

Abstract
Machine learning methods are attracting considerable attention from the pharmaceutical industry for use in drug discovery and applications beyond. In recent studies we and others have applied multiple machine learning algorithms, modeling metrics and in some cases compared molecular descriptors to build models for individual targets or properties on a relatively small scale. Several research groups have used large numbers of datasets from public databases such as ChEMBL in order to evaluate machine learning methods of interest to them. The largest of these types of studies used on the order of 1400 datasets. We have now extracted well over 5000 datasets from CHEMBL for use with the ECFP6 fingerprint and comparison of our proprietary software Assay Central® with random forest, k-Nearest Neighbors, support vector classification, naïve Bayesian, AdaBoosted decision trees, and deep neural networks (3 levels). Model performance was assessed using an array of five-fold cross-validation metrics including area-under-the-curve, F1 score, Cohen’s kappa and Matthews correlation coefficient. Based on ranked normalized scores for the metrics or datasets all methods appeared comparable while the distance from the top indicated Assay Central® and support vector classification were comparable. Unlike prior studies which have placed considerable emphasis on deep neural networks (deep learning), no advantage was seen in this case. If anything, Assay Central® may have been at a slight advantage as the activity cutoff for each of the over 5000 datasets representing over 570,000 unique compounds was based on Assay Central® performance, although support vector classification seems to be a strong competitor. We also applied Assay Central® to perform prospective predictions for the toxicity targets PXR and hERG to further validate these models. This work appears to be the largest scale comparison of these machine learning algorithms to date. Future studies will likely evaluate additional databases, descriptors and machine learning algorithms, as well as further refining the methods for evaluating and comparing such models.
Keywords: Deep learning, drug discovery, machine learning, pharmaceutics, support vector machines
Graphical Abstarct
INTRODUCTION
The pharmaceutical industry is increasingly looking to using machine learning for drug discovery applications to leverage their considerable high throughput screening as well as many other types of data 1. Some recent examples outside of big pharma have demonstrated the speed with which the machine learning and in vitro testing can produce new leads compared to traditional efforts 2. With many thousands of structure activity datasets with data from hundreds to hundreds of thousands of molecules screened against a single target or organism, this represents a potentially valuable starting point for drug discovery as well as understanding molecular properties and predicting toxicity. Accessible public databases like ChEMBL 3, PubChem 4, 5 and many others have spawned numerous studies utilizing such data for a wide array of applications in machine learning with many algorithms such as deep neural networks (DNN) 6–8, support vector machines (SVM) 9–15, k-Nearest Neighbors (kNN) 16, naïve Bayesian 17–21, decision trees 22 and others 23, 24 which have been increasingly used 25–28. We are also observing wider usage of deep learning for pharmaceutical applications 29 and this has warranted comparisons with other methods 30 described as follows.
Large scale analyses of public structure activity relationship datasets have been primarily focused on creating chemogenomic databases which can be used to predict off-target effects as well as compound repurposing. In 2013 TargetHunter was described as a web portal that integrated ECFP6 descriptors with two different algorithms (Targets Associated with its MOst Similar Counterparts (TAMOSIC) and multiple category models (MCM)) for 794 targets in ChEMBL. Seven-fold cross-validation results gave average accuracy of 90.9% for TAMOSIC and 74.8% for MCM 31. 894 human protein targets from ChEMBL were used to train and compare two probabilistic machine-learning algorithms (naïve Bayes and Parzen-Rosenblatt Window) against a test set from the WOMBAT database used as an external test set. Recall was consistently higher for the Parzen-Rosenblatt Window 32. Smaller subsets of ChEMBL have also been used for various studies including extraction of 29 datasets for comparing 11 ligand efficiency scores with pIC50 as well using Morgan fingerprints or physicochemical descriptors with four machine learning algorithms 33. Models were found to have a higher predictive power when based on ligand efficiencies. Viral focused datasets from ChEMBL (including human immunodeficiency, hepatitis C, hepatitis B, and human herpes virus among a selection of 26 viruses) were used to create SVM regression or classification models after generating 18,000 descriptors with Padel software. Ten-fold cross-validation statistics were generated as well as validation with molecules left out of the models. Validation statistics for classification models indicated sensitivity, specificity and accuracy > 80%. These models were subsequently used to generate an integrated webserver called AVCpred 34. A set of G-protein coupled receptor targets for 5-HT2c, melanin concentrating hormone and adenosine A1 were used to build Gaussian process models with CATS2 descriptors which were more predictive for a test set than models developed from a proprietary Boehringer Ingelheim dataset. This was likely due to the fact that the ChEMBL set was larger and more structurally diverse 35. 173 human targets with Ki bioactivity data were extracted from ChEMBL and used to build Naïve Bayes, logistic regression and random forest models using Morgan fingerprints or FP2 that were in turn used for target inference 36. Seven-fold cross-validation and temporal cross-validation demonstrated that cutoffs that were more potent had better accuracy and MCC. These models were then used for multiple target prediction using the most similar ligand with false discovery rate control procedures to correct the p values. Only two molecules were used as examples for which predictions were made 36. 25 datasets from ChEMBL were used to compare random forest, multiple linear regression, ridge regression, similarity searching and random selection of compounds as approaches to identify highly potent molecules 37. Linear and ridge regression were 2–3 times faster than random forest (RF) and similarity searching at finding highly potent molecules. 550 human protein targets from ChEMBL version 22 were used for RF and conformal prediction models and used to predict additional targets from versions 23 and 24. The conformal prediction approach generally outperformed RF on internal and temporal validation whereas for external validation the situation was reversed 38. There are likely many additional published examples like this, but these clearly point to the progress that has been made to date.
As a database like ChEMBL is so large it has become a central resource for many types of machine learning projects 39–48. It also contains a large number of cell lines with cytotoxicity data and cancer related targets and these have been combined and used recently to derive combined perturbation theory machine learning models 49, however the authors did not explore prospective external testing of this method. Natural products are important as they are the basis for many small molecule drugs. Efforts to identify compounds that are natural product-like have curated 265,000 natural products (including those in ChEMBL) alongside 322,000 synthetic molecules to derive RF classification models using Morgan2 fingerprints and MACCS keys. Models were tested using 10-fold cross-validation and an independent test set and in both cases the receiver operator characteristic (ROC) score was 0.996–0.997 50. Subsets of natural products such as macrolactones have also been the recent focus of machine learning. In one case using biological data from ChEMBL models were generated for macrolactones that possessed a range of activities for Plasmodium falciparum, hepatitis C virus and T-cells) using RF, SVM Regression, naïve Bayes, KNN, DNN, as well as consensus and hybrid approaches from the two best algorithms (RF_KNN and RF_DNN, the average prediction from RF and KNN or DNN). These three case studies demonstrated RF was the best predictor across six descriptor sets while consensus modeling or the hybrid slightly increased the predictive power of the quantitative structure activity relationship (QSAR) models 51.
All of the preceding examples have in common that few if any used prospective prediction as a form of validation. The main aim of this study was to evaluate a Bayesian method, Assay Central® which we have previously used in several examples where we have compared it versus other machine learning methods against a relatively small number of datasets or targets such as drug induced liver injury 52, Rat Acute Oral toxicity 53, estrogen receptor 54, 55, androgen receptor 56, GSK3β 57 Mycobacterium tuberculosis 58, 59, non-nucleoside reverse transcriptase and whole cell HIV 60. Our evaluation has now been expanded with this study to over 5000 CHEMBL datasets and provides statistics and data visualization methods in order to assess each algorithm. In contrast to other large-scale machine learning comparisons we also now describe prospective prediction for a selection of ADME/Tox properties and retrospective analysis with infectious disease datasets for Mycobacterium tuberculosis and non-nucleoside reverse transcriptase and whole cell HIV.
EXPERIMENTAL SECTION
Computing
Computational Servers consisted of the following components: Supermicro EATX DDR4 LGA 2011, Intel Computer CPU 2.1 8 BX80660E52620V4, Crucial 64GB Kit (16GBx4) DDR4 2133 (PC42133) DR x4 288 Pin Server Memory CT4K16G4RFD4213 / CT4C16G4RFD4213, 10 x EVGA GeForce GTX 1080 Ti FOUNDERS EDITION GAMING, 11GB GDDR5X, Intel 730 SERIES 2.5Inch Solid State Drive SSDSC2BP480G410, WD Gold 4TB Datacenter Hard Disk Drive 7200 RPM Class SATA 6 Gb/s 128MB Cache 3.5 Inch WD4002FYYZ and Supermicro 920 Watt 4U Server. The following software modules were installed: nltk 3.2.2, scikit-learn 0.18.1, Python 3.5.2, Anaconda 4.2.0 (64-bit), Keras 1.2.1, Tensorflow 0.12.1, Jupyter Notebook 4.3.1.
Datasets
Datasets were generated in the following manner as described previously 61. A list of all molecules present in ChEMBL 62 version 25 was generated from the chembl_25.sdf.gz file downloaded from the ChEMBL ftp server. A list of all the targets present in ChEMBL25 was generated via the interface on the ChEMBL API. Targets of types “ADMET”, “CELL-LINE”, “CHIMERIC PROTEIN”, “MACROMOLECULE”, “METAL”, “OLIGOSACCHARIDE”, “ORGANISM”, “PHENOTYPE”, “PROTEIN COMPLEX”, “PROTEIN COMPLEX GROUP”, “PROTEIN FAMILY”, “PROTEIN NUCLEIC-ACID COMPLEX”, “SINGLE PROTEIN”, “SMALL MOLECULE”, and “TISSUE” were included in the analysis, while targets of types “LIPID”, “NON-MOLECULAR”, “NO TARGET”, “NUCLEIC-ACID”, “SELECTIVITY GROUP”, “SUBCELLULAR”, “UNCHECKED”, and “UNKNOWN”, were excluded. In total, 12,310 targets were present in the initial data collation. Assays which matched the selected targets were collated via the ChEMBL API, producing a list of 915,781 assays. From these assays, all activities expressed in molar units (i.e. units of type “M”, “mM”, “uM” and “nM”) were collected. For each target, the activities were partitioned into datasets based on the type of assay (“Functional”, “Binding”, “ADME”, or “All”) and the type of assay endpoint (“C50” corresponding to combined IC50/EC50/AC50/GI50, “K” corresponding to Ki/Kd, “MIC”, or “All” which combined all possible endpoints). This produced 16,135 datasets, of which 2,915 had only one activity and were excluded from further analysis. From the resulting 13,220, we excluded datasets with fewer than 100 activities so as to compare to our previous work 61, to give 5,279 datasets. Of these, 8 datasets failed to converge when used to train SVM machine learning models and were excluded from further consideration. The binarization of these continuous datasets by our Assay Central® algorithm occasionally picked a threshold so that there were fewer than 5 actives. These datasets have also been excluded from the analysis, resulting in a final total of 5091 datasets with at least 100 activity measurements each, the largest of which has 15,994 molecules. As these datasets were curated in an automated manner, as opposed to manually seeking out specific endpoints or assays, these datasets are referred to herein as ‘autocurated’ datasets and we have provided these files for others to use (Supplemental Datasets).
Machine learning - Assay Central®
Assay Central® is proprietary software for curating high-quality datasets, generating Bayesian machine learning models with extended-connectivity fingerprints (ECFP6) generated from the CDK library 63, and making prospective predictions of potential for bioactivity 1, 54, 55, 58, 60, 64–70. We have previously described this software in detail 1, 55, 58, 60, 64–71 as well as the interpretation of prediction scores 61, 72, and the metrics generated from internal five-fold cross-validation used to evaluate and compare predictive performances. These metrics include, but are not limited to, ROC score 55, Cohen’s kappa (CK) 73, 74, and Matthew’s correlation coefficient (MCC) 75 as described by us previously.
Each of the 5091 ChEMBL version 25 datasets described previously were subjected to the same standardization processes (i.e. removing salts, metal complexes and mixtures, merging finite activities for duplicate compounds) prior to building a Bayesian classification model 72. Classification models such as these require a defined threshold of bioactivity, and Assay Central® implements an automated method to select this threshold to optimize internal model performance metrics 61. After the initial generation of the Bayesian model with Assay Central®, a binary activity column was applied to the output dataset for the accurate comparison of machine learning methods.
Other machine learning methods
The threshold for all the datasets was set by Assay Central and these were output (i.e. with the threshold applied so that the activity is binary, merged duplications, etc.) so that they could then be applied by the alternative machine learning algorithms. Additional machine learning algorithms were used which also utilized ECFP6 molecular descriptors for consistency between methods and were generated from the RDKit (www.rdkit.org) cheminformatics library. These additional algorithms include RF, kNN, SVM classification, naïve Bayesian, AdaBoosted decision trees, and DNN of three layers; these algorithms have been described fully in detail in our earlier publications 55, 58, 60. It should also be noted that the hyperparameters for these different models were determined as previously described 76.
Validation metrics
Internal five-fold cross-validation metrics between algorithms for 5091 datasets were compared with a rank normalized score, which was also used in previous investigations 60, 76, 77 of various machine learning methods. Rank normalized scores 60 can be evaluated in either a pairwise or independent fashion: the former is informative for a comparison of algorithms per training set, while the latter provides a more generalized comparison of algorithms overall. A total of 188 datasets were excluded from this study because one or more of the internal five-fold cross-validation metrics was unable to be calculated (i.e. contained a denominator calculated to be zero).
The rank normalized score takes the mean of the normalized scores of several traditional measurements of model performance including accuracy (ACC), recall, precision, F1 score, ROC AUC, Cohen’s Kappa (CK) and Matthews correlation coefficient (MCC). As ACC, ROC AUC, precision, recall, specificity and F1 scores are fixed from 0 to 1, the raw scores were used directed to calculate the rank normalized score. As CK and MCC range from −1 to 1, the normalized score was calculated as in order to truncate the range from 0 to 1. The rank normalized score is just the mean of these values.
Additionally, a “difference from the top” (∆RNS) metric, where the rank normalized score for each algorithm is subtracted from the highest corresponding score of a specific dataset 76. This metric maintains the pairwise comparison results and enables a direct assessment of the performance of multiple machine learning algorithms while maintaining information from the other machine learning algorithms tested simultaneously.
External validation examples for Assay Central® models
Several recent internal projects have involved using manually curated and specific machine learning models (i.e. single measurement type such as IC50, Ki etc.) to score compounds prior to selection of compounds for testing in vitro with Assay Central®. We have compared the predictions made previously for PXR (ChEMBL target ID 3401) and hERG (ChEMBL target ID 240) using manually curated datasets to those predictions from auto-curated “C50” datasets from ADME or binding assays (described earlier in the “datasets” section). All datasets were subjected to the same standardization steps (i.e. removing salts, neutralizing anions, merging duplicates) as described for generating models with Assay Central®. When structures are predicted they are also desalted and neutralized for compatibility with a model.
Additionally, several test sets from previous studies were subjected to external validation by corresponding auto-curated datasets from ChEMBL. Two HIV datasets from the NIAID ChemDB used previously by us 60 as training datasets were utilized herein as test sets for either whole cell HIV or reverse transcriptase. All whole cell data was from MT-4 cells with a molecular weight limit of 750 g/mol and totalled 13,783 compounds. A test set of three reverse transcriptase training sets from the same publication 60 (from colorimetric, immunological, and incorporation assays) totalled 1578 compounds. A final test set of literature MIC data against M. tuberculosis H37Rv strain was utilized in another previous publication 58 and totalled 155 compounds. All these test sets were re-evaluated to remove compounds overlapping with the corresponding ChEMBL autocurated dataset in order to enable a true external validation.
The HIV whole cell datasets from the NIAID ChemDB were evaluated with the auto-curated “C50” datasets from functional assays from ChEMBL target ID 378 (HIV-1), and the reverse transcriptase dataset with autocurated “C50” datasets from binding assays from ChEMBL target ID 2366516 (HIV-1 reverse transcriptase). The M. tuberculosis test set was evaluated against the autocurated MIC dataset from functional assays from ChEMBL target ID 2111188 (M. tuberculosis H37Rv strain). All external validations were then evaluated at the calculated activity threshold unique to each training model.
Descriptor calculations
To assess the molecular properties of the ChEMBL datasets (570,872 uniquecompounds) versus known drug-like molecules we used the SuperDrug2 library (3992 unique compounds)78 we generated molecular descriptors (AlogP, molecular weight, number of rotatable bonds, number of rings, number of aromatic rings, number of hydrogen bond acceptors, number of hydrogen bond donors, and molecular fractional polar surface area) using Discovery Studio (Biovia, San Diego, CA) for each dataset individually and these property distributions were compared.
Statistics
All statistics were performed using Graphpad Prism 8 for macOS version 8.4.3.
External biological testing
External PXR and hERG testing was performed by ThermoFisher Scientific SelectScreen™ Profiling Service (Life Technologies Corporation, (Chicago, IL 60693). The PXR assay uses a LanthaScreen TR-FRET competitive binding assay and used SR-12813 as a positive control. a Tb-labeled anti-GST antibody is used to indirectly label the receptor by binding to its GST tag. Competitive ligand binding is detected by a test compound’s ability to displace a fluorescent ligand (tracer) from the receptor, which results in a loss of FRET signal between the Tb-anti-GST antibody and the tracer. The hERG screening used a fluorescence polarization assay previously described 79 and E-4031 as a positive control.
RESULTS
Model statistics
Multiple machine learning models were generated and their internal five-fold cross-validation metrics were compared to assess their performance. The distribution of the model metrics for each model approach was not normally distributed, (Figure S1) therefore the median model statistics are used for comparative purposes (Table 1). Even though the magnitudes vary drastically, there is a statistically significant difference between essentially all of the algorithms using the RNS metrics (RNS and ΔRNS). As shown in Figure 1A, the two algorithms that show the most pronounced score superiority with these metrics are the Bayesian algorithm used by Assay Central® and SVC. Even though the RNS between these two algorithms were statistically indifferent from each other (Table 2) there was a statistically significant difference with the ΔRNS between AC and SVC, suggesting AC as the better algorithm based on 5-fold cross validation for these datasets. This difference in the ∆RNS metric is visualized in Figure 1B and quantified in Table 3. Knn and RF lacked statistically significant differences when comparing the RNS or ΔRNS independently, but there was a statically significant win for RF when utilizing a pairwise RNS comparison (Table 2). As the ΔRNS score comparison shows no significance difference and should be a more sensitive metric to compare the algorithms in the same way as the RNS pairwise comparison is intending to do, this difference, while significant, is likely inconsequential. DL showed a similar, but reduced performance as compared to Knn and RF using both the pairwise and independent methods. Ada and Bnb had the most drastic disparity in their performances as compared to all the other algorithms tested (Table 2). This is quantified by the mean rank differences, which shows the extent of these differences (Table 3).
Table 1.
Median values for each model and metric color coded by scale.
| AC | SVC | ADA | BNB | DL | KNN | RF | |
|---|---|---|---|---|---|---|---|
| ACC | 0.827 | 0.841 | 0.811 | 0.801 | 0.825 | 0.829 | 0.832 |
| AUC | 0.887 | 0.826 | 0.772 | 0.766 | 0.796 | 0.810 | 0.807 |
| Cohen’s Kappa | 0.627 | 0.643 | 0.557 | 0.540 | 0.597 | 0.617 | 0.616 |
| MCC | 0.636 | 0.646 | 0.563 | 0.544 | 0.604 | 0.619 | 0.620 |
| Precision | 0.782 | 0.796 | 0.775 | 0.750 | 0.778 | 0.765 | 0.795 |
| Recall | 0.833 | 0.817 | 0.732 | 0.730 | 0.773 | 0.810 | 0.775 |
| Specificity | 0.841 | 0.857 | 0.863 | 0.845 | 0.855 | 0.835 | 0.868 |
| F1-Score | 0.791 | 0.798 | 0.749 | 0.738 | 0.766 | 0.785 | 0.781 |
AC = Assay Central (Bayesian), rf = Random Forest, knn = k-Nearest Neighbors, svc = Support Vector Classification, bnb = Naïve Bayesian, ada = AdaBoosted Decision Trees, DL = Deep Learning.
Figure 1.
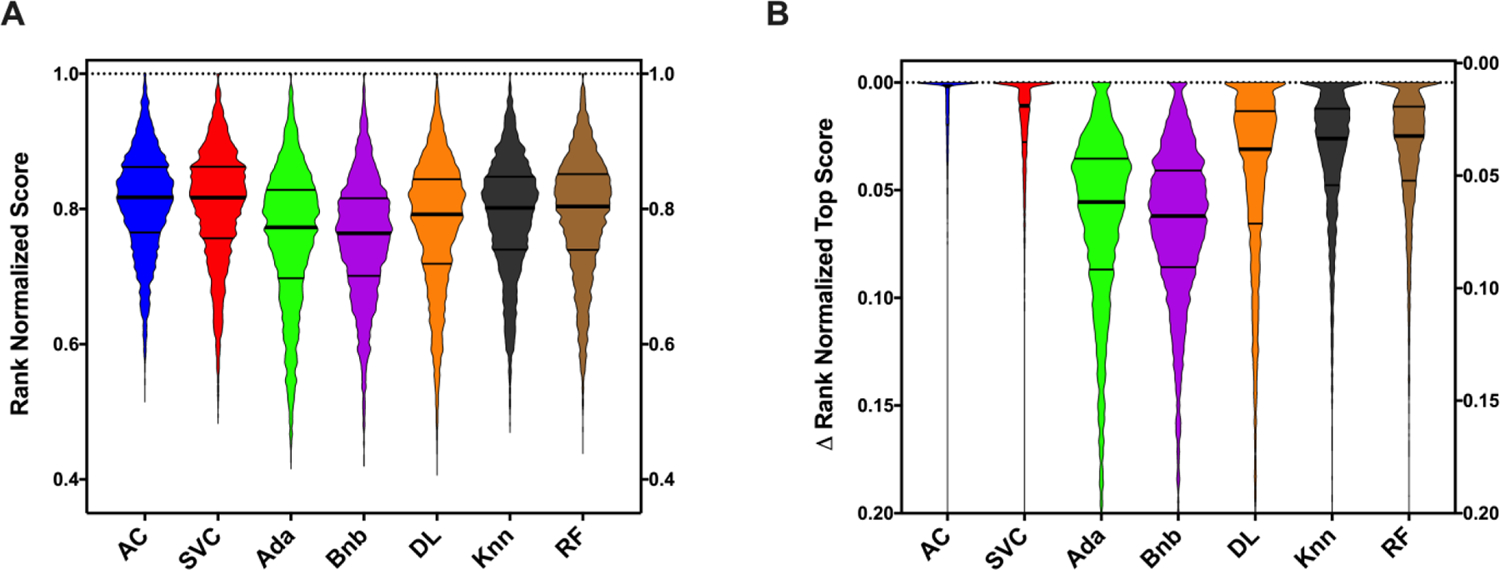
Machine learning algorithm comparisons for ChEMBL datasets across multiple five-fold cross-validation metrics based on the rank normalized scores. A) Rank normalized score and B) ∆RNS distributions. Truncated violin plots are shown with minimal smoothing to retain an accurate distribution representation. The solid central line represents the median with the quarterlies indicated. AC = Assay Central (Bayesian), RF = Random Forest, Knn = k-Nearest Neighbors, SVC = Support Vector Classification, Bnb = Naïve Bayesian, Ada = AdaBoosted Decision Trees, DL = Deep Learning.
Table 2.
Rank normalized score comparisons (Friedman (pairwise comparison) (A) or Kruskal-Wallis test (Independent comparison) (B) with Dunn’s multiple comparisons follow up tests).
| A. | |||||||
|---|---|---|---|---|---|---|---|
| Rank Normalized Score: Pairwise Comparison (Adjusted P-score) | |||||||
| Algorithm | Ada | Bnb | DL | Knn | RF | SVC | AC |
| Ada | N/A | >0.9999 (ns) | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** |
| Bnb | >0.9999 (ns) | N/A | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** |
| DL | <0.0001**** | <0.0001**** | N/A | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** |
| Knn | <0.0001**** | <0.0001**** | <0.0001**** | N/A | 0.0001*** | <0.0001**** | <0.0001**** |
| RF | <0.0001**** | <0.0001**** | <0.0001**** | 0.0001*** | N/A | <0.0001**** | <0.0001**** |
| SVC | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** | N/A | >0.9999 (ns) |
| AC | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** | >0.9999 (ns) | N/A |
| Rank Normalized Score: Pairwise Comparison (Mean Rank Difference) | |||||||
|
Winning Algorithm |
Ada | Bnb | DL | Knn | RF | SVC | AC |
| Ada | N/A | 198.5 (ns) | |||||
| Bnb | N/A | ||||||
| DL | 7585**** | 7784**** | N/A | ||||
| Knn | 9776**** | 9975**** | 2191**** | N/A | |||
| RF | 10763**** | 10961**** | 3178**** | 986.5*** | N/A | ||
| SVC | 16820**** | 17019**** | 9235**** | 7044**** | 6058**** | N/A | |
| AC | 17226**** | 17425**** | 9641**** | 7450**** | 6464**** | 406 (ns) | N/A |
| B. | |||||||
| Rank Normalized Score: Independent Comparison (Adjusted P-score) | |||||||
| Algorithm | Ada | Bnb | DL | Knn | RF | SVC | AC |
| Ada | N/A | 0.0011** | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** |
| Bnb | 0.0011** | N/A | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** |
| DL | <0.0001**** | <0.0001**** | N/A | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** |
| Knn | <0.0001**** | <0.0001**** | <0.0001**** | N/A | >0.9999 (ns) | <0.0001**** | <0.0001**** |
| RF | <0.0001**** | <0.0001**** | <0.0001**** | >0.9999 (ns) | N/A | <0.0001**** | <0.0001**** |
| SVC | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** | N/A | 0.6123 (ns) |
| AC | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** | 0.6123 (ns) | N/A |
| Rank Normalized Score: Independent Comparison (Mean Rank Difference) | |||||||
|
Winning Algorithm |
Ada | Bnb | DL | Knn | RF | SVC | AC |
| Ada | N/A | 825.8** | |||||
| Bnb | N/A | ||||||
| DL | 1996**** | 2822**** | N/A | ||||
| Knn | 3143**** | 3968**** | 1146**** | N/A | |||
| RF | 3390**** | 4216**** | 1394**** | 247.4 (ns) | N/A | ||
| SVC | 5008**** | 5834**** | 3011**** | 1865**** | 1618**** | N/A | |
| AC | 5453**** | 6278**** | 3456**** | 2310**** | 2062**** | 444.8 (ns) | N/A |
AC = Assay Central (Bayesian), rf = Random Forest, knn = k-Nearest Neighbors, svc = Support Vector Classification, bnb = Naïve Bayesian, ada = AdaBoosted Decision Trees, DL = Deep Learning.
Table 3.
Distance from the top normalized score (Kruskal-Wallis test (Independent comparison) with Dunn’s multiple comparisons follow up tests).
| Rank Normalized Score (DFT): Independent Comparison (Adjusted P-score) | |||||||
|---|---|---|---|---|---|---|---|
| Algorithm | Ada | Bnb | DL | Knn | RF | SVC | AC |
| Ada | N/A | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** |
| Bnb | <0.0001**** | N/A | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** |
| DL | <0.0001**** | <0.0001**** | N/A | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** |
| Knn | <0.0001**** | <0.0001**** | <0.0001**** | N/A | 0.662 (ns) | <0.0001**** | <0.0001**** |
| RF | <0.0001**** | <0.0001**** | <0.0001**** | 0.662 (ns) | N/A | <0.0001**** | <0.0001**** |
| SVC | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** | N/A | <0.0001**** |
| AC | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** | <0.0001**** | N/A |
| Rank Normalized Score (DFT): Independent Comparison (Mean Rank Difference) | |||||||
|
Winning Algorithm |
Ada | Bnb | DL | Knn | RF | SVC | AC |
| Ada | N/A | 943.4**** | |||||
| Bnb | N/A | ||||||
| DL | 5781**** | 6725**** | N/A | ||||
| Knn | 7777**** | 8721**** | 1996**** | N/A | |||
| RF | 8215**** | 9158**** | 2434**** | 437.8 (ns) | N/A | ||
| SVC | 13256**** | 14199**** | 7475**** | 5479**** | 5041**** | N/A | |
| AC | 15841**** | 16784**** | 10059**** | 8064**** | 7626**** | 2585**** | N/A |
AC = Assay Central (Bayesian), rf = Random Forest, knn = k-Nearest Neighbors, svc = Support Vector Classification, bnb = Naïve Bayesian, ada = AdaBoosted Decision Trees, DL = Deep Learning.
Overall, the trends seen with the RNS and ΔRNS score comparisons are mimicked in each individual metric comparison (Figure 2, Table S2), with some minor exceptions being highlighted. AC or SVC ranked in the top 2 for every metric examined except for Specificity, where RF scored the best. The most pronounced metric difference between the top scoring algorithms was AUC, with AC and SVC having median scores of 0.887 (95% CI: 0.884–0.888) and 0.826 (95% CI: 0.823–0.829), respectively. While the RNS of Knn and RF were similar, each algorithm had multiple metrics where one dominated over the other. RF had significantly higher Precision and Specificity where Knn was substantially better in Recall. Finally, while Ada had one of the lowest RNS’s, it did have the second-best Specificity score out of all the algorithms tested. The median score for each metric by algorithm is shown in Table 1, which is expanded to show the confidence intervals in Table S1. A full statistical comparison of each metric is shown in Table S2.
Figure 2.
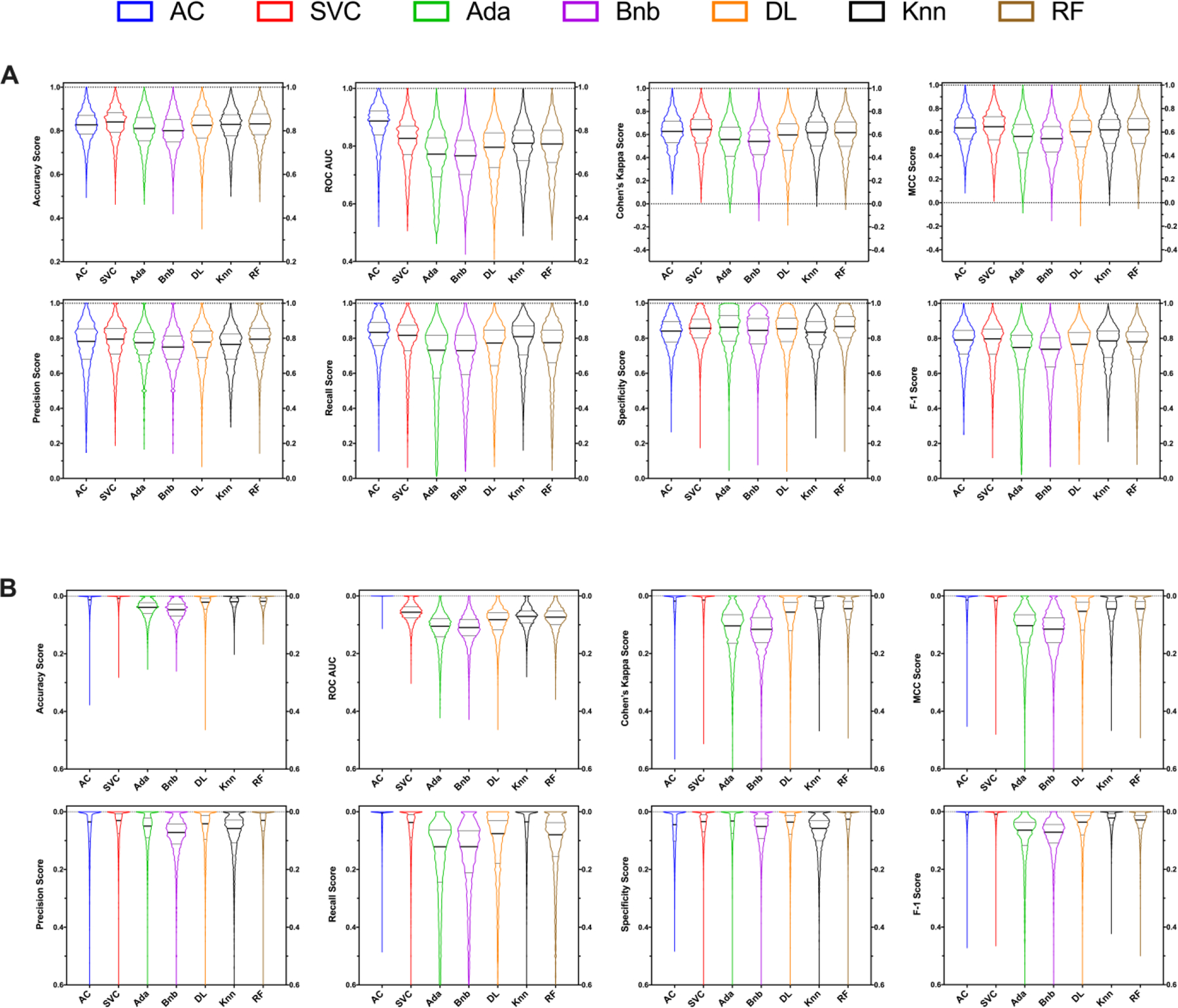
Machine learning algorithm comparisons for ChEMBL datasets across multiple five-fold cross-validation using multiple classical metrics. Distributions are show as either a raw score (A) or as a ‘difference from the top’ metric score (B). Truncated violin plots are shown with minimal smoothing to retain an accurate distribution representation. The solid central line represents the median with the quarterlies indicated. AC = Assay Central (Bayesian), RF = Random Forest, Knn = k-Nearest Neighbors, SVC = Support Vector Classification, Bnb = Naïve Bayesian, Ada = AdaBoosted Decision Trees, DL = Deep Learning.
Molecular properties
As the ChEMBL dataset is proposed to represent drug-like molecules we sought to compare it to actual drugs, using the Superdrug2 library. The distribution of each of the 8 calculated molecular properties selected was assessed for normality and none of these were statistically likely to be normally distributed (D’Agostino & Pearson test, p <0.0001****), therefore the differences were evaluated nonparametrically. The Gaussian fit displayed on each graph is meant only for easier visual discrimination of the two datasets and not for statistical purposes. Both the distributions (Kolmogorov-Smirnov test) and medians (Mann Whitney test) of each chemical library were determined to be statically significantly different for all measured metrics except for the number of hydrogen-bond donors, where the medium was not significantly different between the libraries (p = 0.1404) (Figure 3). The molecular weight, AlogP, number of aromatic rings, number of rings, number of hydrogen bond acceptors and number of rotatable bonds were all higher in the ChEMBL dataset. These results would suggest that the ChEMBL dataset consists of molecules that have properties outside of known drugs. Individual models built with subsets of the ChEMBL data would likely also vary in their overlap with approved drug properties.
Figure 3.
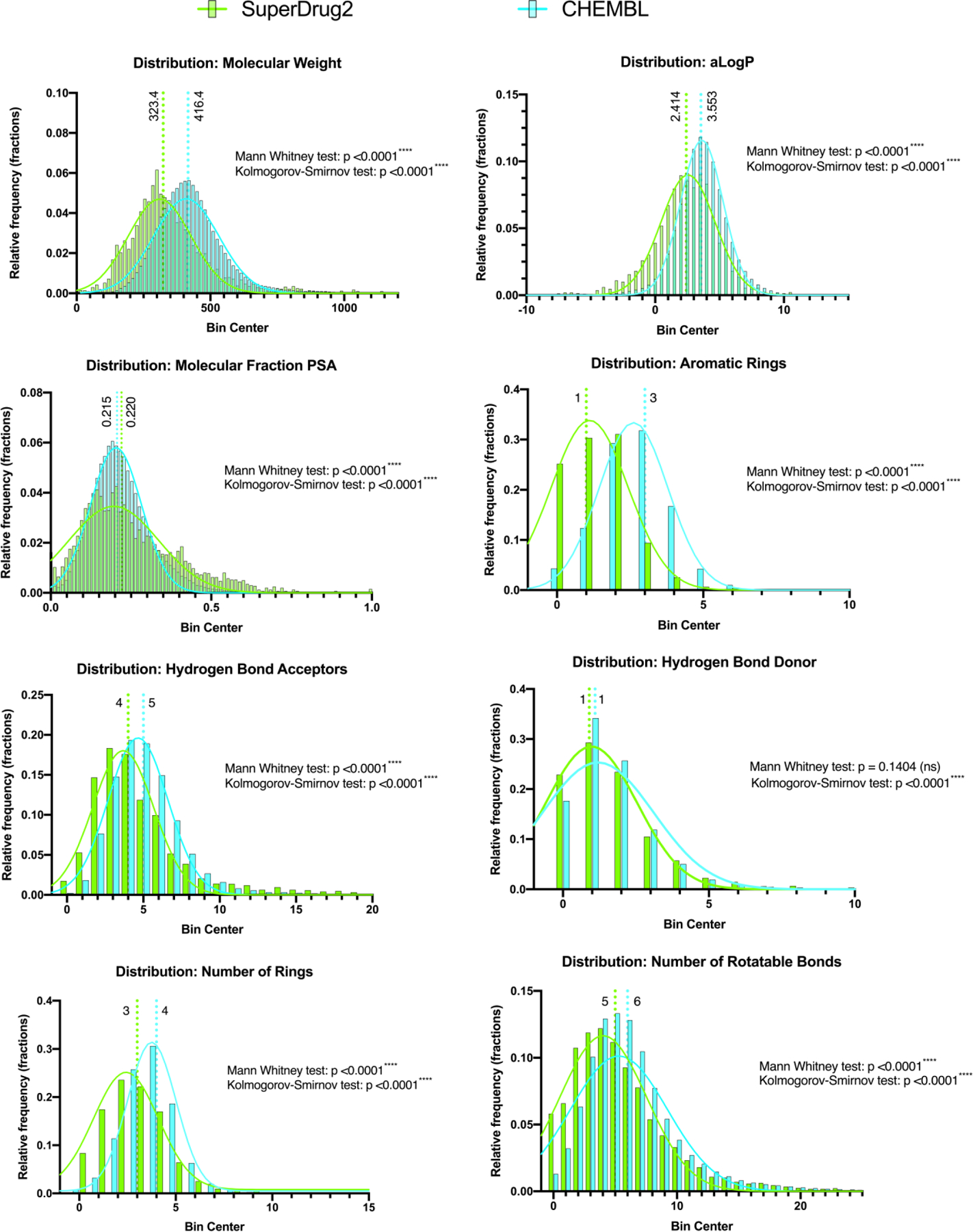
Comparison of the distribution of molecular properties for >570,000 unique compounds in the ChEMBL datasets versus 3992 compounds in the SuperDrug2 library as an example of approved drugs. Median values are highlighted along with statistical analyses.
External test sets
We used machine learning to prospectively score libraries of compounds to test in vitro for several toxicology properties of interest important for drug discovery. We have now collated this unpublished experimental data against the ion channel hERG (N = 10) and the nuclear receptor PXR (N = 14). We have then compared the predictions for these compounds from the autocurated models with additional machine learning models that used manually curated datasets in order to evaluate whether there is a difference between these techniques in the model prediction outcome. These two datasets therefore represent prospective external test sets. We have also anonymized these molecules to maintain confidentiality of our pipeline which consists of multiple infectious disease targets.
The two auto-curated datasets for PXR were quite dissimilar from the manually curated dataset (Table S3a, Figure S2). While the model metrics remained in a reasonably tight range, the calculated model thresholds were ~16 µM and 329 nM for ‘ADME’ and ‘binding’ models, respectively. The manually curated ChEMBL model contained agonism data exclusively and had an activity threshold of 665 nM. This resulted in little agreement between the predictions of PXR activity for the compounds chosen as a prospective test set from our internal library of compounds. Only three of the 14 molecules of the test set were predicted to have the same classification across the PXR models. However, both the manually curated and the auto-curated ‘ADME’ assays datasets generated similar prediction classifications for nine of the molecules in the test set. This is despite the vast threshold difference and less than half the amount of data present in the manually curated model (Table S3b). Additionally, this finding is interesting considering the specificity of the manually curated dataset (agonism assay descriptions) whereas the other models are capturing different assay types. An enrichment curve (Figure 4A) suggests that the manually curated and auto-curated ‘ADME’ model performed similarly, though the manually curated model suggested it has a slight edge.
Figure 4.
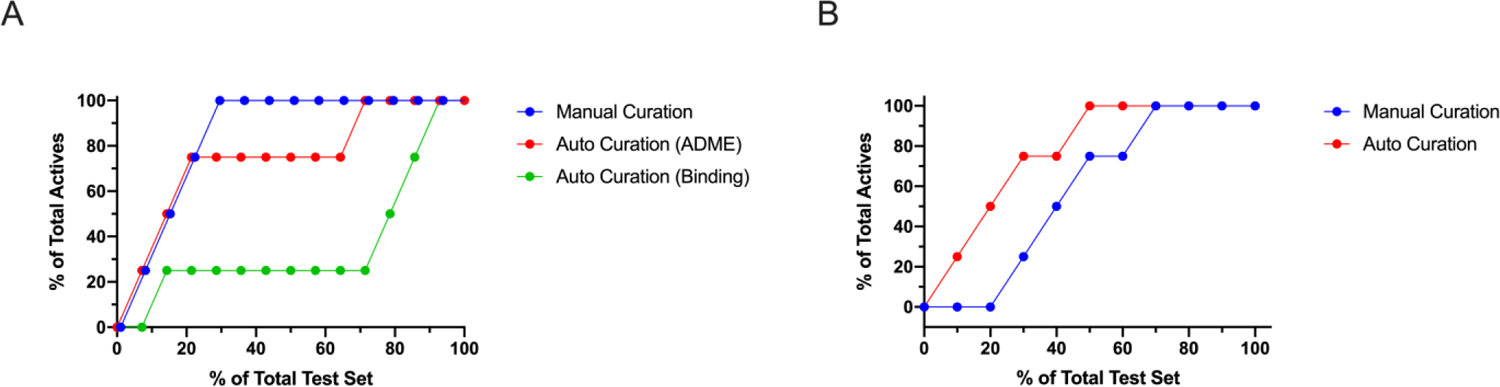
Comparison of enrichment rates of finding actives in the external test sets for A. PXR and B. hERG models using manually curated and auto-curated Assay Central models.
The metrics generated from five-fold cross-validation auto-curated and manually curated models of hERG were largely similar, the number of actives and total size was nearly the same, and the calculated threshold was the same (Table S4a, Figure S3), Unsurprisingly, predictions of hERG inhibition were nearly identical between these two models (Table S4b). While the predicted classifications were identical with each of the models a small difference can be visualized using an enrichment curve (Figure 4B), which appears to favor the auto-curated model.
To further test the predictive performance of auto-curated datasets on much larger external test sets (consisting of thousands of molecules), we utilized data from our previous publications for HIV whole cell and reverse transcriptase from NIAID 60 and M. tuberculosis whole cell data from literature 58. These datasets were predicted against auto-curated training models of HIV, whole cell and reverse transcriptase (Table S5, Figure S4) or M. tuberculosis, respectively (Table S6, Figure S5). All compounds which overlapped with the auto-curated training set were removed from the original published test set, and the activity threshold was set as equivalent to the training set.
The auto-curated HIV whole cell training model was able to more accurately predict the corresponding testing set (ROC = 0.79) in comparison to the reverse transcriptase model (ROC = 0.73) and test set (Table S5, Figure S4). In relation to whole cell validations, most of the reverse transcriptase test set metrics are diminished with the exception of ROC and specificity (Table S5c, Figure S4d). The M. tuberculosis validation (ROC = 0.77) had the highest specificity of all test sets, but all other metrics were comparable to the HIV test sets (Table S6, Figure S5).
DISCUSSION
Several of the largest comparisons of machine learning models relevant to drug discovery to date have used over 1000 models in numerous studies. For example 1227 datasets from ChEMBL version 20 were used to compare naïve Bayes, RF, SVM, logistic regression and DNN using random split and temporal validation 39. Morgan fingerprints were used as descriptors and validation metrics included MCC and Boltzmann Enhanced Discrimination of ROC (BEDROC). The random split used a 70% training and 30% validation; DNN appeared to perform better than other methods when BEDROC was used, while RF was the best for MCC using random split validation. Temporal validation demonstrated DNN outperformed all other methods, but all approaches performed better in the random split validation 39. Another study used 1310 assays from ChEMBL version 20 to compare binary models generated with SVM, kNN, RF, naïve Bayes, and several types of DNN including feed-forward neural networks, convolutional neural networks and recurrent neural networks for target prediction. Models were generated with a wide array of molecular descriptors and evaluated using ROC-AUC. Feed-forward neural networks were found to perform better than all other methods followed by SVM 40. 1067 human targets in ChEMBL version 23 have been selected to train and test multitask and single task DNN models considering sequence similarities of targets. Multitask learning outperformed single task learning when the targets are similar (ROC > 0.8) but when this is not considered or they are diverse then single task learning was superior (ROC 0.76– 0.79) 41. ChEMBL version 22 was used to extract molecules tested in 758 cell lines which was then used for machine learning with SVM and similarity analysis which illustrated comparable data on 10-fold cross-validation (accuracy = 0.65) but SVM was superior on external validation 42. ChEMBL version 22 was used for compound-target interactions and 1720 targets were selected with at least 10 compounds. Various molecular fingerprints and machine learning (naïve Bayes and DNN) were compared with nearest neighbor approaches. It was demonstrated that combining naïve Bayes and nearest neighbors performed the best in terms of recall and in a case study using a TRPV6 inhibitor 43. 1360 datasets from ChEMBL version 19 were used to build RF regression models with ECFP4 descriptors out of which 440 were reliable and in turn were used to create a QSAR-based affinity fingerprint. This was then evaluated for similarity searching and biological activity classification and found to be equivalent to Morgan fingerprints for similarity searching and outperform Morgan fingerprints for scaffold hopping 44. Deep learning has been used to learn the compound protein interactions using data from ChEMBL, BindingDB and DrugBank using latent semantic analysis to compare a method called DeepCPI to other similar approaches. It was found to perform well with AUC ROC of 0.92. Several G-protein coupled receptors (GLP-1R, GCGR, VIPR) were then selected for prospective virtual screening and three positive allosteric modulators for GLP-1R were identified 45. These examples are representative of the types of comparisons performed to date using subsets of ChEMBL.
There has also been considerable discussion about how best to evaluate machine learning models. One approach offered the quantile-activity bootstrap to mimic the extrapolation onto previously unseen areas of molecular space in order to evaluate 25 sets of targets from ChEMBL with multiple machine learning methods. This approach was found to remove any advantage of deep neural networks or RF versus using SVM and ridge regression 46. The work of Mayr et al. was reanalyzed in another study on the optimal way to benchmark studies questioned the AUCROC and instead proposed the area under the precision-recall curve should be used as well. SVM were comparable to feed-forward neural networks 47. It is important to note that not all computational approaches using machine learning models are successful and this likely represents publication bias. One rare example described using ChEMBL and REAXYS to generate SVM models used along with pharmacophores and generative topographic mapping to select compounds as bromodomain BRD4 binders with a 2.6 fold increase in hit rate relative to random screening 48.
We have previously described Assay Central® which uses a previously described Bayesian method and ECFP6 fingerprints that were made open source 61, 72 and the underlying components are therefore publicly available. We have shown that ECFP6 fingerprints compare favorably with other descriptors 54. We have compared Assay Central® with other machine learning methods for a relatively small number of targets such as drug induced liver injury 52, Rat Acute Oral toxicity 53, estrogen receptor 54, 55, androgen receptor 56, GSK3β 57 Mycobacterium tuberculosis 58, 59, non-nucleoside reverse transcriptase and whole cell HIV 60. In general, we have found from our earlier studies that while DNN generally performed the best for five-fold cross validation, but with external test sets this superiority was not observed and generally Assay Central® performed comparably with SVM classification or DNN. Our evaluation of these algorithms is now expanded to well over 5000 CHEMBL datasets in this current study making it the largest performed to date. We find that Assay Central® compares very well to SVM classification using the commonly used five-fold cross-validation when many metrics are compared and visualized using several methods in order to assess each algorithm. While this is only one form of leave out and it has limitations, it is certainly not as optimistic as leave-one out and it provides a useful guide for model utility. Unlike prior studies that have placed their emphasis on demonstrating the capabilities of DNN, we saw little or no advantage here where model tuning was performed as previously described 76. In this study Assay Central® had a slight advantage as the algorithm was used to select the optimal classification cutoff 61 for each of the > 5000 datasets was based on that used for Assay Central®. SVM classification seems a very strong competitor overall producing comparable model metrics. Ideally a standardized threshold would be applied to all datasets, but due to their heterogeneity and uncertainty in how that would skew datasets to be extremely unbalanced (i.e. some datasets at 1μM threshold would be 90% active and others 2%); so in order to simplify the process, we opted for a calculated, albeit non-standardized, threshold. One of the reasons for this performance observed by the Bayesian approach may be that in can handle unbalanced datasets well and in this study no attempts were made to balance datasets. In such cases it is likely other methods would perform better. We have not attempted to understand the effects of different algorithms with models of different sizes or degrees of (im)balance. However, dataset balancing will also reduce the dataset size and in some cases this would likely make them too small to be of use. In our previous multiple dataset comparisons of multiple machine learning methods and metrics, we limited our assessment to just eight datasets ranging in size from hundreds to hundreds of thousands of molecules curated from different sources 76. A rank normalized score across the various model metrics suggested in this case that DNN outperformed SVM during external testing with a validation set held out from model training 76. The current analysis represents a study that is orders of magnitude larger, allowing us to draw statistically significant conclusions. It should also be noted that our prior comparisons of leave out validation groups has reported stability in the ROC obtained when large datasets are used, whether this is leave one out or 30–50% x100 fold validations 80, 81. 5-fold cross validation is one way to obtain a snapshot comparison of how the models perform.
To our knowledge this also appears the largest scale comparison of such machine learning algorithms and metrics that we are aware of. As we have now shown, the ChEMBL data set is also statistically different to drugs that are approved based on important molecular properties (Figure 3). This is important because it illustrates the structural diversity of ChEMBL and also the dataset diversity in general is much larger than previous studies performing Machine learning comparisons. This should therefore provide more confidence in the algorithm comparison results as it would suggest that we are covering more diverse chemical property space. Future studies could certainly evaluate additional descriptors, algorithms and machine learning methods for evaluating the resulting models. Such public datasets and databases as ChEMBL will likely increase in size and they will continue be utilized more extensively to generate machine learning models by many more scientists. In order for this to be the case we need to make such datasets accessible for machine learning and this requires some degree of curation. In this study we have demonstrated that minimally curated datasets (desalted, neutralized and excluding any problematic compounds) can produce useful machine learning models with good statistics across a range of different algorithms. We have termed this approach “auto-curation”. We would naturally expect that it would be advantageous to expend significant effort to further manually curate datasets before using them for prediction, however there may be some value in such minimally curated models, and it may be impractical to manual curate thousands of models. While it is cost prohibitive and virtually impossible to prospectively predict all of the over 5000 ChEMBL models we have selected several to demonstrate their potential. We have therefore tested the utility of these auto-curated and manually curated models for PXR (Table S3) and hERG (Table S4). We noted that the model built with auto-curation (pxr/adme/ic50) predicted several of the most active PXR activators including CPI1072, CPI1073, CPI1077. These molecules were also scored highly with the manually curated model with the lowest cutoff. Similarly, the most potent inhibitors of hERG CPI1071, CPI1223a and CPI1286 were also predicted by the auto-curated ChEMBL model, however other compounds were similarly scored indicating poor prediction capability. Similar results were seen with manually curated models suggesting in these two limited cases (for relatively small datasets), the benefits of different curation approaches. We would certainly benefit from a much larger evaluation using many more examples to see whether there is a statistically relevant improvement in models after curation, however the cost of doing this either internally or through an external organization is currently cost prohibitive. In addition, an alternative approach to validation is to use several of these auto-curated models to predict data that was previously manually curated and used for training or testing models for HIV whole cell, HIV reverse transcriptase and Mtb inhibition 58, 60. In all cases the ROC for these test sets was > 0.7 (Figure S4 and S5). These results also suggest the potential of auto-curated models (Figure 4). Efforts are ongoing to develop more advanced auto-curation approaches which might bring more of the benefits of full manual curation but with limited human input. This work builds on our recent comparisons of different machine learning methods and algorithms for various individual projects 52, 53, 55–60, 82 to enable a very large scale comparison. It also demonstrates that several widely used (and openly accessible) machine learning methods consistently outperform the others at classification. Assay Central® uses a Bayesian approach which appears comparable to SVM classification (Figure 1B) and these in turn outperform all other algorithms used including the much hyped deep learning which is being more widely applied in pharmaceutical research 29. This study may be enlightening as it echoes our earlier findings on individual datasets after extensive manual curation 52, 53, 55–60, 82. It also goes some way further in using external validation with these methods for toxicology and drug discovery properties. Now that we have generated such a vast array of over 5000 machine learning models it presents opportunities for using them for predicting the potential of new molecules to interact with targets of interest or avoiding others (such as PXR and HERG) that may result in undesirable effects. We will also be able to compare the prospective ability of all the machine learning methods which is also rarely undertaken. This current study therefore represents a step towards using large scale machine learning models to assist in creating a drug discovery pipeline1. Our approach with models created with ChEMBL could also be taken using other very large databases such as PubChem or even considered by pharmaceutical companies to leverage their internal databases.
Supplementary Material
ACKNOWLEDGMENTS
Dr. Daniel P. Russo is kindly acknowledged for initially setting up and providing code for large scale machine learning comparisons and considerable discussions on machine learning. Mr. Valery Tkachenko is kindly acknowledged for hardware and software support. Mr. Kushal Batra is kindly acknowledged for providing a script used in data analysis. Dr. Vadim Makarov is acknowledged for providing several compounds that were used in our hERG and PXR test sets. Dr. Alex M. Clark is thanked for Assay Central® support. We have made use of the ChEMBL database and kindly acknowledge the team at EBI that has over several years worked hard to provide this valuable resource to the scientific community.
Grant information
We kindly acknowledge NIH funding: R44GM122196-02A1 from NIGMS, 3R43AT010585-01S1 from NCCAM and 1R43ES031038-01 from NIEHS (PI – Sean Ekins). “Research reported in this publication was supported by the National Institute of Environmental Health Sciences of the National Institutes of Health under Award Number R43ES031038. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.”
ABBREVIATIONS USED
- AUC
area under the curve
- DNN
Deep Neural Networks
- kNN
k-Nearest Neighbors
- QSAR
quantitative structure activity relationships
- RF
Random forest
- ROC
receiver operating characteristic
- SVM
support vector machines
Footnotes
Competing interests:
S.E., D.H.F., E.M., K.M.Z., and T.R.L. work for Collaborations Pharmaceuticals, Inc. F.U. has no conflicts of interest.
SUPPORTING INFORMATION
Supporting further details on the models and structures of public molecules. This material is available free of charge via the Internet at http://pubs.acs.org. The Supplemental Data file exceeds the 350MB limit imposed by ACS and is therefore available from the authors upon request.
Obtaining the HIV dataset in ChemDB requires interested parties to contact NIH NIAID and has restrictions on reuse.
REFERENCES
- 1.Ekins S; Puhl AC; Zorn KM; Lane TR; Russo DP; Klein JJ; Hickey AJ; Clark AM Exploiting machine learning for end-to-end drug discovery and development. Nat Mater 2019, 18, (5), 435–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhavoronkov A; Ivanenkov YA; Aliper A; Veselov MS; Aladinskiy VA; Aladinskaya AV; Terentiev VA; Polykovskiy DA; Kuznetsov MD; Asadulaev A; Volkov Y; Zholus A; Shayakhmetov RR; Zhebrak A; Minaeva LI; Zagribelnyy BA; Lee LH; Soll R; Madge D; Xing L; Guo T; Aspuru-Guzik A Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat Biotechnol 2019, 37, (9), 1038–1040. [DOI] [PubMed] [Google Scholar]
- 3.Gaulton A; Bellis LJ; Bento AP; Chambers J; Davies M; Hersey A; Light Y; McGlinchey S; Michalovich D; Al-Lazikani B; Overington JP ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res 2012, 40, (Database issue), D1100–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim S; Thiessen PA; Bolton EE; Chen J; Fu G; Gindulyte A; Han L; He J; He S; Shoemaker BA; Wang J; Yu B; Zhang J; Bryant SH PubChem Substance and Compound databases. Nucleic Acids Res 2016, 44, (D1), D1202–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anon The PubChem Database. http://pubchem.ncbi.nlm.nih.gov/
- 6.Schmidhuber J Deep learning in neural networks: an overview. Neural Netw 2015, 61, 85–117. [DOI] [PubMed] [Google Scholar]
- 7.Capuzzi SJ; Politi R; Isayev O; Farag S; Tropsha A QSAR Modeling of Tox21 Challenge Stress Response and Nuclear Receptor Signaling Toxicity Assays. Frontiers in Environmental Science 2016, 4, (3). [Google Scholar]
- 8.Russakovsky O; deng J; Su H; Krause J; Satheesh S; Ma S; Huang Z; Karpathy A; Khosla A; Bernstein M; Berg AC; Fei-Fei L ImageNet large scale visual recognition challenge. https://arxiv.org/pdf/1409.0575.pdf
- 9.Bennet KP; Campbell C Support vector machines: Hype or hallelujah? SIGKDD Explorations 2000, 2, 1–13. [Google Scholar]
- 10.Christianini N; Shawe-Taylor J, Support vector machines and other kernel-based learning methods. Cambridge University Press: Cambridge, MA, 2000. [Google Scholar]
- 11.Chang CC; Lin CJ LIBSVM: A library for support vector machines, 2001.
- 12.Lei T; Chen F; Liu H; Sun H; Kang Y; Li D; Li Y; Hou T ADMET Evaluation in Drug Discovery. Part 17: Development of Quantitative and Qualitative Prediction Models for Chemical-Induced Respiratory Toxicity. Mol Pharm 2017, 14, (7), 2407–2421. [DOI] [PubMed] [Google Scholar]
- 13.Kriegl JM; Arnhold T; Beck B; Fox T A support vector machine approach to classify human cytochrome P450 3A4 inhibitors. J Comput Aided Mol Des 2005, 19, (3), 189–201. [DOI] [PubMed] [Google Scholar]
- 14.Guangli M; Yiyu C Predicting Caco-2 permeability using support vector machine and chemistry development kit. J Pharm Pharm Sci 2006, 9, (2), 210–21. [PubMed] [Google Scholar]
- 15.Kortagere S; Chekmarev D; Welsh WJ; Ekins S Hybrid scoring and classification approaches to predict human pregnane X receptor activators. Pharm Res 2009, 26, (4), 1001–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen M; Xiao Y; Golbraikh A; Gombar VK; Tropsha A Development and validation of k-nearest neighbour QSPR models of metabolic stability of drug candidates. J Med Chem 2003, 46, 3013–3020. [DOI] [PubMed] [Google Scholar]
- 17.Wang S; Sun H; Liu H; Li D; Li Y; Hou T ADMET Evaluation in Drug Discovery. 16. Predicting hERG Blockers by Combining Multiple Pharmacophores and Machine Learning Approaches. Mol Pharm 2016, 13, (8), 2855–66. [DOI] [PubMed] [Google Scholar]
- 18.Li D; Chen L; Li Y; Tian S; Sun H; Hou T ADMET evaluation in drug discovery. 13. Development of in silico prediction models for P-glycoprotein substrates. Mol Pharm 2014, 11, (3), 716–26. [DOI] [PubMed] [Google Scholar]
- 19.Nidhi; Glick M; Davies JW; Jenkins JL Prediction of biological targets for compounds using multiple-category Bayesian models trained on chemogenomics databases. J Chem Inf Model 2006, 46, (3), 1124–33. [DOI] [PubMed] [Google Scholar]
- 20.Azzaoui K; Hamon J; Faller B; Whitebread S; Jacoby E; Bender A; Jenkins JL; Urban L Modeling promiscuity based on in vitro safety pharmacology profiling data. ChemMedChem 2007, 2, (6), 874–80. [DOI] [PubMed] [Google Scholar]
- 21.Bender A; Scheiber J; Glick M; Davies JW; Azzaoui K; Hamon J; Urban L; Whitebread S; Jenkins JL Analysis of Pharmacology Data and the Prediction of Adverse Drug Reactions and Off-Target Effects from Chemical Structure. ChemMedChem 2007, 2, (6), 861–873. [DOI] [PubMed] [Google Scholar]
- 22.Susnow RG; Dixon SL Use of robust classification techniques for the prediction of human cytochrome P450 2D6 inhibition. J Chem Inf Comput Sci 2003, 43, (4), 1308–15. [DOI] [PubMed] [Google Scholar]
- 23.Mitchell JB Machine learning methods in chemoinformatics. Wiley Interdiscip Rev Comput Mol Sci 2014, 4, (5), 468–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wacker S; Noskov SY Performance of Machine Learning Algorithms for Qualitative and Quantitative Prediction Drug Blockade of hERG1 channel. Comput Toxicol 2018, 6, 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu H; Zhang J; Kim MT; Boison A; Sedykh A; Moran K Big data in chemical toxicity research: the use of high-throughput screening assays to identify potential toxicants. Chem Res Toxicol 2014, 27, (10), 1643–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clark AM; Ekins S Open Source Bayesian Models: 2. Mining A “big dataset” to create and validate models with ChEMBL. J Chem Inf Model 2015, 55, 1246–1260. [DOI] [PubMed] [Google Scholar]
- 27.Ekins S; Clark AM; Swamidass SJ; Litterman N; Williams AJ Bigger data, collaborative tools and the future of predictive drug discovery. J Comput Aided Mol Des 2014, 28, (10), 997–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ekins S; Freundlich JS; Reynolds RC Are Bigger Data Sets Better for Machine Learning? Fusing Single-Point and Dual-Event Dose Response Data for Mycobacterium tuberculosis. J Chem Inf Model 2014, 54, 2157–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ekins S The Next Era: Deep Learning in Pharmaceutical Research. Pharm Res 2016, 33, (11), 2594–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baskin II; Winkler D; Tetko IV A renaissance of neural networks in drug discovery. Expert Opin Drug Discov 2016, 11, 785–795. [DOI] [PubMed] [Google Scholar]
- 31.Wang L; Ma C; Wipf P; Liu H; Su W; Xie XQ TargetHunter: an in silico target identification tool for predicting therapeutic potential of small organic molecules based on chemogenomic database. AAPS J 2013, 15, (2), 395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koutsoukas A; Lowe R; Kalantarmotamedi Y; Mussa HY; Klaffke W; Mitchell JB; Glen RC; Bender A In silico target predictions: defining a benchmarking data set and comparison of performance of the multiclass Naive Bayes and Parzen-Rosenblatt window. J Chem Inf Model 2013, 53, (8), 1957–66. [DOI] [PubMed] [Google Scholar]
- 33.Cortes-Ciriano I Benchmarking the Predictive Power of Ligand Efficiency Indices in QSAR. J Chem Inf Model 2016, 56, (8), 1576–87. [DOI] [PubMed] [Google Scholar]
- 34.Qureshi A; Kaur G; Kumar M AVCpred: an integrated web server for prediction and design of antiviral compounds. Chem Biol Drug Des 2017, 89, (1), 74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bieler M; Reutlinger M; Rodrigues T; Schneider P; Kriegl JM; Schneider G Designing Multi-target Compound Libraries with Gaussian Process Models. Mol Inform 2016, 35, (5), 192–8. [DOI] [PubMed] [Google Scholar]
- 36.Huang T; Mi H; Lin CY; Zhao L; Zhong LL; Liu FB; Zhang G; Lu AP; Bian ZX; for MG MOST: most-similar ligand based approach to target prediction. BMC Bioinformatics 2017, 18, (1), 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cortes-Ciriano I; Firth NC; Bender A; Watson O Discovering Highly Potent Molecules from an Initial Set of Inactives Using Iterative Screening. J Chem Inf Model 2018, 58, (9), 2000–2014. [DOI] [PubMed] [Google Scholar]
- 38.Bosc N; Atkinson F; Felix E; Gaulton A; Hersey A; Leach AR Large scale comparison of QSAR and conformal prediction methods and their applications in drug discovery. J Cheminform 2019, 11, (1), 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lenselink EB; Ten Dijke N; Bongers B; Papadatos G; van Vlijmen HWT; Kowalczyk W; AP IJ; van Westen GJP Beyond the hype: deep neural networks outperform established methods using a ChEMBL bioactivity benchmark set. J Cheminform 2017, 9, (1), 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mayr A; Klambauer G; Unterthiner T; Steijaert M; Wegner JK; Ceulemans H; Clevert DA; Hochreiter S Large-scale comparison of machine learning methods for drug target prediction on ChEMBL. Chem Sci 2018, 9, (24), 5441–5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee K; Kim D In-Silico Molecular Binding Prediction for Human Drug Targets Using Deep Neural Multi-Task Learning. Genes (Basel) 2019, 10, (11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tejera E; Carrera I; Jimenes-Vargas K; Armijos-Jaramillo V; Sanchez-Rodriguez A; Cruz-Monteagudo M; Perez-Castillo Y Cell fishing: A similarity based approach and machine learning strategy for multiple cell lines-compound sensitivity prediction. PLoS One 2019, 14, (10), e0223276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Awale M; Reymond JL Polypharmacology Browser PPB2: Target Prediction Combining Nearest Neighbors with Machine Learning. J Chem Inf Model 2019, 59, (1), 10–17. [DOI] [PubMed] [Google Scholar]
- 44.Škuta C; Cortés-Ciriano I; Dehaen W; Kříž P; van Westen GJP; Tetko IV; Bender A; Svozil D QSAR-derived affinity fingerprints (part 1): fingerprint construction and modeling performance for similarity searching, bioactivity classification and scaffold hopping. Journal of Cheminformatics 2020, 12, (1), 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wan F; Zhu Y; Hu H; Dai A; Cai X; Chen L; Gong H; Xia T; Yang D; Wang MW; Zeng J DeepCPI: A Deep Learning-based Framework for Large-scale in silico Drug Screening. Genomics Proteomics Bioinformatics 2019, 17, (5), 478–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Watson OP; Cortes-Ciriano I; Taylor AR; Watson JA A decision-theoretic approach to the evaluation of machine learning algorithms in computational drug discovery. Bioinformatics 2019, 35, (22), 4656–4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robinson MC; Glen RC; Lee AA Validating the validation: reanalyzing a large-scale comparison of deep learning and machine learning models for bioactivity prediction. J Comput Aided Mol Des 2020, 34, (7), 717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Casciuc I; Horvath D; Gryniukova A; Tolmachova KA; Vasylchenko OV; Borysko P; Moroz YS; Bajorath J; Varnek A Pros and cons of virtual screening based on public “Big Data”: In silico mining for new bromodomain inhibitors. Eur J Med Chem 2019, 165, 258–272. [DOI] [PubMed] [Google Scholar]
- 49.Bediaga H; Arrasate S; Gonzalez-Diaz H PTML Combinatorial Model of ChEMBL Compounds Assays for Multiple Types of Cancer. ACS Comb Sci 2018, 20, (11), 621–632. [DOI] [PubMed] [Google Scholar]
- 50.Chen Y; Stork C; Hirte S; Kirchmair J NP-Scout: Machine Learning Approach for the Quantification and Visualization of the Natural Product-Likeness of Small Molecules. Biomolecules 2019, 9, (2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zin PPK; Williams GJ; Ekins S Cheminformatics Analysis and Modeling with MacrolactoneDB. Sci Rep 2020, 10, (1), 6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Minerali E; Foil DH; Zorn KM; Lane TR; Ekins S Comparing Machine Learning Algorithms for Predicting Drug-Induced Liver Injury (DILI). Mol Pharm 2020, 17, 2628–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Minerali E; Foil DH; Zorn KM; Ekins S Evaluation of Assay Central® Machine Learning Models for Rat Acute Oral Toxicity Prediction. ACS Sustain Chem Eng 2020, 8, 16020–16027. [Google Scholar]
- 54.Zorn KM; Foil DH; Lane TR; Russo DP; Hillwalker W; Feifarek DJ; Jones F; Klaren WD; Brinkman A; Ekins S Machine Learning Models for Estrogen Receptor Bioactivity and Endocrine Disruption Prediction. Environ Sci Technol 2020, 54, 12202–12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Russo DP; Zorn KM; Clark AM; Zhu H; Ekins S Comparing Multiple Machine Learning Algorithms and Metrics for Estrogen Receptor Binding Prediction. Mol Pharm 2018, 15, (10), 4361–4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zorn KM; Foil DH; Lane TR; Hillwalker W; Feifarek DJ; Jones F; Klaren WD; Brinkman AM; Ekins S Comparison of Machine Learning Models for the Androgen Receptor. Environ Sci Technol 2020, 54, 13690–13700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vignaux P; Minerali E; Foil DH; Puhl AC; Ekins S Machine Learning for Discovery of GSK3β Inhibitors. ACS Omega 2020, 5, 26551–26561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lane T; Russo DP; Zorn KM; Clark AM; Korotcov A; Tkachenko V; Reynolds RC; Perryman AL; Freundlich JS; Ekins S Comparing and Validating Machine Learning Models for Mycobacterium tuberculosis Drug Discovery. Mol Pharm 2018, 15, (10), 4346–4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Puhl AC; Lane TR; Vignaux PA; Zorn KM; Capodagli GC; Neiditch MB; Freundlich JS; Ekins S Computational Approaches to Identify Molecules binding to Mycobacterium Tuberculosis KasA. ACS Omega 2020, In Press. [DOI] [PMC free article] [PubMed]
- 60.Zorn KM; Lane TR; Russo DP; Clark AM; Makarov V; Ekins S Multiple Machine Learning Comparisons of HIV Cell-based and Reverse Transcriptase Data Sets. Mol Pharm 2019, 16, (4), 1620–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Clark AM; Ekins S Open Source Bayesian Models. 2. Mining a “Big Dataset” To Create and Validate Models with ChEMBL. J Chem Inf Model 2015, 55, (6), 1246–60. [DOI] [PubMed] [Google Scholar]
- 62.Gaulton A; Hersey A; Nowotka M; Bento AP; Chambers J; Mendez D; Mutowo P; Atkinson F; Bellis LJ; Cibrian-Uhalte E; Davies M; Dedman N; Karlsson A; Magarinos MP; Overington JP; Papadatos G; Smit I; Leach AR The ChEMBL database in 2017. Nucleic Acids Res 2017, 45, (D1), D945–D954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Willighagen EL; Mayfield JW; Alvarsson J; Berg A; Carlsson L; Jeliazkova N; Kuhn S; Pluskal T; Rojas-Cherto M; Spjuth O; Torrance G; Evelo CT; Guha R; Steinbeck C The Chemistry Development Kit (CDK) v2.0: atom typing, depiction, molecular formulas, and substructure searching. J Cheminform 2017, 9, (1), 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Anantpadma M; Lane T; Zorn KM; Lingerfelt MA; Clark AM; Freundlich JS; Davey RA; Madrid PB; Ekins S Ebola Virus Bayesian Machine Learning Models Enable New in Vitro Leads. ACS Omega 2019, 4, (1), 2353–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dalecki AG; Zorn KM; Clark AM; Ekins S; Narmore WT; Tower N; Rasmussen L; Bostwick R; Kutsch O; Wolschendorf F High-throughput screening and Bayesian machine learning for copper-dependent inhibitors of Staphylococcus aureus. Metallomics 2019, 11, (3), 696–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ekins S; Gerlach J; Zorn KM; Antonio BM; Lin Z; Gerlach A Repurposing Approved Drugs as Inhibitors of Kv7.1 and Nav1.8 to Treat Pitt Hopkins Syndrome. Pharm Res 2019, 36, (9), 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ekins S; Mottin M; Ramos P; Sousa BKP; Neves BJ; Foil DH; Zorn KM; Braga RC; Coffee M; Southan C; Puhl AC; Andrade CH Deja vu: Stimulating open drug discovery for SARS-CoV-2. Drug Discov Today 2020. [DOI] [PMC free article] [PubMed]
- 68.Hernandez HW; Soeung M; Zorn KM; Ashoura N; Mottin M; Andrade CH; Caffrey CR; de Siqueira-Neto JL; Ekins S High Throughput and Computational Repurposing for Neglected Diseases. Pharm Res 2018, 36, (2), 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sandoval PJ; Zorn KM; Clark AM; Ekins S; Wright SH Assessment of Substrate-Dependent Ligand Interactions at the Organic Cation Transporter OCT2 Using Six Model Substrates. Mol Pharmacol 2018, 94, (3), 1057–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang PF; Neiner A; Lane TR; Zorn KM; Ekins S; Kharasch ED Halogen Substitution Influences Ketamine Metabolism by Cytochrome P450 2B6: In Vitro and Computational Approaches. Mol Pharm 2019, 16, (2), 898–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zorn KM; Foil DH; Lane TR; Russo DP; Hillwalker W; Feifarek DJ; Jones F; Klaren WD; Brinkman A; Ekins S Machine Learning Models for Estrogen Receptor Bioactivity and Endocrine Disruption Prediction. Environ Sci Technol 2020, 54, 12202–12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Clark AM; Dole K; Coulon-Spektor A; McNutt A; Grass G; Freundlich JS; Reynolds RC; Ekins S Open Source Bayesian Models. 1. Application to ADME/Tox and Drug Discovery Datasets. J Chem Inf Model 2015, 55, (6), 1231–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Carletta J Assessing agreement on classification tasks: The kappa statistic. Computational linguistics 1996, 22, 249–254. [Google Scholar]
- 74.Cohen J A coefficient of agreement for nominal scales. Education and Psychological Measurement 1960, 20, 37–46. [Google Scholar]
- 75.Matthews BW Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim Biophys Acta 1975, 405, (2), 442–51. [DOI] [PubMed] [Google Scholar]
- 76.Korotcov A; Tkachenko V; Russo DP; Ekins S Comparison of Deep Learning With Multiple Machine Learning Methods and Metrics Using Diverse Drug Discovery Data Sets. Mol Pharm 2017, 14, (12), 4462–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Caruana R; Niculescu-Mizil A, An empirical comparison of supervised learning algorithms. In 23rd International Conference on Machine Learning, Pittsburgh, PA, 2006. [Google Scholar]
- 78.Siramshetty VB; Eckert OA; Gohlke BO; Goede A; Chen Q; Devarakonda P; Preissner S; Preissner R SuperDRUG2: a one stop resource for approved/marketed drugs. Nucleic Acids Res 2018, 46, (D1), D1137–D1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Piper DR; Duff SR; Eliason HC; Frazee WJ; Frey EA; Fuerstenau-Sharp M; Jachec C; Marks BD; Pollok BA; Shekhani MS; Thompson DV; Whitney P; Vogel KW; Hess SD Development of the predictor HERG fluorescence polarization assay using a membrane protein enrichment approach. Assay Drug Dev Technol 2008, 6, (2), 213–23. [DOI] [PubMed] [Google Scholar]
- 80.Ekins S; Williams AJ; Xu JJ A predictive ligand-based Bayesian model for human drug-induced liver injury. Drug Metab Dispos 2010, 38, (12), 2302–8. [DOI] [PubMed] [Google Scholar]
- 81.Zientek M; Stoner C; Ayscue R; Klug-McLeod J; Jiang Y; West M; Collins C; Ekins S Integrated in silico-in vitro strategy for addressing cytochrome P450 3A4 time-dependent inhibition. Chem Res Toxicol 2010, 23, (3), 664–76. [DOI] [PubMed] [Google Scholar]
- 82.Zorn KM; Foil DH; Lane TR; Russo DP; Hillwalker W; Feifarek DJ; Jones F; Klaren WD; Brinkman AM; Ekins S Machine Learning Models for Estrogen Receptor Bioactivity and Endocrine Disruption Prediction. Environ Sci Technol 2020, 54, 12202–12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.