

Abstract
The spread of a novel coronavirus SARS-CoV-2 and a resulting COVID19 disease in late 2019 has transformed into a worldwide pandemic and has effectively brought the world to a halt. Proteases 3CLpro and PLpro, responsible for proteolysis of new virions, represent vital inhibition targets for the COVID19 treatment. Herein, we report an in silico docking study of more than 860 COVID19-related compounds from the PubChem database. Molecular dynamic simulations were carried out to validate the conformation stability of compound-ligand complexes with best docking scores. The MM-PBSA approach was employed to calculate binding free energies. The comparison with ca. 50 previously reported potential SARS-CoV-2′s proteases inhibitors show a number of new compounds with excellent binding affinities. Anti-inflammatory drugs Montelukast, Ebastine and Solumedrol, the anti-migraine drug Vazegepant or the anti-MRSA pro-drug TAK-599, among many others, all show remarkable affinities to 3CLpro and with known side effects present candidates for immediate clinical trials. This study reports thorough docking scores summary of COVID19-related compounds found in the PubChem database and illustrates the asset of computational screening methods in search for possible drug-like candidates. Several yet-untested compounds show affinities on par with reported inhibitors and warrant further attention. Furthermore, the submitted work provides readers with ADME data, ZINC and PubChem IDs, as well as docking scores of all studied compounds for further comparisons.
Keywords: SARS-CoV-2, 3CLpro, PLpro, PubChem, Molecular docking
Graphical abstract

1. Introduction
In the late 2019, the world was taken by storm by a novel type of β coronavirus dubbed SARS-CoV-2 or 2019-nCoV originating in Wuhan, China [1,2]. The disease caused by SARS-CoV-2 was named COVID19 and like diseases caused by other coronaviruses outbreaks, such as SARS-CoV [3] (Severe Acute Respiratory Syndrome Coronavirus) in 2003 and MERS [4] (Middle East Respiratory Syndrome Coronavirus) in 2012, it presents itself with respiratory symptoms including fever, dry cough, fatigue and loss of taste or smell. In more severe cases COVID19 leads to pneumonia, dyspnea and eventually death [5,6].
Key moments for stopping the spread of a virus in the human body, besides vaccination, include: A. virus entrance to the cell; B. inhibition of polyproteins proteolysis, whose products are subsequently employed in new virions production; C. replication of the RNA genome. Thus, potential targets for SARS-CoV-2 treatment include: A. the SARS-CoV-2 receptor-binding spike protein; B. proteases 3CLpro (also denoted as Mpro)/PLpro (3C-like protease/papain-like protease); C. the RNA polymerase (RdRP) [7,8]. SARS-CoV-2 spike protein binds angiotensin-converting enzyme 2 (ACE2) making it an ideal target for vaccine development, preventing the virus from entering the cell. Proteases 3CLpro/PLpro and RNA polymerase display high genome sequence similarities with SARS-CoV (82–96%) and present inhibition options A. and B. for terminating the virus replication in cells [9]. Protease 3CLpro appears to be the most promising inhibition target of SARS-CoV-2 polyproteins proteolysis (B). 3CLpro monomer consists of three domains (domain I – residues 8–101, domain II – residues 102–184, domain III – residues 201–303) and a long loop (residues 185–200) connecting domains II and III.
Its active site forms a cavity located in the cleft between domains I and II with catalytic dyad (Cys145 and His41) that is large enough to bind the active substance forming a drug-protein complex [10,11], see Fig. 1 A. The interior of the cavity is composed of several hydrophobic amino acids (Leu27, Phe140, Gly143, Met165 Pro168), forming hydrophobic environment and polar amino acids (Thr26, Asn142, His163, Gln189) capable of stabilizing the drug-protein complex [10,11]. 3PLpro consists of two distinct domains (ubiquitin-like domain – residues 1–60, thumb-palm-finger domain – residues 61–315) with its active site containing a catalytic triad (Cys111, His272, Asp286) and an important zinc ion coordinated by four cysteine residues [12], see Fig. 1B. The surrounding of a smaller catalytic site is comprised of polar amino acids (Asn109, Asn110, Thr265, Tyr273) and hydrophobic amino acids (Gly266, Gly271, Gly287, Ala288).
Fig. 1.

Three-dimensional ribbon structures of 3CLpro (1A) and PLpro (1B) proteases, with catalytic diad/triad indicated by sticks and balls representations of participating molecules.
In the herein presented in silico work, we have performed molecular docking experiments of potential SARS-CoV-2 inhibitors accessed from external compounds database against two 3CLpro and PLpro structures. Docking protocols were followed by molecular dynamics simulations of most promising compound-protein complexes to ascertain their conformational stability, and by the evaluation of free energy of binding. Several in silico research papers have been published over the previous year focusing either on docking studies [11,[13], [14], [15], [16], [17], [18], [19], [20], [21], [22]] of selected compounds or screening a large number of compounds from external databases utilizing machine learning approaches [23] or virtual screening [24]. Our obtained results were compared with previously published papers [11,[13], [14], [15], [16], [17], [18], [19], [20], [21], [22]] and redocking of several compounds with high affinity towards SARS-Cov-2 proteases from these works was performed to allow for a direct comparison to the presented results.
2. Materials and methods
2.1. Preparation
The 3-dimensional structures of 866 compounds tagged by the keyword “SARS-CoV-2″ [25] were downloaded from the PubChem [26,27] [October 23, 2020] database in the .sdf format and converted to the .mol2 format using the OpenBabel compiler 2.3.2 [28]. Furthermore, 3D structures of 33 best scoring compounds from four different publications [11,16,17,21] were downloaded from the PubChem database, ten best scoring compounds from another paper [13] were downloaded from the ZINC database [29,30] and three geometries of compounds not found in either database were optimized at the DFT level of theory (B3LYP/6–311G*) [31], [32], [33], [34], [35] using the Gaussian09 software [36]. Where possible, compounds were described by their CID number, DB identifier, InChI key and ZINC code as can be found in the ESI files. Polar hydrogens and Gasteiger charges were added to all structures using the AutoDock utility scripts [37].
The structures of SARS-CoV-2 3CLpro and PLpro at 100 K (PDB IDs: 6LU7 [38] and 6WZU [39]) as well as their crystal structures determined at the room temperature (PDB IDs: 6WQF [10] and 7CMD [40]) were downloaded from the RCSB protein data bank [41]. Swiss-PDBViewer [42] was used to fix the missing atoms within the proteins and to remove present ligands (6LU7 and 7CMD). Additional protein strands were removed from the selected crystal structures and the 6LU7 and 6WZU/7CMD structures were stripped of water molecules, while one crystal water was retained within the 6WQF structure. This molecule is situated within the pocket of the protease active site cavity, near residues His41 and Asp187, and it has been previously reported that this water molecule takes part in several interactions leading to charge stabilization on neighbouring residues [10].
Conformational differences between pairs of protein structures were evaluated by template modeling (TM) score ranging from 0 to 1, with 1 indicating a perfect match and 0 indicating no match at all between compared structures [43]. The selected 3CLpro structures have the TM score 0.98, with minor local structural differences at the C-terminus. The TM score further increases to 1 if calculated for the initial 300 amino acids only. The compared PLpro structures have the TM score 0.96, with minor differences throughout the whole 3D structures. Furthermore, 7CMD does not have a completely solved crystal structure, as there are amino acids missing between Val220 and Thr231. This should affect neither TM score, as it is calculated only from pairs of amino acids that are present in both structures nor the docking results, as the sequence is not located in the proximity of the active site cavity.
2.2. Docking
Semi-flexible docking simulations were performed using the software Autdock4.2.6 [37,44]. A grid box of 90×90×90 Å [with a resolution of 0.275 Å and centered at x, y, z = (-20 Å, -5 Å, 15 Å)] and a grid box of 60×60×60 Å [with same resolution and centered at x, y, z = (-8 Å, 82 Å, 37 Å)/(-32 Å, -24 Å, -28 Å)] were used to calculate potential maps within the protein binding cavity of 6LU7/6WQF and 6WZU/7CMD protein units, respectively. In the first approximation, total of 50 Lamarckian genetic algorithm docking runs were performed for each tested molecule, with the initial population containing 300 individuals. The maximum number of energy evaluations was set to 30,000,000 and the maximum number of generations was set to 27,000. Rates of gene mutations and crossovers were set to 0.02 and 0.8, respectively, with one individual to survive to the next generation. The probability of Solis & Wets local search was set to 0.06 with a maximum number of iterations of 300 and a minimum step size of 1% of the grid box. Compounds with docking scores below -11.0 kcal/mol (total of 34 structures) for 6QWF, below -12.0 kcal/mol for 6WQF (total of 63 structures) and below -9.0 kcal/mol for 6WZU/7CMD (total of 47/37 structures) were selected into a second round of docking calculations with the number of the genetic algorithm runs increased to 100 and the number of energy evaluations increased to 50,000,000. All resulting docked poses were clustered with 2.0 Å tolerance and analysed with the AutoDockTools utility [37,45] with one compound (CID: 73774610) omitted from further data evaluation due to unforeseeable errors.
2.3. Molecular dynamics (MD)
In addition, five structures with the lowest docking scores for 6WQF and 6WZU protein units were selected for MD simulations in GROMACS2018.7 [46], [47], [48], [49] using the CHARMM36-july forcefield and the molecular parameters and their topologies were generated with the CgenFF utility [50], [51], [52], [53], [54].
The proteins were solvated in the rhombic dodecahedron with edges of the box distant 1.0 nm with respect to the nearest protein residue using the four-point TIP4P rigid water model [55] and Na+/Cl− ions to ensure electro neutrality. The steepest descent algorithm was employed for energy minimization purposes, with the studied systems reaching their energy minimum within 1500 steps. Subsequently, all systems were equilibrated to a 300 K temperature (using the modified Berendsen thermostat [56]) and a 1 bar pressure (the Parrinello-Rahman coupling [57]) for a total of 200 ps. Finally, a 10,000 ps molecular dynamics simulation was run using the leap-frog integrator with a step of 0.002 ps. Resulting trajectories were analysed using the GROMACS software package calculating parameters such as the short-ranged compound-protein Coulombic interaction energy, the short-ranged compound-protein Lennard-Jones energy, the RMSD (Root-mean-square Deviation) of compound's heavy atoms with respect to the initial structure of the residues’ backbone, distances/angles between compound atoms and protein atoms within the cavity, etc. The formation and propagation of hydrogen bonds were analysed using the VMD 1.9.3 software [58] with a 3.5 Å distance cut-off and a 30° angle cut-off.
Free energies of binding were calculated using the MM-PBSA (Molecular Mechanics – Poisson-Boltzmann Surface Area) approach as implemented in the g_mmpbsa GROMACS package [59,60]. Snapshots at an interval of 10 ps were extracted from MD trajectories and binding free energies of the compound-protein complex were expressed as a difference between ensemble averaged free energies of complexes and their constituents.
The polar solvation calculation was carried out with a 0.1 M salt concentration and a solute dielectric constant of 2. The SASA (Solvent Accessible Surface Area) model was used in calculating non-polar contributions to free energies of binding.
2.4. ADME
Absorption, distribution, metabolism, excretion (ADME) parameters as well as other physicochemical descriptors and druglike properties of studied compounds were computed from a list of SMILES codes using the SwissADME website [61] and can be found in the supporting excel file PubChem_SwissADME.xlsx along with the other identifiers. These data were used to check whether the compounds satisfy criteria of empirical drug likeness rules, such as the Lipinski's et al. rule of five [62] and the Ghose et al. filter [63].
3. Results
Molecular docking analyses are nowadays an important part of drug design and are often used to examine drug-protein interactions to allow for a better understanding of the target active site or the importance of functional group substitutions within a drug structure. Final docked poses can be evaluated by the number of drug-protein interactions (H-bonds, π-π, etc.), as well as by the binding energy (score) which allows for a comparison of the affinity of a set of different compounds to the targeted receptor. Compounds with lowest (most negative) score and a reasonable hydrogen bond pattern can be chosen as perspective drug targets for further studies and derivatizations. Compounds downloaded from the PubChem database achieved in the initial docking runs against 6LU7 (6WQF) docking scores ranging from -2.80 (-3.35) kcal/mol to -13.88 (-14.45) kcal/mol, with a median score of -7.45 (-8.09) kcal/mol and an average score of -7.65 (-8.48) kcal/mol. Most negative docking scores were leaning heavily towards molecules with a higher number of atoms (molecular weight), which can be explained by the size of 3CLpro cavity. An average shift of -0.83 kcal/mol towards lower docking scores was observed, see Fig. 2 , when comparing the 6WQF docking scores against 6LU7 ones, with one outlier (CID: 3793) and a variance 0.45 kcal/mol of the difference between 6WQF and 6LU7 docking scores. The strong correlation of the docking scores is consistent with structural similarity as discussed in the previous section.
Fig. 2.

The correlation of compounds’ docking scores for 6WQF 3CLpro to their respective 6LU7 3CLpro scores.
Docking in silico experiments with PLpro structure 6WZU (7CMD) yielded docking scores ranging from -2.77 (-0.72) kcal/mol to -10.51 (-11.21) kcal/mol, with a median score of -6.42 (-5.91) kcal/mol and an average score -6.61 (-5.98) kcal/mol with significantly lower bias towards compounds with a higher molecular weight. The average shift of -0.63 kcal/mol towards lower docking scores against PLpro structure 6WZU was observed when comparing to the respective docking scores of 7CMD, see Fig. 3 . The differences in selected PLpro crystal structures lead to a higher variance (1.91 kcal/mol) of the difference between 6WZU and 7CMD docking scores compared to the 6WQF and 6LU7 one (compare Figs. 3 and 2, respectively). It is evident that the origin of the targeted crystal structure affects docking score in the semi-flexible docking protocols. Due to a smaller cavity size, the differences in crystallization methods (crystallization temperature and/or liganded vs. un-liganded structure) affect the active site of PLpro, and hence the docking scores, more significantly than for 3CLpro.
Fig. 3.

The correlation of compounds’ docking scores for 7CMD PLpro to their respective 6WZU PLpro scores.
See section “Comparison with other studies” for graphical representations of all obtained docking results. The docking studies against all four protein targets have not indicated any clear pattern between docking scores and the number of formed hydrogen bonds, as well as any of the most common ADME descriptors, with most complexes stabilized by six and less hydrogen bonds, see Fig. 4 . Redocking of top structures lead to a small decrease in docking scores indicating that the initially chosen docking parameters were sufficient for this study. Table 1 contains structures, CID identifiers, trivial names and docking scores of top five compounds studied along with the lowest docking score from the second round of redockings for all four protein targets. Extended version of this table containing top ten compounds for each protein can be found in Table S1.
Fig. 4.

Frequency of predicted H-bonds formed between docked compounds and the respective protein targets.
Table 1.
CID PubChem identifiers, trivial names (in brackets), docking scores in kcal/mol, recognized pharmacological functions and molecular structures [74] of five compounds with highest binding affinities towards 3CLpro/PLpro structures. Molecular structures are downloaded from the PubChem database [26,27]. For an extended version of this table see Table S1.
| № | 6WQF (3CLpro) | 6LU7 (3CLpro) | 6WZU (PLpro) | 7CMD (PLpro) |
|---|---|---|---|---|
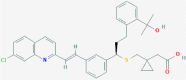
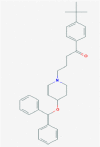
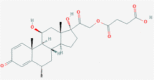
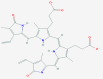
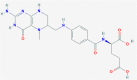
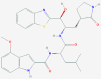
| 1 | 5281040 (Montelukast) | 3191 (Ebastine) | 16923 (Solumedrol) | 121893 (Protoporphyrinogen IX) |
| -14.75 | -14.11 | -10.89 | -11.58 | |
| Anti-inflammatory and bronchodilating activity | Anti-inflammatory activity | Anti-inflammatory and immunosuppressive activity | – | |
 |
 |
 |
 |
|
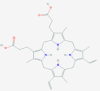
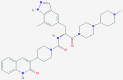
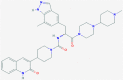
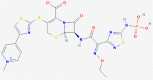
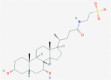
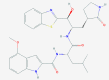
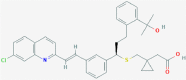
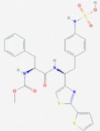
| 2 | 53472683 (Vazegepant/zavegepant) | 53472683 (Vazegepant/zavegepant) | 73425380 (TAK-599) | 9548902 (Taurocheno- deoxycholate(1-)) |
| -14.57 | -13.80 | -10.69 | -11.24 | |
| Anti-migraine activity | Anti-migraine activity | Anti-MRSA activity | Human metabolite | |
 |
 |
 |
 |
|
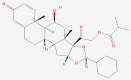
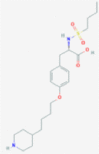
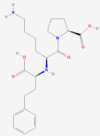
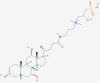
| 3 | 6918155 (Ciclesonide) | 154573806 (GRL-024–20) | 25245769 (Biliverdine(2-)) | 60947 (Tirofiban) |
| -14.39 | -13.58 | -10.67 | -10.97 | |
| Anti-inflammatory and anitiviral activity | - [a] | Human metabolite | Anti-coagulant activity | |
 |
 |
 |
 |
|
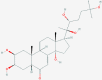
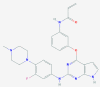
| 4 | 5459840 (20-Hydroxyecdysone) | 5281040 (Montelukast) | 135483998 (5-Methyltetrahydrofolate) | 5362119 (Lisinopril) |
| -14.07 | -12.83 | -10.66 | -10.90 | |
| Protective role in the cardiovascular system | Anti-inflammatory and bronchodilating activity | Anti-neoplastic and antidepressant activity | ACE inhibitor with anti-hypertensive activity | |
 |
 |
 |
 |
|
| 5 | 154573806 (GRL-024–20) | 46700782 (Razuprotafib) | 122146 (-[b]) | 72734520 (Avitinib) |
| -14.05 | -12.83 | -10.61 | -10.77 | |
| - [a] | Potential vasculature stabilizing activity | - [a] | Tyrosine kinase inhibitor | |
 |
 |
 |
 |
(a) No pharmacological functions have been reported.
(b) Compound does not have a trivial name.
3.1. 6WQF
The obtained screening results show that compounds such as the potential inhibitor of SARS-CoV-2 main proteinase 3CLpro Peptidomimetic aldehyde 11a [9] (-13.48 kcal/mol), the cathepsin K inhibitor Relacatib [64] (-13.49 kcal/mol), the pair of vitamin D analogues Calcitriol and Calcifediol (-13.63 and -13.78 kcal/mol, respectively), as well as the novel drug designed to treat acute respiratory distress syndrome Razuprotafib [65] (-13.86 kcal/mol) exhibit high affinities to 3CLpro (6WQF). Thus, indicating their potential in the treatment of SARS-CoV-2. The actual top five structures with the lowest docking scores against 6WQF obtained in this study include the compound GRL-024–20, with docking score -14.05 kcal/mol, whose crystal structure in the complex with 3CLpro protease had already been determined [66]. Another compound with a low docking score is 20-hydroxyecdysone, docking score -14.07 kcal/mol, which is also the only compound that is currently not part of any COVID19 treatment trials. Anti-asthmatic drug Ciclesonide [67] achieved docking score -14.57 kcal/mol with predicted H-bonds to Thr26 and Gln189 amino acids. Potential anti-migraine drug (phase three of clinical trials) Vazegepant/zevagepant [68], -14.57 kcal/mol, also emerged as a possible 3CLpro inhibitor with predicted H-bonds to Thr26, Asn142 and Gly143 amino acid residues. The lowest docking score against 6WQF, -14.75 kcal/mol, was observed for the anti-asthmatic drug Montelukast [69], see Fig. 5 A and B. Its predicted H-bonds pattern includes bonds between His163 and the carbonyl group of Montelukast as well as bonds between both atoms of its’ hydroxyl groups and Thr190/Arg188 residues in 6WQF cavity. Other notable frequently predicted H-bonds to 6WQF structure include bonds to Thr26, Gly143, Glu166 and Gln189 amino acids.
Fig. 5.

Putative bindings sites of Montelukast, Ebastine, Solumedrol and Protoporphyrinogen IX in 6WQF, 6LU7, 6WZU and 7CMD protein cavities with the compounds in purple color and proteins in green, orange, blue and yellow, respectively. (A) Interacting amino acid residues of 6WQF with Montelukast. (B) Binding site of Montelukast in 6WQF 3CLpro. (C) Interacting amino acid residues of 6LU7 with Ebastine. (D) Binding site of Ebastine in 6LU7 3CLpro. (E) Interacting amino acid residues of 6WZU with Solumedrol. (F) Binding site of Solumedrol in 6WZU PLpro. (G) Interacting amino acid residues of 7CMD with Protoporphyrinogen. (H) Binding site of Protoporphyrinogen in 7CMD PLpro (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
3.2. 6LU7
Screening results of the set of studied (PubChem chosen) molecules against the second 3CLpro structure 6LU7 differ from the 6WQF ones in relative positions of high affinity molecules in the top scores list. The lower docking scores to 6LU7 were observed in aforementioned Peptidomimetic aldehyde 11a [9] (-12.50 kcal/mol), anticoagulant Argatroban [70] (-12.76 kcal/mol), broad-spectrum anti-viral protease inhibitor GC-376 (-12.79 kcal/mol) currently under study for its potential for inhibiting SARS-CoV-2 main protease 3CLpro [71], and two compounds whose biological effect has not been described yet, CIDs: 122,146 and 71,316,139 (-12.57 and -12.66 kcal/mol, respectively). One of these compounds (CID: 122146) has also shown high affinity towards PLpro (see below). Five compounds with the lowest docking scores obtained in this study include previously mentioned Razuprotafib [65], with docking score -12.83 kcal/mol, Montelukast [69], score -12.83 kcal/mol, GRL-024–20 [66], score -13.58 kcal/mol, and Vazegepant (zevagepant) [68], with score -13.80 kcal/mol. It should be noted that these molecules exhibit different poses and H-bonds patterns than in the complex with 6WQF, see ESI Table 1A and B. The highest apparent affinity towards 6LU7, -14.11 kcal/mol, was observed for H1 antihistamine Ebastine [72] as already found in Vatansever et al. [73], see Fig. 5C and D. The predicted binding pattern of Ebastine consists of two hydrogen bonds between its oxygens and hydrogens of His163 and Glu166 amino acids. In a similar way to previous 3CLpro structure 6WQF, H-bonds with highest incidence include bonds from Thr26, Glu166 and Gln189 amino acid residues.
3.3. 6WZU
Vazegepant/zevagepant [68] also emerged as one of compounds with highest binding affinity towards PLpro (-10.21 kcal/mol), along Taurochenodeoxycholate (1-) anion (-10.37 kcal/mol), taurochloric acid (-10.40 kcal/mol), immunosuppressant Hydrocortisone hemisuccinate (-10.41 kcal/mol) and folic acid (-10.58 kcal/mol). Top five best scoring compounds include human metabolite Biliverdine(2-), score -10.67 kcal/mol, with predicted H-bonds to Lys105, Trp106, Asp108 and Ala288 amino acids and the novel prodrug TAK-599 designed to treat MRSA (Methicillin-resistant Staphylococcus aureus) infection [75], score -10.69 kcal/mol, with predicted H-bonds to His89, Lys105 and Asp108 amino acid residues. The lowest docking score was observed for the anti-inflammatory immunosuppressant Solumedrol, see Fig. 5E and F, with predicted H-bonds to Lys105, Trp106, Asp286 and Ala288 amino acids. Excellent docking score, -10.61 kcal/mol, was also observed for compound with CID: 122146. This molecule achieved very good scores in both docking rounds against 6WZU and is the prime example of the importance of docking studies, with a good conformational stability within the protein cavity and a stable H-bonds pattern (see the next section) warranting further research. Overall, the predicted H-bond pattern of studied compounds in the complex with PLpro forms a smaller set of interactions found with a higher frequency, most notably H-bonds between Lys105, Trp106, Asp108, Ala288 amino acid residues and compound's carbonyl groups.
3.4. 7CMD
The compounds with the lowest docking scores against PLpro 7CMD crystal structure significantly differ from results obtained for 6WZU structure. The only compound that is found among ten best scoring compounds against both PLpro targets is the prodrug TAK-559 [75] (-10.56 kcal/mol against 7CMD). Other low scoring compounds against 7CMD include yet not-studied compound with CID: 9810132 (-10.55 kcal/mol), angiotensin II antagonist Valsartan (-10.67 kcal/mol), human metabolite Geranylgeranyl diphosphate (-10.71 kcal/mol) and tyrosine kinase inhibitor Mastinib (-10.75 kcal/mol). Five best scoring compounds include another tyrosine kinase inhibitor Avitinib (-10.77 kcal/mol) that has been investigated for use in the treatment of non-small cell lung cancer (NSCLC) and its ability to supress cytokine storm associated with COVID19 made it part of clinical trials for the treatment of COVID19 [76]. Angiotensive enzyme (ACE) inhibitor Lisinopril (-10.90 kcal/mol) has also been already linked with the treatment of COVID19. It has been found that patients with prescribed ACE inhibitors or angiotensin receptor blockers (ARB) are at reduced risk of COVID19 while not increasing risk of ICU care [77]. Anticoagulant Tirofiban (trade name AGGRASTAT) (-10.97 kcal/mol) has also shown promise in reducing thrombotic effects observed in patients suffering from COVID19 and is part of further clinical trials [78]. Its predicted H-bonds include bonds with two amino acids from PLpro catalytic triad (His272 and Asp286), as well as bonds with Cys270 and Ala288. The second best scoring compound against 7CMD PLpro structure was the human metabolite Taurochenodeoxycholate(1-) (-11.24) formed by deprotonation of taurochenodeoxycholic acid. Its predicted H-bond pattern includes bonds with Lys105, Trp106, Asn267 and Cys270. However, its utility in treatment of COVID19 is unlikely (being human metabolite). The compound with highest apparent binding affinity towards 7CMD structure PLpro was Protoporphyrinogen IX (-11.58 kcal/mol), see Fig. 5G and 5H. This compound is a direct precursor of protoporphyrin IX, which plays a critical role in living organisms during production of hemoglobin or chlorophyll. Protoporphyrin has already emerged as a potential antiviral drug for treatment of COVID19 by method interfering with the interaction of ACE2 and the receptor-binding domain of spike protein of SARS-CoV-2 [79]. Its predicted H-bonds include bonds with Lys105 and Trp106. Most frequently predicted H-bonds in compound-protein (7CMD) complexes include bonds with Lys105, Trp106, Asp286 and Ala288.
Docked poses of abovementioned compound-protein complexes, together with the description of the predicted H-bonds can be found in ESI Table S2A–D. The docking results together with evaluation of drug likeness criteria can be found in ESI excel files Docking_6WQF/6LU7/6WZU/7CMD.xlsx.
3.5. Molecular dynamics – docking pose verification
To investigate the conformational stability and to overcome the rigid protein picture, molecular dynamic simulations in water solvent at physiological conditions were carried out on five complexes for the 6WQF 3CLpro structure (CIDs: 5,281,040, 53,472,683, 6,918,155, 5,459,840, 154,573,806) and for the 6WZU PLpro structure (CIDs: 16,923, 73,425,380, 25,245,769, 135,483,998, 122,146) for the time of 10 ns.
The conformational stability of formed compound-protein complexes was validated by calculating the relative RMSD value of compounds’ heavy atoms with respect to the initial structure of the residues’ backbone, see Fig. 6 A and B. Time evolution plot of RMSD for complexes with 3CLpro (6WQF) shows that four structures achieved equilibrium at the start of simulation with averaged RMSD values around 0.3 Å, with Montelukast (CID: 5281040) and Ciclesonide (CID: 6918155) manifesting small conformational changes at 3.2 and 4.5 ns, respectively, suggesting small drifts in the trajectory during the simulation. These values indicate that all four structures are well positioned within the cavity's active site and form stable complexes.
Fig. 6.

MD time plot evolutions of RMSD values of compounds’ heavy atoms with respect to the initial structure of the residues’ backbone of 6WQF (A) and 6WZU (B).
RSMD values of three compounds’ heavy atoms in complexes with PLpro (6WZU) show a higher motion patterns during the simulation period. Based on the visualizations of dynamics trajectories and on the time evolution plot of hydrogen bonds, this increase in the RMSD value is caused by the movement of parts of molecules that do not participate in the formation of H-bonds while rest of the molecule remains firmly attached to the protein. On the other hand, 20-hydroxyecdysone (3CLpro) and Solumedrol (PLpro) have shown no conformational stability in the cavity and a gradual increase in the RMSD value, linked to a decrease in the compound-protein interaction energy, eventually being dislodged from the target proteins. Compared to other high scoring compounds, 20-hydroxyecdysone contains only a single carbonyl group and oxygens participating in formations of H-bonds identified by docking protocols belonged to hydroxyl moiety making it highly hydrophilic. Solumedrol, on the other hand, contains an ideal number of both, H-bond donor and acceptor atoms, and the reason for its compound-protein complex instability is unclear and requires further data collection.
A second parameter that was used to monitor the conformational stability of these complexes, as well as to examine the compound-protein interactions was the evolution of the number of hydrogen bonds formed between the target proteins and the studied compounds during the MD simulation. The MD simulations have shown that the 3CLpro complexes form up to five hydrogen bonds, but these do not appear to be long-lived and the actual number of stable H-bonds is between one and three. In comparison, complexes with PLpro exhibit formation of steadier H-bonds, ranging from one to three, that once formed last throughout the whole simulation period. Time evolution plots of the hydrogen bonds formation and disappearance during the MD simulation along with further data including bonds identification and their occupancy can be found in Fig. S1A, B and Tables S3A–E and S4A–E.
Binding free energies, expressed as a sum of the electrostatic, Van der Waals interactions, polar and non-polar solvation free energies were calculated to better understand the compound-protein interaction during MD simulations, see Fig. S2A and B. In addition, the contribution of each protein residue to the total free binding energy was calculated. Montelukast has shown the lowest binding free energy towards 6WQF with a value of ΔGbind = -30.99 ± 5.75 kJ/mol confirming its lowest docking score among the studied compounds. The binding free energy of Ciclesonide, ΔGbind = -29.31 ± 5.35 kJ/mol, differs only slightly from the lowest value of Montelukast, with other two molecules having significantly higher (less negative) energies. However, the binding free energy of GRL-024–20, ΔGbind = -25.24 ± 5.78 kJ/mol, is slightly lower than that of Vazegepant/zevagepant, ΔGbind = -23.95 ± 5.46 kJ/mol. This is most likely caused by differences between the treatment of compound's atoms by AutoDock's and CHARMM36 forcefields. Binding free energies of complexes with PLpro (6WZU) exhibit higher fluctuations than for 3CLpro (6WQF) suggesting that these systems did not achieve equilibrium within the simulation duration (possibly due to a smaller size of the cavity), hence their averaged ensemble values are not discussed here.
3.6. Comparison with other docking studies
As previously mentioned, acquired docking results were compared to several published papers [11,[13], [14], [15], [16], [17], [18], [19], [20], [21], [22]] in order to better ascertain the compound inhibitory potential against both SARS-CoV-2 proteases. Compounds that were found in these works as well as within the already downloaded set of compounds from the PubChem database include among others well-known antivirals such as Ribavirin, Oseltamivir, Darunavir, Remdesivir or the antimalarial drug Chloroquine etc., see Table 2 . However, utilization of several different docking software with different scoring functions renders a direct comparison of binding energies ineffective.
Table 2.
Docking scores of compounds found within the PubChem set and selected previously published works [11,13,[15], [16], [17], [18], [19], [20], [21]].
| Identifier (CID) | Name | 6LU7 ΔGbind [kcal/mol] | 6WQF ΔGbind [kcal/mol] | 6WZU ΔGbind [kcal/mol] | 7CMD ΔGbind [kcal/mol] | Original score against 6LU7 | Original score against 5R** (3CLpro) i | Original score against 3E9S (PLpro) |
|---|---|---|---|---|---|---|---|---|
| 896 | Melatonin | -7.97 | -7.03 | -6.02 | -6.82 | -10.80 a | ||
| 2719 | Chloroquine | -9.86 | -9.06 | -7.05 | -5.60 | -11.37 b-7.50 c-5.10 f-10.80 f | -13.88 b | |
| 6253 | Cytabarine | -6.74 | -6.19 | -4.96 | -5.79 | -5.43 b | -11.70 b | |
| 37542 | Ribavirin | -7.67 | -6.82 | -5.73 | -5.23 | -38.59 b | -8.30 b | |
| 44093 | Captopril | -6.93 | -6.10 | -6.10 | -5.60 | -4.22 c | ||
| 65028 | Oseltamivir | -10.69 | -9.43 | -7.92 | -7.42 | -4.70 f | -6.891 i | |
| 73115 | Clevudine | -7.02 | -6.96 | -5.09 | -5.89 | -14.52 b | 15.20 b | |
| 119209 | Thymidine-Methyl-T | -7.26 | -6.93 | -6.22 | -5.52 | -18.10 b | -7.57 b | |
| 131411 | Arbidol | -10.40 | -10.03 | -6.47 | -6.30 | -10.12 b | -15.43 b | |
| 213039 | Darunavir | -12.35 | -12.92 | -8.85 | -6.02 | -15.70 a-1.04 b-7.49 c | -8.23 b | |
| 439153 | Dihydrocozymase | -12.60 | -10.79 | -8.69 | -3.40 | -11.016 d-7.0415 e | ||
| 464205 | Tenofovir | -9.05 | -7.07 | -6.06 | -4.69 | -25.06 b | -6.692 i | -8.49 b |
| 492405 | Favipiravir | -5.18 | -4.81 | -4.88 | -4.41 | -19.91 b-5.40 f-6.90 g | -11.89 b | |
| 3002977 | Maraviroc | -12.27 | -12.03 | -8.62 | -8.05 | 0.06 b | -12.61 b | |
| 5280804 | Isoquercetin | -10.87 | -11.43 | -7.78 | -5.18 | -156.08 h | ||
| 9875401 | Rivaroxaban | -10.83 | -9.83 | -8.04 | -9.16 | -14.70 a | ||
| 10182969 | Apixaban | -10.50 | -9.40 | -7.22 | -6.95 | -16.40 a | ||
| 10445549 | Galidesivir | -7.46 | -7.72 | -5.87 | -4.47 | -6.861 i | ||
| 44205240 | Baricitinib | -10.10 | -11.13 | -7.07 | -7.02 | -7.253 i | ||
| 121304016 | Remdesivir | -12.75 | -11.38 | -7.71 | -4.41 | 3.68 b-5.80 c-7.215 d-6.50 f-7.60 g | -7.529 i | -7.75 b |
| 135398641 | Inosine | -7.97 | -7.03 | -6.02 | -4.69 | -10.80 a |
(a) Docking scores from Fischer et al. [13].
(b) Docking scores from Wu et al. [11].
(c) Docking scores from Hosseini and Amanlou [16].
(d) Docking scores from Hall and Ji [15].
(e) Docking scores from Owis et al. [19].
(f) Docking scores from Narkhede et al. [18].
(g) Docking scores from Silva Arouche et al.[20].
(h) Docking scores from Adem et al. [17].
(i) Docking scores from Shah et al. [21], expressed as an average of up to five docking scores against 3CLpro structures (5R7Y, 5R7Z, 5R80, 5R81, 5R82).
Therefore, 45 various best scoring compounds have been selected from the five different publications mentioned [11,13,16,17,21] and redocked using our docking protocols against all three protein targets (original selection of 52 best scoring compounds contained six that have already been evaluated in our set and one compound has been found present in two publications), see Table 3 . Several of these compounds, especially from papers [11,13,21], have indeed the calculated binding energy below -13 kcal/mol in the case of 3CLpro and below -9 kcal/mol in the case of PLpro while having a reasonable low molar mass making them well suited for a medical use, see Table 4. Candidates with the highest potential to bind with 3CLpro protease include four antiretroviral protease inhibitors, Nelfinavir [80], Lopinavir [81] and Indinavir [82]. Human CYP3A protein inhibitor Cobicistat [83], as well as the antivirals Telaprevir [84] and Simeprevir [85], used in the treatment of hepatitis C virus, have also achieved excellent docking scores. Furthermore, several flavonoids, such as Rutin, Apiin and Diosmin, and an anti-neoplastic drug Docetaxel, whose application include treatment of breast, head and neck, stomach, prostate and non-small-cell lung cancers [86] all achieved scores below -14 kcal/mol against either 6WQF or 6LU7 3CLpro structure. The highest affinity towards PLpro 6WZU structure was observed in the case of Suramine, with docking score as low as -18.92 kcal/mol, with gap of almost 8 kcal/mol to next best scoring compound. However, pose of the compound within the active site of the protease suggests cyclisation of the molecule itself, see Fig. S3, which may be the cause of the unusually low docking score and this result should be taken with a grain of salt. High affinity towards PLpro 6WZU structure was observed in the cases of abovementioned Stavudine Triphosphate, Apiin, Docetaxel and Ferristatin II. Compounds from the other studies with lowest docking scores against PLpro 7CMD structure include Diacetylcurcumin, Alectinib, Paritaprevir and two compounds without trivial names, CIDs: 101,502,236 and 101,526,067. Suramine's tendency to form intramolecular interactions was observed in smaller degree for 7CMD protein target as well (-13.74 kcal/mol). Even though, the best scoring compounds for PLpro structures form two distinct sets, the differences in their scores are well within the AutoDock scoring function error and they should be looked upon as one large set. These results have been deposited into previously mentioned excel documents containing docking results of the PubChem set of compounds and their graphical representations can be found in Fig. 7 A–D.
Table 3.
| Identifier | Trivial name | 6WQF ΔGbind [kcal/mol] |
6LU7 ΔGbind [kcal/mol] |
6WZU ΔGbind [kcal/mol] |
7CMD ΔGbind [kcal/mol] |
Reference |
|---|---|---|---|---|---|---|
| 146131763 | - b | -9.41 | -8.41 | -7.39 | -7.89 | [11] |
| CP02 a | - b | -10.54 | -9.06 | -7.52 | -8.29 | |
| CP03 a | - b | -11.31 | -9.62 | -8.13 | -9.16 | |
| 5,750,099 | - b | -11.67 | -10.06 | -8.15 | -8.80 | |
| 4,666,447 | - b | -12.09 | -11.47 | -7.31 | -7.92 | |
| CP06 a | - b | -11.50 | -10.89 | -7.73 | -8.29 | |
| CP07 a | - b | -8.96 | -8.03 | -6.84 | -9.16 | |
| CP08 a | - b | -11.36 | -10.00 | -7.89 | -8.44 | |
| CP09 a | - b | -9.13 | -8.17 | -6.78 | -8.46 | |
| CP10 a | - b | -10.06 | -9.18 | -7.38 | -8.95 | |
| CP11 a | - b | -10.85 | -9.08 | -7.45 | -8.38 | |
| 439533 | (-)-Taxifolin | -9.34 | -8.75 | -6.35 | -7.44 | |
| 4463 | Nevirapine | -7.91 | -7.25 | -6.17 | -6.26 | [13] |
| 5361 | Suramine | -13.36 | -15.53 | -18.92 | -13.74 | |
| 64143 | Nelfinavir | -13.02 | -14.43 | -7.67 | -9.72 | |
| 64993 | 6-Hept | -9.64 | -8.96 | -7.73 | -7.42 | |
| 65015 | Plerixafor | -10.64 | -11.23 | -6.85 | -7.29 | |
| 65355 | Stavudine Triphosphate | -9.73 | -8.38 | -11.40 | -9.25 | |
| 3010818 | Telaprevir | -14.08 | -13.44 | -9.99 | -8.90 | |
| 25151504 | Cobicistat | -13.54 | -14.66 | -9.93 | -8.94 | |
| 49773361 | Beclabuvir | -12.71 | -12.85 | -7.39 | -8.91 | |
| 35314 | O-Methyl-Dcba | -7.40 | -6.86 | -6.88 | -6.98 | [16] |
| 107771 | Pyronaridine | -13.92 | -13.60 | -8.99 | -9.89 | |
| 148124 | Docetaxel | -15.12 | -14.00 | -10.39 | -8.36 | |
| 5330286 | Palbociclib | -12.36 | -12.23 | -9.29 | -9.47 | |
| 9854073 | Cabazitaxel | -14.85 | -14.87 | -9.77 | -9.76 | |
| 24873435 | Simeprevir c | -15.70 | -12.67 | -9.88 | -8.88 | |
| 49806720 | Alectinib | -11.66 | -10.42 | -8.24 | -10.18 | |
| 10621 | Hesperidin | -14.04 | -12.66 | -9.38 | -8.76 | [17] |
| 5280746 | Apiin | -14.15 | -13.28 | -10.41 | -9.01 | |
| 5280805 | Rutin | -14.43 | -13.71 | -9.26 | -8.67 | |
| 5281613 | Diosmin | -13.88 | -13.48 | -9.00 | -8.94 | |
| 5281672 | Myricetin | -11.32 | -10.17 | -7.31 | -8.44 | |
| 6441419 | Diacetylcurcumin | -12.92 | -12.30 | -9.98 | -11.38 | |
| 101502236 | - b | -12.38 | -11.39 | -9.68 | -10.53 | |
| 101526067 | - b | -12.68 | -12.12 | -9.67 | -10.54 | |
| 92727 | Lopinavir | -14.26 | -15.02 | -8.81 | -9.37 | [21] |
| 123794 | CGP42112A | -11.95 | -10.25 | -7.28 | -7.89 | |
| 392622 | Ritonavir | -12.76 | -13.17 | -9.35 | -9.81 | |
| 667492 | Marboran/Methisazone | -7.97 | -7.27 | -7.77 | -7.39 | |
| 5362440 | Indinavir | -15.66 | -14.78 | -9.75 | -8.65 | |
| 5702678 | Ferristatin II | -11.74 | -10.68 | -10.25 | -9.25 | |
| 45110509 | Paritaprevir | -14.36 | -14.27 | -9.17 | -10.28 | |
| 71661251 | Elbasvir | -12.42 | -9.26 | -7.05 | -8.63 | |
| 137967122 | Asunaprevir | -11.99 | -12.5 | -7.61 | -9.47 |
Fig. 7.

Docking scores of initially selected compounds (colored diamonds), redocked compounds from selected publications (empty triangles) [13], (full triangles) [11], (empty squares) [16], (full squares) [17] and (empty circles) [21] against 6WQF (A), 6LU7 (B), 6WZU (C) and 7CMD (D) with respect to their molecular weight. Compounds with molecular weight exceeding 1000 Da, one from Wu et al. [11] (CID: 5361) and one from Shah et al. [21] (CID: 123794), are excluded from these representations.
4. Discussion
Highest affinity towards 3CLpro from PubChem database [26,27] was observed for anti-asthmatic drug Montelukast (CID: 5281040) [69] (6WQF) and for anti-inflammatory drug Ebastine (CID: 3191) [72] (6LU7). Interestingly, while Montelukast has reached top ten against both 3CLpro structures, Ebastine achieved a score of only -12.99 kcal/mol, ranking 18th against 6WQF structure. The anti-inflammatory immunosuppressant Solumedrol (CID: 16923) achieved the lowest docking score against PLpro 6WZU structure but was dislodged from the protein during subsequent MD simulations. Other compounds with low docking scores against both 3CLpro structures include potential anti-migraine drug Vazegepant/zevagepant (CID: 53472683) [68] and the compound GRL-024–20 (CID: 154573806).
The novel prodrug TAK-599 (CID: 73425380) achieved low docking scores against PLpro indicating its’ possible use in the COVID19 therapy. Docking protocols also revealed one compound (CID: 122146) with high affinity towards PLpro and 3CLpro 6LU7 structure that medical use has yet to be examined experimentally. Relatively high affinity towards both protein targets was also observed for Vazegepant/zavegepant, indicating that these compounds could be used as potential multi-targeting drugs. It is worth noting that many drugs that were discussed as possible SARS-CoV-2 Mpro inhibitors, such as Remdesivir, chloroquine and hydroxy-chloroquine have not achieved outstanding docking scores [11,15,16,[18], [19], [20]].
It is important to note, that most of best scoring compounds do not satisfy selected drug likeness empirical rules due to their molar mass, number of hydrogen bond acceptors/donors and/or molar refractivity. However, it is also equally important to note, that these compounds are already approved drugs that were originally proposed for other treatments, thus constituting viable set for drug refurbishment.
Comparison of obtained data with previously published results serves as an illustration of their robustness. Best scoring compounds against 6WQF show the same level of affinity towards the target as the lowest docking score compounds from other publications. Our results are on par with best results from Adem et al. [17] and have comparable molecular weights. On the other hand, redocking these structures against 6LU7 shows a decrease in docking scores compared to our set of compounds but comes at a prize of molecular weights exceeding 700 Da. Admittedly, comparison of compounds with the same molecular weight indicate that chosen set of structures obtained from the PubChem database are lagging behind best scoring compounds from other publications [11,13,[15], [16], [17], [18], [19], [20], [21]] by about 0.5 kcal/mol against 6LU7. Results of redockings against 6WZU show that compounds from the PubChem database achieve comparable docking scores with considerably lower molecular weights making them interesting targets for potential lead-like structures. However, interested reader is advised to judge the compounds not only by their docking score but also by the ligand efficacy. This parameter, expressed as the docking score with respect to the number of non-hydrogen atoms of the compound, attempts to eliminate the shift of docking score towards the more negative values caused by decrease in the Van der Waals interaction energy that goes hand in hand with increase of compound's molecular weight. Upon closer examination of ligand efficacies of the redocked compounds it becomes clear that several of the best scoring compounds are biased towards lower docking scores due to their molecular weights, see Fig. S4A–D.
In addition, we provide a set of previously published redocked compounds with reported half maximal inhibitory concentration (IC50) values against 3CLpro [38,73,87,88] and PLpro [40,[89], [90], [91], [92]] to give the interested reader more detailed view on the docking score vs. dose concentration relation of in vitro tested compounds. Refer to the supplementary materials in respective .xlsx documents, sheets IC50_dockings. However, no direct comparison has been made due to many challenges regarding such relation, e.g. imperfection of docking scores (derived from desolvation and entropy terms in AD scoring function [93]), hand in hand with relatively high uncertainties of experimental IC50 values.
5. Conclusion
We have performed in silico docking studies of over 860 compounds from external PubChem database [26,27] against SARS-CoV-2 proteases. Conformation stability of complexes formed by compounds with the highest affinity towards inhibition targets was validated by MD simulations and by the estimation of free energy of binding using the MM-PBSA approach [59,60]. Acquired results were compared with several previously published works [11,13,[15], [16], [17], [18], [19], [20], [21]] and show that top scoring structures are comparable with hand-picked selection of known antivirals. Docking scores database, along with ADME parameters of studied compounds, can serve for further analysis of the antiviral activity and/or for molecular design of potential drug candidates. Furthermore, the submitted data could serve as a source of validation for other researches in the fight against COVID19 and call for further in vitro verifications of antiviral activity of several best scoring compounds.
Additional supporting information
Supporting_info_README.pdf – detailed information about the supporting information files, including description of individual columns in the excel files.
Supporting_info.pdf – text document containing additional figures and tables referred to in this work.
Docking_6WQF/6LU7/6WZU/7CMD.xlsx – excel documents containing the docking results computed in this work.
PubChem_SwissADME.xlsx – excel document containing physicochemical descriptors and ADME parameters of the studied compounds.
CRediT authorship contribution statement
Marek Štekláč: Methodology, Visualization, Formal analysis, Writing – review & editing. Dávid Zajaček: Investigation, Visualization, Writing – original draft. Lukáš Bučinský: Conceptualization, Resources, Software, Writing – original draft.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the Science and Technology Assistance Agency under the contract no. APVV-19–0024 and APVV-19–0087, by the Slovak Grant Agency VEGA under contract nos. 1/0718/19 and 1/0139/20 and by the Ministry of Education, Science, Research and Sport of the Slovak Republic by funding within the scheme “Excellent research teams”. We thank the HPC center at the Slovak University of Technology in Bratislava, which is a part of the Slovak Infrastructure of High Performance Computing (SIVVP Project, ITMS code 26230120002, funded by the European Region Development Funds), for computing facilities.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.molstruc.2021.130968.
Appendix. Supplementary materials
References
- 1.Chen Y., Liu Q., Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J. Med. Virol. 2020;92:418–423. doi: 10.1002/jmv.25681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G.F., Tan W. A novel coronavirus from patients with pneumonia in China. N. Engl. J. Med. 2019;382:727–733. doi: 10.1056/NEJMoa2001017. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng V.C.C., Lau S.K.P., Woo P.C.Y., Kwok Y.Y. Severe acute respiratory syndrome coronavirus as an agent of emerging and reemerging infection. Clin. Microbiol. Rev. 2007;20:660–694. doi: 10.1128/CMR.00023-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zaki A.M., van Boheemen S., Bestebroer T.M., Osterhaus A.D.M.E., Fouchier R.A.M. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012;367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 5.Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., Cheng Z., Yu T., Xia J., Wei Y., Wu W., Xie X., Yin W., Li H., Liu M., Xiao Y., Gao H., Guo L., Xie J., Wang G., Jiang R., Gao Z., Jin Q., Wang J., Cao B. Clinical features of patients infected with 2019 novel Coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.World Health Organization (2020, March 21). World Health Organization. Coronavirus; (accessed 2020, December 7). Retrieved from https://www.who.int/health-topics/coronavirus#tab=tab_3.
- 7.Morse J.S., Lalonde T., Xu S., Liu W.R. Learning from the past: possible urgent prevention and treatment options for severe acute respiratory infections caused by 2019-nCoV. ChemBioChem. 2020;21:730–738. doi: 10.1002/cbic.202000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367 doi: 10.1126/science.abb2507. (80-.)1260 LP –1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dai W., Zhang B., Jiang X.-M., Su H., Li J., Zhao Y., Xie X., Jin Z., Peng J., Liu F., Li C., Li Y., Bai F., Wang H., Cheng X., Cen X., Hu S., Yang X., Wang J., Liu X., Xiao G., Jiang H., Rao Z., Zhang L.K., Xu Y., Yang H., Liu H. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368 doi: 10.1126/science.abb4489. 80-.1331 LP –1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kneller D.W., Phillips G., O'Neill H.M., Jedrzejczak R., Stols L., Langan P., Joachimiak A., Coates L., Kovalevsky A. Structural plasticity of SARS-CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat. Commun. 2020;11:3202. doi: 10.1038/s41467-020-16954-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu C., Liu Y., Yang Y., Zhang P., Zhong W., Wang Y., Wang Q., Xu Y., Li M., Li X., Zheng M., Chen L., Li H. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B. 2020;10:766–788. doi: 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Osipiuk J., Azizi S.-A., Dvorkin S., Endres M., Jedrzejczak R., Jones K.A., Kang S., Kathayat R.S., Kim Y., Lisnyak V.G., Maki S.L., Nicolaescu V., Taylor C.A., Tesar C., Zhang Y.-A., Zhou Z., Randall G., Michalska K., Snyder S.A., Dickinson B.C., Joachimiak A. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 2020;(12) doi: 10.1038/s41467-021-21060-3. 2020.08.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.A. Fischer, M. Sellner, S. Neranjan, M. Smieško, M.A. Lill, Potential Inhibitors for Novel Coronavirus Protease Identified by Virtual Screening of 606 Million Compounds, Int. J. Mol. Sci. 21 (2020) 3626. doi: 10.3390/ijms21103626. [DOI] [PMC free article] [PubMed]
- 14.Yu R., Chen L., Lan R., Shen R., Li P. Computational screening of antagonists against the SARS-CoV-2 (COVID-19) coronavirus by molecular docking. Int. J. Antimicrob. Agents. 2020;56 doi: 10.1016/j.ijantimicag.2020.106012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hall D.C., Ji H.F. A search for medications to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2 spike glycoprotein and 3CL protease. Travel Med. Infect. Dis. 2020;35 doi: 10.1016/j.tmaid.2020.101646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hosseini F.S., Amanlou M. Anti-HCV and anti-malaria agent, potential candidates to repurpose for coronavirus infection: virtual screening, molecular docking, and molecular dynamics simulation study. Life Sci. 2020;258 doi: 10.1016/j.lfs.2020.118205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.S. Adem, V. Eyupoglu, I. Sarfraz, A. Rasul, M. Ali, Identification of Potent COVID-19 Main Protease (Mpro) Inhibitors from Natural Polyphenols: An in Silico Strategy Unveils a Hope against CORONA. Preprints 2020, 2020030333 (doi:10.20944/preprints202003.0333.v1).. [DOI] [PMC free article] [PubMed]
- 18.Narkhede R.R., Cheke R.S., Ambhore J.P., Shinde S.D. The molecular docking study of potential drug candidates showing anti-COVID-19 activity by exploring of therapeutic targets of SARS-CoV-2. EURASIAN J. Med. Oncol. 2020;4:185–195. [Google Scholar]
- 19.Owis A.I., El-Hawary M.S., El Amir D., Aly O.M., Abdelmohsen U.R., Kamel M.S. Molecular docking reveals the potential of Salvadora persica flavonoids to inhibit COVID-19 virus main protease. RSC Adv. 2020;10:19570–19575. doi: 10.1039/D0RA03582C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.da Silva Arouche T., Reis A.F., Martins A.Y., Costa J.F.S, Carvalho Junior R.N., Neto A.M.J C. Interactions between remdesivir, ribavirin, favipiravir, galidesivir, hydroxychloroquine and chloroquine with fragment molecular of the COVID-19 main protease with inhibitor N3 complex (PDB ID:6LU7) using molecular docking. J. Nanosci. Nanotechnol. 2020;20:7311—7323. doi: 10.1166/jnn.2020.18955. [DOI] [PubMed] [Google Scholar]
- 21.Shah B., Modi P., Sagar S.R. In silico studies on therapeutic agents for COVID-19: drug repurposing approach. Life Sci. 2020;252 doi: 10.1016/j.lfs.2020.117652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.da Silva Hage-Melim L.I., Federico L.B., de Oliveira N.K.S., Francisco V.C.C., Correia L.C., de Lima H.B., Gomes S.Q., Barcelos M.P., Francischini I.A.G., de Paula da Silva C.H.T. Virtual screening, ADME/Tox predictions and the drug repurposing concept for future use of old drugs against the COVID-19. Life Sci. 2020;256 doi: 10.1016/j.lfs.2020.117963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mohapatra S., Nath P., Chatterjee M., Das N., Kalita D., Roy P., Satapathi S. Repurposing therapeutics for COVID-19: rapid prediction of commercially available drugs through machine learning and docking. PLoS ONE. 2020;15 doi: 10.1371/journal.pone.0241543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aghaee E., Ghodrati M., Ghasemi J.B. In silico exploration of novel protease inhibitors against coronavirus 2019 (COVID-19) Inform. Med. Unlocked. 2021;23 doi: 10.1016/j.imu.2021.100516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information (2020, April 11). PubChem Docs. COVID-19/SARS-CoV-2 Data in PubChem; (accessed 2020, October 23). Retrieved from https://pubchemdocs.ncbi.nlm.nih.gov/covid-19.
- 26.Kim S., Chen J., Cheng T., Gindulyte A., He J., He S., Li Q., Shoemaker B.A., Thiessen P.A., Yu B., Zaslavsky L., Zhang J., Bolton E.E. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 2018;47:D1102–D1109. doi: 10.1093/nar/gky1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim S., Chen J., Cheng T., Gindulyte A., He J., He S., Li Q., Shoemaker B.A., Thiessen P.A., Yu B., Zaslavsky L., Zhang J., Bolton E.E. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 2021;49:D1388–D1395. doi: 10.1093/nar/gkaa971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O'Boyle N.M., Banck M., James C.A., Morley C., Vandermeersch T., Hutchison G.R. Open babel: an open chemical toolbox. J. Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sterling T., Irwin J.J. ZINC 15 – ligand discovery for everyone. J. Chem. Inf. Model. 2015;55:2324–2337. doi: 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Irwin J.J., Sterling T., Mysinger M.M., Bolstad E.S., Coleman R.G. ZINC: a free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012;52:1757–1768. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Becke A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993;98:5648–5652. doi: 10.1063/1.464913. [DOI] [Google Scholar]
- 32.Lee C., Yang W., Parr R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- 33.Francl M.M., Pietro W.J., Hehre W.J., Binkley J.S., Gordon M.S., DeFrees D.J., Pople J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982;77:3654–3665. doi: 10.1063/1.444267. [DOI] [Google Scholar]
- 34.Krishnan R., Binkley J.S., Seeger R., Pople J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980;72:650–654. doi: 10.1063/1.438955. [DOI] [Google Scholar]
- 35.McLean A.D., Chandler G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11-18. J. Chem. Phys. 1980;72:5639–5648. doi: 10.1063/1.438980. [DOI] [Google Scholar]
- 36.M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. F. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian 09 (Revision D.01), Gaussian, Inc., Wallingford, CT 2013.
- 37.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L.W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 39.A. (National I. of H.I.O.A. and I.D. Osipiuk, J., Tesar, C., Endres, M., Jedrzejczak, R., Joachimiak, PDB ID: 6WZU the crystal structure of papain-like protease of SARS CoV-2, P3221 space group, 2020. 10.2210/pdb6WZU/pdb.
- 40.Gao X., Qin B., Chen P., Zhu K., Hou P., Wojdyla J.A., Wang M., Cui S. Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm. Sin. B. 2021;11:237–245. doi: 10.1016/j.apsb.2020.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guex N., Peitsch M.C. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 43.Xu J., Zhang Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics. 2010;26:889–895. doi: 10.1093/bioinformatics/btq066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wojciechowski M. Simplified AutoDock force field for hydrated binding sites. J. Mol. Graph. Model. 2017;78:74–80. doi: 10.1016/j.jmgm.2017.09.016. [DOI] [PubMed] [Google Scholar]
- 45.Sanner M.F. Python: a programming language for software integration and development. J. Mol. Gr. Model. 1999;17:57–61. [PubMed] [Google Scholar]
- 46.Lindahl E., Hess B., van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. Mol. Model. Annu. 2001;7:306–317. doi: 10.1007/s008940100045. [DOI] [Google Scholar]
- 47.Bekker H., Berendsen H.J.C., Dijkstra E.J., Achterop S., Vondrumen R., Van Der Spoal D., Sijbers A., Keegstra H., Renardus M.K.R. Proceedings of the 4th International Conference on Computational Physics. 1993. Gromacs - a parallel computer for molecular-dynamics simulations; pp. 252–256. (PC 92)BT-PHYSICS COMPUTING '92. [Google Scholar]
- 48.Berendsen H.J.C., van der Spoel D., van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995;91:43–56. doi: 10.1016/0010-4655(95)00042-E. [DOI] [Google Scholar]
- 49.Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A.E., Berendsen H.J.C. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 50.Vanommeslaeghe K., Raman E.P., MacKerell A.D. Automation of the CHARMM general force field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 2012;52:3155–3168. doi: 10.1021/ci3003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu W., He X., Vanommeslaeghe K., MacKerell A.D.J. Extension of the CHARMM general force field to sulfonyl-containing compounds and its utility in biomolecular simulations. J. Comput. Chem. 2012;33:2451–2468. doi: 10.1002/jcc.23067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vanommeslaeghe K., Hatcher E., Acharya C., Kundu S., Zhong S., Shim J., Darian E., Guvench O., Lopes P., Vorobyov I., Mackerell A.D.J. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010;31:671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vanommeslaeghe K., MacKerell A.D. Automation of the CHARMM general force field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model. 2012;52:3144–3154. doi: 10.1021/ci300363c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Soteras Gutiérrez I., Lin F.Y., Vanommeslaeghe K., Lemkul J.A., Armacost K.A., Brooks C.L., MacKerell A.D. Parametrization of halogen bonds in the CHARMM general force field: improved treatment of ligand–protein interactions. Bioorg. Med. Chem. 2016;24:4812–4825. doi: 10.1016/j.bmc.2016.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abascal J.L.F., Vega C. A general purpose model for the condensed phases of water: TIP4P/2005. J. Chem. Phys. 2005;123 doi: 10.1063/1.2121687. [DOI] [PubMed] [Google Scholar]
- 56.Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F., DiNola A., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. doi: 10.1063/1.448118. [DOI] [Google Scholar]
- 57.Parrinello M., Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 1981;52:7182–7190. doi: 10.1063/1.328693. [DOI] [Google Scholar]
- 58.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 59.Kumari R., Kumar R., Lynn A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014;54:1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 60.Baker N.A., Sept D., Joseph S., Holst M.J., McCammon J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings1PII of original article: S0169-409X(96)00423-1. The article was originally published in advanced drug delivery reviews 23 (1997) 3. Adv. Drug Deliv. Rev. 2001;46:3–26. doi: 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 63.Ghose A.K., Viswanadhan V.N., Wendoloski J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999;1:55–68. doi: 10.1021/cc9800071. [DOI] [PubMed] [Google Scholar]
- 64.Kumar S., Dare L., Vasko-Moser J.A., James I.E., Blake S.M., Rickard D.J., Hwang S.-M., Tomaszek T., Yamashita D.S., Marquis R.W., Oh H., Jeong J.U., Veber D.F., Gowen M., Lark M.W., Stroup G. A highly potent inhibitor of cathepsin K (relacatib) reduces biomarkers of bone resorption both in vitro and in an acute model of elevated bone turnover in vivo in monkeys. Bone. 2007;40:122–131. doi: 10.1016/j.bone.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 65.Aerpio Pharmaceuticals (2020, August 13). Aerpio Pharmaceuticals. Razuprotafib COVID-19/ARDS; (accessed 2020, December 14). Retrieved from https://aerpio.com/pipeline/razuprotafib-covid-19-ards/.
- 66.B. Anson, A.K. Ghosh, A. Mesecar, PDB ID: 6XR3 X-ray Structure of SARS-CoV-2 main protease bound to GRL-024-20 at 1.45 A, to be published doi: 10.2210/pdb6XR3/pdb. [DOI]
- 67.Mutch E., Nave R., McCracken N., Zech K., Williams F.M. The role of esterases in the metabolism of ciclesonide to desisobutyryl-ciclesonide in human tissue. Biochem. Pharmacol. 2007;73:1657–1664. doi: 10.1016/j.bcp.2007.01.031. [DOI] [PubMed] [Google Scholar]
- 68.Biohaven Pharmaceuticals (2018, November 2). Biohaven Pharmaceuticals. Clinical trials; (accessed 2020, December 15). Retrieved from https://www.biohavenpharma.com/science-pipeline/resources/clinical-trials.
- 69.Grainger J., Drake-Lee A. Montelukast in allergic rhinitis: a systematic review and meta-analysis. Clin. Otolaryngol. 2006;31:360–367. doi: 10.1111/j.1749-4486.2006.01276.x. [DOI] [PubMed] [Google Scholar]
- 70.Di Nisio M., Middeldorp S., Büller H.R. Direct thrombin inhibitors. N. Engl. J. Med. 2005;353:1028–1040. doi: 10.1056/NEJMra044440. [DOI] [PubMed] [Google Scholar]
- 71.Ma C., Sacco M.D., Hurst B., Townsend J.A., Hu Y., Szeto T., Zhang X., Tarbet B., Marty M.T., Chen Y., Wang J. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. BioRxiv. 2020 doi: 10.1101/2020.04.20.051581. Prepr. Serv. Biol2020.04.20.051581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tagawa M., Kano M., Okamura N., Higuchi M., Matsuda M., Mizuki Y., Arai H., Iwata R., Fujii T., Komemushi S., Ido T., Itoh M., Sasaki H., Watanabe T., Yanai K. Neuroimaging of histamine H1-receptor occupancy in human brain by positron emission tomography (PET): a comparative study of ebastine, a second-generation antihistamine, and (+)-chlorpheniramine, a classical antihistamine. Br. J. Clin. Pharmacol. 2001;52:501–509. doi: 10.1046/j.1365-2125.2001.01471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vatansever E.C., Yang K., Kratch K.C., Drelich A., Cho C.C., Mellott D.M., Xu S., Tseng C.T.K., Liu W.R. Bepridil is potent against SARS-CoV-2 in vitro. BioRxiv. 2020 doi: 10.1101/2020.05.23.112235. 2020.05.23.112235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information (2004-). PubChem Compound Summary for CID: XXXXX; (accessed 2020, December 1). Available from: https://pubchem.ncbi.nlm.nih.gov/compound/XXXXX.
- 75.Ishikawa T., Matsunaga N., Tawada H., Kuroda N., Nakayama Y., Ishibashi Y., Tomimoto M., Ikeda Y., Tagawa Y., Iizawa Y., Okonogi K., Hashiguchi S., Miyake A. TAK-599, a novel N-phosphono type prodrug of anti-MRSA cephalosporin T-91825: synthesis, physicochemical and pharmacological properties. Bioorg. Med. Chem. 2003;11:2427–2437. doi: 10.1016/s0968-0896(03)00126-3. [DOI] [PubMed] [Google Scholar]
- 76.Wishart D.S., Feunang Y.D., Guo A.C., Lo E.J., Marcu A., Grant J.R., Sajed T., Johnson D., Li C., Sayeeda Z., Assempour N., Iynkkaran I., Liu Y., Maciejewski A., Gale N., Wilson A., Chin L., Cummings R., Le D., Pon A., Knox C., Wilson M. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46:D1074–D1082. doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hippisley-Cox J., Young D., Coupland C., Channon K.M., Tan P.S., Harrison D.A., Rowan K., Aveyard P., Pavord I.D., Watkinson P.J. Risk of severe COVID-19 disease with ACE inhibitors and angiotensin receptor blockers: cohort study including 8.3 million people. Heart. 2020;106:1503. doi: 10.1136/heartjnl-2020-317393. LP –1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Viecca M., Radovanovic D., Forleo G.B., Santus P. Enhanced platelet inhibition treatment improves hypoxemia in patients with severe Covid-19 and hypercoagulability. A case control, proof of concept study. Pharmacol. Res. 2020;158 doi: 10.1016/j.phrs.2020.104950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gu C., Wu Y., Guo H., Zhu Y., Xu W., Wang Y., Zhou Y., Sun Z., Cai X., Li Y., Liu J., Huang Z., Yuan Z., Zhang R., Deng Q., Qu D., Xie Y. Protoporphyrin IX and verteporfin potently inhibit SARS-CoV-2 infection in vitro and in a mouse model expressing human ACE2. Sci. Bull. 2021;66:925–936. doi: 10.1016/j.scib.2020.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bardsley-Elliot A., Plosker G.L. Nelfinavir: an update on its use in HIV infection. Drugs. 2000;59:581–620. doi: 10.2165/00003495-200059030-00014. [DOI] [PubMed] [Google Scholar]
- 81.Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information (2004-). PubChem Compound Summary for CID 92727, Lopinavir; (accessed 2021, February 1). Available from: https://pubchem.ncbi.nlm.nih.gov/compound/92727.
- 82.Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information (2004-). PubChem Compound Summary for CID 5362440, Indinavir; (accessed 2021, February 1). Available from: https://pubchem.ncbi.nlm.nih.gov/compound/5362440.
- 83.Mathias A.A., German P., Murray B.P., Wei L., Jain A., West S., Warren D., Hui J., Kearney B.P. Pharmacokinetics and pharmacodynamics of GS-9350: a novel pharmacokinetic enhancer without anti-HIV activity. Clin. Pharmacol. Ther. 2010;87:322–329. doi: 10.1038/clpt.2009.228. [DOI] [PubMed] [Google Scholar]
- 84.Lin C., Kwong A.D., Perni R.B. Discovery and development of VX-950, a novel, covalent, and reversible inhibitor of hepatitis C virus NS3.4A serine protease. Infect. Disord. Drug Targets. 2006;6:3–16. doi: 10.2174/187152606776056706. [DOI] [PubMed] [Google Scholar]
- 85.Majumdar A., Kitson M.T., Roberts S.K. Systematic review: current concepts and challenges for the direct-acting antiviral era in hepatitis C cirrhosis. Aliment. Pharmacol. Ther. 2016;43:1276–1292. doi: 10.1111/apt.13633. [DOI] [PubMed] [Google Scholar]
- 86.Lyseng-Williamson K.A., Fenton C. Docetaxel. Drugs. 2005;65:2513–2531. doi: 10.2165/00003495-200565170-00007. [DOI] [PubMed] [Google Scholar]
- 87.Coelho C., Gallo G., Campos C.B., Hardy L., Würtele M. Biochemical screening for SARS-CoV-2 main protease inhibitors. PLoS ONE. 2020;15 doi: 10.1371/journal.pone.0240079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li Z., Li X., Huang Y.Y., Wu Y., Liu R., Zhou L., Lin Y., Wu D., Zhang L., Liu H., Xu X., Yu K., Zhang Y., Cui J., Zhan C.G., Wang X., Luo H.B. Identify potent SARS-CoV-2 main protease inhibitors via accelerated free energy perturbation-based virtual screening of existing drugs. Proc. Natl. Acad. Sci. U. S. A. 2020;117:27381–27387. doi: 10.1073/pnas.2010470117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fu Z., Huang B., Tang J., Liu S., Liu M., Ye Y., Liu Z., Xiong Y., Zhu W., Cao D., Li J., Niu X., Zhou H., Zhao Y.J., Zhang G., Huang H. The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat. Commun. 2021;12:488. doi: 10.1038/s41467-020-20718-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shen Z., Ratia K., Cooper L., Kong D., Lee H., Kwon Y., Li Y., Alqarni S., Huang F., Dubrovskyi O., Rong L., Thatcher G.R., Xiong R. Potent, novel SARS-CoV-2 PLpro inhibitors block viral replication in monkey and human cell cultures. BioRxiv. 2021 doi: 10.1101/2021.02.13.431008. Prepr. Serv. Biol. [DOI] [Google Scholar]
- 91.Smith E., Davis-Gardner M.E., Garcia-Ordonez R.D., Nguyen T.-T., Hull M., Chen E., Baillargeon P., Scampavia L., Strutzenberg T., Griffin P.R., Farzan M., Spicer T.P. High-Throughput Screening for Drugs That Inhibit Papain-Like Protease in SARS-CoV-2. SLAS Discov. Adv. Sci. Drug Discov. 2020;25:1152–1161. doi: 10.1177/2472555220963667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chen Z., Cui Q., Cooper L., Zhang P., Lee H., Chen Z., Wang Y., Liu X., Rong L., Du R. Ginkgolic acid and anacardic acid are specific covalent inhibitors of SARS-CoV-2 cysteine proteases. Cell Biosci. 2021;11:45. doi: 10.1186/s13578-021-00564-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Huang S.Y., Zou X. Advances and challenges in protein-ligand docking. Int. J. Mol. Sci. 2010;11:3016–3034. doi: 10.3390/ijms11083016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.