

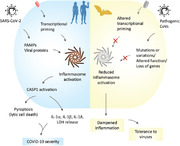
Abstract
Coronaviruses (CoVs) are RNA viruses that cause human respiratory infections. Zoonotic transmission of the SARS‐CoV‐2 virus caused the recent COVID‐19 pandemic, which led to over 2 million deaths worldwide. Elevated inflammatory responses and cytotoxicity in the lungs are associated with COVID‐19 severity in SARS‐CoV‐2‐infected individuals. Bats, which host pathogenic CoVs, operate dampened inflammatory responses and show tolerance to these viruses with mild clinical symptoms. Delineating the mechanisms governing these host‐specific inflammatory responses is essential to understand host–virus interactions determining the outcome of pathogenic CoV infections. Here, we describe the essential role of inflammasome activation in determining COVID‐19 severity in humans and innate immune tolerance in bats that host several pathogenic CoVs. We further discuss mechanisms leading to inflammasome activation in human SARS‐CoV‐2 infection and how bats are molecularly adapted to suppress these inflammasome responses. We also report an analysis of functionally important residues of inflammasome components that provide new clues of bat strategies to suppress inflammasome signaling and innate immune responses. As spillover of bat viruses may cause the emergence of new human disease outbreaks, the inflammasome regulation in bats and humans likely provides specific strategies to combat the pathogenic CoV infections.
Keywords: Bat immunity, Coronaviruses, COVID‐19, Immune tolerance, Inflammasome, NLRP3, SARS‐CoV‐2
Graphical Abstract
Human SARS‐CoV‐2 infection‐induced inflammasome activation is associated with COVID‐19 severity, whereas bats, which host pathogenic CoVs, show dampened inflammasome activation and immune tolerance to viral infections.
Abbreviations
- ACE2
angiotensin‐converting enzyme 2
- AIM2
absent in melanoma 2
- ALRs
AIM2‐like receptors
- ASC
adaptor protein apoptosis‐associated speck‐like protein containing a CARD
- CARD
caspase activation and recruitment domain
- CASP1
caspase‐1
- CASP3
caspase‐3
- cGAMP
cyclic GMP‐AMP
- cGAS
cyclic GMP‐AMP synthase
- COVID‐19
coronavirus disease 2019
- DAMPs
damage‐associated molecular patterns
- E protein
envelope protein
- ER
endoplasmic reticulum
- GSDMD
Gasdermin D
- HMGB‐1
high mobility group box protein 1
- IL‐1Ra
IL‐1 receptor antagonist
- LDH
lactate dehydrogenase
- LRR
leucine‐rich repeat
- MERS‐CoV
Middle East respiratory syndrome coronavirus
- MHV
mouse hepatitis virus
- NINJ1
nerve injury protein 1
- NLRP3
NOD‐like receptor family pyrin domain containing 3
- NLRs
Nod‐like receptors
- ORF3a
open reading frame 3a protein
- ORF7
open reading frame 7 protein
- ORF8
open reading frame 8 protein
- ORF8‐Δ382
a 382‐nucleotide deletion in ORF8 encoding genome
- PANoptosis
pyroptosis, apoptosis and necroptosis
- Poly(I:C)
polyinosinic:polycytidylic acid
- PYD
pyrin domain
- PYHIN
pyrin and HIN domain
- S protein
spike protein
- SARS‐CoV
severe acute respiratory syndrome coronavirus‐1
- SARS‐CoV‐2
severe acute respiratory syndrome coronavirus‐2
- STING
stimulator of IFN genes
- TMPRSS2
transmembrane protease serine 2
- TRAF3
TNFR‐associated factor 3
1. INTRODUCTION
Coronaviruses (CoVs) are single‐stranded RNA viruses in the family of Coronaviridae. 1 Zoonotic transmission of CoVs causes mild to severe human infections. Seven different CoVs have entered humans so far. Human infections of OC43‐CoV, 229E‐CoV, NL63‐CoV, and HKU1‐CoVs cause seasonal colds and are mostly asymptomatic, 1 , 2 , 3 whereas infections of severe acute respiratory syndrome CoV (SARS‐CoV or SARS‐CoV‐1) and Middle East respiratory syndrome CoV (MERS‐CoV) led to severe disease outbreaks. 1 , 2 , 3 SARS‐CoV‐2 infection caused the ongoing pandemic and led to more than 2 million deaths worldwide. SARS‐CoV‐2 infects the upper respiratory tract and binds to host receptor protein, angiotensin‐converting enzyme 2 (ACE2), through its spike protein (S protein). 4 , 5 , 6 The membrane‐bound host serine protease, transmembrane protease serine 2 (TMPRSS2), activates S protein to facilitate the entry of SARS‐CoV‐2 into the host cells (Fig. 1). 4 The RNA genome of the SARS‐CoV‐2 will be released into the cytoplasm, where it replicates to generate new progeny of virions. 1 , 2 New virion particles transport through the secretion pathway to be released into extracellular space. In most cases, the SARS‐CoV‐2 infection is asymptomatic and, in some cases, it spreads to the lower respiratory tract causing pneumonia. 2 , 6 SARS‐CoV‐2 infection in some patients becomes severe and develops into acute respiratory distress syndrome leading to severe respiratory abnormalities and mortality. 2 , 6 The SARS‐CoV‐2‐induced moderate and severe disease in humans is collectively named coronavirus disease 2019 (COVID‐19). 2 , 6
FIGURE 1.
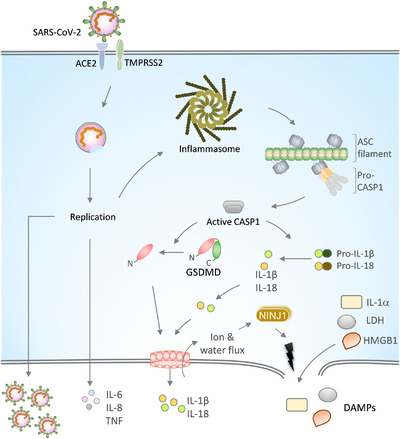
SARS‐CoV‐2 infection, inflammasome activation, and release of proinflammatory cytokines.
Spike (S) protein of the SARS‐CoV‐2 binds to the ACE2 receptor on the host cell membrane for its entry into the cell. The cell surface protease TMPRSS2 cleaves S protein to enable membrane fusion and entry of the virus. The replication of the viral RNA genome generates new SARS‐CoV‐2 virions. SARS‐CoV‐2 replication triggers the activation and assembly of the inflammasome and release of IL‐1α, IL‐1β, and IL‐18 cytokines that are implicated in COVID‐19 severity. Upon inflammasome activation, ASC oligomerizes into large filamentous structures/speck‐like structures that recruit and activate CASP1 via CARD‐mediated homotypic interactions. Active CASP1 cleaves pro‐IL‐1β and pro‐IL‐18 into mature forms. The CASP1 also cleaves GSDMD to liberate its N‐terminal domain that oligomerizes into a pore‐structure on the plasma membrane. GSDMD pores facilitate the passage of ions, water, and small molecules like IL‐1β and IL‐18. GSDMD pore formation licenses plasma membrane rupture through NINJ1 and triggers lytic cell death. Plasma membrane rupture is crucial for releasing large molecular weight DAMPs like IL‐1α, LDH, and HMGB1. Inflammasome‐independent cytokines, IL‐6, IL‐8, and TNF, secreted in response to SARS‐CoV‐2 infection, are associated with CoVID‐19 severity
The SARS‐CoV‐2‐driven pathogenesis in human patients is a complex process. It is increasingly evident that excessive cytokine responses and infiltration of inflammatory cells, particularly inflammatory monocytes and neutrophils, are associated with COVID‐19 severity. 7 , 8 , 9 Virus‐induced cytopathic effects of lung epithelial cells in association with excessive inflammatory cytokines trigger acute lung injury and loss of respiratory function during COVID‐19. 6 , 7 , 8 , 9 , 10 Endothelial barrier damage further leads to viral dissemination and spread to other organs. Dysregulated immune responses and increased inflammatory cytokine signatures also occur in other pathogenic RNA virus infections. 8 , 11 Inflammation‐driven excessive cytokine response in response to SARS‐CoV‐2 infection is highly associated with severe COVID‐19 and a great effort has been put to understand and target these responses for alleviating COVID‐19 severity. Several studies report the association of excessive levels of proinflammatory cytokines, IL‐1α, IL‐1β, IL‐6, IL‐8, and TNF with COVID‐19 severity. 10 , 12 , 13 , 14 , 15 , 16 The mechanisms triggering these specific cytokine responses during SARS‐CoV‐2 remain elusive.
