

Abstract
Biological systems are by nature multiscale, consisting of subsystems that factor into progressively smaller units in a deeply hierarchical structure. At any level of the hierarchy, an ever-increasing diversity of technologies can be applied to characterize the corresponding biological units and their relations, resulting in large networks of physical or functional proximities – e.g. proximities of amino acids within a protein, of proteins within a complex, or of cell types within a tissue. Here, we review general concepts and progress in using network proximity measures as a basis for creation of multiscale hierarchical maps of biological systems. We discuss the functionalization of these maps to create predictive models, including those useful in translation of genotype to phenotype; strategies for model visualization; and challenges faced by multiscale modeling in the near future. Collectively, these approaches enable a unified hierarchical approach to biological data, with application from the molecular to the macroscopic.
Keywords: Multiscale, hierarchical models, systems biology, structure, structural biology, networks, network biology, community detection
Introduction
Biological structure is fundamentally multiscale (Simon, 1991). Atoms make up amino acids which, in turn, are the building blocks of proteins; proteins interact to form protein complexes; and protein complexes interrelate within cellular components such as organelles. The hierarchy can be expanded further to consider interactions among cell types to form tissues, among different tissues in an organ, or among organs in an organism. Determining this multiscale structural hierarchy, as well as its relation to biological function, is one of the ultimate goals of the biological sciences (Box 1).
Box 1: What is Scale?
Many different studies have been described variously as “multiscale,” “large-scale” or “genome-scale”, motivating an important question of what exactly is meant by “scale” in these efforts. In many contexts including this article, scale is used to describe the physical size of the entities under study. Entities of interest in the biological sciences range from single atoms with diameters measured in angstroms, to amino acid residues on the order of a nanometer, to proteins and protein complexes of tens to hundreds of nanometers, to organelles and whole cells several microns in diameter, to tissues, organs and individuals measured in fractions of meters. In this sense, a “multiscale” model is one that includes entities at more than one of these size ranges, along with defined relationships between the entities at different scales (e.g., containment of two proteins within a protein complex; trafficking of a protein complex between the nucleus and cytoplasm). Some contexts measure scale using mass rather than size, especially molecular characterizations by mass spectrometry or centrifugation which typically measure units in Daltons.
The biological entities characterized by an analysis can be described by upper and lower limits in size or mass, determined by the experimental method used to generate the data. The lower limit of scale is known as the resolution, or the ability to distinguish between two entities. The upper limit of scale is called the field of view (FOV), originally based on the literal two-dimensional field of view of a light microscope but applicable to many technologies. For example, a multiscale model of a protein complex generated using cryo-EM analysis typically has near-atomic resolution and a field of view equal to the overall complex size, and thus the scale spans from approximately 0.1 – 10.0 nm. Standard proteomic and genomic analyses on a single cell type result in a field of view at the cellular level, whereas single-cell RNA-sequencing analyses extend the field of view to the tissue level when clustering is performed to form a hierarchy of cell types.
In contrast to these definitions, the phrase “genome-scale” has a very different connotation in that it refers to studies that include measurements for nearly all instances of a single type of entity, the gene. Similarly, proteome-scale analysis refers to studies at the level of the whole proteome in terms of the number of proteins analyzed. Despite these monikers, genome-and proteome-scale analyses are typically narrow in scale as they focus on measurement of a single type of biological entity – genes or proteins.
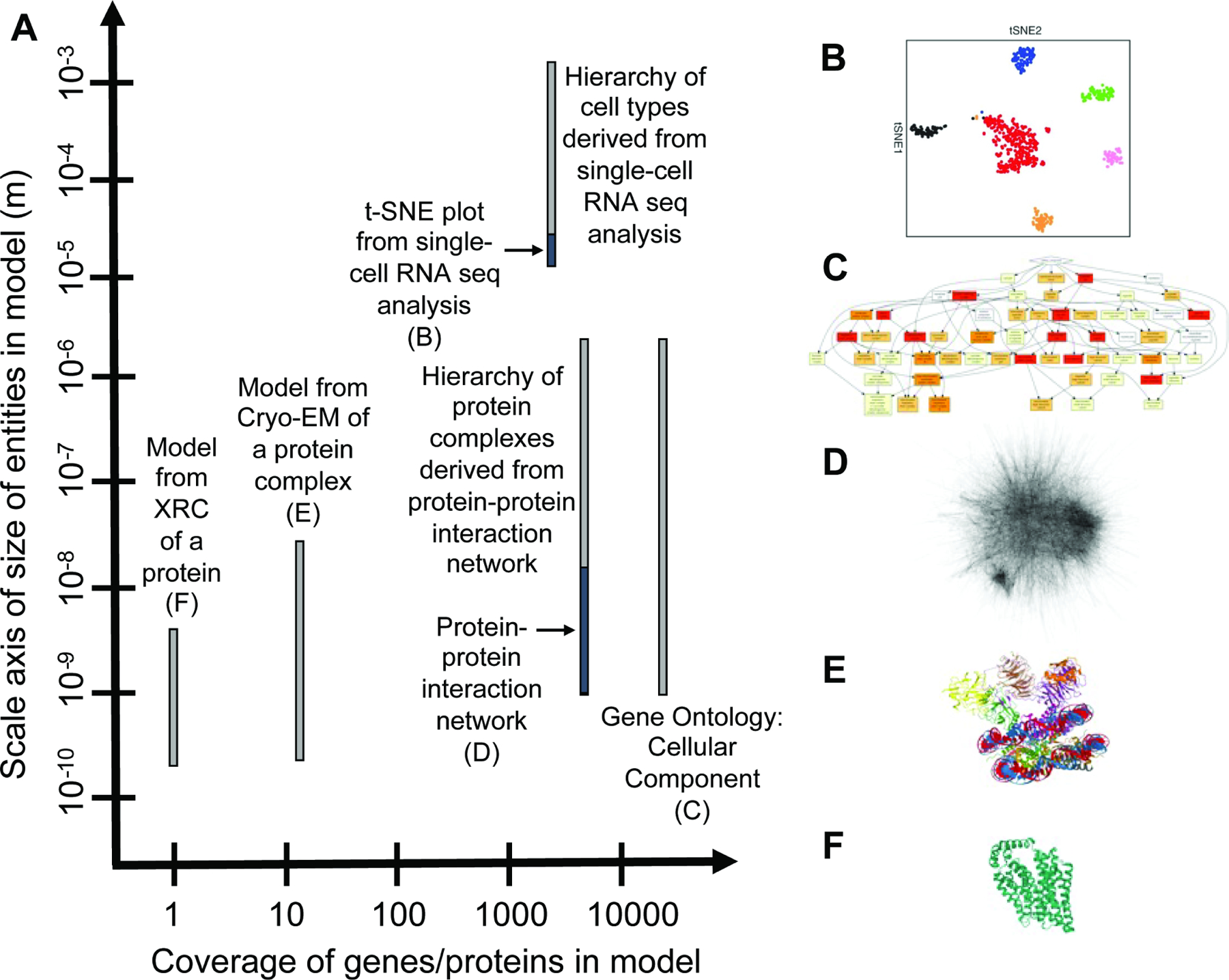
Various biological studies excel at different aspects of encapsulating both a comprehensive analysis (i.e., examining all genes, proteins or other entities) and multiscale dimensionality regarding resolution and field of view (Box 1 Figure). An ideal model of biological structure of a tissue would perhaps be one that captures both the breadth of proteins (i.e. proteome-wide) as well as encapsulates the many different sizes of subsystems (i.e. deeply multiscale).
Box 1 Figure. Scale and coverage of common biological analyses.
(A) Various biological analyses result in models with different coverage of the human proteome (x-axis) that span multiple scales to varying degrees (y-axis). (B) t-SNE plot from single-cell RNA sequencing data of malignant cells. Distinct communities of cells (colored points) are visible in the plot area, with each community containing 1 to 100 cells measuring in the 10–4 to 10–5 m range for a human cell with nominal radius 10–5 m. From Tirosh, I., Izar, B., Prakadan, S.M., Wadsworth, M.H., 2nd, Treacy, D., Trombetta, J.J., Rotem, A., Rodman, C., Lian, C., Murphy, G., et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. Reprinted with permission from AAAS. (C) Enriched Cellular Component GO terms for list of genes present in MitoCarta 2.0 database (Calvo et al., 2016) visualized with GOrilla (Eden et al., 2009). Color coding shows the degree of enrichment for each GO term. (D) Protein interaction network from BioPlex AP-MS data of HEK293 cells (Huttlin et al., 2015). Reprinted with permission from Elsevier. (E) Model of MLL1-ubNCP complex from cryo-EM analysis. Image from the RCSB PDB (rcsb.org) of PDB entry 6KIW (Xue et al., 2019). (F) Model of human glucose transporter GLUT1 from x-ray crystallography. Image from the RCSB PDB (rcsb.org) of PDB entry 4PYP (Deng et al., 2014).
There has been much recent interest in the challenge of multiscale modeling within the field of “integrative” structural biology, whereby data from different measurement approaches are combined to generate a unified, superior model of a biological structure (Alber et al., 2008; Faini et al., 2016; Lasker et al., 2010; Rout and Sali, 2019; Russel et al., 2012; Ward et al., 2013). A core concept is that complementary measurement techniques are not only beneficial in general, but they may ultimately be necessary to gain a full understanding of biological structure across multiple scales. For instance, such an approach was applied to the challenging problem of determining the 552-protein nuclear pore structure in yeast at sub-nanometer precision, illustrating the power of integrating multiple experimental approaches (Kim et al., 2018).
Many of the same principles and analysis frameworks important for modeling the structure of proteins and protein complexes are now emerging in the analysis of larger biological systems – scales usually considered to be in the realm of “systems”, rather than structural, biology (Singla et al., 2018). For instance, gene and protein expression, interaction, and other ‘omics profiles are routinely integrated to elucidate multiscale hierarchies of cellular processes (Jaimovich et al., 2010; Kramer et al., 2017; Ryan et al., 2012; Vaske et al., 2010). At a larger scale still, single-cell RNA sequencing data are now often used to infer multiscale maps of cell types comprising an anatomical tissue along with their developmental trajectories (Durruthy-Durruthy et al., 2014; Farrell et al., 2018; Wagner et al., 2018; Zeisel et al., 2015; Zhong et al., 2018).
Importantly, many of the measurement techniques underlying structural, systems, and developmental biology are based on observations of pairwise proximity, which are naturally represented by biological “proximity” networks. In each of these cases, measurements focus on a particular type of biological entity – atoms, nucleic or amino acids, DNA fragments, peptides, proteins, cells, tissues, and so on – with the goal of determining which groups of entities are in close physical contact or are otherwise related by a high degree of functional similarity. In the representation of these data, the entities are treated as network “nodes”, and proximal or similar entities are connected by network “edges” (connecting pairs of biological entities) or “hyperedges” (used to interconnect larger groups greater than two) (Berge, 1984). Edges may be assigned weights, expressing quantitative proximity relationships, or they may be present/absent, in which they simply mark the entities determined to be physically or functionally close to one another. Thus, networks provide a core foundation for representing many types of experimental evidence – across many different scales – in the inference of biological structure and function.
