

Abstract
Co‐infection with the human pegivirus 1 (HPgV‐1) often has a beneficial effect on disease progression in HIV‐1‐infected individuals. Several HPgV‐1 proteins and peptides, including a 20‐mer peptide (P6‐2) derived from the N‐terminal region of the HPgV‐1 surface protein E2, have been associated with this phenomenon, which is referred to as viral interference. We identified the cysteine residues, the hydrophobic core tetrapeptide, as well as the C‐terminal negative charge as key factors for the HIV‐1 inhibitory activity of P6‐2. Analysis of mutations in P6‐2‐resistant HIV‐1 indicated a binding site for the peptide in the HIV‐1 envelope glycoprotein gp120. In fact, P6‐2 was shown to bind to soluble gp120, as well as to a peptide presenting the gp120 V3 loop. Furthermore, the HIV‐1 inhibitory activity of P6‐2 could be revoked by the V3 loop peptide, thus indicating a molecular mechanism that involves interaction of P6‐2 with the gp120 V3 loop.
Keywords: gp120 V3 loop, HIV-1, human pegivirus, peptides, viral interference
Virus versus virus The HIV‐1 inhibitory activity of P6‐2, a peptide derived from the HPgV‐1 E2 protein, is linked to its cysteine residues, hydrophobic core, and C‐terminal negative charge. Similar to the HIV‐1 coreceptors, P6‐2 binds to the V3 loop of HIV‐1 gp120, which mighty point to a molecular mechanism of viral interference of HPgV‐1 with HIV‐1 infection.
Introduction
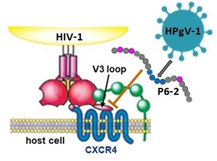
Human Pegivirus 1 (HPgV‐1), previously referred to as GBV−C or hepatitis G virus, is a human blood borne virus that is largely nonpathogenic in healthy individuals.[ 1 , 2 ] HPgV‐1 shares transmission routes and target cells with the human immunodeficiency virus 1 (HIV‐1), facilitating co‐infection with both viruses. The single stranded RNA genome of HPgV‐1 encodes several nonstructural, as well as two envelope proteins, which are presented as heterodimeric spikes on the virus surface. [3] Although the prevalence of HPgV‐1 in the overall population is generally low, it is considerably higher among HIV‐1 infected individuals. Interestingly, such co‐infection with HPgV‐1 has been reported to slow down HIV‐1 disease progression in the patients. [4] This phenomenon, termed viral interference (Figure 1A), manifests itself in reduced HIV‐1 viral load, [5] improved quality of life, [6] enhanced response to antiretroviral therapy, [7] as well as lower morbidity and mortality. [8] At the molecular level, the anti‐HIV‐1 effect of HPVg‐1 could be linked to its envelope proteins E1 and E2,[ 9 , 10 ] as well as to the nonstructural protein NS5 A. [11] Peptides derived from the E2 N terminus were shown to inhibit HIV‐1 infection at low‐micromolar concentrations[ 12 , 13 ] (Figure 1B). The molecular mechanism of this effect, however, as well as its chemical determinants, have not been fully elucidated yet.
Figure 1.
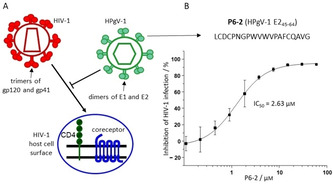
A) Schematic presentation of the viral interference of HPgV‐1 with HIV‐1. B) Sequence and HIV‐1‐neutralizing activity of P6‐2, a peptide derived from the HPgV‐1 surface protein E2. See the Experimental Section for details. Error bars represent SEMs based on three independent experiments.
Results and Discussion
Dissecting the structural requirements for HIV‐1 inhibitory activity of an HPgV‐1‐derived peptide
One of the strongest HIV‐1‐inhibiting peptides derived from the HPgV‐1 E2 protein, termed P6‐2, [12] presents residues 45–64 of the protein (see Table 1 for peptide sequences), and inhibits infection of cells with HIV‐1 at low‐micromolar concentrations (Figure 1B). This peptide, which had been identified using a peptide scan of E2, presenting the protein sequence in the form of overlapping 20‐mer peptides, features a hydrophobic core composed of a WVWV tetrapeptide motif, as well as three cysteine residues. Oxidation of the cysteine thiols can result in a range of different intra‐ and intermolecular disulfides, yielding cyclic, dimeric and oligomeric peptides, respectively. This is illustrated by the heterogeneity of the peptide upon oxidation with hydrogen peroxide (Figure 2A). As complete oxidation with hydrogen peroxide largely abrogates the HIV‐1 inhibitory activity (Table 2), we surmised that at least one of the three cysteine residues must be present in the reduced state, i. e. as a thiol, for the peptide to be active. In order to dissect the role of the different cysteine residues for the HIV‐1 inhibitory activity of P6‐2, we synthesized a range of substitution variants of the peptide, in which one, two, or all three cysteines were replaced with serine (Figure 2B), and tested them for their HIV‐1 inhibitory activity (Table 1). For separate binding assays, some of the peptides were N‐terminally biotinylated, which does not appear to affect the HIV‐1 inhibitory activity of the peptides, as shown by P6‐2 and its biotinylated variant (P6‐2(Bio)), whose IC50 values (2.63 μm and 1.24 μm, respectively) are very similar. Interestingly, of the three cysteine residues, C60 appears to be most important as its individual replacement with serine (P6‐2‐3) had a dramatic effect on the HIV‐1 inhibitory activity, whereas the peptides with serine at C46 (P6‐2‐1) and C48 (P6‐2‐2) still retained some activity. Likewise, in the variants, in which two of the three cysteine residues are replaced with serine (P6‐2‐4, P6‐2‐5 and P6‐2‐6), maintaining C60 (P6‐2‐4) resulted in better preservation of antiviral activity, as compared to the variants in which either C46 or C48 was kept (P6‐2‐5 and P6‐2‐6). Finally, replacing all three cysteines with serine (P6‐2‐7) resulted in complete loss of activity.
