Dimple (Keegan et al., 2015); ligand-binding events were identified using PanDDA (Pearce et al., 2017) (both the released version 0.2 and the pre-release development version (https://github.com/ConorFWild/pandda)); ligands were modelled into PanDDA-calculated event maps using Coot (Emsley et al., 2010); restraints were calculated with GRADE (Long et al., 2017); and structures were refined with BUSTER (Bricogne et al., 2011). Coordinates, structure factors and PanDDA event maps for all data sets are deposited in the Protein Data Bank under PDB IDs 7NW2, 5RGT, 5RH5, 5RH6, 5RH7, 5RH9, 5RL0, 5RL1, 5RL2, 5RL3, 5RL4 and 5RL5. Data collection and refinement statistics are summarised in Data S3E. The ground-state structure and all corresponding datasets are deposited under PDB ID 5R8T.
Quantification and statistical analysis
No statistical analysis was performed. Experimental results are presented as means and with error bars indicating standard deviation over n=2 or 3 as indicated in the figure legends.
Acknowledgments
We thank Prof. Oded Livnah for generously providing us recombinant ERK2. N.L. is the incumbent of the Alan and Laraine Fischer Career Development Chair. N.L. would like to acknowledge funding from the Israel Science Foundation (grant nos. 2462/19 and 3824/19), the Israel Cancer Research Fund, the Israeli Ministry of Science Technology (grant no. 3-14763), the Moross Integrated Cancer Center and the Barry Sherman Institute for Medicinal Chemistry. This research was supported by Nelson P. Sirotsky. N.L. is also supported by the Helen and Martin Kimmel Center for Molecular Design, the Joel and Mady Dukler Fund for Cancer Research, the Estate of Emile Mimran and Virgin JustGiving, and the George Schwartzman Fund. D.Z. was funded in part by the Pearlman student-initiated research award. The SGC is a registered charity (no. 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, the Eshelman Institute for Innovation, Genome Canada, the Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck KGaA Darmstadt, Germany, MSD, Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome [106169/ZZ14/Z]. We would also like to thank Dr. Mihaly Varadi and the rest of the PDBe-KB team for the integration of covalentizer results. Finally, we would like to thank Noa Zaidman for help with the graphical abstract.
Author contributions
D.Z. and N.L. designed the algorithm. D.Z. implemented the algorithm, performed all computational experiments as well as ERK2 LC/MS experiments. P.G. led all organic syntheses, with help from M.F., D.F., and A.D., as well as ERK2 LC/MS experiments. F.v.D. performed Mpro crystallography. R.G. performed ERK2 LC/MS and LC/MS/MS experiments. E.R. performed Mpro LC/MS studies. J.P. implemented the webserver. D.O., C.S.-D., P.L., and M.A.W. took care of Mpro protein production. S.D., G.C., and H.B. performed Mpro biochemical assays. The Covid-Moonshot Consortium contributed Mpro-related reagents and protocols. D.Z., P.G., and N.L. wrote the manuscript. All authors contributed to the final form of the manuscript.
Declaration of interests
N.L. is a member of the SAB of Totus medicines, Monte Rosa Therapeutics and MetaboMed.
Published: June 25, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.chembiol.2021.05.018.
