

Summary
Background:
Upregulated glucose metabolism is a common feature of tumors. Glucose can be broken down by either glycolysis or the oxidative pentose phosphate pathway (oxPPP). The relative usage within tumors of these catabolic pathways remains unclear. Similarly, the extent to which tumors make biomass precursors from glucose, versus take them up from the circulation, is incompletely defined.
Methods:
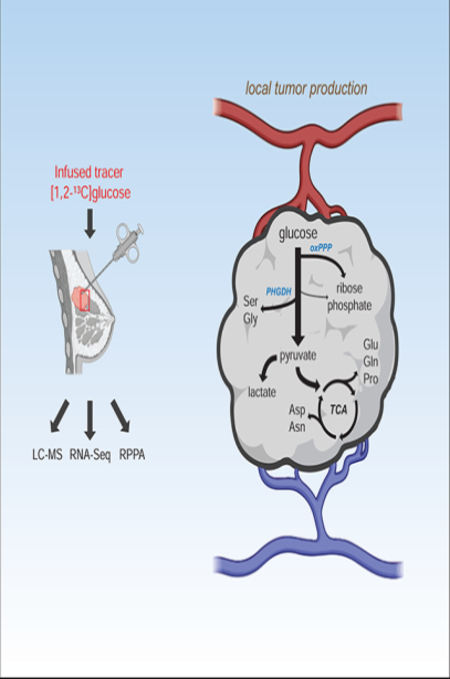
We explore human triple negative breast cancer (TNBC) metabolism by isotope tracing with [1,2-13C]glucose, a tracer that differentiates glycolytic versus oxPPP catabolism and reveals glucose-driven anabolism. Patients enrolled in clinical trial NCT03457779 and received IV infusion of [1,2-13C]glucose during core biopsy of their primary TNBC. Tumor samples were analyzed for metabolite labeling by liquid chromatography-mass spectrometry (LC-MS). Genomic and proteomic analyses were performed and related to observed metabolic fluxes.
Findings:
TNBC ferments glucose to lactate, with glycolysis dominant over the oxPPP. Most ribose phosphate is nevertheless produced by oxPPP. Glucose also feeds amino acid synthesis, including of serine, glycine, aspartate, glutamate, proline and glutamine (but not asparagine). Downstream in glycolysis, tumor pyruvate and lactate labeling exceeds that found in serum, indicating that lactate exchange via monocarboxylic transporters is less prevalent in human TNBC compared with most normal tissues or non-small cell lung cancer.
Conclusions:
Glucose directly feeds ribose phosphate, amino acid synthesis, lactate, and the TCA cycle locally within human breast tumors.
eTOC blurb:
Ghergurovich et al. infused triple negative breast cancer patients with [1,2-13C]glucose to assess tumor metabolism. Metabolite labeling revealed a blend of glucose oxidation, glucose-driven biosynthesis, and classical Warburg metabolism, with most tumor lactate locally produced.
Graphical Abstract
Introduction
Breast cancer afflicts nearly 2 million women annually world-wide 1. The “triple negative” subtype (TNBC), comprising tumors that lack expression of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2), represents ~15% of all breast tumors and is associated with worse patient outcomes 2. Few targeted therapies exist for TNBC and new treatment options are needed 2,4.
Alterations in glucose metabolism are commonly observed in cancer 5. Enhanced tumor glucose uptake provides the biochemical basis for the widely utilized imaging modality 18- fluoro-2-deoxyglucose positron emission tomography (FDG-PET) 6. Even under aerobic conditions, cancer cells often ferment glucose to lactate, a longstanding observation known as the Warburg effect 7. Such metabolism is thought to facilitate synthesis of macromolecular precursors from glucose, including ribose phosphate for nucleotides, nicotinamide adenine dinucleotide phosphate (NADPH) for reductive biochemistry, non-essential amino acids including aspartate, glutamine, serine and glycine for protein and nucleotide synthesis, and fatty acids for assembly of membranes. Tumors require all of these components to grow. It remains unclear, however, the extent to which different tumor types synthesize these components internally de novo from glucose, or instead import them or more closely related precursors (e.g. ribose for ribose phosphate) from the circulation 8,9.
Oncogenes can drive both nutrient uptake and utilization. For example, the master transcriptional regulator MYC promotes glucose uptake (via glucose transporter 1, GLUT1) 10,11, glycolysis 10–13, pyruvate reduction to lactate (via lactate dehydrogenases, LDHA,B) 14,15, lactate and pyruvate transport (via the monocarboxylate transporters, MCT1,4) 16, the pentose phosphate pathway (PPP) 12,13, de novo serine and glycine production 12,17,18, entry of pyruvate into the tricarboxylic acid cycle (TCA) 12,19, glutamine uptake 20,21, glutaminolysis 20,21, de novo glutamine production via glutamine synthetase (GS)22 and the biosynthesis of the glutamate-derivative proline23.
Mounting evidence suggests glucose metabolism is disrupted in TNBC 24. Of the breast tumor subtypes, TNBC and the frequently overlapping “basal-like” subtype 3 have the highest FDG-PET uptake 25,26. FDG-PET activity correlates with MYC, glycolytic enzyme, and pentose phosphate pathway enzyme expression in these tumors 27. Elevated transcript levels of GLUT1, LDHA, and MCT1 are often observed, the latter two being associated with poorer prognoses 28–32. LDHA and MCT1 work in concert to interconvert tumor pyruvate and circulating lactate, and can support lactate secretion as waste, lactate uptake as fuel 33,34, or redox buffering between the tumor and circulating lactate and pyruvate 35,36.
While metabolic upregulation in tumors is mediated mainly through epigenetics and oncogene signaling, upregulation of the serine pathway also occurs through genomic amplification of the enzyme phosphoglycerate dehydrogenase (PHGDH). Such amplification occurs in roughly 39% of melanomas and 6% of breast cancers 37–39. In breast cancers lacking PHGDH amplification, the enzyme is nevertheless often transcriptionally upregulated 37. These observations have led to great interest in targeting PHGDH, although subsequent studies suggest that its suppression per se is insufficient in mouse models to stop the growth of established tumors 40.
