

Abstract
The enzyme butyrylcholinesterase (BChE) represents a promising target for imaging probes to potentially enable early diagnosis of neurodegenerative diseases like Alzheimer's disease (AD) and to monitor disease progression in some forms of cancer. In this study, we present the design, facile synthesis, in vitro and preliminary ex vivo and in vivo evaluation of a morpholine‐based, selective inhibitor of human BChE as a positron emission tomography (PET) tracer with a pseudo‐irreversible binding mode. We demonstrate a novel protecting group strategy for 18F radiolabeling of carbamate precursors and show that the inhibitory potency as well as kinetic properties of our unlabeled reference compound were retained in comparison to the parent compound. In particular, the prolonged duration of enzyme inhibition of such a morpholinocarbamate motivated us to design a PET tracer, possibly enabling a precise mapping of BChE distribution.
Keywords: carbamate, enzyme kinetics, fluorine-18, positron emission tomography, radiotracers
PET tracer: A pseudo‐irreversible butyrylcholinesterase inhibitor with prolonged duration of action, high inhibitory potency and pronounced selectivity was used to design an 18F‐labeled radiotracer for positron emission tomography. Syntheses and a new protecting group strategy for radiolabeling carbamates are presented, binding and kinetics are investigated, and the radiotracer is tested in ex vivo autoradiography and in vivo microPET studies.
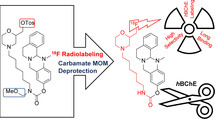
Introduction
Dementia and its most frequent form, Alzheimer's disease (AD), are increasingly challenging health‐care systems all around the world. The number of affected people doubled from 1990 to 2016, meaning that there are up to 50 million patients right now. On the one hand, this can be correlated with a growing and aging world population, on the other hand, there is evidence that avoiding risk factors like high body mass index or smoking can delay the onset and therefore contribute to a reduced future number of affected people, as there is still no chance to cure or even modify the progress of AD. [1] Alzheimer published the first description of histological alterations, [2] namely amyloid β deposits (Aβ plaques) and neurofibrillary tangles (NFTs), which occur during the progression of the disease named after him and are accompanied by memory deficits and cognitive decline. [3] These pathological markers are still used to make a definite diagnosis of AD, which, however, can only take place post mortem. [4] The fatal consequences of the disease can be ascribed to a proceeding loss of cholinergic neurons and a decline of the neurotransmitter acetylcholine, which is metabolized by the serine hydrolase acetylcholinesterase (AChE). [5] Apart from the N‐methyl‐d‐aspartate receptor antagonist memantine, all of the approved drugs on the market to combat AD are AChE inhibitors and can ameliorate symptoms in the early stage of the disease by preventing further degradation of acetylcholine through enzyme hydrolysis. This is far from any disease‐modifying or curative treatment, and, in general, the complex pathologies of AD are yet too little understood.[ 5a , 6 ] However, there is evidence that butyrylcholinesterase (BChE) represents an attractive target for novel imaging probes in AD diagnostics and such imaging probes represent tool compounds to provide further insights into BChE's involvement in neuropathologies and consequently for more powerful drugs to combat the disease. It was shown that in late‐stage AD, the level of AChE dramatically decreases by approximately 90 %, while BChE activity remains unaffected or can be elevated by up to 30 %. [7] Furthermore, in the human brain large amounts of BChE can be found in regions associated with cognition and behavior, which are among the first areas affected by neurodegeneration. [8] Last, there is evidence that BChE is involved in AD pathologies by generating harmful attributes of Aβ plaques. [9] In up to 30 % of cognitively normal older adults, Aβ plaques appear without being colocalized with BChE, which is the case in AD. [10] Consequently, it has been shown in several studies that there is a positive, symptomatic effect on memory deficits and cognitive decline, when BChE is inactivated. [11] Moreover, it is possible to reverse the effect of injected Aβ25–35 in an AD mouse model with highly selective BChE inhibitors exhibiting a pronounced neuroprotective profile, providing first hints that a disease‐modifying treatment can potentially be achieved. [12] Besides, BChE is known to play an important physiological role in serum metabolism by detoxifying xenobiotics or naturally occurring bioactive compounds with its ability to hydrolyze ester or carbamate functionalities. Therefore, the active site exhibits a low substrate specificity compared to AChE. [13] Additionally, there is evidence that BChE contributes to other diseases, including cardiovascular pathologies, obesity or diabetes mellitus type 2. [14] Interestingly, both ChEs are known to be involved in cellular proliferation and differentiation, which has raised attention to their potential role in tumorigenesis. [15] For example, the malignancy grade of tumors in sporadic breast cancer is positively correlated with the total number of alterations in the BChE gene. [16] Furthermore, a biphasic alteration of BChE level was observed in prostate cancer, meaning a downregulation in the early stage and a subsequent upregulation in the late‐stage of the disease, which is additionally correlated with recurrence and reduction of disease‐free survival. [17] Consequently, there is the need for imaging probes selectively targeting BChE not only as potential AD diagnostics to support a diagnosis delimiting other forms of dementia, but also to shed new light on the pathologies of other diseases with related altered BChE activity, expression levels and occurrence. For this reason, positron emission tomography (PET) radiotracers have been developed to gain information about the spatial and temporal distribution of an injected, radiolabeled compound. Ideally, the tracer should address the target of choice with high affinity and selectivity. [18] In case of BChE, one can design either an inhibitor‐ or substrate‐based compound for radiolabeling, while the former can be classified in reversibly or irreversibly acting inhibitors. Even though substrate analogs as BChE PET tracers can usually pass the blood–brain barrier (BBB), their hydrolysis rate is often too high to achieve an accurate estimation of the actual enzyme distribution in the brain.[ 6a , 19 ] 1‐11C‐Methyl‐4‐piperidinyl‐n‐butyrate (11C‐MP4B) and N‐[18F]fluoroethylpiperidin‐4‐ylmethyl butyrate are examples of such tracers with a pronounced rate of hydrolysis in brain and plasma. The biodistribution of 11C‐MP4B in the brains of AD patients most widely represented the neuroanatomy of BChE known from post mortem studies of human brain, apart from a much higher uptake in the striatum than in the cortex. Further studies are required to fully characterize the diagnostic value of both tracers.[ 6a , 20 ] N‐Methylpiperidin‐4‐yl 4‐[123I]iodobenzoate was consequently developed to overcome these problems and indeed it exhibited slower hydrolysis and retained capability of BBB penetration. It was possible to demonstrate increased brain retention in an AD mouse model compared to wild type and single‐photon emission computed tomography (SPECT) studies revealed that radioactivity accumulated in cortical and subcortical brain areas in accordance with BChE activity known from histochemistry. [21] Thereby first preclinical evidence was provided that it is possible to distinguish healthy and AD mice by targeting BChE with imaging probes.
