

Abstract
Chronic inflammation results from excessive pro-inflammatory signaling and the failure to resolve the inflammatory reaction. Lipid mediators orchestrate both the initiation and resolution of inflammation. Switching from pro-inflammatory to pro-resolving lipid mediator biosynthesis is considered as efficient strategy to relieve chronic inflammation, though drug candidates exhibiting such features are unknown. Starting from a library of Vietnamese medical plant extracts, we identified isomers of the biflavanoid 8-methylsocotrin-4′-ol from Dracaena cambodiana, which limit inflammation by targeting 5-lipoxygenase and switching the lipid mediator profile from leukotrienes to specialized pro-resolving mediators (SPM). Elucidation of the absolute configurations of 8-methylsocotrin-4′-ol revealed the 2S,γS-isomer being most active, and molecular docking studies suggest that the compound binds to an allosteric site between the 5-lipoxygenase subdomains. We identified additional subordinate targets within lipid mediator biosynthesis, including microsomal prostaglandin E2 synthase-1. Leukotriene production is efficiently suppressed in activated human neutrophils, macrophages, and blood, while the induction of SPM biosynthesis is restricted to M2 macrophages. The shift from leukotrienes to SPM was also evident in mouse peritonitis in vivo and accompanied by a substantial decrease in immune cell infiltration. In summary, we disclose a promising drug candidate that combines potent 5-lipoxygenase inhibition with the favorable reprogramming of lipid mediator profiles.
KEY WORDS: Lipoxygenase, Inflammation, Resolution, Natural product, Lipidomics, Lipid mediator
Abbreviations: 5-H(p)ETE, 5-hydro(pero)xy-eicosatetraenoic acid; 12-HHT, 12(S)-hydroxy-5-cis-8,10-trans-heptadecatrienoic acid; COX, cyclooxygenase; DAD, diode array detector; DPPH, 2,2-diphenyl-1-picrylhydrazyl; ECD, electronic circular dichroism; ESI, electrospray ionization; FCS, fetal calf serum; HPLC, high performance liquid chromatography; HR, high resolution; IFN, interferon; IL, interleukin; LOX, lipoxygenase; LT, leukotriene; LTC4S, leukotriene C4 synthase; MaR, maresin; mPGES-1, microsomal prostaglandin E2 synthase 1; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; PBMC, peripheral blood mononuclear cells; PD, protectin; PG, prostaglandin; PMNL, polymorphonuclear neutrophils; RP, reversed phase; Rv, resolvin; sEH, soluble epoxide hydrolase; SPE, solid phase extraction; SPM, specialized pro-resolving mediators; TX, thromboxane; UPLC‒MS/MS, ultra-performance liquid chromatography–tandem mass spectrometry
Graphical abstract
From a library of Vietnamese plant extracts, we identified the 2S,γS-isomer of 8-methylsocotrin-4′-ol (compound 2) as a promising anti-inflammatory drug candidate. The biflavonoid induces a lipid mediator class switch from pro-inflammatory leukotrienes to specialized pro-resolving lipid mediators and relieves inflammation in vitro and in vivo.

1. Introduction
Inflammation is a self-defensive process of the host microcirculation against external stimuli like pathogen infection and injury1. Both initiation and termination of inflammation are regulated through a network of oxygenated lipid mediators, which are produced from polyunsaturated fatty acids by a limited set of biosynthetic enzymes that are differentially expressed in innate immune cells and are spatially and temporally controlled2. Inflammation is initiated and maintained by the production of pro-inflammatory mediators, among which leukotrienes (LTs) and prostaglandins (PGs) are dominating3. The physiological inflammatory response is actively terminated in the resolution phase, a homeostatic process that is governed by the biosynthesis of specialized pro-resolving mediators (SPM)4,5. Impaired resolution together with the excessive production of pro-inflammatory mediators and the failure to dampen their signaling allow acute inflammation to turn into a chronic state, which causes permanent tissue damage and is closely related to chronic inflammatory pathologies, such as osteoarthritis, atherosclerosis, asthma, and cancer6. Classical strategies to treat inflammation mainly focus on suppressing pro-inflammatory signaling, while novel approaches aim at promoting resolution programs.
LTs and PGs are produced by lipoxygenases (LOXs) and cyclooxygenases (COXs) in concert with further redox- and non-redox-active enzymes7, whose inhibition relieves inflammation-related diseases like asthma, allergy, and osteoarthritis8,9. However, current anti-inflammatory drugs targeting COX and 5-LOX are associated with gastrointestinal, renal, and cardiovascular side effects10, 11, 12 and were proposed to impair resolution13. Additional targets within eicosanoid biosynthesis, i.e., microsomal prostaglandin E2 synthase 1 (mPGES-1), leukotriene C4 synthase (LTC4S), and soluble epoxide hydrolase (sEH), were reported12,14,15, and polypharmacological strategies were explored16,17, including dual inhibition of LT biosynthesis and mPGES-118. While some of these approaches favorably modulate pro-inflammatory lipid mediator signaling, strategies that alter lipid mediator networks from a pro-inflammatory to a pro-resolving state have not yet been realized, despite their proclaimed high therapeutic potential19,20.
SPM biosynthesis essentially depends on the activity of 12-LOX and 15-LOX, which are abundantly expressed in anti-inflammatory (M2-like) macrophage subsets and other immune cells21,22. 5-LOX also participates in SPM biosynthesis but its role is diffuse. Increasing evidence shows that SPM inhibit neutrophil trafficking, scavenge pro-inflammatory cytokines, stimulate efferocytosis and bacterial clearance, promote tissue regeneration, and protect from oxidative stress4,5.
To identify small molecules that induce a lipid mediator class switch from inflammation to resolution, we started from an in-house library of Vietnamese medical plants to access structurally diverse pharmacophores23,24. Detailed investigations disclose the isomers of 8-methylsocotrin-4′-ol (2) that limit inflammation by suppressing pro-inflammatory lipid mediator formation and inducing SPM biosynthesis, with the 2S,γS-isomer being most active. Compound 2 inhibits LT formation across human innate immune cells while specifically triggering SPM production in macrophages of the M2 phenotype. The lipid mediator class switch by compound 2 was confirmed in vivo using an experimental mouse model of zymosan-induced peritonitis.
2. Materials and methods
2.1. Materials
Solvents used for extract fractionation and compound isolation were purchased from VWR International (Darmstadt, Germany). Solvents for high performance liquid chromatography (HPLC) were obtained from Merck (Darmstadt, Germany). Ultrapure water was produced by a Sartorius Arium 611 UV water purification system (Göttingen, Germany). Deuterated and non-deuterated lipid mediator standards for ultra-performance liquid chromatography‒tandem mass spectrometry (UPLC‒MS/MS) were purchased from Cayman Chemicals (Ann Arbor, MI, USA). Zymosan, indomethacin, staurosporine, and all other reagents were purchased from Merck (Darmstadt, Germany) unless indicated otherwise.
2.2. General experimental procedures for extract fractionation and compound isolation
Optical rotations were determined with a Perkin–Elmer 341 polarimeter (Wellesley, MA, USA) at 20 °C. Electronic circular dichroism (ECD) and UV spectra were measured on a JASCO J-1500 CD spectrometer (Tokyo, Japan). 1D- and 2D-NMR experiments were recorded on a Bruker DRX 300 (Bruker Biospin, Rheinstetten, Germany) operating at 300.13 MHz (1H) and 75.47 MHz (13C) or Bruker Advance II 600 operating at 600.19 MHz (1H) and 150.91 MHz (13C) at 300 K; NMR solvents: methanol-d4/CDCl3/DMSO-d6 with 0.03% TMS (Eurisotop, Gif-Sur-Yvette, France), which was used as internal standard.
HPLC-DAD analysis was carried out using a Prominence UFLC-XR system (Shimadzu, Kyoto, Japan) equipped with degasser, binary pump, autosampler, column thermostat, and diode array detector (DAD). Separations were performed on a HyperClone ODS (C18) 120 Å (150 mm × 4.6 mm i.d., 3 μm, Phenomenex, Torrance, CA, USA) and a Merck (VWR, Darmstadt, Germany) LiChroCART 4-4 guard column with LiChrospher 100 RP18 (5 μm) packing. A mobile phase consisting of 0.02% TFA in H2O (v/v) (solvent A) and methanol (solvent B) was employed with gradient elution (0 min, 30% B; 10 min, 50% B; 25 min, 65% B; 40 min, 70% B; 50 min, 98% B; 60 min, 98% B). The thermostat was set to 35 °C, the flow rate was 0.5 mL/min, the injection volume 10 μL, and the detection wavelength 280 nm. Chiral Analytical separations were carried out at an Agilent 1100 HPLC system (Agilent Austria, Vienna, Austria) equipped with DAD detector, auto-sampler, and column thermostat. LC parameters: stationary phase: Phenomenex Lux 3 μm Cellulose-1, 250 mm × 4.6 mm or Phenomenex Lux 3 μm i-Amylose-3, 250 mm × 4.6 mm, flow: 0.5 mL/min, 20 °C, isocratic elution, detection at 280 nm. Solvent composition, sample concentration, and injection volume for each sample were individually optimized as indicated in Supporting Information Section S1.
For low resolution LC‒electrospray ionization (ESI)-MS experiments, the HPLC system was coupled to a Bruker (Bruker Daltonics, Bremen, Germany) Esquire 3000plus iontrap, replacing solvent A with a solution of 0.5% formic acid in H2O (v/v). LC‒MS‒LC-parameters: HP 1100 system (Agilent, Waldbronn, Germany) equipped with auto-sampler, DAD, and column thermostat. MS-parameters: Esquire 3000plus (Bruker Daltonics, Bremen, Germany); ESI, temperature: 340 °C; dry gas: 10.00 L/min; nebulizer gas 30 psi; full scan mode: m/z 100–1500. High resolution-(HR)-ESI-MS analysis was performed on a Bruker micrOTOF-QII mass spectrometer after LC separation (identical to LC‒MS), using the following parameters: ESI-MS parameters: 1:5 split from HPLC, dry temp.: 220 °C; dry gas: 5.00 L/min; nebulizer 1.6 bar; full scan mode: m/z 100–1500; ion polarity: positive; capillary voltage: 4.5 kV; end plate offset: −0.5 kV. Semi-preparative HPLC was performed on a Dionex UltiMate 3000 preparative HPLC system (Thermo Fisher Scientific, Waltham, MA, USA) equipped with degasser, pump, autosampler, column compartment, variable wavelength detector, fraction collector, and Chromeleon software. Centrifugal partition chromatography was performed using a FCPC C apparatus (Kromaton, Angers, France) with a rotor volume of 55 mL equipped with an LC-10AD pump system (Shimadzu, Kyoto, Japan) and a 5 mL sample loop. Column chromatography was performed with Sephadex LH-20 (Pharmacia Biotech AB, Stockholm, Sweden) or silica gel 60 (0.040–0.063 mm; Merck, VWR, Darmstadt, Germany) as stationary phases. Thin layer chromatography (TLC) was carried out on silica gel 60 F254 plates (VWR, Darmstadt, Germany).
2.3. Plant material
The red wood of D. cambodiana was collected in Tinh Bien (An Giang, Vietnam) in March 2014, identified by Prof. Dr. Tran Hung (Department of Pharmacognosy, Faculty of Pharmacy, University of Medicine and Pharmacy of HoChiMinh city, Vietnam) and air-dried. A voucher specimen (AG3032014) is stored at the Department of Pharmacognosy, Faculty of Pharmacy, University of Medicine and Pharmacy of HoChiMinh City, Vietnam.