Supplemental information
References
- Angst D., Gessier F., Janser P., Vulpetti A., Wälchli R., Beerli C., Littlewood-Evans A., Dawson J., Nuesslein-Hildesheim B., Wieczorek G., et al. Discovery of LOU064 (remibrutinib), a potent and highly selective covalent inhibitor of Bruton’s tyrosine kinase. J. Med. Chem. 2020;63:5102–5118. doi: 10.1021/acs.jmedchem.9b01916. [DOI] [PubMed] [Google Scholar]
- Backus K.M. Applications of reactive cysteine profiling. Curr. Top. Microbiol. Immunol. 2019;420:375–417. doi: 10.1007/82_2018_120. [DOI] [PubMed] [Google Scholar]
- Backus K.M., Correia B.E., Lum K.M., Forli S., Horning B.D., González-Páez G.E., Chatterjee S., Lanning B.R., Teijaro J.R., Olson A.J., et al. Proteome-wide covalent ligand discovery in native biological systems. Nature. 2016;534:570–574. doi: 10.1038/nature18002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay A., Gao J. Targeting biomolecules with reversible covalent chemistry. Curr. Opin. Chem. Biol. 2016;34:110–116. doi: 10.1016/j.cbpa.2016.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bemis G.W., Murcko M.A. The properties of known drugs. 1. Molecular frameworks. J. Med. Chem. 1996;39:2887–2893. doi: 10.1021/jm9602928. [DOI] [PubMed] [Google Scholar]
- Bensinger D., Stubba D., Cremer A., Kohl V., Waßmer T., Stuckert J., Engemann V., Stegmaier K., Schmitz K., Schmidt B. Virtual screening identifies irreversible FMS-like tyrosine kinase 3 inhibitors with activity toward resistance-conferring mutations. J. Med. Chem. 2019;62:2428–2446. doi: 10.1021/acs.jmedchem.8b01714. [DOI] [PubMed] [Google Scholar]
- Bradshaw J.M., McFarland J.M., Paavilainen V.O., Bisconte A., Tam D., Phan V.T., Romanov S., Finkle D., Shu J., Patel V., et al. Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat. Chem. Biol. 2015;11:525–531. doi: 10.1038/nchembio.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek W., Roversi P., Sharff A., Smart O.S., Vonrhein C., et al. Global Phasing Ltd; 2011. BUSTER Version 2.10. 0. [Google Scholar]
- Burdick D.J., Wang S., Heise C., Pan B., Drummond J., Yin J., Goeser L., Magnuson S., Blaney J., Moffat J., et al. Fragment-based discovery of potent ERK2 pyrrolopyrazine inhibitors. Bioorg. Med. Chem. Lett. 2015;25:4728–4732. doi: 10.1016/j.bmcl.2015.08.048. [DOI] [PubMed] [Google Scholar]
- Burger J.A., Buggy J.J. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) Leuk. Lymphoma. 2013;54:2385–2391. doi: 10.3109/10428194.2013.777837. [DOI] [PubMed] [Google Scholar]
- Caldwell R.D., Qiu H., Askew B.C., Bender A.T., Brugger N., Camps M., Dhanabal M., Dutt V., Eichhorn T., Gardberg A.S., et al. Discovery of evobrutinib: an oral, potent, and highly selective, covalent Bruton’s tyrosine kinase (BTK) inhibitor for the treatment of immunological diseases. J. Med. Chem. 2019;62:7643–7655. doi: 10.1021/acs.jmedchem.9b00794. [DOI] [PubMed] [Google Scholar]
- Canon J., Rex K., Saiki A.Y., Mohr C., Cooke K., Bagal D., Gaida K., Holt T., Knutson C.G., Koppada N., et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575:217–223. doi: 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- Chaikuad A., Tacconi E.M.C., Zimmer J., Liang Y., Gray N.S., Tarsounas M., Knapp S. A unique inhibitor binding site in ERK1/2 is associated with slow binding kinetics. Nat. Chem. Biol. 2014;10:853–860. doi: 10.1038/nchembio.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodera J., Lee A.A., London N., von Delft F. Crowdsourcing drug discovery for pandemics. Nat. Chem. 2020;12:581. doi: 10.1038/s41557-020-0496-2. [DOI] [PubMed] [Google Scholar]
- Chowdhury S.R., Kennedy S., Zhu K., Mishra R., Chuong P., Nguyen A.-U., Kathman S.G., Statsyuk A.V. Discovery of covalent enzyme inhibitors using virtual docking of covalent fragments. Bioorg. Med. Chem. Lett. 2019;29:36–39. doi: 10.1016/j.bmcl.2018.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M.S., Zhang C., Shokat K.M., Taunton J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science. 2005;308:1318–1321. doi: 10.1126/science1108367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven G.B., Affron D.P., Allen C.E., Matthies S., Greener J.G., Morgan R.M.L., Tate E.W., Armstrong A., Mann D.J. High-throughput kinetic analysis for target-directed covalent ligand discovery. Angew. Chem. Int. Ed. Engl. 2018;57:5257–5261. doi: 10.1002/anie.201711825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delépine B., Duigou T., Carbonell P., Faulon J.-L. RetroPath2.0: a retrosynthesis workflow for metabolic engineers. Metab. Eng. 2018;45:158–170. doi: 10.1016/j.ymben.2017.12.002. [DOI] [PubMed] [Google Scholar]
- Douangamath A., Fearon D., Gehrtz P., Krojer T., Lukacik P., Owen C.D., Resnick E., Strain-Damerell C., Aimon A., Ábrányi-Balogh P., et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat. Commun. 2020;11:5047. doi: 10.1038/s41467-020-18709-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubiella C., Baur R., Cui H., Huber E.M., Groll M. Selective inhibition of the immunoproteasome by structure-based targeting of a non-catalytic cysteine. Angew. Chem. Int. Ed. 2015;54:15888–15891. doi: 10.1002/anie.201506631. [DOI] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel J., Richters A., Getlik M., Tomassi S., Keul M., Termathe M., Lategahn J., Becker C., Mayer-Wrangowski S., Grütter C., et al. Targeting drug resistance in EGFR with covalent inhibitors: a structure-based design approach. J. Med. Chem. 2015;58:6844–6863. doi: 10.1021/acs.jmedchem.5b01082. [DOI] [PubMed] [Google Scholar]
- Ertl P., Schuffenhauer A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 2009;1:8. doi: 10.1186/1758-2946-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan M.E., Abramite J.A., Anderson D.P., Aulabaugh A., Dahal U.P., Gilbert A.M., Li C., Montgomery J., Oppenheimer S.R., Ryder T., et al. Chemical and computational methods for the characterization of covalent reactive groups for the prospective design of irreversible inhibitors. J. Med. Chem. 2014;57:10072–10079. doi: 10.1021/jm501412a. [DOI] [PubMed] [Google Scholar]
- Gajiwala K.S., Feng J., Ferre R., Ryan K., Brodsky O., Weinrich S., Kath J.C., Stewart A. Insights into the aberrant activity of mutant EGFR kinase domain and drug recognition. Structure. 2013;21:209–219. doi: 10.1016/j.str.2012.11.014. [DOI] [PubMed] [Google Scholar]
- Ghosh A.K., Samanta I., Mondal A., Liu W.R. Covalent inhibition in drug discovery. ChemMedChem. 2019;14:889–906. doi: 10.1002/cmdc.201900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagel M., Miduturu C., Sheets M., Rubin N., Weng W., Stransky N., Bifulco N., Kim J.L., Hodous B., Brooijmans N., et al. First selective small molecule inhibitor of FGFR4 for the treatment of hepatocellular carcinomas with an activated FGFR4 signaling pathway. Cancer Discov. 2015;5:424–437. doi: 10.1158/2159-8290.CD-14-1029. [DOI] [PubMed] [Google Scholar]
- Hoffer L., Voitovich Y.V., Raux B., Carrasco K., Muller C., Fedorov A.Y., Derviaux C., Amouric A., Betzi S., Horvath D., et al. Integrated strategy for lead optimization based on fragment growing: the diversity-oriented-target-focused-synthesis approach. J. Med. Chem. 2018;61:5719–5732. doi: 10.1021/acs.jmedchem.8b00653. [DOI] [PubMed] [Google Scholar]
- Hoffer L., Saez-Ayala M., Horvath D., Varnek A., Morelli X., Roche P. CovaDOTS: in silico chemistry-driven tool to design covalent inhibitors using a linking strategy. J. Chem. Inf. Model. 2019;59:1472–1485. doi: 10.1021/acs.jcim.8b00960. [DOI] [PubMed] [Google Scholar]
- Huang Q., Li L.-L., Yang S.-Y. RASA: a rapid retrosynthesis-based scoring method for the assessment of synthetic accessibility of drug-like molecules. J. Chem. Inf. Model. 2011;51:2768–2777. doi: 10.1021/ci100216g. [DOI] [PubMed] [Google Scholar]
- Jacobs J., Grum-Tokars V., Zhou Y., Turlington M., Adrian Saldanha S., Chase P., Eggler A., Dawson E.S., Baez-Santos Y.M., Tomar S., et al. Discovery, synthesis, and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. J. Med. Chem. 2013;56:534–546. doi: 10.1021/jm301580n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Johansson H., Isabella Tsai Y.-C., Fantom K., Chung C.-W., Kümper S., Martino L., Thomas D.A., Eberl H.C., Muelbaier M., House D., et al. Fragment-based covalent ligand screening enables rapid discovery of inhibitors for the RBR E3 ubiquitin ligase HOIP. J. Am. Chem. Soc. 2019;141:2703–2712. doi: 10.1021/jacs.8b13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 2010;66:133–144. doi: 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathman S.G., Xu Z., Statsyuk A.V. A fragment-based method to discover irreversible covalent inhibitors of cysteine proteases. J. Med. Chem. 2014;57:4969–4974. doi: 10.1021/jm500345q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathman S.G., Span I., Smith A.T., Xu Z., Zhan J., Rosenzweig A.C., Statsyuk A.V. A small molecule that switches a ubiquitin ligase from a processive to a distributive enzymatic mechanism. J. Am. Chem. Soc. 2015;137:12442–12445. doi: 10.1021/jacs.5b06839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan R., Wojdyr M., Winter G., Ashton A. Acta Crystallographica A-Foundation and Advances. Int Union Crystallography 2 Abbey Sq; 2015. DIMPLE: a difference map pipeline for the rapid screening of crystals on the beamline; p. S18. [Google Scholar]
- Krojer T., Talon R., Pearce N., Collins P., Douangamath A., Brandao-Neto J., Dias A., Marsden B., von Delft F. The XChemExplorer graphical workflow tool for routine or large-scale protein-ligand structure determination. Acta Crystallogr. D Struct. Biol. 2017;73:267–278. doi: 10.1107/S2059798316020234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman B., Baker D. Native protein sequences are close to optimal for their structures. Proc. Natl. Acad. Sci. U S A. 2000;97:10383–10388. doi: 10.1073/pnas.97.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung A., Chen Y.-C., Schimpl M., Ni F., Zhu J., Turner M., Molina H., Overman R., Zhang C. Development of specific, irreversible inhibitors for a receptor tyrosine kinase EphB3. J. Am. Chem. Soc. 2016;138:10554–10560. doi: 10.1021/jacs.6b05483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law J., Zsoldos Z., Simon A., Reid D., Liu Y., Khew S.Y., Johnson A.P., Major S., Wade R.A., Ando H.Y. Route Designer: a retrosynthetic analysis tool utilizing automated retrosynthetic rule generation. J. Chem. Inf. Model. 2009;49:593–602. doi: 10.1021/ci800228y. [DOI] [PubMed] [Google Scholar]
- Lewell X.Q., Judd D.B., Watson S.P., Hann M.M. RECAP—retrosynthetic combinatorial analysis procedure: a powerful new technique for identifying privileged molecular fragments with useful applications in combinatorial chemistry. J. Chem. Inf. Comput. Sci. 1998;38:511–522. doi: 10.1021/ci970429i. [DOI] [PubMed] [Google Scholar]
- Liang X., Lv F., Wang B., Yu K., Wu H., Qi Z., Jiang Z., Chen C., Wang A., Miao W., et al. Discovery of 2-((3-acrylamido-4-methylphenyl)amino)-N-(2-methyl-5-(3,4,5-trimethoxybenzamido)phenyl)-4-(methylamino)pyrimidine-5-carboxamide (CHMFL-BMX-078) as a highly potent and selective type II irreversible bone marrow kinase in the X chromosome (BMX) kinase inhibitor. J. Med. Chem. 2017;60:1793–1816. doi: 10.1021/acs.jmedchem.6b01413. [DOI] [PubMed] [Google Scholar]