This approach was extended to an inhibitor‐based, irreversibly acting ChE inhibitor, phenyl 4‐[123I]‐iodophenylcarbamate (123I‐PIP). In ex vivo autoradiography studies using brain tissue of AD patients, cognitively normal, Aβ positive older adults and Aβ negative aged humans, this tracer could discriminate AD tissue from the other two. [22] However, it is not selective towards BChE, ex vivo studies are preliminary due to a limited number of investigated brain tissues and the in vivo assessment of the tracer is still pending. [22] Additionally, 123I‐PIP was investigated in a recent study for its potential ability to detect pathological changes in multiple sclerosis (MS). In vitro autoradiography revealed that the tracer can detect MS lesions in white matter by binding to BChE. [23]
Other irreversible ChE inhibitors are organophosphates and a respective PET tracer was designed to target central nervous system AChE in rats, but these compounds are highly toxic nerve agents. [24] However, due to the lack of results concerning irreversibly acting and selective BChE PET tracers as potential in vivo AD diagnostics, we continued our previous studies, in which we had investigated enzyme kinetics and binding, radiolabeling and preliminary ex vivo brain tissue binding of pseudo‐irreversible, selective BChE inhibitors with high affinity.[ 12 , 19 , 25 ] Starting point of our investigations was heptylcarbamate 1a (Figure 1) bearing a tetracyclic carrier scaffold. Since this compound turned out to be a highly potent carbamate‐based BChE inhibitor with pronounced selectivity over AChE, we performed kinetic studies to gain information about the complex binding mode of this class of BChE inhibitors (Figure 1). In the first binding step of a carbamate‐based BChE inhibitor [C−X] to enzyme [E] the complex [EC−X] forms spontaneously, which may either dissociate or result in a transfer of the carbamate group C onto the enzyme E, which is subsequently inhibited by means of a covalent bond. Under the presumption that the formation of [EC‐X], characterized by the kinetic rate constant k 1, is considerably faster than the chemical reaction of carbamate transfer, the affinity of carbamate‐based BChE inhibitors can be characterized by equilibrium constant K c. Dissociation of [EC‐X] is kinetically characterized by the rate constant k 2. The carbamate transfer from the tetracyclic carrier scaffold [X] onto the enzyme [E] is a nucleophilic substitution reaction, in which the serine in the catalytically active site of BChE attacks the activated carbon of the carbamate group, while the carrier scaffold X is released as free alcohol. This chemical reaction is described by the kinetic rate constant k 3 and results in the formation of the carbamylated enzyme E‐C, that is, the catalytic site in the enzyme is blocked by a covalent bond. Due to the inherent chemical instability of carbamate bonds, the enzyme is not inhibited in an irreversible fashion, but enzyme activity is reconstituted by hydrolysis of the carbamate, that is, a second chemical reaction characterized through rate constant k 4 and half‐life t 1/2 of the carbamylated state.[ 12 , 25 , 26 ] For the reason of their complex binding mode, carbamate‐based PET tracers on the one hand need to be designed particularly carefully with respect to their kinetic behavior, but on the other hand offer great opportunities in their application. The first aspect to be considered is the speed of carbamylation (k 3), that is, the speed at which the enzyme is deactivated and is, hence, radiolabeled through transfer of a carbamate group comprising a positron emitting radionuclide. Nevertheless, the carbamylation rate requires precise tuning: If the rate is too high, the tracer may bind to BChE circulating in the blood immediately hampering the suitability for BChE imaging in the CNS. PET scans thus obtained only reflect the blood flow and highlight highly vascularized areas of the body, such as brain, liver, heart, kidneys, lung and the like, that is, high brain uptakes may be found although not reflecting true BChE distribution throughout the body. If the carbamylation rate is too slow, however, the tracer may be metabolized before binding to BChE occurs to a considerable extent, also rendering the tracer inappropriate. Only if the carbamylation speed is at an appropriate level, a molecular scan representing the true distribution of BChE throughout the body and CNS can be obtained. Once the enzyme is labeled with the radionuclide, however, slow hydrolysis and long half‐life time of the carbamylated enzyme state result in a prolonged retention of radioactivity in the area of interest allowing for increased time of observation and highly flexible study design. Therefore, the ultimate aim for the successful design of a carbamate‐based PET tracer for BChE imaging is the provision of a radiolabeled carbamate that is stably transferred to BChE to a considerable extent within a short period of time, such as 5 minutes, but remains bound to BChE for a preferably long time.
Figure 1.
Design of radiotracer 2 based on potent, selective BChE inhibitors 1a, 1b and 1c with (pseudo‐)irreversible binding mode. The mechanism of enzyme‐inactivation is presented below.[ 12 , 19 ]
Due to the scarcity of highly selective and nanomolar BChE inhibitors, especially with pseudo‐irreversible binding mode meeting the demanding requirements, we developed a first 18F‐ or 11C‐labeled PET tracer by attaching fluorine at the end of the heptyl chain (compound 1b, Figure 1) or introducing 11C as the carbamate carbonyl group ([11C]‐1a, Figure 1). [19]
Even though the affinity of the fluorinated compound was not significantly altered, a harsh and complicated radiolabeling procedure of four steps and a short half‐life of enzyme reactivation forced us to develop a new generation of carbamates with heterocyclic moieties at the end of alkylene chains of different lengths to examine their influence on inhibition times. Thus, we found out that a morpholine substitution at the end of a hexylene chain substantially increases the time of enzyme inactivation and beneficially increases water solubility of this highly lipophilic class of compounds. [12] Consequently, in this study, we present a second generation BChE‐selective 18F‐PET tracer with significantly prolonged duration of action, which most widely retains its inhibitory potency. Additionally, we demonstrate a new radiolabeling strategy for carbamates leading to improved radiochemical yields. Last, we show preliminary ex vivo and in vivo results of the tracer.
Results and Discussion
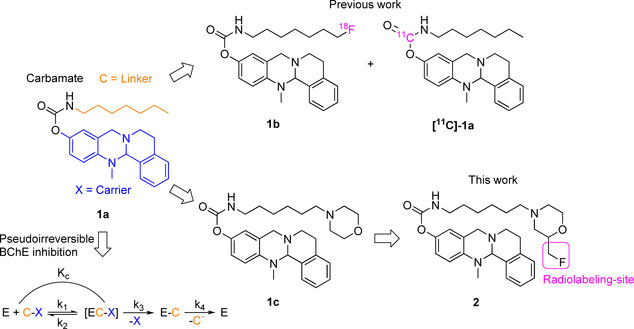
For the design of an advanced, pseudo‐irreversible PET radiotracer, we chose highly selective and potent, carbamate‐based BChE inhibitor 1c (Figure 1, IC50=49.3 nM, residual activity on hAChE at 10 μM=26.1±1.0 %) as scaffold, mainly due to its pronounced decelerated speed of enzyme regeneration (1c: t 1/2=16.50 h, 1a: t 1/2=1.12 h, 1b: t 1/2=0.86 h).[ 12 , 19 ] Our aim was to take advantage of a preferably long carbamoylated enzyme, since this would presumably result in representative, precise in vivo distribution of BChEs by PET imaging, when the radionuclide is part of the transferred carbamate moiety (Figure 1). Previously described structure‐activity relationships revealed that a hexylene‐sized linker between the carbamate and morpholine is best for both retaining inhibitory potency and a prolonged duration of action. [12] In contrast, the unsubstituted carbamate bearing a heptyl chain attached at the carbamate nitrogen exhibited a much shorter half‐life of enzyme inhibition (t 1/2=1.12 h). [25] This obvious influence of the morpholine moiety on the duration of enzyme inhibition has been well investigated using ChE inhibitors carrying either a morpholine or dimethylmorpholine moiety at the end of alkylene chain. [27] The carrier X is responsible for generating selectivity over AChE and contributes to BChE binding. [12] Considering the design of the radiotracer, we chose to introduce 18F in the compound, because it has advantageous properties over other radioisotopes commonly used for nuclear imaging techniques like PET. These include a long half‐life (t 1/2=110 min, t 1/2 (68Ga)=68 min), high positron yields (96.86 %, 68Ga: 89.14 %) and a good spatial resolution, which is a result of relatively low positron energies (0.65 MeV, 68Ga: 1.90 MeV). [28] Consequently, we aimed at the synthesis of the cold, fluorinated inhibitor 2 at first to investigate its binding properties and kinetics. Moreover, this compound was essential as reference during radiolabeling. Our first investigated radiotracer [18F]‐1b (Figure 1) required an inconvenient radiolabeling procedure of four steps and the respective cold reference had an even shorter half‐life of enzyme reactivation (t 1/2=0.86 h) than heptyl carbamate 1a (t 1/2=1.12 h). The synthetic procedure is shown in Scheme 1. The synthesis started from N‐benzylaminoethanol 3, which was reacted with epifluorohydrin and cyclized in an acid‐mediated intramolecular ether formation to give 4‐benzylated 2‐fluoromethylmorpholine 4 in satisfying yield (Scheme 1). [29] Subsequent deprotection of the benzylamine by catalytic hydrogenation, [30] and Gabriel synthesis with 2‐(6‐bromohexyl)isoindoline‐1,3‐dione, were followed by a hydrazinolysis, which gave hexylamine 7 in very good yields. The resulting amine was activated with p‐nitrophenyl chloroformate and subsequently transferred onto the 13‐methyl‐5,8,13,13a‐tetrahydro‐6H‐isoquinolino[1,2‐b]quinazolin‐10‐ol carrier scaffold to yield the non‐radioactive compound 2 as a racemic mixture, which was used for in vitro evaluation of hAChE and hBChE binding properties and as a reference compound for analysis and purification after 18F‐labeling. Synthesis of the carrier scaffold has been described before in detail. [31] Because radiolabeling with 18F requires a precursor bearing a suitable leaving group to be replaced by the radionuclide, we developed a similar synthesis as presented in Scheme 1 leading to a respective tosylated compound 14 (Scheme 2). N‐benzylaminoethanol 3 was reacted with epichlorohydrin, followed by an acid‐mediated ether condensation giving 4‐benzyl‐2‐(chloromethyl)morpholine in satisfying yield. [29] After that, the chlorine atom was replaced by a hydroxy group in a nucleophilic substitution reaction with excellent yield, [30] and the benzyl protecting group was subsequently removed by catalytic hydrogenation to yield morpholine‐2‐yl methanol 9 quantitatively (Scheme 2). Compound 9 was N‐alkylated with 2‐(6‐bromohexyl)isoindoline‐1,3‐dione in satisfying yields for an upcoming hydrazinolysis (Gabriel synthesis). Then the hydroxy group on the morpholine moiety was activated by tosylation with 4‐toluenesulfonyl chloride (TosCl) in good yield. The resulting tosylate 10 was subjected to a hydrazinolysis, and the amine 11 was subsequently activated with p‐nitrophenyl chloroformate and transferred onto 13‐methyl‐5,8,13,13a‐tetrahydro‐6H‐isoquinolino[1,2‐b]quinazolin‐10‐ol to give the desired tosyl carbamate 12 as precursor again in excellent yields (Scheme 2). As carbamates can easily decompose under the harsh conditions of 18F labeling, it is necessary to employ a protecting group strategy. [19] We adopted a facile method by introducing a methoxymethyl ether (MOM) group at the carbamate nitrogen, without using highly cancerogenic methoxymethyl chloride. [32] In the first step, a chloromethyl group was attached in situ at the carbamate yielding compound 13. The reaction mixture was subsequently quenched with methanol to give the desired MOM‐protected precursor in very good yields (Scheme 2). However, this inevitable modification of our precursor forced us to perform an additional deprotection step after radiolabeling, which might be disadvantageous due to additional time investment for reaction and work‐up. Even though the introduction of a fluoromethyl group might represent just a minor modification on the parent BChE inhibitor 1c (Figure 1), we fully characterized cold reference compound 2 for its retained inhibitory potency in colorimetric Ellman's assay and performed an additional thorough kinetic study to compare the results.[ 12 , 19 , 33 ] Thus, we have proven that both the binding properties and kinetic characteristics are in excellent accordance with the parent compound 1c and greatly exceed the properties of heptylcarbamate 1a (Table 1). The colorimetric assay (Ellman's assay) was performed by incubation of a solution of 5,5′‐dithiobis‐(2‐nitrobenzoic acid) (DTNB) and the respective human ChE with the inhibitor. After 20 min, enzyme activity was measured by adding the synthetic substrate of each ChE (acetylthiocholine for AChE and butyrylthiocholine for BChE). After reaction of the hydrolyzed thiols with DTNB and their photometric quantification, enzyme activity was calculated. The evaluation in kinetic studies revealed that the pseudo‐irreversible binding of carbamate 2 is comparable to that of compound 1c. K c and k 3 representing kinetic values for the carbamoylation step appear to be in the suitable range to allow for molecular scans reflecting the true BChE distribution among the whole body. The decarbamoylation step is kinetically characterized by k 4 and the half‐life of enzyme reactivation, t 1/2. Dilution experiments (Figure 2)[ 12 , 19 , 34 ] revealed a time‐dependent plot of enzyme activity characterized by a first‐order exponential curve and the related Equation (1), out of which t 1/2 and k 4 were calculated (Figure 2).
| (1) |
Scheme 1.
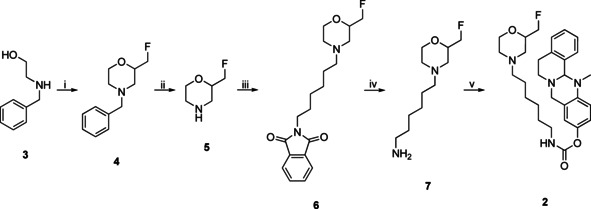
Synthesis of nonradioactive compound 2 for in vitro ChE testing and as reference compound for HPLC purification after 18F labeling. i) 1. epifluorohydrine, RT, 3 h; 2. H2SO4, 140 °C, 1 h, 67 %; ii) H2, Pd/C, MeOH, RT, 1 h, quant.; iii) 2‐(6‐bromohexyl)isoindoline‐1,3‐dione, NEt3, DMF, 105 °C, 4 h, 63 %; iv) H4N2⋅H2O, EtOH, 85 °C, 4 h, 77 %; v) 1. p‐nitrophenyl chloroformate, NEt3, CH2Cl2, 2 h, RT, 67 %; 2. 13‐methyl‐5,8,13,13a‐tetrahydro‐6H‐isoquinolino[1,2‐b]quinazolin‐10‐ol, NaH, CH2Cl2, 1 h, RT, 75 %.
Scheme 2.
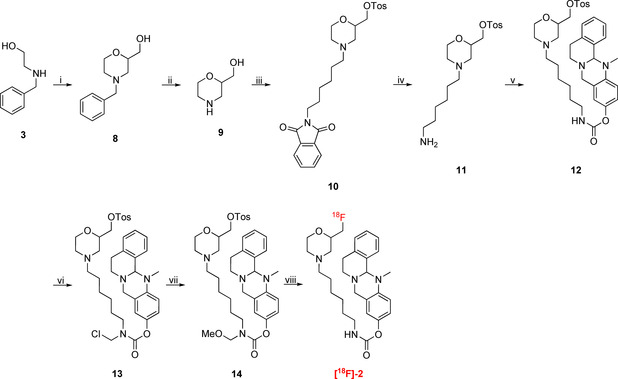
Synthesis of MOM‐protected, tosylated precursor 14 for 18F labeling to [18F]‐2. i) 1. epichlorohydrine, RT, 3 h; 2. H2SO4, 140 °C, 1 h, 67 % over two steps; 3. H2O, CHONH2, 145 °C, 20 h, 93 %; ii) H2, Pd/C, MeOH, RT, 1 h, quant.; iii) 1. 2‐(6‐bromohexyl)isoindoline‐1,3‐dione, N,N‐diisopropylethylamine (DIPEA), DMF, 100 °C, 20 h, 66 %; 2. TosCl, NEt3, CH2Cl2, RT, 20 h, 71 %; iv) H4N2⋅H2O, EtOH, 80 °C, 1.5 h, 88 %; v) 1. p‐nitrophenyl chloroformate, NEt3, CH2Cl2, 3 h, RT, 55 %; 2. 13‐methyl‐5,8,13,13a‐tetrahydro‐6H‐isoquinolino[1,2‐b]quinazolin‐10‐ol, NaH, CH2Cl2, 2 h, RT, 94 %; vi) TMS−Cl, para‐formaldehyde, CH2Cl2, RT, 18 h; vii) MeOH, RT, 1 h, 85 % (two steps); viii) 1. [18F]KF, K222, MeCN, 110 °C, 10 min; 2. 6 M HClaq, 90 °C, 5 min.
Table 1.
Inhibitory potencies and kinetic parameters of heptylcarbamate 1a, and cold reference compound 2. K C, k 3 and k 4 are presented with SEM (standard error of the mean) for triplicates of the respective experiments (n=3).
|
Cmpd. |
IC50 (hBChE [nM]) |
IC50 (hAChE [nM]) |
K c ± SEM (hBChE [nM]) |
|---|---|---|---|
|
1a |
|||
|
1b |
5.2 [19] |
3600 [19] |
24.3±19.3 [19] |
|
1c |
49.3 [12] |
>100000 [12] |
202±55 [12] |
|
2 |
66.6 |
>100000 |
199.9±160.4 |
Figure 2.
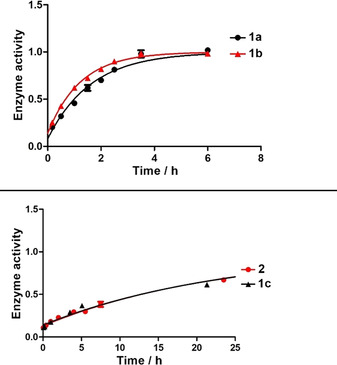
Recovery of hBChE activity after pseudo‐irreversible inhibition with compounds 1a, 1b, 1c, and 2 and subsequent dilution. Normalized enzyme activity (1=full activity) was measured in triplicate for each time point (mean±SD).
In summary, we found that our cold inhibitor 2 shows a very minor decrease in its inhibitory potency compared to parent compound 1c (Table 1 and Figure 2), but still exhibits binding affinity in the two‐digit nanomolar range. Importantly, it retains long duration of action considering the half‐life (t 1/2=16.13 h) and rate constant k 4 (Table 1, Figure 2), which met our expectations. Altogether, the pronounced selectivity over hAChE, the sufficiently fast transfer of the carbamate onto the enzyme and long binding represent promising attributes for a suitable BChE PET tracer. Consequently, we used precursor 14 (Scheme 2) for 18F radiolabeling to generate the respective radiotracer. In the first step, we adopted an established procedure for 18F labeling of tosylates by nucleophilic substitution. [18F]KF was reacted with the precursor at elevated temperatures in the presence of a [2.2.2.]cryptand (K2.2.2). Finally, the MOM group was deprotected with 6 M hydrochloric acid. Purification of the tracer was achieved by semi‐preparative high‐performance liquid chromatography (HPLC). Thus, we obtained the tracer in a time frame of 180 min and a radiochemical yield of 13 % after decay correction (specific activity a=191 GBq/mmol). Autoradiography of a thin‐layer chromatogram (TLC) indicated sufficient purity (95.3 %) of our tracer for subsequent preliminary ex vivo and in vivo experiments.
In the next step, we characterized the radiotracer's in vitro properties in protein binding assays on hBChE. First, the time course of binding was investigated by incubating a mixture of hBChE and radiotracer [18F]‐2 in PBS for 5, 10 and 20 min at 37 °C. A maximum of tracer‐bound hBChE was reached after 20 min (Figure 3). This demonstrates the specificity of tracer binding to hBChE and the maximum after 20 min is in good agreement with previous kinetic studies of compounds 1a, 1b and 1c (Figure 1), where equilibrium constant K C and rate constant of carbamoylation k 3 were determined.[ 12 , 19 , 25 ] Additionally, we studied the blocking of tracer binding in vitro by the known reversible BChE‐selective inhibitor ethopropazine and nonselective, reversible ChE inhibitor tacrine. [35] Therefore, a solution of hBChE in PBS was preincubated with ethopropazine hydrochloride (60 μM) or tacrine (1 μM), respectively. Subsequently, [18F]‐2 was added to each of the inhibitors and incubated for 20 min at 37 °C (cf. Figure S1 in the Supporting Information). Next, we investigated the binding properties of [18F]‐2 to mice brain tissue using autoradiography. After incubation of healthy mouse brain slices with the tracer, autoradiography images exhibited high accumulation of radioactivity in relevant brain areas (Figure 4), proving a good
Figure 3.
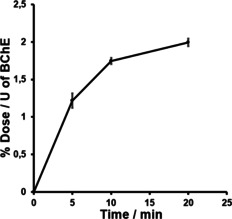
Time‐dependent in vitro hBChE binding assay with radiotracer [18F]‐2. Enzyme‐bound radioactivity was measured in triplicate after 5, 10 and 20 min (mean±SD). % Dose: the percentage of binding dose according to overall radioactivity used, U: the enzyme unit for BChE added in each vial.
Figure 4.
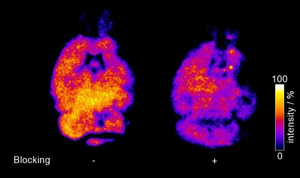
Autoradiography of mice brain slices incubated with radiotracer [18F]‐2. Left: without and right: with ethopropazine hydrochloride. Bright areas represent high binding of the tracer. Tracer intensity [arbitrary units] is shown as a percentage of the maximum tracer activity.
binding of [18F]‐2 to brain tissue. [36] In parallel, we performed blocking experiments on mice brain tissue preincubated with ethopropazine hydrochloride, a selective inhibitor of BChE that prevents binding of other BChE inhibitors and substrates due to its blocking of the active site. [35a] Consequently, we found a significantly decreased binding of [18F]‐2 to the preincubated tissue, which demonstrates the specificity of the tracer towards BChE binding in areas with high BChE activity like white matter bundles, thalamus, upper brainstem and cerebral cortex. [36] It has to be mentioned that a higher level of displaceable binding was observed in autoradiography experiments with [18F]‐1b, which is likely due to its higher binding affinity (IC50=5.2 nM). [19] Nevertheless, the complicated radiolabeling procedure and unfavorable kinetic parameters of this tracer are disadvantageous compared to [18F]‐2. Due to the successful radiolabeling and promising in vitro and ex vivo results, we felt motivated to initially evaluate the radiotracers in vivo characteristics in PET studies. Therefore, a solution of the tracer (5 % ethanol in saline) was injected via tail vein to a healthy male Wistar rat, which was directly scanned in a small animal PET system to obtain dynamic images and generate time‐activity curves of regions of interest (Figure 5). From both dynamic images and time activity curves only limited brain uptake of the tracer could be observed, which could indicate limited BBB penetration. After initial blood‐pool circulation and a rapid enrichment in heart and lung, the tracer accumulates in liver tissue. BChE occurs in the above‐mentioned organs in an elevated manner. [37] It is noteworthy that parent compound 1c (and closely related compounds) exhibited pronounced activity in vivo. [12] Duration of action at hBChE in vitro (t 1/2) correlates well with neuroprotective effects in vivo. However, since the tracer reaches heart, lung and liver with increased activity, a potential application is given in prognosis, diagnosis and monitoring disease state and progress or malignancy grade in certain types of cancer, which can be associated with down‐ and/or upregulation of BChE. [38] In particular, BChE expression has been found to decrease significantly in large‐cell‐ and squamous‐cell lung carcinoma. [39] Therefore, an application of the tracer in diseases monitoring and prognosis in lung cancer might be conceivable. Further studies in animal models are warranted based on the current initial evaluation to prove its feasibility.
Figure 5.
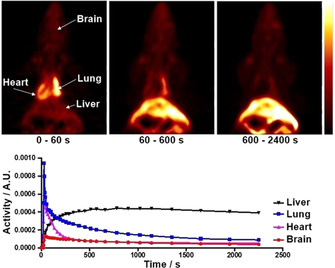
Preliminary in vivo evaluation of the tracer. Top: Dynamic coronal PET images after tail vein injection of a solution of [18F]‐2 to a male Wistar rat and subsequent microPET scanning over 40 min. Bottom: Time–activity curves of regions of interest obtained from the above images (A.U=arbitrary unit).
Conclusion
In this study, we present design and synthesis of an 18F‐labeled radiotracer for PET imaging of BChE. A potent and selective BChE inhibitor with pseudo‐irreversible binding mode served as parent compound due to its long duration of action. We modified the carbamate moiety by attaching a fluoromethyl group to the morpholine moiety, which is responsible for the prolonged enzyme inhibition. We present a facile synthesis of both cold reference compound and a MOM‐protected tosylate precursor, which is very suitable for 18F‐radiolabeling in good radiochemical yields. Thus, we provide access to a new method to radiolabel sensitive carbamates. Colorimetric in vitro studies revealed that the compound widely retains the inhibitory potency of the parent compound, and kinetic investigations exhibited that the half‐life of enzyme reactivation due to slow hydrolysis of the carbamate is in excellent accordance with the parent compound as well. Radiolabeling of the tosylate precursor was achieved in two steps through an established procedure with subsequent MOM deprotection in a reasonable time frame and good radiochemical yields. Subsequently, we investigated the tracer's properties in protein binding studies and found that the time course of binding was in accordance with our expectations. Consequently, we applied the radiotracer in preliminary ex vivo autoradiography studies on mice brain slices, where we could observe good binding to brain tissue. Additionally, we demonstrated the specificity of binding to BChE in blocking experiments with the selective BChE inhibitor ethopropazine. Finally, we present a preliminary in vivo PET study on a healthy male rat, where we found only limited brain uptake. The tracer rapidly addressed heart and lung, accompanied by long‐term accumulation in liver tissue ‐ organs with BChE expression. Future studies will build upon these results and elucidate the precise biodistribution of the tracer to clarify the remaining question of its limited brain uptake, which is likely due to the inability to penetrate the blood‐brain‐barrier efficiently or extrusion by P‐glycoprotein. A potential application of this tracer in diagnosis and monitoring of diseases with altered BChE activity in the addressed organs, such as certain types of lung cancer, is conceivable.
Experimental Section
Chemistry
Reagents and solvents were obtained from commercial suppliers in reagent grade and were used without further purification unless stated otherwise. Dichloromethane as solvent was distilled from CaH2 under argon. The reaction progress was controlled with analytical thin‐layer chromatography (TLC) on precoated silica gel GF254 plates (Macherey Nagel, Düren, Germany). Compounds were detected under UV light (254 and 366 nm) or through staining with iodine, KMnO4, or Ehrlich's reagent. Crude products after work‐up were purified by manual flash column chromatography. Silica gel (particle size of 40–63 μM; VWR chemicals, Leuven, Belgium) was used as the stationary phase and mixtures from petroleum ether/ethyl acetate or dichloromethane/methanol as eluent systems. The pure compounds were submitted for nuclear magnetic resonance spectra on a AV‐400 NMR instrument (Bruker) in deuterated solvents (DMSO‐d6, CDCl3, CD2Cl2, CD3OD). Chemical shifts are expressed in ppm relative to DMSO‐d6, CDCl3, CD2Cl2, or CD3OD (2.50/7.26/5.32/3.31 for 1H; 39.5/77.2/53.5/49.0 for 13C). Purity of compounds was controlled with analytical HPLC on a Shimadzu system equipped with a DGU‐20A3R controller, LC20AB liquid chromatograph, and SPD‐20 A UV/vis detector (Shimadzu): Stationary phase: Synergi 4 U fusion RP (Synergi, Aschaffenburg, Germany) column; mobile phase: water+0.1 % formic acid (phase A) and methanol+0.1 % formic acid (phase B) with a flow of 1.0 mL/min. Method: conc. B, gradient 5→90 % from 0 to 8 min, 90 % isocratic from 8 to 13 min, gradient 90→5 % from 13 to 15 min, 5 % isocratic from 15 to 18 min. Compounds were detected at λ=254 nm, and target compounds were ≥95 % pure.
4‐Benzyl‐2‐(fluoromethyl)morpholine (4): In a flask, 2‐(benzylamino)ethan‐1‐ol (3; 1.866 mL, 1.988 g, 13.15 mmol, 1 equiv.) and epifluorohydrin (1 g, 13.15 mmol, 1 equiv.) were mixed together and stirred for 3 h at room temperature. Then concentrated H2SO4 (5 mL) was added carefully and the mixture was heated to 140 °C for 1 h. Afterwards, the reaction mixture was allowed to cool down to room temperature and was poured onto ice. The aqueous layer was basified with aqueous sodium hydroxide (10 M) and extracted with three portions of ethyl acetate. The combined organic layers were washed with water and brine, dried over sodium sulphate, filtered, and evaporated to dryness under reduced pressure. The residue was purified with flash column chromatography (petroleum ether/ethyl acetate 3 : 1, R f=0.35) to yield 4‐benzyl‐2‐(fluoroethyl)morpholine (4; 1.31 g, 6.26 mmol, 48 %) as colorless oil. 1H NMR (400 MHz, CDCl3): δ=7.29–7.14 (m, 5H), 4.41–4.31 (m, 1H), 4.29–4.21 (m, 1H), 3.89–3.58 (m, 3H), 3.53–3.35 (m, 2H), 2.74–2.55 (m, 2H), 2.14 (td, J=11.4, 3.3 Hz, 1H), 2.04–1.86 ppm (m, 1H); 13C NMR (101 MHz, CDCl3): δ=137.7 (1 C), 129.3 (2 C), 128.5 (2 C), 127.4 (1 C), 85.0 (1 C), 83.3 (1 C), 74.5 (0.5 C), 74.3 (0.5 C), 66.9 (1 C), 63.5 (1 C), 53.9 (1 C), 53.8 (1 C), 53.0 ppm (1 C); MS (ESI): [M+H]+ calcd for C12H16NOF=210.13, found 210.10.
2‐(Fluoromethyl)morpholine hydroformiate (5): A solution of 4‐benzyl‐2‐(fluoromethyl)morpholine (4; 1.3 g, 6.21 mmol, 1 equiv.) in MeOH (30 mL) was prepared and formic acid (0.5 mL) was added. No precipitation was observed. Then palladium on activated charcoal (10 wt/%, 130 mg) was added and the atmosphere was replaced with hydrogen. The mixture was stirred for 1 h at room temperature. After that, the catalyst was filtered off by a pad of celite, which was washed with methanol. The filtrate was evaporated to dryness in vacuo to yield 2‐(fluoromethyl)morpholine hydroformiate (5; 1.025 g, 6.21 mmol, quant.) as colorless oil. 1H NMR (400 MHz, CD3OD): δ=8.37 (s, 1H), 4.57–4.49 (m, 1H), 4.44–4.37 (m, 1H), 4.15–4.06 (m, 1H), 4.04–3.90 (m, 1H), 3.90–3.80 (m, 1H), 3.39–3.32 (m, 1H), 3.29–3.26 (m, 1H), 3.25 (dt, J=2.5, 1.3 Hz, 1H), 3.18–3.12 (m, 1H), 3.10–2.99 ppm (m, 1H); 13C NMR (101 MHz, CD3OD): δ=168.2 (1 C), 84.8 (1 C), 83.1 (1 C), 73.6 (0.5 C), 73.4 (0.5 C), 64.9 (1 C), 43.8 ppm (1 C); MS (ESI): [M+H]+ calcd for C5H10FNO=120.08; found 120.15.
2‐{6‐[2‐(Fluoromethyl)morpholino]hexyl}isoindoline‐1,3‐dione (6): 2‐(Fluoromethyl)morpholine hydroformiate (5; 1001 mg, 6.06 mmol, 1 equiv.) was dissolved in dry dimethylformamide (10 mL) and triethylamine (3 mL) was added. Then 2‐(6‐bromohexyl)isoindoline‐1,3‐dione (2 g, 6,45 mmol, 1.06 equiv.) was added and the resulting solution was heated to 105 °C for 4 h. The mixture was allowed to cool down to room temperature and water was added. The aqueous layer was extracted with ethyl acetate (3x). Then the combined organic layers were washed with water and brine, dried over sodium sulphate, filtered, and the solvent was evaporated under reduced pressure. The residue was purified with flash column chromatography (CH2Cl2/MeOH 98 : 2, R f=0.33) to yield 2‐(6‐[2‐(fluoromethyl)morpholino]hexyl}isoindoline‐1,3‐dione (6; 1.334 g, 3.83 mmol, 63 %) as colorless oil. 1H NMR (400 MHz, CDCl3): δ=7.80–7.74 (m, 2H), 7.66–7.61 (m, 2H), 4.41–4.36 (m, 1H), 4.29–4.24 (m, 1H), 3.83 (ddd, J=11.3, 3.3, 1.7 Hz, 1H), 3.66–3.50 (m, 4H), 2.74–2.67 (m, 1H), 2.61 (dd, J=11.5, 1.9 Hz, 1H), 2.34–2.22 (m, 2H), 2.05 (td, J=11.4, 3.3 Hz, 1H), 1.94–1.82 (m, 1H), 1.69–1.56 (m, 3H), 1.50–1.36 (m, 3H), 1.37–1.24 ppm (m, 4H); 13C NMR (101 MHz, CDCl3): δ=168.4 (1 C), 133.9 (2 C), 132.2 (1 C), 123.3 (2 C), 84.9 (1 C), 83.1 (1 C), 74.3 (0.5 C), 74.1 (0.5 C), 66.8 (1 C), 58.8 (1 C), 54.0 (1 C), 52.9 (1 C), 37.9 (1 C), 28.5 (1 C), 27.0 (1 C), 26.7 (1 C), 26.4 ppm (1 C); MS (ESI): [M+H]+ calcd for C19H26N2O4=349.19, found 349.15.
6‐[2‐(Fluoromethyl)morpholino]hexan‐1‐amine (7): To a solution of 2‐{6‐[2‐(fluoromethyl)morpholino]hexyl}isoindoline‐1,3‐dione (6; 1.2 g, 3.44 mmol, 1 equiv.) in ethanol (50 mL), hydrazine hydrate (1 mL, >5 equiv.) was added. The solution was heated to 85 °C for 4 h. Then the mixture was allowed to cool down to room temperature and the precipitate was filtered off. The filtrate was evaporated to dryness under reduced pressure. The residue was triturated with CH2Cl2, filtered, and the filtrate was evaporated under reduced pressure to yield 6‐[2‐(fluoromethyl)morpholino]hexan‐1‐amine (7; 577 mg, 2.64 mmol, 77 %) as colorless oil without further purification. 1H NMR (400 MHz, CDCl3): δ=4.48–4.42 (m, 1H), 3.95–3.86 (m, 1H), 3.72–3.64 (m, 1H), 3.64–3.58 (m, 1H), 2.80–2.74 (m, 1H), 2.71–2.63 (m, 3H), 2.37–2.29 (m, 2H), 2.12 (td, J=11.5, 3.4 Hz, 1H), 2.01–1.89 (m, 1H), 1.55–1.41 (m, 4H), 1.39–1.27 ppm (m, 4H); 13C NMR (101 MHz, CDCl3): δ=85.0 (1 C), 83.3 (1 C), 74.4 (0.5 C), 74.2 (0.5 C), 67.0 (1 C), 59.1 (1 C), 54.2 (1 C), 54.1 (1 C), 53.1 (1 C), 42.2 (1 C), 33.7 (1 C), 27.4 (1 C), 26.9 (1 C), 26.6 ppm (1 C); MS (ESI): [M+H]+ calcd for C11H23FN2OH=219.19; found 219.20.
4‐Nitrophenyl‐{6‐[2‐(fluoromethyl)morpholino]hexyl}‐carbamate: To a solution of 6‐[2‐(fluoromethyl)morpholino]hexan‐1‐amine (7; 500 mg, 2.29 mmol) in dry dichloromethane (10 mL), triethylamine (0.5 mL) and 4‐nitrophenylchloroformiate (508 mg, 2.52 mmol) were added. The resulting solution was stirred for 2 h at room temperature. Then the solvent was evaporated under reduced pressure and the residue was purified by flash column chromatography (CH2Cl2/MeOH 99 : 1, R f=0.11) to yield 4‐nitrophenyl {6‐[2‐(fluoromethyl)morpholino]hexyl}carbamate (588 mg, 1.53 mmol, 67 %) as yellow oil. MS (ESI): [M+H]+ calcd for C18H26FN3O5=384.20, found 384.05.
Due to high instability of the title compound only LCMS data was measured, before directly reacting it in the next step.
13‐Methyl‐5,8,13,13a‐tetrahydro‐6H‐isoquinolino[1,2‐b]quinazolin‐10‐yl‐{6‐[2‐(fluoromethyl)morpholino]‐hexyl}carbamate (2): To a solution of 4‐nitrophenyl [6‐(2‐(fluoromethyl)morpholino)hexyl]carbamate (86 mg, 0.23 mmol, 1.2 equiv.) and 13‐methyl‐5,8,13,13a‐tetrahydro‐6H‐isoquinolino[1,2‐b]quinazolin‐10‐ol (50 mg, 0.19 mmol, 1 equiv.), sodium hydride (60 % suspension in paraffin oil, 9 mg, 0.23 mmol, 1 equiv.) was added. The resulting mixture was stirred for 1 h at room temperature. Then water was added, and the aqueous layer was extracted with three portions of dichloromethane. The combined organic layers were washed with water, saturated aqueous sodium hydrogen carbonate solution and brine, dried over sodium sulphate, filtered, and evaporated to dryness under reduced pressure. The residue was purified by flash column chromatography (CH2Cl2/MeOH 98 : 2, R f=0.14) to yield 13‐methyl‐5,8,13,13a‐tetrahydro‐6H‐isoquinolino[1,2‐b]quinazolin‐10‐yl{6‐[2‐(fluoromethyl)morpholino]hexyl}carbamate (2; 72 mg, 0.14 μmol, 75 %) as colorless oil. 1H NMR (400 MHz, CD2Cl2): δ=7.38–7.32 (m, 1H), 7.22–7.15 (m, 2H), 7.14–7.09 (m, 1H), 6.89–6.81 (m, 2H), 6.74–6.68 (m, 1H), 4.77 (s, 1H), 4.42–4.35 (m, 1H), 4.30–4.22 (m, 1H), 3.94 (d, J=15.6 Hz, 1H), 3.89–3.79 (m, 2H), 3.78–3.58 (m, 2H), 3.24–3.11 (m, 3H), 3.10–2.98 (m, 1H), 2.83–2.60 (m, 4H), 2.51 (s, 3H), 2.34–2.26 (m, 2H), 2.06 (td, J=11.4, 3.3 Hz, 1H), 1.94–1.82 (m, 1H), 1.55–1.39 (m, 4H), 1.37–1.26 ppm (m, 4H); 13C NMR (101 MHz, CD2Cl2): δ=155.5 (1 C), 146.3 (1 C), 144.9 (1 C), 136.7 (1 C), 134.5 (1 C), 129.1 (1 C), 128.9 (1 C), 127.7 (1 C), 126.1 (1 C), 120.7 (1 C), 120.5 (1 C), 120.0 (1 C), 85.5 (1 C), 83.8 (1 C), 76.8 (1 C), 74.8 (0.5 C), 74.6 (0.5 C), 67.0 (1 C), 56.5 (1 C), 54.38 (1 C), 54.33 (1 C), 54.26 (1 C), 53.8 (1 C), 53.4 (1 C), 48.3 (1 C), 41.5 (1 C), 38.2 (1 C), 30.2 (1 C), 29.0 (1 C), 27.4 (1 C), 27.0 (1 C), 26.8 ppm (1 C); MS (ESI): [M+H]+ calcd for C29H39FN4O3=511.31, found 511.15; HPLC: t R=6.74 min, 97.5 % purity.
Experimental procedures for the synthesis of MOM‐protected precursor 14 (Scheme 2) are described in the Supporting Information.
Enzyme inhibition
hAChE (EC 3.1.1.7, from human erythrocytes), DTNB (Ellman's reagent), ATC and BTC iodides were purchased from Sigma–Aldrich. hBChE (E.C. 3.1.1.8) was kindly donated by Dr. Oksana Lockridge, Nebraska Medical Centre. For the assays, the inhibitors were dissolved in ethanol (absolute, reag. Ph. Eur.) to give a concentration of 3.33 mM (100 μM in the assay) and stepwise diluted to 3.33 nM (0.1 nM in the assay). Buffer was prepared from 3.12 g of potassium dihydrogen phosphate in 500 mL of doubly distilled water. After the potassium dihydrogen phosphate was dissolved, the pH value was adjusted to pH 8.0 with 0.1 M sodium hydroxide solution. Both enzymes were dissolved in assay buffer and diluted to 2.5 units/mL. The solutions were stabilized with 1 mg/mL of bovine serum albumin (Sigma–Aldrich) and were stored at 7 °C until usage. DTNB was dissolved in buffer at 10 mM (0.3 mM in the assay). The substrates ATC and BTC were prepared with a concentration of 75 mM (452 μM in the assay) in assay buffer and kept frozen until usage. The absorbance of probes was measured with a Shimadzu UVmini‐1240 spectrometer at 412 nm.
IC50 determination: The assay was carried out at room temperature (25 °C). Thereby, 900 μL of the buffer, 30 μL of DTNB solution (10 mM, 0.3 mM in the assay) and 30 μL of enzyme solution (hAChE or hBChE, 2.5 units/mL) were mixed in a cuvette. The incubation was started directly after the addition of 30 μL of inhibitor 2 (Scheme 1) solutions with different concentrations. The solutions were mixed well by manual stirring. After 20 min incubation, 6 μL of substrate solution (ATC or BTC; 75 mM, 452 μM in the assay) were added. The mixture was left for 2.5 min to allow substrate hydrolysis, and the absorbance was measured at λ=412 nm, whereas enzyme activity was determined three times for every concentration with at least seven different concentrations. A blank value was determined by replacing the enzyme solution with buffer; the compound solution was replaced with ethanol. The maximum enzyme activity was determined with 30 μL of ethanol instead of the compound solution. 10 % of ethanol did not reduce enzyme activity. The enzyme activity in percent of maximum activity was plotted against the logarithmic inhibitor concentration, from which IC50 values were calculated with the software GraphPad Prism 5.
Kinetic studies: For determination of K c and k 3 values, the same general setup was used (900 μL of the assay buffer, 30 μL of DTNB solution (10 mM, 0.3 mM in the assay), and 30 μL of hBChE solution (2.5 units/mL) were mixed at room temperature). Then, 30 μL of inhibitor 2 (Scheme 1) solutions with different concentrations were added, but enzyme activity was determined after 1, 2, 4, 6, 10, 15, 20, 30, and 40 min of incubation by addition of 6 μL of BTC solution (75 mM, 452 μM in the assay). The absorbance at λ=412 nm was measured after 2.5 min. All concentrations were measured three times, at least five different concentrations were measured for at least seven time points. The obtained enzyme activities in percent of maximum enzyme activity were plotted in a time‐dependent manner and fitted to Equation (2) to determine the rate constant k obs using the software GraphPad Prism 5.
| (2) |
A: enzyme activity at time t, A 0: enzyme activity at time t=0, 100 %; A ∞: enzyme activity at infinite time.
The obtained inverse rate constants k obs were plotted against the reciprocal concentration [I]−1, and k 3 was calculated from the y‐intercept of the resulting curve, and K c from the slope of the resulting linearization according to Equation (3) using the software GraphPad Prism 5.
| (3) |
[I]: concentration of BChE inhibitor 2 (Scheme 1).
For the measurement of decarbamylation kinetics, the enzyme was incubated with inhibitor 2 to carbamylate >85 % of the enzyme. After 1 h, the solution was diluted 1000‐fold so that no enzyme was carbamylated anymore. The enzyme activity was measured at several (at least 8) time points, as described above. For the determination of full enzyme activity, a batch of the enzyme was treated with ethanol instead of inhibitor solution and diluted 1000‐fold in the same manner. The enzyme activity in percent was plotted against time after dilution to give first‐order rate constant k 4 according to Equation (1) using the software GraphPad Prism 5. All experiments were carried out in triplicates.
Radiochemistry
Solvents and chemicals were purchased from Aldrich and directly used without further purification. [18F]F− was produced by a cyclotron (GE Medical Systems, Uppsala) at the Department of Nuclear Medicine of the University Hospital of Würzburg. Enriched [18O]H2O was irradiated with protons to produce [18F]F− and radiofluorination was carried out manually. HPLC was used for purification and analyses of radioactive products (Shimadzu system equipped with UV detector, λ=220 and 254 nm, and γ‐detector). Purified radiotracer 2 (Scheme 1) was diluted with either PBS or saline to the corresponding concentration for further evaluation.
13‐Methyl‐5,8,13,13a‐tetrahydro‐6H‐isoquinolino[1,2‐b] quinazolin‐10‐yl‐{6‐[2‐([18F]‐fluoromethyl)morpholino]‐hexyl}carbamate ([18F]‐2): [18F]F− was separated from [18O]H2O by an anion exchange cartridge (Sep‐Pak QMA Cartridge) and eluted with 0.5 mL of 25 mM potassium carbonate and 50 mM Kryptofix222 solution in MeCN/H2O (6 : 2) into a V‐vial. The solution was dried at 120 °C under a nitrogen flow, which was repeated twice with 500 μL of anhydrous MeCN. Labeling was carried out using 1.3 mg of precursor 14 (Scheme 2) dissolved in 0.4 mL of anhydrous MeCN at 110 °C for 20 min, followed by addition of 100 μL of 6 M hydrochloric acid with further 5 min heating at 90 °C. After addition of 500 μL of 1 M aqueous sodium hydrogencarbonate solution and 10 mL water to quench the reaction, the crude product was trapped on a Sep‐Pak light C18, which was washed with 5 mL of water and eluted with 0.5 mL of ethanol. The crude radiotracer was purified via semi‐preparative HPLC. Column: 10×100 mm C18 Phenomenex Onyx Monolithic, Mobile phase: Phase A: H2O, Phase B: MeCN, 0–20 min, 20 %→80 % B, 20–26 min, 95 % B, 28–30 min 20 % B, Flow rate: 1.5 ml/min. The collected fraction was diluted with 10 mL water and trapped on a Sep‐Pak light C18, which was dried with 5 mL air and eluted with 0.3 mL ethanol. The total radiolabeling procedure was feasible in 180 min in a radiochemical yield of 13 % (corrected for decay). Sufficient radiochemical purity was measured by TLC autoradiography (95.3 %).
Experimental procedures for protein binding assays with tracer [18F]‐2 (time course and blocking of binding) are described in the Supporting Information.
Tissue binding and in vivo PET imaging
Ex vivo tissue binding studies were carried out with one C57BL/6 N mouse from Charles River. Two series of horizontal brain slices with 20 μm thickness were prepared for either control or blocking group. A buffer (150 mM NaCl, 5 mM EDTA, 50 mM Na2HPO4, pH 8.0) containing [18F]‐2 (A=1.44 MBq) with or without ethopropazine hydrochloride (60 μM) as blocking agent was prepared for incubation with the mice brain slices. Slices were incubated for 30 min at 25 °C and then rinsed five times in PBS buffer (1 min each time). The slices were dried at room temperature and exposed to a phosphor imaging plate (GE healthcare, BAS IP MS 2025 E, Munich, Germany). Images were produced with a digital autoradiographic system (Typhoon FLA 7000).
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. Animal protocols were approved by the local Animal Care and Use Committee (approval no. 15063) and conducted according to the Guide for the Care and Use of Laboratory Animals. A healthy, male Wistar rat was anaesthetized and maintained with isoflurane for in vivo PET imaging using a micro PET system (FOCUS, Siemens, Erlangen, Germany). A 60‐min dynamic imaging protocol was started directly after the injection of a solution of [18F]‐2 (5 % ethanol in saline, 6.3 MBq). Analysis of the obtained PET images was performed with the public domain tool AMIDE imaging software (A Medical Imaging Data Examiner, version 1.01) and time‐activity curves of regions of interest were generated.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the German Research Council (Deutsche Forschungsgemeinschaft DFG; grants DE 1546/6‐3 for M.D. and HI 1789/3‐3 for T.H.). M.H. was supported by the German Academic Scholarship Foundation (“Studienstiftung des deutschen Volkes”) with a PhD scholarship. C. G. and M. H. were supported by the MuTaLig COST Action (active participation at the 3rd Working Group Meeting, Paris). We gratefully acknowledge Professor Oksana Lockridge (University of Nebraska Medical Center) for providing hBChE. Open access funding enabled and organized by Projekt DEAL.
C. Gentzsch, M. Hoffmann, Y. Ohshima, N. Nose, X. Chen, T. Higuchi, M. Decker, ChemMedChem 2021, 16, 1427.
Contributor Information
Prof. Dr. Takahiro Higuchi, Email: higuchi_t@ukw.de.
Prof. Dr. Michael Decker, Email: michael.decker@uni-wuerzburg.de.
References
- 1.
- 1a. Nichols E., Szoeke C. E. I., Vollset S. E., Abbasi N., Abd-Allah F., et al., Lancet Neurol. 2019, 18, 88–106; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Crous-Bou M., Minguillón C., Gramunt N., Molinuevo J., Alzheimer's Res. Ther. 2017, 9, 71; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Alzheimer′s Dementia 2019, 15, 321–387. [Google Scholar]
- 2. Alzheimer A., Neurologisches Centralblatt 1906, 25, 1134. [Google Scholar]
- 3. Katzman R., Arch. Neurol. 1976, 33, 217–218. [DOI] [PubMed] [Google Scholar]
- 4. Sengoku R., Neuropathology 2020, 40, 22–29. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Davies P., Maloney A. J. F., Lancet 1976, 308, 1403; [DOI] [PubMed] [Google Scholar]
- 5b. Perry E., Perry R., Blessed G., Tomlinson B., Lancet 1977, 309, 189; [DOI] [PubMed] [Google Scholar]
- 5c. Coyle J., Price D., DeLong M., Science 1983, 219, 1184–1190; [DOI] [PubMed] [Google Scholar]
- 5d. Bartus R., Dean R., Beer B., Lippa A., Science 1982, 217, 408–414. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Darvesh S., Curr. Alzheimer Res. 2016, 13, 1–5; [DOI] [PubMed] [Google Scholar]
- 6b. Rountree S. D., Chan W., Pavlik V. N., Darby E. J., Siddiqui S., Doody R. S., Alzheimer's Res. Ther. 2009, 1, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Greig N. H., Utsuki T., Yu Q., Zhu X., Holloway H. W., et al., Curr. Med. Res. Opin. 2001, 17, 159–165. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Darvesh S., Grantham D. L., Hopkins D. A., J. Comp. Neurol. 1998, 393, 374–390; [PubMed] [Google Scholar]
- 8b. Darvesh S., Hopkins D. A., J. Comp. Neurol. 2003, 463, 25–43. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Guillozet A. L., Smiley J. F., Mash D. C., Mesulam M. M., Ann. Neurol. 1997, 42, 909–918; [DOI] [PubMed] [Google Scholar]
- 9b. Mesulam M., Geula C., Ann. Neurol. 1994, 36, 722–727. [DOI] [PubMed] [Google Scholar]
- 10. Macdonald I. R., Maxwell S. P., Reid G. A., Cash M. K., DeBay D. R., et al., J. Alzheimer's Dis. 2017, 58, 491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Mesulam M. M., Guillozet A., Shaw P., Levey A., Duysen E. G., et al., Neuroscience 2002, 110, 627–639; [DOI] [PubMed] [Google Scholar]
- 11b. Hartmann J., Kiewert C., Duysen E. G., Lockridge O., Greig N. H., et al., J. Neurochem. 2007, 100, 1421–1429; [DOI] [PubMed] [Google Scholar]
- 11c. Furukawa-Hibi Y., Alkam T., Nitta A., Matsuyama A., Mizoguchi H., et al., Behav. Brain Res. 2011, 225, 222–229; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11d. Greig N. H., Utsuki T., Ingram D. K., Wang Y., Pepeu G., et al., Proc. Natl. Acad. Sci. USA 2005, 102, 17213–17218; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11e. Maurice T., Strehaiano M., Siméon N., Bertrand C., Chatonnet A., Behav. Brain Res. 2015, 296, 351–360; [DOI] [PubMed] [Google Scholar]
- 11f. Naik R. S., Hartmann J., Kiewert C., Duysen E. G., Lockridge O., et al., J. Pharm. Pharm. Sci. 2009, 12, 79–85; [DOI] [PubMed] [Google Scholar]
- 11g. Darreh-Shori T., Brimijoin S., Kadir A., Almkvist O., Nordberg A., Neurobiol. Dis. 2006, 24, 326–333. [DOI] [PubMed] [Google Scholar]
- 12. Hoffmann M., Stiller C., Endres E., Scheiner M., Gunesch S., et al., J. Med. Chem. 2019, 62, 9116–9140. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Lockridge O., Pharmacol. Ther. 2015, 148, 34–46; [DOI] [PubMed] [Google Scholar]
- 13b. Ashani Y., Drug Dev. Res. 2000, 50, 298–308; [Google Scholar]
- 13c. Li B., Sedlacek M., Manoharan I., Boopathy R., Duysen E. G., et al., Biochem. Pharmacol. 2005, 70, 1673–1684. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Li B., Duysen E. G., Lockridge O., Chem.-Biol. Interact. 2008, 175, 88–91; [DOI] [PubMed] [Google Scholar]
- 14b. Iwasaki T., Yoneda M., Nakajima A., Terauchi Y., Intern. Med. J. 2007, 46, 1633–1639; [DOI] [PubMed] [Google Scholar]
- 14c. Sato K. K., Hayashi T., Maeda I., Koh H., Harita N., et al., Clin. Endocrinol. (Oxf) 2014, 80, 362–367; [DOI] [PubMed] [Google Scholar]
- 14d. Stojanov M., Stefanović A., Džingalašević G., Mandić-Radić S., Prostran M., Clin. Biochem. 2011, 44, 623–626; [DOI] [PubMed] [Google Scholar]
- 14e. Alcântara V. M., Chautard-Freire-Maia E. A., Scartezini M., Cerci M. S. J., Braun-Prado K., et al., Scand. J. Clin. Lab. Invest. 2002, 62, 399–404. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Layer P. G., Cell. Mol. Neurobiol. 1991, 11, 7–33; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Aziz-Aloya R. B., Sternfeld M., Soreq H., Progress in Brain Research, Vol. 98 (Ed.: A. C. Cuello), Elsevier, 1993, pp. 147–153; [DOI] [PubMed] [Google Scholar]
- 15c. Layer P. G., Willbold E., Prog. Histochem. Cytochem. 1994, 29, III–92. [DOI] [PubMed] [Google Scholar]
- 16. Bernardi C. C., Ribeiro E. d. S. F., Cavalli I. J., Chautard-Freire-Maia E. A., Souza R. L. R., Cancer Genet. Cytogenet. 2010, 197, 158–165. [DOI] [PubMed] [Google Scholar]
- 17. Gu Y., Chow M. J., Kapoor A., Mei W., Jiang Y., et al., Transl. Oncol. 2018, 11, 1012–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McCluskey S. P., Plisson C., Rabiner E. A., Howes O., Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 451–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sawatzky E., Al-Momani E., Kobayashi R., Higuchi T., Samnick S., et al., ChemMedChem 2016, 11, 1540–1550. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Roivainen A., Rinne J., Virta J., Järvenpää T., Salomäki S., et al., J. Nucl. Med. 2004, 45, 2032–2039; [PubMed] [Google Scholar]
- 20b. Kikuchi T., Zhang M.-R., Ikota N., Fukushi K., Okamura T., et al., Bioorg. Med. Chem. Lett. 2004, 14, 1927–1930. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. DeBay D. R., Reid G. A., Pottie I. R., Martin E., Bowen C. V., et al., Alzheimers Dement. (N Y) 2017, 3, 166–176; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21b. Macdonald I. R., Reid G. A., Joy E. E., Pottie I. R., Matte G., et al., Mol. Imaging Biol. 2011, 13, 1250–1261. [DOI] [PubMed] [Google Scholar]
- 22. Macdonald I. R., Reid G. A., Pottie I. R., Martin E., Darvesh S., J. Nucl. Med. 2016, 57, 297–302. [DOI] [PubMed] [Google Scholar]
- 23. Thorne M. W. D., Cash M. K., Reid G. A., Burley D. E., Luke D., Pottie I. R., Darvesh S., Mol. Imaging Biol. 2021, 23, 127–138. [DOI] [PubMed] [Google Scholar]
- 24. James S. L., Ahmed S. K., Murphy S., Braden M. R., Belabassi Y., et al., ACS Chem. Neurosci. 2014, 5, 519–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Darras F. H., Kling B., Heilmann J., Decker M., ACS Med. Chem. Lett. 2012, 3, 914–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.
- 26a. Sawatzky E., Wehle S., Kling B., Wendrich J., Bringmann G., et al., J. Med. Chem. 2016, 59, 2067–2082; [DOI] [PubMed] [Google Scholar]
- 26b. Huang G., Kling B., Darras F. H., Heilmann J., Decker M., Eur. J. Med. Chem. 2014, 81, 15–21. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Bartolucci C., Perola E., Cellai L., Brufani M., Lamba D., Biochemistry 1999, 38, 5714–5719; [DOI] [PubMed] [Google Scholar]
- 27b. Perola E., Cellai L., Lamba D., Filocamo L., Brufani M., Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1997, 1343, 41–50; [DOI] [PubMed] [Google Scholar]
- 27c. Rampa A., Piazzi L., Belluti F., Gobbi S., Bisi A., et al., J. Med. Chem. 2001, 44, 3810–3820; [DOI] [PubMed] [Google Scholar]
- 27d. Rampa A., Bartolini M., Bisi A., Belluti F., Gobbi S., et al., ACS Med. Chem. Lett. 2012, 3, 182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.
- 28a. Sanchez-Crespo A., Appl. Radiat. Isot. 2013, 76, 55–62; [DOI] [PubMed] [Google Scholar]
- 28b. Kesch C., Kratochwil C., Mier W., Kopka K., Giesel F. L., J. Nucl. Med. 2017, 58, 687–688. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Fehler S. K., Maschauer S., Höfling S. B., Bartuschat A. L., Tschammer N., et al., Chem. Eur. J. 2014, 20, 370–375; [DOI] [PubMed] [Google Scholar]
- 29b. Kato S., Morie T., Hino K., Kon T., Naruto S., et al., J. Med. Chem. 1990, 33, 1406–1413. [DOI] [PubMed] [Google Scholar]
- 30. Lainton J. A. H., Allen M. C., Burton M., Cameron S., Edwards T. R. G., et al., J. Comb. Chem. 2003, 5, 400–407. [DOI] [PubMed] [Google Scholar]
- 31. Sawatzky E., Bukowczan J., Decker M., Tetrahedron Lett. 2014, 55, 2973–2976. [Google Scholar]
- 32. Barnes D. M., Barkalow J., Plata D. J., Org. Lett. 2009, 11, 273–275. [DOI] [PubMed] [Google Scholar]
- 33. Ellman G. L., Courtney K. D., Andres V., Featherstone R. M., Biochem. Pharmacol. 1961, 7, 88–95. [DOI] [PubMed] [Google Scholar]
- 34. Bartolucci C., Stojan J., Yu Q.-s., Greig N. H., Lamba D., Biochem. J. 2012, 444, 269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.
- 35a. Meuling W. J., Jongen M. J., van Hemmen J. J., Am. J. Ind. Med. 1992, 22, 231–241; [DOI] [PubMed] [Google Scholar]
- 35b. Ahmed M., Rocha J. B. T., Corrêa M., Mazzanti C. M., Zanin R. F., et al., Chem.-Biol. Interact. 2006, 162, 165–171. [DOI] [PubMed] [Google Scholar]
- 36. Reid G. A., Darvesh S., Neuroscience 2015, 298, 424–435. [DOI] [PubMed] [Google Scholar]
- 37. Uhlén M., Fagerberg L., Hallström B. M., Lindskog C., Oksvold P., et al., Science 2015, 347, 1260419 (http://www.proteinatlas.org).25613900 [Google Scholar]
- 38. Santarpia L., Grandone I., Contaldo F., Pasanisi F., J. Cachexia Sarcopenia Muscle 2013, 4, 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Martínez-Moreno P., Nieto-Cerón S., Torres-Lanzas J., Ruiz-Espejo F., Tovar-Zapata I., et al., Carcinogenesis 2005, 27, 429–436. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary