

Abstract
The suppression of types I and III interferon (IFN) responses by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) contributes to the pathogenesis of coronavirus disease 2019 (COVID‐19). The strategy used by SARS‐CoV‐2 to evade antiviral immunity needs further investigation. Here, we reported that SARS‐CoV‐2 ORF9b inhibited types I and III IFN production by targeting multiple molecules of innate antiviral signaling pathways. SARS‐CoV‐2 ORF9b impaired the induction of types I and III IFNs by Sendai virus and poly (I:C). SARS‐CoV‐2 ORF9b inhibited the activation of types I and III IFNs induced by the components of cytosolic dsRNA‐sensing pathways of RIG‐I/MDA5‐MAVS signaling, including RIG‐I, MDA‐5, MAVS, TBK1, and IKKε, rather than IRF3‐5D, which is the active form of IRF3. SARS‐CoV‐2 ORF9b also suppressed the induction of types I and III IFNs by TRIF and STING, which are the adaptor protein of the endosome RNA‐sensing pathway of TLR3‐TRIF signaling and the adaptor protein of the cytosolic DNA‐sensing pathway of cGAS–STING signaling, respectively. A mechanistic analysis revealed that the SARS‐CoV‐2 ORF9b protein interacted with RIG‐I, MDA‐5, MAVS, TRIF, STING, and TBK1 and impeded the phosphorylation and nuclear translocation of IRF3. In addition, SARS‐CoV‐2 ORF9b facilitated the replication of the vesicular stomatitis virus. Therefore, the results showed that SARS‐CoV‐2 ORF9b negatively regulates antiviral immunity and thus facilitates viral replication. This study contributes to our understanding of the molecular mechanism through which SARS‐CoV‐2 impairs antiviral immunity and provides an essential clue to the pathogenesis of COVID‐19.
Keywords: antiviral immunity, COVID‐19, IFNs, ORF9b, SARS‐CoV‐2
1. INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), which causes coronavirus disease 2019 (COVID‐19), is a novel emerging coronavirus that is spreading globally, might be lethal to humans and other animals, and thus poses significant threats to public health worldwide.1, 2, 3 SARS‐CoV‐1, Middle East respiratory syndrome human (MERS‐CoV), and SARS‐CoV‐2, which might cause severe pulmonary disease with acute respiratory distress syndrome and systemic inflammation and might be fatal, have successively emerged in the human population in the 21st century. The genome of SARS‐CoV‐2 is approximately 30 kb in length and encodes 14 putative open reading frames, including the large replicase genes expressing two replicative polyproteins (pp1a and pp1ab) that are cleaved into NSP1‐16 by viral proteases, the structural genes expressing the spike (S), membrane (M), envelope (E), and nucleocapsid (N) proteins, and the accessory genes expressing ORF3a, ORF3b, ORF6, ORF7a, ORF7b, ORF8, ORF9b, ORF9c, and ORF10. 4 Although the accessory proteins of coronaviruses are not essential for viral replication and virion assembly, they contribute to virulence by affecting the release, stability, and pathogenesis of the virus. 5 To date, the function of SARS‐CoV‐2 accessory proteins in immune evasion still needs to be addressed.
Although the SARS‐CoV‐2 infection‐mediated dysregulation of the immune system, which involves the suppression of antiviral immunity and the elevation of inflammatory responses, contributes to the pathogenesis of COVID‐19,6, 7, 8, 9, 10, 11 the mechanism through which the recently emerged SARS‐CoV‐2 is recognized by innate immunity has not been clarified. Double‐stranded RNA (dsRNA), which is produced by many viruses during replication, is a common viral pathogen‐associated molecular pattern that is sensed by pattern recognition receptors. 12 Cytosolic retinoic acid‐inducible gene (RIG)‐I‐like receptors, including RIG‐I and MDA‐5, and endosomal Toll‐like receptor 3 (TLR3) recognize dsRNAs from intermediates generated during viral replication, and this recognition results in the serial activation of innate antiviral signaling cascades via induction of the production of types I and III IFNs.12, 13 The coronaviruses have homologous genomes, similar replication intermediates, and the same lifecycles; thus, it appears that SARS‐CoV‐2 can be recognized by RNA sensors similarly to other coronaviruses to elicit innate antiviral immunity. 14 RIG‐I participates in the immune sensing of murine coronavirus mouse hepatitis virus (MHV) in oligodendrocyte cells. 15 MDA5 can recognize MHV in brain macrophages, microglial cells, and oligodendrocyte cells.15, 16 The sensing of cytosolic dsRNA by RIG‐I/MDA‐5 recruits the adaptor protein MAVS (also known as VISA, Cardiff, or IPS‐1), which activates TANK‐binding kinase 1 (TBK1)/inhibitor of κB kinase epsilon (IKKε) and then induces the phosphorylation and subsequent nuclear translocation of the transcription factor IFN regulatory factor 3 (IRF3), and nuclear IRF3 together with nuclear factor‐κB (NF‐κB), which is also activated by RIG‐I/MDA‐5 signaling, initiates the transcription of types I and III IFNs and other proinflammatory cytokines, which lead to antiviral immune responses. 12 TLR3 is involved in the defense against SARS‐CoV‐1 infection. 17 dsRNA‐activated TLR3 activates IRF3 and NF‐κB signaling via TIR‐domain‐containing adapter‐inducing interferon‐β (TRIF)‐TBK1/IKKε signaling cascades, which results in the production of types I and III IFNs and other pro‐inflammatory cytokines. 12 Although the involvement of the cytosolic DNA‐sensing pathway of cGAS‐stimulator of IFN genes (STING) signaling in the recognition of coronaviruses has not been elucidated, the papain‐like protease domain from SARS‐CoV‐1 can act as an antagonist of IFNs by targeting STING,18, 19 which suggests that the cGAS‐STING pathway should play a vital role in the defense against certain coronaviruses. STING is activated by the second messenger 2ʹ−3ʹcGAMP produced by DNA‐activated cGAS. 20 Subsequently, STING recruits TBK1, which phosphorylates IRF3, and this phosphorylation leads to the translocation of IRF3 into the nucleus to induce the expression of types I and III IFNs and other proinflammatory cytokines. 20 The RIG‐I/MDA‐5–MAVS, TLR3–TRIF, and cGAS–STING signaling pathways converge at TBK1/IKKε, which catalyzes IRF3 phosphorylation and the subsequent transcription of types I and III IFNs. 21 Secreted type I and III IFNs bind to their receptors and then activate Janus kinase/signal transducers and activators of transcription signaling to drive the expression of IFN‐stimulated genes (ISGs), which can initiate antiviral states by suppressing viral replication and spreading, activating immune cells, and causing the death of infected cells.13, 22
The types I and III IFN response is the essential action of host antiviral immunity in the clearance of virus infection.13, 22 To establish a successful infection of host cells, viruses, including coronaviruses, have developed various strategies to antagonize the IFN response. 14 Previous studies have proposed that the accessory proteins of SARS‐CoV‐1, such as open reading frame 3b (ORF3b), ORF6, and ORF9b, inhibit the production of type I IFNs. 14 In COVID‐19 patients, the induction of types I and III IFNs is suppressed.7, 8, 23 The replenishment of types I or III IFNs can significantly contribute to the clearance of SARS‐CoV‐2 and to COVID‐19 symptom relief.24, 25, 26 Compared with type I IFNs, type III IFNs exhibit some advantages in COVID‐19 treatment regarding the induction of a longer‐lasting antiviral state and a less proinflammatory response. 27 Although SARS‐CoV‐2 infection impairs the antiviral immunity elicited by types I and III IFNs in COVID‐19 patients and cell models,7, 8, 23 the mechanism through which the recently emerged SARS‐CoV‐2 blocks the induction of types I and III IFNs remains elusive. Therefore, dissecting the molecular mechanism through which SARS‐CoV‐2 evades types I and III IFN responses will improve the understanding of the pathogenesis of COVID19 and provide therapeutic strategies for counteracting SARS‐CoV‐2 infections. SARS‐CoV‐1 ORF9b was reported to suppress IFN production; however, whether SARS‐CoV‐2 ORF9b could evade host antiviral innate immunity is still unknown; thus, we explored the effect of SARS‐CoV‐2 ORF9b on IFN production and the potential mechanism. We found that the SARS‐CoV‐2 accessory protein ORF9b, which is encoded by an alternative ORF within the N gene, can remarkably suppress RIG‐I/MDA‐5–MAVS, TLR3–TRIF, and cGAS–STING signaling‐activated types I and III IFN production by targeting multiple molecules of these innate antiviral pathways.
2. MATERIALS AND METHODS
2.1. Reagents and antibodies
Protein A/G beads were purchased from Santa Cruz Biotechnology, and the anti‐Flag magnetic beads were purchased from Bimake. Poly (I:C) and 2ʹ3ʹ‐cGAMP were purchased from Invivogen. Rabbit anti‐Myc‐tag (71D10), rabbit anti‐IRF3 (D83B9), rabbit anti‐pIRF3 (4D46), rabbit anti‐TBK1 (3031S), rabbit anti‐pTBK1 (D52C2) were purchased from Cell Signaling Technology. Mouse anti‐MAVS was purchased from Santa Cruz Biotechnology. Mouse anti‐actin, mouse anti‐V5, and rabbit anti‐calnexin antibodies were purchased from proteintech. Mouse anti‐Flag M2 antibody was purchased from Sigma‐Aldrich. Mouse anti‐Myc‐tag (9E10) was purchased from Origene. Rabbit anti‐GM130 was purchased from Abcam. Rabbit anti‐Tom20 antibody was purchased from Abclonal. Mouse anti‐HA was purchased from MDL Biotech.
2.2. Constructs and plasmids
Plasmids expressing RIG‐I, RIG‐IN, MDA‐5, MAVS, TBK1, IKKε, IRF3‐5D, TRIF, and STING were cloned into mammalian expression vectors, and the luciferase reporter plasmids including pGL3‐IFN‐β‐Luc (IFN‐β luciferase reporter) and pGL3‐IFN‐λ1‐Luc (IFN‐λ1 luciferase reporter) were constructed by inserting the promoter region into pGL3‐Basic by standard molecular cloning methods as described in our previous publications.28, 29, 30 pISRE‐Luc (the luciferase reporter of ISGs) was purchased from Clontec. The SARS‐CoV‐2 ORF9b gene was synthesized according to the genome sequence of the SARS‐CoV‐2 Wuhan‐Hu‐1 strain (NC_045512.2) at General Biol. The coding region of the SARS‐CoV‐2 ORF9b gene was amplified using primers list in Table S1 by polymerase chain reaction (PCR) and cloned into the pCAG mammalian expression vector with a C‐terminal Flag‐tag.
2.3. Cell culture
HEK‐293T, HeLa, and Vero E6 cells were obtained from the American Type Culture Collection (ATCC) and maintained according to the culture methods provided by the ATCC. All these cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% heat‐inactivated fetal bovine serum (FBS) at 37°C in a humidified incubator with 5% CO2.
2.4. Transfection
The plasmids were transiently transfected into the cells using Lipofectamine 3000 (Invitrogen) or Polyethylenimine “Max” (Polysciences, Inc.) following the manufacturer's instruction. Poly (I:C) and 2′−3′ cGAMP were delivered into cells using Lipofectamine 2000 (Invitrogen) as described previously. 28
2.5. RNA extraction and real‐time quantitative PCR
Total RNA was extracted with TRIzol reagent (Invitrogen) and then was reverse‐transcribed into first‐strand cDNA with the HiScript III 1st Strand cDNA Synthesis Kit with gDNA wiper (Vazyme) following the manufacturer's protocol. The SYBR Green‐based RT‐qPCR kit UltraSYBR Mixture (CWBIO) was used to perform real‐time quantitative PCR (RT‐qPCR) assays using primers of each gene (Table S1) by a Roche LightCycler96 system according to the manufacturer's instructions. The relative expression of the indicated genes was normalized to the mRNA level of glyceraldehyde 3‐phosphate dehydrogenase, one of the internal housekeeping genes in human cells. A comparative C t method (ΔΔC t method) was used to calculate the fold changes by normalizing to that of genes expressed in the control group as described previously. 31
2.6. Enzyme‐linked immunosorbent assay (ELISA)
The concentration of secreted IFN‐β in culture supernatants was measured by enzyme‐linked immunosorbent assay (ELISA) kits (R&D Systems) according to the manufacturer′s instructions.
2.7. Dual‐luciferase reporter assays
HEK‐293T cells (approximately 0.5 × 105/well) were seeded in 48‐well plates 12 h before transfection. The luciferase reporter plasmids and the gene expression plasmids were cotransfected into HEK‐293T cells as indicated in each figure. The pRL‐TK Renilla luciferase reporter (Promega) was transfected to serve as an internal control. Thirty‐six hours later, the cells were harvested to assess the luciferase activities using the Dual‐Luciferase Reporter Assay Kit (Vazyme) as described in our previous studies.31, 32, 33 The luciferase activity was measured in a Centro XS3 LB 960 microplate luminometer (Berthold Technologies). The activity of firefly luciferase was normalized to that of Renilla luciferase to calculate the relative luciferase activity.
2.8. Viral infection
Vesicular stomatitis virus (VSV)‐enhanced green fluorescent protein (eGFP) and SeV were used to infect HeLa or HEK‐293T cells as described in our previous publications.28, 29, 30 Before infection, the target cells were washed with serum‐free DMEM prewarmed at 37°C. The virus was then diluted to the desired multiplicity of infection with serum‐free DMEM and incubated with the target cells for 1–2 h. At the end of the infection period, the virus‐medium complexes were discarded, and DMEM containing 10% FBS was added.
2.9. Immunoblot analysis and immunoprecipitation
For coimmunoprecipitation assay, HEK‐293T cells were first transfected with the indicated plasmids in each figure for 24 h and further lysed in lysis buffer [1.0% (vol/vol) NP‐40, 50 mM Tris‐HCl, pH 7.4, 50 mM EDTA, 150 mM NaCl] complemented with a protease inhibitor cocktail (Sigma Aldrich), and a phosphatase inhibitor cocktail (Sigma Aldrich). Supernatants were transferred into new tubes after centrifugation for 10 min at 14,000g and further incubated with the indicated antibodies for 3 h at 4°C followed by the addition of protein A/G beads (Santa Cruz), or with anti‐Flag magnetic beads (Bimake), anti‐Myc magnetic beads (Bimake). After incubation overnight at 4°C, beads were subject to washing four times with lysis buffer. After washing, the beads were boiled by boiling with 2X SDS loading buffer containing 100 mM Tris‐HCl pH 6.8, 4% (wt/vol) SDS, 20% (vol/vol) glycerol, 0.2% (wt/vol) bromophenol blue, and 1% (vol/vol) 2‐mercaptoethanol to collect the immunoprecipitates.
For immunoblot analysis, cell pellets were lysed with the M‐PER Protein Extraction Reagent (Pierce) complemented with a protease inhibitor cocktail (Sigma Aldrich) and a phosphatase inhibitor cocktail (Sigma Aldrich). A bicinchoninic acid assay (Pierce) was used to measure the protein concentrations in the supernatant. The prepared total cell lysates or immunoprecipitates were electrophoretically separated by SDS‐PAGE, transferred onto a polyvinylidene difluoride membrane (Millipore), blocked with 3% (wt/vol) bovine serum albumin, probed with indicated primary antibodies and corresponding secondary antibodies, and visualized by ECL Western blot analysis detection reagent (Pierce).
2.10. Confocal microscopy
HeLa cells were seeded on 12‐well slides 24 h before transfection. Each well was transfected with the indicated plasmids (1 µg each). Transfected or infected HeLa cells were subject to fix, permeabilization, and blocking as described in the previous paper. The fixation, permeabilization, and blocking buffer were all purchased from Beyotime Biotechnology. The cells were then incubated with indicated primary antibodies at 4°C overnight, rinsed, and incubated with corresponding secondary antibodies (Invitrogen). Nuclei were counterstained with 4′,6‐diamidino‐2‐phenylindole (Abcam). Images were captured with a Zeiss LSM880 confocal microscope.
2.11. Flow cytometry analysis
VSV–eGFP infected cells was assessed by a Beckman Coulter Gallios flow cytometer with at least 10,000 cells per sample as described in our previous publication. 34
2.12. Plaque assays
Vero‐E6 cells were used to perform plaque assays to determine the titer of VSV–eGFP. Vero cells at approximately 100% confluency cultured in 24‐well plates were infected with serial dilutions of VSV–eGFP. 0.5 h later, the culture medium was discarded, and then DMEM containing 0.5% agar and 2% FBS overlaid. After 20 h culture, the cells were fixed with a 1:1 methanol–ethanol mixture and then visualized with 0.05% crystal violet. The plaques on the monolayer were used to determine the titer of VSV–eGFP as described in our previous publication. 34
2.13. Statistical analysis
The statistical analyses were performed using two‐tailed unpaired Student's t tests with GraphPad Prism 8.0 and Microsoft Excel. Unless otherwise specified, the results, which are representative of three independent experiments, are presented as the mean ± SD. A value of p<.05 was considered statistically significant, as indicated in each figure.
3. RESULTS
3.1. SARS‐CoV‐2 ORF9b antagonizes types I and III IFNs
To explore the function of SARS‐CoV‐2 ORF9b in viral infection, HEK‐293T cells expressing SARS‐CoV‐2 ORF9b were infected with SeV. RT‐qPCR analysis revealed that the induction of IFN‐β, IFN‐λ1, and two ISGs called ISG56 and CXCL10 after SeV infection was suppressed in SARS‐CoV‐2 ORF9b‐expressing cells compared with the control HEK‐293T cells that did not express any viral protein (Figure 1A). The ELISA assays showed that less IFN‐β is released into the culture supernatant from SARS‐CoV‐2 ORF9b‐expressing cells than that from the control cells (Figure 2). Similar results were observed with HEK‐293T cells that were transfected with the dsRNA mimic poly (I:C) to stimulate antiviral immunity (Figure 1B). However, SARS‐CoV‐2 ORF9c, another alternative ORF within the N gene, exerted no effect on either SeV infection‐ or poly (I:C) transfection‐induced types I and III IFN production (Figures S1 and S2).
Figure 1.
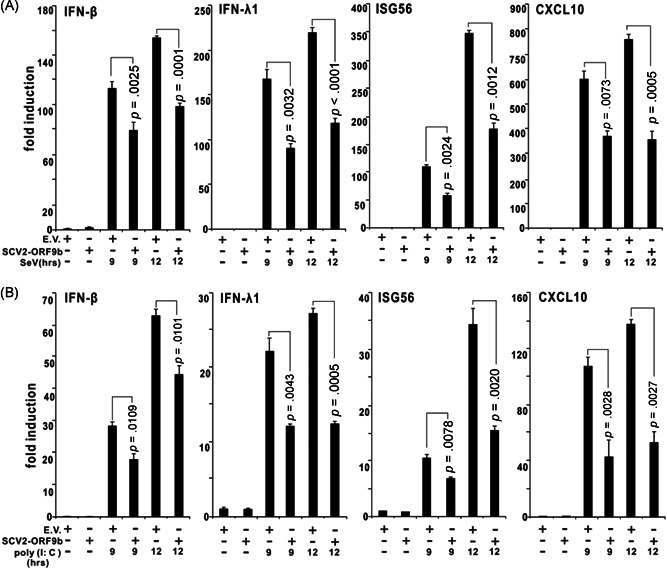
SARS‐CoV‐2 ORF9b suppresses SeV‐ or poly (I:C)‐induced interferon β (IFN‐ β), IFN‐λ1, ISG56, and CLXL10 production. Plasmids of the pcDNA6B empty vector (500 ng) or SCV2‐ORF9b (500 ng) were transfected into HEK‐293T cells. Twenty‐four hours later, the cells were infected with SeV (A) or transfected with poly (I:C) (B) as indicated, and 9 and 12 h after stimulation, the expression of IFN‐β, IFN‐λ1, ISG56, and CLXL10 in these cells was determined by RT‐qPCR analysis. The results from one representative experiment are shown to represent three independent biological replicates. The error bars indicate the SD. EV, empty vector; RT‐qPCR, real‐time quantitative polymerase chain reaction; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2; SCV2, SARS‐CoV‐2
Figure 2.
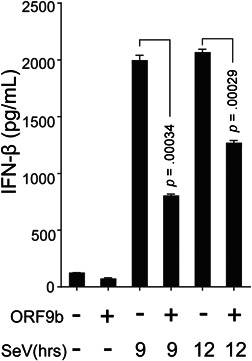
SARS‐CoV‐2 ORF9b inhibits SeV‐induced IFN‐β secretion. HEK‐293T cells were transfected with plasmids of the pcDNA6B empty vector (500 ng) or SCV2‐ORF9b (500 ng). Twenty‐four hours later, the cells were infected with SeV as indicated, and 9 and 12 h after stimulation, the culture supernatant was collected for ELISA assays. The results from one representative experiment are shown to represent three independent biological replicates. The error bars indicate the SD. ELISA, enzyme‐linked immunosorbent assay; IFN, interferon; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
We subsequently attempted to map the layer where SARS‐CoV‐2 ORF9b exerts its inhibitory effect on IFN production. We cotransfected SARS‐CoV‐2 ORF9b with RIG‐IN, RIG‐I, MDA‐5, MAVS, TBK1, and IRF3‐5D into HEK‐293T cells. Luciferase reporter assays showed that SARS‐CoV‐2 ORF9b clearly inhibited the activities of types I (IFN‐β‐Luc) and III (IFN‐λ1‐Luc) IFN and ISG (ISRE‐Luc) luciferase reporters activated by RIG‐IN, RIG‐I, MDA‐5, MAVS, and TBK1 but not by IRF3‐5D, which suggested that SARS‐CoV‐2 ORF9b inhibited RIG‐I/MDA‐5–MAVS signaling‐activated IFN production upstream of IRF3 (Figure 3). We also assessed the effect of SARS‐CoV‐2 ORF9b on the TLR3–TRIF and cGAS–STING signaling pathways by coexpressing SARS‐CoV‐2 ORF9b with TRIF or STING. The results indicated that SARS‐CoV‐2 ORF9b also suppressed the TRIF‐ and STING‐induced activation of the luciferase reporter of IFN‐β, IFN‐λ1, and ISGs (Figure 3). Thus, it appears that SARS‐CoV‐2 ORF9b exerts its IFN inhibitory function upstream of IRF3 but downstream of MAVS, TRIF, and STING.
Figure 3.
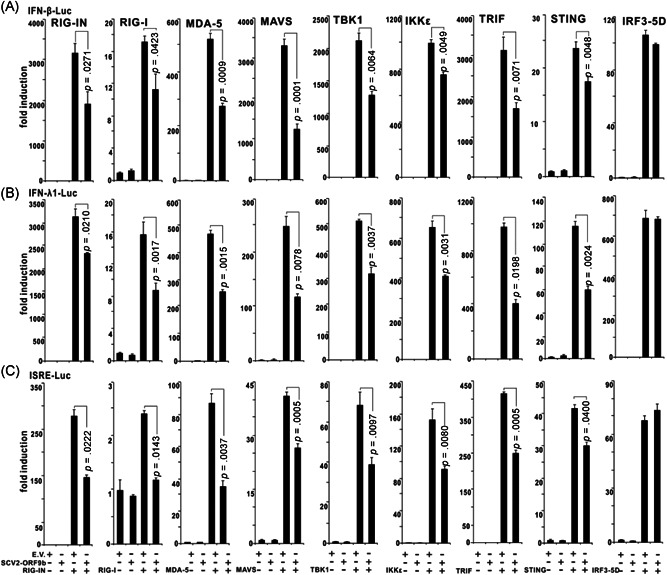
SARS‐CoV‐2 ORF9b inhibits the activation of luciferase reporters of types I and III IFNs and ISGs. RIG‐IN (100 ng, an active form of RIG‐I), MDA‐5 (100 ng), TANK‐binding kinase 1 (TBK1) (100 ng), IKKε (100 ng), IFN regulatory factor 3 (IRF3)‐5D (100 ng, an active form of IRF3), TIR‐domain‐containing adapter‐inducing interferon‐β (TRIF) (100 ng, the adaptor of the TLR3–TRIF pathway), or STING (100 ng, the adaptor of the cGAS–STING pathway) were transfected alone or together with a plasmid expressing SARS‐CoV‐2 ORF9b into HEK‐293T cells cultured in 48‐well plates as indicated. The plasmids of IFN‐β‐Luc (45 ng), IFN‐λ1‐Luc (45 ng), or ISRE‐Luc (45 ng) were cotransfected with the above plasmids to assess the activation of (A) type I IFNs, (B) type III IFNs, or (C) ISGs, respectively. pRL‐TK (5 ng) was also cotransfected into each well as an internal control. The pcDNA6 empty vector was used to balance the total amount of plasmid DNA transfected into each well. Dual‐luciferase assays were performed 36 h after transfection. Error bars indicate SD. IFN, interferon; IKKε, inhibitor of κB kinase epsilon; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2; TLR3, Toll‐like receptor 3
3.2. SARS‐CoV‐2 ORF9b targets multiple proteins of antiviral signaling pathways
To further identify the location where SARS‐CoV‐2 ORF9b performs its function, we first explored its subcellular localization by confocal microscopy. A plasmid expressing SARS‐CoV‐2 ORF9b was transfected into HeLa cells. Twenty hours after transfection, the cells were incubated with the primary antibody and then stained with fluorescently labeled secondary antibody as indicated (Figure 4). The mitochondria, endoplasmic reticulum (ER), and Golgi were visualized with corresponding markers. The results showed that SARS‐CoV‐2 ORF9b was strongly colocalized with mitochondria but only weakly colocalized with the ER and Golgi (Figure 4A–C). We subsequently studied the colocalization of SARS‐CoV‐2 ORF9b with components of the innate antiviral signal pathways. Plasmids expressing RIG‐I, MDA‐5, MAVS, TBK1, TRIF, or STING were cotransfected with the SARS‐CoV‐2 ORF9b plasmid into HeLa cells, and these proteins were stained with fluorescently labeled secondary bodies after incubation with the primary antibodies as indicated (Figure 4). Confocal microscopy observations showed that SARS‐CoV‐2 ORF9b strongly colocalizes with MASV but partially localizes with RIG‐I, MDA‐5, TBK1, TRIF, and STING (Figure 4D–I).
Figure 4.
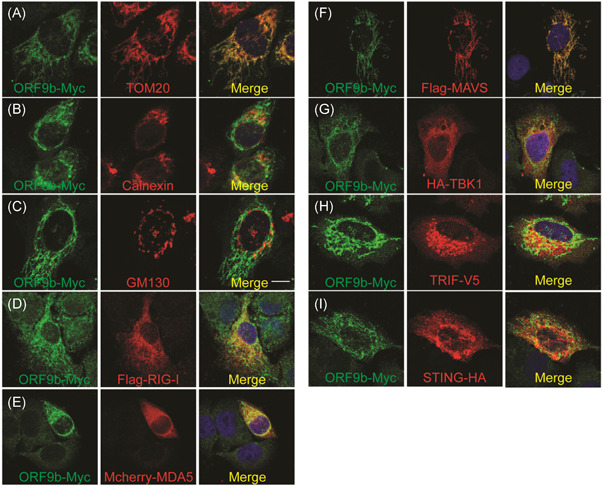
(A–C) Subcellular localization of SARS‐CoV‐2 ORF9b. HeLa cells seeded on 12‐well coverslips were transfected with the indicated plasmids. Twenty hours after transfection, the cells were subjected to immunofluorescence staining with mouse anti‐Myc antibody and rabbit antibodies against the corresponding organelle marker. Scale bar = 10 μm. (D–I) Relative localization of SARS‐CoV‐2 ORF9b protein with signaling molecules, including RIG‐I, MDA5, MAVS, TBK1, TRIF, and STING. The seeding and transfection of HeLa cells were performed as described in (A). After transfection, ORF9b was stained with a rabbit anti‐Myc antibody, and the signaling molecules were reacted with mouse antibodies against the indicated tags. Scale bar = 10 μm. TOM20, Mitochondria marker; Calnerxin, ER marker; GM130, Golgi marker. SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2; TBK1, TANK‐binding kinase 1; TRIF, TIR‐domain‐containing adapter‐inducing interferon‐β
The results from luciferase reporter assays suggest that the action of SARS‐CoV‐2 ORF9b might occur upstream of IRF3 (Figure 3). We postulated that this protein might interact with molecules upstream of IRF3 that are involved in the innate antiviral signaling pathways. We performed coimmunoprecipitation experiments to determine which protein is the target of SARS‐CoV‐2 ORF9b. The plasmids expressing Myc‐tagged SARS‐CoV‐2 ORF9b were individually cotransfected with plasmids of RIG‐I, MDA‐5, MAVS, TBK1, TRIF, STING, or IRF3. Whole‐cell lysates were subjected to immunoprecipitation using antibodies as indicated (Figure 5). The immunoprecipitation results indicated that SARS‐CoV‐2 ORF9b associated with RIG‐I (Figure 5A), MDA‐5 (Figure 5B), MAVS (Figure 5C), TBK1 (Figure 5D), STING (Figure 5E), and TRIF (Figure 5G) but not IRF3 (Figure 5F), which is consistent with the results from the colocalization studies (Figure 4). SARS‐CoV‐2 ORF9b prevents the association between TRIF and TBK1 but has no effect on the interactions between TBK1 and IRF3 (Figure 5H and I). These data showed that SARS‐CoV‐2 ORF9b might target multiple molecules of the RIG‐I/MDA‐5–MAVS, TLR3–TRIF, and cGAS–STING signaling pathways to suppress IFN production.
Figure 5.
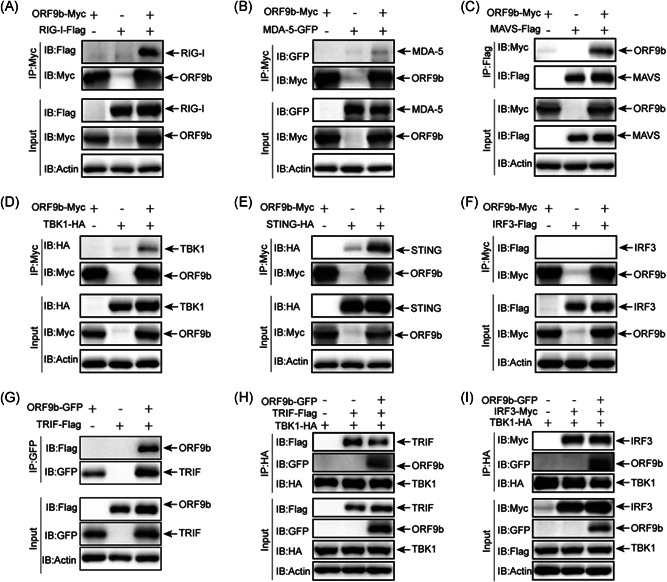
SARS‐CoV‐2 ORF9b interacts with RIG‐I, MDA‐5, MAVS, TBK1, STING, and TRIF but not IRF3. HEK‐293T cells were transfected with the indicated plasmids for 24 h before coimmunoprecipitation by the indicated antibody‐conjugated beads. The input and immunoprecipitates were reacted with the indicated antibodies. IRF3, IFN regulatory factor 3; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2; TBK1, TANK‐binding kinase 1; TRIF, TIR‐domain‐containing adapter‐inducing interferon‐β
3.3. SARS‐CoV‐2 ORF9b decreases TBK1 phosphorylation
The RIG‐I/MDA‐5–MAVS, TLR3–TRIF, and cGAS–STING signaling pathways converge at TBK1, and SARS‐CoV‐2 ORF9b colocalizes and interacts with TBK1; thus, the affection of TBK1 activity might be important to block signaling transduced from molecules of the RIG‐I/MDA‐5–MAVS, TLR3–TRIF, and cGAS–STING signaling pathways. Thus, we investigated the effect of SARS‐CoV‐2 ORF9b on TBK1 phosphorylation, which is an important biochemical process for IRF3 activation and subsequent IFN transcription. The phosphorylation of TBK1 is induced by the overexpression of RIG‐IN (Figure 6A), MAVS (Figure 6B), or TRIF (Figure 6C) and by STING activated by its ligand 2′−3′cGAMP (Figure 6D). The cotransfection of the SARS‐CoV‐2 ORF9b plasmid markedly reduced the phosphorylation of TBK1 induced by RIG‐IN, MAVS, or STING but not by TRIF (Figure 6A‐D). Thus, SARS‐CoV‐2 ORF9b targets TBK1 and prevents its phosphorylation induced by signaling molecules of RIG‐IN, MAVS, and STING but not TRIF. Furthermore, we also observed that SARS‐CoV‐2 ORF9b can also impair TBK1 phosphorylation induced by SeV infection (Figure 7A).
Figure 6.
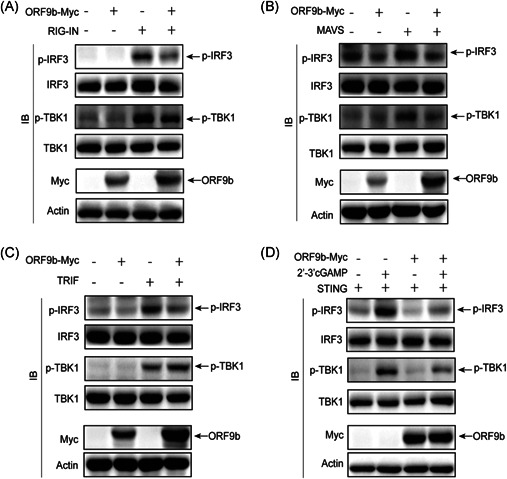
SARS‐CoV‐2 ORF9b suppresses the phosphorylation of TBK1 and IRF3. HEK‐293T cells were transfected with plasmids of (A) RIG‐IN (A), (B) MAVS, (C) TRIF, or (D) STING in the presence or absence of ORF9b for 24 h. The cells transfected with STING plasmid were further stimulated by the transfection of 2′3′‐cGAMP for 8 h. The transfected or stimulated cells were lysed and subjected to immunoblot analysis with the indicated antibodies. IRF3, IFN regulatory factor 3; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2; TBK1, TANK‐binding kinase 1; TRIF, TIR‐domain‐containing adapter‐inducing interferon‐β
Figure 7.
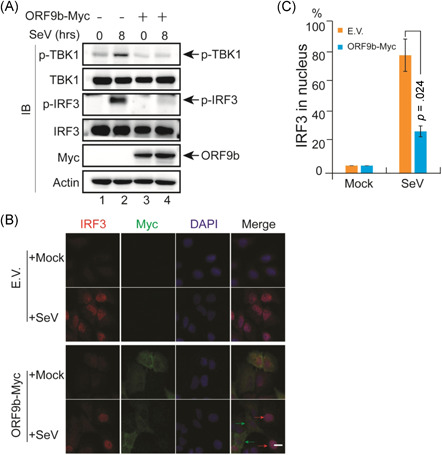
SARS‐CoV‐2 ORF9b suppressed IRF3 phosphorylation and nuclear translocation. (A) SARS‐CoV‐2 ORF9b protein affects the phosphorylation of IRF3 upon SeV infection. HeLa cells seeded on six‐well plates (5 × 105 cells per well) were transfected with the Myc empty vector or ORF9b‐Myc plasmid and infected with SeV 20 h after transfection. At the indicated time points, the cells were scraped and processed for immunoblotting with the indicated antibodies. (B) HeLa cells were seeded on 12‐well coverslips (5 × 104 cells per well) one day before transfection with the Myc E.V. or ORF9b‐Myc plasmids. Twenty hours after transfection, the cells were infected with SeV. Eight hours after infection, the slides were harvested and processed for immunofluorescence staining with mouse anti‐Myc antibody and rabbit anti‐IRF3 antibody. (C) Quantification of the percentage of IRF3 in the nucleus after SeV infection. The localization of IRF3 in 50 cells within each group was determined and calculated before and after SeV infection. DAPI, 4′,6‐diamidino‐2‐phenylindole; E.V., empty vector; IRF3, IFN regulatory factor 3; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
3.4. SARS‐CoV‐2 ORF9b suppresses the phosphorylation and nuclear translocation of IRF3
TBK1 phosphorylation is a crucial step for IRF3 phosphorylation and nuclear translocation. As SARS‐CoV‐2 ORF9b blocks TBK1 phosphorylation induced by all three critical antiviral pathways, that is, the RIG‐I/MDA‐5–MAVS, TLR3–TRIF, and cGAS–STING signaling pathways, determining whether SARS‐CoV‐2 ORF9b exerts any effect on the phosphorylation and nuclear translocation of IRF3 would be interesting. The expression of RIG‐IN, MAVS, TRIF, or STING in HEK‐293T cells strongly induced the phosphorylation of IRF3, whereas the coexpression of SARS‐CoV‐2 ORF9b in these cells markedly impaired IRF3 phosphorylation (Figure 6). To determine whether SARS‐CoV‐2 ORF9b can still affect IRF3 phosphorylation during viral infection, we performed virus infection studies using the RNA virus SeV as a surrogate for SARS‐CoV‐2 (Figure 7A). The control HeLa cells transfected with an empty vector and HeLa cells expressing SARS‐CoV‐2 ORF9b were infected with SeV. Twenty hours later, the cells were lysed for SDS‐PAGE and immunoblotting analyses. The results indicated that SeV infection induced the phosphorylation of both TBK1 and IRF3. The overexpression of SARS‐CoV‐2 ORF9b in these cells significantly suppressed SeV‐induced TBK1 and IRF3 phosphorylation (Figure 7A). Thus, SARS‐CoV‐2 ORF9b can inhibit SeV infection‐stimulated TBK1 and IRF3 phosphorylation.
The transcription of IFNs is initiated by phosphorylated IRF3 after its translocation into the nucleus. The retention of IRF3 in the cytosol and thus the inhibition of its nuclear translocation arrests its action on IFN induction. As SARS‐CoV‐2 ORF9b inhibits IRF3 phosphorylation, we subsequently examined the effect of SARS‐CoV‐2 ORF9b on SeV‐induced IRF3 nuclear translocation. In resting cells, IRF3 was primarily distributed in the cytosol regardless of the expression of SARS‐CoV‐2 ORF9b (Figure 7B). After SeV infection, IRF3 was translocated into the nucleus of the control cells; however, IRF3 was restricted in the cytosol of cells expressing SARS‐CoV‐2 ORF9b (Figure 7B). These data support the hypothesis that SARS‐CoV‐2 ORF9b inhibits the nuclear translocation of IRF3 upon SeV infection (Figure 7C).
3.5. SARS‐CoV‐2 ORF9b impairs antiviral immunity
Although SARS‐CoV‐2 ORF9b suppresses IFN production, whether it can antagonize antiviral immunity during viral infection remains unknown. Thus, we activated the antiviral signaling pathways by transfecting TBK1 and explored whether SARS‐CoV‐2 ORF9b can affect antiviral immunity. HEK‐293T cells expressing TBK1 alone and HEK‐293T cells expressing both TBK1 and SARS‐CoV‐2 ORF9b were infected with VSV–eGFP, which is commonly used as a model virus to study the effect of IFNs on viral replication. The infection of the virus was determined through the examination of GFP‐positive cells by fluorescence microscopy and flow cytometry (Figure 8A). Viral replication was determined by measuring the titer of the virus released into the culture medium (Figure 8B). The results showed that HEK‐293T cells expressing TBK1 and SARS‐CoV‐2 ORF9b presented a stronger GFP‐positive signal than those expressing TBK1 alone, which suggested that the overexpression of SARS‐CoV‐2 ORF9b in HEK‐293T cells might promote VSV–eGFP infection (Figure 8A). The viral titer in the culture medium of HEK‐293T cells expressing both TBK1 and SARS‐CoV‐2 ORF9b was higher than that in the culture medium of HEK‐293T cells expressing TBK1 alone; thus, SARS‐CoV‐2 ORF9b might facilitate VSV–eGFP replication (Figure 8B). These data indicated that overexpressing SARS‐CoV‐2 ORF9b can enhance virus infection and replication by blunting TBK1‐induced antiviral immunity.
Figure 8.
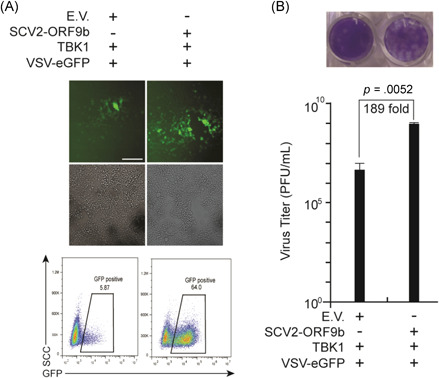
SARS‐CoV‐2 ORF9b overexpression impairs TBK1‐dependent antiviral immunity. Plasmids were transfected into HEK‐293 cells as indicated. Twenty‐four hours after transfection, the cells were infected with vesicular stomatitis virus (VSV)–enhanced green fluorescent protein (eGFP) (MOI = 0.001). Ten hours after infection, (A) the GFP‐positive cells were observed and analyzed with fluorescence microscopy and flow cytometry, and (B) the culture supernatant (20 h postinfection) was harvested for plaque assays to measure the titer of extracellular VSV–eGFP. The fluorescence imaging results are representative of two independent experiments. Scale bar = 50 μm. In Panel b, the results from one representative experiment are shown, three independent biological replicates were analyzed, and the error bars indicate the SD. The statistical significance is shown as indicated. E.V., empty vector; IRF3, IFN regulatory factor 3; MOI, multiplicity of infection; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2; TBK1, TANK‐binding kinase 1
4. DISCUSSION
The COVID‐19 pandemic is affecting the economy, transport, and relationships of countries as well as people′s lives and health worldwide, and researchers worldwide are attempting to find various strategies for the treatment of COVID‐19. The immune system is essential for defense against virus infection; unfortunately, SARS‐CoV‐2 infection subverts this system by suppressing types I and III responses and elevating the proinflammatory response, which will accelerate viral replication and damage the host tissues and organs. 8 Types I and III IFN responses play a critical role in human antiviral immunity against SARS‐CoV‐2, and clinical trials have shown that the restoration of types I and III IFNs in COVID‐19 patients is an effective therapeutic option; thus, further investigation of the mechanism through which SARS‐CoV‐2 evades antiviral immunity is warranted.13, 25, 26, 35 Here, we found that SARS‐CoV‐2 ORF9b antagonizes types I and III IFNs and impairs host antiviral immunity. The overexpression of SARS‐CoV‐2 ORF9b inhibits the production of type I and III IFNs induced by SeV and poly (I:C) and the activation of the RIG‐I/MDA‐5–MAVS, TLR3–TRIF, and cGAS–STING signaling pathways. Molecular mechanism studies showed that SARS‐CoV‐2 ORF9b interacts with RIG‐I, MDA‐5, MAVS, TRIF, STING, and TBK1. Moreover, SARS‐CoV‐2 ORF9b inhibits TBK1 phosphorylation induced by RIG‐/MDA‐5‐MAVS and cGAS–STING signaling and consequently inhibits the phosphorylation and nuclear translocation of IRF3 and types I and III IFN transcription. Furthermore, ectopic expression of the SARS‐CoV‐2 ORF9b facilitated the infection and replication of vesicular stomatitis virus. Thus, SARS‐CoV‐2 ORF9b antagonizes types I and III IFNs and contributes to the pathogenesis of COVID‐19.
Among coronaviruses, a homologous gene of SARS‐CoV‐2 ORF9b has only been found in SARS‐CoV‐1. 36 SARS‐CoV‐1 ORF9b is characterized as an IFN antagonist because it targets mitochondria to enhance the proteasomal degradation of MAVS via its K48‐linked ubiquitination. 36 SARS‐CoV‐1 and SARS‐CoV‐2 ORF9b show 72.4% amino acid identity. Surprisingly, a recent screening study showed that SARS‐CoV‐2 ORF9b does not affect IFN activation induced by RIG‐I signaling. 37 Thus, further investigation of whether SARS‐CoV‐2 ORF9b is also involved in the suppression of IFNs would be interesting. During the preparation of this manuscript, another study showed that SARS‐CoV‐2 ORF9b suppresses type I IFN production by targeting TOM70. 38 Although the molecular mechanism through which SARS‐CoV‐2 ORF9b inhibits the type I IFN response by interacting with TOM70 has not been investigated, it has been proposed that SARS‐CoV‐2 ORF9b might compete with HSP90 for binding to TOM70 or might induce the production of lactic acid, which has been proven to inhibit the IFN response. 38 Consistent with the above findings, we found that SARS‐CoV‐2 ORF9b inhibits the production of types I and III IFNs induced by SeV infection, poly (I:C) stimulation, and activation of the RIG‐I/MDA‐5–MAVS, TLR3–TRIF, and cGAS–STING signaling pathways.
Due to our lack of a biosafety level‐3 laboratory, we had to use another RNA virus, SeV, in the viral infection studies, and we found that the overexpression of SARS‐CoV‐2 ORF9b significantly reduced the production of IFN‐β, IFN‐λ1, ISG56, and CXCL10 stimulated by SeV infection (Figure 1). We also found that SARS‐CoV‐2 ORF9b inhibited the SeV‐induced phosphorylation and nuclear translocation of IRF3 (Figure 7). Moreover, the overexpression of SARS‐CoV‐2 ORF9b in HEK‐293T cells facilitated the replication of VSV–eGFP, which is sensitive to the activation of IFN signaling; thus, SARS‐CoV‐2 ORF9b might enhance VSV–eGFP replication by suppressing IFN production (Figure 8). Although SARS‐CoV‐1 ORF9b reportedly inhibits IFN production, its role in viral infection is currently unknown; thus, this report provides the first demonstration that coronavirus ORF9b suppresses SeV‐induced type I and III IFN production and promotes VSV–eGFP replication.
Luciferase reporter assays showed that SARS‐CoV‐2 ORF9b inhibited the promoter activities of IFN‐β, IFN‐λ1, and ISGs induced by multiple molecules of the RIG‐I/MDA‐5–MAVS, TLR3–TRIF, and cGAS–STING signaling pathways (Figure 3). We experimentally validated the interactions between SARS‐CoV‐2 ORF9b and numerous components of the RIG‐I/MDA‐5 signaling pathways, such as RIG‐I, MDA‐5, and MAVS. Although we found that SARS‐CoV‐2 ORF9b associates with MAVS (Figure 5) and inhibits MASV‐induced IFN activation (Figure 3), this finding cannot explain why SARS‐CoV‐2 also inhibits TRIF‐ and STING‐induced IFN production. Thus, we hypothesize that SARS‐COV‐2 might target TRIF and STING directly or target signaling molecules parallel to or downstream of the point at which the RIG‐I/MDA‐5–MAVS, TLR3–TRIF, and cGAS–STING signaling pathways converge. Thus, we first evaluated whether SARS‐CoV‐2 ORF9b can directly target TRIF and STING. Coimmunoprecipitation results indicated that SARS‐CoV‐2 ORF9b associates with TRIF and STING (Figure 5), which is consistent with the colocalization between SARS‐CoV‐2 ORF9b and TRIF or STING (Figure 4). Although these results can explain why SARS‐CoV‐2 ORF9b inhibits TRIF‐ and STING‐activated IFN signaling pathways, we questioned whether it can target TBK1, which is the molecule at which the RIG‐I/MDA‐5–MAVS, TLR3–TRIF, and cGAS–STING signaling pathways converge.
Coimmunoprecipitation results indicated that SARS‐CoV‐2 ORF9b interacts with TBK1; thus, SARS‐CoV‐2 ORF9b might exert an inhibitory effect on IFN production at the layer of TBK1. Combined with the result that SARS‐CoV‐2 ORF9b inhibits the induction of types I and III IFNs byTBK1 but not IRF3‐5D, we propose that ORF9b fulfills this role at the layer or upstream of TBK1. We then explored whether SARS‐CoV‐2 ORF9b can affect the phosphorylation of TBK1 and IRF3, and immunoblotting results showed that the overexpression of SARS‐CoV‐2 ORF9b inhibits TBK1 phosphorylation induced by RIG‐IN, MAVS, and SeV infection (Figures 6 and 7A). These findings suggest that SARS‐CoV‐2 ORF9b inhibits RIG‐I/MDA‐5–MAVS signaling not only by interacting with RIG‐I, MDA‐5, and MAVS but also by perturbing TBK1 phosphorylation. Similarly, SARS‐CoV‐2 ORF9b suppresses cGAS–STING signaling by interacting with STING and inhibiting TBK1 phosphorylation. For the inhibition of TLR3–TRIF signaling, SARS‐CoV‐2 ORF9b targets TRIF but does not have any effect on TRIF‐induced TBK1 phosphorylation (Figure 6C); however, it can prevent TRIF and TBK1 interactions, which is important for IRF3 phosphorylation and activation, suggesting that the inhibitory effect of SARS‐CoV‐2 ORF9b on TRIF‐induced IFN signaling activation is achieved by directly disturbing TRIF‐TBK1 interaction but not by affecting TBK1 phosphorylation. In addition, the M proteins of both SARS‐COV‐1 and SARS‐COV‐2 can inhibit IFN production by targeting RIG‐I/MDA‐5 signaling but have no effect on TBK1 phosphorylation34, 39; thus, the inhibition of TBK1 phosphorylation is not required for the antagonizing of IFNs by these viral proteins. The overexpression of SARS‐CoV‐2 ORF9b inhibits IRF3 phosphorylation induced by RIG‐IN, MAVS, STING, TRIF, and SeV infection; thus, the inhibition of IRF3 phosphorylation might be indispensable for the antagonization of IFN by these viral proteins. Therefore, SARS‐CoV‐2 ORF9b targets and interacts with RIG‐I, MDA‐5, MAVS, TRIF, and STING and impairs TBK1 phosphorylation activated by the RIG‐I/MDA‐5–MAVS and cGAS–STING signaling pathways but not the TLR3–TRIF signaling pathway. Overall, the following finding of our study is novel compared with those obtained in previous studies: in addition to MAVS, ORF9b might also associate with the dsRNA receptors RIG‐I and MDA‐5, TBK1, TRIF, and STING. Importantly, to our knowledge, we provide the first evidence showing that coronavirus ORF9b might associate TBK1 and inhibit TBK1 phosphorylation induced by signaling from the RIG‐I/MDA‐5 and cGAS–STING pathways. Finally, we showed that coronavirus ORF9b could inhibit TLR3–TRIF signaling pathway by preventing TRIF‐TBK1 interaction.
The localization of SARS‐CoV‐1 ORF9b in the mitochondria, where it degrades the MAVS signalosome, is essential for its IFN inhibitory function. We found that in addition to its mitochondrial localization, SARS‐CoV‐2 also localizes to the ER and Golgi (Figure 4). The ER is an important platform for TRIF and STING, whereas the Golgi is an important platform for TBK1.40, 41, 42 Thus, these findings explain the colocalization and association of SARS‐COV‐2 ORF9b with TRIF, STING, and TBK1. The localization of SARS‐CoV‐2 ORF9b in the ER and Golgi might provide a platform for its inhibitory effects on TRIF‐, STING‐, and TBK1‐induced IFN production. Thus, this study extends our understanding of the molecular mechanisms through which coronavirus ORF9b mediates the antagonizing of IFN.
SARS‐CoV‐2 is more sensitive to IFN treatment than other coronaviruses. 26 Multiple viral proteins that suppress IFN production at different steps become more critical to ensure that the production and function of IFNs are minimized during SARS‐CoV‐2 infection. SARS‐COV‐2 ORF9b targets multiple proteins of distinct antiviral signaling pathways and thus suppresses IFN signaling at different steps. Similarly, SARS‐CoV‐2 ORF6 and MERS‐CoV ORF4b are capable of perturbing multiple antiviral signaling pathways by targeting various components of these pathways.37, 43, 44 Although cGAS‐STING is a cytosolic dsDNA sensing pathway, coronaviruses, a family of RNA viruses, also encode viral proteins such as papain‐like protease to impair STING function; thus, this pathway is essential for defense against coronavirus infection.18, 19 The inhibition of the cGAS‐STING pathway by SARS‐CoV‐2 ORF9b may suggest that this pathway may play a role in SARS‐CoV‐2 clearance; thus, drugs or chemicals, such as 2′−3′cGAMP that activate this pathway may be considered to be used in COVID‐19 treatment.
Distinct methods have shown that SARS‐CoV‐2 ORF9b inhibits type I and III IFN production by targeting multiple proteins of antiviral signaling pathways. We should be conscious that the transfection system might differ from real viral infection. Therefore, further studies should be conducted in the context of real SARS‐CoV‐2 infection experiments. The accessory proteins of coronaviruses have been proposed to be not essential for viral replication. These proteins are not directly involved in viral assembly 5 ; thus, theoretically, ORF9b‐null SARS‐CoV‐2 might be available if it cannot affect the translation and expression of the N protein, which is a structural protein needed for virion assembly. However, whether this ORF9b‐null SARS‐CoV‐2 virus is accessible needs experimental validation. Once this mutant virus is available, the results from real viral infection studies should contribute to our understanding of the role of ORF9b in IFN antagonization.
Although the administration of exogenous IFNs has been shown to be valid for SARS‐CoV2 clearance in both SARS‐CoV‐2 patients and cell models24, 27, 35, 45 full evaluation of these treatments requires extensive studies on the relative importance of all IFN‐antagonizing viral proteins encoded by SAR‐CoV‐2. Thus, our finding that SARS‐COV‐2 ORF9b suppresses type I and III IFN production contributes to our understanding of the pathogenesis of COVID‐19, and the identification of multiple protein targets might provide more precise targets for COVID‐19 treatment.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
Chengjiang Gao and Pei‐Hui Wang conceptualized the study. Lulu Han, Meng‐Wei Zhuang, Jian Deng, Yi Zheng, Jing Zhang, Mei‐Ling Nan, and Xue‐Jing Zhang performed the experiments. Pei‐Hui Wang wrote the first draft of the manuscript. All of the authors contributed to revising the manuscript and approved the final version for publication.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1002/jmv.27050
Supporting information
Supporting information.
ACKNOWLEDGEMENTS
This study was supported by grants from the COVID‐19 emergency tackling research project of Shandong University (Grant No. 2020XGB03 to Pei‐Hui Wang), grants from the Natural Science Foundation of Jiangsu Province (BK20200225 to Pei‐Hui Wang), grants from the Natural Science Foundation of Shandong Province (ZR2020QC085 to Pei‐Hui Wang), grants from the Key Research and Development Program of Shandong Province (2020CXGC011305 to Pei‐Hui Wang), grants from the Natural Science Foundation of China (81930039, 31730026, 81525012) awarded to Chengjiang Gao, and the Fundamental Research Funds of Shandong University (21510078614099), the Fundamental Research Funds of Cheeloo College of Medicine (21510089393109), the China Postdoctoral Science Foundation (2018M642662), and the Natural Science Foundation of China (81901604) awarded to Yi Zheng. We thank the Translational Medicine Core Facility of Shandong University for consultation and instrument availability that supported this study.
Han L, Zhuang M, Deng J, et al. SARS‐CoV‐2 ORF9b antagonizes type I and III interferons by targeting multiple components of the RIG‐I/MDA‐5–MAVS, TLR3–TRIF, and cGAS–STING signaling pathways. J Med Virol. 2021;93:5376–5389. 10.1002/jmv.27050
Lulu Han, Meng‐Wei Zhuang, and Jian Deng contributed equally to this study.
Contributor Information
Chengjiang Gao, Email: cgao@sdu.edu.cn.
Pei‐Hui Wang, Email: pei-hui.wang@connect.hku.hk.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382(8):727‐733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kim D, Lee JY, Yang JS, Kim JW, Kim VN, Chang H. The architecture of SARS‐CoV‐2 transcriptome. Cell. 2020;181(4):914‐921 e910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu DX, Fung TS, Chong KK, Shukla A, Hilgenfeld R. Accessory proteins of SARS‐CoV and other coronaviruses. Antiviral Res. 2014;109:97‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee JS, Park S, Jeong HW, et al. Immunophenotyping of COVID‐19 and influenza highlights the role of type I interferons in development of severe COVID‐19. Sci Immunol. 2020;5(49). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blanco‐Melo D, Nilsson‐Payant BE, Liu WC, et al. Imbalanced host response to SARS‐CoV‐2 drives development of COVID‐19. Cell. 2020;181(5):1036‐1045 e1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hadjadj J, Yatim N, Barnabei L, et al. Impaired type I interferon activity and inflammatory responses in severe COVID‐19 patients. Science. 2020;369:718‐724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Broggi A, Ghosh S, Sposito B, et al. Type III interferons disrupt the lung epithelial barrier upon viral recognition. Science. 2020;369(6504):706‐712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Grajales‐Reyes GE, Colonna M. Interferon responses in viral pneumonias. Science. 2020;369(6504):626‐627. [DOI] [PubMed] [Google Scholar]
- 11. Major J, Crotta S, Llorian M, et al. Type I and III interferons disrupt lung epithelial repair during recovery from viral infection. Science. 2020;369(6504):712‐717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805‐820. [DOI] [PubMed] [Google Scholar]
- 13. Park A, Iwasaki A. Type I and type III interferons—induction, signaling, evasion, and application to combat COVID‐19. Cell Host Microbe. 2020;27(6):870‐878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Totura AL, Baric RS. SARS coronavirus pathogenesis: host innate immune responses and viral antagonism of interferon. Curr Opin Virol. 2012;2(3):264‐275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li J, Liu Y, Zhang X. Murine coronavirus induces type I interferon in oligodendrocytes through recognition by RIG‐I and MDA5. J Virol. 2010;84(13):6472‐6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roth‐Cross JK, Bender SJ, Weiss SR. Murine coronavirus mouse hepatitis virus is recognized by MDA5 and induces type I interferon in brain macrophages/microglia. J Virol. 2008;82(20):9829‐9838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhao J, Zhao J, Van Rooijen N, Perlman S. Evasion by stealth: inefficient immune activation underlies poor T cell response and severe disease in SARS‐CoV‐infected mice. PLOS Pathog. 2009;5(10):e1000636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen X, Yang X, Zheng Y, Yang Y, Xing Y, Chen Z. SARS coronavirus papain‐like protease inhibits the type I interferon signaling pathway through interaction with the STING‐TRAF3‐TBK1 complex. Protein Cell. 2014;5(5):369‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sun L, Xing Y, Chen X, et al. Coronavirus papain‐like proteases negatively regulate antiviral innate immune response through disruption of STING‐mediated signaling. PLOS One. 2012;7(2):e30802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang X, Bai XC, Chen ZJ. Structures and mechanisms in the cGAS‐STING innate immunity pathway. Immunity. 2020;53(1):43‐53. [DOI] [PubMed] [Google Scholar]
- 21. Liu S, Cai X, Wu J, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. 2015;347(6227):aaa2630. [DOI] [PubMed] [Google Scholar]
- 22. Lazear HM, Schoggins JW, Diamond MS. Shared and distinct functions of type I and type III interferons. Immunity. 2019;50(4):907‐923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. O′Brien TR, Thomas DL, Jackson SS, Prokunina‐Olsson L, Donnelly RP, Hartmann R. Weak induction of interferon expression by SARS‐CoV‐2 supports clinical trials of interferon lambda to treat early COVID‐19. Clin Infect Dis. 2020;71:1410‐1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hung IF, Lung KC, Tso EY, et al. Triple combination of interferon beta‐1b, lopinavir‐ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID‐19: an open‐label, randomised, phase 2 trial. Lancet. 2020;395(10238):1695‐1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Abigail Vanderheiden PR, Chirkova Tatiana, Upadhyay AmitA, et al. Type I and type III IFN restrict SARS‐CoV‐2 infection of human airway epithelial cultures. bioRxiv. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lokugamage KG, Hage A, Schindewolf C, Rajsbaum R, Menachery VD. SARS‐CoV‐2 is sensitive to type I interferon pretreatment. bioRxiv. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stanifer ML, Kee C, Cortese M, et al. Critical role of type III interferon in controlling SARS‐CoV‐2 infection in human intestinal epithelial cells. Cell Rep. 2020;32:107863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang PH, Fung SY, Gao WW, et al. A novel transcript isoform of STING that sequesters cGAMP and dominantly inhibits innate nucleic acid sensing. Nucleic Acids Res. 2018;46(8):4054‐4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Song G, Liu B, Li Z, et al. E3 ubiquitin ligase RNF128 promotes innate antiviral immunity through K63‐linked ubiquitination of TBK1. Nat Immunol. 2016;17(12):1342‐1351. [DOI] [PubMed] [Google Scholar]
- 30. Liu B, Zhang M, Chu H, et al. The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63‐linked polyubiquitination. Nat Immunol. 2017;18(2):214‐224. [DOI] [PubMed] [Google Scholar]
- 31. Wang PH, Ye ZW, Deng JJ, et al. Inhibition of AIM2 inflammasome activation by a novel transcript isoform of IFI16. EMBO Rep. 2018;19(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zheng Y, Zhuang M‐W, Han L, et al. Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) membrane (M) protein inhibits type I and III interferon production by targeting RIG‐I/MDA‐5 signaling. bioRxiv. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhuang MW, Cheng Y, Zhang J, et al. Increasing host cellular receptor‐angiotensin‐converting enzyme 2 expression by coronavirus may facilitate 2019‐nCoV (or SARS‐CoV‐2) infection. J Med Virol. 2020;92:2693‐2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zheng Y, Zhuang MW, Han L, et al. Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) membrane (M) protein inhibits type I and III interferon production by targeting RIG‐I/MDA‐5 signaling. Signal Transduct Target Ther. 2020;5(1):299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Felgenhauer U, Schoen A, Gad HH, et al. Inhibition of SARS‐CoV‐2 by type I and type III interferons. J Biol Chem. 2020;295:13958‐13964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shi CS, Qi HY, Boularan C, et al. SARS‐coronavirus open reading frame‐9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J Immunol. 2014;193(6):3080‐3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yuen CK, Lam JY, Wong WM, et al. SARS‐CoV‐2 nsp13, nsp14, nsp15, and orf6 function as potent interferon antagonists. Emerg Microbes Infect. 2020;9(1):1418‐1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jiang HW, Zhang HN, Meng QF, et al. SARS‐CoV‐2 Orf9b suppresses type I interferon responses by targeting TOM70. Cell Mol Immunol. 2020;17:998‐1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Siu KL, Kok KH, Ng MJ, et al. Severe acute respiratory syndrome coronavirus M protein inhibits type I interferon production by impeding the formation of TRAF3.TANK.TBK1/IKKepsilon complex. J Biol Chem. 2009;284(24):16202‐16209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pourcelot M, Zemirli N, Silva Da Costa L, et al. The Golgi apparatus acts as a platform for TBK1 activation after viral RNA sensing. BMC Biol. 2016;14:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang BC, Nandakumar R, Reinert LS, et al. STEEP mediates STING ER exit and activation of signaling. Nat Immunol. 2020;21(8):868‐879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fan S, Chen S, Liu Y, et al. Zebrafish TRIF, a Golgi‐localized protein, participates in IFN induction and NF‐kappaB activation. J Immunol. 2008;180(8):5373‐5383. [DOI] [PubMed] [Google Scholar]
- 43. Lei X, Dong X, Ma R, et al. Activation and evasion of type I interferon responses by SARS‐CoV‐2. Nat Commun. 2020;11(1):3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang Y, Ye F, Zhu N, et al. Middle East respiratory syndrome coronavirus ORF4b protein inhibits type I interferon production through both cytoplasmic and nuclear targets. Sci Rep. 2015;5:17554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhongji Meng TW, Chen Li, Chen Xinhe, et al. An experimental trial of recombinant human interferon alpha nasal drops to prevent coronavirus disease 2019 in medical staff in an epidemic area. medRxiv. 2020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.