Here, we explore the extent to which proximity networks and their analysis can provide a general, unifying framework to map the hierarchical structures and functions that make up an organism. In what follows, we survey the specific network proximity measurements most commonly used to characterize biological systems at different size scales. We then review general methods by which multiscale hierarchical maps can be assembled from network proximity measures, and we discuss the similarities and differences between these assembled maps and biological ontologies. Next, we explore the functionalization and visualization of multiscale maps and models. Finally, we explore current challenges facing the field along with potential solutions. Collectively, these techniques provide the opportunity to create unified hierarchical models across multiple relevant biological scales, from the very small (angstroms) to the very large (meters).
Proximity networks as a general data structure across scales
While examples of proximity networks abound at every scale of biology, perhaps the largest diversity of biological network research has been in proteomics, where nodes represent proteins and edges represent physical proximity of a protein pair. Such protein interaction networks are produced by a variety of methods including affinity purification mass spectrometry (AP-MS) (Gordon et al., 2020; Huttlin et al., 2015, 2017), co-elution mass spectrometry (Bludau et al., 2020; Havugimana et al., 2012; Salas et al., 2020), peptide crosslinking (Liu and Heck, 2015; Vasilescu et al., 2004), yeast two-hybrid assays (Luck et al., 2020; Rolland et al., 2014; Stelzl et al., 2005), and proximity labeling (Go et al., 2019; Lobingier et al., 2017; Roux et al., 2012). Protein interactions of these various types are documented in online databases such as BioGRID (Stark et al., 2006) and STRING (Szklarczyk et al., 2018). The PrePPI database documents protein-protein interactions predicted using high resolution structural data as well as functional, evolutionary, and expression data (Zhang et al., 2012b).
Closely related to physical protein interactions are so-called “genetic” interactions, which indicate a close functional, rather than physical, relationship among pairs of genes. A genetic interaction occurs when mutations to two genes produce a phenotype that is unexpected from mutations to each gene individually (Dixon et al., 2009). Large genetic interaction screens, in which panels of genes are mutated or otherwise perturbed in combination, were originally reserved for model species (Bandyopadhyay et al., 2010; Costanzo et al., 2010; van Leeuwen et al., 2016; Tong et al., 2001) but are increasingly applied to humans using combinatorial CRISPR techniques (Behan et al., 2019; Han et al., 2017; Shen et al., 2017). The phenotype of each combination can be measured using static growth assays to characterize fitness (Byrne et al., 2007; Luo et al., 2008; Tsherniak et al., 2017) or more complex or dynamic readouts (Díaz-Mejía et al., 2018; Gonatopoulos-Pournatzis et al., 2020; Jaffe et al., 2019; Norman et al., 2019; Tsherniak et al., 2017). Although genes with similar genetic interaction profiles can encode proteins in close physical proximity, this does not always have to be the case, as genetic interactions can also be observed among genes participating in distinct parallel biological pathways (Gonatopoulos-Pournatzis et al., 2020).
Beyond genes and proteins, networks are convenient data structures for representing relations among entities at many other levels of biological hierarchy (Figure 1). At the atomic scale, electron density images generated by x-ray crystallography (Adams et al., 2002; Kleywegt et al., 2004; Yang et al., 2016) or cryo-electron microscopy (cryo-EM) (Chen et al., 2016; Frenz et al., 2017; Hryc et al., 2017) produce proximity constraints that can be represented as networks of atoms (nodes) linked to other atoms determined to be in close spatial proximity (edges). Moving from atoms to residues, cross-linking MS has recently been very successful in generating networks of proximal amino acids in a protein, or between proteins in a protein complex (Holding, 2015). In these networks, amino acid residues (nodes) are interconnected to others within a certain distance of one another (edges), where this maximum interresidue distance depends on the length of the cross-linker utilized. Such amino-acid proximity networks, whether experimentally generated or inferred, are central to many efforts to model protein structure. They are also central to protein structure prediction. For instance, AlphaFold, a recent major achievement in protein folding prediction (Senior et al., 2019, 2020), uses a deep neural network to generate (initial AlphaFold version) and/or interpret (most recent implementation) the amino-acid proximity network as a central data structure. Here, proximity can be due to either covalent bonds that link atoms or weaker chemical interactions (including hydrogen bonds, hydrophobic interactions, and steric occlusion), which are typically the relevant ones for biological systems.
Figure 1. Biological entities and proximity measures ranging in scale from angstroms to meters.
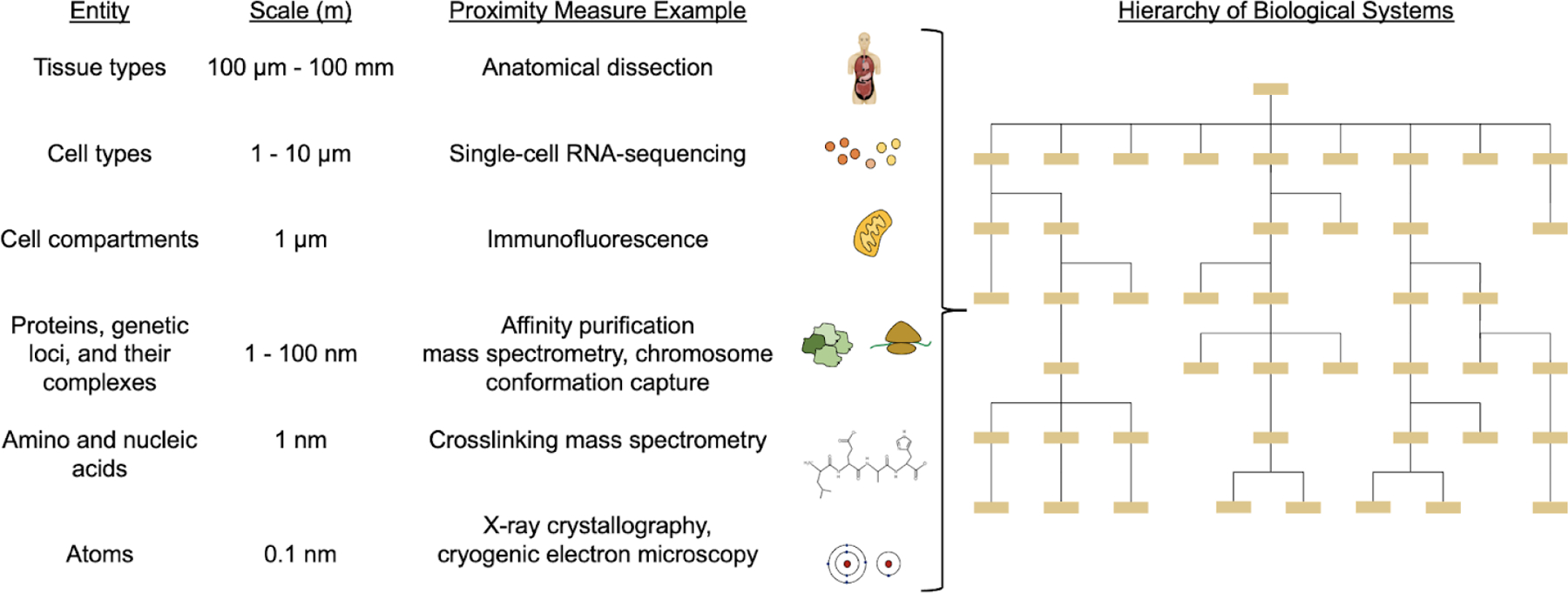
A wide range of analytical methods provide proximity or distance measurements among entities at successive layers of biological structure. A unified hierarchy of biological systems would ideally span all scales of biological structure and function within an organism.
Moving from proteins to DNA, the family of techniques known as chromosome conformation capture, including 3C (Dekker et al., 2002) and Hi-C (Lieberman-Aiden et al., 2009), record pairs of genomic loci that are at close proximity. In Hi-C studies, certain pairs of loci emerge at close proximity in three dimensions although some of these loci are separated at large nucleotide distances along the linear chromosome; such linkages can implicate regulatory interactions among enhancers, promoters, and gene bodies. Similar to how atomic or amino-acid distances are used in determination of protein structure, HiC networks can be used to reconstruct the 3D structure of chromatin and its spatial distribution within the nucleus (Varoquaux et al., 2014).
Above the level of individual proteins and genomic loci, many recent studies use the technique of single-cell RNA sequencing to generate cell-cell networks, in which nodes represent the RNA expression state of an individual cell, and edges connect pairs of cells that are highly similar (Bendall et al., 2014; Trapnell et al., 2014). Edges are typically determined by drawing the shortest cell-cell Euclidean distances from a reduced representation of the RNA expression data, such as a t-Distributed Stochastic Neighbor Embedding (t-SNE) (van der Maaten and Hinton, 2008) or Uniform Manifold Approximation and Projection (UMAP) (Becht et al., 2018). The resulting cell-cell similarity network can then be analyzed to track cell developmental trajectories over time (Farrell et al., 2018; Wagner et al., 2018; Zhong et al., 2018) or to recognize groups of cells forming a distinct cell type or tissue (Jaitin et al., 2014; Zeisel et al., 2015). Finally, at the highest level in the structural hierarchy of an organism, results from anatomical dissection can be thought of as generating a network of tissue types interrelated to one another by physical or functional proximity. This concept can be extended even further to consider interactions at and above the level of organisms, such as social networks interconnecting individuals of a species (Sah et al., 2019), or host-pathogen (Gordon et al., 2020; Penn et al., 2018) or symbiotic interactions interconnecting different species within an ecosystem (Pringle and Tarnita, 2017; Widder et al., 2016). These examples portray how different methods result in networks of biological entities at different levels of biological hierarchy, from angstroms to meters.
Inferring multiscale hierarchical maps from proximity networks
Developing a multiscale representation requires analyzing communities in biological networks at more than one scale. Communities (clusters of highly interconnected nodes) are first defined and identified at each of multiple scales; subsequently, communities that are contained within other communities, in whole or in part, can be traced to infer a hierarchical map (Figure 2) (Dotan-Cohen et al., 2009; Jaimovich et al., 2010; Kelley and Ideker, 2005; Park and Bader, 2011; Ravasz et al., 2002; Tanay et al., 2004). Such a community hierarchy is also a type of network (formally, a directed acyclic graph or DAG) (Thulasiraman and Swamy, 1992) in which hierarchy nodes represent communities and directed edges in the hierarchy (A → B) represent containment or partial containment of one community (A) by another (B). An example containment relation would be a set of protein subunits (A1, A2, …) contained by a multimeric protein complex B. Note that in the hierarchical DAG structure, a system can be assigned (receive edges from) multiple subsystems, and it can participate in (have edges to) multiple overarching supersystems.
Figure 2. Community detection in a biological network.