Isotopically enriched (e.g. 13C, 2H, 15N) nutrient tracers coupled with liquid chromatography mass spectrometry (LC-MS) or nuclear magnetic resonance (NMR) detection can provide insight into pathway fluxes in complex metabolic systems 41,42. Recent work has employed this methodology in human tumors. Infusion of [U-13C]glucose during surgical brain tumor resection proved simultaneous occurrence of both TCA cycle-mediated glucose oxidation and pyruvate reduction to lactate 43. Infusion of [U-13C]glucose and [U-13C]lactate in patients with non-small cell lung adenocarcinoma (NSCLC) 44,45 and related murine GEMM models 46 exposed local uncoupling of glycolysis and TCA cycles in tumors, with rapid mixing of tumor and circulating pyruvate and lactate pools. In these cases, circulating lactate directly supplies carbon to the TCA cycle, with glucose contributing to metabolism downstream of pyruvate and lactate in part indirectly via circulating lactate 44–46. Infusion of [U-13C]glucose in patients with renal cell carcinoma undergoing nephrectomy demonstrated enhanced glucose contribution to glycolysis and lactate but decreased contribution to downstream TCA metabolism, consistent with renal cell carcinoma tumors being highly glycolytic with impaired glucose oxidation 47. Most recently, infusion of [U-13C]glucose in pediatric patients with various childhood malignancies revealed glycolytic and TCA activity across these tumors48.
Here, patients with newly diagnosed, palpable stage II or III TNBC received an IV infusion of [1,2-13C]glucose prior to biopsy of their primary breast cancer. This tracer was chosen because the positional labeling reveals the magnitude of oxidative pentose phosphate pathway (oxPPP) flux relative to glycolysis and the source of the key nucleotide component ribose-5-phosphate 49. Specifically, the oxPPP releases the first carbon (C1) of glucose as carbon dioxide and thereby generates M+1 (i.e. containing one 13C) labeled ribulose-5-phosphate (and by extension, ribose-5-phosphate), lower glycolytic intermediates, pyruvate, and lactate from [1,2-13C]glucose. In contrast, glycolysis and the non-oxPPP generate only M+2 forms (i.e. containing two 13C atoms). We find that TNBCs have modest oxPPP flux relative to glycolysis, but nevertheless generate most of their ribose-5-phosphate by the oxPPP. In addition, glucose flows locally within breast tumors into serine, glycine, pyruvate, lactate, the TCA cycle, aspartate, glutamate, proline and glutamine. Compared to lung tumors, exchange between tumor lactate and circulating lactate is less in TNBCs. Thus, compared to lung cancers, TNBCs have a more isolated metabolic microenvironment.
Results
Glycolysis predominates over the oxPPP in breast tumors
Twelve patients received an IV infusion of [1,2-13C]glucose for 1 h prior to needle core biopsy of their untreated primary TNBCs (Figure 1A). Individual patient clinical characteristics are provided in Table S1 and, together with whole exome sequencing-based driver mutation calls, are summarized in Figure S1A and Table S2. Similar to other stable isotope tracing studies in human cancer patients, participants received an IV bolus (6 g) followed by a continuous infusion (6 g/h) of isotopically enriched glucose 43–45,47 This approach led to ~ 35% serum isotopic enrichment of glucose at the time of biopsy of the primary breast cancer mass (Figure 1B, Figure S1B). Substantial labeling was observed also in circulating lactate and pyruvate (Figure 1B, Figure S1B). In line with other human trials in which labeled glucose was infused, about 40% of circulating lactate came from glucose, compared to about 65% in similar studies in mice (Figure 1C, Figure S1C) 44–47,50,51. This interspecies difference may reflect methodological issues (i.e. length of infusion, use of anesthesia) and/or a greater contribution from (unlabeled) muscle glycogen to circulating lactate pools in humans 51,52. For all TNBC patients, the M+2 isotopomer was the dominant labeled lactate form (Figure S1D), indicating little tracer scrambling mediated by gluconeogenesis during the infusion and dominance of glycolysis over the oxPPP at the whole-body level (Figure 2A).
Figure 1. Infusion of [1,2-13C]glucose in patients with TNBC.

a) Infusion of [1,2-13C]glucose. Blue circles represent 13C, red arrows indicate serum collection and black arrow biopsy collection. b) Serum isotopic labeling of glucose, pyruvate and lactate from [1,2-13C]glucose during infusion (mean, ± 95% confidence interval, n = 12 at 30 min, n = 11 at 60 min, 3 technical replicates per patient). c) Average per carbon labeling of circulating lactate normalized to that of circulating glucose in indicated human and mouse infusion studies (mean ± SEM, n = number of individuals infused). See also Figure S1.
Figure 2. Glycolytic flux exceeds pentose phosphate activity in human breast tumors.

a) Tracer strategy for comparing glycolysis and pentose phosphate pathway flux using [1,2-13C]glucose. Blue circles depict glycolytic metabolites produced exclusively by glycolysis (M+2 labeling); orange circles depict glycolytic intermediates arising from glucose that first passed through the oxPPP (M+1). b) Individual patient M+1 and M+2 labeling of tumor lactate (mean ± SEM, n = 2 needle biopsy samples per patient), and c) M+1 and M+2 labeling of tumor glycolytic species (mean ± SEM, n = 24 total biopsy samples collected from twelve patients) from [1,2-13C]glucose. Dominance of M+2 fraction means that lower glycolytic intermediates are mainly from glycolysis, not oxPPP flux. G6P, glucose-6-phosphate; 3PG, 3-phosphoglycerate; R5P, ribose-5-phosphate and its isomers ribulose-5-phosphate and xyulose-5-phosphate. See also Figure S2.
Similarly, in TNBCs, M+2 labeling greatly exceeds M+1 labeling throughout glycolysis, pyruvate, and lactate (Figure 2B–C, Figure S2A–C), demonstrating low oxPPP flux relative to glycolysis in the production of these metabolites. While M+1 labeling in tumor lower glycolytic intermediates and lactate was low, it was clearly detectable, with a ratio of M+1/M+2 of 6–9% implying that tumor lower glycolytic intermediates come roughly 90–95% from glycolysis and 5–10% from the oxPPP.
