

Abstract
Hydrogels are useful delivery vehicles for therapeutic proteins. The ability to control the rate of protein release is paramount to a gel’s utility and, in part, defines its clinical application. Electrostatic interactions made between encapsulated protein and a gel’s network represents one modality in which protein motility can be controlled. For many gels this strategy works well under low ionic strength solution conditions, but dramatically less so in solutions of physiologically relevant ionic strength where electrostatic interactions are more effectively screened. Herein, we find that highly charged self-assembling peptides can be used to prepare fibrillar hydrogels of sufficient electropotential to allow electrostatic-based control over protein release under physiological buffer conditions. Rheology shows that proteins, differing significantly in their isoelectric point, can be directly encapsulated within negatively- or positively-charged peptide hydrogel networks during the peptide self-assembly event leading to gelation. Bulk adsorption studies coupled with transmission electron microscopy shows that electrostatic interactions drive the association of protein to oppositely charged fibrils in the final gel state, which in turn, dictates the diffusion and retention of these macromolecules in the hydrogel network.
Graphical Abstract

Highly charged hydrogel networks from self-assembling peptides allow electrostatic-based control over protein release under physiological buffer conditions.
Introduction
Injectable hydrogels prepared from self-assembling peptides have utility as delivery depots for protein therapeutics due to their facile fabrication, ability to homogeneously encapsulate protein, and potential for localized delivery to tissue at controlled rates.1–6 Protein therapeutics present a wide range of benefits and challenges to the biomedical community.7 Proteins can offer excellent specificity in their action and minimize off-target effects.8,9 However, their formulation can be problematic wherein significant issues can arise in protein solubility and stability.10,11
Encapsulation of proteins within delivery vehicles, such as hydrogels, can enhance solubility and protect the payload from degradation and denaturation. Furthermore, hydrogel material properties can be easily tailored to influence the release rate of the encapsulated payload.12 Indeed, simple adjustment of hydrogel pore size can sterically hinder the diffusion of macromolecules within and out of the gel.13,14 Additional control over protein release can be obtained through designing covalent or physical interactions between the protein payload and the hydrogel network15,16 or employing protease-sensitive linkers to release tethered proteins as a function of the rate of enzymatic action.17–19
Electrostatics can also be used to control the release of proteins. Depending on their isoelectric point (pI) and the solution pH, proteins will be characterized by a net-positive (pI > pH) or net-negative (pI < pH) charge, or be net-neutral (pI ≈ pH). Electrostatic interactions made between a protein and a material’s network can potentially retard or promote release. For example, Shillemans et al. have shown that the release of proteins from positively and negatively charged dextran-based networks was dependent on protein charge, where proteins having opposite charge to that of the material network were largely immobilized.20 Typically, protein-material electrostatic interactions are strongest in solutions of low ionic strength, but much weaker in higher ionic strength physiological solutions as a result of screening. Thus, many materials capable of slow release and/or protein retention under model low ionic strength solution conditions rapidly release their payload when subjected to physiological solutions. One possible way to mitigate this problem is to drastically increase the density of charge within the material network, making screening much more difficult. Our previous work studying macromolecular release from peptide-based hydrogels suggested that this would be the case.1,14 Herein, we expand on this initial work to systematically test the influence of highly charged networks on protein release. We employ hydrogels prepared from self-assembling β-hairpin peptides that contain a large number of charged residues in their primary sequence. Self-assembly of highly charged monomers lead to the formation of hydrogel networks of exceedingly high electrostatic potential. We show that proteins can be directly encapsulated during peptide self-assembly and that the resulting gel networks can effectively influence the release profile of encapsulated protein mainly via electrostatics even at physiological NaCl concentrations.
Experimental Methods
Materials
PL-Rink amide resin was purchased from Polymer Laboratories. Fmoc-protected valine, threonine, proline, glutamic acid, glutamine and leucine were purchased through EMD Chemicals. Fmoc-protected lysine was purchased from Bachem. Diisopropylethylamine, methanol, acetic anhydride, piperidine, 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU), spectroscopic grade trifluoroacetic acid (TFA), ammonium bicarbonate, and HPLC grade ammonium hydroxide were purchased through Sigma-Aldrich. α-lactalbumin, myoglobin, and lactoferrin were also purchased through Sigma-Aldrich. 2-(6-Chloro-1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate (HCTU) was purchased from Peptides International. N-methylpyrolidinone, acetonitrile, dimethylformamide (DMF), trifluoroacetic acid, thioanisole, ethanedithiol, anisole, BIS-TRIS propane (BTP), sodium chloride (NaCl), diethyl ether, and hydrochloric acid were purchased through Fisher.
Peptide Synthesis and Purification
All peptides were synthesized using standard Fmoc solid phase peptide synthesis with HCTU activation on an ABI 433A automated peptide synthesizer. Resin bound peptide was cleaved and side-chain deprotected using trifluoroacetic acid/thioanisole/ethanedithiol/anisole (90:5:3:2) for two hours under inert gas. Crude peptides were precipitated with cold diethyl ether after separation of resin by filtration. HLT2 was purified by RP-HPLC (Vydac C18 Column) at 40 °C using an isocratic gradient from 0 – 2 minutes at 0 % standard B, then utilizing a linear gradient from 0 – 24 % standard B for 4 minutes followed by a gradient of 24 – 100 % standard B over 153 minutes. Here, standard A is 0.1 % TFA in water and standard B is 90 % MeCN, 9.9 % H2O, and 0.1 % TFA. HLT2 elutes at 28 minutes. VEQ3 was purified by RP-HPLC (apHera C18 Column) at 40 °C using an isocratic gradient from 0 – 2 minutes at 0 % standard D, then utilizing a linear gradient from 0 – 18 % standard D for 5 minutes followed by a gradient of 18 – 100 % standard D over 164 minutes. Here, HPLC solvents consisted of standard C: 20 mM ammonium bicarbonate 5 mM ammonium hydroxide in water and standard D: 20 mM ammonium bicarbonate 5 mM ammonium hydroxide in 8:2 acetonitrile:water. VEQ3 elutes at 25 minutes. Purified peptide solutions were lyophilized, resulting in pure peptide powders that were utilized in all assays. Purity of each peptide was confirmed by analytical HPLC and electrospray ionization - mass spectrometry, Figures S1 and S2.
