

Abstract
Upon infection, mycobacteria, such as Mycobacterium tuberculosis (Mtb) and nontuberculous mycobacteria (NTM), are recognized by host innate immune cells, triggering a series of intracellular processes that promote mycobacterial killing. Mycobacteria, however, have developed multiple counter‐strategies to persist and survive inside host cells. By manipulating host effector mechanisms, including phagosome maturation, vacuolar escape, autophagy, antigen presentation, and metabolic pathways, pathogenic mycobacteria are able to establish long‐lasting infection. Counteracting these mycobacteria‐induced host modifying mechanisms can be accomplished by host‐directed therapeutic (HDT) strategies. HDTs offer several major advantages compared to conventional antibiotics: (a) HDTs can be effective against both drug‐resistant and drug‐susceptible bacteria, as well as potentially dormant mycobacteria; (b) HDTs are less likely to induce bacterial drug resistance; and (c) HDTs could synergize with, or shorten antibiotic treatment by targeting different pathways. In this review, we will explore host‐pathogen interactions that have been identified for Mtb for which potential HDTs impacting both innate and adaptive immunity are available, and outline those worthy of future research. We will also discuss possibilities to target NTM infection by HDT, although current knowledge regarding host‐pathogen interactions for NTM is limited compared to Mtb. Finally, we speculate that combinatorial HDT strategies can potentially synergize to achieve optimal mycobacterial host immune control.
Keywords: drug resistance, host‐directed therapy, Mycobacterium avium, Mycobacterium tuberculosis, nontuberculous mycobacteria
1. INTRODUCTION
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb), remains a major health problem. With an estimated 10 million disease cases and 1.4 million deaths in 2019, Mtb is the deadliest infectious agent worldwide and TB is one of the top‐10 leading causes of deaths globally. 1 Approximately a quarter of the world's population is infected with Mtb and in most cases, progression toward TB disease is prevented by an efficient host immune response, often resulting in a latent TB infection (LTBI). 1 Five to fifteen percent of LTBI individuals will develop TB disease during their life‐time, often concomitant with host immunocompromising conditions, including HIV infection and use of immunosuppressive medication. Treatment of patients with active TB has largely remained unchanged for over 30 years, 1 and due to its lengthiness (6‐24 months) and considerable side effects, treatment‐adherence is low fueling development of multi‐drug and extensive‐drug resistance (MDR and XDR). The large TB disease burden and the increasing incidence of drug resistance make alternative treatment solutions imperative.
While the number of TB cases is slowly declining, a trend that may well be broken as a result of the COVID‐19 pandemic, 2 the prevalence of infections known to be caused by nontuberculous mycobacteria (NTM) is increasing at an alarming rate, currently reaching 0.2‐9.8 per 100.000 individuals. 3 NTM represent a group of opportunistic mycobacterial pathogens that mostly cause pulmonary diseases (PD), predominantly in vulnerable populations due to immunodeficiencies and/or pre‐existing lung conditions. Mycobacterium avium (Mav) complex (MAC) and Mycobacterium abscessus (Mab) account for the large majority of reported cases. 3 Despite extended treatment regimens, clinical outcome is poor, with cure rates of approximately 50%‐88% among MAC‐PD patients and 25%‐58% among Mab‐infected individuals, 3 urging the development of novel treatment modalities.
Mycobacteria are well known for their capability to manipulate intracellular signaling pathways to escape from host‐defense mechanisms in human cells. Mtb is best studied in this regard, but NTM have also been shown to modulate host immune responses, including preventing phagosome acidification and maturation or escaping from phagosomes into the nutrient‐rich cytosol. Counteracting pathogen‐induced immune modulation by host‐directed therapy (HDT) is a promising adjunct therapy to antibiotic therapy to combat intracellular mycobacterial infections, with several major advantages over current antibiotics. First, HDT can also be effective against MDR/XDR mycobacteria that are insensitive to current standard antibiotics. Second, because there is no direct selection pressure on mycobacteria, host‐targeting compounds are less likely to result in drug resistance. Third, host‐targeting compounds have the potential to target metabolically inactive, non‐replicating bacilli during LTBI, which are tolerant or resistant to conventional therapies. Fourth, HDT may allow shortening of current lengthy TB/NTM‐treatment regimens, thereby increasing compliance. Fifth, HDT may permit dose lowering of standard antibiotics, thus reducing toxicity without impacting efficacy. Finally, as HDT and mycobacterium‐targeting compounds (ie, antibiotics) by definition act on different pathways, combinatorial regimens would be expected to synergize. In this review, we will provide a comprehensive overview of host‐pathogen interactions that have been identified in Mtb infections and that are amenable to targeting by HDTs (summarized in Figure 1 and Table 1). Furthermore, despite a limited number of reports, we will also discuss NTM‐mediated host modulation and speculate whether HDTs could also be of interest to combat these mycobacterial infections. Finally, we will discuss the possibility of combinatorial HDTs that target distinct host signaling pathways to promote possible synergistic treatment effects.
Figure 1.
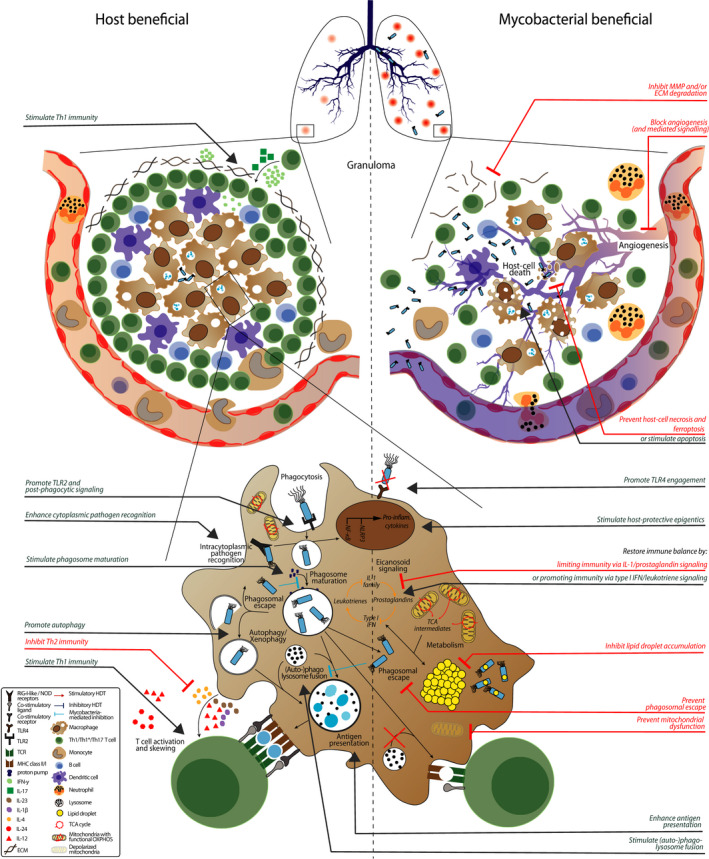
Host‐pathogen interactions and potential host‐directed therapies (HDT). Granulomas are characteristic for tuberculosis and mycobacterial infections in general. Pathologic granulomas are poorly vascularized due to ineffective angiogenesis, leading to hypoxia and concomitant host‐cell necrosis and bacterial dissemination. Blocking angiogenesis, preventing host‐cell necrosis (or stimulating apoptosis) or inhibiting extracellular matrix (ECM) degradation improves granuloma structure and concomitant disease outcome. Macrophages, key cells in the anti‐mycobacterial response, initiate phagocytosis after toll‐like receptor (TLR) recognition, which is prevented and/or modulated by mycobacteria. Promoting TLR4 engagement, TLR2 signaling and post‐phagocytic signaling via receptor tyrosine kinase are all potential targets for HDT to improve host immunity during mycobacterial infection. After internalization, mycobacteria are located to phagosomes that slowly mature and ultimately fuse with lysosomes, which are all inhibited by mycobacteria. Alternatively, mycobacteria escape to the cytosol where they can be recognized by cytoplasmic pathogen recognition receptor (PRR) and “recaptured” using autophagy, which again is inhibited by mycobacteria. HDTs that (1) prevent phagosomal escape, (2) alleviate blockage of (auto‐)phagosome maturation, (3) promote autophagy and/or (4) stimulate (auto‐)phagolysosome fusion all enhance mycobacterial killing. HDT that enhance cytoplasmic recognition of mycobacteria also improve the anti‐mycobacterial immune response. Mycobacteria that remain in the cytosol impair host metabolic pathways by stimulating tricarboxylic acid (TCA) cycle intermediates from mitochondria to be expelled into the cytosol to form lipid droplets and induce mitochondrial membrane depolarization. HDTs that (1) impair lipid droplet accumulation, (2) prevent mitochondrial membrane depolarization, and/or (3) stimulate TCA cycle intermediates being allocated in eicosanoid signaling maintain macrophage functionality which leads to better mycobacterial control. Finally, mycobacteria prevent the host from mounting an effective adaptive immune response by inhibiting antigen presentation and impairing T‐cell skewing. HDTs that promote adaptive immunity by enhancing antigen presentation, stimulating Th1 skewing or inhibiting Th2/Treg immunity all improve disease outcome. Compounds that can correct the above processes are represented in red for inhibitory/blocking therapies and in green for stimulatory therapies and summarized in Table 1, ordered per physiological process
Table 1.
HDT compounds and their biological activity against mycobacterial infections
| Compound | Biological activity | Model | Pathogen | Reference | Compound | Biological activity | Model | Pathogen | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Phagocytosis and phagosome maturation | Antigen presentation and priming | ||||||||
| PRR agonists | ↑ PRR‐signaling | In vitro | Mtb | 13, 14, 15, 16, 17 | MiRNA‐106b inhib. | ↑ Antigen presentation | In vitro | Mtb | 121 |
| miRNA‐125 inhib. a | ↑ TLR2‐signaling | In vitro | Mtb | 19 | AGK2 | ↑ Antigen presentation | Ex vivo, in vivo | Mtb | 116 |
| miRNA‐23a‐5p inhib. a | ↑ TLR2‐signaling | In vitro | Mtb | 20 | G1‐4A | ↑ Th1 immunity (TLR4‐signaling) | In vitro, in vivo | Mtb | 122 |
| Imatinib a | ↑ Phagosomal acidification | In vitro, in vivo, (R)CT | Mtb, Mm | 26, 27 | LPS + CD40 agonist | ↑ Th1 immunity (TLR4‐signaling) | In vitro, in vivo | Mtb | 13 |
| ↓ Phagocytosis | Bergenin | ↑ Th1 immunity | Ex vivo, in vivo | Mtb | 124 | ||||
| AZD0530 | ↑ Phagosomal acidification | In vivo | Mtb | 28 | D‐1MT a | ↑ Th1 immunity | Ex vivo, in vivo | Mtb | 129 |
| AT9283 | Unknown | In vitro | Mtb | 29 | Skewing of T‐cells | ||||
| ENMD‐2076 | Unknown | In vitro | Mtb | 29 | IFN‐γ | ↑ Th1 immunity | In vitro, (R)CT | Mtb, Mav | 133, 134, 137, 138, 139, 140 |
| Dovitinib | Unknown | In vitro | Mtb | 29 | IL‐12 | ↑ Th1 immunity | In vivo | Mtb | 141 |
| Nitazoxanide | ↑ Cytoplasmic pathogen recognition | In vitro, (R)CT | Mtb | 33, 34 | IL‐24 | ↑ Th1 immunity | In vivo | Mtb | 142 |
| Metformin a | ↑ Phagosome maturation | In vitro, in vivo, (R)CT | Mtb | 71, 72, 73, 74, 75, 76, 77 | FPS‐ZM1 | ↓ Immune response | In vivo | Mtb | 145 |
| Gefitinib a | ↑ Lysosomal biogenesis | In vitro, in vivo | Mtb | 78 | Blocking IL‐4 | ↓ Th2 immunity | In vitro | Mtb | 131 |
| Bedaquilline a | ↑ (Auto)‐phagolysosome fusion | In vitro, in vivo, (R)CT | Mtb | 84, 221, 222, 223 | IL‐2 | ↑ Th1 immunity | In vivo, (R)CT | Mtb, Mav | 146, 147, 148, 149 |
| 2062 a | ↑ (Auto)‐phagolysosome fusion | In vitro, in vivo | Mtb | 85 | IL‐2 + HMBPP | ↑ Th1 immunity | In vivo | Mtb | 151 |
| GM‐CSF a |
↑ Phagocytosis ↑ (Auto)‐phagolysosome fusion |
In vitro, in vivo, CR | Mtb, Mav | 86, 87, 88, 89, 90, 91, 92 | PBMCs + cytokines | ↑ Th1 immunity | CR | Mtb | 152 |
| Prednisolone | ↓ Immune response | (R)CT | Mtb | 158, 159, 160 | |||||
| Resveratrol a | ↑ (Auto)‐phagolysosome fusion | In vitro | Mtb | 70 | (Programmed) cell death | ||||
| SRT1720 a | ↑ (auto)‐phagolysosome fusion | In vitro, in vivo | Mtb | 70 | Mcl‐1 inhib. | ↑ Apoptosis | In vitro | Mtb | 155 |
| Celecoxib a | ↑ Phagocytosis | In vivo, (R)CT | Mtb | 187 | Nicotinamide | ↓ Necrosis | In vitro | Mtb | 99 |
| Statins a | ↑ Phagosome maturation | In vitro, in vivo, (R)CT | Mtb | 181, 184, 185, 186 | SQ22536 | ↑ TNF‐α | In vitro | Mtb | 161 |
| Autophagy | H‐89 | ↑ TNF‐α | In vitro | Mtb | 161 | ||||
| Imatinib a | ↑ Autophagy | In vitro | Mtb | 26, 27 | Dexamethasone | ↓ Necrosis | In vitro, in vivo, R(CT) | Mtb | 157, 158, 159, 160, 188, 191 |
| Vitamin‐D | ↑ Autophagy | In vitro, (R)CT | Mtb | 40, 41, 42, 43, 44, 45, 46, 47, 52 | ↓ Immune response | ||||
| PBA | ↑ Autophagy | In vitro, (R)CT | Mtb | 49, 50, 51, 52, 53 | Doramapimod | ↓ Necrosis | In vitro | Mtb | 157 |
| Vitamin‐A (± Zn2+) | ↑ Autophagy | In vitro, in vivo, (R)CT | Mtb | 55, 56, 57, 58, 59, 60, 61, 62 | Alisporivir |
↓ Necrosis ↓ TNF‐α |
In vivo | Mm | 163 |
| MiRNA‐27a antagomir | ↑ Autophagy | In vivo | Mtb | 66 | |||||
| Everolimus | ↑ Autophagy | In vitro, (R)CT | Mtb | 68 | Desiparamine | ↓ Necrosis | In vivo | Mm | 163 |
| Ibrutinib | ↑ Autophagy | In vitro, ex vivo | Mtb | 69 | Cilostazol | ↓ TNF‐α | In vivo | Mtb | 164, 165 |
| Metformin a | ↑ Autophagy, | In vitro, in vivo, (R)CT | Mtb | 71, 72, 73, 74, 75, 76, 77 | Sildenafil | ↓ TNF‐α | In vivo | Mtb | 164, 165 |
| ↓ Mitochondrial dysfunction | Ferrostatin 1 | ↓ Ferroptosis | In vitro, in vivo | Mtb | 171 | ||||
| Gefitinib a | ↑ Autophagy | In vitro, in vivo | Mtb | 78 | D‐1MT a | ↑ Apoptosis | Ex vivo, in vivo | Mtb | 129 |
| Bazedoxifene a | ↑ Autophagy | In vitro | Mtb | 79 | ATP administration a | ↑ Apoptosis | In vitro | Mtb, Mav | 176, 177 |
| Loperamide | ↑ Autophagy | In vitro, ex vivo | Mtb | 80 | Cabrohydrate and lipids | ||||
| Bedaquilline a | ↑ Autophagy | In vitro, in vivo, (R)CT | Mtb | 84, 221, 222, 223 | 2‐DG | ↓ Glycolysis | In vitro | Mtb | 174 |
| 2062 a | ↑ Autophagy | In vitro, in vivo | Mtb | FX11 | ↓ Glycolysis | In vivo | Mtb | 175 | |
| Resveratrol a | ↑ Autophagy | In vitro | Mtb | 70 | ATP administration a | ↑ Iron chelation | In vitro | Mtb, Mav | 176, 177 |
| SRT1720 a | ↑ Autophagy | In vitro, in vivo | Mtb | 70 | Clemastine | ↑ Immune response? | In vivo | Mm | |
| miRNA‐125 inhib. a | ↑ Autophagy | In vitro | Mtb | 19 | M1 | ↓ Mitochondrial dysfunction | In vitro | Mtb | 180 |
| miRNA‐23a‐5p inhib. a | ↑ Autophagy | In vitro | Mtb | 20 | Ezetimibe | ↓ Lipid droplet accumulation | In vitro | Mtb | 179 |
| Statins a | ↑ Autophagy | In vitro, in vivo, (R)CT | Mtb | 181, 184, 185, 186 | Statins a | ↓ Membranal cholesterol incorporation | In vitro, in vivo, (R)CT | Mtb | 181, 184, 185, 186 |
| Intracellular killing mechanisms | Eicosanoids | ||||||||
| CD157 | ↑ Reactive oxygen species | In vitro | Mtb | 95, 97 | PGE2 and/or Zileuton | ↑ IL‐1β/prostaglandin signaling | In vivo | Mtb | 189, 190 |
| N‐acetyl‐cysteine | ↓ Reactive oxygen species | In vitro, in vivo, (R)CT | Mtb | 99, 100, 101 | Celecoxib a | ↓ IL‐1β/prostaglandin signaling | In vivo, (R)CT | Mtb | 187 |
| L‐arginine | ↑ Reactive nitrogen species | (R)CT | Mtb | 107, 108, 109 | LTB4 | ↑ IL‐1β/prostaglandin signaling | In vivo | Mtb | 187 |
| Bazedoxifene a | ↑ Reactive oxygen species | In vitro | Mtb | 79 | Ibuprofen | ↓ IL‐1β/prostaglandin signaling | In vivo, (R)CT | Mtb | 192, 193 |
| Metformin a | ↑ Reactive oxygen species | In vitro, in vivo, (R)CT | Mtb | 71, 72, 73, 74, 75, 76, 77 | Aspirin | ↓ IL‐1β/prostaglandin signaling | In vivo, (R)CT | Mtb | 193, 194, 195, 196, 197, 198 |
| Celecoxib a | ↑ Reactive oxygen species | In vivo, (R)CT | Mtb | 187 | Granuloma: Formation, angiogenesis and hypoxia | ||||
| Epigenetic regulation | Cipemastat | ↓ MMP/ECM degradation | In vivo | Mtb | 208 | ||||
| TMP195 | ↑ Host‐protective epigenetics | In vitro, in vivo | Mtb, Mm | 115 | MMP‐9 inhib. | ↓ MMP/ECM degradation | In vivo | Mm | 25 |
| TMP269 | ↑ Host‐protective epigenetics | In vitro, in vivo | Mtb, Mm | 115 | Sb‐3ct | ↓ MMP/ECM degradation | In vitro, in vivo | Mtb | 212, 213 |
| Trichostatin A | ↑ Host‐protective epigenetics | In vitro, in vivo | Mtb, Mm | 115 | AB0046 | ↓ MMP/ECM degradation | In vivo | Mtb | 166 |
| Resveratrol a | ↑ Host‐protective epigenetics | In vitro | Mtb | 70 | Doxycycline | ↓ MMP/ECM degradation | In vitro, in vivo, (R)CT | Mtb | 210, 214 |
| SRT1720 a | ↑ Host‐protective epigenetics | In vitro, in vivo | Mtb | 70 | Marimastat | ↓ MMP/ECM degradation | In vitro, in vivo | Mtb | 203, 205 |
| Valproic acid | ↑ Host‐protective epigenetics | In vitro | Mtb | 117 | Batimastat | ↓ MMP/ECM degradation | In vivo | Mtb | 205 |
| SAHA | ↑ Host‐protective epigenetics | In vitro | Mtb | 117 | MMP‐9 inhib. | ↓ MMP/ECM degradation | In vivo | Mtb | 205 |
| Bevacizumab | ↓ Angiogenesis | In vivo | Mtb | 217 | |||||
| SU5416 | ↓ Angiogenesis | In vivo | Mm | 218 | |||||
| Pazopanib | ↓ Angiogenesis | In vivo | Mm | 218 | |||||
| AKB‐9785 | ↓ Angiogenesis | In vivo | Mm | 219 | |||||
Abbreviations: (R)CT, (randomized) clinical trial; 2‐DG, 2‐deoxy‐D‐glucose; ATP, adenosine triphosphatases; CR, case report; GM‐CSF, granulocyte‐macrophage colony‐stimulating factor; HMBPP, (E)‐4‐hydroxy‐3‐methyl‐but‐2‐enyl pyrophosphate; IFN‐γ, interferon‐γ; IL, interleukin; inhib., inhibitor; LTB4, leukotriene‐B4; Mav, Mycobacterium avium; Mm, Mycobacterium marinum; MMP, matrix metalloprotease; Mtb, Mycobacterium tuberculosis; PBA, phenylbutyrate; PBMCs, peripheral blood mononuclear cells; PRR, pathogen recognition receptor; SAHA, suberoylanilide hydroxamic acid; TNF‐α, tumor necrosis factor‐α.
Compounds targeting distinct host intracellular pathways are categorized under multiple sections.
[Correction added on 07 March 2021, after first online publication: the Table 1 has been updated in this version.]
2. HDT MODULATING INNATE IMMUNE CELL FUNCTION
2.1. Phagocytosis and phagosome maturation
The first potential target for HDT to interfere with host‐pathogen interactions is to modulate mycobacterial host‐cell entry. Mycobacteria infect host cells, predominantly alveolar macrophages and epithelial cells, in the lower respiratory tract, following inhalation of small bacteria‐containing aerosols. Mycobacteria express pathogen‐associated molecular patterns (PAMPs) that are recognized by pattern recognition receptors (PRRs), such as Toll‐like receptors (TLRs), C‐type lectin receptors, and other scavenger receptors expressed on the surface of host cells to initiate phagocytosis. 4 Especially TLR2, which forms heterodimers with TLR1 and TLR6 to recognize lipoproteins or lipopeptides (eg, lipomannan), and TLR4, which recognizes cell wall lipids, glycoproteins, and secreted proteins, are known to mediate Mtb‐induced cellular activation. 5 Mice lacking TLR2 are more susceptible to infections with virulent Mtb and Mav strains. 6 , 7 , 8 NTM, including Mav and Mab, express a class of glycolipids known as glycopeptidolipids (GPLs) which mask underlying cell wall phosphatidyl‐myo‐inositol mannosides, thereby limiting interactions with TLR2. 9 , 10 Moreover, the acetylation state of lipomannan modulates TLR2‐mediated macrophage activation, and subversion of the TLR2‐MyD88 pathway has been linked to phagolysosome escape of virulent Mtb to the cytosol, 11 , 12 indicating a crucial role for TLR signaling pathways in the control of intracellular mycobacteria. Several PRR agonists, including TLR2 agonist Pam2Cys, have been identified that activate both innate and adaptive immune responses against Mtb, positioning PPRs as potential HDT targets to combat mycobacterial infections. 13 , 14 , 15 , 16 , 17 Furthermore, also downstream PRR signaling might be modulated by HDT. TLR2‐dependent expression of miRNA‐125 hampered autophagy in murine macrophages. 18 , 19 TLR2‐MyD88 signaling in Mtb‐infected murine macrophages was also repressed by upregulation of miRNA‐23a‐5p, restricting Mtb infection–induced autophagy and thus increasing intracellular Mtb survival. 20 Inhibition of miRNA‐125 18 , 19 or miRNA‐23a 20 both reduced Mtb survival, identifying miRNA‐125 and miRNA‐23a as potential targets for HDTs.
After phagocytosis, mycobacteria become localized in phagosomes that are initially non‐degradative, but slowly mature into increasingly hostile organelles. This so‐called phagosome maturation hinges upon fusion with lysosomes that contain antimicrobial peptides and induce intravesicular acidification enhancing lysosomal enzyme activity. 21 While initially thought to be simply transport vehicles, phagosomes have appeared to be highly dynamic structures that are regulated by several membrane markers, such as PI3P, acidifying proton adenosine triphosphatases (ATPases), and Rab‐GTPases. 21 , 22 As GTPases are also involved in autophagy induction, these enzymes could be interesting targets for HDT, but as of yet have not been investigated in this context. To prevent (auto)phagosome maturation, Mtb secretes proteins such as SapM and PknG, which inhibit PI3P‐phosphorylation, dissociation of early endosomal protein Rab5 and acquisition of late endosomal protein Rab7. 23 In addition, Mtb prevents recruitment of the proton pumping enzyme vacuolar‐type H+‐ATPase (v‐ATPase) by phagosomes, thus further arresting phagosome acidification. 24 Several receptor tyrosine kinases (RKTs) that are activated upon internalization of Mtb and NTM are involved in both bacterial uptake and intracellular trafficking, and could be exploited as targets for HDT. 25 Abelson tyrosine kinase (Abl), involved in bacterial uptake by regulating cytoskeletal dynamics in host cells, can be chemically inhibited by imatinib, 24 and indeed, imatinib treatment impaired internalization of Mtb by human macrophages. 26 Furthermore, Abl also modulates the expression of v‐ATPase, and inhibiting RTKs with imatinib induced expression of several v‐ATPase pump‐subunits and their colocalization with Mtb‐containing phagosomes, promoting phagosomal acidification and enhancing bacterial killing in human macrophages. 27 In line with this, imatinib treatment of mice infected with Mtb or Mycobacterium marinum (Mm) decreased mycobacterial load and combining imatinib with first‐line anti‐TB drug rifampicin synergistically reduced mycobacterial load in mice and in a murine macrophage‐like cell line. 25 , 26 , 27 The potential of inhibiting host tyrosine kinases to impair intracellular mycobacterial survival is further highlighted with AZD0530 treatment, a Src‐inhibitor, that lowered disease burden in Mtb‐infected guinea pigs by promoting phagosomal acidification. 28 Moreover, Korbee et al showed that inhibiting RTK signaling with the repurposed drugs AT9283, ENMD‐2076 and dovitinib significantly reduced intracellular Mtb in primary human macrophages. 29 Being repurposed, these compounds are already FDA‐approved or in phase II and III clinical trials, thus accelerating potential clinical application as adjunct therapy in treating (MDR)‐TB and treatment of refractory NTM infections.
Formation and subsequent acidification of phagolysosomes is also inhibited by Mtb‐secreted protein 1‐tuberculosinyladenosineantacid (1‐TbAd). 30 Accumulation of 1‐TbAd in acidic intracellular compartments resulted in swelling and ultimately bursting of phagosomes, permitting mycobacterial escape into the cytosol. To further impair phagosome integrity, Mtb permeabilizes phagosomal membranes using the bacterial ESX‐1 secretion system (ie, ESAT‐6), 31 which leads to leakage of phagosomal cargo into the cytosol, allowing phagosomal escape of mycobacteria. Although the cytosol contains an abundance of nutrients to support bacterial growth, translocation into the cytosol also activates DNA‐ and RNA‐sensing pathways via intracellular recognition of PAMPs and danger‐associated molecular patterns (DAMPs) to induce anti‐mycobacterial host effector mechanisms. Retinoid acid–inducible gene I (RIG‐I)–like receptors are cytosolic PRRs recognizing single‐ and double‐stranded RNA and upon ligation induce the type‐I IFN pathway, among others. 32 Nucleotide‐binding oligomerization domain (NOD)–like receptors are intracellular sensors for several DAMPs and PAMPs, including bacterial RNA, that can induce both type‐I IFN and IL‐1 responses. 32 Enhancing expression levels of RIG‐I‐like receptors using nitazoxanide treatment during mycobacterial infection increased IFN‐β levels and concomitantly reduced mycobacterial loads in an in vitro TB model, 33 but did not show efficacy in TB patients, possibly due to negligible concentrations at the site of infection. 34
2.2. Autophagy
Autophagy is a mechanism mediating self‐maintenance and cellular homeostasis and is induced under stress such as hypoxia, starvation but also microbial infection. 35 Autophagy is crucial during Mtb and NTM infections and inhibition of autophagy using azithromycin increased susceptibility of cystic fibrosis patients to NTM infection. 36
Autophagy is initiated by formation of a double‐membraned phagophore that, under stringent control of ubiquitin‐like protein conjugation systems, expands around the intracytoplasmic cargo to form autophagosomes, which ultimately fuse with lysosomes to mediate degradation. Two autophagic pathways are important for mycobacterial degradation: LC3‐associated phagocytosis (LAP) and the STING‐dependent cytosolic pathway. 37 LAP is initiated by downstream signaling of numerous receptors, including TLRs, 37 after which the phagosome becomes decorated with PI3P produced by the PI3KC3 complex, that includes Beclin‐1 and Rubicon. PI3P and Rubicon are required for the generation of reactive oxygen species (ROS) and conjugation of lipidated LC3‐II to the membrane to enhance phagosomal maturation. 37 The STING‐dependent pathway is triggered by mycobacterial DNA released into the cytosol through the bacterial ESX‐1 system. When mycobacterial DNA is sensed by a STING‐dependent DNA sensor, cytosolic Mtb is ubiquitinated by the ubiquitin‐ligating (E3) ligase, bound to autophagic receptors including p62/sequestosome 1, NDP52 protein and TBK1, and subsequently delivered to autophagosomes by engagement of membrane‐associated LC3. 32
Numerous drugs have been identified that promote autophagy by targeting different components of the autophagic pathways. Beclin‐1 is induced by human antimicrobial peptide (AMP) LL‐37, also known as cathelicidin. 38 Cathelicidin is able to suppress Mtb growth and can be induced by pathogens after TLR2/TLR1 ligation, and also by vitamin‐D. 39 In vitro experiments identified calcitriol, the bioactive metabolite of vitamin‐D, to exert antimicrobial activity by mediating intracellular killing of Mtb through cathelicidin. 40 Calcitriol has also been linked to nitric oxide (NO) production and suppression of matrix metalloproteases (MMPs) which may further protect the host from TB immunopathology. 41 , 42 The efficacy of vitamin‐D as HDT during TB disease has been investigated in multiple randomized controlled trials (RCTs). Vitamin‐D administration corrected any vitamin‐D deficiencies and was safe in use but did not show consistent beneficial outcomes during mycobacterial infections in meta‐analyses. 43 , 44 , 45 Acceleration of Mtb clearance from sputum was mainly observed in MDR‐TB cases or patients with a specific genotype, such as polymorphisms in the vitamin‐D receptor‐gene. 46 , 47 Furthermore, low levels of vitamin‐D have been linked to a higher susceptibility to develop TB disease. 48 Some studies combined vitamin‐D therapy with phenylbutyrate (PBA), which stimulates cathelicidin‐induced autophagy and also inhibits bacterial growth directly. 49 , 50 Combining vitamin‐D and PBA treatment further increased expression of cathelicidin in healthy volunteers, but the augmented expression level was constrained to a defined dose range of PBA. 51 The narrow therapeutic window of PBA might clarify why certain RCTs failed to detect accelerated sputum‐smear conversion by co‐administering vitamin‐D and PBA 52 and only showed accelerated sputum‐smear conversion at week 4 following combined treatment, but not at week 8 in vitamin‐D‐deficient patients. 53 Due to these inconsistencies, progression of vitamin‐D as potential HDT in TB treatment regimens has not been successful.
Vitamin‐A‐deficiency has also been correlated with an increased risk to develop TB disease. 54 STING‐dependent autophagy can be targeted by the active metabolite of vitamin‐A, that is, all‐trans retinoic acid (ATRA), which promotes TBK1‐mediated enhancement of autophagy which reduces Mtb survival in human macrophages. 55 ATRA is also known to increase CD1d receptor expression on innate immune cells, 56 and treatment with non‐mycobacterial CD1d ligand α‐galactosylceramide (α‐GalCer) reduced mycobacterial load and improved survival of mice with TB, 57 and while α‐GalCer combined with ATRA and vitamin‐D did not clear the infection in mice, it improved containment of the infection. 58 In patients, vitamin‐A supplementation combined with Zn2+ or vitamin‐D gave inconsistent results. 59 , 60 , 61 , 62 Thus, although vitamin‐A reduced Mtb loads in vitro and in vivo, evidence for its efficacy in patients is inconsistent. An additional regulator of the selective STING‐dependent autophagy pathway is DNA‐damage regulated autophagy‐modulator protein 1 (DRAM1). DRAM1 was found to trigger autophagy in both Mtb‐infected human macrophages and Mm‐infected zebrafish larvae, whereas DRAM1‐deficiency resulted in host‐detrimental cell death, underscoring DRAM1 as an interesting target for HDT. 63 , 64
In addition to Beclin‐1 and TBK1, other components of the autophagic pathways have also been targeted to promote mycobacterial clearance. Ca2+ signaling is pivotal in inducing autophagy by activating the Ca2+/calmodulin‐dependent serine/threonine‐kinase (CaMKK2)/ULK1 complex. 65 CaMKK2‐mediated autophagy and killing of intracellular Mtb requires Ca2+ transporter CACNA2D3 which is, however, suppressed by Mtb‐induced miRNA27a. 66 Intracellular survival of Mtb could be impaired by inhibiting miRNA‐27, providing a new HDT target.
Another important negative regulator of autophagy is the PI3K‐Akt‐mTOR signaling pathway, which is robustly activated by Mtb to facilitate its intracellular survival. 67 Everolimus, an improved analog of mTOR inhibitor rapamycin, was able to reduce Mtb burden in a human granuloma model and these effects were additive to first‐line TB drugs, possibly by HDT activity and/or by inhibiting mycobacterial growth directly. 68 Inhibition of protein‐kinase C‐beta (PKC‐B), another important regulator of the PI3K‐Akt‐mTOR pathway, by ibrutinib also enhanced autophagy and restricted intracellular growth of Mtb in macrophages and mice in the spleen, although not in the lungs. 69 Alleviating the Mtb‐mediated suppression of sirtuin‐1, a class‐III histone deacetylase that also modulates autophagy via 5’AMP‐activated protein‐kinase (AMPK), using resveratrol restricted intracellular Mtb growth by stimulating autophagy and phagosome‐lysosome fusion. 70 Metformin, a well‐established stimulator of AMPK‐mediated inhibition of mTOR signaling, is widely used for the treatment of type‐2 diabetes, but also induces ROS production, phagosome maturation, and autophagy in vitro and prevents mitochondrial membrane depolarization. 71 , 72 , 73 In non‐diabetic healthy volunteers, metformin treatment downregulated genes involved in Mtb‐mediated modulation of autophagy, as well as type‐I IFN signaling, while upregulating genes involved in phagocytosis and ROS production. 74 Several clinical trials have shown that metformin treatment reduces the risk of latent TB reactivation and TB mortality, and in patients with cavitary TB, improves sputum culture conversion. 75 , 76 , 77
Like metformin, repurposing of drugs that are clinically approved in the context of other diseases has been shown to enhance autophagy and to reduce intracellular bacterial growth, suggesting these drugs may be considered as HDT candidates. The anti‐cancer drug gefitinib, an inhibitor of epidermal growth factor receptor (EGFR), promotes intracellular Mtb killing by alleviating the STAT3‐dependent repression of effective immune responses in Mtb‐infected mice and by enhancing lysosomal biogenesis and targeting of mycobacteria to lysosomes in Mtb‐infected macrophages. 78 Gefitinib also induced autophagy, 78 but since no specific targeting of mycobacteria to the autophagic pathway was observed, this activity has not been formally linked to restricting intracellular Mtb survival. Bazedoxifene, a selective estrogen receptor modulator (SERM) used for breast cancer treatment, was also shown to inhibit intracellular Mtb growth in macrophages through enhanced ROS‐dependent autophagy 79 and to inhibit Mtb growth in liquid culture. Furthermore, one study showed that loperamide, an anti‐diarrheal drug, promoted autophagy as indicated by p62 degradation and decreased mycobacterial burden in vitro and ex vivo in murine macrophages. 80
Mtb not only inhibits the initiation of autophagy, but also fusion of autophagosomes with lysosomes via protein P2‐PGRS47. 23 , 81 Furthermore, the Mtb secretion‐factor SapM inhibits Rab7‐recruitment to prevent autophagosome‐lysosome fusion. 82 Mtb‐expressed mannosylated lipoarabinomannan (ManLAM) also inhibits maturation of autophagosomes, by blocking LC3‐translocation to autophagosome membranes. 32 , 83 Releasing such blockades in autophagosome‐lysosome fusion could represent potential HDT strategies. Bedaquiline, a novel antibiotic now in use for MDR‐TB, has also been shown to induce phagosome‐lysosome fusion and autophagy via activation of TFEB, possibly contributing to it successful application as a new TB drug. 84 In line with this, a small molecule called 2062 improved autophagy and lysosomal pathway activity via activation of TFEB when administered with suboptimal doses of rifampicin. 85
Although autophagy‐targeting HDTs have been investigated mainly in the context of Mtb infections, several case reports have been published for (disseminated) Mav infections in patients who received granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) or IFN‐γ. GM‐CSF treatment during Mtb infection reduced bacterial burden by promoting phagosome‐lysosome fusion and increased expression of TNF‐α, IFN‐γ, and inducible nitric oxide synthase (iNOS). 86 , 87 , 88 GM‐CSF treatment during Mav infection increased phagocytosis and impaired bacterial growth in vitro in human macrophages and in Mav‐infected patients with or without HIV infection (partially) improved clinical outcome. 89 , 90 , 91 , 92 Thus, autophagy likely plays an important role also in NTM immunity, and could represent an attractive target for HDT in severe NTM infections.
2.3. Intracellular killing mechanisms: reactive oxygen and nitrogen species
To eliminate mycobacteria during infection, host cells trigger the production of reactive oxygen species (ROS) and reactive nitrogen species (RNS), via NADPH oxidase 2 (NOX2) 93 and inducible nitric oxide synthase 2 (iNOS), respectively. iNOS catalyzes production of nitric oxide (NO) by converting L‐arginine into L‐citrulline, which is subsequently converted into RNS. 94 Once expressed, both ROS and RNS interact with the phagosome to destroy bacterial components. 95 Mycobacterium‐induced ROS production occurs via the TLR(2)‐MyD88 signaling axis and impairments in this pathway increase susceptibility to Mtb infection. 7 , 96 Recently, TLR2‐dependent ROS production and bactericidal activity was found to be impaired in CD157‐deficient murine macrophages which could be rescued by administration of soluble CD157. 95 , 97 Moreover, expression levels of CD157, an enzyme important for leukocyte migration and involved in nicotinamide‐adenine‐dinucleotide (NAD+) metabolism, are elevated in patients with active TB compared to LTBI and lowered when patients are treated with TB chemotherapy, indicating an important role of CD157 in host immunity, biomarker profiling and also providing a potential HDT. 97
Although ROS production is important for host resistance against mycobacterial infection, modulating ROS as HDT strategy requires careful monitoring as excess ROS leads to oxidative stress and concomitant necrosis. 98 Corroborating this view, reducing ROS accumulation in Mtb‐infected macrophages with ROS scavenger N‐acetyl‐cysteine in fact restricted Mtb replication and restored macrophage cell viability, 99 and in a guinea pig Mtb infection model N‐acetyl‐cysteine administration was also shown to be efficacious. 100 Moreover, N‐acetyl‐cysteine was found to be safe in a cohort of TB‐HIV co‐infected individuals, 101 although its impact on culture conversion remains to be determined. 101 Nevertheless, ROS production is important for the bactericidal activity of macrophages 102 and the critical balance in ROS production and its regulation is important in restricting intracellular mycobacterial growth without harming the host.
Multiple studies in mice and humans have shown antimicrobial effects of NO, but the exact underlying mechanisms remain unclear. 103 , 104 Macrophages from LTBI patients were shown to control Mtb growth via NO production, and human macrophages required iNOS for intracellular killing of Mtb. 105 Moreover, compared to wildtype murine macrophages, protein‐kinase R (PKR)–deficient macrophages induced higher levels of iNOS during Mtb infection, 106 and PKR‐deficient mice had lower Mtb loads and less severe lung pathology compared to infected wildtype mice, highlighting the potential of PKR as HDT target. Despite its importance as substrate for NO production, supplementing L‐arginine did not consistently improve clinical outcomes such as cure rate or (sputum) smear conversion in several clinical trials. 107 , 108 , 109
Several Mtb‐associated proteins have been identified that protect Mtb from RNS, but Mav naturally tolerates intracellular NO levels and may even benefit from host NO. 110 , 111 , 112 Mice that cannot produce NO were more resistant to Mav infections, while being more prone to Mtb infections. 113 In agreement with this, compared to wildtype mice, NOS2‐deficient mice showed higher IFN‐γ responses during Mav infection and increased accumulation of especially CD4+ T‐cells. 114 Enhancing NO production can thus be beneficial in combatting mycobacterial infections such as Mtb, but not Mav.
2.4. Epigenetic regulation
Macrophage polarization is an important mechanism of the immune system to respond adequately to the plethora of pathogens, which is partly mediated by epigenetic regulation of gene expression using histone acetylation. The level of histone acetylation is regulated by the balanced activity of histone acetyltransferases (HATs) and histone deacetylases (HDACs). HDACs are divided into four classes, three of which are Zn2+‐dependent (class I, IIa/IIb and IV), while class‐III is NAD+‐dependent. 115 Mtb infection actively modulates the acetylation status of host histones by (a) suppressing expression of class‐II HDACs (ie, HDAC 3, 5, 7, and 10) in macrophages, with anti‐inflammatory M2 macrophages being more affected than pro‐inflammatory M1 macrophages, 115 (b) inhibiting the expression of class‐III HDAC sirtuin‐1 both in vitro and in human tissues from TB patients, 70 and (c) upregulating expression of sirtuin‐2, another class‐III HDAC that regulates cell cycle and metabolism. 116 Type IIa–specific HDAC inhibitors (HDACi) TMP195 or TMP269 reduced bacterial loads in M2, but not M1 macrophages, while broad‐spectrum HDACi trichostatin A reduced bacterial loads in both M1 and M2 macrophages. Interestingly, combining HDACi with AKT1 kinase inhibitor H‐89 resulted in cumulative reduction in bacterial loads. Importantly, in a Mm zebrafish infection model, both class‐IIa and pan‐HDAC inhibition reduced bacterial loads, confirming the in vivo potential of HDAC inhibition as HDT to treat TB. 115 Sirtuin‐1, a class‐III HDAC important during (viral) infections, regulates stress responses and cellular metabolism. Resveratrol or SRT1720, a natural and synthetic activator of sirtuin‐1, enhanced clearance of drug‐sensitive and drug‐resistant Mtb. 70 Both compounds stimulate autophagy and phagolysosome fusion in THP‐1 cells, which likely accounts for the enhanced bacterial killing, while reducing pathology in a TB mouse model, possibly by inhibiting expression of IL‐1β, IL‐6, MCP‐1, and TNF‐α. 70
The Mtb genome encodes Rv1151c, a sirtuin‐like NAD‐dependent deacetylase, allowing Mtb to produce acetyl coenzyme A (acetyl‐CoA) synthetase, a critical enzyme in energy metabolism of both host cells and bacteria. Targeting this pathway using HDACi valproic acid directly inhibited bacterial growth, likely by inhibiting acetyl‐CoA production by Mtb itself, while co‐treatment of valproic acid and rifampicin/isoniazid therapy resulted in cumulative effects. 117 By contrast, FDA‐approved HDACi suberoylanilide hydroxamic acid (SAHA) had no direct effect on Mtb growth, but it reduced mycobacterial growth via host‐directed mechanisms and synergized with rifampicin/isoniazid therapy. 117 As Rv1151c is well conserved across different mycobacterial species including Mav, 118 the above therapies may also be efficacious against NTM.
3. HDT MODULATING ADAPTIVE IMMUNE RESPONSES
3.1. Antigen presentation and priming
Upon phagocytosis, pathogens are processed and degraded, such that pathogen‐derived peptides can be loaded and presented in MHC‐class I and II molecules to initiate adaptive T‐cell responses. One strategy of mycobacteria to evade host adaptive immune responses is to impair presentation of mycobacterial peptides by evading phagosomal degradation. Improving mycobacterial degradation by promoting phagosomal maturation and/or autophagy induction as discussed above likely enhances both antigen presentation and concomitant adaptive immunity. Another strategy of mycobacteria to evade host adaptive immune responses is to predominantly infect macrophages instead of dendritic cells, the former requiring stronger activation before being able to efficiently process and present antigens for priming naive T‐cells. 119 Macrophage activation is required to induce expression of CIITA, a major positive regulator of MHC‐class II. By actively engaging TLR2 rather than other TLRs, Mtb (and to a lesser extent M smegmatis) minimizes upregulation of CIITA and concomitant MHC‐class II expression. In addition, TLR2 (among all TLRs) most potently induces an innate (IL‐6) response, 120 leading to upregulation of suppressor‐of‐cytokine‐signaling‐1 (SOCS1) that in turn inhibits signal‐transducer‐and‐activator‐of‐transcription 1 (STAT1) phosphorylation and antigen presentation, further impairing the adaptive host immune response. 23 MiR106b, which degrades mRNA encoding cathepsin S, a protein that modulates MHC‐class II molecules to allow peptide loading, is significantly upregulated during Mtb infection. 121 Inhibition of miR106b using miRIDIAN hairpin inhibitors upregulated expression of both cathepsin S and HLA‐DR and enhanced subsequent CD4 T‐cell proliferation. 121 Alternatively, inhibition of sirtuin‐2 activity in macrophages using AGK2 modulated gene expression‐promoted antigen presentation. 116 AGK2 treatment of mice resulted in upregulation of MHC‐class II expression but also of co‐stimulatory molecules and markers of activation, leading to enhanced priming of T‐cells and improved Mtb killing. 116
Since Mtb limits activation of antigen‐presenting cells (APCs), which precludes the host from mounting an effective adaptive immune response, proper activation of APCs could be an interesting HDT. A possible strategy for HDT could be administration of G1‐4A, a polysaccharide from Tinospora cordifolia that presumably signals via TLR4, or TLR4 ligand LPS combined with a CD40 agonistic antibody. 13 , 122 Both treatments induced vast cytokine production (IFN‐γ/IL‐12, TNF‐α, IL‐6) and upregulation of co‐stimulatory molecules by dendritic cells in vitro. 122 Furthermore, both treatments reduced bacterial loads in murine TB infection models which was, at least in part, T‐cell‐mediated. 122 However, systemic administration of TLR ligands is known to cause significant side effects, 123 and may only be applicable via local administration. Bergenin, a phytochemical extracted from tender leaves, enhanced macrophage activation, as evidenced by enhanced CD11b expression as well as augmented NO, TNF‐α, and IL‐12 production, through activating the MAPK/ERK pathway. The resulting increased IL‐12 production induced a robust Th1 response with concomitant IFN‐γ production by T‐cells. Bergenin therapy reduced bacterial loads as well as lung pathology in a murine TB infection model. 124 Of note, vaccination could also be an interesting HDT approach to activate APCs or reprogram an effective adaptive immune response. This, however, falls outside the scope of this review and is excellently reviewed elsewhere. 125
Due to the chronic immune stimulation during persisting mycobacterial infections, including LTBI, T‐cells and APCs upregulate inhibitory receptors such as PD‐1/PD‐L1, which can impair T‐cell effector functions, 23 and may be interesting targets for HDT. Expression of exhaustion‐associated markers by T‐cells during active disease, however, is rather ambiguous: Despite successes in anti‐cancer therapies by inhibiting immune checkpoint molecules with anti‐PD‐1/PD‐L1 antibodies, PD1/PDL1‐directed experimental therapies in in vitro and in vivo TB models resulted in impaired intracellular control of Mtb and TB exacerbation rather than improved resolution, 126 , 127 suggesting PD‐1 may be a T‐cell activation rather than exhaustion marker during TB. Moreover, reports of LTBI‐reactivation in cancer patients treated with anti‐PD‐1/PD‐L1 128 warrant a cautionary note against this therapy in TB.
Indoleamine 2,3‐dioxygenase (IDO) expression is actively induced by mycobacteria in animal (macaques and mice) models of acute TB, but not LTBI, and IDO levels correlated with bacterial burden. IDO catabolizes tryptophan into kynurenine, which in turn suppresses IFN‐γ production by CD4 T‐cells, a cytokine pivotal in the anti‐TB response, identifying IDO as potential target for HDT. In vivo inhibition of IDO activity using D‐1MT one week after mycobacterial infection enhanced T‐cell proliferation and differentiation in effector and memory cells while apoptosis was enhanced. 129 Furthermore, D‐1MT treatment improved penetration of T‐cells into granulomas, likely allowing protective T‐cell‐mediated granuloma reorganization, and reduced bacterial loads and lung pathology. 129
3.2. Skewing of T‐cells
Th1‐responses, characterized by high IFN‐γ secretion, are crucial in effective anti‐Mtb immune responses. 130 , 131 , 132 Nevertheless, Mav and Mtb reduce cellular responses to IFN‐γ and deficiencies in the IL‐12/IL‐23/IFN‐γ‐axis increase susceptibility to Mav infections. 133 , 134 In several patients suffering from pulmonary TB, direct administration of IFN‐γ accelerated sputum‐smear conversion and improved chest radiograph. 135 , 136 Administration of IFN‐γ also reduced Mav growth in murine macrophages, 137 and improved clinical outcome (ie, decreased respiratory symptoms and mortality) in several but not all Mav‐infected individuals, 138 , 139 , 140 suggesting potential of IFN‐γ as HDT in both Mtb and Mav infections. In vivo administration of IL‐12, a key cytokine that drives Th1 skewing, enhanced IFN‐γ and TNF‐α responses and significantly reduced bacterial burden in an acute mouse TB model. 141 Similarly, restoring IL‐24 expression in a mouse TB model enhanced Th1‐responses and IFN‐γ production, with concomitant improved survival and reduced bacterial loads. 142 A large proportion of human Mtb‐specific CD4 Th1‐cells expresses CCR6 and co‐produces IFN‐γ/IL‐17, often depicted as Th1* or Th1‐17 cells, and being associated with LTBI suggest their importance in protection against active TB. 143 However, IL‐17 responses during TB need to be carefully regulated to prevent neutrophil‐driven lung pathology, which is mediated by regulatory T‐cells as well as so‐called regulatory CD4 Th17‐cells that co‐produce IL‐17 and IL‐10. 144 In case of disbalanced Th17 responses with concomitant excessive neutrophil recruitment, RAGE receptor inhibition may be an interesting HDT. RAGE receptor is upregulated during active TB disease and after ligation with S100A8/A9 mediates neutrophil recruitment. 145 In a TB model, mice deficient in S100A8/A9 had reduced bacterial loads, neutrophil influx and pathology compared to wildtype. Moreover, inhibition of the RAGE receptor using FPS‐ZM1 improved outcome comparably as S100A8/A9‐deficiency. 145
Th2‐responses have been associated with active cavitary TB disease or TB treatment failure, 130 , 131 and administration of IL‐4, a hallmark Th2‐cytokine, impaired mycobacterial control by human macrophages and enhanced the proportion of regulatory T‐cells in vitro. 131 Blocking IL‐4 completely alleviated these effects and improved bacterial control, 131 suggesting Th skewing could be an interesting target for HDT.
Alternatively, administration of IL‐2, which stimulates T‐cell proliferation while inhibiting T‐cell anergy, in patients infected with drug‐resistant Mtb has been investigated in five RCTs and compared in a meta‐analysis. 146 While CD4 T‐cell numbers increased and time to culture conversion improved, radiographic changes were not observed compared to standard chemotherapy. 146 In mice infected with Mav, IL‐2 therapy resulted in decreased bacterial burden, 147 whereas mixed results were described in case reports. 148 , 149 The limited effect of IL‐2 therapy may be due to immune suppression caused by expansion of regulatory T‐cells and myeloid‐derived suppressor cells (MDSC), both expressing elevated levels of the high affinity IL‐2 receptor, as depletion of these suppressor cells improved outcome in a mouse TB model. 150 Combining IL‐2 therapy with mycobacterial phosphoantigen (E)‐4‐hydroxy‐3‐methyl‐but‐2‐enyl pyrophosphate (HMBPP) in non‐human primates induced significant expansion of Vγ2Vδ2 T‐cells that migrated to the lungs, evoking a Th1‐response that significantly reduced mycobacterial burden as well as lung pathology. 151 Rather than systemic administration of cytokines, which frequently results in systemic side effects, ex vivo stimulation of autologous peripheral blood mononuclear cells (PBMCs) with a cocktail of IFN‐γ, IL‐2, IL‐1α, and anti‐CD3 before reinfusion yielded positive results with minimal side effects in a case report with MDR‐TB. 152 This, however, requires further clinical investigation.
4. (PROGRAMMED) CELL DEATH
Several types of cell death can follow mycobacterial infection of macrophages: apoptosis, necrosis, and ferroptosis. 32 During apoptosis, bacteria remain encapsulated, which facilitates bacterial clearance; however, pathogenic mycobacteria have developed strategies to limit apoptosis. 153 Activation of transcriptional regulator peroxisome proliferator‐activated receptor gamma (PPARγ) by ManLAM, stimulating mannose receptors, upregulated (pro‐host‐cell survival) Mcl‐1 and repressed (pro‐apoptotic) Bax without Bak and improved host‐cell survival. 154 , 155 In agreement with the PPARγ‐dependent inhibition of host‐cell apoptosis and concomitant anti‐mycobacterial immunity, direct pharmacological inhibition of Mcl‐1 resulted in reduced intracellular Mtb growth in human macrophages. 155
Besides inhibiting apoptosis, virulent Mtb stimulates host‐cell necrosis, which allows infection to disseminate to neighboring cells. 32 Mtb can induce necrosis via the virulence factor tuberculosis necrotizing toxin (TNT), which is secreted into the cytosol where its NAD+ glycohydrolase activity depletes the host cell from NAD+, 156 leading to permeabilization of mitochondrial membranes, decreasing ATP‐production and activating necrosis. Nicotinamide‐based HDT alleviated necrosis‐induced host‐cell cytotoxicity in Mtb‐infected cells by replenishing NAD+. 99 Mtb can furthermore induce necrosis mediated by mitochondrial membrane permeability transition via p38‐MAPK phosphorylation, which can be inhibited by corticosteroids dexamethasone and doramapimod. 157 In addition, corticosteroids dexamethasone and prednisolone, both well‐known general immunosuppressants, have also been investigated as HDT during mycobacterial infections. With some reports of improved survival, 158 , 159 likely by limiting secretion of pro‐inflammatory cytokines, meta‐analysis failed to show a significant improvement in clinical outcome after corticosteroid therapy in patients with TB. 160 Interestingly, while promoting pro‐inflammatory cytokine levels of TNF‐α by adenylate cyclase inhibitor (SQ22536) or a PKA inhibitor (H‐89) has been shown to improve control of infection by stimulating mitochondrial ROS production, 161 excess TNF‐α leads to membrane disruption and ATP depletion via mitochondrial enzyme cyclophilin D, which together with lysosomal enzyme acid sphingomyelinase induced necrosis. 162 , 163 Alisporivir and desipramine, two clinically approved drugs that inhibit cyclophilin D and acid sphingomyelinase, respectively, prevented TNF‐α‐induced necrosis without compromising TNF‐α‐induced ROS‐dependent mycobacterial killing. 163 Correspondingly, upregulation of cAMP levels by phosphodiesterase (PDE) inhibitors cilostazol and sildenafil decreased TNF‐α levels, resulting in reduced immunopathology and fastened bacterial clearance in Mtb‐infected mice. 164 , 165 Blocking TNF‐α, which facilitates necrotizing granulomas during active TB, displayed promising results in preclinical animal models. 166 , 167 However, blocking TNF‐α also leads to disease reactivation and concomitant dissemination in LTBI patients and in the absence of standard TB chemotherapy exacerbated disease severity, 168 , 169 , 170 precluding clinical application of TNF‐α inhibition as HDT in TB. The balance between TNF‐α‐mediated beneficial and detrimental effects on host control of TB and likely other mycobacterial infections including NTM is thus delicate. Taken together, these data indicate that Mtb‐induced host‐cell necrosis favors mycobacterial survival and this can be effectively counteracted by HDT, while the double‐edge sword of modulating TNF‐α levels currently prohibits clinical application.
Ferroptosis is a type of necrosis characterized by accumulation of free iron and toxic lipid peroxides. 171 In Mtb‐infected cells expression of glutathione peroxidase‐4 (GPX4) is reduced, leading to failure of glutathione‐dependent antioxidant defenses and cell death. 172 Inhibiting ferroptosis by ferrostatin 1 reduced bacterial burden both in vitro in human macrophages and in vivo in Mtb‐infected mice. 171 Furthermore, ferroptosis could also be inhibited by increasing GPX4 levels with selenium, a protein involved in GPX4 catalysis, 173 showing that targeting this host pathway is a potential HDT strategy.
5. METABOLISM
5.1. Carbohydrate and lipids
Mtb has developed numerous strategies to modulate host metabolic pathways, which are broadly divided into glycolysis, oxidative phosphorylation (OXPHOS), and lipid metabolism. Glycolysis conditions an environment favoring Mtb growth, and inhibition of glycolysis using 2‐deoxy‐D‐glucose (2‐DG) reduced Mtb viability in one study, and as a result of ATP depletion induced macrophage apoptosis. 174 An important enzyme during glycolysis, lactate dehydrogenase (LDH), which converts pyruvate into lactate, is significantly upregulated during Mtb infection. 175 Although the pathophysiology of LDH upregulation remains to be addressed, pharmacological inhibition of LDH using FX11 reduced bacterial load and development of necrotic lesions in granulomas in a murine TB model, suggesting a significant role of LDH in driving disease and a potential target for HDT. 175 Interestingly, while ATP depletion can induce macrophage apoptosis (considered host protective), exogenous ATP activates macrophages via the P2RX7/P2X7 receptor and also directly inhibits growth of mycobacteria, including Mtb and Mav, due to chelation of iron. 176 , 177 ATP treatment has already been shown to synergize with standard Mav antibiotic treatment, making ATP an interesting adjunctive HDT molecule to enhance chemotherapeutic efficacy against mycobacterial infections. 177 In addition, an FDA‐approved potentiator of P2RX7/P2X7, clemastine enhanced mycobacterial killing in a zebrafish model. 178 This may provide a potentially attractive avenue to explore synergistic effects between ATP and clemastine treatment in future studies.
Conversion of pyruvate into acetyl coenzyme A (Ac‐CoA) initiates the tricarboxylic acid (TCA) cycle which produces energy using OXPHOS. During Mtb infection, several enzymes important in the TCA cycle are downregulated and TCA cycle intermediates, such as citrate, are translocated from mitochondria into the cytosol. Typically, citrate is converted into itaconate which dampens tissue hyperinflammation by suppressing both ROS production and production of pro‐inflammatory cytokines such as IL‐1β, IL‐6, and IL‐12. 174 In the cytosol, however, citrate is cleaved into Ac‐CoA, which is either converted into arachidonic acid or into mevalonate and malonyl‐CoA. This leads to synthesis of eicosanoids, cholesterol, and free fatty acids, respectively, of which the latter two are stored intracellularly in lipid droplets. 174 Hypercholesterolemia results in spontaneous formation of lipid droplets in macrophages. Further accumulation of intracellular lipid droplets is actively stimulated by Mtb 174 , 179 and enhanced by both IL‐6 and TNF‐α signaling, while IL‐17 and IFN‐γ limit intracellular lipid accumulation. 174 Lipid‐loaded macrophages are impaired in killing intracellular mycobacteria (ie, Mtb, Mav, and BCG) 180 and ultimately transform into foamy macrophages, which are associated with necrotic granulomas and tissue pathology. 174 The impaired functionality of lipid‐loaded macrophages involves mitochondrial dysfunction and could be restored using small molecule mitochondrial fusion promoter M1, which also restored macrophage bactericidal activity. 180 In addition, ezetimibe, a cholesterol absorption inhibitor, prevented intracellular lipid accumulation and concomitantly reduced intracellular growth in Mtb‐infected macrophages. 179 The effects of standard antibiotic treatment improved and perhaps even synergized with ezetimibe treatment, 179 and investigating the in vivo efficacy of ezetimibe as well as M1 could be promising.
Statins, currently clinically used to reduce cholesterol levels, could be interesting drugs to prevent lipid accumulation in macrophages. Comparing eight different statins, simvastatin, pravastatin and fluvastatin were most efficacious in enhancing mycobacterial killing without affecting cell viability in vitro. 181 Mechanistically, while (simva)statin inhibits phagosomal acidification and degradation, 181 cholesterol incorporation in (auto‐)phagosomal membranes is prevented. The presence of cholesterol in phagosomal membranes facilitates prolonged survival of Mtb and Mav within host cells due to blockage of phagolysosome fusion by mechanisms not fully understood. 182 , 183 , 184 Preventing phagosomal escape ultimately enhances delivery of mycobacteria to (auto‐)phagolysosomes and thereby bacterial degradation. 185 , 186 In vivo treatment with either pravastatin or simvastatin in a mouse TB model reduced mycobacterial loads both as a single therapy 181 , 185 or combined with standard antibiotic treatment. 181 , 184
5.2. Eicosanoids
Eicosanoids are lipid mediators involved in regulating inflammatory responses and are categorized into prostaglandins (PG), leukotrienes (LT), thromboxanes, lipoxins, and hydroxy eicosatetraenoic acids, all of which are produced from arachidonic acid by a competing network of enzymes, including cyclooxygenases (COX) and lipoxygenases. 187 , 188 During Mtb infection, the expression of eicosanoids is significantly altered, with prostaglandin‐E2 (PGE2) and leukotriene‐B4 (LTB4) mostly upregulated. 187 Being an immune suppressor and immune stimulator, respectively, the balance between these eicosanoids is highly important in regulating immunity to clear the infection, without causing tissue pathology due to excessive inflammation. Important in this regulation are IL‐1β and type‐I IFN signaling. IL‐1β signaling stimulates production of prostaglandin‐E2, which is necessary to dampen the inflammation mediated by pro‐inflammatory leukotrienes A4 and B4 (LTA4 is the precursor of LTB4) that are induced upon type‐I IFN signaling. In severe TB, the PGE2/LTA4 ratio is reduced, suggesting potential benefit of enhancing PGE2 signaling. Indeed, both increasing PGE2 levels using administration of exogenous PGE2 or reducing LTA4/LTB4 production with zileuton improved host survival, while reducing bacterial loads and necrotic lung pathology in Mtb‐infected mice. 189 Moreover, combinatory therapy of zileuton with PGE2 further restricted Mtb replication. 190
A single nucleotide polymorphism in the promotor of the gene encoding LTA4‐hydrolase (rs17525495), the enzyme that converts LTA4 into LTB4, has been shown to affect expression of LTA4 hydrolase, with homozygous individuals having a high (T/T) or low (C/C) expression. 188 , 191 Both homozygous genotypes have poorer survival compared to heterozygous individuals, showing the delicateness of the immune balance during mycobacterial infection. 188 Depending on the genotype, different treatment regimens will be required, as general immune suppression using dexamethasone favored outcome in T/T individuals, while being detrimental in C/C individuals, 188 , 191 suggesting the necessity of personalized HDT‐based medicine targeting eicosanoid metabolism. Mice deficient in 5‐lipoxygenase, an enzyme that stimulates production of LTA4 and thus LTB4 (thereby being a model for C/C individuals), were impaired in controlling mycobacterial infection due to absence of LTB4. Treatment with celecoxib, a COX inhibitor that prevents PGE2 production and thereby stimulates LTB4 production, or directly supplementing LTB4, restored mycobacterial control. 187 Furthermore, COX inhibitors ibuprofen and aspirin administered as single therapy or combined with conventional TB antibiotics were shown to limit bacterial burden in Mtb‐infected mice, 192 , 193 , 194 and low‐dose aspirin treatment also reduced bacterial loads in a Mm zebrafish infection model. 195 Aspirin treatment of TB or TB meningitis patients improved survival, 196 , 197 but may impair conventional treatment regimens by reducing efficacy of isoniazid, 198 but not pyrazinamide. 193 Both ibuprofen and aspirin are currently tested in clinical trials as adjunct therapy for treating (drug‐resistant) TB. 190
5.3. GRANULOMA: FORMATION, ANGIOGENESIS and HYPOXIA
One hallmark of TB is the extensive formation of granulomas. Granulomas are highly heterogenous and dynamic structures which differ significantly in the level of hypoxia and available nutrients. Granuloma formation is actively initiated by Mtb to stimulate matrix metalloprotease 9 (MMP9) production. Granulomas are also induced during NTM infections, including Mav 199 , 200 and Mm. 201 During initial granuloma formation non‐activated macrophages are recruited to the site of infection and serve as feeder cells for the granuloma. 25 , 202 In addition to MMP9, upregulation of several other MMPs has been observed in lung samples from individuals infected with Mtb, and other mycobacteria including Mav, which may suggest that similar mechanisms are involved. 202 , 203 , 204 , 205 , 206 MMPs are enzymes that degrade and modulate extracellular matrix and are therefore key in the development of granulomas. 204 Their expression and activity has multiple layers of regulation. Many MMPs require Zn2+ for their activation, potent MMPs require activation by other MMPs, and their activation is inhibited by tissue inhibitors of metalloproteinases (TIMPs). Expression of MMPs is stimulated by pro‐inflammatory cytokines, including IFN‐γ, TNF‐α, IL‐12, and IL‐17 and because enhanced MMP‐activity is associated with extensive tissue damage during TB, 203 MMPs are promising targets for HDTs.
MMP1, a collagenase that degrades collagen in the extracellular matrix, is upregulated after TLR2‐ligation and due to its high potency may drive granuloma formation during TB. 207 In transgenic mice expressing human MMP1, Mtb infection promoted alveolar destruction and collagen breakdown in lung granulomas, identifying MMP1 as a therapeutic target to limit immunopathology. 207 MMP7, which is highly expressed in the cavitary wall and hypoxic granulomas, stimulates epithelial proliferation and promotes activity of other MMPs. Inhibition of MMP7 and MMP1 using cipemastat, a drug originally registered to prevent lung fibrosis, surprisingly increased cavitation, immunopathology and mortality in mice, 208 suggesting either a protective role of MMP1 or MMP7 during TB or off‐target effects of the drug. The role of MMP8 is more controversial with high interindividual variation, 203 , 209 , 210 which may relate to the presence of neutrophils in granulomas. MMP8 is more readily detectable in HIV‐associated TB, 210 suggesting that neutrophils are recruited preferentially in settings of impaired adaptive immunity. Mice deficient in MMP9 have less granuloma formation and reduced bacterial loads, 211 suggesting a prominent role of MMP9 in driving disease pathology. Indeed, inhibition of MMP9 expression using morpholinos reduced granuloma formation and bacterial growth in a zebrafish Mm‐model. 25 In agreement with this concept, treatment with Sb‐3ct, a specific MMP2 and MMP9 inhibitor, combined with frontline TB antibiotics potentiated bacterial clearance both in vitro and in vivo in a TB meningitis mouse model. 212 , 213 Blocking MMP9 using monoclonal antibody AB0046 did not affect bacterial burden, but the rate of relapse was reduced in a necrotic granuloma TB mouse model, by mechanisms not yet fully clarified. 166 Using an in vitro model for extracellular matrix degradation, treatment with doxycycline, an FDA‐approved antibiotic that non‐selectively inhibits human MMPs, strongly abolished Mtb‐induced matrix degradation. 210 In addition, doxycycline reduced granuloma formation in a guinea pig model, likely resulting from abolishing Mtb‐enhanced promotor activity of MMP1 and by directly inhibiting bacterial growth. 214 Pan‐MMP inhibitor marimastat (BB‐2516), a collagen‐peptidomimetic binding the active Zn2+ site contained in many MMPs, reduced granuloma size and bacterial burden during Mtb infection in lung tissue models. 203 Interestingly, treatment of Mtb‐infected mice with a panel of MMP inhibitors, including marimastat, as solo therapy was not effective, while all 4 small molecules enhanced in vivo potency of frontline TB drugs isoniazide and rifampicin, likely by blocking MMP‐mediated cleavage of collagen and by improving vascular integrity, resulting in enhanced delivery of isoniazide and rifampicin to the lungs. The finding that batimastat (a pan‐MMP inhibitor), Sb‐3ct (a MMP2 and MMP9 inhibitor) and MMP9 inhibitor‐I yielded similar results highlights the importance of MMP9 in driving these effects. 205 Augmenting TIMP1 activity to inhibit activity of multiple MMPs may also be an interesting HDT target. To our knowledge, however, modulating the activity of TIMPs has not been investigated yet in the context of HDT.
Central hypoxia in granulomas may initially favor host immunity as low oxygen tension increases granulysin expression in T‐cells and NK‐cells, enhancing bacterial killing in an in vitro co‐culture system of Mtb‐specific T‐cells and macrophages. 215 However, due to poor vasculature within granulomas and hyperactive IFN‐γ or possible superimposed IL‐4/IL‐13 released by activated T‐cells, full blown central necrosis leads to cavity formation and concomitant bacterial dissemination within the host. 202 , 211 , 215 Due to the hypoxic, acidic and nutrient‐poor conditions in granulomas, mycobacterial dormancy is promoted, 25 and while this effectively inhibits bacterial replication, eradication of mycobacteria is greatly hampered because most antibiotics only affect replicating and metabolically active bacteria. Furthermore, poor vascularization hampers drug delivery in granulomas, which is further impaired due to fibrosis and scarring of lung tissue caused by the disease. 25 Trehalose dimycolate, a mycolic acid expressed on mycobacterial cell walls, directly induces vascular endothelial growth factor (VEGF) expression in host cells to stimulate angiogenesis. 216 Although angiogenesis could potentially increase host‐cell viability, the net effect likely favors bacterial replication and dissemination. Blocking angiogenesis may therefore be an interesting HDT. Indeed, inhibition of VEGF using FDA‐approved bevacizumab in Mtb‐infected rabbits, reduced the total number of vessels but improved both structurally and functionally the remaining vessels, leading to enhanced drug targeting to granulomatous lesions and diminished hypoxia. 217 Corroborating these findings, treatment of Mm‐infected zebrafish with VEGF pathway inhibitors SU5416, a tyrosine kinase inhibitor, or pazopanib, a VEGF receptor inhibitor, reduced bacterial loads and dissemination. Both drugs also synergized with first‐line anti‐mycobacterial drugs rifampicin and metronidazole, a drug that targets hypoxic bacteria. 218 Inhibiting vascular leakage rather than angiogenesis may be equally efficacious to limit nutrient supply to mycobacteria. During Mm infection, angiopoietin‐2 (ANG2) is robustly induced in granulomatous lesions. ANG2 antagonizes ANG1, which promotes vessel stability while limiting angiogenesis and vascular leakage. Indeed, AKB‐9785, a molecule that mimics functions of ANG1, reduced vascular leakage and bacterial burden in a Mm zebrafish infection model. 219 Thus, inhibition of angiogenesis is an interesting target for HDT to enhance drug delivery to the site of infection and combined with other therapies is likely to be even more potent.
6. PERSONALIZED AND COMBINATORIAL HDT
Although HDT could be considered as stand‐alone therapy, for example, in patients suffering from total drug‐resistant TB, HDT is primarily envisaged as adjunct therapy in combination with classical antibiotics. HDT might be co‐administered for a limited duration at the initiation of the standard of care regimens to shorten treatment length and reduce dosage of antibiotics to minimize side effects, or toward the end of treatment to boost host immunity to prevent potential relapse. Consequently, investigating the interactions between HDT and conventional chemotherapy is pivotal, but has only been reported for a limited number of HDTs. Furthermore, in case of undesired interactions between TB drugs and drugs for TB comorbidities (eg, between rifampicin and anti‐HIV therapy or anti‐diabetic drugs), 220 HDT might be used to shorten current treatment regimens or possibly partially replace components of the conventional chemotherapy cocktail. In line with this, interactions between HDT and drugs used to treat TB comorbidities should also be investigated thoroughly.
Rather than targeting one specific aspect of the inflammatory response during mycobacterial infections, we hypothesize that correcting the overall immunological disbalance likely is most promising. Type‐I IFN and IL‐1β signaling, regulating levels of anti‐inflammatory prostaglandins and pro‐inflammatory leukotrienes, respectively, play an important role in regulating the immune balance during mycobacterial infections. 189 At the time of writing, multiple randomized controlled trials investigate targeting of (one of) these pathways by HDTs. As some TB patients suffer from overactive type‐I IFN/leukotriene signaling while others are characterized by overactive IL‐1/prostaglandin activity, we postulate that in this context personalized HDT would be safest and most efficacious. However, this will increase therapeutic costs, which could make such therapy stratifications less attractive and feasible in lower resourced settings. To be able to predict whether patients would benefit from a certain HDT, biomarkers monitoring the (immunological) status of patients may need to be identified and developed. This, however, may not be required for all HDTs as some HDT may improve anti‐mycobacterial immunity in all patients. As mycobacteria modulate host immunity via many different pathways, a multi‐targeted approach could be necessary to fully counteract mycobacteria‐mediated host modulation. To our knowledge, however, only two combinations of HDT treatments have been published; combining vitamin‐D with PBA did not mediate additive effects compared to solo therapy, 51 , 52 , 53 likely because both compounds target the same pathway, while in another in vitro study combining protein‐kinase A/B inhibitors H‐89 or 97i with HDAC inhibitors revealed additive effects in vitro in reducing bacterial load in primary human macrophages. 115
Modulating (auto‐)phagosome maturation using receptor tyrosine kinase inhibitors including imatinib, 25 AZD0530, 28 and multiple repurposed drugs recently identified in our own group 29 has been shown to improve mycobacterial clearance by human macrophages in vitro. Importantly, releasing the mycobacteria‐mediated arrest in (auto‐)phagosome maturation likely benefits both patients with active disease as well as individuals with latent infection. Above, we have reviewed multiple HDT candidates that enhance autophagy‐mediated bacterial clearance. Which of these will be most efficacious against mycobacteria should ideally be determined in head‐to‐head comparisons. Metformin, being the most frequently investigated, has already been shown to reduce TB recurrence and bacterial loads in patients, 75 , 76 , 77 and in addition to its effects on autophagy, also enhances mitochondrial membrane polarization, 221 which could further enhance its efficacy.
As discussed above, host‐cell death pathways are actively exploited by mycobacteria to promote their survival and dissemination and have been shown to be a potential target for HDT in multiple in vitro and animal studies. Active clinical modulation of (programmed) cell death in patients, however, could lead to significant adverse effects given the complex time‐ and context‐dependency of this mechanism during mycobacterial infection.
Targeting metabolic pathways has been shown to be feasible and represents an attractive target for HDT. While most metabolic pathways are also necessary for host‐cell energy production, intracellular lipid accumulation in lipid droplets seems to mainly benefit the intracellular survival of mycobacteria. Preventing or reducing lipid droplet formation in macrophages and concomitant impaired immunity can be mediated by (a) limiting oxidative phosphorylation by, for example, stimulating polarization of macrophages toward pro‐inflammatory M1‐macrophages, 174 (b) improving/maintaining mitochondrial membrane potential using small molecule M1 180 or NAD 156 and/or (c) blocking cellular cholesterol uptake using, for example, ezetimibe, 179 which also inhibits phagosomal escape by mycobacteria. Targeting metabolism with HDT may also help correcting the balance between prostaglandins and thromboxanes, as lipid droplets and cytosolic TCA intermediates are the most important sources of eicosanoids.
Irrespective of what causes defective mycobacterial clearance, improving drug delivery to the site of infection likely benefits all TB patients. Angiogenesis in granulomas is significantly impaired and further enhances hypoxia and nutrient‐limitation. Targeting angiogenesis during TB by (a) inhibiting VEGF (bevacizumab 217 ), (b) inhibiting VEGF‐mediated signaling (SU5416 or pazopanib 218 ), or (c) antagonizing pro‐angiogenesis growth factor ANG2 (AKB‐9785 219 ) has all been shown to enhance both drug delivery and oxygenation within granulomas in animal models of TB, and may be promising HDTs in combination with other therapies. Despite being most frequently investigated in combination with antibiotics, efflux pump inhibitors could also improve drug delivery of HDTs. To our knowledge, however, this has not been investigated so far but verapamil, known to enhance the efficacy of rifampicin and bedaquiline against different mycobacterial infections, both in vitro and in mice, 222 , 223 , 224 and also chloroquine 225 and piperine 226 are interesting molecules for combinatorial HDT.
Given their central and important role in orchestrating a functional anti‐mycobacterial immune response, restoring (CD4 Th1/17) T‐cell immunity has been pursued in many investigations. In addition to enhancing activation of antigen‐presenting cells, HDTs that promote phagosomal bacterial degradation (ie, stimulating autophagy, enhancing phagosome maturation and promoting (auto‐)phagolysosome fusion) are all expected to enhance presentation of bacterial‐derived peptides and thereby improve adaptive immunity. Modulating T‐cell responses to restore immunity can be mediated by vaccination or T‐cell cytokine therapies. Administration of IL‐12 141 or IL‐24 142 or blocking Th2 cytokine IL‐4, 131 promotes Th1 responses with lasting IFN‐γ production that may be preferred over IFN‐γ administration. Which of these strategies is (most) efficacious and which patients benefit most from this therapy remains to be addressed.
While most of the evidence available for host‐pathogen interactions and HDT are from TB studies, the limited number of NTM experimental models investigating host modulation and/or HDT emphasizes the need and urgency to understand NTM pathogenesis as well as identify potentially relevant host targets. Together, these studies will help assess the safety and efficacy of HDT, paving the way for the introduction of HDT against a wide range of mycobacteria.
7. SEARCH STRATEGY AND SELECTION CRITERIA
We searched PubMed (MEDLINE) for all relevant studies published from 1 January 2000 until 1 October 2020. The medical subject headings used were “host directed”, “HDT”, “adjunctive”, “immunotherapeutic” or “immunomodulation” combined with “mycobacterium”, “mycobacteria”, “tuberculosis”, “nontuberculous” or “NTM”. All relevant abstracts were screened independently by two researchers. The final reference list was generated based on relevance to the topics covered in this review. Only papers published in English were included.
CONFLICT OF INTEREST
All authors declare no competing or conflicting interests.
ACKNOWLEDGEMENTS
This work was supported by grants from the Netherlands Organisation for Scientific Research (NWO‐TTW) within the Novel Antibacterial Compounds and Therapies Antagonising Resistance (NACTAR) program (Grant No. 16444), European Union's Horizon 2020 research and innovation program (SMA‐TB Grant No. 847762), and the Innovative Medicines Initiative 2 Joint Undertaking (IMI2 JU) under the RespiriNTM (Grant No. 853932) and RespiriTB (Grant No. 853903) projects within the IMI AntiMicrobial Resistance (AMR) Accelerator program. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Kilinç G, Saris A, Ottenhoff THM, Haks MC. Host‐directed therapy to combat mycobacterial infections. Immunol Rev. 2021;301:62–83. 10.1111/imr.12951
This article is part of a series of reviews covering Immunity to Mycobacteria appearing in Volume 301 of Immunological Reviews.
Kilinç, Saris, Ottenhoff, and Haks authors are Contributed equally.
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analysed during the current study
REFERENCES
- 1. World Health Organization . Tuberculosis. https://www.who.int/news‐room/fact‐sheets/detail/tuberculosis. 2020. [Google Scholar]
- 2. Cilloni L, Fu H, Vesga JF, et al. The potential impact of the COVID‐19 pandemic on the tuberculosis epidemic a modelling analysis. EClinicalMedicine. 2020;28:100603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wu ML, Aziz DB, Dartois V, Dick T. NTM drug discovery: status, gaps and the way forward. Drug Discov Today. 2018;23(8):1502‐1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schafer G, Jacobs M, Wilkinson RJ, Brown GD. Non‐opsonic recognition of Mycobacterium tuberculosis by phagocytes. J Innate Immun. 2009;1(3):231‐243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kawai T, Akira S. TLR signaling. Cell Death Differ. 2006;13(5):816‐825. [DOI] [PubMed] [Google Scholar]
- 6. Gomes MS, Florido M, Cordeiro JV, et al. Limited role of the Toll‐like receptor‐2 in resistance to Mycobacterium avium. Immunology. 2004;111(2):179‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Drennan MB, Nicolle D, Quesniaux VJ, et al. Toll‐like receptor 2‐deficient mice succumb to Mycobacterium tuberculosis infection. Am J Pathol. 2004;164(1):49‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Orme IM, Ordway DJ. Host response to nontuberculous mycobacterial infections of current clinical importance. Infect Immun. 2014;82(9):3516‐3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bhatnagar S, Schorey JS. Elevated mitogen‐activated protein kinase signalling and increased macrophage activation in cells infected with a glycopeptidolipid‐deficient Mycobacterium avium. Cell Microbiol. 2006;8(1):85‐96. [DOI] [PubMed] [Google Scholar]
- 10. Rhoades ER, Archambault AS, Greendyke R, Hsu FF, Streeter C, Byrd TF. Mycobacterium abscessus Glycopeptidolipids mask underlying cell wall phosphatidyl‐myo‐inositol mannosides blocking induction of human macrophage TNF‐alpha by preventing interaction with TLR2. J Immunol. 2009;183(3):1997‐2007. [DOI] [PubMed] [Google Scholar]
- 11. Rahman A, Sobia P, Gupta N, Kaer LV, Das G. Mycobacterium tuberculosis subverts the TLR‐2‐MyD88 pathway to facilitate its translocation into the cytosol. PLoS One. 2014;9(1):e86886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gilleron M, Nigou J, Nicolle D, Quesniaux V, Puzo G. The acylation state of mycobacterial lipomannans modulates innate immunity response through toll‐like receptor 2. Chem Biol. 2006;13(1):39‐47. [DOI] [PubMed] [Google Scholar]
- 13. Khan N, Pahari S, Vidyarthi A, Aqdas M, Agrewala JN. Stimulation through CD40 and TLR‐4 is an effective host directed therapy against Mycobacterium tuberculosis . Front Immunol. 2016;7:386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Khan N, Pahari S, Vidyarthi A, Aqdas M, Agrewala JN. NOD‐2 and TLR‐4 signaling reinforces the efficacy of dendritic cells and reduces the dose of TB drugs against Mycobacterium tuberculosis . J Innate Immun. 2016;8(3):228‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gowthaman U, Singh V, Zeng W, et al. Promiscuous peptide of 16 kDa antigen linked to Pam2Cys protects against Mycobacterium tuberculosis by evoking enduring memory T‐cell response. J Infect Dis. 2011;204(9):1328‐1338. [DOI] [PubMed] [Google Scholar]
- 16. Chodisetti SB, Gowthaman U, Rai PK, Vidyarthi A, Khan N, Agrewala JN. Triggering through Toll‐like receptor 2 limits chronically stimulated T‐helper type 1 cells from undergoing exhaustion. J Infect Dis. 2015;211(3):486‐496. [DOI] [PubMed] [Google Scholar]
- 17. Pahari S, Negi S, Aqdas M, Arnett E, Schlesinger LS, Agrewala JN. Induction of autophagy through CLEC4E in combination with TLR4: an innovative strategy to restrict the survival of Mycobacterium tuberculosis . Autophagy. 2020;16(6):1021‐1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harding CV, Boom WH. Regulation of antigen presentation by Mycobacterium tuberculosis: a role for Toll‐like receptors. Nat Rev Microbiol. 2010;8(4):296‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim JK, Yuk JM, Kim SY, et al. MicroRNA‐125a inhibits autophagy activation and antimicrobial responses during mycobacterial infection. J Immunol. 2015;194(11):5355‐5365. [DOI] [PubMed] [Google Scholar]
- 20. Gu X, Gao Y, Mu DG, Fu EQ. MiR‐23a‐5p modulates mycobacterial survival and autophagy during mycobacterium tuberculosis infection through TLR2/MyD88/NF‐kappaB pathway by targeting TLR2. Exp Cell Res. 2017;354(2):71‐77. [DOI] [PubMed] [Google Scholar]
- 21. Jefferies KC, Cipriano DJ, Forgac M. Function, structure and regulation of the vacuolar (H+)‐ATPases. Arch Biochem Biophys. 2008;476(1):33‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lawrence RE, Zoncu R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat Cell Biol. 2019;21(2):133‐142. [DOI] [PubMed] [Google Scholar]
- 23. Ankley L, Thomas S, Olive AJ. Fighting persistence: how chronic infections with Mycobacterium tuberculosis Evade T cell‐mediated clearance and new strategies to defeat them. Infect Immun. 2020;88(7):e00916‐00919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wong D, Bach H, Sun J, Hmama Z, Av‐Gay Y. Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar‐H+‐ATPase to inhibit phagosome acidification. Proc Natl Acad Sci U S A. 2011;108(48):19371‐19376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hawn TR, Matheson AI, Maley SN, Vandal O. Host‐directed therapeutics for tuberculosis: can we harness the host? Microbiol Mol Biol Rev. 2013;77(4):608‐627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Napier RJ, Rafi W, Cheruvu M, et al. Imatinib‐sensitive tyrosine kinases regulate mycobacterial pathogenesis and represent therapeutic targets against tuberculosis. Cell Host Microbe. 2011;10(5):475‐485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bruns H, Stegelmann F, Fabri M, et al. Abelson tyrosine kinase controls phagosomal acidification required for killing of Mycobacterium tuberculosis in human macrophages. J Immunol. 2012;189(8):4069‐4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chandra P, Rajmani RS, Verma G, Bhavesh NS, Kumar D, Stallings CL. Targeting drug‐sensitive and ‐resistant strains of Mycobacterium tuberculosis by Inhibition of Src family kinases lowers disease burden and pathology. mSphere.. 2016;1(2):e00043‐e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Korbee CJ, Heemskerk MT, Kocev D, et al. Combined chemical genetics and data‐driven bioinformatics approach identifies receptor tyrosine kinase inhibitors as host‐directed antimicrobials. Nat Commun. 2018;9(1):358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Buter J, Cheng TY, Ghanem M, et al. Mycobacterium tuberculosis releases an antacid that remodels phagosomes. Nat Chem Biol. 2019;15(9):889‐899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Houben D, Demangel C, van Ingen J, et al. ESX‐1‐mediated translocation to the cytosol controls virulence of mycobacteria. Cell Microbiol. 2012;14(8):1287‐1298. [DOI] [PubMed] [Google Scholar]
- 32. Chai Q, Wang L, Liu CH, Ge B. New insights into the evasion of host innate immunity by Mycobacterium tuberculosis. Cell Mol Immunol. 2020;17(9):901‐913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ranjbar S, Haridas V, Nambu A, et al. Cytoplasmic RNA sensor pathways and nitazoxanide broadly inhibit intracellular Mycobacterium tuberculosis growth. iScience. 2019;22:299‐313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Walsh KF, McAulay K, Lee MH, et al. Early bactericidal activity trial of nitazoxanide for pulmonary tuberculosis. Antimicrob Agents Chemother. 2020;64(5):e01956‐e1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469(7330):323‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Renna M, Schaffner C, Brown K, et al. Azithromycin blocks autophagy and may predispose cystic fibrosis patients to mycobacterial infection. Journal of Clinical Investigation. 2011;121(9):3554‐3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Paik S, Kim JK, Chung C, Jo EK. Autophagy: A new strategy for host‐directed therapy of tuberculosis. Virulence. 2019;10(1):448‐459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yuk JM, Shin DM, Lee HM, et al. Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe. 2009;6(3):231‐243. [DOI] [PubMed] [Google Scholar]
- 39. Liu PT, Stenger S, Li HY, et al. Toll‐like receptor triggering of a vitamin D‐mediated human antimicrobial response. Science. 2006;311(5768):1770‐1773. [DOI] [PubMed] [Google Scholar]
- 40. Liu PT, Stenger S, Tang DH, Modlin RL. Cutting edge: Vitamin D‐mediated human antimicrobial activity against Mycobacterium tuberculosis is dependent on the induction of cathelicidin. Journal of Immunology. 2007;179(4):2060‐2063. [DOI] [PubMed] [Google Scholar]
- 41. Rockett KA, Brookes R, Udalova I, Vidal V, Hill AV, Kwiatkowski D. 1,25‐Dihydroxyvitamin D3 induces nitric oxide synthase and suppresses growth of Mycobacterium tuberculosis in a human macrophage‐like cell line. Infect Immun. 1998;66(11):5314‐5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Coussens A, Timms PM, Boucher BJ, et al. 1alpha,25‐dihydroxyvitamin D3 inhibits matrix metalloproteinases induced by Mycobacterium tuberculosis infection. Immunology. 2009;127(4):539‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jolliffe DA, Ganmaa D, Wejse C, et al. Adjunctive vitamin D in tuberculosis treatment: meta‐analysis of individual participant data. Eur Respir J. 2019;53(3):1802003. [DOI] [PubMed] [Google Scholar]
- 44. Tang XZ, Huang T, Ma Y, et al. Meta‐analysis of efficacy and safety of vitamin D supplementation in the treatment of pulmonary tuberculosis. Zhonghua Yi Xue Za Zhi. 2020;100(32):2525‐2531. [DOI] [PubMed] [Google Scholar]
- 45. Wu HX, Xiong XF, Zhu M, Wei J, Zhuo KQ, Cheng DY. Effects of vitamin D supplementation on the outcomes of patients with pulmonary tuberculosis: a systematic review and meta‐analysis. Bmc Pulm Med. 2018;18(1):108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tukvadze N, Sanikidze E, Kipiani M, et al. High‐dose vitamin D3 in adults with pulmonary tuberculosis: a double‐blind randomized controlled trial. Am J Clin Nutr. 2015;102(5):1059‐1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ganmaa D, Munkhsul B, Bromage S, Buyankhishig B, Martineau AR. High‐dose vitamin D3 during intensive phase treatment of pulmonary tuberculosis in mongolia: a double‐blind randomised controlled trial. Thorax. 2016;71:A67‐A68. [Google Scholar]
- 48. Huang SJ, Wang XH, Liu ZD, et al. Vitamin D deficiency and the risk of tuberculosis: a meta‐analysis. Drug Des Dev Ther. 2017;11:91‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Coussens AK, Wilkinson RJ, Martineau AR. Phenylbutyrate is bacteriostatic against Mycobacterium tuberculosis and regulates the macrophage response to infection, synergistically with 25‐hydroxy‐vitamin D3. PLoS Pathog. 2015;11(7):e1005007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rekha RS, Muvva SSVJR, Wan M, et al. Phenylbutyrate induces LL‐37‐dependent autophagy and intracellular killing of Mycobacterium tuberculosis in human macrophages. Autophagy. 2015;11(9):1688‐1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mily A, Rekha RS, Kamal SMM, et al. Oral intake of phenylbutyrate with or without vitamin D‐3 upregulates the cathelicidin LL‐37 in human macrophages: a dose finding study for treatment of tuberculosis. Bmc Pulm Med. 2013;13:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bekele A, Gebreselassie N, Ashenafi S, et al. Daily adjunctive therapy with vitamin D3 and phenylbutyrate supports clinical recovery from pulmonary tuberculosis: a randomized controlled trial in Ethiopia. J Intern Med. 2018;284(3):292‐306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mily A, Rekha RS, Kamal SM, et al. Significant effects of oral phenylbutyrate and vitamin D3 adjunctive therapy in pulmonary tuberculosis: a randomized controlled trial. PLoS One. 2015;10(9):e0138340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tenforde MW, Yadav A, Dowdy DW, et al. Vitamin A and D deficiencies associated with incident tuberculosis in HIV‐infected patients initiating antiretroviral therapy in multinational case‐cohort study. Jaids‐J Acq Imm Def. 2017;75(3):E71‐E79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Coleman MM, Basdeo SA, Coleman AM, et al. All‐trans retinoic acid augments autophagy during intracellular bacterial infection. Am J Respir Cell Mol Biol. 2018;59(5):548‐556. [DOI] [PubMed] [Google Scholar]
- 56. Chen QY, Mosovsky KL, Ross AC. Retinoic acid and alpha‐galactosylceramide regulate the expression of costimulatory receptors and transcription factors responsible for B cell activation and differentiation. Immunobiology. 2013;218(12):1477‐1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sada‐Ovalle I, Skold M, Tian T, Besra GS, Behar SM. alpha‐Galactosylceramide as a therapeutic agent for pulmonary mycobacterium tuberculosis infection. Am J Resp Crit Care. 2010;182(6):841‐847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mourik BC, Leenen PJ, de Knegt GJ, et al. Immunotherapy added to antibiotic treatment reduces relapse of disease in a mouse model of tuberculosis. Am J Respir Cell Mol Biol. 2017;56(2):233‐241. [DOI] [PubMed] [Google Scholar]
- 59. Karyadi E, West CE, Schultink W, et al. A double‐blind, placebo‐controlled study of vitamin A and zinc supplementation in persons with tuberculosis in Indonesia: effects on clinical response and nutritional status. Am J Clin Nutr. 2002;75(4):720‐727. [DOI] [PubMed] [Google Scholar]
- 60. Visser ME, Grewal HM, Swart EC, et al. The effect of vitamin A and zinc supplementation on treatment outcomes in pulmonary tuberculosis: a randomized controlled trial. Am J Clin Nutr. 2011;93(1):93‐100. [DOI] [PubMed] [Google Scholar]
- 61. Wang J, Xiong K, Wang Q, Zhao S, Liu Y, Ma A. Adjunctive vitamin A and D during pulmonary tuberculosis treatment: a randomized controlled trial with a 2 × 2 factorial design. Food Function. 2020;11(5):4672‐4681. [DOI] [PubMed] [Google Scholar]
- 62. Lawson L, Thacher TD, Yassin MA, et al. Randomized controlled trial of zinc and vitamin A as co‐adjuvants for the treatment of pulmonary tuberculosis. Trop Med Int Health. 2010;15(12):1481‐1490. [DOI] [PubMed] [Google Scholar]
- 63. van der Vaart M, Korbee CJ, Lamers GE, et al. The DNA damage‐regulated autophagy modulator DRAM1 links mycobacterial recognition via TLR‐MYD88 to autophagic defense. Cell Host Microbe. 2014;15(6):753‐767. [DOI] [PubMed] [Google Scholar]
- 64. Zhang R, Varela M, Forn‐Cuni G, Torraca V, van der Vaart M, Meijer AH. Deficiency in the autophagy modulator Dram1 exacerbates pyroptotic cell death of Mycobacteria‐infected macrophages. Cell Death Dis. 2020;11(4):277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Racioppi L, Means AR. Calcium/calmodulin‐dependent protein kinase Kinase 2: roles in signaling and pathophysiology. J Biol Chem. 2012;287(38):31658‐31665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Liu F, Chen J, Wang P, et al. MicroRNA‐27a controls the intracellular survival of Mycobacterium tuberculosis by regulating calcium‐associated autophagy. Nat Commun. 2018;9(1):4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Singh P, Subbian S. Harnessing the mTOR pathway for tuberculosis treatment. Front Microbiol. 2018;9:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ashley D, Hernandez J, Cao R, et al. Antimycobacterial effects of everolimus in a human granuloma model. J Clin Med. 2020;9(7):2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hu Y, Wen Z, Liu S, et al. Ibrutinib suppresses intracellular mycobacterium tuberculosis growth by inducing macrophage autophagy. J Infect. 2020;80(6):e19‐e26. [DOI] [PubMed] [Google Scholar]
- 70. Cheng CY, Gutierrez NM, Marzuki MB, et al. Host sirtuin 1 regulates mycobacterial immunopathogenesis and represents a therapeutic target against tuberculosis. Sci Immunol. 2017;2(9):eaaj1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Singhal A, Jie L, Kumar P, et al. Metformin as adjunct antituberculosis therapy. Sci Transl Med 2014;6(263):263ra159. [DOI] [PubMed] [Google Scholar]
- 72. Wallis RS, Hafner R. Advancing host‐directed therapy for tuberculosis. Nat Rev Immunol. 2015;15(4):255‐263. [DOI] [PubMed] [Google Scholar]
- 73. Russell SL, Lamprecht DA, Mandizvo T, et al. Compromised metabolic reprogramming is an early indicator of CD8(+) T Cell dysfunction during chronic Mycobacterium tuberculosis infection. Cell Rep. 2019;29(11):3564‐3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lachmandas E, Eckold C, Bohme J, et al. Metformin alters human host responses to Mycobacterium tuberculosis in healthy subjects. J Infect Dis. 2019;220(1):139‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Degner NR, Wang JY, Golub JE, Karakousis PC. Metformin use reverses the increased mortality associated with diabetes mellitus during tuberculosis treatment. Clin Infect Dis. 2018;66(2):198‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lee MC, Chiang CY, Lee CH, et al. Metformin use is associated with a low risk of tuberculosis among newly diagnosed diabetes mellitus patients with normal renal function: A nationwide cohort study with validated diagnostic criteria. PLoS One. 2018;13(10):e0205807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lee YJ, Han SK, Park JH, et al. The effect of metformin on culture conversion in tuberculosis patients with diabetes mellitus. Korean J Intern Med. 2018;33(5):933‐940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sogi KM, Lien KA, Johnson JR, Krogan NJ, Stanley SA. The tyrosine kinase inhibitor gefitinib restricts Mycobacterium tuberculosis growth through increased lysosomal biogenesis and modulation of cytokine signaling. ACS Infect Dis. 2017;3(8):564‐574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ouyang Q, Zhang K, Lin D, Feng CG, Cai Y, Chen X. Bazedoxifene suppresses intracellular Mycobacterium tuberculosis growth by enhancing autophagy. mSphere. 2020;5(2):e00124‐e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Juarez E, Carranza C, Sanchez G, et al. Loperamide restricts intracellular growth of Mycobacterium tuberculosis in lung macrophages. Am J Respir Cell Mol Biol. 2016;55(6):837‐847. [DOI] [PubMed] [Google Scholar]
- 81. Saini NK, Baena A, Ng TW, et al. Suppression of autophagy and antigen presentation by Mycobacterium tuberculosis PE_PGRS47. Nat Microbiol. 2016;1(9):16133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chandra P, Ghanwat S, Matta SK, et al. Mycobacterium tuberculosis inhibits RAB7 recruitment to selectively modulate autophagy flux in macrophages. Sci Rep. 2015;5:16320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Shui W, Petzold CJ, Redding A, et al. Organelle membrane proteomics reveals differential influence of mycobacterial lipoglycans on macrophage phagosome maturation and autophagosome accumulation. J Proteome Res. 2011;10(1):339‐348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Giraud‐Gatineau A, Coya JM, Maure A, et al. The antibiotic bedaquiline activates host macrophage innate immune resistance to bacterial infection. eLife. 2020;9:e55692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bryk R, Mundhra S, Jiang X, et al. Potentiation of rifampin activity in a mouse model of tuberculosis by activation of host transcription factor EB. PLoS Pathog. 2020;16(6):e1008567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Francisco‐Cruz A, Mata‐Espinosa D, Estrada‐Parra S, Xing Z, Hernandez‐Pando R. Immunotherapeutic effects of recombinant adenovirus encoding granulocyte‐macrophage colony‐stimulating factor in experimental pulmonary tuberculosis. Clin Exp Immunol. 2013;171(3):283‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Pasula R, Azad AK, Gardner JC, Schlesinger LS, McCormack FX. Keratinocyte growth factor administration attenuates murine pulmonary mycobacterium tuberculosis infection through granulocyte‐macrophage colony‐stimulating factor (GM‐CSF)‐dependent macrophage activation and phagolysosome fusion. J Biol Chem. 2015;290(11):7151‐7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Benmerzoug S, Marinho FV, Rose S, et al. GM‐CSF targeted immunomodulation affects host response to M tuberculosis infection. Sci Rep. 2018;8(1):8652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hariadi NI, Blackwood RA. Disseminated mycobacterium avium complex in an adolescent with perinatally‐acquired HIV infection. Infect Dis Rep. 2017;9(2):6884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Nannini EC, Keating M, Binstock P, Samonis G, Kontoyiannis DP. Successful treatment of refractory disseminated Mycobacterium avium complex infection with the addition of linezolid and mefloquine. J Infect. 2002;44(3):201‐203. [DOI] [PubMed] [Google Scholar]
- 91. de Silva TI, Cope A, Goepel J, Greig JM. The use of adjuvant granulocyte‐macrophage colony‐stimulating factor in HIV‐related disseminated atypical mycobacterial infection. J Infect. 2007;54(4):e207‐210. [DOI] [PubMed] [Google Scholar]
- 92. Kedzierska K, Mak J, Mijch A, et al. Granulocyte‐macrophage colony‐stimulating factor augments phagocytosis of Mycobacterium avium complex by human immunodeficiency virus type 1‐infected monocytes/macrophages in vitro and in vivo. J Infect Dis. 2000;181(1):390‐394. [DOI] [PubMed] [Google Scholar]
- 93. Nathan C. Specificity of a third kind: reactive oxygen and nitrogen intermediates in cell signaling. J Clin Invest. 2003;111(6):769‐778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bogdan C, Rollinghoff M, Diefenbach A. The role of nitric oxide in innate immunity. Immunol Rev. 2000;173:17‐26. [DOI] [PubMed] [Google Scholar]
- 95. Awuh JA, Flo TH. Molecular basis of mycobacterial survival in macrophages. Cell Mol Life Sci. 2017;74(9):1625‐1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Fremond CM, Yeremeev V, Nicolle DM, Jacobs M, Quesniaux VF, Ryffel B. Fatal Mycobacterium tuberculosis infection despite adaptive immune response in the absence of MyD88. J Clin Invest. 2004;114(12):1790‐1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Yang Q, Liao M, Wang W, et al. CD157 confers host resistance to Mycobacterium tuberculosis via TLR2‐CD157‐PKCzeta‐induced reactive oxygen species production. MBio. 2019;10(4):e01949–01919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Zhang Y, Su SS, Zhao S, et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun. 2017;8:14329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Pajuelo D, Gonzalez‐Juarbe N, Niederweis M. NAD hydrolysis by the tuberculosis necrotizing toxin induces lethal oxidative stress in macrophages. Cell Microbiol. 2020;22(1):e13115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Palanisamy GS, Kirk NM, Ackart DF, Shanley CA, Orme IM, Basaraba RJ. Evidence for oxidative stress and defective antioxidant response in guinea pigs with tuberculosis. PLoS One. 2011;6(10):e26254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Safe IP, Lacerda MVG, Printes VS, et al. Safety and efficacy of N‐acetylcysteine in hospitalized patients with HIV‐associated tuberculosis: An open‐label, randomized, phase II trial (RIPENACTB Study). PLoS One. 2020;15(6):e0235381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Yang CS, Shin DM, Kim KH, et al. NADPH oxidase 2 interaction with TLR2 is required for efficient innate immune responses to mycobacteria via cathelicidin expression. J Immunol. 2009;182(6):3696‐3705. [DOI] [PubMed] [Google Scholar]
- 103. Chan ED, Chan J, Schluger NW. What is the role of nitric oxide in murine and human host defense against tuberculosis? Current knowledge. Am J Resp Cell Mol. 2001;25(5):606‐612. [DOI] [PubMed] [Google Scholar]
- 104. Pearl JE, Torrado E, Tighe M, et al. Nitric oxide inhibits the accumulation of CD4+CD44hiTbet+CD69lo T cells in mycobacterial infection. Eur J Immunol. 2012;42(12):3267‐3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Yang CS, Yuk JM, Jo EK. The role of nitric oxide in mycobacterial infections. Immune Netw. 2009;9(2):46‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Wu K, Koo J, Jiang X, Chen R, Cohen SN, Nathan C. Improved control of tuberculosis and activation of macrophages in mice lacking protein kinase R. PLoS One. 2012;7(2):e30512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Schon T, Elias D, Moges F, et al. Arginine as an adjuvant to chemotherapy improves clinical outcome in active tuberculosis. Eur Respir J. 2003;21(3):483‐488. [DOI] [PubMed] [Google Scholar]
- 108. Ralph AP, Waramori G, Pontororing GJ, et al. L‐arginine and vitamin D adjunctive therapies in pulmonary tuberculosis: a randomised, double‐blind, placebo‐controlled trial. PLoS One. 2013;8(8):e70032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Schon T, Idh J, Westman A, et al. Effects of a food supplement rich in arginine in patients with smear positive pulmonary tuberculosis–a randomised trial. Tuberculosis. 2011;91(5):370‐377. [DOI] [PubMed] [Google Scholar]
- 110. Ruan J, St John G, Ehrt S, Riley L, Nathan C. noxR3, a novel gene from Mycobacterium tuberculosis, protects Salmonella typhimurium from nitrosative and oxidative stress. Infect Immun. 1999;67(7):3276‐3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Chen L, Xie QW, Nathan C. Alkyl hydroperoxide reductase subunit C (AhpC) protects bacterial and human cells against reactive nitrogen intermediates. Mol Cell. 1998;1(6):795‐805. [DOI] [PubMed] [Google Scholar]
- 112. McGarvey J, Bermudez LE. Pathogenesis of nontuberculous mycobacteria infections. Clin Chest Med. 2002;23(3):569‐583. [DOI] [PubMed] [Google Scholar]
- 113. Gomes MS, Florido M, Pais TF, Appelberg R. Improved clearance of Mycobacterium avium upon disruption of the inducible nitric oxide synthase gene. J Immunol. 1999;162(11):6734‐6739. [PubMed] [Google Scholar]
- 114. Pearl JE, Torrado E, Tighe M, et al. Nitric oxide inhibits the accumulation of CD4+CD44hiTbet+CD69loT cells in mycobacterial infection. Eur J Immunol. 2012;42(12):3267‐3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Moreira JD, Koch BEV, van Veen S, et al. Functional inhibition of host Histone Deacetylases (HDACs) enhances in vitro and in vivo anti‐mycobacterial activity in human macrophages and in zebrafish. Front Immunol. 2020;11:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Bhaskar A, Kumar S, Khan MZ, Singh A, Dwivedi VP, Nandicoori VK. Host sirtuin 2 as an immunotherapeutic target against tuberculosis. eLife. 2020;9:e55415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Rao M, Valentini D, Zumla A, Maeurer M. Evaluation of the efficacy of valproic acid and suberoylanilide hydroxamic acid (vorinostat) in enhancing the effects of first‐line tuberculosis drugs against intracellular Mycobacterium tuberculosis . Int J Infect Dis. 2018;69:78‐84. [DOI] [PubMed] [Google Scholar]
- 118. Hegde SR. Computational identification of the proteins associated with quorum sensing and biofilm formation in Mycobacterium tuberculosis . Front Microbiol. 2019;10:3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Cohen SB, Gern BH, Delahaye JL, et al. Alveolar macrophages provide an early Mycobacterium tuberculosis niche and initiate dissemination. Cell Host Microbe. 2018;24(3):439‐446 e434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Flynn CM, Garbers Y, Lokau J, et al. Activation of Toll‐like Receptor 2 (TLR2) induces Interleukin‐6 trans‐signaling. Sci Rep. 2019;9(1):7306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Pires D, Bernard EM, Pombo JP, et al. Mycobacterium tuberculosis modulates miR‐106b‐5p to control cathepsin S expression resulting in higher pathogen survival and poor T‐cell activation. Front Immunol. 2017;8:1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Gupta PK, Chakraborty P, Kumar S, et al. G1–4A, a Polysaccharide from tinospora cordifolia inhibits the survival of Mycobacterium tuberculosis by modulating host immune responses in TLR4 dependent manner. PLoS One. 2016;11(5):e0154725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Anwar MA, Shah M, Kim J, Choi S. Recent clinical trends in Toll‐like receptor targeting therapeutics. Med Res Rev. 2019;39(3):1053‐1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Dwivedi VP, Bhattacharya D, Yadav V, et al. The phytochemical bergenin enhances T Helper 1 responses and anti‐mycobacterial immunity by activating the MAP kinase pathway in macrophages. Front Cell Infect Microbiol. 2017;7:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Ottenhoff TH, Kaufmann SH. Vaccines against tuberculosis: where are we and where do we need to go? PLoS Pathog. 2012;8(5):e1002607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Khan N, Vidyarthi A, Amir M, Mushtaq K, Agrewala JN. T‐cell exhaustion in tuberculosis: pitfalls and prospects. Crit Rev Microbiol. 2017;43(2):133‐141. [DOI] [PubMed] [Google Scholar]
- 127. Tezera LB, Bielecka MK, Ogongo P, et al. Anti‐PD‐1 immunotherapy leads to tuberculosis reactivation via dysregulation of TNF‐alpha. eLife. 2020;9:e52668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Anastasopoulou A, Ziogas DC, Samarkos M, Kirkwood JM, Gogas H. Reactivation of tuberculosis in cancer patients following administration of immune checkpoint inhibitors: current evidence and clinical practice recommendations. J Immunother Cancer. 2019;7(1):239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Gautam US, Foreman TW, Bucsan AN, et al. In vivo inhibition of tryptophan catabolism reorganizes the tuberculoma and augments immune‐mediated control of Mycobacterium tuberculosis . Proc Natl Acad Sci U S A. 2018;115(1):E62‐E71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Marchant A, Amedei A, Azzurri A, et al. Polarization of PPD‐specific T‐cell response of patients with tuberculosis from Th0 to Th1 profile after successful antimycobacterial therapy or in vitro conditioning with interferon‐alpha or interleukin‐12. Am J Respir Cell Mol Biol. 2001;24(2):187‐194. [DOI] [PubMed] [Google Scholar]
- 131. Pooran A, Davids M, Nel A, Shoko A, Blackburn J, Dheda K. IL‐4 subverts mycobacterial containment in Mycobacterium tuberculosis‐infected human macrophages. Eur Respir J. 2019;54(2):1802242. [DOI] [PubMed] [Google Scholar]
- 132. Esmail H, Riou C, Bruyn ED, et al. The immune response to Mycobacterium tuberculosis in HIV‐1‐coinfected persons. Annu Rev Immunol. 2018;36:603‐638. [DOI] [PubMed] [Google Scholar]
- 133. Vazquez N, Greenwell‐Wild T, Rekka S, Orenstein JM, Wahl SM. Mycobacterium avium‐induced SOCS contributes to resistance to IFN‐gamma‐mediated mycobactericidal activity in human macrophages. J Leukoc Biol. 2006;80(5):1136‐1144. [DOI] [PubMed] [Google Scholar]
- 134. Ottenhoff TH, Verreck FA, Lichtenauer‐Kaligis EG, Hoeve MA, Sanal O, van Dissel JT. Genetics, cytokines and human infectious disease: lessons from weakly pathogenic mycobacteria and salmonellae. Nat Genet. 2002;32(1):97‐105. [DOI] [PubMed] [Google Scholar]
- 135. Dawson R, Condos R, Tse D, et al. Immunomodulation with recombinant interferon‐gamma1b in pulmonary tuberculosis. PLoS One. 2009;4(9):e6984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Gao XF, Yang ZW, Li J. Adjunctive therapy with interferon‐gamma for the treatment of pulmonary tuberculosis: a systematic review. Int J Infect Dis. 2011;15(9):e594‐600. [DOI] [PubMed] [Google Scholar]
- 137. Appelberg R, Orme IM. Effector mechanisms involved in cytokine‐mediated bacteriostasis of Mycobacterium avium infections in murine macrophages. Immunology. 1993;80(3):352‐359. [PMC free article] [PubMed] [Google Scholar]
- 138. Holland SM, Eisenstein EM, Kuhns DB, et al. Treatment of refractory disseminated nontuberculous mycobacterial infection with interferon gamma. A preliminary report. N Engl J Med. 1994;330(19):1348‐1355. [DOI] [PubMed] [Google Scholar]
- 139. Milanes‐Virelles MT, Garcia‐Garcia I, Santos‐Herrera Y, et al. Adjuvant interferon gamma in patients with pulmonary atypical Mycobacteriosis: a randomized, double‐blind, placebo‐controlled study. BMC Infect Dis. 2008;8:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Lauw FN, van Der Meer JT, de Metz J, Danner SA, van Der Poll T. No beneficial effect of interferon‐gamma treatment in 2 human immunodeficiency virus‐infected patients with Mycobacterium avium complex infection. Clin Infect Dis. 2001;32(4):e81‐82. [DOI] [PubMed] [Google Scholar]
- 141. Mata‐Espinosa D, Francisco‐Cruz A, Marquina‐Castillo B, et al. Immunotherapeutic effects of recombinant adenovirus encoding interleukin 12 in experimental pulmonary tuberculosis. Scandinavian J. Immunol. 2018;89:e12743. [DOI] [PubMed] [Google Scholar]
- 142. Ma Y, Chen HD, Wang Y, et al. Interleukin 24 as a novel potential cytokine immunotherapy for the treatment of Mycobacterium tuberculosis infection. Microbes Infect. 2011;13(12–13):1099‐1110. [DOI] [PubMed] [Google Scholar]
- 143. Lindestam Arlehamn CS, Gerasimova A, Mele F, et al. Memory T cells in latent Mycobacterium tuberculosis infection are directed against three antigenic islands and largely contained in a CXCR3+CCR6+ Th1 subset. PLoS Pathog. 2013;9(1):e1003130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Rakshit S, Hingankar N, Alampalli SV, et al. HIV skews a balanced Mtb‐specific Th17 response in latent tuberculosis subjects to a pro‐inflammatory profile independent of viral load. Cell Rep. 2020;33(9):108451. [DOI] [PubMed] [Google Scholar]
- 145. Scott NR, Swanson RV, Al‐Hammadi N, et al. S100A8/A9 regulates CD11b expression and neutrophil recruitment during chronic tuberculosis. J Clin Invest. 2020;130(6):3098‐3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Zhang R, Xi X, Wang C, et al. Therapeutic effects of recombinant human interleukin 2 as adjunctive immunotherapy against tuberculosis: A systematic review and meta‐analysis. PLoS One. 2018;13(7):e0201025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Bermudez LE, Stevens P, Kolonoski P, Wu M, Young LS. Treatment of experimental disseminated Mycobacterium avium complex infection in mice with recombinant IL‐2 and tumor necrosis factor. J Immunol. 1989;143(9):2996‐3000. [PubMed] [Google Scholar]
- 148. Sekiguchi Y, Yasui K, Yamazaki T, Agematsu K, Kobayashi N, Koike K. Effective combination therapy using interferon‐gamma and interleukin‐2 for disseminated Mycobacterium avium complex infection in a pediatric patient with AIDS. Clin Infect Dis. 2005;41(11):e104‐106. [DOI] [PubMed] [Google Scholar]
- 149. Trojan T, Collins R, Khan DA. Safety and efficacy of treatment using interleukin‐2 in a patient with idiopathic CD4(+) lymphopenia and Mycobacterium avium‐intracellulare. Clin Exp Immunol. 2009;156(3):440‐445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Gupta S, Cheung L, Pokkali S, et al. Suppressor cell‐depleting immunotherapy with denileukin diftitox is an effective host‐directed therapy for tuberculosis. J Infect Dis. 2017;215(12):1883‐1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Chen CY, Yao S, Huang D, et al. Phosphoantigen/IL2 expansion and differentiation of Vgamma2Vdelta2 T cells increase resistance to tuberculosis in nonhuman primates. PLoS Pathog. 2013;9(8):e1003501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Xu P, Pang Y, Xu J, Chen H, Tang P, Wu M. Cytokine‐induced killer cell therapy as a promising adjunctive immunotherapy for multidrug‐resistant pulmonary TB: a case report. Immunotherapy. 2018;10(10):827‐830. [DOI] [PubMed] [Google Scholar]
- 153. Mohareer K, Asalla S, Banerjee S. Cell death at the cross roads of host‐pathogen interaction in Mycobacterium tuberculosis infection. Tuberculosis. 2018;113:99‐121. [DOI] [PubMed] [Google Scholar]
- 154. Rajaram MV, Brooks MN, Morris JD, Torrelles JB, Azad AK, Schlesinger LS Mycobacterium tuberculosis activates human macrophage peroxisome proliferator‐activated receptor gamma linking mannose receptor recognition to regulation of immune responses. J Immunol. 2010;185(2):929‐942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Arnett E, Weaver AM, Woodyard KC, et al. PPARgamma is critical for Mycobacterium tuberculosis induction of Mcl‐1 and limitation of human macrophage apoptosis. PLoS Pathog. 2018;14(6):e1007100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Pajuelo D, Gonzalez‐Juarbe N, Tak U, Sun J, Orihuela CJ, Niederweis M. NAD(+) depletion triggers macrophage necroptosis, a cell death pathway exploited by Mycobacterium tuberculosis . Cell Rep. 2018;24(2):429‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Grab J, Suarez I, van Gumpel E, et al. Corticosteroids inhibit Mycobacterium tuberculosis‐induced necrotic host cell death by abrogating mitochondrial membrane permeability transition. Nat Commun. 2019;10(1):688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Kim YJ, Pack KM, Jeong E, et al. Pulmonary tuberculosis with acute respiratory failure. Eur Respir J. 2008;32(6):1625‐1630. [DOI] [PubMed] [Google Scholar]
- 159. Malhotra HS, Garg RK, Singh MK, Agarwal A, Verma R. Corticosteroids (dexamethasone versus intravenous methylprednisolone) in patients with tuberculous meningitis. Ann Trop Med Parasitol. 2009;103(7):625‐634. [DOI] [PubMed] [Google Scholar]
- 160. Critchley JA, Orton LC, Pearson F. Adjunctive steroid therapy for managing pulmonary tuberculosis. Cochrane Database Syst Rev. 2014(11):CD011370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Kalamidas SA, Kuehnel MP, Peyron P, et al. cAMP synthesis and degradation by phagosomes regulate actin assembly and fusion events: consequences for mycobacteria. J Cell Sci. 2006;119(Pt 17):3686‐3694. [DOI] [PubMed] [Google Scholar]
- 162. Roca FJ, Whitworth LJ, Redmond S, Jones AA, Ramakrishnan L. TNF induces pathogenic programmed macrophage necrosis in tuberculosis through a mitochondrial‐lysosomal‐endoplasmic reticulum circuit. Cell. 2019;178(6):1344‐1361 e1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Roca FJ, Ramakrishnan L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell. 2013;153(3):521‐534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Maiga M, Agarwal N, Ammerman NC, et al. Successful shortening of tuberculosis treatment using adjuvant host‐directed therapy with FDA‐approved phosphodiesterase inhibitors in the mouse model. PLoS One. 2012;7(2):e30749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Maiga M, Ammerman NC, Maiga MC, et al. Adjuvant host‐directed therapy with types 3 and 5 but not type 4 phosphodiesterase inhibitors shortens the duration of tuberculosis treatment. J Infect Dis. 2013;208(3):512‐519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Ordonez AA, Pokkali S, Kim S, et al. Adjunct antibody administration with standard treatment reduces relapse rates in a murine tuberculosis model of necrotic granulomas. PLoS One. 2018;13(5):e0197474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Skerry C, Harper J, Klunk M, Bishai WR, Jain SK. Adjunctive TNF inhibition with standard treatment enhances bacterial clearance in a murine model of necrotic TB granulomas. PLoS One. 2012;7(6):e39680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Kolloli A, Subbian S. Host‐directed therapeutic strategies for tuberculosis. Front Med (Lausanne). 2017;4:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Zhang Z, Fan W, Yang G, et al. Risk of tuberculosis in patients treated with TNF‐alpha antagonists: a systematic review and meta‐analysis of randomised controlled trials. BMJ Open. 2017;7(3):e012567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Danko JR, Gilliland WR, Miller RS, Decker CF. Disseminated Mycobacterium marinum infection in a patient with rheumatoid arthritis receiving infliximab therapy. Scand J Infect Dis. 2009;41(4):252‐255. [DOI] [PubMed] [Google Scholar]
- 171. Amaral EP, Costa DL, Namasivayam S, et al. A major role for ferroptosis in Mycobacterium tuberculosis‐induced cell death and tissue necrosis. J Exp Med. 2019;216(3):556‐570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Conrad M, Kagan VE, Bayir H, et al. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. 2018;32(9–10):602‐619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Ingold I, Berndt C, Schmitt S, et al. Selenium utilization by GPX4 Is required to prevent hydroperoxide‐induced ferroptosis. Cell. 2018;172(3):409‐422 e421. [DOI] [PubMed] [Google Scholar]
- 174. Howard NC, Khader SA. Immunometabolism during Mycobacterium tuberculosis Infection. Trends Microbiol. 2020;28(10):832‐850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Krishnamoorthy G, Kaiser P, Abu Abed U, et al. FX11 limits Mycobacterium tuberculosis growth and potentiates bactericidal activity of isoniazid through host‐directed activity. Dis Model Mech. 2020;13(3):dmm041954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Tatano Y, Kanehiro Y, Sano C, Shimizu T, Tomioka H. ATP exhibits antimicrobial action by inhibiting bacterial utilization of ferric ions. Sci Rep. 2015;5:8610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Tatano Y, Yamabe S, Sano C, Tomioka H. Anti‐Mycobacterium avium complex activity of clarithromycin, rifampin, rifabutin, and ethambutol in combination with adenosine 5'‐triphosphate. Diagn Microbiol Infect Dis. 2017;88(3):241‐246. [DOI] [PubMed] [Google Scholar]
- 178. Matty MA, Knudsen DR, Walton EM, et al. Potentiation of P2RX7 as a host‐directed strategy for control of mycobacterial infection. Elife. 2019;8:e39123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Tsai IF, Kuo CP, Lin AB, et al. Potential effect of ezetimibe against Mycobacterium tuberculosis infection in type II diabetes. Respirology. 2017;22(3):559‐566. [DOI] [PubMed] [Google Scholar]
- 180. Asalla S, Mohareer K, Banerjee S. Small molecule mediated restoration of mitochondrial function augments anti‐mycobacterial activity of human macrophages subjected to cholesterol induced asymptomatic dyslipidemia. Front Cell Infect Microbiol. 2017;7:439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Dutta NK, Bruiners N, Zimmerman MD, et al. Adjunctive host‐directed therapy with statins improves tuberculosis‐related outcomes in mice. J Infect Dis. 2020;221(7):1079‐1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Pieters J. Mycobacterium tuberculosis and the macrophage: maintaining a balance. Cell Host Microbe. 2008;3(6):399‐407. [DOI] [PubMed] [Google Scholar]
- 183. de Chastellier C, Thilo L. Cholesterol depletion in Mycobacterium avium‐infected macrophages overcomes the block in phagosome maturation and leads to the reversible sequestration of viable mycobacteria in phagolysosome‐derived autophagic vacuoles. Cell Microbiol. 2006;8(2):242‐256. [DOI] [PubMed] [Google Scholar]
- 184. Dutta NK, Bruiners N, Pinn ML, et al. Statin adjunctive therapy shortens the duration of TB treatment in mice. J Antimicrob Chemother. 2016;71(6):1570‐1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Parihar SP, Guler R, Khutlang R, et al. Statin therapy reduces the mycobacterium tuberculosis burden in human macrophages and in mice by enhancing autophagy and phagosome maturation. J Infect Dis. 2014;209(5):754‐763. [DOI] [PubMed] [Google Scholar]
- 186. Guerra‐De‐Blas PDC, Bobadilla‐Del‐Valle M, Sada‐Ovalle I, et al. Simvastatin enhances the immune response against Mycobacterium tuberculosis . Front Microbiol. 2019;10:2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Sorgi CA, Soares EM, Rosada RS, et al. Eicosanoid pathway on host resistance and inflammation during Mycobacterium tuberculosis infection is comprised by LTB4 reduction but not PGE2 increment. Biochim Biophys Acta Mol Basis Dis. 2020;1866(3):165574. [DOI] [PubMed] [Google Scholar]
- 188. Tobin DM, Roca FJ, Oh SF, et al. Host genotype‐specific therapies can optimize the inflammatory response to mycobacterial infections. Cell. 2012;148(3):434‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Mayer‐Barber KD, Andrade BB, Oland SD, et al. Host‐directed therapy of tuberculosis based on interleukin‐1 and type I interferon crosstalk. Nature. 2014;511(7507):99‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Kaufmann SHE, Dorhoi A, Hotchkiss RS, Bartenschlager R. Host‐directed therapies for bacterial and viral infections. Nat Rev Drug Discov. 2018;17(1):35‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Thuong NTT, Heemskerk D, Tram TTB, et al. Leukotriene A4 hydrolase genotype and HIV infection influence intracerebral inflammation and survival from tuberculous meningitis. J Infect Dis. 2017;215(7):1020‐1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Vilaplana C, Marzo E, Tapia G, Diaz J, Garcia V, Cardona PJ. Ibuprofen therapy resulted in significantly decreased tissue bacillary loads and increased survival in a new murine experimental model of active tuberculosis. J Infect Dis. 2013;208(2):199‐202. [DOI] [PubMed] [Google Scholar]
- 193. Byrne ST, Denkin SM, Zhang Y. Aspirin and ibuprofen enhance pyrazinamide treatment of murine tuberculosis. J Antimicrob Chemother. 2007;59(2):313‐316. [DOI] [PubMed] [Google Scholar]
- 194. Kroesen VM, Rodriguez‐Martinez P, Garcia E, et al. A beneficial effect of low‐dose aspirin in a murine model of active tuberculosis. Front Immunol. 2018;9:798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Hortle E, Johnson KE, Johansen MD, et al. Thrombocyte inhibition restores protective immunity to mycobacterial infection in zebrafish. J Infect Dis. 2019;220(3):524‐534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Lee MR, Lee MC, Chang CH, et al. Use of antiplatelet agents and survival of tuberculosis patients: a population‐based cohort study. J Clin Med. 2019;8(7):923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Misra UK, Kalita J, Nair PP. Role of aspirin in tuberculous meningitis: a randomized open label placebo controlled trial. J Neurol Sci. 2010;293(1–2):12‐17. [DOI] [PubMed] [Google Scholar]
- 198. Byrne ST, Denkin SM, Zhang Y. Aspirin antagonism in isoniazid treatment of tuberculosis in mice. Antimicrob Agents Chemother. 2007;51(2):794‐795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Smith D, Hänsch H, Bancroft G, Ehlers S. T‐cell‐independent granuloma formation in response to Mycobacterium avium: role of tumour necrosis factor‐alpha and interferon‐gamma. Immunology. 1997;92(4):413‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Hänsch HC, Smith DA, Mielke ME, Hahn H, Bancroft GJ, Ehlers S. Mechanisms of granuloma formation in murine Mycobacterium avium infection: the contribution of CD4+ T cells. Int Immunol. 1996;8(8):299‐310. [DOI] [PubMed] [Google Scholar]
- 201. Streit M, Bregenzer T, Heinzer I. Cutaneous infections due to atypical mycobacteria. Hautarzt. 2008;59(1):59‐70. [DOI] [PubMed] [Google Scholar]
- 202. Ehlers S, Schaible UE. The granuloma in tuberculosis: dynamics of a host‐pathogen collusion. Front Immunol. 2012;3:411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Parasa VR, Muvva JR, Rose JF, Braian C, Brighenti S, Lerm M. Inhibition of tissue matrix metalloproteinases interferes with mycobacterium tuberculosis‐induced granuloma formation and reduces bacterial load in a human lung tissue model. Front Microbiol. 2017;8:2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Sabir N, Hussain T, Mangi MH, Zhao D, Zhou X. Matrix metalloproteinases: Expression, regulation and role in the immunopathology of tuberculosis. Cell Prolif. 2019;52(4):e12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Xu Y, Wang L, Zimmerman MD, et al. Matrix metalloproteinase inhibitors enhance the efficacy of frontline drugs against Mycobacterium tuberculosis . PLoS Pathog. 2018;14(4):e1006974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Singh S, Kubler A, Singh UK, et al. Antimycobacterial drugs modulate immunopathogenic matrix metalloproteinases in a cellular model of pulmonary tuberculosis. Antimicrob Agents Chemother. 2014;58(8):4657‐4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Elkington P, Shiomi T, Breen R, et al. MMP‐1 drives immunopathology in human tuberculosis and transgenic mice. J Clin Invest. 2011;121(5):1827‐1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Ordonez AA, Pokkali S, Sanchez‐Bautista J, et al. Matrix metalloproteinase inhibition in a murine model of cavitary tuberculosis paradoxically worsens pathology. J Infect Dis. 2019;219(4):633‐636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Ong CW, Elkington PT, Brilha S, et al. Neutrophil‐derived MMP‐8 drives AMPK‐dependent matrix destruction in human pulmonary tuberculosis. PLoS Pathog. 2015;11(5):e1004917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Walker NF, Wilkinson KA, Meintjes G, et al. Matrix degradation in human immunodeficiency virus type 1‐associated tuberculosis and tuberculosis immune reconstitution inflammatory syndrome: a prospective observational study. Clin Infect Dis. 2017;65(1):121‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Machelart A, Song OR, Hoffmann E, Brodin P. Host‐directed therapies offer novel opportunities for the fight against tuberculosis. Drug Discov Today. 2017;22(8):1250‐1257. [DOI] [PubMed] [Google Scholar]
- 212. Majeed S, Singh P, Sharma N, Sharma S. Title: role of matrix metalloproteinase ‐9 in progression of tuberculous meningitis: a pilot study in patients at different stages of the disease. BMC Infect Dis. 2016;16(1):722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Majeed S, Radotra BD, Sharma S. Adjunctive role of MMP‐9 inhibition along with conventional anti‐tubercular drugs against experimental tuberculous meningitis. Int J Exp Pathol. 2016;97(3):230‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Walker NF, Clark SO, Oni T, et al. Doxycycline and HIV infection suppress tuberculosis‐induced matrix metalloproteinases. Am J Respir Crit Care Med. 2012;185(9):989‐997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Zenk SF, Vollmer M, Schercher E, Kallert S, Kubis J, Stenger S. Hypoxia promotes Mycobacterium tuberculosis‐specific up‐regulation of granulysin in human T cells. Med Microbiol Immunol. 2016;205(3):219‐229. [DOI] [PubMed] [Google Scholar]
- 216. Walton EM, Cronan MR, Cambier CJ, et al. Cyclopropane modification of trehalose dimycolate drives granuloma angiogenesis and mycobacterial growth through vegf signaling. Cell Host Microbe. 2018;24(4):514‐525 e516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Datta M, Via LE, Kamoun WS, et al. Anti‐vascular endothelial growth factor treatment normalizes tuberculosis granuloma vasculature and improves small molecule delivery. Proc Natl Acad Sci U S A. 2015;112(6):1827‐1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Oehlers SH, Cronan MR, Scott NR, et al. Interception of host angiogenic signalling limits mycobacterial growth. Nature. 2015;517(7536):612‐615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Oehlers SH, Cronan MR, Beerman RW, et al. Infection‐induced vascular permeability aids mycobacterial growth. J Infect Dis. 2017;215(5):813‐817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Niemi M, Backman JT, Fromm MF, Neuvonen PJ, Kivisto KT. Pharmacokinetic interactions with rifampicin : clinical relevance. Clin Pharmacokinet. 2003;42(9):819‐850. [DOI] [PubMed] [Google Scholar]
- 221. Russell SL, Lamprecht DA, Mandizvo T, et al. Compromised metabolic reprogramming is an early indicator of CD8(+) T cell dysfunction during Chronic Mycobacterium tuberculosis Infection. Cell Rep. 2019;29(11):3564‐3579 e3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Gupta S, Tyagi S, Bishai WR. Verapamil increases the bactericidal activity of bedaquiline against Mycobacterium tuberculosis in a mouse model. Antimicrob Agents Chemother. 2015;59(1):673‐676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Martin A, Bouyakoub Y, Soumillion K, Mantu EON, Colmant A, Rodriguez‐Villalobos H. Targeting bedaquiline mycobacterial efflux pump to potentially enhance therapy in Mycobacterium abscessus . Int J Mycobacteriol. 2020;9(1):71‐75. [DOI] [PubMed] [Google Scholar]
- 224. Viljoen A, Raynaud C, Johansen MD, et al. Verapamil improves the activity of bedaquiline against Mycobacterium abscessus in vitro and in macrophages. Antimicrob Agents Chemother. 2019;63(9):e00705‐00719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Matt U, Selchow P, Dal Molin M, et al. Chloroquine enhances the antimycobacterial activity of isoniazid and pyrazinamide by reversing inflammation‐induced macrophage efflux. Int J Antimicrob Agents. 2017;50(1):55‐62. [DOI] [PubMed] [Google Scholar]
- 226. Hegeto LA, Caleffi‐Ferracioli KR, Nakamura‐Vasconcelos SS, et al. In vitro combinatory activity of piperine and anti‐tuberculosis drugs in Mycobacterium tuberculosis . Tuberculosis. 2018;111:35‐40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study