Most tumor ribose-5-phosphate comes from the oxPPP
We next sought to determine the main source of ribose-5-phosphate in TNBC, which could come from the circulating ribose, the oxPPP, or the non-oxPPP (Figure 3A). Ribose-5-phosphate was not detected in the circulation, and circulating ribose labeling was low (undetectable in most patients) (Figure S2D). In contrast, tumor ribose-5-phosphate became substantially labeled (Figure 3B), indicating synthesis within TNBCs. Total tumor glucose-6-phosphate and ribose-5-phosphate labeling were tightly correlated (Figure 3C), with ribose-5-phosphate labeling consistently about half of glucose-6-phosphate labeling, suggesting that about half of ribose-5-phosphate comes from an alternative source, likely nucleotide recycling.
Figure 3. oxPPP is primary source of ribose phosphate in TNBCs.

a) Tracer strategy for comparing oxPPP and non-oxPPP activity using [1,2-13C]glucose. Blue circles depict the path tracer 13C carbons take to produce R5P via the non-oxPPP; orange circles depict the path taken via the oxPPP. Since one glucose carbon is lost as CO2 during oxPPP catabolism, M+1 labeling of R5P indicates oxPPP production, while M+2 labeling indicates non-oxPPP production. b) Individual patient labeling of R5P in TNBCs tumors (mean ± SEM, n = 2 biopsy samples per patient). c) Total labeling (the sum of M+1 and M+2) of R5P correlates with that of G6P. Fit using linear regression. d) Average tumor labeling of G6P and R5P (mean ± SEM, n = 24 total biopsy samples collected from twelve patients). Dominance of M+1 fraction indicates the oxPPP is the primary PPP arm for producing R5P. G6P, glucose-6-phosphate; R5P, ribose-5-phosphate and its isomers ribulose-5-phosphate and xyulose-5-phosphate.
Do tumors use the oxPPP or non-oxPPP to make their ribose? Comparison of M+1 and M+2 labeling provides an estimate of relative oxPPP to non-oxPPP contribution to ribose-5-phosphate and thus nucleotide synthesis (Figure 3A). In each TNBC sample, labeled ribose-5-phosphate was majority M+1, indicating production by the oxPPP. A considerable M+2 fraction, indicating production also by the non-oxPPP, was observed in some patients (Figure 3B, D). Downstream nucleotide labeling itself was minimal (Figure S2E–F), which together with the above low contribution of oxPPP to lower glycolytic intermediates implies low overall oxPPP flux relative to glycolysis. Thus, glycolysis predominates over the oxPPP, but the oxPPP is active and responsible for making most ribose-5-phosphate in TNBCs.
TNBCs produce serine and glycine from glucose
We next explored TNBC production of serine and glycine. Like ribose, these can come from the circulation or synthesis within the tumor. The synthetic pathway starts with the glycolytic intermediate 3-phosphoglycerate via the enzyme PHGDH, which is genomically amplified in some TNBCs 37. No carbon bonds are broken or formed in making serine from 3-phosphoglycerate, thus newly synthesized serine should have mainly M+2 labeling (Figure 4A). Subsequent catabolism by serine hydroxymethyltransferase (SHMT) yields M+1 labeled glycine. Reverse flux from glycine to serine via SHMT will generate M+1 labeled serine.
Figure 4. TNBCs produce serine and glycine from glucose.

a) Tracing of de novo serine/ glycine production using [1,2-13C]glucose. Blue circles (M+2 labeling) depict 13C carbons arising directly from [1,2-13C]glucose by de novo serine production; orange circles (M+1 labeling) depict labeled glycine and serine arising from subsequent reversible SHMT activity. b) Individual patient labeling of serine in TNBCs (mean ± SEM, n = 2 biopsy samples per patient). c) Fractional carbon labeling of tumor glycine correlates with that of tumor serine. d) Fractional carbon tumor serine labeling normalized to that of 3PG in individual patients (mean ± SEM, n = 2 biopsy samples per patient). This ratio serves as an estimate of de novo serine production; purple bars indicate patients designated as having “high” de novo serine production. e) PHGDH protein expression (as determined by RPPA analysis) correlates with de novo serine production (mean, n = 2 biopsy samples per patient). Purple dots represent same patients as purple bars in d). f) Fractional carbon tumor serine labeling normalized to that of 3PG in indicated cancers (mean ± SEM, each point represents a single tumor sample of several collected from each of 5 (brain tumors), 5 (RCC) or 12 (TNBC) patients; one-way ANOVA w/Tukey’s correction). 3PG, 3-phosphoglycerate; PHGDH, phosphoglycerate dehydrogenase; SHMT, serine hydroxymethyl transferase * = p < 0.05, *** = p < 0.001. See also Figure S3.
Across patients, none showed detectable serine labeling in the circulation (Figure S3A). In contrast, all showed detectable serine and glycine labeling within the tumors (Figure 4B, Figure S3B). The total fraction of labeled serine and glycine varied markedly across patients, with serine and glycine fractional per carbon labeling strongly correlated (Figure 4C). Glycine labeling was about half that of serine labeling, suggesting that TNBCs get about half of their glycine from serine (and the rest likely from the circulation). The relative abundance of M+1 and M+2 serine was similar in many patients, indicating substantial reversible SHMT flux.
The labeling fraction of serine to 3-phosphoglycerate (3PG) roughly corresponds to the fraction of serine synthesized de novo, which was up to 50% in some patients’ tumors (Figure 4D), indicating that some TNBCs synthesize a majority of their serine de novo. These measurements are a lower bound on the net contribution of internal tumor serine synthesis: the actual contribution might be bigger but diluted by exchange with unlabeled circulating serine. To better contextualize these findings, we assessed serine production from glucose across various murine tissues (Figure S3C). TNBCs with high serine synthesis had activity comparable to that of the pancreas and brain, the mouse tissues with the highest de novo serine production.
To relate these metabolic fluxes to enzyme expression, we conducted RNA-Seq and proteomic analysis (RPPA) on core biopsies from these patients’ primary breast cancers (see Table S3 for processed RNA-Seq data and Table S4 for normalized RPPA data). We observed a strong correlation between de novo serine production and PHGDH protein (but not RNA) expression in the tumor compartment of these biopsies (Figure 4E, Figure S3D). To compare this finding with other tumor types, we analyzed de novo serine production from glucose in RCC and brain tumors 43 (serine labeling was not available for NSCLC). As expected given the impermeability of the blood-brain barrier to serine 53, brain tumors had the highest fraction of serine from de novo synthesis. The TNBCs on average derived more serine from glucose than RCC, due largely to a few individual TNBC samples with high serine synthesis (Figure 4D,F). In line with this observation, analysis of de novo serine biosynthesis genes from The Cancer Genome Atlas (TCGA) demonstrated lower expression in RCC compared with TNBC (Figure S3E). Thus, de novo serine and glycine synthesis is a metabolic hallmark of TNBCs that highly express PHGDH.