Oscillatory Shear Rheology
Rheological assessment was conducted on a Texas Instruments AR-G2 rheometer using a 25 mm stainless steel parallel geometry at a gap height of 0.5 mm. For VEQ3, a 1 wt % peptide stock solution was prepared in chilled 20 mM NaOH. To this solution, an equal volume of chilled gelation buffer composed of 100 mM BTP, 300 mM NaCl at pH 7.4 was added. For HLT2, a 2 wt % peptide stock was prepared in chilled water. This solution was then diluted in a 3:1 ratio with the chilled gelation buffer to afford a 0.5 wt % HLT2 solution containing 75 mM BTP 225 mM NaCl at pH 7.4. Immediately following the addition of gelation buffer, 300 μL of the 0.5 wt % gel was added to the center of the plate and the upper geometry was lowered to a gap height of 0.5 mm. The temperature of the system was then increased from 5 °C to 37 °C over 100 seconds at a constant angular frequency of 6 rad/s and 0.2 % strain. The storage and loss moduli were then monitored for 1 hour at a constant angular frequency of 6 rad/s and 0.2 % strain at 37 °C. After the time sweep, dynamic frequency sweeps (0.1–100 rad/s at constant 0.2% strain) and strain sweeps (0.1–1000% strain at constant 6 rad/sec) were performed to ensure that time sweep data were collected in the linear viscoelastic regime, Figure S3. Mesh sizes for each gel were determined as previously described using experimentally derived plateau moduli derived from the frequency-sweep data (Figure S3A, 170 Pa (HLT2) and 190 Pa (VEQ3). Mesh size determination assumes similar estimated fibril persistence lengths (~55 nm) for both peptide-based fibrils.14
Bulk Protein Release Studies
For the protein release studies, 0.5 wt % VEQ3 and HLT2 peptide hydrogels were prepared as in the oscillatory rheology experiments. In this case, however, the buffer used in initiate gelation contains the respective protein to be encapsulated. The gelation buffer for VEQ3 contained 2 mg/mL protein and the gelation buffer for HLT2 hydrogels contained 1.33 mg/mL protein such that all gels contained 1 mg/mL protein in their final formulation. After the initiation of gelation, 300 μL hydrogels containing 1 mg/mL protein were placed in an incubator at 37 °C where gelation occurs within minutes. However, the gels were allowed to incubate for 3 hours to ensure complete self-assembly. After 3 hours, 1 mL of 50 mM BTP 150 mM NaCl buffer at pH 7.4 was gently added to the top of each gel. The addition of buffer marked T=0 min. Over the course of 28 days the entire volume of buffer above the gel (1 mL) was removed and replaced with fresh buffer at designated time points: 1 hour, 2 hour, 3 hour, 4.5 hour, 6 hour, 24 hour, 2 day, 4 day, 7 day, 14 day, 21 day, and 28 day. The concentration of protein within the supernatant was determined by absorbance at 280 nm by UV-Vis spectroscopy and was compared to a calibration curve.
Electron Microscopy
Micrographs of diluted hydrogel samples to report on local fibril morphology in the absence and presence of encapsulated protein were obtained using a Hitachi H-7650 transmission electron microscopy at a voltage of 80 kV. Fibrils imaged by TEM were isolated from hydrogels that were prepared following the procedure in the rheology and bulk release studies. Hydrogels were prepared the night before each TEM experiment was to occur. After overnight incubation, an aliquot of the gel was removed, diluted 40X with water and mixed thoroughly. 2 μL of the diluted gel was added to a 200 mesh carbon coated copper grid with excess liquid blotted away with filter paper. 1 % uranyl acetate solution was then added to the grid as a negative stain to enhance image contrast. Excess stain was blotted away and the grids were imaged immediately. Fibril widths were measured with ImageJ.
Protein Partition and Retention Studies
0.5 wt % VEQ3 and HLT2 hydrogels were prepared in glass vials as outlined in the oscillatory rheology experiments. Upon the initiation of gelation, 300 μL hydrogels were placed in an incubator at 37 °C for 3 hours. After 3 hours, 300 μL solution containing 50 mM BTP, 150 mM NaCl and 1 mg/mL protein at pH 7.4 was gently added to the top of the gel and placed back in the incubator for 48 hours. After 48 hours, the entire volume above the gel (300 μL) was removed and replaced with 1 mL of fresh buffer free of protein. This buffer wash was repeated every 48 hours for an additional two time points at day 4 and day 6. The concentration of protein within the supernatant was determined by absorbance at 280 nm by UV-Vis spectroscopy and was compared to a calibration curve.
Circular Dichroism Studies
0.5 wt% gels (300 μL total volume) were prepared encapsulating 1 mg/mL of each protein in a glass vial. Buffer (1 mL) was added to the top of each gel. Gels were shaken at 100 rpm at 37 °C for 1 week to allow protein release. Supernatants were collected and filter with a 0.22 μm spin filter. The elutants were collected and filter again (3X) via ultrafiltration with a 10K cutoff to remove any soluble peptide that may have co-eluted during protein release. Protein concentrations were then determined by UV at 280 nm using published extinction coefficients [myoglobin (13980); α-lactalbumin (28341); lactoferrin (111650) M−1cm−1]. CD wavelength spectra were measured from 200 to 260 nm at 37 °C using a 1 mm path length quartz cell. The mean residue ellipticity, [θ], was calculated from the equation [θ] = (θobs/10lc)/r, where θobs is the measured ellipticity (mdeg), l is the length of the cell (cm), c is the molar concentration, and r is the number of residues. CD spectra were collected on an AVIV model 420 circular dichroism spectrometer (AVIV Biomedical, Lakewood, NJ). Control spectra were also collected of protein that had never been encapsulated in gel. Each gel-protein combination was studied expect for lactoferrin released from the VEQ3 gel; here, too little of the protein was released to accurately collect a spectrum.
Results and Discussion
Hydrogel Design
Two self-assembling peptides were used in this study to prepare both negatively and positively charged hydrogel networks, namely the anionic peptide VEQ3 and the cationic peptide HLT2, Table 1.
Table 1.
Sequences of HLT2 and VEQ3
| Hydrogel | Sequence | Charge |
|---|---|---|
| HLT2 | VLTKVKTKVDPLPTKVEVKVLV-NH2 | +5 |
| VEQ3 | VEVQVEVEVDPLPTEVQVEVEV-NH2 | −5 |
HLT2 was previously reported and originally designed to form fibrillar gels used to deliver chondrocytes for tissue engineering applications.21 This peptide has a net charge of +5 per monomer at neutral solution pH and forms positively charged networks. Also shown in Table 1, VEQ3 is a 20 amino acid peptide with a net formal charge of −5 per monomer at neutral solution pH. Fibrillar networks formed from this peptide are highly electronegative. Similar to previous generations of β-hairpin peptides developed in the Schneider and Pochan laboratories21–28, HLT2 and VEQ3 undergo triggered hydrogelation in response to adjusting the solution conditions. In pure water, each of the peptides remains unfolded and soluble. Adjusting the ionic strength by the addition of buffer and slightly raising the solution temperature triggers peptide folding, self-assembly and gelation. Solid state NMR performed on a similar β-hairpin peptide, MAX1, shows that this class of peptide assembles into a fibrillar network where each fibril is comprised of a bilayer of folded β-hairpin peptides that hydrogen bond down the long axis of the fibril.29 For MAX1, hairpins assemble with an in-register Syn orientation within each monolayer and with an Anti orientation between layers. Importantly, each peptide projects its charged side chains into the solvent away from the fibril interior. Thus, each fibril in the gel network is characterized by a high copy number of solvent-accessible point charges defined by the net charge of the constitutive peptide. When proteins are present during gelation they become encapsulated within the fibrillar network. Depending on the charge state of the protein and the fibrils comprising gel, a protein will partition either within the pore space or become tightly associated with the fibrils. Each scenario defines the release profile of the protein, Figure 1.
Figure 1.