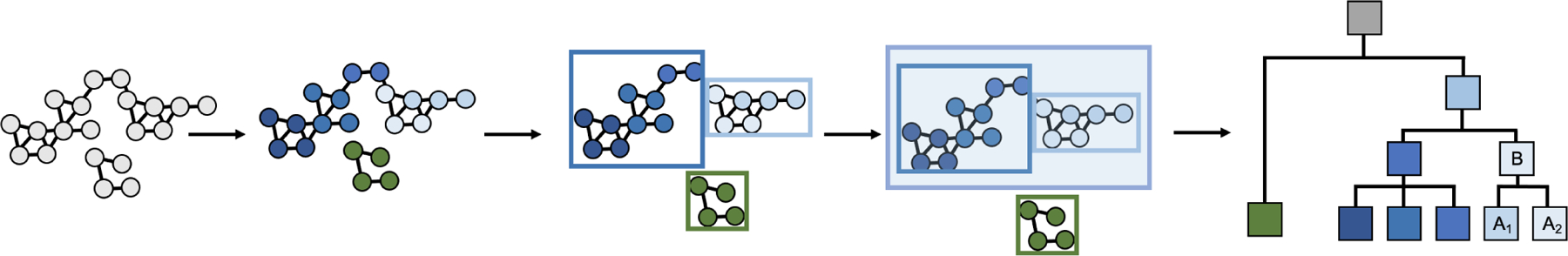
Community detection algorithms define communities (different color circle nodes) in a biological network. Subsequently, communities that are contained within other communities (boxes around circle nodes) can be determined to create a multiscale hierarchical map, where hierarchy nodes (square nodes) represent communities and directed edges in the hierarchy (A → B) represent containment or partial containment of one community (A) by another (B).
In network science, the clustering process that generates the hierarchy is known as community detection, for which a great many algorithms have been developed (Newman, 2018). Specific methods include modularity-based clustering (Newman, 2004), clique percolation (Palla et al., 2005), Louvain clustering (Blondel et al., 2008), Infomap (Rosvall and Bergstrom, 2008, 2011), Order Statistics Local Optimization Method (OSLOM) (Lancichinetti et al., 2011), Clique eXtracted Ontologies (CliXO) (Kramer et al., 2014), HiDef (Zheng et al., 2021), and many others. The Community Detection APplication and Service (CDAPS) (Singhal et al., 2020) integrates end-to-end hierarchical analysis of network communities in the Cytoscape biological network analysis tool (Shannon et al., 2003; Smoot et al., 2011). Functionality includes detection of hierarchical communities with a selection of methods; annotation by alignment with a known ontology such as the Gene Ontology (GO) (Ashburner et al., 2000); visualization of the hierarchical structure; and analysis of the resulting hierarchical communities.
The community detection approach was well illustrated in recent analyses of protein-protein interaction networks in yeast (Dutkowski et al., 2013) and human (Huttlin et al., 2015). In the human study, the authors first performed clique percolation to determine high-level protein communities; subsequently, they ran modularity-based clustering to further subdivide these communities into more granular subsystems, resulting in a multi-layer hierarchical structure (Figure 3A). One of the large communities following clique percolation corresponded to the proteasome, which modularity-based clustering was able to further divide into catalytic and regulatory subunits alongside a cluster of kinetochore components. The result of this data analysis was a multiscale model of the proteasome, including entities such as the protein complex as a whole, subunits of the complex, and individual proteins in each subunit. A very similar hierarchical structure was reported in the corresponding study in yeast (Dutkowski et al., 2013).
Figure 3. Analyzing biological networks to reveal a hierarchy of communities.
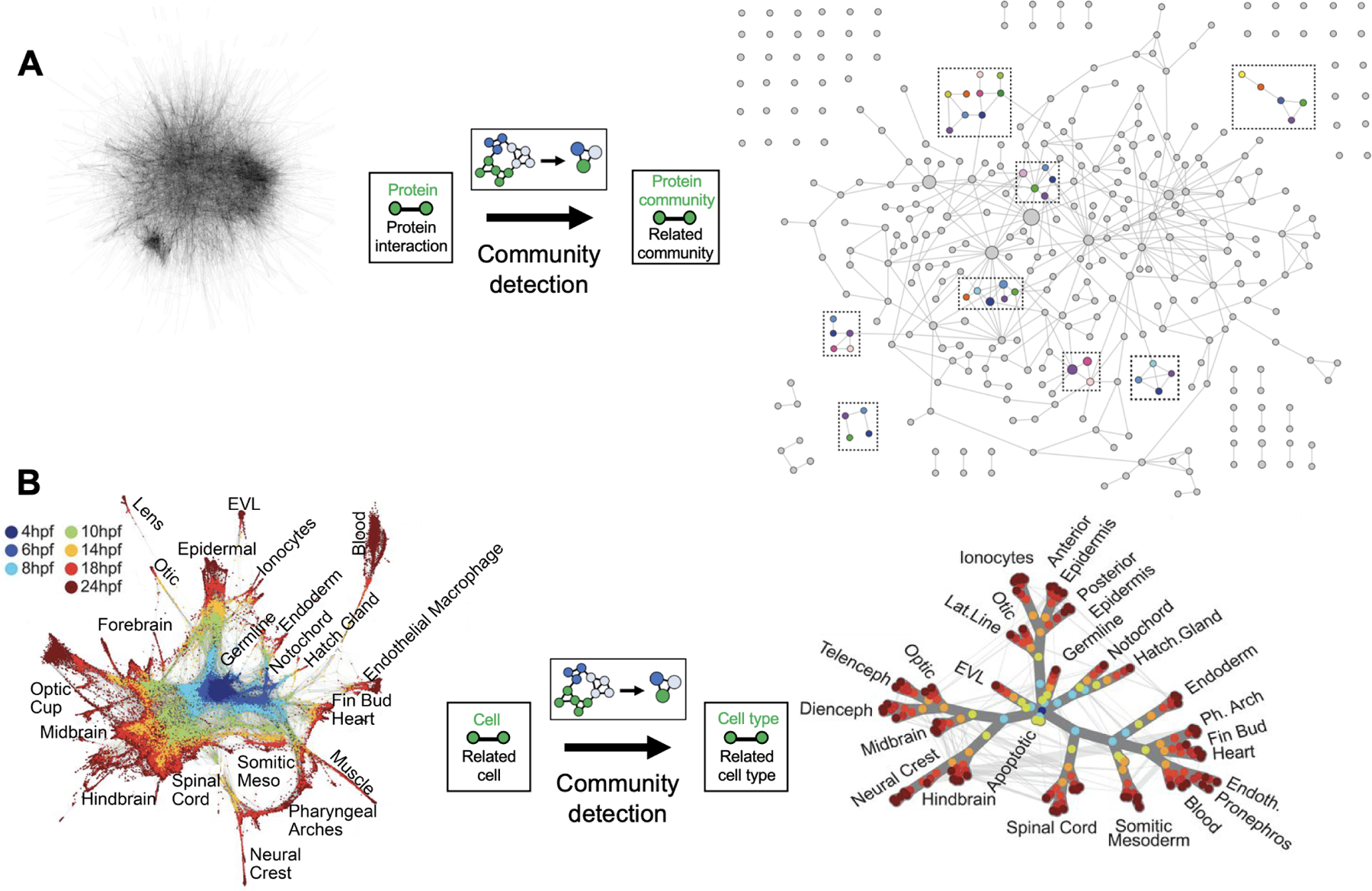
(A) The BioPlex resource uses affinity purification mass spectrometry (AP-MS) experiments to define a large network of interacting pairs of proteins. In the raw network data (left), nodes are proteins; edges are measured protein interactions. Community detection is applied to subdivide communities into increasingly smaller units. In the resulting multiscale model (right), the dotted boxes highlight modules of multiple protein communities, and nodes represent protein communities consisting of proteins; edges run between communities that share a protein or that were subdivided using modularity-based clustering. Figure 3A adapted from Huttlin, E.L., Ting, L., Bruckner, R.J., Gebreab, F., Gygi, M.P., Szpyt, J., Tam, S., Zarraga, G., Colby, G., Baltier, K., et al. (2015). The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell 162, 425–440. Reprinted with permission from Elsevier. (B) Single-cell network from single-cell RNA sequencing analysis of zebrafish embryo development at different hours post fertilization (hpf). In the raw network data (left), nodes represent single cells colored by time of collection; edges represent pairs of cells that are neighbors in a low-dimensional projection of RNA expression data. Community detection is applied to determine a hierarchy of cell types. In the resulting multiscale model, nodes are cell types (consisting of clusters of cells), and edges are weighted based on the number of original single-cell edges. Figure 3B adapted from Wagner, D.E., Weinreb, C., Collins, Z.M., Briggs, J.A., Megason, S.G., and Klein, A.M. (2018). Single-cell mapping of gene expression landscapes and lineage in the zebrafish embryo. Science 360, 981–987. Reprinted with permission from AAAS.
As another example, Costanzo et al. assembled a hierarchy of biological processes using data from a large scale screen of genetic interactions, which examined cell growth for most double mutants of yeast genes, identifying ~1 million genetic interactions (Costanzo et al., 2016, 2019). The lower levels of the hierarchy contained many small but highly related communities of genes, representing focused biological processes and protein complexes. In contrast, the top levels contained a small number of large gene communities with weakly similar profiles, representing pathways and whole organelles. In another recent study, similarities between mutational profiles were converted into an upper bound on distance between mutational residues to create multiscale models of protein complex structures (Braberg et al., 2020). This approach was used to determine the structure of the yeast histone H3-H4 complex by crossing 350 mutations in histones H3 and H4 against 1370 gene deletions.
Moving to higher scales in the biological hierarchy, conceptually similar methods were recently used to build a compelling multiscale map of zebrafish embryo cell types from single-cell RNA sequencing data (Figure 3B) (Wagner et al., 2018). A cell-cell similarity network was constructed using a k-nearest-neighbor graph, where each node represents a cell and edges connect cells to their k nearest neighbors based on their RNA expression profiles. A hierarchy of cell types was constructed from this network: nodes annotated to the same tSNE density cluster were grouped into cell type nodes; edges between cell type nodes were weighted based on the number of edges between grouped single-cell nodes in the original k-nearest-neighbor graph. The result was a hierarchical model of the zebrafish embryo cell types, containing clusters of single cells, with weighted edges connecting similar cell types (either within or between time points). In this example, the scale of the hierarchy is in the time dimension as opposed to physical space, under the assumption that cells related in developmental lineage will have similar expression profiles; it is a challenge in single-cell analyses to deconvolve the relationship between cell type and time, resulting in the development of a diversity of algorithms to infer the trajectory of cell differentiation across time (Saelens et al., 2019). These examples illustrate a process by which biological networks provide the starting points towards building multiscale hierarchical models (Bechtel, 2017, 2020).
Generating multiscale models from multiple network datasets requires combining information from different data types. One approach is to treat the multiple datasets as different “features” annotated to each edge in a single unified network; these multiple features are combined to estimate a single interaction likelihood score for each node pair using supervised machine learning (Kramer et al., 2017) or Bayesian inference (Franceschini et al., 2013; Wong et al., 2004). In a related method, the Mashup algorithm analyzes the topology of each network separately and learns a single weighted network representation that best explains the topological patterns observed across all networks (Cho et al., 2016). Once the data have been combined into a unified network, community detection algorithms may then be run on this network to build hierarchical maps as described above.