Table 1.
Sequences and HIV‐1 inhibitory activity of peptides.
|
Peptide |
Sequence |
IC50±SEM[b] [μm] |
|---|---|---|
|
P6‐2 |
Ac[c]‐45LCDCPNGPWVWVPAFCQAVG64‐OH |
2.63±0.33 (>60[d]) |
|
P6‐2(Bio) |
Biotin‐Ahx[e]‐LCDCPNGPWVWVPAFCQAVG‐OH |
1.24±0.03 |
|
P6‐2 cysteine exchange variants | ||
|
P6‐2‐1 |
Ac‐LSDCPNGPWVWVPAFCQAVG‐OH |
27.29±0.52 |
|
P6‐2‐2 |
Ac‐LCDSPNGPWVWVPAFCQAVG‐OH |
16.01±6.10 |
|
P6‐2‐3 |
Ac‐LCDCPNGPWVWVPAFSQAVG‐OH |
>60 |
|
P6‐2‐4 |
Biotin‐Ahx‐LSDSPNGPWVWVPAFCQAVG‐OH |
10.93±2.28 |
|
P6‐2‐5 |
Biotin‐Ahx‐LCDSPNGPWVWVPAFSQAVG‐OH |
54.64±10.04 |
|
P6‐2‐6 |
Biotin‐Ahx‐LSDCPNGPWVWVPAFSQAVG‐OH |
>60 |
|
P6‐2‐7 |
Ac‐LSDSPNGPWVWVPAFSQAVG‐OH |
>60 |
|
P6‐2‐7(Bio) |
Biotin‐Ahx‐LSDSPNGPWVWVPAFSQAVG‐OH |
n.d. |
|
P6‐2 hydrophobic core exchange variants | ||
|
P6‐2‐8 |
Ac‐LCDCPNGPAVWVPAFCQAVG‐OH |
>60 |
|
P6‐2‐9 |
Ac‐LCDCPNGPWAWVPAFCQAVG‐OH |
19.03±6.83 |
|
P6‐2‐10 |
Ac‐LCDCPNGPWVAVPAFCQAVG‐OH |
>60 |
|
P6‐2‐11 |
Ac‐LCDCPNGPWVWAPAFCQAVG‐OH |
27.48±14.16 |
|
P6‐2 C‐terminal variants | ||
|
P6‐2‐12 |
Ac‐LCDCPNGPWVWVPAFCQAVG‐NH2 |
>60 |
|
P6‐2‐13 |
Ac‐LCDCPNGPWVWVPAFCQAVD‐NH2 |
4.61±1.06 |
|
P6‐2‐14 |
Ac‐LCDCPNGPWVWVPAFCQAVGWGD‐NH2 |
7.56±0.75 |
|
V3 loop peptide | ||
|
V3(Bio) |
Biotin‐Ahx‐G‐Ahx‐[296CTRPNNNTRKRIRIQRGPGRAFVTIGKIGNMRQAHC331][f]‐NH2 |
|
|
V3(Fluo) |
Fluo[g]‐Aoa[h]‐G‐Aoa‐[CTRPNNNTRKRIRIQRGPGRAFVTIGKIGNMRQAHC]‐NH2 |
|
[a] Position numbers are based on the HPgV‐1 E2 protein (P6‐2 variants) and HIV‐1 gp120HxBc2 (V3 loop peptides). [b] Calculated from data from three independent experiments. [c] Ac: acetyl. [d] After oxidation with hydrogen peroxide. [e] Ahx: 6‐aminohexanoic acid. [f] Brackets indicate a disulfide bridge between cysteine residues. [g] Fluo, carboxyfluorescein. [h] Aoa, 8‐amino‐3,6‐dioxaoctanoic acid.
Figure 2.
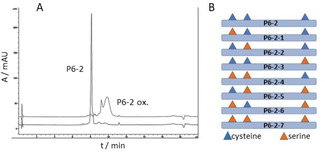
A) HPLC chromatogram of P6‐2 before and after oxidation with hydrogen peroxide. B) Addressing the role of the cysteine residues in P6‐2 through systematic replacement with serine (see Table 1 for peptide sequences and HIV‐1 inhibitory activities).
Table 2.
