

Abstract
Background
Several cancer-susceptibility syndromes are reported to underlie pediatric rhabdomyosarcoma (RMS); however, to our knowledge there have been no systematic efforts to characterize the heterogeneous genetic etiologies of this often-fatal malignancy.
Methods
We performed exome-sequencing on germline DNA from 615 patients with newly diagnosed RMS consented through the Children’s Oncology Group. We compared the prevalence of cancer predisposition variants in 63 autosomal-dominant cancer predisposition genes in these patients with population controls (n = 9963). All statistical tests were 2-sided.
Results
We identified germline cancer predisposition variants in 45 RMS patients (7.3%; all FOXO1 fusion negative) across 15 autosomal dominant genes, which was statistically significantly enriched compared with controls (1.4%, P = 1.3 × 10–22). Specifically, 73.3% of the predisposition variants were found in predisposition syndrome genes previously associated with pediatric RMS risk, such as Li-Fraumeni syndrome (TP53) and neurofibromatosis type I (NF1). Notably, 5 patients had well-described oncogenic missense variants in HRAS (p.G12V and p.G12S) associated with Costello syndrome. Also, genetic etiology differed with histology, as germline variants were more frequent in embryonal vs alveolar RMS patients (10.0% vs 3.0%, P = .02). Although patients with a cancer predisposition variant tended to be younger at diagnosis (P = 9.9 × 10–4), 40.0% of germline variants were identified in those older than 3 years of age, which is in contrast to current genetic testing recommendations based on early age at diagnosis.
Conclusions
These findings demonstrate that genetic risk of RMS results from germline predisposition variants associated with a wide spectrum of cancer susceptibility syndromes. Germline genetic testing for children with RMS should be informed by RMS subtypes and not be limited to only young patients.
Rhabdomyosarcoma (RMS) is a highly malignant tumor believed to arise from developing skeletal muscle cells (myoblasts) and is the most common soft-tissue sarcoma in children and adolescents, with an overall incidence of 4.5 cases per million among those younger than 20 years of age (1). Relative to other pediatric cancers, the prognosis for many children with RMS remains poor. For example, treatment of children with intermediate-risk disease using maximal intensive therapy and application of new agents has only led to modest improvements in 5-year survival rates since the 1970s, which remain at only 43% to 67% by different estimates (2). Furthermore, for those RMS patients with metastatic disease, the 5-year survival rate is less than 30%, even with aggressive therapy (3).
The vast majority of RMS tumors have been classified by histology into embryonal RMS (ERMS), representing 70% of patients, and alveolar RMS (ARMS), representing approximately 20% (3,4). Further, nearly 80% of ARMS cases are driven by a characteristic balanced chromosomal translocation between either PAX3 or PAX7 and FOXO1 (5‐7). FOXO1 fusion-positive tumors have decreased tumor mutation burden compared with FOXO1 fusion-negative tumors (8), consistent with the driving role of FOXO1 fusions in disease pathogenicity. Also, FOXO1 fusion-negative ARMS tumors have gene expression profiles similar to those from ERMS (7,9,10). Notably, a wide range of somatic alterations have been identified in ERMS, including mutations in TP53, genes within the RAS family (NRAS, KRAS, HRAS), PIK3CA, CTNNB1, and FGFR4 (8,11‐14), as well as loss of heterozygosity at 11p15.5 (15).
Numerous reports and studies of individual genetic disease cohorts consistently highlight that children with certain genetic syndromes develop RMS more frequently than their unaffected peers (16). Cancer susceptibility syndromes that are most commonly reported among those with RMS include Li-Fraumeni (LFS) (17), neurofibromatosis type 1 (18,19), Costello (20,21), Noonan (20), and DICER1 (22). In an assessment by Zhang et al. (23) of germline mutations in cancer predisposition genes (CPGs) among 1120 pediatric cancer patients, only 43 patients in the cohort were diagnosed with RMS, of which 3 (7.0%) had a pathogenic variant in a known CPG. However, given the small sample size, it was difficult to evaluate the spectrum of cancer predisposition variants in CPGs among children with RMS. Additionally, there was no further stratification by RMS histology or fusion status. Thus, the burden of cancer susceptibility syndromes among children with RMS is still unclear. Delineating the germline risk for RMS could help to inform germline genetic testing strategies in this population. Therefore, we sought to determine the prevalence of predisposition variants in CPGs among a large, unselected cohort of children with newly diagnosed RMS. We also evaluated differences by histologic subtypes and compared the prevalence of these variants with a large population-based cohort.
Methods
Study Cohorts
We obtained germline DNA from diagnostic blood samples of 616 pediatric patients with RMS (age <25 years; 1 sample was removed due to quality, see below) from the Children’s Oncology Group (COG) Biopathology Center. This study was approved by the institutional review board of Baylor College of Medicine. RMS patients were consented as part of the COG Soft Tissue Sarcoma Biology and Banking Protocol (COG Protocol D9902). There were no selection criteria in place for inclusion in D9902; therefore, these patients represent the demographic and clinical characteristics seen in previous population-based assessments of RMS (24,25). Additionally, all risk groups (low, intermediate, and high) were included in this analysis. The histologic diagnosis of RMS and histologic subtype status were confirmed by COG central review. Demographic and clinical characteristics of these patients were provided by COG.
For the population control cohort, we used exome-sequencing data from 10 853 European American and African American individuals included in the Atherosclerosis Risk in Communities (ARIC) cohort (dbGaP: phs000280.v4.p1) and 1030 Hispanic individuals from the Viva la Familia (VIVA, dbGaP: phs000616.v2.p2) study (26).
Exome-sequencing of the RMS cases and controls was performed at the Human Genome Sequencing Center at Baylor College of Medicine (see Supplementary Methods, available online). After quality control measurements and filtering, we included 615 RMS patients, 9663 controls from ARIC (73.7% of European Americans and 26.3% of African Americans), and 300 controls from VIVA (all of whom were Hispanic) in the final analyses.
Identification of Candidate Genes and Predisposition Variants
We precurated a set of autosomal-dominant CPGs (n = 24) from cancer susceptibility syndromes that have been specifically implicated in RMS predisposition as well as a set of additional autosomal dominant CPGs (n = 39) from the report by Zhang et al. (23). We also evaluated 7 genes underlying autosomal recessive disorders that are known to be associated with sarcomas (see Supplementary Methods for gene selection criteria). We annotated all the filtered variants using ANNOVAR (v2018.04.16) (27) in candidate CPGs for further analyses.
To identify germline predisposition variants in RMS, we focused on rare variants with minor allele frequency less than 0.01 in all of the following population databases: 1000 Genome Phase 3 (28), gnomAD (v2.1.1) (29), and ExAC (v0.3) (30). We defined germline predisposition variants as either 1) known pathogenic or likely pathogenic variants [reported in ClinVar (31) with 1 or more “stars” describing the level of support of clinical significance (version 20190305)]; or 2) novel, rare, potential loss-of-function variants (splicing variants, frameshift insertions or deletions, nonsense single-nucleotide variants, or stoploss variants).
Statistical Analyses
Logistic regression models were used to test associations between the occurrence of pathogenic variants and the status of the individuals while adjusting for the following covariates: ERMS vs ARMS: sex, age at diagnosis, tumor stage, and the first 10 principle components (PCs); case vs control: sex and the first 10 PCs. We used P less than .05/3 (Bonferroni correction) as a statistical significance threshold for both cohort-level (3 groups) and gene-level (3 genes) analyses. A linear regression model was used to determine the association between age at diagnosis and the pathogenic variant status within the ERMS subtype while using sex, tumor stage, and the first 10 PCs as covariates. All statistical tests were 2-sided and were performed using the statsmodels package (32). Odds ratios were calculated as the exponential of the estimated coefficient of the tested independent variable.
Results
Study Participants
To estimate the prevalence of germline predisposition variants among pediatric RMS, we analyzed samples from a total of 615 RMS patients who were consented to the COG D9902 protocol. These patients were newly diagnosed and were not specifically selected for the existence of known germline pathogenic variants or family history of cancer or any other diagnostic characteristics. Within this cohort, the patients were mostly of European American, Hispanic, and African American populations (Table 1). A majority of the patients were diagnosed with ERMS (56.4%, n = 347; including 38 botryoid and 30 spindle cell subtypes), followed by ARMS (27.2%, n = 167) (Table 1). Notably, among the ARMS patients whose FOXO1 fusion status was known, 82.5% (66 of 80) were FOXO1 fusion-positive. Both ERMS and ARMS subtypes showed bimodal peaks regarding age at diagnosis (Supplementary Figure 1, available online). The male-to-female ratio among RMS patients was approximately 1.5 to 1, which is largely driven by ERMS (Table 1) and consistent with previous reports (25). Also, ERMS patients were younger (74.9% were <10 years of age) compared with ARMS patients (53.9% were <10 years of age; Table 1). In addition, a larger proportion of the ARMS patients were diagnosed at a later stage (stage 4) compared with ERMS patients (37.7% ARMS vs 13.8% ERMS; Table 1). In summary, this unselected childhood RMS cohort had epidemiological features (such as sex ratio, subtype composition, and age distribution) that were representative of previous population-based studies (4,24,25).
Table 1.
Epidemiological features of the RMS cohorta
| Characteristics | Embryonal RMS (n = 347) | Alveolar RMS (n = 167) | Other subtypes or not otherwise specified (n = 101) | Total (n = 615) |
|---|---|---|---|---|
| Sex, No. (%) | ||||
| Female | 124 (35.7) | 86 (51.5) | 33 (32.7) | 243 (39.5) |
| Male | 223 (64.3) | 81 (48.5) | 68 (67.3) | 372 (60.5) |
| Population, No. (%) | ||||
| European American | 235 (67.7) | 105 (62.9) | 65 (64.4) | 405 (65.9) |
| Hispanic | 45 (13.0) | 34 (20.4) | 17 (16.8) | 96 (15.6) |
| African American | 45 (13.0) | 18 (10.8) | 15 (14.9) | 78 (12.7) |
| East Asian | 17 (4.9) | 8 (4.8) | 3 (3.0) | 28 (4.6) |
| South Asian | 5 (1.4) | 2 (1.2) | 1 (1.0) | 8 (1.3) |
| Age at diagnosis, No. (%), y | ||||
| 0-5 | 171 (49.3) | 62 (37.1) | 31 (30.7) | 264 (42.9) |
| 5-10 | 89 (25.6) | 28 (16.8) | 23 (22.8) | 140 (22.8) |
| 10-15 | 51 (14.7) | 44 (26.3) | 17 (16.8) | 112 (18.2) |
| 15-20 | 33 (9.5) | 25 (15.0) | 28 (27.7) | 86 (14.0) |
| 20-25 | 3 (0.9) | 8 (4.8) | 2 (2.0) | 13 (2.1) |
| Median, (interquartile range) | 5 (7) | 9 (11) | 9 (12) | 6 (10) |
| Cancer stage, No. (%) | ||||
| 1 | 135 (38.9) | 21 (12.6) | 24 (35.8) | 180 (31.0) |
| 2 | 47 (13.5) | 30 (18.0) | 4 (6.0) | 81 (13.9) |
| 3 | 117 (33.7) | 53 (31.7) | 16 (23.9) | 186 (32.0) |
| 4 | 48 (13.8) | 63 (37.7) | 23 (34.3) | 134 (23.1) |
| NA | 0 | 0 | 34 | 34 |
NA = not available; RMS = rhabdomyosarcoma.
Evaluation of Germline Predisposition Variants Among RMS Patients
We performed exome sequencing of DNA from blood samples of the 615 RMS patients. We curated 2 sets of autosomal-dominant genes for investigation: 1) RMS-associated CPGs (n = 24) that have been specifically associated in the medical literature with RMS as part of cancer susceptibility syndromes, such as LFS (TP53), neurofibromatosis type 1 (NF1), and Costello Syndrome (HRAS), as well as the 4 mismatch repair genes associated with both recessive constitutional mismatch repair deficiency syndrome and dominant Lynch Syndrome (Table 2); and 2) other autosomal dominant CPGs (n = 39) curated from previous studies (23).
Table 2.
Candidate CPGsa
| Genes | Cancer susceptibility syndrome associated with RMS |
|---|---|
| RMS-associated CPGs | |
| TP53, CHEK2 | Li-Fraumeni syndrome |
| NF1 | Neurofibromatosis type 1 |
| HRAS | Costello syndrome |
| DICER1 | DICER1 syndrome |
| PTCH1, SUFU | Gorlin syndrome |
| SOS1, PTPN11, RAF1, CBL, KRAS, NRAS, RIT1, SHOC2 | Noonan syndrome |
| BRAF, MAP2K1, MAP2K2 | Cardiofaciocutaneous syndrome |
| MLH1, MSH2, MSH6, PMS2 | Constitutional mismatch repair deficiency |
| CDKN1C | Beckwith-Wiedemann syndrome |
| CREBBP | Rubinstein-Taybi syndrome |
| Other CPGs (from non-RMS–associated cancer susceptibility syndromes) | |
| ALK, APC, BAP1, BMPR1A, BRCA1, BRCA2, CDC73, CDH1, CDK4, CDKN2A, CEBPA, EPCAM, FH, GATA2, MAX, MEN1, NF2, PALB2, PAX5, PHOX2B, PRKAR1A, PTEN, RB1, RET, RUNX1, SDHA, SDHAF2, SDHB, SDHC, SDHD, SMAD4, SMARCA4, SMARCB1, STK11, TMEM127, TSC1, TSC2, VHL, WT1 | NA |
CPG = cancer predisposition gene; NA = not applicable; RMS = rhabdomyosarcoma.
Altogether, we identified germline predisposition variants in 45 out of 615 RMS patients (7.3%), 6 of which have not been reported in ClinVar previously (Table 3). Among them, 33 patients carried predisposition variants in RMS-associated CPGs, and the most common findings were in TP53 (n = 11), NF1 (n = 9), and HRAS (n = 5) (Table 3; Figure 1, A-C). Only 12 patients had a predisposition variant identified within the other general CPGs, among which BRCA2 pathogenic variants were the most prevalent (n = 6; Table 3; Figure 1, D). Taken together, 7.3% of the childhood RMS patients have a molecular finding consistent with cancer predisposition, and the majority of those (33 of 45, 73.3%) are due to known RMS-associated cancer susceptibility syndrome genes.
Table 3.
Cancer predisposition variants identified from RMS patients
| Variant information |
ClinVar P or LP variant |
Patient histology | |||
|---|---|---|---|---|---|
| Gene and genomic change (GRCh37) | Variant type | ClinVar status | Variation ID | Review status | |
| RMS-associated CPGs | |||||
| TP53 (n = 11) | |||||
| 17:7577046 C > A | Stopgain | P | 93323 | Criteria provided, multiple submitters, no conflicts | ERMS |
| 17:7577120 C > T | Missense SNV | P/LP | 12366 | Criteria provided, multiple submitters, no conflicts | Spindle cell |
| 17:7577144 A > G | Missense SNV | P/LP | 245777 | Criteria provided, multiple submitters, no conflicts | ARMS |
| 17:7577538 C > T | Missense SNV | P/LP | 12356 | Criteria provided, multiple submitters, no conflicts | ERMS |
| 17:7577551 C > T | Missense SNV | P/LP | 376600 | Criteria provided, multiple submitters, no conflicts | BRMS |
| 17:7578290 C > G | Splicing | — | — | — | Spindle cell |
| 17:7578457 C > T | Missense SNV | P/LP | 141963 | Criteria provided, multiple submitters, no conflicts | Mixed RMS |
| 17:7578479 G > A | Missense SNV | P/LP | 12370 | Criteria provided, multiple submitters, no conflicts | ERMS |
| 17:7578479 G > T | Missense SNV | LP | 12369 | Criteria provided, multiple submitters, no conflicts | ERMS |
| 17:7579312 C > T | Synonymous SNV | P | 177825 | Criteria provided, multiple submitters, no conflicts | ERMS |
| 17:7579320 TCA > T | Frameshift deletion | P | 127809 | Criteria provided, multiple submitters, no conflicts | BRMS |
| NF1 (n = 9) | |||||
| 17:29541542 A > G | Missense SNV | P | 354 | Criteria provided, multiple submitters, no conflicts | ERMS |
| 17:29553492 C > T | Stopgain | P | 188280 | Criteria provided, multiple submitters, no conflicts | BRMS |
| 17:29560043 C > T | Stopgain | — | — | — | ERMS |
| 17:29562746 C > T | Stopgain | P | 237556 | Criteria provided, multiple submitters, no conflicts | ERMS |
| 17:29652987 G > A | Stopgain | P | 573015 | Criteria provided, single submitter | ERMS |
| 17:29654553 C > T | Stopgain | P | 228381 | Criteria provided, multiple submitters, no conflicts | ERMS |
| 17:29654857 G > A | Missense SNV | P | 185354 | Criteria provided, multiple submitters, no conflicts | ERMS |
| 17:29664899 G > T | Splicing | P | 547680 | Criteria provided, multiple submitters, no conflicts | ERMS |
| 17:29670115 CTAACTT > C | Nonframeshift deletion | P/LP | 220715 | Criteria provided, multiple submitters, no conflicts | ERMS |
| HRAS (n = 5) | |||||
| 11:534288 C > G | Missense SNV | P | 12603 | Criteria provided, multiple submitters, no conflicts | ERMS |
| 11:534289 C > T | Missense SNV | P | 12602 | Reviewed by expert panel | ERMS (n = 4) |
| CBL (n = 2) | |||||
| 11:119149251 G > A | Missense SNV | P | 13810 | Criteria provided, single submitter | ERMS |
| 11:119155742 C > T | Stopgain | — | — | — | ERMS |
| DICER1 (n = 2) | |||||
| 14:95579443 G > A | Stopgain | P | 242054 | Criteria provided, multiple submitters, no conflicts | ERMS |
| 14:95590735 G > A | Stopgain | P | 429141 | Criteria provided, multiple submitters, no conflicts | RMS-NOS |
| MSH2 | |||||
| 2:47656951 C > T | Stopgain | P | 90554 | Reviewed by expert panel | ARMS |
| MSH6 | |||||
| 2:48027422 C > T | Missense SNV | P | 141058 | Reviewed by expert panel | RMS-NOS |
| PMS2 | |||||
| 7:6017260 G > A | Stopgain | P | 9237 | Reviewed by expert panel | ERMS |
| PTCH1 | |||||
| 9:98279010 G > C | Stopgain | — | — | — | RMS-NOS |
| Other CPGs | |||||
| BRCA2 (n = 6) | |||||
| 13:32900272 CAA > C | Frameshift deletion | P | 51684 | Reviewed by expert panel | ERMS |
| 13:32911595 G > T | Stopgain | P | 51400 | Reviewed by expert panel | Spindle cell |
| 13:32912089 CTG > C | Frameshift deletion | P | 51493 | Reviewed by expert panel | Spindle cell |
| 13:32912337 CTG > C | Frameshift deletion | P | 37859 | Reviewed by expert panel | ARMS |
| 13:32929123 C > G | Stopgain | P | 38085 | Reviewed by expert panel | ERMS |
| . 13:32936711 G > A | Stopgain | P | 38122 | Reviewed by expert panel | ARMS |
| SDHA (n = 2) | |||||
| 5:218472 T > G | Missense SNV | P/LP | 422382 | Criteria provided, multiple submitters, no conflicts | ERMS |
| 5:223624 C > T | Stopgain | P | 142601 | Criteria provided, multiple submitters, no conflicts | ARMS |
| BRCA1 | |||||
| 17:41215362 TTTTC > T | Frameshift deletion | P | 37644 | Reviewed by expert panel | ERMS |
| PTEN | |||||
| 10:89692904 C > T | Stopgain | P | 7819 | Criteria provided, multiple submitters, no conflicts | RMS-NOS |
| SDHC | |||||
| 1:161326611 G > A | Stopgain | — | — | — | ERMS |
| ALK | |||||
| 2:29416655 TC > T | Frameshift deletion | — | — | — | ERMS |
— = new pathogenic variants found in our cohort; ARMS = alveolar rhabdomyosarcoma; BRMS = botryoid rhabdomyosarcoma; CPG = cancer predisposition gene; ERMS = embryonal rhabdomyosarcoma; LP = likely pathogenic; NOS = not otherwise specified; P = pathogenic; RMS = rhabdomyosarcoma; SNV = single-nucleotide variant.
Figure 1.

Predisposition variants identified in the rhabdomyosarcoma cohort. The location and type of the predisposition variants identified in (A) TP53, (B) NF1, (C) HRAS, and (D) BRCA2. Each colored box represents a protein domain in the gene. Each colored circle represents a variant of a specific type.
Five ERMS patients from this cohort had germline HRAS codon 12 missense variants that correspond to well-described oncogenic variants: 4 patients had p.G12S and 1 had p.G12V variant (Table 3). Germline pathogenic variants of this codon, especially p.G12S, have been associated with Costello syndrome, a very rare multiple congenital anomaly and neurodevelopmental condition previously reported to be associated with increased risk of RMS and other cancers (20,21). We observed that 0.8% of patients with RMS, more specifically 1.4% of patients with ERMS, carried a germline pathogenic HRAS allele. Data collection on the COG D9902 protocol specifically asks about a list of syndrome diagnoses (including Costello syndrome). However, this was notated in only 1 of the 5 patients with the HRAS variant.
Prevalence of Cancer Predisposition Variants in RMS Subtypes
We compared the prevalence of germline predisposition variants between different RMS subtypes across the 63 CPGs. Notably, 10.0% (35 of 347) of children with ERMS (including the botryoid and spindle cell subtypes) harbored a predisposition variant compared with 3.0% (5 of 167) of those diagnosed with ARMS (odds ratio [OR] = 3.26, 95% confidence interval [CI] = 1.18 to 9.00, P = .02; logistic regression model). None of the FOXO1 fusion-positive ARMS patients (n = 66) had a predisposition variant in genes we investigated. Within the ERMS samples, patients who were positive for a predisposition variant were younger at diagnosis (median age = 3.0 years, all but 1 diagnosed before age 10 years) than those without a predisposition variant (median age = 5.5 years; β = 3.07, 95% CI = 1.26 to 4.87, P = 9.9 × 10–4; linear regression model; Figure 2, A). We did not observe the same trend in ARMS patients, which in part could be due to the small number of patients who carried a predisposition variant (Figure 2, B).
Figure 2.
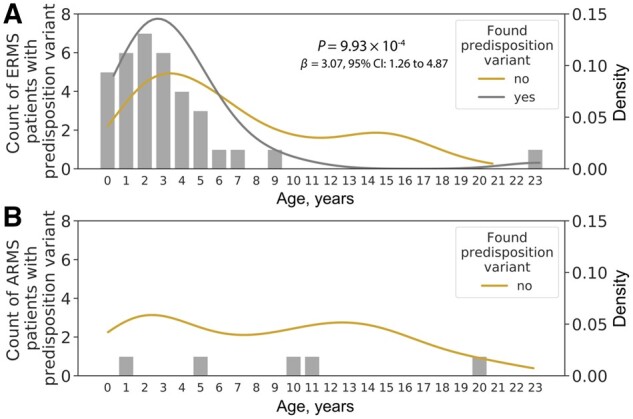
Age distribution of patients with and without a predisposition variant. Bar plots indicate the number of patients with a predisposition variant. (A) Age density among embryonal rhabdomyosarcoma (ERMS) patients with (grey) and without (golden) a predisposition variant. The P value was calculated from a linear regression model using age at diagnosis as the independent variable. (B) Age density among alveolar rhabdomyosarcoma (ARMS) patients without a pathogenic variant (golden). Density plot for ARMS patients who have predisposition variants (n = 5) was not generated due to the small patient count.
Prevalence of Cancer Predisposition Variants in Cases and Controls
To compare the prevalence of germline predisposition variants between RMS cases and the general population, we used a set of unrelated population controls consisting of 9963 samples with similar ethnicity characteristics (Supplementary Table 1, available online), sequenced at the same sequencing center and platform as the RMS cases and analyzed using the same bioinformatic pipelines. We evaluated the data for predisposition variants using the same approach in case and control cohorts.
We found that 143 samples (1.4%) from the population controls carried at least 1 predisposition variant from the 63 candidate genes, which is statistically significantly lower compared with RMS cases (OR = 6.34, 95% CI = 4.38 to 9.18, P = 1.3 × 10–22; logistic regression model, same below). This difference is largely driven by the RMS-associated CPGs (OR = 14.20, 95% CI = 8.72 to 23.11, P = 1.3 × 10–26) than by the other CPGs (OR = 2.32, 95% CI = 1.22 to 4.45, P = .01; Figure 3, A). In particular, we observed a 109-fold and 40-fold increase in the prevalence of predisposition variants among RMS cases compared with controls in TP53 (OR = 109.42, 95% CI = 23.07 to 518.99, P = 3.4 × 10–9) and NF1 (OR = 39.64, 95% CI = 11.49 to 136.81, P = 5.8 × 10–9), respectively (Figure 3, B). The most common germline findings in controls were from BRCA2 (n = 28, 0.29%; Figure 3, B), yet we still observed an increase for BRCA2 predisposition variants among RMS cases (n = 6, 0.98%) compared with controls (OR = 3.55, 95% CI = 1.34 to 9.40; P = .01). Notably, all the above-mentioned statistical results surpassed the Bonferroni correction threshold (P < .0166). Therefore, the prevalence of germline predisposition variants was statistically significantly more common in patients with RMS and also represented a different spectrum of germline variants in autosomal-dominant CPGs between childhood RMS patients and population controls.
Figure 3.

Prevalence of predisposition variants in cancer predisposition genes (CPGs) among rhabdomyosarcoma (RMS) patients and controls. (A) Comparison of the prevalence of germline predisposition variants between RMS patients and population controls stratified by gene types on the x-axis. (B) Percentage of samples in RMS cases and controls that carry a predisposition variant. This figure only shows genes containing predisposition variants in greater than 1 RMS cases. Numbers on top of the RMS bars indicate patient count.
In a separate analysis, we evaluated the prevalence of pathogenic variants among 7 genes underlying autosomal recessive disorders that are known to be associated with sarcomas. We did not observe any individuals with a biallelic pathogenic variant in these genes among our cases or controls. We identified carriers of pathogenic variants in 4 autosomal recessive genes in 1.14% (7 of 615) of RMS patients (RECQL4 n = 3; BLM n = 2; BUB1B n = 1; TRIP13 n = 1) and 0.75% (75 of 9963) of controls, which were not statistically different (OR = 1.37, 95% CI = 0.59 to 3.20, P = .47; logistic regression model).
Discussion
In one of the largest unselected cohorts of RMS patients assembled, we found that 7.3% of pediatric RMS patients carried a cancer predisposition variant in a known CPG. This is compared with 1.4% of the population-based controls included in our assessment. Notably, this is consistent with an independent assessment in a set of 394 RMS patients, which concluded that 6.6%-7.7% of patients carried a dominant-acting cancer predisposition variant in a known CPG (33). While there were no statistically significant differences in the overall frequency of pathogenic variants by ethnicity in RMS cases, relative differences compared with controls were consistent and statistically significant across groups (Supplementary Table 2, available online). As this study focused on newly diagnosed RMS patients rather than individuals from a survivor cohort, we limited the potential for survival bias. Previous genomic studies focusing on heterogeneous pediatric cancer populations report that 8%-10% of patients carry a germline predisposition variant (23,34,35). It is unclear if this is true for pediatric sarcomas, where there have been few large-scale assessments of CPGs. However, Mirabello et al. (36) recently reported that 28.0% (281 of 1004) of osteosarcoma patients had a pathogenic or likely pathogenic variant in a CPG, although they analyzed a larger set of genes and variants, including several autosomal recessive disorders. We also evaluated the impact of autosomal recessive disorders in the context of RMS and showed that RMS is not strongly driven by autosomal recessive disorders, similar to other studies of pediatric cancer cohorts (23,35). Importantly, these results suggest distinct germline genetic susceptibility patterns underlying different types of sarcomas.
Of note, the majority of the molecular findings in RMS patients from the COG cohort (5.3% in total) are within CPGs previously reported in pediatric RMS patients. This prevalence is relatively consistent with previous clinic-based estimations for individual syndromes, where 2%-9% of RMS patients were thought to have a certain comorbid germline cancer susceptibility syndrome [eg, 2.0% with neurofibromatosis type I (19), 3.8% with DICER1 syndrome (22), and 9.1% with LFS (17)]. Additionally, individuals with a predisposition variant from RMS-associated CPG have higher odds (OR = 14.20) of developing RMS compared with individuals carrying a predisposition variant from CPGs not previously associated with RMS (OR = 2.32).
An important aspect of this study is the differences demonstrated in the frequency of cancer predisposition variants by demographic and clinical features of RMS. For example, predisposition variants were more common in ERMS patients compared with ARMS patients and were completely absent in the 66 FOXO1 fusion-positive patients in our cohort. This further supports the idea that there are etiologic differences based on histology and fusion status. Notably, we did not observe associations between predisposition variants and tumor stage (data not shown). Additionally, consistent with previous smaller reports (37), cancer predisposition variants were more frequent in younger patients. However, our findings also indicate that these variants are still present in older children diagnosed with RMS. For instance, 11 of the 35 (31.4%) ERMS patients (and 18 of the 45 [40.0%] RMS patients regardless of histology type) with predisposition variants were older than 3 years of age, suggesting that genetic testing should not be limited to only very young patients.
Within the genes where we discovered germline pathogenic variants in RMS, some of the same genes are frequently reported to also have somatic mutations in RMS tumors, such as TP53, NF1, and HRAS (8,11‐13). For example, Shern et al. (8) evaluated the frequency of somatic mutations across 147 RMS patients using paired germline and tumor samples, and somatic mutations in TP53 (3.4%) and NF1 (3.4%) were found in the RMS patients. In our study, 1.7% and 1.5% of our pediatric RMS patients had a germline pathogenic variant in TP53 and NF1, respectively. Therefore, patients with a somatic pathogenic variant in these genes should have further germline analysis to determine the origin of the variant and implications for family members (38,39).
Five ERMS patients had a well-described oncogenic variant in codon 12 of HRAS. The p.G12S pathogenic variant seen in 4 patients represents the only recurrent germline pathogenic variant we found in this cohort. In addition, germline mutations in HRAS (more commonly p.G12S than p.G12V) are known to cause Costello syndrome, a rare multiple congenital anomaly and neurodevelopmental syndrome associated with an increased risk of developing childhood RMS and other cancers (21,40). Although 1 patient was noted to have Costello syndrome in the case report form, additional phenotypic information was not available for the other patients. Further study is required to determine if children with these pathogenic alleles develop RMS without the many other features of Costello syndrome.
We observed 6 RMS patients with a pathogenic variant in BRCA2, which was statistically significantly higher than our control population (OR = 3.55, 95% CI = 1.34 to 9.40, P = .01). Furthermore, 5 of the 6 were diagnosed before 6 years old. We did not expect such an enrichment in BRCA2 as it has not traditionally been thought of as an RMS-associated gene and is usually implicated in adult-onset cancers. Interestingly, 1 of the 3 pathogenic variants identified in RMS patients by Zhang et al. (23) was also in BRCA2. Also, in a recent study of germline pathogenic variants across 1162 adult sarcoma patients, Ballinger and Goode et al. (41) reported an enrichment for pathogenic variants in BRCA2. In addition, an excess number of breast cancer cases have been reported in mothers of children with sarcomas (42). Similarly, first-degree relatives of women with breast cancer are found to have increased prevalence of soft tissue and bone sarcomas (43,44). These studies suggest that, while LFS with underlying TP53 pathogenic variants have been known to play a role in the aggregation of breast cancer and sarcomas (45), breast and ovarian cancer genes, such as BRCA2, may also play a role in this aggregation.
Notably, our analysis did not include pathogenic variation that may result from intragenic or whole-gene deletions, gene imprinting, or epigenetic alterations. Therefore, the overall prevalence of germline pathogenic genetic variation among children diagnosed with RMS is likely to be higher when considering these other types of variants. Additionally, our assessment did not include an evaluation of outcomes for those with pathogenic variants in CPGs but rather focused on the prevalence of cancer predisposition syndromes associated with RMS in an unselected pediatric RMS cohort at diagnosis. Finally, while we did use population-based controls for our assessment, future assessment should leverage other noncancer populations to evaluate the impact of underlying characteristics, which could influence effect estimates.
In this study, we investigated the role of germline predisposition variants in one of the largest pediatric RMS cohorts. Notably, the prevalence of the predisposition variants differs by histology and age at diagnosis as well as by the type of cancer susceptibility syndromes with which the genes are associated. These results shed light on the spectrum of CPGs that may cause RMS. These findings warrant further discussion about the clinical management of children newly diagnosed with RMS and the modification of guidelines for when to consider germline genetic testing. Additionally, there is a growing awareness that leveraging germline information of cancer predisposition could provide innovative therapeutic strategies, thereby improving patient care (46). Finally, future studies should evaluate novel genes, through the assessment of both common and rare variants on RMS susceptibility, to yield new insights into the mechanisms underlying pediatric RMS.
Funding
This work was supported by the Cancer Prevention and Research Institute of Texas (RP170071). This work was also supported by grants from the WW WW (QuadW) Foundation, the Children’s Oncology Group Foundation, and the Isabella Santos Foundation. This work was also supported by U10CA098543, U10CA098413, U10CA180899, and U10CA180886 from the National Cancer Institute to the Children’s Oncology Group, and 1UM1HG008898 from the National Human Genome Research Institute to the Human Genome Sequencing Center at Balor College of Medicine. SDS is supported by Cancer Prevention and Research Institute of Texas (CPRIT) RP160283 - Baylor College of Medicine Comprehensive Cancer Training Program.
Notes
Role of the funder: The funders had no role in the design of the study; the collection, analysis, and interpretation of the data; the writing of the manuscript; or the decision to submit the manuscript for publication.
Disclosures: The authors have no conflicts of interest to disclose.
Acknowledgements: We thank all patients, general practitioners, and health professionals who participated in the study. The authors thank the staff and participants of the ARIC study for their important contributions. The Atherosclerosis Risk in Communities study has been funded in whole or in part with Federal funds from the National Heart, Lung, and Blood Institute, National Institutes of Health, Department of Health and Human Services, under Contract nos. (HHSN268201700001I, HHSN268201700002I, HHSN268201700003I, HHSN268201700005I, HHSN268201700004I). The VIVA cohort collection and phenotyping was supported by National Institutes of Health (NIH) (R01 DK59264, DK092238 and DK080457) and United States Department of Agriculture, Agricultural Research Service (USDA/ARS) under Cooperative Agreement 58–6250-51000.
Author contributions: He Li: Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Software; Validation; Visualization; Writing – original draft; Writing – review & editing. Saumya D. Sisoudiya: Conceptualization; Data curation; Methodology. Bailey A. Martin-Giacalone: Data curation; Formal analysis. Michael M. Khayat: Data curation; Visualization; Writing – review & editing. Shannon Dugan-Perez: Project administration; Resources. Deborah A. Marquez-Do: Project administration; Resources. Michael E. Scheurer: Project administration; Resources. Donna Muzny: Project administration; Resources. Eric Boerwinkle: Data curation. Richard A. Gibbs: Data curation; Resources. Yueh-Yun Chi: Data curation. Donald A. Barkauskas: Data curation. Tammy Lo: Data curation. David Hall: Data curation. Douglas R. Stewart: Resources; Writing – review & editing. Joshua D. Schiffman: Resources; Writing – review & editing. Stephen X. Skapek: Resources. Douglas S. Hawkins: Resources; Writing – review & editing. Sharon E. Plon: Conceptualization; Data curation; Funding acquisition; Investigation; Methodology; Resources; Supervision; Writing – original draft; Writing – review & editing. Aniko Sabo: Conceptualization; Data curation; Funding acquisition; Investigation; Methodology; Resources; Supervision; Validation; Writing – original draft; Writing – review & editing. Philip J. Lupo: Conceptualization; Data curation; Funding acquisition; Investigation; Methodology; Resources; Supervision; Validation; Writing – original draft; Writing – review & editing.
Data Availability
All of the pathogenic and likely pathogenic variants observed in RMS patients from this manuscript have been submitted to and are searchable in ClinVar. The individual sequencing and phenotypic data for the 615 RMS cases are available through dbGaP requests (accession ID phs002304.v1.p1).
Supplementary Material
References
- 1. Ognjanovic S, Linabery AM, Charbonneau B, Ross JA.. Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975-2005. Cancer. 2009;115(18):4218–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ries LS, Gurney JG, Linet M, Tamra T, Young JL, Bunin GR.. Cancer Incidence and Survival Among Children and Adolescents: United States SEER Program 1975-1995, National Cancer Institute, SEER Program. Bethesda, MD: NIH Pub. No. 99-4649; 1999.
- 3. Hawkins DS, Spunt SL, Skapek SX on behalf of the COG Soft Tissue Sarcoma Committee. Children's Oncology Group's 2013 blueprint for research: soft tissue sarcomas. Pediatr Blood Cancer. 2013;60(6):1001–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Skapek SX, Ferrari A, Gupta AA, et al. Rhabdomyosarcoma. Nat Rev Dis Primers. 2019;5(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Newton WA Jr., Gehan EA, Webber BL, et al. Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification--an intergroup rhabdomyosarcoma study. Cancer. 1995;76(6):1073–1085. [DOI] [PubMed] [Google Scholar]
- 6. Barr FG. Gene fusions involving PAX and FOX family members in alveolar rhabdomyosarcoma. Oncogene. 2001;20(40):5736–5746. [DOI] [PubMed] [Google Scholar]
- 7. Williamson D, Missiaglia E, de Reynies A, et al. Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J Clin Oncol. 2010;28(13):2151–2158. [DOI] [PubMed] [Google Scholar]
- 8. Shern JF, Chen L, Chmielecki J, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014;4(2):216–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wachtel M, Dettling M, Koscielniak E, et al. Gene expression signatures identify rhabdomyosarcoma subtypes and detect a novel t(2; 2)(q35; p23) translocation fusing PAX3 to NCOA1. Cancer Res. 2004;64(16):5539–5545. [DOI] [PubMed] [Google Scholar]
- 10. Davicioni E, Anderson MJ, Finckenstein FG, et al. Molecular classification of rhabdomyosarcoma--genotypic and phenotypic determinants of diagnosis: a report from the Children's Oncology Group. Am J Pathol. 2009;174(2):550–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Taylor AC, Shu L, Danks MK, et al. P53 mutation and MDM2 amplification frequency in pediatric rhabdomyosarcoma tumors and cell lines. Med Pediatr Oncol. 2000;35(2):96–103. [DOI] [PubMed] [Google Scholar]
- 12. Stratton MR, Fisher C, Gusterson BA, Cooper CS.. Detection of point mutations in N-ras and K-ras genes of human embryonal rhabdomyosarcomas using oligonucleotide probes and the polymerase chain reaction. Cancer Res. 1989;49(22):6324–6327. [PubMed] [Google Scholar]
- 13. Shukla N, Ameur N, Yilmaz I, et al. Oncogene mutation profiling of pediatric solid tumors reveals significant subsets of embryonal rhabdomyosarcoma and neuroblastoma with mutated genes in growth signaling pathways. Clin Cancer Res. 2012;18(3):748–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Taylor JG, Cheuk AT, Tsang PS, et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J Clin Invest. 2009;119(11):3395–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Seki M, Nishimura R, Yoshida K, et al. Integrated genetic and epigenetic analysis defines novel molecular subgroups in rhabdomyosarcoma. Nat Commun. 2015;6(1):7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zimmerman R, Schimmenti L, Spector L.. A catalog of genetic syndromes in childhood cancer. Pediatr Blood Cancer. 2015;62(12):2071–2075. [DOI] [PubMed] [Google Scholar]
- 17. Diller L, Sexsmith E, Gottlieb A, Li FP, Malkin D.. Germline p53 mutations are frequently detected in young children with rhabdomyosarcoma. J Clin Invest. 1995;95(4):1606–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hartley AL, Birch JM, Marsden HB, Harris M, Blair V.. Neurofibromatosis in children with soft tissue sarcoma. Pediatr Hematol Oncol. 1988;5(1):7–16. [DOI] [PubMed] [Google Scholar]
- 19. Yang P, Grufferman S, Khoury MJ, et al. Association of childhood rhabdomyosarcoma with neurofibromatosis type I and birth defects. Genet Epidemiol. 1995;12(5):467–474. [DOI] [PubMed] [Google Scholar]
- 20. Kratz CP, Rapisuwon S, Reed H, Hasle H, Rosenberg PS.. Cancer in Noonan, Costello, cardiofaciocutaneous and LEOPARD syndromes. Am J Med Genet C Genet. 2011;157(2):83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Estep AL, Tidyman WE, Teitell MA, Cotter PD, Rauen KA.. HRAS mutations in Costello syndrome: detection of constitutional activating mutations in codon 12 and 13 and loss of wild-type allele in malignancy. Am J Med Genet A. 2006;140A(1):8–16. [DOI] [PubMed] [Google Scholar]
- 22. Doros L, Yang J, Dehner L, et al. DICER1 mutations in embryonal rhabdomyosarcomas from children with and without familial PPB-tumor predisposition syndrome. Pediatr Blood Cancer. 2012;59(3):558–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang J, Walsh MF, Wu G, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. 2015;373(24):2336–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Malempati S, Hawkins DS.. Rhabdomyosarcoma: review of the Children's Oncology Group (COG) Soft-Tissue Sarcoma Committee experience and rationale for current COG studies. Pediatr Blood Cancer. 2012;59(1):5–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sultan I, Qaddoumi I, Yaser S, Rodriguez-Galindo C, Ferrari A.. Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: an analysis of 2,600 patients. J Clin Oncol. 2009;27(20):3391–3397. [DOI] [PubMed] [Google Scholar]
- 26. Butte NF, Cai G, Cole SA, Comuzzie AG.. Viva la Familia Study: genetic and environmental contributions to childhood obesity and its comorbidities in the Hispanic population. Am J Clin Nutr. 2006;84(3):646–654. quiz 673–644. [DOI] [PubMed] [Google Scholar]
- 27. Wang K, Li M, Hakonarson H.. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164–e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Auton A, Brooks LD, Durbin RM, et al. ; 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature2020;581(7809):434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lek M, Karczewski KJ, Minikel EV, et al. ; Exome Aggregation Consortium. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46(D1):D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Seabold S, Perktold J. Statsmodels: Econometric and statistical modeling with python. In. 9th Python in Science Conference; June 28, 2010; Austin, Texas, USA.
- 33. Kim J, Light N, Subasri V, et al. Pathogenic germline variants in cancer-susceptibility genes in children and young adults with rhabdomyosarcoma. JCO Precis Oncol. 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mody RJ, Wu YM, Lonigro RJ, et al. Integrative clinical sequencing in the management of refractory or relapsed cancer in youth. JAMA. 2015;314(9):913–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Parsons DW, Roy A, Yang Y, et al. Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol. 2016;2(5):616–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mirabello L, Zhu B, Koster R, et al. Frequency of pathogenic germline variants in cancer-susceptibility genes in patients with osteosarcoma. JAMA Oncol. 2020;6(5):724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lupo PJ, Danysh HE, Plon SE, et al. Family history of cancer and childhood rhabdomyosarcoma: a report from the Children's Oncology Group and the Utah Population Database. Cancer Med. 2015;4(5):781–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. DeLeonardis K, Hogan L, Cannistra SA, Rangachari D, Tung N.. When should tumor genomic profiling prompt consideration of germline testing? J Oncol Pract. 2019;15(9):465–473. [DOI] [PubMed] [Google Scholar]
- 39. Li MM, Chao E, Esplin ED, et al. ; ACMG Professional Practice and Guidelines Committee. Points to consider for reporting of germline variation in patients undergoing tumor testing: a statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2020;22(7):1142–1148. [DOI] [PubMed] [Google Scholar]
- 40. Aoki Y, Niihori T, Kawame H, et al. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet. 2005;37(10):1038–1040. [DOI] [PubMed] [Google Scholar]
- 41. Ballinger ML, Goode DL, Ray-Coquard I, et al. Monogenic and polygenic determinants of sarcoma risk: an international genetic study. Lancet Oncol. 2016;17(9):1261–1271. [DOI] [PubMed] [Google Scholar]
- 42. Hartley AL, Birch JM, Blair V.. Malignant disease in the mothers of a population-based series of young adults with bone and soft tissue sarcomas. Br J Cancer. 1991;63(3):416–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bennett KE, Howell A, Evans DG, Birch JM.. A follow-up study of breast and other cancers in families of an unselected series of breast cancer patients. Br J Cancer. 2002;86(5):718–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ji J, Hemminki K.. Familial risk for histology-specific bone cancers: an updated study in Sweden. Eur J Cancer. 2006;42(14):2343–2349. [DOI] [PubMed] [Google Scholar]
- 45. Malkin D, Li FP, Strong LC, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250(4985):1233–1238. [DOI] [PubMed] [Google Scholar]
- 46. Thavaneswaran S, Rath E, Tucker K, et al. Therapeutic implications of germline genetic findings in cancer. Nat Rev Clin Oncol. 2019;16(6):386–396. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All of the pathogenic and likely pathogenic variants observed in RMS patients from this manuscript have been submitted to and are searchable in ClinVar. The individual sequencing and phenotypic data for the 615 RMS cases are available through dbGaP requests (accession ID phs002304.v1.p1).