Interplay of data-driven hierarchies and literature curated ontologies
There is a notable relation of the multiscale hierarchies described above and biological ontologies, in which a biological entity or concept is recursively factored into a set of subconcepts, sub-subconcepts, and so on (Gruber, 1995). The Gene Ontology (GO) (Ashburner et al., 2000), Cell Ontology (CL) (Bard et al., 2005; Diehl et al., 2016), Human Phenotype Ontology (HPO) (Robinson et al., 2008) and related projects provide a set of widely used, manually curated ontologies that attempt to hierarchically factor entities such as biological processes, components and functions (GO), cell types (CL), and human phenotypes (HPO) into progressively more specific subunits. These ontologies are therefore prime examples of multiscale maps, albeit ones that are manually curated rather than data-driven. Conversely, hierarchical maps generated from proximity networks can be thought of as a type of ontology, as demonstrated in the NeXO project (Dutkowski et al., 2013), albeit one that is driven by direct data analysis rather than manual curation of literature. Regardless, in both cases, entities are interrelated by nested relationships in a hierarchical structure.
The existence of a curated hierarchy of cellular systems like GO greatly facilitates analysis of data-driven maps, by identifying systems in the data-driven hierarchy that correspond to well-known biological components and processes documented in GO. Conversely, systems not found in GO may correspond to novel discoveries. For instance, the multiscale models of cellular components constructed by the NeXO and Mashup efforts (see above) found that the majority of cellular components catalogued in GO could be recovered from hierarchical detection of communities in protein network data (Cho et al., 2016; Dutkowski et al., 2013). Likewise, hierarchies of cell communities inferred from single-cell RNA seq data are typically compared to the CL to determine which communities correspond to known versus novel cell types (Bernstein et al., 2021; Hou et al., 2019). For this purpose, the area known as ”ontology alignment” seeks to address the challenge of determining entities in different hierarchies that represent the same concept, including how to handle multiple overlapping entities and differences in ontology organization. Aligning biological ontologies is also computationally challenging due to their complexity and large size; the GO catalogs tens of thousands of entities for example. Methods for alignment of hierarchies and ontologies are under active research in computer science, including the evaluation of various algorithms for their performance (Ehrig, 2006; Mohammadi and Rezaei, 2020; Otero-Cerdeira et al., 2015). Many of these ontology alignment algorithms may turn out to have useful application in biology, provided the equivalence between multiscale analyses of data and biological ontologies is fully realized.
Functionalization and application of multiscale models
Multiscale models have great potential for the applications of understanding function and predicting outcomes in biological systems, since knowledge of the higher-order protein assemblies in which proteins participate provides evidence for their function and relevance to particular diseases. One important motivation for multiscale biological modeling is to provide a framework for understanding the complex mechanisms by which genotype is expressed as phenotype, including states of health and disease. Entities at very different scales can have significant functional effects on a phenotype under study (Figure 4A). For example, genetic mutations in a genotype may exert their effects at the level of the gene, the protein complex, or the broader pathway or organelle; determining the relevant scale in the biological hierarchy can enable accurate prediction of phenotype, revealing the importance of multiple scales in modeling biological function.
Figure 4. Functionalization of multiscale hierarchies.
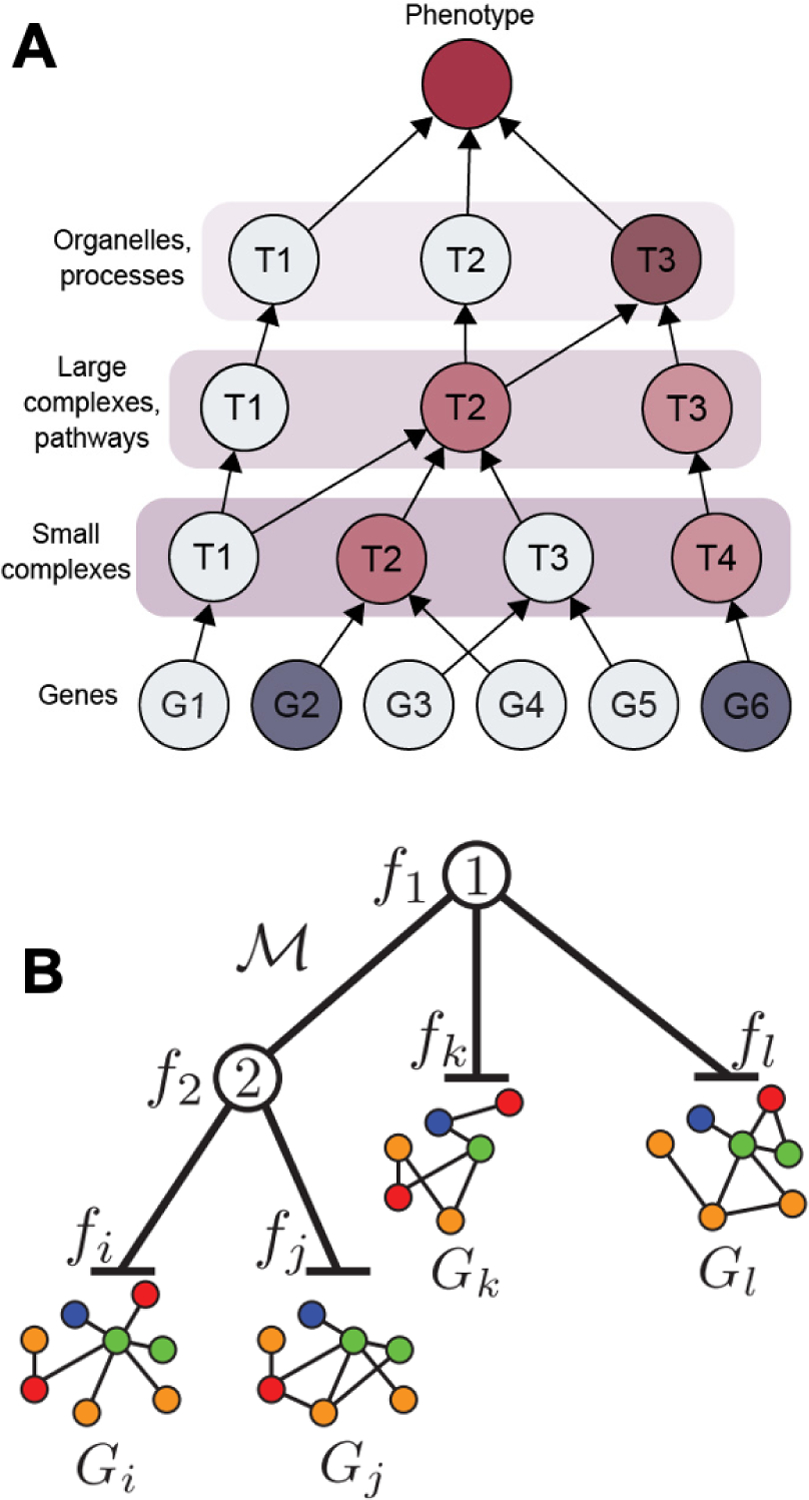
(A) Translation of genotype to phenotype through a hierarchy of subcellular systems. Gene deletions are represented by dark blue. Resulting changes in state for affected systems are shown in red, with darker colors representing effects that are larger or more likely. (B) The OhmNet algorithm learns tissue-specific interactions in a multi-layer network by taking into account tissue hierarchy. OhmNet learns function fi given tissue-specific network graphs Gi in hierarchical model M. Figure 4B reprinted from Zitnik, M., and Leskovec, J. (2017). Predicting multicellular function through multi-layer tissue networks. Bioinformatics 33, i190–i198, by permission of Oxford University Press.
An explicit hierarchical approach for translation of genotype to phenotype was proposed by (Yu et al., 2016), in which mutations were propagated upward through a hierarchy of cellular components and processes. The mutation states of these entities (i.e. entities containing mutated genes) were then used as features to explain phenotype, enabling the appropriate scale in the cell to be used for accurate prediction. This hierarchical approach was used to accurately predict yeast growth phenotypes from pairwise deletions of non-essential yeast genes, determining affected areas in the hierarchy. For example, many genetic interactions were predicted within the oxidative phosphorylation subsystem; negative interactions (worse than expected growth) occurred between the two subsystems of electron and protein transport, whereas positive interactions (better than expected growth) occurred solely within electron transport.
Even with a multiscale model, the translation of genotype to phenotype can be complex, and the use of standard “black-box” machine learning algorithms prevents a straightforward biological interpretation (Rudin, 2019). In this respect, recent work has shown that using a biological hierarchy to guide the machine learning model can help with interpretability (Ma et al., 2018). In this study, the structure of a deep neural network (i.e. configuration of artificial neurons) was constrained to mirror the hierarchy of cellular components recorded by GO or, alternatively, a multiscale map of cellular components derived from molecular interaction data. This model, called DCell, was able to learn not only to translate genotype to phenotype, but also to capture the functional state of cell subsystems throughout the hierarchy. This thread was recently extended through the development of DrugCell, an interpretable deep learning model of cancer cell proliferation (Kuenzi et al., 2020). Here, the model was trained on the response of tumor cell lines to a large panel of drugs, combining genotype and drug structure to predict the drug response along with the specific cellular subsystems mediating that response. In a related study, Gaudelet et al. modeled a neural network structure on the multiscale functional organization of a cell by integrating gene-pathway annotations in the Reactome database; their model was able to accurately determine patient diagnoses based on gene expression data. Importantly, the trained neural network enabled extraction of biological knowledge by determining novel disease-pathway and disease-gene associations (Gaudelet et al., 2020). The concept of interpretable neural networks has also been applied to determine how genotype governs brain phenotypes, by embedding a regulatory network into a multilayer neural network model including intermediate phenotypes (such as gene expression), gene groupings, and observed traits such as psychiatric disorders (Wang et al., 2018). Related work has used neural networks to perform interpretable deep learning on biological networks to differentiate cell types and determine regulatory differences in single cell RNA sequencing data (Fortelny and Bock, 2020). Here, the authors describe their neural networks as “knowledge-primed”: each neuron is a protein or gene and each edge is an annotated regulatory relationship, with regulated gene expression levels as input neurons (measured by single-cell RNA-seq), signaling protein and transcription factors as hidden neurons, and receptors as output neurons to reflect the cell type phenotype. The use of multiscale maps to guide machine learning systems has potential applications outside of biology (Lipton, 2018), for example in business or finance in which interpretable machine learning systems are necessary to ensure unbiased predictions, meet regulatory requirements, and increase confidence in the model.
Multiscale maps have also facilitated determination of novel gene functions. For example, Gaudelet et al. modeled pairwise protein interactions, complexes, and pathways as multiscale hypernetworks; analysis of these hypergraphs resulted in determining protein function for many uncharacterized genes (Gaudelet et al., 2018). Another study generated a multiscale hierarchical map from a co-expression network of a pathogenic fungus; this map was used to identify clusters in the hierarchy associated with plant cell infection, determining genes not previously reported to function in these pathways (Ames, 2017). The Ohmnet algorithm was developed to perform hierarchy-aware unsupervised feature learning in multi-layer networks, with a focus on the hierarchical relationship between different tissues (e.g. cell types within tissues; organs within organ systems) (Zitnik and Leskovec, 2017). Each layer in the multi-layer protein interaction network represents interactions between proteins in a different human tissue (Figure 4B); the tissue hierarchical relationships were determined by the BRENDA Tissue Ontology (Gremse et al., 2011). This multiscale approach at the level of biological tissues enabled prediction of tissue-specific cellular functions for different proteins, outperforming competing methods which did not account for the multiscale hierarchical organization of tissues. DeepGO used features from protein sequences and interactions to predict protein function; the features were used to build a hierarchical neural network classifier based on GO, so that predictions and features could be refined at each level of the biological hierarchy, resulting in a model optimized for function prediction (Kulmanov et al., 2018). Finally, Knowledge- and Context-driven Machine Learning (KCML) is a recently developed strategy for using a hierarchical approach to predict gene functions from high-throughput gene perturbation screening (Sailem et al., 2020). In this approach, a support vector machine (SVM) classifier for each GO biological process learned to distinguish between phenotypic profiles of genes annotated to that process versus random subsets of other genes. The study found examples where genes annotated to the same higher-scale process perform different functions at lower scales, reflecting the hierarchical organization of cellular subsystems.
Visualization of multiscale models
A critical aspect of model construction is visualization, which enables researchers to determine biological subsystems of interest and perform further exploration or validation. While many applications of clustering and community detection show the detected communities as a flat list or network (Figure 3A), visualization of results as a hierarchy (Figure 5A) communicates the hierarchical relationships among subsystems across multiple levels. Visualization of hierarchical structures is challenging if the containment relations among communities cannot be well-described by a tree but have the aforementioned directed acyclic graph (DAG) structure, i.e. with each system potentially having not only multiple subsystems but also multiple supersystems that contain it. As a result, attempts to visualize the hierarchy can take on the appearance of an uninterpretable “hairball”, with many edges crossing one another (Di Battista et al., 1999). In this common case, there is an important tradeoff between conveying the complete information represented in the hierarchy while avoiding a graphic that is too complex to interpret or investigate.
Figure 5. Visualization of multiscale models as a hierarchy.
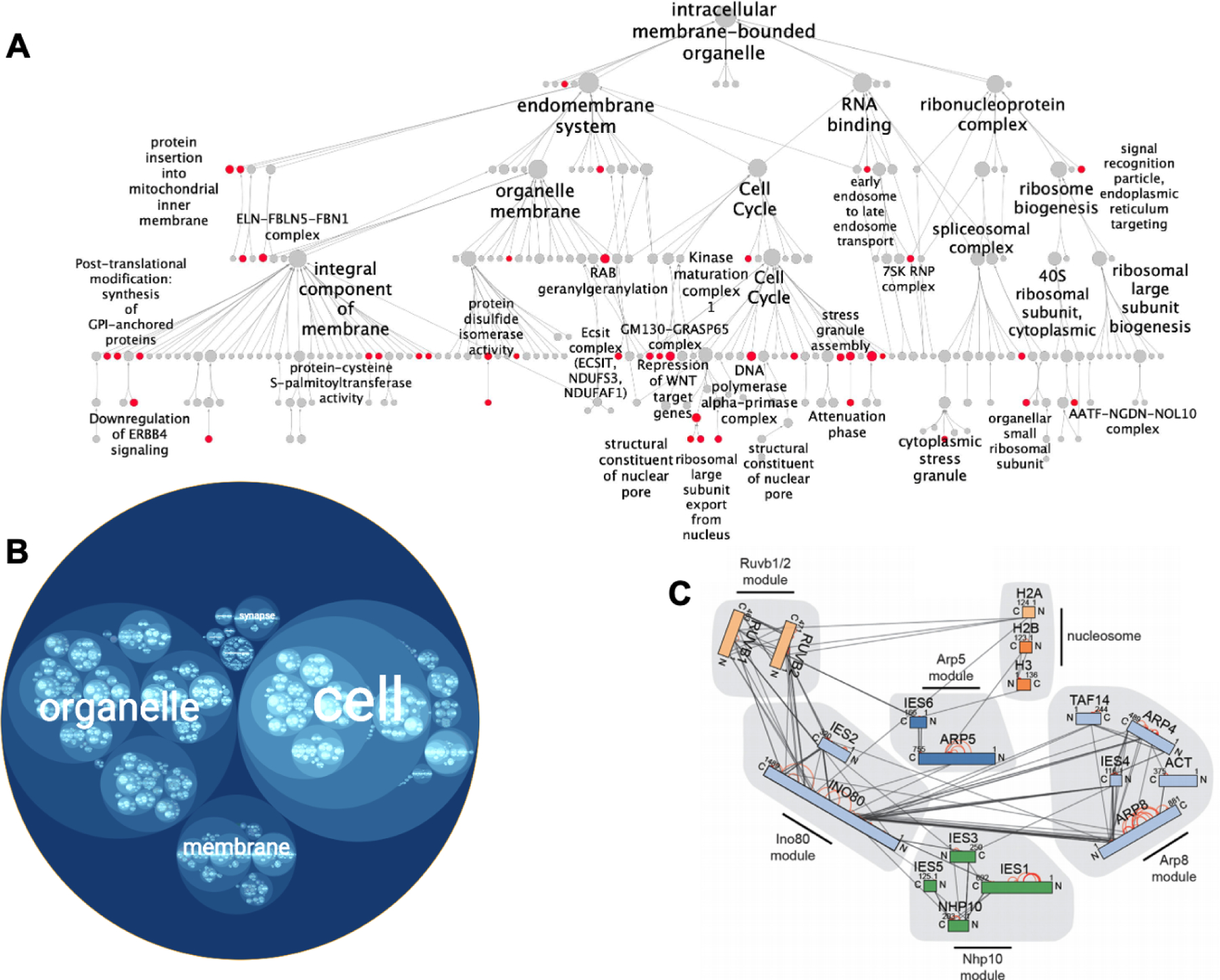
(A) Node-link hierarchy of human protein assemblies interacting with SARS-CoV-2. Red communities are enriched for human proteins that interact with SARS-CoV-2 viral proteins. Figure 5A adapted from Zheng, F., Zhang, S., Churas, C., Pratt, D., Bahar, I., and Ideker, T. (2021). HiDeF: identifying persistent structures in multiscale ‘omics data. Genome Biol. 22, 21., with permission (https://creativecommons.org/licenses/by/4.0/). (B) Circle-packing diagram showing an alternative visualization of the same SARS-CoV-2 hierarchy, generated with HiView. Protein assemblies are represented as circles, with hierarchical containment of one assembly by another represented as nesting of one circle inside another. (C) Hierarchy of protein complexes involving INO80 and the nucleosome, assembled using data from peptide cross-linking analysis. Protein complex modules are highlighted in grey. Rectangle nodes represent proteins, and edges represent crosslinks. Figure 5C reprinted from Grimm, M., Zimniak, T., Kahraman, A., and Herzog, F. (2015). xVis: a web server for the schematic visualization and interpretation of crosslink-derived spatial restraints. Nucleic Acids Res. 43, W362–W369, by permission of Oxford University Press.
Biological ontologies are large hierarchies which also have a DAG structure; as such, existing tools for visualizing ontologies can also have direct application to data-driven multiscale models (Binns et al., 2009; Eden et al., 2009; Sealfon et al., 2006; Supek et al., 2011; Zhu et al., 2019). For example, the Augmented Exploration of GO with Interactive Simulations (AEGIS) platform implements a focus-and-context framework, where a focus graph shows the structure of a selected sub-hierarchy, and a context view is color-coded to show the overall number of nodes related to the sub-hierarchy (Zhu et al., 2019). NaviGO uses an interactive visualization of the hierarchy named GO Visualizer (Khan et al., 2015), allowing users to expand GO terms in the hierarchy and change the view. Similarly, VisANT uses a metagraph (Hu et al., 2004, 2007) to display hierarchies, containing metanodes that can be collapsed or expanded to show subnodes (Hu et al., 2009). HiVis clusters biological networks; to simplify visualization, nodes are only visible in the current level and the next level of the hierarchy (Qiang et al., 2018). The HiView web application contains different options for transforming a DAG hierarchy into a tree (Figure 5A); in each method, a tradeoff exists between information included and the size (Yu et al., 2019). In HiView, the hierarchy can also be visualized as a “circle-packing” diagram (Wang et al., 2006), where subsystems lower in the hierarchy are represented as circles within larger circles (systems) higher in the hierarchy (Figure 5B).
A separate consideration in the visualization of hierarchical models is how to integrate geometric and spatial localization information when it is available. Visualization methods that attend only to the containment relations within the system, as described above, do not necessarily capture spatial subcellular localizations or the number, shape, or positions of subcellular systems, which are important geometric aspects of cellular suborganization. On the other hand, the hierarchical multiscale representations have the major advantages of generality across analytical methods (and their scales) and a comprehensive scope (genome-wide, see Box 1). One strategy for integrating hierarchical maps with spatial organization is to overlay the localization coordinates of each entity in 2D or 3D. For example, programs have been developed to analyze and visualize cross-linking data as a network of amino acid nodes, where edges represent a detected cross-link (Combe et al., 2015; Courcelles et al., 2017; Grimm et al., 2015; Riffle et al., 2016); organizing clusters of nodes spatially according to subunit is one approach that has been applied in these networks (Figure 5C). At a larger scale, studies have integrated protein-protein interaction networks with subcellular organization by partitioning a network into subnetworks according to subcellular location (Barsky et al., 2007; Heberle et al., 2017; Nagasaki et al., 2011; Ofran et al., 2006; Zhang et al., 2012a). A complementary multiscale visualization strategy is to integrate 3D structural information with associated nodes in a hierarchy; software tools have been developed that display available 3D structural data for selected proteins and physical pairwise interactions in protein-protein interaction networks (Morris et al., 2007; Mosca et al., 2013; Nepomnyachiy et al., 2015).
Outstanding Challenges in Multiscale Modeling
There are still many challenges related to the construction and application of multiscale models as they seek to accurately capture biological structure and exhibit high predictive power. First, multiscale modeling carries with it all of the challenges of general community detection, including determining the optimal number of communities, since this number is typically not known in biological analysis. Community detection algorithms such as Louvain (Blondel et al., 2008) or Newman Girvan (Girvan and Newman, 2002) determine the approximate number of communities by optimizing modularity, or the density of links within communities versus links between communities. When performing community detection at multiple scales to develop a hierarchy, there is the additional challenge of determining a suitable number of levels for the hierarchy, as this number is also typically not known beforehand. Another well-known problem in community detection is the tradeoff between quality control and completeness, since not all biological entities of interest may be detected or meet a quality threshold due to experimental challenges. Clustering analysis of networks is sensitive to network noise (Stacey et al., 2020), and biochemical experimental challenges as well as cell-to-cell variation can contribute to this issue. One way this issue may be addressed is through fuzzy or overlapping communities, where nodes can belong to more than one (Yazdanparast et al., 2020).
A second open question regards the best way to evaluate multiscale models, a question that is also vital to optimization of the number of communities and levels. One way to evaluate multiscale models is to align hierarchies with known gold-standard ontologies and determine how many known cellular subsystems are recovered. As discussed above, however, alignment of hierarchies has its own set of challenges. Another option is to determine how many communities in the hierarchy can be validated with experimental evidence from another data set, with the caveat that comprehensive datasets may not exist for the biological contexts or conditions under study.
A third set of challenges relates to integrating diverse modalities of data, such as protein interaction networks with single cell sequencing. How to actually connect communities across these broader scales to generate a single unified hierarchy is an open question to be explored. Some approaches have used machine learning to integrate pairwise distance measurements at different scales (Qin et al., 2020) or to integrate single cell data with context-independent data to create cell-type specific interactomes (Mohammadi et al., 2019). In integrative structural biology, data from multiple methods are collected and translated to spatial restraints, which are used in scoring sampled structures (Rout and Sali, 2019); this type of wholly integrative constraints-based approach may also prove useful when constructing broader multiscale models.
