

Abstract
The twin cycle hypothesis postulated that type 2 diabetes was a result of excess liver fat causing excess supply of fat to the pancreas with resulting dysfunction of both organs. If this was so, the condition should be able to be returned to normal by calorie restriction. The Counterpoint study tested this prediction in short‐duration type 2 diabetes and showed that liver glucose handling returned to normal within 7 days and that beta‐cell function returned close to normal over 8 weeks. Subsequent studies have demonstrated the durability of remission from type 2 diabetes. Remarkably, during the first 12 months of remission, the maximum functional beta‐cell mass returns completely to normal and remains so for at least 24 months, consistent with regain of insulin secretory function of beta cells which had dedifferentiated in the face of chronic nutrient oversupply. The likelihood of achieving remission after 15% weight loss has been shown to be mainly determined by the duration of diabetes, with responders having better beta‐cell function at baseline. Remission is independent of BMI, underscoring the personal fat threshold concept that type 2 diabetes develops when an individual acquires more fat than can be individually tolerated even at a BMI which in the nonobese range. Observations on people of South Asian or Afro‐American ethnicity confirm that substantial weight loss achieves remission in the same way as in the largely White Europeans studied in detail. Diagnosis of type 2 diabetes can now be regarded as an urgent signal that weight loss must be achieved to avoid a progressive decline of health.
Keywords: aetiology, liver fat, pancreas fat, personal fat threshold, remission, type 2 diabetes, weight loss

The twin cycle hypothesis
The prevalence of type 2 diabetes in populations depends upon food availability [1, 2, 3]. Given that condition is associated with both insulin resistance and pancreatic endocrine dysfunction, the application of Occam’s razor suggested that both underlying abnormalities were likely to be caused by the same factor. Specifically, excess intra‐organ fat could potentially explain dysfunction of both liver and pancreas, and this underpinned the twin cycle hypothesis of the aetiology of type 2 diabetes [4]. Fig. 1 illustrates the postulated sequence of events. It had already been established that hepatic insulin resistance was proportional to liver fat content, and it was known that both occur before onset of type 2 diabetes [5, 6]. Hence, over a long period of time, the intake of more food energy than required would lead to increased liver fat, with steadily increasing hepatic insulin resistance [7, 8, 9]. The major insulin‐responsive role of the liver is to maintain glucose production, and hepatic insulin resistance would bring about a tendency to increase plasma glucose levels and then compensatory elevation of fasting plasma insulin levels. This would stimulate the process of converting glucose to triglyceride would accelerate, creating a vicious cycle in the liver.
Fig. 1.
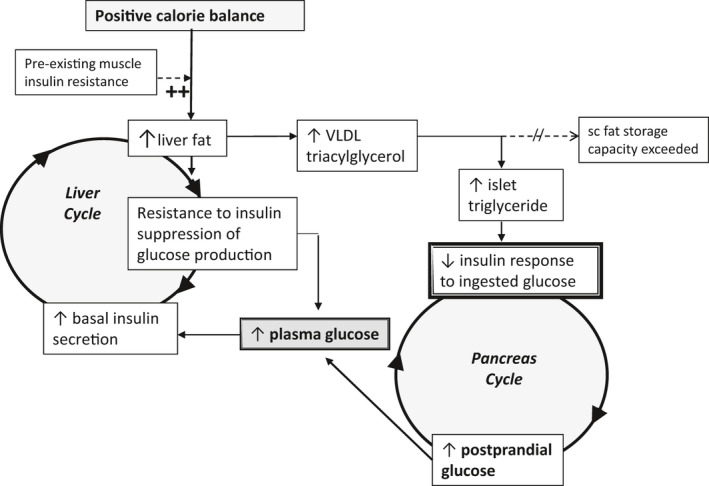
The twin cycle hypothesis of the aetiology of type 2 diabetes: Any excess carbohydrate must undergo de novo lipogenesis, and this promotes fat accumulation in the liver. As insulin stimulates de novo lipogenesis, individuals with relative insulin resistance in muscle (determined by genetic or lifestyle factors) will accumulate liver fat more readily because of higher plasma insulin levels. Resistance to insulin suppression of hepatic glucose production will result from the increased liver fat. Over many years, the slight increase in fasting plasma glucose level will stimulate increased basal insulin secretion rates. The resulting hyperinsulinaemia will speed the conversion of excess calories into liver fat. A vicious cycle of hyperinsulinemia and blunted suppression of hepatic glucose production becomes established. At the same time, export of very‐low‐density lipoprotein triglyceride will increase fat delivery to all tissues including the islets. The increased fatty acid availability in and around pancreatic islets impairs the acute insulin secretion in response to ingested food, and at a certain point, postprandial hyperglycaemia will develop. Day‐long hyperglycaemia will further increase insulin secretion rates, resulting in increased hepatic lipogenesis, spinning the liver cycle faster and driving on the pancreas cycle. Eventually, the fatty acid and glucose inhibitory effects on the islets reach a trigger level leading to a relatively sudden onset of clinical diabetes. Figure adapted from [4].
As liver fat levels increased, the rate of export of triglyceride to the rest of the body would increase. Once subcutaneous fat stores were relatively replete, this would increase deposition of fat in ectopic sites, including the pancreas. By 2006, it was known that chronic exposure to fat would inhibit glucose‐stimulated insulin secretion and that human beta cells avidly take up fatty acids and store these as triglyceride [10, 11, 12]. Failure of postprandial insulin secretion would lead to prolonged hyperglycaemia and further increase the de novo lipogenesis. The twin cycle hypothesis could thus explain why beta‐cell function deteriorated, albeit with a different time course to that of the liver vicious cycle.
Critically, the hypothesis could be tested. If this concept of aetiology of type 2 diabetes was correct, then sudden induction of negative calorie balance would cause the cycles to reverse with resulting normoglycaemia. A diet delivering around 800 kcal/day which would be acceptable to most people had to be devised. That was the first step in launching the Counterpoint study [13].
The acute metabolic response to calorie restriction
The rapidity of metabolic adaptation to sudden drop in calorie intake in nondiabetic people is striking [14, 15]. In type 2 diabetes, such dietary restriction also causes a major decrease in hepatic glucose production, which can normalize fasting blood glucose within 7 days (Fig. 2) [13, 16]. The major underlying factor is the sudden reversal of hepatic insulin resistance [7, 13]. As fasting plasma insulin levels are not deficient at this stage of type 2 diabetes but rather supra‐normal, complete normalization of hepatic glucose production can be achieved. Unlike insulin resistance in other tissues, hepatic insulin resistance in respect to glucose metabolism is entirely determined by the presence of excess fat [7, 8, 17]. Intra‐hepatic fat content decreased by 30% during the first 7 days of the 800kcal/day diet and to low normal levels after 8 weeks (Fig. 3) [13]. Taken at face value, it may be thought that this extent of decrease might be insufficient to account for the return of normal insulin sensitivity. However, the measurement of triglyceride content of the liver even by the accurate and precise magnetic resonance methods does not reflect the levels of the lipid intermediaries, which actually inhibit transmission of the insulin signal [18]. Intracellular triglyceride is itself metabolically inert (reflecting its biological purpose for safe storage of energy). The induced accumulation of diacylglycerol and potentially other intermediaries would change rapidly with change in substrate supply. Drop in food intake and a sudden call on endogenous energy stores would rapidly drain the intracellular ‘swamp’ of toxic intracellular lipid intermediaries and convert this into a rapid transit system from production to utilization. Hence, the 30% drop in intra‐hepatic triglyceride content indicates a dramatically sudden removal of those moieties actually causing inhibition of insulin action. The result of this is resumption of normal control of hepatic glucose output and complete normalization of fasting plasma glucose.
Fig. 2.
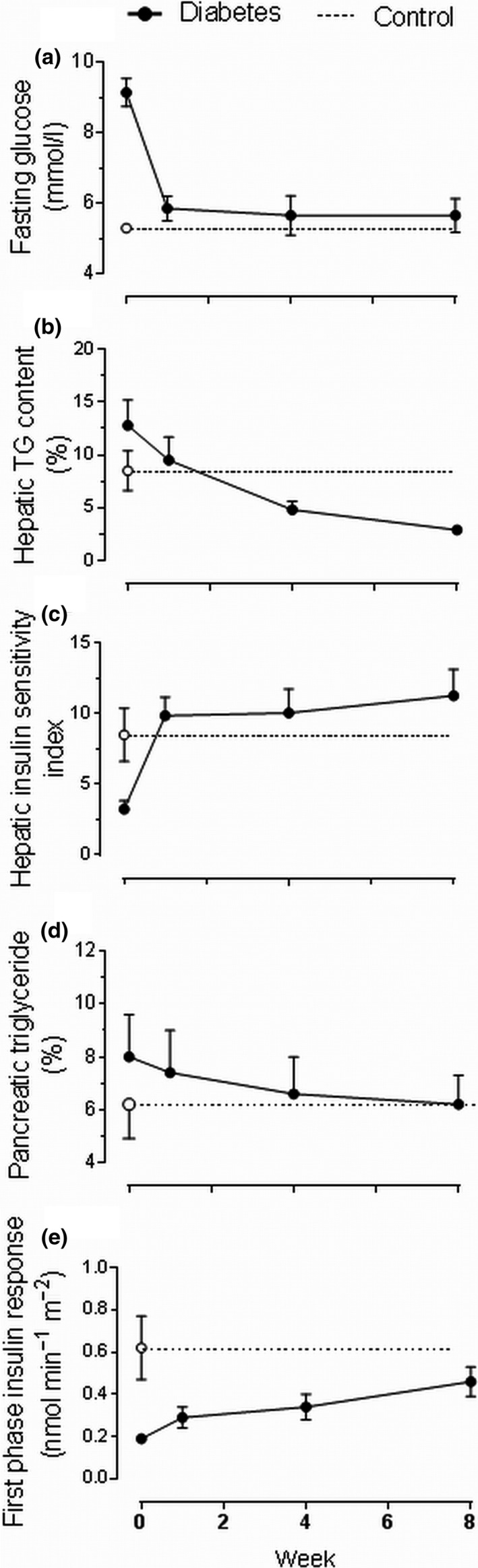
The Counterpoint study. Metabolic changes after initiation of a very‐low‐energy diet (~700 kcal per day) and withdrawal of metformin therapy in people with type 2 diabetes (red) and matched nondiabetic controls (studied at a single time‐point) (blue). Figure shows changes in (a) fasting plasma glucose, (b) hepatic triglyceride content, (c) hepatic insulin sensitivity index, (d) pancreatic triglyceride content and (e) immediate insulin response (known as ‘first‐phase’) to a 3 mmol/L increase in plasma glucose concentration. Figure adapted from Lim et al [13].
Fig. 3.
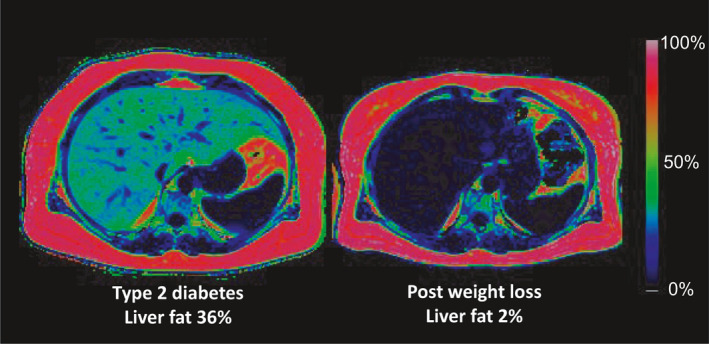
Colour‐coded MRI scans showing the change in regional fat content before and after 8 weeks of an 800kcal/day diet. In this individual, no liver pathology has been identified, but liver fat content was very high (left panel). The extent of hepatic steatosis in most people with type 2 diabetes has not widely been appreciated. In the whole DiRECT cohort at baseline, liver fat content averaged 16% [78], hugely raised above the accepted normal upper level of 5.5%. Dietary weight loss restores normality (right panel).
In contrast to the sudden normalization of hepatic glucose production, first‐phase insulin secretion (i.e. the immediate beta‐cell response to a rise in plasma glucose) changed little in the first 7 days despite the removal of any suppressive effect of the raised plasma glucose levels [13]. Very gradually, over the remainder of the 8‐week low‐calorie diet period, the level of intra‐pancreatic fat decreased. In step with this, the first‐phase insulin response gradually increased (Fig. 2). The twin cycle hypothesis had predicted that the decreased VLDL‐TG‐mediated delivery of fat to the pancreas would bring about release of beta cells from the suppressive effect of excess intracellular fatty acids and their intermediaries [4]. More recently, evidence has emerged of fat‐induced dedifferentiation of beta cells with loss of specialist function but continued viability [19, 20, 21, 22].
The 8‐week time course of recovery of beta cells observed in Counterpoint is consistent with switch on of genes controlling insulin production (re‐differentiation) as the underlying mechanism. Given that there is no capacity for beta‐cell neogenesis in adult humans outside pregnancy [23, 24], beta‐cell death clearly had not occurred. Although apoptosis has been suggested to explain the 50% decrease in beta‐cell number or functional capacity seen at diagnosis of type 2 diabetes, the absence of lipofuscin bodies in the pancreases of people with type 2 diabetes is strong evidence against such a mechanism [24]. The apparent absence of around 50% of beta cells on immunostaining of pancreas sections [25] is consistent with the lack of insulin production from those cells that had lost their specialized function. Just in the same way that diabetes never occurs without beta‐cell dysfunction, recovery of function is essential for return to nondiabetic glucose homeostasis.
Much confusion has resulted from the assessment of whole‐body insulin resistance using either calculated indices or the hyperinsulinaemic–euglycaemic clamp. Both methods measure whole‐body insulin sensitivity without distinguishing between the major components of liver and muscle. Attempts to understand the pathophysiological mechanisms of acute response to calorie restriction using whole‐body estimation of insulin sensitivity have continued, even though all studies using appropriate isotopic methods have demonstrated the normalization of liver insulin sensitivity and absence of improvement in muscle insulin sensitivity during the early weeks of calorie restriction in type 2 diabetes [7, 13, 26, 27]. Although this may appear surprising, this is in the context of longstanding acceptance of the concept that muscle insulin resistance is a major factor underlying continued hyperglycaemia. Detailed consideration reveals the lack of logic or data to support this belief. For instance, mice genetically engineered to lack insulin receptors in skeletal muscle do not develop diabetes [28]. In humans, muscle contributes only modestly to postprandial glucose homeostasis and hardly at all to overnight glucose homeostasis [29, 30]. Glycogen storage in muscle after meals is almost absent in people with the genetic variant of glycogen synthase PPP1R3A, yet this is compatible with normal glucose tolerance [31]. Furthermore, the range of muscle insulin sensitivity is very large in the population, and many normoglycaemic individuals have a similar degree of muscle insulin resistance to those with type 2 diabetes or impaired glucose tolerance [32].
Whilst it is certain that lower muscle insulin sensitivity is the earliest feature predicting onset of type 2 diabetes, this must be distinguished from it being a driver of the hyperglycaemia once the disease has developed [33]. Prior to onset of type 2 diabetes, ingestion of carbohydrate in excess of any 24‐hour requirement is less able to be stored muscle glycogen depots and the carbon energy has to be stored fat by de novo lipogenesis. This process only happens in the liver in humans, and triglyceride synthesized by this route is particularly likely to be stored in hepatocytes rather than transported for storage in subcutaneous adipose tissue where it is metabolically inert. As de novo lipogenesis is stimulated by insulin, those people who are relatively insulin resistant in muscle – and who therefore have a raised plasma insulin level – are especially likely to accumulate fat in the liver [29, 34]. This could explain the reason why muscle insulin resistance is the first detectable signal of risk for type 2 diabetes [33, 35].
Duration of type 2 diabetes is the major determinant of return to nondiabetic glucose control
The Counterpoint study also opened up a further vital question. In order to carry out the initial test of the twin cycle hypothesis on a well‐characterized, homogenous group of people with type 2 diabetes, only those with a duration of diabetes of less than 4 years and on therapy with only diet or diet plus metformin were included. The Counterbalance study examined whether normalization of glucose control would also occur in longer‐duration type 2 diabetes. It also included those treated with sulphonylurea or insulin [26]. All antidiabetic agents were withdrawn on the first day of the diet. In the short‐duration group (<4 years), all exhibited a rapid fall in fasting plasma glucose, but unlike in Counterpoint, not all achieved nondiabetic fasting plasma glucose, presumably reflecting a more advanced state of diabetes requiring multiple antidiabetic agents. However, in the longer‐duration groups (>8 years) there was a wide range of plasma glucose responses with only 50% returning to normal. Some behaved like the short‐duration group; some very gradually returned to nondiabetic levels; and some hardly responded. Overall, there was clear relationship of achieved fasting plasma glucose at the end of the 8‐week low‐calorie diet period with duration of type 2 diabetes (r = 0.50, P < 0.0006).
The nonresponders in the Counterbalance study, mainly with 8–23 years of duration of type 2 diabetes, demonstrated similar hepatic response to weight loss as did the responders (Fig. 4). It is now clear that the improvement of hepatic insulin sensitivity can achieve normalization of hepatic lipid handling whatever the duration of type 2 diabetes [36]. However, the beta‐cell response is very different, and the capacity to recover decreases with increasing duration of diabetes. Those people with type 2 diabetes who were able to return to nondiabetic fasting plasma glucose levels tend to have higher fasting plasma insulin levels at baseline [37, 38]. Nonresponders and responders could be distinguished at baseline as the nonresponders had an absent first‐phase insulin response, which was significantly different from the small response of responders (Fig. 4). This reflection of beta‐cell competence suggests that return to normal fasting plasma glucose also requires a degree of residual beta‐cell function but that acute change in hepatic fat metabolism will occur as a basic response to sudden calorie restriction. Recently, this has been confirmed with beta‐cell capacity to recover being determinative for remission of type 2 diabetes [38].
Fig. 4.
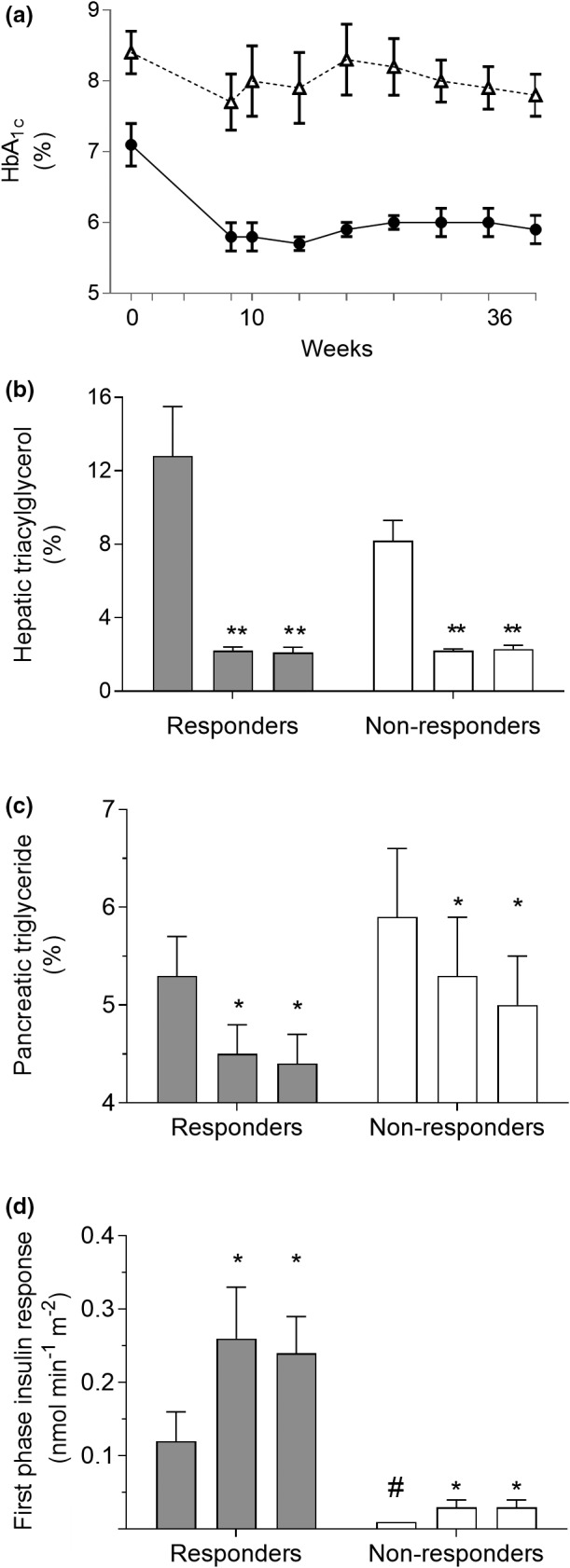
The Counterbalance study. Thirty people with type 2 diabetes of up to 23 years of duration lost approximately 15 kg in weight and then maintained steady weight for 6 months. Those achieving nondiabetic fasting plasma glucose levels were classified as responders and those remaining in the diabetic range as nonresponders. Panel (a): change in HbA1c in responders (closed symbols) and nonresponders (open symbols). Panel (b): decrease in liver fat content in both groups despite ongoing overweight or obesity. Bars show data at baseline, postweight loss and after 6 months of weight stability within the responder and nonresponder groups. Panel (c): decrease in intra‐pancreatic fat. Panel (d): At baseline, the first‐phase insulin response was higher in responders and increased to normal levels, whereas the grossly deficient baseline level in nonresponders hardly changed. Figure adapted with permission from the American Diabetes Association [45]. *** P < 0.0001 vs. baseline (responders); ††† P < 0.0001, †† P < 0.01 vs. baseline (non‐responders); ‡‡‡ P < 0.0001, ‡‡ P < 0.001, ‡ P < 0.05 vs. baseline (relapsers); ### P < 0.0001, # P < 0.01 vs. 5 months (relapsers).
Additional information can be gained from the Look AHEAD study. Compared with diabetes duration of 0–2 years, the chance of achieving remission was less than 50% after 2–7 years of diabetes and around 20% with duration greater than 7 years [39]. Similar data have been observed 10 years after bariatric surgery with remission rates of 60% for those with type 2 diabetes duration of less than one year compared with just over 20% for duration of 1–3 years and around 15% for duration of over 4 years [40]. The author has observed remission of type 2 diabetes with withdrawal of more than two oral hypoglycaemic agents in two individuals with diabetes duration 24 years, but it may be concluded that the chance of achieving remission after adequate weight loss becomes very small with long‐duration disease. These observations suggest that individuals have differing degrees of beta‐cell resilience to ongoing metabolic stress, presumably genetically determined.
Further insight into the physiology of continuing remission of type 2 diabetes was provided by detailed study of a cohort within the DiRECT study [36, 38]. This study involved people with type 2 diabetes of up to 6 years of duration and of BMI 27–45 kg/m2. In the group randomized to weight loss and then weight maintenance, those remaining in remission were termed ‘responders’ as defined by HbA1c of <6.5% or >48 mmol/mol and fasting plasma glucose of < or >7 mmol/l. During the initial phase of weight loss, responders lost 16.2 ± 1.2 kg compared with nonresponders who lost slightly less (13.4 ± 1.4 kg) (Fig. 5). The weight loss brought about a dramatic fall to normal in liver fat content in both groups, and this remained substantially decreased during follow‐up. Clearly, weight loss is dramatically effective in abolishing nonalcoholic fatty liver disease.
Fig. 5.
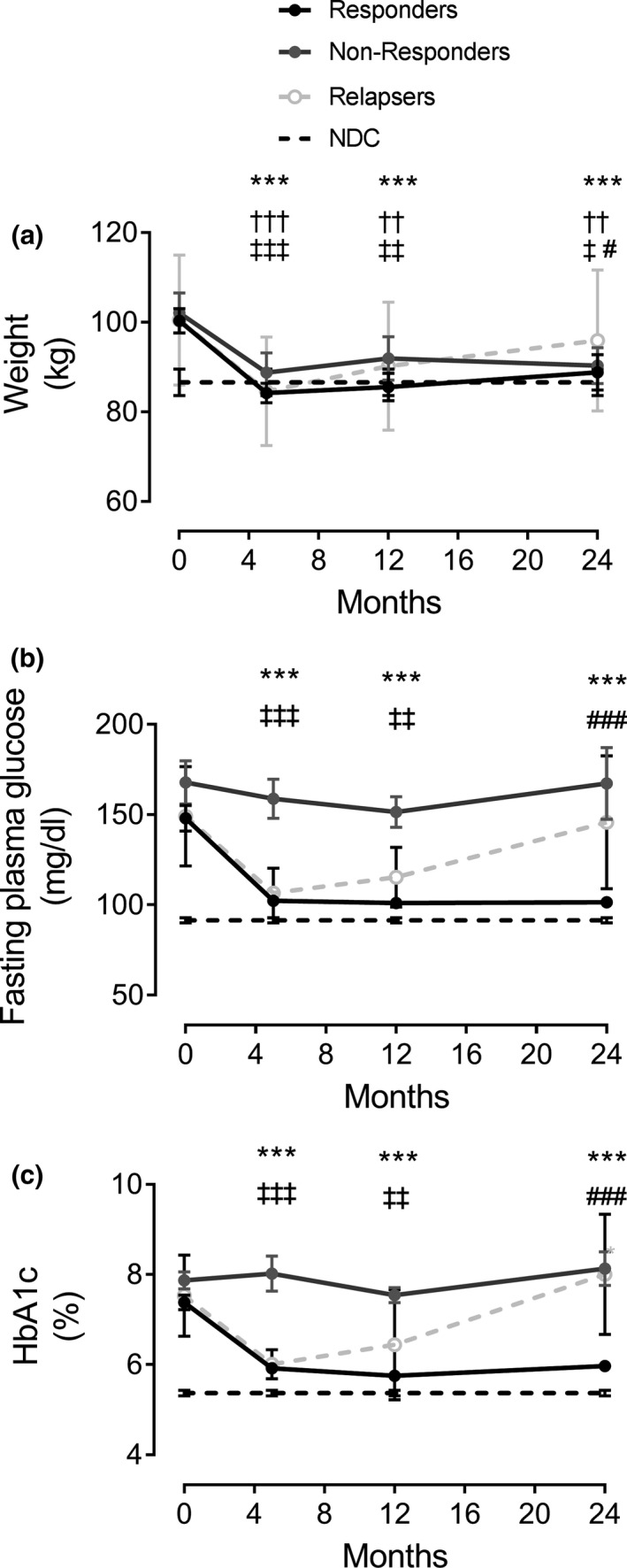
Change in weight and glucose control in DiRECT. Changes are shown for weight (panel a), fasting plasma glucose (panel b) and HbA1c (panel c) responders (solid circles) and nonresponders (open circles). Data on matched nondiabetic controls (NDC) studied at one time‐point are shown as a dotted line. Figure reproduced with permission from Taylor et al 2018 [38] with 24 month data from Al‐Mrabeh et al 2020 [36]. *** P < 0.001 responders vs. baseline; ††† P < 0.0001, †† P < 0.01 nonresponders vs. baseline; ‡‡ P < 0.0001, ‡‡ P < 0.001, ‡ P < 0.05 vs. baseline (relapsers); ### P < 0.0001, # P < 0.05, ### P < 0.001 relapsers vs. 5 months.
Longer‐term changes following weight loss
If weight regain is avoided after achieving reversal of the mechanisms underlying type 2 diabetes, both the mechanisms and the resulting plasma glucose control remain normal. The first study to demonstrate this was Counterbalance, which observed the effect of 6 months of weight stability after remission of type 2 diabetes [26]. Individualized eating advice (based upon goal setting, action planning and identification of potential barriers to success) was supported by monthly reviews. This achieved complete weight stability after the initial acute weight loss phase. Fig. 4 shows the maintenance of the acute fall in liver fat content, normalization of hepatic insulin resistance and stability of the recovered first‐phase insulin response. These data demonstrated that the profound pathophysiological changes underlying reversal of type 2 diabetes do not depend upon a continued hypocaloric state but rather are durable during normal eating.
The twin cycle hypothesis postulated that the link between excess fat in the liver and excess fat in the pancreas would be an increased production rate of very‐low‐density lipoprotein triglyceride (VLDL1‐TG) by the liver due largely to de novo lipogenesis. After 24 months of follow‐up in DiRECT, responders remained 10.5 ± 1.5 kg lighter than baseline and nonresponders 8.4 ± 1.4 kg lighter (nonsignificantly different at both 12 and 24 months) (Fig. 5). VLDL‐TG production rate fell during the weight loss phase and remained suppressed [26, 38] (Fig. 6). This was associated with corresponding decrease in plasma VLDL‐TG [36]. As the sole product of de novo lipogenesis is the saturated fat palmitic acid, and as VLDL‐triglyceride is produced only in the liver, the specific prediction of the twin cycle hypothesis could be tested by the measurement of palmitic acid content of plasma VLDL‐TG. This was observed to be elevated at baseline in type 2 diabetes compared with nondiabetic controls (52.0 ± 4.1 vs. 28.4 ± 3.2 µmol/L) [36]. Those who returned to and then maintained nondiabetic glucose control after weight loss were characterized by a decrease in VLDL‐TG palmitic acid content to normal, with maintenance at 31.6 ± 5.42 µmol/L at 24 months. The longer‐term, apparently steady state of remission of type 2 diabetes was thus associated with a decrease in overall hepatic export of triglyceride as VLDL‐TG.
Fig. 6.
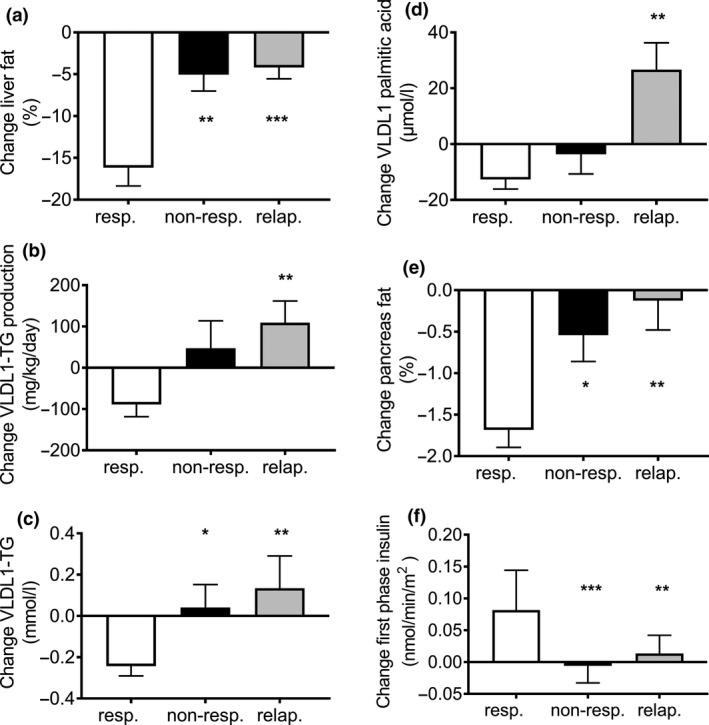
Changes over 2 years in DiRECT. Change in liver fat (a), hepatic VLDL1‐TG production (b), fasting plasma VLDL1‐TG (c), VLDL1 palmitic acid (d), intra‐pancreatic fat (e) and beta‐cell function (f) between baseline and 24 months in responders (white), nonresponders(black) and relapsers (grey). Relapsers show significant increase in liver fat, VLDL1‐TG production rate, VLDL‐TG plasma concentration and VLDL1‐TG palmitic acid with return to baseline levels of pancreas fat and first‐phase insulin secretion. Data are presented as mean ± SEM except for first‐phase insulin (median with IQ range). Figure reproduced with permission from Al‐Mrabeh A et al 2020 [36]. *P ≤ 0.05 vs. responders, **P ≤ 0.01 vs. responders, ***P ≤ 0.001 vs. responders.
Observation of type 2 diabetes over time has unequivocally shown that beta‐cell capacity falls with increasing duration of type 2 diabetes [41]. The impression of inevitable decline following diagnosis was reinforced by postmortem histological studies showing that beta‐cell number was decreased by 24–65% in type 2 diabetes. This became believed to be due to beta‐cell death or apoptosis [25, 42, 43]. However, the recent studies of pathogenesis draw attention to an overlooked aspect of these studies. The apparently irreversible beta‐cell loss in such observational studies is characterized by no weight loss but rather steady weight gain [41]. The Counterpoint, Counterbalance and DiRECT studies all demonstrate that the factor driving the processes of type 2 diabetes is the continued excess fat accumulation [13, 26, 36, 38]. If the body weight achieved by the time of onset of type 2 diabetes is maintained or increased, then type 2 diabetes indeed is inexorably progressive. Conversely, if the excess intra‐organ fat is removed by weight loss then the underlying processes are reversed to normal with no progression.
The detailed beta‐cell studies undertaken during 24 months of remission of type 2 diabetes have shown the very different time course of recovery of first‐phase insulin response and that of the maximal functional capacity of beta cells (Fig. 6). Following the gradual recovery of first‐phase response during the weeks of weight loss [13], the recovery process was shown to be complete by 5 months [38]. Thereafter, the recovered first‐phase response remained constant. In sharp contrast, recovery of maximal functional beta‐cell capacity was only modest at 5 months but was not only complete but also back to normal by 12 months. This full recovery was maintained steadily to 24 of remission [36].
The Look AHEAD study reported remission of type 2 diabetes after 4 years of follow‐up in 7.3% of participants (median duration of diabetes: 5 years) even though this was not a planned aim of the study, and this was associated with achieved mean weight loss of 8.6% [39]. The individuals who are successful in avoiding weight regain after remission remain nondiabetic [44].
After weight loss, some people are returned to a state of glucose metabolism that is clearly normal, whilst others achieve an HbA1c which is nondiabetic (<48mmol/mol), but in the range conventionally referral to as ‘prediabetic’. Even so, blood pressure and plasma lipids improve markedly, and such individuals have substantially improved their overall likelihood of long‐term good health. The Counterbalance study group were followed up for 6 months with the aim of avoiding weight regain, and mean blood pressure and lipids improved substantially [45]. The average ten‐year cardiovascular risk score (QRISK) [46] fell from 23% to 7% during the study and calculated heart age fell from 71 to 56 years (average chronological age: 55 years). The unfavourable cardiovascular and overall prognosis associated with ‘prediabetes’ clearly does not apply to people who lost sufficient weight to achieve remission of type 2 diabetes.
A term is now required to describe the HbA1c levels of those who have achieved stable weight, with normalized intra‐hepatic and intra‐pancreatic fat and reversed their type 2 diabetes. At 2 years, in DiRECT approximately one third of the responders achieved normal fasting plasma glucose and HbA1c. For the one third who remained below the diabetic range but not normal, the term ‘prediabetes’, which implies high cardiovascular risk, is inappropriate. It has been proposed that their metabolic state would most appropriately be termed ‘postdiabetes’, retaining the implication that they remain susceptible to diabetes if weight regain occurs [47]. However, if the UK consensus definition of remission is adopted [48] such a separate category would not be needed, and individuals in remission can be managed according to their personal results. They do not have diabetes, an important point for insurance purposes and also a motivating factor to avoid weight regain. Weight regain, which exceeds the personal fat threshold, will certainly bring about return of type 2 diabetes, and hence, ongoing support and follow‐up are required. In UK primary care, they would appropriately be assigned SNOMED code 703136005, indicating resolution of diabetes but need for ongoing annual checks [49].
The personal fat threshold
The personal fat threshold concept was developed to explain the clinical observation that individuals of ‘normal’ BMI could develop type 2 diabetes and that this would remit on weight loss. Fig. 7 illustrates how population norms are inappropriate to apply to individuals [50]. BMI is only a very general guide to a healthy metabolic weight for an individual.
Fig. 7.
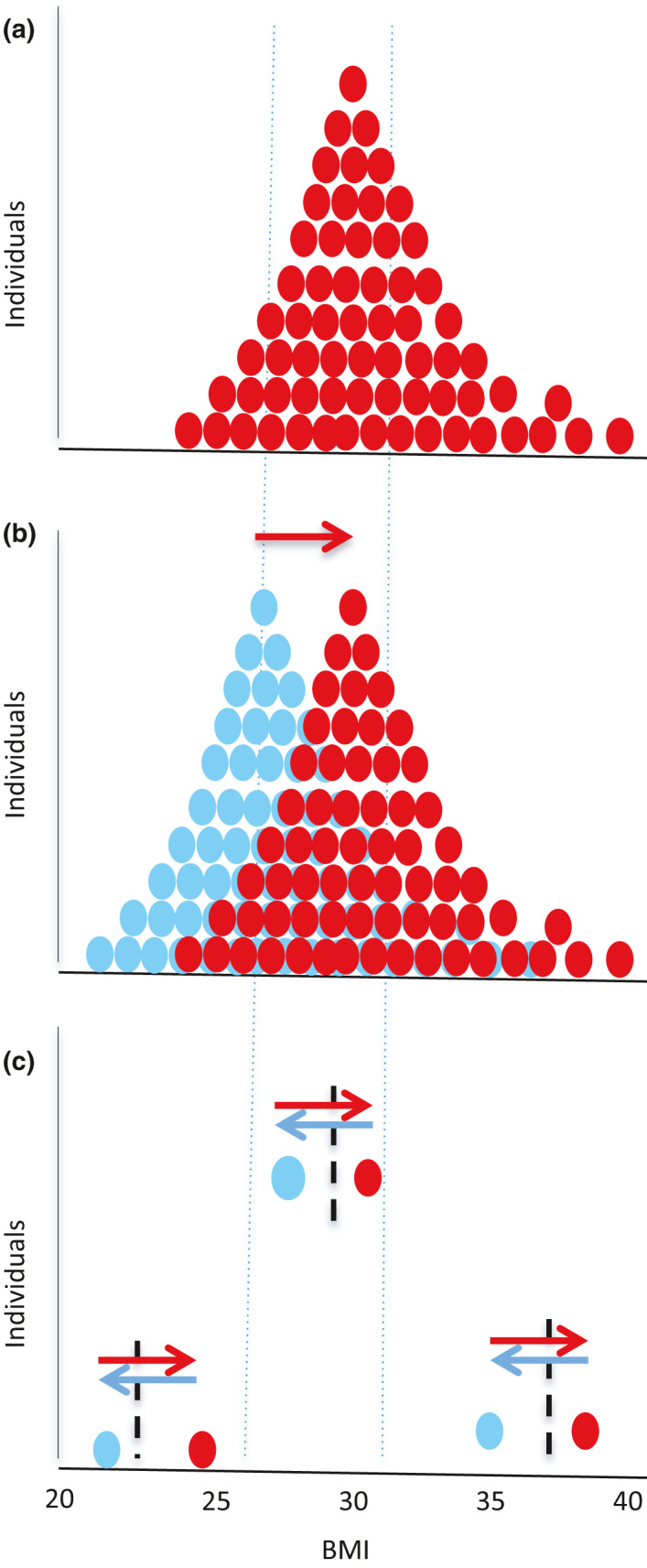
The personal fat threshold. (a) Frequency distribution of BMI for a typical group of individuals (red dotes) with type 2 diabetes (T2DM). (b) Frequency distribution of BMIs in blue for the individuals depicted in panel A before they gained weight. The red frequency distribution, when diabetes had developed, is right‐shifted (red arrow). This is commonly interpreted as a higher prevalence of obesity. (c) Three individuals from panel B are shown, one obese, one overweight and one ‘normal’ weight. Weight loss of 15 kg in each case results in return to normal glucose metabolism – although classification by the population measure of BMI remains the same. Each individual has a personal fat threshold (vertical dotted lines) above which excess fat is stored within liver and pancreas, and diabetes will develop if the individual also has beta cells susceptible to fat‐induced metabolic stress. For each such individual, moving to the right of their personal fat threshold triggers T2DM (red arrows), and moving to the left of the line restores normal glucose tolerance (blue arrows).
Type 2 diabetes is widely regarded as being caused by ‘obesity’. But in the 1970s, the Whitehall study showed only a small association between obesity and type 2 diabetes [51]. At the time, it was considered that obesity had no major effect on the development of type 2 diabetes [51, 52, 53]. Why was expert opinion so different then? It is known that the risk of type 2 diabetes rises most rapidly at very high BMIs. In 1980, only 7% of the population had a BMI > 30 kg/m2 [54], and the effect of high BMI upon development of diabetes was simply not detectable. This association with high BMI is now clear, although it has not generally been recognized that diabetes risk rises steadily throughout the population weight distribution and is not restriction to BMIs over 30. The Nurses’ Health Study showed a fourfold increase in type 2 diabetes prevalence in women with BMI 23–25 compared with those who maintained their BMI less than 22 kg/m2 [55]. In the UK Prospective Diabetes Study, which recruited between 1977 and 1991, 36% of individuals newly diagnosed with type 2 diabetes had a BMI < 25 kg/m2 [56], yet the initial weight loss phase achieved fasting plasma glucose of less than 6 mmol/L in 16% of the whole cohort [57]. Conversely, in the background population, most people (72%) of BMI > 40 kg/m2 currently do not have diabetes [58].
The misconception that type 2 diabetes is caused by having a BMI over 30 kg/m2 needs to be widely recognized. Many people continue to have BMI over 30 kg/m2 despite remission of type 2 diabetes, and the mean BMI after weight loss in Counterbalance and DiRECT was 28.6 and 31.5 kg/m2, respectively. Therefore, approximately half of each group remained obese by definition. Would the remaining excess fat redistribute into liver and pancreas despite keeping weight steady? This question can now be answered. The mean liver fat was 12.8% at baseline in Counterbalance, and weight loss brought about decrease to 2% (low normal) and this level was unchanged at 6 months. In DiRECT, liver fat was 16.0% and mean liver fat fell to 3.3% in responders and remained normal over 2 years (Fig. 6). A parallel pattern of change was observed in the pancreas fat. Once individuals had lost approximately 3 BMI units of weight, whatever the baseline BMI, type 2 diabetes could go into remission and remain in remission provided weight gain was avoided. Then, overspill of fat into ectopic sites including pancreas did not happen.
Case reports illustrate that nonobese individuals with type 2 diabetes can achieve remission after significant weight loss [59]. The concept that the pathogenesis of type 2 diabetes differs between people who are above or below certain BMI boundaries can readily be shown to be an artefact of inappropriate comparisons [50].
Type 2 diabetes in ethnic groups
Most studies of weight loss‐induced remission of type 2 diabetes have been carried out in Western countries and involved few non‐White participants [60, 61]. One exception to this is Look AHEAD, which included ~38% non‐White (mainly Hispanic and African American). This study achieved remission as expected from degree of weight loss achieved (11.5% and 7.3% remission at years 1 and 4, with weight loss of 8.6% and 4.7%, respectively), with no association of ethnicity [39]. A community‐based study showed slightly higher likelihood of remission in African Americans compared with the White population of California, with overall 7‐year remission of 4.6% in those with < 2 years of duration of type 2 diabetes [62]. A similar retrospective survey of older people observed slightly higher 8‐year rates of remission after dietary weight loss in Asian and Hispanic compared with White and African American groups [63]. A predominantly African Caribbean population in Barbados achieved comparable rates of weight loss‐induced remission to those documented in DiRECT with nine of 11 (82%) of those who lost more than 10 kg achieving remission [64].
Studies in South Asian and Middle Eastern populations have shown similar rates of remission for similar weight loss compared with White Europeans [65, 66, 67]. In India, remission of prediabetes by weight loss and exercise did not differ between ethnic groups [68, 69]. Further information on South Asians from the ongoing large prospective STANDby and DIADEM‐1 studies, using the same low‐calorie liquid diet as DiRECT, will soon be available.
The high level of acceptance of liquid formula diets to achieve rapid weight loss appears to be similar in non‐Europeans and Europeans [70, 71, 72].
African Americans typically have lower levels of intra‐hepatic and intra‐pancreatic fat compared with other ethnicities, and it has been suggested that this implies differing aetiology [73]. However, those with type 2 diabetes have higher levels than those without, and it would appear unlikely that the aetiology of the commonest metabolic disease of our species would differ between subgroups. The achievement of weight loss‐induced remission of type 2 diabetes in African Americans such as observed in Look AHEAD and in the Barbados study underscores this point [39, 64].
Physical activity, weight loss and weight maintenance
There is good evidence that long‐term weight maintenance is optimized by sustainable, daily physical activity combined with the moderate restraint of food intake [74]. However, the phenomenon of compensatory eating has received little attention [75, 76, 77]. When overweight or obese individuals commence an exercise regimen, weight does not fall and may increase, due to a partly subconscious increase in energy intake [76]. During the phase of dietary weight loss, it is important that new exercise programmes are not commenced. However, once weight loss has been achieved, a gradual sustainable increase in daily activity should be encouraged. Lack of recognition of the need for temporal separation of commencing a new exercise programme and initial weight loss is a potent reason for failure of advice to lose weight. This is particularly the case in the group who typically develop type 2 diabetes, who are no longer young and overweight.
As discussed above, muscle insulin resistance is the first detectable signal of risk for type 2 diabetes [33, 35], but it does not change during weight loss‐induced transition from diabetes to normal glucose metabolism [13, 26, 38, 78]. Before type 2 diabetes develops in an individual, muscle insulin resistance and hence postprandial storage of glucose as muscle glycogen are negligible [29, 34]. This drives meal‐derived glucose into de novo lipogenesis, and vigorous exercise training can ameliorate this [79, 80] with modest non‐weight‐related decrease in liver fat [81, 82]. Conversely, low physical activity promotes initiation of the twin cycles and ultimately increases the rate of development of type 2 diabetes. It was the investigation of muscle glycogen metabolism and relationship with liver fat, which set the scene for postulation of the twin cycle hypothesis [4].
The relationship between habitual exercise and the twin cycle predictions was examined in the IMI DIRECT Study [83]. This cross‐sectional study of people with type 2 diabetes demonstrated associations of exercise with both liver fat and liver insulin sensitivity according to the postulated liver cycle. Removing fat from intra‐organ sites is necessary – but not sufficient – to achieve remission of type 2 diabetes, and weight loss always achieves major decrease in liver fat and small decrease in pancreas fat with or without remission [38]. However, the very wide range of both pancreas fat and beta‐cell function in health and in type 2 diabetes inevitably prevents relationships such as with exercise being observed in cross‐sectional studies [26, 38, 41]. Only longitudinal study of individuals over time is likely to detect the small changes in intracytoplasmic fat in the pancreas, given the high background level of fat in adipocytes scattered throughout the parenchyma of the pancreas. In DiRECT, mean pancreas fat decreased from 8.7 to 7.8% (P < 0.0001), whereas liver fat decreased from 16.7 to 3.3% (P < 0.0001). Consequently, IMI DIRECT was unable to test any association of exercise with pancreas fat or calculated beta‐cell function.
Practical delivery of remission
Conventional dietetic approaches to achieving weight loss are unsuccessful in achieving the 10–15% weight loss required for remission of type 2 diabetes [84, 85]. Prolonged dieting with the associated hunger and daily decisions about what and how much to eat are major drawbacks. These adverse factors can be avoided by rapid weight loss using a liquid formula diet supplied in individual meal sachets [13, 26, 61, 86]. The very different activity of longer‐term avoidance of weight is then required. To induce weight loss to test the twin cycle hypothesis initially, an approach had to be devised, which would be feasible and acceptable in everyday normal life. As research tool, a liquid formula diet was combined with up to 240 g/day of nonstarchy vegetables to minimize constipation. The total energy intake in the Counterpoint and Counterbalance studied was therefore approximately 700 kcal/day [13, 26]. Notable well‐being was experienced by most people, and there was almost complete lack of hunger after the first 35 h [72]. The common belief that weight loss is more difficult to achieve in people with type 2 diabetes is contradicted by experimental evidence [87].
Rapid weight loss is not inevitably followed by rapid weight regain [87]. However, if weight loss by any regimen is not followed up by a supportive programme it is unlikely to be sustained. In Counterbalance, complete avoidance of weight regain was achieved monthly follow‐up for 6 months. DiRECT was designed to evaluate the success of this approach as applied by routine NHS primary care staff, as widespread application has to be affordable. During the 2‐year follow‐up, modest weight regain occurred over 2 years and 36% of the intervention group remained in by remission intention to treat [70]. Importantly, individuals were provided with clear information in advance that remission of their diabetes was possible, although not certain, provided weight loss was achieved and maintained, and they were advised to expect after weight loss to eat only two‐thirds of the amount of food previously consumed to maintain a steady body weight.
The nature of the ongoing support required after reversal of diabetes is important to consider. In contrast to the reassuringly simple liquid low‐energy diet, the return to normal eating after the liquid diet, with decisions about what and how much to eat, is a challenging time. A gradual, stepwise re‐introduction of normal foods was found to be effective in Counterbalance, and evidence from RCTs confirms that gradual transition to an isocaloric diet over a period of weeks is associated with improved weight maintenance at 12 months [88, 89]. During this phase of Counterbalance, weekly face‐to‐face review was undertaken, with specification of what and how much to eat. Individual dietary preferences were followed, but with avoidance of calorie‐dense foods. The calorie intake for weight stabilization was estimated from predictive equations [90]. Monthly review and weekly self‐weighing are important for weight stability [91]. Family support was observed to be critical for success. Spouses/partners typically report losing weight as well, and changing the obesogenic microenvironment of the home is a vital factor. Whilst some individuals can successfully maintain a new lower body weight under their own direction [92], most are likely to require ongoing support to limit weight regain over time [93].
Long‐term avoidance of weight regain is the most challenging aspect of weight management [94], although with appropriate support, long‐term success is possible for at least one in five people [74, 95]. The curious idea that one type of macronutrient content will suit all individuals is now displaced in official guidelines by a more person‐centred approach [96]. Mediterranean, low carbohydrate, low fat and intermittent energy restriction have each been shown to be effective [97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109]. Regular checking and recording of body weight are important [74].
Finally, the role of anorectic drugs in assisting long‐term weight stability requires to be examined. Although 87% of people who originally had lost more than 13.6kg (30 lb) were found to maintain more than 10% weight loss at 10 years [110], for the majority who were not so successful initially, appetite or habit may underlie the typical gradual weight regain. It is possible that the central aspect of type 2 diabetes may be addressed in future by supplementation of long‐term support after dietary weight loss with anorectic drug therapy. Recent data on GLP agonists are encouraging in this respect [111].
Acceptability and psychological impact
Following media interest in the Counterpoint results in 2011, a previously unrecognized aspect of the attitudes of people with type 2 diabetes became apparent. A considerable influx of emails and letters requested how‐to‐do‐it information, and this was placed on a website [112]. In contrast to the common medical view that ‘none of my patients with type 2 diabetes would want to do this’, it became clear that a sizable proportion of people with diabetes would go to any length to escape from what they regarded as a life sentence. A second wave of emails reported personal outcomes, and analysis of these showed a mean weight loss of 15 kg. Approximately half had used a liquid diet replacement, and half had used small portions of ordinary foods [92]. Weight loss was similar in both groups. When provided with clear information, motivated people could achieve remission of type 2 diabetes without medical help.
Formal psychological evaluation of people undertaking the low‐calorie diet and weight maintenance underscored the misperception of healthcare professionals in estimating the likely acceptability of this approach [72]. This group of people reported that the low‐calorie phase was easier than they had expected (although still challenging). The increased well‐being and energy levels provided ongoing motivation. Clearly, volunteers in a research study are a self‐selected group, and acceptability to the whole population of people with type 2 diabetes must be considered. Information on this was obtained in the recruitment phase of DiRECT. Letters were sent to all eligible people on the lists of participating primary care centres, and 28% volunteered for the study (compared with around 6% for most primary care research studies). Only 14% dropped out of the study in the first year [61]. Hence, for people in the first 6 years of type 2 diabetes, well over a quarter were motivated to participate despite the considerable time input required for research tests.
Twin cycle hypothesis revisited
Since publication of the hypothesis in 2008, magnetic resonance studies of weight loss have confirmed the predicted changes in liver and intra‐pancreatic fat along with abolition of liver insulin resistance and restoration of beta‐cell function in most people with short‐duration type 2 diabetes as described above [13, 26, 36, 78]. Additionally, the role of VLDL‐TG and especially palmitic acid, the product of de novo lipogenesis, is supported by these data. Demonstration that first‐phase insulin response returns over a period of months after calorie restriction and that the maximal functional capacity of beta cells returns completely to normal by 12 months confirms the original predictions [113]. Overturning the belief that type 2 diabetes was inevitably a lifelong, progressive condition has been a gradual process. Acceptance of the possibility of remission of type 2 diabetes increased from almost zero in 2008 to becoming recognized in the American Diabetes Association clinical practice guidelines in 2018 [114]. In the UK, an NHS pilot programme for remission of type 2 diabetes in the community was launched in 2020.
One feature of the twin cycle hypothesis, which was initially regarded as contentious, was the seminal role excess of fat supply to the pancreas. In order to establish whether weight loss itself brought about a decrease in intra‐pancreatic fat or whether this was specific to type 2 diabetes, a study of weight loss in matched groups of nondiabetic and type 2 diabetes was carried out [27]. With similar weight loss over 8 weeks, those with normal glucose tolerance had no change in intra‐pancreatic triglyceride, whereas those with type 2 diabetes decreased significantly from the higher baseline levels, demonstrating that there is an larger, labile pool of triglyceride within the pancreas in people with type 2 diabetes. Histologically, intracellular fat droplets are present in endocrine and exocrine cells of the pancreas and the fat content of each has been shown to correlate [115]. This is relevant as the MR measurement of intra‐pancreatic fat reflects the overall environment and predominantly parenchymal cell fat content. Human beta cells avidly take up fatty acids and store these as triglyceride, with loss of insulin secretory response to glucose [12]. In the Zucker diabetic fatty rat, a genetic model of spontaneous type 2 diabetes, the onset of hyperglycaemia is preceded by a rapid increase in pancreatic fat [11].
New information on loss of specialized function of the beta cell points to dedifferentiation as being the most likely mechanism [116, 117, 118, 119, 120]. The metabolic stress of chronic nutrient oversupply is particularly evident in vitro following palmitate exposure [121]. Impaired beta‐cell function in type 2 diabetes cannot be accounted for by increased apoptosis [122, 123]. The altered beta‐cell phenotype of type 2 diabetes may be associated with degranulation, explaining the decreased islet insulin‐positive area. There is a 350% increase in the number of dedifferentiated endocrine cells in type 2 diabetes [124]. In an ex vivo human islet lipotoxicity model, incubation with palmitate led to beta‐cell dysfunction associated with loss end‐differentiated phenotype evidenced by reduced levels of key beta‐cell transcription factors [121].
The twin cycle hypothesis provides a workable explanation of how long‐term remission of type 2 diabetes can be achieved. It can simply be explained, doctor to patient. Observations to date demonstrate that it explains the basic pathophysiological mechanisms involved and it has been the vehicle for rollout of clinical practice studies. Overall, the hypothesis carries major implications for clinical practice and specifically the approach to the management of this potentially reversible condition.
Conflict of interest
RT reports lecture fees from Lilly and Novartis, consultancy fees from Wilmington Healthcare, membership of the UK Government Scientific Committee on Nutrition working group on low‐carbohydrate diets and is author of the book ‘Life Without Diabetes’.
Acknowledgements
The author is enormously grateful to the research participants whose input made possible the clinical research described above. It would not have been possible without the hard work, skill and determination of many research associates, especially Ee Lim Lin, Sarah Steven, Ahmad Al‐Mrabeh, Kieren Hollingsworth, Carl Peters, Alison Barnes, Sviatlana Zhyzyneuskaya, Mavin Macauley, Mike Lean and Lucia Rehackova. This review is based on a lecture given to the 2019 Swedish Endocrine Society Annual Meeting.
Taylor R (Newcastle University, Newcastle, UK). Type 2 diabetes and remission: practical management guided by pathophysiology (Review). J Intern Med 2021;289:754–770. 10.1111/joim.13214
References
- 1. Himsworth HP. Diet in the aetiology of human diabetes. Proc Royal Soc Med 1949; 42: 323–6. [Google Scholar]
- 2. Franco M, Bilal U, Ordunez P et al. Population‐wide weight loss and regain in relation to diabetes burden and cardiovascular mortality in Cuba 1980–2010: repeated cross sectional surveys and ecological comparison of secular trends. Brit Med J 2013; 346: f1515. [DOI] [PubMed] [Google Scholar]
- 3. Schulz LO, Bennett PH, Ravussin E et al. Effects of traditional and western environments on prevalence of type 2 diabetes in Pima Indians in Mexico and the U.S. Diabetes Care 2006; 29: 1866–71. [DOI] [PubMed] [Google Scholar]
- 4. Taylor R. Pathogenesis of Type 2 diabetes: Tracing the reverse route from cure to cause. Diabetologia 2008; 51: 1781–9. [DOI] [PubMed] [Google Scholar]
- 5. Sattar N, McConnachie A, Ford I et al. Serial metabolic measurements and conversion to type 2 diabetes in the west of Scotland coronary prevention study: specific elevations in alanine aminotransferase and triglycerides suggest hepatic fat accumulation as a potential contributing factor. Diabetes 2007; 56: 984–91. [DOI] [PubMed] [Google Scholar]
- 6. Shibata M, Kihara Y, Taguchi M, Tashiro M, Otsuki M. Nonalcoholic fatty liver disease is a risk factor for type 2 diabetes in middle‐aged Japanese men. Diabetes Care 2007; 30: 2940–4. [DOI] [PubMed] [Google Scholar]
- 7. Petersen KF, Dufour S, Befroy D, Lehrke M, Hendler RE, Shulman GI. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes 2005; 54: 603–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ravikumar B, Gerrard J, Dalla Man C et al. Pioglitazone decreases fasting and postprandial endogenous glucose production in proportion to decrease in hepatic triglyceride content. Diabetes 2008; 57: 2288–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Seppala‐Lindroos A, Vehkavaara S, Hakkinen AM et al. Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. J Clin Endocrinol Metab 2002; 87: 3023–8. [DOI] [PubMed] [Google Scholar]
- 10. Boucher A, Lu D, Burgess SC et al. Biochemical mechanism of lipid‐induced impairment of glucose‐stimulated insulin secretion and reversal with a malate analogue. J Biol Chem 2004; 279: 27263–71. [DOI] [PubMed] [Google Scholar]
- 11. Lee Y, Hirose H, Ohneda M, Johnson JH, McGarry JD, Unger RH. Beta‐cell lipotoxicity in the pathogenesis of non‐insulin‐dependent diabetes mellitus of obese rats: impairment in adipocyte‐beta‐cell relationships. Proc Natl Acad Sci USA 1994; 91: 10878–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lalloyer F, Vandewalle B, Percevault F et al. Peroxisome proliferator‐activated receptor alpha improves pancreatic adaptation to insulin resistance in obese mice and reduces lipotoxicity in human islets. Diabetes 2006; 55: 1605–13. [DOI] [PubMed] [Google Scholar]
- 13. Lim EL, Hollingsworth KG, Aribisala BS, Chen MJ, Mathers JC, Taylor R. Reversal of type 2 diabetes: normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia 2011; 54: 2506–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hoffer LJ, Bistrian BR, Young VR, Blackburn GL, Matthews DE. Metabolic effects of very low calorie weight reduction diets. J Clin Invest 1984; 73: 750–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sevastianova K, Santos A, Kotronen A et al. Effect of short‐term carbohydrate overfeeding and long‐term weight loss on liver fat in overweight humans. Am J Clin Nutr 2012; 96: 427–34. [DOI] [PubMed] [Google Scholar]
- 16. Kelley DE, Wing R, Buonocore C, Sturis J, Polonsky K, Fitzsimmons M. Relative effects of calorie restriction and weight loss in noninsulin‐dependent diabetes mellitus. J Clin Endocrinol Metab 1993; 77: 1287–93. [DOI] [PubMed] [Google Scholar]
- 17. Yki‐Jarvinen H. Fat in the liver and insulin resistance. Ann Med. 2005; 37: 347–56. [DOI] [PubMed] [Google Scholar]
- 18. Erion DM, Shulman GI. Diacylglycerol‐mediated insulin resistance. Nat Med 2010; 16: 400–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cinti F, Bouchi R, Kim‐Muller JY et al. Evidence of beta‐Cell Dedifferentiation in Human Type 2 Diabetes. J Clin Endocrinol Metab 2016; 101: 1044–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pinnick K, Neville M, Clark A, Fielding B. Reversibility of metabolic and morphological changes associated with chronic exposure of pancreatic islet beta‐cells to fatty acids. J Cell Biochem 2010; 109: 683–92. [DOI] [PubMed] [Google Scholar]
- 21. Pinnick KE, Collins SC, Londos C, Gauguier D, Clark A, Fielding BA. Pancreatic ectopic fat is characterized by adipocyte infiltration and altered lipid composition. Obesity. 2008; 16: 522–30. [DOI] [PubMed] [Google Scholar]
- 22. White MG, Marshall HL, Rigby R et al. Expression of mesenchymal and alpha‐cell phenotypic markers in islet beta‐cells in recently diagnosed diabetes. Diabetes Care 2013; 36: 3818–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Butler AE, Cao‐Minh L, Galasso R et al. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia 2010; 53: 2167–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cnop M, Hughes SJ, Igoillo‐Esteve M et al. The long lifespan and low turnover of human islet beta cells estimated by mathematical modelling of lipofuscin accumulation. Diabetologia 2010; 53: 321–30. [DOI] [PubMed] [Google Scholar]
- 25. Butler AE, Janson J, Bonner‐Weir S, Ritzel R, Rizza RA, Butler PC. Beta‐cell deficit and increased beta‐cell apoptosis in humans with type 2 diabetes. Diabetes 2003; 52: 102–10. [DOI] [PubMed] [Google Scholar]
- 26. Steven S, Hollingsworth KG, Al‐Mrabeh A et al. Very low‐calorie diet and 6 months of weight stability in type 2 diabetes: pathophysiological changes in responders and nonresponders. Diabetes Care 2016; 39: 808–15. [DOI] [PubMed] [Google Scholar]
- 27. Steven S, Hollingsworth KG, Small P et al. Weight loss decreases excess pancreatic triacylglycerol specifically in type 2 diabetes. Diabetes Care 2016; 39: 158–65. [DOI] [PubMed] [Google Scholar]
- 28. Bruning JC, Michael MD, Winnay JN et al. A muscle‐specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell 1998; 2: 559–69. [DOI] [PubMed] [Google Scholar]
- 29. Carey PE, Halliday J, Snaar JEM, Morris PG, Taylor R. Direct assessment of muscle glycogen storage after mixed meals in normal and type 2 diabetic subjects. Am J Physiol 2003; 284: E286–94. [DOI] [PubMed] [Google Scholar]
- 30. Yki‐Jarvinen H, Helve E, Sane T, Nurjhan N, Taskinen MR. Insulin inhibition of overnight glucose production and gluconeogenesis from lactate in NIDDM. Am J Physiol 1989; 256: E732–9. [DOI] [PubMed] [Google Scholar]
- 31. Savage DB, Zhai L, Ravikumar B et al. A prevalent variant in PPP1R3A impairs glycogen synthesis and reduces muscle glycogen content in humans and mice. PLoS Med 2008; 5: e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Taylor R. Insulin resistance and type 2 diabetes. Diabetes 2012; 61: 778–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Petersen KF, Dufour S, Savage DB et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci USA 2007; 104: 12587–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Taylor R, Price TB, Katz LD, Shulman RG, Shulman GI. Direct measurement of change in muscle glycogen concentration after a mixed meal in normal subjects. Am J Physiol 1993; 265: E224–9. [DOI] [PubMed] [Google Scholar]
- 35. Lillioja S, Mott DM, Howard BV et al. Impaired glucose tolerance as a disorder of insulin action: Longitudinal and cross sectional studies in Pima Indians. N Engl J Med 1988; 318: 1217–25. [DOI] [PubMed] [Google Scholar]
- 36. Al‐Mrabeh A, Zhyzhneuskaya SV, Peters C et al. Hepatic Lipoprotein Export and Remission of Human Type 2 Diabetes after Weight Loss. Cell Metab 2020; 31: 233–49. [DOI] [PubMed] [Google Scholar]
- 37. Steven S, Taylor R. Restoring normoglycaemia by use of a very low calorie diet in long‐ and short‐duration Type 2 diabetes. Diabet Med 2015; 32: 1149–55. [DOI] [PubMed] [Google Scholar]
- 38. Taylor R, Al‐Mrabeh A, Zhyzhneuskaya S et al. Remission of human type 2 diabetes requires decrease in liver and pancreas fat content but is dependent upon capacity for beta cell recovery. Cell Metab. 2018; 28: 547–56. [DOI] [PubMed] [Google Scholar]
- 39. Gregg EW, Chen H, Wagenknecht LE et al. Association of an intensive lifestyle intervention with remission of type 2 diabetes. J Am Med Assoc 2012; 308: 2489–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sjostrom L, Peltonen M, Jacobson P et al. Association of bariatric surgery with long‐term remission of type 2 diabetes and with microvascular and macrovascular complications. J Am Med Assoc 2014; 311: 2297–304. [DOI] [PubMed] [Google Scholar]
- 41. U.K. prospective diabetes study 16 . Overview of 6 years' therapy of type II diabetes: a progressive disease. U.K. Prospective Diabetes Study Group. Diabetes 1995; 44: 1249–58. [PubMed] [Google Scholar]
- 42. Marchetti P, Bugliani M, Lupi R et al. The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia 2007; 50: 2486–94. [DOI] [PubMed] [Google Scholar]
- 43. Shimabukuro M, Zhou YT, Levi M, Unger RH. Fatty acid‐induced B‐cell apoptosis: a link between obesity and diabetes. Proc Nat Acad Sci USA 1998; 95: 2498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Peters C, Steven S, Taylor R. Reversal of type 2 diabetes by weight loss despite presence of macro‐and micro‐vascular complications. In: Draznin B, ed. Diabetes Case Studies: Real Problems, Practical Solutions. Alexandria, VA: American Diabetes Association, 2015. [Google Scholar]
- 45. Steven S, Hollingsworth KG, Al‐Mrabeh A et al. Very low calorie diet and 6 months of weight stability in type 2 diabetes: pathophysiological changes in responders and nonresponders. Diabetes Care 2016; 39: 158–65. [DOI] [PubMed] [Google Scholar]
- 46. The QRISK®2‐2016 risk calculator 2016. [Available from: https://qrisk.org/lifetime/.
- 47. Taylor R, Barnes AC. Can type 2 diabetes be reversed and how can this best be achieved? James Lind Alliance research priority number one. Diabet Med 2019; 36: 308–15. 10.1111/dme.13851. [DOI] [PubMed] [Google Scholar]
- 48. Nagi D, Hambling C, Taylor R. Remission of type 2 diabetes: a position statement from the association of british clinical diabetologists (ABCD) and the primary care diabetes society (PCDS). Br J Diabet. 2019; 19: 73–6. [Google Scholar]
- 49. McCombie L, Leslie W, Taylor R, Kennon B, Sattar N, Lean MEJ. Beating type 2 diabetes into remission. BMJ 2017; 358: j4030. [DOI] [PubMed] [Google Scholar]
- 50. Taylor R, Holman R. Normal weight individuals who develop Type 2 diabetes: the personal fat threshold. Clin Sci 2015; 128: 405–10. [DOI] [PubMed] [Google Scholar]
- 51. Jarrett RJ, Keen H, Fuller JH, McCartney M. Worsening to diabetes in men with impaired glucose tolerance ("borderline diabetes"). Diabetologia 1979; 16: 25–30. [DOI] [PubMed] [Google Scholar]
- 52. Taylor R. Aetiology of non‐insulin dependent diabetes. Br Med Bull 1989; 45: 73–91. [DOI] [PubMed] [Google Scholar]
- 53. Leslie RDG, Pyke DA. Genetics of Diabetes. In: Alberti KGMM, Krall LP, eds. Diabetes Annual. Amsterdam: Elsevier, 1985; 53–66. [Google Scholar]
- 54. Rosenbaum S, Skinner RK, Knight IB, Garrow JS. A survey of heights and weights of adults in Great Britain, 1980. Ann Hum Biol. 1985; 12: 115–27. [DOI] [PubMed] [Google Scholar]
- 55. Hu FB, Manson JE, Stampfer MJ et al. Diet, lifestyle, and the risk of type 2 diabetes mellitus in women. N Engl J Med. 2001; 345: 790–7. [DOI] [PubMed] [Google Scholar]
- 56. UKPDS . UK Prospective Diabetes Study (UKPDS). VIII. Study design, progress and performance. Diabetologia 1991; 34: 877–90. [PubMed] [Google Scholar]
- 57. UKPDS . UK Prospective Diabetes Study 7: response of fasting plasma glucose to diet therapy in newly presenting type II diabetic patients, UKPDS Group. Metabolism 1990; 39: 905–12. [PubMed] [Google Scholar]
- 58. Gregg EW, Cheng YJ, Narayan KM, Thompson TJ, Williamson DF. The relative contributions of different levels of overweight and obesity to the increased prevalence of diabetes in the United States: 1976–2004. Prev Med. 2007; 45: 348–52. [DOI] [PubMed] [Google Scholar]
- 59. Doughty R.Type 2 diabetes and the diet that cured me 2013. Available from: http://www.theguardian.com/lifeandstyle/2013/may/12/type‐2‐diabetes‐diet‐cure
- 60. Dambha‐Miller H, Day AJ, Strelitz J, Irving G, Griffin SJ. Behaviour change, weight loss and remission of Type 2 diabetes: a community‐based prospective cohort study. Diabetic Med. 2020; 37: 681–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lean ME, Leslie WS, Barnes AC et al. Primary care‐led weight management for remission of type 2 diabetes (DiRECT): an open‐label, cluster‐randomised trial. Lancet 2018; 391: 541–51. [DOI] [PubMed] [Google Scholar]
- 62. Karter AJ, Nundy S, Parker MM, Moffet HH, Huang ES. Incidence of remission in adults with type 2 diabetes: the diabetes & aging study. Diabetes Care 2014; 37: 3188–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tangelloju S, Little BB, Esterhay RJ, Brock G, LaJoie AS. Type 2 Diabetes Mellitus (T2DM) "Remission" in Non‐bariatric Patients 65 Years and Older. Front Public Health. 2019; 7: 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bynoe K, Unwin N, Taylor C et al. Inducing remission of Type 2 diabetes in the Caribbean: findings from a mixed methods feasibility study of a low‐calorie liquid diet‐based intervention in Barbados. Diabetic Med 2020; 37: 1816–1824. [DOI] [PubMed] [Google Scholar]
- 65. Bhatt AA, Choudhari PK, Mahajan RR et al. Effect of a low‐calorie diet on restoration of normoglycemia in obese subjects with type 2 diabetes. Indian Med J 2017; 21: 776–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sarathi V, Kolly A, Chaithanya HB, Dwarakanath CS. High rates of diabetes reversal in newly diagnosed Asian Indian young adults with type 2 diabetes mellitus with intensive lifestyle therapy. J Nat Sci Biol Med. 2017; 8: 60–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Umphonsathien M, Prutanopajai P, Aiam ORJ et al. Immediate and long‐term effects of a very‐low‐calorie diet on diabetes remission and glycemic control in obese Thai patients with type 2 diabetes mellitus. Food Sci Nutr 2019; 7: 1113–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Snehalatha C, Mary S, Selvam S et al. Changes in insulin secretion and insulin sensitivity in relation to the glycemic outcomes in subjects with impaired glucose tolerance in the Indian Diabetes Prevention Programme‐1 (IDPP‐1). Diabetes Care 2009; 32: 1796–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Nanditha A, Ram J, Snehalatha C et al. Early improvement predicts reduced risk of incident diabetes and improved cardiovascular risk in prediabetic Asian Indian men participating in a 2‐year lifestyle intervention program. Diabetes Care 2014; 37: 3009–15. [DOI] [PubMed] [Google Scholar]
- 70. Lean MEJ, Leslie WS, Barnes AC et al. Durability of a primary care‐led weight‐management intervention for remission of type 2 diabetes: 2‐year results of the DiRECT open‐label, cluster‐randomised trial. Lancet Diabet Endocrinol 2019; 7: 344–55. [DOI] [PubMed] [Google Scholar]
- 71. Rehackova L, Araujo‐Soares V, Steven S, Adamson AJ, Taylor R, Sniehotta FF. Behaviour change during dietary Type 2 diabetes remission: a longitudinal qualitative evaluation of an intervention using a very low energy diet. Diabetic Med 2020; 37: 953–62. [DOI] [PubMed] [Google Scholar]
- 72. Rehackova L, Araújo‐Soares V, Adamson AJ, Stevens S, Taylor R, Sniehotta FF. Acceptability of a very low energy diet in type 2 diabetes: patient experiences and behaviour regulation. Diabet Med 2017; 34: 1554–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Szczepaniak LS, Victor RG, Mathur R et al. Pancreatic Steatosis and Its Relationship to beta‐Cell Dysfunction in Humans: Racial and Ethnic Variations. Diabetes Care 2012; 35: 2377–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wing RR, Phelan S. Long‐term weight loss maintenance. Am J Clin Nutr. 2005; 82(1 Suppl): 222S–5S. [DOI] [PubMed] [Google Scholar]
- 75. Finlayson G, Bryant E, Blundell JE, King NA. Acute compensatory eating following exercise is associated with implicit hedonic wanting for food. Physiol Behav. 2009; 97: 62–7. [DOI] [PubMed] [Google Scholar]
- 76. Hopkins M, Blundell JE, King NA. Individual variability in compensatory eating following acute exercise in overweight and obese women. Br J Sports Med. 2014; 48: 1472–6. [DOI] [PubMed] [Google Scholar]
- 77. King NA, Horner K, Hills AP et al. Exercise, appetite and weight management: understanding the compensatory responses in eating behaviour and how they contribute to variability in exercise‐induced weight loss. Br J Sports Med. 2012; 46: 315–22. [DOI] [PubMed] [Google Scholar]
- 78. Taylor R, Al‐Mrabeh A, Sattar N. Understanding the mechanisms of reversal of type 2 diabetes. Lancet Diabetes Endocrinol. 2019; 7: 726–736. [DOI] [PubMed] [Google Scholar]
- 79. Rabol R, Petersen KF, Dufour S, Flannery C, Shulman GI. Reversal of muscle insulin resistance with exercise reduces postprandial hepatic de novo lipogenesis in insulin resistant individuals. Proc Natl Acad Sci USA. 2011; 108: 13705–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Flannery C, Dufour S, Rabol R, Shulman GI, Petersen KF. Skeletal muscle insulin resistance promotes increased hepatic de novo lipogenesis, hyperlipidemia, and hepatic steatosis in the elderly. Diabetes 2012; 61: 2711–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Cassidy S, Thoma C, Hallsworth K et al. High intensity intermittent exercise improves cardiac structure and function and reduces liver fat in patients with type 2 diabetes: a randomised controlled trial. Diabetologia 2016; 59: 56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hallsworth K, Fattakhova G, Hollingsworth KG et al. Resistance exercise reduces liver fat and its mediators in non‐alcoholic fatty liver disease independent of weight loss. Gut 2011; 60: 1278–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Koivula RW, Atabaki‐Pasdar N, Giordano GN et al. The role of physical activity in metabolic homeostasis before and after the onset of type 2 diabetes: an IMI DIRECT study. Diabetologia 2020; 63: 744–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Shai I, Schwarzfuchs D, Henkin Y et al. Weight loss with a low‐carbohydrate, Mediterranean, or low‐fat diet. N Engl J Med 2008; 359: 229–41. [DOI] [PubMed] [Google Scholar]
- 85. van Zuuren EJ, Fedorowicz Z, Kuijpers T, Pijl H. Effects of low‐carbohydrate‐ compared with low‐fat‐diet interventions on metabolic control in people with type 2 diabetes: a systematic review including GRADE assessments. Am J Clin Nutr. 2018; 108: 300–31. [DOI] [PubMed] [Google Scholar]
- 86. Henry RR, Wallace P, Olefsky JM. Effects of weight loss on mechanisms of hyperglycaemia in obese non‐insulin dependent diabetes mellitus. Diabetes 1986; 35: 990–8. [DOI] [PubMed] [Google Scholar]
- 87. Leslie WS, Taylor R, Harris L, Lean ME. Weight losses with low‐energy formula diets in obese patients with and without type 2 diabetes: systematic review and meta‐analysis. Int J Obes (Lond). 2017; 41: 96–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Johansson K, Neovius M, Hemmingsson E. Effects of anti‐obesity drugs, diet, and exercise on weight‐loss maintenance after a very‐low‐calorie diet or low‐calorie diet: a systematic review and meta‐analysis of randomized controlled trials. Am J Clin Nutr 2014; 99: 14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Gripeteg L, Torgerson J, Karlsson J, Lindroos AK. Prolonged refeeding improves weight maintenance after weight loss with very‐low‐energy diets. Br J Nutr 2010; 103: 141–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Mifflin MD, St Jeor ST, Hill LA, Scott BJ, Daugherty SA, Koh YO. A new predictive equation for resting energy expenditure in healthy individuals. Am J Clin Nutr. 1990; 51: 241–7. [DOI] [PubMed] [Google Scholar]
- 91. Shieh C, Knisely MR, Clark D, Carpenter JS. Self‐weighing in weight management interventions: A systematic review of literature. Obesity Res Clin Pract 2016; 10: 493–519. [DOI] [PubMed] [Google Scholar]
- 92. Steven S, Lim E, Taylor R. Population response to information on reversibility of type 2 diabetes. Diabet Med 2013; 30: e135–8. [DOI] [PubMed] [Google Scholar]
- 93. Middleton KMR, Patidar SM, Perri MG. The impact of extended care on the long‐term maintenance of weight loss: a systematic review and meta‐analysis. Obes Rev 2012; 13: 509–17. [DOI] [PubMed] [Google Scholar]
- 94. MacLean PS, Wing RR, Davidson T et al. NIH Working Group Report: Innovative Research to Improve Maintenance of Weight Loss. Obesity. 2015; 23: 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. van Wyk HJ, Davis RE, Davies JS. A critical review of low‐carbohydrate diets in people with Type 2 diabetes. Diabet Med 2016; 33: 148–57. [DOI] [PubMed] [Google Scholar]
- 96. Dyson PA, Kelly T, Deakin T et al. Diabetes UK evidence‐based nutrition guidelines for the prevention and management of diabetes. Diabet Med 2011; 28: 1282–8. [DOI] [PubMed] [Google Scholar]
- 97. Feinman RD, Pogozelski WK, Astrup A et al. Dietary carbohydrate restriction as the first approach in diabetes management: critical review and evidence base. Nutrition 2015; 31: 1–13. [DOI] [PubMed] [Google Scholar]
- 98. Spiro A, Stanner S. The National Obesity Forum report is an opinion piece not a scientific review. Nutr Bullet 2016; 41: 257–69. [Google Scholar]
- 99. Estruch R, Martinez‐Gonzalez MA, Corella D et al. Effect of a high‐fat Mediterranean diet on bodyweight and waist circumference: a prespecified secondary outcomes analysis of the PREDIMED randomised controlled trial. Lancet Diabetes Endocrinol. 2016; 4: 666–76. [DOI] [PubMed] [Google Scholar]
- 100. Garcia‐Fernandez E, Rico‐Cabanas L, Rosgaard N, Estruch R, Bach‐Faig A. Mediterranean diet and cardiodiabesity: a review. Nutrients. 2014; 6: 3474–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Martinez‐Gonzalez MA, Martin‐Calvo N. Mediterranean diet and life expectancy; beyond olive oil, fruits, and vegetables. Curr Opin Clin Nutr Metab Care 2016; 19: 401–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Esposito K, Maiorino MI, Petrizzo M, Bellastella G, Giugliano D. The effects of a Mediterranean diet on the need for diabetes drugs and remission of newly diagnosed type 2 diabetes: follow‐up of a randomized trial. Diabetes Care 2014; 37: 1824–30. [DOI] [PubMed] [Google Scholar]
- 103. Davis CS, Clarke RE, Coulter SN et al. Intermittent energy restriction and weight loss: a systematic review. Eur J Clin Nutr 2016; 70: 292–9. [DOI] [PubMed] [Google Scholar]
- 104. Harvie M, Wright C, Pegington M et al. The effect of intermittent energy and carbohydrate restriction v. daily energy restriction on weight loss and metabolic disease risk markers in overweight women. Br J Nutr. 2013; 110: 1534–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Carter S, Clifton PM, Keogh JB. The effects of intermittent compared to continuous energy restriction on glycaemic control in type 2 diabetes; a pragmatic pilot trial. Diabetes Res Clin Pract 2016; 122: 106–12. [DOI] [PubMed] [Google Scholar]
- 106. Harvie MN, Howell T. Could intermittent energy restriction and intermittent fasting reduce rates of cancer in obese, overweight, and normal‐weight subjects? A summary of evidence. Adv Nutr 2016; 7: 690–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Clayton DJ, Stensel DJ, James LJ. Effect of breakfast omission on subjective appetite, metabolism, acylated ghrelin and GLP‐17‐36 during rest and exercise. Nutrition 2016; 32: 179–85. [DOI] [PubMed] [Google Scholar]
- 108. Kealey T. Breakfast is a Dangerous Meal. London: 4th Estate; 2016. [Google Scholar]
- 109. Athinarayanan SJ, Adams RN, Hallberg SJ et al. Long‐term effects of a novel continuous remote care intervention including nutritional ketosis for the management of type 2 diabetes: a 2‐year non‐randomized clinical trial. Front Endocrinol 2019; 10: 348–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Thomas JG, Bond DS, Phelan S, Hill JO, Wing RR. Weight‐loss maintenance for 10 years in the National Weight Control Registry. Am J Prev Med. 2014; 46: 17–23. [DOI] [PubMed] [Google Scholar]
- 111. O'Neil PM, Birkenfeld AL, McGowan B et al. Efficacy and safety of semaglutide compared with liraglutide and placebo for weight loss in patients with obesity: a randomised, double‐blind, placebo and active controlled, dose‐ranging, phase 2 trial. Lancet 2018; 392(10148): 637–49. [DOI] [PubMed] [Google Scholar]
- 112. Taylor R.Reversing Type 2 diabetes 2011. Available from: http://go.ncl.ac.uk/diabetes‐reversal
- 113. Zhyzhneuskaya SV, Al‐Mrabeh A, Peters C et al. Time course of normalization of functional beta‐cell capacity in the diabetes remission clinical trial after weight loss in type 2 diabetes. Diabetes Care 2020; 43: 813–20. [DOI] [PubMed] [Google Scholar]
- 114. Davies MJ, D'Alessio DA, Fradkin J et al. Management of hyperglycemia in type 2 diabetes, 2018. a consensus report by the American Diabetes Association (ADA) and the European Association for the. Study of Diabetes (EASD). Diabetes Care 2018; 41: 2669–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Lee Y, Lingvay I, Szczepaniak LS, Ravazzola M, Orci L, Unger RH. Pancreatic steatosis: harbinger of type 2 diabetes in obese rodents. Int J Obes. 2009; 34: 396–400. [DOI] [PubMed] [Google Scholar]
- 116. Brereton MF, Iberl M, Shimomura K et al. Reversible changes in pancreatic islet structure and function produced by elevated blood glucose. Nat Commun. 2014; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Spijker HS, Song H, Ellenbroek JH et al. Loss of β‐Cell Identity Occurs in Type 2 Diabetes and Is Associated With Islet Amyloid Deposits. Diabetes 2015; 64: 2928–38. [DOI] [PubMed] [Google Scholar]
- 118. Talchai C, Xuan S, Lin Hua V, Sussel L, Accili D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 2012; 150: 1223–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Wang Z, York Nathaniel W, Nichols Colin G, Remedi Maria S. Pancreatic β cell dedifferentiation in diabetes and redifferentiation following insulin therapy. Cell Metab 2014; 19: 872–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. White MG, Marshall HL, Rigby R et al. Expression of mesenchymal and α‐cell phenotypic markers in islet β‐cells in recently diagnosed diabetes. Diabetes Care 2013; 36: 3818–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Cnop M, Abdulkarim B, Bottu G et al. RNA sequencing identifies dysregulation of the human pancreatic islet transcriptome by the saturated fatty acid palmitate. Diabetes 2014; 63: 1978–93. [DOI] [PubMed] [Google Scholar]
- 122. Marselli L, Suleiman M, Masini M et al. Are we overestimating the loss of beta cells in type 2 diabetes? Diabetologia 2013; 57: 362–5. [DOI] [PubMed] [Google Scholar]
- 123. Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC. Pancreatic β‐cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab 2008; 10: 32–42. [DOI] [PubMed] [Google Scholar]
- 124. Cinti F, Bouchi R, Kim‐Muller JY, Ohmura Y, Sandoval PR, Masini M et al. Evidence of β‐cell dedifferentiation in human type 2 diabetes. J Clin Endocrinol Metab 2016; 101: 1044–54. [DOI] [PMC free article] [PubMed] [Google Scholar]