2.4. Extraction, isolation and chiral separation
Milled and air-dried red wood of D. cambodiana (5 kg) was extracted with 20 L of 96% ethanol under reflux. The plant material was recovered by filtration and the extraction process was repeated twice with 20 and 10 L of 96% ethanol, respectively. The filtrates were combined and the obtained solution was evaporated to dryness under reduced pressure at a temperature of 40 °C yielding 628 g crude extract. The initial separation was performed by means of liquid–liquid extraction; 300 g of crude extract were suspended in 1.0 L water and extracted with dichloromethane (500 mL × 4), ethyl acetate (500 mL × 6), followed by n-butanol (500 mL × 4). Individually combined organic solutions as well as aqueous solution were evaporated to dryness, affording dichloromethane (44.2 g), ethyl acetate (104.3 g), n-butanol (38.5 g) and water subfractions (128.2 g). A part of the ethyl acetate subfraction (52.4 g) was subjected to silica gel column chromatography (CC) using a stepwise gradient of dichloromethane and methanol (100:0 to 0:100, v/v) as mobile phase to obtain subfractions 1 to 20. Subfraction 5 (1.04 g) was subjected to silica gel CC (petroleum ether/acetone, 95.5:0.5 to 85:15, v/v) to afford compound 9 (179.8 mg). Subfraction 7 (3.7 g) was separated by silica gel CC (petroleum ether/acetone, 95:5 to 70:30, v/v) to afford compound 10 (30.4 mg) and a subfraction (7-9), which was purified by Sephadex-LH-20 CC using methanol as a mobile phase to yield compound 8 (27.4 mg). A further subfraction (7-15) was separated by semi-preparative HPLC (Phenomenex Aqua 5 μm C18 125 Å; 250 mm × 10.0 mm, gradient acetonitrile/water, 70:30 to 86:14 in 20 min; flow rate 1.2 mL/min, 35 times) to obtain compound 3 (11.7 mg). Subfraction 8 (1.4 g) was combined with subfraction 9 (1.0 g) and subjected to silica gel CC (petroleum ether/acetone, 90:10 to 70:30, v/v) to yield compound 14 (317.8 mg). Subfraction 10 (840 mg) was separated by Sephadex LH-20 CC with methanol as eluent to afford 9 fractions (10-1 to 10-9). Compound 15 (17 mg) precipitated from the methanolic solution of subfraction 10–3. Subfraction 10-4 was separated by Sephadex LH-20 CC (dichloromethane/acetone, 85:15, v/v) to yield compounds 5 (24.7 mg) and 6 (23.9 mg); a further subfraction of this step, 10-4-6, was further purified by silica gel CC (petroleum ether/ethyl acetate, 6:4) to obtain compounds 11 (10 mg) and 16 (6.5 mg). Subfraction 11 (786.0 mg) was subjected to silica gel CC (petroleum ether/acetone, 7:3) to yield 6 fractions (11-1 to 11-6). Subfraction 11-6 (549.8 mg) was purified by Sephadex LH-20 CC (methanol as mobile phase) and then separated with semi-preparative HPLC (Phenomenex Aqua 5 μm C18 125 Å; 250 mm × 10.0 mm, gradient methanol/water, 70:30 to 85:15 in 30 min; flow rate 1.2 mL/min, 80 times) to afford compounds 1 (21.7 mg) and 2 (25.3 mg). Preparative chiral separation of compound 1 and 2 was carried out at a Prominence UFLC-XR HPLC system (Shimadzu) equipped with auto-sampler, DAD, and column thermostat using the following conditions: stationary phase: Phenomenex Lux 3 μm Cellulose-1, 250 mm × 4.6 mm, flow: 0.5 mL/min, 30 °C, isocratic elution with ethanol abs. and n-hexane (3:7, v/v), detection at 280 nm injection volume: 2 μL; sample concentration: 9.0 mg/mL mobile phase for compound 1 and 5.6 mg/mL mobile phase for compound 2. Collected fractions of 80 injections were evaporated to dryness and passed through Sephadex LH-20 (diameter 0.5 cm, bed high 3 cm, swollen in mobile phase; flow rate 0.5 mL/min, methanol as solvent) to remove stationary material yielding enantiomeric pure compound 1a (1.18 mg), 1b (1.00 mg), 2a (0.80 mg), and 2b (1.26 mg). Compound 18 (11.2 mg) was isolated from subfraction 12 (830.2 mg) by Sephadex LH-20 CC (methanol as mobile phase) and then purified by silica gel CC (petroleum ether/ethyl acetate, 6:4). Subfraction 13 (1.12 g) was separated by silica gel CC (petroleum ether/acetone, 10:0 to 6:4) to obtain 13 subfractions (13-1 to 13-13). Sub-fractions 13-4 and 13-8 were purified by Sephadex LH-20 CC to yield compounds 13 (119.2 mg), 17 (6.6 mg), and 4 (20.4 mg). Subfraction 16 was separated by FCPC (dichloromethane/methanol/water, 4:3:3, v/v; lower phase: mobile phase) and then purified by Sephadex LH-20 CC (methanol as mobile phase) to afford compounds 7 (60.5 mg) and 12 (4.0 mg). Structure elucidation of known compounds isolated was carried out by means of mass spectrometry (LC‒(HR)-ESI-MS) and NMR spectroscopy as well as by comparison of physical and spectral data with those of reference compounds reported in the literature. Structure elucidations of new compounds are described in Supporting Information Sections S2–4.
2.5. Computational methods for ECD calculation
3D structures of 1 and 2 were drawn and subjected to a conformational search by using OPLS-3 force field in MacroModel v.9 (Schrödinger Ltd., New York, NY, USA) in water. The number of steps was set high enough to include all low energy conformers. Conformers occurring in the energy window of 10 kcal/mol from the global minima were subjected to geometrical minimization and optimization in the DFT/B3LYP/6-31+G(d,p) basis set and level of theory and using the conductor-like polarizable continuum model (CPCM) in ethanol, using Gaussian 16 v. A.03 software (Gaussian, Inc., Wallingford, CT, USA) (Supporting Information Fig. S1). Frequency calculations were performed at the same level and no imaginary frequencies were observed. Calculation of the excitation energy (nm), rotatory strength dipole velocity (Rvel) and dipole length (Rlen) were performed by using time-dependent density-functional theory (TD-DFT) at PBEPBE/def2-SV/6-31+G(d,p)/CPCM level in ethanol. ECD curves were simulated on the basis of rotatory strengths using SpecDis v1.7 with half-band of 0.2 eV and UV shift of 10–30 nm.
2.6. Docking simulation
The molecular docking simulation was conducted with GOLD 5.2 (CCDC, Cambridge, UK). Scoring was calculated using the CHEMPLP (https://www.ccdc.cam.ac.uk/support-and-resources/support/case/?caseid=5d1a2fc0-c93a-49c3-a8e2-f95c472dcff0) scoring function.
Pdb entry 6ncf25, stable 5-LOX, co-crystalized with 3-O-acetyl-11-keto-β-boswellic acid (AKBA) at the allosteric binding site was used as a target structure. The protein was energetically minimized in Discovery Studio (version 2018; Biovia, San Diego, CA, USA). Hydrogens were added with GOLD, using default settings. The binding site was defined on the B chain in a 9 Å radius at the site of the co-crystalized ligand. Protein‒ligand interactions of the docking poses were analyzed using LigandScout 4.2 (www.inteligand.com/ligandscout). A re-docking of AKBA was conducted to optimize the docking settings. The best ranked pose with the final docking settings had an RMSD of 1.061 compared to the co-crystallized ligand.
2.7. Isolation of primary cells from human blood
Human blood (3.13% sodium citrate) and human leukocyte concentrates from freshly withdrawn venous blood (16 I.E. heparin/mL blood) were provided by the Institute for Transfusion Medicine of the University Hospital Jena (Germany) with informed consent of the volunteers26. The selected fasted (12 h) healthy male and female blood donors (18–65 years) were registered and regularly donated blood every 8–12 weeks. They were physically inspected by a clinician and free of obvious infections and inflammatory or allergic disorders and neither had taken antibiotics nor anti-inflammatory drugs within 10 days before blood collection.
Human polymorphonuclear leukocytes (PMNL), peripheral blood mononuclear cells (PBMC) and platelets were freshly isolated from leukocyte concentrates as described26. Briefly, PMNL, PBMC and platelets were separated by dextran sedimentation, followed by density gradient centrifugation using lymphocyte separation medium (LSM 1077, GE Healthcare, Freiburg, Germany). Platelets and PBMC were collected from the supernatant and the intermediate layer, respectively, whereas PMNL were obtained from the pelleted cells after removal of erythrocytes by hypotonic lysis. To enrich monocytes, PBMC were seeded in RPMI 1640 medium plus 5% heat-inactivated fetal calf serum (FCS), 2 mmol/L l-glutamine, 100 U/mL penicillin and 100 μg/mL streptomycin (GE Healthcare) in culture flasks (Greiner Bio-one, Frickenhausen, Germany) and incubated for 1.5 h at 37 °C and 5% CO2. Adherent monocytes were recovered, and their purity (>85%) was defined by flow cytometry26. Experimental procedures on human blood and blood cells were approved by the ethical commission of the University Hospital Jena.
2.8. Monocyte differentiation and macrophage polarization
For differentiation of monocytes to macrophages and further polarization towards M1 and M2 phenotypes, human primary monocytes were first stimulated with 20 ng/mL GM-CSF or M-CSF (Peprotech, Hamburg, Germany) for 6 days in macrophage medium (RPMI 1640 supplemented with 10% FCS, 2 mmol/L l-glutamine, 100 U/mL penicillin and 100 μg/mL streptomycin) to obtain monocyte-derived macrophages. These macrophages were further polarized for 48 h in macrophage medium with 100 ng/mL lipopolysaccharide (LPS) and 20 ng/mL interferon (IFN)-γ (Peprotech, Hamburg, Germany) to obtain M1 or with 20 ng/mL interleukin (IL)-4 (Peprotech) to obtain M2 phenotypes22.
2.9. Determination of cell-free 5-LOX activity
Human recombinant 5-LOX enzyme was purified by affinity chromatography on an ATP-agarose column26. In brief, Escherichia coli Bl21 (DE3) was transformed with pT3-5LO plasmid and lysed in lysis buffer containing 50 mmol/L triethanolamine, 5 mmol/L EDTA, 60 μg/mL soybean trypsin inhibitor, 1 mmol/L phenylmethanesulphonyl fluoride, 1 mmol/L dithiothreitol, and 1 mg/mL lysozyme. Lysates were sonicated (3 times, 15 s, each) and centrifuged twice (first at 10,000×g, 15 min, 4 °C; then the supernatant again at 40,000×g, 70 min, 4 °C). The supernatant was applied to an ATP-agarose column, which was extensively washed with PBS pH 7.4 plus 1 mmol/L EDTA, phosphate buffer (50 mmol/L, containing 0.5 mol/L NaCl and 1 mmol/L EDTA) and phosphate buffer (50 mmol/L plus 1 mmol/L EDTA). Semi-purified 5-LOX enzyme was eluted with phosphate buffer (50 mmol/L) containing 1 mmol/L EDTA and 20 mmol/L ATP.
For determination of 5-LOX activity, semi-purified enzyme (0.5 μg) was pre-treated with vehicle (DMSO) or test compounds in PBS pH 7.4 containing 1 mmol/L EDTA and 1 mmol/L ATP for 10 min at 4 °C. The mixture was pre-warmed for 30 s at 37 °C before arachidonic acid (20 μmol/L, if not indicated otherwise) and CaCl2 (2 mmol/L) were added. After 10 min at 37 °C, the reaction was stopped by addition of an equal volume of ice-cold methanol. Formed lipid mediators were extracted by solid phase extraction (SPE) using Sep-Pak C18 35 cc Vac Cartridges (Waters, Milford, MA, USA). 5-LOX products (all-trans isomers of LTB4 and 5-hydro(pero)xy-eicosatetraenoic acid (5-H(p)ETE) were separated by reversed phase (RP)-HPLC on a Nova-Pak C18 Radial-Pak Column (4 μm, 100 mm × 5 mm, Waters) under isocratic conditions (73% methanol/27% water/0.007% trifluoroacetic acid) at a flow rate of 1.2 mL/min and detected at 235 and 280 nm. The selective 5-LOX inhibitors zileuton (3 μmol/L; Tocris Bioscience, Bristol, UK) and BWA4C (0.5 μmol/L, Merck) used as reference suppressed 5-LOX product formation by 70.7 ± 2.7% and 59.1 ± 6.9%, respectively.
To investigate whether compounds 2 and 5 reversibly inhibit 5-LOX activity, the purified enzyme was pre-incubated with the test compounds at 30 or 3 μmol/L for 15 min and then 10-fold diluted to a final compound concentration of 3 or 0.3 μmol/L, respectively. Arachidonic acid (20 μmol/L) and CaCl2 (2 mmol/L) were added and after 10 min at 37 °C 5-LOX products were extracted and analyzed as described above.
2.10. Determination of cellular 5-LOX, 15-LOX, and 12-LOX activity
Freshly isolated human PMNL (1 × 107 cells/mL) were resuspended in PBS pH 7.4 plus 1 mg/mL glucose and 1 mmol/L CaCl2. PMNL were pre-treated with vehicle (DMSO) or test compounds for 10 min before addition of arachidonic acid (20 μmol/L) and/or calcium ionophore A23187 (2.5 μmol/L) and incubation for another 10 min at 37 °C. The reaction was terminated by addition of an equal volume of ice-cold methanol. Lipid mediators were extracted by solid phase extraction using Sep-Pak C18 35 cc Vac Cartridges, and 5-LOX products (LTB4, its all-trans isomers and 5-H(p)ETE), 15-HETE (15-LOX product), and 12-HETE (12-LOX product) were analyzed by RP-UV‒HPLC as described for the determination of 5-LOX metabolites from cell-free assays. The reference inhibitors zileuton (3 μmol/L) and BWA4C (0.5 μmol/L) inhibited the biosynthesis of 5-LOX metabolites by 64.1 ± 6.3% and 78.6 ± 9.7%, respectively.