A fourth challenge relates to the capture of dynamics. While many of the multiscale models that have been described herein are presented as a static snapshot of biological structure, cellular processes are highly dynamic, involving transient regulatory interactions and control of protein interactions via post-translational modifications. Limiting one’s study to a static snapshot of protein interactions may limit predictive power for determining biological consequences of chemical drugs or genetic variations, as the resulting variations may be context dependent. An important strategy moving forward will be to determine hierarchical models for each context under study, then implement hierarchy or ontology alignment algorithms to compare the various context-dependent hierarchies.
Finally, many networks and subsequent multiscale models have a one gene-per-node representation, when in reality protein diversity (including sequence isoforms and post-translational modifications) controls interactions between proteins and individual protein function (Aebersold et al., 2018). Additionally, small molecular interactions are critical for biological regulation in both intracellular processes and cell-to-cell communications, up to the even broader coordination between organs in an organism (Schreiber, 2005). Thus, significantly broadening the set of biological entities included in a modeling project is a significant future direction.
Conclusions
Biological structure can be effectively modeled as a hierarchy of subsystems that, collectively, span scales of at least ten orders of magnitude, from angstroms to meters. Various experimental data types and analytical methods generate networks of proximity between biological entities at different levels of the hierarchy and thus at different scales. With ongoing technological developments in data collection and analytical methodology, we expect biological networks and the resulting multiscale models to vastly improve in both scope (field of view) and level of detail (resolution), towards ever-larger and more complete hierarchical models that enable unprecedented understanding of biological function.
In this review, Schaffer and Ideker discuss concepts and progress towards the goal of creating unified multiscale models of biological structure and function. Many experimental technologies measure physical proximity or functional similarity among biological entities at different scales– including amino acids within a protein, proteins within an enzymatic complex, and cells within a tissue. This review discusses use of these proximity networks to create multiscale models, along with major applications, visualization techniques, and current challenges.
Acknowledgements
We are indebted to Adrian Anderson for her indispensable work with preparation of figures. We thank Dexter Pratt, Jing Chen, and Fan Zheng for their contributions to the HiView visualization figure. This work was supported by grants from the National Institutes of Health to T.I. (HG009979, ES014811).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of Interests
T.I. is a cofounder of Data4Cure and has an equity interest. T.I. is a compensated scientific advisor of Ideaya BioSciences and has an equity interest. The terms of this arrangement have been reviewed and approved by the University of California, San Diego, in accordance with its conflict of interest policies.
References
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, and Terwilliger TC (2002). PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr 58, 1948–1954. [DOI] [PubMed] [Google Scholar]
- Aebersold R, Agar JN, Amster IJ, Baker MS, Bertozzi CR, Boja ES, Costello CE, Cravatt BF, Fenselau C, Garcia BA, et al. (2018). How many human proteoforms are there? Nat. Chem. Biol 14, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alber F, Förster F, Korkin D, Topf M, and Sali A (2008). Integrating diverse data for structure determination of macromolecular assemblies. Annu. Rev. Biochem 77, 443–477. [DOI] [PubMed] [Google Scholar]
- Ames RM (2017). Using Network Extracted Ontologies to Identify Novel Genes with Roles in Appressorium Development in the Rice Blast Fungus Magnaporthe oryzae. Microorganisms 5, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene Ontology: tool for the unification of biology. Nat. Genet 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay S, Mehta M, Kuo D, Sung M-K, Chuang R, Jaehnig EJ, Bodenmiller B, Licon K, Copeland W, Shales M, et al. (2010). Rewiring of genetic networks in response to DNA damage. Science 330, 1385–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard J, Rhee SY, and Ashburner M (2005). An ontology for cell types. Genome Biol 6, R21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsky A, Gardy JL, Hancock REW, and Munzner T (2007). Cerebral: a Cytoscape plugin for layout of and interaction with biological networks using subcellular localization annotation. Bioinformatics 23, 1040–1042. [DOI] [PubMed] [Google Scholar]
- Becht E, McInnes L, Healy J, Dutertre C-A, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2018). Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol [DOI] [PubMed] [Google Scholar]
- Bechtel W (2017). Using the hierarchy of biological ontologies to identify mechanisms in flat networks. Biol. Philos 32, 627–649. [Google Scholar]
- Bechtel W (2020). Hierarchy and levels: analysing networks to study mechanisms in molecular biology. Philos. Trans. R. Soc. Lond. B Biol. Sci 375, 20190320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behan FM, Iorio F, Picco G, Gonçalves E, Beaver CM, Migliardi G, Santos R, Rao Y, Sassi F, Pinnelli M, et al. (2019). Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature 568, 511–516. [DOI] [PubMed] [Google Scholar]
- Bendall SC, Davis KL, Amir E-AD, Tadmor MD, Simonds EF, Chen TJ, Shenfeld DK, Nolan GP, and Pe’er D (2014). Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell 157, 714–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berge C (1984). Hypergraphs: Combinatorics of Finite Sets (Elsevier) [Google Scholar]
- Bernstein MN, Ma Z, Gleicher M, and Dewey CN (2021). CellO: comprehensive and hierarchical cell type classification of human cells with the Cell Ontology. iScience 24, 101913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binns D, Dimmer E, Huntley R, Barrell D, O’Donovan C, and Apweiler R (2009). QuickGO: a web-based tool for Gene Ontology searching. Bioinformatics 25, 3045–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondel VD, Guillaume J-L, Lambiotte R, and Lefebvre E (2008). Fast unfolding of communities in large networks. J. Stat. Mech 2008, P10008. [Google Scholar]
- Bludau I, Heusel M, Frank M, Rosenberger G, Hafen R, Banaei-Esfahani A, van Drogen A, Collins BC, Gstaiger M, and Aebersold R (2020). Complex-centric proteome profiling by SEC-SWATH-MS for the parallel detection of hundreds of protein complexes. Nat. Protoc 15, 2341–2386. [DOI] [PubMed] [Google Scholar]
- Braberg H, Echeverria I, Bohn S, Cimermancic P, Shiver A, Alexander R, Xu J, Shales M, Dronamraju R, Jiang S, et al. (2020). Genetic interaction mapping informs integrative structure determination of protein complexes. Science 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne AB, Weirauch MT, Wong V, Koeva M, Dixon SJ, Stuart JM, and Roy PJ (2007). A global analysis of genetic interactions in Caenorhabditis elegans. J. Biol 6, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo SE, Clauser KR, and Mootha VK (2016). MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res 44, D1251–D1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Baldwin PR, Ludtke SJ, and Baker ML (2016). De Novo modeling in cryo-EM density maps with Pathwalking. J. Struct. Biol 196, 289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Berger B, and Peng J (2016). Compact Integration of Multi-Network Topology for Functional Analysis of Genes. Cell Syst 3, 540–548.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combe CW, Fischer L, and Rappsilber J (2015). xiNET: cross-link network maps with residue resolution. Mol. Cell. Proteomics 14, 1137–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JLY, Toufighi K, Mostafavi S, et al. (2010). The Genetic Landscape of a Cell. Science 327, 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, VanderSluis B, Koch EN, Baryshnikova A, Pons C, Tan G, Wang W, Usaj M, Hanchard J, Lee SD, et al. (2016). A global genetic interaction network maps a wiring diagram of cellular function. Science 353, aaf1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, Kuzmin E, van Leeuwen J, Mair B, Moffat J, Boone C, and Andrews B (2019). Global Genetic Networks and the Genotype-to-Phenotype Relationship. Cell 177, 85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courcelles M, Coulombe-Huntington J, Cossette É, Gingras A-C, Thibault P, and Tyers M (2017). CLMSVault: A Software Suite for Protein Cross-Linking Mass-Spectrometry Data Analysis and Visualization. J. Proteome Res 16, 2645–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J, Rippe K, Dekker M, and Kleckner N (2002). Capturing chromosome conformation. Science 295, 1306–1311. [DOI] [PubMed] [Google Scholar]
- Deng D, Xu C, Sun P, Wu J, Yan C, Hu M, and Yan N (2014). Crystal structure of the human glucose transporter GLUT1. Nature 510, 121–125. [DOI] [PubMed] [Google Scholar]
- Díaz-Mejía JJ, Celaj A, Mellor JC, Coté A, Balint A, Ho B, Bansal P, Shaeri F, Gebbia M, Weile J, et al. (2018). Mapping DNA damage-dependent genetic interactions in yeast via party mating and barcode fusion genetics. Mol. Syst. Biol 14, e7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Battista G, Tollis IG, Eades P, and Tamassia R (1999). Graph Drawing: Algorithms for the Visualization of Graphs (Prentice Hall) [Google Scholar]
- Diehl AD, Meehan TF, Bradford YM, Brush MH, Dahdul WM, Dougall DS, He Y, Osumi-Sutherland D, Ruttenberg A, Sarntivijai S, et al. (2016). The Cell Ontology 2016: enhanced content, modularization, and ontology interoperability. J. Biomed. Semantics 7, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Costanzo M, Baryshnikova A, Andrews B, and Boone C (2009). Systematic mapping of genetic interaction networks. Annu. Rev. Genet 43, 601–625. [DOI] [PubMed] [Google Scholar]
- Dotan-Cohen D, Letovsky S, Melkman AA, and Kasif S (2009). Biological process linkage networks. PLoS One 4, e5313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durruthy-Durruthy R, Gottlieb A, Hartman BH, Waldhaus J, Laske RD, Altman R, and Heller S (2014). Reconstruction of the mouse otocyst and early neuroblast lineage at single-cell resolution. Cell 157, 964–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutkowski J, Kramer M, Surma MA, Balakrishnan R, Cherry JM, Krogan NJ, and Ideker T (2013). A gene ontology inferred from molecular networks. Nat. Biotechnol 31, 38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, and Yakhini Z (2009). GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrig M (2006). Ontology Alignment: Bridging the Semantic Gap (Springer Science & Business Media) [Google Scholar]
