

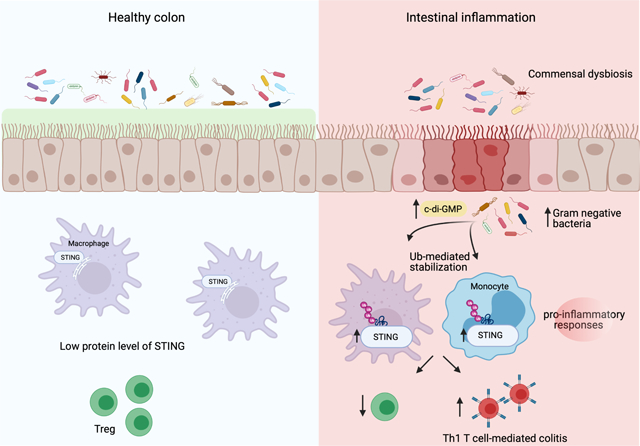
Summary
Alterations in the cGAS-STING DNA sensing pathway affect intestinal homeostasis. We sought to delineate the functional role of STING in intestinal inflammation. Increased STING expression was a feature of intestinal inflammation in mice with colitis and in humans afflicted with inflammatory bowel disease. Mice bearing an allele rendering STING constitutively active exhibited spontaneous colitis, dysbiosis and progressive chronic intestinal inflammation and fibrosis. Bone marrow chimera experiments revealed STING accumulation in intestinal macrophages and monocytes as the initial driver of inflammation. Depletion of Gram-negative bacteria prevented STING accumulation in these cells and alleviated intestinal inflammation. STING accumulation occurred at the protein rather than transcript level, suggesting posttranslational stabilization. We found that STING was ubiquitinated in myeloid cells, and this K63-linked ubiquitination could be elicited by bacterial products, including c-di-GMP. Our findings suggest a positive feedback loop wherein dysbiosis foments the accumulation of STING in intestinal myeloid cells, driving intestinal inflammation.
Keywords: STING, SAVI, Dysbiosis, Colon, Intestinal inflammation, Colitis, Microbiome, Commensal, Ubiquitination, Myeloid cells, T cells
Graphical Abstract
eTOC
To understand the function of the DNA sensing protein, STING, in intestinal inflammation, Shmeul-Galia et al. utilize a constitutively active STING mouse model that exhibits dysbiosis, severe colitis and fibrosis. Bacterial nucleotides like cyclic di-GMP initiate a feed-forward loop through the ubiquitination and stabilization of STING in intestinal myeloid cells to promote T cell-dependent colitis.
Introduction
Nucleic acid sensing is a cornerstone of host-defense. Foreign and host-DNA is detected by cytosolic DNA sensors leading to acute inflammatory responses. Cyclic GMP-AMP (cGAMP) synthase (cGAS) is a critical DNA sensor with important roles in anti-viral immunity. However, activation of cGAS by self-DNA has also been linked to sterile inflammation. Upon DNA binding, cGAS converts ATP and GTP into the non-canonical cyclic dinucleotide cGAMP, which in turn binds and activates an Endoplasmic Reticulum (ER) resident adapter protein STING (Stimulator of Interferon inducing Genes) (Chen et al., 2016, Shang et al., 2019, Ishikawa and Barber, 2008). In addition to cGAMP, STING is also activated by cyclic dinucleotides produced by bacteria, including cyclic-di-GMP (Marinho et al., 2017). STING activation leads to oligomerization and downstream activation of IRF3-dependent type I IFN responses, NF-kB-dependent proinflammatory cytokine responses, and autophagy. Activation of STING also leads to apoptosis and necroptosis in certain cell types (Zhang et al., 2019, Gui et al., 2019, Liu et al., 2015, Abe and Barber, 2014, Fitzgerald et al., 2003, Tanaka and Chen, 2012).Thus, STING can be activated by an array of signals and trigger a plethora of downstream inflammatory responses.
Given the non-discriminatory detection of host and foreign DNA by the cGAS–STING pathway, a number of homeostatic control mechanisms exist to limit excessive cGAS–STING activation. For example, nucleases such as Trex1 degrade cytosolic DNA preventing cGAS ligation and downstream activation of IFN responses. Loss of function mutations in Trex1 result in Aicardi Goutières syndrome (AGS), an interferonopathy characterized by high levels of Interferon Stimulated Gene (ISG) expression (Gray et al., 2015). Excessive activation of STING has also been linked to familial chilblain lupus, and STING-associated vasculopathy with onset in infancy (SAVI) (Jeremiah et al., 2014, Liu et al., 2014). SAVI is a rare genetic disease caused by heterozygous gain-of-function mutations in STING (Picard et al., 2015, Picard et al., 2016, Chia et al., 2016). SAVI-associated mutations result in constitutive activation of STING that is independent of cGAMP-binding (Shang et al., 2019). Murine models of SAVI, (e.g. Tmem173+/N153s, hereafter referred to as N153s) are useful in vivo tools to explore STING function and elucidate new tissue and cell-type specific roles for the STING pathway (Motwani et al., 2019, Bennion et al., 2019).
STING plays important roles in maintaining intestinal homeostasis. STING-deficient mice display an altered commensal composition and reduced intestinal epithelial cell integrity, rendering animals more susceptible to dextran sodium sulfate (DSS) or Salmonella enterica serovar Typhimurium (S. typhimurium) induced intestinal inflammation (Canesso et al., 2018). In addition, STING-deficient mice are highly susceptible to colitis-associated colorectal cancer (Zhu et al., 2014) and acute intestinal tissue injury (Fischer et al., 2017). Conversely, STING activation is linked to the exacerbation of colitis (Ahn et al., 2017, Martin et al., 2019, Ma et al., 2020). Spontaneous colitis in IL-10 deficient mice is abrogated when crossed to STING deficient mice (Ahn et al., 2017). Furthermore, colitis is greatly exacerbated in mice treated with DSS in the presence of a STING agonist23. Given these conflicting reports, the role of STING in intestinal inflammation remains unclear.
Here, we sought to delineate the functional role of STING in intestinal inflammation using a constitutively active mouse model of STING. Increased protein levels of STING correlated with the severity of intestinal inflammation in both mice with colitis and in IBD patients. Constitutive activation of STING in mice resulted in spontaneous colitis, dysbiosis and progressive chronic intestinal inflammation that led to fibrosis. Intestinal inflammation in these mice was dependent on T cell expansion in the intestinal tract. Bone marrow chimera approaches revealed STING activation and accumulation in intestinal macrophages and monocytes as the initial driver of inflammation. Accumulation of STING is these cells was prevented by depletion of Gram-negative bacteria, including Helicobacter typholonlus, which also alleviated intestinal inflammation. STING protein but not mRNA levels increased, suggesting posttranslational stabilization. We found that STING was ubiquitinated in myeloid cells, and this K63-linked ubiquitination could be elicited by bacterial products, including c-di-GMP. Our findings suggest a positive feedback loop whereby dysbiosis results in K63-linked ubiquitination and concomitant stabilization of STING in the colon, thereby supporting STING accumulation and exacerbation of inflammation.
Results
Intestinal inflammation licenses STING accumulation in the colon
To better understand the role of the STING pathway in intestinal inflammation and regulation of microbiota composition (Ahn et al., 2017, Canesso et al., 2018), we examined protein and transcript levels of STING in the colons of mice and human biopsies under conditions of intestinal inflammation. The protein levels of STING were low to undetectable in healthy mouse colons (Figure 1A–D). In contrast, mice challenged with Salmonella enterica serovar Typhimurium SL1344 (S. Typhimurium) (Figure 1A), Citrobacter rodentium (C. Rodentium) (Figure 1B) or subjected to DSS (Figure 1C) had elevated levels of STING protein in the colon. High protein levels of STING were also found in the T cell transfer model of chronic colitis (Figure 1D). STING mRNA was unchanged between treated groups and healthy controls in all cases, demonstrating that the increase in STING protein was not due to increased transcription (Figure 1E–H). In all cases, IL-1β and RANTES mRNA were increased in treated conditions. Consistent with this finding, STING expression was low in colonic tissue from healthy human patients as well as in uninvolved areas from colitis patients. In contrast, STING expression was elevated in regions of the colon affected by colitis (Figure 1I). Collectively, these findings indicate that accumulation of STING represents a feature of intestinal inflammation in both mice and humans.
Figure 1. Intestinal inflammation licenses STING accumulation in the colon.
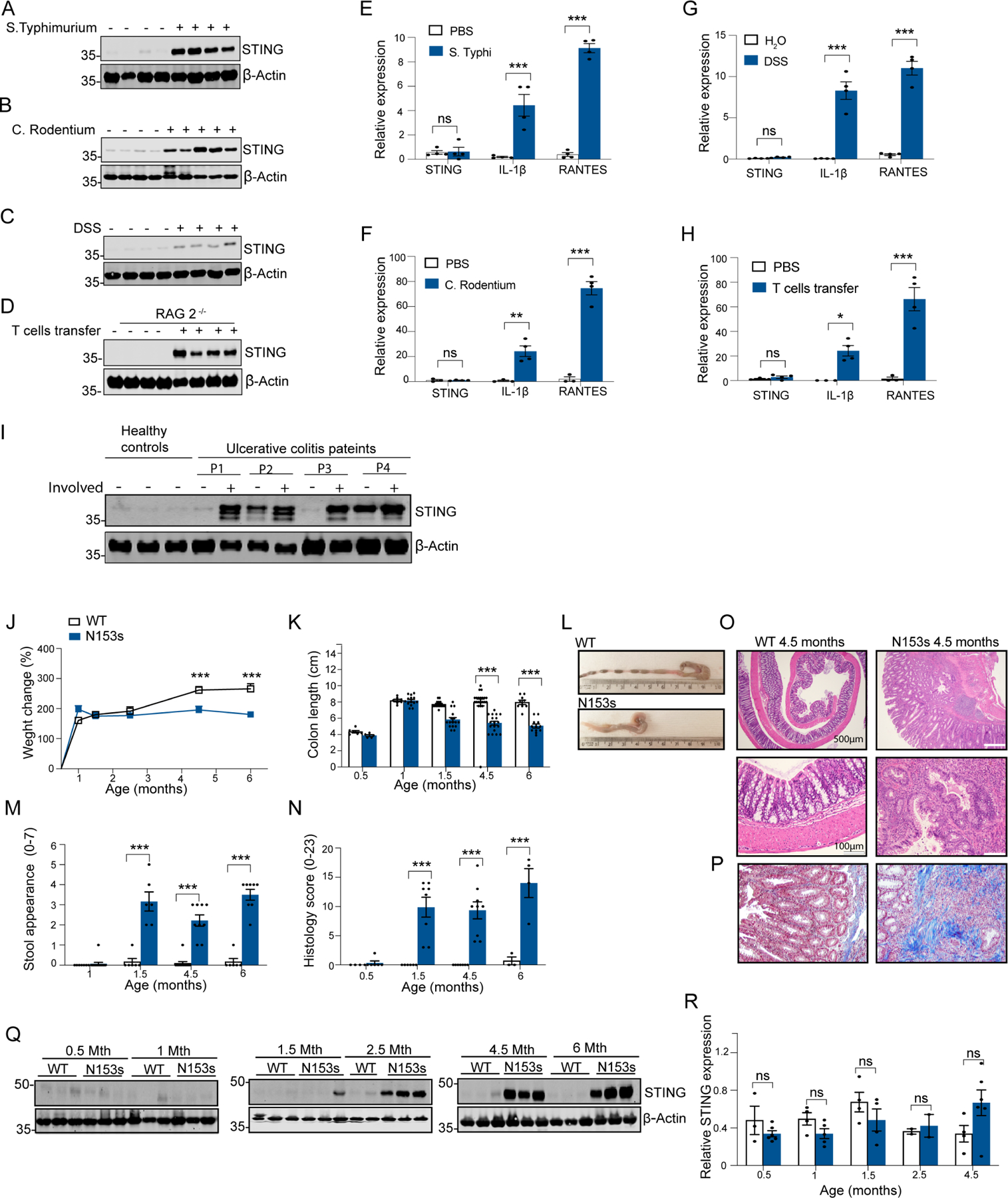
Representative images of Western blotting detecting STING (A-D) and qPCR analysis of colonic STING, IL-1β and RANTES mRNA (E-H) in mice challenged with (A, E) Salmonella typhimurium, (B, F) Citrobacter rodentium, (C, G) DSS and (D,H) adoptive CD45R+ T cells transfer (*P<0.05, **P<0.01, ***P<0.001, Data are represented as mean ±SEM of n=2–4 from 2 independent experiments). Representative image of Western blotting detecting STING in (I) healthy patients and affected and unaffected areas of colon from colitis patients. Data are represented as mean ±SEM of n=3–5). Western blot equal loading was detected by measuring β-actin and qPCR gene expression levels were normalized to TBP. Tmem173 heterozygous mice, N153s, develop spontaneous colitis and exhibit accumulation of STING and immune cells alterations in the colon. (J) Percentage weight change, (K-L) colon length, (M) stool appearance and (N) pathological score of H&E stained colon sections from N153s and WT littermate control mice at the indicated age. (***P<0.001, Data are represented as mean ±SEM of n=4–20). Representative (O) H&E and (P) Trichome stained colon sections of 4.5-month-old N153s and WT littermate control mice. (Q) Representative images of Western blotting detecting STING in N153s and WT littermate control mice at the indicated age. Equal loading was detected by measuring β-actin. (R) QPCR analysis of colonic STING mRNA in N153s and WT littermate control mice at the indicated age. Gene expression levels were normalized to TBP. (Data are represented as mean ±SEM of n=2–3 from 2 independent experiments).
STING activation promotes spontaneous and progressive intestinal inflammation and fibrosis
We next wanted to address how STING would impact intestinal inflammation. We took advantage of a STING gain-of-function mouse model in which STING is constitutively active (N153s)(Motwani et al., 2019). In addition to the lung disease previously described in these mice (Motwani et al., 2019), constitutive activation of STING resulted in spontaneous colitis with onset in 1.5-month-old mice that progressed with age (Figure 1J–P, S1A). Over a 6-month period, N153s mice demonstrated reduced body weight (Figure 1J), colon shortening (Figure 1K–L) and developed progressive diarrhea and rectal bleeding (Figure 1M). Histological analysis of the colons of N153s mice revealed goblet cell loss, crypt abscesses, erosion, hyperplasia, ulceration and lymphoplasmacytic aggregates in the mucosa, submucosa, muscularis propria and subserosa layers (Figure 1N–O, S1A). Furthermore, trichrome staining revealed fibrosis in the colons of N153s mice (Figure 1P) N153s mice exhibited colon permeability, as assessed by elevated serum concentration of FITC dextran (Figure S1B), decreased expression of colonic tight junction proteins (Figure S1C) and decreased goblet cell number (Figure S1D), compared to their WT littermate counterparts.
STING protein expression was low to undetectable in whole colon tissue of WT mice but accumulated and stabilized in N153s mice as disease progressed, as detected by Western blotting (Figure 1Q). In some circumstances, STING is an ISG that can be elevated following Type I Interferon receptor (IFNAR)-Signal transducer and activator of transcription 1(STAT1) signaling (Ma et al., 2015). However, protein levels of STING were comparable between N153s and N153s/Ifnar−/− mice (Figure S1E). In addition, the elevated protein expression of STING was independent of transcription, as STING mRNA was comparable between N153s and WT littermate controls (Figure 1R). These observations suggested that STING is regulated post-transcriptionally under conditions of intestinal inflammation. IFNAR deficiency failed to rescue intestinal inflammation in N153s mice and N153s/Ifnar−/− mice displayed comparable intestinal inflammation to N153s mice (Figure S1F–I). Therefore, STING accumulation and STING-driven intestinal inflammation occurs independently of IFNAR signaling.