Inhibition of antibody‐gp120/gp41 interactions by P6‐2.
|
Antibody |
Antibody target |
IC50±SEM[a] [μm] of P6‐2 |
|---|---|---|
|
2G12 |
gp120 glycan |
>300 |
|
VRC01 |
gp120 CD4 binding site |
ca. 50[b] |
|
F425 B4e8 |
gp120 V3 loop |
1.84±0.19 |
|
ID6 |
gp120 N terminus |
>300 |
|
F240 |
gp41 disulfide loop |
93.36±6.47 |
[a] Calculated from data from two independent experiments; [b] IC50 value could not be calculated due to incorrect curve fitting.
Similar to the cysteine residues, the amino acids making up the hydrophobic core of P6‐2, in particular the two tryptophan residues, were shown to be important for the HIV‐1 inhibitory activity. While replacing the two valine residues with alanine (P6‐2‐9 and P6‐2‐11) resulted in an approximately tenfold loss of P6‐activity, the W53A and W55A variants (P6‐2‐8 and P6‐2‐10) were essentially inactive.
The peptide P6‐2 was originally synthesized as a C‐terminal acid, introducing a negative charge that is not present in the E2 protein, from which the peptide was derived. Surprisingly, changing that C‐terminal acid to an amide (P6‐2‐12) abrogates HIV‐1 inhibitory activity, which, however, can be largely restored by replacing G64 with aspartate (P6‐2‐13), re‐introducing a negative charge at the C terminus of the peptide, while maintaining the C‐terminal amide. Apparently, a negative charge at the C terminus is required for HIV‐1 inhibitory activity of the peptide. In the E2 protein, this negative charge may be provided by an aspartate residue (D67) three positions downstream, as a C‐terminally extended peptide amide including this residue (P6‐2‐14) is equally effective as P6‐2‐13.
In summary, using a range of amino acid exchange variants of P6‐2, the cysteine residues, in particular C60, the hydrophobic core, in particular the tryptophan residues, as well as a C‐terminal negative charge, were identified as key determinants for the HIV‐1 inhibitory activity of the peptide.
Exploring the molecular mechanism of HIV‐1 inhibition by P6‐2
Conceptually, the HIV‐1 inhibitory activity of P6‐2 may be linked to a range of molecular interactions involved in the entry of HIV‐1 into its target cell and its replication cycle, respectively. Considering the molecular size and chemical nature of the peptide, however, P6‐2 cannot be expected to passively penetrate membranes, eliminating interference with intracellular replication processes, such as reverse transcription, integration and assembly, as a probable molecular mechanism. Therefore, a more likely mechanism of action is interference of P6‐2 with extracellular virus ‐ host cell protein interactions that result in virus entry into the cell. These interactions are located in the protein spikes on the viral surface, composed of glycoproteins gp120 and gp41, which contact the cellular receptors, that is, CD4 and coreceptors CCR5 and CXCR4, respectively (Figure 1A). Previous studies had shown that P6‐2 does not bind to cellular receptors, but that it interferes with the interaction of an antibody with viral gp41. [13] This antibody recognizes the disulfide loop, which is located between the N‐terminal and C‐terminal heptad repeat regions of gp41. These regions are essential for the fusion of the viral and cellular membranes as a key step in virus entry. Furthermore, a peptide presenting the gp41 disulfide loop had been shown to bind to the HPgV‐1 E2 protein. Based on these data, P6‐2 was postulated to inhibit HIV‐1 infection through interaction with the gp41 disulfide loop, preventing fusion of viral and cellular membranes.
In order to probe that notion, we generated, through long‐term incubation with P6‐2, HIV‐1 variants that can no longer be inhibited by the peptide (IC50>60 μm). The locations of mutations in such resistant viruses often indicate binding sites of the inhibitor. Based on the postulated binding site for P6‐2, that is, the gp41 disulfide loop, we had expected to see mutations in that area in the P6‐2 resistant viruses. Surprisingly, however, most mutations we identified, after 23 rounds of passaging, were located in the second envelope glycoprotein of HIV‐1, that is, gp120, namely in the N terminus (K33N), the C1 region (V120E, S128N), as well as in the V3 loop (A327T) of gp120. Only one mutation (R696K) was found in gp41, namely in the transmembrane region of the protein, which is unlikely to serve as the binding site for a peptide like P6‐2. As this result indicated a binding site for P6‐2 in gp120, rather than in gp41, we probed this interaction and could show that indeed P6‐2 selectively and dose‐dependently binds to recombinant gp120, while the triple serine variant P6‐2‐7, which does not inhibit HIV‐1 infection, does not bind to gp120 either (Figure 3C). In order to identify the binding site for P6‐2 on gp120, we tested the peptide's ability to interfere with the interaction of a range of antibodies with gp120. The epitopes of these antibodies had been mapped to the N terminus (ID6 [14] ), the CD4 binding site (VRC01 [15] ), the glycan shield (2G12 [16] ) and the V3 loop (F425 B4e8 [17] ), respectively, of gp120. Additionally, we also included the gp41 disulfide antibody (F240 [18] ) that had previously been used for inhibition studies with P6‐2. [13] Interestingly, the strongest inhibition by P6‐2 was found for the interaction of gp120 with F425 B4e8 (Table 2, Figure 3A), indicating an interaction of P6‐2 with the V3 loop of HIV‐1 gp120. Inhibition of the interaction of gp120 with VRC01, and of gp41 with F240, respectively, was considerably weaker, and no inhibition was found for the interaction of gp120 with ID6 and 2G12, respectively. Inhibition by P6‐2 could also be shown for the interaction of F425 B4e8 with a peptide presenting the antibody target, that is, the V3 loop of gp120 (Figure 3B), corroborating the notion of an interaction of P6‐2 with the V3 loop. This was further confirmed in direct binding experiments, in which P6‐2, but not the inactive triple serine variant (P6‐2‐7), was shown to dose‐dependently bind to a peptide presenting the gp120 V3 loop (Figure 3D). Notably, this is a further example of a peptide‐peptide interaction, [19] in which two intrinsically unfolded peptides are able to bind to each other, presumably through an induced fit type of binding mechanism.
Figure 3.
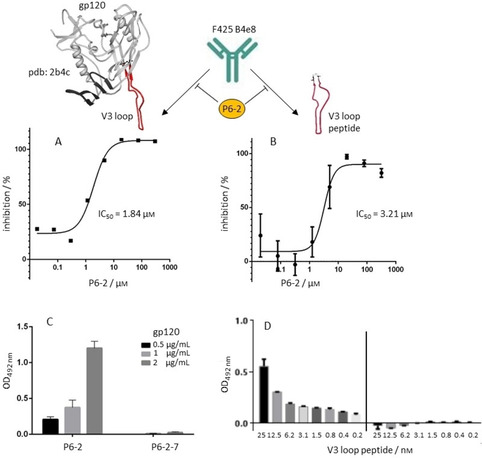
P6‐2 inhibits the interaction of mAb F425 B4e8A) with HIV‐1 gp120 and B) with the V3 loop peptide. P6‐2 binds selectively C) to HIV‐1 gp120 and D) to the V3 loop peptide. See the Experimental Section for experimental details. Error bars represent SEMs based on two experiments.
The V3 loop is the principal component of the coreceptor binding site of gp120, as its sequence governs the coreceptor selectivity (CXCR4 vs. CCR5) of HIV‐1 strains. [20] Binding of P6‐2 to the V3 loop would compete with coreceptor‐V3 loop interaction, preventing attachment of the virus to the coreceptor, and, consequently, entry into the cell. The V3 loop of X4 tropic HIV‐1, which uses CXCR4 as the coreceptor, contacts the second extracellular loop (ECL2) of the receptor, which is also part of a CXCR4 mimetic peptide we have previously published,[ 19 , 21 , 22 ] and whose HIV‐1 inhibitory activity is also based on interaction with the gp120 V3 loop. Interestingly, the CXCR4 ECL2 (182DRYICDRFYPNDLWV196) bears some resemblance with P6‐2. In both peptides, negative charges are important for the interaction with the positively charged V3 loop. It has previously been shown for CXCR4, [23] as well as for the receptor mimetic peptide, [22] that replacing the aspartate residues in ECL2 with alanine results in dramatic loss of binding to gp120, as well as of HIV‐1 inhibitory activity. Likewise, P6‐2 needs a C‐terminal negative charge in order to be active. Furthermore, in the process of HIV‐1 entry, interaction of CXCR4 with the gp120 V3 loop is preceded by contact of gp120 with CD4, which triggers a conformational change within gp120 that exposes its hitherto shielded coreceptor binding site. We have previously shown that soluble CD4 (sCD4) also boosts binding of the CXCR4 mimetic peptide CX4‐M1 to gp120,[ 19 , 22 ] illustrating the functional mimicry of CXCR4 through CX4‐M1. Interestingly, this enhancing effect of sCD4 was now also demonstrated for the interaction of P6‐2 with gp120 (Figure 4A), suggesting that P6‐2 interacts with the gp120 V3 loop in a fashion related to the V3 loop‐CXCR4 interaction. This notion is further supported by the observation that P6‐2 is able to inhibit the interaction of gp120 with mAb 17 (Figure 4B), which belongs to the group of CD4 induced (CD4i) antibodies that recognize the coreceptor binding site of gp120 upon prior contact of gp120 with CD4. It should be noted, however, that, in addition to the V3 loop, the coreceptor binding site of gp120 also comprises its bridging sheet, [24] which hence may be involved in the interaction of gp120 with P6‐2 as well.
Figure 4.
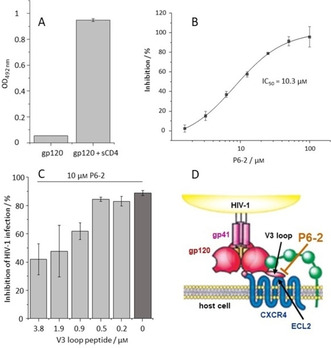
Functional evidence of the P6‐2‐V3 loop interaction. A) The interaction of P6‐2 with HIV‐1 gp120 is enhanced by sCD4. B) P6‐2 inhibits binding of the coreceptor antibody 17B to HIV‐1 gp120. C) The V3 loop peptide counteracts the P6‐2‐induced inhibition of HIV‐1 infection. D) Proposed mechanism for the interference of P6‐2 with HIV‐1 entry into the host cell (modified from ref. 25). See the Experimental Section for experimental details. Error bars represent SEMs based on two experiments.
The interaction of P6‐2 with the gp120 V3 loop could also be shown functionally, that is, in the context of HIV‐1 infection. For that, we tested the V3 loop peptide for its ability to interfere with the infection inhibition effected by P6‐2. As shown in Figure 4C, P6‐2, at 10 μm, inhibits the infection of SEAP cells with HIV‐1 by 86 %. This nearly complete inhibition could be counteracted by the V3 loop peptide, restoring infection of cells with HIV‐1. This effect corresponds well with the notion of P6‐2 binding to the V3 loop peptide, which would interfere with the interaction of P6‐2 with viral gp120, and, consequently, result in recovery of infection.
Based on these data, we are now proposing a mechanism for HIV‐1 inhibition by P6‐2 that involves interaction of the peptide with the V3 loop of HIV‐1 gp120, preventing its interaction with the coreceptor binding site, in particular with ECL2 of CXCR4 (Figure 4D). It should be noted, however, that this is not the sole possible mechanism for the established viral interference of HPgV‐1 with HIV‐1 infection, as other regions of HPgV‐1, including peptides derived from the E1 protein, [26] the non‐structural protein NS5 A, [11] as well as chimera of peptides derived from E1 and E2, [27] have also been shown to interfere with HIV‐1 infection.
Conclusion
The HIV‐1 inhibitory activity of P6‐2, a peptide derived from the HPgV‐1 E2 protein, is linked to its cysteine residues, hydrophobic core, and C‐terminal negative charge. Similar to the cellular coreceptors for HIV‐1, P6‐2 binds to the V3 loop of HIV‐1 gp120, which may point to a molecular mechanism of viral interference of HPgV‐1 with HIV‐1 infection, as well as to a possible novel strategy for the development of HIV‐1 entry inhibitors. Ongoing studies are aimed at improving the metabolic stability of P6‐2 through systematic chemical modification, which includes the incorporation of nonproteinogenic amino acids and other building blocks. Furthermore, strategies to improve the HIV‐1 neutralizing capacity of P6‐2 will include fusion to other HIV‐1 neutralizing peptides (and other molecules) that target alternative regions of the HIV‐1 envelope proteins, such as the CD4 binding site of gp120, or the heptad repeat regions (NHR and CHR) of gp41, generating bispecific molecules.
Experimental Section
Materials. gp120HxBc2 and gp41HxBc2 were obtained from Immune Technology, sCD4 from Sino Biological, Fmoc‐amino acids from Iris Biotech, and synthesis supports from Rapp Polymere. MAbs 2G12,VRC01, F425 B4e8, ID6, 17B and F240 were obtained through the NIH AIDS Research and Reference Reagent Program.
Peptide synthesis. Peptides (see Table 1 for sequences and Supporting Information for analytical data) were synthesized by Fmoc/tBu‐based solid‐phase synthesis, essentially as previously described, [19] on TentaGel® S RAM Resin (peptide amides) or on pre‐loaded Fmoc‐amino acid‐TentaGel® S PHB resins (peptide acids). The N‐terminal amino groups were acetylated with a mixture of acetic anhydride/pyridine/DMF (1 : 2 : 3) for 30 min. The biotin moiety in P6‐2(Bio) and P6‐2‐7(Bio) was introduced by coupling 3 equiv. of biotin/DIC/HOBt in DMF overnight. Fluorescein was introduced into V3(Fluo) by coupling 2 equiv. fluorescein‐N‐hydroxysuccinimide/5 % DIPEA in DMF in the dark overnight. Peptides were cleaved from the resin using Reagent K (TFA/water/phenol/thioanisole/ethane‐1,2‐dithiol; 82.5 : 5 : 5 : 2.5), precipitated in cold cyclohexane/tert‐butyl methyl ether (1 : 1), extracted with water, and lyophilized. V3(Bio) and V3(Fluo) were cyclized by air oxidation at 0.3 mg/mL in 50 % acetonitrile in 0.1 m ammonium acetate, pH 8, for three days. Absence of free SH groups was confirmed by a negative Ellman's test. Peptides were purified by preparative HPLC (conditions: column: Dr. Maisch Reprosil 100, 250×25 mm, flow rate: 9 mL/min, gradient: 20–50 % acetonitrile in H2O (both containing 0.1 % TFA) in 60 min and UV detection at 214 und 280 nm). Peptides were characterized by analytical HPLC with online ESI mass spectrometry detection (LC–MS). Conditions: column: Phenomenex Kinetex 2.6 μm C18 100 Å, 50×2.1 mm, flow rate: 0.4 mL/min, gradient: 5–95 % acetonitrile in H2O (both containing 0.1 % TFA) in 15 min. Stock solutions of purified peptides were prepared at 10 mm in DMSO. For Figure 2A, P6‐2 (67 μm) was oxidized for 30 min. with 2 equiv. H2O2 (134 μm) in 0.1 m phosphate buffer (pH 7.2). See the Supporting Information for analytical data (HPLC chromatograms and ESI‐MS data).
Generation of viral stocks. The plasmid HIV‐1pBRNL4‐3, kindly provided by F. Kirchhoff (University Hospital Ulm), was propagated in XL‐1 Blue competent cells (Stratagene, Agilent Technologies) and transfected into 293T cells using FuGENE HD (Roche). After 48 h, supernatants were cleared by filtering through 0.22 μm Millex‐GS units (Millipore), and stored in aliquots at −80 °C.
SEAP reporter cell HIV‐1 infection assay (Figures 1B and 4C, Table 1). The indicator cell line CEMx174, which was kindly provided by R. E. Means and R. C. Desrosiers (Harvard Medical School, Boston), contained the gene for the secreted alkaline phosphatase (SEAP) under the control of the simian immunodeficiency virus long terminal repeat. [27] The suitability of this cell line for HIV‐1 drug resistance testing has been evaluated extensively. Cells were propagated in RPMI 1640 medium supplemented with 10 % heat‐inactivated (56 °C, 60 min) fetal calf serum, 0.3 mg/mL glutamine, 200 U/mL penicillin, and 90 U/mL streptomycin. Peptides were added to the cells prior to addition of HIV‐1NL4‐3. Three days after infection, cell culture supernatants were removed and analyzed for SEAP activity using the Phospha‐Light kit (Life Technologies) according to the manufacturer's recommendations. Data represent mean values and SEMs of at least three independently performed experiments. Curves were fitted and IC50 values calculated using the program SigmaPlot 11.0.
Selection of resistant virus. HIV‐1NL4‐3 was cultivated on CEMx174 SEAP cells in the presence of P6‐2 peptides at increasing concentrations (0.11–60 μm). After an incubation period of three to six days, depending on the degree of virus replication, 25 μL of cell culture supernatants were transferred to another plate. To select for resistant viruses, supernatants were repeatedly passaged onto wells containing increasing concentrations of the inhibitor, until resistant viruses emerged after several weeks of passaging. Control wells contained virus but no inhibitor. Before viral stocks were frozen, they were passaged once onto MT‐2 cells, kindly provided by the NIH AIDS Research and Reference Reagent program, in the absence of inhibitor.
Mutational analysis. Viral RNA was isolated from cell culture supernatants using the QIAamp DNA Blood Mini Kit (Qiagen) according to the manufacturer's recommendations. The eluted RNA was denatured at 65 °C for 3 min, chilled on ice, and transcribed into cDNA using random hexamers (MWG Biotech) with MuLV reverse transcriptase (Applied Biosystems/Life Technologies) at 42 °C for 30 min and 95 °C for 5 min. HIV‐1 gp120 sequences were amplified using Expand High Fidelity taq polymerase (Roche) to generate four overlapping PCR fragments with primers pairs env01 (5’‐AGAAAGAGCAGAAGACAGTG‐3’) and env02 (5’‐ATCATTCTCCCGCTACTACT‐3’), env1 (5’‐GACATGGTAGAACAGATGCA‐3’) and env2 (5’‐TTTAGAATCGCAAAACCAGC‐3’), env3 (5’‐TATCCTTTGAGCCAATTCCC‐3’) and env4 (5’‐CTCCTGAGGATTGCTTAAAGA‐3’), and env5 (5’‐CAGTTTTAATTGTGGAGGGG‐3’) or env5a (5’‐GCCAATTTCACAGACAATGCT‐3’) and env6 (5’‐TTCACTTCTCCAATTGTCCC‐3’). For these purposes, samples were denatured at 95 °C for 5 min, followed by 40 cycles of 95 °C for 30 s, 50 °C for 30 s, and 72 °C for 75 s, and a final elongation step at 72 °C for 5 min. PCR products were visualized on 2 % agarose gels and further purified using the MinElute PCR Purification kit following the manufacturer's instructions (Qiagen). DNA concentrations were determined using NanoDrop‐ND‐1000 (Peqlab Biotechnologie). Each PCR fragment was sequenced in forward and reverse direction, and analyzed using the software SeqMan (DNASTAR, Madison, WI).
P6‐2–gp120 binding assay (Figure 3C and 4A). High binding Immulon microtiter plates were coated overnight at 4 °C with streptavidin (4.0 μg/mL) in 0.1 M sodium carbonate buffer pH 9.5. Unspecific binding was blocked with 1 % BSA in 0.1 m phosphate buffer pH 7.2, 200 μL/well for 1 hour. The streptavidin‐coated plate was incubated with 100 μL biotinylated P6‐2(Bio) and P6‐2‐7(Bio), respectively, at 5 μm for 2 h. Plates were then incubated for 3 h with gp120HxBc2 at 2, 1 and 0.5 μg/mL, respectively. For the data shown in Figure 4A, sCD4 (0.125 μg/mL) was added to the gp120 solution. Bound gp120 was detected by incubation with sheep anti‐gp120 (D7324) from Aalto Bio Reagents (0.2 μg/mL), followed by rabbit anti‐sheep HRP conjugate from Dianova (3 μg/mL), both 100 μL/well for one hour. After each incubation, plates were washed four times with 200 μL 0.1 m phosphate buffer pH 7.2. Plates were developed with 100 μL/well OPD (1 mg/mL) in the presence of 0.03 % H2O2 for approximately 10 minutes in the dark. After the reaction was stopped with 50 μL/well 2 m H2SO4, absorbance was read at 492 nm. All data points present means of duplicates, error bars present SEMs.
P6‐2‐V3 loop binding assay (Figure 3D). High binding Immulon microtiter plates were coated with streptavidin and biotinylated peptides as described above. Plates were then incubated for 3 h with V3(Fluo) at twofold serial dilutions starting at 25 nm. Bound V3(Fluo) was detected by incubation with anti‐fluorescein‐HRP (0.2 μg/mL). After each incubation, plates were washed four times with 200 μL 0.1 m phosphate buffer pH 7.2. Plates were developed as described above. All data points present means of duplicates, error bars present SEMs.
Competitive antibody–gp120/gp41/V3 loop peptide binding assays (Figure 3A and B, Table 2). High binding Immulon microtiter plates were coated overnight at 4 °C with gp120HxBc2 (0.5 μg/mL) in 0.1 m sodium carbonate buffer pH 9.5. For the F425 B4e8‐V3 loop peptide assay, plates were incubated with streptavidin as described above, followed by 100 μL 5 μm V3(Bio). Unspecific binding was blocked with 1 % BSA in 0.1 m phosphate buffer pH 7.2, 200 μL/well for 1 h. Plates were then incubated for 18 h at 4 °C with 50 μL P6‐2 at fourfold serial dilutions, starting at 300 μm, together with 50 μL of the respective antibody (ID6: dilution 1 : 1000, 2G12: 0.05 μg/mL, VRC01: 0.1 μg/mL, F425 B4e8: 1.5 μg/mL). Bound antibody was detected by incubation with anti‐human IgG‐HRP (Sigma‐Aldrich, 1 : 5000 dilution, 100 μL/well) for one hour. For the competitive F240‐gp41 binding assay, plates were coated overnight at 4 °C with anti‐human IgG antibody (Sigma‐Aldrich, 1 : 2500 dilution, 100 μL/well). Unspecific binding was blocked with 1 % BSA in 0.1 m phosphate buffer pH 7.2, 200 μL/well for 1 h. Plates were then incubated with F240 (100 μL/well, 0.016 μg/mL) for 2.5 h, followed by incubation for 18 h at 4 °C with 50 μL P6‐2 at fourfold serial dilutions, starting at 300 μm, along with 50 μL of gp41HxBc2 (0.2 μg/mL). Bound gp41 was detected by incubation with anti‐HIS‐HRP (Sigma‐Aldrich 100 μL/well, dilution 1 : 5000). After each incubation, plates were washed four times with 200 μL 0.1 m phosphate buffer pH 7.2. Plates were developed as described above. IC50 values were determined using the regression wizard of GraphPad Prism. All data points present means of duplicates, error bars present SEMs.
Competitive 17B‐gp120 binding assay (Figure 4B) A high binding Costar microtiter half area plate was coated overnight at 4 °C with mAb 17B (0.5 μg/mL) in 0.1 m sodium carbonate buffer pH 9.5. Unspecific binding was blocked with 1 % BSA in 0.1 m phosphate buffer pH 7.2, 110 μL/well for 1 h. The plate was then incubated for 5 h with 50 μL P6‐2 at twofold serial dilutions, starting at 100 μm, together with 50 μL gp120HxBc2 (0.25 μg/mL) containing sCD4 (0.125 μg/mL). Bound gp120 was detected via its His tag by incubation with 100 μL anti‐His‐HRP (dilution 1 : 20 000) for 1 h. After each incubation, plates were washed four times with 200 μL 0.1 m phosphate buffer (pH 7.2). Plates were developed as described above. All data points present means of duplicates. IC50 values were determined as described above.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
T.R. was supported by the ReForM A program of the Faculty of Medicine, University of Regensburg. K.M.F. was supported by GRK 1910, funded by the German Research Foundation. Monoclonal antibodies (2G12, VRC01, F425 B4e8, ID6, F240, 17B) were obtained through the NIH AIDS Research and Reference Reagent Program. The indicator cell line CEMx174 was kindly provided by R. E. Means and R. C. Desrosiers, gp12096ZM by R. Wagner (University of Regensburg), and the plasmid HIV‐1NL4‐3 by F. Kirchhoff (Ulm University Hospital). B.S. thanks André Gessner for continuous support. Open access funding enabled and organized by Projekt DEAL.
R. Hoffmann, T. Ruegamer, J. Schaubächer, A. Rohrhofer, P. Kirmeß, K. M. Fiebig, B. Schmidt, J. Eichler, ChemMedChem 2021, 16, 1290.