2.11. Sample preparation and metabololipidomics analysis of lipid mediators
Lipid mediator biosynthesis in human PMNL, M1/M2 macrophages or citrated blood were treated with A23187 (30 μmol/L, PMNL or blood, 15 min), pathogenic E. coli (serotype O6:K2:H1, ratio 1:50, 90 min for macrophages; 1 × 109/mL blood, 180 min for blood) or Staphylococcus aureus (6850)-conditioned medium (SACM, 1%, 180 min). SACM was prepared by growing the bacteria for 17 h at 37 °C in brain heart infusion medium (Carl Roth, Karlsruhe, Germany), followed by centrifugation at 3350×g for 10 min and sterile-filtration through a Millex-GP filter unit (0.22 μm; Millipore) for the indicated time.
Supernatants of treated cells (PMNL or M1/M2 macrophages in 1 mL PBS pH 7.4 plus 1 mmol/L CaCl2), blood (1 mL) or exudates from murine peritonitis (1 mL) were mixed with ice-cold methanol (2 mL) supplemented with deuterium-labeled internal standards (10 μL; 200 nmol/L d8-5S-HETE, d4-LTB4, d5-LXA4, d5-RvD2, d4-PGE2, and 10 μmol/L d8-arachidonic acid). Lipid mediators were extracted by SPE on Sep-Pak C18 35 cc Vac Cartridges (Waters)22. In brief, samples were placed at −20 °C for at least 45 min to allow protein precipitation. After centrifugation (1200×g, 4 °C, 10 min), 8 mL acidified H2O (pH = 3.5) was added to the supernatants, and the samples were loaded onto equilibrated RP-columns. After extensive washing with H2O and n-hexane, lipid mediators were eluted with methyl formate. Eluates were brought to dryness (TurboVap LV, Biotage, Uppsala, Sweden) and dissolved in methanol/water (50/50) for UPLC‒MS/MS analysis. Lipid mediators were analyzed by UPLC‒MS/MS using an Acquity UPLC system (Waters) coupled to a QTRAP 5500 mass spectrometer (Sciex, Darmstadt, Germany), which was equipped with a Turbo V™ electrospray ionization source22. Analytes were separated on an Acquity UPLC BEH C18 column (1.7 μm, 2.1 mm × 100 mm; Waters) at 50 °C with a flow rate of 0.3 mL/min. The mobile phase consisting of methanol, water, and acetic acid (42:58:0.01) was ramped to 86:14:0.01 over 12.5 min and then switched to 98:2:0.01 for isocratic elution (3 min). Diagnostic ions were detected in the negative ionization mode by scheduled multiple reaction monitoring using optimized MS parameters for each lipid mediator22. Retention times were confirmed using external standards (Cayman Chemicals). Automatic peak integration was performed with Analyst 1.6 software (Sciex) using IntelliQuan default settings. Lipid mediator amounts were calculated from signal intensities normalized to internal standards using external calibration curves.
2.12. Determination of cell viability
Mitochondrial dehydrogenase activity was measured by MTT assay to assess cell viability27. Briefly, monocytes were pre-incubated with vehicle (DMSO), test compounds or staurosporine (1 μmol/L, cytotoxic reference) for 15 min and incubated for 24 h at 37 °C and 5% CO2. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was added, and cells were incubated for 2–4 h. After cells were lysed and the formazan crystals solubilized with SDS (10% in 20 mmol/L HCl), the absorption was read at 570 nm using a Multiskan Spectrum Microplate Reader (Thermo Fisher Scientific).
2.13. Determination of radical scavenging activity
2,2-Diphenyl-1-picrylhydrazyl (DPPH, Merck; 50 mmol/L) was incubated with vehicle (DMSO), test compounds or ascorbic acid (50 μmol/L, reference compound for radical scavenging) in ethanol for 30 min27. Absorbance was measured at 520 nm using a Multiskan Spectrum Microplate Reader.
2.14. Determination of COX-1 and COX-2 activity
For the determination of cell-free COX activity, isolated ovine COX-1 (50 units, Cayman Chemicals) or human recombinant COX-2 (20 units, Cayman Chemicals) were pre-treated with vehicle (DMSO) or test compounds in 100 mmol/L Tris pH 8 (supplemented with 5 mmol/L glutathione, 5 μmol/L hemoglobin and 100 μmol/L EDTA) for 5 min at 4 °C. The mixture was pre-warmed for 1 min at 37 °C, and arachidonic acid was added (5 μmol/L for COX-1 and 2 μmol/L for COX-2). The reaction was stopped by addition of an equal volume of ice-cold methanol, and COX-derived 12(S)-hydroxy-5-cis-8,10-trans-heptadecatrienoic acid (12-HHT) was extracted by SPE using Sep-Pak C18 35 cc Vac Cartridges (Waters) and analyzed by RP-UV‒HPLC on a Nova-Pak C18 Radial-Pak Column as described for 5-LOX products. The COX-1/2 inhibitor indomethacin (10 μmol/L) was used as control.
For the determination of cellular COX-1 activity, freshly isolated human platelets (1 × 108) were pre-treated with vehicle (DMSO), test compounds or indomethacin (10 μmol/L) for 5 min at room temperature, and arachidonic acid (5 μmol/L) was added. After 5 min at 37 °C, the reaction was stopped by addition of an equal volume of ice-cold methanol, and formed 12-HHT was extracted and analyzed as described above.
2.15. Determination of LTC4S activity
Human embryonic kidney (HEK)293 cells (ATCC, Manassas, VA, USA) were cultivated in DMEM/high glucose (4.5 g/L; GE Healthcare) supplemented with heat-inactivated FCS (10%) and penicillin/streptomycin at 37 °C and 5% CO2. Confluent cells were reseeded at 1 × 104/cm2 after being detached with trypsin/EDTA (GE Healthcare). Microsomal membranes (2.5 μg total protein) from HEK293 cells that stably express LTC4S were pre-treated with vehicle (DMSO) or test compounds in potassium phosphate buffer (0.1 mol/L, pH 7.4, containing 5 mmol/L glutathione) for 10 min28. The reaction was started by addition of LTA4 methyl ester (1 μmol/L, Cayman Chemicals) and stopped after 10 min at 4 °C by addition of an equal volume of ice-cold methanol. LTC4-d5 methyl ester (5 ng, Cayman Chemicals) was used as internal standard. Formed LTC4 was extracted by SPE and analyzed by UPLC‒MS/MS28. MK886 (10 μmol/L, Cayman Chemicals) was used as reference for LTC4S inhibition.
2.16. Determination of mPGES-1 activity
Human lung adenocarcinoma epithelial A549 cells (ATCC) were cultivated in DMEM/high glucose (4.5 g/L) supplemented with heat-inactivated FCS (10%) and penicillin/streptomycin at 37 °C and 5% CO2. Confluent cells were reseeded at 1 × 104/cm2 after being detached with trypsin/EDTA. mPGES-1 activity was determined in microsomal membranes of A549 lung carcinoma cells that were first incubated with IL-1β (2 ng/mL) for 48 h29. Then, cells were harvested, frozen in liquid nitrogen, and incubated with ice-cold homogenization buffer (0.1 mol/L potassium phosphate buffer pH 7.4 plus 1 mmol/L phenylmethanesulphonyl fluoride, 60 μg/mL soybean trypsin inhibitor, 1 μg/mL leupeptin, 2.5 mmol/L glutathione, and 250 mmol/L sucrose) for 15 min at 4 °C. After sonication (3 times, 20 s, each, at 4 °C), the microsomal membrane fraction was obtained by differential centrifugation (first 10,000×g, 10 min, 4 °C; then 174,000×g, 1 h, 4 °C). Microsomal membranes (containing 2.5–5 μg total protein) were suspended in potassium phosphate buffer (0.1 mol/L, pH 7.4, plus 2.5 mmol/L glutathione) and pre-treated with vehicle (DMSO), test compounds or the reference inhibitor MK886 (10 μmol/L) for 15 min at 4 °C. PGH2 (20 μmol/L, final concentration) was added and the reaction terminated after 1 min on ice by addition of stop solution (40 mmol/L FeCl2, 80 mmol/L citric acid) supplemented with 1 nmol 11β-PGE2 as internal standard (Cayman Chemicals). Formed PGE2 was extracted by SPE using Sep-Pak C18 35 cc Vac Cartridges, separated by RP-UV‒HPLC on a Nova-Pak C18 Radial-Pak column (4 μmol/L, 5 mm × 100 mm, Waters) under isocratic conditions (30% acetonitrile, 70% water, 0.007% trifluoroacetic acid) at a flow rate of 1 mL/min and detected at 195 nm.
2.17. Determination of sEH activity
Human recombinant sEH enzyme was purified from baculovirus-infected Sf9 insect cells by benzylthio-sepharose affinity chromatography28. Briefly, cells were lysed in 50 mmol/L NaHPO4 pH 8 containing 300 mmol/L NaCl, 10% glycerol, 1 mmol/L EDTA, 1 mmol/L phenylmethanensulfonyl fluoride, 10 μg/mL leupeptin, and 60 μg/mL soybean trypsin inhibitor. After sonication (3 times, 10 s, each, at 4 °C) and centrifugation (100,000×g, 60 min, 4 °C), supernatants were collected and subjected to benzylthio-sepharose affinity chromatography. The sEH enzyme was eluted with 4-fluorochalcone oxide in PBS pH 7.4 containing 1 mmol/L dithiothreitol and 1 mmol/L EDTA. Purified sEH (60 ng) was dialysed, concentrated and pre-treated with vehicle (DMSO), test compounds or the reference inhibitor AUDA (100 nmol/L, Cayman Chemicals) for 10 min in 25 mmol/L Tris buffer, pH 7 plus 0.1 mg/mL BSA. PHOME (50 μmol/L, Cayman Chemicals) was added and converted by sEH to fluorescent 6-methoxy-naphtaldehyde within 1 min at room temperature in the dark. The reaction was stopped by 200 mmol/L ZnSO4, and the fluorescence was determined using a NOVOstar fluorescence microplate reader (BMG Labtech, excitation: 330 nm, emission: 465 nm).
2.18. Flow cytometry
Macrophages were stained in PBS pH 7.4 containing 0.5% BSA, 2 mmol/L EDTA and 0.1% sodium azide by Zombie Aqua™ Fixable Viability Kit (Biolegend, San Diego, CA, USA) to determine cell viability (5 min, 4 °C) and by fluorochrome-labelled antibodies (20 min, 4 °C): FITC anti-human CD14 (clone M5E2, #555397, BD Bioscience, San Jose, CA), APC-H7 anti-human CD80 (clone L307.4, #561134, BD Bioscience), PE-Cy7 anti-human CD54 (clone HA58, #353115, Biolegend), PE anti-human CD163 (clone GHI/61, #556018, BD Bioscience), APC anti-human CD206 (clone 19.2, #550889, BD Bioscience) to determine M1 and M2 surface marker expression using a LSRFortessa™ cell analyzer (BD Bioscience), and data were analyzed using FlowJo X Software (BD Bioscience)21.
2.19. SDS-PAGE and Westernblot
M2 macrophages were treated with vehicle (DMSO) or compound 2 for 24 h at 37 °C and 5% CO2 and then lysed in ice-cold lysis buffer containing Tris-HCl (20 mmol/L, pH 7.4), NaCl (150 mmol/L), EDTA (2 mmol/L), NaF (5 mmol/L), phenylmethanesulphonyl fluoride (1 mmol/L), leupeptin (1 mg/mL), soybean trypsin inhibitor (60 μg/mL), sodium vanadate (1 mmol/L), and sodium pyrophosphate (1 mmol/L). After lysates have been sonicated (3 × 3 s, on ice) and centrifuged (21,100×g, 5 min, 4 °C), cell supernatants were collected and their protein concentration determined by DC-protein assay kit (Bio-Rad Laboratories GmbH, Munich, Germany). Aliquots were mixed (1:1, v/v) with 1 × SDS-PAGE loading buffer containing Tris-HCl (125 mmol/L, pH 6.5), 25% sucrose, 5% SDS, 0.25% bromophenol blue, and 5% β-mercaptoethanol, and heated at 95 °C for 5 min. Equal aliquots (corresponding to 12.5 μg protein) were loaded and separated on 10% SDS-PAGE gels and then transferred onto hybond ECL nitrocellulose membranes (GE Healthcare, Freiburg, Germany). The membranes were incubated with the following primary antibodies: mouse monoclonal anti-15-LOX-1, 1:500 (ab119774, Abcam, Cambridge, UK); rabbit polyclonal anti-15-LOX-2, 1:1000 (ab23691, Abcam); mouse monoclonal anti-5-LOX, 1:1000 (610,695, BD Biosciences, NJ, USA), and mouse monoclonal anti-β-actin, 1:1000 (3700 S, Cell Signaling). Immunoreactive bands were stained with the following fluorescence-labeled antibodies: DyLight® 680 Cross-Adsorbed Goat anti-Mouse IgG (H + L) (10,797,775, Invitrogen, Thermo Fisher Scientifics, MA, USA), DyLight® 800 Cross-Adsorbed Goat anti-Mouse IgG (H + L) (13,477,327, Invitrogen), DyLight® 680 Cross-Adsorbed Goat anti-Rabbit IgG (H + L) (10,517,364, Invitrogen), DyLight® 800 Cross-Adsorbed Goat anti-Rabbit IgG (H + L) (13,477,187, Invitrogen), and visualized by a Fusion FX imaging system (Vilber Lourmat, Marne-la-Vallée, France). Data from densitometric analysis were background-corrected.
2.20. Zymosan-induced peritonitis in mice
Male CD-1 mice (33–39 g, 6–8 weeks, Charles River Laboratories, Calco, Italy) were housed at 21 ± 2 °C, provided with standard rodent chow and water and subjected to a 12 h light/12 h dark cycle. Mice were randomly assigned for the experiments and allowed to acclimate for four days. The experimental procedures comply with the ARRIVE guidelines and were approved by the Italian Ministry (744/2019) and conducted according to European and Italian regulations for animal protection (directives 2010/63/EU and DL 26/2014).
Mice (n = 6 per experimental group) received vehicle (0.5 mL, 2% DMSO in saline), compound 2 (10 mg/kg) or MK886 (3 mg/kg) by intraperitoneal injection (i.p.) 30 min prior to peritonitis induction by zymosan (0.5 mL, 2 mg/mL suspension in 0.9% saline, i.p.)30. Mice were euthanized 120 min post zymosan injection with CO2, followed by a peritoneal lavage with 3 mL of cold PBS pH 7.4. Exudates were collected from the peritoneal cavity, and cells were immediately counted by vital trypan blue staining. After centrifugation (18,000×g, 5 min, 4 °C), supernatants were collected, and lipid mediators were extracted by SPE and analyzed by UPLC‒MS/MS. LTC4 levels in the exudates were analyzed by enzyme immunoassays (Enzo Life Sciences, Lörrach, Germany) according to manufacturer's instructions. To measure myeloperoxidase activity, cell pellets were sonicated in PBS buffer (50 mmol/L, pH 6) with 0.5% hexadecyltrimethylammonium bromide, freeze-thawed three times, and centrifuged (32,000×g, 20 min, 4 °C). Cell supernatants were collected, and the reaction was initiated by addition of o-dianisidine (0.167 mg/mL) and 0.0005% hydrogen peroxide. Changes in absorbance were time-dependently monitored using an iMark microplate Reader (Bio-Rad, Segrate, Milan, Italy). Myeloperoxidase activity was determined from the calibration curve using human neutrophil myeloperoxidase as reference standard and is given in U/mL31.
2.21. Statistical analysis
Data are presented as mean ± SEM of n observations, where n represents the number of independent experiments (including independent immune cell preparations) or the number of animals per group, as indicated. GraphPad Prism 8 software (San Diego, CA, USA) was used for statistical calculations. Normal distribution of the data was addressed by Kolmogorov–Smirnov tests. Samples were blinded for screening of plant extracts, fractions and natural products and for counting infiltrated cells in the mouse peritoneum. Data were log-transformed for statistical analysis where indicated. Outliers were determined using a Grubb's test. For multiple comparison, ordinary or repeated-measures one-way ANOVA with Tukey or Dunnett's post hoc tests or ordinary two-way ANOVA with Tukey post hoc tests were applied. For comparison of two groups, paired t-tests were used. The criterion for statistical significance was ∗P < 0.05. IC50 values were calculated by non-linear regression using GraphPad Prism 8.
3. Results
3.1. Targeting LOX isoenzymes by 8-methylsocotrin-4′-ol from Vietnamese plant extracts
To identify novel natural product-derived pharmacological agents that induce a lipid mediator class switch from inflammation to resolution, we evaluated an in-house library of 94 extracts from 31 Vietnamese medicinal plants in activated PMNL (Fig. 1A). Our aim was to identify extracts, which potently suppress 5-LOX-dependent LT biosynthesis (IC50 ≤ 10 μg/mL) while enhancing 15-LOX (residual activity ≥ 100%) and optionally 12-LOX product formation (Supporting Information Table S132,33). The methanolic red wood extract of Dracaena cambodiana Pierre ex Gagnep. (Asparagaceae) showed the most favorable pharmacological profile. It inhibited the formation of LTB4 and other 5-LOX products with an IC50 of 0.4 μg/mL (Fig. 1B), increased the biosynthesis of the 15-LOX metabolite 15-HETE (Fig. 1C), and marginally decreased 12-HETE formation, the major 12-LOX metabolite of platelets in the PMNL fraction (Fig. 1D).
Figure 1.

Bioactivity-guided fractionation of D. cambodiana extract. (A) Schematic overview about the strategy to identify potential drug candidates from 31 Vietnamese medicinal plants, which suppress 5-LOX-dependent inflammation and trigger 15-LOX-dependent resolution; EtOAc, ethyl acetate; NP, natural product; AA, arachidonic acid. (B–D) Human PMNL were treated with a methanolic extract of the red wood of D. cambodiana. LOX product formation was initiated by AA plus A23187, and metabolites were analyzed by RP-UV‒HPLC. 5-LOX (B), 15-LOX (C), and 12-LOX product biosynthesis (D). (C) Interconnected lines indicate data from the same independent experiment. Results are given as means ± or + SEM (B–D), percentage of vehicle control, or paired data (C), ng/5 × 106 cells, n = 3. Data were log-transformed for statistical analysis. ∗∗∗P < 0.001, ∗∗P < 0.01, ∗P < 0.05 compared to vehicle control; repeated measures one-way ANOVA plus Tukey post hoc tests (B–D) or paired t-test (C). (E–G) D. cambodiana red wood extract (ethanol, EtOH, 96%) and its subfractions (dichloromethane subfraction (DCM), ethyl acetate subfraction (EtOAc), 1-butanol subfraction (BuOH)) (E), subfractions of the separation of the EtOAc fraction (F), and subfraction 12 (G) inhibit 5-LOX product synthesis in activated PMNL as analyzed by RP-UV‒HPLC. Results are given as means ± SEM, percentage of vehicle control, n = 3. Data were log-transformed for statistical analysis. ∗∗∗P < 0.001, ∗∗P < 0.01, ∗P < 0.05, compared to vehicle control; repeated measures one-way ANOVA (E, G) or ordinary one-way ANOVA (F) plus Tukey post hoc tests.
Aiming at the identification of the active principle(s) responsible for this favorable pharmacological effect, we prepared an ethanolic extract from the D. cambodiana red wood material and fractionated it by liquid–liquid extraction with dichloromethane, ethyl acetate, and n-butanol to afford fractions of different polarity. Biological testing confirmed the potent bioactivity of the ethanolic extract on 5-LOX product formation in PMNL. Evaluation of the subfractions revealed strong 5-LOX-inhibitory activity of the dichloromethane and the ethyl acetate subfraction, while the n-butanol fraction turned out to be inactive (Fig. 1E). Therefore, the ethyl acetate subfraction, which showed a strong 5-LOX-inhibitory potential, was selected for further bioactivity-guided fractionation. Separation by silica gel column chromatography yielded 20 subfractions, which were analyzed for their 5-LOX-inhibitory activity in PMNL. At 3 μg/mL, subfractions 5–18 showed a statistically significant 5-LOX inhibition, with subfractions 11 and 12 being most active (Fig. 1F and G). By using different chromatographic techniques, 18 (poly)phenolic compounds were isolated from the bioactive subfractions (Table 1, Fig. 1F), among them three new natural products (3–5) and another three (9, 10, 16) that have not yet been reported in Dracaena species (Supporting Information Section S5). Details on the elucidation of the chemical structures and the obtained physical and spectral data of the new compounds 3–5 are given in Sections S2–4. Due to the observed small optical rotation values of all chiral compounds (1–11), with the exception of compound 10, we assumed the presence of racemic/scalemic mixtures of the respective compounds. This assumption could be verified by chiral HPLC analysis, which revealed for all chiral compounds, except for 10, at least two peaks (Section S1).
Table 1.
Inhibitory activity of natural products isolated from D. cambodiana against 5-LOX.
| Compd. | Activated PMNL | Human recombinant 5-LOX | Compd. | Activated PMNL | ||
|---|---|---|---|---|---|---|
| 1 | 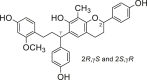 |
0.34 ± 0.03a | 0.50 ± 0.17a | 10 | 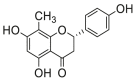 |
65.7 ± 5.7b |
| 2 | 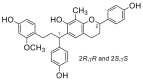 |
0.28 ± 0.06a | 0.50 ± 0.23a | 11 | 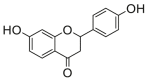 |
89.2 ± 6.7b |
| 3 | 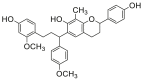 |
0.80 ± 0.14a | 0.30 ± 0.07a | 12 | 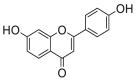 |
5.0 ± 1.4a |
| 4 | 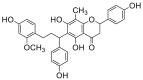 |
0.46 ± 0.12a | 0.90 ± 0.26a | 13 | 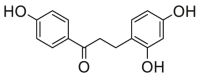 |
60.1 ± 8.2b |
| 5 | 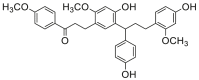 |
0.51 ± 0.12a | 1.10 ± 0.28a | 14 | 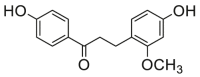 |
8.9 ± 1.6a |
| 6 | 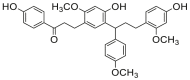 |
0.85 ± 0.15a | 0.30 ± 0.09a | 15 | 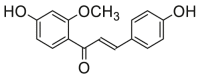 |
6.5 ± 0.4a |
| 7 | 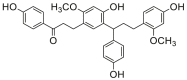 |
0.59 ± 0.16a | 0.60 ± 0.16a | 16 | 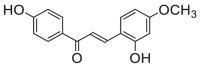 |
4.4 ± 0.7a |
| 8 | 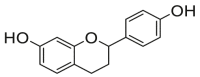 |
67.4 ± 6.2b | n.d. | 17 | 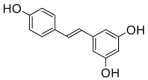 |
7.2 ± 0.9a |
| 9 | 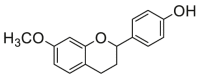 |
5.7 ± 0.9a | n.d. | 18 | 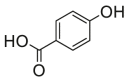 |
n.i. |
IC50 values (μmol/L).
Residual activities at 10 μmol/L (% control) are given as means ± SEM, n = 3. n.i., no inhibition; n.d., not determined.
Potent 5-LOX inhibition in PMNL was observed for two series of dimeric compounds, either comprising a 2-(4-hydroxyphenyl)chromanol (1–4) or 4,4′-propane-1,3-diyldiphenol moiety (5–7) (Table 1). Compounds within these series differed only slightly in their substitution pattern with methoxy- or hydroxy-groups. The mixtures of the 8-methylsocotrin-4′-ol isomers 1 and 2 were most active and inhibited the biosynthesis of 5-LOX products in PMNL with an IC50 of 0.3 μmol/L. Interestingly, the formation of 15-LOX products was markedly enhanced by the dimeric compounds 1–7 (Fig. 2A), whereas 12-LOX product formation was not affected or slightly reduced (Fig. 2B). The monomeric compounds 8–16 were considerably less potent 5-LOX inhibitors (Table 1).
Figure 2.

Dimeric phenolic compounds induce a shift from 5- to 15-LOX metabolites. (A, B) Human PMNL were treated with compounds 1–7 isolated from the D. cambodiana ethyl acetate extract subfraction. LOX product formation was initiated by arachidonic acid plus A23187, and metabolites were analyzed by RP-UV‒HPLC. 15-LOX (A) and 12-LOX product formation (B). Results are given as means ± SEM, percentage of vehicle control, ng/5 × 106 cells, n = 3. Data were log-transformed for statistical analysis. ∗∗∗P < 0.001, ∗P < 0.05 compared to vehicle control; repeated measures one-way ANOVA plus Dunnett's post hoc test. (C) Cell viability of monocytes upon 24 h of treatment with compounds 1–7; STS, staurosporine (1 μmol/L). (D) Scavenging of DPPH radicals by compounds 1–7; ascorbic acid (50 μmol/L). (C, D) Results are given as means ± SEM, percentage of vehicle control, n = 3. ∗∗P < 0.01, ∗P < 0.05, compared to vehicle control, compound 1–7: repeated measures one-way ANOVA plus Tukey post hoc tests; control inhibitors (STS or ascorbic acid): paired t-test. (E) Dependency of 5-LOX inhibition on the substrate (arachidonic acid, AA) concentration. (F) Reversibility of 5-LOX inhibition. Human recombinant 5-LOX was pre-incubated with vehicle or compound and then 10-fold diluted before AA was added. Numbers in brackets indicate the diluted compound concentration after pre-incubation. (E, F) Products of human recombinant 5-LOX were quantified by RP-UV-HPLC. Results are given as means ± SEM, percentage of vehicle control, n = 3; ∗∗P < 0.01, ∗P < 0.05; paired t-test.
All seven dimeric compounds 1–7 directly inhibited human recombinant 5-LOX in a cell-free assay (IC50 = 0.3–1.1 μmol/L) (Table 1). Neither cytotoxic (Fig. 2C) nor radical scavenging activities (Fig. 2D) were observed at relevant concentrations. The two compound series, represented by compound 2 and 5, inhibited 5-LOX independently of the substrate concentration (Fig. 2E) in a reversible manner (Fig. 2F). Together, we identified a group of potent 5-LOX inhibitors from a library of medicinal plant extracts, which shift the balance from 5-LOX to 15-LOX product formation in PMNL.
3.2. Stereoisomeric requirements for 5-LOX inhibition
As major constituents of the most active subfractions 11 and 12 from activity-guided fractionation (Supporting Information Fig. S2), we separated the 8-methylsocotrin-4′-ol isomers (1 and 2) by chiral chromatography and determined their absolute configuration by comparing simulated and experimental ECD spectra (Fig. 3A). The isomers 1a, 1b, 2a, and 2b have 2R,γS, 2S,γR, 2R,γR, and 2S,γS configuration, respectively, and were investigated individually for their potency to inhibit 5-LOX.
Figure 3.

5-LOX inhibition by 8-methylsocotrin-4′-ol isomers depends on their stereochemistry. (A) Comparison of predicted and experimental ECD spectra of the individual isomers of the scalemic mixtures 1 and 2 and chemical structures drawn with the assigned absolute configurations. (B) Effects of the purified isomers 1a, 1b, 2a, and 2b on cell-free 5-LOX and 5-LOX product formation in PMNL that were treated with A23187 alone or in combination with arachidonic acid. 5-LOX products were analyzed by RP-UV-HPLC. Results are given as means + SEM, percentage of vehicle control, n = 3. Data were log-transformed for statistical analysis. ∗∗∗P < 0.001, ∗∗P < 0.01; ordinary two-way ANOVA. (C, D) Docking poses of compound 2b (C) or compounds 1a and 1b (green), 2b (light green), and 2a (red) (D) at an allosteric binding site at 5-LOX, suggesting key interactions with Lys140, Glu136 and Arg101. Compound 2a is turned within the binding pocket, which might explain the weaker activity (D).
The two stereoisomers 1a and 1b of the scalemic mixture 1 (Section S1) inhibited human recombinant 5-LOX (IC50 = 0.7 μmol/L) (Fig. 3B) and suppressed 5-LOX product formation in PMNL with comparable potency [IC50 (A23187) = 1.7 μmol/L, IC50 (A23187 plus arachidonic acid) = 2.1 μmol/L] (Fig. 3B). In contrast, the scalemic mixture 2 consists of the potent stereoisomer 2b [5-LOX enzyme: IC50 = 0.5 μmol/L; PMNL: IC50 (A23187) = 0.4 μmol/L, IC50 (A23187 plus arachidonic acid) = 0.8 μmol/L] and the considerably less active 2a [5-LOX enzyme: IC50 = 1.5 μmol/L; PMNL: IC50 (A23187) = 11.3 μmol/L, IC50 (A23187 plus arachidonic acid) = 13.4 μmol/L]. The ratio of 2b to 2a in mixture 2 is 62.8/37.2 (Section S1). While the isomers 1a, 1b and 2b comparably inhibit 5-LOX in the cell-free assay, only compound 2b maintains its high inhibitory potency in PMNL (Fig. 3B).
Molecular docking studies on the allosteric binding site of 5-LOX suggest that the most active stereoisomer 2b forms hydrogen bonds with Lys140, Glu136, and Arg101 (Fig. 3C, Supporting Information Table S2). While the first two bonds are also formed by 1a and 1b, 2a is turned around in the binding site, due to steric hindrance, and forms other interactions, which could explain the loss of activity (Fig. 3D).
Due to the poor accessibility of the purified stereoisomers, we selected the racemic/scalemic mixtures 2 and 5 (as representatives of the two series of dimeric compounds) for further pharmacological evaluations. Note that lower effective concentrations would be expected for the purified isomer 2b.
3.3. Subordinate modulation of lipid mediator biosynthesis
Multiple enzymes besides LOXs cooperate in accomplishing the complex lipid mediator networks, thereby regulating initiation, maintenance, and resolution of inflammation2,20. Compounds 2 and 5 substantially inhibited mPGES-1 in a cell-free assay (IC50 ∼ 1 μmol/L) (Fig. 4A) and to some extend weakly COX-1, both as purified enzyme (IC50 > 10 μmol/L) (Fig. 4B) and in activated human platelets (IC50 > 1 μmol/L) (Fig. 4C). sEH activity was slightly suppressed at 10 μmol/L, exclusively by compound 5 (Fig. 4D), and both 2 and 5 showed a minor inhibitory effect on LTC4S by trend, again only at 10 μmol/L (Fig. 4E). Neither of the two compounds affected COX-2 activity (Fig. 4B). Together, in addition to prominent 5-LOX inhibition and the efficient increase of 15-LOX product formation in PMNL, we identified mPGES-1 and COX-1 as subordinated targets of 2 and 5.
Figure 4.

Subordinate effects of compounds 2 and 5 on lipid mediator biosynthesis. Effects of compound 2 and 5 on (A) mPGES-1 in microsomal preparations of IL-1β-treated A549 cells; (B) isolated ovine COX-1 and human recombinant COX-2; (C) COX-1-dependent 12-HHT formation in platelets; (D) human recombinant sEH; (E) human recombinant LTC4S. MK886 (10 μmol/L), indomethacin (Indo, 10 μmol/L) or AUDA (1 μmol/L) were used as control. Results are given as means ± SEM, percentage of vehicle control, n = 3, except n = 2 (E, LTC4S, 10 μmol/L). Data were log-transformed for statistical analysis. ∗∗∗P < 0.001, ∗∗P < 0.01, ∗P < 0.05, compound 2 or 5 compared to vehicle control, repeated measures one-way ANOVA plus Tukey post hoc tests; control inhibitors (MK886, Indo or AUDA) compared to vehicle control, paired t-test.
3.4. Efficient inhibition of 5-LOX in blood and in activated PMNL
To investigate the effect of compounds 2 and 5 on more complex lipid mediator profiles, we performed metabololipidomics studies on human blood, which contains PMNL and monocytes as major cells contributing to large lipid mediator networks34. When lipid mediator production was initiated by A23187, compound 2 efficiently suppressed the biosynthesis of LTs (LTB4, LTB4 isomers, 20-OH-LTB4) and other 5-LOX products (5-HETE, 5,6-diHETE, 5-HEPE) (Fig. 5A and Supporting Information Table S3). The formation of the 15-LOX product 15-HETE and to a minor extend the 12-LOX-derived 12-HETE, 12-HEPE, and 14-HDHA was moderately elevated, with strong inter-individual differences, whereas the generation of COX-derived prostanoids (PGE2, PGD2, thromboxane (TXB2)), and the release of fatty acid substrates (arachidonic acid, eicosapentaenoic acid, docosahexaenoic acid) was not influenced. Comparable effects on 5-LOX production were observed when pathogenic E. coli was used as physiological stimulus (Fig. 5B and Supporting Information Table S4). Compound 5, on the other hand, failed to suppress the formation of 5-LOX products in human blood, even at the highest concentration tested (30 μmol/L), potentially due to unfavorable plasma protein binding.
Figure 5.

Effect of compound 2 on the lipid mediator profile of activated blood. Lipid mediator profiles of human blood elicited by A23187 (A) or pathogenic E. coli (B) as determined by UPLC‒MS/MS. Heatmaps show mean percentage changes upon treatment with compound 2 or 5 relative to the vehicle control; zileuton (3 μmol/L). 20-OH-LTB4, 20-hydroxy-LTB4; HEPE, hydroxy-eicosapentaenoic acid; (di)HETE, (di)hydroxy-eicosatetraenoic acid; HDHA, hydroxy-docosahexaenoic acid; AA, arachidonic acid; EPA, eicosapentaenoic acid; DHA, docosahexaenoic acid. Line charts present data in pg/mL blood for selected lipid mediators as means + SEM, n = 3 (A), n = 4 (B). Data were log-transformed for statistical analysis. ∗∗∗P < 0.001 compared to vehicle control; repeated measures one-way ANOVA plus Tukey post hoc tests.
PMNL, which are the major 5-LOX-expressing cells in blood, predominantly produce LTs and other pro-inflammatory lipid mediators2. Compound 2 potently suppressed the biosynthesis of 5-LOX products in activated PMNL and moderately elevated the formation of the 15-LOX products 15-HETE and 15-HEPE at concentrations in the range of 0.3 and 3 μmol/L (Fig. 6). SPM biosynthesis was instead not upregulated. Compound 2 equally inhibited the formation of resolvins, lipoxins, and 5-LOX products. Comparable changes in the lipid mediator profile were evident for the 5-LOX reference inhibitor zileuton, which underlines a role for 5-LOX in SPM biosynthesis. It should be noted that PMNL dominate during the initial phase of inflammation while SPM are mainly produced by cells related to the resolution phase, i.e., macrophages2.
Figure 6.

Effect of compound 2 on the lipid mediator profile of A23187-activated PMNL. Lipid mediator profiles were analyzed by UPLC‒MS/MS. The heatmap shows mean percentage changes upon treatment with compound 2 relative to the vehicle control; zileuton (3 μmol/L). (di)HEPE, (di)hydroxy-eicosapentaenoic acid; 20-OH-LTB4, 20-hydroxy-LTB4; (di)HETE, (di)hydroxy-eicosatetraenoic acid; HDHA, hydroxy-docosahexaenoic acid; LX, lipoxin; AA, arachidonic acid; EPA, eicosapentaenoic acid; DHA, docosahexaenoic acid. Line charts present data in pg/5 × 106 cells for selected lipid mediators as means + SEM, n = 3. Data were log-transformed for statistical analysis. ∗∗∗P < 0.001, ∗∗P < 0.01, ∗P < 0.05 compared to vehicle control; repeated measures one-way ANOVA plus Tukey post hoc tests.
3.5. Induction of SPM biosynthesis in M2 macrophages
Macrophages with M2-like phenotype strongly express 15-LOX-1 and are the major innate immune cells responsible for SPM production at sites of inflammation during resolution21,22,35. The M1 subtype, on the other hand, excels in generating pro-inflammatory lipid mediators, including PGs and LTs, and is associated with acute and unresolved inflammation21,22,34. To obtain M1 and M2 subsets, we differentiated human primary monocytes into macrophages and subsequently induced their polarization. By monitoring the expression of typical M1 (CD54 and CD80) and M2 surface markers (CD163 and CD206), we could exclude that compound 2 affects macrophage polarization (Supporting Information Fig. S3). We then investigated the two subsets for the effect of compound 2 on the lipid mediator network and confirmed that the formation of LTs and other 5-LOX products is strongly attenuated, regardless of whether cells were stimulated with E. coli (Fig. 7 and Supporting Information Table S5) or S. aureus (Supporting Information Table S6). Most remarkably, M2-like macrophages produced substantial amounts of SPM, including resolvins (RvE3, RvD5), protectins (PD1, AT-PD1, PDX), and maresins (MaR1), as well as monohydroxylated precursors derived from 12- and 15-LOX, when treated with compound 2. In M1 macrophages, compound 2 moderately increased the biosynthesis of 15-LOX products (i.e., 15-HETE, 15-HEPE, 17-HDHA) without substantially influencing the minute SPM generation (Fig. 7 and Table S5). Neither protein expression of 15-LOX-1, 15-LOX-2 nor 5-LOX were affected by compound 2 in M2 macrophages within 24 h (Supporting Information Fig. S4). Together, compound 2 efficiently suppresses pro-inflammatory LT biosynthesis throughout innate immune cells and selectively stimulates M2 macrophages to generate SPM.
Figure 7.

Modulation of lipid mediator profiles in macrophages by compound 2. Lipid mediator profiles of human macrophages of the M1 and M2 phenotype stimulated with pathogenic E. coli as determined by UPLC‒MS/MS. (A) Heatmaps show mean percentage changes upon treatment with compound 2 relative to the vehicle control. HEPE, hydroxy-eicosapentaenoic acid; (di)HETE, (di)hydroxy-eicosatetraenoic acid; HDHA, hydroxy-docosahexaenoic acid; AA, arachidonic acid; EPA, eicosapentaenoic acid; DHA, docosahexaenoic acid. Line charts present data in pg/2 × 106 cells for selected lipid mediators as single data with values from the same independent experiment connected by lines, n = 3. Data were log-transformed for statistical analysis. ∗∗∗P < 0.001, ∗∗P < 0.01, ∗P < 0.05 compared to vehicle control; repeated measures one-way ANOVA plus Tukey post hoc tests. (B) Quantitative illustration of the lipid mediator network in M2 macrophages treated with compound 2 (3 μmol/L) and stimulated with pathogenic E. coli as compared to vehicle control. The node size represents the mean concentration in pg and the color intensity denotes the fold increase for each lipid mediator; n = 3. HpEPE, hydroperoxy-eicosapentaenoic acid; HpETE, hydroperoxy-eicosatetraenoic acid; HpDHA, hydroperoxy-docosahexaenoic acid.
3.6. Lipid mediator class switch to relieve inflammation in murine peritonitis
Whether the lipid mediator class switch from LTs to SPM, which we observed in vitro, also occurs at inflammatory sites in vivo, was investigated for zymosan-induced mouse peritonitis—a self-resolving model of acute inflammation that is initiated by LTs (Fig. 8A)36. Mice were sacrificed at 2 h after zymosan administration to allow the simultaneous analysis of LTs and SPM. Intraperitoneal administration of compound 2 prior to zymosan effectively reduced 5-LOX product levels, including LTs, in peritoneal exudates but neither substantially altered the release of PGs nor of polyunsaturated fatty acid substrates (Fig. 8B). Resolvin (RvE3 and RvD5) concentrations decreased, though this 2- to 3-fold drop was overcompensated by the strong increase of 12-LOX- and 15-LOX-derived SPM precursors and the 2.5- to 15-fold upregulation of protectins (PD1 and PDX) and MaR1 (Fig. 8B). MK886, an inhibitor of 5-LOX-activating protein (FLAP), had a similar but less prominent effect. The vehicle control (0.5 mL, 2% DMSO in saline) did not substantially affect the inflammatory reaction in the peritoneal cavity within 30 min to 4 h after zymosan injection, as suggested from the barely altered cell infiltration, myeloperoxidase activity (as marker for infiltrated neutrophils), and peritoneal LTC4 levels (Supporting Information Fig. S5). Together, compound 2 shifts the lipid mediator profile in experimental peritonitis from a pro-inflammatory to a pro-resolving signature, thereby attenuating inflammation, as indicated by the drop of immune cell infiltration into the peritoneal cavity (Fig. 8C).
Figure 8.

Compound 2 induces a lipid mediator class switch and limits peritoneal inflammation. Mice received vehicle (DMSO), compound 2 (10 mg/kg) or MK886 (3 mg/kg) by i.p. administration 30 min prior zymosan and were euthanized 120 min after zymosan injection. (A) Time-scale for zymosan-induced murine peritonitis. (B) Lipid mediator profiles of the exudates were analyzed by UPLC‒MS/MS and their concentration is given in pg/mL exudate as means ± SEM, n = 6 mice. Data were log-transformed for statistical analysis. ∗∗∗P < 0.001, ∗∗P < 0.01, ∗P < 0.05 compared to vehicle control; ordinary one-way ANOVA plus Dunnett's post hoc tests. The heatmap shows percentage changes upon administration of compound 2 relative to the vehicle control. HEPE, hydroxy-eicosapentaenoic acid; (di)HETE, (di)hydroxy-eicosatetraenoic acid; HDHA, hydroxy-docosahexaenoic acid; AA, arachidonic acid; EPA, eicosapentaenoic acid; DHA, docosahexaenoic acid. (C) Cell infiltration into the peritoneal cavity. Data are shown as mean plus single data points, n = 6 mice. ∗∗P < 0.01, ∗P < 0.05 compared to vehicle control; ordinary one-way ANOVA plus Dunnett's post hoc tests.
4. Discussion
Chronic inflammation and inflammation-related diseases are accomplished by pro-inflammatory signaling, which essentially orchestrates the acute phase of inflammation but becomes pathological when being persistent6. Current pharmacological strategies to combat inflammatory disorders, i.e., NSAIDs, focus on the pro-inflammatory branch of inflammation, but are confronted with major challenges, including i) lack of selectivity and efficacy, ii) undesired gastrointestinal, renal, and cardiovascular safety issues, and iii) impaired resolution10,11,13,14. By identifying a unique natural product that induces the lipid mediator class switch from pro-inflammatory LTs to SPM, our study gives access to a novel anti-inflammatory strategy that, in addition to suppressing LT-driven inflammation, may actively promote resolution and the return to homeostasis.
We took advantage of a comprehensive library of 94 Vietnamese medical plant extracts (31 different plants), from which we identified two structural different series of dimeric phenolic compounds that i) are present in D. cambodiana, ii) potently inhibit 5-LOX, and iii) trigger 15-LOX product formation. D. cambodiana is an important source for the preparation of ‘Dragon's Blood’, a dark red plant resin, which is used in many cultures as antimicrobial, antiviral, antitumor, and anti-inflammatory remedy37,38. The promising anti-inflammatory and analgesic activities of ‘Dragon's Blood’ were confirmed in pre-clinical and clinical studies39, 40, 41, 42, 43, 44, 45, 46. Li and co-workers showed reduction of chronic inflammatory and neuropathic pain in rats, which they ascribed to an interference with Ca2+ signaling, COX-2 expression, and substance P release46 by components of ‘Dragon's Blood’ that relieve central pain39,40,46. The impact on lipid mediators was not evaluated, despite the central role of PGs and related mediators in pain sensitization47. ‘Dragon's Blood’ further exhibits protective effects against radiation-induced oxidative stress, brain injury and myelosuppression in rodents41, 42, 43, 44, and as a cream, improved wound healing in a randomized, double-blind and placebo-controlled trial on 60 patients with removed skin tags45. We speculate that the bioflavonoids identified in this study might contribute to these activities by increasing the biosynthesis of SPM that actively terminate neutrophil transmigration, suppress oxidative stress, stimulate phagocytosis and promote tissue regeneration4,48,49 including dermal tissue repair50, 51, 52.
For the natural products 1, 2, and 6, which are representatives of the two identified series of dimeric phenols, anti-Helicobacter pylori, anti-thrombin, and anti-diabetic activities have been reported in vitro53,54. However, their anti-inflammatory potential remained elusive. Our findings suggest that especially the biflavonoid isomer 2b contributes to the anti-inflammatory activity of D. cambodiana by suppressing the biosynthesis of pro-inflammatory 5-LOX-derived LTs and causing a lipid mediator class switch towards SPM that promote resolution. Flavonoids are excellent scaffolds for 5-LOX inhibition55,56, but neither were adducts of them reported (as realized in the two biflavonoid series 1–7 of this study) nor effects on SPM biosynthesis addressed. Strong differences between the two biflavonoid series seem to exist for plasma protein binding. While the IC50 value of compound 2 for 5-LOX inhibition was only slightly shifted to higher values in human blood as compared to PMNL, compound 5, which is comparably effective to 2 in PMNL, failed to inhibit 5-LOX. Our finding is in agreement with a previous study, showing that compound 6, which differs from 5 only in its substitution pattern, is prone to human serum albumin binding57.
5-LOX inhibitors can target the active site iron (redox-active compounds and iron chelators) or compete with arachidonic acid or fatty acid peroxides for binding to 5-LOX (non-redox type inhibitors)9,58, 59, 60. Higher efficacy under clinical settings is expected for allosteric inhibitors that interact with the ATP or membrane binding site61. Recently, we identified a series of allosteric 5-LOX inhibitors26 and proposed that other reported inhibitors actually bind to this site. We confirmed this theory by co-crystalizing 5-LOX with the pentacyclic triterpene acid AKBA25. Since compound 2 inhibits 5-LOX like AKBA in a substrate concentration-independent manner and exhibits poor radical scavenging activity, we explored the structure–activity relationship at this allosteric binding site. The scoring function did not directly correlate with the 5-LOX-inhibitory activity of compounds 1–18, but the analysis of the docking simulation suggests three key interaction partners on the protein side, Lys140, Glu134, and Glu136, which form hydrogen bonds to those compounds that display IC50 values in the low or even submicromolar range (Table S2).
While strong 5-LOX expression in PMNL and the pro-inflammatory macrophage subtype M1 enables efficient LT biosynthesis22,62,63, macrophages of the M2 phenotype instead express substantial 15-LOX-1, generate high amounts of SPM4,21,22, and promote the resolution phase of inflammation as well as tissue repair64. Compound 2 inhibits LT biosynthesis throughout the cellular systems investigated in this study, i.e., PMNL, the macrophage subtypes M1 and M2, and blood, and slightly enhanced the formation of 15-LOX products (e.g., 15-HETE, 15-HEPE) in most settings. However, a strong increase of both 12- and 15-LOX products was mainly evident for M2 macrophages. Accordingly, the formation of distinct SPM and their precursors was exclusively elevated in this macrophage subtype.
Substrate redirection of polyunsaturated fatty acids might partially explain the moderately increased biosynthesis of 15-LOX products throughout innate immune cells and blood. In support of this hypothesis, compound 2 also increased the production of PGE2 and other prostanoids in vitro and in vivo. However, this effect is weak, and we hypothesize that the shunting of arachidonic acid towards prostanoid biosynthesis is buffered by the inhibition of the low-affinity targets COX-1 and mPGES-1. Compound 2 (COX-1: IC50 = 6.8 μmol/L; mPGES-1: IC50 = 1.6 μmol/L) inhibits COX-1 less potent than indomethacin (IC50 = 0.02 μmol/L)65 but is comparably efficient in inhibiting mPGES-1 as the control inhibitor MK886 (IC50 = 1.6 μmol/L)66. Accordingly, PGE2 formation slightly increased at low concentrations (≤3 μmol/L) of compound 2 in PMNL and again decreased at effective concentrations to inhibit COX-1 and mPGES-1 (10 μmol/L). Depending on the spatial and temporal distribution, PGE2 and other prostanoids display pro-inflammatory or anti-inflammatory activities during the course of inflammation3. Helminth larval products have been proposed to exploit this bias by lowering LT and enhancing PG production67. However, the balance between pro- and anti-inflammatory PG activities is sensitive, and we therefore consider the switch from pro-inflammatory LT to pro-resolving SPM as more robust and safer for pharmacotherapy.
Pharmacological strategies that trigger a lipid mediator class switch from inflammation to resolution are still rare. Endogenous metabolites such as PGE268 and oxidized phosphatidylcholine69 inhibit LT formation while enhancing the biosynthesis of lipoxins, a class of 5-LOX-derived SPM70, though they do qualify for therapeutic application. The allosteric 5-LOX inhibitor AKBA (IC50 = 3 μmol/L71) increases the formation of 12-LOX (270%, at 10 μmol/L) and less 15-LOX metabolites (210%, at 10 μmol/L) in human neutrophils25 with unknown consequences on SPM production. Endogenous long-chain vitamin E metabolites excel in 5-LOX inhibition (IC50 = 0.035–0.075 μmol/L) but the increase in SPM is mainly restricted to systemic RvE3 levels during the resolution of murine peritonitis26. Other small molecules that trigger SPM formation in human neutrophils or macrophages include benzenesulfonamide-derivatives (110%–140% at 10 μmol/L)72, ginkgolic acid (110%–240% at 10 μmol/L)73, benzoxanthene lignans (130%–170% at 10 μmol/L)74, and oxymetazoline (130% at 400 μmol/L)75. While oxymetazoline failed to inhibit 5-LOX product formation (at 1000 μmol/L), the other compounds inhibited 5-LOX with IC50 values of 1.2–2.3 μmol/L, being considerably less potent than compound 2 (IC50 = 0.3 μmol/L), except for ginkgolic acid (IC50 = 0.2 μmol/L). Evidence for a lipid mediator class switch in vivo was limited to the benzoxanthene ligans 1 and 2, which were less effective in raising SPM (precursor) levels in inflamed peritoneal exudates than compound 2. Additional mechanisms besides substrate redirection are needed to explain the extraordinarily strong increase in 12-LOX and 15-LOX metabolites as well as SPM in activated M2 subsets both in vitro and in the inflamed peritoneal cavity in vivo. On the one hand, binding of AKBA to the allosteric site that is also addressed by compound 2 changes the regioselectivity of 5-LOX from C5- to C12-hydroperoxidation of arachidonic acid25. It is therefore tempting to speculate that compound 2 exploits a similar mechanism to reprogram the 5-LOX enzyme, thereby triggering the generation of 15-LOX products in innate immune cells. On the other hand, compound 2 might indirectly act on signaling pathways that are specific for M2 macrophages. Along these lines, we have recently shown that 5-LOX inhibition by zileuton neither enhanced the formation of SPM (precursors) in M2 macrophages76 nor in mouse peritonitis26. FLAP inhibition by MK886, however, manipulated the lipid mediator profile similar to compound 2 (Fig. 8B), although upregulation of SPM (precursor) was less pronounced. This finding is surprising because substantial FLAP inhibition can be excluded for compound 2, which suppressed cellular 5-LOX product formation in presence of excess substrate, thereby circumventing the need for FLAP in LT biosynthesis77. How macrophages switch between LT and SPM biosynthesis was recently ascribed to HMGB1-C1q complexes that crosslink receptor for advanced glycation end product (RAGE) and leukocyte-associated immunoglobulin-like receptor (LAIR)-178. Whether compound 2 targets this pathway besides direct inhibition of 5-LOX needs further investigation. However, such a mechanism would explain why SPM upregulation is limited to macrophages, while 5-LOX inhibition occurs also in other immune cells.
The compound 2-induced switch from LT to SPM biosynthesis is not equally efficient for all SPM. In zymosan-induced mouse peritonitis, which is driven by infiltrated PMNL and peritoneal macrophages36, compound 2 lowered LT and elevated protectin (PD1, PDX) and MaR1 levels, whereas resolvin (RvD5, RvD3) concentrations even decreased; an effect that was overcompensated by the strong increase of PDX. It should be noted that mouse 12/15-LOX differs in regiospecificity to the human orthologues 15-LOX-1 and 15-LOX-279. In line with these findings, 5-LOX has been reported to participate in resolvin biosynthesis80. On the other hand, compound 2 induces the production of resolvins along with protectins and maresins in human M2 macrophages in vitro. This apparent discrepancy likely depends on our focus on the early phase of peritonitis, for which PMNL are the dominating immune cells in the peritoneal cavity36. Compound 2 decreases resolvin biosynthesis in PMNL, and although they do not substantially contribute to SPM formation during resolution, they seem to be the major cell type that generates the low resolvin levels at 2 h after administration of the inflammatory stimulus. When PMNL are replaced by infiltrated macrophages as major innate immune cells in the peritoneal cavity during the late acute and resolution phase of inflammation4, we rather expect compound 2 to increase resolvin levels along with other SPM.
5. Conclusions
Promoting signaling pathways that resolve inflammation is considered as promising complementary or even superior therapeutic approach to treat chronic inflammatory diseases over simply suppressing pro-inflammatory signals19. Starting from the investigation of an extract library of Vietnamese medicinal plants, bioactivity-guided extract fractionation led to the identification of the potent biflavonoid 2 as promising drug candidate that relieves inflammation by inducing the lipid mediator class switch from inflammation to resolution in anti-inflammatory macrophage subsets. Our study provides a molecular basis for the anti-inflammatory, wound-healing, and protective activities of D. cambodiana and gives access to a unique pharmacological tool that might open the door to a more efficient and safe strategy to fight chronic inflammation.
Acknowledgments
The authors thank Katrin Fischer, Vo Van Leo, Stefanie Liening, Monika Listing, Nguyen Minh Luan, Heidi Traber, Tran Thuy Van, and Maria Völkel for technical assistance in performing experimental work as well as Dr. Ma Chi Thanh for supporting the plant material collection. This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) [project number 316213987, SFB 1278 PolyTarget (projects A04, C02), SFB 1127/2 ChemBioSys-239748522 (project A04)], Europäischer Fonds für regionale Entwicklung (EFRE, “NanoCARE4skin” 2019FGR0095), the Austrian Science Fund (FWF) [grant number S107 “Drugs from Nature Targeting Inflammation”], the Tiroler Zukunftsstiftung [grant number AP740021, Austria]. Tran Thi Van Anh was supported by the OEAD [Ernst Mach Scholarship, Austria]. Veronika Temml is funded by the FWF [Hertha Firnberg fellowship, grant number T 942-B30, Austria] and thanks Inte:Ligand for the academic software licenses.
Author contributions
Tran Thi Van Anh, Alilou Mostafa, Simona Pace and Antonietta Rossi: investigation, methodology, writing - review & editing. Zhigang Rao: investigation, visualization, writing - original draft, writing - review & editing. Christian Kretzer, Carsten Giesel, Paul M. Jordan, Rossella Bilancia: investigation, writing - review & editing. Veronika Temml: formal analysis, writing - review & editing. Stefan Schwaiger: investigation, methodology, conceptualization, data curation, writing - review & editing, supervision. Birgit Waltenberger: conceptualization, writing - review & editing. Hermann Stuppner: conceptualization, resources, data curation, writing - review & editing, project administration, funding acquisition. Christina Weinigel, Silke Rummler, Tran Hung: resources. Oliver Werz: conceptualization, writing - review & editing, supervision, project administration, funding acquisition. Andreas Koeberle: project administration, conceptualization, data curation, writing - original draft, writing - review & editing, supervision, funding acquisition.
Conflicts of interest
The authors declare no conflicts of interest. The above mentioned funding sources were neither involved in study design, data collection, analysis, and interpretation nor in writing and submission of the manuscript.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting information to this article can be found online at https://doi.org/10.1016/j.apsb.2021.04.011.
Contributor Information
Oliver Werz, Email: oliver.werz@uni-jena.de.
Andreas Koeberle, Email: andreas.koeberle@uibk.ac.at.
Appendix A. Supporting information
The following is the Supporting information to this article:
References
- 1.Pober J.S., Sessa W.C. Inflammation and the blood microvascular system. Cold Spring Harb Perspect Biol. 2014;7:a016345. doi: 10.1101/cshperspect.a016345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bennett M., Gilroy D. Lipid mediators in inflammation. In: Gordon S., editor. Myeloid cells in health and disease. ASM Press; Washington DC: 2017. pp. 343–366. [Google Scholar]
- 3.Funk C.D. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 4.Serhan C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510:92–101. doi: 10.1038/nature13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Serhan C.N., Levy B.D. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J Clin Invest. 2018;128:2657–2669. doi: 10.1172/JCI97943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 7.Haeggström J.Z. Leukotriene biosynthetic enzymes as therapeutic targets. J Clin Invest. 2018;128:2680–2690. doi: 10.1172/JCI97945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turini M.E., DuBois R.N. Cyclooxygenase-2: a therapeutic target. Annu Rev Med. 2002;53:35–57. doi: 10.1146/annurev.med.53.082901.103952. [DOI] [PubMed] [Google Scholar]
- 9.Sinha S., Doble M., Manju S.L. 5-Lipoxygenase as a drug target: a review on trends in inhibitors structural design, SAR and mechanism based approach. Bioorg Med Chem. 2019;27:3745–3759. doi: 10.1016/j.bmc.2019.06.040. [DOI] [PubMed] [Google Scholar]
- 10.Castellsague J., Riera-Guardia N., Calingaert B., Varas-Lorenzo C., Fourrier-Reglat A., Nicotra F. Individual NSAIDs and upper gastrointestinal complications. Drug Saf. 2012;35:1127–1146. doi: 10.1007/BF03261999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grosser T., Yu Y., FitzGerald G.A. Emotion recollected in tranquility: lessons learned from the COX-2 saga. Annu Rev Med. 2010;61:17–33. doi: 10.1146/annurev-med-011209-153129. [DOI] [PubMed] [Google Scholar]
- 12.Koeberle A., Werz O. Inhibitors of the microsomal prostaglandin E2 synthase-1 as alternative to non steroidal anti-inflammatory drugs (NSAIDs)a critical review. Curr Med Chem. 2009;16:4274–4296. doi: 10.2174/092986709789578178. [DOI] [PubMed] [Google Scholar]
- 13.Su W.H., Cheng M.H., Lee W.L., Tsou T.S., Chang W.H., Chen C.S. Nonsteroidal anti-inflammatory drugs for wounds: pain relief or excessive scar formation?. Mediat Inflamm. 2010;2010:413238. doi: 10.1155/2010/413238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koeberle A., Laufer S.A., Werz O. Design and development of microsomal prostaglandin E2 synthase-1 inhibitors: challenges and future directions. J Med Chem. 2016;59:5970–5986. doi: 10.1021/acs.jmedchem.5b01750. [DOI] [PubMed] [Google Scholar]
- 15.Samuelsson B., Morgenstern R., Jakobsson P.J. Membrane prostaglandin E synthase-1: a novel therapeutic target. Pharmacol Rev. 2007;59:207–224. doi: 10.1124/pr.59.3.1. [DOI] [PubMed] [Google Scholar]
- 16.Ramsay R.R., Popovic-Nikolic M.R., Nikolic K., Uliassi E., Bolognesi M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin Transl Med. 2018;7:3. doi: 10.1186/s40169-017-0181-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koeberle A., Werz O. Multi-target approach for natural products in inflammation. Drug Discov Today. 2014;19:1871–1882. doi: 10.1016/j.drudis.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 18.Koeberle A., Werz O. Natural products as inhibitors of prostaglandin E2 and pro-inflammatory 5-lipoxygenase-derived lipid mediator biosynthesis. Biotechnol Adv. 2018;36:1709–1723. doi: 10.1016/j.biotechadv.2018.02.010. [DOI] [PubMed] [Google Scholar]
- 19.Fullerton J.N., Gilroy D.W. Resolution of inflammation: a new therapeutic frontier. Nat Rev Drug Discov. 2016;15:551–567. doi: 10.1038/nrd.2016.39. [DOI] [PubMed] [Google Scholar]
- 20.Serhan C.N., Chiang N., Dalli J., Levy B.D. Lipid mediators in the resolution of inflammation. Cold Spring Harb Perspect Biol. 2014;7:a016311. doi: 10.1101/cshperspect.a016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rao Z., Pace S., Jordan P.M., Bilancia R., Troisi F., Borner F. Vacuolar (H+)-ATPase critically regulates specialized proresolving mediator pathways in human M2-like monocyte-derived macrophages and has a crucial role in resolution of inflammation. J Immunol. 2019;203:1031–1043. doi: 10.4049/jimmunol.1900236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Werz O., Gerstmeier J., Libreros S., De la Rosa X., Werner M., Norris P.C. Human macrophages differentially produce specific resolvin or leukotriene signals that depend on bacterial pathogenicity. Nat Commun. 2018;9:59. doi: 10.1038/s41467-017-02538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Newman D.J., Cragg G.M. Natural products as sources of new drugs from 1981 to 2014. J Nat Prod. 2016;79:629–661. doi: 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- 24.Harvey A.L., Edrada-Ebel R., Quinn R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat Rev Drug Discov. 2015;14:111–129. doi: 10.1038/nrd4510. [DOI] [PubMed] [Google Scholar]
- 25.Gilbert N.C., Gerstmeier J., Schexnaydre E.E., Börner F., Garscha U., Neau D.B. Structural and mechanistic insights into 5-lipoxygenase inhibition by natural products. Nat Chem Biol. 2020;16:783–790. doi: 10.1038/s41589-020-0544-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pein H., Ville A., Pace S., Temml V., Garscha U., Raasch M. Endogenous metabolites of vitamin E limit inflammation by targeting 5-lipoxygenase. Nat Commun. 2018;9:3834. doi: 10.1038/s41467-018-06158-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koeberle A., Munoz E., Appendino G.B., Minassi A., Pace S., Rossi A. SAR studies on curcumin's pro-inflammatory targets: discovery of prenylated pyrazolocurcuminoids as potent and selective novel inhibitors of 5-lipoxygenase. J Med Chem. 2014;57:5638–5648. doi: 10.1021/jm500308c. [DOI] [PubMed] [Google Scholar]
- 28.Garscha U., Romp E., Pace S., Rossi A., Temml V., Schuster D. Pharmacological profile and efficiency in vivo of diflapolin, the first dual inhibitor of 5-lipoxygenase-activating protein and soluble epoxide hydrolase. Sci Rep. 2017;7:9398. doi: 10.1038/s41598-017-09795-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koeberle A., Siemoneit U., Buhring U., Northoff H., Laufer S., Albrecht W. Licofelone suppresses prostaglandin E2 formation by interference with the inducible microsomal prostaglandin E2 synthase-1. J Pharmacol Exp Therapeut. 2008;326:975–982. doi: 10.1124/jpet.108.139444. [DOI] [PubMed] [Google Scholar]
- 30.Hanke T., Dehm F., Liening S., Popella S.D., Maczewsky J., Pillong M. Aminothiazole-featured pirinixic acid derivatives as dual 5-lipoxygenase and microsomal prostaglandin E2 synthase-1 inhibitors with improved potency and efficiency in vivo. J Med Chem. 2013;56:9031–9044. doi: 10.1021/jm401557w. [DOI] [PubMed] [Google Scholar]
- 31.Rossi A., Pergola C., Pace S., Rådmark O., Werz O., Sautebin L. In vivo sex differences in leukotriene biosynthesis in zymosan-induced peritonitis. Pharmacol Res. 2014;87:1–7. doi: 10.1016/j.phrs.2014.05.011. [DOI] [PubMed] [Google Scholar]
- 32.Waltenberger B., Atanasov A.G., Heiss E.H., Bernhard D., Rollinger J.M., Breuss J.M. Drugs from nature targeting inflammation (DNTI): a successful Austrian interdisciplinary network project. Monatsh Chem. 2016;147:479–491. doi: 10.1007/s00706-015-1653-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tran T.V.A., Malainer C., Schwaiger S., Hung T., Atanasov A.G., Heiss E.H. Screening of Vietnamese medicinal plants for NF-κB signaling inhibitors: assessing the activity of flavonoids from the stem bark of Oroxylum indicum. J Ethnopharmacol. 2015;159:36–42. doi: 10.1016/j.jep.2014.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dalli J., Serhan C.N. Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood. 2012;120:60–72. doi: 10.1182/blood-2012-04-423525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramon S., Dalli J., Sanger J.M., Winkler J.W., Aursnes M., Tungen J.E. The Protectin PCTR1 is produced by human M2 macrophages and enhances resolution of infectious inflammation. Am J Pathol. 2016;186:962–973. doi: 10.1016/j.ajpath.2015.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cash J.L., White G.E., Greaves D.R. Chapter 17. Zymosan-induced peritonitis as a simple experimental system for the study of inflammation. Methods Enzymol. 2009;461:379–396. doi: 10.1016/S0076-6879(09)05417-2. [DOI] [PubMed] [Google Scholar]
- 37.Gupta D., Bleakley B., Gupta R.K. Dragon's blood: botany, chemistry and therapeutic uses. J Ethnopharmacol. 2008;115:361–380. doi: 10.1016/j.jep.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 38.Sun J., Liu J.N., Fan B., Chen X.N., Pang D.R., Zheng J. Phenolic constituents, pharmacological activities, quality control, and metabolism of Dracaena species: a review. J Ethnopharmacol. 2019;244:112138. doi: 10.1016/j.jep.2019.112138. [DOI] [PubMed] [Google Scholar]
- 39.Wang S.T., Chen S., Guo M., Liu X.M. Inhibitory effect of cochinchinenin B on capsaicin-activated responses in rat dorsal root ganglion neurons. Brain Res. 2008;1201:34–40. doi: 10.1016/j.brainres.2007.12.068. [DOI] [PubMed] [Google Scholar]
- 40.Guo M., Chen S., Liu X. Material basis for inhibition of Dragon's Blood on evoked discharges of wide dynamic range neurons in spinal dorsal horn of rats. Sci China C Life Sci. 2008;51:1025–1038. doi: 10.1007/s11427-008-0133-6. [DOI] [PubMed] [Google Scholar]
- 41.Ran Y., Wang R., Gao Q., Jia Q., Hasan M., Awan M.U. Dragon's blood and its extracts attenuate radiation-induced oxidative stress in mice. J Radiat Res. 2014;55:699–706. doi: 10.1093/jrr/rru013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xin N., Li Y.J., Li X., Wang X., Li Y., Zhang X. Dragon's blood may have radioprotective effects in radiation-induced rat brain injury. Radiat Res. 2012;178:75–85. doi: 10.1667/rr2739.1. [DOI] [PubMed] [Google Scholar]
- 43.Ran Y., Wang R., Hasan M., Jia Q., Tang B., Shan S. Radioprotective effects of dragon's blood and its extracts on radiation-induced myelosuppressive mice. J Ethnopharmacol. 2014;154:624–634. doi: 10.1016/j.jep.2014.04.036. [DOI] [PubMed] [Google Scholar]
- 44.Ran Y., Wang R., Lin F., Hasan M., Jia Q., Tang B. Radioprotective effects of Dragon's blood and its extract against gamma irradiation in mouse bone marrow cells. Phys Med. 2014;30:427–431. doi: 10.1016/j.ejmp.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 45.Namjoyan F., Kiashi F., Moosavi Z.B., Saffari F., Makhmalzadeh B.S. Efficacy of Dragon's blood cream on wound healing: a randomized, double-blind, placebo-controlled clinical trial. J Tradit Complement Med. 2015;6:37–40. doi: 10.1016/j.jtcme.2014.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Y.S., Wang J.X., Jia M.M., Liu M., Li X.J., Tang H.B. Dragon's blood inhibits chronic inflammatory and neuropathic pain responses by blocking the synthesis and release of substance P in rats. J Pharmacol Sci. 2012;118:43–54. doi: 10.1254/jphs.11160fp. [DOI] [PubMed] [Google Scholar]
- 47.Park K.A., Vasko M.R. Lipid mediators of sensitivity in sensory neurons. Trends Pharmacol Sci. 2005;26:571–577. doi: 10.1016/j.tips.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 48.Chiang N., Fredman G., Backhed F., Oh S.F., Vickery T., Schmidt B.A. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature. 2012;484:524–528. doi: 10.1038/nature11042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gobbetti T., Dalli J., Colas R.A., Federici Canova D., Aursnes M., Bonnet D. Protectin D1n-3 DPA and resolvin D5n-3 DPA are effectors of intestinal protection. Proc Natl Acad Sci U S A. 2017;114:3963–3968. doi: 10.1073/pnas.1617290114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hellmann J., Sansbury B.E., Wong B., Li X., Singh M., Nuutila K. Biosynthesis of D-series resolvins in skin provides insights into their role in tissue repair. J Invest Dermatol. 2018;138:2051–2060. doi: 10.1016/j.jid.2018.03.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Menon R., Krzyszczyk P., Berthiaume F. Pro-resolution potency of eesolvins D1, D2 and E1 on neutrophil migration and in dermal wound healing. Nano LIFE. 2017;7:1750002. doi: 10.1142/S1793984417500027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Quiros M., Feier D., Birkl D., Agarwal R., Zhou D.W., Garcia A.J. Resolvin E1 is a pro-repair molecule that promotes intestinal epithelial wound healing. Proc Natl Acad Sci U S A. 2020;117:9477–9482. doi: 10.1073/pnas.1921335117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu Y., Zhang P., Yu H., Li J., Wang M.W., Zhao W. Anti-Helicobacter pylori and thrombin inhibitory components from Chinese dragon's blood, Dracaena cochinchinensis. J Nat Prod. 2007;70:1570–1577. doi: 10.1021/np070260v. [DOI] [PubMed] [Google Scholar]
- 54.Sha Y., Shi Y., Niu B., Chen Q., Cochinchinenin C. A potential nonpolypeptide anti-diabetic drug, targets a glucagon-like peptide-1 receptor. RSC Adv. 2017;7:49015–49023. [Google Scholar]
- 55.Mahapatra D.K., Bharti S.K., Asati V. Chalcone Derivatives: anti-inflammatory potential and molecular targets perspectives. Curr Top Med Chem. 2017;17:3146–3169. doi: 10.2174/1568026617666170914160446. [DOI] [PubMed] [Google Scholar]
- 56.Werz O. Inhibition of 5-lipoxygenase product synthesis by natural compounds of plant origin. Planta Med. 2007;73:1331–1357. doi: 10.1055/s-2007-990242. [DOI] [PubMed] [Google Scholar]
- 57.Chen X., Qian K., Chen Q. Comparison between loureirin A and cochinchinenin C on the interaction with human serum albumin. Eur J Med Chem. 2015;93:492–500. doi: 10.1016/j.ejmech.2015.02.025. [DOI] [PubMed] [Google Scholar]
- 58.Werz O., Gerstmeier J., Garscha U. Novel leukotriene biosynthesis inhibitors (2012‒2016) as anti-inflammatory agents. Expert Opin Ther Pat. 2017;27:607–620. doi: 10.1080/13543776.2017.1276568. [DOI] [PubMed] [Google Scholar]
- 59.Pergola C., Werz O. 5-Lipoxygenase inhibitors: a review of recent developments and patents. Expert Opin Ther Pat. 2010;20:355–375. doi: 10.1517/13543771003602012. [DOI] [PubMed] [Google Scholar]
- 60.Zhou Y., Liu J., Zheng M., Zheng S., Jiang C., Zhou X. Structural optimization and biological evaluation of 1,5-disubstituted pyrazole-3-carboxamines as potent inhibitors of human 5-lipoxygenase. Acta Pharm Sin B. 2016;6:32–45. doi: 10.1016/j.apsb.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bruno F., Spaziano G., Liparulo A., Roviezzo F., Nabavi S.M., Sureda A. Recent advances in the search for novel 5-lipoxygenase inhibitors for the treatment of asthma. Eur J Med Chem. 2018;153:65–72. doi: 10.1016/j.ejmech.2017.10.020. [DOI] [PubMed] [Google Scholar]
- 62.Sorgi C.A., Zarini S., Martin S.A., Sanchez R.L., Scandiuzzi R.F., Gijón M.A. Dormant 5-lipoxygenase in inflammatory macrophages is triggered by exogenous arachidonic acid. Sci Rep. 2017;7:10981. doi: 10.1038/s41598-017-11496-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Peters-Golden M., Henderson W.R., Jr. Leukotrienes. N Engl J Med. 2007;357:1841–1854. doi: 10.1056/NEJMra071371. [DOI] [PubMed] [Google Scholar]
- 64.Wynn T.A., Vannella K.M. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44:450–462. doi: 10.1016/j.immuni.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Riendeau D., Percival M.D., Boyce S., Brideau C., Charleson S., Cromlish W. Biochemical and pharmacological profile of a tetrasubstituted furanone as a highly selective COX-2 inhibitor. Br J Pharmacol. 1997;121:105–117. doi: 10.1038/sj.bjp.0701076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Riendeau D., Aspiotis R., Ethier D., Gareau Y., Grimm E.L., Guay J. Inhibitors of the inducible microsomal prostaglandin E2 synthase (mPGES-1) derived from MK-886. Bioorg Med Chem Lett. 2005;15:3352–3355. doi: 10.1016/j.bmcl.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 67.de los Reyes Jiménez M., Lechner A., Alessandrini F., Bohnacker S., Schindela S., Trompette A. An anti-inflammatory eicosanoid switch mediates the suppression of type-2 inflammation by helminth larval products. Sci Transl Med. 2020;12 doi: 10.1126/scitranslmed.aay0605. [DOI] [PubMed] [Google Scholar]
- 68.Levy B.D., Clish C.B., Schmidt B., Gronert K., Serhan C.N. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol. 2001;2:612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 69.Ke Y., Zebda N., Oskolkova O., Afonyushkin T., Berdyshev E., Tian Y. Anti-inflammatory effects of OxPAPC involve endothelial cell-mediated generation of LXA4. Circ Res. 2017;121:244–257. doi: 10.1161/CIRCRESAHA.116.310308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Samuelsson B., Dahlen S., Lindgren J., Rouzer C., Serhan C. Leukotrienes and lipoxins: structures, biosynthesis, and biological effects. Science. 1987;237:1171–1176. doi: 10.1126/science.2820055. [DOI] [PubMed] [Google Scholar]
- 71.Siemoneit U., Pergola C., Jazzar B., Northoff H., Skarke C., Jauch J. On the interference of boswellic acids with 5-lipoxygenase: mechanistic studies in vitro and pharmacological relevance. Eur J Pharmacol. 2009;606:246–254. doi: 10.1016/j.ejphar.2009.01.044. [DOI] [PubMed] [Google Scholar]
- 72.Cheung S.Y., Werner M., Esposito L., Troisi F., Cantone V., Liening S. Discovery of a benzenesulfonamide-based dual inhibitor of microsomal prostaglandin E2 synthase-1 and 5-lipoxygenase that favorably modulates lipid mediator biosynthesis in inflammation. Eur J Med Chem. 2018;156:815–830. doi: 10.1016/j.ejmech.2018.07.031. [DOI] [PubMed] [Google Scholar]
- 73.Gerstmeier J., Seegers J., Witt F., Waltenberger B., Temml V., Rollinger J.M. Ginkgolic acid is a multi-target inhibitor of key enzymes in pro-inflammatory lipid mediator biosynthesis. Front Pharmacol. 2019;10:797. doi: 10.3389/fphar.2019.00797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gerstmeier J., Kretzer C., Di Micco S., Miek L., Butschek H., Cantone V. Novel benzoxanthene lignans that favorably modulate lipid mediator biosynthesis: a promising pharmacological strategy for anti-inflammatory therapy. Biochem Pharmacol. 2019;165:263–274. doi: 10.1016/j.bcp.2019.03.003. [DOI] [PubMed] [Google Scholar]
- 75.Beck-Speier I., Oswald B., Maier K.L., Karg E., Ramseger R. Oxymetazoline inhibits and resolves inflammatory reactions in human neutrophils. J Pharmacol Sci. 2009;110:276–284. doi: 10.1254/jphs.09012FP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Werner M., Jordan P.M., Romp E., Czapka A., Rao Z., Kretzer C. Targeting biosynthetic networks of the proinflammatory and proresolving lipid metabolome. Faseb J. 2019;33:6140–6153. doi: 10.1096/fj.201802509R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Radmark O., Werz O., Steinhilber D., Samuelsson B. 5-Lipoxygenase: regulation of expression and enzyme activity. Trends Biochem Sci. 2007;32:332–341. doi: 10.1016/j.tibs.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 78.Liu T., Xiang A., Peng T., Doran A.C., Tracey K.J., Barnes B.J. HMGB1–C1q complexes regulate macrophage function by switching between leukotriene and specialized proresolving mediator biosynthesis. Proc Natl Acad Sci U S A. 2019;116:23254–23263. doi: 10.1073/pnas.1907490116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mashima R., Okuyama T. The role of lipoxygenases in pathophysiology; new insights and future perspectives. Redox Biol. 2015;6:297–310. doi: 10.1016/j.redox.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Serhan C.N., Petasis N.A. Resolvins and protectins in inflammation resolution. Chem Rev. 2011;111:5922–5943. doi: 10.1021/cr100396c. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.