Intestinal inflammation from constitutive STING activation is associated with T cell accumulation in the colon, but independent of epithelial cell death
Characterization of cell populations in the spleen of N153s mice revealed neutrophilia when compared to WT littermate controls (Figure S2A). In line with previous studies (Motwani et al., 2019, Warner et al., 2017, Wu et al., 2019), we also observed splenic T cell and B cell cytopenia (Figure S2A). To further dissect the colonic phenotype observed in N153s mice, we profiled the immune cell populations in the lamina propria (LP) and intra epithelial lymphocyte (IEL) compartments of 4.5-month-old N153s and WT littermate control mice using flow cytometry (Figure 2A–F, S2B–E). A CD45+ immune cell infiltrate was observed in the N153s mice (Figure 2A–B). Specifically, neutrophils and monocytes represented the largest proportion of myeloid cell infiltrates in the colonic LP (Figure 2C, S2C) and IE immune cells compartment (Figure 2D, S2D). As opposed to the T cell cytopenia observed in the spleen, accumulation of TCRβ CD4+ T cells was observed in the LP of N153s mice (Figure 2E, S2C). By contrast, the frequency of T cells in the IELs was reduced in N153s mice, but CD4+ T cells replaced the normally dominant CD8+ T cell subset (Figure 2F, S2D) The N153s LP CD4+ T cells included an increased frequency of IFN-γ-producing Th1 cells, reduced frequency of Treg cells and minimal numbers of Th17 cells (Figure S2E). EpCAM+ intestinal epithelial cells (IECs) were unchanged between N153s and WT littermate controls (Figure 2G). N153s mice exhibit activation of the unfolded protein response, ER stress and cell death in splenic T cells (Wu et al., 2019). We observed direct activation of ER-stress and cell death in splenic CD4+ T cells from N153s mice when measuring the ER-stress markers Bip and CHOP. Cleaved PARP was also detected in splenic CD4+ T cells from N153s, indicting active cell death in these cells. (Figure S2F). However, colonic LP CD4+ T cells from N153s mice displayed no activation of the unfolded protein response, ER-stress or apoptotic response (Figure S2F). These results suggest that colonic LP CD4+ T cells do not undergo chronic activation of ER stress and instead accumulate in the colon. Despite having no signs of intestinal inflammation at 1-month of age, N153s mice begin to accumulate colonic myeloid cells and had comparable numbers of infiltrating monocytes when compared to 4.5 month old N153s mice (Figure 2G–H, S2H–I). However, expansion of colonic CD4+ T cells had not yet occurred. Furthermore, T and B cell cytopenia was retained in the colon of these younger mice (Figure 2G–H, S2H–I). These data suggest that intrinsic activation of STING in T and B cells may induce cell death at an early stage (Wu et al., 2019), with Th1-skewed effector cells accumulating in the colon where they are spared from ER stress induced death as disease progresses.
Figure 2. N153s mice exhibit immune cell alterations in the colon.
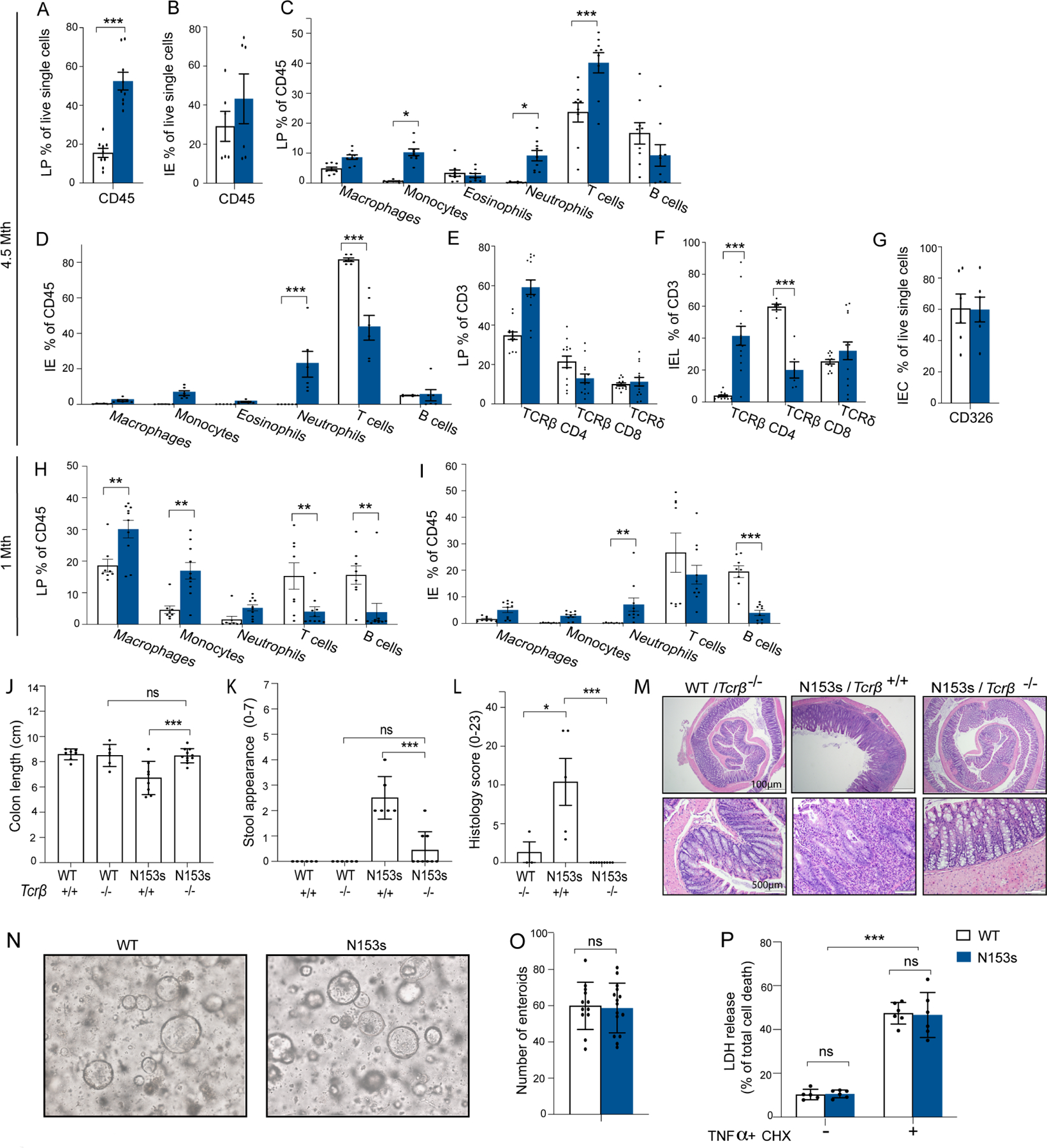
Graphical summary of flow cytometry analysis of colonic LP (A) and IE (B) of N153s and WT mice at the age of 4.5-month-old mice. CD45+ cells presented as % out of live single cells. Graphical summary of LP (C) and IE (D) immune cell populations from 4.5-month-old N153s and WT littermate control mice presented as % out of CD45+ cells. Graphical summary of LP (E) and IE (F) TCRβ CD4, TCRβ CD8 and TCRδ T cells presented as % out of CD3+ cells. (G) CD326 positive IE cells presented as % out of live single cells. (Data are represented as mean ±SEM, for J-K n=3 from 3 independent experiments, for M-N, P n=3 from 2 independent experiments, for L, O n=1–4 from 5 independent experiments). LP (H) and IE (I) immune cell populations presented as % out of CD45+ cells from 1-month-old N153s and WT mice. (*P<0.05, **P<0.01, ***P<0.001. Data are represented as mean ±SEM of n=2–3 from 5 independent experiments). Intestinal inflammation in N153s mice is dependent on TCRβ T-cells not on intestinal epithelial cells. (J) colon length colitis score, (K) stool appearance, (L) pathology score and (M) representative H&E images of colon sections of the indicated mice at 2.5 to 4.5-months of age. (*P<0.05, ***P<0.001. Data are represented as mean ±SEM of n=2–6 from 5 independent experiments). (N) Representative images of mature enteroids and (O) enteroid numbers cultured from N153s and WT intestinal epithelial stem cells for 5 days (Data are represented as mean ±SEM of n=5–6 from 2 independent experiments). (P) Percentage LDH release of single layer intestinal epithelial cells (5×10^4) treated with or without TNFα (50ng/ml) and CHX (10μg/ml) for 24 hours (***P<0.001. Data are represented as mean ±SEM of n=3 of 2 independent experiments).
Given the expansion of T cells in the colons of adult N153s mice, we next sought to define the contribution of T cells to the colitis phenotype. As such, we intercrossed N153s mice with TCRβ- or TCRδ-deficient mice (N153s/Tcrb−/− and N153s/Tcrd−/−, respectively). N153s/Tcrb−/− mice were protected from intestinal inflammation, with colon lengths (Figure 2J), stool appearance, (Figure 2K) and pathology scores (Figure 2L–M) comparable to WT/Tcrb−/− mice. Colon tissue from N153s/Tcrb−/− mice displayed low to undetectable amounts of STING (Figure S3A). Conversely, N153s/Tcrd−/− did not display ameliorated intestinal inflammation (Figure S3B–E), indicating that TCRβ, but not TCRδ, T cells are essential for intestinal inflammation in N153s mice.
STING activation is linked to epithelial cell death (Aden et al., 2018). STING-mediated cell death in IECs may initially drive barrier dysfunction, dysbiosis and expansion of intestinal T cells. Thus, we next assessed intrinsic activation of STING in ex-vivo cultured colonic enteroids. Colonic epithelial stem cells were isolated from 1-month-old mice and embedded in basement membrane matrix (Matrigel) with 50% L-WRN media until completely matured (Figure 2N). Both WT and N153s epithelial stem cells developed into mature enteroids 5 days post culture and displayed comparable numbers of enteroids (Figure 2O). To determine if N153s IECs were more sensitive to cell death, a 2D monolayer of enteroid-derived cells were treated with TNFα and cycloheximide (CHX) to induce apoptosis. Cells from WT and N153s mice showed comparable levels of lactate dehydrogenase (LDH) release both basally and in response to TNFα/CHX (Figure 2P). These results suggest that intrinsic activation of STING in IECs does not elicit cell death, in this context. Elevated levels of antimicrobial peptides were detected in colons of N153s mice as compared to WT littermate control mice (Figure S3F), further supporting normal IEC function in N153s mice. Taken together, these data show that CD4 T cell-dependent colitis in N153s mice occurring independently of intrinsic activation of STING in IECs.
STING accumulates predominantly in myeloid cells during colitis
In order to determine which intestinal cells express STING under homeostatic or inflammatory conditions, we performed immunofluorescence-immunohistochemistry (IF-IHC) on colon tissues collected from N153s and WT littermate control mice harboring intestinal inflammation. STING expression was higher in colonic WT CD11b+ cells (Figure 3A) compared to CD3+ T cells (Figure 3B), or IECs (Figure 3C). There was an accumulation of STING in CD11b+ cells from N153s mice (Figure 3A) that was not observed in CD3+ T cells (Figure 3B) or IECs (Figure 3C). This observation was further confirmed by immunoblotting. STING expression and accumulation was observed in colonic LP CD11b+ myeloid cells, but not CD11b− lymphocytes (Figure 3D). WT IECs and IELs showed low STING expression, with no accumulation observed in N153s IECs or IELs (Figure 3E). Expression and accumulation of STING in LP CD11b+ cells, but not CD11b− lymphocytes, was also observed in mice treated with DSS (Figure 3F). Thus, STING accumulates in colonic myeloid cells under conditions of intestinal inflammation.
Figure 3. STING protein primarily expresses and accumulates in resident intestinal macrophages and proinflammatory monocytes during intestinal inflammation.
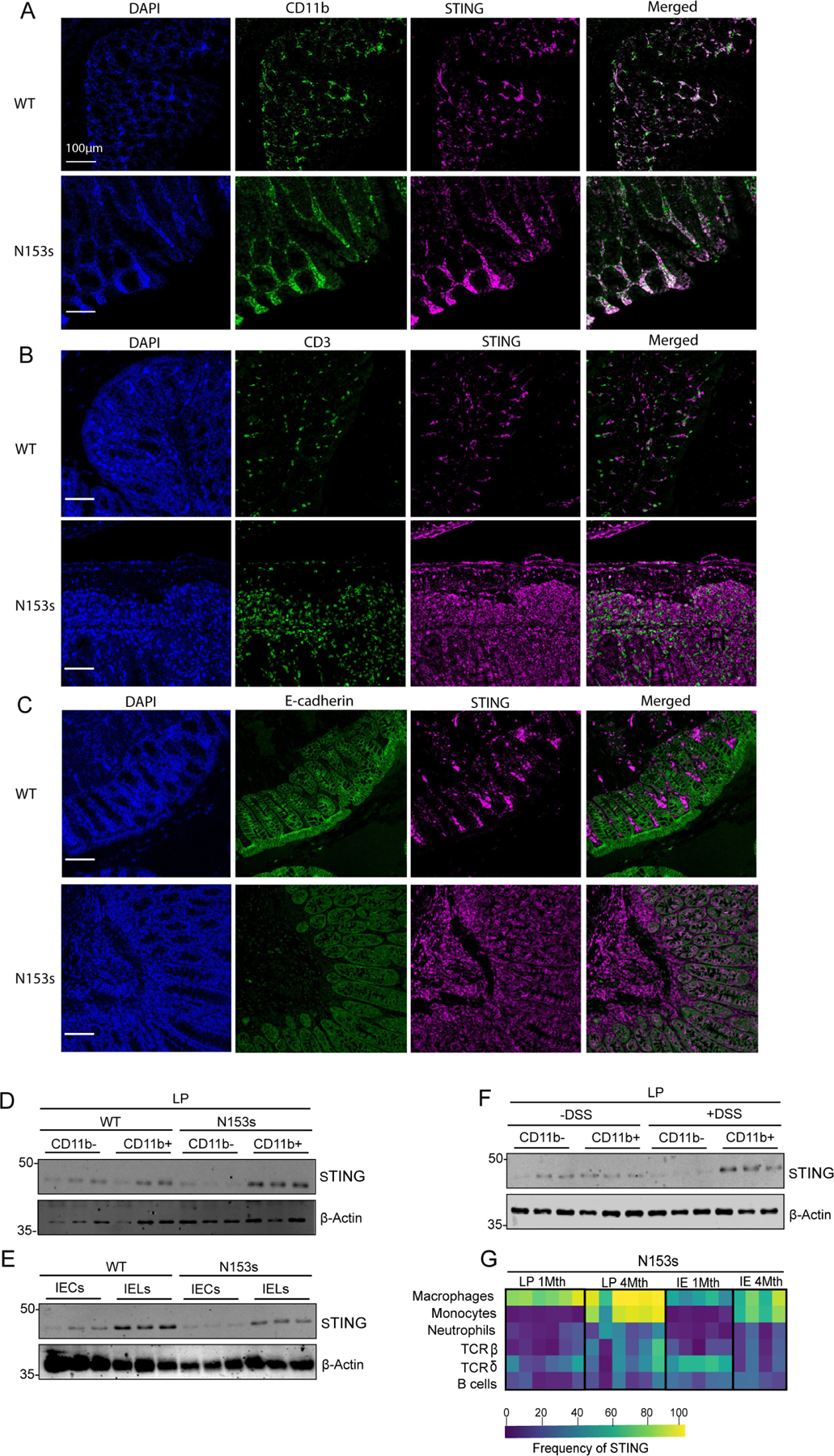
(A-C) Representative images of cellular localization of STING and (A) CD11b+ cells, (B) CD3 cells and (C) IECs (E-cadherin) in WT and N153s mice observed by confocal microscopy. Cells were probed with DAPI (left panels, blue), Opal 480-labeled CD11b+, Opal 480-labeled CD3 or FITC conjugated E-cadherin antibody as indicated (second left panels, green). Cells were co-stained with Opal 690-labeled STING (magenta, second right). Merged images of STING and the indicated cell populations are shown in the right panels. Scale bars, 100μm. Representative images of Western blotting detecting STING in indicated colonic cell populations isolated from (D-E) N153s and WT littermate control mice at 4.5 months of age and (F) WT mice challenged with 2% DSS for 7 days. Equal loading was detected by measuring β-actin. (G) Heat-map representing frequency of cells expressing STING in colonic LP and IE immune cells of each cell subset of N153s mice at the indicated age, quantified by flow cytometry (Data are represented as mean ±SEM of n=2–3 from 3–4 independent experiments).
We next characterized the specific cell types expressing STING during colitis. Given that colons from WT mice do not contain any infiltrating proinflammatory monocytes or neutrophils, we utilized colons from 1-month-old N153s mice, which have infiltrating monocytes and neutrophils but still exhibited a healthy colon. Cells expressing STING in colonic LP and IEL cells of N153s mice at 1 and 4-months of age were assessed by flow cytometry. STING-expressing cells were predominantly detected in N153s myeloid cell populations, specifically intestinal resident macrophages at 1 month of age. In addition, intestinal macrophages had significantly elevated STING expression by 4-months of age when these mice had clear intestinal inflammation (Figure 3G). Monocytes derived from healthy colons expressed low amounts of STING, however high levels of STING protein were detected in monocytes from N153s mice monitored at 4-months of age (Figure 3G, S4A–B). Neutrophils, T cells and B cells expressed low levels of STING in healthy colons and had no elevated levels of STING in inflamed colons (Figure 3G, S4A–B). Comparable numbers of resident macrophages and infiltrating proinflammatory monocytes were detected in 1- and 4-month-old N153s mice (Figure S2C,D,G,H), suggesting that increased STING staining is not due to infiltration of STING-expressing cells, but rather that there is increased expression of STING protein in macrophages and monocytes.
Intrinsic activation of STING in myeloid cells triggers intestinal inflammation
Given that STING selectively accumulated in myeloid cells, we next determined if myeloid cells were the initial driver of colitis. We assessed if STING in hematopoietic or nonhematopoietic cells initiates intestinal inflammation by performing bone-marrow chimeras in which lethally irradiated WT mice were reconstituted with bone marrow (BM) cells from WT or N153s mice (WT > WT and N153s > WT, respectively). Additionally, irradiated N153s mice were reconstituted with WT or N153s BM cells (WT > N153s and N153s > N153s, respectively). Disparate CD45 alleles were used to distinguish donor BM-derived cells from recipient radioresistant cells (Figure 4A). Successful reconstitution of the recipient mice was confirmed by expression of the donor CD45 allele by splenocytes of the recipient mice (Figure S4C). WT mice expressing CD45.1 allele and N153s mice expressing CD45.2 allele mice were used throughout the experiments, with the exception of WT mice expressing CD45.2 allele which were used as donors in WT >WT chimeras. Due to severe intestinal inflammation and weight loss (Figure 4B–F) in the N153s >WT chimeras, the experiments were terminated 6 weeks post reconstitution.
Figure 4. Intrinsic activation of STING in myeloid cells drives intestinal inflammation.
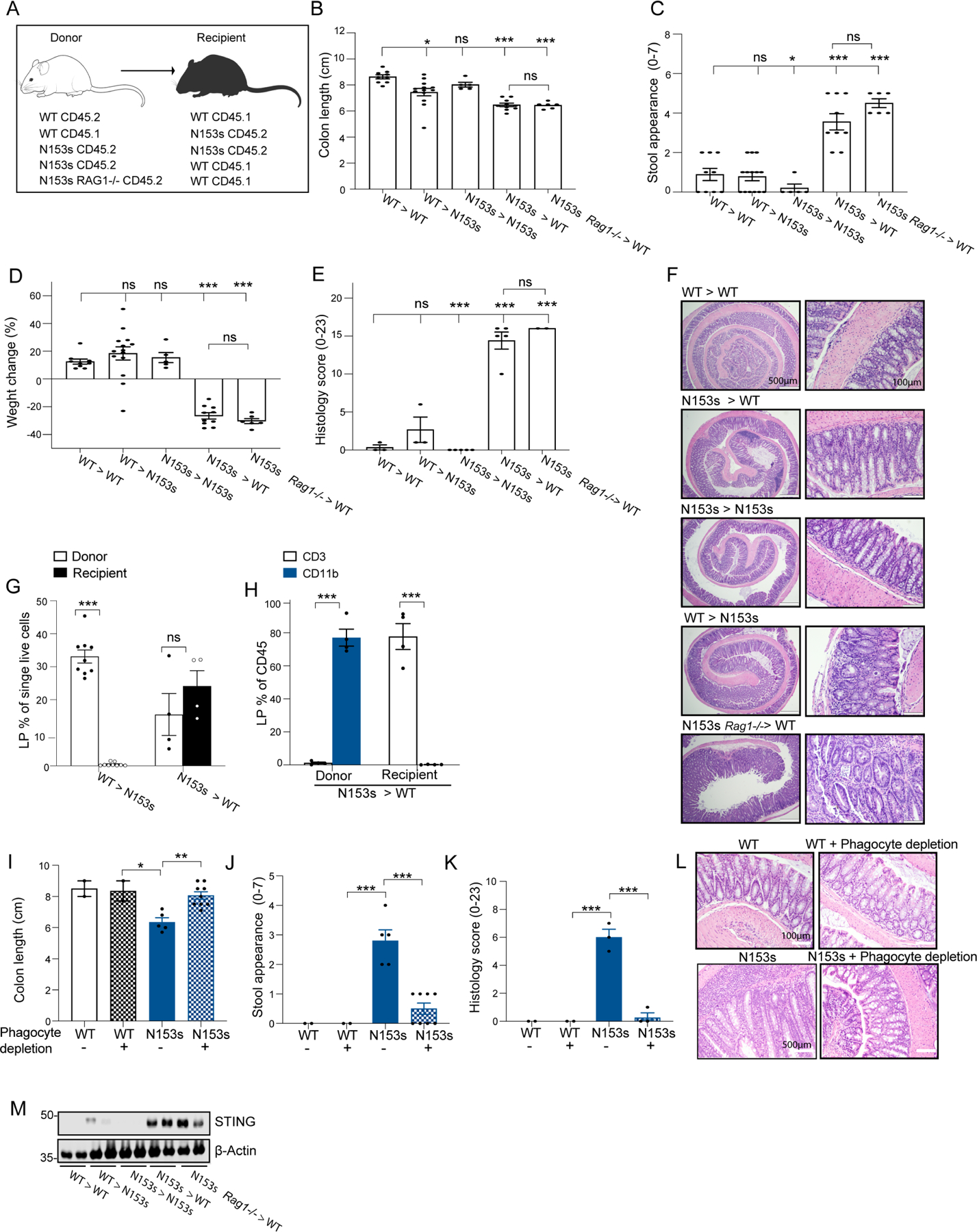
(A-I) Bone marrow chimera experiments of the indicated irradiated (900R) mice reconstituted with 107 bone marrow cells from donor mice. WT CD45.2 > WT CD45.1, WT CD45.1 > N153s CD45.2, N153s CD45.2 > N153s CD45.2, N153s CD45.2 > WT CD45.1 and N153s RAG−/− CD45.2 > WT CD45.1 mice. (A) Schematic image illustrating chimera experiment design. Graphical summary of (B) colon length, (C) stool appearance, (D) weight change and (E) pathology score of the indicated mice 6 weeks post-reconstitution (*P<0.05, **P<0.01, ***P<0.001. Data are represented as mean ±SEM of n=2–9 from 2 independent experiments). (F) Representative images of H&E-stained colon sections from the indicated mice. (G) Flow cytometry analysis of LP donor and recipient cells presented as % out of single live cells of the indicated chimera mice. (H) Flow cytometry analysis of LP CD3+ and CD11b+ cells presenting as % out of donor and recipient cells of N153s > WT chimera mice. (***P<0.001. Data are represented as mean ±SEM of n=2–5 from 2 independent experiments). (I) Colon length, (J) stool appearance, (K) pathology score and (L) representative H&E images of colon sections of the indicated mice at 2-months of age. (*P<0.05, **P<0.01, ***P<0.001. Data are represented as mean ±SEM of n=2–4 from 2 independent experiments). (M) Immunoblot analysis of STING and β-actin in colon tissue lysates of the indicated mice.
WT > WT chimeras did not develop intestinal inflammation as demonstrated by normal colon length (Figure 4B), normal stool appearance, (Figure 4C), weight gain (Figure 4D) and low pathology score (Figure 4E–F). WT > N153s chimeras also did not develop intestinal inflammation, suggesting that intrinsic activation of STING in nonhemopoietic cells was insufficient to drive intestinal inflammation. These mice mainly expressed WT donor cells with all recipient bone marrow-derived cells dying as a result of irradiation (Figure 4G, S4D). N153s > WT chimera mice developed severe colitis, further supporting the role of STING signaling in hemopoietic cells as initiators of intestinal inflammation. While CD11b+ cells in these mice were all derived from the donor, the T cells were exclusively from recipients (Figure 4G–H, S4D). These results demonstrate the persistence of WT radioresistant T cells in the N153s > WT, as documented previously (Motwani et al., 2019). Donor N153s T cells failed to reconstitute in recipient mice (Figure 4H, S4D), presumably reflecting a heightened ER stress response due to constitutive STING activation (Wu et al., 2019). These data suggest that intrinsic activation of STING in CD11b expressing myeloid cells drive intestinal inflammation irrespective of the genotype of T cells. This is further supported by our observation that depletion of colonic phagocytes using clodronate liposomes abolished colitis in N153s mice (Figure 4I–L, S4E).
We generated additional chimeras by reconstituting irradiated WT recipient mice with N153s/Rag1−/− BM. Given that the N153s/Rag1−/− mice lack mature B and T cells, CD3+ T cells in the recipient mice must be derived from radioresistant host cells. N153s/Rag1−/− > WT chimeras developed severe intestinal inflammation comparable to that seen in the N153s > WT chimeras (Figure 4A–F). The N153s > N153s chimera mice failed to develop colitis and were comparable to WT > WT chimeras. Since N153s mice were already T cell lymphopenic and N153s recipient T cells did not survive lethal irradiation, while N153s donor cells failed to engraft the T cell compartment, N153s > N153s presumably did not possess the T cells required for the development of colitis. These findings are consistent with TCR deficiency abrogating intestinal inflammation in N153s mice. WT >WT, WT >N153s and N153s >N153s chimera mice displayed healthy colons and had low to undetectable levels of STING by immunoblotting, whereas N153s > WT and N153s/Rag1−/− > WT harboring intestinal inflammation had high STING expression (Figure 4M).
Collectively, these results underscore the importance of STING activation in myeloid cells as an initial driver of intestinal inflammation and highlight how STING protein levels increase with time in colonic myeloid cells during intestinal inflammation.
Commensal bacteria stabilize STING and promote intestinal inflammation in N153s mice
Commensal dysbiosis correlates with intestinal inflammation (Levy et al., 2015) To assess if N153s mice develop dysbiosis, we performed 16S rDNA sequencing of stool samples from N153s and WT co-housed littermate controls. The N153s mice exhibited β-diversity differences associated with genotype but not by gender (Figure S5A). The relative abundance of Enterobacteriaceae (Gram-negative), Helicobacteraceae (Gram-negative), Lactobacillaceae (Gram-positive) and Peptostreptococcaceae (Gram-negative) were significantly increased in N153s mice compared to co-housed littermate controls at an early age of 1.5-months (Figure 5A, S5A–B). These bacterial families are frequently associated with inflammatory bowel disease (IBD) in humans and in murine models of intestinal inflammation (Lupp et al., 2007, Guarner, 2008, Man et al., 2008, Hold et al., 2014, Lavelle et al., 2015, Alam et al., 2020). In addition, N153s mice also had reduced relative abundance of specific bacteria including Lachnospiraceae and Rikenellaceae (Figure 5A, S5A–B), which are implicated in protection from intestinal inflammation (Dziarski et al., 2016, Shen et al., 2018).
Figure 5. Commensal dysbiosis drives colitis through accumulation of STING.
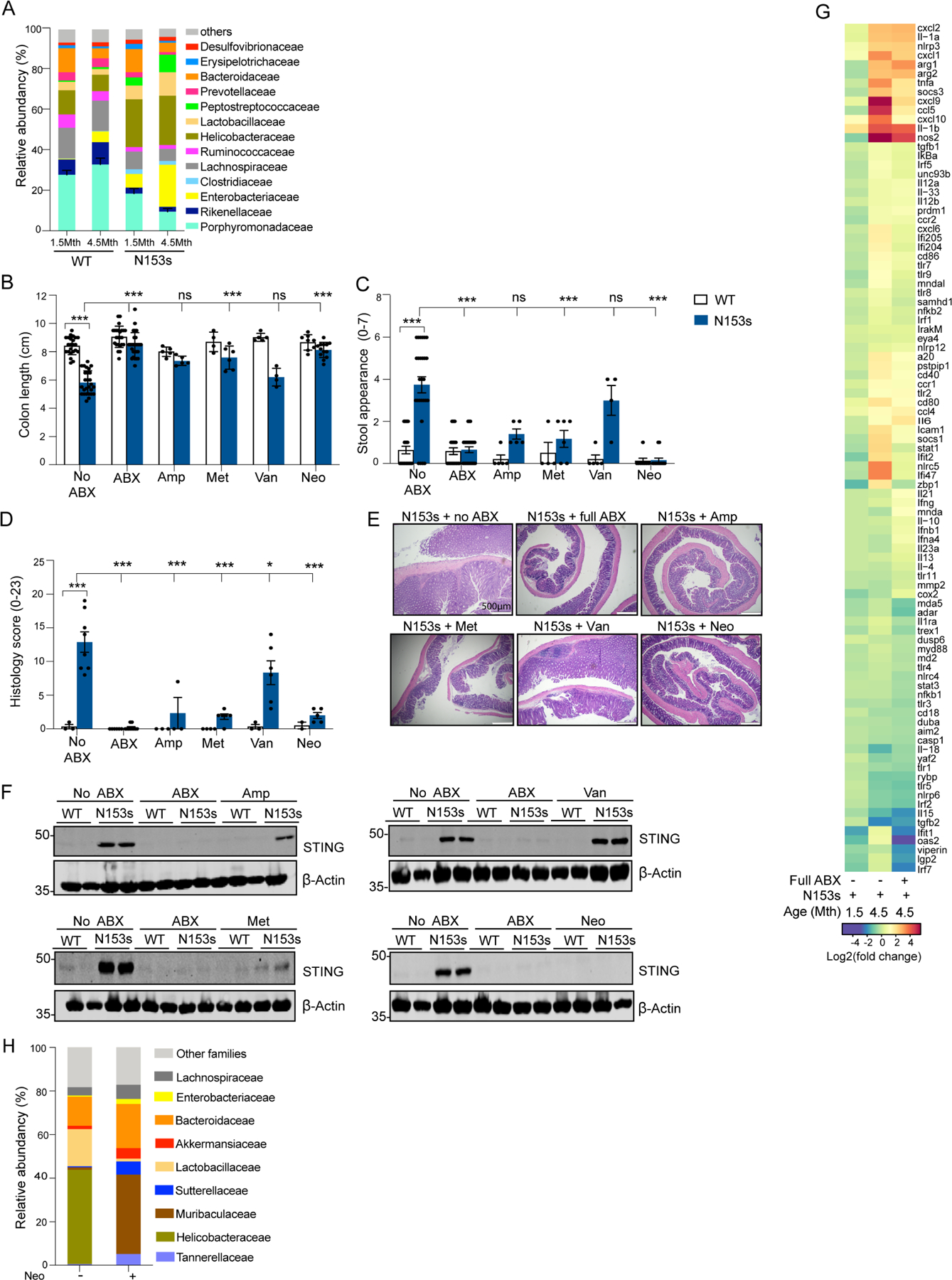
(A) Taxonomic profiling by 16S rDNA sequencing of the V3-V4 region showing relative abundances at the family levels of fecal commensals from N153s and WT cohoused littermate control mice at 1.5 and 4.5-months of age (Data are represented as mean ±SEM of n=22–24). Significance was measured by MaAsLin multivariate statistical framework. Data represent the mean relative abundance of bacterial families higher than 5% and sorted from highest significance on the bottom to lowest on the top. (B) colon length, (C) stool appearance and (D) pathology score of WT and N153s mice treated for 4-weeks with a full spectrum antibiotic (ABX) cocktail or the indicated single antibiotic, ampicillin (1gr/L), metronidazole (1gr/L), vancomycin (0.5gr/L) and neomycin (1gr/L). (***P<0.001. Data are represented as mean ±SEM of n=2–5 from 3–8 independent experiments). (E) Representative images of H&E stained colon sections from N153s mice following the indicated treatments. (F) Immunoblot analysis of STING and β-actin in colon tissue lysates isolated from WT and N153s mice following the indicated treatments. (G) Heat map of Nanostring analysis of inflammatory cytokines signatures in RNA extracted from colons of N153s and WT littermate control mice treated with full spectrum ABX cocktail calculated as fold reads in N153s over reads of the related WT mice. (Data are represented as mean of n=4). (H) Family-level relative abundance of fecal commensal flora by neomycin treatment of N153s mice as determined by WGS. Only taxa whose average relative abundance was greater than 5% are named, all other taxa are indicated as “Other families”. Significance as assessed using MaAsLin2 multivariate statistical framework. Neomycin treated mice received 1gr/L neomycin for 4 weeks.
To characterize the contribution of dysbiosis to the progression of disease, we performed antibiotic-induced microbiota depletion experiments (Hill et al., 2010, Ubeda et al., 2010). After receiving a broad-spectrum antibiotic cocktail (ampicillin, vancomycin, neomycin and metronidazole) for 4 weeks, N153s mice had normal colon length (Figure 5B), normal stool appearance (Figure 5C), and no histological evidence of colonic inflammation (Figure 5D–E). These data emphasize the fundamental role of commensals in driving disease manifestations due to STING activation. Quantitative real-time PCR with 16s rDNA gene-based specific primers targeting the V3-V4 region (Graspeuntner et al., 2018, Drewes et al., 2017) was used to assess successful depletion of intestinal bacteria treated with broad spectrum antibiotics (Figure S5C). N153s mice treated with a broad-spectrum antibiotic cocktail had a reduction in the frequency of CD45+ immune cells (Figure S6A, D). Antibiotic-treated N153s mice had lower levels of proinflammatory monocytes, neutrophils and B cells comparable to WT littermate control mice (Figure S6B, E). The expanded population of CD4+ T cells was retained in antibiotic treated N153s mice (Figure S6C, F). These data further highlight the essential role myeloid cells play in initiating STING-induced colitis in our model.
To further elucidate the role of commensals in mediating intestinal inflammation under conditions of STING activation, we administered individual antibiotics to deplete specific bacterial families. Treatment of N153s mice with neomycin, which largely targets Gram-negative bacteria, was sufficient to rescue intestinal inflammation in N153s mice (Figure 5B–E). Metronidazole, which targets anaerobic bacteria, and ampicillin which targets a broad spectrum of bacteria, both displayed partial rescue of the colonic phenotype. However, vancomycin, largely targeting Gram-positive bacteria, had no impact on disease phenotypes and was comparable to untreated N153s mice (Figure 5B–E).
We also assessed if STING protein expression was impacted by the dysbiosis observed in N153s mice. STING was undetectable by western blot in the colon of N153s mice treated with either the broad-spectrum antibiotic cocktail or neomycin (Figure 5F). Ampicillin and metronidazole partially reduced STING protein and vancomycin treatment had no impact (Figure 5F). Expression analysis of inflammatory genes in the colon of N153s mice at early (1.5-months) and late (4.5-months) timepoints revealed an early-stage inflammatory gene signature which further increased with age (Figure 5G). High levels of certain cytokine and immune response genes including TNF-α, Cxcl9, Ccl5 and Cxcl10 were reduced in 4.5-month-old N153s mice treated with broad spectrum antibiotics (Figure 5G).
Given the complete restoration of intestinal health in neomycin treated N153s mice, we next performed shotgun sequencing on stool samples from N153s mice with or without a 4-week course of neomycin treatment. Shotgun sequencing revealed a significant reduction of Helicobacteraceae, but not Enterobacteriaceae or Peptostreptococcaceae, in N153s mice that received neomycin (Figure 5H, S7A). In addition, neomycin treatment reduced the abundance of Lactobacillaceae, indicating that Gram-negative bacteria are not the sole target of neomycin. Helicobacteraceae and Lactobacillaceae were the only families reduced by neomycin. Species-level analysis of whole genome sequencing (WGS) data indicated a significant reduction of Helicobacter typholonlus in N153s mice that received neomycin (Figure S7B). Helicobacter typholonlus can induce intestinal inflammation in murine models of colitis (Chichlowski et al., 2008, Franklin et al., 2001, Kullberg et al., 2003). A 6-week course of anti-Helicobacter treatment (Sharp et al., 2008) alleviated colitis in N153s mice (Figure S7C–F). STING was undetectable in N153s depleted of Helicobacter (Figure S7G). Depletion of Helicobacter typholonlus following treatment was confirmed by qPCR (Figure S7H). Expansion of Helicobacter typholonlus was also observed in mice subjected to DSS (Figure S7I) and S. typhimurium infection (Figure S7J).
Fecal microbiota transplantation impairs intestinal inflammation and accumulation of STING in N153s mice
To further consider the role of dysbiosis as a driver of disease, we hypothesized that fecal microbiota transplantation (FMT) from WT litter mate control mice may alleviate intestinal inflammation in N153s mice (Figure 6A). FMT from WT controls administered to N153s mice for 2 weeks resulted in a significant improvement in colon length (Figure 6B), stool appearance (Figure 6C) and pathology score (Figure 6D–E). FMT eliminated the elevated STING expression in N153s mice (Figure 6F). These observations provide evidence that the microbiota regulated STING stability in the colon. While N153s mice demonstrated expansion of Helicobacter typholonlus in their stool, N153s receiving WT FMT had low Helicobacter typholonlus, which were comparable to the levels observed in WT mice (Figure S7K).
Figure 6. Fecal Microbiota Transplantation (FMT) regulates STING protein levels in the colon altering intestinal inflammation.
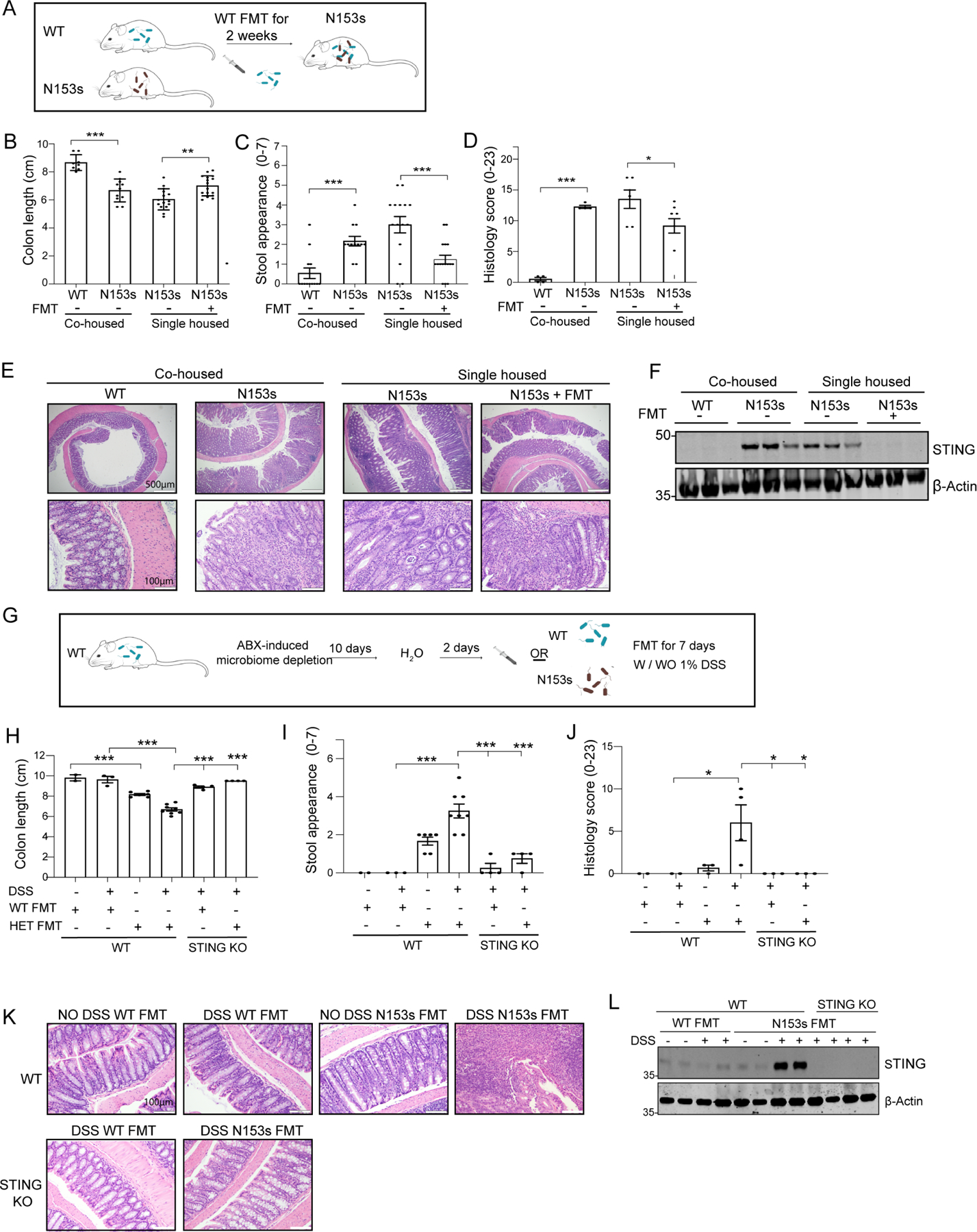
(A-F) WT FMT alleviates intestinal inflammation and STING protein levels in N153s mice. (A) Schematic image illustrating FMT experimental design. WT FMT (aerobic and anaerobic) was administered via oral gavage to N153s mice every other day for 2 weeks. (B) colon length, (C) stool appearance and (D) pathology score of the indicated mice and treatments (*P<0.05, **P<0.01, ***P<0.001. Data are represented as mean ±SEM of n=3–13 from 2 independent experiments). (E) Representative images of H&E-stained colon sections from the indicated mice. (F) Immunoblot analysis of STING and β-actin in colon tissue lysates isolated from WT and N153s mice with the indicated treatments. (G-L) N153s FMT stabilizes STING protein in the colon and promote colitis in WT mice. (G) Schematic image illustrating FMT experimental design. WT mice were treated with full spectrum antibiotic cocktail for 10 days to deplete commensal bacteria, following 2 days of sterile water. WT or N153s FMT was administrated every other day (aerobic and anaerobic) for 7 days with/wo low concentration of DSS (1%). (H) colon length, (I) stool appearance and (J) pathology score of the indicated mice and treatments (*P<0.05,***P<0.001. Data are represented as mean ±SEM of n=2–5 from 2 independent experiments). (K) Representative images of H&E-stained colon sections from the indicated mice. (L) Immunoblot analysis of STING and β-actin in colon tissue lysates isolated from WT and N153s mice with the indicated treatments.
We next assessed whether FMT from N153s mice could lead to colitis in WT mice (Figure 6G–K). To that end, WT mice were treated with broad spectrum antibiotics for 10 days to deplete commensal bacteria, followed by 2 days of regular water. Thereafter, FMT from N153s or WT littermate control mice was administrated for 7 days (Figure 6G). WT mice receiving N153s FMT demonstrated mild colon shortening (Figure 6H), normal stool appearance (Figure 6I) and normal pathology score (Figure 6J–K). Administration of FMT in conjunction with low doses of DSS, was sufficient to cause minimal gut barrier disruption and local inflammation, but not to cause colitis. WT mice receiving WT FMT maintained healthy colons with no sign of colitis (Figure 6H–K), whereas mice receiving N153s FMT developed colitis (Figure 6H–K). WT mice received N153s FMT in the presence of DSS, harboring intestinal inflammation, expressed high protein levels of STING (Figure 6L). STING-deficient mice administered N153s FMT did not develop intestinal inflammation in the presence of DSS (Figure 6H–L). Thus, reconstitution of N153s mice with a healthy WT microbiota suppresses dysbiosis and alleviates intestinal inflammation in N153s mice.
Bacterial products promote K63-linked ubiquitination and stabilization of STING during intestinal inflammation
Since the increase in STING levels was post-transcriptional, we hypothesized that STING was stabilized in the colon through a post-translational modification. K63-linked ubiquitination of proteins serves as a scaffold for recruitment of other signaling complexes but also promotes protein stability(Yang et al., 2014, Chen, 2012). Thus, we hypothesized that STING stabilization could be achieved through ubiquitination. STING was immunoprecipitated (IP) from colonic lysates under conditions of intestinal inflammation and assessed for ubiquitination. STING from N153s colons was highly ubiquitinated, while no ubiquitination of STING detected in WT healthy colons (Figure 7A). STING ubiquitination in the colon was further confirmed by detection of STING in Tandem Ubiquitin Binding Entity (TUBEs) enriched for total ubiquitin (Figure 7B). K63-linked specific TUBEs revealed that STING undergoes K63-linked ubiquitination under conditions of intestinal inflammation (Figure 7C).
Figure 7. Dysbiosis-mediated STING ubiquitination stabilizes STING in the colon.
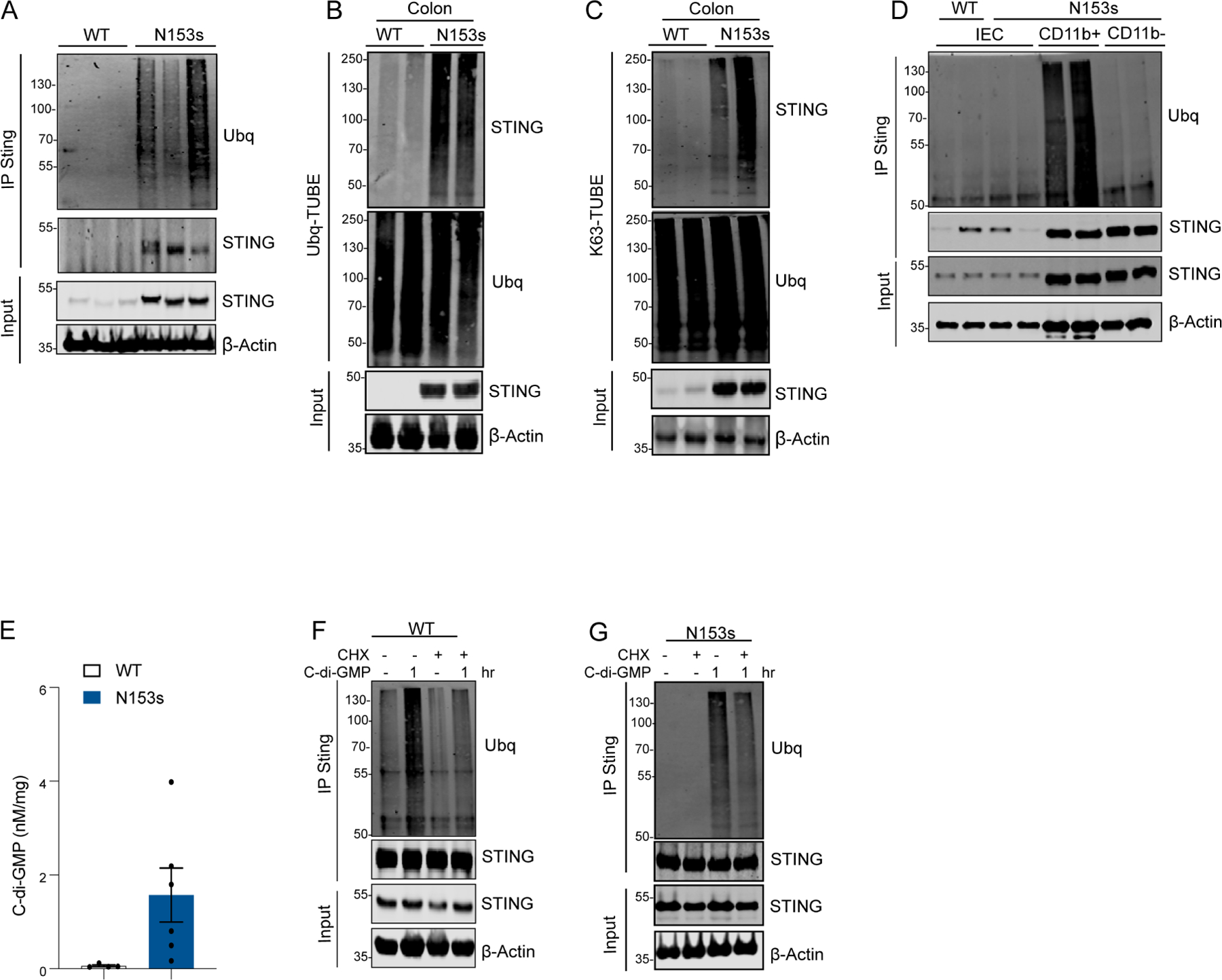
Representative images of western blotting detecting STING ubiquitination. (A) Immunoblot analysis of Ubiquitin (Ubq) and STING in lysate and immunoprecipitated (IP) samples from N153s mice. (B) STING and Ubq in TUBE-Ubq elution and cell lysates from colon tissue of the indicated mice at the age of 4.5-month-old. (C) STING and K63-Ubq in K63-TUBE-FLAG elution and cell lysates from colon tissue of the indicated mice (pooled from 4–6 mice) at the age of 4.5-month-old. Immunoblot analysis of Ubiquitin (Ubq) and STING in lysate and immunoprecipitated (IP) samples from (D) isolated colonic IECs, CD11b+ myeloid cells and CD11b- lymphocytes from N153s (for all cell types) and WT littermate control mice (only for IECs). (E) Mass spectrometry measurements of colonic C-di-GMP concentration (*P<0.05. Data are represented as mean ±SEM of 6 N153s and 4 WT littermate control mice). Immunoblot analysis of Ubiquitin (Ubq) and STING in lysate and immunoprecipitated (IP) samples from (F) WT and (G) N153s BMDM with the indicated treatments. Equal loading was detected by measuring β-actin.
We next measured ubiquitination of STING in isolated colonic IECs from N153s and WT littermate control mice (Figure 7D). As reported in Figure 4, IECs from both WT and N153s expressed low levels of STING and no ubiquitination of STING was observed (Figure 7D). We next measured STING ubiquitination in isolated colonic CD11b+ myeloid cells and CD11b− lymphocytes from N153s mice (Figure 7D). Due to low cell number in WT mice, STING ubiquitination was only assessed in CD11b+ myeloid cells and CD11b− lymphocytes isolated from N153s mice. STING ubiquitination was detected in CD11b+ myeloid cells from N153s but not CD11b− lymphocytes. Thus, ubiquitin-mediated STING stabilization and subsequent accumulation occurs primarily in myeloid cells (Figure 7D).
Given the essential role of the microbiota in driving STING stabilization and intestinal inflammation, we hypothesized that bacterial products rather than the organism itself may play a role in stabilizing STING and potentiating intestinal inflammation. Thus, we developed a quantitative mass spectrometry (MS) method for the analysis of the bacterial second messenger STING-ligand cyclic di-GMP (c-di-GMP). MS analysis revealed a significant increase of c-di-GMP in the colon tissue of N153s compared to WT littermate controls (Figure 7E). In addition, macrophages treated with c-di-GMP resulted in high levels of ubiquitinated STING (Figure 7F). In order to determine if c-di-GMP induced ubiquitination of STING enhancing its stability we utilized cycloheximide (CHX). CHX treatment blocks new protein synthesis enabling the comparison of steady state levels of STING before and after c-di-GMP treatment (Schneider-Poetsch et al., 2010). STING decreased following CHX treatment, however macrophages co-treated with CHX and c-di-GMP retained slightly higher levels of STING when compared to CHX alone (Figure 7F). STING stabilization in part correlated with c-di-GMP induced STING ubiquitination (Figure 7F). Thus, c-di-GMP-mediates STING ubiquitination and subsequent stabilization. To further confirm this, we assessed c-di-GMP induced ubiquitination of STING in macrophages from N153s mice (Figure 7G). Under resting conditions STING ubiquitination was not observed in N153s macrophages (Figure 7G). Treatment of N153s macrophages with c-di-GMP resulted in ubiquitination and stabilization of STING. Thus, c-di-GMP induced ubiquitination of STING correlates with enhanced protein stability.
Taken together, these results suggest that the microbiota, directly or through their products (e.g. c-di-GMP), can promote STING stabilization in colonic myeloid cells through K63-linked ubiquitination following intestinal dysbiosis.
Discussion
Loss of function studies in STING-deficient mice illustrate a role for STING in controlling the composition of the commensal flora. Here, we report that under homeostatic conditions STING levels are low in the colon but are elevated during intestinal inflammation. We show that commensal dysbiosis drives stabilization and accumulation of STING under conditions of intestinal inflammation with STING levels directly correlating with the severity of intestinal inflammation. These findings reveal mutual commensal-innate immune system crosstalk and emphasize the fundamental role of STING in homeostasis and intestinal inflammation.
Constitutive activation of STING in vivo leads to immune abnormalities, lung inflammation (Motwani et al., 2019) and, as we show here, resulted in the early onset of spontaneous intestinal inflammation and progression to fibrosis. Type I IFNs produced downstream of STING activation did not contribute to intestinal disease, as N153s animals had comparable colitis relative to their IFNAR-deficient counterparts. However, TCRβ-deficiency rescued intestinal inflammation in N153s mice, suggesting a fundamental role of TCRβ T cells in the progression of disease. N153s mice exhibited expansion of colonic Th1 T cells, which are directly associated with intestinal disease (Imam et al., 2018). Despite their fundamental role in the progression of intestinal inflammation in N153s mice, bone marrow chimeras as well as clodronate-liposome phagocyte depletion experiments revealed STING activation in myeloid cells—but not T cells—was the essential trigger in initiating intestinal inflammation. In addition, intestinal inflammation in N153s mice was independent of intrinsic STING activation in IECs. Thus, STING activation in intestinal myeloid cells represented an initial inflammatory signal that triggered a T cell-dependent feed-forward loop leading to dysbiosis, expansion of Gram-negative bacteria such as Helicobacter typholonlus and stabilization of STING.
Immune cells and commensal colonization of the gut mucosa have coevolved through mutual regulation (Hooper et al., 2012, Zheng et al., 2020). In line with this, STING is a key mediator of intestinal homeostasis, with STING-deficient mice displaying altered commensal composition and immune cell abnormalities (Canesso et al., 2018). Further, STING co-operates with gut commensals to produce both pro- and anti-inflammatory cytokines whilst maintaining gut homeostasis (Ahn et al., 2017). In the context of constitutive STING activation however, altered commensal bacteria profiles were identified allowing for dominance of specific families of bacteria. Why specific bacteria such as Helicobacter typholonlus benefit from prolonged STING activation and by which mechanism these species expand remains to be further elucidated. While protein levels of STING remain low in healthy colons, dysbiosis and colitis resulted in STING accumulation in the colon and potentiation of intestinal inflammation. STING was predominantly expressed in resident macrophages in healthy colons and accumulated in macrophages and infiltrating pro-inflammatory monocytes during colitis. The protein levels of STING were extremely low in intestinal IECs and lymphocytes under homeostatic conditions and remained unchanged in different models of colitis. Stabilization of STING during dysbiosis was preceded by K63-linked ubiquitination of STING, which correlated with its enhanced stability in colonic myeloid cells during intestinal inflammation.
FMT from WT mice significantly alleviated colitis in N153s mice and abrogated the accumulation of STING in the colon. Additionally, administration of FMT from N153s mice to WT mice treated with low concentrations of DSS resulted in intestinal inflammation and accumulation of STING in the colon of WT mice. These observations further emphasize the role of commensal bacteria in regulating STING in the colon. Treatment with neomycin alleviated intestinal inflammation in the N153s mice. Furthermore, N153s mice treated with neomycin had no detectable STING in the colon, suggesting that neomycin-sensitive bacteria can regulate the protein levels of STING in the gut. The Helicobacteraceae bacterial family, in particular Helicobacter typhlonius, was significantly expanded in the gut of N153s mice and was undetectable following neomycin treatment and in the WT FMT transfer experiments. Helicobacter typhlonius induces intestinal inflammation in murine models of colitis (Chichlowski et al., 2008, Franklin et al., 2001, Kullberg et al., 2003) and was elevated in mice subjected to DSS and S. typhimurium. Treatment with a therapy mainly targeting Helicobacteraceae alleviated colitis in N153s mice. However, given the expansion of other genera of bacteria in the N153s colon, it is likely this is not the sole species of bacteria contributing to the development of the disease.
Given that STING was stabilized in a range of models of colitis, we hypothesized that bacterial products rather than the bacteria itself might contribute to the stabilization of STING to potentiate intestinal inflammation. MS analysis detected high levels of c-di-GMP in the colon of N153s mice. Furthermore, treatment of macrophages with c-di-GMP resulted in high levels of ubiquitinated STING indicating that pathogen produced c-di-GMP may trigger the K63-linked ubiquitination and thus stabilization of STING in the gut.
The expansion of certain Gram-negative bacteria coupled with the loss of intestinal barrier integrity may facilitate the entry of bacterial products such as c-di-GMP to and uptake by c-di-GMP transporter expressing myeloid cells. Differential expression of c-di-GMP membrane transporters may also explain cell specific stabilization of STING in myeloid cells during intestinal inflammation. Additionally, infiltration of bacteria into the colon tissue and uptake by phagocytic cells may also facilitate c-di-GMP access to STING.
The STING pathway is also associated with inflammatory bowel disease (IBD) in humans. Genome-wide association studies (GWAS) have identified single nucleotide polymorphisms (SNPs) in signaling molecules linked to the STING pathway as risk factors for IBD. An IBD SNP, rs12654812, is associated with the promoter of DDX41, a helicase associated with the STING-TBK1-IRF3 pathway (Jiang et al., 2017, Zhang et al., 2011). Additionally, the IBD risk gene, Atg16l1, is capable of orchestrating IL-22 signaling in intestinal epithelial cells (IECs) via modulation of the cGAS-STING pathway (Aden et al., 2018). IL-22 stimulation induces transient ER stress, thus activating STING in intestinal organoids to promote TNF-induced epithelial cell death. In addition, Herpes Simplex viral (HSV) infection is also associated with IBD in some settings (Schunter et al., 2007, Nahar et al., 2019).
S. Typhimurium, C. rodentium, DSS and T cell transfer-induced colitis treatments resulted in accumulation of STING in the colon, indicating that STING can be stabilized in multiple contexts where intestinal inflammation occurs. Measurements of STING levels in the colon tissue biopsies from IBD patients further revealed stabilization of STING in the areas of the colon with active disease compared to adjacent unaffected tissue and tissue from healthy patients. Thus, stabilization of STING may represent a key marker of intestinal inflammation.
Given its role in sensing the microbial environment in the colon, we propose that STING serves as a sentinel of intestinal homeostasis. Our findings unveil a commensal-STING signaling axis that orchestrates intestinal inflammation and suggests that balancing STING function during infection or intestinal epithelial barrier dysfunction could limit the progression of IBD. Further, we propose that the N153s mouse represents a new genetic model of intestinal inflammation and fibrosis that may be further leveraged to study the contributing factors that lead to the progression of IBD and aid in the development of new therapeutics for patients.
LIMITATIONS OF THE STUDY
The current study proposes a commensal bacteria-innate immune axis that orchestrates intestinal inflammation via regulation of STING stabilization in the colon. Given the important role the microbiota plays in regulating STING during intestinal inflammation, it is important to highlight the variation that may exist across mouse vivaria. The abundances of different bacterial species may alter the severity of intestinal inflammation observed. In our study, enhanced levels of STING correlated with an increase in the levels of c-di-GMP and ubiquitination of STING. Although c-di-GMP is a STING ligand we cannot rule out other indirect stimuli which may also contribute to enhanced STING stability under conditions of intestinal inflammation. In addition, the precise E3 ligase that catalyzes K63-linked ubiquintation of STING in intestinal myeloid cells is unknown. In our study, STING accumulation was observed in all of the experimental animal models of colitis. In addition, we also observed STING accumulation in colon biopsies from ulcerative colitis patients, relative to healthy control tissue. Due to limitations in selecting what treatments the ulcerative colitis patients received, we were unable to directly show that stabilization of STING in ulcerative colitis patients was microbiome dependent.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Katherine A Fitzgerald (Kate.Fitzgerald@umassmed.edu).
Material availability
This study did not generate new unique reagents.
Data and Code availability
Whole-genome shotgun (WGS) DNA sequencing and 16s rDNA profiling generated in this study are available at NCBI BioProject, BioProject ID: PRJNA642084.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal studies.
All animal experiments were approved by the Institutional Animal Care Use Committees at the University of Massachusetts Medical School. Animal were kept in specific pathogen free (SPF) environment. N153s mice were generated as previously described (Motwani et al., 2019) and heterozygous mice were generated by crossing heterozygous male mice with WT littermate control female mice. Co-housed WT littermate control mice were used throughout the study unless mentioned otherwise. No gender bias was observed for intestinal inflammation and both male and female mice were used. Ifnar−/− mice were kindly provided by Prof. Jonathan Sprent, from Scripps Research, CA. Tcrδ−/−, Tcrβ−/− and Rag1−/− mice were kindly provided by Prof. Joonsoo Kang, Prof. Stuart M Levitz and Prof. Andrea Reboldi from UMASS medical school, MA, respectively. CD45.1 B6 and C57BL/GL mice were purchased from Jackson Laboratory and were back crossed for two generations prior to experimental use. No gender bias was detected with both males and females were used throughout the study. Mice at different age were used for the assessment of colitis at different stages of the disease. Group sizes are specified in the figure legends. Animal were used solely for experiments described in this study and were not subjected to any treatment unless mentioned otherwise. Mice were co-housed with no more than 5 mice per cage.
Bone Marrow Derived Macrophage (BMDM) maturation.
Tibia and femur bones were extracted from 6–8 weeks old mice and bone marrow was flushed using PBS. BM cells were cultured in DMEM supplemented with 10% FCS, 1% penicillin and streptomycin and 20% L929 conditioned media for 7 days.
Human subjects
4 colitis and 3 healthy control patients were used. Biopsis were collected and stored at −80°C until used. samples were processed and and used for Western blot together.
METHOD DETAILS
Immunoblotting and Immunoprecipitation.
For immunoblot analysis cells or were lysed in NP-40 lysis buffer (50mM Tris-HCl, pH 7.4, containing 150mM NaCl, 0.5% (w/v) IgePal, 50mM NaF, 1mM Na3VO4, 1mM dithiothreitol) containing 1mM phenylmethylsulfonyl fluoride and protease inhibitor cocktail (ThermoFisher). For analysis of whole colon tissue was first sonicated in lysis buffer and protein concentration determined by DC protein assay kit (Bio Rad). For immunoprecipitation of STING, cells were treated as indicated and collected in 250μl of NP-40 lysis buffer, followed by incubation for 15 min on ice. Prior to immunoprecipitation lysates were incubated in 10% SDS at 95°C to remove any STING- associated proteins for ubiquitin analysis. Lysates were incubated with STING antibody and protein A/G FLUS-agarose (Santa Cruz) was added to each sample, followed by incubation overnight at 4°C. Immunoprecipitates were collected by centrifugation and washed three times with 1ml of lysis buffer. Immunoprecipitates were eluted from beads using 1X Lamelli sample buffer. Samples were resolved by using SDS-PAGE and transferred to nitrocellulose membranes and analyzed by immunoblot with the indicated antibodies. Immunoreactivity was visualized by the Odyssey Imaging System (LICOR Biosciences).
Tandem Ubiquitin Binding Entity Analysis.
Colon tissues were sonicated in TUBE lysis buffer (100 mM Tris HCL pH 7.5, 150 mM NaCl, 5 mM EDTA, 0.5% NP-40, 0.5% Triton) containing 1,10-phenanthroline (5 mM), PR-619 (100 μM) and N-Ethylmaleimide (5 mM). Protein concentration was determined by DC protein assay kit (Bio Rad). For enrichment of ubiquitinated proteins, Ubq-TUBE-Agarose (UM-401) or K63-TUBE-FLAG (UM-604) (LifeSensors) were used according to manufacturer instructions. Briefly, cell lysates were incubated for 1 hour on ice to allow TUBE binding. For enrichment of FLAG-tagged TUBEs, ANTI-FLAGM2-affinity gel (A220, Sigma) was equilibrated in TBST for 5 min then added to samples and incubated at 4 °C for 2 hours with rotation. TUBE-enriched FLAG affinity gel was then collected by centrifugation for 5 minutes at 5000 rpm at 4 °C and washed three times with 1ml of wash buffer (TUBE lysis buffer without Triton). An aliquot (25 μl) of SDS-PAGE sample buffer (62.5 mM TrisHCl, pH 6.8, 10% (w/v) glycerol, 2% (w/v) SDS, 0.7 M β-mercaptoethanol and 0.001% (w/v) bromophenol blue) was added to the beads. Samples were resolved by SDS-PAGE, transferred to nitrocellulose membranes and analyzed by immunoblot with the following antibodies as appropriate: anti-STING, anti-ubiquitin. Immunoreactivity was visualized by the Odyssey Imaging System (LICOR Biosciences).
Lamina propria and Intestinal epithelial cells extraction.
For extraction of IECs, colon samples were added to Hanks’ Balanced Salt Solution (HBSS) containing 2.5mM EDTA, 1mM DTT and 5% of fetal calf serum and incubated at 37°C with 300RPM for 30 minutes. Remaining colon pieces were then washed with PBS and further digested for LP cell extraction in HBSS containing 10% fecal calf serum, 0.5mg/ml collagenase I (Gibco), 0.5mg/ml collagenase IV (Millipore Sigma) and 0.1mg/ml DNase I (Roche) at 37°C with 300RPM for 30 minutes. Samples were then passed through 100μm mesh and cells were collected by centrifugation at 900(xg) for 5 minutes.
NanoString.
Total RNA was isolated from whole colon using Aurum™ Total RNA Mini kit (Bio Rad) and quantitated by a Nanodrop ND-1000 spectrophotometer (Thermo Scientific). 50ng of RNA was then hybridized with a custom probe set and data was analyzed using the NanoString nSolver analysis system (NanoString technology). Gene expression data was normalized to internal positive and negative controls. Heat map was generated using R-software.
cDNA synthesis and real time PCR.
Total RNA was extracted from whole colon and 1μg of RNA was reverse transcribed using the iScript cDNA synthesis kit (Bio Rad). 5ng of cDNA was then subjected to qPCR analysis using iQ SYBR Green super-mix reagent (Bio Rad). Gene expression levels were normalized to TATA-binding protein (TBP). Relative mRNA expression was calculated by a change in cycling threshold method as 2−ddC(t). Specificity of RT-qPCR amplification was assessed by melting curve analysis. The sequences of primers used in this study are as follows: STING forward: GGTCACCGCTCCAAATATGTAG, STING reverse: CAGTAGTCCAAGTTCGTGCGA, TBP forward: GAAGCTGCGGTACAATTCCAG, TBP reverse: CCCTTGTACCCTTCACCAAT.
Colitis score.
Colitis scoring was carried out on freshly collected fecal samples using the following criteria: stool consistency: 0- well-formed pellets, 1- changed form pellets, 2- loose stool, 3- diarrhea, occult: 0-no blood, 1- traces of blood, 2-moderate, 3- severe, 4-bleeding from anus.
Histology.
Colon tissues were fixed in 10% neutral buffered formalin for 24–48 hours before being processed and embedded in paraffin. Five micrometer thin sections were stained by H&E or PAS in an automated stainer (Leica Autostainer XL) or stained using a trichome staining kit. Histomorphology of each H&E slide was evaluated by Applied Pathology Systems at low and high-power field on an Olympus BX40 microscope, and the images were captured with Olympus cellSens Entry software at X4 magnifications. Grading of histology scores was performed by Applied Pathology Systems under the following scoring criteria. Inflammatory cell infiltrate was evaluated by the number of leukocyte foci: 0- no significant change, 1- mild, infiltrated leukocytes in focal or occasional, 2- moderate, infiltrated leukocytes with more than one focus and 3- severe, infiltrated leukocytes diffuse or continuous. Epithelial changes in goblet cell loss were evaluated by the reduction of goblet cell numbers relative to baseline goblet cell numbers per crypt: 0- no significant change, 1- goblet cell loss of 10% of normal amount, 2- goblet cell loss of 10–50% and 3- goblet cell loss higher than 50%. Crypt abscesses were evaluated by the neutrophils in crypt lumen: 0- no crypt abscess, 1- rare crypt abscesses, 2- multiple crypt abscesses and 3- continuous crypt abscesses. Erosion was evaluated by the loss of surface epithelium: 0- no erosion, 1- one focus, 2- multiple foci and 3- continuous surface loss. Hyperplasia was evaluated by the increase in crypt number or length relative to baseline, visible as crypt elongation or thickened crypt layers: 0- no hyperplasia, 1- mild, increase 50%, 2- moderate, increase of 50–100% and 3- marked, increase 100%. Ulceration was evaluated by crypt loss reaching beyond muscularis mucosae: 0- no ulceration, 1- one focus, 2- multiple foci and 3- continuous. Lymphoplasmacytic aggregates were evaluated by a score based on the location of lymphoplasmacytic aggregates reach: 0- no lymphoplasmacytic aggregates, 1- lymphoplasmacytic aggregates reach mucosa, 2- lymphoplasmacytic aggregates reach submucosa and 3- lymphoplasmacytic aggregates reach muscularis propria or subserosa.
Flow cytometry.
LP and IE cells were stained with anti-CD45.2 BV650, anti-CD11b BV510, anti-Ly6C APC, anti-TCRß PerCP-cy5.5, anti-CD8 Alexa 700, anti-CD11c eF450, anti-CD64 BV711, anti-Ly6G PE-cy7, anti-Ly6G FITC, anti-CD3 PerCP-ef710, anti-CD4 APC-Cy7, anti-TCRδ PE, anti-B220 PE-Cy5, anti-CD326 APC, anti-CD45.1 APC-Cy7. Cells were acquired on a CytekTM Aurora technology. Flow cytometry analysis was done with the FlowJo software. For intracellular staining of STING, cells were fixed and permeabilized using eBioscience Foxp3/Transcription Factor Staining Buffer Set and stained with anti-STING FITC. For Th1 and Th17 cells, colonic LP cells were fractionated on a Percoll density gradient (40% and 80%). For intracellular cytokine staining, 1.5×106 cells were washed in RPMI supplemented with 10% fecal calf serum, 1% penicillin/streptomycin cocktail and 0.1% ß-Mercaptoethanol and treated with 50ng/ml PMA, 1μg/ml ionomycin and Brefeldin A (Biolegend) for 3.5 hours at 37°C. Cells were then washed with MACS buffer and stained with anti-CD3 PerCP-ef710, anti-CD4 APC-Cy7, anti-CD8 Alexa 700, anti-TCRδ PE and Ghost violet 540 viability dye. Following, cells were washed with MACS buffer, fixed and permeabilized by the eBioscience Foxp3/Transcription Factor Staining Buffer Set and stained intracellularly with anti-FoxP3 FITC, anti- IL-17 PE Cy7, anti-IFNγ eF450 and anti-RORγt PE eFluor610. For sorting of colonic and splenic cells, cells were extracted as described under sterile conditions and sorted directly into lysis buffer. Sorting was performed on FACSAria III or FACSAria Fusion.
Bone marrow chimera.
Lethally irradiated (900R) 8-week-old recipient mice were reconstituted by retro-orbital injection of 1×107 bone marrow cells from age and gender matched donor mice. Extent of reconstitution was assessed by flow cytometry for CD45.1/CD45.2 allele-expressing cells in the spleen.
Antibiotic-induced microbiota depletion.
3-month-old N153s and co-housed WT littermate controls mice were treated with autoclaved drinking water or autoclaved drinking water supplemented with 1 gr/L neomycin, 1gr/L metronidazole, 0.5 gr/L vancomycin and 1 gr/L ampicillin for 1 month. Antibiotics were given individually or as a cocktail containing all 4 antibiotics.
16S rDNA quantification.
Fecal pellets were collected, and total DNA was extracted using DNeasy PowerSoil Kit (Qiagen) and subjected to qPCR using universal primers referred to as the 319F and 806R that hybridize to the V3-V4 region of the rRNA gene.
Fecal Microbiota transplantation.
WT or N153s FMT into N153s mice: N153s and co-housed WT littermate controls mice were separated when weaned and aged until 2–3 months of age. FMT was performed with fresh feces from WT donor mice and administered to N153s recipient mice every other day via oral gavage. To prevent opportunistic organism growth, fecal samples were collected freshly, processed at room temperature and immediately gavaged to the recipient mice. Anaerobic FMT were prepared under anaerobic conditions, using an anaerobic chamber. WT FMT into WT or N153s mice: WT mice were treated with broad spectrum antibiotics for 10 days followed by regular water for 2 days. Thereafter, mice FMT from WT or N153s mice was performed w/wo the presence of 1% DSS for a period of 7 days.
Enteroids.
Enteroids were prepared as previously described (Miyoshi and Stappenbeck, 2013). Briefly, 4-week-old N153s and WT mice were euthanized, and colons were extracted and washed with PBS and all fat and connective tissue was removed. 3cm of distal colon was digested in collagenase I solution for 30 minutes with shaking. Cells were then filtered using a 70um cell strainer and washed twice in washing medium (DMEM/F12). Cells were then resuspended in matrigel in 24 well plates. Plates were inverted in culture to polymerize the Matrigel. After polymerization, 500ul of 50% L-WRN conditioned media containing 10μM Y27632 and 10μM SB431542 were added to each well. After 3 days, media was replaced with DMEM/F12 containing 20% fecal bovine serum, 100U Penicillin/Streptomycin and 2mM L-Glutamine and enteroids were evaluated after 5 days. For 2D monolayers, enteroids were disrupted by trypsin and cells were cultured in 96 well plates (30,000 cells/well).
DSS-induced acute colitis.
C57BL/GL mice were treated with 3% DSS in the drinking water for 7 days followed by 2 days water. Control C57BL/GL mice were given regular drinking water.
Salmonella-induced colitis.
C57BL/GL mice were pretreated with streptomycin for 1 day following by administration of S. typhimurium (SL1344) by gavage (1.8*10^8 CFU). Colon samples were extracted 2 days post infection.
Citrobacter-induced colitis.
C57BL/GL mice were infected with Citrobacter rodentium by gavage (4.4*10^8 CFU). colon samples were collected 2 weeks post infection.
T cell adoptive transfer.
B6 CD4 splenocytes were negatively selected by MicroBeads (Miltenyi Biotec) and were sorted by gating of naïve CD4+ CD45Rhi live T cells. Cells were then transferred by IP injection (0.5*10^6 cells/mouse into immunodeficiency RAG2−/− mice and colon samples were extracted 5 weeks post transfer.
Microbiome sequencing:
Whole-genome shotgun (WGS) DNA sequencing libraries were constructed using Nextera XT DNA Library Prep Kits (Illumina Inc., San Diego, CA) and sequenced on a NextSeq500 Sequencing System as 150-base paired-end reads. Reads were trimmed reads and removed host decontamination using Trimmomatic and Bowtie2 (Bolger et al., 2014, Langmead and Salzberg, 2012) using Mus musculus genome assembly mm10 as the host reference. Reads were profiled for bacterial spices abundances using MetaPhlAn2 version 2.9.14 and database mpa_v292_CHOCOPhlAn_201901 (Truong et al., 2015). Sequencing libraries for 16S rDNA profiling were constructed following methods previously described (Kozich et al., 2013) using the 341F and 806R universal primers to amplify the V3-V4 region. 300nt paired-end sequences were generated on the Illumina MiSeq platform. Reads were assembled and clustered, and an OTU table was generated using the UPARSE pipeline in USEARCH version v10.0.240 (Edgar, 2013). Taxonomic classifications were determined using SINTAX (Edgar, 2013) and RDP training set v16 (with species names) (https://drive5.com/usearch/manual/sintax_downloads.html).
Colon permeability analysis:
Mice were water and food restricted. The next morning, mice were administrated with 450mg/kg fluorescein isothiocyanate 4KDa conjugated dextran, FITC-dextran, by oral gavage (SIGMA) and euthanized 4 hours post administration for serum collection. FITC-dextran concentration in serum was calculated by standard concentrations prepared in PBS ranging from 0 to 12μg/ml. Emission signal in plasma from mice receiving PBS was subtracted.
Phagocyte depletion experiment:
1.5- to 2-month-old mice were injected IP with 200μl clodronate liposomes inducing apoptosis of all phagocytic cells every other day for 3 weeks. Untreated mice received control liposome.
Anti- Helicobater therapy:
2–3-month-old age mice were subjected to anti- Helicobacter therapy treatment (BioServ) containing 3 mg amoxicillin, 0.5 mg clarithromycin, 1 mg metronidazole, and 20 μg omeprazole per 5g of food. Mice were maintained on the anti- Helicobacter therapy for 6 weeks.
MACS beads separation of colonic cells:
Colonic cells were extracted as described above and subjected to cell debris depletion kit (Miltenyi Biotec) followed by column based magnetic cells separation. For LP and IE cells isolation, cells were subjected to EasySep™ Mouse CD11b positive selection kit (StemCell) and CD11b+ cells were collected. Negative fraction containing CD11b− cells were further subjected to CD45 MACS MicroBeads (Miltenyi Biotec) for CD11b− lymphocytes isolation.
Immunohistochemistry (IHC) & immunofluorescent (IF) staining:
Immunofluorescence staining of mouse colons was performed by Applied Pathology Systems, LLC. Formalin-Fixed Paraffin-Embedded (FFPE) colon tissues were sectioned and mounted to glass slides. Tissue sections were dewaxed, rehydrated, and subjected to the antigen retrieval in microwave with AR9 Buffer (Akoya Biosciences). Slides were then blocked with Blox All blocking buffer (Vector Laboratories) and Opal antibody block reagent (Akoya Biosciences) prior to the co-staining. The co-staining of STING (CST) with CD3 (Genetex) or CD11b (Abcam) was performed using the Opal 4 color manual IHC kit (NEL810001KT). STING was labeled with Opal 690 reagent while CD3 or CD11b was labeled with Opal 480 reagent FITC conjugated E-Cadherin antibody (BD Biosciences) was used in the STING and E-Cadherin co-staining. Fluorescence co-stained slides were cover slipped with VectorSheild Mounting media (Vector Laboratories) before imaging with Leica SP8 Lightning Confocal Microscope.
Quantification of cyclic guanosine monophosphate (Cyclic-di-GMP) by LC-MS/MS
Mouse colon tissue was homogenized, and protein precipitation was carried out by adding 100 μl of ice-cold 80% acetonitrile, 20% water and centrifuging at 16,000 g for 5 minutes. The supernatant was collected, spiked with 2 μl of 5μM 13C10, 15N5 cyclic guanosine monophosphate-adenosine monophosphate (2’,3’-c-GAMP), and analyzed by liquid chromatography–tandem mass spectrometry (LC-MS/MS). The analysis was performed by injecting 10 μl extract in technical triplicate onto a 2.1 × 50mm(1.8μm) iHILIC-Fusion (HILICON AB, Umeå, Sweden) column using an UltiMate 3000 UPLC (Thermo Fisher Scientific, Waltham, MA) interfaced to a TSQ Quantiva (Thermo) triple quadrupole mass spectrometer operating in the positive ion electrospray mode. LC separation was carried out at a flow rate of 0.3 ml/min with mobile phase (A) containing 20 mM ammonium acetate in water (pH 5.0) and mobile phase B containing 20mM ammonium acetate, 0.1% (v/v) formic acid, 80% acetonitrile in water. The gradient conditions were as follows: 0–2 min (100% B), 2–15 min (100-37% B), 15–16 min (37–100% B), and 16–20 min (100%B). For 3’,5’ cyclic-diGMP, data was collected in selected reaction monitoring (SRM) mode using m/z 691.2>97.1, 691.2>135, 691.2>152, 691.2>248, and 691.2>540 transitions. For 13C10,15N5-2’,3’c-GAMP m/z 690.1>146.1, 690.1>152, 690.1>327.1, 690.1>491, 690.1>521 SRM transitions were monitored. Quantitative analysis of 3’,5’-cyclic-diGMP was performed using peak area of SRM transition at m/z 691.2>152, and 13C10, 15N5 2’,3’-c-GAMP using summed peak area of SRM transitions at m/z 690.1>146.1 and 690.1>521. A 9-point calibration curve in the range of 0.002 – 0.5 μM was constructed by making two-fold serial dilutions of 0.5 μM cyclic 3’,5’-diguanylate monophosphate (InvivoGen, San Diego, CA) in a solution containing 80% acetonitrile in water, each spiked with 0.1 μM of 13C10, 15N5 2’, 3’-c-GAMP. Integration of peak areas and further data analysis was carried out in Xcalibur Quan Browser (Thermo). Sample concentrations were determined from the calibration curve (with 1/X weight, R2 = 0.99). The relative standard deviations for the triplicate analyses were all within 5%.
QUANTIFICATION AND STATISTICAL ANALYSIS
Samples sizes were chosen with adequate statistical power based on previously published comparable studies. Differences between group means were tested with a t-test when the experiment contained two groups, or one-way ANOVA (followed by a Tukey post hoc test) when the experiment contained more than two groups. In cases where the experiment was running in two batches, “batch” was entered as a random categorical factor into a 2-way ANOVA. All pathology evaluation and takedowns were performed blindly. Data are presented as mean ± SEM. For in vivo and in vitro experiments, n represent number of animal or number of cells, respectively. Values of p < 0.05 were considered statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001.
For microbiome sequencing, ecological diversity metrics were generated using QIIME2 software version 2020.2 (Bolyen et al., 2019). Beta diversity was evaluated using the weighted and unweighted UniFrac metrics for 16S rDNA data (Lozupone and Knight, 2005) and Bray-Curtis dissimilarity for WGS data; significance was tested using pairwise PERMANOVA. Differential abundance of taxa was evaluated using MaAsLin2 version 0.99.12 (Multivariate Analysis by Linear Models 2; https://huttenhower.sph.harvard.edu/maaslin2).
Supplementary Material
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-CD45.2 Brilliant Violent 650 | Biolegend | Cat# 109836, clone 104 |
| Rat anti-CD11b Brilliant Violent 510 | Biolegend | Cat# 101245, clone M1/70 |
| Rat anti-Ly6C APC | eBioscience | Cat# 17-5932-82, clone HK1.4 |
| Armenian hamster anti-TCRß PerCP Cy5.5 | Invitrogen | Cat# 45596182, clone H57-597 |
| Rat anti-CD8a Alexa Flour 700 | eBioscience | Cat# 56-0081-82, clone 53-6.7 |
| Armenian hamster anti-CD11c eFluor450 | eBioscience | Cat# 48-0114-82, clone N418 |
| Mouse anti-CD64 Brilliant Violent 711 | Biolegend | Cat# 139311, clone X54-5/7.1 |
| Rat anti-Ly6G/LyGC PE Cy7 | eBioscience | Cat# 25-593182, clone RB6-8C5 |
| Rat anti-Ly6G FITC | BD Biosciences | Cat# 551460, clone 1A8 |
| Rat anti-CD3 PerCPef710 | Invitrogen | Cat# 46003280, clone 17A2 |
| Rat anti-CD4 APC Cy7 | TONBO bioscience | Cat# 25-0042-U025, clone RM4-5 |
| Mouse anti-TCRδ PE | Biolegend | Cat# 331209, clone B1 |
| Rat anti-B220 PE Cy5 | BD Biosciences | Cat# 553091, clone RA3-6B2 |
| Rat anti-CD326 APC | ThermoFisher | Cat# 17-5791-82, clone G8.8 |
| Mouse anti-CD45.1 APC Cy7 | TONBO bioscience | Cat# 25-0453-U100, clone A20 |
| Rat anti-SiglecF (CD170) Alexa Flour 700 | ThermoFisher | Cat# 56-1702-82, clone 1RNM44N |
| Rat anti-MHC class II (I-A/I-E) APC Cyanine7 | Biolegend | Cat# 107628, clone M5/114.15.2 |
| Rat anti-FoxP3 FITC | ThermoFisher | Cat# 11-5773-82, clone FJK-16s |
| Rat anti-IL-17 PE Cy7 | ThermoFisher | Cat# 25-7177-82, clone eBio17B7 |
| Rat anti-IFNγ eFluor450 | ThermoFisher | Cat# 48-7311-82, clone XMG1.2 |
| Rat anti-RORγt PE eFluor610 | eBioscience | Cat# 61-6981-82, clone B2D |
| Mouse anti-TCRδ FITC | eBioscience | Cat# 11-5711-82, clone GL-33 |
| Rabbit anti-STING FITC | Novus | Cat# NBP2-24683F, polyclonal |
| Rabbit anti-STING | Cell Signaling | Cat# 13647, clone D2P2F |
| Mouse anti-ubiquitin | Santa Cruz | Cat# cd-8017 |
| Mouse anti E-cadherin FITC | BD Biosciences | Cat # 612131 |
| Rabbit anti Bip | Cell Signaling | Cat# 3177T |
| Mouse anti CHOP | Cell Signaling | Cat# 2895T |
| Rabbit anti PARP | Cell Signaling | Cat# 9542S |
| Rabbit anti CD11b | Abcam | Cat# ab133357 |
| Rabbit anti CD3 | GeneTex | Cat# GTX16669 |
| Rabbit anti CD11b | Abcam | Cat# ab133357 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dextran sulfate sodium salt (DSS) | Termo Scientific | Cat# AAJ1448922 |
| Clodronate and control liposomes | Liposoma | Cat# CP-010-010 |
| Cyclic di-GMP | Invitrogen | Cat# tlrl-nacda |
| Fluorescein isothiocyanate-dextran 3–5KD | SIGMA | Cat# FD4-250mg |
| Mouse Diet, Helicobacter 4 Drug Combo | Bio-Serv | Cat# S4998 |
| Critical Commercial Assays | ||
| DC protein assay kit | Bio Rad | Cat# 5000112 |
| iScript cDNA synthesis kit | Bio Rad | Cat# 1708891 |
| Foxp3/Transcription Factor Staining Buffer Set | eBioscience | Cat# 00-5523-00 |
| Aurum™ Total RNA Mini kit | Bio Rad | Cat# 732-6820 |
| Nextera XT DNA Library Prep Kits | Illumina | Cat# FC-131-1096 |
| EasySep™ Mouse CD11b Positive Selection Kit II | StemCell | Cat# 18970 |
| Debris removal solution | Miltenyi Biotec | Cat# 130-109-398 |
| CD45 MicroBeads, Mouse | Miltenyi Biotec | Cat# 130-052-301 |
| DNeasy PowerSoil Kit | Qiagen | Cat# 12888-100 |
| AR9 Buffer | Akoya Biosciences | Cat# AR900 |
| Opal 4 color manual IHC kit | Akoya Biosciences | Cat# NEL810001KT |
| Opal antibody block reagent | Akoya Biosciences | Cat# ARD1001EA |
| Opal 690 reagent | Akoya Biosciences | Cat# FP1497001KT |
| Opal 480 reagent | Akoya Biosciences | Cat# FP1500001KT |
| VectorSheild Mounting media | Vector Laboratories | Cat# H-1500 |
| Deposited Data | ||
| Gut microbiome of murine model of intestinal inflammation and interaction with ER-membrane protein STING | NCBI BioProject | BioProject ID# PRJNA642084 |
| Experimental Models: Organisms/Strains | ||
| Mouse: N153s | Prof. Katherine A Fitzgerald | (Motwani et al., 2019) |
| Mouse: Ifnar/− | Prof. Prof. Jonathan Sprent, Scripps Research, CA | (Kolumam et al., 2005) |
| Mouse: Tcrδ−/− | Prof. Joonsoo Kang, UMASS medical school, MA | NA |
| Mouse: Tcrβ−/− | Prof. Stuart M Levitz, UMASS medical school, MA | NA |
| Mouse: Rag1−/− | Prof. Andrea Reboldi, UMASS medical school, MA | NA |
| Mouse: C57BL/GL | Jackson Laboratory | Stock# 000664 |
| Mouse: CD45.1 B6 | Jackson Laboratory | Stock# 002014 |
| Oligonucleotides | ||
| Primer for STING, forward GGTCACCGCTCCAAATATGTAG | Prof. Katherine A Fitzgerald | This paper |
| Primer for STING, reverse CAGTAGTCCAAGTTCGTGCGA | Prof. Katherine A Fitzgerald | This paper |
| Primer for TBP, forward GAAGCTGCGGTACAATTCCAG | Prof. Katherine A Fitzgerald | (Shmuel-Galia et al., 2016) |
| Primer for TBP, reverse CCCTTGTACCCTTCACCAAT | Prof. Katherine A Fitzgerald | (Shmuel-Galia et al., 2016) |
| 319, forward ACTCCTACGGGAGGCAGCAG | Prof. Katherine A Fitzgerald | (Drewes et al., 2017) |
| 806, reverse GGACTACHVGGGTWTCTAAT | Prof. Katherine A Fitzgerald | (Drewes et al., 2017) |
| ZO-1, forward CCACCTCTGTCCAGCTCTTC | Prof. Katherine A Fitzgerald | This paper |
| ZO-1, reverse CACCGGAGTGATGGTTTTCT | Prof. Katherine A Fitzgerald | This paper |
| Mucin-2, forward GATGGCACCTACCTCGTTGT | Prof. Katherine A Fitzgerald | This paper |
| Mucin-2, reverse GTCCTGGCACTTGTGGA AT | Prof. Katherine A Fitzgerald | This paper |
| Claudin-2, forward GTCATCGCCCATCAGAAGAT | Prof. Katherine A Fitzgerald | This paper |
| Claudin-2, reverse ACTGTTGGACAGGGAACCAG | Prof. Katherine A Fitzgerald | This paper |
| Tff3, forward TCTGGCTAATGCTGTTGGTG | Prof. Katherine A Fitzgerald | This paper |
| Tff3, reverse CTCCTGCAGAGGTTTGAAGC | Prof. Katherine A Fitzgerald | This paper |
| DEAF-1, forward TCAAGAGGCTGCAAAGGAAGAGAAC | Prof. Katherine A Fitzgerald | This paper |
| DEAF-1, reverse TGGTCTCCATGTTCAGCGACAGC | Prof. Katherine A Fitzgerald | This paper |
| H. typhlonius, forward CACTTGGAATCTTGGCTCATC | Prof. Katherine A Fitzgerald | This paper |
| H. typhlonius, reverse CCAATGCGAATACCAAGCAC | Prof. Katherine A Fitzgerald | This paper |
| REG3G, forward ATGCTTCCCCGTATAACCATCA | Prof. Katherine A Fitzgerald | This paper |
| REG3G, reverse GGCCATATCTGCATCATACCAG | Prof. Katherine A Fitzgerald | This paper |
| A-DEF25, forward AGTGAAGATCTGATATGCTATTG | Prof. Katherine A Fitzgerald | This paper |
| A-DEF25, reverse ACCAGAGCATGTACATTAAATG | Prof. Katherine A Fitzgerald | This paper |
| Software and Algorithms | ||
| NanoString nSolver analysis system | (Motwani et al., 2019) | https://nanostring.com/ |
| R-software | (Motwani et al., 2019) | https://r-project.org/ |
| FlowJo software | (Shmuel-Galia et al., 2016) | https://flowjo.com/ |
| MetaPhlAn2 version 2.9.14 | (Truong et al., 2015) | https://segatalab.cibio.unitn.it/tools/metaphlan2/ |
| database mpa_v292_CHOCOPhlAn_201901 | (Truong et al., 2015) | https://bitbucket.org/biobakery/metaphlan2/downloads/ |
| UPARSE pipeline in USEARCH version v10.0.240 | (Edgar, 2013) | https://drive5.com/uprase/ |
| SINTAX | (Edgar, 2013) | https://drive5.com/usearch/manual/sintax_downlopads.html |
| QIIME2 software version 2020.2 | (Bolyen et al., 2019) | https://qiime2.org |
| MaAsLin2 version 0.99.12 | Mallick et al. (2020+). “Multivariable Association Discovery in Population-scale Meta-omics Studies” (In Submission) | https://huttenhower.sph.harvard.edu/maaslin2 |
| Other | ||
| A/G FLUS-agarose | Santa Cruz | Cat# sc-2003 |
| Ghost violet 540 viability dye | TONBO | Cat# 13-0879-t100 |
| Collagenase type I | Gibco | Cat# 17100017 |
| Collagenase type IV | Millipore Sigma | Cat# c5138 |
| DNase I | Millipore Sigma | Cat#10104159001 |
| iTaq Universal SYBR Green supermix | Bio Rad | Cat# 1725124 |
| Ubq-TUBE-Agarose (UM-401) | LifeSensors | Cat# UM-0401-100 |
| K63-TUBE-FLAG (UM-604) | LifeSensors | Cat# UM-0604-0050 |
| ANTI-FLAGM2-affinity gel | Millipore Sigma | Cat# A2220 |
| Basement Membrane Matrix (Matrigel) | BD Biosciences | Cat#356235 |
| Y27632 | R&D Systems | Cat# 1254 |
| SB431542 | R&D Systems | Cat# 1614 |
Highlights.
Constitutive activation of STING initiates commensal dysbiosis
STING accumulates predominantly in intestinal myeloid cells during colitis
STING activation in myeloid cells results in progressive and chronic colitis
Colitis-promoting bacterial products trigger ubiquitination and stabilization of STING
Acknowledgments
We thank all the members of the Fitzgerald laboratory for helpful discussion. We further thank the staff of the Umass Medical School Animal facility and the Flow Cytometry Core for expert advice and technical assistance. We also thank Prof. Joonsoo Kang, Dr. Mor Gross-Vered and Prof. Pranoti Mandrekar for helpful suggestions and insightful discussions. This study was funded by a Target Identification in Lupus grant from the Lupus Research Alliance and by grants from the NIH (AI128358) awarded to KAF. LS-G is a recipient of the EMBO-long term postdoctoral fellowship (ALTF 534-2017) and the Crohn’s and Colitis Foundation, award number 708946 (title: SAVI mutations in STING lead to intestinal inflammation and fibrosis in mice), FH is a recipient of a GSK-postdoctoral fellowship and NK-C is a Cancer Research Irvington fellow supported by the Cancer Research Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data and Code availability
The 16s rDNA and Whole Genome Shotgun sequencing datasets generated during this study are available at NCBI BioProject ID# PRJNA642084.
Competing Interests
Authors declare no financial interest.
References
- ABE T & BARBER GN 2014. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-kappaB activation through TBK1. J Virol, 88, 5328–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ADEN K, TRAN F, ITO G, SHEIBANI-TEZERJI R, LIPINSKI S, KUIPER JW, TSCHURTSCHENTHALER M, SAVELJEVA S, BHATTACHARYYA J, HASLER R, BARTSCH K, LUZIUS A, JENTZSCH M, FALK-PAULSEN M, STENGEL ST, WELZ L, SCHWARZER R, RABE B, BARCHET W, KRAUTWALD S, HARTMANN G, PASPARAKIS M, BLUMBERG RS, SCHREIBER S, KASER A & ROSENSTIEL P 2018. ATG16L1 orchestrates interleukin-22 signaling in the intestinal epithelium via cGAS-STING. J Exp Med, 215, 2868–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AHN J, SON S, OLIVEIRA SC & BARBER GN 2017. STING-Dependent Signaling Underlies IL-10 Controlled Inflammatory Colitis. Cell Rep, 21, 3873–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALAM MT, AMOS GCA, MURPHY ARJ, MURCH S, WELLINGTON EMH & ARASARADNAM RP 2020. Microbial imbalance in inflammatory bowel disease patients at different taxonomic levels. Gut Pathog, 12, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENNION BG, INGLE H, AI TL, MINER CA, PLATT DJ, SMITH AM, BALDRIDGE MT & MINER JJ 2019. A Human Gain-of-Function STING Mutation Causes Immunodeficiency and Gammaherpesvirus-Induced Pulmonary Fibrosis in Mice. J Virol, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOLGER AM, LOHSE M & USADEL B 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOLYEN E, RIDEOUT JR, DILLON MR, BOKULICH NA, ABNET CC, AL-GHALITH GA, ALEXANDER H, ALM EJ, ARUMUGAM M, ASNICAR F, BAI Y, BISANZ JE, BITTINGER K, BREJNROD A, BRISLAWN CJ, BROWN CT, CALLAHAN BJ, CARABALLO-RODRIGUEZ AM, CHASE J, COPE EK, DA SILVA R, DIENER C, DORRESTEIN PC, DOUGLAS GM, DURALL DM, DUVALLET C, EDWARDSON CF, ERNST M, ESTAKI M, FOUQUIER J, GAUGLITZ JM, GIBBONS SM, GIBSON DL, GONZALEZ A, GORLICK K, GUO J, HILLMANN B, HOLMES S, HOLSTE H, HUTTENHOWER C, HUTTLEY GA, JANSSEN S, JARMUSCH AK, JIANG L, KAEHLER BD, KANG KB, KEEFE CR, KEIM P, KELLEY ST, KNIGHTS D, KOESTER I, KOSCIOLEK T, KREPS J, LANGILLE MGI, LEE J, LEY R, LIU YX, LOFTFIELD E, LOZUPONE C, MAHER M, MAROTZ C, MARTIN BD, MCDONALD D, MCIVER LJ, MELNIK AV, METCALF JL, MORGAN SC, MORTON JT, NAIMEY AT, NAVAS-MOLINA JA, NOTHIAS LF, ORCHANIAN SB, PEARSON T, PEOPLES SL, PETRAS D, PREUSS ML, PRUESSE E, RASMUSSEN LB, RIVERS A, ROBESON MS 2ND, ROSENTHAL P, SEGATA N, SHAFFER M, SHIFFER A, SINHA R, SONG SJ, SPEAR JR, SWAFFORD AD, THOMPSON LR, TORRES PJ, TRINH P, TRIPATHI A, TURNBAUGH PJ, UL-HASAN S, VAN DER HOOFT JJJ, VARGAS F, VAZQUEZ-BAEZA Y, VOGTMANN E, VON HIPPEL M, WALTERS W, et al. 2019. Author Correction: Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol, 37, 1091. [DOI] [PubMed] [Google Scholar]
- CANESSO MCC, LEMOS L, NEVES TC, MARIM FM, CASTRO TBR, VELOSO ES, QUEIROZ CP, AHN J, SANTIAGO HC, MARTINS FS, ALVES-SILVA J, FERREIRA E, CARA DC, VIEIRA AT, BARBER GN, OLIVEIRA SC & FARIA AMC 2018. The cytosolic sensor STING is required for intestinal homeostasis and control of inflammation. Mucosal Immunol, 11, 820–834. [DOI] [PubMed] [Google Scholar]
- CHEN Q, SUN L & CHEN ZJ 2016. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol, 17, 1142–9. [DOI] [PubMed] [Google Scholar]
- CHEN ZJ 2012. Ubiquitination in signaling to and activation of IKK. Immunol Rev, 246, 95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHIA J, EROGLU FK, OZEN S, ORHAN D, MONTEALEGRE-SANCHEZ G, DE JESUS AA, GOLDBACH-MANSKY R & COWEN EW 2016. Failure to thrive, interstitial lung disease, and progressive digital necrosis with onset in infancy. J Am Acad Dermatol, 74, 186–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHICHLOWSKI M, SHARP JM, VANDERFORD DA, MYLES MH & HALE LP 2008. Helicobacter typhlonius and Helicobacter rodentium differentially affect the severity of colon inflammation and inflammation-associated neoplasia in IL10-deficient mice. Comp Med, 58, 534–41. [PMC free article] [PubMed] [Google Scholar]
- DREWES JL, WHITE JR, DEJEA CM, FATHI P, IYADORAI T, VADIVELU J, ROSLANI AC, WICK EC, MONGODIN EF, LOKE MF, THULASI K, GAN HM, GOH KL, CHONG HY, KUMAR S, WANYIRI JW & SEARS CL 2017. High-resolution bacterial 16S rRNA gene profile meta-analysis and biofilm status reveal common colorectal cancer consortia. NPJ Biofilms Microbiomes, 3, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DZIARSKI R, PARK SY, KASHYAP DR, DOWD SE & GUPTA D 2016. Pglyrp-Regulated Gut Microflora Prevotella falsenii, Parabacteroides distasonis and Bacteroides eggerthii Enhance and Alistipes finegoldii Attenuates Colitis in Mice. PLoS One, 11, e0146162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDGAR RC 2013. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods, 10, 996–8. [DOI] [PubMed] [Google Scholar]
- FISCHER JC, BSCHEIDER M, EISENKOLB G, LIN CC, WINTGES A, OTTEN V, LINDEMANS CA, HEIDEGGER S, RUDELIUS M, MONETTE S, POROSNICU RODRIGUEZ KA, CALAFIORE M, LIEBERMANN S, LIU C, LIENENKLAUS S, WEISS S, KALINKE U, RULAND J, PESCHEL C, SHONO Y, DOCAMPO M, VELARDI E, JENQ RR, HANASH AM, DUDAKOV JA, HAAS T, VAN DEN BRINK MRM & POECK H 2017. RIG-I/MAVS and STING signaling promote gut integrity during irradiation- and immune-mediated tissue injury. Sci Transl Med, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FITZGERALD KA, MCWHIRTER SM, FAIA KL, ROWE DC, LATZ E, GOLENBOCK DT, COYLE AJ, LIAO SM & MANIATIS T 2003. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol, 4, 491–6. [DOI] [PubMed] [Google Scholar]
- FRANKLIN CL, GORELICK PL, RILEY LK, DEWHIRST FE, LIVINGSTON RS, WARD JM, BECKWITH CS & FOX JG 2001. Helicobacter typhlonius sp. nov., a Novel Murine Urease-Negative Helicobacter Species. J Clin Microbiol, 39, 3920–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRASPEUNTNER S, LOEPER N, KUNZEL S, BAINES JF & RUPP J 2018. Selection of validated hypervariable regions is crucial in 16S-based microbiota studies of the female genital tract. Sci Rep, 8, 9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRAY EE, TREUTING PM, WOODWARD JJ & STETSON DB 2015. Cutting Edge: cGAS Is Required for Lethal Autoimmune Disease in the Trex1-Deficient Mouse Model of Aicardi-Goutieres Syndrome. J Immunol, 195, 1939–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUARNER F 2008. What is the role of the enteric commensal flora in IBD? Inflamm Bowel Dis, 14 Suppl 2, S83–4. [DOI] [PubMed] [Google Scholar]
- GUI X, YANG H, LI T, TAN X, SHI P, LI M, DU F & CHEN ZJ 2019. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature, 567, 262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HILL DA, HOFFMANN C, ABT MC, DU Y, KOBULEY D, KIRN TJ, BUSHMAN FD & ARTIS D 2010. Metagenomic analyses reveal antibiotic-induced temporal and spatial changes in intestinal microbiota with associated alterations in immune cell homeostasis. Mucosal Immunol, 3, 148–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOLD GL, SMITH M, GRANGE C, WATT ER, EL-OMAR EM & MUKHOPADHYA I 2014. Role of the gut microbiota in inflammatory bowel disease pathogenesis: what have we learnt in the past 10 years? World J Gastroenterol, 20, 1192–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOOPER LV, LITTMAN DR & MACPHERSON AJ 2012. Interactions between the microbiota and the immune system. Science, 336, 1268–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IMAM T, PARK S, KAPLAN MH & OLSON MR 2018. Effector T Helper Cell Subsets in Inflammatory Bowel Diseases. Front Immunol, 9, 1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ISHIKAWA H & BARBER GN 2008. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature, 455, 674–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JEREMIAH N, NEVEN B, GENTILI M, CALLEBAUT I, MASCHALIDI S, STOLZENBERG MC, GOUDIN N, FREMOND ML, NITSCHKE P, MOLINA TJ, BLANCHE S, PICARD C, RICE GI, CROW YJ, MANEL N, FISCHER A, BADER-MEUNIER B & RIEUX-LAUCAT F 2014. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J Clin Invest, 124, 5516–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JIANG Y, ZHU Y, QIU W, LIU YJ, CHENG G, LIU ZJ & OUYANG S 2017. Structural and functional analyses of human DDX41 DEAD domain. Protein Cell, 8, 72–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOLUMAM GA, THOMAS S, THOMPSON LJ, SPRENT J & MURALI-KRISHNA K 2005. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med, 202, 637–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOZICH JJ, WESTCOTT SL, BAXTER NT, HIGHLANDER SK & SCHLOSS PD 2013. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol, 79, 5112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KULLBERG MC, ANDERSEN JF, GORELICK PL, CASPAR P, SUERBAUM S, FOX JG, CHEEVER AW, JANKOVIC D & SHER A 2003. Induction of colitis by a CD4+ T cell clone specific for a bacterial epitope. Proc Natl Acad Sci U S A, 100, 15830–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANGMEAD B & SALZBERG SL 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods, 9, 357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAVELLE A, LENNON G, O’SULLIVAN O, DOCHERTY N, BALFE A, MAGUIRE A, MULCAHY HE, DOHERTY G, O’DONOGHUE D, HYLAND J, ROSS RP, COFFEY JC, SHEAHAN K, COTTER PD, SHANAHAN F, WINTER DC & O’CONNELL PR 2015. Spatial variation of the colonic microbiota in patients with ulcerative colitis and control volunteers. Gut, 64, 1553–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEVY M, THAISS CA, ZEEVI D, DOHNALOVA L, ZILBERMAN-SCHAPIRA G, MAHDI JA, DAVID E, SAVIDOR A, KOREM T, HERZIG Y, PEVSNER-FISCHER M, SHAPIRO H, CHRIST A, HARMELIN A, HALPERN Z, LATZ E, FLAVELL RA, AMIT I, SEGAL E & ELINAV E 2015. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell, 163, 1428–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU S, CAI X, WU J, CONG Q, CHEN X, LI T, DU F, REN J, WU YT, GRISHIN NV & CHEN ZJ 2015. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science, 347, aaa2630. [DOI] [PubMed] [Google Scholar]
- LIU Y, JESUS AA, MARRERO B, YANG D, RAMSEY SE, SANCHEZ GAM, TENBROCK K, WITTKOWSKI H, JONES OY, KUEHN HS, LEE CR, DIMATTIA MA, COWEN EW, GONZALEZ B, PALMER I, DIGIOVANNA JJ, BIANCOTTO A, KIM H, TSAI WL, TRIER AM, HUANG Y, STONE DL, HILL S, KIM HJ, ST HILAIRE C, GURPRASAD S, PLASS N, CHAPELLE D, HORKAYNE-SZAKALY I, FOELL D, BARYSENKA A, CANDOTTI F, HOLLAND SM, HUGHES JD, MEHMET H, ISSEKUTZ AC, RAFFELD M, MCELWEE J, FONTANA JR, MINNITI CP, MOIR S, KASTNER DL, GADINA M, STEVEN AC, WINGFIELD PT, BROOKS SR, ROSENZWEIG SD, FLEISHER TA, DENG Z, BOEHM M, PALLER AS & GOLDBACH-MANSKY R 2014. Activated STING in a vascular and pulmonary syndrome. N Engl J Med, 371, 507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOZUPONE C & KNIGHT R 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol, 71, 8228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUPP C, ROBERTSON ML, WICKHAM ME, SEKIROV I, CHAMPION OL, GAYNOR EC & FINLAY BB 2007. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe, 2, 119–29. [DOI] [PubMed] [Google Scholar]
- MA C, YANG D, WANG B, WU C, WU Y, LI S, LIU X, LASSEN K, DAI L & YANG S 2020. Gasdermin D in macrophages restrains colitis by controlling cGAS-mediated inflammation. Science Advances, 6, eaaz6717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MA F, LI B, YU Y, IYER SS, SUN M & CHENG G 2015. Positive feedback regulation of type I interferon by the interferon-stimulated gene STING. EMBO Rep, 16, 202–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAN SM, ZHANG L, DAY AS, LEACH S & MITCHELL H 2008. Detection of enterohepatic and gastric helicobacter species in fecal specimens of children with Crohn’s disease. Helicobacter, 13, 234–8. [DOI] [PubMed] [Google Scholar]
- MARINHO FV, BENMERZOUG S, OLIVEIRA SC, RYFFEL B & QUESNIAUX VFJ 2017. The Emerging Roles of STING in Bacterial Infections. Trends Microbiol, 25, 906–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARTIN GR, BLOMQUIST CM, HENARE KL & JIRIK FR 2019. Stimulator of interferon genes (STING) activation exacerbates experimental colitis in mice. Sci Rep, 9, 14281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIYOSHI H & STAPPENBECK TS 2013. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat Protoc, 8, 2471–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOTWANI M, PAWARIA S, BERNIER J, MOSES S, HENRY K, FANG T, BURKLY L, MARSHAK-ROTHSTEIN A & FITZGERALD KA 2019. Hierarchy of clinical manifestations in SAVI N153S and V154M mouse models. Proc Natl Acad Sci U S A, 116, 7941–7950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAHAR S, HOKAMA A & FUJITA J 2019. Clinical significance of cytomegalovirus and other herpes virus infections in ulcerative colitis. Pol Arch Intern Med, 129, 620–626. [DOI] [PubMed] [Google Scholar]
- PICARD C, MATHIEU AL, HASAN U, HENRY T, JAMILLOUX Y, WALZER T & BELOT A 2015. Inherited anomalies of innate immune receptors in pediatric-onset inflammatory diseases. Autoimmun Rev, 14, 1147–53. [DOI] [PubMed] [Google Scholar]
- PICARD C, THOUVENIN G, KANNENGIESSER C, DUBUS JC, JEREMIAH N, RIEUX-LAUCAT F, CRESTANI B, BELOT A, THIVOLET-BEJUI F, SECQ V, MENARD C, REYNAUD-GAUBERT M & REIX P 2016. Severe Pulmonary Fibrosis as the First Manifestation of Interferonopathy (TMEM173 Mutation). Chest, 150, e65–71. [DOI] [PubMed] [Google Scholar]
- SCHNEIDER-POETSCH T, JU J, EYLER DE, DANG Y, BHAT S, MERRICK WC, GREEN R, SHEN B & LIU JO 2010. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol, 6, 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHUNTER MO, WALLES T, FRITZ P, MEYDING-LAMADE U, THON KP, FELLERMANN K, STANGE EF & LAMADE W 2007. Herpes simplex virus colitis complicating ulcerative colitis: A case report and brief review on superinfections. J Crohns Colitis, 1, 41–6. [DOI] [PubMed] [Google Scholar]
- SHANG G, ZHANG C, CHEN ZJ, BAI XC & ZHANG X 2019. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature, 567, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHARP JM, VANDERFORD DA, CHICHLOWSKI M, MYLES MH & HALE LP 2008. Helicobacter infection decreases reproductive performance of IL10-deficient mice. Comp Med, 58, 447–53. [PMC free article] [PubMed] [Google Scholar]
- SHEN ZH, ZHU CX, QUAN YS, YANG ZY, WU S, LUO WW, TAN B & WANG XY 2018. Relationship between intestinal microbiota and ulcerative colitis: Mechanisms and clinical application of probiotics and fecal microbiota transplantation. World J Gastroenterol, 24, 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHMUEL-GALIA L, AYCHEK T, FINK A, PORAT Z, ZARMI B, BERNSHTEIN B, BRENNER O, JUNG S & SHAI Y 2016. Neutralization of pro-inflammatory monocytes by targeting TLR2 dimerization ameliorates colitis. EMBO J, 35, 685–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANAKA Y & CHEN ZJ 2012. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal, 5, ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRUONG DT, FRANZOSA EA, TICKLE TL, SCHOLZ M, WEINGART G, PASOLLI E, TETT A, HUTTENHOWER C & SEGATA N 2015. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods, 12, 902–3. [DOI] [PubMed] [Google Scholar]
- UBEDA C, TAUR Y, JENQ RR, EQUINDA MJ, SON T, SAMSTEIN M, VIALE A, SOCCI ND, VAN DEN BRINK MR, KAMBOJ M & PAMER EG 2010. Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. J Clin Invest, 120, 4332–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WARNER JD, IRIZARRY-CARO RA, BENNION BG, AI TL, SMITH AM, MINER CA, SAKAI T, GONUGUNTA VK, WU J, PLATT DJ, YAN N & MINER JJ 2017. STING-associated vasculopathy develops independently of IRF3 in mice. J Exp Med, 214, 3279–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WU J, CHEN YJ, DOBBS N, SAKAI T, LIOU J, MINER JJ & YAN N 2019. STING-mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell death. J Exp Med, 216, 867–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG S, WANG B, HUMPHRIES F, HOGAN AE, O’SHEA D & MOYNAGH PN 2014. The E3 ubiquitin ligase Pellino3 protects against obesity-induced inflammation and insulin resistance. Immunity, 41, 973–87. [DOI] [PubMed] [Google Scholar]
- ZHANG C, SHANG G, GUI X, ZHANG X, BAI XC & CHEN ZJ 2019. Structural basis of STING binding with and phosphorylation by TBK1. Nature, 567, 394–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG Z, YUAN B, BAO M, LU N, KIM T & LIU YJ 2011. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol, 12, 959–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHENG D, LIWINSKI T & ELINAV E 2020. Interaction between microbiota and immunity in health and disease. Cell Research, 30, 492–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHU Q, MAN SM, GURUNG P, LIU Z, VOGEL P, LAMKANFI M & KANNEGANTI TD 2014. Cutting edge: STING mediates protection against colorectal tumorigenesis by governing the magnitude of intestinal inflammation. J Immunol, 193, 4779–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Whole-genome shotgun (WGS) DNA sequencing and 16s rDNA profiling generated in this study are available at NCBI BioProject, BioProject ID: PRJNA642084.