Locally-produced lactate accumulates in TNBC
Recently, circulating lactate has been recognized as a nearly universal tissue fuel, with glucose feeding the TCA cycle substantially via circulating lactate. In genetically engineered mouse models of both lung and pancreas cancer, 13C-lactate infusion contributed substantial carbon to TCA metabolites 46. Lactate also labeled the TCA cycle in human lung tumors 44. Such labeling does not necessarily reflect net tumor lactate uptake, but can also come from rapid mixing between cellular pyruvate and circulating lactate catalyzed by lactate dehydrogenase (LDH) and MCT transporters (Figure 5A). Such exchange requires effective tumor perfusion, sufficient to equilibrate interstitial lactate with the circulation. If such exchange is limited by low perfusion or MCT activity, then glucose-derived lactate will accumulate in the tumor.
Figure 5. Local lactate fermentation from glucose in TNBCs.

a) Schematic showing the potential for glucose to label tumor pyruvate/label either via tumor glycolysis or via circulating lactate. Blue circles represent 13C. Glucose, pyruvate, and lactate can exchange between the systemic circulation and tumor. b) Fractional carbon labeling of tumor glucose, pyruvate and lactate relative to the same metabolites in serum at time of biopsy (mean ± SEM, n = 2 biopsies per patient, 3 technical replicates for serum). Patient 17 data are missing due to insufficient serum collection. c) Fractional carbon labeling of glucose, pyruvate and lactate in serum and tumor at time of biopsy (mean ± SEM, n = 11 patients, 3 technical replicates per patient for serum and 2 biopsy samples per patient for tumor, Student’s t-test). d) Fractional carbon labeling of tumor lactate relative to that of the circulation in indicated cancers (mean ± SEM, each point represents a single tumor sample of several collected from each of 30 (NSCLC), 4 (RCC) or 11 (TNBC) patients, one-way ANOVA w/ Tukey’s correction). TNBC patient 17 data are missing due to insufficient serum collection; data for one RCC patient are missing due to poor plasma detection of labeled lactate. Glc, glucose; G6P, glucose-6-phosphate; F6P, fructose-6-phosphate; GAP, glyceraldehyde-3-phosphate; 3PG, 3-phosphoglycerate; GLUT, glucose transporter; LDH, lactate dehydrogenase; MCT, monocarboxylic acid transporter. * = p < 0.05, **** = p < 0.0001. See also Figures S4,S5.
From 13C-glucose infusion, this would manifest as tumor pyruvate and lactate labeling exceeding circulating labeling. In TNBC, following [l,2-13C]glucose infusion, tumor lactate and pyruvate enrichment often exceeded that of serum (Figure 5B–C). We did not detect any other highly labeled circulating glycolytic precursors (Figure S2G–H). Accordingly, the high tumor lactate labeling implies its local synthesis from glucose within the tumor microenvironment.
The extent of lactate label enrichment in the tumor varied substantially across patients. One potential explanation is differences in expression of the relevant transporters. Accordingly, we measured glucose (GLUT) and lactate (MCT) transporter expression in these tumors at both the RNA and protein level, but observed no correlation between transporter expression and tumor-serum exchange of lactate (Figure S4A–B). Higher GAPDH expression, but not expression of several other glycolytic enzymes, aligned with increased tumor/serum lactate labeling (Figure S4C–D).
The Cancer Genome Atlas (TCGA) data showed that MCT and LDH transcript levels are on average highest in RCC, intermediate in NSCLC, and lowest on TNBC (Figure S4E). This suggested TNBC might be distinct in local lactate coming mainly from glycolysis, rather than from exchange with circulation. To explore this possibility, we reanalyzed previously published glucose infusion data for human NSCLC and RCC (circulating lactate labeling was not available for prior brain tumor studies) 44,47 (Figure 5D). Compared with lung and kidney tumors (Figure S5A–B), TNBCs demonstrated a significantly higher ratio of tumor to circulating lactate labeling (Figure 5D). Thus, lactate and pyruvate pools in TNBC are relatively isolated from the systemic circulation.
Glucose is converted into TCA intermediates and associated amino acids
We next examined the flow of 13C tracer into the TCA cycle (Figure 6A). To estimate the relative contribution of carbohydrate to tumor TCA pools, we normalized the fractional carbon labeling in tumor malate, succinate and α-ketoglutarate (αKG) to that of tumor lactate/pyruvate (which closely correlate, Figure S5C). The fractional carbohydrate contribution to TCA pools ranged between 10–40% (Figure 6B). This contribution occurs locally in the tumors, as serum TCA intermediates were minimally labeled (Figure S6B). Similar analysis on available data from brain tumors, NSCLC and RCC revealed that the carbohydrate contribution to the TCA cycle in TNBC was less than brain and lung tumors, but markedly higher than renal tumors (Figure 6C). Since TNBC pyruvate is substantially produced directly by glycolysis, this reflects a considerable direct glucose contribution to the TCA cycle.
Figure 6. Glucose carbon makes TCA intermediates and associated amino acids in TNBC.

a) Pathways from circulating [1,2-13C]glucose to M+2 TCA intermediates and associated amino acids. Blue circles represent 13C. Glucose, pyruvate, lactate, glutamine, glutamate, proline and asparagine can exchange between the systemic circulation and tumor. b) Fractional carbon labeling of tumor succinate, malate, and α-ketoglutarate (αKG) to that of tumor lactate (mean ± SEM, n = 2 biopsies per patient). c) Fractional carbon labeling of tumor malate to that of tumor lactate in the indicated cancers (mean ± SEM, each point represents a single tumor sample of several collected from each of 5 (brain), 30 (NSCLC), 5 (RCC) or 12 (TNBC) patients, one-way ANOVA w/ Tukey’s correction). d) Fractional carbon labeling of indicated tumor amino acid relative to that of respective metabolic precursor in TNBC (mean ± SEM, n = 12 patients, 2 technical replicates per patient). e) Fractional carbon labeling of tumor glutamine relative to that of tumor glutamate in indicated cancers (mean ± SEM, each point represents a single tumor sample of several collected from each of 5 (brain), 30 (NSCLC), 5 (RCC) or 12 (TNBC) patients, one-way ANOVA w/ Tukey’s correction). Three fragments (two from NSCLC, one from RCC) where ratios exceeded 1 were removed as outliers. * = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001. See also Figure S6.
TCA intermediates serve as substrates for producing the non-essential amino acids glutamate, glutamine, proline, aspartate, and asparagine—critical building blocks for protein and (in the case of aspartate and glutamine) nucleic acid synthesis. With the possible exception of aspartate, which is present at particularly low levels in serum 54, tumors can alternatively acquire these nutrients from the circulation or internal synthesis (Figure 6A). To determine their source in TNBC, we examined tumor and serum labeling of these amino acids, finding substantial tumor but not serum labeling for glutamate, glutamine, and proline (Figure S6C–D). To estimate the fractional contribution from de novo synthesis to these tumor amino acid pools, tumor amino acid labeling was normalized to that of their respective precursors (Figure 6D). Consistent with local production, tumor glutamate labeling closely matched that of its precursor α-KG (Figure S6A,C), and tumor aspartate labeling mirrored that of tumor malate (Figure S6A,E). In contrast, tumor proline labeling was substantially lower than that of tumor glutamate (Figure S6C), and asparagine labeling was virtually undetectable (Figure 6E).
Interestingly, tumor glutamine was labeled about half as much as tumor glutamate (Figure 6D, Figure S6C), consistent with its coming roughly equally from circulation and from intratumoral production. Analysis of existing data from brain tumors, NSCLC and RCC demonstrated that glutamine production from glutamate in TNBC was less than brain or NSCLC but substantially higher than renal tumors (Figure 6E). Thus, TNBCs synthesize much of their own glutamate, aspartate and glutamine, some proline, and minimal asparagine.
Tumor lactate is more labeled than tumor 3-phosphoglycerate or circulating lactate
Tumor pyruvate and lactate labeling correlated closely, consistent with rapid LDH-mediated interconversion (Figure S5C). To quantify their source, we constructed a simple model that relies solely on data from labeled glucose infusion (Figure 7A). Mathematically, lactate labeling in the tumor can be approximated as the sum of the fractional contributions from local glycolysis, which we call “α”, and from circulating lactate (which, when there are no other inputs, is simply 1 – α). Negative α values are feasible if there are other contributors beyond glucose and lactate, such as amino acids and macropinocytosis, which are not included in the model. This analysis revealed substantial direct glucose contributions to the lactate and pyruvate pools in most TNBCs (Figure 7B). Across TNBC tumors, the fraction of locally produced lactate (α) and the tumor lactate concentration tended to positively correlate, consistent with accumulation of lactate made by glycolysis within the high α tumors (Figure S7A). Analogous analysis of previous glucose infusion data in NSCLC revealed a much smaller direct glucose contribution (lower α), reflecting pyruvate/lactate coming mainly from exchange with the circulation (Figure 7C, Figure S7B–C). Together, these findings further support that TNBCs, unlike lung cancer tumors, tend to have a local metabolic microenvironment enriched in lactate produced by tumor fermentation.
Figure 7. Evidence for local lactate production through a subset of tumor cells.

a) Model depicting sources of tumor lactate and associated equations for calculating the direct contribution of circulating glucose to tumor lactate, reflecting lactate made locally from glucose (α). b) Direct contribution of circulating glucose to tumor lactate (α) in TNBC (mean ± SEM, n = 2 biopsy samples per patient, 3 technical replicates for serum). c) Direct contribution of circulating glucose to tumor lactate (α) in the indicated cancers (mean ± SEM, each point represents a single tumor sample of several collected from each of 5 (brain), 30 (NSCLC), 5 (RCC) or 11 (TNBC) patients, one-way ANOVA w/ Tukey’s correction). d) Lactate/3PG labeling ratio in the indicated cancers (mean ± SEM, each point represents a single tumor sample of several collected from each of 5 (brain), 30 (NSCLC), 5 (RCC) or 12 (TNBC) patients). In line with published work, the lactate/3PG labeling ratio is calculated using the highest labeled forms of lactate and 3PG observed in each study (M+3 in NSCLC, brain tumors, and RCC; M+2 in TNBC). e) Schematic depicting proposed compartmentation of tumor glycolysis. See also Figure S7. ** = p < 0.01.
Previously, DeBerardinis and colleagues 44,45,47 observed more extensive lactate labeling than labeling of upstream glycolytic intermediates in NSCLC and brain tumors. For example, in many patients, tumor lactate is more heavily labeled than the last readily measured intermediate in glycolysis, 3-phosphoglycerate (3PG) (Figure S7D–E). One logical explanation, in line with the low fraction of lactate coming from local glycolysis in NSCLC, is that tumor lactate comes substantially from the circulation, with locally produced lactate matching 3PG labeling, i.e. less labeled than circulating lactate. Consistent with this interpretation, in NSCLC, high tumor lactate/3PG labeling ratio correlates with high MCT and LDH expression and thus seems to reflect rapid tumor-circulation lactate exchange 44. Conversely, intratumoral lactate and 3PG labeling were roughly equal in RCC (i.e. lactate/ 3PG ratio ~ 1; Figure S7F), consistent with local lactate production from glucose, although similar lactate labeling from glucose was also observed in the circulation of RCC patients, with α intermediate between NSCLC and TNBC.
Based on the high direct contribution of circulating glucose to tumor lactate in TNBC, we expected TNBCs to manifest 3PG labeling comparable to or exceeding lactate (lactate/3PG labeling ratio ≤ 1). Most TNBCs, however, showed tumor lactate labeling exceeding tumor 3PG labeling (Figure S7G), with an average ratio trending higher in TNBC than in RCC and NSCLC, but lower than in brain tumors (Figure 7D). As tumor lactate labeling also exceeds circulating lactate labeling (Figure 5B,C), tumor lactate is more labeled than either of its logical precursors, suggesting compartmentalization of tumor glycolysis (Figure 7E).
Discussion
A tumor’s metabolic character reflects interactions between its cancer, stromal, and immune compartments. In addition, metabolism is impacted by perfusion, which is heterogeneous within tumors. Replicating this complexity in the laboratory or animal models is challenging. Thus, study of human cancer in situ is crucial. Stable isotope-labeled nutrient infusion studies in human cancer patients, dating back to the 1980s 55 and revitalized by DeBerardinis and colleagues, have revealed active aerobic metabolism of glucose in brain and lung cancers, impaired glucose oxidation in renal cancer, and lactate uptake and catabolism in human lung tumors. Here we carry out a similar investigation of human TNBC, capitalizing on the distinct arrangement of isotopes in [1,2-13C]glucose to gain additional insights into pentose phosphate pathway activity.
We show glycolytic flux far exceeds that of the PPP in these tumors. Nonetheless, the oxidative branch of the PPP is the primary route for the generation of ribose phosphate, as key substrate for nucleotide biosynthesis. These findings clarify the relative fluxes through these pathways—arguing against the PPP being a dominant carbon consumer or having high enough flux to materially “steal” carbon from glycolysis. Cell culture studies using [1,2-13C]glucose have similarly found low PPP flux relative to glycolysis, while claiming rapid non-oxPPP flux between the hexose- and ribose phosphate pools 49. We show that oxPPP exceeds non-oxPPP flux in human TNBCs.
Amino acid biosynthesis
Serine and glycine can be synthesized de novo from the glycolytic intermediate 3-phosphoglycerate. Serine synthesis activity correlates with protein expression of PHGDH, the first and committed step of de novo serine production. Previous studies have highlighted the penchant for TNBC cells with high PHGDH expression to synthesize serine from glucose when cultured or implanted as xenografts into mice 56,57. The observation of strongly elevated serine and glycine synthesis in select intact human tumors argues for further efforts to pharmacologically target these pathways, perhaps using PHGDH expression and/or isotope tracing as a patient selection criterion 56,58,59.
We also observed TNBC synthesis of amino acids branching from the TCA cycle: aspartate, glutamate, glutamine and proline (but not asparagine). Among the standard 20 amino acids, aspartate is the least abundant in the circulation, and its internal synthesis is accordingly a necessity 54,60,61. Glutamate is also relatively low in the bloodstream, and in tumors is both made and consumed through rapid exchange with TCA intermediates. In contrast, glutamine is the most abundant and highest flux circulating amino acid. Its catabolism via glutaminase, a MYC induced gene, is a main source of carbon and energy in cultured cancer cells. TNBC cells grown in vitro or as patient-derived xenografts in mice are sensitive to pharmacological glutaminase inhibition 62. Thus, glutamine is classically viewed as a tumor fuel, not product. Due to its importance for nucleotide and amino-sugar synthesis, however, glutamine can become limiting for tumors 63. Indeed, recently glutamine synthesis has been shown to support pancreatic cancer tumorigenesis in mice 22. Our analyses here, carried out using both our own TNBC data and literature NSCLC and RCC data, reveal that highly active glutamine synthesis, often accounting for a majority of tumor glutamine, is a common feature of intact human tumors.
Like glutamine, asparagine can potentially be a limiting amino acid for tumors. Yet in TNBC we observed only trace asparagine synthesis. Acute lymphoblastic leukemia similarly lacks asparagine synthetase capacity and is accordingly susceptible to asparaginase therapy 64. Asparaginase is currently being tested clinically as a potential therapy for TNBC (NCT03674242) 65. One consideration is whether certain rare TNBC tumor cells might be able to make asparagine. For example, recent work in mice suggests that acquisition of asparagine synthetase capacity gates TNBC metastasis 66. Moreover, studies using TCGA data show TNBCs possess higher expression of asparagine synthetase than other breast cancer subtypes, with higher expression associated with metastasis to the brain and worse distal metastasis-free survival 67. Accordingly, isotope tracing analysis of TNBC metastases would be valuable.
Local tumor lactate production
A notable difference between our observations in TNBC, and prior work in NSCLC and RCC, is that we observe locally produced pyruvate and lactate pools that do not readily exchange with the systemic circulation. Elevated tumor lactate labeling provides human evidence for intratumor glucose fermentation. Such fermentation has long been expected based on the Warburg effect, FDG-PET, and intratumoral lactate accumulation, but proof for rapid local flux through the full glycolytic pathway in human tumors has been elusive. Moreover, the predominance of local tumor lactate production has been called into question by the emerging evidence for rapid exchange between tumor and circulating lactate 44,46. Lactate import from the systemic circulation can only produce tumor labeling less than or equal to that of the serum, and this is frequently observed in NSCLC. In contrast, in TNBC, we find tumor labeling of lactate exceeding that of serum, demonstrating local production from glucose as a tumor lactate source.
To quantify local tumor lactate sources, we applied a reductionist form of metabolic flux analysis (MFA). MFA is routinely used to quantify pathway activity in controlled experimental settings such as microbial cultures, where complete accounting of system inputs, outputs, and internal labeling is often possible. Its use in in vivo tracing studies, however, has been limited. Our MFA model quantifies tumor lactate sources, finding a predominance of local production in TNBC, and lactate uptake from (or exchange with) the circulation in NSCLC. The existence of a locally produced tumor lactate pool reflects TNBC having a distinct metabolic microenvironment, relatively isolated from the circulation. This may reflect relatively limited tumor perfusion. In NSCLC, glucose utilization has been shown to vary within tumors based on the degree of tumor perfusion as determined by dynamic contrast enhanced MRI (DCE-MRI) 45, and DCE-MRI has also revealed heterogeneous TNBC perfusion 68.
Compartmentalization of tumor glycolysis
The propensity for local lactate production, as measured by our simple quantitative model, provides complementary information to assessment of the labeling of lactate relative to lower glycolytic intermediates, a metric used productively by DeBerardinis, Morrison, and colleagues. This latter measure (lactate/3PG labeling ratio) correlates in NSCLC with MCT and LDH expression and in melanoma with metastatic propensity 69. Thus, it is undeniably medically relevant. But its metabolic interpretation remains unclear.
In a well-mixed system, downstream products never label more than their upstream precursors. Yet DeBerardinis found that tumor lactate is more labeled than its putative precursor 3PG. In NSCLC, this was logically interpreted as tumor lactate coming from circulating lactate, rather than tumor glycolytic intermediates. In TNBC, we see the same surprising greater labeling of lactate than 3PG, but here tumor lactate is also more labeled than circulating lactate. Thus, in TNBC, tumor lactate is more labeled than both of its potential precursors. Given only the canonical metabolic pathways, this cannot happen in a well-mixed system.
A potential explanation is compartmentalization of lactate production (Figure 7E). Specifically, tumor lactate could be produced preferentially from circulating glucose by a subset of tumor cells with high glycolytic flux but relatively low total 3PG content. 3PG could reside mainly in other tumor cells, which use glycogen as a major metabolic input (as occurs in many normal tissues 51). Such tumor metabolic heterogeneity could potentially presage metastasis or therapy resistance, explaining the correlation of high lactate/3PG labeling ratio with bad outcomes. Elucidating the identity of the high and low glycolysis cell types in heterogeneous tumors is an important future objective.
Limitations of Study
This study involved a relatively small cohort of TNBC patients and a single isotope tracer [1,2-13C]glucose. Metabolite concentration and labeling measurements were from venous blood samples and ground core biopsies of primary tumors, and represent averages across the biopsied region. Study power was limited by the small patient population, which precluded effectively correlating molecular phenotypes (transcriptional, proteomic, and metabolomic) with each other or with clinical outcomes. Comparison of tumor lactate labeling to circulating lactate labeling and tumor glycolytic intermediate labeling revealed that tumor lactate was more labeled than either of its logical precursors, arguing for compartmentalization of glycolysis within the tumor. But this is an indirect inference, and we did not directly measure tumor metabolic heterogeneity. Comparisons of isotope labeling across tumor types revealed that TNBCs stood out for high tumor lactate labeling from circulating glucose, consistent with TNBC having a relatively isolated metabolic microenvironment. We did not apply other tracers, such as 13C-lactate, which could offer an orthogonal perspective on metabolite exchange between TNBC tumors and the circulation, nor did we directly measure extracellular metabolites in the tumor microenvironment. Moreover, comparisons across tumor types may be confounded by differences across study sites, investigators, patient populations, and tissue collection procedures. For example, the breast biopsies here were collected from awake patients using local anesthetic, whereas previous work on lung and kidney tumors involved bulk resections under general anesthesia. Expanded studies, with better controlled comparisons across tumor types, additional tracers, and more direct measurements of metabolic heterogeneity are merited.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Joshua D. Rabinowitz (joshr@princeton.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
There are IRB restrictions regarding next generation sequencing data sharing for this study and patients were not consented for public genomic data sharing. Select somatic variants and processed RNA-Seq data (TPM), RPPA data, metabolomics and TNBC isotope labeling data are available in Tables S2–S7. Isotope labeling data for NSCLC were obtained from Faubert et al 44. Isotope labeling data for RCC and brain tumors were obtained from Courtney et al47. Further access to data may be made by contacting the corresponding author.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Subjects
Seventeen female patients with TNBC were enrolled in a Baylor Scott and White Healthcare System IRB-approved protocol (Baylor 017–396) after obtaining informed consent. The first four patients enrolled did not receive glucose infusions; their primary TNBC biopsy samples were collected and processed as a pilot study to ensure adequate biopsy mass and LC-MS signal quality. These pilot data are not included in this manuscript. Thereafter, thirteen patients were consented to receive infusions with [1,2-13C]glucose; however, one patient withdrew consent before the infusion. Patients were considered eligible if they were ≥18 years of age and had TNBC defined as follows: invasive ductal cancer: ER-negative with <10% of tumor nuclei; immunoreactive; PR-negative with <10% of tumor nuclei immunoreactive; HER2-negative defined as follows: i. FISH-negative (FISH ratio <2.0), or IHC 0–1+, or iii. IHC 2+ AND FISH-negative (FISH ratio<2.0). Patients also had to be willing to undergo a core biopsy (4–8 passes) for research purposes and to understand the investigational nature of the study. Clinical and pathological characteristics about the patients and their TNBCs are summarized in Table S1.
Animal studies
Mouse studies followed protocols approved by the Animal Care and Use Committee for Princeton University. In vivo infusions were performed on 12–14 week old male C57BL/6 mice pre-catheterized in the right jugular vein (Charles River Laboratories, Wilmington, MA). Animals received a normal AA purified chow diet (TestDiet Modified Baker AA Diet with 8% Fat and 15% sucrose, 5WA1 St. Louis, MO). Mice were allowed at least 5 days of acclimation to the facilities prior to experimentation and were randomly chosen for infusion studies. No blinding was implemented. The mice were on normal light cycle (8 AM – 8 PM). Mice were monitored regularly and determined to be healthy by the veterinary staff.
METHOD DETAILS
Patient infusions and biopsy collection
On day of biopsy, patients undergoing infusion received a 6g bolus of [1,2-13C]glucose in normal saline through a peripheral IV over 10 min followed by 6 g/h continuous infusion until they underwent needle biopsy of their primary breast cancer. Prior to infusion, the [1,2-13C]glucose (Sigma-Aldrich) underwent sterility and endotoxin testing. The target interval from start of bolus to biopsy was 60 min.
For percutaneous core needle biopsies of primary TNBCs, breast area was prepped adjacent to the mass. Using ultrasound for localization, the skin and peritumoral area was infiltrated with local anesthetic and a small dermal incision was made, through which six 14-gauge core biopsies were obtained with ultrasound guidance. Biopsies were immediately dispensed into crytotubes pre-cooled on dry ice. Tubes were sealed and stored at −80°C.
Patient blood collection
Research blood samples were collected via peripheral blood draw at 30 min (10 mL) and approximately 60 min (just prior to biopsy collections; 10 mL) after initiation of the bolus. Blood samples were collected in red-top (no additives) tubes, allowed to clot at room temperature for 15–30 min, and processed into serum by the Baylor Biobank and Project Management (BPM) Core. Red-top tubes were centrifuged at 1,200g for 15 min at room temperature (25°C). Using a sterile pipette, the serum layer was removed and aliquoted into 2 mL cryovials. Serum was stored at −80°C prior to metabolite analysis.
In addition, 40 mL of whole blood was collected for germline exome sequencing to compliment next generation sequencing that was performed on the biopsy tissue. This blood was collected after the patient had consented, with no time frame, into lavender-top, EDTA tubes. Tubes were inverted several times and processed into plasma and buffy coat by the Baylor BPM Core. Briefly, lavender-top tubes were centrifuged at 1,300g for 10 min at room temperature (25°C). Using a sterile pipette, the upper layer (plasma) was removed and aliquoted into 1.2 mL cryovials. The thin interface (buffy coat) was then extracted and transferred into a 1.2 mL cryovial. Both plasma and buffy coat were stored at −80°C until analysis.
