

Abstract
Background:
Canakinumab is a human anti-IL-1β blocking agent that effectively neutralizes IL-1β mediated signaling for treatment of systemic juvenile idiopathic arthritis (sJIA). While many patients have dramatic clinical response to IL-1 blockade, approximately one-third fail to respond, but there currently exist no validated clinical or immunologic predictors of response. Here, we characterize distinct gene signatures for treatment response and non-response to canakinumab in sJIA patients.
Methods:
We performed a secondary analysis of whole blood gene expression microarrays of healthy controls and sJIA patients during baseline and day 3 after treatment [GEO:GSE80060]. Strong clinical responders were based on the JIA American College of Rheumatology response criteria and defined as ≥ACR90 (≤ACR30 for non-responders). A random effects model with patient identity as the random variable was used for differential expression analysis.
Results:
We identified a distinct gene expression signature in patients with a strong clinical response to canakinumab treatment as compared to non-responders, mediated by upregulation of neutrophil and IL-1 associated genes, and characterized by increasing divergence from control transcriptomes with increasing clinical response. We also identify a signature including upregulated CD163 expression associated with canakinumab non-response. Intriguingly, canakinumab treatment either induces up- or downregulation of type I IFN genes, independent of clinical response.
Conclusion:
Here, we identify a gene signature which distinguishes strong responders to canakinumab from non-responders before treatment onset. Further prospective study is needed to assess the utility of these insights for treatment decisions in sJIA and tracking the association of upregulated type I IFN signatures with sJIA complications.
Introduction
Systemic juvenile idiopathic arthritis (sJIA) is a chronic inflammatory arthropathy characterized by quotidian fevers, rash, arthritis, and hepatosplenomegaly (1). Gene expression studies of the immune response in sJIA have revealed key features of this disorder including the prominence of autoinflammation (2–4). While some findings diverge, there is consensus about the upregulation of innate immune system processes including IL-1 signaling (2), Toll-like receptor signaling (2,3), IL-6 signaling (4), inflammasome related genes (2,3) and neutrophil activation (2,3,5,6).
sJIA can be accompanied by severe complications, including the life-threatening cytokine storm syndrome macrophage activation syndrome (MAS) which can lead to multi-organ dysfunction and death. This complication is pathogenically driven by upregulated type II Interferon (IFNγ) and IL-18, and recent evidence additionally points at a role of type I IFNs as drivers of IL-18 expression in sJIA-MAS (7,8). Children with sJIA are also at risk for chronic lung disease (sJIA-LD), which has only emerged over the last few years and is temporally associated with the introduction of biologic treatments (9,10).
Based on the role for IL-1 and IL-6 in sJIA, biologics that target these upregulated pro-inflammatory cytokines have been tested and validated, with beneficial outcomes in a majority of patients (11,12). First-line therapy for sJIA includes anakinra (a recombinant human IL-1 receptor antagonist) and canakinumab (a selective monoclonal antibody that binds to IL-1β), which effectively neutralize downstream IL-1 signaling pathways (13). However, some patients fail to respond to IL-1 blockade but do respond to IL-6 blockade, and given the emerging evidence for a “window of opportunity” in the treatment of sJIA, tools such as gene expression profiling to predict response (or non-response) is of crucial importance.
The previously published clinical trial of canakinumab provides a unique opportunity to define gene expression signatures in a large cohort of patients with a range of clinical responses (14). Previous analysis of these gene expression microarrays by Brachat and colleagues highlighted a strong upregulation of innate immune system genes at baseline, with more severely dysregulated profiles that rapidly declined in some canakinumab responders, while non-responders in general had more muted dysregulation not affected by treatment (15). However, this finding was not consistent and was not further characterized. Here, we reanalyzed the gene expression data examining extreme phenotypes of canakinumab non-response (ACR≤30 on day 15 after treatment) compared to strong clinical response (ACR≥90).
Based on this we were able to characterize a baseline gene signature specific for strong clinical response that was consistent with proinflammatory and neutrophil dysregulation. We also defined a signature specific for canakinumab non-response, including CD163, a marker of regulatory monocytes and macrophages. Finally, we identified a bi-modal IFN response gene signature that was activated in a subset of canakinumab treated patients. Together these signatures provide key clues to molecular predictors of treatment response in sJIA patients.
Methods
Patients and study design
This study is a secondary analysis of gene expression profiles from patients with sJIA from two phase 3-trials evaluating canakinumab treatment (ClinicalTrials.gov, NCT00886769 (trial 1) and NCT00889863 (trial 2)), conducted by the members of the Pediatric Rheumatology International Trials Organization (PRINTO) and the Pediatric Rheumatology Collaborative Study Group (PRCSG). Study design including eligibility criteria and ethics approval have been reported previously (14,15). Eighty-six sJIA patient and 22 healthy control blood samples for RNA isolation were collected at baseline and at day 3. Neutrophil counts from whole blood and serum IL-18 were collected at baseline and on day 15 or 29, respectively.
Gene transcription profiles and MDS plots
Raw CEL files were downloaded from the Gene Expression Omnibus (GSE80060) and were preprocessed and annotated using the crossmeta Bioconductor package (16). Mixed effect differential expression analyses were performed using the duplicate correlation function from the limma Bioconductor package with subject as a random effect (17,18). Differentially expressed probes were identified as those with an FDR ≤0.05. Over-represented gene ontologies were identified using the goana function from limma (unadjusted p-value < 10−5) and further summarized with REVIGO (19). For limma MDS plots, the remef R package was used to remove the subject effect from limma preprocessed log-expression value (20).
Transcriptional response to canakinumab
The early transcriptional response to canakinumab was evaluated by comparing gene expression values in patients with sJIA at day 3 with the values measured at baseline.
Patient response was previously measured by using the Pediatric ACR response on day 15 (15). Strong clinical responders were defined as ACR≥90, with ≥90% improvement in at least 3 of 6 core criteria, >30% worsening in no more than 1 core criterion and no intermittent fever during the last 7 days. Non-responders were defined as ≤30% improvement in at least 3 of 6 core criteria and >30% worsening in no more than 1 core criterion (21). In total, 86 patients were analyzed of which 26 were categorized as strong clinical responders, 34 intermediate responders, and 26 non-responders.
Results
A distinct gene expression signature characterizes canakinumab responders
Children with sJIA and a strong clinical response (ACR≥90) to canakinumab have a baseline gene signature distinct from non-responders (ACR≤30), which highlights molecular differences in these two groups before and after treatment. As previously suggested (15), strong clinical responders have a more dysregulated transcriptional profile prior to treatment as compared to healthy controls (9629 differentially expressed genes (DEGs) with FDR ≤ 0.05). On the other hand, sJIA patients who did not respond well to canakinumab had only 6017 DEGs prior to treatment.
At baseline, the gene expression signature of strong canakinumab responders as compared to non-responders was defined by significant upregulation of transcripts related to neutrophil activation (such as CD177, CXCL1), IL-1 signaling (IL1B, IL1R1, IL1RAP, IL1RN) and TNFA, Toll-like receptor (TLR) signaling (TLR5, LRG1, TLR8, TLR9), and the inflammasome (NLRC4, AIM2, CASP5). The top 100 significantly upregulated genes are shown in supplementary table 1. These pro-inflammatory immune genes are also reflected in the top over-represented gene ontology terms, which include myeloid leukocyte activation (GO:0002274), inflammatory response (GO:0006954) and cytokine production (GO:0001816) (Figure 1).
Figure 1:

REVIGO gene ontology treemap highlighting upregulated pathways in the strong clinical responders compared to non-responders. Colors indicate superclusters, and size of boxes indicate p-value (19).
After treatment, strong clinical responders experienced a significant decrease in the expression of this gene signature composed of genes related to IL-1, TLRs, inflammasome and neutrophil activation. Three days after canakinumab treatment, their gene profile reduces to a level closer to healthy controls (Figure 2A). The strongly overrepresented neutrophil activation pathways on day 1 (supplementary table 2) were no longer among the top upregulated GO pathways on day 3 (not shown). Non-responders presented with a much more modest change of gene expression, which is barely affected by canakinumab treatment. Moderate responders (ACR=50–70), correspondingly, showed an intermediate displacement of their gene expression profile towards healthy controls upon treatment (Figure 2A).
Figure 2:

Canakinumab responders strongly shift their gene signature towards healthy controls. A) MDS plots for ACR score 0&30 (yellow, 26 subjects), 50&70 (red, 34 subjects) or 90&100 (blue, 26 subjects) vs healthy (gray, 22 subjects). Arrows indicate the change in MDS coordinates after treatment. B) Absolute neutrophil counts measured at baseline and on day 15 after treatment shown across ACR response from 0 (yellow) to 100 (blue). C) Serum levels of total IL-18 in pg/ml measured on day 3 and day 29 after treatment shown across ACR response from 0 (yellow) to 100 (blue). D) CD163 gene expression measured by microarray for healthy controls (gray) and for canakinumab treated patients at baseline and on day 3, shown across ACR response from 0 (yellow) to 100 (blue).
Neutrophil counts and IL-18 serum levels are strongly increased in strong clinical responders
Strong clinical responders had significantly higher numbers of absolute neutrophils compared to non-responders at baseline, which was strongly decreased upon day 15 after canakinumab (Figure 2B). The neutrophil counts varied linearly with ACR score both at baseline and after treatment.
As noted above, neutrophil activity was also highly represented within the top 15 GO pathways observed in strong clinical responders vs non-responders at baseline (supplementary table 2). In line with previous results (15), IL-18, thought to be a key driver of sJIA-MAS and sJIA-LD, was also significantly increased in serum (though IL18 gene expression was unchanged; data not shown) of responders compared to non-responders at day 3, and remained significantly elevated even after 29 days of treatment (Figure 2C).
CD163 gene expression is upregulated in non-responders at baseline and remains upregulated compared to strong clinical responders
Next, we defined the transcriptional signature associated with canakinumab non-response by identifying genes differentially expressed in the same direction in non-responders as compared to strong responders at baseline and day 3, i.e. that do not change with canakinumab treatment. This approach identified 14 probes, corresponding to 3 genes that were consistently downregulated in non-responders and 8 genes which were consistently upregulated in non-responders vs strong clinical responders (supplementary table 3). The most notable of these was CD163 (Figure 2D), which is expressed on regulatory monocytes induced by IL-10, and also a key marker for hemophagocytic macrophage differentiation present in sJIA-MAS (22). In addition, and in marked contrast to the neutrophil signature discussed above, expression of CD163 did not significantly change after canakinumab treatment. Together, this supports detection of myeloid phenotypes present in sJIA that can distinguish canakinumab non-response from strong response.
Canakinumab treated sJIA patients either up- or downregulate a type I IFN gene signature
Finally, we examined changes in the IFN response signature upon canakinumab treatment from a previously established module representing type I IFN genes (23). Previous studies have suggested a link between IL-1 blockade and upregulation of type I IFN genes (2,24). Here, IL-1 blockade did not significantly upregulate IFN response genes when looking at all patients compared to untreated healthy controls (not shown). However, when we examine this more granularly, we found that irrespective of treatment response individual patients responses diverged, with a subset upregulating the IFN signature (clusters A+C) and others downregulating the IFN signature (clusters B+D) (Figure 3). While both upregulation and downregulation was seen in canakinumab non-responders and strong responders, the effect was more robust in both directions in the strong clinical responders (Figure 3). The persistence of this upregulation, and any association with disease complications, is unknown.
Figure 3:
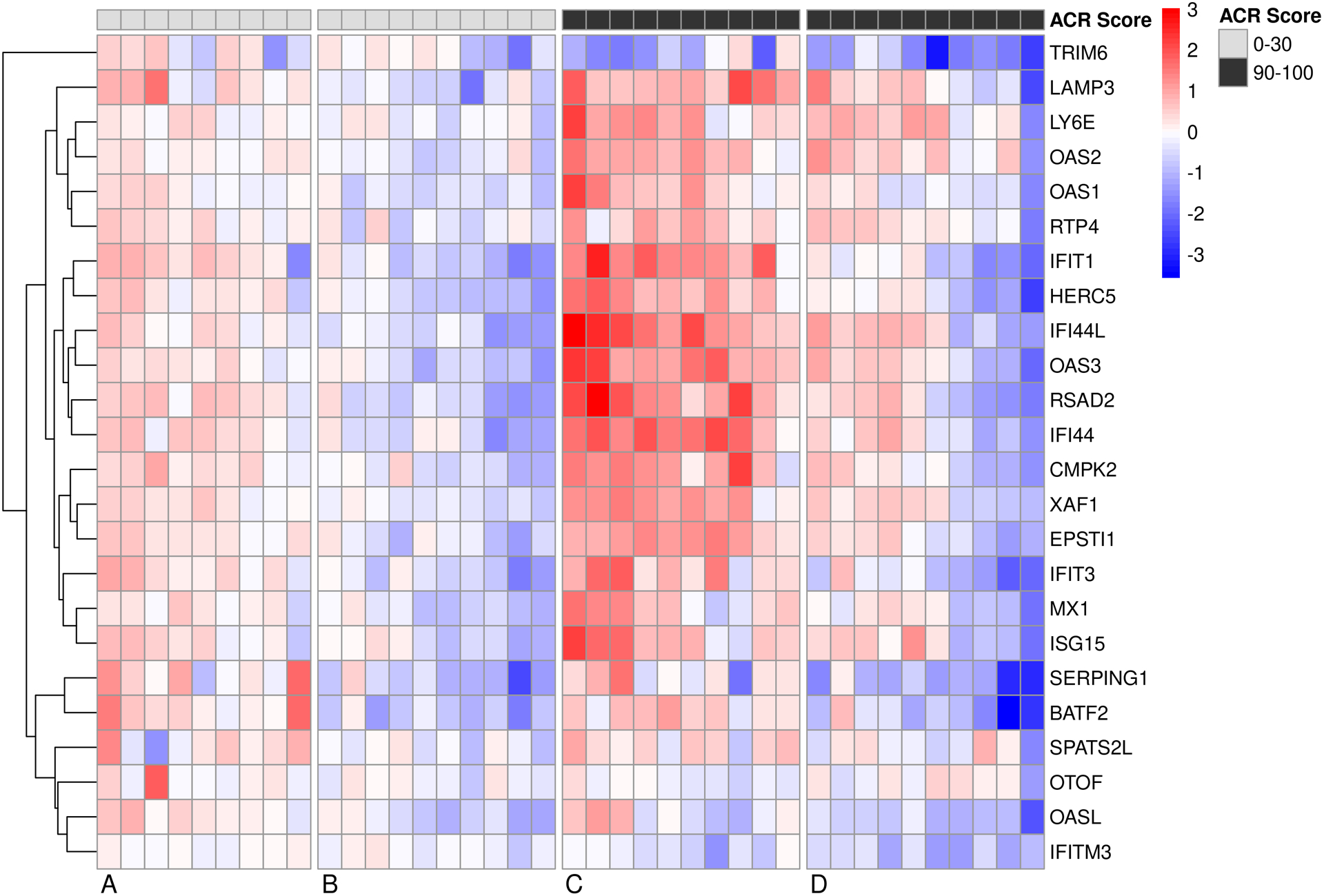
Canakinumab treated sJIA patients either up- or downregulate a type I IFN gene signature. Heatmap shows the fold change (+3 to −3) of type I IFN genes from day 0 to day 3 in non-responders and strong clinical responders. Most patients, regardless of response, either up- or downregulate this signature.
Discussion
Use of patient gene expression profiling can be a powerful tool in identifying molecular mechanisms associated with disease. More so, comparison of expression profiles in patients before and after treatment onset with a cytokine inhibitor such as canakinumab allows unique insight into the molecular effects of such drugs, and the potential to identify predictive signatures of strong clinical response or failure. Here, we re-analyzed previously collected gene expression data from two phase-3 trials evaluating canakinumab in sJIA patients to characterize the signature associated with extreme phenotypes of clinical response – strong response (ACR≥90) and non-response (ACR≤30) (15).
As previously noted by Brachat and others, we observed the baseline (pre-treatment) blood transcriptional profile of sJIA patients is strongly enriched for the IL-1 pathway, various TLRs, inflammasome and neutrophil activation pathways (15). However, this gene expression profile was significantly more dysregulated in strong canakinumab responders than non-responders pre-treatment. In contrast to the findings by Brachat and colleagues, the transcriptional effects observed here from strong clinical responders (ACR≥90) are consistent and significant throughout this group. Strong clinical responders also showed striking movement of this gene signature towards levels of healthy controls, while non-responders barely shifted upon canakinumab treatment. These observations highlight that IL-1 blockade quickly normalizes expression in sJIA patients with the most severely dysregulated baseline IL-1 and neutrophil-predominant blood gene expression profile.
In contrast to the IL-1 driven signature characterizing strong clinical response, we found 11 genes that characterized non-response, which have not been identified by Brachat (15). These genes were significantly differently expressed in the non-responders compared to the strong clinical responders and did not change expression after canakinumab treatment. Most genes we found to be functionally unrevealing, however we identified CD163 to be significantly higher expressed in the non-responders compared to strong clinical responders and to healthy controls at baseline. CD163 is upregulated in sJIA, and known as marker for IL-10 polarized regulatory macrophages as well as hemophagocytic macrophages associated with sJIA-MAS (22). The upregulation of CD163 in those sJIA patients suggests IL-1 independent pathways induce CD163, including the presence of a resolution monocyte phenotype and potentially contributing to hemophagocyte differentiation as a defining feature of sJIA-MAS (25). In fact, previous results have shown that CD163 mRNA correlates negatively with IL-1 (26). Taken together this supports a hypothesis that patients defined by this alternative gene signature may rather benefit from other biologic treatments that target alternative pathways, such as tocilizumab (anti-IL-6R) or JAK/STAT inhibitors (27).
Neutrophils and neutrophil gene signatures are known to be elevated in sJIA patients (2,3,5), and elevated neutrophil count has been previously suggested to predict treatment response to anti IL-1 therapy with anakinra (6). Our cohort also showed higher blood neutrophil counts were associated with stronger clinical response to canakinumab. Consistent with previous findings in anakinra treated sJIA patients (28), responders had significantly increased upregulated neutrophil genes (particularly CD177) compared to non-responders. However, it is unlikely that increased cell numbers are solely responsible for driving the whole blood transcriptional profile in sJIA, as we and others have shown that purified neutrophils demonstrate a TLR-driven proinflammatory gene expression signature (3).
We next investigated IL-18, which is strongly expressed in neutrophils and monocytes from sJIA patients, with corresponding increased serum levels of IL-18 (29). Interestingly, IL-18 serum levels were upregulated in responders compared to non-responders both on day 3 and 29 after treatment, but this was not reflected by increased IL18 gene expression in either the responders or non-responders vs. healthy controls, suggesting IL-18 may be produced primarily by non-hematopoietic cells. Strong IL-18 elevation in serum particularly distinguishes the severe complications sJIA-MAS and sJIA-LD (8,10). It is unclear to date which pathways drive the overproduction of IL-18 in sJIA, though recent evidence points at type I IFNs regulating IL-18 gene and protein expression (2,7,24).
When we investigated a type I IFN signature composed of 24 genes (23), we found that patients separated evenly into two clusters: regardless of clinical response, canakinumab treatment induced either up- or downregulation of this IFN signature. However, strong clinical responders showed a much stronger reaction either way upon treatment compared to non-responders, which was in line with the low overall movement observed in non-responders. We did not observe differences between healthy controls and sJIA patients at baseline (data not shown).
There is significant cross-talk between IL-1 and type I IFNs, and IL-1 has been shown to potently antagonize type I IFN transcription and translation (30). Thus, it is conceivable that IL-1 blockade, which is commonly used to treat sJIA, in some patients enables increased signaling of type I IFN pathways which further increases IL-18 and IFNγ reactivity.
In fact, previous studies have observed increased type I IFN gene expression in patients treated with IL-1 blockade (2,24). Similar to our findings, Rice and colleagues reported only 6/10 sJIA patients treated with IL-1 blockade had an upregulated type I IFN signature. Why we and others only found some patients reacting by increased IFN response remains to be evaluated. As recent evidence associates the development of sJIA-LD with the use of biologics treatment and increased IFN-related gene expression in the lungs (10,31), understanding why some patients react to canakinumab by upregulating the type I IFN signature and whether this primes them for developing sJIA complications will need to be evaluated in future studies.
Taken together, our findings highlight a role for severe dysregulation of pro-inflammatory genes and immune pathways that present a target for canakinumab in canakinumab responders. However, our findings are limited by the lack of a suitable independent validation cohort, and thus further studies are required to validate and define this profile for prospective utility in a clinical setting. Further studies will also need to shed light on the association of type I IFN upregulation upon canakinumab treatment and the onset of sJIA-MAS and sJIA-LD.
Supplementary Material
Acknowledgements
This work was funded by the Systemic JIA Foundation. Dr. Verweyen is supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Projectnumber 448863690. Dr. Schulert is supported by the National Institute of Arthritis, Musculoskeletal, and Skin Disorders at the National Institutes of Health (NIAMS/NIH) K08-AR072075.
Author disclosures
Dr. Schulert has received consulting fees from Novartis and Sobi of less than $10.000. Dr. Grom has served as a consultant for Juno and Novartis, and has received research support from NovImmune and AB2Bio of less than $10.000. All other authors declare no conflicts of interest.
References
- 1.Mellins ED, Macaubas C, Grom AA. Pathogenesis of systemic juvenile idiopathic arthritis: some answers, more questions. NatRev Rheumatol 2011;7:416–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Quartier P, Allantaz F, Cimaz R, Pillet P, Messiaen C, Bardin C, et al. A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial). Ann Rheum Dis 2011;70:747–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown RA, Henderlight M, Do T, Yasin S, Grom AA, DeLay M, et al. Neutrophils From Children With Systemic Juvenile Idiopathic Arthritis Exhibit Persistent Proinflammatory Activation Despite Long-Standing Clinically Inactive Disease. Front Immunol 2018;9:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ogilvie EM, Khan A, Hubank M, Kellam P, Woo P. Specific gene expression profiles in systemic juvenile idiopathic arthritis. Arthritis Rheum 2007;56:1954–1965. [DOI] [PubMed] [Google Scholar]
- 5.Allantaz F, Chaussabel D, Stichweh D, Bennett L, Allman W, Mejias A, et al. Blood leukocyte microarrays to diagnose systemic onset juvenile idiopathic arthritis and follow the response to IL-1 blockade. J Exp Med 2007;204:2131–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haar NM ter, Dijkhuizen EHP van, Swart JF, Royen-Kerkhof A van, Idrissi A el, Leek AP, et al. Treatment to Target Using Recombinant Interleukin-1 Receptor Antagonist as First-Line Monotherapy in New-Onset Systemic Juvenile Idiopathic Arthritis: Results From a Five-Year Follow-Up Study. Arthritis Rheumatol 2019;71:1163–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verweyen E, Holzinger D, Weinhage T, Hinze C, Wittkowski H, Pickkers P, et al. Synergistic signaling of TLR and IFNα/β facilitates escape of IL-18 expression from endotoxin tolerance Supplement. Am J Respir Crit Care Med 2020;201:526–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weiss ES, Girard-Guyonvarc’h C, Holzinger D, Jesus AA de, Tariq Z, Picarsic J, et al. Interleukin-18 diagnostically distinguishes and pathogenically promotes human and murine macrophage activation syndrome. Blood 2018:blood-2017-12–820852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kimura Y, Weiss JE, Haroldson KL, Lee T, Punaro M, Oliveira S, et al. Pulmonary hypertension and other potentially fatal pulmonary complications in systemic juvenile idiopathic arthritis. Arthritis Care Res 2013;65:745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schulert GS, Yasin S, Carey B, Chalk C, Do T, Schapiro AH, et al. Systemic Juvenile Idiopathic Arthritis–Associated Lung Disease: Characterization and Risk Factors. Arthritis Rheumatol 2019;71:1943–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benedetti F De Brunner HI, Ruperto N, Kenwright A, Wright S, Calvo I, et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med 2012;367:2385–2395. [DOI] [PubMed] [Google Scholar]
- 12.Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med 2005;201:1479–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, Leslie KS, Hachulla E, Quartier P, et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med 2009;360:2416–2425. [DOI] [PubMed] [Google Scholar]
- 14.Ruperto N, Brunner HI, Quartier P, Constantin T, Wulffraat N, Horneff G, et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med 2012;367:2396–2406. [DOI] [PubMed] [Google Scholar]
- 15.Brachat AH, Grom AA, Wulffraat N, Brunner HI, Quartier P, Brik R, et al. Early changes in gene expression and inflammatory proteins in systemic juvenile idiopathic arthritis patients on canakinumab therapy. Arthritis Res Ther 2017;19:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pickering A crossmeta: Cross Platform Meta-Analysis of Microarray Data . R package version 1.14.0 2020. [Google Scholar]
- 17.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smyth GK, Michaud J, Scott HS. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics 2005;21:2067–2075. [DOI] [PubMed] [Google Scholar]
- 19.Supek F, Bošnjak M, Škunca N, Šmuc T. Revigo summarizes and visualizes long lists of gene ontology terms. PLoS One 2011;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hohenstein S, Kliegl R. remef: Remove Partial Effects. R package version 1.0.7.https://github.com/hohenstein/remef/. 2020.
- 21.Wallace CA, Giannini EH, Huang B, Itert L, Ruperto N. American College of Rheumatology provisional criteria for defining clinical inactive disease in select categories of juvenile idiopathic arthritis. Arthritis Care Res 2011;63:929–936. [DOI] [PubMed] [Google Scholar]
- 22.Thornton S, Tan R, Sproles A, Do T, Schick J, Grom AA, et al. A Multiparameter Flow Cytometry Analysis Panel to Assess CD163 mRNA and Protein in Monocyte and Macrophage Populations in Hyperinflammatory Diseases. J Immunol 2019;202:1635–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chaussabel D, Quinn C, Shen J, Patel P, Glaser C, Baldwin N, et al. A Modular Analysis Framework for Blood Genomics Studies: Application to Systemic Lupus Erythematosus. Immunity 2008;29:150–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rice GI, Melki I, Frémond M-L, Briggs TA, Rodero MP, Kitabayashi N, et al. Assessment of Type I Interferon Signaling in Pediatric Inflammatory Disease. J Clin Immunol 2017;37:123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakumura N, Shimizu M, Mizuta M, Inoue N, Nakagishi Y, Yachie A. Soluble CD163, a unique biomarker to evaluate the disease activity, exhibits macrophage activation in systemic juvenile idiopathic arthritis. Cytokine 2018;110:459–465. [DOI] [PubMed] [Google Scholar]
- 26.Liu B, Hu B, Shao S, Wu W, Fan L, Bai G, et al. CD163/Hemoglobin Oxygenase-1 Pathway Regulates Inflammation in Hematoma Surrounding Tissues after Intracerebral Hemorrhage. J Stroke Cerebrovasc Dis 2015;24:2800–2809. [DOI] [PubMed] [Google Scholar]
- 27.Hausmann JS. Targeting cytokines to treat autoinflammatory diseases. Clin Immunol 2019;206:23–32. [DOI] [PubMed] [Google Scholar]
- 28.Gattorno M, Piccini A, Lasigliè D, Tassi S, Brisca G, Carta S, et al. The pattern of response to anti-interleukin-1 treatment distinguishes two subsets of patients with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum 2008;58:1505–1515. [DOI] [PubMed] [Google Scholar]
- 29.Yasin S, Schulert GS. Systemic juvenile idiopathic arthritis and macrophage activation syndrome. Curr Opin Rheumatol 2018;30:1. [DOI] [PubMed] [Google Scholar]
- 30.Tian Z, Shen X, Feng H, Gao B. IL-1β Attenuates IFN-αβ-Induced Antiviral Activity and STAT1 Activation in the Liver: Involvement of Proteasome-Dependent Pathway. J Immunol 2000;165:3959–3965. [DOI] [PubMed] [Google Scholar]
- 31.Saper VE, Chen G, Deutsch GH, Guillerman PR, Birgmeier, Johannes Jagadeesh K, Canna S, et al. Emergent high fatality lung disease in systemic juvenile arthritis. Ann Rheum Dis 2019;78:1722–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.