

Summary
Searching for factors to improve knock-in efficiency for therapeutic applications, biotechnology, and generation of non-human primate (NHP) models of disease, we found that the strand exchange protein RAD51 can significantly increase Cas9-mediated homozygous knock-in in mouse embryos through an interhomolog repair (IHR) mechanism. IHR a hallmark of meiosis, but only occurs at low frequencies in somatic cells and its occurrence in zygotes is controversial. Using multiple approaches, we provide evidence for an endogenous IHR mechanism in the early embryo that can be enhanced by RAD51. This process can be harnessed to generate homozygotes from wildtype zygotes using exogenous donors and to convert heterozygous alleles into homozygous alleles without exogenous templates. Furthermore, we identify additional IHR-promoting factors and describe features of IHR events. Together, our findings show conclusive evidence for IHR in mouse embryos and describe an efficient method for enhanced gene conversion.
In Brief:
Genes of mouse embryos can be efficiently edited by enhancing interhomolog repair via co-injection of RAD51 with Cas9.
Graphical Abstract
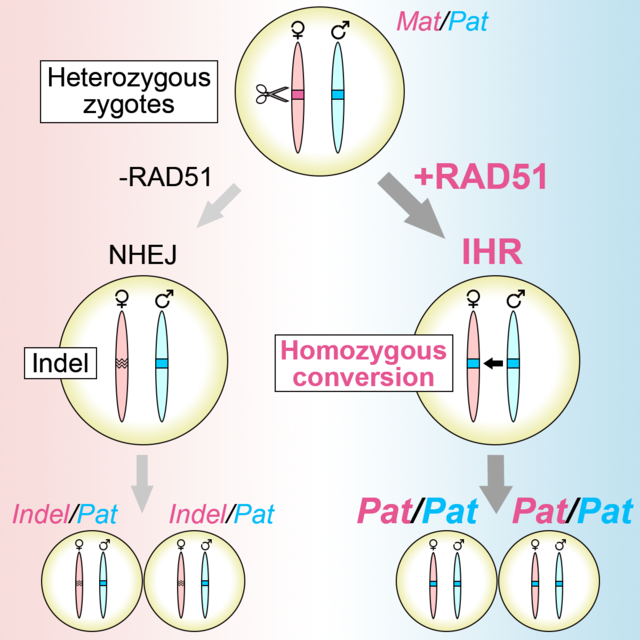
Introduction
CRISPR-associated (Cas) proteins and other targeted nucleases have revolutionized our ability to manipulate the genome, but the precise knock-in of exogenous DNA at targeted double-strand breaks can still be difficult. Successful knock-in requires both efficient cutting by a targeted endonuclease and recruitment of endogenous DNA repair factors to incorporate the desired edit into the host genome. However, while numerous improvements in our ability to make targeted breaks in the genome have been made, our current understanding of how to leverage endogenous DNA repair proteins to promote successful knock-in is still incomplete.
Attempts to enhance knock-in efficiency have primarily focused on three distinct approaches: inhibition of non-homologous end-joining, promotion of end resection, and enhancement of homology search and strand exchange. While methods for suppressing NHEJ through inhibition of the NHEJ-promoting protein 53BP1 have yielded promising results (Canny et al., 2018; Jayavaradhan et al., 2019), the mechanism and its direct effect on homologous recombination remains unclear. Additional methods to promote end resection, which is thought to be a definitive step toward homologous recombination, have given modest results (Charpentier et al., 2018; Nakade et al., 2018; Tran et al., 2019) and suggest a need to target additional molecules and deepen our understanding of end resection and its consequences.
Given the potential dangers of NHEJ inhibition and our poor understanding of end-resection and its ability to promote HR, directly targeting the homologous recombination machinery is an appealing method for enhancing knock-in. In 2016, a study in rabbit embryos demonstrated significant enhancement of knock-in using RS-1, a chemical agonist of the strand-exchange factor RAD51 (Song et al., 2016), a strand exchange protein critical for HR. In cells lacking DNA damage, RAD51 forms small, enzymatically inactive heptameric rings (Yu et al., 2003). In response to double-strand breaks (DSBs), the protein BCCIP promotes BRCA2-mediated disruption of RAD51 self-association, allowing RAD51 recruitment to resected DSBs (Lu et al., 2005). RAD51 then forms long nucleofilaments on single-stranded overhangs produced by end resection and actively performs the homology search and strand invasion steps that are the basis of homologous recombination (Krejčí et al., 2012). Reinforcing the central role of RAD51 in HR and knock-in events following targeted double-strand breaks, fusion of RAD51 to Cas9, which has a dominant-negative effect on endogenous RAD51 function, inhibits HDR (Rees et al., 2019).
Based on the aforementioned findings and the key role of RAD51 in homologous recombination we sought to further explore the utility of RAD51 for Cas9-based knock-in events. Unexpectedly, we find that RAD51 can increase homozygosity without any effects on initial knock-in efficiency through enhancement of interhomolog repair (IHR). After demonstrating increased homozygosity in knock-in experiments using single-strand oligonucleotide donors, we demonstrate frequent homozygous conversion at multiple sites on different chromosomes without the use of donor DNA templates. We then identify additional factors that can enhance IHR, including a RAD51 variant capable of nearly indel-free IHR, and describe mechanistic properties of IHR events. In summary, our study provides conclusive evidence of IHR in the early mouse embryo and shows that RAD51 and homologous recombination-associated factors can be used to enhance the process for efficient homozygous knock-in and template-free conversion of heterozygous alleles to homozygous alleles.
Results
Co-Injection of RAD51 with Cas9 RNPs Enhances Homozygous Knock-in
Based on our previous success using recombinant Cas9 ribonucleoproteins (RNPs) to enhance HDR in mouse embryos (Aida et al., 2015; Quadros et al., 2017) and the reported enhancement of HDR by the RAD51 agonist RS-1, we began by testing whether recombinant RAD51 could efficiently stimulate knock-in of an autism-associated point mutation in Chd2 (c.5051G>A; R1685H in human, R1684H in mouse; hereon referred to as Chd2RH; Figure 1A). Previous studies have shown that KI efficiency is affected by the proximity of the Cas9 cut site to the insertion site (Paquet et al., 2016), so we chose a guide positioned where the cut site is directly adjacent to the desired G>A point mutation. We performed both pronuclear (PNI) and cytoplasmic (CPI) injections of RNPs containing crRNA, tracrRNA, and SpyCas9 along with a single-stranded DNA donor (30 ng/μL for PNI and 100 ng/μL for CPI) and RAD51 (10 ng/μL) in mouse zygotes and observed overall KI efficiencies of 78% (7/9) and 56% (5/9) for CPI and PNI, respectively (Figure 1B,C). Both CPI and PNI resulted in high homozygous KI efficiencies of 71% and 100% for CPI- and PNI-derived KI embryos, respectively (Figure 1D) and on-target editing was confirmed with primers sitting outside of the donor template. We further confirmed homozygosity by breeding putative homozygotes with wild-type mice and genotyping the resulting F1 pups, which found that 100% (36/36) of animals from 6 successful mating pairs had a Chd2RH/+ genotype (Figure 1E).
Figure 1. Efficient homozygous knock-in with RAD51.
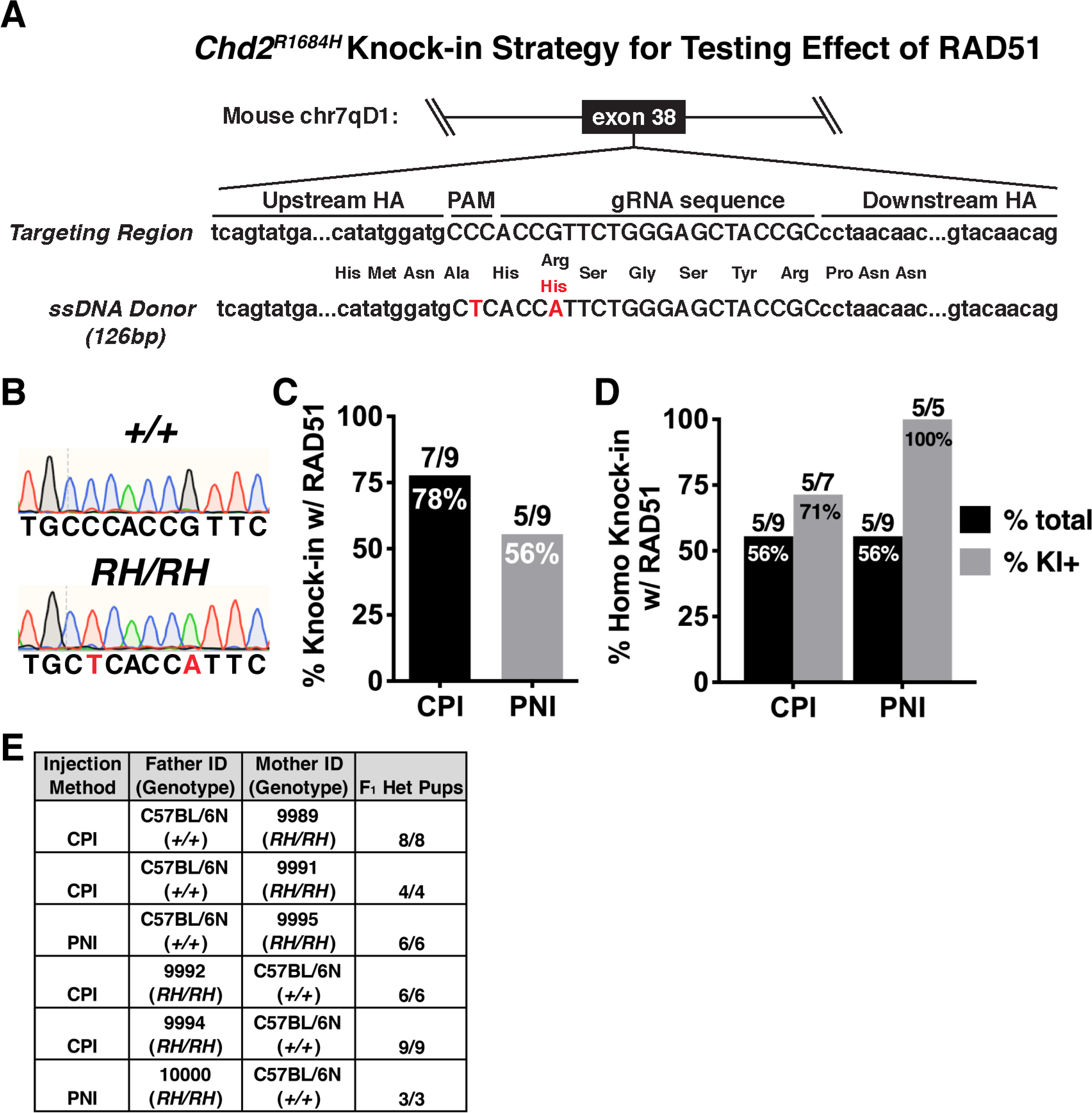
(A) Schematic of the targeted locus and HR donor for generating Chd2RH mutant mice. Donor contains both the c.5051G>A point mutation and a synonymous mutation in the relevant PAM site (HA=homology arm). (B) Example chromatograms of wildtype (top) and Chd2RH/RH animals (bottom). (C) Overall KI efficiency observed in F0 pups derived from either cytoplasmic (CPI) or pronuclear (PNI) injection (pups with ≥1 KI allele/total pups). (D) Homozygous KI rates observed in F0 pups generated by either CPI or PNI, shown as percentage of all pups or pups positive for KI. (E) Genotyping results from F1 pups derived from crosses between F0 Chd2RH/RH animals and wild-type animals.
Since we did not initially generate Chd2RH mice from injections lacking RAD51, we performed injections to directly test the impact of RAD51 on KI efficiency at the Chd2RH locus. Since we did not observe differences in editing efficiency or homozygosity rates between CPI and PNI (Figures 1C and 1D and Table S1), we utilized PNI for the rest of this study, as this is our standard approach for generating mouse models. Pronuclear injection was performed with or without recombinant RAD51 and embryos were cultured for 2–3 days until the morula or blastocyst stage before DNA purification and nested PCR for genotyping. Sanger sequencing showed that RAD51 only slightly increased overall knock-in efficiency (Figure 2A, left), but strongly increased homozygous knock-in efficiency (Figure 2A, middle, right). To test whether the observed effects of RAD51 were locus-dependent, we used the same strategy to knock in an albinism-associated mutation (c.265T>A; C89S; hereon referred to as TyrC89S; schematic in Figure 2B) in tyrosinase (Tyr), the rate-limiting enzyme in melanin production (Ghodsinejad Kalahroudi et al., 2014). Homozygous disruption of Tyr results in mice completely lacking pigment (Figure 2C), but because homozygous indels or compound disruption of Tyr can result in albinism, all F0 animals were genotyped (Figure 2D) and phenotyped (Table S1) to assess the effects of RAD51 on knock-in efficiency. In pups with visible mosaic albinism, genotyping was performed using samples from tissue completely lacking melanin to assess the number of animals with at least one homozygous KI event (Figure 2E, middle). Injections without RAD51 showed a high overall KI efficiency that was not significantly increased by the addition of RAD51 (Figure 2E, left), but sequencing found that RAD51 significantly enhanced homozygous KI efficiency, with 44% of pups (including mosaics) having an albino phenotype resulting from homozygous KI of the TyrC89S allele with RAD51, as compared to only 12% without (Figure 2E, middle). Furthermore, RAD51 co-injection increased the rate of pure, non-mosaic homozygous knock-in (Figure 2E, right). Together, these data demonstrate that RAD51 can increase homozygous knock-in rates.
Figure 2. RAD51 enhances homozygous KI efficiency at multiple loci.
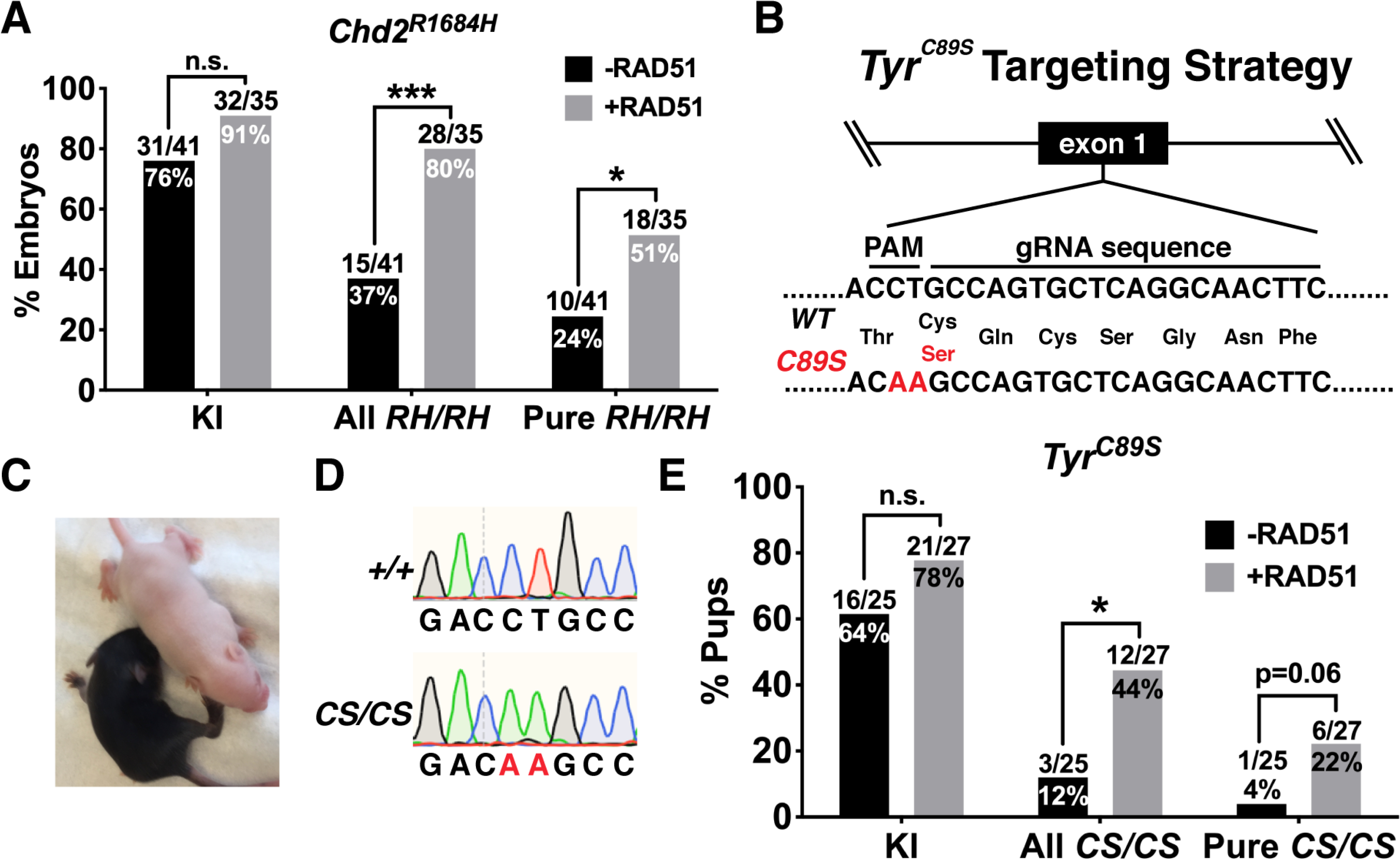
(A) Quantification of KI efficiency (left) and homozygous KI efficiency in embryos generated by Chd2RH PNI with or without RAD51. All RH/RH includes mosaic embryos and denotes embryos with ≥3:2 ratio of mutant allele to wildtype allele (one-sided Fisher’s exact test). (B) Schematic of knock-in strategy for the albinism-associated TyrC89S mutation. (C) Representative examples of Tyr+/+ (black) and TyrC89S/C89S (white) littermates derived from injections using exogenous RAD51 protein. (D) Representative chromatograms from Tyr+/+ (top) and TyrC89S/C89S animals. (E) Genotyping of F0 animals from injections targeting the TyrC89S locus with or without RAD51 (one-sided Fisher’s exact test). *p<0.05, ***p<0.001, n.s. p>0.05
We were intrigued to see that RAD51 co-injection more than doubled the observed homozygosity at both the Chd2 and Tyr loci, regardless of whether or not we took mosaicism into account (Figure 2A,E). At the onset of this study, previous attempts to increase HDR through pharmacological or genetic mechanisms had not shown such high homozygosity rates (Maruyama et al., 2015; Paquet et al., 2016; Takayama et al., 2017), leading us to investigate the mechanism by which RAD51 can promote such an outcome. Our findings that RAD51 did not increase overall knock-in efficiency (Figure 2A,E) agree with more recent work showing that RAD51 is not required for single-stranded template repair (SSTR) (Richardson et al., 2018) and therefore suggest that RAD51-mediated effects on homozygosity do not arise from the stimulation of independent knock-in events on both alleles. Additionally, we found it surprising that we observed multiple embryos from both Chd2RH and TyrC89S injections whose genotyping results suggested the presence of the same indel on both alleles (Figure S1 and Table S1). Genotyping of F1 pups derived from a Chd2-targeted female with such a homozygous indel revealed that all pups were heterozygous for the same indel, confirming homozygosity of the founder (Figure S1). Although we could not completely rule out that these deletions resulted from stereotyped generation of the same indel on both alleles or microhomology-mediated end-joining (MMEJ) (Aida et al., 2016; McVey and Lee, 2008), the variety of observed indels and lack of corresponding MMEJ sequence motifs suggest that the homozygosity could have arisen from direct transfer of genetic information between homologous chromosomes.
RAD51 Promotes Interhomolog Repair in the Early Embryo
More than 20 years ago, experiments in mouse ES cells found evidence of recombination events between homologous chromosomes (Moynahan and Jasin, 1997; Richardson et al., 1998). While these IHR events are common and well-understood in meiotic cells, their occurrence in mitotic cell types is poorly described. In light of several recent studies that have described embryonic IHR after the induction of a targeted double-strand break (DSB) by Cas9 (Ma et al., 2017; Wu et al., 2013), we hypothesized that RAD51 can promote homozygosity through the same mechanism. To directly test this, we attempted to generate Chd2RH/RH embryos from Chd2RH/+ embryos without the use of an exogenous donor template. To do this, we generated Chd2RH/+ zygotes by in vitro fertilization of wild-type oocytes with sperm from Chd2RH/RH donors and then specifically targeted the wild-type maternal allele with Cas9. It should be noted that this strategy rules out the possibility of false-positive results from parthenogenetic embryos, as the mutant allele to be copied is carried by the sperm. As illustrated in Figure 3A, PNI was performed on zygotes ~8 hours post fertilization (hpf) using an RNP mixture of crRNA, tracrRNA, and SpyCas9 with or without RAD51. The Chd2RH allele contains mutations in both the protospacer and its associated PAM and we confirmed through an in vitro digestion assay that Cas9 is only capable of cutting the wild-type maternal allele (Figure S2). Consistent with the rates observed in previous work (Ma et al., 2017; Wu et al., 2013), we saw IHR events in 26% of control-injected embryos, including mosaics (Figure 3B,C). Co-injection of RAD51 significantly increased this rate to 74% (Figure 3C, left). We also observed a similar increase in pure, non-mosaic IHR rates in RAD51-injected embryos (Figure 3C, right), supporting the hypothesis that RAD51 can promote IHR in mouse embryos.
Figure 3. RAD51 enhances interhomolog repair.
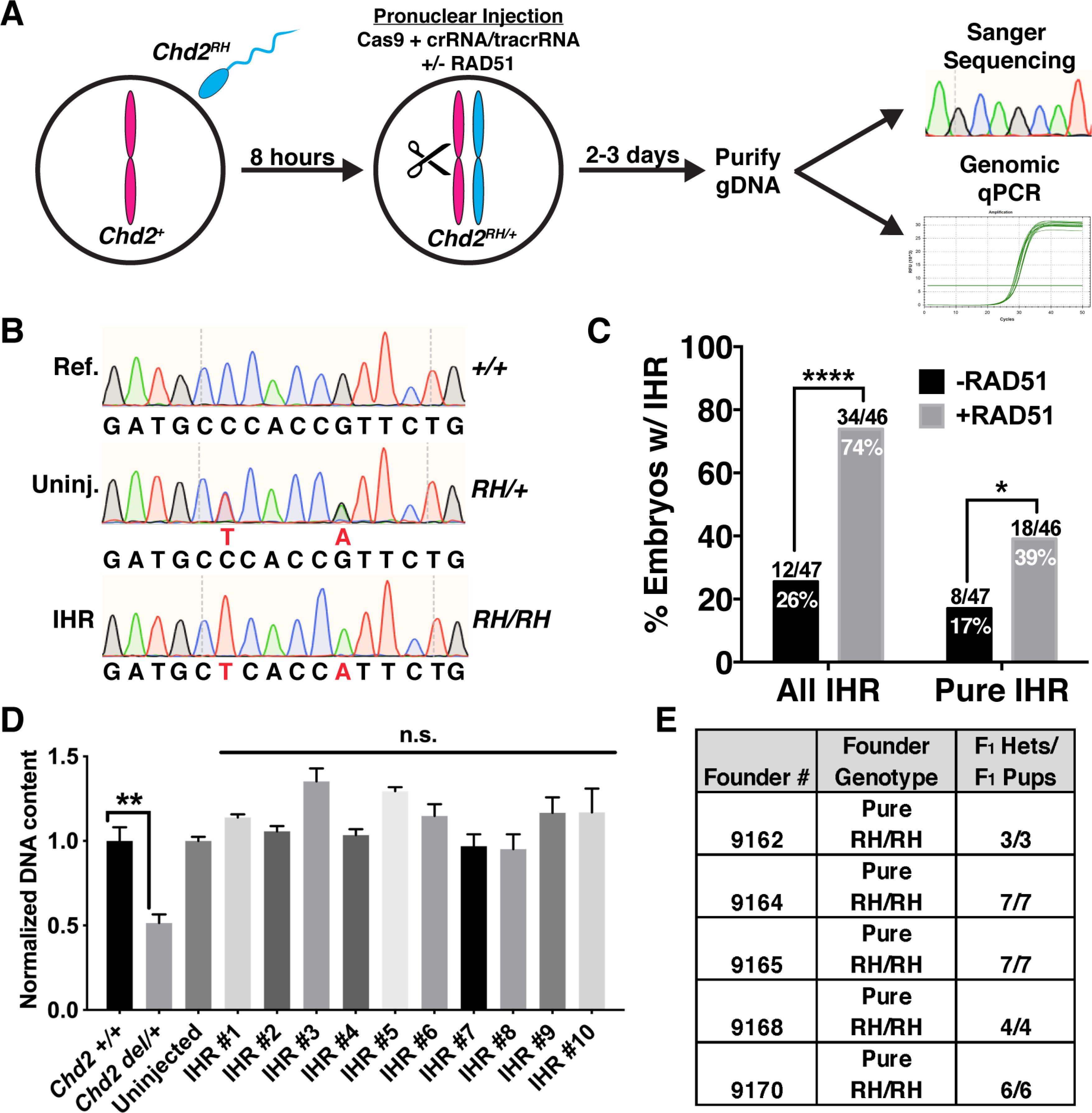
(A) Schematic of strategy for testing evaluating embryonic IHR. Wild-type oocytes were fertilized in vitro by sperm collected from Chd2RH/RH males and cultured for ~8 hours. PNI was then performed with Cas9 protein, tracrRNA, and crRNA specifically targeting the wild-type maternal allele. Injected embryos were then cultured for 2–3 days and collected at the morula or blastocyst stage. Half of the purified DNA was used for nested PCR and Sanger sequencing and the other half was used for multiplex PCR and subsequent qPCR to analyze genomic copy number at the Chd2 editing locus. (B) Representative chromatograms showing the wild-type reference sequence (Ref., top), an uninjected Chd2RH/+ embryo generated by IVF (Uninj., middle), and a pure Chd2RH/RH homozygous mutant (IHR, bottom) generated through IHR. (C) Quantification of Sanger sequencing results from IVF-derived embryos edited with or without RAD51. ‘All IHR’ includes mosaic embryos and was determined by identifying embryos with ≥3:2 ratio of R1684H allele to all other alleles (one-sided Fisher’s exact test). Pure IHR denotes embryos without mosaicism (p=0.018, one-tailed chi-square test). (D) Genomic qPCR targeting the Chd2RH locus in embryos carrying a large heterozygous deletion (Chd2del/+) as a control for copy-number sensitivity (lanes 1–2, unpaired t-test, n=3 embryos, error bars=SEM). Lanes 3–13 show genomic qPCR for the Chd2RH locus using DNA from an uninjected embryo and 10 randomly selected pure homozygous Chd2RH embryos (p>0.05, unpaired t-test, n=4 technical replicates per sample, error bars=SEM). (E) F1 genotyping results of litters derived from crosses between wild-type mice and 5 pure Chd2RH/RH F0 animals generated by the strategy described in (A), including RAD51. *p<0.05, **p<0.01, ****p<0.0001
To validate our Sanger sequencing-based genotyping strategy, we performed an independent set of injections with Cas9 and RAD51 using the same strategy described in Figure 3, then split the DNA from the lysed embryos into two aliquots and used one aliquot for nested PCR and Sanger sequencing and the second aliquot for an independent nested PCR reaction and NGS-based amplicon sequencing (see STAR Methods). Because this strategy uses unique sets of PCR primers for amplification of two pools of DNA from the same embryo, the likelihood of false-positives due to PCR bias or allele dropout is greatly decreased. After analyzing the Sanger sequencing traces, we chose a batch of embryos with different RH allele levels to analyze by amplicon sequencing. Comparison of the Sanger and amplicon sequencing results demonstrated a high correlation of the RH allele percentage derived from the two methods (r2=0.9672; Figure S2, Table S2), validating the accuracy of our Sanger-based analysis strategy and our findings of both endogenous and enhanced IHR rates at the Chd2 locus.
Comprehensive studies of indel properties in single sgRNA-based CRISPR studies have found indels large enough to disrupt our genotyping strategy (>250bp in either direction), but show that they are too infrequent to account for the observed rates of homozygosity in our experiments (Adikusuma et al., 2018; Kosicki et al., 2018; Shin et al., 2017). Nonetheless, we performed multiple experiments to directly rule out the possibility of widespread false positives resulting from large deletions on the Cas-targeted maternal allele. First, we developed and performed a multiplex genomic qPCR assay to analyze copy number at the target locus (see STAR Methods). To confirm that our method was capable of identifying large heterozygous deletions in Chd2, we generated a mouse line carrying a 1630bp deletion encompassing the RH locus (Figure S2) and performed genomic qPCR on DNA extracted from embryos generated by IVF with either a wild-type or homozygous mutant sperm donor. Analysis of normalized DNA content at the Chd2 locus in embryos heterozygous for the deletion confirmed our assay’s ability to identify heterozygous genomic deletions (Figure 3D). Using this qPCR strategy, we were able to confirm a normal copy number at the Chd2 locus in 10/10 randomly selected pure homozygous mutants (Figure 3D). Next, in an attempt to more definitively rule out large heterozygous deletions, we generated pups using the IVF strategy described in Figure 3A (including RAD51), identified potential pure homozygotes, and crossed them with wild-type mice to generate F1 offspring. Genotyping of F1 animals confirmed that 100% were heterozygous for the R1684H mutation (Figure 3E), proving that the F0 animals were true homozygotes generated by interhomolog repair. Thus, our results strongly support the conclusion that interhomolog repair occurs in the early mouse embryo and that RAD51 is capable of enhancing this process.
RAD51 Localizes to Cas9-targeted Regions in the Zygote
After fertilization, the maternal and paternal genomes remain physically separated in their own pronuclei until the completion of S-phase, at which time the nuclear envelopes break down in preparation for mitosis. This poses a significant hurdle to IHR in the zygote and has led researchers to question whether zygotic IHR is possible, given that it requires direct physical interaction between maternal and paternal homologs (Egli et al., 2018). Because alleles harboring an unresolved DSB cannot be amplified by PCR, accurate genotyping of one-cell embryos is not possible and thus, we chose to investigate the occurrence of IHR in zygotes via immunocytochemistry (ICC) for RAD51 in uninjected, RAD51-injected, Cas9-injected (with Chd2-targeting crRNA), and Cas9- and RAD51-injected zygotes at three stages of the cell cycle: G1/S, late S/G2, and M-phase. RAD51 staining is diffuse in the absence of DSBs, but strong RAD51 puncta that are indicative of nucleofilament formation appear in response to DNA damage, allowing direct visualization of RAD51-mediated repair events (Ladstätter and Tachibana-Konwalski, 2016). We did not see RAD51 puncta in uninjected embryos, but we observed a significant increase in RAD51 puncta solely during the G2 phase of the cell cycles in Cas9-injected embryos (Figure 4A,B). In agreement with our genotyping, co-injection of RAD51 with Cas9 resulted in the highest percentage of RAD51-positive embryos (Figure 4B). Additionally, we performed dual ICC/DNA-FISH with a RAD51 antibody and FISH probe specific to the Chd2 genomic region to visualize the localization of RAD51 puncta in Cas9-injected embryos. Although this method was not robust enough to perform quantitative experiments, we exclusively visualized RAD51 nucleofilament formation at the Chd2 genomic locus targeted by Cas9, suggesting a RAD51-dependent repair mechanism at Cas9-induced breaks in Chd2RH/+ embryos (Figure S3A).
Figure 4. RAD51 Localizes to Cas9-Induced DSBs During G2 in Zygotes.
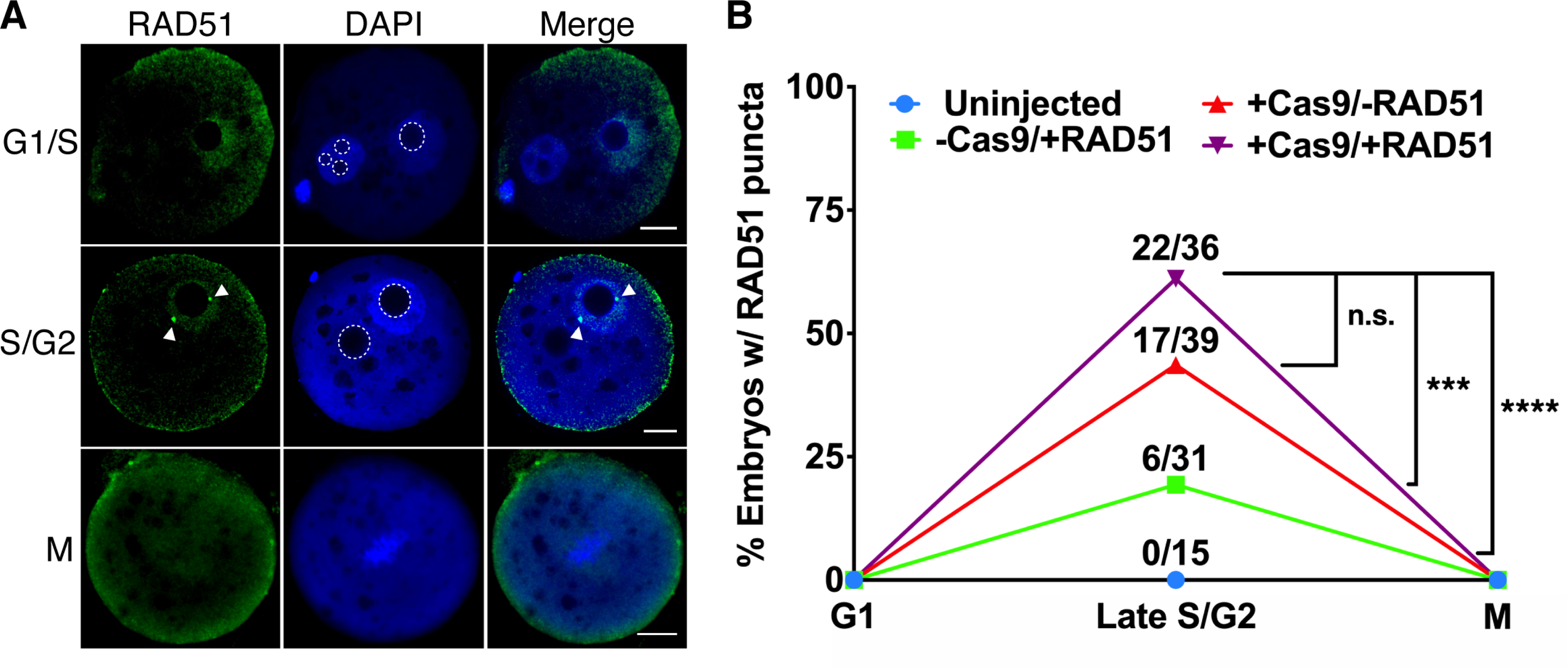
(A) Representative images of RAD51 immunocytochemistry in G1/S, S/G2, and M-phase zygotes injected with Cas9 and RAD51. Arrowheads indicate RAD51 puncta. Dotted lines highlight pronuclear nucleoli. (Scale bar=10μm) (B) Quantification of zygotes positive for RAD51 puncta (one-sided Fisher’s exact test). ***p<0.001, ****p<0.0001, n.s. p>0.05
Though we did not observe RAD51 puncta in any uninjected embryos, we were surprised to see RAD51 foci in several embryos injected solely with RAD51 (Figure 4B). Embryos injected with RAD51 alone only exhibited single RAD51 puncta, but Cas-injected embryos were often observed with 2 or more puncta (Figure S3B,C), indicating the co-occurrence of RAD51-mediated repair events at the Cas9 cut site(s) and either off-target DSBs or sites of endogenous DNA damage. To determine whether RAD51 could be localizing to off-target DNA damage induced by Cas9, we performed targeted analysis of 24 loci with ≤4 mismatches to the Chd2 crRNA. After pronuclear injection and whole-genome amplification of blastocyst DNA, we performed NGS-based amplicon sequencing of these loci in uninjected embryos, embryos only injected with Cas9, and embryos injected with Cas9 RNPs. Targeted NGS of the potential off-target found no evidence of off-target indel formation, demonstrating high specificity of the Chd2 crRNA and indicating that the increased number of RAD51 puncta in some embryos was not due to off-target genomic modification by Cas9 (Figure S3D). This was further confirmed using genome-wide off-target analysis in two-cell injected embryos (GOTI; Zuo et al., 2019, 2020), which also failed to identify mutations at potential off-target sites (Figure S3E, Table S2). These findings and the observation that RAD51 nucleofilaments also form in RAD51-injected embryos, but not uninjected embryos, suggest that both Cas9-induced DSBs, endogenous DSBs, and DSBs caused by injection-related cellular stress are repaired by RAD51-mediated mechanisms. Taken as a whole, our imaging and sequencing studies demonstrate that RAD51 can be recruited to Cas9-induced DSBs during late S/G2 and that RAD51 nucleofilament formation can be stimulated by addition of exogenous RAD51.
Enhancement of Interhomolog Repair Across Loci and Chromosomes
In support of our finding that IHR can occur in mouse zygotes during G2, a study that used light sheet microscopy to visualize the zygotic cell cycle in mice identified a short period of overlap between the maternal and paternal genomes during the late S/G2/M transition (Reichmann et al., 2018). However, only partial overlap is observed, suggesting that some chromosomal loci could be more permissible to IHR than others. Although we found that IHR is robust at both the Chd2 and Tyr loci (Figures 2 and 3), both are located on mouse chromosome 7. This led us to question whether IHR occurs and can be enhanced on other chromosomes. First, we designed an IHR assay for the X chromosome. Using sperm from hemizygous Mecp2 mutant mice (c.882_886>A), we performed IVF with wild-type oocytes and targeted the wild-type maternal allele with Cas9. After culture and DNA purification, we used 80% of the genomic DNA as input for nested PCR to genotype the Mecp2 locus and used the remaining 20% of genomic DNA for Sry PCR to determine sex. In female embryos injected only with Cas9, we observed a baseline IHR rate of 33% (16/49), but all 16 IHR-positive embryos were mosaic (Figure 5A). Co-injection of RAD51 increased the total IHR rate in females to 62% (24/39) and also increased the pure, non-mosaic IHR rate to 13% (5/39), demonstrating the ability of RAD51 to increase IHR rates on the X chromosome.
Figure 5. RAD51 Enhances Interhomolog Repair at Loci on Multiple Chromosomes.
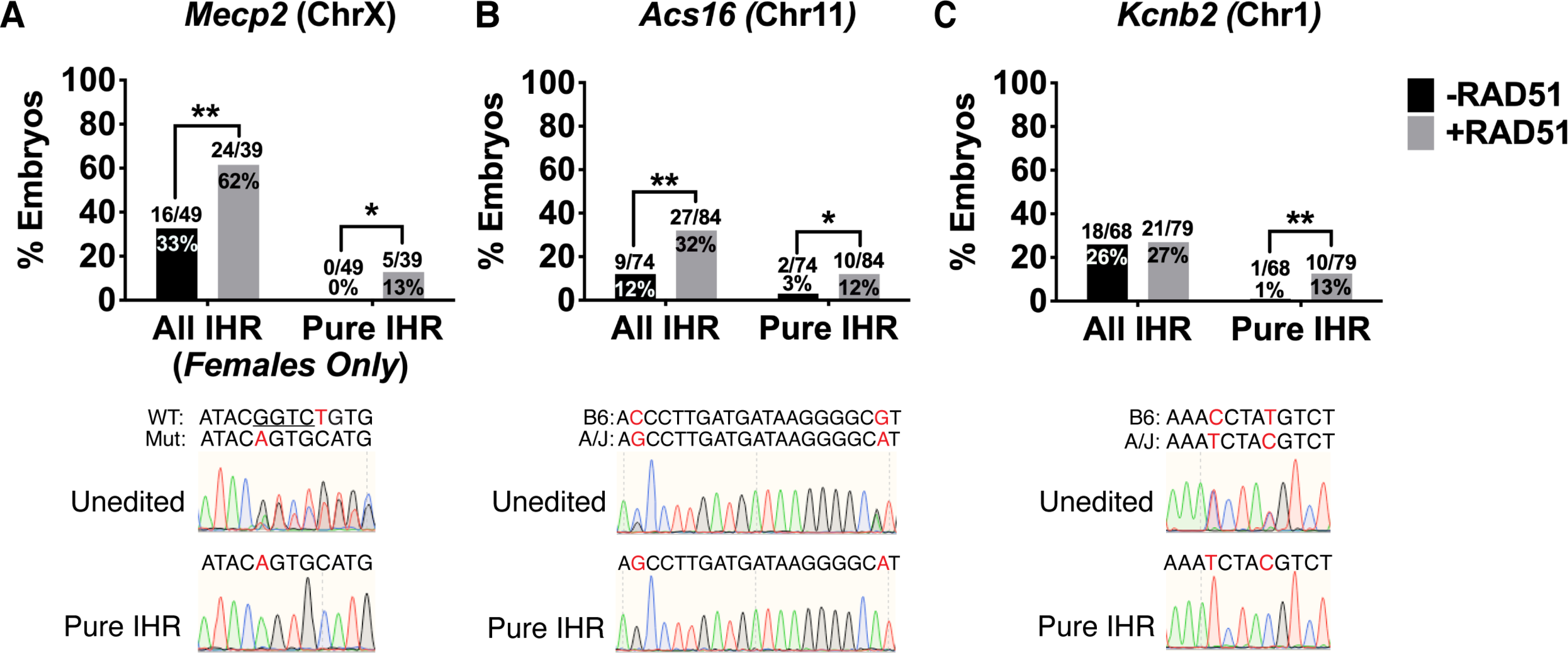
(A-C) Genotyping of embryos at the Mecp, Acs16, and Kcnb2 loci. Representative chromatograms of unedited and pure IHR embryos shown below graphs. Red letters denote SNVs and underlined bases represent deletions. All IHR denotes embryos with ≥3:2 ratio of the IHR allele to all other alleles. Pure IHR denotes non-mosaic IHR (one-sided Fisher’s exact test). *p<0.05, **p<0.01
To design additional IHR assays for different autosomes we used the MGI SNP database to find SNP-dense loci between C57BL/6J and A/J mice, confirmed the SNP genotypes by sequencing, designed allele-specific sgRNAs, and performed in vivo screening to identify sgRNAs with high efficiency and allele specificity (data not shown). We settled on two loci for analysis: an intronic region of Acs16 on chromosome 11 and an intronic region of Kcnb2 on chromosome 1. We then performed IVF with sperm from A/J males and oocytes from C57Bl/6J females, injected maternally-targeted Cas9 with or without RAD51, and genotyped blastocysts via Sanger sequencing. At the Acs16 locus we observed a low overall IHR rate (12%; 9/74) in embryos injected with Cas9 alone, and a pure IHR rate of only 3% (2/74; Figure 5B). However, co-injection of RAD51 increased these rates to 32% (27/84) and 12% (10/84), respectively (Figure 5B). At the Kcnb2 locus, the baseline IHR rate in Cas9-only embryos was 26% (18/68), but only 1 out of 68 embryos showed non-mosaic IHR (Figure 5C). Surprisingly, although RAD51 co-injection significantly increased the pure IHR rate to 13% (10/79), it had no effect on the overall IHR rate (27%, 21/79; Figure 5C), suggesting that the baseline IHR rate observed at this site is at its maximum. In total, our results at 4 sites on 3 autosomes and 1 site on the X chromosome demonstrate that endogenous IHR mechanisms are broadly active in the early embryo, but that the ability of RAD51 to enhance IHR is site-specific.
Identification of Additional IHR Enhancers
Previous studies have not given significant insight into the molecular pathways governing interhomolog repair, but the low IHR rates in cells that efficiently perform HR suggests distinct mechanisms (Stark and Jasin, 2003a). Thus, we sought to identify additional IHR-associated proteins by performing a series of injections in Chd2RH/+ zygotes with Cas9 and proteins associated with different DSB repair pathways: USP1/WDR48 (Fanconi anemia (FA) pathway regulator (Murai et al., 2011)), BCCIP (promotes BRCA2-mediated HR (Kelso et al., 2017; Lu et al., 2005; Wray et al., 2008)), participates in FA pathway (Moldovan and D’Andrea, 2009)), XRCC1 (promotes MMEJ and co-localizes with RAD51 (Dutta et al., 2017; Taylor et al., 2000)), and XRCC4 (promotes NHEJ and regulates V(D)J recombination (Critchlow et al., 1997; Normanno et al., 2017)). Supporting a role for canonical HR proteins in IHR, co-injection of USP1/WDR48 was capable of promoting IHR (Figure 6A) and BCCIP exhibited a striking effect on IHR, with 95% of edited embryos showing some degree of IHR (Figure 6A). Conversely, XRCC1 and XRCC4 were not capable stimulating IHR (Figure 6A). Together, these data indicate that IHR in the early embryo is driven by canonical HR pathways.
Figure 6. RAD51 G151D and BCCIP promote zygotic IHR and suppress indel formation.
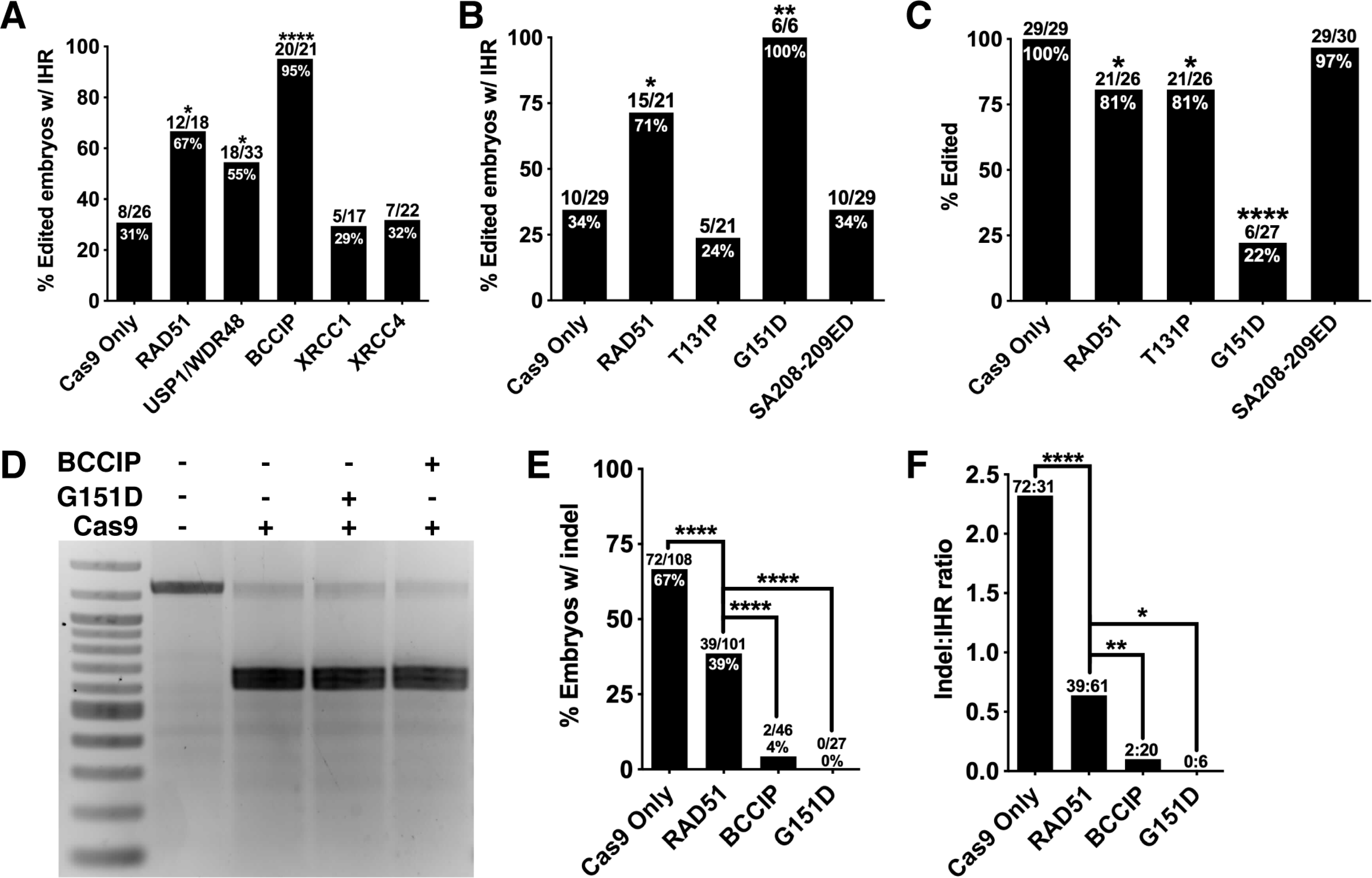
(A) Quantification of IHR efficiency in blastocysts derived Chd2RH/+ zygotes injected with Cas9/crRNA/tracrRNA and the indicated proteins. Quantification includes mosaic embryos (one-sided Fisher’s exact test). (B) Quantification of IHR efficiency in blastocysts derived Chd2RH/+ zygotes injected with Cas9/crRNA/tracrRNA and the indicated RAD51 variants (one-sided Fisher’s exact test). (C) Quantification of editing efficiency at the Chd2RH locus in embryos injected with Cas9/crRNA/tracrRNA and the indicated RAD51 variants (one-sided Fisher’s exact test). (D) In vitro Cas9 nuclease activity assay using a PCR amplimer of the Chd2RH locus and Cas9 alone or co-incubated with either RAD51 G151D or BCCIP. (E) Quantification of the percent of total injected embryos carrying an indel after injection with the specified proteins (one-sided Fisher’s exact test). (F) Quantification of the ratio of indel-positive embryos to IHR-positive embryos derived from injections with the specified proteins (one-sided Fisher’s exact test). *p<0.05, **p<0.01, ****p<0.0001
RAD51 mutations have been found in Fanconi anemia and multiple cancers and a number of these mutations have been studied in depth. To gain additional mechanistic insights into IHR, we co-injected three different RAD51 mutants with Cas9 and Chd2 crRNA into Chd2RH/+ embryos: T131P (lacks FA pathway-associated function, but not canonical HR (Wang et al., 2015; Zadorozhny et al., 2017)), SA208–209ED (disrupted BRCA2-association (Yu et al., 2003)), and G151D (gain-of-function mutant (Marsden et al., 2016)). T131P injection did not increase IHR frequency (Figure 6B), indicating that FA pathway-related function of RAD51 function is critical for its role in interhomolog repair. Similarly, SA208–209ED did not increase IHR rates (Figure 6B), reinforcing the importance of BRCA2-mediated RAD51 activation for IHR. With the G151D variant, we were surprised to find that in 27 zygotes injected we did not record a single indel event and observed some degree of interhomolog repair in all edited cells (Figure 6B). However, the apparent editing efficiency dropped to 22% (Figure 6C), suggesting that G151D either directly inhibits Cas9 nuclease activity or stimulates HR so robustly that the majority of DSBs are repaired through inter-sister HR. Co-incubation of Cas9 with G151D did not inhibit in vitro digestion of target (Figure 6D), supporting the hypothesis that G151D stimulates high levels of HR. We noted a similar decrease in apparent editing efficiency in BCCIP-injected embryos that was also not due to Cas9 inhibition (Figures 6D and S4), suggesting an inverse relationship between HR/IHR rates and indel rates in BCCIP- and G151D-injected embryos. Additional analyses of indel rates and indel-to-IHR ratios in embryos injected with RAD51, BCCIP, or RAD51 G151D revealed a drastic decrease in both measures compared to control embryos (Figure 6E and F). Furthermore, both BCCIP- and G151D-injected embryos showed significant decreases in these measures compared to embryos injected with wildtype RAD51. Together, our data argue that BCCIP and the RAD51 G151D mutant are highly efficient at stimulating both HR and IHR events at the expense of NHEJ.
Properties of Interhomolog Repair Events
Observations in germ cells and somatic cells have found that the properties of meiotic IHR events are distinct from those of somatic HR, particularly in terms of the recombination tract length. Specifically, meiotic non-crossover IHR events tend to have recombination tracts of less than 100bp (Li et al., 2019), while recombination tracts in somatic cells often span hundreds to thousands of bases, depending on context (Donoho et al., 1998; Ertl et al., 2017; Liskay and Stachelek, 1986; Mansai et al., 2011; Neuwirth et al., 2007; Rukść et al., 2008; Smith et al., 2007). However, a study of IHR events in somatic cells found that ~90% had recombination tracts of less than 300bp, though some spanned as far as 6kb (Stark and Jasin, 2003b). We found that IHR in embryos is far more efficient than in somatic cells (LaRocque et al., 2011; Stark and Jasin, 2003b; Yusa et al., 2004) and may therefore be similar mechanistically to the process observed in germ cells, particularly considering their close temporal proximity.
To gain a better understanding of IHR properties in the early embryo, we made a strategy to analyze recombination tract lengths at the Acs16 and Kcnb2 loci. First, we performed IVF with A/J sperm and C57BL/6J oocytes and injected resulting zygotes with RAD51 and Cas9 RNPs targeting either locus. Next, we transferred 2-cell embryos to pseudopregnant females and allowed them to develop in utero for 10 days before harvesting the embryos and collecting genomic DNA. This protocol was designed to identify embryos that had undergone pure IHR and provide sufficient amounts of gDNA for tiled SNP genotyping upstream and downstream of the cut sites, as well as long-range PCR and TOPO cloning for analysis SNP genotypes on individual alleles (Figure 7A). We identified 7 Acs16 and 5 Kcnb2 embryos that had undergone pure IHR (Figure 7B) and performed targeted PCR and sequencing of SNPs located between 3kb and 20kb upstream and downstream of the Cas9 target sites. This revealed heterozygosity in all Cas9-injected embryos and controls (Figure 7B), ruling out widespread loss of heterozygosity. Our analysis strategy for cloned alleles was limited by the presence of repeat elements and/or the presence of SNPs, but we were able to screen a 2.6kb region (2kb upstream + 600bp downstream of cut site) region at the Acs16 locus and a 3.1kb region (1.5kb upstream and downstream of cut site) at the Kcnb2 locus. In all embryos, PCR amplification of the target region failed to identify large deletions indicated by the presence of multiple PCR products (Figure S5). After TOPO cloning, we screened at least 16 clones from each embryo by Sanger sequencing all SNP-containing loci. At the Acs16 locus, 5/7 embryos were homozygous for the A/J allele at all SNPs in 100% of clones, suggesting recombination tract lengths >2kb upstream of the cut site and >304bp downstream of the cut site (Figure 7C). In 2/7 embryos, we identified alleles carrying the A/J SNPs at the cut site, but C57BL/6J SNPs 2kb upstream (Figure 7C). In one of these embryos, the switch back to the C57BL/6J genotype occurred between the cut site and the SNP 178bp upstream (Figures 7C and S5–6). In both of these embryos, the alleles with quantifiable recombination tract lengths represented a small fraction of the total alleles (2/22 in embryo #5 and 3/19 in embryo #7), indicating that these embryos may be mosaic for multiple IHR events. We observed a similar pattern at the Kcnb2 locus, with 3/5 embryos completely converted to A/J across the 3kb window and 2/5 carrying alleles converted to A/J at the cut site, but carrying C57BL/6J SNPs at sites ~1.5kb upstream and/or downstream (Figure 7C). Of these two embryos, one had an upstream conversion tract >1.5kb and a downstream conversion tract <1.5kb, while the second had an upstream conversion tract <1.5kb and a downstream conversion tract <213bp (Figures 6C and S5–6). At this site we observed evidence of mosaicism, with >2 alleles present in embryos #1 and #4, but we also observed >2 alleles in control embryos as a result of template switching during the initial PCR reaction. Importantly, this template switching does not alter our conclusions, as it can only be observed at sites that are heterozygous for a given SNP in the input pool of DNA. Thus, our results show that the conversion tracts for IHR in the early embryo are generally 1.5–3kb in both directions, but that the properties of individual IHR events can vary, with some conversion tracts as short as several hundred base pairs from the DSB. Together, these findings indicate that zygotic IHR utilizes canonical HR pathways and has features similar to that of somatic HR.
Figure 7. IHR Induces Conversion of Long Recombination Tracts.
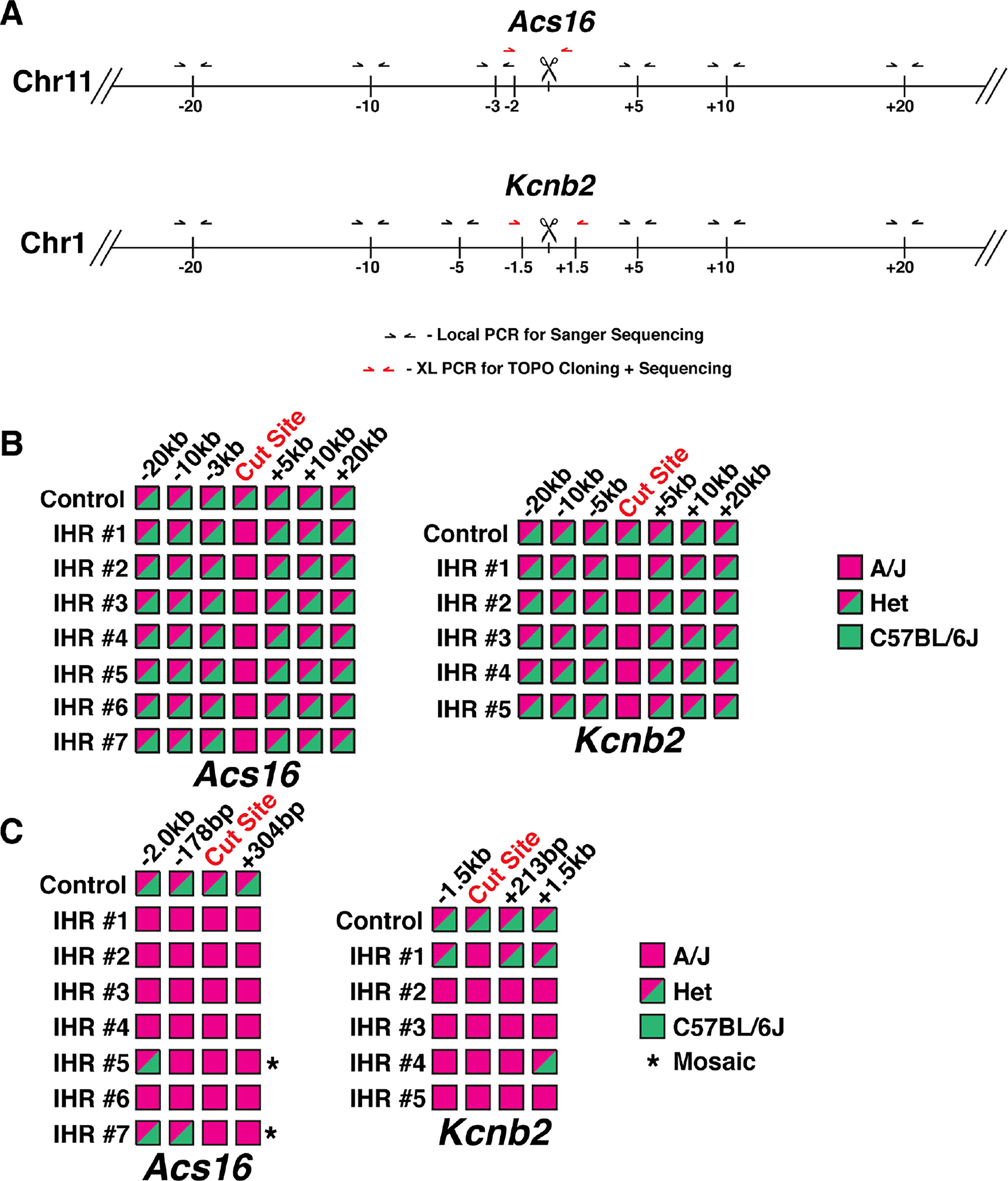
(A) Schematic of genotyping strategies for Acs16 and Kcnb2 loci. Black arrows denote locations of primers for local PCR of SNPs tiled across regions. Red arrows denote locations of primers for long-range PCR and TOPO cloning. Vertical lines denote approximate locations of SNPs. (B) Summary of genotyping results from local PCR of tiled SNPs. Pink boxes denote homozygous A/J alleles and mixed-color boxes denote heterozygous alleles with A/J and C57BL/6J SNPs. (C) Summary of genotyping results from TOPO cloning of long-range PCR products. Mixed-color boxes are used for any sites with at least one clone of the indicated allele. Asterisks denote embryos with low level of an allele, indicating mosaicism (see also Figure S5).
Discussion
Previously, Ma et al. reported efficient interhomolog repair in human zygotes, leading others to question how such a process is possible given the physical separation of homologous chromosomes into separate pronuclei (Egli et al., 2018). This near complete separation of the maternal and paternal genomes was elegantly described in a study from Reichmann et al. that used time-lapse light sheet microscopy to follow mitosis in mouse zygotes. However, their data clearly show that there is a window of approximately 45 minutes following nuclear envelope breakdown when there is consistent overlap between the maternal and paternal genomes (Reichmann et al., 2018). Furthermore, this overlap occurs in the absence of a targeted DSB and recent studies have found that DNA damage induces reorganization of the genome and relocation of the broken chromosome prior to repair (Caridi et al., 2018; Oshidari et al., 2018). Our ICC experiments found that RAD51 localizes to Cas9 cut sites at or slightly before the time of nuclear envelope breakdown (Figure 4), suggesting that IHR occurs during this window. However, Sanger sequencing, amplicon sequencing, and sequencing of TOPO-cloned alleles all uncovered evidence of mosaicism at rates consistent with IHR during the 2- and 4-cell stages (Figures S2, S5, Table S2), meaning that IHR is a common method of DSB repair in the early embryo. This is further supported by our observation that although embryos injected with both Cas9 and RAD51 display a higher frequency of RAD51 puncta than embryos injected with Cas9 alone, the difference is not nearly as significant as the difference observed by genotyping, which captures IHR events occurring any time before embryo collection. Altogether, our data suggest that the early embryo is uniquely permissive to IHR because of high baseline HR rates and frequent availability of the homolog as a template for repair.
Understanding whether there is a unique mechanism for IHR is critical for future efforts to induce and/or enhance IHR. We found that RAD51, the BRCA2-interacting protein BCCIP, and the Fanconi anemia pathway-associated protein complex of USP1/WDR48 are all capable of increasing the frequency of interhomolog repair in the early embryo (Figures 3, 5). RAD51 and BCCIP participate in multiple homologous recombination pathways (D’Andrea and Grompe, 2003; Lu et al., 2005; Miele et al., 2015) and although USP1/WDR48 is most well-known for its regulation of the Fanconi anemia pathway through deubiquitylation of FANCD2/I, it can also promote HR through FA-independent pathways (Cukras et al., 2016; Liang et al., 2016; Lu et al., 2005; van Twest et al., 2017; Yang et al., 2011). Based on this knowledge, IHR enhancement by RAD51, BCCIP, and USP1/WDR48 does not clearly implicate a specific pathway in the process. However, the observation that the RAD51 T131P mutant, which is specifically defective in the FA pathway-associated function of RAD51 (Wang et al., 2015; Zadorozhny et al., 2017) suggests that IHR in the early embryo could be mediated by an FA-associated pathway.
Though applications of IHR in the early embryo are limited in mice, this study provides the basis for future work to enhance IHR in somatic cells for therapeutic purposes. While recent years have seen a rapid expansion of clinical gene editing efforts, HDR-based strategies still suffer from low efficiency and carry the risk of unwanted integration of donor DNA throughout the genome. IHR has the potential to overcome these problems because it relies upon endogenous pathways and the patient’s own wild-type allele for correction of the deleterious mutation. We show that IHR can copy deletions, point mutations, and combinations of both between alleles, demonstrating its potential use for correcting a wide variety of genetic lesions. Additionally, we find that BCCIP and the RAD51 G151D mutant are factors that can both enhance IHR and reduce indel rates, which are of concern for many clinical gene therapy approaches. One concern regarding IHR and its clinical use is the induction of widespread loss of heterozygosity or genomic rearrangements. However, we identified single alleles in embryo #1 of the Kcnb2 group where the genotype was converted to A/J at the Cas9 cut site but remained C57BL/6J on both sides (Figures 7 and S5–6), indicating that IHR occurs through gene conversion without crossover. Importantly, we found that IHR recombination tract lengths are generally several hundred to several thousand base pairs long, but less than 5kb, suggesting that widespread loss of heterozygosity is not a major risk. Of note, however, recent studies have found that intrachromosomal recombination can occur between sites with highly homologous sequences, meaning that genome-wide homology must be considered before designing IHR-based strategies for gene conversion (Javidi-Parsijani et al., 2020; Liang et al., 2015). If methods to induce IHR in somatic cells can be developed, the recombination tract lengths associated with IHR enable the use of guides targeted to intronic regions adjacent to disease-causing mutations, decreasing the risk of unintended indels and offering more potential sites for the allele-specific targeting required to induce IHR. Looking forward, studies designed to probe the endogenous mechanisms of IHR, identify factors limiting its occurrence in somatic cells, and develop methods to safely and efficiently induce IHR in diverse cell types are necessary, but IHR represents a promising approach for applications in molecular biology, biotechnology, and gene therapy.
Limitations of the Study
Although we have definitive evidence that interhomolog repair occurs in mouse embryos and can be enhanced by multiple proteins, we cannot rule out that a small fraction of the IHR-positive embryos are false-positives resulting from large deletions. Additionally, we cannot differentiate between error-free NHEJ and RAD51-stimulated inter-sister HR and thus cannot definitively conclude that decreases in observed editing efficiency are due to solely to increased rates of inter-sister HR. Last, without additional experiments in both species, findings from our study cannot be directly compared to those in human embryos.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Guoping Feng (fengg@mit.edu).
Materials Availability
Chd2R1684H mice will be available from The Jackson Laboratory after initial characterization has been completed for a separate study.
Data and Code Availability
The amplicon sequencing included in this study has been submitted to the Sequence Read Archive and can be found under accession number PRJNA614534. Additional raw data for the figures in this manuscript can be obtained from the Lead Contact or corresponding authors upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All mouse work was performed with the supervision of the Massachusetts Institute for Technology Division of Comparative Medicine (DCM) under protocol 0416–024-19, which was approved by the Committee for Animal Care (CAC). All procedures were in accordance with the guidelines set forth by the Guide for Care and Use of Laboratory Animals, National Research Council, 1996 (institutional animal assurance no. A-3125–01). For all experiments except those targeting the Mecp2 locus, we did not perform sex genotyping and therefore used males and females. At the Mecp2 locus we genotyped all embryos, but only analyzed female embryos. Embryonic experiments analyzed morula and blastocyst-stage embryos. Analysis of IHR conversion tract length was performed using E10.5 embryos. Genotyping of F0 and F1 animals was performed using tissue from animals collected at ~P10.
METHOD DETAILS
Preparation of Injection Mixtures:
tracrRNA, crRNAs, and ssODNs were synthesized by Integrated DNA Technologies and modified sgRNAs were synthesized by Synthego (see Table S3 for sequences). All injection mixtures were prepared in a final volume of 50μL according to the following protocol. Using RNase-free water, reagents, and consumables, crRNA (final concentration 0.61μM), tracrRNA (final concentration 0.61μM), and ultrapure Tris-HCl, pH 7.39 (final concentration 10mM, ThermoFisher) were mixed and incubated at 95°C for 5 minutes. The mixtures were cooled to room temperature for 10 minutes on the benchtop and then EnGen Cas9 NLS, S. pyogenes (New England Biolabs) was added to a final concentration of 30ng/μL. The mixtures were incubated at 37°C for 15 minutes before adding any remaining components: ssODN (final concentration 30ng/μL), RAD51 (Creative Biomart, 134H, final concentration 10ng/μL), or additional recombinant proteins (described below, 10ng/μL). Injection mixtures were stored on ice and briefly heated to 37°C prior to injection. Injection mixtures utilizing sgRNAs were prepared at the same final concentration in the same buffer, but the 95°C incubation step was omitted. For experiments utilizing RS-1, embryos were cultured in KSOM-AA (EMD Millipore MR-121-D) with 7.5μM RS-1 (Sigma) dissolved in DMSO or DMSO (Sigma, 1:1000) for 24 hours, washed, and cultured in EmbryoMax FHM HEPES Buffered Medium (Sigma) until collection for genotyping.
Recombinant Proteins:
For experiments using wild-type recombinant proteins, the following proteins were used: RAD51 (Creative Biomart RAD51–134H), USP1/WDR48 (Creative Biomart USP1&WDR48–1067H), BCCIP (Origene TP303061), XRCC1 (TP304952), XRCC4 (Origene TP312684). For experiments using mutant RAD51, custom RAD51 preparations were produced by Creative Biomart according to their standard protocol for producing the wildtype RAD51 used in all other experiments. DNA sequences used for cloning the mutant forms of Rad51 are described in Table S3. All RAD51 mutants were modeled based on human RAD51 transcript variant 4 (NCBI accession NM_001164269), as this is the transcript variant of the wildtype RAD51–134H protein used in all other experiments. Identity and/or purity of all recombinant proteins used in this study are shown in Figure S7A–C (methods described below). Biochemical activity of recombinant RAD51 used in this study is shown in Figure S7D,E (methods described below).
Total Protein Staining and Western Blotting:
1μg of each recombinant protein was mixed with 2X Laemmli Sample Buffer (Bio-Rad) and heated to 95°C for 5 minutes. After brief centrifugation, samples were loaded into 4–15% Mini-PROTEAN TGX Pre-Cast gels (Bio-Rad) along with 5μL PrecisionPlus Protein Kaleidoscope Prestained Protein Standards (Bio-Rad) and run at 200V for 40 minutes in 1X Tris/Glycine/SDS running buffer. Protein was transferred to nitrocellulose at 100V for 1 hour at 4°C and total protein was visualized using REVERT Total Protein Stain (LI-COR Biosciences) according to the manufacturer’s protocol. For RAD51 Western blotting in Figure S7A, REVERT Total Protein Stain was reversed according to the manufacturer’s protocol, the membrane was blocked at room temperature for 1 hour in TBST containing 5% non-fat dry milk. Primary antibody (Rb anti-RAD51, Abcam ab63801, 1:500) was diluted in blocking buffer and applied to the membrane overnight at 4°C. Following washes with TBST, secondary antibody (IRDye 800CW Gt anti-rabbit IgG H+L, LI-COR 925–32211, 1:15,000) was diluted in blocking buffer and applied to the membrane for 1 hour at room temperature in the dark. The membrane was then washed and imaged. For all experiments, imaging was performed on an Odyssey CLx Imaging System with ImageStudio (LI-COR Biosciences). Coomassie staining and Western blots in Figure S7B were performed by Creative Biomart.
ssDNA Binding Assay:
To test the activity of the RAD51 variants utilized in this study, binding to ssDNA was assayed based upon a previously described protocol (Chen et al., 2015). 1μg recombinant RAD51 protein was mixed with 20μM φX174 ssDNA (NEB, N3023) in fresh 1X reaction buffer containing 25mM HEPES (pH7.5), 1mM MgCl2, 30mM NaCl, 1mM DTT, 0.4mM 2-mercaptoethanol, 0.03mM EDTA, 0.1mg/mL BSA, 2% glycerol, and 1mM ATP. The mixtures were incubated for 30 minutes at 37°C to form complexes. Following incubation, mixtures were loaded onto 0.8% TAE agarose gel and run at 25V for 16 hours. Gels were stained with 1X GelStar Nucleic Acid Gel Stain (Lonza, Cat. No. 50535) in TAE for 2 hours in the dark and then trans-illuminated with UV light for imaging. Wild-type RAD51 and all assayed mutants of RAD51 functioned as expected based on previously published data (Chen et al., 2015; Marsden et al., 2016; Wang et al., 2015; Yu et al., 2003).
Near-Infrared Strand Exchange Assay:
Recombinant RAD51 strand exchange activity was assessed using a modified version of a previously described strand exchange assay (Wang et al., 2015). The assay used three oligos, described in Table S3: an unlabeled 40-mer oligo, a complementary 40-mer oligo labeled on its 5’ end with IRDYE-800, and a 167-mer oligo containing the sequence of the unlabeled 40-mer (complementary to the labeled 40-mer). A 1:1 solution of the 40-mer oligos (40nM final concentration for each) was heated to 95°C for 5 minutes in a thermal cycler and allowed to cool 4°C at a ramp speed of 0.1°C/second. The strand exchange reaction mixture was assembled on ice, containing the unlabeled 167-mer (final concentration 4nM), RAD51 (final concentration 440nM), and final concentration 1X reaction buffer (25mM TrisOAc pH7.5, 1mM MgCl2, 2mM CaCl2, 0.1mg/mL BSA, 2mM ATP, and 1mM DTT). The reaction was incubated at 37°C for 10 minutes to allow RAD51 nucleofilament formation and then the annealed 40-mer oligos were added to a final concentration of 4nM. The reaction was incubated at 37°C for 30 minutes and then terminated via the addition of SDS (final concentration 0.25%) and proteinase K (final concentration 0.5μg/μL) and further incubation at 37°C for 10 minutes. Samples were then separated on a 5% TBE gel run in 1X TBE at 60V for 70 minutes and imaged on a LI-COR Odyssey CLx imaging system.
Natural Mating for Zygotic Injections:
Female mice (4–5 weeks old, C57BL/6NTac) were superovulated by IP injection of PMS (5 IU/mouse, three days prior to microinjection) and hCG (5 IU/mouse, 47 hours after PMS injection) and then paired with males. Pregnant females were sacrificed by cervical dislocation at day 0.5pcd and zygotes were collected into 0.1% hyaluronidase/FHM (Sigma). Zygotes were washed in drops of FHM and cumulus cells were removed. Zygotes were cultured in KSOM-AA for one hour and then used for microinjection.
In Vitro Fertilization for Zygotic Injections:
In vitro fertilization was performed using FERTIUP Mouse Preincubation Medium and CARD MEDIA (Kyudo Company) according to the manufacturer’s protocol. Non-virgin Chd2RH/RH, Mecp2del/Y, and A/J (The Jackson Laboratory, Stock No. 000646) males that were >8 weeks old were used as sperm donors. For experiments using Chd2 or Mecp2 sperm, oocytes from C57Bl/6NTac females (Taconic) were used. For experiments using A/J sperm, oocytes from C57Bl/6J females (The Jackson Laboratory, Stock No. 000664) were used. Following IVF, embryos were cultured for ~8 hours and then injected using the PNI protocol described below.
Zygotic Microinjections:
Except for Figure 1, all experiments described in this manuscript show data from a minimum of two days of injections. For all experiments except the CPI experiments described in Figure 1, the male pronucleus was injected. Injections performed for the CPI portion of Figure 1 were targeted to the cytoplasm. All microinjections were performed using a Narishige Micromanipulator, Nikon Eclipse TE2000-S microscope, and Eppendorf 5242 microinjector. Individual zygotes were injected with 1–2pL of injection mixture using an “automatic” injection mode set according to needle size and adjusted for clear increase in pronuclear volume. Following injections, cells were cultured in KSOM-AA until collection for genotyping. For experiments giving rise to F0 animals, embryos were surgically implanted into pseudopregnant CD-1 females (Charles River Laboratories, Strain Code 022) 24-hours post-injection and allowed to develop normally until natural birth.
Conditions for Embryo Collection, DNA Purification, and Genotyping:
To avoid contamination and ensure accuracy and consistency of genotyping results, embryo collection, DNA purification, and genotyping were all performed using special protocols and conditions. For both embryo collection and PCR, all plates, tubes, filter tips, strip caps, plate holders, ice buckets, and aliquots of nuclease-free water (not DEPC-treated) were UV-sterilized for >15 minutes in a laminar flow hood. All embryo manipulation and PCR was performed in a laminar flow hood. For PCR, all reagents (buffer, MgCl2, dNTPs, primers, polymerase) were aliquoted in the laminar flow hood upon receipt from the manufacturer and were solely used for embryo genotyping. No template controls were included with all PCR runs to rule out the presence of background DNA that could influence genotyping results.
Embryo Collection and DNA Purification:
Embryos were collected between the morula and blastocyst stage (3–4 days post injection) into 4μL nuclease-free water. After collection, 4μL of 2X embryo digestion buffer was added to each sample (final concentrations: 125μg/mL proteinase K, 100mM Tris-HCl pH 8.0, 100mM KCl, 0.02% gelatin, 0.45% Tween-20, 60μg/mL yeast tRNA) and embryos were lysed for 1 hour at 56°C. Proteinase K was inactivated via incubation at 95°C for 10 minutes and DNA was stored at −20°C until use.
Mouse Tail DNA Purification:
Tail tissue (~0.5cm length) was collected from individual animals, placed in 75μL alkaline lysis buffer (25mM NaOH, 0.2mM EDTA), and incubated at 95°C for 30 minutes. Digestion was stopped via addition of 75μL neutralization buffer (40mM Tris-HCl pH 5.0). Samples were stored at 4°C until genotyping.
Chd2RH PCR:
Chd2RH genotyping was performed via a two-step, nested approach. Except for experiments described in Figure 3, all 8μL of embryo DNA were used as input for the initial, long round of PCR. Genotyping of mouse pups and adult animals was performed using 2μL purified tail DNA. For all embryo genotyping, reaction mixtures were set up in a UV-sterilized laminar flow hood using sterile reagents and filter tips to avoid contamination. All PCR was performed using Biolase DNA Polymerase (Bioline), fresh aliquots of dNTPs (New England Biolabs), and 2% DMSO (final concentration). The first PCR reaction of the nested PCR was performed using the C2RH-Long_F/R primer pairs listed in Table S3. Primer pairs C2RH-Long_F1/R1 and C2RH-short_F1/R1 were used for Figures 1–3, as well as Supplementary Figures 1 and 2. All other experiments utilized primer pairs C2RH-Long_F2/R2 and C2RH-short_F2/R2, which was designed to amplify a larger genomic region as a way to further rule out large deletions that could disrupt PCR primer binding sites. 20 cycles of amplification were performed using 30 second extension and annealing times and an annealing temperature of 67°C. Nested PCR was then performed using 2μL of the initial 25μL PCR reaction as input. The C2RH-Short_F/R primer pairs listed in Table S3 were used for this PCR, with sets 1 and 2 being used as described above. Thirty-five cycles of amplification were performed using 30 second extension and annealing times and an annealing temperature of 68°C.
TyrC89S PCR:
Initial PCR was performed using 2μL purified tail DNA. The longer amplification of the nested PCR reaction was performed using the Tyr-Long_F/R primer pair listed in Table S3. PCR was performed using Biolase DNA polymerase (Bioline) and a final concentration of 2% DMSO. Twenty cycles of PCR were run with 30 second annealing and extension steps and an annealing temperature of 66°C. Nested PCR was performed using 2μL of the initial 25μL PCR reaction and the Tyr-Short_F/R primer pair listed in Table S3. The same PCR conditions were used for the nested PCR as were used for the initial PCR.
Sanger Sequencing:
For all sequencing reactions, 5μL of PCR product was mixed with 3μL dH2O and 2μL ExoSAP (Exonuclease I, NEB #M0293 and rSAP, NEB #M0371). Products were incubated at 37°C for 30 minutes to degrade primers and dephosphorylate dNTPs and enzymes were then heat inactivated at 80°C for 15 minutes. 5μL 5μM sequencing primer was then added to each mixture and samples were submitted to Genewiz for Sanger sequencing. Sequence files (.ab1) were blinded using a freely-available Perl script (https://github.com/jimsalterjrs/blindanalysis) and analyzed using SnapGene. To identify IHR events in mosaic samples, we compared the major and minor peak heights at the relevant SNPs using 4Peaks (Nucleobytes), analyzed traces with ICE (Synthego), or a combination of both. Sequences showing ≥3:2 ratio of major allele to minor allele at both sites were called IHR-positive.
On-Target Next-Generation Amplicon Sequencing:
Blastocyst DNA from uninjected embryos and embryos injected with Cas9, Chd2 crRNA, and tracrRNA was collected as described above. Half of the DNA was used for the previously described Sanger sequencing and analysis protocol. After analysis, 10 uninjected, 10 “pure IHR,” and 32 embryos with varying levels of Chd2RH allele were selected for subsequent next-generation sequencing. For these embryos, the second half of the genomic DNA was used as input for nested PCR (primers described in Table S3) using Biolase DNA Polymerase (Bioline). To ensure capture of all PCR products, PCR products were purified using the DNA Clean & Concentrator Kit (Zymo Research, D4033) according to the manufacturer’s instructions. Purified PCR products were then submitted for sequencing with Genewiz’s Amplicon-EZ service. Fastq files were then batch analyzed using the CRISPResso2 command line tool (Clement et al. 2019) in HDR mode using default settings and the Chd2RH allele sequence as the HDR template. Since the Chd2RH allele contains two point mutations in close proximity to the Cas9 cut site, certain indels can obscure those sites, making the program unable to definitively call an outcome and classifying those reads as ambiguous. Total indels were therefore characterized as the sum of NHEJ and ambiguous reads. PCR-induced mutations or errors during IHR can create “imperfect” knock-in alleles and thus, total Chd2RH allele frequency was calculated as the sum percentage of Chd2RH and imperfect Chd2RH reads. Sanger sequencing traces and estimated allele frequencies, as well as read counts and NGS-derived allele frequencies can be found in Table S2. All sequencing data was deposited in the NCBI Sequence Read Archive (SRA) under project accession PRJNA614534.
In vitro Cas9 Digestion Assays:
Wild-type or Chd2RH/RH genomic DNA was used as input for PCR using the conditions described above and the primers specified in Table S3. After confirmation of a single band via gel electrophoresis, the PCR reactions were purified using the DNA Clean & Concentrator kit (Zymo) according to the manufacturer’s instructions. crRNA and tracrRNA were diluted 1μM in Nuclease-free Duplex Buffer (IDT) and incubated at 95°C for 5 minutes before being allowed to cool to room temperature on the bench. Digestion mixtures were made in separate tubes by combining nuclease-free water, 10X Cas9 Nuclease Reaction Buffer (final concentration 1X, New England Biolabs), 1μL of the cooled crRNA/tracrRNA duplex, and, if necessary, 1μL of EnGen Cas9 NLS, S. pyogenes (New England Biolabs). These mixtures were heated to 37°C for 10 minutes before adding 250ng PCR product and, if necessary, RAD51 G151D (10ng/μL final concentration, custom-produced by Creative Biomart) or BCCIP (10ng/μL final concentration, Origene TP303061). Digestion was performed at 37°C for 1 hour, Cas9 was denatured at 95°C, 6X Purple Gel Loading Dye (New England Biolabs) was added to a final concentration of 1X, and the samples were allowed to passively cool to room temperature. After cooling, reactions were separated by electrophoresis in 2% agarose gel, post-stained with GelRed Nucleic Acid Gel Stain (Biotium) according to manufacturer’s instructions and visualized using an InGenius Gel Documentation System (Syngene).
Genomic Copy-Number qPCR:
To determine copy number at specific genomic loci, we developed a strategy utilizing multiplex nested qPCR. We first performed multiplex amplification of both the edited Chd2 region (Chr7) and a region of the Gapdh promoter (Chr6) that we have previously targeted for ChIP experiments (Wilde et al., 2017) in a short round of PCR (10 cycles). We then used this PCR product as input for two separate nested qPCR reactions, one targeting the Gapdh control locus (2N) and one targeting the Chd2 locus (unknown copy number). Chd2 signal was then normalized to the Gapdh signal to determine copy number at the Chd2 locus. As a 2N control, we used DNA from uninjected embryos. As a 1N control we generated a mouse line harboring a large deletion at the Chd2 locus (see below) and used heterozygous embryos for qPCR. To test for large deletions in Cas9-injected embryos presumably having undergone IHR, we isolated genomic DNA from Cas9/Chd2 crRNA/tracrRNA-injected blastocysts, used 50% for genotyping as described above, and then randomly-selected 10 “pure IHR” blastocysts for genomic qPCR. Initial nested PCR was performed using Biolase DNA Polymerase (Bioline). qPCR was performed using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad). Primers are listed in Table S3.
Generation of the Chd2del Mouse Line:
Zygotes were prepared for injection via natural mating and pronuclear injections were performed as described above. Embryos were cultured overnight in KSOM-AA and embryos that failed to undergo cleavage were discarded. Two-cell embryos were surgically implanted into pseudopregnant CD-1 females (Charles River Laboratories, Strain Code 022) and allowed to develop until natural birth. Pups were genotyped at postnatal day 21 using the primers described in Table S3.
Immunocytochemistry:
First, embryos were blinded before staining and analysis. Zygotes were collected during G1 (~9hpf, ~1hpi, PN2), G2 (~14.5hpf, ~6.5hpi, PN5), or M-phase (~16hpf, ~8hpi) and fixed overnight at 4°C in 4% PFA + 0.1% Tween-20 in PBS and then briefly transferred to acidified Tyrode’s (Sigma T1788) to remove the zona. Collection times and cell cycle stages were determined empirically based on morphology (Santos et al., 2013). Specifically, G2 embryos were fixed when maternal and paternal pronuclei had overlapped and nuclear envelopes were undergoing clear breakdown (at least one no longer visible). M-phase embryos were selected ~1 hour after complete breakdown of the nuclear envelopes. After washing in PBS + 0.1% Tween-20 (PBST), zygotes were permeabilized in PBS + 1% Triton X-100 for 1 hour at 4°C and then blocked for 1 hour at room temperature in blocking solution (PBS + 3% BSA + 5% normal goat serum). Primary antibody (rabbit anti-RAD51, Abcam ab63801, 1:500) was diluted in blocking solution and applied to coverslips overnight at 4°C. Cells were washed 3 times in PBST before application of secondary antibody (goat anti-rabbit IgG conjugate, Alexa Fluor 488, ThermoFisher A-11008) diluted in blocking solution for 1 hour at room temperature. Nuclei were counterstained with DAPI and coverslips were mounted to slides with Fluoromount Aqueous Mounting Medium (Sigma F4680). Cells were imaged on an Olympus Fluoview FV1000 confocal microscope with a 60X oil immersion objective and variable digital zoom. For all cells, we acquired z-stacks incorporating both pronuclei. Raw image files were exported to FIJI (Schindelin et al., 2012) for Z-projection (sum slices), channel splitting, and scale bar generation.
Off-Target Next-Generation Amplicon Sequencing:
Off-target analysis by NGS was performed as previously described (Li et al., 2020). Briefly, pre-genotyped blastocyst stage embryos were subjected to whole genome amplification using REPLI-g Single Cell kit (Qiagen) according to manufacturer’s instructions. Equal amounts of amplified DNA were mixed and 3 pools were prepared: an uninjected pool from 8 embryos, a Cas9-only pool from 12 embryos injected with Cas9 protein but no crRNA/tracrRNA, and a pool from 19 embryos injected with Cas9 protein, Chd2 crRNA, and tracrRNA. Twenty-six off-target candidate sites for crChd2 containing up to 4 mismatches were predicted and primer pairs were designed using CRISPOR (Concordet and Haeussler, 2018). All sites were PCR amplified from each pool using Herculase II Fusion DNA Polymerases (Agilent) and gel purified using Zymoclean Gel DNA Recovery kit (Zymo Research). Equimolar amounts of the twenty-six PCR amplicons of each pool were mixed and subjected to library construction for Illumina sequencing with reagents from TruSeq Stranded mRNA Sample Preparation kit (Illumina) according to the manufacturer’s instructions. Barcoded libraries were mixed and sequenced on a MiSeq (Illumina) with 150bp paired-end reads and a 6-base Index 1 read to an averaged depth of 200,000 reads per locus per pool. Sequencing was analyzed in three batches using CRISPResso2 (Clement et al. 2019). For each batch, we ran CRISPResso’s CRISPRessoPooled tool. Since the amplicon sites that we considered for analysis were relatively short (minimum of 66 bp, maximum 145 bp, median 126 bp), CRISPResso failed to analyze some amplicons due to the low number of reads that aligned properly on the amplicon. Furthermore, the poor mapping to the amplicons caused some artifactual deletions in the reads. Therefore, we extended all amplicon sequences to 250 bases. We also trimmed the reads 5 bases from the start and 55 bases from the end, and decreased the minimum alignment score from the default of 60 to 10, in order to allow more mapping of reads (allowing more reads in the analysis negatively biases our results). Even with these modifications, 5 amplicons still failed the CRISPResso analysis. We discarded two amplicon regions since they are on L1 repeat regions (OT21 and OT22), and we performed another round of deep sequencing on 3 amplicons (OT15, OT18, and OT20), this time using a longer region (303, 311 and 332bp). These amplicons were sequenced using the Amplicon-EZ service from Genewiz and we re-analyzed them using the web-based CRISPResso2 tool (http://crispresso.pinellolab.partners.org). All sequencing data was deposited in the NCBI Sequence Read Archive (SRA) under project accession PRJNA614534.
Genome-wide Off-Target Analysis by Two-Cell Embryo Injection (GOTI):
GOTI was performed as previously described (Zuo et al., 2019, 2020). Briefly, IVF was performed as described above using homozygous Ai14 (The Jackson Laboratory, Stock No. 007908) males and C57Bl/6NTac females. A mixture of Cas9 protein (final concentration 30 ng/μl) and Cre mRNA (final concentration 2 ng/μl), Cas9 protein (final concentration 30 ng/μl), Chd2 crRNA (final concentration 0.61 μM), tracrRNA (final concentration 0.61 μM) and Cre mRNA (final concentration 2 ng/μl), or Cas9 protein (final concentration 30 ng/μl), Chd2 crRNA (final concentration 0.61 μM), tracrRNA (final concentration 0.61 μM), RAD51 (Abcam ab63808, final concentration 10 ng/μL) and Cre mRNA (final concentration 2 ng/μl) was injected into both the nucleus and cytoplasm of one blastomere of 2-cell stage embryos. The injected embryos were transferred into pseudopregnant CD-1 females as described above.
Mosaic fetuses were harvested at E14.5, minced, and enzymatically dissociated in 0.05% 5 mL Trypsin-EDTA at 37°C for 30 minutes. Digestion was then stopped by the addition of 5mL DMEM/10% FBS. Digested tissues were further homogenized by pipetting 30–40 times using 1ml pipette tips and were then spun down and pellets were resuspended in DMEM/10% FBS and filtered through a 40μm cell strainer. The tdTomato-positive/tdTomato-negative cells were then sorted by FACS. The purity of sorted cells was confirmed to be >95% by fluorescence microscopy and PCR cloning analyses.
Genomic DNA was extracted from sorted cells using the NucleoSpin Tissue kit (Macherey-Nagel). The libraries for whole genome sequencing (WGS) were prepared using the Illumina DNA PCR-Free Prep kit (Illumina) with barcodes, pooled, and run on a NovaSeq 6000 (Illumina) to generate mean coverages from 30X to 83X.
The off-target indels and SNVs were identified by comparing the tdTomato-positive cells with tdTomato-negative cells from the same embryo using three variant calling algorithms, as detailed in the GOTI method (Zuo et al., 2019, 2020). In detail, BWA (v0.7.12) was used to map quality-filtered sequencing reads to the reference genome (mm10). Picard tools (v2.3.0) was then applied to sort and mark duplicates of the mapped BAM files. To identify the genome-wide de novo single nucleotide variants with high confidence, we separately ran three algorithms that detect single nucleotide variants present in the tdTomato-positive cells but not present in tdTomato-negative cells: Mutect2 v3.5 (Benjamin et al., 2019), Lofreq v2.1.2 (Wilm et al., 2012)and Strelka v2.7.1 (Kim et al., 2018). Only SNVs called by all three SNV-detection algorithms were considered to be true variants. To detect indels present in tdTomato-positive cells but not in tdTomato-negative cells, we used Mutect2 v3.5, Strelka v2.7.1, and Scalpel v0.5.3 (Fang et al., 2016). Once again, only indels called by all three algorithms were considered to be true de novo indels. As previously done (Zuo et al., 2019, 2020), we annotated the SNVs and indels using ANNOVAR v2020–06-08 (Wang et al., 2010) and performed additional filtering on the variants by choosing variants with allele frequencies at least 10%. The off-target SNVs and indels were compared to the Chd2 guide RNA sequence by blasting the genomic sequence of the SNV/indel 17 bases upstream and 5 bases downstream with the 23-base Chd2 guide RNA sequence. We also obtained the sequence in the reverse strand, 5 bases upstream and 17 bases downstream of the SNV/indel and compared using blast with the Chd2 guide RNA sequence. To further analyze the off-target SNVs and indels, we overlapped them with predicted off target sites using two algorithms: Cas-OFFinder v2.4 (Bae et al., 2014) and CRISPOR v4.98 (Concordet and Haeussler, 2018). For CRISPOR, we utilized the web-based tool with default settings. We ran Cas-OFFinder twice, first with parameter 4 mismatches (MM) and second with parameter 9 MM. We then looked for overlap between the genomic regions 18 bases upstream and 18 bases downstream of each SNV/indel and the off-target sites predicted by CRISPOR and Cas-OFFinder. We did not observe any indels overlapping with putative off-target sites, but for SNVs overlapping with potential off-target sites we ruled them out as true off-targets based on the following observations: 1) one overlapping SNV was found in the Cas9-only condition (8 mismatches), 2) all SNVs founds in the Cas9 + crRNA-injected embryos contained at least 9 mismatches with the Chd2 crRNA, 3) 2 of the 3 SNVs in Cas9 + crRNA-injected embryos were adjected to non-canonical NAG PAM sequences, 4) the third SNV in a Cas9 + crRNA-injected embryo had 3 mismatches at the positions immediately adjacent to the PAM. All sequencing data was deposited in the NCBI Sequence Read Archive (SRA) under project accession PRJNA614534.
Dual DNA-FISH/Immunocytochemistry:
Late S/G2 stage zygotes were used for immunocytochemistry as described above. Zygotes were stained with primary antibody (Rb anti-RAD51, Abcam, ab63801, 1:100) followed by tyramide signal amplification (Biotin XX Tyramide SuperBoost Kit, goat anti-rabbit IgG (ThermoFisher, B40921) and streptavidin, Alexa Fluor 488 Conjugate (ThermoFisher, S32354)) according to manufacturer’s instructions. Zygotes were fixed again with 4% PFA, and processed for FISH as previously described (Nakaya et al., 2017). Zygotes were mounted in EASI-FISH chambers (poly-L-lysine hydrobromide (Sigma P1399)-coated coverslip (VWR 48393–106) mounted with double-layered clear patches (KOKUYO TA-3N) and fixed with 4% PFA. EASI-FISH chambers were treated with 0.5% saponin/0.5% TritonX-100 in PBS (PBST) for 30 minutes to permeabilize cells, incubated in 20% glycerol at room temperature in PBS for 30 minutes, and transferred to 50% glycerol in PBS before storing at −20°C overnight. The chambers were then frozen in liquid nitrogen for 30 seconds followed by freezing-thawing in 20% glycerol five times. They were then washed in PBS, fixed with 4% PFA, incubated in 0.1N HCl for 10 minutes, washed with PBS, fixed with 4% PFA, incubated with PBST for 1 hour, fixed with 4% PFA, washed in 2x SSC, and incubated in 2X SSC/50% formamide until hybridization. FISH probe was prepared as previously described (Inoue et al., 2017, 2018). Briefly, Cy3-labled FISH probe was prepared from a BAC clone containing the Chd2 locus (BACPAC, clone RP24–290C18) using the Nick Translation Reagent Kit (Abbott Molecular 07J00–001) with Cy3-dCTP (GE Healthcare PA53021) according to manufacturer’s instructions. The Cy3-labeled probe was then ethanol-precipitated with 10μg of Mouse Cot-1 DNA (ThermoFisher 18440–016) using GlycoBlue (ThermoFisher AM9515), and dissolved in 50% formamide/10% dextran sulfate/2X SSC. The probe mixture was heat denatured at 80.5°C for 4 minutes, cooled on ice, and applied to the EASI-FISH chamber. The EASI-FISH chamber was then heat denatured at 80.5°C for 4 minutes and then incubated at 37°C for at least for 5 days. After hybridization, the EASI-FISH chamber was washed once in 2X SSC, followed by three times in 0.1X SSC at 62.5°C for 5 minutes once in 4X SSC with 0.2% Tween20 (SSCT). The chamber was then subjected to immunocytochemistry a second time as described above, mounted with Fluoromount-G with DAPI (ThermoFisher 00–4959-52), and imaged and analyzed as described above.
Embryonic Tissue Collection and DNA Purification:
After pronuclear injection, zygotes were cultured overnight and two-cell embryos were transferred to pseudopregnant CD-1 females as described above. Embryos were allowed to develop in utero for 9 days before pregnant females were sacrificed. Embryos were microdissected into cold, sterile PBS and washed twice before transfer to UV-sterilized 1.5mL snap-cap tubes. Embryo DNA was then purified using the NucleoSpin Tissue Genomic DNA Purification Kit (Takara, Cat. No. 740952) according to manufacturer’s instructions. Genomic DNA was eluted with sterile TE buffer and split into aliquots of either 10ng/μL or 100ng/μL for use in downstream applications and stored at −20°C.
TOPO Cloning and Screening of Individual Alleles:
Primers for TOPO cloning and sequencing of clones are described in Table S3. For TOPO cloning, 100ng of genomic DNA per sample was used as input for PCR using Phusion High-Fidelity DNA Polymerase (New England Biolabs, M0350) using the standard buffer according to the manufacturer’s instructions. Samples were then run on a 1.5% agarose gel and, after confirming the presence of a single band, the target band was gel-purified using the ZymoClean Gel DNA Recovery Kit (Zymo Research, D4007). Products were eluted with 8μL of elution buffer and 4μL of the eluate was used as input for TOPO cloning with the Zero Blunt TOPO PCR Cloning Kit (ThermoFisher, K280020) according to the manufacturer’s instructions. After transformation and plating, colonies were screened by colony PCR using the M13F/R primer pair. Colonies positive for the insert were then grown overnight and plasmids were purified using the ZymoPure Plasmid Miniprep Kit (Zymo Research, D4208) according to the manufacturer’s instructions. Plasmids were then sequenced and sequences were aligned and analyzed using SnapGene (GSL Biotech).
QUANTIFICATION AND STATISTICAL ANALYSIS
For all experiments testing homozygosity rates and rates of interhomolog repair a one-sided Fisher’s exact test was used. Because we found in Figure 1 that RAD51 appeared to increase homozygosity and thus interhomolog repair, we explicitly tested this hypothesis and this was our justification for using a one-sided analysis.
For the qPCR experiments described in Figure 3D a two-sided Student’s t-test was utilized.
For the correlation analysis in Figure S3B, we performed a simple linear regression and calculated Pearson’s correlation coefficient.
For the calculation of mosaicism rates in Figure S4C, we utilized as a two-sided Fisher’s exact test, as we did not have a predetermined expectation of the effects of the given enhancers on mosaicism.
All statistical analyses were performed using Prism 8 (GraphPad). All graphs display mean±SEM. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001
Supplementary Material
Table S1. Genotyping Results for Individual F0 Pups from CPI and PNI Experiments, Related to Figures 1 and 2.
Table S2. Sanger Sequencing, Amplicon Sequencing, and GOTI Results, Related to Figures 3, 4, S2, and S3. Table shows approximated RH% based on Sanger chromatograms and results of CRISPRESSO2 analysis of NGS results, including read counts and percentages. It also includes tabs for the sequencing statistics, SNV analysis, and indel analysis for GOTI performed in Figure S3E.
Table S3. Sequences of RNAs, Oligonucleotides, and Proteins Used in this Study, Related to STAR Methods.
Figure S1. RAD51 stimulates the generation of homozygous indels, Related to Figure 1. A, Representative Sanger sequencing traces of putative homozygous indels observed in F0 animals obtained from Chd2RH knock-in experiments. B, Representative Sanger sequencing traces of putative homozygous indels observed in F0 animals obtained from TyrC89S knock-in experiments. c, Genotyping traces for a putative homozygous Chd2 indel and an F1 offspring from a cross between the founder and a wildtype C57BL/6N mate. The F1 genotyping, which was consistent for all 7 pups in the litter, indicates heterozygous presence of the indel observed in the founder.
Figure S2. Chd2RH crRNA Allele Specificity, Concordance Between Sanger Sequencing and Amplicon Sequencing, and Production of the Chddel Allele, Related to Figure 3. (A) PCR was performed using the Chd2-RH_F/R primer pair with DNA purified from either wildtype or Chd2RH/RH mice and the PCR product was used as a template for in vitro digestion assays with or without Cas9. (B) Comparisons of individual RH allele percentages between Sanger sequencing estimates and amplimer sequencing in uninjected, pure IHR, and mosaic embryos. (C) Correlation plot of all calculated RH allele percentages from Sanger sequencing and amplimer sequencing. (D) Schematic of Chd2del locus on chromosome 7. A combination of 4 sgRNAs, 2 on each side of exon 38, were co-injected to generate a large deletion for use as a qPCR control for Figure 3D. (E) Close-up of sequencing at the deletion junctions in the Chd2del founder animal showing a large deletion originating from Chd2-Int37-g1 and Chd2-Int38-g1.
Figure S3. Cas9 injection alters the number of RAD51 puncta in G2, Related to Figure 4. (A) Representative image of immunocytochemistry for RAD51 paired with DNA-FISH for the Chd2 genomic locus showing RAD51 nucleofilament formation specifically at the Cas9 cut site. (B) Quantification of RAD51 puncta number observed in control G2 zygotes and cells injected with RAD51 alone, Cas9/crRNA/tracrRNA alone, or Cas9/crRNA/tracrRNA and RAD51. Each point represents a single cell. (C) Representative images of cells with one (top left), two (top right), three (bottom left), or four (bottom right) RAD51 puncta (arrowheads indicate puncta; DAPI, blue; RAD51, green; scale=10μm). (D) Summary of amplimer sequencing results showing observed indel percentages at genomic loci with 4 mismatches to the Chd2RH crRNA. Lower-case letters in putative off-target sites denote mismatches with the crRNA sequence. (E) Summary of GOTI data. For read depth, TdTomato- cells are in white and TdTomato+ cells are in red. ‘SNVs Found’ describes the number of SNVs overlapping from analysis with Mutect2, Strelka, and Lofreq. ‘Indels Found’ describes the number indels overlapping from analysis with Mutect2, Strelka, and Scalpel. Complete GOTI analysis data can be found in Table S2.
Figure S4. Effects of IHR enhancers on editing efficiency and mosaicism, Related to Figure 6. (A) Editing efficiency observed in IVF-derived embryos injected with Cas9/crRNA/tracrRNA only (no enhancer) or the indicated HR- and DSB repair-associated proteins (one-side Fisher’s exact test). (B) Representative chromatograms showing examples traces for genotypes characterized as unedited (top left), 100% IHR (top right), mosaic with wildtype (bottom left), and mosaic with indel (bottom right). (C) Quantification of mosaicism in embryos displaying IHR for all conditions analyzed. Data includes all IVF-derived embryos (Figures 3C, 6A, and 6B; two-sided Fisher’s exact test). (D) Distribution of types of mosaicism observed in mosaic embryos described in panel b (two-sided Fisher’s exact test). (E) Numbers for observed mosaicism used to generate panel C.
Figure S5. Summary of SNP Genotyping of TOPO-Cloned Alleles in Acs16 and Kcnb2 Embryos, Related to Figure 7. Gels show PCR products used as input for TOPO cloning. Each box represents the genotype at a SNP-containing region on the cloned allele. Magenta boxes are positive for the A/J SNP and green boxes are positive for the C57BL/6J SNP.
Figure S6. Examples of Recombination Tracts Identified by TOPO Cloning of Individual Alleles, Related to Figure 7. (A) Top track shows the Acs16 locus spanning from the sgRNA target site on the right to the last A/J-specific sequence on the left (a 4bp insertion). The top chromatogram is from a pure A/J allele and the bottom is from an allele that is A/J at the Cas9 cut site (red arrowhead) and C57BL/6J 178bp upstream at the site of the 4bp insertion. (B) Top track shows the Kcnb2 locus spanning from the sgRNA target site on the left to the last A/J-specific SNP on the right. The top chromatogram is from a pure A/J allele and the bottom is from an allele that is A/J at the Cas9 cut site (red arrowhead) and C57BL/6J 211bp downstream at the site of the SNP. (C) Top track shows the Kcnb2 sgRNA target site in the middle, with two distal SNPs located ~±1.5kb from the cut site on the left and right sides. The top chromatograms are from a pure A/J clone and the bottom chromatograms are from a single allele that carries the A/J sequences at the cut site and upstream locus, but the C57BL/6J SNP at the 1.5kb downstream locus.
Figure S7. Verification of Recombinant Protein Identity, Purity, and Activity, Related to STAR Methods. (A) Total protein staining and Western blotting of recombinant RAD51. Membrane was stained for total protein, imaged, then stripped and probed with an anti-RAD51 antibody. (B) Coomassie staining (left, prior to His-tag removal; right, after tag removal) and His-tag Western blotting of recombinant RAD51 mutants produced for injections described in Figure 4. (C) Total protein staining of recombinant DSB repair- and HR-associated proteins used in Figure 6B (protein marker on far-right). (D) Single-stranded DNA binding assay to assess the function of wild-type and mutant RAD51 recombinant protein.
Interhomolog repair (IHR) occurs throughout the genome in the early mouse embryo
RAD51, BCCIP, and USP1/WDR48 enhance IHR at sites of Cas9-induced DNA breaks
Embryonic IHR occurs via gene conversion without crossover
Enhanced IHR can be used for homozygous knock-in and donor-free allelic conversion
Acknowledgements
The experiments described in this manuscript were funded by the Hock E. Tan and K. Lisa Yang Center for Autism Research at MIT, the Poitras Center for Affective Disorders Research at MIT, NIH/NIMH Conte Center Grant (P50 MH094271) and NIH Office of the Director (U24 OD026638). We would like to thank Yuanyuan Hou (MIT) for technical assistance, Tetsushi Sakuma and Takashi Yamamoto (Hiroshima University) for sharing their knock-in enhancer screening data, Masataka Nakaya (Shiga Medical University), Hideyuki Tanabe (SOKENDAI), Zhiyuan Chen (Boston Children’s Hospital), Yi Zhang (Boston Children’s Hospital), and Azusa Inoue (RIKEN) for instruction and sharing protocols and materials for embryo FISH experiments, The Genomics platform at The Broad Institute of MIT and Harvard, Xian Adiconis (Broad Institute) and Yinqing Li (Tsinghua University) for helping with NGS experiments, and Hui Yang, Erwei Zuo, and Yidi Sun (Institute of Neuroscience) for additional instructions for GOTI experiments. Finally, we are grateful for the helpful discussions and valuable insights provided by Charles Jennings, Sabbi Lall, and members of the Feng lab.
Footnotes
Declaration of Interests
MIT and J.J.W, T.A., M.W., Q.Z. and G.F. have submitted a patent application (U.S. Patent Application No.: 16/260,630) related to the use of enhanced interhomolog repair for gene editing purposes.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adikusuma F, Piltz S, Corbett MA, Turvey M, McColl SR, Helbig KJ, Beard MR, Hughes J, Pomerantz RT, and Thomas PQ (2018). Large deletions induced by Cas9 cleavage. Nature 560, E8–E9. [DOI] [PubMed] [Google Scholar]
- Aida T, Chiyo K, Usami T, Ishikubo H, Imahashi R, Wada Y, Tanaka KF, Sakuma T, Yamamoto T, and Tanaka K (2015). Cloning-free CRISPR/Cas system facilitates functional cassette knock-in in mice. Genome Biol 16, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aida T, Nakade S, Sakuma T, Izu Y, Oishi A, Mochida K, Ishikubo H, Usami T, Aizawa H, Yamamoto T, et al. (2016). Gene cassette knock-in in mammalian cells and zygotes by enhanced MMEJ. BMC Genomics 17, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae S, Park J, and Kim J-S (2014). Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin D, Sato T, Cibulskis K, Getz G, Stewart C, and Lichtenstein L (2019). Calling Somatic SNVs and Indels with Mutect2. [Google Scholar]
- Canny MD, Moatti N, Wan LCK, Fradet-Turcotte A, Krasner D, Mateos-Gomez PA, Zimmermann M, Orthwein A, Juang Y-C, Zhang W, et al. (2018). Inhibition of 53BP1 favors homology-dependent DNA repair and increases CRISPR-Cas9 genome-editing efficiency. Nat. Biotechnol 36, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caridi CP, Agostino C.D. x., Ryu T, Zapotoczny G, Delabaere L, Li X, Khodaverdian VY, Amaral N, Lin E, Rau AR, et al. (2018). Nuclear F-actin and myosins drive relocalization of heterochromatic breaks. Nature 559, 1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpentier M, Khedher AHY, Menoret S, Brion A, Lamribet K, Dardillac E, Boix C, Perrouault L, Tesson L, Geny S, et al. (2018). CtIP fusion to Cas9 enhances transgene integration by homology-dependent repair. Nat. Commun 9, 1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Morrical MD, Donigan KA, Weidhaas JB, Sweasy JB, Averill AM, Tomczak JA, and Morrical SW (2015). Tumor-associated mutations in a conserved structural motif alter physical and biochemical properties of human RAD51 recombinase. Nucleic Acids Res 43, 1098–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concordet J-P, and Haeussler M (2018). CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res 46, W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Critchlow SE, Bowater RP, and Jackson SP (1997). Mammalian DNA double-strand break repair protein XRCC4 interacts with DNA ligase IV. Curr. Biol 7, 588–598. [DOI] [PubMed] [Google Scholar]
- Cukras S, Lee E, Palumbo E, Benavidez P, Moldovan G-L, and Kee Y (2016). The USP1-UAF1 complex interacts with RAD51AP1 to promote homologous recombination repair. Cell Cycle 15, 2636–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Andrea AD, and Grompe M (2003). The Fanconi anaemia/BRCA pathway. Nat. Rev. Cancer 3, 23–34. [DOI] [PubMed] [Google Scholar]
- Deng Q, Ramsköld D, Reinius B, and Sandberg R (2014). Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 343, 193–196. [DOI] [PubMed] [Google Scholar]
- Donoho G, Jasin M, and Berg P (1998). Analysis of gene targeting and intrachromosomal homologous recombination stimulated by genomic double-strand breaks in mouse embryonic stem cells. Mol. Cell. Biol 18, 4070–4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta A, Eckelmann B, Adhikari S, Ahmed KM, Sengupta S, Pandey A, Hegde PM, Tsai M-S, Tainer JA, Weinfeld M, et al. (2017). Microhomology-mediated end joining is activated in irradiated human cells due to phosphorylation-dependent formation of the XRCC1 repair complex. Nucleic Acids Res 45, 2585–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egli D, Zuccaro MV, Kosicki M, Church GM, Bradley A, and Jasin M (2018). Inter-homologue repair in fertilized human eggs? Nature 560, E5–E7. [DOI] [PubMed] [Google Scholar]
- Ertl HA, Russo DP, Srivastava N, Brooks JT, Dao TN, and LaRocque JR (2017). The Role of Blm Helicase in Homologous Recombination, Gene Conversion Tract Length, and Recombination Between Diverged Sequences in Drosophilamelanogaster. Genetics 207, 923–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang H, Bergmann EA, Arora K, Vacic V, Zody MC, Iossifov I, O’Rawe JA, Wu Y, Jimenez Barron LT, Rosenbaum J, et al. (2016). Indel variant analysis of short-read sequencing data with Scalpel. Nat. Protoc. 11, 2529–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghodsinejad Kalahroudi V, Kamalidehghan B, Arasteh Kani A, Aryani O, Tondar M, Ahmadipour F, Chung LY, and Houshmand M (2014). Two Novel Tyrosinase (TYR) Gene Mutations with Pathogenic Impact on Oculocutaneous Albinism Type 1 (OCA1). PLoS One 9, e106656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Jiang L, Lu F, and Zhang Y (2017). Genomic imprinting of Xist by maternal H3K27me3. Genes Dev 31, 1927–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Chen Z, Yin Q, and Zhang Y (2018). Maternal Eed knockout causes loss of H3K27me3 imprinting and random X inactivation in the extraembryonic cells. Genes Dev 32, 1525–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javidi-Parsijani P, Lyu P, Makani V, Sarhan WM, Yoo KW, El-Korashi L, Atala A, and Lu B (2020). CRISPR/Cas9 increases mitotic gene conversion in human cells. Gene Ther [DOI] [PubMed] [Google Scholar]
- Jayavaradhan R, Pillis DM, Goodman M, Zhang F, Zhang Y, Andreassen PR, and Malik P (2019). CRISPR-Cas9 fusion to dominant-negative 53BP1 enhances HDR and inhibits NHEJ specifically at Cas9 target sites. Nat. Commun 10, 2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelso AA, Goodson SD, Watts LE, Ledford LL, Waldvogel SM, Diehl JN, Shah SB, Say AF, White JD, and Sehorn MG (2017). The β-isoform of BCCIP promotes ADP release from the RAD51 presynaptic filament and enhances homologous DNA pairing. Nucleic Acids Res 45, 711–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Källberg M, Chen X, Kim Y, Beyter D, Krusche P, et al. (2018). Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods 15, 591–594. [DOI] [PubMed] [Google Scholar]
- Kosicki M, Tomberg K, and Bradley A (2018). Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejčí L, Altmannova V, Špírek M, and Zhao X (2012). Homologous recombination and its regulation. Nucleic Acids Res 40, 5795–5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladstätter S, and Tachibana-Konwalski K (2016). A Surveillance Mechanism Ensures Repair of DNA Lesions during Zygotic Reprogramming. Cell 167, 1774–1787.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRocque JR, Stark JM, Oh J, Bojilova E, Yusa K, Horie K, Takeda J, and Jasin M (2011). Interhomolog recombination and loss of heterozygosity in wild-type and Bloom syndrome helicase (BLM)-deficient mammalian cells. Proc. Natl. Acad. Sci. U. S. A 108, 11971–11976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Bitoun E, Altemose N, Davies RW, Davies B, and Myers SR (2019). A high-resolution map of non-crossover events reveals impacts of genetic diversity on mammalian meiotic recombination. Nat. Commun 10, 3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Lopez-Huerta VG, Adiconis X, Levandowski K, Choi S, Simmons SK, Arias-Garcia MA, Guo B, Yao AY, Blosser TR, et al. (2020). Distinct subnetworks of the thalamic reticular nucleus. Nature 583, 819–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang F, Longerich S, Miller AS, Tang C, Buzovetsky O, Xiong Y, Maranon DG, Wiese C, Kupfer GM, and Sung P (2016). Promotion of RAD51-Mediated Homologous DNA Pairing by the RAD51AP1-UAF1 Complex. Cell Rep 15, 2118–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang P, Xu Y, Zhang X, Ding C, Huang R, Zhang Z, Lv J, Xie X, Chen Y, Li Y, et al. (2015). CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell 6, 363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liskay RM, and Stachelek JL (1986). Information transfer between duplicated chromosomal sequences in mammalian cells involves contiguous regions of DNA. Proc. Natl. Acad. Sci. U. S. A 83, 1802–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Guo X, Meng X, Liu J, Allen C, Wray J, Nickoloff JA, and Shen Z (2005). The BRCA2-Interacting Protein BCCIP Functions in RAD51 and BRCA2 Focus Formation and Homologous Recombinational Repair. Mol. Cell. Biol 25, 1949–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Marti-Gutierrez N, Park S-W, Wu J, Lee Y, Suzuki K, Koski A, Ji D, Hayama T, Ahmed R, et al. (2017). Correction of a pathogenic gene mutation in human embryos. Nature 548, 413–419. [DOI] [PubMed] [Google Scholar]
- Mansai SP, Kado T, and Innan H (2011). The Rate and Tract Length of Gene Conversion between Duplicated Genes. Genes 2, 313–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden CG, Jensen RB, Zagelbaum J, Rothenberg E, Morrical SW, Wallace SS, and Sweasy JB (2016). The Tumor-Associated Variant RAD51 G151D Induces a Hyper-Recombination Phenotype. PLoS Genet 12, e1006208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama T, Dougan SK, Truttmann MC, Bilate AM, Ingram JR, and Ploegh HL (2015). Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol 33, 538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVey M, and Lee SE (2008). MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends Genet 24, 529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miele E, Mastronuzzi A, Po A, Carai A, Alfano V, Serra A, Colafati GS, Strocchio L, Antonelli M, Buttarelli FR, et al. (2015). Characterization of medulloblastoma in Fanconi Anemia: a novel mutation in the BRCA2 gene and SHH molecular subgroup. Biomarker Research 3, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldovan G-L, and D’Andrea AD (2009). How the fanconi anemia pathway guards the genome. Annu. Rev. Genet 43, 223–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynahan ME, and Jasin M (1997). Loss of heterozygosity induced by a chromosomal double-strand break. Proc. Natl. Acad. Sci. U. S. A 94, 8988–8993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J, Yang K, Dejsuphong D, Hirota K, Takeda S, and D’Andrea AD (2011). The USP1/UAF1 complex promotes double-strand break repair through homologous recombination. Mol. Cell. Biol 31, 2462–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakade S, Mochida K, Kunii A, Nakamae K, Aida T, Tanaka K, Sakamoto N, Sakuma T, and Yamamoto T (2018). Biased genome editing using the local accumulation of DSB repair molecules system. Nat. Commun 9, 3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakaya M, Tanabe H, Takamatsu S, Hosokawa M, and Mitani T (2017). Visualization of the spatial arrangement of nuclear organization using three-dimensional fluorescence in situ hybridization in early mouse embryos: A new “EASI-FISH chamber glass” for mammalian embryos. J. Reprod. Dev 63, 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuwirth EAH, Honma M, and Grosovsky AJ (2007). Interchromosomal crossover in human cells is associated with long gene conversion tracts. Mol. Cell. Biol 27, 5261–5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normanno D, Négrel A, de Melo AJ, Betzi S, Meek K, and Modesti M (2017). Mutational phospho-mimicry reveals a regulatory role for the XRCC4 and XLF C-terminal tails in modulating DNA bridging during classical non-homologous end joining. Elife 6, 1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshidari R, Strecker J, Chung DKC, Abraham KJ, Chan JNY, Damaren CJ, and Mekhail K (2018). Nuclear microtubule filaments mediate non-linear directional motion of chromatin and promote DNA repair. Nat. Commun 9, 2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet D, Kwart D, Chen A, Sproul A, Jacob S, Teo S, Olsen KM, Gregg A, Noggle S, and Tessier-Lavigne M (2016). Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 533, 125–129. [DOI] [PubMed] [Google Scholar]
- Quadros RM, Miura H, Harms DW, Akatsuka H, Sato T, Aida T, Redder R, Richardson GP, Inagaki Y, Sakai D, et al. (2017). Easi-CRISPR: a robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biol 18, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees HA, Yeh W-H, and Liu DR (2019). Development of hRad51-Cas9 nickase fusions that mediate HDR without double-stranded breaks. Nat. Commun 10, 2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichmann J, Nijmeijer B, Hossain MJ, Eguren M, Schneider I, Politi AZ, Roberti MJ, Hufnagel L, Hiiragi T, and Ellenberg J (2018). Dual-spindle formation in zygotes keeps parental genomes apart in early mammalian embryos. Science 361, 189–193. [DOI] [PubMed] [Google Scholar]
- Richardson C, Moynahan ME, and Jasin M (1998). Double-strand break repair by interchromosomal recombination: suppression of chromosomal translocations. Genes Dev 12, 3831–3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson CD, Kazane KR, Feng SJ, Zelin E, Bray NL, Schäfer AJ, Floor SN, and Corn JE (2018). CRISPR-Cas9 genome editing in human cells occurs via the Fanconi anemia pathway. Nat. Genet 50, 1132–1139. [DOI] [PubMed] [Google Scholar]
- Rukść A, Bell-Rogers PL, Smith JDL, and Baker MD (2008). Analysis of spontaneous gene conversion tracts within and between mammalian chromosomes. J. Mol. Biol 377, 337–351. [DOI] [PubMed] [Google Scholar]
- Santos F, Peat J, Burgess H, Rada C, Reik W, and Dean W (2013). Active demethylation in mouse zygotes involves cytosine deamination and base excision repair. Epigenetics Chromatin 6, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin HY, Wang C, Lee HK, Yoo KH, Zeng X, Kuhns T, Yang CM, Mohr T, Liu C, and Hennighausen L (2017). CRISPR/Cas9 targeting events cause complex deletions and insertions at 17 sites in the mouse genome. Nat. Commun 8, 15464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JA, Bannister LA, Bhattacharjee V, Wang Y, Waldman BC, and Waldman AS (2007). Accurate homologous recombination is a prominent double-strand break repair pathway in mammalian chromosomes and is modulated by mismatch repair protein Msh2. Mol. Cell. Biol 27, 7816–7827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Yang D, Xu J, Zhu T, Chen YE, and Zhang J (2016). RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nat. Commun 7, 10548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark JM, and Jasin M (2003a). Extensive loss of heterozygosity is suppressed during homologous repair of chromosomal breaks. Mol. Cell. Biol 23, 733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark JM, and Jasin M (2003b). Extensive loss of heterozygosity is suppressed during homologous repair of chromosomal breaks. Mol. Cell. Biol 23, 733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama K, Igai K, Hagihara Y, Hashimoto R, Hanawa M, Sakuma T, Tachibana M, Sakurai F, Yamamoto T, and Mizuguchi H (2017). Highly efficient biallelic genome editing of human ES/iPS cells using a CRISPR/Cas9 or TALEN system. Nucleic Acids Res 45, 5198–5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RM, Moore DJ, Whitehouse J, Johnson P, and Caldecott KW (2000). A cell cycle-specific requirement for the XRCC1 BRCT II domain during mammalian DNA strand break repair. Mol. Cell. Biol 20, 735–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran N-T, Bashir S, Li X, Rossius J, Chu VT, Rajewsky K, and Kühn R (2019). Enhancement of Precise Gene Editing by the Association of Cas9 With Homologous Recombination Factors. Front. Genet 10, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Twest S, Murphy VJ, Hodson C, Tan W, Swuec P, O’Rourke JJ, Heierhorst J, Crismani W, and Deans AJ (2017). Mechanism of Ubiquitination and Deubiquitination in the Fanconi Anemia Pathway. Mol. Cell 65, 247–259. [DOI] [PubMed] [Google Scholar]
- Wang AT, Kim T, Wagner JE, Conti BA, Lach FP, Huang AL, Molina H, Sanborn EM, Zierhut H, Cornes BK, et al. (2015). A Dominant Mutation in Human RAD51 Reveals Its Function in DNA Interstrand Crosslink Repair Independent of Homologous Recombination. Mol. Cell 59, 478–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, and Hakonarson H (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilde JJ, Siegenthaler JA, Dent SYR, and Niswander LA (2017). Diencephalic Size is Restricted by a Novel Interplay Between GCN5 Acetyltransferase Activity and Retinoic Acid Signaling. Journal of Neuroscience 37, 2121–16 – 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilm A, Aw PPK, Bertrand D, Yeo GHT, Ong SH, Wong CH, Khor CC, Petric R, Hibberd ML, and Nagarajan N (2012). LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res 40, 11189–11201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray J, Liu J, Nickoloff JA, and Shen Z (2008). Distinct RAD51 Associations with RAD52 and BCCIP in Response to DNA Damage and Replication Stress. Cancer Res 68, 2699–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Liang D, Wang Y, Bai M, Tang W, Bao S, Yan Z, Li D, and Li J (2013). Correction of a Genetic Disease in Mouse via Use of CRISPR-Cas9. Cell Stem Cell 13, 659–662. [DOI] [PubMed] [Google Scholar]
- Yang K, Moldovan GL, Vinciguerra P, Murai J, Takeda S, and D’Andrea AD (2011). Regulation of the Fanconi anemia pathway by a SUMO-like delivery network. Genes Dev 25, 1847–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu DS, Sonoda E, Takeda S, Huang CLH, Pellegrini L, Blundell TL, and Venkitaraman AR (2003). Dynamic control of Rad51 recombinase by self-association and interaction with BRCA2. Mol. Cell 12, 1029–1041. [DOI] [PubMed] [Google Scholar]
- Yusa K, Horie K, Kondoh G, Kouno M, Maeda Y, Kinoshita T, and Takeda J (2004). Genome-wide phenotype analysis in ES cells by regulated disruption of Bloom’s syndrome gene. Nature 429, 896–899. [DOI] [PubMed] [Google Scholar]
- Zadorozhny K, Sannino V, Beláň O, Mlčoušková J, Špírek M, Costanzo V, and Krejčí L (2017). Fanconi-Anemia-Associated Mutations Destabilize RAD51 Filaments and Impair Replication Fork Protection. Cell Rep 21, 333–340. [DOI] [PubMed] [Google Scholar]
- Zuo E, Sun Y, Wei W, Yuan T, Ying W, Sun H, Yuan L, Steinmetz LM, Li Y, and Yang H (2019). Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science 364, 289–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo E, Sun Y, Wei W, Yuan T, Ying W, Sun H, Yuan L, Steinmetz LM, Li Y, and Yang H (2020). GOTI, a method to identify genome-wide off-target effects of genome editing in mouse embryos. Nat. Protoc 15, 3009–3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genotyping Results for Individual F0 Pups from CPI and PNI Experiments, Related to Figures 1 and 2.
Table S2. Sanger Sequencing, Amplicon Sequencing, and GOTI Results, Related to Figures 3, 4, S2, and S3. Table shows approximated RH% based on Sanger chromatograms and results of CRISPRESSO2 analysis of NGS results, including read counts and percentages. It also includes tabs for the sequencing statistics, SNV analysis, and indel analysis for GOTI performed in Figure S3E.
Table S3. Sequences of RNAs, Oligonucleotides, and Proteins Used in this Study, Related to STAR Methods.
Figure S1. RAD51 stimulates the generation of homozygous indels, Related to Figure 1. A, Representative Sanger sequencing traces of putative homozygous indels observed in F0 animals obtained from Chd2RH knock-in experiments. B, Representative Sanger sequencing traces of putative homozygous indels observed in F0 animals obtained from TyrC89S knock-in experiments. c, Genotyping traces for a putative homozygous Chd2 indel and an F1 offspring from a cross between the founder and a wildtype C57BL/6N mate. The F1 genotyping, which was consistent for all 7 pups in the litter, indicates heterozygous presence of the indel observed in the founder.
Figure S2. Chd2RH crRNA Allele Specificity, Concordance Between Sanger Sequencing and Amplicon Sequencing, and Production of the Chddel Allele, Related to Figure 3. (A) PCR was performed using the Chd2-RH_F/R primer pair with DNA purified from either wildtype or Chd2RH/RH mice and the PCR product was used as a template for in vitro digestion assays with or without Cas9. (B) Comparisons of individual RH allele percentages between Sanger sequencing estimates and amplimer sequencing in uninjected, pure IHR, and mosaic embryos. (C) Correlation plot of all calculated RH allele percentages from Sanger sequencing and amplimer sequencing. (D) Schematic of Chd2del locus on chromosome 7. A combination of 4 sgRNAs, 2 on each side of exon 38, were co-injected to generate a large deletion for use as a qPCR control for Figure 3D. (E) Close-up of sequencing at the deletion junctions in the Chd2del founder animal showing a large deletion originating from Chd2-Int37-g1 and Chd2-Int38-g1.
Figure S3. Cas9 injection alters the number of RAD51 puncta in G2, Related to Figure 4. (A) Representative image of immunocytochemistry for RAD51 paired with DNA-FISH for the Chd2 genomic locus showing RAD51 nucleofilament formation specifically at the Cas9 cut site. (B) Quantification of RAD51 puncta number observed in control G2 zygotes and cells injected with RAD51 alone, Cas9/crRNA/tracrRNA alone, or Cas9/crRNA/tracrRNA and RAD51. Each point represents a single cell. (C) Representative images of cells with one (top left), two (top right), three (bottom left), or four (bottom right) RAD51 puncta (arrowheads indicate puncta; DAPI, blue; RAD51, green; scale=10μm). (D) Summary of amplimer sequencing results showing observed indel percentages at genomic loci with 4 mismatches to the Chd2RH crRNA. Lower-case letters in putative off-target sites denote mismatches with the crRNA sequence. (E) Summary of GOTI data. For read depth, TdTomato- cells are in white and TdTomato+ cells are in red. ‘SNVs Found’ describes the number of SNVs overlapping from analysis with Mutect2, Strelka, and Lofreq. ‘Indels Found’ describes the number indels overlapping from analysis with Mutect2, Strelka, and Scalpel. Complete GOTI analysis data can be found in Table S2.
Figure S4. Effects of IHR enhancers on editing efficiency and mosaicism, Related to Figure 6. (A) Editing efficiency observed in IVF-derived embryos injected with Cas9/crRNA/tracrRNA only (no enhancer) or the indicated HR- and DSB repair-associated proteins (one-side Fisher’s exact test). (B) Representative chromatograms showing examples traces for genotypes characterized as unedited (top left), 100% IHR (top right), mosaic with wildtype (bottom left), and mosaic with indel (bottom right). (C) Quantification of mosaicism in embryos displaying IHR for all conditions analyzed. Data includes all IVF-derived embryos (Figures 3C, 6A, and 6B; two-sided Fisher’s exact test). (D) Distribution of types of mosaicism observed in mosaic embryos described in panel b (two-sided Fisher’s exact test). (E) Numbers for observed mosaicism used to generate panel C.
Figure S5. Summary of SNP Genotyping of TOPO-Cloned Alleles in Acs16 and Kcnb2 Embryos, Related to Figure 7. Gels show PCR products used as input for TOPO cloning. Each box represents the genotype at a SNP-containing region on the cloned allele. Magenta boxes are positive for the A/J SNP and green boxes are positive for the C57BL/6J SNP.
Figure S6. Examples of Recombination Tracts Identified by TOPO Cloning of Individual Alleles, Related to Figure 7. (A) Top track shows the Acs16 locus spanning from the sgRNA target site on the right to the last A/J-specific sequence on the left (a 4bp insertion). The top chromatogram is from a pure A/J allele and the bottom is from an allele that is A/J at the Cas9 cut site (red arrowhead) and C57BL/6J 178bp upstream at the site of the 4bp insertion. (B) Top track shows the Kcnb2 locus spanning from the sgRNA target site on the left to the last A/J-specific SNP on the right. The top chromatogram is from a pure A/J allele and the bottom is from an allele that is A/J at the Cas9 cut site (red arrowhead) and C57BL/6J 211bp downstream at the site of the SNP. (C) Top track shows the Kcnb2 sgRNA target site in the middle, with two distal SNPs located ~±1.5kb from the cut site on the left and right sides. The top chromatograms are from a pure A/J clone and the bottom chromatograms are from a single allele that carries the A/J sequences at the cut site and upstream locus, but the C57BL/6J SNP at the 1.5kb downstream locus.
Figure S7. Verification of Recombinant Protein Identity, Purity, and Activity, Related to STAR Methods. (A) Total protein staining and Western blotting of recombinant RAD51. Membrane was stained for total protein, imaged, then stripped and probed with an anti-RAD51 antibody. (B) Coomassie staining (left, prior to His-tag removal; right, after tag removal) and His-tag Western blotting of recombinant RAD51 mutants produced for injections described in Figure 4. (C) Total protein staining of recombinant DSB repair- and HR-associated proteins used in Figure 6B (protein marker on far-right). (D) Single-stranded DNA binding assay to assess the function of wild-type and mutant RAD51 recombinant protein.
Data Availability Statement
The amplicon sequencing included in this study has been submitted to the Sequence Read Archive and can be found under accession number PRJNA614534. Additional raw data for the figures in this manuscript can be obtained from the Lead Contact or corresponding authors upon request.