

SUMMARY
As genome engineering advances cell-based therapies, a versatile approach to introducing both CRISPR-Cas9 ribonucleoproteins (RNPs) and therapeutic transgenes into specific cells would be transformative. Autologous T cells expressing a chimeric antigen receptor (CAR) manufactured by viral transduction are approved to treat multiple blood cancers, but additional genetic modifications to alter cell programs will likely be required to treat solid tumors and for allogeneic cellular therapies. We have developed a one-step strategy using engineered lentiviral particles to introduce Cas9 RNPs and a CAR transgene into primary human T cells without electroporation. Furthermore, programming particle tropism allows us to target a specific cell type within a mixed cell population. As a proof-of-concept, we show that HIV-1 envelope targeted particles to edit CD4+ cells while sparing co-cultured CD8+ cells. This adaptable approach to immune cell engineering ex vivo provides a strategy applicable to the genetic modification of targeted somatic cells in vivo.
Graphical Abstract
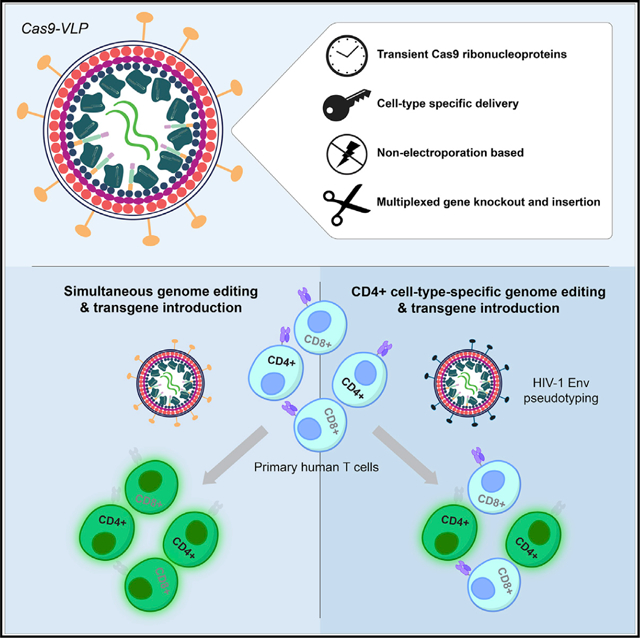
In brief
Hamilton et al. demonstrate that engineered virus-like particles enable simultaneous CRISPR-Cas9 genetic knockout and the anti-tumor reprogramming of T cells. By outwardly making virus-like particles resemble HIV-1, the authors additionally demonstrate specific targeting of CD4+ T cells for genome editing within a mixed cell population.
INTRODUCTION
Engineering target specificity into immune cells enables the antigen-specific elimination of cells expressing cancer-associated epitopes (Lim and June, 2017). Currently approved cell therapies require the isolation of patient T cells, the viral introduction of a chimeric antigen receptor (CAR) to redirect cytotoxic activity toward target cells, and subsequent reintroduction into the body. These autologous immune cell therapies can effectively combat blood cancers but remain largely ineffective for the treatment of solid tumors (Newick et al., 2017; Wagner et al., 2020). However, genome editing may have the potential to unleash engineered cell activity against solid tumors: knockout of transforming growth factor-β (TGF-β) receptor or programmed cell death protein-1 (PD-1) have both shown some promise for enhancing the activity of cell therapies (Bailey and Maus, 2019; Hu et al., 2019; Rupp et al., 2017; Tang et al., 2020). In addition, genome editing may increase the accessibility of engineered cell therapies by enabling the production of “off-the-shelf” allogeneic cells with disrupted major histocompatibility complex class I (MHC class I) and endogenous T cell receptor expression, thereby minimizing the risks of rejection and graft-versus-host disease (GVHD) (Weber et al., 2020). Future immune cell therapies must therefore incorporate complex modifications of both targeted genetic disruption and stable gene addition. Current genetic engineering approaches, either to prevent premature exhaustion or enable allogeneic adoptive cell transfer, generally require viral transduction to program antigen specificity combined with the electroporation of nucleases to produce targeted genetic disruptions. Streamlining the generation of engineered immune cells with enhanced cytotoxic activity, resistance to cellular exhaustion, and minimized risk of rejection or GVHD would facilitate the next generation of universally accessible cellular therapies against solid tumor malignancies.
CRISPR-Cas9 genome editing enables the disruption of targeted genes but requires the effective delivery of genome editing tools into target cells (Doudna, 2020; van Haasteren et al., 2020; Jinek et al., 2012; Porteus, 2019; Wilson and Gilbert, 2018). The modification of Cas9 ribonucleoprotein (RNP) complexes with cell-penetrating and endosomolytic peptides has improved direct cellular uptake (Del’Guidice et al., 2018; Ramakrishna et al., 2014; Staahl et al., 2017; Wang et al., 2018); however, electroporation remains the predominant strategy for delivering Cas9 RNPs into the intracellular environment. Retroviral virus-like particles (VLPs) packaging Cas9 protein have been produced by fusing Cas9 directly to the group-specific antigen (Gag) structural protein (Choi et al., 2016; Mangeot et al., 2019). This has proved an efficient strategy to promote Cas9 particle encapsidation and to couple the cell-entry mechanisms of an enveloped virus to the transient genome editing activity of Cas9 RNPs. However, VLPs are an unexplored strategy for linking the delivery of pre-formed Cas9 RNPs with a clinically relevant transgene and leveraging viral glycoprotein pseudotyping to direct genome editing to specific cell types.
Here, we optimize and demonstrate that engineered lentiviral particles can deliver Cas9 RNP complexes for genome editing, either tracelessly or while simultaneously integrating a lentiviral-encoded transgene (Cas9-VLPs) in immortalized cell lines and primary human T cells. Treatment of primary human T cells with Cas9-VLPs packaging a lentiviral-encoded CAR resulted in the simultaneous knockout of genetic targets relevant to allogeneic T cell production while effectively mediating CAR expression, an approach that was amenable to multiplexing. In addition, the treatment of T cells with broadly transducing Cas9-VLPs resulted in targeted genetic knockout in CD4+ and CD8+ T cells, while treatment with Cas9-VLPs pseudotyped with the CD4-tropic HIV-1 envelope glycoprotein drove the exclusive transduction and genome editing of CD4+ T cells within a mixed cell population. These data establish Cas9-VLPs as an effective approach for mediating cell-type-specific genome editing using Cas9 RNPs. As Cas9-VLPs circumvent the requirement for ex vivo Cas9 RNP delivery via electroporation, this strategy suggests a path forward for leveraging the tropism of viral glycoproteins in targeting specific cell types for genome engineering in vivo.
RESULTS
Engineering lentivirus-based VLPs for the controlled delivery of Cas9 RNP complexes
Lentiviral production involves the co-transfection of producer cells with plasmids encoding the viral structural components, viral glycoprotein, and lentiviral transfer plasmid with a transgenic sequence flanked by long terminal repeat (LTR) sequences. To promote the packaging of Cas9 protein in HIV-1 VLPs (Cas9-VLPs), we constructed a plasmid to express Streptococcus pyogenes Cas9 fused to the C terminus of the Gag polypeptide and included this during lentiviral production. A lentiviral transfer plasmid encoding the expression cassettes for both an mNeonGreen fluorescent reporter and a single guide RNA (sgRNA) was included (Figure 1A). To promote the separation of Cas9 from Gag during proteolytic virion maturation, we inserted an HIV-1 protease-cleavable linker between Gag and Cas9 (Figure 1B). We produced Cas9-VLPs pseudotyped with the broadly transducing vesicular stomatitis virus glycoprotein (VSV-G) and varied the ratio of Gag-pol and Gag-Cas9 plasmids to optimize Cas9 incorporation in budded particles. Bands corresponding to the expected size of Cas9 fused to Gag (55 kDa + 160 kDa = 215 kDa) and proteolytically released Cas9 (160 kDa) were detectable by western blot in all of the Cas9-VLP formulations tested (Figure 1C). A component of the Gag polypeptide, capsid (CA), was used for quantifying Cas9-VLP production by ELISA. CA-containing particles were detected for all of the formulations except for Cas9-VLP formulation F (Figure 1D). Formulation F is composed entirely of Gag-Cas9 polypeptides, which may interfere with the successful budding of Cas9-VLPs from transfected cells.
Figure 1. Production and characterization of Cas9-VLPs.
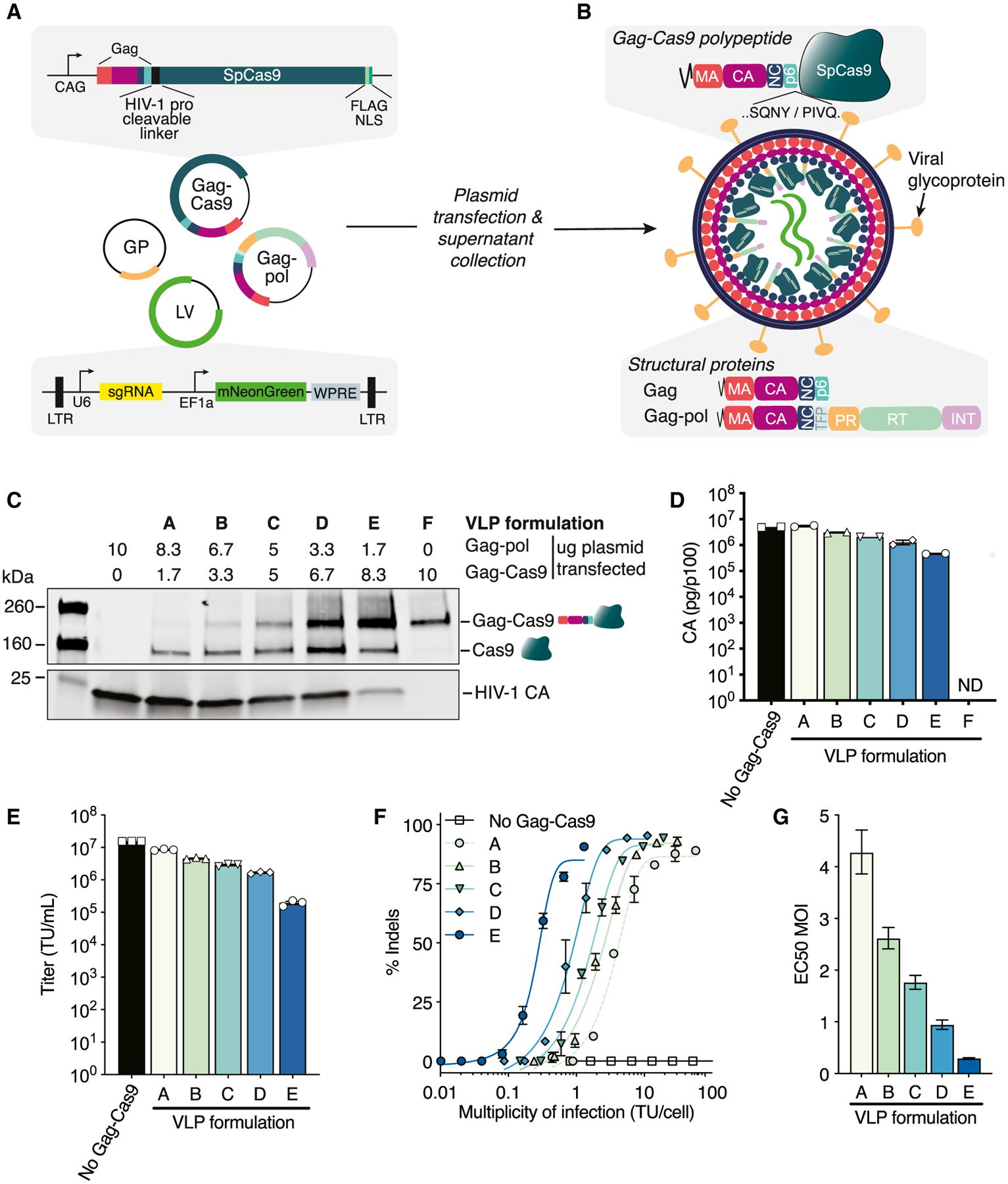
(A) Schematic of plasmids for Cas9-VLP production. GP, glycoprotein; LTR, long terminal repeat; LV, lentiviral transfer plasmid.
(B) Schematic of an immature Cas9-VLP produced through transient transfection. An HIV-1 protease cleavable linker (SQNYPIVQ) was inserted between Gag and Cas9.
(C) Western blot of Cas9-VLP content with various ratios of Gag-pol and Gag-Cas9 plasmids used for production. Anti-FLAG (Cas9) and anti-p24 (capsid, CA) antibodies were used for detection.
(D) Quantification of Cas9-VLPs produced per transfected p100 dish by CA ELISA; n = 2 technical replicates.
(E) Jurkats were treated with B2M-targeting Cas9-VLPs and the transducing units (TUs) per milliliter titer was calculated.
(F) Percentage of B2M indels plotted against the multiplicity of infection (MOI) using a sigmoidal 4-parameter logistic fit. Indels were quantified using Synthego’s ICE analysis tool.
(G) The predicted MOI for each Cas9-VLP formulation to achieve 50% indels, interpolated from (F). EC50, effective concentration at which a drug gives a half-maximal response.
n = 3 technical replicates (E and F), except for formulation A (n = 2) (F). Error bars indicate standard error of the mean (D–F) and 95% confidence interval (G). ND, not detected.
We hypothesized that Cas9-VLPs packaging relatively high Gag-Cas9 polypeptide content would require fewer individual Cas9-VLPs to deliver a sufficient quantity of Cas9 RNPs for successful genome editing. To assess genome editing activity, Cas9-VLPs were produced with a lentiviral transfer plasmid expressing a sgRNA targeting the β-2 microglobulin (B2M) gene. The transduction-competent Cas9-VLP titer (transducing units TU/mL) was quantified for each Cas9-VLP preparation (Figure 1E) and used to calculate the multiplicity of infection (MOI, TU/cell) required to achieve 50% editing (effective concentration 50 [EC50] MOI) in the Jurkat cells (Figure 1F). We confirmed that as increasing amounts of Gag-Cas9 are packaged per particle, a lower EC50 MOI is needed to achieve genome editing (Figure 1G), with an approximate MOI of 2.6 required to achieve 50% indels using Cas9-VLP formulation B and an MOI of 0.9 using Cas9-VLP formulation D.
Characterization of Cas9-VLPs for genome editing
We next assessed the kinetics of genome editing following Cas9-VLP treatment. Jurkat and A549 cells were treated with formulation D B2M-targeting Cas9-VLPs, and cell-surface-expressed B2M protein was assessed by flow cytometry at 3, 6, and 8 days post-treatment. We observed dose-dependent knockout of B2M protein by day 3 (Figure 2A), with a maximum loss of protein expression achieved by day 6. We further confirmed genetic knockout by next-generation sequencing and observed B2M-guide-specific indels at the B2M locus (Figure 2B). Similar to what has been observed for Cas9-packaging MLV VLPs (Mangeot et al., 2019), mixing Cas9-VLPs with a DNA template was sufficient to mediate homology-directed repair (HDR) in a fluorescence reporter assay (Richardson et al., 2016). We found that Cas9-VLP-directed HDR activity could be further enhanced by electroporating Cas9-VLPs with the DNA template before the treatment of target cells, which may promote the complexing of Cas9-VLPs with the HDR template (Figure S1).
Figure 2. Cas9-VLPs efficiently mediate genome editing.

B2M-targeting Cas9-VLP treatment results in genome editing of Jurkat and A549 cells.
(A) Flow cytometry quantification of B2M expression at 3, 6, and 8 days post-treatment (dpt). Heatmaps represent the mean of technical replicates; n = 3, except for A549 at 8 dpt (n = 2). The highest treatment dose = 10% of Cas9-VLPs produced in a p100 dish.
(B) Amplicon sequencing quantification of indels 3 dpt. Control = tdTom298 sgRNA. n = 3 technical replicates, except for A549 treated with 10 × 104 pg CA (n = 2).
(C) Flow cytometry quantification of B2M expression and transduction (mNeonGreen+) 6 dpt. Non-targeting control = guideless Cas9-VLP. n = 3 technical replicates.
(D) Treatment with Cas9-VLPs that target B2M but do not co-package a lentiviral genome. Amplicon sequencing quantification of indels 3 dpt. Control = tdTom298 sgRNA. n = 3 technical replicates, except for Jurkats treated with 10 × 104 pg CA traceless B2M Cas9-VLPs (n = 2).
(E) Schematic of hypothetical lentiviral insertion at the Cas9 RNP-induced DNA break.
(F) PCR assessment of targeted lentiviral integration. DNA was isolated from 293T cells 3 dpt with B2M-targeting or non-targeting Cas9-VLPs, and indicated primer pairs were used for analysis. Error bars indicate standard error of the mean.
Current RNP-based genome editing approaches have not permitted the quantification of cells edited as a function of the number of cells receiving RNPs. We reasoned that Cas9-VLPs co-delivering Cas9 RNPs and a lentiviral genome may enable tracking cells that receive Cas9 RNPs. To assess whether transduction is a marker of RNP-edited cells, we treated A549s and Jurkats with serial dilutions of B2M-Cas9-VLPs delivering the mNeonGreen transgene and quantified B2M expression at day 6 post-treatment (Figures 2C and S2). For Jurkats, successfully edited cells largely correlated with the transduction marker mNeonGreen; however, we did observe a population of B2M-knockout cells that did not express mNeonGreen. We hypothesized that this discordance could be explained by a proportion of Cas9-VLPs not co-packaging both the lentiviral genome and Cas9 RNPs. However, in A549 cells treated with the same Cas9-VLP preparation, cells lacking B2M overwhelmingly expressed the transduction marker. This suggests that in the A549 cell line, transduction is a reliable marker for identifying the cell population edited by Cas9 RNPs and that Jurkats may use a cell-intrinsic mechanism restricting reverse transcription of the lentiviral genome, nuclear import, or integration independent of Cas9-mediated genome editing.
As the sgRNA expression cassette is embedded within the lentiviral genome, sgRNA transcription could occur both in the packaging cell line during Cas9-VLP production and in transduced cells. To assess whether Cas9 RNP formation occurs predominantly in the packaging cells or in the treated cells, we produced Cas9-VLPs lacking a lentiviral genome and instead expressed the B2M sgRNA from an orthogonal expression plasmid. We found that treatment with “traceless” Cas9-VLPs mediated high levels of editing (Figures 2D and S3A–S3D), suggesting that the majority of Cas9 RNPs are formed within the packaging cell line. We noted a slight increase in editing efficiency when a lentiviral genome including the sgRNA expression cassette was co-packaged within the Cas9-VLP (compare Figure 2B versus Figure 2D), suggesting, at this concentration, that a fraction of VLP-packaged Cas9 may remain in the guideless apo-Cas9 state until sgRNA transcription occurs in treated cells. It was also possible to generate hybrid Cas9-VLPs that co-package a lentiviral genome but do not require a lentiviral-encoded guide RNA expression cassette (Figures S3E and S3F). The ability of Cas9-VLPs to deliver Cas9 RNPs without co-packaging a lentiviral genome enables Cas9-VLPs to mediate genome editing in the absence of transgene integration, which may be advantageous for clinical applications.
As Cas9-VLPs deliver the reverse-transcribed viral genome concomitantly with double-stranded DNA (dsDNA) break-inducing Cas9 RNPs, we reasoned that targeted lentiviral insertion may occur at the genomic site targeted for genome editing. To investigate this possibility, we isolated DNA from cells treated with either B2M-targeting or non-targeting Cas9-VLPs co-packaging a lentiviral genome. Amplification of cellular genomic DNA with primers specific to the B2M locus and the lentiviral LTR resulted in detectable viral insertion at the Cas9-targeted region (Figures 2E and 2F). This was further validated using primers specific to the B2M locus and the lentiviral Psi sequence, and next-generation sequencing confirmed targeted lentiviral integration (Figure S4).
Cas9-VLPs efficiently edit primary human T cells
Engineered T cell therapies are transforming the treatment of certain cancers by retargeting T cell activity through the introduction of antigen-specific receptors such as CARs (Fesnak et al., 2016; Sadelain et al., 2017). We next tested whether Cas9-VLPs could mediate genome editing in primary human T cells. Transducing bulk CD4+ and CD8+ primary human T cells with Cas9-VLPs resulted in B2M knockout levels comparable to Cas9 RNP electroporation, the current clinical standard (Figures 3A, 3B, and S5). Cas9-VLP-mediated transduction and B2M knockout was dose dependent and cellular viability (as measured by relative cell count) was improved compared to previous reports using Cas9 RNP nucleofection (Roth et al., 2018) (Figure 3B).
Figure 3. Generation of highly engineered CAR-expressing primary human T cells using Cas9-VLPs.
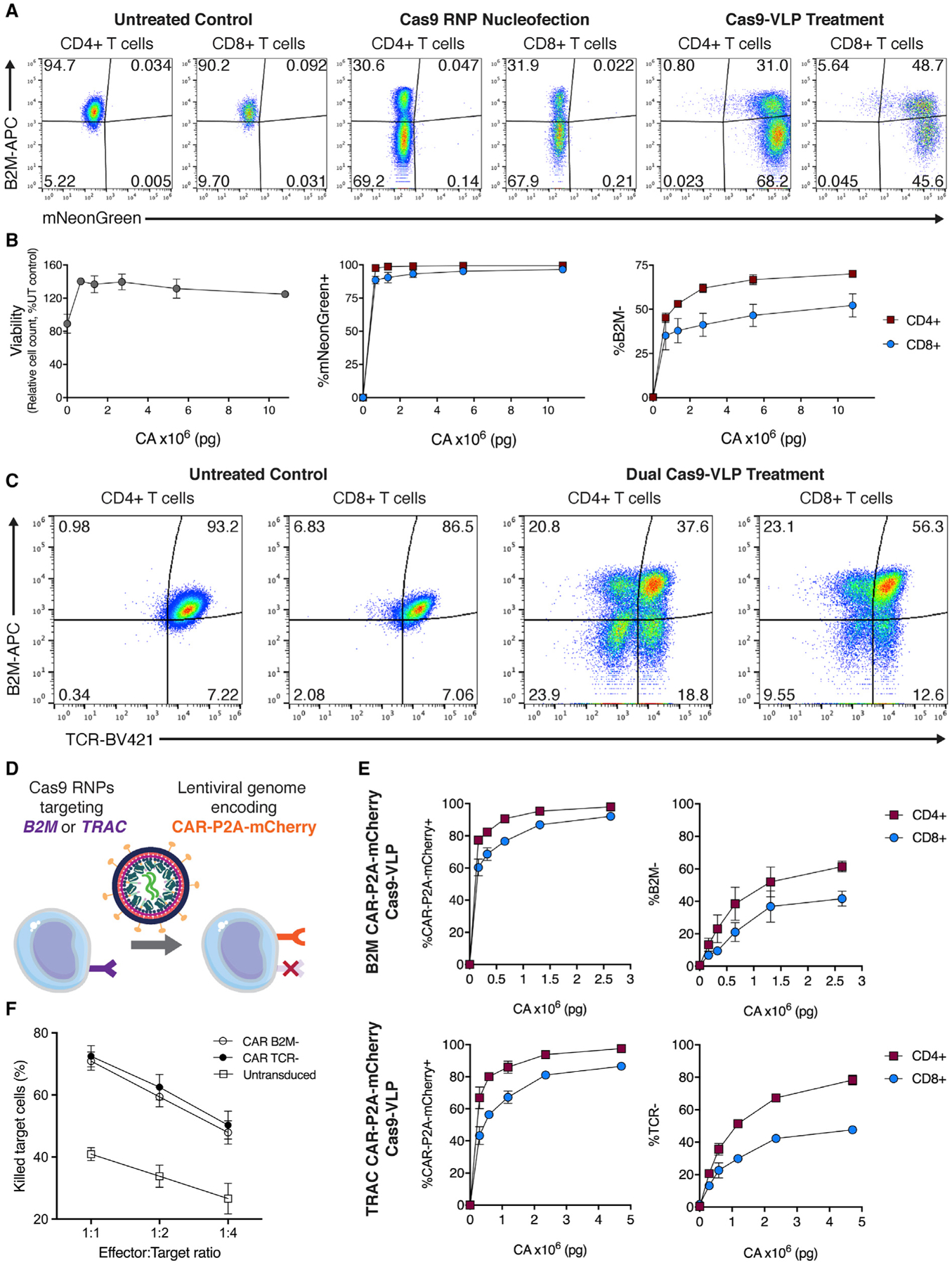
(A) Cas9 RNP nucleofection and Cas9-VLP treatment of primary human T cells. Flow cytometry quantification of the mNeonGreen transduction marker and B2M expression 7 dpt.
(B) Viability, transduction, and B2M expression in primary human T cells treated with Cas9-VLPs. B2M expression is plotted for CD4+ (red squares) and CD8+ (blue circles) subpopulations.
(C) Simultaneous treatment with two Cas9-VLP preparations results in multiplexed genome editing. Cas9-VLPs targeting B2M and Cas9-VLPs targeting TRAC were used to co-treat primary human T cells. Surface expression of B2M and TCR was assessed by flow cytometry 13 dpt. n = 2 biological replicates from independent donors were used (A–C), and representative flow cytometry plots are shown for 1 donor (A and C).
(D) Schematic of a single-step method to generate highly engineered CAR-T cells.
(E) Primary human T cells were treated with CAR-Cas9-VLPs targeting B2M (top panels) or TRAC (bottom panels). Knockout was assessed for both CD4+ (red squares) and CD8+ (blue circles) subpopulations 12 dpt.
(F) CAR-T cells generated by Cas9-VLP treatment, or untreated primary human T cells, were co-cultured with CD19+ Nalm-6 cells, and cytotoxic killing activity was measured at 24 h. Error bars indicate standard error of the mean.
Recently, transgenic T cell receptor (TCR) T cells modified by CRISPR-Cas9 were tested in the first phase I clinical trial (Stadtmauer et al., 2020). The engineered T cell product was produced by the electroporation of Cas9 RNPs to first disrupt expression of PD-1 and the endogenous TCR (by targeting PDCD1 and TRAC, respectively), followed by subsequent lentiviral transduction to integrate an exogenous TCR for retargeting antigen specificity. We hypothesized that Cas9-VLPs could simplify the production of multiply edited engineered T cells by simultaneously delivering Cas9 RNPs and a lentiviral genome encoding a transgenic TCR or CAR (Figure 3D). To test this, we assessed whether it was possible to multiplex genetic knockout by treating primary human T cells with Cas9-VLPs targeting two genetic loci for disruption. Treatment of primary human T cells with separate Cas9-VLPs targeting B2M or TRAC resulted in 23.9% CD4+ and 9.55% CD8+ double-knockout cells by 13 days post-treatment (Figures 3C and S6A). We next optimized the production of Cas9-VLPs to maximize the simultaneous integration of a lentiviral-encoded CAR and knockout of B2M expression in Jurkats (“CAR-Cas9-VLPs”) (Figure S6B). To determine how capsid quantity correlates to MOI in primary T cells, we next assessed genome editing levels generated using both mNeonGreen and CAR Cas9-VLPs. An approximate MOI of 20 for mNeonGreen Cas9-VLPs resulted in ~7% of cells lacking B2M protein while the equivalent MOI for CAR-Cas9-VLPs resulted in ~28% B2M− cells (Figure S7). The enhanced editing efficiency of CAR-Cas9-VLPs may be explained by a higher proportion of VLP-packaged Cas9 being associated with guide RNA, as the optimized CAR-Cas9-VLP production involves the overexpression of guide RNA in VLP producer cells (Figure S6B). Finally, we generated CAR-Cas9-VLPs packaging Cas9 RNPs targeting either B2M or TRAC for disruption; the treatment of primary T cells exhibited dose-dependent CAR-P2A-mCherry expression and reduction in surface-expressed B2M or TCR (Figures 3E, S6C, and S6D). In addition, Cas9-VLP-engineered CAR-T cells were functionalized to kill CD19+ Nalm-6 target cells (Figure 3F) and stimulation resulted in effector profiles for cytokine production and activation marker expression (Figure S8). Cas9-VLPs provide a simplified workflow for manufacturing complex CRISPR-modified CAR-T cells in a single step, which compares favorably to current clinical manufacturing methods.
Cell-type-specific editing via pseudotyping of Cas9-VLPs
Virus and VLP cell-type specificity may be altered through pseudotyping with varied surface glycoproteins (Cronin et al., 2005). To test whether the Cas9-VLP glycoproteins were essential for the genome editing of mammalian cells, we produced Cas9-VLPs lacking viral glycoproteins (“bald” Cas9-VLPs) and assessed their ability to mediate genome editing. Bald Cas9-VLPs were effectively produced (Figures S9A–S9D), but cellular treatment resulted in <0.1% of reads containing indels by deep sequencing, a 3-log reduction in genome editing compared to treatment with VSV-G pseudotyped Cas9-VLPs (Figure 4A). Efficient delivery of VLP-packaged Cas9 RNPs is therefore dependent upon the expression of viral glycoproteins. To test whether we could engineer Cas9-VLPs to target a specific cell type for genome editing, we produced Cas9-VLPs pseudotyped with the HIV-1 envelope glycoprotein (Env), the viral determinant for the CD4+ T cell tropism of HIV-1 (Clapham and McKnight, 2001) (Figures S9E and S9F). Env-Cas9-VLPs were produced packaging Cas9 RNPs targeting the human B2M locus and an mNeonGreen-expressing lentiviral genome. A mixture of CD4+ and CD8+ T cells were treated with Env-Cas9-VLPs, and transduction and B2M protein expression were assessed. At the highest treatment dose, Env-Cas9-VLPs preferentially transduced CD4+ cells over CD8+ cells (53.20% versus 2.51%, respectively) (Figures 4B, 4C, and S9G). Concomitantly, Env-Cas9-VLP treatment resulted in the knockout of B2M in CD4+ T cells while co-cultured CD8+ T cells remained unmodified. This establishes Cas9-VLP pseudotyping as a promising approach to specifically retarget Cas9 RNP-mediated genome editing to predetermined cell types within a mixed cell population without the unintended modification of bystander cells.
Figure 4. HIV-1 envelope pseudotyping targets Cas9-VLP genome editing to CD4+ T cells.
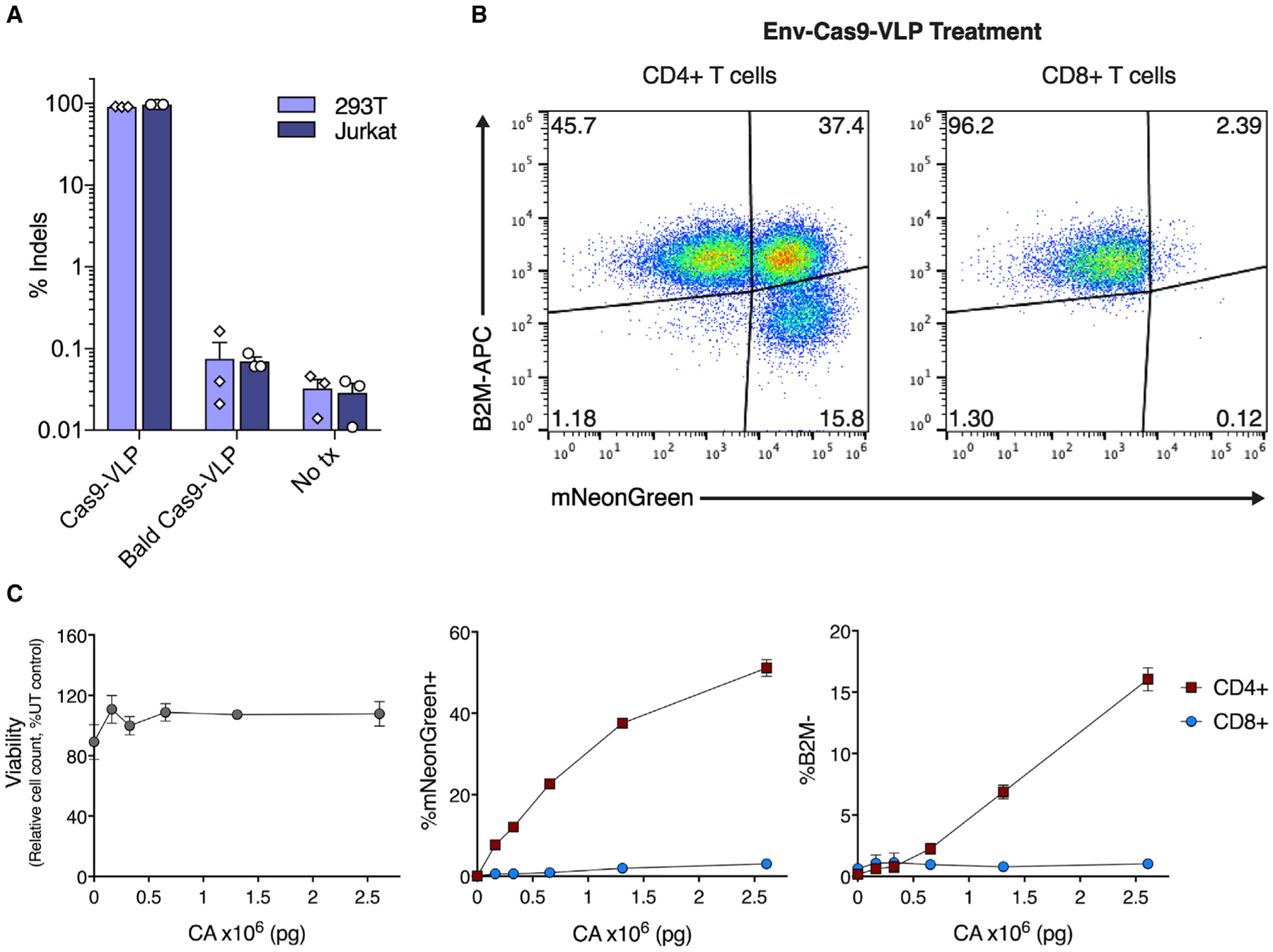
(A) A viral glycoprotein is essential for Cas9-VLP-mediated genome editing. 293T and Jurkat cells were treated with B2M-targeting Cas9-VLPs pseudotyped with VSV-G (Cas9-VLP), without VSV-G (bald Cas9-VLP) or were left untreated (No tx). Indels were quantified by amplicon sequencing 3 dpt, n = 3.
(B) B2M-targeting Cas9-VLPs pseudotyped with the HIV-1 envelope glycoprotein (Env-Cas9-VLPs) were used to treat primary human T cells (a mixture of CD4+ and CD8+ cells).
(C) Viability, transduction (mNeonGreen), and B2M expression were assessed for CD4+ (red squares) and CD8+ (blue circles) subpopulations 7 dpt.
n = 2 biological replicates from independent donors were used (B and C) and representative flow cytometry plots are shown for 1 donor (B). Error bars indicate standard error of the mean.
DISCUSSION
The therapeutic translation of genome editing requires the safe and effective delivery of CRISPR-Cas9 genome editing tools to therapeutically relevant cell types, either ex vivo or in vivo. Established viral delivery strategies generally result in the expression of genome editing tools for the lifetime of the cell, thereby increasing the risk of off-target genomic damage, malignant transformation, and potentially invoking adaptive immune responses against the edited cells in vivo. Most strategies for the non-viral delivery of genome editing tools rely upon electroporation, thus limiting therapeutic applications of genome editing to cells that can be manipulated outside the body. We sought to couple the delivery efficiency and cell type specificity of a virus with the transient genome editing activity of preassembled Cas9 RNPs. Such a delivery tool would enable laboratories to perform Cas9 RNP-mediated genome editing, targetable to any cell type susceptible to glycoprotein-mediated viral transduction, without the need for an electroporator, and amenable to both ex vivo and in vivo applications.
In this study, we engineered Cas9-VLPs as a delivery vehicle for Cas9 RNP complexes, with or without the co-delivery of a lentiviral genome for permanent transgene expression in treated cells. We demonstrate that Cas9-VLPs are a modular delivery system in which the genome editing efficiency, ability to codeliver a lentiviral-encoded transgene, and cellular tropism are programmable elements. Retroviral gag, integrase, Vpx, and Vpr have previously been engineered to direct particle packaging and delivery of enzymatic and reporter proteins, as well as I-SceI, zinc finger, and TAL effector nucleases (Aoki et al., 2011; Cai et al., 2014a, 2014b; Izmiryan et al., 2011; Michel et al., 2010; Miyauchi et al., 2012; Schenkwein et al., 2010; Wu et al., 1995). Here, we directly fused Cas9 protein to the Gag structural protein to promote Cas9 incorporation during VLP assembly, an approach that has been successful at promoting the packaging of Cas9 protein in other engineered retroviruses. Specifically, lentiviral VLPs packaging a Cas9-N-terminal Gag fusion induced modest genome editing (14%–28%) but required guide RNA expression in target cells (Choi et al., 2016). In contrast, murine leukemia virus-like particles (MLV VLPs) have also been engineered by fusing Cas9 to the C terminus of MLV Gag (Mangeot et al., 2019). This protein fusion orientation allowed guide RNA to associate with fused Cas9 within the MLV VLPs, thereby allowing for the delivery of pre-formed Cas9 RNP complexes capable of mediating robust levels of genome editing in target cells. Recently it has been demonstrated that fusing Cas9 to the lentiviral accessory protein Vpr also promotes Cas9 packaging in budding particles (Indikova and Indik, 2020). Vpr-Cas9-containing lentivirus was highly effective at mediating genome editing in immortalized cell lines but only modestly effective (2.7%–15% indels) at mediating genome editing in primary human CD4+ T cells. The reduced efficiency of genome editing in T cells may be due to Cas9-Vpr being packaged within the lentiviral core. While this intravirion localization may promote the delivery of Cas9 RNPs directly to the nuclear pore complex, antiviral restriction factors expressed by T cells may inhibit viral uncoating and, concomitantly, Cas9 RNP access to the nucleus. It has also been shown that Cas9 RNPs can be packaged into lentivirus by encoding RNA aptamers in the sgRNA tetraloop and directly fusing cognate aptamer binding proteins to the C terminus of Gag (Lyu et al., 2019). By expressing Cas9 during VLP production, aptamer-sgRNA-Cas9 complexes were incorporated, a strategy that ensures that all particle-packaged Cas9 protein is bound by sgRNA. Future studies will be needed to directly compare the effectiveness of viral engineering strategies, as VLPs are an emerging strategy for coupling the cell-targeting and cell-fusion capabilities of enveloped viruses to the transient delivery of CRISPR-Cas9 tools.
By producing Cas9-VLPs simultaneously packaging Cas9 RNPs and a lentiviral-encoded CAR, we demonstrate a streamlined strategy for mediating gene knockout (of either the therapeutically relevant B2M or TRAC genes) and simultaneous CAR transgene integration for the production of genetically modified CAR-T cells. Recently, lentiviral transduction combined with multiplexed CRISPR-Cas9 genome editing was used for the first time to treat three patients with refractory cancer in a phase I study (Stadtmauer et al., 2020). Production of the infused T cell product required Cas9 RNP electroporation to mediate genetic knockouts, followed by cellular expansion and subsequent lentiviral transduction to introduce the NY-ESO-1 T cell receptor transgene. This multistep process significantly increases the complexity of clinical-grade manufacturing and the mixture of cellular products obtained highlights the challenge of generating a consistent outcome when combining multiple independent genome modifications. In contrast, by combining the genome editing and transduction capabilities into a single particle, Cas9-VLPs can couple lentiviral genome integration and Cas9-mediated knockout into one high-efficiency genome editing step allowing for coordinated transgene addition and endogenous gene knockout. This streamlined approach simplifies manufacturing, reduces the requirements for good manufacturing practice reagents and equipment, and may improve the consistency of the final therapeutic product.
Lastly, we leverage viral pseudotyping to target Cas9 RNP-mediated genome editing activity to a specific cell type within a mixed-cell population. By pseudotyping Cas9-VLPs with the HIV-1 viral glycoprotein Env, it was possible to exclusively direct Cas9 RNP genome editing to CD4+ T cells, while leaving bystander CD8+ T cells unmodified. Strategies for mediating cell-type-specific genome editing with Cas9 RNPs remain limited and Cas9-VLPs offer an approach for linking a payload of pre-formed Cas9 RNP complexes to the tropism of a viral glycoprotein. Our group has previously demonstrated that directly modifying the Cas9 RNP with asialoglycoprotein receptor ligands promotes Cas9 RNP uptake into hepatic cell types in vitro (Rouet et al., 2018). In addition, work has also been done to direct Cas9 RNP-loaded nanoparticles to specific cell types and tissues via targeting moieties. Decorating nanoparticles with the targeting ligand all-trans retinoic acid promotes genome editing in the retinal pigment epithelium of the eye (Chen et al., 2019), and directly altering the nanoparticle lipid composition could target genome editing activity to either the liver or lung in vivo (Wei et al., 2020). Pseudotyping with viral glycoproteins is a strategy that has been successful for re-targeting retroviral/lentiviral transgene delivery to predetermined cell types; Cas9-VLPs can leverage this well-established field to achieve cell-type-specific delivery of Cas9 RNPs. Future Cas9-VLP studies will investigate additional viral glycoproteins and other targeting strategies for mediating specific genome editing of additional cell types, introduction of transgenes by homology-directed repair, and whether the targeting of CD4+ T cells by Env-pseudotyped Cas9-VLPs will promote the generation of CAR-T cells in vivo.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jennifer Doudna (doudna@berkeley.edu).
Materials availability
Plasmids generated in this study have been deposited to Addgene or are available upon request.
Data and code availability
Amplicon sequencing data have been deposited in the National Institutes of Health NCBI SRA (Bioproject PRJNA680046). Flow cytometry raw data files are available upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Culture of human cell lines
Lenti-X, 293T, A549, CCRF-CEM, HuT 78 and Jurkat cell lines were obtained from the UC Berkeley Cell Culture Facility. All cells were cultured with 10% fetal bovine sera (VWR) and 100 U/mL penicillin-streptomycin (GIBCO). Lenti-X, 293T, and A549 cells were cultured in DMEM (Corning), Jurkat and CCRF-CEM cells were cultured in RPMI 1640 (Thermo Fisher) and 1 mM sodium pyruvate, while HuT 78 cells were cultured in IMEM (Thermo Fisher). Cell lines were routinely checked for mycoplasma using the MycoAlert mycoplasma detection kit (Lonza) according to the manufacturer’s instructions.
Isolation and culture of human primary T cells
Primary adult blood cells were obtained from anonymous healthy human donors as a leukoreduction pack purchased from StemCell Technologies, Inc. or Allcells Inc, or as a Trima residual from Vitalant, under a protocol approved by the University of California, San Francisco Institutional Review Board (IRB). If needed, peripheral blood mononuclear cells were isolated by Ficoll-Paque (GE Healthcare) centrifugation. Bulk CD3+ T lymphocytes were then further isolated by magnetic negative selection using an EasySep magnetic Cell Isolation kit (STEMCELL, as per the manufacturer’s instructions). 96-well flat bottom plates were primed for stimulation by incubating with anti-human CD3 (10 μg/mL) and anti-human CD28 (5 μg/mL) antibodies in PBS for 1 hour at 37°C prior to washing. Primary T cells were activated by plating at 250,000 cells/mL and culturing for one day in XVivo15 medium (Lonza) containing fetal bovine serum (5%), 2-mercaptoethanol (50 μM), N-acetyl L-cysteine (10 mM), IL-2 (300 U/mL), IL-7 (5 ng/mL), and IL-15 (5 ng/mL). Cas9-VLPs in RPMI 1640 were added to primary human T cells 24 hours later along with IL-2 (500 U/mL) and protamine sulfate (4 μg/mL). Media and growth factors were replaced as needed, approximately every 5–6 days. The number of unique primary human T cell donors used for each experiment is listed in Table S4.
METHOD DETAILS
Plasmid construction
The Gag-pol expression plasmid psPax2 was a gift from Didier Trono (Addgene plasmid #12260). pCMV-VSV-G was a gift from Bob Weinberg (Addgene plasmid #8454). Gag-Cas9 was constructed by amplifying Gag from psPax2 and Cas9 from pMJ920 (Addgene plasmid #42234). HIV-1 Env amino acid sequence was obtained from UniProt (P04578), human codon optimized (IDT), and ordered as a gBlock (IDT). In-Fusion (Takara Bio) cloning was used to clone Gag-Cas9 and Env into the pCAGGS expression vector. pCF221 (Addgene plasmid #121669) was modified to express mNeonGreen (Allele Biotechnology) or the ɑ-CD19-4-1BBζ-P2A-mCherry (CAR-P2A-mCherry) construct (Hill et al., 2018; Muller et al., 2020) in place of mCherry and was used as the sgRNA-expressing lentiviral transfer plasmid. For generation of hybrid Cas9-VLPs, the guide RNA expression cassette was removed from the CAR-P2A-mCherry lentiviral plasmid via digestion with EcoRI and KpnI (NEB). The following primers (IDT) were phosphorylated, annealed, and ligated into the digested vector: 5′- cATCGATCTTAAGTCGCGACTCGAg and 5′ - aattcTCGAGTCGCGACTTAAGATCGATggtac. The U6-sgRNA CAG-mTagBFP2 expression plasmid used for traceless Cas9-VLP and CAR-Cas9 VLP production was a gift from Benjamin Oakes. Oligos encoding guide RNA spacers were ordered from IDT, phosphorylated, annealed and ligated into digested sgRNA expression vectors.
Cas9-VLP production
Cas9-VLPs were produced in mammalian cell culture by transient transfection of Lenti-X cells (Takara Bio). 3.5–4 million cells were seeded into 10 cm tissue culture dishes (Corning). The following day, cells were transfected with psPax2, Gag-Cas9, 1 μg pCMV-VSV-G or 0.2 μg HIV Env glycoprotein, and 10 μg of plasmid encoding the sgRNA-expression cassette (either transiently or in the context of a lentiviral transfer plasmid). Plasmids were diluted in Opti-MEM (GIBCO) and mixed with polyethylenimine (PEI, Polysciences Inc.) at a 3:1 PEI:plasmid ratio. Quantities of transfected Gag-Cas9 and psPax2 plasmid are listed in Figure 1C for VLP formulations A-F. A549 and Jurkat experiments used Cas9-VLP formulation D, unless indicated otherwise, and supernatant was harvested at 48 hours post transfection. Cas9-VLP experiments with primary human T cells used Cas9-VLP formulation B, where the Lenti-X media was replaced with Opti-MEM 6–18 hours post transfection. Cas9-VLP-containing Opti-MEM was collected at 48 and 96 hours post media change, with fresh Opti-MEM being added to the cells after 48 hours. Harvested supernatants were centrifuged at 1,500 rpm for 10 minutes and filtered through a 0.45 μM PES membrane bottle top filter (Thermo Fisher) or syringe filter (VWR). Cas9-VLPs were concentrated via ultracentrifugation by floating Cas9-VLP-containing supernatant on top of a cushioning buffer of 30% (w/v) sucrose in 100 mM NaCl, 10 mM Tris-HCl pH 7.5, 1 mM EDTA pH 8.0, at 25,000 rpm with a SW28 or SW41 Ti rotor (Beckman Coulter) for 2 hours at 4°C in polypropylene tubes (Beckman Coulter). After ultracentrifugation, the Cas9-VLP pellet was resuspended in RPMI 1640 (GIBCO) or XVivo15 (Lonza) for treatment of primary T cells or Opti-MEM. Cas9-VLPs were either stored at 4°C or frozen at −80°C within a isopropanol-filled freezing container until use.
Cas9-VLP quantification
Western blots were performed to assess protein components of Cas9-VLPs. Cas9-VLPs were denatured by mixing with Laemmli buffer with 10% 2-mercaptoethanol and heating at 90°C for 5 minutes. Samples were run on 4%–20% SDS-PAGE gels (Bio-Rad) prior to transfer onto a methanol soaked polyvinylidene difluoride (PVDF, Bio-Rad) membrane. PVDF membranes were blocked with 5% non-fat milk (Apex) in PBS (GIBCO) with 0.1% Tween (Sigma) (PBS-T) for one hour at room temperature (~22–25°C). The solution was replaced with 1% non-fat milk in PBS-T and a 1:5000 primary antibody dilution containing anti-FLAG (Sigma) or a 1:2000 dilution of anti-p24 (Abcam) antibodies prior to shaking at 4°C overnight. The following day, the solution was replaced with 1% non-fat milk in PBS-T and a 1:5000 secondary antibody dilution containing IR680 or IR800 conjugated antibodies (LI-COR) and shaken for 1 hour. Western blot membranes were washed with PBS-T three times prior to imaging on a LI-COR OdysseyCLx.
Lenti-X p24 rapid titer kits (Takara Bio) were used to quantify the titer of Cas9-VLPs after concentration. Cas9-VLPs were diluted 1:1,000–100,000 and the ELISA was performed according to the manufacturer’s directions. Absorbance was measured at 450 nm on a BioTek plate reader. Cas9-VLP p24 content was calculated by comparison to serial dilution of a p24 standard (Takara Bio). To calculate transducing units per mL (TU/mL), Cas9-VLP preps were serially diluted and used to treat 15k Jurkats or 25k primary T cells in 96-well u-bottom plates. The percent of cells transduced (mNeonGreen+) was quantified at 6–7 days post treatment using an Attune NxT flow cytometer with a 96-well autosampler (Thermo Fisher Scientific) and titer was quantified as TU/mL = (number of cells transduced × percent mNeonGreen+)/(virus treatment volume). Wells where Cas9-VLP transduction was < 25% were used for titer calculation. MOI was plotted against indels and a sigmoidal four parameter logistic fit was applied to each dataset to interpolate the MOI at which 50% indels would be expected, using a 95% confidence interval.
Cas-VLP homology-directed repair
Cas9-VLPs targeting BFP were produced as previously described (see Table S1 for guide sequence). Cas9-VLPs were mixed with a single-stranded DNA template (IDT) in either DPBS (Thermo Fisher Scientific), Opti-MEM (Thermo Fisher Scientific), or SE/SF/SG buffer (Lonza). Unless otherwise noted, SE buffer (Lonza) with pulse code CM-150 was utilized. The mixture was electroporated using a 4D-nucleofector (Lonza) before immediately adding to 293T cells stably expressing a BFP-to-GFP reporter (Addgene plasmid #71825). A three nucleotide conversion within the BFP gene results in GFP expression. Cells were analyzed for loss of BFP (non-homologous end joining) and gain of GFP (homology-directed repair) expression after 5–7 days on a Attune NxT flow cytometer with a 96-well autosampler (Thermo Fisher Scientific).
Targeted integration analysis
15k 293T cells treated with B2M-targeting or non-targeting Cas9-VLPs and DNA was isolated 3 days post treatment by resuspending in Quick Extract (Lucigen) and heating at 65°C for 20 minutes followed by 95°C for 20 minutes before storing at −20°C. A nested PCR approach using PrimeStar GXL DNA polymerase (Takara Bio) was used to detect integration of the lentiviral genome into the B2M genomic site targeted by Cas9. For PCR analysis of lentiviral integration, the B2M targeted region was first amplified using nested primer set #1 and cleaned up (NucleoSpin Gel and PCR Clean-Up kit, Takara Bio) followed by amplification with primer sets a-g (Table S2). For MiSeq next generation sequencing analysis of targeted integration, the B2M targeted region was first amplified with nested primer set #2 and cleaned up (SPRI beads, UC Berkeley Sequencing Core) followed by amplification with primer sets to detect both integration orientations (primer pairs NGS Fwd and NGS Rev, Table S2). Pair-end reads were merged, trimmed, and aligned to the expected sequence of lentiviral insertion into the expected Cas9 target site in the B2M gene (Geneious).
RNP nucleofection
Cas9 RNPs were formed as previously described (Nguyen et al., 2020) at a 1:2 molar ratio between Cas9-NLS (UC Berkeley QB3 MacroLab) and annealed crRNA and tracrRNA (Horizon Discovery) in IDT duplex buffer with a polyglutamic acid electroporation enhancer, aliquoted, and stored frozen at −80°C until use. Cas9 RNPs (50 pmol) were electroporated into primary human T cells using a 96-well format 4D-nucleofector (Lonza) with the P3 buffer and the EH-115 pulse code. Immediately after electroporation cells were rescued by adding growth media and incubating for 20 minutes prior to diluting to 0.5 to 1e6 cells/mL for culturing.
Flow cytometry
All flow cytometry was performed on an Attune NxT flow cytometer with a 96-well autosampler (Thermo Fisher Scientific). Cells were resuspended in FACS buffer (1%–2% BSA in PBS) and stained with the surface marker-targeting antibodies: B2M-FITC (Biolegend), B2M-PE (Biolegend), B2M-APC (Biolegend), CD4-FITC (Biolegend), CD8-PeCy7 (BD Biosciences), and TCRa/b-BV421 (Biolegend) and live/dead stains Ghost Dye red 780 (Tonbo) or Ghost Dye violet 450 (Tonbo), prior to analyzing. All analysis was done using the FlowJo v10 software. The gating strategy for flow cytometry can be seen in Figures S5, S6, and S9.
Cytotoxicity assay
Nalm-6 target cells were labeled using CellTrace Violet Cell Proliferation Kit (Thermo Fisher Scientific) according to the supplier’s information. T cells were co-cultured with labeled target cells at various Effector:Target ratios for 16–24 hours. The percent of transduced cells were normalized by adding untransduced T cells. Absolute count of remaining living target cells was analyzed and percent killing was calculated by comparing to control wells (target cells only). Measurement was performed on an Attune NxT Flow Cytometer (Thermo Fisher Scientific).
Intracellular cytokine and activation assay
Cells were stimulated with Nalm-6 target cells at an E:T ratio of 1:1. Transduction rates were normalized by adding untransduced T cells. 24 hours later, eBioscience Brefeldin A Solution (1000X) was added and incubated for 4 hours at 37°C. Cells were stained with extracellular antibodies eBioscience Fixable Viability Dye eFluor 780 (Thermo Fisher Scientific), CD25 PE-Cy7 (BD), CD69 PerCP (BioLegend), 4-1BB BV711 (BioLegend) and intracellular antibodies TNF-a Pacific Blue (BioLegend), IL-2 APC (BD) and IFN-g FITC (BioLegend) using the FIX & PERM Cell Fixation & Cell Permeabilization Kit (Thermo Fisher Scientific). CAR samples were gated on mCherry+ cells. Measurement was performed on an Attune NxT Flow Cytometer (Thermo Fisher Scientific).
Amplicon sequencing
Genome editing was determined either by Sanger sequencing or next-generation sequencing; in both cases, the presence of insertions or deletions around the Cas9-targeted sequence was used to determine genome editing efficiency. Cells were pelleted and resuspended in QuickExtract (Lucigen) and heated at 65°C for 20 minutes followed by 95°C for 20 minutes before storing at −80°C. An amplicon containing the target sequence was amplified via PCR with Q5 polymerase (NEB) or PrimeStar GXL DNA polymerase (Takara Bio) and the resulting sample was cleaned with magnetic SPRI beads (UC Berkeley Sequencing Core). PCR amplicons were analyzed via Sanger sequencing (UC Berkeley Sequencing Core) and the resulting traces were deconvolved with Synthego’s Inference of CRISPR Edits (ICE) program (https://ice.synthego.com). NGS sequencing was prepared similarly, but with PCR primers containing Illumina adaptor sequences. PCR amplicons were analyzed on an Illumina MiSeq by QB3 Genomics at UC Berkeley. Paired-end NGS reads were analyzed for indels with CRISPResso2 (http://crispresso.pinellolab.partners.org/login).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
Statistical analysis was performed in Prism v7,v8, and v9. Statistical details for all experiments, including value and definition of n, error bars, and significance thresholds can be found in the Figure Legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| B2M-FITC | Biolegend | 316304; RRID:AB_492837 |
| B2M-PE | Biolegend | 316305; RRID:AB_492840 |
| B2M-APC | Biolegend | 316311; RRID:AB_10643412 |
| CD8-PeCy7 | BD Biosciences | 557746; RRID:AB_396852 |
| CD4-FITC | Biolegend | 300505; RRID:AB_314073 |
| TCRa/b-BV421 | Biolegend | 306721; RRID:AB_2562077 |
| Ghost Dye red 780 | Tonbo | 13-0865-T100 |
| Ghost Dye violet 450 | Tonbo | 13-0863-T100 |
| eBioscience Fixable Viability Dye eFluor 780 | Thermo Fisher Scientific | 65-0865-14 |
| CD25 PE-Cy7 | BD | 557741; RRID:AB_396847 |
| CD69 PerCP | BioLegend | 310927; RRID:AB_10696423 |
| 4-1BB BV711 | BioLegend | 309831; RRID:AB_2650990 |
| TNF-a Pacific Blue | BioLegend | 502920; RRID:AB_528965 |
| IL-2 APC | BD | 341116; RRID:AB_400574 |
| IFN-g FITC | BioLegend | 502505; RRID:AB_315230 |
| anti-FLAG | Sigma | F3165; RRID:AB_259529 |
| anti-p24 | Abcam | Ab9071; RRID:AB_306981 |
| IR680RD IgG | LI-COR | 926-68070; RRID:AB_1095658 |
| IR800CW IgG | LI-COR | 925-32211; RRID:ab_2651127 |
| Bacterial and virus strains | ||
| Mach1 chemically competent E. Coli | UC Berkeley QB3 MacroLab | https://qb3.berkeley.edu/facility/qb3-macrolab/services/#services-molecular-biology-reagents |
| Biological samples | ||
| Human PBMCs | Blood Centers of the Pacific, University of California, San Francisco | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Cas9-NLS recombinant protein | UC Berkeley QB3 MacroLab | https://qb3.berkeley.edu/facility/qb3-macrolab/projects/#projects-cas9-nls-purified-protein |
| Cas9 crRNA and tracrRNA | Horizon Discovery | https://horizondiscovery.com/en/gene-editing/gene-editing-reagents/crispr-guide-rna |
| Polyethylenimine | Polysciences Inc. | 24314 |
| P3 Primary Cell Nucleofector Solution | Lonza | V4SP-3096 |
| SE. Nucleofector Solution | Lonza | V4SC-1096 |
| eBioscience Brefeldin A Solution | Invitrogen | 00-4506-51 |
| QuickExtract | Lucigen | QE09050 |
| Q5 polymerase | NEB | M0491S |
| PrimeStar GXL DNA polymerase | Takara Bio | R050B |
| SPRI beads | UC Berkeley Sequencing Core | https://ucberkeleydnasequencing.com/reagents-for-sale |
| Critical commercial assays | ||
| EasySep magnetic Cell Isolation kit | STEMCELL | 17951 |
| Lenti-X p24 rapid titer kits | Takara Bio | 632200 |
| Cell Line Optimization 4D-Nucleofector X Kit | Lonza | V4XC-9064 |
| MycoAlert mycoplasma detection kit | Lonza | LT07-118 |
| CellTrace Violet Cell Proliferation Kit | Thermo Fisher Scientific | C34557 |
| FIX & PERM Cell Fixation & Cell Permeabilization Kit | Thermo Fisher Scientific | GAS003 |
| Deposited data | ||
| Raw MiSeq sequencing files for analysis of genome editing | This paper | NIH NCBI Bioproject PRJNA680046 |
| Experimental models: Cell lines | ||
| Jurkats | UC Berkeley Cell Culture Facility | https://bds.berkeley.edu/facilities/cell-culture |
| A549 | UC Berkeley Cell Culture Facility | https://bds.berkeley.edu/facilities/cell-culture |
| 293T | UC Berkeley Cell Culture Facility | https://bds.berkeley.edu/facilities/cell-culture |
| Lenti-X | UC Berkeley Cell Culture Facility | https://bds.berkeley.edu/facilities/cell-culture |
| CCRF-CEM | UC Berkeley Cell Culture Facility | https://bds.berkeley.edu/facilities/cell-culture |
| HuT 78 | UC Berkeley Cell Culture Facility | https://bds.berkeley.edu/facilities/cell-culture |
| Oligonucleotides | ||
| See Tables S1–S3 | IDT | N/A |
| Recombinant DNA | ||
| Plasmids generated for this paper (Gag-Cas9, HIV-1 envelope, lentiviral expression of guide RNA plasmids transient expression of guide RNA plasmids) | This paper | Addgene plasmid #171060, 171061, 171625, 171628, 171632, 171633, 171634, 171635, 171636, 171637 |
| psPax2 | psPAX2 was a gift from Didier Trono | Addgene plasmid #12260 |
| pCMV-VSV-G | pCMV-VSV-G was a gift from Bob Weinberg | Addgene plasmid #8454 |
| pCF221 | pCF221 was a gift from David Savage | Addgene plasmid #121669 |
| Software and algorithms | ||
| Prism v7 or v8 | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| Inference of CRISPR Edits (ICE) | Synthego | https://ice.synthego.com |
| CRISPResso2 | https://github.com/pinellolab/CRISPResso2 | |
Highlights.
Lentivirus-like particles transiently deliver Cas9-guide RNA complexes (Cas9-VLPs)
Cas9-VLPs mediate genome editing, with or without co-delivery of a transgene
Cas9-VLPs enable simultaneous gene insertion and knockout for T cell reprogramming
Pseudotyping Cas9-VLPs drives cell-type-specific genome editing of CD4+ T cells
ACKNOWLEDGMENTS
We thank Yannick D. Muller for providing the α-CD19-4-1BBζ-P2A-mCherry plasmid, as well as Stacia Wyman for bioinformatic analysis of NGS amplicons. We thank all of the members of the Doudna and Marson laboratories for their thoughtful input and technical assistance. This research was supported by the Centers for Excellence in Genomic Science of the NIH (award no. RM1HG009490) and the Somatic Cell Genome Editing Program of the Common Fund of the NIH (award no. U01AI142817-02). J.R.H. is a Fellow of The Jane Coffin Childs Memorial Fund for Medical Research. C.A.T. is supported by Campus Executive Grants 2101705 and 1655264 through Sandia National Laboratories. D.N.N. is supported by NIH grants L40AI140341 and K08AI153767 and the CIRM Alpha Stem Cell Clinic Fellowship. B.R.S. is supported by the UCSF Herbert Perkins Cellular Therapy and Transfusion Medicine Fellowship. F.B. was supported by the Care-for-Rare Foundation and the German Research Foundation (DFG). A.M. holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund, is an investigator at the Chan Zuckerberg Biohub, and is a recipient of The Cancer Research Institute (CRI) Lloyd J. Old STAR grant. The Marson lab has received funds from the Innovative Genomics Institute (IGI), the Simons Foundation, and the Parker Institute for Cancer Immunotherapy (PICI). J.A.D. is an investigator of the Howard Hughes Medical Institute (HHMI).
DECLARATION OF INTERESTS
The Regents of the University of California have patents issued and pending for CRISPR technologies on which the authors are co-inventors. J.A.D. is a cofounder of Caribou Biosciences, Editas Medicine, Scribe Therapeutics, Intellia Therapeutics, and Mammoth Biosciences. J.A.D. is a scientific advisory board member of Caribou Biosciences, Intellia Therapeutics, eFFECTOR Therapeutics, Scribe Therapeutics, Mammoth Biosciences, Synthego, Algen Biotechnologies, Felix Biosciences, and Inari. J.A.D. is a Director at Johnson & Johnson and Tempus and has research projects sponsored by Biogen, Pfizer, AppleTree Partners, and Roche. A.M. is a compensated co-founder, member of the boards of directors, and a member of the scientific advisory boards of Spotlight Therapeutics and Arsenal Biosciences. A.M. was a compensated member of the scientific advisory board at PACT Pharma and was a compensated advisor to Juno Therapeutics and Trizell. A.M. owns stock in Arsenal Biosciences, Spotlight Therapeutics, and PACT Pharma. A.M. has received honoraria from Merck and Vertex, a consulting fee from AlphaSights, and is an investor in and informal advisor to Offline Ventures. The Marson lab has received research support from Juno Therapeutics, Epinomics, Sanofi, GlaxoSmithKline, Gilead, and Anthem. J.R.H. has consulted for Scribe Therapeutics. All of the other authors have no competing interests.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109207.
REFERENCES
- Aoki T, Miyauchi K, Urano E, Ichikawa R, and Komano J (2011). Protein transduction by pseudotyped lentivirus-like nanoparticles. Gene Ther. 18, 936–941. [DOI] [PubMed] [Google Scholar]
- Bailey SR, and Maus MV (2019). Gene editing for immune cell therapies. Nat. Biotechnol 37, 1425–1434. [DOI] [PubMed] [Google Scholar]
- Cai Y, Bak RO, Krogh LB, Staunstrup NH, Moldt B, Corydon TJ, Schrøder LD, and Mikkelsen JG (2014a). DNA transposition by protein transduction of the piggyBac transposase from lentiviral Gag precursors. Nucleic Acids Res 42, e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Bak RO, and Mikkelsen JG (2014b). Targeted genome editing by lentiviral protein transduction of zinc-finger and TAL-effector nucleases. eLife 3, e01911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Abdeen AA, Wang Y, Shahi PK, Robertson S, Xie R, Suzuki M, Pattnaik BR, Saha K, and Gong S (2019). A biodegradable nanocapsule delivers a Cas9 ribonucleoprotein complex for in vivo genome editing. Nat. Nanotechnol 14, 974–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JG, Dang Y, Abraham S, Ma H, Zhang J, Guo H, Cai Y, Mikkelsen JG, Wu H, Shankar P, and Manjunath N (2016). Lentivirus prepacked with Cas9 protein for safer gene editing. Gene Ther. 23, 627–633. [DOI] [PubMed] [Google Scholar]
- Clapham PR, and McKnight A (2001). HIV-1 receptors and cell tropism. Br. Med. Bull 58, 43–59. [DOI] [PubMed] [Google Scholar]
- Cronin J, Zhang X-Y, and Reiser J (2005). Altering the tropism of lentiviral vectors through pseudotyping. Curr. Gene Ther 5, 387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del’Guidice T, Lepetit-Stoffaes J-P, Bordeleau L-J, Roberge J, Thé-berge V, Lauvaux C, Barbeau X, Trottier J, Dave V, Roy D-C, et al. (2018). Membrane permeabilizing amphiphilic peptide delivers recombinant transcription factor and CRISPR-Cas9/Cpf1 ribonucleoproteins in hard-to-modify cells. PLoS ONE 13, e0195558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna JA (2020). The promise and challenge of therapeutic genome editing. Nature 578, 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fesnak AD, June CH, and Levine BL (2016). Engineered T cells: the promise and challenges of cancer immunotherapy. Nat. Rev. Cancer 16, 566–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill ZB, Martinko AJ, Nguyen DP, and Wells JA (2018). Human antibody-based chemically induced dimerizers for cell therapeutic applications. Nat. Chem. Biol 14, 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Zi Z, Jin Y, Li G, Shao K, Cai Q, Ma X, and Wei F (2019). CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol. Immunother 68, 365–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indikova I, and Indik S (2020). Highly efficient ‘hit-and-run’ genome editing with unconcentrated lentivectors carrying Vpr.Prot.Cas9 protein produced from RRE-containing transcripts. Nucleic Acids Res. 48, 8178–8187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izmiryan A, Basmaciogullari S, Henry A, Paques F, and Danos O (2011). Efficient gene targeting mediated by a lentiviral vector-associated meganuclease. Nucleic Acids Res. 39, 7610–7619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, and Charpentier E (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim WA, and June CH (2017). The Principles of Engineering Immune Cells to Treat Cancer. Cell 168, 724–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu P, Javidi-Parsijani P, Atala A, and Lu B (2019). Delivering Cas9/sgRNA ribonucleoprotein (RNP) by lentiviral capsid-based bionanoparticles for efficient ‘hit-and-run’ genome editing. Nucleic Acids Res. 47, e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangeot PE, Risson V, Fusil F, Marnef A, Laurent E, Blin J, Mournetas V, Massouridès E, Sohier TJM, Corbin A, et al. (2019). Genome editing in primary cells and in vivo using viral-derived Nanoblades loaded with Cas9-sgRNA ribonucleoproteins. Nat. Commun 10, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel G, Yu Y, Chang T, and Yee J-K (2010). Site-specific gene insertion mediated by a Cre-loxP-carrying lentiviral vector. Mol. Ther 18, 1814–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyauchi K, Urano E, Takizawa M, Ichikawa R, and Komano J (2012). Therapeutic potential of HIV protease-activable CASP3. Sci. Rep 2, 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller YD, Nguyen DP, Ferreira LMR, Ho P, and Raffin C (2020). The CD28-transmembrane domain mediates chimeric antigen receptor heterodimerization with CD28. Front. Immunol 12, 639818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newick K, O’Brien S, Moon E, and Albelda SM (2017). CAR T Cell Therapy for Solid Tumors. Annu. Rev. Med 68, 139–152. [DOI] [PubMed] [Google Scholar]
- Nguyen DN, Roth TL, Li PJ, Chen PA, Apathy R, Mamedov MR, Vo LT, Tobin VR, Goodman D, Shifrut E, et al. (2020). Polymer-stabilized Cas9 nanoparticles and modified repair templates increase genome editing efficiency. Nat. Biotechnol 38, 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porteus MH (2019). A New Class of Medicines through DNA Editing. N. Engl. J. Med 380, 947–959. [DOI] [PubMed] [Google Scholar]
- Ramakrishna S, Kwaku Dad AB, Beloor J, Gopalappa R, Lee SK, and Kim H (2014). Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res. 24, 1020–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson CD, Ray GJ, DeWitt MA, Curie GL, and Corn JE (2016). Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol 34, 339–344. [DOI] [PubMed] [Google Scholar]
- Roth TL, Puig-Saus C, Yu R, Shifrut E, Carnevale J, Li PJ, Hiatt J, Saco J, Krystofinski P, Li H, et al. (2018). Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 559, 405–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouet R, Thuma BA, Roy MD, Lintner NG, Rubitski DM, Finley JE, Wisniewska HM, Mendonsa R, Hirsh A, de Oñate L, et al. (2018). Receptor-Mediated Delivery of CRISPR-Cas9 Endonuclease for Cell-Type-Specific Gene Editing. J. Am. Chem. Soc 140, 6596–6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, and Marson A (2017). CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep 7, 737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadelain M, Rivière I, and Riddell S (2017). Therapeutic T cell engineering. Nature 545, 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenkwein D, Turkki V, Kärkkäinen H-R, Airenne K, and Ylä-Herttuala S (2010). Production of HIV-1 integrase fusion protein-carrying lentiviral vectors for gene therapy and protein transduction. Hum. Gene Ther 21, 589–602. [DOI] [PubMed] [Google Scholar]
- Staahl BT, Benekareddy M, Coulon-Bainier C, Banfal AA, Floor SN, Sabo JK, Urnes C, Munares GA, Ghosh A, and Doudna JA (2017). Efficient genome editing in the mouse brain by local delivery of engineered Cas9 ribonucleoprotein complexes. Nat. Biotechnol 35, 431–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, Mangan PA, Kulikovskaya I, Gupta M, Chen F, et al. (2020). CRISPR-engineered T cells in patients with refractory cancer. Science 367, eaba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang N, Cheng C, Zhang X, Qiao M, Li N, Mu W, Wei X-F, Han W, and Wang H (2020). TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight 5, e133977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Haasteren J, Li J, Scheideler OJ, Murthy N, and Schaffer DV (2020). The delivery challenge: fulfilling the promise of therapeutic genome editing. Nat. Biotechnol 38, 845–855. [DOI] [PubMed] [Google Scholar]
- Wagner J, Wickman E, DeRenzo C, and Gottschalk S (2020). CAR T Cell Therapy for Solid Tumors: Bright Future or Dark Reality? Mol. Ther 28, 2320–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H-X, Song Z, Lao Y-H, Xu X, Gong J, Cheng D, Chakraborty S, Park JS, Li M, Huang D, et al. (2018). Nonviral gene editing via CRISPR/Cas9 delivery by membrane-disruptive and endosomolytic helical polypeptide. Proc. Natl. Acad. Sci. USA 115, 4903–4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber EW, Maus MV, and Mackall CL (2020). The Emerging Landscape of Immune Cell Therapies. Cell 181, 46–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei T, Cheng Q, Min Y-L, Olson EN, and Siegwart DJ (2020). Systemic nanoparticle delivery of CRISPR-Cas9 ribonucleoproteins for effective tissue specific genome editing. Nat. Commun 11, 3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RC, and Gilbert LA (2018). The Promise and Challenge of In Vivo Delivery for Genome Therapeutics. ACS Chem. Biol 13, 376–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Liu H, Xiao H, Kim J, Seshaiah P, Natsoulis G, Boeke JD, Hahn BH, and Kappes JC (1995). Targeting foreign proteins to human immunodeficiency virus particles via fusion with Vpr and Vpx. J. Virol 69, 3389–3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Amplicon sequencing data have been deposited in the National Institutes of Health NCBI SRA (Bioproject PRJNA680046). Flow cytometry raw data files are available upon request.