References
- 1. Leary T. P., Muerhoff A. S., Simons J. N., Pilot-Matias T. J., Erker J. C., Chalmers M. L., Schlauder G. G., Dawson G. J., Desai S. M., Mushahwar I. K., J. Med. Virol. 1996, 48, 60–67. [DOI] [PubMed] [Google Scholar]
- 2. Simons J. N., Leary T. P., Dawson G. J., Pilot-Matias T. J., Muerhoff A. S., Schlauder G. G., Desai S. M., Mushahwar I. K., Nat. Med. 1995, 1, 564–569. [DOI] [PubMed] [Google Scholar]
- 3. Stapleton J. T., Semin. Liver Dis. 2003, 23, 137–148. [DOI] [PubMed] [Google Scholar]
- 4. Toyoda H., Fukuda Y., Hayakawa T., Takamatsu J., Saito H., J. Acquired Immune Defic. Syndr. Hum. Retrovirol. 1998, 17, 209–213. [DOI] [PubMed] [Google Scholar]
- 5. Hattori J., Okumura N., Yamazaki Y., Uchiyama M., Hamaguchi M., Nishiyama Y., Kaneda T., Microbiol. Immunol. 2007, 51, 193–200. [DOI] [PubMed] [Google Scholar]
- 6. Tillmann H. L., Manns M. P., Claes C., Heiken H., Schmidt R. E., Stoll M., AIDS Care 2004, 16, 736–743. [DOI] [PubMed] [Google Scholar]
- 7. Souza I. E., Zhang W., Diaz R. S., Chaloner K., Klinzman D., Stapleton J. T., HIV Med. 2006, 7, 25–31. [DOI] [PubMed] [Google Scholar]
- 8. Tillmann H. L., Heiken H., Knapik-Botor A., Heringlake S., Ockenga J., Wilber J. C., Goergen B., Detmer J., McMorrow M., Stoll M., Schmidt R. E., Manns M. P., N. Engl. J. Med. 2001, 345, 715–724. [DOI] [PubMed] [Google Scholar]
- 9. Jung S., Eichenmuller M., Donhauser N., Neipel F., Engel A. M., Hess G., Fleckenstein B., Reil H., AIDS 2007, 21, 645–647. [DOI] [PubMed] [Google Scholar]
- 10. Mohr E. L., Stapleton J. T., J. Viral. Hepat. 2009, 16, 757–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xiang J., McLinden J. H., Chang Q., Kaufman T. M., Stapleton J. T., Proc. Natl. Acad. Sci. USA 2006, 103, 15570–15575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Koedel Y., Eissmann K., Wend H., Fleckenstein B., Reil H., J. Virol. 2011, 85, 7037–7047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eissmann K., Mueller S., Sticht H., Jung S., Zou P., Jiang S., Gross A., Eichler J., Fleckenstein B., Reil H., PLoS One 2013, 8, e54452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dickey C., Ziegner U., Agadjanyan M. G., Srikantan V., Refaeli Y., Prabhu A., Sato A., Williams W. V., Weiner D. B., Ugen K. E., DNA Cell Biol. 2000, 19, 243–252. [DOI] [PubMed] [Google Scholar]
- 15. Wu X., Yang Z. Y., Li Y., Hogerkorp C. M., Schief W. R., Seaman M. S., Zhou T., Schmidt S. D., Wu L., Xu L., Longo N. S., McKee K., O′Dell S., Louder M. K., Wycuff D. L., Feng Y., Nason M., Doria-Rose N., Connors M., Kwong P. D., Roederer M., Wyatt R. T., Nabel G. J., Mascola J. R., Science 2010, 329, 856–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Buchacher A., Predl R., Strutzenberger K., Steinfellner W., Trkola A., Purtscher M., Gruber G., Tauer C., Steindl F., Jungbauer A., et al., AIDS Res. Hum. Retroviruses 1994, 10, 359–369. [DOI] [PubMed] [Google Scholar]
- 17. Pantophlet R., Aguilar-Sino R. O., Wrin T., Cavacini L. A., Burton D. R., Virology 2007, 364, 441–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cavacini L. A., Emes C. L., Wisnewski A. V., Power J., Lewis G., Montefiori D., Posner M. R., AIDS Res. Hum. Retroviruses 1998, 14, 1271–1280. [DOI] [PubMed] [Google Scholar]
- 19. Gross A., Möbius K., Haussner C., Donhauser N., Schmidt B., Eichler J., Front. Immunol. 2013, 4, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hwang S. S., Boyle T. J., Lyerly H. K., Cullen B. R., Science 1991, 253, 71–74. [DOI] [PubMed] [Google Scholar]
- 21. Gross A., Brox R., Damm D., Tschammer N., Schmidt B., Eichler J., Bioorg. Med. Chem. 2015, 23, 4050–4055. [DOI] [PubMed] [Google Scholar]
- 22. Möbius K., Durr R., Haussner C., Dietrich U., Eichler J., Chem. Eur. J. 2012, 18, 8292–8295. [DOI] [PubMed] [Google Scholar]
- 23. Brelot A., Heveker N., Adema K., Hosie M. J., Willett B., Alizon M., J. Virol. 1999, 73, 2576–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shaik M. M., Peng H., Lu J., Rits-Volloch S., Xu C., Liao M., Chen B., Nature 2019, 565, 318–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Doranz B. J., Baik S. S., Doms R. W., J. Virol. 1999, 73, 10346–10358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gomara M. J., Sanchez-Merino V., Paus A., Merino-Mansilla A., Gatell J. M., Yuste E., Haro I., Biochim. Biophys. Acta 2016, 1860, 1139–1148. [DOI] [PubMed] [Google Scholar]
- 27. Gomara M. J., Perez Y., Martinez J. P., Barnadas-Rodriguez R., Schultz A., von Briesen H., Peralvarez-Marin A., Meyerhans A., Haro I., Sci. Rep. 2019, 9, 3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary