

Abstract
Aim
Peroxisomes play a key role in lipid metabolism, and peroxisome defects have been associated with neurodegenerative diseases such as X‐adrenoleukodystrophy and Alzheimer's disease. This study aims to elucidate the contribution of peroxisomes in lipid alterations of area 8 of the frontal cortex in the spectrum of TDP43‐proteinopathies. Cases of frontotemporal lobar degeneration‐TDP43 (FTLD‐TDP), manifested as sporadic (sFTLD‐TDP) or linked to mutations in various genes including expansions of the non‐coding region of C9ORF72 (c9FTLD), and of sporadic amyotrophic lateral sclerosis (sALS) as the most common TDP43 proteinopathies, were analysed.
Methods
We used transcriptomics and lipidomics methods to define the steady‐state levels of gene expression and lipid profiles.
Results
Our results show alterations in gene expression of some components of peroxisomes and related lipid pathways in frontal cortex area 8 in sALS, sFTLD‐TDP and c9FTLD. Additionally, we identify a lipidomic pattern associated with the ALS‐FTLD‐TDP43 proteinopathy spectrum, notably characterised by down‐regulation of ether lipids and acylcarnitine among other lipid species, as well as alterations in the lipidome of each phenotype of TDP43 proteinopathy, which reveals commonalities and disease‐dependent differences in lipid composition.
Conclusion
Globally, lipid alterations in the human frontal cortex of the ALS‐FTLD‐TDP43 proteinopathy spectrum, which involve cell membrane composition and signalling, vulnerability against cellular stress and possible glucose metabolism, are partly related to peroxisome impairment.
Keywords: fatty acid profiling, human frontal cortex, lipidomics, peroxisomes, plasmalogens, transcriptomics
3lipid alterations in human frontal cortex in ALS‐FTLD‐TDP43 proteinopathy spectrum are partly related to peroxisome impairment.
INTRODUCTION
Peroxisomes are single membrane‐bound cytoplasmic organelles in eukaryotic cells which harbour a variety of biochemical reactions and metabolic pathways involved in oxidative stress homeostasis, and carbohydrate, amino acid and lipid metabolism. In the lipid field, peroxisomes play a key role in α‐oxidation of branched‐chain fatty acids and β‐oxidation of very long fatty acids (>C20), as well as biosynthesis of ether lipids, bile acids and docosahexaenoic acid [1, 2, 3, 4, 5, 6, 7, 8, 9]. Peroxisomes are found ubiquitously, but their number, shape and enzymatic content respond rapidly to cellular and environmental factors [6, 8, 10, 11]. Peroxisomes have close functional relationships with the endoplasmic reticulum, mitochondria and lipid droplets [12, 13, 14, 15, 16, 17]. The biogenesis of peroxisomes requires a group of proteins named peroxins, encoded by PEX genes, which participate in the formation of peroxisomal membranes incorporating peroxisomal membrane proteins, peroxisome proliferator‐activated receptors (encoded by PPARS) that modulate peroxisomal biogenesis and regulate lipid metabolism, and dynamin‐related proteins [6, 7, 18, 19, 20, 21].
Primary peroxisomal defects of lipid metabolism are genetically determined disorders linked to mutations of specific peroxisomal genes that lead to distinct diseases with neurologic and systemic manifestations and invariably poor outcomes. These may affect fatty acid β‐oxidation, ether lipid biosynthesis and fatty acid α‐oxidation [11, 31]. In addition, peroxisomes contribute to cellular ageing and redox balance, under the control of peroxisome/mitochondria function, which are altered in age‐related diseases such as diabetes, hypertension, cancer and neurodegenerative diseases [32, 33, 34, 35]. Impaired peroxisomal function occurs in Alzheimer's disease (AD) and related transgenic mouse models [36, 37, 38]. More precisely, accumulation of C22:0 and very long‐chain fatty acids, and decreased levels of plasmalogens, together with increased volume density and loss of peroxisomes in neurons with neurofibrillary tangles, are all observed with AD progression [39]. These alterations have prompted the study of several specific therapeutic tools directed to curbing altered peroxisomal function in AD [40, 41, 42, 43, 44, 45].
Amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD) are two fatal neurodegenerative disorders with considerable clinical, pathological and genetic overlap. ALS is a fatal neurodegenerative disorder characterised by the progressive degeneration of both upper and lower motor neurons, resulting in a multitude of motor symptoms, including muscle weakness, fasciculations, spasticity, dysphagia and, eventually, respiratory dysfunction [46]. FTLD is a pathological diagnosis that manifests clinically in the form of frontotemporal dementia (FTD), characterised by cognitive, behavioural and linguistic dysfunction. The link between these disorders is made clear by the fact that almost 50% of ALS patients show cognitive impairment of the type observed in FTD, and also that 15% of ALS cases meet the diagnostic criteria for FTD at the time ALS is diagnosed [47]. In addition, 15% of FTLD cases have clinically detectable motor symptoms [48]. Both disorders are characterised by the accumulation of pathological protein aggregates that contain a number of proteins, most notably TAR DNA‐binding protein 43 kDa (TDP‐43).
This study is aimed at elucidating the contribution of peroxisomal alterations to lipid metabolism in frontal cortex area 8 within the spectrum of TDP‐43 proteinopathies. FTLD‐TDP, manifested as sporadic (sFTLD‐TDP) or linked to mutations in various genes including expansions of the non‐coding region of C9ORF72 (c9FTLD), as well as sporadic ALS (sALS), are the most common TDP‐43 proteinopathies. To this end, we used transcriptomic and lipidomic methods to define the steady‐state levels of gene expression and lipid profiles. Our results show alterations in gene expression of some components of peroxisomes and related lipid pathways in frontal cortex (FC) area 8 in sALS, sFTLD‐TDP and c9FTLD.
MATERIALS AND METHODS
Human cases
Post‐mortem samples of fresh‐frozen FC area 8 were obtained from the Institute of Neuropathology HUB‐ICO‐IDIBELL Biobank and the Hospital Clinic‐IDIBAPS Biobank following the guidelines of Spanish legislation on this matter and approval of the local ethics committees and in accordance with criteria of sample quality [49, 50, 51]. The post‐mortem delay varied from 2 h 15 min to 18 h. This post‐mortem delay does not compromise the quality of the sample [49, 50, 51]. One hemisphere was immediately cut in coronal sections, 1 cm thick, and selected areas of the encephalon were rapidly dissected, frozen on metal plates over dry ice, placed in individual air‐tight plastic bags and stored at −80°C until the use for biochemical studies. The other hemisphere was fixed by immersion in 4% buffered formalin for 3 weeks for morphological studies.
The neuropathological study was carried out on 20 selected 4‐μm‐thick de‐waxed paraffin sections of representative regions of the brain. Sections were stained with haematoxylin and eosin, Klüver‐Barrera, or processed for immunohistochemistry with anti‐β‐amyloid, phospho‐tau (clone AT8), α‐synuclein, αB‐crystallin, TDP‐43, ubiquitin, p62, glial fibrillary acidic protein, CD68 and Iba1 antibodies [52]. Sporadic FTLD‐TDP (sFTLD‐TDP) cases were diagnosed following well‐established criteria: frontotemporal atrophy, loss of neurons and variable spongiosis in the upper cortical layers, astrocytic gliosis and presence of TDP‐43‐immunoreactive inclusions in the cytoplasm or in the nucleus of neurons, and in dendrites (NCIs, NIIs and DNs respectively), and were then categorised as type A, B or C.[53, 54] Cases with familial frontotemporal lobar degeneration linked to C9ORF72 expansion (henceforth referred to as c9FTLD for practical purposes), all of them carrying more than 30 intronic hexanucleotide repeats, were classified as type A or B. All these cases showed a sequential pattern II or III [55]. The frontal cortex of sporadic ALS (sALS) cases showed variable alterations; TDP‐43‐immunoreactive small dystrophic neurites and/or TDP‐43‐positive granules and/or small cytoplasmic globules in neurons were observed in 11 of 18 cases, but they were abundant in only three cases (cases 56, 57 and 58) (Table 1); spongiosis in the upper cortical layers was found in only one case (case 55). The whole series included 16 sFTLD‐TDP (71.6 ± 9.6 years; 11 men and 3 women), 19 c9FTLD (mean age 70 years; 10 men and 9 women), 15 sALS (mean age 54 years; 11 men and 4 women) and 17 control cases (64.7 ± 8.9 years; 11 men and 6 women), as summarised in Table 1. Although there are significant differences in the average age of the groups analysed, the age range of the study subjects is between 60 and 70 years. Previous studies on brain (and particularly in frontal cortex) lipid composition showed that lipids remain stable in adulthood; minimal changes appear in older ages than those analysed here [56, 57].
TABLE 1.
Summary of cases
| Case | Sex | Age | Diagnosis | PMD | RIN | TDP‐43 |
|---|---|---|---|---|---|---|
| 1 | M | 66 | Control | 18 h 00 min | 6.4 | – |
| 2 | M | 61 | Control | 03 h 40 min | 7.0 | – |
| 3 | M | 62 | Control | 05 h 45 min | 5.0 | – |
| 4 | M | 74 | Control | 06 h 40 min | 7.2 | – |
| 5 | M | 65 | Control | 05 h 15 min | 6.8 | – |
| 6 | F | 64 | Control | 02 h 15 min | 5.0 | – |
| 7 | M | 63 | Control | 08 h 05 min | 7.1 | – |
| 8 | F | 79 | Control | 03 h 35 min | 6.8 | – |
| 9 | F | 67 | Control | 05 h 20 min | 6.2 | – |
| 10 | M | 70 | Control | 03 h 45 min | 7.2 | – |
| 11 | M | 52 | Control | 04 h 40 min | 7.2 | – |
| 12 | F | 52 | Control | 05 h 45 min | 5.1 | – |
| 13 | F | 82 | Control | 07 h 35 min | 5.2 | – |
| 14 | F | 74 | Control | 02 h 45 min | 5.7 | – |
| 15 | M | 55 | Control | 05 h 40 min | 7.7 | – |
| 16 | M | 59 | Control | 07 h 05 min | 7.8 | – |
| 17 | M | 56 | Control | 03 h 50 min | 7.6 | – |
| 18 | M | 76 | sFTLD‐TDP | 05 h 00 min | 6.2 | A |
| 19 | F | 82 | sFTLD‐TDP | 03 h 40 min | 6.4 | A |
| 20 | M | 71 | sFTLD‐TDP | 04 h 00 min | 6.1 | A |
| 21 | F | 77 | sFTLD‐TDP | 16 h 00 min | 6.9 | C |
| 22 | M | 73 | sFTLD‐TDP | 05 h 00 min | 6.7 | C |
| 23 | M | 63 | sFTLD‐TDP | 09 h 30 min | 5.0 | A |
| 24 | F | 77 | sFTLD‐TDP | 07 h 39 min | 7.0 | A |
| 25 | M | 65 | sFTLD‐TDP | 13 h 00 min | 7.4 | A |
| 26 | F | 88 | sFTLD‐TDP | 06 h 30 min | 5.4 | A |
| 27 | M | 59 | sFTLD‐TDP | 08 h 00 min | 7.4 | A |
| 28 | M | 58 | sFTLD‐TDP | 04 h 00 min | 7.3 | A |
| 29 | M | 56 | sFTLD‐TDP | 08 h 00 min | 5.0 | A |
| 30 | F | 84 | sFTLD‐TDP | 06 h 00 min | 5.9 | B |
| 31 | M | 78 | sFTLD‐TDP | 07 h 15 min | 6.7 | C |
| 32 | M | 66 | sFTLD‐TDP | 05 h 15 min | 7.2 | A |
| 33 | M | 74 | sFTLD‐TDP | 15 h 00 min | 6.4 | C |
| 34 | M | 69 | c9FTLD | 11 h 30 min | 6.5 | A‐B |
| 35 | F | 69 | c9FTLD | 13 h 15 min | 5.4 | A‐B |
| 36 | M | 68 | c9FTLD | 02 h 30 min | 6.8 | A‐B |
| 37 | M | 61 | c9FTLD | 07 h 45 min | 6.9 | A‐B |
| 38 | M | 66 | c9FTLD | 15 h 15 min | 7.9 | A‐B |
| 39 | F | 55 | c9FTLD | 03 h 15 min | 8.7 | A‐B |
| 40 | M | 69 | c9FTLD | 05 h 00 min | 6.1 | A‐B |
| 41 | F | 75 | c9FTLD | 17 h 30 min | 7.5 | A‐B |
| 42 | F | 92 | c9FTLD | 09 h 15 min | 7.1 | A‐B |
| 43 | F | 58 | c9FTLD | 11 h 00 min | 8.4 | A‐B |
| 44 | F | 66 | c9FTLD | 11 h 30 min | 8.1 | A‐B |
| 45 | M | 73 | c9FTLD | 15 h 30 min | 6.2 | A‐B |
| 46 | F | 69 | c9FTLD | 12 h 30 min | 5.9 | A‐B |
| 47 | F | 57 | c9FTLD | 03 h 40 min | 7.2 | A‐B |
| 48 | M | 80 | c9FTLD | 12 h 00 min | 8.0 | A‐B |
| 49 | F | 57 | c9FTLD | 08 h 00 min | 6.9 | A‐B |
| 50 | M | 88 | c9FTLD | 05 h 00 min | 7.3 | A‐B |
| 51 | M | 69 | c9FTLD | 05 h 45 min | 7.1 | A‐B |
| 52 | M | 80 | c9FTLD | 08 h 30 min | 6.5 | A‐B |
| 53 | M | 70 | sALS | 03 h 00 min | 7.0 | – |
| 54 | F | 56 | sALS | 03 h 45 min | 7.7 | – |
| 55 | M | 59 | sALS | 03 h 15 min | 7.7 | – |
| 56 | F | 63 | sALS | 13 h 50 min | 8.2 | – |
| 57 | F | 59 | sALS | 14 h 15 min | 6.7 | – |
| 58 | M | 54 | sALS | 04 h 50 min | 7.8 | – |
| 59 | M | 76 | sALS | 12 h 40 min | 7.4 | – |
| 60 | M | 64 | sALS | 16 h 30 min | 7.3 | – |
| 61 | F | 57 | sALS | 04 h 00 min | 8.6 | – |
| 62 | F | 75 | sALS | 04 h 05 min | 6.8 | – |
| 63 | F | 57 | sALS | 10 h 00 min | 7.1 | – |
| 64 | M | 50 | sALS | 10 h 10 min | 5.9 | – |
| 65 | F | 59 | sALS | 02 h 30 min | 7.5 | – |
| 66 | M | 46 | sALS | 07 h 00 min | 8.0 | – |
| 67 | F | 69 | sALS | 17 h 00 min | 6.3 | – |
Sixty‐seven cases corresponding to 17 controls, 16 sFTLD‐TDP cases, 19 c9FTLD and 15 sALS were used for this study.
A, B, C, Classification of FTLD according to [53]; F, female; M, male; PMD, post‐mortem delay (hours, minutes); RIN, RNA integrity number; TDP‐43, TDP‐43 abnormal inclusions in neurons and dendrites in frontal cortex.
Patients with additional associated pathologies of the nervous system, excepting early stages of neurofibrillary tangle pathology in the entorhinal cortex and hippocampus, and those with the presence of mild small blood vessel disease, were excluded, as were those cases with infectious, inflammatory or autoimmune diseases. Clinically, patients with FTLD had suffered from variable cognitive deficits, reporting parkinsonism and progressive aphasia in some cases. sALS cases had no cognitive alterations. Age‐matched control cases had not suffered from neurologic or psychiatric disorders, or systemic diseases, and had no neuropathological lesions other than those permitted in included disease cases.
RNA extraction and RT‐qPCR validation
RNA from frozen FC area 8 was extracted following the instructions of the supplier (RNeasy Mini Kit, Qiagen® GmbH, Hilden, Germany). RNA integrity and 28S/18S ratios were determined with the Agilent Bioanalyzer (Agilent Technologies Inc, Santa Clara, CA, USA) to assess RNA quality, and the RNA concentration was evaluated using a NanoDrop™ Spectrophotometer (Thermo Fisher Scientific). Complementary DNA (cDNA) preparation used a High‐Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA, USA) following the protocol provided by the supplier. TaqMan RT‐qPCR assays were performed in duplicate for each gene on cDNA samples in 384‐well optical plates using an ABI Prism 7900 Sequence Detection system (Applied Biosystems, Life Technologies, Waltham, MA, USA). For each 10 μl TaqMan reaction, 2.25 μl cDNA was mixed with 0.25 μl 20× TaqMan Gene Expression Assays and 2.50 μl of 2× TaqMan Universal PCR Master Mix (Applied Biosystems). The identification numbers and names of TaqMan probes are shown in Table S1. The values for β‐glucuronidase (GUS‐β) were used as internal controls for normalisation purposes [58]. The parameters of the reactions were 50°C for 2 min, 95°C for 10 min and 40 cycles of 95°C for 15 s and 60°C for 1 min. Finally, Sequence Detection Software (SDS version 2.2.2, Applied Biosystems) was used to capture TaqMan PCR data. The double‐delta cycle threshold (ΔΔCT) method was utilised to analyse the data. The statistical study was performed using the T‐student test or ANOVA‐one way when necessary. The significance level was set at *p < 0.05, **p < 0.01 and ***p < 0.001 versus control group; # p < 0.05, ## p < 0.01 and ### p < 0.001 versus sALS; and $ p < 0.05, $$ p < 0.01 and $$$ p < 0.001 versus sFTLD‐TDP.
Fatty acid profiling
Briefly, samples were incubated for lipid extraction and FAs transesterification in 2 ml of 5% methanolic HCL at 75 °C for 90 min. FAs methyl esters were extracted by adding 2 ml of n‐pentane and 1 ml of saturated NaCl solution. Samples were separated and evaporated under N2 gas n‐pentane phase and finally dissolved in 80 µl of carbon disulphide. Gas chromatography (GC) analysis was then performed.
The GC method was used for separation with a DBWAX capillary column (30 m × 0.25 mm ×0.20 μm) in a GC System 7890 A with a Series Injector 7683B and an FID detector (Agilent Technologies, Barcelona, Spain). The temperature of the injector was 220 °C using the splitless mode. A constant rate (1.8 ml/min) of helium (99.99%) was maintained. The column temperature was held at 145°C for 5 min; subsequently, the column temperature was increased by 2°C/min to 245°C for 50 min, and held at 245°C for 10 min, with a post‐run of 250°C for 10 min as previously described [59, 60, 61]. Based on FA composition, different indexes were calculated, and elongase and desaturase activity was estimated from specific product/substrate ratios [61, 62].
Non‐targeted lipidomic analysis
A previously validated method was used for lipid extraction [63]. Briefly, 5 μl of miliQ water and 20 μl of methanol were added to 10 μl of homogenised tissue. Samples were then shaken vigorously for 2 min. Following this, methyl tert‐butyl ether (MTBE) containing isotopically labelled lipid standards was added. Samples were then immersed in a water bath (ATU Ultrasonidos, Valencia, Spain) with an ultrasound frequency of 40 kHz and power of 100 W, at 10°C for 30 min. After this, 25 μl of miliQ water was added to the mixture, which was centrifuged at 300 rpm at 4°C for 10 min to separate the organic phase. Finally, the upper phase was collected and stored for mass‐spectrometry analysis. A pool (20 µl of each sample) of all lipid extracts was prepared and used as quality control [64].
Lipid extracts were analysed by LC‐MS according to the method described [59]. An Agilent UPLC 1290 system coupled to an ESI‐Q‐TOF MS/MS 6545 (Agilent Technologies, Barcelona, Spain) was used. Two runs were performed to collect positive and negative electrospray ionised lipid species. Data pre‐processing was done as published before [65, 66, 67]. Finally, identities were confirmed by searching experimental MS/MS spectra against in silico libraries, using HMDB and LipidMatch, an R‐based tool for lipid identification [68, 69]. Multivariate and univariate statistics were calculated using Metaboanalyst [70].
RESULTS
Peroxisome‐related genes
Peroxisome biogenesis
When compared with controls, only PPARG expression was significantly increased in sALS (p < 0.01) and significantly decreased in c9FTLD cases (p = 0.018), whereas PPARGC1A was significantly increased in c9FTLD (p = 0.033). However, differences were also identified when comparing expression levels among the three disease groups. Thus, PEX14 was significantly increased in c9FTLD when compared with sALS (p < 0.001) and sFTLD‐TDP (p < 0.001); PPARD was decreased in sFTLD‐TDP when compared with sALS (p < 0.001); PPARG was decreased in sFTLD‐TDP and c9FTLD when compared with sALS (p = 0.006 and p < 0.001 respectively); PPARGC1A was down‐regulated in sFLTD‐TDP when compared with sALS (p < 0.005) and up‐regulated in c9FTLD when compared with sFTLD‐TDP (p < 0.001); and finally, DNM1L mRNA expression was significantly increased in c9FTLD when compared with sFTLD‐TDP (p < 0.001). No differences were detected for PPARA. See Figure 1A.
FIGURE 1.
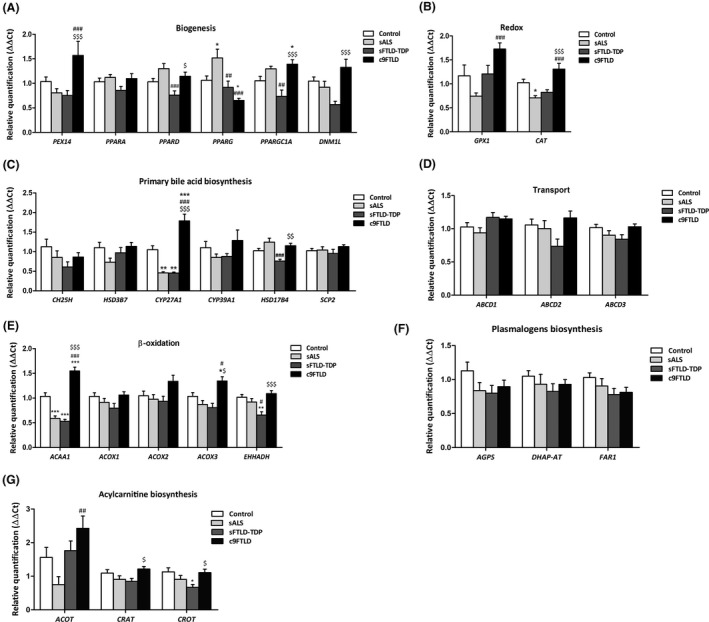
mRNA expression levels of peroxisome‐related genes in frontal cortex area 8 in controls, sALS, sFTLD‐TDP and c9FTLD cases assessed with TaqMan RT‐qPCR assays. (A) Genes implicated in peroxisome biogenesis. (B) Genes coding for redox mechanisms. (C) Genes involved in peroxisomal primary bile acid biosynthesis. (D) Genes related with peroxisome substrate transport. (E) Genes involved in peroxisomal β‐oxidation. Genes linked to plasmalogens biosynthesis (F) and acylcarnitine biosynthesis (G). Data are expressed as the mean values±SEM. The significance level was set at *p < 0.05, **p < 0.01 and ***p < 0.001 versus control group; # p < 0.05, ## p < 0.01 and ### p < 0.001 versus sALS; and $ p < 0.05, $$ p < 0.01 and $$$ p < 0.001 versus sFTLD‐TDP
Peroxisome redox mechanisms
GPX1 mRNA levels were increased in c9FTLD when compared with sALS (p < 0.001), whereas CAT transcript levels were down‐regulated in sALS when compared with controls (p = 0.011), and up‐regulated in c9FTLD when compared with sALS and sFTLD‐TDP (p < 0.001 and p < 0.001 respectively) (Figure 1B).
Primary bile acid metabolism
CYP27A1 mRNA levels were down‐regulated in sALS and sFTLD‐TDP when compared with controls (p = 0.002 and p = 0.006 respectively). In contrast, CYP27A1 gene expression was up‐regulated in c9FTLD when compared with controls, sALS and FTLD‐TDP (p < 0.001). Reduced HSD17B4 expression was limited to sFTLD‐TDP when compared with sALS and c9FTLD (p = 0.001, and p = 0.006 respectively). No differences in the expression of CH25H, HSD3B7, CYP39A1 or SCP2 were found between controls and disease cases, nor among the three pathological groups (Figure 1C).
Transport
No modifications in the expression of genes coding for β‐oxidation ATP‐binding cassette (ABC) transporters ABCD1, ABCD2 and ABCD3 were observed in sALS, sFTLD‐TDP or c9FTLD when compared with controls. Nor were significant differences observed among the three disease groups (Figure 1D).
β‐oxidation components
ACAA1 gene expression was decreased in sALS and sFTLD‐TDP when compared with controls (p < 0.001 and p < 0.001 respectively), but it was increased in c9FTLD, not only when compared with controls (p < 0.001), but also with respect to sALS (p < 0.001) and sFTLD‐TDP cases (p < 0.001). Following a similar trend, levels of ACOX3 were increased in c9FTLD when compared with controls (p < 0.001), sALS (p < 0.001) and sFTLD‐TDP (p < 0.001). EHHADH transcript levels were down‐regulated in sFTLD‐TDP when compared with controls and sALS (p = 0.004 and p = 0.049 respectively), and up‐regulated in c9FTLD when compared with sFTLD‐TDP (p < 0.001). No differences in the expression of ACOX1 and ACOX2 were found between controls and disease cases, nor among the three pathological groups (Figure 1E).
Plasmalogen biosynthesis
Expression levels of genes coding for components of the plasmalogen biosynthesis pathway, AGPS, DHAP‐AT and FAR1, were evaluated, but no differences in the expression of these genes were found between controls and disease cases, or among the three pathological groups (Figure 1F).
Acylcarnitine biosynthesis
Acylcarnitine biosynthesis components revealed few differences in the expression of ACOT, CRAT and CROT transcripts. ACOT gene expression was increased in c9FTLD when compared with sALS (p = 0.003), whereas CRAT and CROT were significantly increased in c9FTLD when compared with sFTLD cases (p = 0.034 and p = 0.029 respectively). Regarding control cases, only CROT mRNA expression levels were significantly decreased in sFTLD cases when compared to controls (p = 0.021) (Figure 1G).
Gene expression linked to fatty acid metabolism
The expression of fifteen genes was assessed. No modifications in the mRNA expression levels of FASN, ELOVL2, ELOVL5, SCD1, SCD5, ACSL1 or ACSL5 were identified in the three diseases when compared with controls, nor among the pathological groups. In sALS, only ACACA and ACSL6 were significantly increased, and ELOVL7 significantly decreased when compared with controls (p = 0.026, p = 0.04 and p = 0.05 respectively). Regarding sFTLD‐TDP, ELOVL6, ELOVL7, ACSL3 and ACSL4 were significantly decreased when compared with controls (p = 0.04, p = 0.028, p = 0.031 and p = 0.028 respectively). In addition, ACACA, ELOVL4, ELOVL6 and ACSL6 were significantly decreased in sFTLD‐TDP when compared with sALS (p < 0.001, p = 0.009, p = 0.032 and p = 0.003 respectively). Finally, expression levels of ELOVL1 and ELOVL7 mRNA were significantly decreased in c9FTLD when compared with controls (p = 0.01, and p = 0.041 respectively), but ACACA, ELOVL4, ELOVL6, ACSL3, ACSL4 and ACSL6 significantly increased in c9FTLD when compared with sFTLD‐TDP (p = 0.001 p = 0.002, p = 0.018, p = 0.002, p = 0.018 and p = 0.038 respectively) (Figure 2).
FIGURE 2.
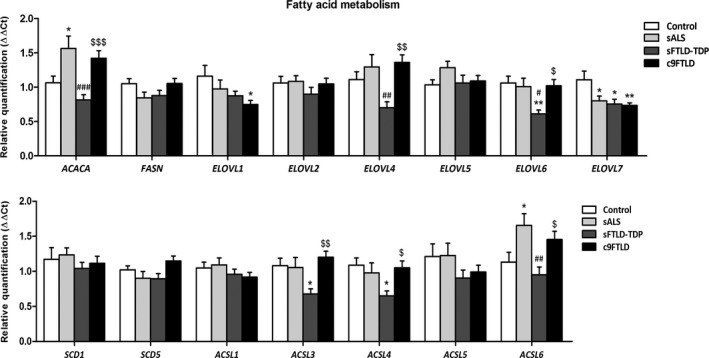
mRNA expression levels of genes linked to fatty acid metabolism in frontal cortex area 8 in controls, sALS, sFTLD‐TDP and c9FTLD cases assessed with TaqMan RT‐qPCR assays. Data are expressed as the mean values±SEM. The significance level was set at *p < 0.05, **p < 0.01 and ***p < 0.001 versus control group; # p < 0.05, ## p < 0.01 and ### p < 0.001 versus sALS; and $ p < 0.05, $$ p < 0.01 and $$$ p < 0.001 versus sFTLD‐TDP
Fatty acid profiling
Since the biosynthesis of highly unsaturated fatty acids is dependent on peroxisomal beta‐oxidation activity, fatty acid composition of total lipids from frontal cortex area 8 was analysed (Table 2). This analysis showed no significant changes in fatty acid profiles among the FTLD‐TDP/ALS spectrum pathologies. Notably, the content of 22:5n6 and 22:6n3, highly PUFAs resulting from peroxisomal beta‐oxidation, was sustained among the FTLD‐TDP/ALS spectrum at equivalent levels as observed in the control group. As a result of this, the total number of double bonds and the global susceptibility to peroxidation, estimated as the global fatty acid indexes DBI and PI, as well as other indexes such as average chain length, SFA and UFA content, were not modified by the pathological condition, nor were desaturase and elongase activities calculated from fatty acid profiles. Only minor changes were observed for total PUFAn6 content (p = 0.039) and Elovl 2 (n3) estimated activity. Specifically, statistically significant differences were found in total PUFAn‐6 in sFTLD‐TDP when compared with control and c9FTLD groups, and in Elovl2(n‐3) between the control and pathogenic groups.
TABLE 2.
Fatty acid compositional profiles of total lipids from frontal cortex area 8 in controls, sALS, sFTLD‐TDP and c9FTLD cases assessed with gas chromatography
| Control | sALS | sFTLD‐TDP | c9FTLD | p | |
|---|---|---|---|---|---|
| Fatty acids | |||||
| 14:0 | 1.44 ± 0.09 | 1.47 ± 0.17 | 1.51 ± 0.08 | 1.54 ± 0.12 | 0.716 |
| 16:0 | 24.73 ± 1.13 | 24.34 ± 2.06 | 24.91 ± 1.55 | 24.04 ± 1.51 | 0.946 |
| 16:1n7 | 1.61 ± 0.15 | 1.87 ± 0.39 | 1.92 ± 0.22 | 2.02 ± 0.23 | 0.643 |
| 18:0 | 24.8 ± 0.53 | 28.28 ± 2.62 | 24.3 ± 0.54 | 23.52 ± 0.55 | 0.05 |
| 18:1n9 | 21.77 ± 1.41 | 19.8 ± 3.05 | 23.07 ± 1.84 | 23.89 ± 2 | 0.586 |
| 18:1n7 | 4.58 ± 0.21 | 3.63 ± 0.63 | 4.94 ± 0.19 | 4.72 ± 0.19 | 0.105 |
| 18:2n6 | 0.77 ± 0.12 | 0.7 ± 0.13 | 0.61 ± 0.05 | 0.59 ± 0.02 | 0.475 |
| 18:3n3 | 0.07 ± 0.01 | 0.08 ± 0.01 | 0.07 ± 0.01 | 0.08 ± 0.01 | 0.962 |
| 18:4n3 | 0.94 ± 0.06 | 1.18 ± 0.25 | 1.06 ± 0.09 | 0.88 ± 0.08 | 0.512 |
| 20:0 | 0.24 ± 0.00 | 0.3 ± 0.03 | 0.25 ± 0 | 0.24 ± 0.01 | 0.073 |
| 20:1n9 | 1.32 ± 0.17 | 1.93 ± 0.66 | 1.41 ± 0.25 | 1.35 ± 0.23 | 0.993 |
| 20:2n6 | 0.45 ± 0.04 | 0.54 ± 0.19 | 0.46 ± 0.08 | 0.42 ± 0.06 | 0.954 |
| 20:3n3 | 0.69 ± 0.03 | 0.67 ± 0.04 | 0.61 ± 0.02 | 0.63 ± 0.08 | 0.282 |
| 20:4n6 | 4.89 ± 0.21 | 5.02 ± 0.51 | 4.71 ± 0.32 | 4.49 ± 0.33 | 0.764 |
| 20:3n6 | 0.27 ± 0.05 | 0.17 ± 0.02 | 0.16 ± 0.03 | 0.16 ± 0.04 | 0.161 |
| 22:0 | 0.03 ± 0.001 | 0.03 ± 0.01 | 0.03 ± 0.001 | 0.03 ± 0.001 | 0.113 |
| 20:5n3 | 0.61 ± 0.13 | 0.59 ± 0.1 | 0.51 ± 0.04 | 0.42 ± 0.03 | 0.278 |
| 22:1 | 0.06 ± 0.01 | 0.08 ± 0.03 | 0.07 ± 0.01 | 0.07 ± 0.01 | 0.964 |
| 22:4n6 | 2.83 ± 0.19 | 2.58 ± 0.21 | 2.53 ± 0.16 | 2.32 ± 0.06 | 0.225 |
| 22:5n6 | 0.52 ± 0.06 | 0.39 ± 0.06 | 0.49 ± 0.09 | 0.40 ± 0.08 | 0.578 |
| 22:5n3 | 0.11 ± 0.01 | 0.14 ± 0.03 | 0.11 ± 0.02 | 0.12 ± 0.02 | 0.99 |
| 24:0 | 0.48 ± 0.07 | 0.51 ± 0.1 | 0.42 ± 0.04 | 0.48 ± 0.08 | 0.985 |
| 22:6n3 | 4.17 ± 0.47 | 3.22 ± 0.64 | 3.6 ± 0.46 | 3.33 ± 0.52 | 0.704 |
| 24:1n7 | 1.1 ± 0.22 | 1.41 ± 0.5 | 1.18 ± 0.25 | 1.29 ± 0.3 | 0.992 |
| 24:5n3 | 1.3 ± 0.22 | 0.82 ± 0.16 | 0.81 ± 0.13 | 2.74 ± 1.4 | 0.329 |
| 24:6n3 | 0.21 ± 0.05 | 0.26 ± 0.08 | 0.25 ± 0.06 | 0.24 ± 0.05 | 0.978 |
| Fatty acid indexes | |||||
| SFA | 51.71 ± 1.48 | 54.92 ± 2.96 | 51.44 ± 1.95 | 49.85 ± 1.89 | 0.533 |
| UFA | 48.29 ± 1.48 | 45.08 ± 2.96 | 48.56 ± 1.95 | 50.15 ± 1.89 | 0.513 |
| PUFA | 17.83 ± 0.65 | 16.36 ± 1.25 | 15.97 ± 0.79 | 16.81 ± 1.66 | 0.892 |
| MUFA | 30.46 ± 2.03 | 28.72 ± 3.71 | 32.59 ± 2.72 | 33.34 ± 2.88 | 0.441 |
| PUFAn3 | 8.1 ± 0.54 | 6.97 ± 0.94 | 7.01 ± 0.64 | 8.43 ± 1.55 | 0.092 |
| PUFAn6 | 9.73 ± 0.33 | 9.39 ± 0.51 | 8.96 ± 0.26 | 8.38 ± 0.35 | 0.039 b , d |
| ACL | 18.13 ± 0.02 | 18.06 ± 0.03 | 18.02 ± 0.01 | 18.12 ± 0.1 | 0.964 |
| DBI | 109.63 ± 1.72 | 99.69 ± 4.97 | 103.11 ± 1.66 | 108.54 ± 6.55 | 0.691 |
| PI | 89.01 ± 4.24 | 78.44 ± 7.69 | 79.03 ± 5.25 | 84.98 ± 10.49 | 0.809 |
| Estimated desaturase and elongase activities | |||||
| Δ9(n−7) | 0.07 ± 0.01 | 0.09 ± 0.03 | 0.08 ± 0.01 | 0.09 ± 0.02 | 0.506 |
| Δ9(n−9) | 0.89 ± 0.08 | 0.77 ± 0.15 | 0.96 ± 0.1 | 1.03 ± 0.11 | 0.433 |
| Δ5(n−6) | 22.08 ± 4.98 | 30.13 ± 2.64 | 32.52 ± 3.9 | 42.36 ± 12.89 | 0.768 |
| Δ6(n−3) | 15.14 ± 2.36 | 18.94 ± 5.96 | 16.19 ± 2.51 | 12.78 ± 2.01 | 0.302 |
| Δ6(n−3) | 0.21 ± 0.07 | 0.52 ± 0.24 | 0.42 ± 0.14 | 0.25 ± 0.09 | 0.444 |
| Elovl3(n−9) | 0.06 ± 0.01 | 0.14 ± 0.09 | 0.06 ± 0.01 | 0.05 ± 0.01 | 0.928 |
| Elovl6 | 1.01 ± 0.03 | 1.23 ± 0.2 | 0.99 ± 0.04 | 1 ± 0.05 | 0.997 |
| Elovl1‐3‐7a | 0.01 ± 0 | 0.01 ± 0 | 0.01 ± 0 | 0.01 ± 0 | 0.346 |
| Elovl1‐3‐7b | 0.1 ± 0.01 | 0.1 ± 0.02 | 0.14 ± 0.01 | 0.11 ± 0.01 | 0.164 |
| Elovl1‐3‐7c | 19.55 ± 3.24 | 29.26 ± 11.07 | 13.45 ± 2.66 | 21.54 ± 5.25 | 0.105 |
| Elovl5(n−6) | 0.66 ± 0.12 | 1.12 ± 0.5 | 0.79 ± 0.16 | 0.73 ± 0.11 | 0.672 |
| Elovl2‐5 (n−6) | 0.59 ± 0.05 | 0.56 ± 0.09 | 0.56 ± 0.07 | 0.54 ± 0.05 | 0.42 |
| Elovl 2–5(n−3) | 0.2 ± 0.03 | 0.23 ± 0.04 | 0.21 ± 0.03 | 0.29 ± 0.06 | 1 |
| Elovl 2(n−3) | 12.17 ± 2.04 | 6.38 ± 0.5 | 8.24 ± 1.93 | 36.21 ± 20.44 | 0.028 a , b , c |
| Peroxisome β‐oxidation | 1.54 ± 0.22 | 1.36 ± 0.29 | 1.52 ± 0.27 | 1.47 ± 0.24 | 0.691 |
Values are reported as mean±SEM from 6 to 8 cases and are expressed as mol%. Inter‐group differences were measured by pairwise Kruskal–Wallis test applying a Bonferroni correction. Minimum significance level is set at p < 0.05. Estimated desaturase and elongase activities from specific product/substrate ratios: Δ9(n−7), ratio 16:1n−9/16:0; Δ9(n−9), ratio 18:1n−9/18:0; Δ5(n−6), ratio 20:4n−6/20:3n−6; Δ6(n−3), ratio 18:4n−3/18:3n−3; Δ6(n−3), ratio 24:6n−3/24:5n−3; Elovl3(n−9), ratio 20:1n−9/18:1n−9; Elovl6, ratio 18:0/16:0; Elovl1‐3‐7a, ratio 20:0/18:0; Elovl1‐3‐7b, ratio 22:0/20:0; Elovl1‐3‐7c, ratio 24:0/22:0; Elovl5(n−6), ratio 20:2n−6/18:2n−6; Elovl2‐5 (n−6), ratio 22:4n−6/20:4n−6; Elovl 2–5(n−3), 22:5n−3/20:5n−3; Elovl 2(n−3), 24:5n−3/22:5n−3; Peroxisome β‐oxidation, ratio 22:6n−3/24:6n−3.
Control versus sALS.
Control versus sFTLD‐TDP.
Control versus c9FTLD.
sFTLD‐TDP versus c9FTLD.
Lipidomic profiling
In order to gain an overview of whole lipidome, an untargeted lipidomic approach was applied. Baseline correction, peak picking and peak alignment were performed on acquired data, resulting in a total of 7951 molecules from both ionisation modes (negative and positive). After quality control assessment, filtering and signal correction, 1119 features remained, which were log‐transformed and auto‐scaled (mean centering/standard deviation) and used for multivariate and univariate statistical analysis.
A lipidomic pattern is associated with ALS‐FTLD‐TDP43 proteinopathy spectrum
To investigate whether there was a common pattern for TDP‐43 proteinopathies, an untargeted lipidomic analysis was performed in tissue samples from frontal cortex region 8 with the three neurological disorders grouped together. Unsupervised methods such as Principal Component Analysis (PCA) and Hierarchical Clustering visualised as a heatmap were used to find patterns in the samples. A PCA analysis was performed using the whole detected lipidome; no differences were found between the diseased (DIS) group and healthy controls (CTL) (Figure 3A ). However, when the 25 lipid species with the lowest p ‐values were represented using hierarchical clustering analyses, as shown in a heatmap (Figure 3B), a clear separation between groups was observed revealing a specific shared trend in patients with neurodegenerative diseases within the ALS‐FTLD‐TDP43 proteinopathy spectrum. Finally, the Wilcoxon test on all acquired data was performed to determine whether there were any significant lipid feature differences between healthy and diseased groups (p < 0.05). Dunn's test was used to correct for multiple comparisons. The statistical test resulted in 63 differential molecules with p < 0.05 (Table 3), of which 30 were identified based on exact mass, retention time and/or MSMS spectrum.
FIGURE 3.
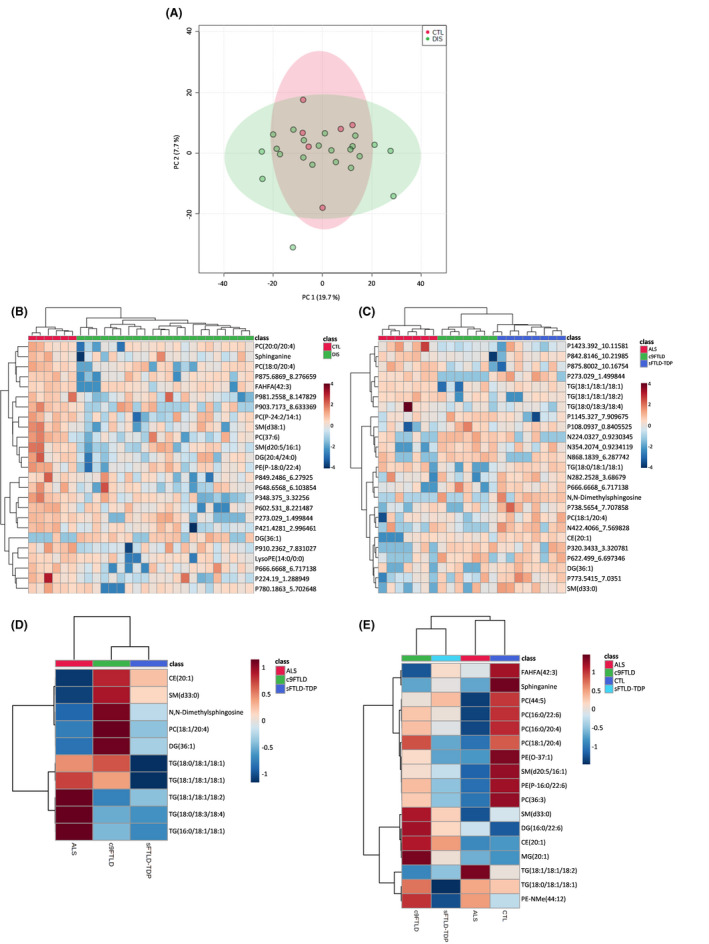
Multivariate statistics and learning methods reveal a specific lipidome shared by TDP‐43 proteinopathies. (A) Principal component analysis (PCA) 2D plot of samples lipidome. (B–C) Heatmap representation of hierarchical clustering of individual samples according to the top 25 statistically significantly different lipid species between (B) neurological disorders versus controls and (C) among groups of TDP‐43 proteinopathies. (D–E) Heatmap representation of hierarchical clustering of groups according to the identified significantly different lipid species (D) among groups of TDP‐43 proteinopathies and (E) between neurological disorders and controls
TABLE 3.
Lipidomic features associated with ALS‐FTLD‐TDP43 proteinopathy spectrum
| Lipid category | Lipidomic feature | Ionisation | mz value | Retention time | p value | Regulation |
|---|---|---|---|---|---|---|
| Fatty acyls | AcCar(8:1DC) a | Positive | 315.118 | 0.9 | 0.031093 | Down |
| FAHFA(42:3) b | Negative | 643.5684 | 9.1 | 0.012598 | Down | |
| FAHFA(5:0/22:3) a | Negative | 433.3377 | 5.4 | 0.033121 | Down | |
| Glycerolipids | DG(20:4/24:0) a | Positive | 746.711 | 7.2 | 0.019979 | Down |
| DG(36:1) c | Positive | 640.5837 | 7.5 | 0.018745 | Up | |
| DG(36:4) c | Positive | 627.5308 | 7.3 | 0.031093 | Down | |
| DG(40:6) c | Positive | 651.5308 | 6.4 | 0.046311 | Down | |
| Glycerophospholipids | PC(16:0/22:6) a | Positive | 806.5692 | 6.8 | 0.033121 | Down |
| PC(16:1/18:2) a | Positive | 756.5511 | 6.7 | 0.021125 | Down | |
| PC(18:0/20:4) a | Positive | 810.5987 | 7.3 | 0.012477 | Down | |
| PC(20:0/20:4) a | Positive | 838.6325 | 7.9 | 0.014856 | Down | |
| PC(37:6) b | Positive | 784.5814 | 7.2 | 0.0097892 | Down | |
| PC(42:7) b | Positive | 860.6127 | 7.3 | 0.046713 | Down | |
| PC(44:5) b | Positive | 874.6672 | 8 | 0.021786 | Down | |
| PC(P‐18:1/22:5) a | Positive | 818.6436 | 7.8 | 0.020481 | Down | |
| PC(P‐24:2/14:1) a | Positive | 796.5834 | 7.7 | 0.0052875 | Down | |
| LysoPE(14:0/0:0) a | Negative | 425.2602 | 0.9 | 0.010806 | Down | |
| PE(18:0/20:4) a | Positive | 768.5532 | 7.3 | 0.020132 | Down | |
| PE(18:0/22:4) a | Positive | 796.577 | 7.2 | 0.04434 | Down | |
| PE(18:0/22:6) a | Positive | 792.5538 | 7.2 | 0.044758 | Down | |
| PE(O‐37:1) b | Positive | 746.5681 | 7.7 | 0.0080321 | Down | |
| PE(P‐16:0/22:6) a | Positive | 748.5247 | 7 | 0.024038 | Down | |
| PE(P‐18:0/22:4) a | Positive | 780.5886 | 7.9 | 0.017083 | Down | |
| PG(14:1/14:1) a | Positive | 663.4529 | 7.8 | 0.033121 | Down | |
| Sphingolipids | Cer(d18:1/17:0) a | Positive | 548.4996 | 8.3 | 0.019979 | Down |
| SM(d20:5/16:1) a | Positive | 721.5057 | 7.8 | 0.01427 | Down | |
| SM(d38:1) b | Positive | 759.6368 | 7.9 | 0.0077921 | Down | |
| Sphinganine c | Positive | 284.2891 | 4.4 | 0.017319 | Down | |
| Unknown | 112.0521_0.8908437 | Positive | 113.0521 | 0.9 | 0.029833 | Down |
| 1145.327_7.909675 | Positive | 1146.327 | 7.9 | 0.024038 | Down | |
| 1168.27_8.160657 | Positive | 1169.27 | 8.2 | 0.020311 | Down | |
| 1387.036_0.9344499 | Positive | 1388.036 | 0.9 | 0.033121 | Down | |
| 1445.068_0.9473143 | Positive | 1446.068 | 0.9 | 0.035663 | Down | |
| 224.19_1.288949 | Positive | 225.19 | 1.3 | 0.00089717 | Down | |
| 266.152_0.9234737 | Negative | 265.152 | 0.9 | 0.035739 | Down | |
| 268.1307_0.840175 | Positive | 269.1307 | 0.8 | 0.024038 | Down | |
| 273.029_1.499844 | Positive | 274.029 | 1.5 | 0.0073599 | Down | |
| 278.1875_0.9220529 | Negative | 277.1875 | 0.9 | 0.046734 | Down | |
| 311.3201_5.293459 | Positive | 312.3201 | 5.3 | 0.035663 | Down | |
| 322.1781_0.9135807 | Positive | 323.1781 | 0.9 | 0.045145 | Down | |
| 348.375_3.32256 | Positive | 349.375 | 3.3 | 0.0020598 | Down | |
| 377.316_1.459708 | Positive | 378.316 | 1.5 | 0.041782 | Down | |
| 421.4281_2.996461 | Positive | 422.4281 | 3 | 0.01427 | Down | |
| 488.4371_6.214781 | Positive | 489.4371 | 6.2 | 0.020311 | Down | |
| 598.5024_8.868424 | Positive | 599.5024 | 8.9 | 0.019663 | Down | |
| 602.531_8.221487 | Positive | 603.531 | 8.2 | 0.0032711 | Down | |
| 604.5456_7.708292 | Positive | 605.5456 | 7.7 | 0.033121 | Down | |
| 608.4662_2.848297 | Positive | 609.4662 | 2.8 | 0.020311 | Down | |
| 610.5369_7.541813 | Positive | 611.5369 | 7.5 | 0.034782 | Down | |
| 648.6568_6.103854 | Positive | 649.6568 | 6.1 | 0.01427 | Down | |
| 650.5316_7.211145 | Positive | 651.5316 | 7.2 | 0.033121 | Down | |
| 666.6668_6.717138 | Positive | 667.6668 | 6.7 | 0.014815 | Down | |
| 683.1998_8.170667 | Positive | 684.1998 | 8.2 | 0.044758 | Down | |
| 703.5196_6.254575 | Positive | 704.5196 | 6.3 | 0.033121 | Down | |
| 761.5996_8.059616 | Positive | 762.5996 | 8.1 | 0.020311 | Down | |
| 780.1863_5.702648 | Positive | 781.1863 | 5.7 | 0.0091894 | Down | |
| 849.2486_6.27925 | Positive | 850.2486 | 6.3 | 0.011865 | Down | |
| 856.5926_2.848731 | Positive | 857.5926 | 2.8 | 0.020311 | Down | |
| 875.6869_8.276659 | Positive | 876.6869 | 8.3 | 0.010806 | Down | |
| 891.6605_8.298165 | Positive | 892.6605 | 8.3 | 0.046253 | Down | |
| 903.7173_8.633369 | Positive | 904.7173 | 8.6 | 0.018183 | Down | |
| 910.2362_7.831027 | Positive | 911.2362 | 7.8 | 0.0052875 | Down | |
| 981.2558_8.147829 | Positive | 982.2558 | 8.1 | 0.01427 | Down |
Significant molecular features after Wilcoxon rank‐sum test with p value <0.05 are shown as LC‐ESI Ionisation mode (P for positive and N for negative) followed by neutral mass and retention time. Potential identities based on MS1 and MS2 data for each feature are found lipidomic feature. Unidentified compounds are labelled as neutral mass and retention time. Ether phospholipids are highlighted in bold.
Abbreviations: AcCar, acylcarnitine; DG, diacylglycerol; FAHFA, fatty acid ester of hydroxyl fatty acid; FDR, false discovery rate; mz value, mass‐to‐charge ratio; PC, phosphocholine; PE, phophoethanolamine; SM, sphingomyelin.
Represents confirmation by data‐dependent MS2.
Represents confirmation by data‐dependent MS2 by class.
Represents confirmation by MS1 exact mass and retention time.
Most of the identified lipids were glycerophospholipids, but a number of glycerolipids and sphingolipids were also found. Also, an acylcarnitine and two fatty acid esters of hydroxyl fatty acids were identified. Noteworthy were the phosphocholines (PC) because many of them, seven to be specific, were significantly different, with four identified at the compound level. Two of them contained 20:4 n6 (arachidonic acid, AA), one contained 22:6 n3 (docosahexaenoic acid, DHA), and one contained 18:2n6 (linoleic acid, LA). AA and DHA were also found in two of the three significantly changed phosphoethanolamines (PE). Notably, we found five ether lipids down‐regulated in the diseased samples, two of them plasmenyl phosphocholines and three plasmenyl phosphoethanolamines, again containing AA and DHA. Two sphingomyelins, one ceramide and sphinganine were identified as ALS‐FTLD‐TDP43 proteinopathy spectrum‐associated molecular features. Among the identified compounds, no bile acid was detected as different.
Differences between neurological diseases within the ALS‐FTLD‐TDP43 proteinopathy spectrum
To determine whether there was a real difference between the distinct TDP43 proteinopathy phenotypes characterised as sALS, sFTLD‐TDP and c9FTLD, the three diseases were compared with each other. Frontal cortex region 8 samples were used to uncover characteristic lipidomic trends and features for each disorder. The heatmap representing the hierarchical clustering of the individual samples (Figure 3D) showed perfectly arranged samples in disease groups when the top differential metabolites obtained with the Kruskal–Wallis test were used. Interestingly, FTLD‐TDP patients (both sporadic and c9) clustered together, indicating that these groups are more similar to each other than they are to the ALS group.
The Kruskal–Wallis test revealed changes in glycerolipids, glycerophospholipids, sphingolipids and sterol lipids listed in Table 4. Most of the compounds identified in this analysis (Figure 3E) of frontal cortex region 8 were TGs and most contained 18:1 oleic acid; these TGs were higher in the ALS group and lower in the sFTLD‐TDP group. Interestingly, CE (20:1) was increased in c9FTLD compared with the other groups.
TABLE 4.
Lipidomic features associated with different neurological diseases within the ALS‐FTLD‐TDP43 proteinopathy spectrum
| Lipid category | Lipidomic feature | Ionisation | mz value | Retention time | chi squared | p value | post hoc |
|---|---|---|---|---|---|---|---|
| Glycerolipids | DG(36:1) c | Positive | 640.5837 | 7.5 | 6.111 | 0.047099 | sALS ‐ c9FTLD, sFTLD‐TDP ‐ c9FTLD |
| TG(16:0/18:1/18:1) a | Positive | 876.8002 | 10.2 | 6.0758 | 0.047935 | sALS ‐ sFTLD‐TDP, sALS ‐ c9FTLD | |
| TG(18:0/18:1/18:1) a | Positive | 904.8296 | 10.3 | 8.8888 | 0.011744 | sALS ‐ sFTLD‐TDP, sFTLD‐TDP ‐ c9FTLD | |
| TG(18:0/18:3/18:4) a | Positive | 894.7523 | 9.6 | 6.2676 | 0.043551 | sALS ‐ sFTLD‐TDP, sALS ‐ c9FTLD | |
| TG(18:1/18:1/18:1) a | Positive | 907.7709 | 10.2 | 7.1797 | 0.027603 | sALS ‐ sFTLD‐TDP | |
| TG(18:1/18:1/18:2) a | Positive | 900.8005 | 10 | 8.618 | 0.013447 | sALS ‐ sFTLD‐TDP, sALS ‐ c9FTLD | |
| Glycerophospholipids | PC(18:1/20:4) a | Positive | 808.5845 | 7 | 7.763 | 0.02062 | sALS ‐ c9FTLD, sFTLD‐TDP ‐ c9FTLD |
| Sphingolipids | N,N‐Dimethylsphingosine c | Positive | 328.3157 | 3.8 | 6.7079 | 0.034946 | sALS ‐ c9FTLD, sFTLD‐TDP ‐ c9FTLD |
| SM(d33:0) b | Positive | 689.555 | 7.2 | 8.0711 | 0.017676 | sALS ‐ c9FTLD | |
| Sterol lipids | CE(20:1) a | Positive | 696.6463 | 8.5 | 9.5299 | 0.0085233 | sALS ‐ c9FTLD, sFTLD‐TDP ‐ c9FTLD |
| Unknown | 108.0937_0.8405525 | Positive | 109.0937 | 0.8 | 6.0373 | 0.048868 | sALS ‐ sFTLD‐TDP, sALS ‐ c9FTLD |
| 1145.327_7.909675 | Positive | 1146.327 | 7.9 | 8.0582 | 0.017791 | sALS ‐ sFTLD‐TDP, sFTLD‐TDP ‐ c9FTLD | |
| 1423.392_10.11581 | Positive | 1424.392 | 10.1 | 10.714 | 0.0047144 | sALS ‐ sFTLD‐TDP, sALS ‐ c9FTLD | |
| 224.0327_0.9230345 | Negative | 223.0327 | 0.9 | 11.061 | 0.0039639 | sALS ‐ sFTLD‐TDP, sFTLD‐TDP ‐ c9FTLD | |
| 273.029_1.499844 | Positive | 274.029 | 1.5 | 10.672 | 0.0048154 | sALS ‐ sFTLD‐TDP, sFTLD‐TDP ‐ c9FTLD | |
| 282.2528_3.68679 | Negative | 281.2528 | 3.7 | 5.9962 | 0.049882 | sALS ‐ c9FTLD, sFTLD‐TDP ‐ c9FTLD | |
| 320.3433_3.320781 | Positive | 321.3433 | 3.3 | 6.6419 | 0.036119 | sALS ‐ sFTLD‐TDP, sALS ‐ c9FTLD | |
| 354.2074_0.9234119 | Negative | 353.2074 | 0.9 | 8.5586 | 0.013853 | sALS ‐ sFTLD‐TDP | |
| 666.6668_6.717138 | Positive | 667.6668 | 6.7 | 6.8608 | 0.032374 | sALS ‐ c9FTLD | |
| 738.5654_7.707858 | Positive | 739.5654 | 7.7 | 6.1141 | 0.047025 | sALS ‐ c9FTLD, sFTLD‐TDP ‐ c9FTLD | |
| 773.5415_7.0351 | Positive | 774.5415 | 7 | 9.2808 | 0.009654 | sALS ‐ c9FTLD | |
| 842.8146_10.21985 | Positive | 843.8146 | 10.2 | 9.1588 | 0.010261 | sALS ‐ sFTLD‐TDP, sFTLD‐TDP ‐ c9FTLD | |
| 868.1839_6.287742 | Negative | 867.1839 | 6.3 | 7.2157 | 0.027109 | sALS ‐ sFTLD‐TDP, sFTLD‐TDP ‐ c9FTLD |
Significant molecular features Kruskal–Wallis test with p value <0.05 are shown as LC‐ESI ionisation mode (P for positive and N for negative) followed by neutral mass and retention time. Potential identities based on MS1 and MS2 data for each feature are found lipidomic feature. Unidentified compounds are labelled as neutral mass and retention time. Ether phospholipids are highlighted in bold.
Abbreviations: CE, cholesterol ester; DG, diacylglycerol; FDR, false discovery rate; mz value, mass‐to‐charge ratio; PC, phosphocholine; SM, sphingomyelin; TG, triacylglycerol.
Represents confirmation by data‐dependent MS2.
Represents confirmation by data‐dependent MS2 by class.
Represents confirmation by MS1 exact mass and retention time.
Important features associated with different phenotypes
To identify the differential lipid molecules in each disorder, we performed a Kruskal–Wallis test with a post hoc comparison using Dunn's test on frontal cortex region 8 samples from sALS, sFTLD‐TDP, c9FTLD and controls. The differential molecules are listed in Table 5. Globally, phospholipid species (mainly PC) presented the greatest differences between ALS (lower) and CTL (higher) groups. Interestingly, FAHFA (42:3) was decreased in c9FTLD and ALS compared with the control group. Furthermore, we found MG(20:1), CE(20:1) and DG(16:0/22:6) to be elevated in sFTLD‐TDP and c9FTLD compared to the control group. PC(36:3) was down‐regulated in ALS and sFTLD‐TDP compared to controls. PC(18:1/20:4) was down‐regulated in sFTLD‐TDP compared to CTL and c9FTLD. Sphinganine was down‐regulated in ALS and sFTLD‐TDP compared to control samples (Figure 3E).
TABLE 5.
Lipidomic features associated with neurological disease of the TDP‐43 spectrum
| Lipid category | Lipidomic feature | Ionisation | mz value | Retention time | chi squared | p value | post hoc |
|---|---|---|---|---|---|---|---|
| Fatty acyls | FAHFA(42:3) b | Negative | 643.5684 | 9.1 | 10.955 | 0.01197 | sALS ‐ CTL, CTL ‐ c9FTLD, sFTLD‐TDP ‐ c9FTLD |
| Glycerolipids | DG(16:0/22:6) a | Positive | 640.5837 | 7.5 | 11.308 | 0.010173 | CTL ‐ sFTLD‐TDP, CTL ‐ c9FTLD, sFTLD‐TDP ‐ c9FTLD |
| MG(20:1) c | Positive | 402.3529 | 3.1 | 8.9668 | 0.029736 | CTL ‐ sFTLD‐TDP, CTL ‐ c9FTLD | |
| TG(18:0/18:1/18:1) a | Positive | 904.8296 | 10.3 | 9.3931 | 0.024496 | sALS ‐ sFTLD‐TDP, sFTLD‐TDP ‐ c9FTLD | |
| TG(18:1/18:1/18:2) a | Positive | 900.8005 | 10 | 8.1153 | 0.043688 | sALS ‐ sFTLD‐TDP, CTL ‐ sFTLD‐TDP | |
| Glycerophospholipids | PC(16:0/20:4) a | Positive | 782.5691 | 6.9 | 8.3459 | 0.039379 | sALS ‐ CTL, CTL ‐ sFTLD‐TDP |
| PC(16:0/22:6) a | Positive | 806.5692 | 6.8 | 9.1774 | 0.027023 | sALS ‐ CTL, sALS ‐ c9FTLD, CTL ‐ sFTLD‐TDP | |
| PC(18:1/20:4) a | Positive | 808.5845 | 7 | 9.7619 | 0.020703 | sALS ‐ CTL, sALS ‐ c9FTLD, CTL ‐ sFTLD‐TDP, sFTLD‐TDP ‐ c9FTLD | |
| PC(36:3) b | Positive | 784.5814 | 7.2 | 8.278 | 0.040603 | sALS ‐ CTL, sALS ‐ c9FTLD | |
| PC(44:5) b | Positive | 846.6285 | 7.7 | 8.8661 | 0.031125 | sALS ‐ CTL, sALS ‐ sFTLD‐TDP, CTL ‐ sFTLD‐TDP | |
| PE(O‐37:1) b | Positive | 746.5681 | 7.7 | 8.1337 | 0.043328 | sALS ‐ CTL, sALS ‐ c9FTLD | |
| PE(P−16:0/22:6) a | Positive | 748.5247 | 7 | 8.7502 | 0.032804 | sALS ‐ CTL, sALS ‐ c9FTLD, CTL ‐ sFTLD‐TDP | |
| PE‐NMe(44:12) c | Negative | 830.5276 | 7.2 | 8.0631 | 0.044726 | CTL ‐ c9FTLD, sFTLD‐TDP ‐ c9FTLD | |
| Sphingolipids | SM(d20:5/16:1) a | Positive | 721.5057 | 7.8 | 9.4698 | 0.023655 | sALS ‐ CTL, sALS ‐ c9FTLD, CTL ‐ sFTLD‐TDP |
| SM(d33:0) b | Positive | 689.555 | 7.2 | 9.1091 | 0.027875 | CTL ‐ sFTLD‐TDP, CTL ‐ c9FTLD | |
| Sphinganine c | Positive | 284.2891 | 4.4 | 7.8587 | 0.049024 | sALS ‐ CTL, CTL ‐ c9FTLD | |
| Sterol lipids | CE(20:1) a | Positive | 696.6463 | 8.5 | 11.753 | 0.0082796 | CTL ‐ sFTLD‐TDP, CTL ‐ c9FTLD, sFTLD‐TDP ‐ c9FTLD |
| Unknown | 1145.327_7.909675 | Positive | 1146.327 | 7.9 | 13.69 | 0.0033584 | sALS ‐ CTL, sALS ‐ sFTLD‐TDP, CTL ‐ c9FTLD, sFTLD‐TDP ‐ c9FTLD |
| 1423.392_10.11581 | Positive | 1424.392 | 10.1 | 11.061 | 0.011402 | sALS ‐ CTL, sALS ‐ sFTLD‐TDP, CTL ‐ sFTLD‐TDP | |
| 224.0327_0.9230345 | Negative | 223.0327 | 0.9 | 11.841 | 0.0079478 | sALS ‐ sFTLD‐TDP, sALS ‐ c9FTLD, sFTLD‐TDP ‐ c9FTLD | |
| 224.19_1.288949 | Positive | 225.19 | 1.3 | 9.4867 | 0.023473 | sALS ‐ CTL, sALS ‐ c9FTLD, CTL ‐ c9FTLD | |
| 273.029_1.499844 | Positive | 274.029 | 1.5 | 15.853 | 0.0012154 | sALS ‐ CTL, sALS ‐ sFTLD‐TDP, sALS ‐ c9FTLD, sFTLD‐TDP ‐ c9FTLD | |
| 320.3433_3.320781 | Positive | 321.3433 | 3.3 | 8.6829 | 0.033819 | sALS ‐ CTL, sALS ‐ sFTLD‐TDP, CTL ‐ sFTLD‐TDP | |
| 348.375_3.32256 | Positive | 349.375 | 3.3 | 8.86 | 0.031212 | sALS ‐ CTL, sALS ‐ c9FTLD, CTL ‐ c9FTLD | |
| 422.4066_7.569828 | Negative | 421.4066 | 7.6 | 8.9619 | 0.029801 | sALS ‐ CTL, sALS ‐ c9FTLD, CTL ‐ sFTLD‐TDP, sFTLD‐TDP ‐ c9FTLD | |
| 598.5024_8.868424 | Positive | 599.5024 | 8.9 | 7.9038 | 0.048042 | sALS ‐ CTL, sALS ‐ c9FTLD | |
| 602.531_8.221487 | Positive | 603.531 | 8.2 | 9.6105 | 0.022185 | sALS ‐ CTL, sALS ‐ c9FTLD, CTL ‐ c9FTLD | |
| 648.6568_6.103854 | Positive | 649.6568 | 6.1 | 9.563 | 0.022671 | sALS ‐ CTL, CTL ‐ c9FTLD, sFTLD‐TDP ‐ c9FTLD | |
| 666.6668_6.717138 | Positive | 667.6668 | 6.7 | 11.872 | 0.0078361 | sALS ‐ CTL, sALS ‐ c9FTLD, CTL ‐ sFTLD‐TDP | |
| 667.059_0.9157435 | Negative | 666.059 | 0.9 | 8.1549 | 0.042917 | sALS ‐ CTL, sALS ‐ sFTLD‐TDP, CTL ‐ sFTLD‐TDP | |
| 703.5196_6.254575 | Positive | 704.5196 | 6.3 | 9.1774 | 0.027023 | sALS ‐ CTL, sALS ‐ c9FTLD, CTL ‐ sFTLD‐TDP | |
| 773.5415_7.0351 | Positive | 774.5415 | 7 | 10.63 | 0.013903 | sALS ‐ CTL, CTL ‐ sFTLD‐TDP, | |
| 780.1863_5.702648 | Positive | 781.1863 | 5.7 | 9.9159 | 0.019295 | sALS ‐ CTL, sALS ‐ c9FTLD, CTL ‐ c9FTLD, sFTLD‐TDP ‐ c9FTLD | |
| 842.8146_10.21985 | Positive | 843.8146 | 10.2 | 8.0448 | 0.045095 | sALS ‐ sFTLD‐TDP, sALS ‐ c9FTLD, sFTLD‐TDP ‐ c9FTLD | |
| 868.1839_6.287742 | Negative | 867.1839 | 6.3 | 9.5946 | 0.022346 | sALS ‐ CTL, sALS ‐ sFTLD‐TDP, CTL ‐ c9FTLD, sFTLD‐TDP ‐ c9FTLD | |
| 981.2558_8.147829 | Positive | 982.2558 | 8.1 | 9.6517 | 0.021772 | sALS ‐ sFTLD‐TDP, sALS ‐ c9FTLD, CTL ‐ c9FTLD |
Significant molecular features Kruskal–Wallis test with p value <0.05 are shown as LC‐ESI ionisation mode (P for positive and N for negative) followed by neutral mass and retention time. Potential identities based on MS1 and MS2 data for each feature are found lipidomic feature. Unidentified compounds are labelled as neutral mass and retention time. Ether phospholipids are highlighted in bold.
Abbreviations: CE, cholesterol ester; DG, diacylglycerol; FDR, false discovery rate; MG, monoacylglycerol; mz value, mass‐to‐charge ratio; PC, phosphocholine; PE, phophoethanolamine; SM, sphingomyelin; TG, triacylglycerol.
Represents confirmation by data‐dependent MS2.
Represents confirmation by data‐dependent MS2 by class.
Represents confirmation by MS1 exact mass and retention time.
DISCUSSION
ALS and FTLD are two neurodegenerative disorders that share a number of genetic, pathological and clinical features. One shared molecular trait is the accumulation of pathological protein aggregates including, among others, the protein TAR DNA‐binding protein 43 kDa (TDP‐43).
Previous reports on altered levels of lipids in the central nervous system and peripheral systems in ageing [59, 71] and neurodegenerative disorders [59, 61, 72, 73, 74, 75, 76] led us to hypothesise that impaired peroxisomal function contributes to the progression of neurodegeneration in TDP‐43 proteinopathies. Peroxisomes harbour a variety of enzymes, which either serve to catalyse a single chemical reaction or cooperate with other peroxisomal enzymes in a series of coupled reactions constituting a complete metabolic pathway [11]. A prominent example of these metabolic pathways that links lipid metabolism and peroxisome functioning is the metabolism of diverse fatty acids by α/β‐oxidation, bile acids, docosahexaenoic acid and ether lipid biosynthesis [77]. The present observations show altered gene expression profiles of different components involved in peroxisomal machinery and lipid metabolism that require peroxisomal activity in frontal cortex area 8 of post‐mortem samples of different TDP‐43 proteinopathies.
Our transcriptomic data revealed slight changes in the expression profiles of the studied peroxisomal genes. Gene expression alterations were mainly in genes in peroxisome biogenesis and β‐oxidation, fatty acid metabolism and acylcarnitine biosynthesis mainly found in FTLD cases. Changes in two of six genes linked to primary bile acid biosynthesis do not match with any pattern associated with ALS‐FTLD‐TDP43 proteinopathy. This suggests that the disparate alteration in the biosynthesis of primary bile acids does not have a definite role in FTLD‐TDP43.
In agreement with the slight transcriptomic alterations, no changes in metabolites of bile acid metabolism or alpha/beta fatty acid oxidation were detected with our lipidomic approach, suggesting that the changes in gene expression are insufficient to induce changes at the level of metabolite concentrations, which are maintained within a physiological range. Reinforcing this idea, transcriptomic changes in fatty acid metabolism do not affect the fatty acid profiles of any of the TDP‐43 proteinopathies, suggesting that the activity of the operating machinery, despite being hampered, is sufficient to sustain the optimal fatty acid profile needed to support neuronal survival and function. Thus, fatty acid profile maintains an ACL of about 18 carbon atoms, and a relative distribution between saturated (SFA) and unsaturated (UFA, being UFA = PUFA series n‐3+PUFA series n‐6) fatty acids of around 45:55, with MUFAs being the predominant UFAs, and AA and DHA the main PUFAs, as previously described in healthy adult frontal cortex [59]. Interestingly, the lack of changes in very long chain saturated fatty acids (VLCFA) 22:0 and 24:0 reinforces the idea that the light peroxisomal dysfunction in gene expression associated with TDP‐43 proteinopathies is not sufficient to affect VLCFA content, in contrast to other neuropathological conditions primary of peroxisomal origin such as X‐adrenoleukodystrophy, or even Alzheimer's disease [39, 78].
A sustained fatty acid profile does not exclude, however, potential changes in the content of lipid species that can be uncovered with a lipidomics approach. An adult human brain contains the largest amount and diversity of lipids (in terms of classes and molecular species) including glycerolipids, glycerophospholipids, sphingolipids and cholesterol. Glycerophospholipids are the major phospholipid components ubiquitously found in neural cell membranes [59, 79, 80]. In the human brain, phospholipids constitute 4.2% of the wet weight of the grey matter [79, 81, 82]. Phosphatidic acid occurs in low concentrations in brain (about 2% of total phospholipids). The predominant form of choline phosphoglycerides is PC (32.8%), with PC (16:0/18:1) being the major molecular species; the choline plasmalogen (PC(P‐)) and the alkyl analogue (PC(O‐)) account for only about 2% of total choline phosphoglycerides. Ethanolamine phosphoglycerides are quantitatively the major phospholipid in human brain (35.6%), and the predominant form is the ethanolamine plasmalogen (PE(P‐)), accounting for 50–60% of the ethanolamine phosphoglyceride lipid class in the whole human brain [83]. The alkylacyl analogue content is relatively low (3–7% of the ethanolamine phosphoglyceride class), whereas PE makes up the remaining amount of ethanolamine phosphoglycerides. Their total fatty acid composition indicates a large content of PUFAs, mainly 20:4n6 and 22:6n3 at position‐2 of sn‐glycerol, with position‐1 occupied primarily by 16:0, 18:0 and 18:1 groups in grey matter [83]. The concentration of serine phosphoglycerides is about 16.6%, which is particularly rich in 22:6; and inositol phosphoglycerides account for about 2.6% of the total phospholipids in the human brain [84, 85]. The brain contains the highest concentrations of phosphoinositides among animal tissues. Finally, 0.2% of phospholipids are present as diphosphatidylglycerol in the human brain. Sphingolipids are a complex lipid group, derived from N‐acylsphingosine (ceramide), which occurs in large concentrations in the human brain. This group of lipids consists of sphingomyelin, cerebrosides, sulfatides and gangliosides. Sphingomyelin (N‐acylsphingosine‐1‐phosphocholine) accounts for about 14.8% of the phospholipid content of the human brain.
Our lipidomics approach shows that the ALS‐FTLD‐TDP43 proteinopathy spectrum is associated with minor but significant changes in lipidomic profile, based on the fact that only 63 of 1119 molecular species (5.6%) were significantly different. Interestingly, this lipidomic pattern defines the ALS‐FTLD‐TDP43 proteinopathy spectrum; and with the exception of one molecular species (DG(36:1)), all lipid species (62 out of 63) were decreased. Notably, among the molecules recorded based on exact mass, retention time, and/or MSMS spectrum, no molecular species belong to the phosphatidic acid, phosphatidylserine or phosphatidylinositol class. The differentially identified lipid species belong mainly to glycerophospholipids (PC and PE, and their plasmalogen forms) and sphingolipid lipid families.
Glycerophospholipids are important building blocks of cell membranes that provide an optimal environment for protein interactions, trafficking and function. In the ageing process and in the pathological context of neurodegeneration, decreased brain phospholipid levels and alterations in brain phospholipid metabolism appear, as observed in brain post‐mortem tissue, CSF and blood [86]. This study demonstrates a down‐regulation in PC and PE levels in the ALS‐FTLD spectrum, suggesting alterations in the architecture of the neural cells [87, 88]. In addition, PC is an important source for the formation of second messengers and lipid mediators [89, 90]. Disturbance of its production interferes with cell proliferation and differentiation, and membrane movement throughout the cell [91]. Furthermore, PE plays essential roles in autophagy, cell division and protein folding, representing a precursor for the synthesis of several protein modifications [88]. In addition, both PC and PE are intermediates in the synthesis of other glycerophospholipid classes [92, 93]. In line with our results, a recent study in cells [94] revealed that TDP‐43‐mediated toxicity induced lower levels of glycerophospholipids (especially glycerophosphocholines) and sphingolipids. Decrease levels of glycerophospholipids were also described in ALS animal models [95]. Globally, our results suggest a minor but crucial disturbance of phospholipid metabolism in TDP‐43 proteinopathies. Decreased levels of glycerophospholipids are consistent with altered cell membranes and altered signal transduction via reduced second messengers at the cell membrane. Since the main cellular alteration in the FC of FTLD (and in sALs) affects neurons, it can be inferred that phospholipid alterations here observed reflect abnormal neuronal membrane lipid composition. Whether this is accompanied by altered protein content, indicating cell membranes as putative subcellular targets in TDP‐43 proteinopathies, will be a subject of future studies.
Ether lipids are a subclass of glycerophospholipids that have two chemical forms: as ‘plasmanyl’ (also termed alkyl ethers and represented by the ‘O‐‘ prefix), and as ‘plasmenyl’ (also termed alkenyl ethers or plasmalogens, and represented by the ‘P‐‘ prefix) [96, 97]. Ether lipids are mostly present as PC and PE species [96]. At the cellular level, ether lipid biosynthesis begins in the peroxisome and is completed in the endoplasmic reticulum [96, 97, 98]. The physiological role of ether lipids is linked to their function as membrane components (fluidity, formation of lipid rafts and a source of second messengers). Other functions in which ether lipids are involved are cholesterol transport, G‐protein‐mediated signal transduction, membrane fusion events, transmembrane protein function and vesicular function [96, 97, 98]. Interestingly, an antioxidant effect has also been assigned to plasmalogens [99]. Available evidence reveals that ether lipids are inversely associated with genetic peroxisomal disorders, and also suggests that they are negatively associated with prevalent disease states such as cancer, cardiovascular diseases and Alzheimer disease, among others [100]. Notably, these pathological conditions share as a common trait, increased oxidative stress, and a potential mechanistic role for plasmalogens. Our study clearly demonstrates down‐regulation of the ether lipid content in frontal cortex area 8 of TDP‐43 proteinopathies. Thus, a reduction in its levels may confer vulnerability against oxidative stress insults potentially contributing to neurodegeneration in these disorders, in addition to acting as a marker of impaired peroxisomal function. Reinforcing this idea, the lack of support at the transcriptional level (no changes were observed in the present work) suggests the existence of alterations at the translational level or, more probably, functional defects mediated by potential oxidative stress conditions. Further studies are needed to obtain a more detailed mechanistic view.
A major category of lipids is the sphingolipids [101] that play a key role in the formation of lipid rafts in cell membranes [102]. The metabolism of sphingolipids is a complex network with ceramide at the core [103]. The result is a wide diversity of lipid species with structural (e.g. sphingomyelins) and bioactive/messenger (e.g. sphingosines, dihydroceramides and ceramides) functions [101]. Sphingolipids regulate membrane physiology (fluidity, geometry, membrane trafficking and clustering of plasma membrane receptors and ion channels, among others) and cell biology (e.g. oxidative stress, apoptosis and cell survival), and they have been seen to be involved in several pathological conditions such as cardiovascular diseases and neurodegeneration [103, 104, 105, 106]. Our lipidomic study showed a down‐regulation of structural sphingolipids such as sphingomyelin, as well as bioactive compounds such as sphinganine and ceramides in TDP‐43 proteinopathies, reinforcing the view that there are alterations in lipid metabolism in these neurodegenerative disorders. Therefore, studies focused on lipid rafts, as analysed in detail in Alzheimer's disease, Parkinson's disease and Dementia with Lewy bodies [107, 108, 109, 110] would be extremely useful to refine possible alterations in these cellular domains in which the main cell to cell interactions take place.
Another observation that argues for a peroxisomal dysfunction in TDP‐43 proteinopathies is the down‐regulation of acylcarnitine. Acylcarnitine is a metabolite that plays a relevant role in enabling long‐chain fatty acid exchanges between peroxisomes and mitochondria for beta‐oxidation [111]. Surprisingly, the down‐regulation of acylcarnitines does not seem to be mediated by defects at the transcriptional level. In fact, the contrary is observed, with an increase in mRNA content of the main components of the biosynthesis pathway. Therefore, it is plausible to hypothesise that this apparent contradiction between phenotype and genotype is caused by translational alterations or, analogously to the postulated for plasmalogens biosynthesis, by functional defects at the protein level leading to a reduced peroxisomal biosynthestic capacity. Further studies are, however, also needed to consolidate these new ideas.
Finally, a special mention should be made of the detection of two branched fatty acid esters of hydroxy fatty acids (FAHFAs) for the first time, to our knowledge, in human brain. Although to date their biosynthesis and metabolism are not well elucidated, FAHFAs are a novel class of endogenous lipids that present beneficial effects on glucose homeostasis and anti‐inflammatory activities. Interestingly, there is a possible link between endogenous FAHFA levels and nuclear factor erythroid 2‐related factor 2 (Nrf2), which is involved in cell antioxidant defences, although the mechanisms of this are unknown [112]. Considering the down‐regulation of these compounds in TDP‐43 proteinopathies, it may be suggested that there is impairment of glucose homeostasis, inflammation and oxidative stress in frontal cortex area 8 in ALS‐FTLD‐TDP cases.
Globally, we have demonstrated minor changes in peroxisome‐related gene expression that is mostly involved in lipid metabolism with phenotypic changes affecting ether lipids and acylcarnitine, whereas fatty acid metabolism seems to be preserved in the human frontal cortex in the ALS‐FTLD‐TDP43 proteinopathy spectrum. Additional changes in glycerophospholipids, sphingolipids and specific fatty acid species like FAHFA suggest a more profound impact of these proteinopathies in lipid metabolism with possible consequences on abnormal neuronal membrane composition and impaired cell signalling at the membranes.
At present, little is known about the role of specific lipid species in TDP‐43 proteinopathies and associated neurodegeneration. The development of cell lines and animal models with defective lipid metabolism may help to clarify the role of these lipid species in TDP‐43 proteinopathies thus facilitate better understanding of the mechanisms responsible for neurodegeneration and allowing better diagnosis and treatment.
CONFLICT OF INTEREST
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1111/nan.12681.
Supporting information
Table S1
Andrés‐Benito P, Gelpi E, Jové M, et al. Lipid alterations in human frontal cortex in ALS‐FTLD‐TDP43 proteinopathy spectrum are partly related to peroxisome impairment. Neuropathol Appl Neurobiol. 2021;47:544–563. 10.1111/nan.12681
FUNDING INFORMATION
Research by the authors was supported by the Spanish Ministry of Economy and Competitiveness, Institute of Health Carlos III (PI 17–00134) to M.P‐O; the Spanish Ministry of Economy and Competitiveness, Institute of Health Carlos III (FIS grants FISPI17/000809) to IF; and by the Spanish Ministry of Science, Innovation, and Universities (Ministerio de Ciencia, Innovación y Universidades, RTI2018‐099200‐BI00) and the Generalitat of Catalonia: Agency for Management of University and Research Grants (2017SGR696) and Department of Health (SLT002/16/00250) to RP. This study was co‐financed by FEDER funds from the European Union (“A way to build Europe”). Support was also received in the form of a FUNDELA Grant, RedELA‐Plataforma Investigación and the Fundació Miquel Valls (Jack Van den Hoek donation). IRBLleida and IDIBELL are CERCA Programmes of the Generalitat of Catalonia.
Contributor Information
Reinald Pamplona, Email: reinald.pamplona@udl.cat.
Isidro Ferrer, Email: reinald.pamplona@udl.cat, Email: 8082ifa@gmail.com.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon request.
REFERENCES
- 1. De Duve C, Baudhuin P. Peroxisomes (microbodies and related particles). Physiol Rev. 1996;46:323‐327. [DOI] [PubMed] [Google Scholar]
- 2. Pedersen JI, Eggertsen G, Hellman U, Andersson U, Björkhem I. Molecular cloning and expression of cDNA encoding 3alpha, 7alpha,12alpha‐trihydroxy‐5beta‐chole stanoyl‐CoA oxidase from rabbit liver. J Biol Chem. 1997;272:18481‐18489. [DOI] [PubMed] [Google Scholar]
- 3. Wanders RJ, Waterham HR. Biochemistry of mammalian peroxisomes revisited. Annu Rev Biochem. 2006;75:295‐332. [DOI] [PubMed] [Google Scholar]
- 4. Islinger M, Cardoso MJ, Schrader M. Be different—the diversity of peroxisomes in the animal kingdom. Biochim Biophys Acta. 2010;1803:881‐887. [DOI] [PubMed] [Google Scholar]
- 5. Wanders RJ. Peroxisomes in human health and disease: metabolic pathways, metabolite transport, interplay with other organelles and signal transduction. Subcell Biochem. 2013;69:23‐44. [DOI] [PubMed] [Google Scholar]
- 6. Smith JJ, Aitchinson JD. Peroxisomes take shape. Nat Rev Mol Cell Biol. 2013;14:803‐817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lodhi IF, Semenkovich CF. Peroxisomes: a nexus for lipid metabolism and cellular signalling. Cell Metab. 2014;19:380‐392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Islinger M, Voelkl A, Fahimi HD, Schrader M. The peroxisome: an update on mysteries 2.0. Histochem Cell Biol. 2018;150:443‐471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Walker CL, Pomatto LCD, Tripathi DN, Davies KJA. Redox regulation of homeostasis and proteostasis in peroxisomes. Physiol Rev. 2018;98:89‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schrader M, Costello JL, Godinho LF, Azadi AS, Islinger M. Proliferation and fission of peroxisomes ‐ an update. Biochim Biophys Acta. 2016;1863:971‐983. [DOI] [PubMed] [Google Scholar]
- 11. Berger J, Dorninger F, Forss‐Petter S, Kunze M. Peroxisomes in brain development and function. Biochim Biophys Acta. 2006;1863:934‐955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pu J, Ha CW, Zhang S, Jung JP, Huh WK, Liu P. Interactomic study on interaction between lipid droplets and mitochondria. Protein Cell. 2011;2:487‐496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. van der Zand A, Gent J, Braakman I, Tabak HF. Biochemically distinct vesicles from the endoplasmic reticulum fuse to form peroxisomes. Cell. 2012;149:397‐409. [DOI] [PubMed] [Google Scholar]
- 14. Schrader M, Grille S, Fahimi HD, Islinger M. Peroxisome interactions and cross‐talk with other subcellular compartments in animal cells. Subcell Biochem. 2013;69:1‐22. [DOI] [PubMed] [Google Scholar]
- 15. Dimitrov L, Lam SK, Schekman R. The role of the endoplasmic reticulum in peroxisome biogenesis. Cold Spring Harb Perspect Biol. 2013;5:a013243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wanders RJA, Waterham HR, Ferdinandusse S. Metabolic interplay between peroxisomes and other subcellular organelles including mitochondria and the endoplasmic reticulum. Front Cell Develop Biol. 2016; 3: 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Walther TC, Chung J, Farese RV. Lipid droplet biogenesis. Annu Rev Cell Dev Biol. 2017;33:491‐510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang YX. PPARs: diverse regulators in energy metabolism and metabolic diseases. Cell Res. 2010;20:124‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schrader M, Bonekamp NA, Islinger M. Fission and proliferation of peroxisomes. Biochim Biophys Acta. 2012;1822:1343‐1357. [DOI] [PubMed] [Google Scholar]
- 20. Ahmadian M, Suh JM, Hah N, et al. PPARg signaling and metabolism: the good, the bad and the future. Nat Med. 2013;19:557‐566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Motley AM, Galvin PC, Ekal L, Nuttall JM, Hettema EH. Re‐evaluation of the role of Pex1 and dynamin‐related proteins in peroxisome membrane biogenesis. J Cell Biol. 2015;211:1041‐1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Powers JM, Moser HV. Peroxisomal disorders: genotype, phenotype, major neuropathologic lesions, and pathogenesis. Brain Pathol. 1998;8:101‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jansen GA, Wanders RJA, Watkins PA, Mihalik SJ. Phytanoyl‐coenzyme A hydroxylase deficiency—the enzyme defect in Refsum's disease. N Engl J Med. 1997;337:133‐134. [DOI] [PubMed] [Google Scholar]
- 24. Faust PL, Banka D, Siriratsivawong R, Ng VG, Wikander TM. Peroxisome biogenesis disorders: the role of peroxisomes and metabolic dysfunction in developing brain. J Inherit Metab Dis. 2005;28:369‐383. [DOI] [PubMed] [Google Scholar]
- 25. Steinberg SJ, Dodt G, Raymond GV, Braverman NE, Moser AB, Moser HW. Peroxisome biogenesis disorders. Biochim Biophys Acta. 2006;1763:1733‐1748. [DOI] [PubMed] [Google Scholar]
- 26. Ferdinandusse S, Denis S, Mooyer PA, et al. Clinical and biochemical spectrum of D‐bifunctional protein deficiency. Ann Neurol. 2006;59: 92‐104. [DOI] [PubMed] [Google Scholar]
- 27. Ferdinandusse S, Denis S, Hogenhout EM, et al. Clinical, biochemical, and mutational spectrum of peroxisomal acyl‐coenzyme A oxidase deficiency. Hum Mutat. 2007;28:904‐912. [DOI] [PubMed] [Google Scholar]
- 28. Wanders RJA, Ferdinandusse S, Brites P, Kemp S. Peroxisomes, lipid metabolism and lipotoxicity. Biochem Biophys Acta. 2010;1801:272‐280. [DOI] [PubMed] [Google Scholar]
- 29. Trompier D, Vejux A, Zarrouk A, et al. Brain peroxisomes. Biochimie. 2014;98:102‐110. [DOI] [PubMed] [Google Scholar]
- 30. Fujiki Y. Peroxisome biogenesis and human peroxisome‐deficiency disorders. Proc Jpn Acad Ser B. 2016;92:463‐477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Waterham HR, Ferdinandusse S, Wanders RJA. Human disorders of peroxisome metabolism and biogenesis. Biochim Biophys Acta. 2016;1863:922‐933. [DOI] [PubMed] [Google Scholar]
- 32. Giordano CR, Terlecky SR. Peroxisomes, cell senescence, and rates of aging. Biochim Biophys Acta. 2012;1822:1358‐1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fransen M, Nordgren M, Wang B, Apanasets O, Van Veldhoven PP. Aging, age‐related diseases and peroxisomes. Subcell Biochem. 2013;69:45‐65. [DOI] [PubMed] [Google Scholar]
- 34. Dorninger F, Forss‐Petter S, Berger J. From peroxisomal disorders to common neurodegenerative diseases – the role of ether phospholipids in the nervous system. FEBS Lett. 2017;591:2761‐2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Deori DM, Kale A, Maurya PK, Nagotu S. Peroxisomes: role in cellular ageing and age related disorders. Biogerontology. 2018;19:303‐324. [DOI] [PubMed] [Google Scholar]
- 36. Cimini A, Moreno S, D’Amelio M, et al. Early biochemical and morphological modifications in the brain of a transgenic mouse model of Alzheimer’s disease: a role for peroxisomes. J Alzheimers Dis. 2009;18:935‐952. [DOI] [PubMed] [Google Scholar]
- 37. Fanelli F, Sepe S, D’Amelio M, et al. Age‐dependent roles of peroxisomes in the hippocampus of a transgenic mouse model of Alzheimer’s disease. Mol Neurodegener. 2013;8:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Igarashi M, Ma K, Gao F, Kim HW, Rapoport SI, Rao JS. Disturbed choline plasmalogen and phospholipid fatty acid concentrations in Alzheimer’s disease prefrontal cortex. J Alzheimers Dis. 2011;24:507‐517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kou J, Kovacs GG, Hoftberger R, et al. Peroxisomal alterations in Alzheimer’s disease. Acta Neuropathol. 2011;122:271‐283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Searcy JL, Phelps JT, Pancani T, et al. Long‐term pioglitazone treatment improves learning and attenuates pathological markers in a mouse model of Alzheimer's disease. J Alzheimers Dis. 2012;30:943‐961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Corbett GT, Gonzalez FJ, Pahan K. Activation of peroxisome proliferator‐activated receptor α stimulates ADAM10‐mediated proteolysis of APP. Proc Natl Acad Sci USA. 2015;112:8445‐8450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vallée A, Lecarpentier Y. Alzheimer disease: crosstalk between the canonical Wnt/β‐catenin pathway and PPARs α and γ. Front Neurosci. 2016;10:459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Silva‐Abreu M, Calpena AC, Andrés‐Benito P, et al. PPARγ agonist‐loaded PLGA‐PEG nanocarriers as a potential treatment for Alzheimer's disease: in vitro and in vivo studies. Int J Nanomed. 2018;13:5577‐5590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. D'Orio B, Fracassi A, Ceru MP, Moreno S. Targeting PPARα in Alzheimer's disease. Curr Alzheimer Res. 2018;15:345‐354. [DOI] [PubMed] [Google Scholar]
- 45. Khan MA, Alam Q, Haque A, et al. Current Progress on peroxisome proliferator‐activated receptor gamma agonist as an emerging therapeutic approach for the treatment of Alzheimer's disease: an update. Curr Neuropharmacol. 2019;17:232‐246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Taylor JP, Brown RH Jr, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539:197‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology. 2005;65:586‐590. [DOI] [PubMed] [Google Scholar]
- 48. Ling S‐C, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79:416‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ferrer I, Santpere G, Arzberger T, et al. Brain protein preservation largely depends on the postmortem storage temperature: implications for study of proteins in human neurologic diseases and management of brain banks: a BrainNet Europe Study. J Neuropathol Exp Neurol. 2007;66:35‐46. [DOI] [PubMed] [Google Scholar]
- 50. Ferrer I, Martinez A, Boluda S, Parchi P, Barrachina M. Brain banks: benefits, limitations and cautions concerning the use of post‐mortem brain tissue for molecular studies. Cell Tissue Bank. 2008;9:181‐194. [DOI] [PubMed] [Google Scholar]
- 51. Ferrer I. Selection of controls in the study of human neurodegenerative diseases in old age. J Neural Transm. 2015;122:941‐947. [DOI] [PubMed] [Google Scholar]
- 52. Ferrer I. Brain banking. In: Aminoff MJ, Daroff RB, eds. Encyclopedia of the Neurological Sciences, 2nd ed. Vol. 1. Oxford: Academic Press; 2014;467‐473. [Google Scholar]
- 53. Mackenzie IR, Neumann M, Baborie A, et al. A harmonized classification system for FTLD‐TDP pathology. Acta Neuropathol. 2011;22:111‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hortobagyi T, Cairns NJ. Amyotrophic lateral sclerosis and frontotemporal lobar degeneration. In: Kovacs Gabor G, ed. Neuropathology of Neurodegenerative Diseases: A Practical Guide. Cambridge: Cambridge University Press; 2015:209‐248. [Google Scholar]
- 55. Brettschneider J, Del Tredici K, Irwin DJ, et al. Sequential distribution of pTDP‐43 pathology in behavioral variant frontotemporal dementia (bvFTD). Acta Neuropathol. 2014;127:423‐439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cabré R, Naudí A, Dominguez‐Gonzalez M, et al. Lipid profile in human frontal cortex is sustained throughout healthy adult life span to decay at advanced ages. J Gerontol A Biol Sci Med Sci. 2018;73:703‐710. [DOI] [PubMed] [Google Scholar]
- 57. Hancock SE, Friedrich MG, Mitchell TW, Truscott RJ, Else PL. The phospholipid composition of the human entorhinal cortex remains relatively stable over 80 years of adult aging. Geroscience. 2017;39:73‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Barrachina M, Castaño E, Ferrer I. TaqMan PCR assay in the control of RNA normalization in human post‐mortem brain tissue. Neurochem Int. 2006;49:276‐284. [DOI] [PubMed] [Google Scholar]
- 59. Naudí A, Cabré R, Jové M, et al. Lipidomics of human brain aging and Alzheimer's disease pathology. Int Rev Neurobiol. 2015;122:133‐189. [DOI] [PubMed] [Google Scholar]
- 60. Caro P, Gomez J, Sanchez I, et al. Forty percent methionine restriction decreases mitochondrial oxygen radical production and leak at complex I during forward electron flow and lowers oxidative damage to proteins and mitochondrial DNA in rat kidney and brain mitochondria. Rejuv Res. 2009;12:421‐434. [DOI] [PubMed] [Google Scholar]
- 61. Naudí A, Cabré R, Dominguez‐Gonzalez M, et al. Region‐specific vulnerability to lipid peroxidation and evidence of neuronal mechanisms for polyunsaturated fatty acid biosynthesis in the healthy adult human central nervous system. Biochim Biophys Acta Mol Cell Biol Lipids. 2017;1862:485‐495. [DOI] [PubMed] [Google Scholar]
- 62. Guillou H, Zadravec D, Martin PG, Jacobsson A. The key roles of elongases and desaturases in mammalian fatty acid metabolism: insights from transgenic mice. Prog Lipid Res. 2010;49:186‐199. [DOI] [PubMed] [Google Scholar]
- 63. Pizarro C, Arenzana‐Rámila I, Pérez‐del‐Notario N, Pérez‐Matute P, González‐Sáiz JM. Plasma lipidomic profiling method based on ultrasound extraction and liquid chromatography mass spectrometry. Anal Chem. 2013;85:12085‐12092. [DOI] [PubMed] [Google Scholar]
- 64. Pradas I, Rovira‐Llopis S, Naudí A, et al. Metformin induces lipid changes on sphingolipid species and oxidized lipids in polycystic ovary syndrome women. Sci Rep. 2019;9:16033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dunn WB, Broadhurst D, Begley P, et al. Procedures for large‐scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat Protoc. 2011;6:1060‐1083. [DOI] [PubMed] [Google Scholar]
- 66. Broadhurst D, Begley P, Zelena E, et al. Guidelines and considerations for the use of system suitability and quality control samples in mass spectrometry assays applied in untargeted clinical metabolomic studies. Metabolomics. 2018;14:1‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hernández‐Alvarez MI, Sebastián D, Vives S, et al. Deficient endoplasmic reticulum‐mitochondrial phosphatidylserine transfer causes liver disease. Cell. 2019;177:881‐895. [DOI] [PubMed] [Google Scholar]
- 68. Koelmel JP, Kroeger N, Ulmer CZ, et al. LipidMatch: an automated workflow for rule‐based lipid identification using untargeted high‐resolution tandem mass spectrometry data. BMC Bioinformatics. 2017;18:331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wishart DS, Feunang YD, Marcu A, et al. HMDB 4.0 — the human metabolome database for 2018. Nucleic Acids Res. 2018;46:D608‐D617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pang Z, Chong J, Li S, Xia J. MetaboAnalystR 3.0: toward an optimized workflow for global metabolomics. Metabolites. 2020;10:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jové M, Pradas I, Dominguez‐Gonzalez M, Ferrer I, Pamplona R. Lipids and lipoxidation in human brain aging. Mitochondrial ATP‐synthase as a key lipoxidation target. Redox Biol. 2019;23:101082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sultana R, Perluigi M, Butterfield DA. Lipid peroxidation triggers neurodegeneration: a redox proteomics view into the Alzheimer disease brain. Free Radic Biol Med. 2013;62:157‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sonnino S, Aureli M, Grassi S, Mauri L, Prioni S, Prinetti A. Lipid rafts in neurodegeneration and neuroprotection. Mol Neurobiol. 2014;50:130‐148. [DOI] [PubMed] [Google Scholar]
- 74. Wood PL, Medicherla S, Sheikh N, et al. Targeted lipidomics of fontal cortex and plasma diacylglycerols (DAG) in mild cognitive impairment and Alzheimer's disease: validation of DAG accumulation early in the pathophysiology of Alzheimer's disease. J Alzheimers Dis. 2015;48:537‐546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wang M, Han X. Advanced shotgun lipidomics for characterization of altered lipid patterns in neurodegenerative diseases and brain injury. Methods Mol Biol. 2016;1303:405‐422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. González‐Domínguez R, García‐Barrera T, Gómez‐Ariza JL. Combination of metabolomic and phospholipid‐profiling approaches for the study of Alzheimer's disease. J Proteomics. 2014;104:37‐47. [DOI] [PubMed] [Google Scholar]
- 77. Van Veldhoven PP. Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J Lipid Res. 2010;51:2863‐2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kemp S, Wanders R. Biochemical aspects of X‐linked adrenoleukodystrophy. Brain Pathol. 2010;20:831‐837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Sastry PS. Lipids of nervous tissue: composition and metabolism. Prog Lipid Res. 1985;24:69‐176. [DOI] [PubMed] [Google Scholar]
- 80. Han X. Neurolipidomics: challenges and developments. Front Biosci. 2007;12:2601‐2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rouser G, Galli C, Kritchevsky G. Lipid class composition of normal human brain and variations in metachromatic leukodystrophy, Tay‐Sachs, Niemann‐Pick, Chronic Gaucher’s and Alzheimer’s diseases. J Am Oil Chem Soc. 1965;42:404‐410. [DOI] [PubMed] [Google Scholar]
- 82. O'Brien JS, Sampson EL. Lipid composition of the normal human brain: gray matter, white matter, and myelin. J Lipid Res. 1965;6:537‐544. [PubMed] [Google Scholar]
- 83. Panganamala RV, Horrocks LA, Geer JC, Cornwell DG. Positions of double bonds in the monounsaturated alk‐1‐enyl groups from the plasmalogens of human heart and brain. Chem Phys Lipids. 1971;6:97‐102. [DOI] [PubMed] [Google Scholar]
- 84. Ando S, Chang NC, Yu RK. High‐performance thin‐layer chromatography and densitometric determination of brain ganglioside compositions of several species. Anal Biochem. 1978;89:437‐450. [DOI] [PubMed] [Google Scholar]
- 85. Merrill AH Jr, Sullards MC, Allegood JC, Kelly S, Wang E. Sphingolipidomics: high‐throughput, structure‐specific, and quantitative analysis of sphingolipids by liquid chromatography tandem mass spectrometry. Methods. 2005;36:207‐224. [DOI] [PubMed] [Google Scholar]
- 86. Kosicek M, Hecimovic S. Phospholipids and Alzheimer's disease: alterations, mechanisms and potential biomarkers. Int J Mol Sci. 2013;14:1310‐1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kanno K, Wu MK, Scapa EF, Roderick SL, Cohen DE. Structure and function of phosphatidylcholine transfer protein (PC‐TP)/StarD2. Biochim Biophys Acta. 2007;1771:654‐662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Farine L, Niemann M, Schneider A, Bütikofer P. Phosphatidylethanolamine and phosphatidylcholine biosynthesis by the Kennedy pathway occurs at different sites in Trypanosoma brucei. Sci Rep. 2015;5:16787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Exton JH. Signaling through phosphatidylcholine breakdown. J Biol Chem. 1990;65:1‐4. [PubMed] [Google Scholar]
- 90. Billah MM, Anthes JC. The regulation and cellular functions of phosphatidylcholine hydrolysis. Biochem J. 1990;269:281‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Fagone P, Jackowski S. Phosphatidylcholine and the CDP‐choline cycle. Biochim Biophys Acta. 2013;1831:523‐532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Stone SJ, Vance JE. Cloning and expression of murine liver phosphatidylserine synthase (PSS)‐2: differential regulation of phospholipid metabolism by PSS1 and PSS2. Biochem J. 1999;342:57‐64. [PMC free article] [PubMed] [Google Scholar]
- 93. Li Z, Vance DE. Phosphatidylcholine and choline homeostasis. J Lipid Res. 2008;9:1187‐1194. [DOI] [PubMed] [Google Scholar]
- 94. Lanznaster D, Bourgeais J, Bruno C, et al. TDP‐43‐mediated toxicity in HEK293T cells: a fast and reproducible protocol to be employed in the search of new therapeutic options against amyotrophic lateral sclerosis. Cells. 2019;9:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Arima H, Omura T, Hayasaka T, et al. Reductions of docosahexaenoic acid‐containing phosphatidylcholine levels in the anterior horn of an ALS mouse model. Neuroscience. 2015;297:127‐136. [DOI] [PubMed] [Google Scholar]
- 96. Dean JM, Lodhi IJ. Structural and functional roles of ether lipids. Protein Cell. 2018;9:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Wallner S, Schmitz G. Plasmalogens the neglected regulatory and scavenging lipid Species. Chem Phys Lipids. 2011;164:573‐589. [DOI] [PubMed] [Google Scholar]
- 98. Braverman NE, Moser AB. Functions of plasmalogen lipids in health and disease. Biochim Biophys Acta ‐ Mol Basis Dis. 2012;1822:1442‐1452. [DOI] [PubMed] [Google Scholar]
- 99. Goldfine H. The appearance, disappearance and reappearance of plasmalogens in evolution. Prog Lipid Res. 2010;49:493‐498. [DOI] [PubMed] [Google Scholar]
- 100. Huynh K, Martins RN, Meikle PJ. Lipidomic profiles in diabetes and dementia. J Alzheimers Dis. 2017;59:433‐444. [DOI] [PubMed] [Google Scholar]
- 101. Fahy E, Subramaniam S, Murphy R, et al. Update of the LIPID MAPS comprehensive classification system for lipids. J Lipid Res. 2009;50:S9‐S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Posse de Chaves E, Sipione S. Sphingolipids and gangliosides of the nervous system in membrane function and dysfunction. FEBS Lett. 2010;584:1748‐1759. [DOI] [PubMed] [Google Scholar]
- 103. Trayssac M, Hannun YA, Obeid LM. Role of sphingolipids in senescence: implication in aging and age‐related diseases. J Clin Invest. 2018;128:2702‐2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Rietveld A, Simons K. The differential miscibility of lipids as the basis for the formation of functional membrane rafts. Biochim Biophys Acta. 1998;1376:467‐479. [DOI] [PubMed] [Google Scholar]
- 105. Meikle PJ, Summers SA. Sphingolipids and phospholipids in insulin resistance and related metabolic disorders. Nat Rev Endocrinol. 2017;13:79‐91. [DOI] [PubMed] [Google Scholar]
- 106. Hannun YA, Obeid LM. Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Biol. 2018;19:175‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Martín V, Fabelo N, Santpere G, et al. Lipid alterations in lipid rafts from Alzheimer's disease human brain cortex. J Alzheimers Dis. 2010;19:489‐502. [DOI] [PubMed] [Google Scholar]
- 108. Fabelo N, Martín V, Santpere G, et al. Severe alterations in lipid composition of frontal cortex lipid rafts from Parkinson's disease and incidental Parkinson's disease. Mol Med. 2011;17:1107‐1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Marin R, Fabelo N, Martín V, et al. Anomalies occurring in lipiod profiles and protein distribution in frontal cortex lipoid rafts in dementia with Lewy bodies disclose neurochemical traits partailly shared by Alzheimer’s disease and Parkinson’s disease. Neurobiol Aging. 2017; 49:52‐59. [DOI] [PubMed] [Google Scholar]
- 110. Díaz M, Fabelo N, Ferrer I, Marín R. "Lipid raft aging" in the human frontal cortex during nonpathological aging: gender influences and potential implications in Alzheimer's disease. Neurobiol Aging. 2018;67:42‐52. [DOI] [PubMed] [Google Scholar]
- 111. Houten SM, Wanders RJA, Ranea‐Robles P. Metabolic interactions between peroxisomes and mitochondria with a special focus on acylcarnitine metabolism. Biochim Biophys Acta Mol Basis Dis. 2020;1866:165720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Kuda O, Brezinova M, Silhavy J, et al. Nrf2‐mediated antioxidant defense and peroxiredoxin 6 are linked to biosynthesis of palmitic acid ester of 9‐hydroxystearic acid. Diabetes. 2018;67:1190‐1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request.