2. INFLAMMASOME ACTIVATION
Inflammasomes are heteromeric multiprotein complexes that promote the activation of caspase‐1 (CASP1) protease. 17 , 18 The CASP1 activation in turn triggers the release of proinflammatory IL cytokines, IL‐1β and IL‐18, and damage‐associated molecular patterns (DAMPs) like IL‐1α, high mobility group box protein 1 (HMGB1), and lactate dehydrogenase (LDH) 19 , 20 (Fig. 1). Activation of innate immune receptors, such as Nod‐like receptors (NLRs) and AIM2‐like receptors (ALRs), recruits an adaptor protein, apoptosis‐associated speck‐like protein containing a CARD (ASC), to assemble the inflammasome complex and trigger proximity‐induced CASP1 activation 18 , 21 (Fig. 1). The widely studied NOD‐like receptor family pyrin domain (PYD) containing 3 (NLRP3) inflammasome requires 2 signals for its activation. The first signal provides priming of the NLRP3 inflammasome through inflammatory stimuli (like TLR or TNF receptor (TNFR)) engagement that promote NF‐kB dependent expression of the NLRP3 and IL‐1β. 22 , 23 The priming signal also facilitates NLRP3 post‐translational modifications favoring inflammasome assembly. 24 , 25 , 26 The second signal, known as the activation signal, is provided by pathogen‐associated molecular patterns or cellular damage signals. 17 , 27 , 28 The second signal triggers NLRP3 inflammasome assembly, ASC recruitment, and activation of the CASP1. The ASC recruitment to NLRs or ALRs is mediated by PYD‐mediated homotypic interactions. ASC polymerizes into large filamentous/speck‐like structures, which further recruit CASP1 through caspase activation and recruitment domain (CARD)‐mediated homotypic interactions between them 18 , 29 (Fig. 1). CASP1 cleaves pro‐IL‐1β and pro‐IL‐18 into mature IL‐1β and IL‐18 forms. CASP1 also cleaves a pore‐forming protein Gasdermin D (GSDMD) at the linker region of its N‐ and C‐terminal domains to liberate it from its autoinhibitory conformation 30 , 31 , 32 (Fig. 1). The N‐terminal domain of the GSDMD is transported to the plasma membrane and oligomerize into a pore structure whose pore size is sufficient to enable the passage of small molecules and mature IL‐1β and IL‐18. 18 , 19 The formation of GSDMD pores in turn promotes osmotic lysis and a lytic form of cell death called pyroptosis 30 , 31 , 32 (Fig. 1). Earlier it was considered that the plasma membrane rupture and osmotic lysis that occur after GSDMD pore formation is a passive event. A recent study challenged this assumption and demonstrates that the plasma membrane rupture during pyroptosis is a programmed phenomenon regulated by nerve injury protein 1 (NINJ1) 33 (Fig. 1). Thus, inflammasome activation triggers GSDMD pore formation and plasma membrane rupture for releasing large molecular weight DAMPs like HMGB1 and LDH. A recent study demonstrates that a lectin protein, Galectin‐1, is also a bona fide DAMP released in inflammatory cell death and promotes inflammation. 34 Serum levels of DAMPs are considered as a measure of inflammatory cell death activation in disease settings. Inflammasome activation facilitates antimicrobial host defense during microbial infections and compromised activation leads to severe pathology. However, inflammasomes also drive chronic inflammatory responses in autoinflammatory and metabolic diseases. 28 , 35 Dysregulated or excessive inflammasome activation accelerates microbial pathogenesis through elevated proinflammatory responses or cytokine storm. 21 , 23 , 28 , 29 , 30 , 35
3. THE INFLAMMASOME ACTIVATION AND COVID‐19 SEVERITY
The NLRP3 inflammasome is a widely studied complex and is known to be activated by RNA viruses. 17 , 18 , 23 , 36 Inflammasome responses play an essential role in clearing viruses and virus‐infected cells and promote tissue repair responses. 27 , 36 , 37 , 38 , 39 , 40 In many RNA virus infection cases, viral RNAs generated during the replication process are sufficient to activate the NLRP3 inflammasome. 27 , 36 In some cases, viral proteins also regulate the activation of the NLRP3 inflammasome. The inflammasome‐induced cell death and the release of proinflammatory IL‐1α, IL‐1β, and IL‐18 cytokines drive the recruitment of inflammatory immune cells to the infection site and promote viral clearance and tissue repair processes. 11 , 27 , 36 , 37 , 38 , 39 , 40
The increased levels of specific cytokines during severe COVID‐19 suggest dysregulated inflammatory responses in pathogenesis progression. 7 , 10 Immune response profiling of moderate and severe COVID‐19 patients indicates a correlation of inflammasome‐associated proinflammatory cytokines IL‐1α and IL‐1β with the severity of the disease. 8 , 10 , 12 , 13 , 41 Although several studies indicate the inflammasome role in severe COVID‐19 disease progression, the activation of inflammasome in response to SARS‐CoV‐2 infection was obscure. A recent study demonstrated that the SARS‐CoV‐2 infection in primary human monocytes triggers CASP1 activation and IL‐1β production, providing direct evidence for the activation of the inflammasome and proinflammatory cytokine release. 41 Similar to other RNA virus infections, replication of the SARS‐CoV‐2 is critical for the activation of the inflammasome. 39 , 40 , 41 , 42 Experiments with MCC950, an inhibitor specific to NLRP3, suggest the specific role of NLRP3 inflammasome in SARS‐CoV‐2 induced CASP1 activation and IL‐1β release. 41 Importantly, increased levels of LDH were seen in severe COVID‐19 disease individuals indicating the lytic cell death programs (such as necroptosis and pyroptosis) after SARS‐CoV‐2 infection. 41 SARS‐CoV‐2 infection in primary human monocytes triggers LDH release; however, MCC950 fails to suppress its release. This suggests the possibility of the NLRP3 inflammasome‐independent mechanisms triggering the lytic form of cell death and the release of DAMPs, like LDH, in SARS‐CoV2 infection (Fig. 2). 41 , 43 The activation of NLRP3 inflammasome and LDH release indicates potential activation of the pyroptosis along with other lytic cell death programs during SARS‐CoV‐2 infection and in COVID‐19. Additional studies monitoring the activation of GSDMD and testing SARS‐CoV2 infection in Gsdmd –/– cells deliver the specific contribution of pyroptosis and other cell death mechanisms in mediating the release of LDH and other DAMPs. Mouse hepatitis virus (MHV) is a mouse‐adapted lab strain, extensively used to understand host responses during CoV infections. 3 A recent study demonstrates that the MHV infection in primary macrophages triggers pyroptosis, apoptosis, and necroptosis (PANoptosis), suggesting the ability of CoVs to engage multiple inflammatory lytic cell death programs. 44 It remains to be established whether pathogenic human CoVs, like SARS‐CoV‐2, engage PANoptosis. The prominent activation of the NLRP3 inflammasome is observed in response to the SARS‐CoV‐2 infection and is active in COVID‐19 patients who require hospitalization. 41 , 43 Higher levels of CASP1, IL‐1β, and IL‐18 are seen in COVID‐19 patients compared with healthy individuals. 12 , 15 , 41 , 45 Analyzing peripheral blood mononuclear cells isolated from COVID‐19 patients indicates the active inflammasome and CASP1 in lethal cases of COVID‐19. 41 Also, NLRP3 inflammasome and ASC speck formation were observed in the area of lung injury and postmortem tissues from COVID‐19 patients. 41 Thus, SARS‐CoV‐2 infection in human lungs engages the inflammasome activation, which may be a likely therapeutic target for alleviating inflammation and severity of COVID‐19. 10 , 41 , 45 , 46 The excessive release of inflammasome cytokines may trigger uncontrolled immune cell infiltration and severe pathology in the lung. Furthermore, these findings corroborate other studies supporting the use of inflammasome activation as a marker of COVID‐19 severity.
FIGURE 2.
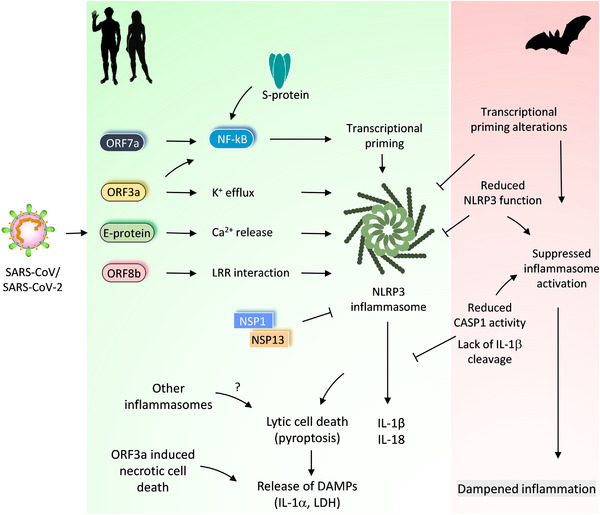
Differential regulation of inflammasome activation in bats and humans and its impact on CoV‐induced immune responses.
Pathogenic CoVs (SARS‐CoV and SARS‐CoV‐2) trigger activation of the NLRP3 inflammasome in host cells. SARS‐CoV‐2 viroporins, ORF3a and E protein promote potassium (K+) efflux or calcium (Ca2+) release into the cytosol, which activates the NLRP3 inflammasome. These viroporins also stimulate NF‐kB activation and transcriptional priming of NLRP3, IL‐1b and IL‐18. ORF7a and S proteins also promote priming and NF‐kB activation. ORF8b of the SARS‐CoV interacts with the LRR domain of the NLRP3 to promote its activation. The ORF3a and S protein of SARS‐CoV‐2 also engage NLRP3 inflammasome activation. NSP1 and NSP13 proteins of the SARS‐CoV‐2 abolish NLRP3 inflammasome activation and CASP1 mediated IL‐1β release. The inflammasome activation and release of IL‐1β and IL‐18 correlate with COVID‐19 severity in SARS‐CoV‐2 infected patients. Other inflammasomes or lytic cell death routes (ORF3a induced necrotic cell death) may also promote the release of DAMPs and excessive inflammatory responses. Bats have adapted to suppress NLRP3 inflammasome activation through regulating transcriptional priming and assembly of the inflammasome complex. This inflammasome suppression in bats dampens inflammation and confer immune tolerance to pathogenic CoVs
Older males with comorbidities show severe COVID‐19 pathology after SARS‐CoV‐2 infection. 47 The comorbidities, like diabetes, obesity, cardiovascular diseases, cancers, digestive and respiratory system disease, and neurologic disorders, make older males more susceptible to COVID‐19. 47 , 48 , 49 Physiologic aging is associated with chronic low‐grade inflammation, which increases the risk of age‐related diseases. 49 , 50 The ligands that appear in aging promote increased NLRP3 inflammasome activation and chronic low‐grade inflammation in older individuals. 50 Thus, NLRP3 inflammasome and chronic low‐grade inflammation in older individuals predispose them to COVID‐19 severity. 51 The higher risk of males compared with females indicates the role of steroid hormones in determining COVID‐19 severity. 52 Female reproductive hormones, estrogen and progesterone, suppress inflammatory immune responses and NLRP3 inflammasome function. 52 , 53 The steroid hormone‐induced anti‐inflammatory responses in females may have a protective role in COVID‐19.
4. MECHANISMS OF PATHOGENIC CORONAVIRUS MEDIATED NLRP3 INFLAMMASOME ACTIVATION
How SARS‐CoV‐2 activate inflammasome, pyroptosis, and other lytic forms of cell death? Currently, the precise mechanisms of SARS‐CoV‐2 induced inflammasome activation are unclear. Potassium (K+) efflux from cytosol or endoplasmic reticulum (ER) stress‐triggered calcium (Ca2+) leakage into the cytosol promotes NLRP3 inflammasome activation. 18 , 21 , 23 RNA virus proteins that act like viroporins or ion channels are linked to the activation of NLRP3 inflammasome and release of IL‐1β. 27 , 46 Also, SARS‐CoV viroporins are associated with the activation of the NLRP3 inflammasome. SARS‐CoV envelope protein (E protein) is a viroporin that is localized to the ER and the Golgi complex and functions as an ion channel to promote Ca2+ leakage and NLRP3 activation 54 , 55 (Fig. 2). SARS‐CoV open reading frame 3a protein (ORF3a) is also a viroporin that promotes K+ efflux. 56 This further promotes mitochondrial reactive oxygen species and activation of the NLRP3 inflammasome and release of IL‐1β. 56 Both E protein and ORF3a of SARS‐CoV provide a priming signal to activate NF‐kB signaling, which up‐regulates the transcription of NLRP3, IL‐1b, and other proinflammatory cytokines 57 , 58 (Fig. 2). In addition to K+ efflux and NF‐kB activation, ORF3a also facilitates the TNFR‐associated factor 3 (TRAF3)‐mediated ubiquitination of the ASC to engage the NLRP3 inflammasome. 59 , 60 The amino acid residues of E protein and ORF3a, critical for ion channel activity, NF‐kB activation, and NLRP3 inflammasome assembly, are conserved in SARS‐CoV and SARS‐CoV‐2 (Supplemental Fig. S1). E protein and ORF3a may likely trigger NLRP3 inflammasome activation in response to SARS‐CoV‐2 infection and promote COVID‐19 severity. A new study demonstrates that the ORF3a of SARS‐CoV‐2 activates the NLRP3 inflammasome by regulating its expression and ASC‐mediated inflammasome assembly. 61 The role of SARS‐CoV‐2 ORF3a in inflammasome‐induced pathogenicity remains to be studied. Nevertheless, viroporins of pathogenic CoVs, which form ion channels, retain the ability to activate the NLRP3 inflammasome like other RNA viruses (Fig. 2). In addition to promoting the NLRP3 inflammasome, the SARS‐CoV ORF‐3a can also trigger necrotic cell death. 62 The dual function of ORF‐3a in promoting inflammatory cell death pathways (pyroptosis and necroptosis) may explain the inflammasome‐independent release of LDH during SARS‐CoV‐2 infection. 41 , 62 In addition to E protein and ORF3a, the spike protein (S protein) and ORF7a of the SARS‐CoV promote NF‐kB activation and inflammasome priming (Fig. 2). 57 , 63 The SARS‐CoV‐2 S protein also triggers inflammasome priming in COVID‐19 patient‐derived macrophages. 64 It appears that SARS‐CoV‐2‐induced inflammasome activation is a complex process that involves multiple viral components. A recent study demonstrates that nonstructural protein (NSP) 1 and NSP13 of SARS‐CoV‐2 abolish NLRP3 inflammasome‐mediated CASP1 activation and IL‐1β release (Fig. 2). 65 Thus, the SARS‐CoV‐2 also operates viral mechanisms counteracting inflammasome activation.
ORF8b protein of SARS‐CoV acts like a virulence factor and is reported to interfere with IFN responses. 66 , 67 SARS‐CoV ORF8 exists in 3 different forms, ORF8ab, ORF8a, and ORF8b. A single ORF encodes ORF8ab; however, a 29‐nucleotide deletion in ORF8ab results in the generation of overlapping ORF8a and ORF8b. Deletion mutants of ORF8 in SARS‐CoV associate with decreased replicative fitness. 1 , 68 , 69 SARS‐CoV ORF8b protein forms insoluble aggregates enabling altered cellular functions. 70 SARS‐CoV ORF8b promotes NLRP3 inflammasome activation in macrophages and triggers cell death in epithelial cells and macrophages. 70 The ORF8b directly interacts with leucine‐rich repeat (LRR) domain of the NLRP3 and localizes to NLRP3‐ASC inflammasome compartments. 70 Whether SARS‐CoV‐2 ORF8 activates the inflammasome and IL‐1β release is not investigated so far. A circulating variant of SARS‐CoV‐2 with a 382‐nucleotide deletion in its ORF8 encoding genome (ORF8‐Δ382) is reported recently, which affects its expression. 71 , 72 Patients infected with ORF8‐Δ382 variant of SARS‐CoV‐2 show milder symptoms compared with wild‐type (WT)‐virus infected individuals. 71 Also, lower systemic levels of proinflammatory cytokines are observed in individuals infected with the ORF8‐Δ382 variant. 71 In vitro studies in nasal epithelial cells show similar transcription patterns after WT SARS‐CoV‐2 and ORF8‐Δ382 mutant infection. 73 Thus, the ORF8 function appeared to be associated with the regulation of proinflammatory responses originated from immune cells during SARS‐CoV‐2 infection. Of note, SARS‐CoV and SARS‐CoV‐2 display striking variation in ORF8 protein sequences (Supplemental Fig. S1). Bat CoVs, which are phylogenetically related to SARS‐CoV, also show variation in the ORF8 region. 1 It appears that the SARS‐CoV‐2 variations associate with host proinflammatory responses and may determine the outcome of COVID‐19 severity. It remains to be established the precise role of ORF8 in SARS‐CoV‐2 inflammasome activation and its impact on host proinflammatory responses. Except ORF8, SARS‐CoV and SARS‐CoV‐2 proteins that promote inflammasome activation are conserved. As several proteins of the SARS‐CoV‐2 are associated with inflammasome regulation, the precise contribution of each viral protein or combination of proteins that determine SARS‐CoV‐2‐induced inflammasome activation or suppression needs to be investigated. Defining specific signatures in SARS‐CoV and SARS‐CoV‐2 proteins, which are critical for triggering inflammasome activation, may facilitate the prediction of the pathogenesis of CoVs in bats that have not infected humans so far. In vivo functional studies with SARS‐CoV‐2 mutant viruses, such as ORF3 mutant restricting its binding to TRAF3, in animal models (mice, hamsters or monkeys) will help to annotate the key elements of SARS‐CoV‐2 orchestrating inflammasome activation and COVID‐19 severity.
Type I IFNs promote efficient activation of the inflammasome and pyroptosis. 74 , 75 Virus‐induced type I IFNs promote the activation of inflammasome responses in various RNA virus infections. 27 , 74 Type I IFNs at the acute stage of the infection or before peak SARS‐CoV or MERS‐CoV viral titers robustly inhibit the dysregulated inflammatory response and lung damage and marginally affect viral titers. 76 , 77 MERS‐CoV, SARS‐CoV, and SARS‐CoV‐2 viruses employ highly evolved viral mechanisms to restrict type I IFN secretion in infected host cells. 76 , 77 , 78 , 79 , 80 This early on type I IFN antagonism during CoV infection helps increase in viral titers and spread. In lethal SARS‐CoV infection, a robust increase in viral titers and delayed type I IFN signaling drive pathologic inflammatory response and lung damage. 76 , 77 In particular, delayed type I IFNs promote the recruitment of inflammatory monocyte and macrophages, resulting in an uncontrolled release of inflammatory cytokines and chemokines in the lung. 76 , 77 The subsequent loss of epithelial integrity and endothelial damage results in lung pathology and viral spread to other organs. Delayed type I IFNs during lethal SARS‐CoV‐2 infection may trigger elevated inflammasome activation, pyroptosis, and release of IL‐1β and IL‐18. Both pyroptosis and proinflammatory cytokines may drive the loss of epithelial and endothelial integrity and lung damage.
Dysregulated cytokine responses or cytokine storms occur in systemic bacterial infections that lead to organ failure and mortality. 81 , 82 RNA virus infections like influenza, Zika, and Ebola are associated with dysregulated inflammatory responses and severe pathology. 83 , 84 , 85 , 86 , 87 , 88 , 89 , 90 , 91 , 92 , 93 Excessive cytokine responses during highly pathogenic influenza infections, H1N1 and H5N1, are associated with severe pulmonary pathology. Do SARS‐CoV‐2 inflammatory cytokine signature mimics other pathogenic RNA virus infections? Recent studies show that the levels of IL‐1α and IL‐1β are significantly lower in COVID‐19‐infected patients when compared with influenza‐infected individuals. 14 , 41 , 80 , 94 In support of these studies, the analysis of gene expression profiles in the upper respiratory tract of COVID‐19 patients with other respiratory virus infections further suggests a striking decrease in the expression of inflammasome genes. 13 However, IL‐1α and IL‐1β levels were higher in COVID‐19 patients compared with healthy controls suggesting a correlation of inflammasome activation with SARS‐CoV‐2‐mediated disease severity. 10 , 12 , 15 Even though the cytokine storm is considered a critical feature of COVID‐19, these new studies reveal a specific set of inflammatory cytokine signatures responsible for the pathogenesis and inflammasome cytokines are among them. Proinflammatory IL‐1 cytokine (IL‐1α and IL‐1β) signaling often promotes host defense responses during RNA virus infections. IL‐1 receptor antagonist (IL‐1Ra) is a natural inhibitor of IL‐1 cytokine signaling. 95 IL‐1Ra‐mediated regulation of IL‐1 cytokines enables balanced control of inflammation and retain homeostasis. Like other pathogenic RNA virus infections, increased levels of IL‐1Ra were reported in individuals with severe COVID‐19 disease. 10 , 12 , 14 , 96 It is not clear yet why inflammation dampening IL‐1Ra is elevated in SARS‐CoV‐2 infection. In the case of Ebola and Zika virus infections, where increased IL‐1Ra levels are reported, IL‐1Ra is required for protective immune responses and its deletion leads to severe proinflammatory responses. 86 , 97
5. BAT CORONAVIRUSES, INFLAMMASOME REGULATION, AND IMMUNE TOLERANCE
Bats are the only mammals that fly and host several pathogenic RNA viruses like CoVs, Ebola, and Nipah viruses. Bats exhibit mild clinical symptoms or often asymptomatic despite high viral titers. 98 , 99 , 100 Pathogenic human CoVs, SARS‐CoV, SARS‐CoV‐2, and MERS‐CoV, likely originated from bats and zoonotic transmission of these CoVs caused severe human outbreaks and pandemics. 1 , 6 , 101 , 102 , 103 Bats host diverse CoVs that are phylogenetically related to the pathogenic human CoVs. 1 , 101 , 102 This indicates a potential threat of a spillover of bat‐originated CoVs that may cause future outbreaks. Bats display high metabolic rates, however, their life span is long. 104 , 105 , 106 Bats appear to show efficient antiviral mechanisms mainly because of constitutive expression of type I IFNs and dampened inflammatory responses. 103 , 105 , 106 , 107 As bats are mammals, they express innate immune receptors and inflammasome‐associated proteins like humans. How bats, unlike humans, alleviate inflammatory responses while hosting several RNA viruses? Do CoVs engage inflammasome activation in bats? Despite the difficulty in performing experiments in the bat system, recent studies attempted to understand the inflammasome activation in bats and provide a basis for regulating inflammation in bats.
The bat immune cells show a striking variation in NLRP3 inflammasome activation compared with human or mouse cells. 108 Stimulations with various NLRP3 ligands show diminished activation of the NLRP3 inflammasome in bat immune cells. 108 Reduced priming/transcriptional up‐regulation of NLRP3 is a characteristic feature of bat immune cells 108 (Fig. 2). Also, altered LRR domain function in bats contributes to the dampened NLRP3 inflammasome function in bats. 108 However, expression of ASC, CASP1, and IL‐1β appeared to be not affected in bat immune cells like human cells. 108 , 109 Because of NLRP3 priming and altered LRR domain defects, bat immune cells show lesser formation ASC speck assemblies and reduced release of IL‐1β cytokine. 108 Besides, bat cells show impaired pyroptosis and LDH release compared with human cells. 108 Infection of bat immune cells with MERS‐CoV fails to induce NLRP3 inflammasome activation and IL‐1β release with unaltered viral loads. 108 , 110 Thus, the bat's immune system is adapted to repress NLRP3 inflammasome activation and proinflammatory responses (Fig. 2).
Flight in bats show high metabolic demand and mitochondrial oxidative phosphorylation activity. This results in DNA damage and the release of mitochondrial or nuclear self‐DNA into the cytoplasm. Absent in melanoma 2 (AIM2) is a cytosolic DNA sensor that assembles inflammasome and triggers pyroptosis. Bats show complete loss of AIM2 and related Pyrin and HIN domain (PYHIN) gene family that play critical roles in nucleic acid sensing at multiple intracellular compartments. 104 , 109 , 111 Cyclic GMP‐AMP synthase (cGAS) is also a cytosolic DNA sensor that produces cyclic GMP‐AMP (cGAMP) upon DNA sensing. cGAS binds to stimulator of IFN genes (STING) protein that activates type I IFN responses. 112 Bats show dampened STING mediated IFN activation due to a mutation at the S358 phosphorylation site of the STING, which is essential for type I IFN response. 113 Thus, bat DNA sensing system is attenuated at multiple signaling pathways to curtail inflammasome activation and type I IFN responses. In addition, a recent study unravels variation in amino acid residues of bat CASP1 (D365N and R371Q) that reduces its enzymatic activity essential for IL‐1β processing and pyroptosis. 109 A recent study by Goh et al. 109 identifies that some of the bat species, which show intact CASP1 function, show abolished IL‐1β cleavage as a complementary strategy to dampen inflammation. Identification of these mutations in bat CASP1 indicates a likely adaptation of inflammasome assembling proteins in bats to dampen inflammatory responses to RNA viruses.
Polyinosinic:polycytidylic acid (poly(I:C)) is a double‐stranded RNA ligand that mimics viral RNAs. Poly(I:C) stimulation experiments indicate that bat and human cells trigger antiviral responses by up‐regulating IFN‐β; however, only human cells express robust TNF cytokine levels. 114 The bat cells utilize a unique repression system to reduce TNF expression in response to viral RNA mimics. 114 TNF signaling facilitates activation of the inflammasome, inflammatory responses, and lytic forms of cell death. Thus, TNF regulating mechanisms of bats may further limit excessive inflammasome activation and release of proinflammatory cytokines and DAMPs compared with humans.
6. BAT VARIATIONS IN KEY INFLAMMASOME MEDIATORS—ASC, CASP1, GSDMD, AND IL‐1β
Though the bats show reduced NLRP3 expression, they appear to show expression of the ASC, CASP1, and GSDMD proteins critical for the inflammasome activation by various innate immune receptors. 104 , 108 , 109 To fully delineate the bat adaptations in the inflammasome pathway, we mapped functionally critical residues of ASC, CASP1, and GSDMD in bats to comprehensively examine variations (Fig. 3). The ASC consists of PYD and CARD domains that facilitate homotypic interactions and filamentous/speck‐like structures. 115 , 116 , 117 , 118 , 119 The assembly of ASC and its polymerization is essential to recruit and activate the CASP1. 18 , 117 , 119 Our functional residue mapping analysis shows that the key residues essential for ASC oligomerization (PYD) and CASP1 recruitment (CARD) 117 , 118 , 119 appeared to be conserved with a notable exception at F59 and E130 residues (Fig. 3). Bats that belong to Yangochiroptera suborder (M. natalensis, M.davidii, and E. fuscus) show variation at one of the critical CARD (E130N) and PYD (F59S) residues required for ASC filament formation and CASP1 interaction (Fig. 3C). We also observed a critical variation at one of the CARD residues (Y82H) of the CASP1 of multiple bat species (Fig. 3D). INCA (inhibitory CARD) is a natural human CARD only protein that shares high similarity with the CARD domain of the CASP1 and interferes with its function. Interestingly, the INCA protein shows Y82H mutation (like bat CASP1), making it unsuitable for CARD‐mediated polymerization, but interfering with the CASP1 function. 118 These variations in ASC and CASP1 may interfere in their interaction and assembly of inflammasome complexes.
FIGURE 3.
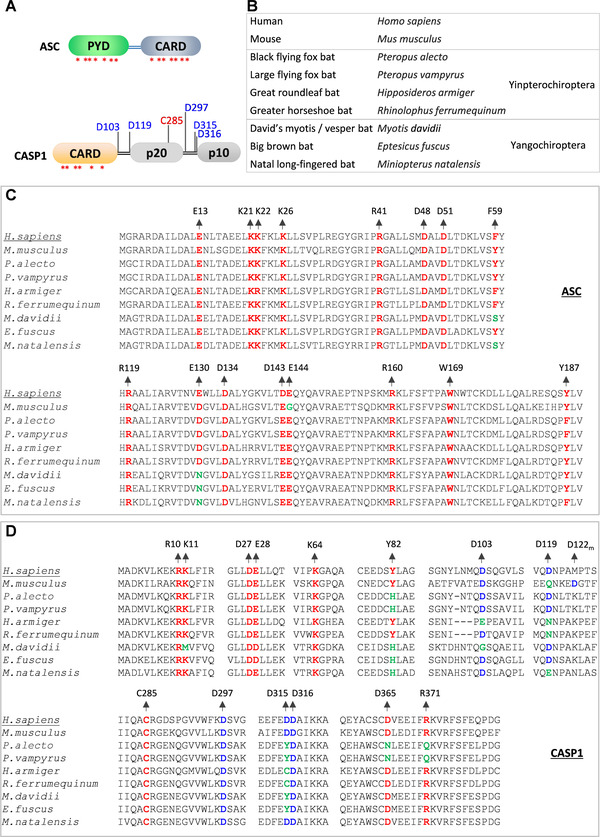
Bats show variation in functionally essential residues of inflammasome components.
(A) Domain architectures of the ASC and CASP1 proteins. Asterisks indicate critical residue positions promoting homotypic interactions and oligomerization of ASC and CASP1; cleavage site residues of CASP1 are indicated in blue; the catalytically important residue of the CASP1 are indicated in red. (B) Common and scientific names of organisms that are used in panels C and D for comparing the conservation of inflammasomes components. Bat species are categorized into Yangochiroptera and Yinpterochiroptera suborders. (C and D) Multiple sequence alignments of specific regions of ASC (C) and CASP1 (D) from human, mouse, and bat species. The human ASC and CASP1 protein sequences are used as references to define the numbers of amino acid residues. The critical residue positions are colored as represented in panel A. The conserved residue positions are highlighted in red, the conserved cleavage site residues are in blue, and the mutated residue positions in green. The D122m in panel D represents the cleavage site in mice that correspond to the D119 cleavage site of the human CASP1
CASP1 contains a CARD domain, a large (p20) and small (p10) subunits. Interdomain linker regions separate these domains. 120 , 121 , 122 , 123 Once activated, CASP1 is self‐cleaved at aspartic acid residues of the linker regions to generate enzymatically active p20 and p10 subunits. 120 , 121 , 122 , 123 Mouse CASP1 shows variation at D119; however, D122 of the mouse CASP1, which is in close vicinity, compensates this variation and retains self‐cleavage in linker regions 123 (Fig. 3D). Our analysis indicates a notable variation at several cleavage site residues located in linker regions of bat CASP1 of both Yangochiroptera and Yinpterochiroptera suborders compared with human CASP1 (Fig. 3D). We also did not observe any compensatory aspartic acid residues in adjacent amino acid sequences suggesting a possible exclusion of self‐cleavage at these CASP1 regions. However, the residue crucial for CASP1 catalytic activity (C285) is highly conserved in bats and humans. We observed D365N and R371Q mutations in P. vampyrus and P. alecto, which were recently reported by Goh et al., 109 to affect the catalytic activity of the bat CASP1 (Fig. 3D). Thus, it appears that a wide range of mutations may interfere with the bat CASP1 function following inflammasome activation.
GSDMD is a pore‐forming protein that consists of a CASP1 cleavage site (D275) at the linker region of its N and C‐terminal domains 31 , 32 (Figs. 4A and 4B). CASP1‐mediated cleavage of the GSDMD liberates the N‐terminal domain that assembles into a pore structure on the cell membrane and triggers pyroptosis. 19 , 30 , 31 , 32 Caspase‐3 (CASP3) also cleaves the GSDMD N‐terminal domain at D87, which abolishes pore‐forming activity 30 , 124 (Figs. 4A and 4B). The positively charged residues in α1‐helix of the GSDMD N‐terminal domain (residues R7, R10, and R11) are critical for its interaction with the cell membrane and subsequent oligomerization into a pore structure 125 , 126 , 127 (Figs. 4A and 4B). A recent study by Goh et al. 109 demonstrated the cleavage of bat GSDMD into N‐ and C‐termini and pro‐IL‐1β into a mature form of IL‐1β by caspases. Consistent with this study, we observed the cleavage sites of CASP1 and CASP3 in GSDMD are highly conserved in bats and humans (Fig. 4B). However, R7 and R11 residues in α1‐helix of the bat GSDMD are mutated to uncharged residues suggesting a potential restriction of pore formation and pyroptosis (Fig. 4B). The caspase cleavage sites of IL‐1β are conserved in bats and humans (Figs. 4A and 4C). Goh et al. 109 identified that the amino acid residues (residue positions 110, 111, and 118) surrounding the cleavage site D116 determine the IL‐1β cleavage ability in M. davidii and P.alecto by CASP1. The cleavage ability of the IL‐1β in these bat species shows an inverse complementary relationship with their CASP1 activity. 109 Previous studies indicate critical residues of IL‐1β, which are necessary for binding to IL‐1 receptor. 128 , 129 , 130 The IL‐1 receptor binding residues of bat IL‐1β are largely conserved; however, few variations were noted at I172, K209, and E221 residues in some of the bat species (Fig. 4C). Overall, the analysis of functionally essential residues in ASC, CASP1, GSDMD, and IL‐1β of bats reveals molecular adaptation of bats to restrict inflammasome mediated proinflammatory responses. Unlike the NLRP3, the expression of ASC, CASP1, and GSDMD is seen in bats, which might have forced them to acquire mutations at crucial residues to restrict inflammasome functions. What evolutionary pressures drive bat variations that dampen inflammasome activation? The causative triggers for the evolution of bat variations remain to be studied to suppress innate immune responses like inflammasome and STING activation. The dampened inflammasome in bat immune tolerance and inflammasome activation in driving COVID‐19 severity suggests inflammasome as a target for alleviating disease severity in humans. Also, monitoring circulating inflammasome cytokines in humans may assist in identifying individuals who are likely predisposed for COVID‐19 severity.
FIGURE 4.
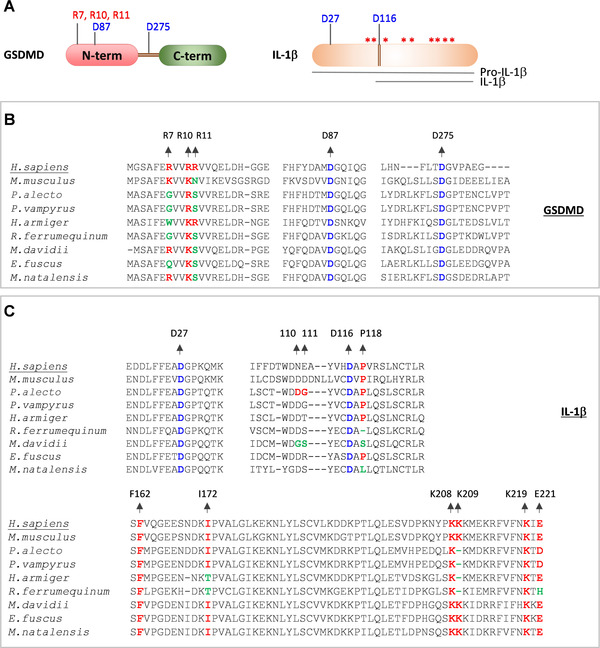
Variations in functionally important residues of bat CASP1 substrates, GSDMD and IL‐1β proteins.
(A) Cartoon representing GSDMD, pro‐IL‐1β and mature form of IL‐1β. Asterisks indicate critical residue positions required for IL‐1β function; cleavage site residues are indicated in blue; critical lipid membrane binding residues of the GSDMD are indicated in red. (B and C) Multiple sequence alignments showing critical residues of GSDMD (B) and IL‐1β protein (C). The human (H.sapiens) GSDMD and IL‐1β protein sequences are used as a reference sequence to represent the amino acid residue numbers. The conserved amino acid residues are highlighted in red. The conserved cleavage site residue positions are highlighted in blue. The mutated amino acid residue positions are highlighted in green
7. CONCLUSIONS
Human cells operate diverse innate immune receptors, ALRs and NLRs, to engage inflammasome assembly and pyroptosis. Thus, human respiratory infections, like SARS‐CoV‐2, readily trigger inflammasome‐mediated proinflammatory responses. The virus‐induced elevated inflammatory responses ultimately drive human immunopathology despite type I IFN responses. Consistent findings showing elevated IL‐1α, IL‐1β, and LDH in COVID‐19 patients substantiate the role of the inflammasome in promoting disease severity. This makes the inflammasome pathway a suitable target to alleviate COVID‐19 severity and prevent immune overreaction. Unlike humans, bats are highly adapted to operate type I IFN responses and suppress the activation of the inflammasome and proinflammatory responses. The robust suppression of the NLRP3 inflammasome in bat immune cells is consistent with the ability to host several RNA viruses that primarily engage this NLRP3 inflammasome. Identification of bat ASC, CASP1, and GSDMD mutations suggest the possible evolution of bat inflammasome proteins that favor dampened inflammasome activation by innate immune receptors. It is likely that bat adaptation to restrict inflammasome activation allows them to tolerate RNA viruses like SARS‐CoV, SARS‐CoV2, and MERS‐CoV (beta‐CoVs) without inducing adverse inflammatory responses and tissue damage. However, the SARS‐CoV‐2‐induced inflammasome activation drives disease pathogenesis in human infections. We still do not understand why only some of the CoVs, particularly bat‐beta CoVs, that infected humans are highly pathogenic but not others. Do these infected human CoVs differ in their ability to activate the inflammasome in human cells? Do inflammasome activation signatures of the CoVs help predict the pathogenic potential of bat CoVs that are prone to spillover and cause future outbreaks? Overall, inflammasome activation in bats and humans provides the basis for species‐specific innate immune activation, viral tolerance, and immunopathology in pathogenic CoV infections. Further exploration of bat and human immune activation mechanisms unravel fundamental mechanisms determining protective and pathologic outcomes in CoV infections.
AUTHORSHIP
S.K. conceptualized and performed protein sequence analysis. S.N., D.J., and S.K wrote and edited the manuscript.
DISCLOSURES
The authors declare no conflicts of interest.
Supporting information
Supplementry meterial
ACKNOWLEDGMENTS
The authors of this manuscript thank all the research groups who made substantial contributions to understanding inflammasome biology and its impact on human and bat inflammatory responses. S.K. laboratory is supported by funds from the Indian Institute of Science (IISc) and Infosys Foundation, Bengaluru, India.
Nagaraja S, Jain D, Kesavardhana S. Inflammasome regulation in driving COVID‐19 severity in humans and immune tolerance in bats. J Leukoc Biol. 2022;111:497‐510. 10.1002/JLB.4COVHR0221-093RR
Summary sentence: Human SARS‐CoV‐2 infection‐induced inflammasome activation is associated with COVID‐19 severity, whereas bats, which host pathogenic CoVs, show dampened inflammasome activation and immune tolerance to viral infections.
REFERENCES
- 1. Cui J, Li F, Shi Z‐Li. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. 2019;17:181‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. V'kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus biology and replication: implications for SARS‐CoV‐2. Nat Rev Microbiol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Weiss SR. Forty years with coronaviruses. J Exp Med. 2020;217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hoffmann M, Kleine‐Weber H, Schroeder S, et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271‐280 e278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lan J, Ge J, Yu J, et al. Structure of the SARS‐CoV‐2 spike receptor‐binding domain bound to the ACE2 receptor. Nature. 2020;581:215‐220. [DOI] [PubMed] [Google Scholar]
- 6. Zhou P, Yang X‐L, Wang X‐G, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Merad M, Martin JC. Pathological inflammation in patients with COVID‐19: a key role for monocytes and macrophages. Nat Rev Immunol. 2020;20:355‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tay MZ, Poh CM, Rénia L, Macary PA, Ng LFP. The trinity of COVID‐19: immunity, inflammation and intervention. Nat Rev Immunol. 2020;20:363‐374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang J, Jiang M, Chen X, Montaner LJ. Cytokine storm and leukocyte changes in mild versus severe SARS‐CoV‐2 infection: review of 3939 COVID‐19 patients in China and emerging pathogenesis and therapy concepts. J Leukoc Biol. 2020;108:17‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Perlman S. COVID‐19 poses a riddle for the immune system. Nature. 2020;584:345‐346. [DOI] [PubMed] [Google Scholar]
- 11. Thomas PG, Shubina M, Balachandran S. ZBP1/DAI‐dependent cell death pathways in influenza A virus immunity and pathogenesis. Curr Top Microbiol Immunol. 2020. [DOI] [PubMed] [Google Scholar]
- 12. Lucas C, Wong P, Klein J, et al. Longitudinal analyses reveal immunological misfiring in severe COVID‐19. Nature. 2020;584:463‐469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mick E, Kamm J, Pisco AO, et al. Upper airway gene expression reveals suppressed immune responses to SARS‐CoV‐2 compared with other respiratory viruses. Nat Commun. 2020;11:5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mudd PA, Crawford JC, Turner JS, et al. Distinct inflammatory profiles distinguish COVID‐19 from influenza with limited contributions from cytokine storm. Sci Adv. 2020;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhou Z, Ren L, Zhang Li, et al. Heightened innate immune responses in the respiratory tract of COVID‐19 patients. Cell Host Microbe. 2020;27:883‐890 e882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Karki R, Sharma BR, Tuladhar S, et al. Synergism of TNF‐alpha and IFN‐gamma triggers inflammatory cell death, tissue damage, and mortality in SARS‐CoV‐2 infection and cytokine shock syndromes. Cell. 2021;184:149‐168 e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lamkanfi M, Dixit VM. Retrospect: the inflammasome turns 15. Nature. 2017;548:534‐535. [DOI] [PubMed] [Google Scholar]
- 18. Xue Y, Enosi Tuipulotu D, Tan WH, Kay C, Man SiM. Emerging activators and regulators of inflammasomes and pyroptosis. Trends Immunol. 2019;40:1035‐1052. [DOI] [PubMed] [Google Scholar]
- 19. Kesavardhana S, Malireddi RKS, Kanneganti T‐D. Caspases in cell death, inflammation, and pyroptosis. Annu Rev Immunol. 2020;38:567‐595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chauhan D, Vande Walle L, Lamkanfi M. Therapeutic modulation of inflammasome pathways. Immunol Rev. 2020;297:123‐138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Poudel B, Gurung P. An update on cell intrinsic negative regulators of the NLRP3 inflammasome. J Leukoc Biol. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bauernfeind FG, Horvath G, Stutz A, et al. Cutting edge: nF‐kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183:787‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mckee CM, Coll RC. NLRP3 inflammasome priming: a riddle wrapped in a mystery inside an enigma. J Leukoc Biol. 2020;108:937‐952. [DOI] [PubMed] [Google Scholar]
- 24. Juliana C, Fernandes‐Alnemri T, Kang S, Farias A, Qin F, Alnemri ES. Non‐transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem. 2012;287:36617‐36622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Py BF, Kim Mi‐S, Vakifahmetoglu‐Norberg H, Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell. 2013;49:331‐338. [DOI] [PubMed] [Google Scholar]
- 26. Yang J, Liu Z, Xiao TS. Post‐translational regulation of inflammasomes. Cell Mol Immunol. 2017;14:65‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hayward JA, Mathur A, Ngo C, Man SiM. Cytosolic recognition of microbes and pathogens: inflammasomes in action. Microbiol Mol Biol Rev. 2018;82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Man SiM. Inflammasomes in the gastrointestinal tract: infection, cancer and gut microbiota homeostasis. Nat Rev Gastroenterol Hepatol. 2018;15:721‐737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16:407‐420. [DOI] [PubMed] [Google Scholar]
- 30. Broz P, Pelegrín P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol. 2020;20:143‐157. [DOI] [PubMed] [Google Scholar]
- 31. Kayagaki N, Stowe IB, Lee BL, et al. Caspase‐11 cleaves gasdermin D for non‐canonical inflammasome signalling. Nature. 2015;526:666‐671. [DOI] [PubMed] [Google Scholar]
- 32. Shi J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660‐665. [DOI] [PubMed] [Google Scholar]
- 33. Kayagaki N, Kornfeld OS, Lee BL, et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature. 2021. [DOI] [PubMed] [Google Scholar]
- 34. Russo AJ, Vasudevan SO, Méndez‐Huergo SP, et al. Intracellular immune sensing promotes inflammation via gasdermin D‐driven release of a lectin alarmin. Nat Immunol. 2021;22:154‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Place DE, Kanneganti T‐D. The innate immune system and cell death in autoinflammatory and autoimmune disease. Curr Opin Immunol. 2020;67:95‐105. [DOI] [PubMed] [Google Scholar]
- 36. Zhao C, Zhao W. NLRP3 inflammasome: a key player in antiviral responses. Front Immunol. 2020;11:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med. 2009;206:79‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thomas PG, Dash P, Aldridge JR, et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase‐1. Immunity. 2009;30:566‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Allen IC, Scull MA, Moore CB, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. 2009;30:556‐565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ramos HJ, Lanteri MC, Blahnik G, et al. IL‐1beta signaling promotes CNS‐intrinsic immune control of West Nile virus infection. PLoS Pathog. 2012;8:e1003039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rodrigues TS, De Sá KSG, Ishimoto AY, et al. Inflammasomes are activated in response to SARS‐CoV‐2 infection and are associated with COVID‐19 severity in patients. J Exp Med. 2021;218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Da Costa LS, Outlioua A, Anginot A, Akarid K, Arnoult D. RNA viruses promote activation of the NLRP3 inflammasome through cytopathogenic effect‐induced potassium efflux. Cell Death Dis. 2019;10:346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bryant C. COVID‐19 stokes inflammasomes. J Exp Med. 2021;218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zheng M, Williams EP, Malireddi RKS, et al. Impaired NLRP3 inflammasome activation/pyroptosis leads to robust inflammatory cell death via caspase‐8/RIPK3 during coronavirus infection. J Biol Chem. 2020;295:14040‐14052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wauters E, Van Mol P, Garg AD, et al. Discriminating mild from critical COVID‐19 by innate and adaptive immune single‐cell profiling of bronchoalveolar lavages. Cell Res. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yap JKY, Moriyama M, Iwasaki A. Inflammasomes and pyroptosis as therapeutic targets for COVID‐19. J Immunol. 2020;205:307‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen N, Zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507‐513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Carvalho T, Krammer F, Iwasaki A. The first 12 months of COVID‐19: a timeline of immunological insights. Nat Rev Immunol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bajaj V, Gadi N, Spihlman AP, Wu SC, Choi CH, Moulton VR. Aging, immunity, and COVID‐19: how age influences the host immune response to coronavirus infections?. Front Physiol. 2020;11:571416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Latz E, Duewell P. NLRP3 inflammasome activation in inflammaging. Semin Immunol. 2018;40:61‐73. [DOI] [PubMed] [Google Scholar]
- 51. Lara PC, Macías‐Verde D, Burgos‐Burgos J. Age‐induced NLRP3 inflammasome over‐activation increases lethality of SARS‐CoV‐2 pneumonia in elderly patients. Aging Dis. 2020;11:756‐762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pinna G. Sex and COVID‐19: a protective role for reproductive steroids. Trends Endocrinol Metab. 2021;32:3‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Slowik A, Lammerding L, Zendedel A, Habib P, Beyer C. Impact of steroid hormones E2 and P on the NLRP3/ASC/Casp1 axis in primary mouse astroglia and BV‐2 cells after in vitro hypoxia. J Steroid Biochem Mol Biol. 2018;183:18‐26. [DOI] [PubMed] [Google Scholar]
- 54. Nieto‐Torres JL, Dediego ML, Verdiá‐Báguena C, et al. Severe acute respiratory syndrome coronavirus envelope protein ion channel activity promotes virus fitness and pathogenesis. PLoS Pathog. 2014;10:e1004077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nieto‐Torres JL, Verdiá‐Báguena C, Jimenez‐Guardeño JM, et al. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology. 2015;485:330‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chen I‐Y, Moriyama M, Chang M‐Fu, Ichinohe T. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front Microbiol. 2019;10:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kanzawa N, Nishigaki K, Hayashi T, et al. Augmentation of chemokine production by severe acute respiratory syndrome coronavirus 3a/X1 and 7a/X4 proteins through NF‐kappaB activation. FEBS Lett. 2006;580:6807‐6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dediego ML, Nieto‐Torres JL, Regla‐Nava JA, et al. Inhibition of NF‐kappaB‐mediated inflammation in severe acute respiratory syndrome coronavirus‐infected mice increases survival. J Virol. 2014;88:913‐924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Siu K‐L, Yuen K‐S, Castano‐Rodriguez C, et al. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3‐dependent ubiquitination of ASC. FASEB J. 2019;33:8865‐8877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lee S, Channappanavar R, Kanneganti T‐D. Coronaviruses: innate immunity, inflammasome activation, inflammatory cell death, and cytokines. Trends Immunol. 2020;41:1083‐1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Xu H, et al. SARS‐CoV‐2 viroporin triggers the NLRP3 inflammatory pathway. bioRxiv, 2020.2010.2027.357731, 10.1101/2020.10.27.357731/J_bioRxiv (2020). [DOI]
- 62. Yue Y, Nabar NR, Shi C‐S, et al. SARS‐Coronavirus Open Reading Frame‐3a drives multimodal necrotic cell death. Cell Death Dis. 2018;9:904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dosch SF, Mahajan SD, Collins AR. SARS coronavirus spike protein‐induced innate immune response occurs via activation of the NF‐kappaB pathway in human monocyte macrophages in vitro. Virus Res. 2009;142:19‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Theobald SJ, A S, Kreer C, et al. The SARS‐CoV‐2 spike protein primes inflammasome‐mediated interleukin‐1‐ beta secretion in COVID‐19 patient‐derived macrophages. Preprint from Research Square. 2020. doi:10.21203/rs.3.rs‐30407/v1. [Google Scholar]
- 65. Kim Na‐E, Kim D‐K, Song Y‐J, SARS‐CoV‐2 nonstructural proteins 1 and 13 suppress caspase‐1 and the NLRP3 inflammasome activation. Microorganisms 2021;9:494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kumar P, Gunalan V, Liu B, et al. The nonstructural protein 8 (nsp8) of the SARS coronavirus interacts with its ORF6 accessory protein. Virology. 2007;366:293‐303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wong HH, Fung ToS, Fang S, Huang M, Le MyT, Liu DX. Accessory proteins 8b and 8ab of severe acute respiratory syndrome coronavirus suppress the interferon signaling pathway by mediating ubiquitin‐dependent rapid degradation of interferon regulatory factor 3. Virology. 2018;515:165‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chiu RWK, Chim SSC, Tong Yu‐K, et al. Tracing SARS‐coronavirus variant with large genomic deletion. Emerg Infect Dis. 2005;11:168‐170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Muth D, Corman VM, Roth H, et al. Attenuation of replication by a 29 nucleotide deletion in SARS‐coronavirus acquired during the early stages of human‐to‐human transmission. Sci Rep. 2018;8:15177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Shi C‐S, Nabar NR, Huang N‐Na, Kehrl JH. SARS‐Coronavirus Open Reading Frame‐8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 2019;5:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Young BE, Fong S‐W, Chan Yi‐H, et al. Effects of a major deletion in the SARS‐CoV‐2 genome on the severity of infection and the inflammatory response: an observational cohort study. Lancet. 2020;396:603‐611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Su YCF, Anderson DE, Young BE, et al. Discovery and genomic characterization of a 382‐nucleotide deletion in ORF7b and ORF8 during the early evolution of SARS‐CoV‐2. mBio. 2020;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gamage AM, Tan KS, Chan WOY, et al. Infection of human nasal epithelial cells with SARS‐CoV‐2 and a 382‐nt deletion isolate lacking ORF8 reveals similar viral kinetics and host transcriptional profiles. PLoS Pathog. 2020;16:e1009130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Malireddi RKS, Kanneganti T‐D. Role of type I interferons in inflammasome activation, cell death, and disease during microbial infection. Front Cell Infect Microbiol. 2013;3:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Schreiber G. The role of type I interferons in the pathogenesis and treatment of COVID‐19. Front Immunol. 2020;11:595739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Channappanavar R, Fehr AR, Vijay R, et al. Dysregulated type I interferon and inflammatory monocyte‐macrophage responses cause lethal pneumonia in SARS‐CoV‐infected mice. Cell Host Microbe. 2016;19:181‐193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Channappanavar R, Fehr AR, Zheng J, et al. IFN‐I response timing relative to virus replication determines MERS coronavirus infection outcomes. J Clin Invest. 2019;129:3625‐3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Xia H, Cao Z, Xie X, et al. Evasion of type I interferon by SARS‐CoV‐2. Cell Rep. 2020;33:108234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lei X, Dong X, Ma R, et al. Activation and evasion of type I interferon responses by SARS‐CoV‐2. Nat Commun. 2020;11:3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Blanco‐Melo D, Nilsson‐Payant BE, Liu W‐C, et al. Imbalanced Host Response to SARS‐CoV‐2 Drives Development of COVID‐19. Cell. 2020;181:1036‐1045 e1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Chousterman BG, Swirski FK, Weber GF. Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol. 2017;39:517‐528. [DOI] [PubMed] [Google Scholar]
- 82. Fajgenbaum DC, June CH. Cytokine storm. N Engl J Med. 2020;383:2255‐2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Baize S, Leroy EM, Georges AJ, et al. Inflammatory responses in Ebola virus‐infected patients. Clin Exp Immunol. 2002;128:163‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Leroy Em, Baize S, Volchkov Ve, et al. Human asymptomatic Ebola infection and strong inflammatory response. Lancet. 2000;355:2210‐2215. [DOI] [PubMed] [Google Scholar]
- 85. Wauquier N, Becquart P, Padilla C, Baize S, Leroy EM. Human fatal zaire ebola virus infection is associated with an aberrant innate immunity and with massive lymphocyte apoptosis. PLoS Negl Trop Dis. 2010;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lei J, Vermillion MS, Jia B, et al. IL‐1 receptor antagonist therapy mitigates placental dysfunction and perinatal injury following Zika virus infection. JCI Insight. 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ornelas AMM, Pezzuto P, Silveira PP, et al. Immune activation in amniotic fluid from Zika virus‐associated microcephaly. Ann Neurol. 2017;81:152‐156. [DOI] [PubMed] [Google Scholar]
- 88. Wen Z, Song H, Ming G‐Li. How does Zika virus cause microcephaly?. Genes Dev. 2017;31:849‐861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Gambotto A, Barratt‐Boyes SM, De Jong MD, Neumann G, Kawaoka Y. Human infection with highly pathogenic H5N1 influenza virus. Lancet. 2008;371:1464‐1475. [DOI] [PubMed] [Google Scholar]
- 90. Kobasa D, Jones SM, Shinya K, et al. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature. 2007;445:319‐323. [DOI] [PubMed] [Google Scholar]
- 91. Liu Q, Zhou Y‐H, Yang Z‐Q. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell Mol Immunol. 2016;13:3‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Peiris JSM, Cheung CY, Leung CYH, Nicholls JM. Innate immune responses to influenza A H5N1: friend or foe?. Trends Immunol. 2009;30:574‐584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Writing Committee of the, W. H. O. C. o. C. A. o. P. I . et al. Clinical aspects of pandemic 2009 influenza A (H1N1) virus infection. N Engl J Med 362, 1708‐1719, 10.1056/NEJMra1000449 (2010). [DOI] [PubMed] [Google Scholar]
- 94. Liu Y, Zhang C, Huang F, et al. Elevated plasma levels of selective cytokines in COVID‐19 patients reflect viral load and lung injury. Natl Sci Rev. 2020;7:1003‐1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Gabay C, Lamacchia C, Palmer G. IL‐1 pathways in inflammation and human diseases. Nat Rev Rheumatol. 2010;6:232‐241. [DOI] [PubMed] [Google Scholar]
- 96. Zhao Y, Qin L, Zhang P, et al. Longitudinal COVID‐19 profiling associates IL‐1RA and IL‐10 with disease severity and RANTES with mild disease. JCI Insight. 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Hill‐Batorski L, Halfmann P, Marzi A, et al. Loss of interleukin 1 receptor antagonist enhances susceptibility to Ebola virus infection. J Infect Dis. 2015;212:S329‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T. Bats: important reservoir hosts of emerging viruses. Clin Microbiol Rev. 2006;19:531‐545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Smith I, Wang L‐Fa. Bats and their virome: an important source of emerging viruses capable of infecting humans. Curr Opin Virol. 2013;3:84‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Wang L‐Fa, Anderson DE. Viruses in bats and potential spillover to animals and humans. Curr Opin Virol. 2019;34:79‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Menachery VD, Yount BL, Debbink K, et al. A SARS‐like cluster of circulating bat coronaviruses shows potential for human emergence. Nat Med. 2015;21:1508‐1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Olival KJ, Cryan PM, Amman BR, et al. Possibility for reverse zoonotic transmission of SARS‐CoV‐2 to free‐ranging wildlife: a case study of bats. PLoS Pathog. 2020;16:e1008758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Banerjee A, Doxey AC, Mossman K, Irving AT. Unraveling the zoonotic origin and transmission of SARS‐CoV‐2. Trends Ecol Evol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Ahn M, Cui J, Irving AT, Wang L‐Fa. Unique loss of the PYHIN gene family in bats amongst mammals: implications for inflammasome sensing. Sci Rep. 2016;6:21722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Mandl JN, Ahmed R, Barreiro LB, et al. Reservoir host immune responses to emerging zoonotic viruses. Cell. 2015;160:20‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Pavlovich SS, Lovett SP, Koroleva G, et al. The Egyptian Rousette genome reveals unexpected features of bat antiviral immunity. Cell. 2018;173: 1098‐1110 e1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Zhou P, Tachedjian M, Wynne JW, et al. Contraction of the type I IFN locus and unusual constitutive expression of IFN‐alpha in bats. Proc Natl Acad Sci USA. 2016;113:2696‐2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Ahn M, Anderson DE, Zhang Q, et al. Dampened NLRP3‐mediated inflammation in bats and implications for a special viral reservoir host. Nat Microbiol. 2019;4:789‐799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Goh G, Ahn M, Zhu F, et al. Complementary regulation of caspase‐1 and IL‐1beta reveals additional mechanisms of dampened inflammation in bats. Proc Natl Acad Sci USA. 2020;117:28939‐28949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Hayman DTS. Bat tolerance to viral infections. Nat Microbiol. 2019;4:728‐729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Zhang G, Cowled C, Shi Z, et al. Comparative analysis of bat genomes provides insight into the evolution of flight and immunity. Science. 2013;339:456‐460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS‐STING pathway in health and disease. Nat Rev Genet. 2019;20:657‐674. [DOI] [PubMed] [Google Scholar]
- 113. Xie J, Li Y, Shen X, et al. Dampened STING‐dependent interferon activation in bats. Cell Host Microbe. 2018;23:297‐301 e294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Banerjee A, Rapin N, Bollinger T, Misra V. Lack of inflammatory gene expression in bats: a unique role for a transcription repressor. Sci Rep. 2017;7:2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Hauenstein AV, Zhang L, Wu H. The hierarchical structural architecture of inflammasomes, supramolecular inflammatory machines. Curr Opin Struct Biol. 2015;31:75‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wu H, Fuxreiter M. The structure and dynamics of higher‐order assemblies: amyloids, signalosomes, and granules. Cell. 2016;165:1055‐1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Li Y, Fu T‐M, Lu A, et al. Cryo‐EM structures of ASC and NLRC4 CARD filaments reveal a unified mechanism of nucleation and activation of caspase‐1. Proc Natl Acad Sci USA. 2018;115:10845‐10852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Lu A, Li Y, Schmidt FI, et al. Molecular basis of caspase‐1 polymerization and its inhibition by a new capping mechanism. Nat Struct Mol Biol. 2016;23:416‐425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Lu A, Magupalli VG, Ruan J, et al. Unified polymerization mechanism for the assembly of ASC‐dependent inflammasomes. Cell. 2014;156:1193‐1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Ball DP, Taabazuing CY, Griswold AR, et al. Caspase‐1 interdomain linker cleavage is required for pyroptosis. Life Sci Alliance. 2020;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Boucher D, Monteleone M, Coll RC, et al. Caspase‐1 self‐cleavage is an intrinsic mechanism to terminate inflammasome activity. J Exp Med. 2018;215:827‐840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Elliott JM, Rouge L, Wiesmann C, Scheer JM. Crystal structure of procaspase‐1 zymogen domain reveals insight into inflammatory caspase autoactivation. J Biol Chem. 2009;284:6546‐6553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Broz P, Von Moltke J, Jones JW, Vance RE, Monack DM. Differential requirement for Caspase‐1 autoproteolysis in pathogen‐induced cell death and cytokine processing. Cell Host Microbe. 2010;8:471‐483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Rogers C, Fernandes‐Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase‐3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017;8:14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Chen X, He W‐T, Hu L, et al. Pyroptosis is driven by non‐selective gasdermin‐D pore and its morphology is different from MLKL channel‐mediated necroptosis. Cell Res. 2016;26:1007‐1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Ding J, Wang K, Liu W, et al. Pore‐forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111‐116. [DOI] [PubMed] [Google Scholar]
- 127. Ruan J, Xia S, Liu X, Lieberman J, Wu H. Cryo‐EM structure of the gasdermin A3 membrane pore. Nature. 2018;557:62‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Evans RJ, Bray J, Childs JD, et al. Mapping receptor binding sites in interleukin (IL)‐1 receptor antagonist and IL‐1 beta by site‐directed mutagenesis. Identification of a single site in IL‐1ra and two sites in IL‐1 beta. J Biol Chem. 1995;270:11477‐11483. [DOI] [PubMed] [Google Scholar]
- 129. Labriola‐Tompkins E, Chandran C, Kaffka KL, et al. Identification of the discontinuous binding site in human interleukin 1 beta for the type I interleukin 1 receptor. Proc Natl Acad Sci USA. 1991;88:11182‐11186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Vigers GPA, Anderson LJ, Caffes P, Brandhuber BJ. Crystal structure of the type‐I interleukin‐1 receptor complexed with interleukin‐1beta. Nature. 1997;386:190‐194. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementry meterial