- Faini M, Stengel F, and Aebersold R (2016). The Evolving Contribution of Mass Spectrometry to Integrative Structural Biology. J. Am. Soc. Mass Spectrom 27, 966–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell JA, Wang Y, Riesenfeld SJ, Shekhar K, Regev A, and Schier AF (2018). Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis. Science 360, eaar3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortelny N, and Bock C (2020). Knowledge-primed neural networks enable biologically interpretable deep learning on single-cell sequencing data. Genome Biol 21, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, et al. (2013). STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res 41, D808–D815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenz B, Walls AC, Egelman EH, Veesler D, and DiMaio F (2017). RosettaES: a sampling strategy enabling automated interpretation of difficult cryo-EM maps. Nat. Methods 14, 797–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudelet T, Malod-Dognin N, and Pržulj N (2018). Higher-order molecular organization as a source of biological function. Bioinformatics 34, i944–i953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudelet T, Malod-Dognin N, Sánchez-Valle J, Pancaldi V, Valencia A, and Pržulj N (2020). Unveiling new disease, pathway, and gene associations via multiscale neural network. PLoS One 15, e0231059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girvan M, and Newman MEJ (2002). Community structure in social and biological networks. Proc. Natl. Acad. Sci. U. S. A 99, 7821–7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go CD, Knight JDR, Rajasekharan A, Rathod B, Hesketh GG, Abe KT, Youn J-Y, Samavarchi-Tehrani P, Zhang H, Zhu LY, et al. (2019). A proximity biotinylation map of a human cell. bioRxiv [Google Scholar]
- Gonatopoulos-Pournatzis T, Aregger M, Brown KR, Farhangmehr S, Braunschweig U, Ward HN, Ha KCH, Weiss A, Billmann M, Durbic T, et al. (2020). Genetic interaction mapping and exon-resolution functional genomics with a hybrid Cas9–Cas12a platform. Nat. Biotechnol 38, 638–648. [DOI] [PubMed] [Google Scholar]
- Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, O’Meara MJ, Rezelj VV, Guo JZ, Swaney DL, et al. (2020). A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583, 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gremse M, Chang A, Schomburg I, Grote A, Scheer M, Ebeling C, and Schomburg D (2011). The BRENDA Tissue Ontology (BTO): the first all-integrating ontology of all organisms for enzyme sources. Nucleic Acids Res 39, D507–D513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm M, Zimniak T, Kahraman A, and Herzog F (2015). xVis: a web server for the schematic visualization and interpretation of crosslink-derived spatial restraints. Nucleic Acids Res 43, W362–W369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber TR (1995). Toward principles for the design of ontologies used for knowledge sharing? Int. J. Hum. Comput. Stud 43, 907–928. [Google Scholar]
- Han K, Jeng EE, Hess GT, Morgens DW, Li A, and Bassik MC (2017). Synergistic drug combinations for cancer identified in a CRISPR screen for pairwise genetic interactions. Nat. Biotechnol 35, 463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havugimana PC, Hart GT, Nepusz T, Yang H, Turinsky AL, Li Z, Wang PI, Boutz DR, Fong V, Phanse S, et al. (2012). A census of human soluble protein complexes. Cell 150, 1068–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heberle H, Carazzolle MF, Telles GP, Meirelles GV, and Minghim R (2017). CellNetVis: a web tool for visualization of biological networks using force-directed layout constrained by cellular components. BMC Bioinformatics 18, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holding AN (2015). XL-MS: Protein cross-linking coupled with mass spectrometry. Methods 89, 54–63. [DOI] [PubMed] [Google Scholar]
- Hou R, Denisenko E, and Forrest ARR (2019). scMatch: a single-cell gene expression profile annotation tool using reference datasets. Bioinformatics 35, 4688–4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hryc CF, Chen D-H, Afonine PV, Jakana J, Wang Z, Haase-Pettingell C, Jiang W, Adams PD, King JA, Schmid MF, et al. (2017). Accurate model annotation of a near-atomic resolution cryo-EM map. Proc. Natl. Acad. Sci. U. S. A 114, 3103–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Mellor J, Wu J, and DeLisi C (2004). VisANT: an online visualization and analysis tool for biological interaction data. BMC Bioinformatics 5, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Mellor J, Wu J, Kanehisa M, Stuart JM, and DeLisi C (2007). Towards zoomable multidimensional maps of the cell. Nat. Biotechnol 25, 547–554. [DOI] [PubMed] [Google Scholar]
- Hu Z, Hung J-H, Wang Y, Chang Y-C, Huang C-L, Huyck M, and DeLisi C (2009). VisANT 3.5: multiscale network visualization, analysis and inference based on the gene ontology. Nucleic Acids Res 37, W115–W121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, et al. (2015). The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell 162, 425–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H, et al. (2017). Architecture of the human interactome defines protein communities and disease networks. Nature 545, 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe M, Dziulko A, Smith JD, St Onge RP, Levy SF, and Sherlock G (2019). Improved discovery of genetic interactions using CRISPRiSeq across multiple environments. Genome Res. 29, 668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaimovich A, Rinott R, Schuldiner M, Margalit H, and Friedman N (2010). Modularity and directionality in genetic interaction maps. Bioinformatics 26, i228–i236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, Zaretsky I, Mildner A, Cohen N, Jung S, Tanay A, et al. (2014). Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 343, 776–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley R, and Ideker T (2005). Systematic interpretation of genetic interactions using protein networks. Nat. Biotechnol 23, 561–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan IK, Wei Q, Chitale M, and Kihara D (2015). PFP/ESG: automated protein function prediction servers enhanced with Gene Ontology visualization tool. Bioinformatics 31, 271–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Fernandez-Martinez J, Nudelman I, Shi Y, Zhang W, Raveh B, Herricks T, Slaughter BD, Hogan JA, Upla P, et al. (2018). Integrative structure and functional anatomy of a nuclear pore complex. Nature 555, 475–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleywegt GJ, Harris MR, Zou JY, Taylor TC, Wählby A, and Jones TA (2004). The Uppsala Electron-Density Server. Acta Crystallogr. D Biol. Crystallogr 60, 2240–2249. [DOI] [PubMed] [Google Scholar]
- Kramer M, Dutkowski J, Yu M, Bafna V, and Ideker T (2014). Inferring gene ontologies from pairwise similarity data. Bioinformatics 30, i34–i42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer MH, Farré J-C, Mitra K, Yu MK, Ono K, Demchak B, Licon K, Flagg M, Balakrishnan R, Cherry JM, et al. (2017). Active Interaction Mapping Reveals the Hierarchical Organization of Autophagy. Mol. Cell 65, 761–774.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuenzi BM, Park J, Fong SH, Sanchez KS, Lee J, Kreisberg JF, Ma J, and Ideker T (2020). Predicting Drug Response and Synergy Using a Deep Learning Model of Human Cancer Cells. Cancer Cell 38, 672–684.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulmanov M, Khan MA, Hoehndorf R, and Wren J (2018). DeepGO: predicting protein functions from sequence and interactions using a deep ontology-aware classifier. Bioinformatics 34, 660–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancichinetti A, Radicchi F, Ramasco JJ, and Fortunato S (2011). Finding Statistically Significant Communities in Networks. PLoS One 6, e18961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasker K, Phillips JL, Russel D, Velázquez-Muriel J, Schneidman-Duhovny D, Tjioe E, Webb B, Schlessinger A, and Sali A (2010). Integrative structure modeling of macromolecular assemblies from proteomics data. Mol. Cell. Proteomics 9, 1689–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen J, Pons C, Mellor JC, Yamaguchi TN, Friesen H, Koschwanez J, Ušaj MM, Pechlaner M, Takar M, Ušaj M, et al. (2016). Exploring genetic suppression interactions on a global scale. Science 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton ZC (2018). The Mythos of Model Interpretability: In machine learning, the concept of interpretability is both important and slippery. Queueing Syst 16, 31–57. [Google Scholar]
- Liu F, and Heck AJR (2015). Interrogating the architecture of protein assemblies and protein interaction networks by cross-linking mass spectrometry. Curr. Opin. Struct. Biol 35, 100–108. [DOI] [PubMed] [Google Scholar]
- Lobingier BT, Hüttenhain R, Eichel K, Miller KB, Ting AY, von Zastrow M, and Krogan NJ (2017). An Approach to Spatiotemporally Resolve Protein Interaction Networks in Living Cells. Cell 169, 350–360.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luck K, Kim D-K, Lambourne L, Spirohn K, Begg BE, Bian W, Brignall R, Cafarelli T, Campos-Laborie FJ, Charloteaux B, et al. (2020). A reference map of the human binary protein interactome. Nature 580, 402–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo B, Cheung HW, Subramanian A, Sharifnia T, Okamoto M, Yang X, Hinkle G, Boehm JS, Beroukhim R, Weir BA, et al. (2008). Highly parallel identification of essential genes in cancer cells. Proc. Natl. Acad. Sci. U. S. A 105, 20380–20385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Yu MK, Fong S, Ono K, Sage E, Demchak B, Sharan R, and Ideker T (2018). Using deep learning to model the hierarchical structure and function of a cell. Nat. Methods 15, 290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maaten L, and Hinton G (2008). Visualizing Data using t-SNE. J. Mach. Learn. Res 9, 2579–2605. [Google Scholar]
- Mohammadi M, and Rezaei J (2020). Evaluating and comparing ontology alignment systems: An MCDM approach. Journal of Web Semantics 64, 100592. [Google Scholar]
- Mohammadi S, Davila-Velderrain J, and Kellis M (2019). Reconstruction of Cell-type-Specific Interactomes at Single-Cell Resolution. Cell Syst 9, 559–568.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JH, Huang CC, Babbitt PC, and Ferrin TE (2007). structureViz: linking Cytoscape and UCSF Chimera. Bioinformatics 23, 2345–2347. [DOI] [PubMed] [Google Scholar]
- Mosca R, Céol A, and Aloy P (2013). Interactome3D: adding structural details to protein networks. Nat. Methods 10, 47–53. [DOI] [PubMed] [Google Scholar]
- Nagasaki M, Saito A, Jeong E, Li C, Kojima K, Ikeda E, and Miyano S (2011). Cell illustrator 4.0: a computational platform for systems biology. Stud. Health Technol. Inform 162, 160–181. [PubMed] [Google Scholar]
- Nepomnyachiy S, Ben-Tal N, and Kolodny R (2015). CyToStruct: Augmenting the Network Visualization of Cytoscape with the Power of Molecular Viewers. Structure 23, 941–948. [DOI] [PubMed] [Google Scholar]
- Newman M (2018). Networks (Oxford University Press) [Google Scholar]
- Newman MEJ (2004). Fast algorithm for detecting community structure in networks. Phys. Rev. E Stat. Nonlin. Soft Matter Phys 69, 066133. [DOI] [PubMed] [Google Scholar]
- Norman TM, Horlbeck MA, Replogle JM, Ge AY, Xu A, Jost M, Gilbert LA, and Weissman JS (2019). Exploring genetic interaction manifolds constructed from rich single-cell phenotypes. Science 365, 786–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofran Y, Yachdav G, Mozes E, Soong T-T, Nair R, and Rost B (2006). Create and assess protein networks through molecular characteristics of individual proteins. Bioinformatics 22, e402–e407. [DOI] [PubMed] [Google Scholar]
- Otero-Cerdeira L, Rodríguez-Martínez FJ, and Gómez-Rodríguez A (2015). Ontology matching: A literature review. Expert Syst. Appl 42, 949–971. [Google Scholar]
- Palla G, Derényi I, Farkas I, and Vicsek T (2005). Uncovering the overlapping community structure of complex networks in nature and society. Nature 435, 814–818. [DOI] [PubMed] [Google Scholar]
- Park Y, and Bader JS (2011). Resolving the structure of interactomes with hierarchical agglomerative clustering. BMC Bioinformatics 12, S44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn BH, Netter Z, Johnson JR, Von Dollen J, Jang GM, Johnson T, Ohol YM, Maher C, Bell SL, Geiger K, et al. (2018). An Mtb-Human Protein-Protein Interaction Map Identifies a Switch between Host Antiviral and Antibacterial Responses. Mol. Cell 71, 637–648.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringle RM, and Tarnita CE (2017). Spatial Self-Organization of Ecosystems: Integrating Multiple Mechanisms of Regular-Pattern Formation. Annu. Rev. Entomol 62, 359–377. [DOI] [PubMed] [Google Scholar]
- Qiang Z, Chen H, Zhu X, and Tu S (2018). HiVis: a portable, scalable tool for hierarchical visualization and analysis of biological networks. Applied Informatics 5, 3. [Google Scholar]
- Qin Y, Winsnes CF, Huttlin EL, Zheng F, Ouyang W, Park J, Pitea A, Kreisberg JF, Gygi SP, Harper JW, et al. (2020). Mapping cell structure across scales by fusing protein images and interactions. bioRxiv 2020.06.21.163709 [Google Scholar]
- Ravasz E, Somera AL, Mongru DA, Oltvai ZN, and Barabási A-L (2002). Hierarchical Organization of Modularity in Metabolic Networks. Science 297, 1551–1555. [DOI] [PubMed] [Google Scholar]
- Riffle M, Jaschob D, Zelter A, and Davis TN (2016). ProXL (Protein Cross-Linking Database): A Platform for Analysis, Visualization, and Sharing of Protein Cross-Linking Mass Spectrometry Data. J. Proteome Res 15, 2863–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson PN, Köhler S, Bauer S, Seelow D, Horn D, and Mundlos S (2008). The Human Phenotype Ontology: a tool for annotating and analyzing human hereditary disease. Am. J. Hum. Genet 83, 610–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland T, Taşan M, Charloteaux B, Pevzner SJ, Zhong Q, Sahni N, Yi S, Lemmens I, Fontanillo C, Mosca R, et al. (2014). A proteome-scale map of the human interactome network. Cell 159, 1212–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosvall M, and Bergstrom CT (2008). Maps of random walks on complex networks reveal community structure. Proc. Natl. Acad. Sci. U. S. A 105, 1118–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosvall M, and Bergstrom CT (2011). Multilevel Compression of Random Walks on Networks Reveals Hierarchical Organization in Large Integrated Systems. PLoS One 6, e18209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout MP, and Sali A (2019). Principles for Integrative Structural Biology Studies. Cell 177, 1384–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Raida M, and Burke B (2012). A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol 196, 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin C (2019). Stop explaining black box machine learning models for high stakes decisions and use interpretable models instead. Nature Machine Intelligence 1, 206–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russel D, Lasker K, Webb B, Velázquez-Muriel J, Tjioe E, Schneidman-Duhovny D, Peterson B, and Sali A (2012). Putting the pieces together: integrative modeling platform software for structure determination of macromolecular assemblies. PLoS Biol 10, e1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan CJ, Roguev A, Patrick K, Xu J, Jahari H, Tong Z, Beltrao P, Shales M, Qu H, Collins SR, et al. (2012). Hierarchical modularity and the evolution of genetic interactomes across species. Mol. Cell 46, 691–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saelens W, Cannoodt R, Todorov H, and Saeys Y (2019). A comparison of single-cell trajectory inference methods. Nat. Biotechnol 37, 547–554. [DOI] [PubMed] [Google Scholar]
- Sah P, Méndez JD, and Bansal S (2019). A multi-species repository of social networks. Sci Data 6, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sailem HZ, Rittscher J, and Pelkmans L (2020). KCML: a machine-learning framework for inference of multiscale gene functions from genetic perturbation screens. Mol. Syst. Biol 16, e9083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas D, Stacey RG, Akinlaja M, and Foster LJ (2020). Next-generation Interactomics: Considerations for the Use of Co-elution to Measure Protein Interaction Networks. Mol. Cell. Proteomics 19, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber SL (2005). Small molecules: the missing link in the central dogma. Nat. Chem. Biol 1, 64–66. [DOI] [PubMed] [Google Scholar]
- Sealfon RSG, Hibbs MA, Huttenhower C, Myers CL, and Troyanskaya OG (2006). GOLEM: an interactive graph-based gene-ontology navigation and analysis tool. BMC Bioinformatics 7, 443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senior AW, Evans R, Jumper J, Kirkpatrick J, Sifre L, Green T, Qin C, Žídek A, Nelson AWR, Bridgland A, et al. (2019). Protein structure prediction using multiple deep neural networks in the 13th Critical Assessment of Protein Structure Prediction (CASP13). Proteins 87, 1141–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senior AW, Evans R, Jumper J, Kirkpatrick J, Sifre L, Green T, Qin C, Žídek A, Nelson AWR, Bridgland A, et al. (2020). Improved protein structure prediction using potentials from deep learning. Nature 577, 706–710. [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen JP, Zhao D, Sasik R, Luebeck J, Birmingham A, Bojorquez-Gomez A, Licon K, Klepper K, Pekin D, Beckett AN, et al. (2017). Combinatorial CRISPR–Cas9 screens for de novo mapping of genetic interactions. Nat. Methods 14, 573–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon HA (1991). The Architecture of Complexity. In Facets of Systems Science, Klir GJ, ed. (Boston, MA: Springer US; ), pp. 457–476. [Google Scholar]
- Singhal A, Cao S, Churas C, Pratt D, Fortunato S, Zheng F, and Ideker T (2020). Multiscale community detection in Cytoscape. PLoS Comput. Biol 16, e1008239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singla J, McClary KM, White KL, Alber F, Sali A, and Stevens RC (2018). Opportunities and Challenges in Building a Spatiotemporal Multi-scale Model of the Human Pancreatic β Cell. Cell 173, 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoot ME, Ono K, Ruscheinski J, Wang P-L, and Ideker T (2011). Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacey RG, Skinnider MA, and Foster LJ (2020). On the Robustness of Graph-Based Clustering to Random Network Alterations. Mol. Cell. Proteomics 20, 100002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark C, Breitkreutz B-J, Reguly T, Boucher L, Breitkreutz A, and Tyers M (2006). BioGRID: a general repository for interaction datasets. Nucleic Acids Res 34, D535–D539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, Stroedicke M, Zenkner M, Schoenherr A, Koeppen S, et al. (2005). A human protein-protein interaction network: a resource for annotating the proteome. Cell 122, 957–968. [DOI] [PubMed] [Google Scholar]
- Supek F, Bošnjak M, Škunca N, and Šmuc T (2011). REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6, e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. (2018). STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47, D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanay A, Sharan R, Kupiec M, and Shamir R (2004). Revealing modularity and organization in the yeast molecular network by integrated analysis of highly heterogeneous genomewide data. Proc. Natl. Acad. Sci. U. S. A 101, 2981–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thulasiraman K, and Swamy MNS (1992). 5.7 acyclic directed graphs. Graphs: Theory and Algorithms 118. [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, Pagé N, Robinson M, Raghibizadeh S, Hogue CW, Bussey H, et al. (2001). Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294, 2364–2368. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. (2017). Defining a Cancer Dependency Map. Cell 170, 564–576.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoquaux N, Ay F, Noble WS, and Vert J-P (2014). A statistical approach for inferring the 3D structure of the genome. Bioinformatics 30, i26–i33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilescu J, Guo X, and Kast J (2004). Identification of protein-protein interactions using in vivo cross-linking and mass spectrometry. Proteomics 4, 3845–3854. [DOI] [PubMed] [Google Scholar]
- Vaske CJ, Benz SC, Sanborn JZ, Earl D, Szeto C, Zhu J, Haussler D, and Stuart JM (2010). Inference of patient-specific pathway activities from multidimensional cancer genomics data using PARADIGM. Bioinformatics 26, i237–i245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner DE, Weinreb C, Collins ZM, Briggs JA, Megason SG, and Klein AM (2018). Single-cell mapping of gene expression landscapes and lineage in the zebrafish embryo. Science 360, 981–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Liu S, Warrell J, Won H, Shi X, Navarro FCP, Clarke D, Gu M, Emani P, Yang YT, et al. (2018). Comprehensive functional genomic resource and integrative model for the human brain. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Wang H, Dai G, and Wang H (2006). Visualization of large hierarchical data by circle packing. Of the SIGCHI Conference on Human … [Google Scholar]
- Ward AB, Sali A, and Wilson IA (2013). Biochemistry. Integrative structural biology. Science 339, 913–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widder S, Allen RJ, Pfeiffer T, Curtis TP, Wiuf C, Sloan WT, Cordero OX, Brown SP, Momeni B, Shou W, et al. (2016). Challenges in microbial ecology: building predictive understanding of community function and dynamics. ISME J 10, 2557–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SL, Zhang LV, Tong AHY, Li Z, Goldberg DS, King OD, Lesage G, Vidal M, Andrews B, Bussey H, et al. (2004). Combining biological networks to predict genetic interactions. Proc. Natl. Acad. Sci. U. S. A 101, 15682–15687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue H, Yao T, Cao M, Zhu G, Li Y, Yuan G, Chen Y, Lei M, and Huang J (2019). Structural basis of nucleosome recognition and modification by MLL methyltransferases. Nature 573, 445–449. [DOI] [PubMed] [Google Scholar]
- Yang H, Peisach E, Westbrook JD, Young J, Berman HM, and Burley SK (2016). DCC: a Swiss army knife for structure factor analysis and validation. J. Appl. Crystallogr 49, 1081–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdanparast S, Havens TC, and Jamalabdollahi M (2020). Soft Overlapping Community Detection in Large-Scale Networks via Fast Fuzzy Modularity Maximization. IEEE Trans. Fuzzy Syst 1–1. [Google Scholar]
- Yu MK, Kramer M, Dutkowski J, Srivas R, Licon K, Kreisberg JF, Ng CT, Krogan N, Sharan R, and Ideker T (2016). Translation of Genotype to Phenotype by a Hierarchy of Cell Subsystems. Cell Systems 2, 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu MK, Ma J, Ono K, Zheng F, Fong SH, Gary A, Chen J, Demchak B, Pratt D, and Ideker T (2019). DDOT: A Swiss Army Knife for Investigating Data-Driven Biological Ontologies. Cell Systems 8, 267–273.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, Muñoz-Manchado AB, Codeluppi S, Lönnerberg P, La Manno G, Juréus A, Marques S, Munguba H, He L, Betsholtz C, et al. (2015). Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142. [DOI] [PubMed] [Google Scholar]
- Zhang C, Hanspers K, Kuchinsky A, Salomonis N, Xu D, and Pico AR (2012a). Mosaic: making biological sense of complex networks. Bioinformatics 28, 1943–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QC, Petrey D, Garzón JI, Deng L, and Honig B (2012b). PrePPI: a structure-informed database of protein–protein interactions. Nucleic Acids Res 41, D828–D833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng F, Zhang S, Churas C, Pratt D, Bahar I, and Ideker T (2021). HiDeF: identifying persistent structures in multiscale ‘omics data. Genome Biol 22, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S, Zhang S, Fan X, Wu Q, Yan L, Dong J, Zhang H, Li L, Sun L, Pan N, et al. (2018). A single-cell RNA-seq survey of the developmental landscape of the human prefrontal cortex. Nature 555, 524–528. [DOI] [PubMed] [Google Scholar]
- Zhu J, Zhao Q, Katsevich E, and Sabatti C (2019). Exploratory Gene Ontology Analysis with Interactive Visualization. Sci. Rep 9, 7793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitnik M, and Leskovec J (2017). Predicting multicellular function through multi-layer tissue networks. Bioinformatics 33, i190–i198. [DOI] [PMC free article] [PubMed] [Google Scholar]