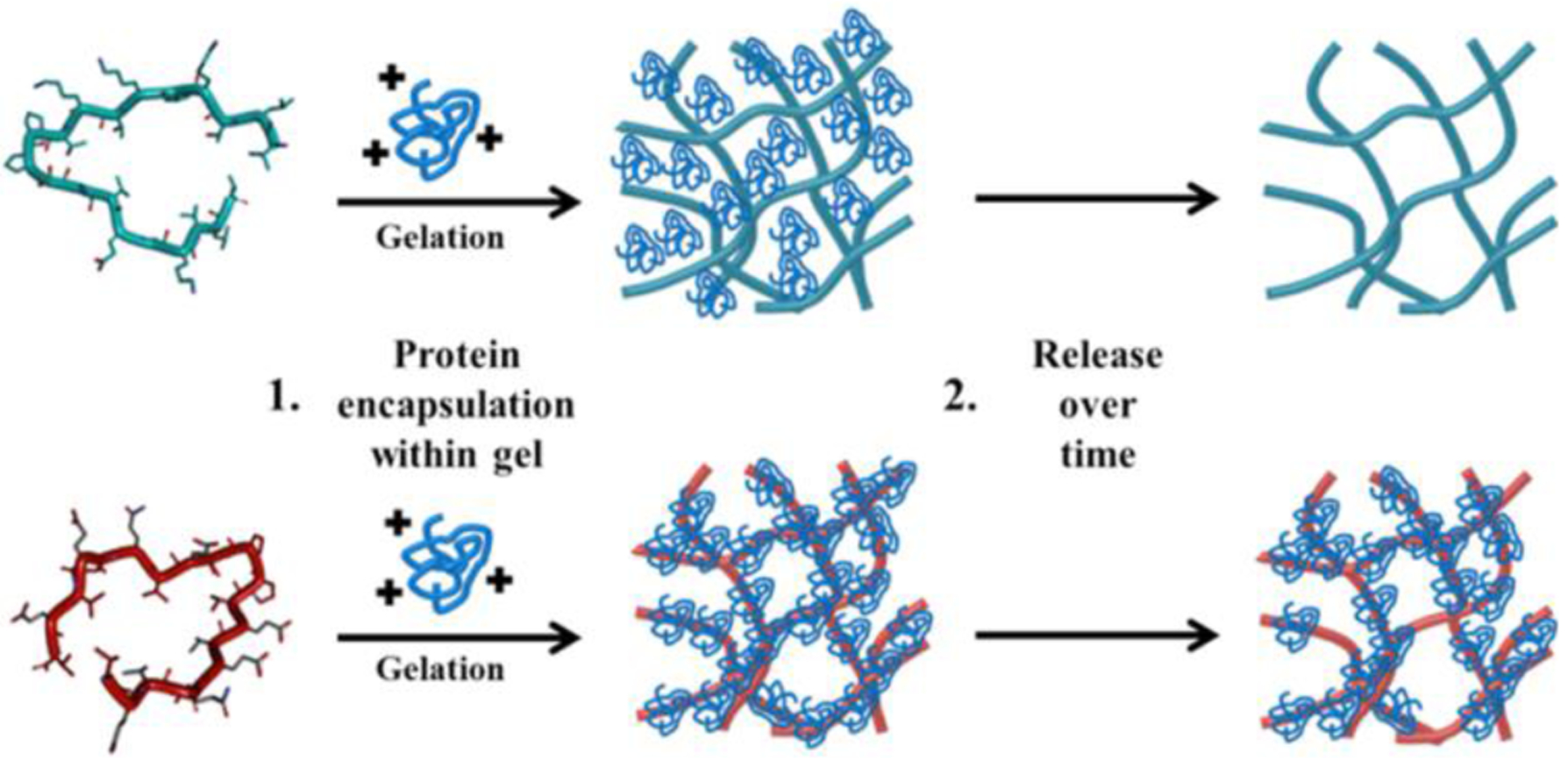
Positively charged protein is encapsulated within an electropositive HLT2 (teal) or electronegative VEQ3 (red) network during gel formation. Protein release is governed, in large part, by network charge
Inspection of the local morphology of fibrils isolated from HLT2 and VEQ3 hydrogels by TEM indicates both peptides form high aspect ratio fibrils of similar width (~ 3.5 nm), Figure 2, as expected from previous studies of other fibrils formed by this class of peptides.28,30 In addition to electrostatic effects, the mesh size of a hydrogel network can also influence the bulk diffusion of encapsulated macromolecules via sterics.14 The mesh size of each network was determined via rheology and found to be similar, ~37 and ~38 nm for VEQ3 and HLT2, respectively. Thus, differences in observed release profiles between the two gels should be mainly due to electrostatic character.
Figure 2.
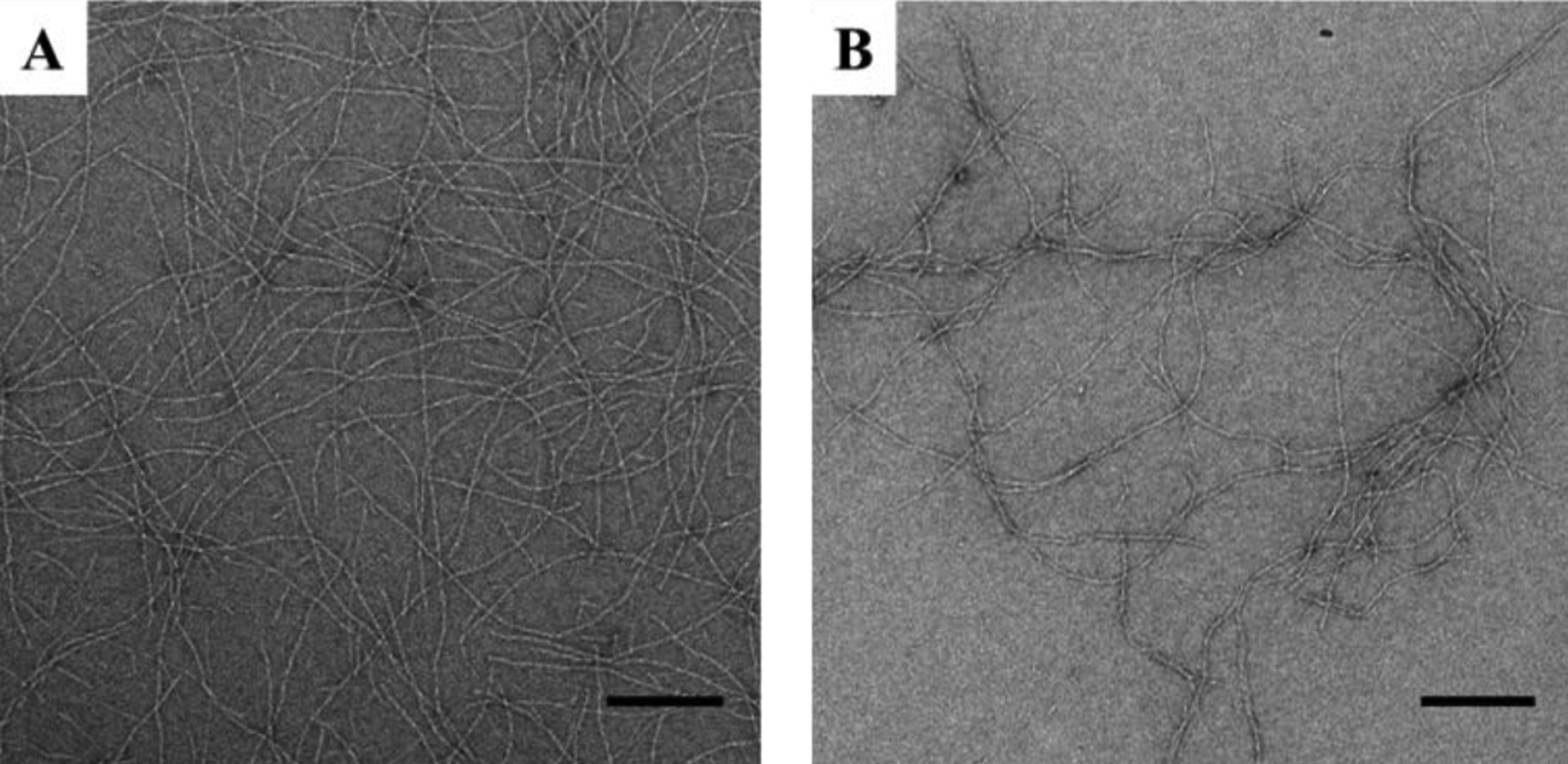
TEM micrographs of fibrils from HLT2 (A) and VEQ3 (B) hydrogels in the absence of encapsulated protein. Scale bar = 200 nm
Encapsulation of Protein into HLT2 and VEQ3 Hydrogel Networks
Proteins can be directly incorporated into HLT2 and VEQ3 gels directly during material formation. For example, adding a buffered solution of protein (2mg/mL protein, 100 mM BTP, 300 mM NaCl at pH 7.4) to a 1 wt % aqueous solution of chilled unfolded VEQ3 in a 1:1 ratio and raising the temperature to 37 °C affords a 0.5 wt % gel (50 mM BTP 150 mM NaCl at pH 7.4) containing 1 mg/mL protein. Three model proteins, specifically α-lactalbumin, myoglobin, and lactoferrin, were encapsulated into both gel networks to investigate the effect of protein charge on material formation, protein partitioning and release profile. These proteins vary significantly in their isoelectric point but are similar in hydrodynamic diameter, Table 2.1,31 α-lactalbumin is negatively charged at pH 7.4 whereas lactoferrin is positively charged, and myoglobin is nearly neutral.
Table 2.
Physical Properties of Model Proteins
| Protein | Molecular Weight (kD) | Hydrodynamic Diameter (nm) | Isoelectric Point, pI | Net Charge at pH 7.4 |
|---|---|---|---|---|
| α-lactalbumin | 14.1 | 3.2 | 4.2–4.5 | − |
| Myoglobin | 14.7 | 4.1 | 7.0 | ∅ |
| Lactoferrin | 77 | 6.1 | 8.4–9.0 | + |
Figure 3 shows the effect of protein encapsulation on the gels’ mechanical rigidity. In the absence of encapsulated protein, the 0.5 wt % HLT2 hydrogel as a storage modulus (a measure of the material’s elastic response to stress, i.e., rigidity) of ~ 200 Pa, 1 hour after the initiation of gelation (solid blue bar). When proteins are present at the onset of gelation (blue checked bars), a slight increase in the rigidity of the resulting hydrogels is observed and is likely due to interactions between the proteins and the evolving peptide network. 0.5 wt % VEQ3 hydrogel formed in the absence of proteins (solid red bar) has a storage modulus of ~ 150 Pa, similar to that of the 0.5 wt % HLT2 hydrogel. VEQ3 hydrogels containing encapsulated α-lactalbumin or myoglobin (checked red bars) yield similar storage moduli values to the empty VEQ3 hydrogel. However, in the case of VEQ3 with lactoferrin, where favorable electrostatic interactions between the peptide network and protein might be expected, a significantly higher hydrogel rigidity is realized (vide infra).
Figure 3.
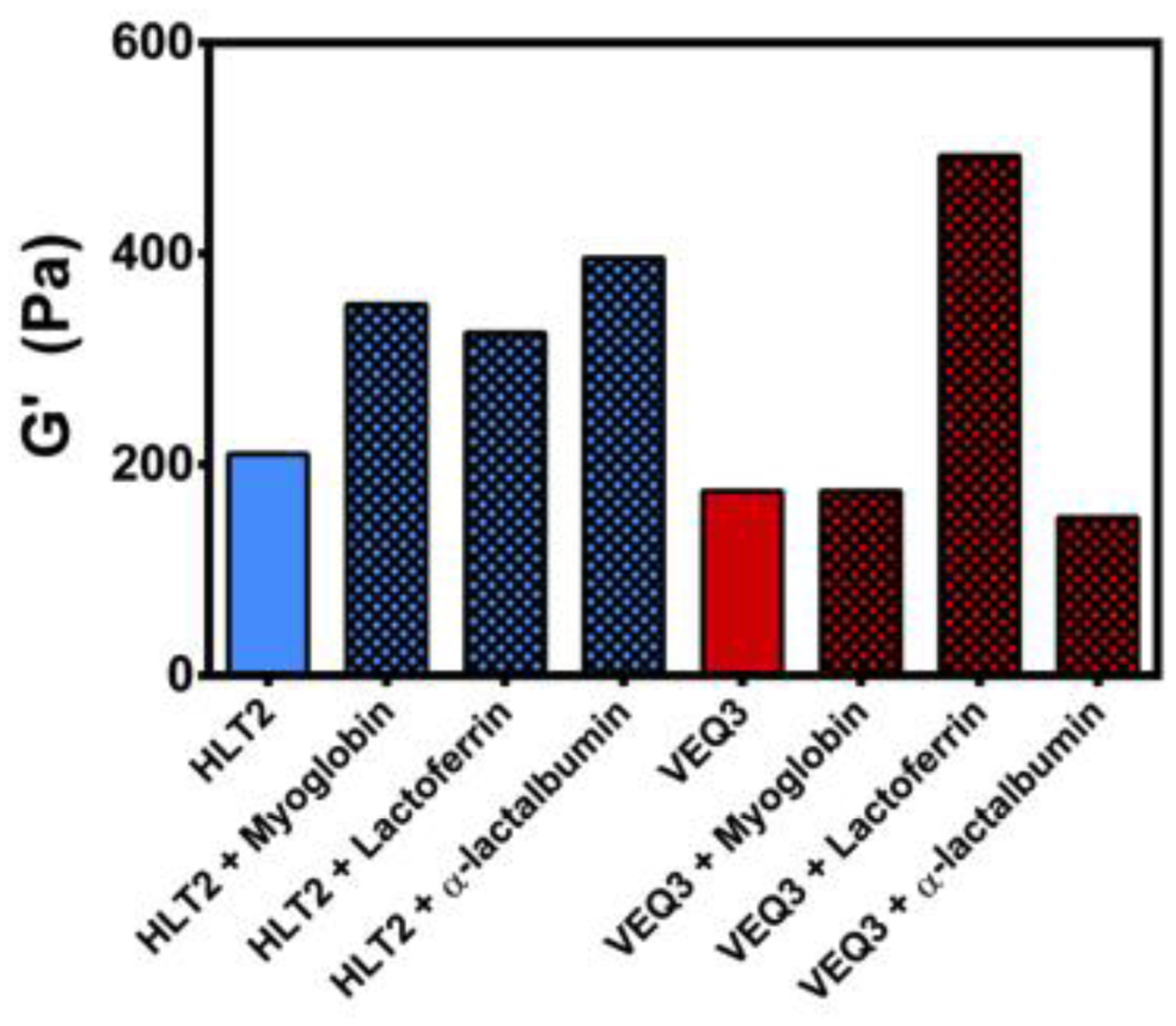
Storage moduli of 0.5 wt % HLT2 and VEQ3 hydrogels 60 minutes after the initiation of gelation in the absence (solid bars) or presence (checkered bars) of encapsulated proteins
Cumulative Release of Proteins from HLT2 and VEQ3 Hydrogels
The release of model proteins α-lactalbumin (−), myoglobin (Ø) and lactoferrin (+) after direct encapsulation within 0.5 wt % HLT2 and VEQ3 hydrogels was monitored over 28 days. Figure 4 shows the amount of protein released from the gels (Mt) normalized to the initial amount of encapsulated protein (M0) with respect to time. For proteins encapsulated in hydrogels of like charge (i.e. lactoferrin within HLT2 hydrogels and α-lactalbumin within VEQ3 hydrogels) over 80 % of the protein was released within the first 4 days. In contrast, those proteins encapsulated in networks of opposite charge (i.e. lactoferrin within VEQ3 hydrogels and α-lactalbumin within HLT2 hydrogels) are largely retained within the respective hydrogel network. Indeed, after 28 days, only 7 % of lactoferrin and 29 % of α-lactalbumin were released from VEQ3 and HLT2 hydrogels respectively. Net-neutral myoglobin was released to similar degrees from both HLT2 and VEQ3 hydrogels (90 and 95 % respectively) as myoglobin is neither attracted to nor repelled by either hydrogel network. These bulk release studies suggest that electrostatics are at play in defining the behavior of protein within the HLT2 and VEQ3 networks even under physiologically relevant ionic strength solutions conditions. Proteins encapsulated within hydrogel networks of opposite charge are largely retained while proteins encapsulated within hydrogel networks of similar charge are fully released.
Figure 4.
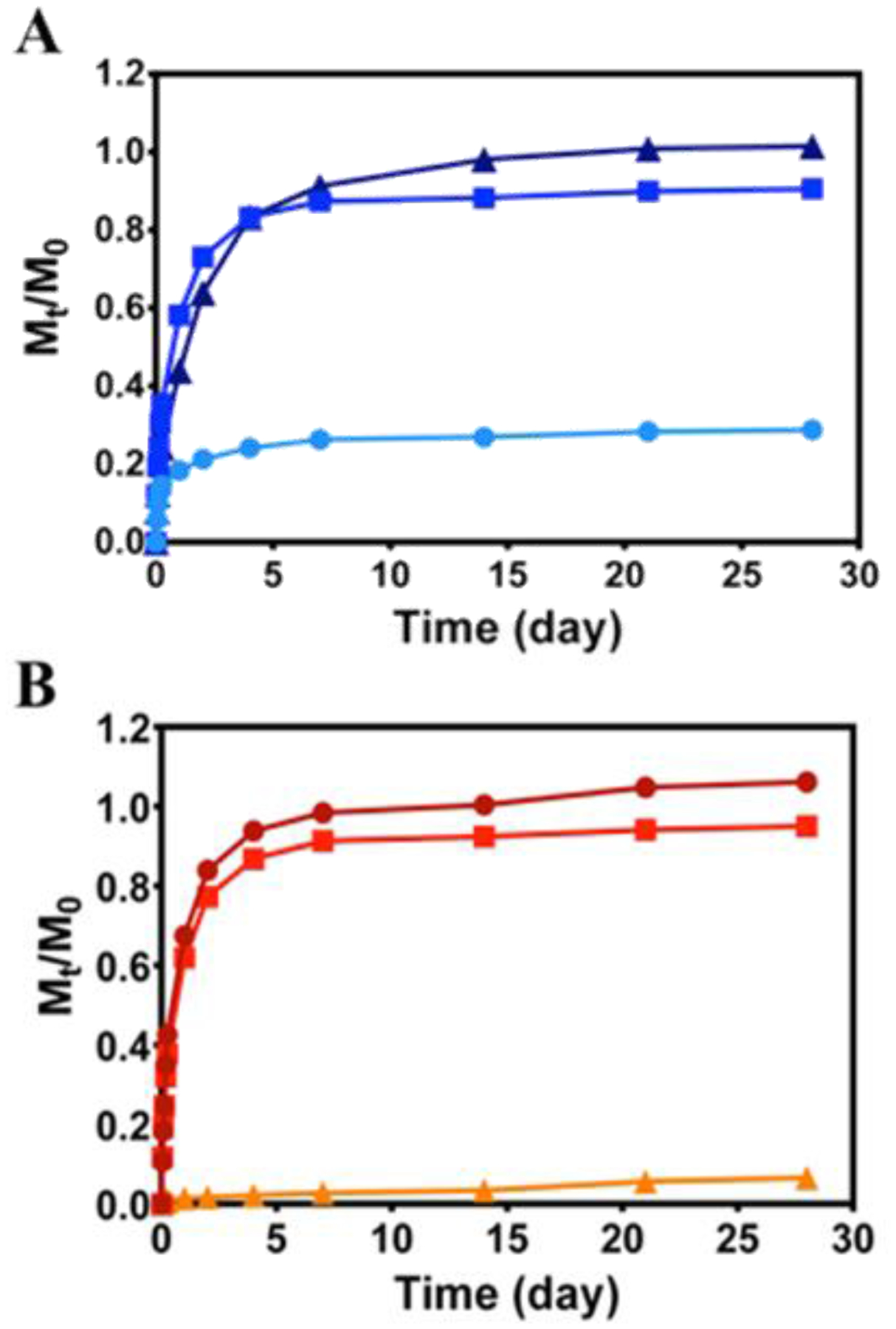
Cumulative release profiles of α-lactalbumin (●), myoglobin (■), and lactoferrin (▲) from 0.5 wt % HLT2 (A) and VEQ3 (B) hydrogels. Error bars are smaller than symbol where not evident. Lines are included to guide the eye.
CD spectroscopy was used to investigate the folded state of each protein after its release from the gels networks. The spectra in Figure S4 show that each protein retains their structure after having been released. Transmission electron microscopy (TEM) was used to access whether or not direct physical interactions between protein and the fibrils could occur as a function of electrostatics. Here, proteins were encapsulated within VEQ3 and HLT2 hydrogels as in the release studies. Aliquots of each hydrogel network were removed at Day 0 and Day 28 of protein release, diluted with water and added to a TEM grid for staining and imaging. Figures 5 & 6 show TEM micrographs of diluted hydrogel networks from both HLT2 and VEQ3 hydrogels containing each model protein. Micrographs were analyzed and fibril widths were measured for comparison to fibrils formed in the absence of encapsulated protein. For both networks at Day 0, charged proteins are found to be adsorbed to the surface of oppositely charged fibrils as indicated by an increase in apparent fibril width compared to fibrils isolated from peptide networks in the absence of encapsulated protein (Panels G-I in Figures 5 & 6).
Figure 5.
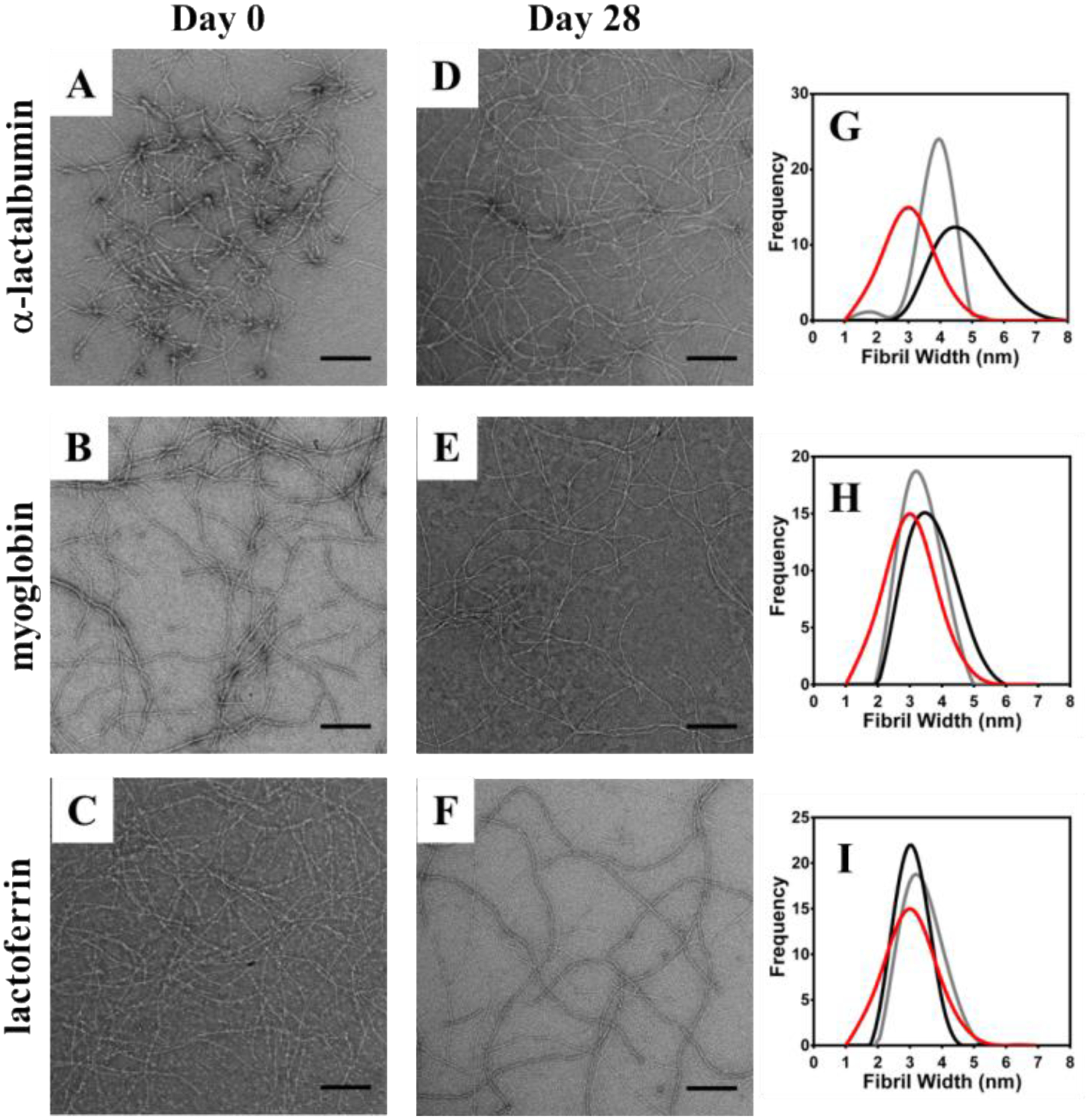
TEM micrographs of α-lactalbumin (A&D) myoglobin (B&E) and lactoferrin (C&F) interaction with fibrils at 0 (A-C) and 28 (D-F) days after encapsulation in 0.5 wt % HLT2 hydrogels. Measured fibril widths provide a direct indication of protein-fibril interactions (G-I) by comparing fibrils in the absence of protein (red) with samples from day 0 (black) and day 28 (gray) after protein release. Scale bar = 200 nm.
Figure 6.
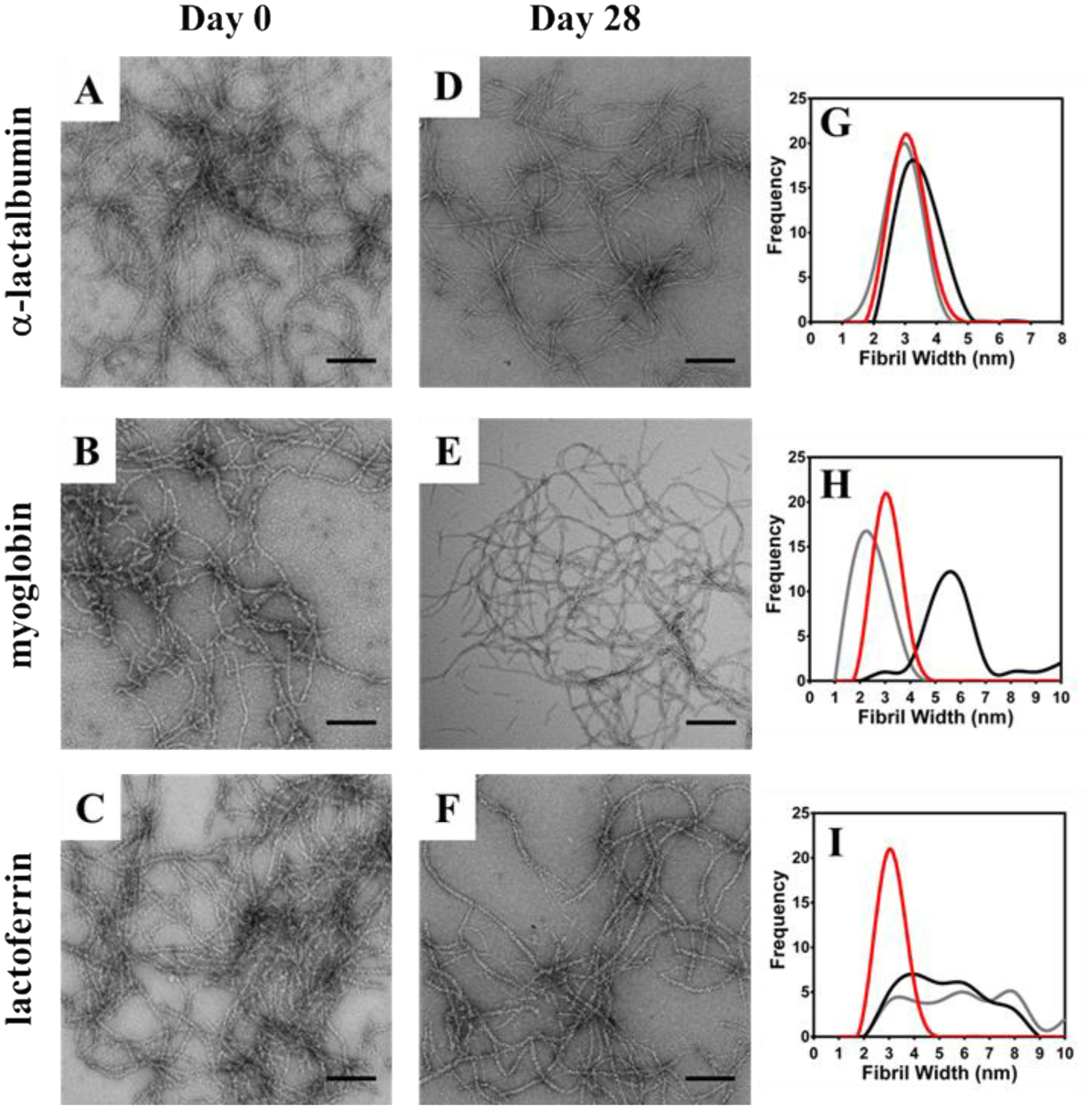
TEM micrographs of α-lactalbumin (A&D) myoglobin (B&E) and lactoferrin (C&F) interaction with fibrils at 0 (A-C) and 28 (D-F) days after encapsulation in 0.5 wt % VEQ3 hydrogels. Measured fibril widths provide a direct indication of protein-fibril interactions (G-I) by comparing fibrils in the absence of protein (red) with samples from day 0 (black) and day 28 (gray) after protein release. Scale bar = 200 nm.
As seen in Figure 5G, when negatively charged α-lactalbumin is encapsulated within the positively charged network of HLT2 the protein readily adsorbs to the fibrils. The average widths of the fibrils constituting the gel significantly increase from ~ 3 nm to ~ 5 nm. In contrast, when neutral myoglobin (panel H) and positively charged lactoferrin (panel I) are encapsulated there is little change in fibril width. A similar trend is seen for the negatively charged VEQ3 fibril network, Figure 6. For example, when lactoferrin (+) is encapsulated, the fibrils are decorated with protein as indicated by an increase in fibril width (Figure 6I). When similarly charged α-lactalbumin is encapsulated, there is little change in fibril dimension indicating that the protein does not significantly interact with the fibrils. Interestingly, myoglobin, although neutral, has a propensity to adsorb to the negatively charged fibrils. After 28 days of release, the micrographs show that for a given adsorbed protein, a significant fraction remains sequestered the fibrils. For example, Figure 5G shows HLT2 fibrils coated with α-lactalbumin (−) decrease in size, but are still larger than naked fibrils. This is consistent with the release profile for α-lactalbumin from the HLT2 gels shown in Figure 4A where, after 28 days, ~ 70 % of the protein remains entrapped in the network bound to fibrils. The TEM data in Figure 5H & 5I are also consistent with the release data in Figure 4A. For example, under conditions where myoglobin (Ø) and lactoferrin (+) are completely released from the gel, little difference in fibril dimensions is observed. Likewise, the TEM data in Figure 6 for VEQ3 hydrogels is also consistent with the release profiles of the proteins in Figure 4B. Both α-lactalbumin (−) and myoglobin (Ø) are fully released from the VEQ3 hydrogels after 28 days and measured fibril widths are similar to that of naked fibrils, Figures 6G and H. In contrast, positively charged lactoferrin is highly retained within the gels and can still be clearly seen adsorbed to the negatively charged VEQ3 fibril network after 28 days with no apparent decrease in width from the onset of the release experiment (Figure 6C, F, & I). These images are in agreement with the cumulative release studies where proteins are retained within hydrogels of opposite charge and fully released from those of the same charge.
To further confirm that these observations are indeed due to attractive electrostatic interactions between the protein and peptide fibrils, surface adsorption studies were performed (Figure 7). Here, 0.5 wt % hydrogels of HLT2 and VEQ3 were prepared and allowed to cure for three hours. Next, solutions containing 1 mg/mL protein were added to the top of each gel. After 48 hours, the solutions above the gels were removed and the amount of protein within the supernatant was quantified to determine the amount, by difference, of protein that had partitioned into the hydrogel. All proteins, regardless of charge or the electrostatic nature of the encapsulating network, partition into the hydrogels. If there are no specific interactions driving the partitioning of protein into the network, one would expect by mass action that approximately 50 % of the protein passively partitions into the gel. As can be seen in Figure 7A, 42 % of lactoferrin (+) and 39 % of myoglobin (Ø) partition in the HLT2 gel (compare black solid bars to checkered bars). These values are close to 50 % indicating that the proteins can freely diffuse into the gel network. However, over 70 % of α-lactalbumin (−) partitions into the HLT2 hydrogel suggesting that preferential electrostatic interactions promote the diffusion of the protein into the hydrogel network. Figure 7B shows that 47 % of negatively charged α-lactalbumin and 44 % of neutral myoglobin diffuses into the negatively charged VEQ3 hydrogels. Convincingly, 87 % of positively charged lactoferrin partitions into the gels with only 13 % protein remaining in the supernatant. Thus, preferential electrostatic-based affinities largely define the amount of protein that initially diffuses into the hydrogel networks when the protein is added to pre-formed gels. This data suggests that similar interactions are responsible for the retention of protein within the network when the protein is directly encapsulated during gel formation.
Figure 7.
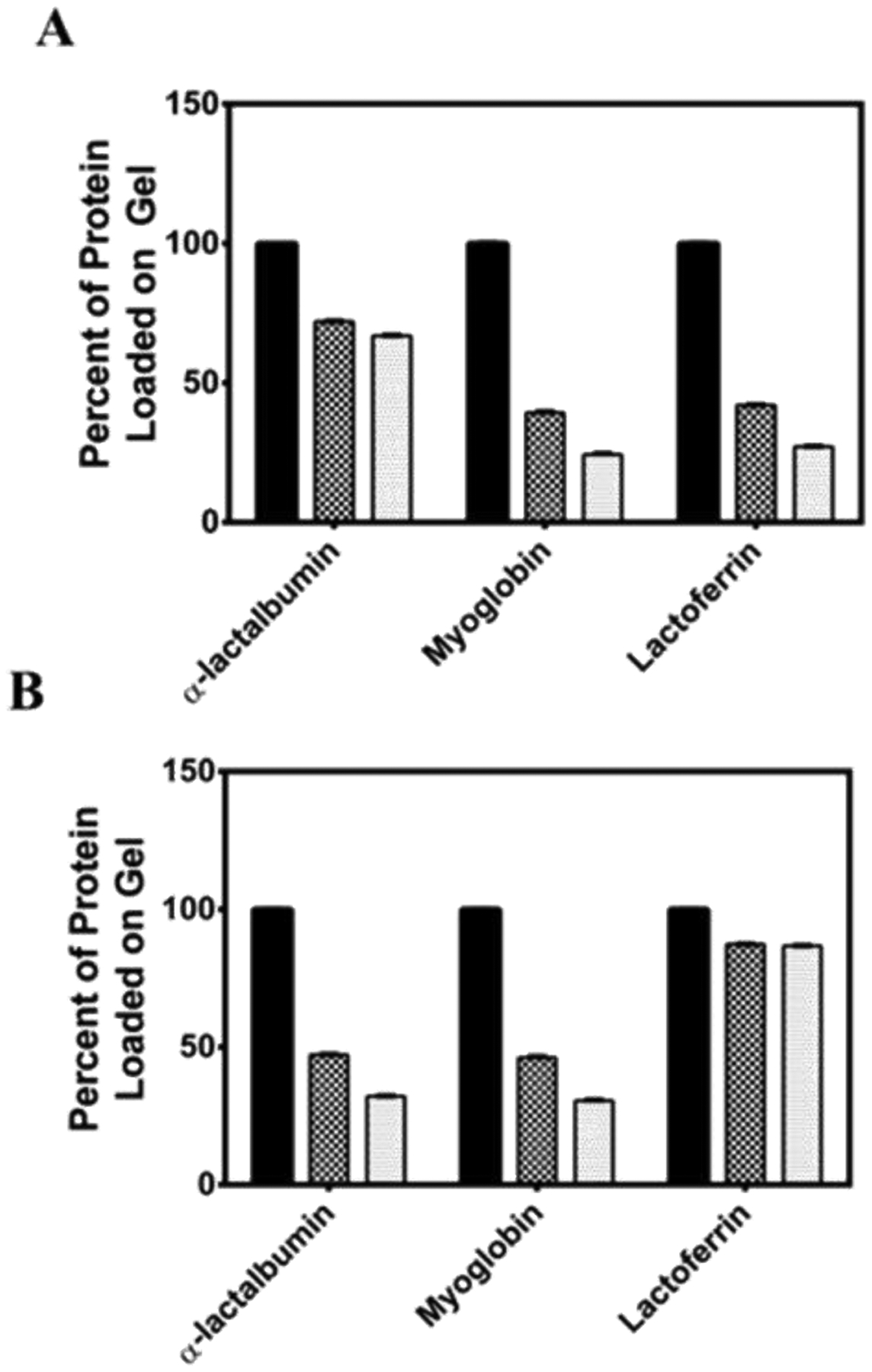
Partition and retention studies of α -lactalbumin, myoglobin, and lactoferrin into HLT2 (A) and VEQ3 (B) hydrogels. Black bar: Initial amount of protein added above gel (~ 0.3 mg); Checkered bar: Percent of protein partitioned into the hydrogel after 24 hours; Gray bar: Percent of protein retained within the gel after 1 week.
Next, we sought to determine to what extent electrostatic interactions could keep proteins immobilized after they had diffused into the network. In this phase of the adsorption experiment, gels containing partitioned protein are extensively washed over the course of a week and subsequently analyzed for the amount of protein remaining in the network. After successive washes of the HLT2 hydrogel (Figure 7A), negatively charged α-lactalbumin is highly retained within the network with 95 % of the immobilized protein remaining adsorbed to the peptide network (compare gray bar to checkered bar). This degree of protein retention to the network is consistent with the formation of strong electrostatic interactions between the positively-charged network and negatively charged α-lactalbumin. In contrast, myoglobin (Ø) and lactoferrin (+) were retained to a significantly lower degree, 67 % and 62 %, respectively. This suggests that these proteins diffuse into and within the gel without significantly interacting locally with the fibrils in the network. For the VEQ3 hydrogels, 99 % of the adsorbed lactoferrin remains in the gel indicating the formation of attractive electrostatic interactions between the negatively charged network and the positively charged protein, Figure 7B (data at far right of panel). As expected, α-lactalbumin is not as well retained in the gel with 68 % of the encapsulated protein remaining within the gel. Neutral myoglobin is retained to a similar degree as α-lactalbumin (−) with approximately 66 % of the encapsulated protein remaining within the VEQ3 hydrogel. The data in Figure 7 is consistent with the release data in Figure 4 where encapsulated proteins that are not adsorbed to the network are capable of quantitative release with time. However, for those proteins capable of electrostatic adsorption to the fibrils within the network, only a fraction of the population is released from the gel, with the remainder being tightly adsorbed. Taken together, the TEM and protein retention studies establish the strong effect of attractive electrostatic interactions between the peptide fibrillar network and encapsulated model proteins. The decoration of the peptide fibrils by adsorbed protein is likely responsible for the increase in storage modulus for gels composed of HLT2/α-lactalbumin and VEQ3/lactoferrin where protein adsorptions serves to effectively stiffen the fibrils that comprise the network leading to a more rigid gel32.
Conclusions
Electrostatics can be used as a materials design principle to modulate the release profile of proteins. However, electrostatic interactions made between a material network and its protein payload can be potentially screened under physiological solution conditions, decreasing the effectiveness of this design strategy. We show that electrostatic interactions made between proteins and peptide-based hydrogel networks of high charge density are not effectively screened under physiological buffer conditions. Peptide HLT2 has a net charge of +5 per monomer at neutral solution pH and forms a highly positively charged fibrillar network. VEQ3 has a net formal charge of −5 per peptide monomer and assembles into a highly negatively charged fibrillar network. The rate and total amount of protein released over a given time period are significantly effected by these highly charged networks. Negatively charged proteins are retained within positively charged HLT2 hydrogels but fully released from negatively charged VEQ3 hydrogels over the course of days. Net-neutral proteins are also fully released from both hydrogels over days, as they are neither repelled nor attracted to the peptide fibrils that constitute each hydrogel. Positively charged proteins are similarly fully released from the electropositive HLT2 hydrogels but are highly retained within the electronegative VEQ3 network.
Supplementary Material
Acknowledgements
This work was supported by the Intramural Research Programs of the National Cancer Institute of the National Institutes of Health.
Footnotes
Electronic Supplementary Information (ESI) available: [Analytical HPLS and ESI-mass spectra of peptides used in this study]. See DOI: 10.1039/c5tb02137e
References
- 1.Branco MC; Pochan DJ; Wagner NJ and Schneider JP Biomaterials 2010, 31, 9527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Branco MC and Schneider JP Acta Biomater. 2009, 5, 817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peng ZP; She YQ and Chen L J. Biomater. Sci.: Poly. Ed 2015, 26, 111. [DOI] [PubMed] [Google Scholar]
- 4.Park JW; Kang YD; Kim JS; Lee JH and Kim HW Biochem. Biophys. Res. Commun 2014, 447, 400. [DOI] [PubMed] [Google Scholar]
- 5.Kopesky PW; Byun S; Vanderploeg EJ; Kisiday JD; Frisbie DD and Grodzinsky AJ J. Biomed. Mater. Res., Part A 2014, 102, 1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang HM and Yang ZM Nanoscale 2012, 4, 5259. [DOI] [PubMed] [Google Scholar]
- 7.Kumar TRS; Soppimath K and Nachaegari SK Curr. Pharm. Biotechnol 2006, 7, 261. [DOI] [PubMed] [Google Scholar]
- 8.Crommelin DJA; Storm G; Verrijk R; de Leede L; Jiskoot W and Hennink WE Int. J. Pharm 2003, 266, 3. [DOI] [PubMed] [Google Scholar]
- 9.Kane C; O’Neil K; Conk M and Picha K Inflamm. Allergy Drug Targets 2013, 12, 81. [DOI] [PubMed] [Google Scholar]
- 10.Vaishya R; Khurana V; Patel S and Mitra AK Expert Opin. Drug Delivery 2015, 12, 415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caon T; Jin L; Simoes CMO; Norton RS and Nicolazzo JA Pharm. Res 2015, 32, 1. [DOI] [PubMed] [Google Scholar]
- 12.Huang WW; Rollett A and Kaplan DL Expert Opin. Drug Delivery 2015, 12, 779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bertz A; Wohl-Bruhn S; Miethe S; Tiersch B; Koetz J; Hust M; Bunjes H and Menzel H J. Biotechnol 2013, 163, 243. [DOI] [PubMed] [Google Scholar]
- 14.Branco MC; Pochan DJ; Wagner NJ and Schneider JP Biomaterials 2009, 30, 1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu Y and Chau Y Biomacromolecules 2015, 16, 56. [DOI] [PubMed] [Google Scholar]
- 16.Pakulska MM; Vulic K and Shoichet MS J. Controlled Release 2013, 171, 11. [DOI] [PubMed] [Google Scholar]
- 17.Van Hove AH; Beltejar MJG and Benoit DSW Biomaterials 2014, 35, 9719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu S; Lam J; Trachtenberg JE; Lee EJ; Seyednejad H; van den Beucken J; Tabata Y; Wong ME; Jansen JA; Mikos AG and Kasper FK Biomaterials 2014, 35, 8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shekaran A; Garcia JR; Clark AY; Kavanaugh TE; Lin AS; Guldberg RE and Garcia AJ Biomaterials 2014, 35, 5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schillemans JP; Hennink WE and van Nostrum CF Eur. J. Pharm. Biopharm 2010, 76, 329. [DOI] [PubMed] [Google Scholar]
- 21.Sinthuvanich C; Haines-Butterick LA; Nagy KJ and Schneider JP Biomaterials 2012, 33, 7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giano MC; Pochan DJ and Schneider JP Biomaterials 2011, 32, 6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schneider JP; Pochan DJ; Ozbas B; Rajagopal K; Pakstis L and Kretsinger J J. Am. Chem. Soc 2002, 124, 15030. [DOI] [PubMed] [Google Scholar]
- 24.Pochan DJ; Schneider JP; Kretsinger J; Ozbas B; Rajagopal K and Haines L J. Am. Chem. Soc 2003, 125, 11802. [DOI] [PubMed] [Google Scholar]
- 25.Rajagopal K; Lamm MS; Haines-Butterick LA; Pochan DJ and Schneider JP Biomacromolecules 2009, 10, 2619. [DOI] [PubMed] [Google Scholar]
- 26.Ozbas B; Kretsinger J; Rajagopal K; Schneider JP and Pochan DJ Macromolecules 2004, 37, 7331. [Google Scholar]
- 27.Haines-Butterick L; Rajagopal K; Branco M; Salick D; Rughani R; Pilarz M; Lamm MS; Pochan DJ and Schneider JP Proc. Nat. Acad. Sci. U.S.A 2007, 104, 7791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagy KJ; Giano MC; Jin A; Pochan DJ and Schneider JP J. Am. Chem. Soc 2011, 133, 14975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagy-Smith K; Moore E; Schneider J and Tycko R Proc. Nat. Acad. Sci. U.S.A 2015, 112, 9816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ozbas B; Rajagopal K; Schneider JP and Pochan DJ Phys. Rev. Lett 2004, 93, 268106. [DOI] [PubMed] [Google Scholar]
- 31.Baker EN and Baker HM Cell. Mol. Life Sci 2005, 62, 2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mackintosh FC; Kas J and Janmey PA Phys. Rev. Lett 1995, 75, 4425. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.