- Lockbaum G.J., Reyes A.C., Lee J.M., Tilvawala R., Nalivaika E.A., Ali A., Kurt Yilmaz N., Thompson P.R., Schiffer C.A. Crystal structure of SARS-CoV-2 main protease in complex with the non-covalent inhibitor ML188. Viruses. 2021;13:174. doi: 10.3390/v13020174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London N., Miller R.M., Krishnan S., Uchida K., Irwin J.J., Eidam O., Gibold L., Cimermančič P., Bonnet R., Shoichet B.K., et al. Covalent docking of large libraries for the discovery of chemical probes. Nat. Chem. Biol. 2014;10:1066–1072. doi: 10.1038/nchembio.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long F., Nicholls R.A., Emsley P., Graǽulis S., Merkys A., Vaitkus A., Murshudov G.N. AceDRG: a stereochemical description generator for ligands. Acta Crystallogr. D Struct. Biol. 2017;73:112–122. doi: 10.1107/S2059798317000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonsdale R., Ward R.A. Structure-based design of targeted covalent inhibitors. Chem. Soc. Rev. 2018;47:3816–3830. doi: 10.1039/c7cs00220c. [DOI] [PubMed] [Google Scholar]
- Lonsdale R., Burgess J., Colclough N., Davies N.L., Lenz E.M., Orton A.L., Ward R.A. Expanding the armory: predicting and tuning covalent warhead reactivity. J. Chem. Inf. Model. 2017;57:3124–3137. doi: 10.1021/acs.jcim.7b00553. [DOI] [PubMed] [Google Scholar]
- Lotesta S.D., Marcus A.P., Zheng Y., Leftheris K., Noto P.B., Meng S., Kandpal G., Chen G., Zhou J., McKeever B., et al. Identification of spirooxindole and dibenzoxazepine motifs as potent mineralocorticoid receptor antagonists. Bioorg. Med. Chem. 2016;24:1384–1391. doi: 10.1016/j.bmc.2016.02.014. [DOI] [PubMed] [Google Scholar]
- Martín-Gago P., Olsen C.A. Arylfluorosulfate-based electrophiles for covalent protein labeling: a new addition to the arsenal. Angew. Chem. Int. Ed. 2019;58:957–966. doi: 10.1002/anie.201806037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalczyk A., Klüter S., Rode H.B., Simard J.R., Grütter C., Rabiller M., Rauh D. Structural insights into how irreversible inhibitors can overcome drug resistance in EGFR. Bioorg. Med. Chem. 2008;16:3482–3488. doi: 10.1016/j.bmc.2008.02.053. [DOI] [PubMed] [Google Scholar]
- Nnadi C.I., Jenkins M.L., Gentile D.R., Bateman L.A., Zaidman D., Balius T.E., Nomura D.K., Burke J.E., Shokat K.M., London N. Novel K-Ras G12C switch-II covalent binders destabilize Ras and accelerate nucleotide exchange. J. Chem. Inf. Model. 2018;58:464–471. doi: 10.1021/acs.jcim.7b00399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxnard G.R., Thress K.S., Alden R.S., Lawrance R., Paweletz C.P., Cantarini M., Yang J.C.-H., Barrett J.C., Jänne P.A. Association between plasma genotyping and outcomes of treatment with osimertinib (AZD9291) in advanced non-small-cell lung cancer. J. Clin. Oncol. 2016;34:3375–3382. doi: 10.1200/JCO.2016.66.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazzesi F., Grundl M.A., Pautsch A., Weber A., Tautermann C.S. A fast ab initio predictor tool for covalent reactivity estimation of acrylamides. J. Chem. Inf. Model. 2019;59:3565–3571. doi: 10.1021/acs.jcim.9b00316. [DOI] [PubMed] [Google Scholar]
- Palazzesi F., Hermann M.R., Grundl M.A., Pautsch A., Seeliger D., Tautermann C.S., Weber A. BIreactive: a machine-learning model to estimate covalent warhead reactivity. J. Chem. Inf. Model. 2020;60:2915–2923. doi: 10.1021/acs.jcim.9b01058. [DOI] [PubMed] [Google Scholar]
- Parker C.G., Galmozzi A., Wang Y., Correia B.E., Sasaki K., Joslyn C.M., Kim A.S., Cavallaro C.L., Lawrence R.M., Johnson S.R., et al. Ligand and target discovery by fragment-based screening in human cells. Cell. 2017;168:527–541.e29. doi: 10.1016/j.cell.2016.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PDBe-KB consortium PDBe-KB: a community-driven resource for structural and functional annotations. Nucleic Acids Res. 2020;48:D344–D353. doi: 10.1093/nar/gkz853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce N.M., Krojer T., Bradley A.R., Collins P., Nowak R.P., Talon R., Marsden B.D., Kelm S., Shi J., Deane C.M., et al. A multi-crystal method for extracting obscured crystallographic states from conventionally uninterpretable electron density. Nat. Commun. 2017;8:15123. doi: 10.1038/ncomms15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podolyan Y., Walters M.A., Karypis G. Assessing synthetic accessibility of chemical compounds using machine learning methods. J. Chem. Inf. Model. 2010;50:979–991. doi: 10.1021/ci900301v. [DOI] [PubMed] [Google Scholar]
- Rachman M., Scarpino A., Bajusz D., Pálfy G., Vida I., Perczel A., Barril X., Keserű G.M. DUckCov: a dynamic undocking-based virtual screening protocol for covalent binders. ChemMedChem. 2019;14:1011–1021. doi: 10.1002/cmdc.201900078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray S., Murkin A.S. New electrophiles and strategies for mechanism-based and targeted covalent inhibitor design. Biochemistry. 2019;58:5234–5244. doi: 10.1021/acs.biochem.9b00293. [DOI] [PubMed] [Google Scholar]
- Resnick E., Bradley A., Gan J., Douangamath A., Krojer T., Sethi R., Geurink P.P., Aimon A., Amitai G., Bellini D., et al. Rapid covalent-probe discovery by electrophile-fragment screening. J. Am. Chem. Soc. 2019;141:8951–8968. doi: 10.1021/jacs.9b02822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpino A., Ferenczy G.G., Keserű G.M. Comparative evaluation of covalent docking tools. J. Chem. Inf. Model. 2018;58:1441–1458. doi: 10.1021/acs.jcim.8b00228. [DOI] [PubMed] [Google Scholar]
- Sequist L.V., Yang J.C.-H., Yamamoto N., O’Byrne K., Hirsh V., Mok T., Geater S.L., Orlov S., Tsai C.-M., Boyer M., et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J. Clin. Oncol. 2013;31:3327–3334. doi: 10.1200/JCO.2012.44.2806. [DOI] [PubMed] [Google Scholar]
- Shannon D.A., Weerapana E. Covalent protein modification: the current landscape of residue-specific electrophiles. Curr. Opin. Chem. Biol. 2015;24:18–26. doi: 10.1016/j.cbpa.2014.10.021. [DOI] [PubMed] [Google Scholar]
- Shi L., Zhong Z., Li X., Zhou Y., Pan Z. Discovery of an orally available janus kinase 3 selective covalent inhibitor. J. Med. Chem. 2019;62:1054–1066. doi: 10.1021/acs.jmedchem.8b01823. [DOI] [PubMed] [Google Scholar]
- Shindo N., Fuchida H., Sato M., Watari K., Shibata T., Kuwata K., Miura C., Okamoto K., Hatsuyama Y., Tokunaga K., et al. Selective and reversible modification of kinase cysteines with chlorofluoroacetamides. Nat. Chem. Biol. 2019;15:250–258. doi: 10.1038/s41589-018-0204-3. [DOI] [PubMed] [Google Scholar]
- Shraga A., Olshvang E., Davidzohn N., Khoshkenar P., Germain N., Shurrush K., Carvalho S., Avram L., Albeck S., Unger T., et al. Covalent docking identifies a potent and selective MKK7 inhibitor. Cell Chem. Biol. 2019;26:98–108.e5. doi: 10.1016/j.chembiol.2018.10.011. [DOI] [PubMed] [Google Scholar]
- Sutanto F., Konstantinidou M., Dömling A. Covalent inhibitors: a rational approach to drug discovery. RSC Med. Chem. 2020;11:876–884. doi: 10.1039/d0md00154f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telliez J.-B., Dowty M.E., Wang L., Jussif J., Lin T., Li L., Moy E., Balbo P., Li W., Zhao Y., et al. Discovery of a JAK3-selective inhibitor: functional differentiation of JAK3-selective inhibition over pan-JAK or JAK1-selective inhibition. ACS Chem. Biol. 2016;11:3442–3451. doi: 10.1021/acschembio.6b00677. [DOI] [PubMed] [Google Scholar]
- Toledo Warshaviak D., Golan G., Borrelli K.W., Zhu K., Kalid O. Structure-based virtual screening approach for discovery of covalently bound ligands. J. Chem. Inf. Model. 2014;54:1941–1950. doi: 10.1021/ci500175r. [DOI] [PubMed] [Google Scholar]
- Touré B.B., Giraldes J., Smith T., Sprague E.R., Wang Y., Mathieu S., Chen Z., Mishina Y., Feng Y., Yan-Neale Y., et al. Toward the validation of maternal embryonic leucine zipper kinase: discovery, optimization of highly potent and selective inhibitors, and preliminary biology insight. J. Med. Chem. 2016;59:4711–4723. doi: 10.1021/acs.jmedchem.6b00052. [DOI] [PubMed] [Google Scholar]
- Vazquez-Rodriguez S., Wright M. Design, synthesis and characterization of covalent KDM5 inhibitors. Angew. Chem. Int. Ed. Engl. 2019;58:515–519. doi: 10.1002/anie.201810179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonrhein C., Flensburg C., Keller P., Sharff A., Smart O., Paciorek W., Womack T., Bricogne G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr. 2011;67:293–302. doi: 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward R.A., Colclough N., Challinor M., Debreczeni J.E., Eckersley K., Fairley G., Feron L., Flemington V., Graham M.A., Greenwood R., et al. Structure-guided design of highly selective and potent covalent inhibitors of ERK1/2. J. Med. Chem. 2015;58:4790–4801. doi: 10.1021/acs.jmedchem.5b00466. [DOI] [PubMed] [Google Scholar]
- Watson I.A., Wang J., Nicolaou C.A. A retrosynthetic analysis algorithm implementation. J. Cheminform. 2019;11:1. doi: 10.1186/s13321-018-0323-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L., Wen W., Rao L., Huang Y., Lei M., Liu K., Hu S., Song R., Ren Y., Wan J. Cov_FB3D: a de novo covalent drug design protocol integrating the BA-SAMP strategy and machine-learning-based synthetic tractability evaluation. J. Chem. Inf. Model. 2020;60:4388–4402. doi: 10.1021/acs.jcim.9b01197. [DOI] [PubMed] [Google Scholar]
- Weisner J., Gontla R., van der Westhuizen L., Oeck S., Ketzer J., Janning P., Richters A., Mühlenberg T., Fang Z., Taher A., et al. Covalent-allosteric kinase inhibitors. Angew. Chem. Int. Ed. 2015;54:10313–10316. doi: 10.1002/anie.201502142. [DOI] [PubMed] [Google Scholar]
- Williams S.P., Sigler P.B. Atomic structure of progesterone complexed with its receptor. Nature. 1998;393:392–396. doi: 10.1038/30775. [DOI] [PubMed] [Google Scholar]
- Winter G., Lobley C.M.C., Prince S.M. Decision making in xia2. Acta Crystallogr. D Biol. Crystallogr. 2013;69:1260–1273. doi: 10.1107/S0907444913015308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G., Waterman D.G., Parkhurst J.M., Brewster A.S., Gildea R.J., Gerstel M., Fuentes-Montero L., Vollmar M., Michels-Clark T., Young I.D., et al. DIALS: implementation and evaluation of a new integration package. Acta Crystallogr. D Struct. Biol. 2018;74:85–97. doi: 10.1107/S2059798317017235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F., Zhao S., Yu B., Chen Y.-M., Wang W., Song Z.-G., Hu Y., Tao Z.-W., Tian J.-H., Pei Y.-Y., et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing L., Klug-Mcleod J., Rai B., Lunney E.A. Kinase hinge binding scaffolds and their hydrogen bond patterns. Bioorg. Med. Chem. 2015;23:6520–6527. doi: 10.1016/j.bmc.2015.08.006. [DOI] [PubMed] [Google Scholar]
- You C., Liu C., Li Y., Jiang P., Ma Q. Structural and enzymatic analysis of the cytochrome b5 reductase domain of Ulva prolifera nitrate reductase. Int. J. Biol. Macromol. 2018;111:1175–1182. doi: 10.1016/j.ijbiomac.2018.01.140. [DOI] [PubMed] [Google Scholar]
- Yun C.-H., Mengwasser K.E., Toms A.V., Woo M.S., Greulich H., Wong K.-K., Meyerson M., Eck M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. U S A. 2008;105:2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng M., Lu J., Li L., Feru F., Quan C., Gero T.W., Ficarro S.B., Xiong Y., Ambrogio C., Paranal R.M., et al. Potent and selective covalent quinazoline inhibitors of KRAS G12C. Cell Chem. Biol. 2017;24:1005–1016.e3. doi: 10.1016/j.chembiol.2017.06.017. [DOI] [PubMed] [Google Scholar]
- Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The code generated in this study is available at GitHub: https://covalentizer.weizmann.ac.il/
https://github.com/LondonLab/Covalentizer.
The Covalentizer database and webserver is available online: http://covalentizer.weizmann.ac.il/