Metabolite extraction from serum and tumor biopsy samples
Aqueous metabolites were extracted from serum (thawed on ice) through addition of 40 μl methanol (precooled on dry ice) to 10 μl of sample. The mixture was vortexed and incubated on dry ice for 20 min, followed by centrifugation (16,000g at 4°C). For tumor samples, two frozen biopsy samples per patient were weighed (~5–10-mg tissue each) and pulverized using a Cryomill (Retsch). The resulting powder was then triturated with ice-cold 40:40:20 acetonitrile:methanol:water w/ 0.5% formic acid (1 ml solvent / 20-mg tissue) followed by neutralization w/ 15% w/v ammonium bicarbonate. Solids were precipitated by centrifugation (16,000g at 4°C). Serum sample supernatants were utilized directly for LC-MS analysis (on HILIC LC-MS); tumor supernatants were either utilized directly (on HILIC LC-MS) or were dried under nitrogen stream and resuspended in water (for reversed-phase LC-MS).
Mouse infusions
On the day of infusion experiment, mice were transferred to new cages without food around 9 AM (beginning of their sleep cycle), were without food until 7:30 PM, at which time chow was placed back in the cages and the 2.5 h infusion was initiated at 9 PM. The infusion setup (Instech Laboratories) included a swivel and tether to allow the mouse to move around the cage freely. A solution of [U-13C]glucose (800 mM, CIL-1396, Cambridge Isotope Laboratories, Tewksbury, MA) was prepared in sterile normal saline and was infused at a rate of 0.1 μl g−1min−1. Tissue harvest was performed at the end of the infusion after euthanasia by cervical dislocation. Tissues were quickly dissected, snap frozen in liquid nitrogen with a pre-cooled Wollenberger clamp, and stored at −80°C until analysis.
LC-MS analysis
Metabolites were analyzed using a quadrupole-orbitrap mass spectrometer (Q Exactive Plus, Thermo Fisher Scientific, Waltham, MA) coupled to hydrophilic interaction chromatography (HILIC) with LC separation by either a XBridge BEH Amide column (Waters) or a Vanquish Horizon UHPLC system with an XBridge BEH Amide column (150 mm × 2.1 mm, 2.5 mM particle size, Waters, Milford, MA) with the corresponding XP VanGuard Cartridge. All mass spectrometers were operating in negative ion mode and were coupled to their respective liquid chromatography methods via electrospray-ionization. Selected ion monitoring was utilized to measure tumor 3-phosphoglycerate, ribose phosphate, serine and glycine. Detailed analytical conditions have been previously described 70. Data were analyzed using the ElMaven software (v 0.2.4, Elucidata), with compounds identified based on exact mass and retention time match to commercial standards. Isotopic labeling of metabolites arising from 13C labeled glucose was corrected for natural abundance, as previously described 71. See Table S5 for mouse tissue metabolite labeling, Table S6 for TNBC tumor and serum metabolite labeling, and Table S7 for TNBC tumor metabolomics.
Nucleic acid extraction and NGS library preparation and sequencing
DNA was extracted from buffy coat using DNeasy Blood & Tissue kit (Qiagen) per manufacturer’s protocol. Fresh frozen tumor biopsies were disrupted and homogenized with BulletBlender Gold tissue homogenizer (Next Advance) and divided for DNA and RNA extraction using DNeasy Blood & Tissue kit (Qiagen) and RNeasy kit (Qiagen), respectively, per manufacturer’s protocols. DNA was eluted into low EDTA TE buffer (10 mM Tris-HCl, pH 8.0, 0.1 mM EDTA), and RNA was eluted into water.
Isolated DNA (250 ng) was used to generate libraries for whole exome sequencing. DNA was fragmented to target peaks of 200bp by sonication on a Covaris E220. DNA samples were prepared for next generation exome sequencing with SureSelect XT (Agilent, Santa Clara, CA, USA), according to manufacturer’s protocol with the following modifications: Library amplification conditions: initial denaturation at 98°C for 30sec, 8 cycles of 98°C for 10sec, 60°C for 30sec, and 72°C for 60sec, then final extension at 72°C for 5min. Whole exome libraries were captured using a custom Agilent SureSelect exome design that encompasses exonic regions along with common structural variations in cancer (Strexome v2; 75Mb). The samples were sequenced on a NovaSeq6000 (Paired end × 150 bp; Illumina, San Diego, CA, USA) to an average depth of 154 ± 29× coverage.
Isolated RNA (500ng) was used to generate libraries for RNAseq. RNA samples were prepared for next generation sequencing with KAPA RNA HyperPrep Kit with RiboErase HMR (Roche) with KAPA Unique Dual Index adapters (Roche), as per manufacturer’s protocol with the following modifications: fragmentation at 94°C for 8 min; 15 cycles of library amplification; purifications with AMPure XP beads (Beckman Coulter Genomics). The libraries were sequenced on a single lane on a NovaSeq6000 (Paired end × 100 bp; Illumina, San Diego, CA, USA) to an average total reads of 8.39E+07 ± 0.95 reads.
NGS data analysis
Primary analysis was performed using the Translational Genomics Research Institute (TGen) Jetstream pipeline Pegasus72. BCL files were converted to FASTQs with BCLConverter (Illumina, San Diego, CA, USA). DNA FASTQs were aligned to the human reference genome (build 37) with BWA-MEM (bwa v0.7.8)73. Base recalibration was performed with GATK v3.1–1 74. Duplicates were marked using Picard v1.111 (http://broadinstitute.github.io/picard/). Joint indel realignment was performed using GATK v3.1–174. BAMs were used for the identification of somatic mutations (point mutations, insertions, deletions), structural variants (SVs), and copy number variants (CNVs). Variant calling was performed using Seurat (quality score > 30), Strelka, and MuTect, and annotated using GENCODE version 3 (Ensembl) and build 37.1 75–77. Final somatic SNVs were called by at least 2 out of 3 callers. CNVs were predicted with tCoNuT (https://github.com/tgen/tCoNuT) with default thresholds: 0.58 for amplification and −0.99 for deletion. Somatic SVs and CNVs were filtered by removing variants predicted to have no protein coding sequence consequence, as well as removal of genes flagged as LOWQC (https://github.com/tgen/GemDb/wiki/Filtering). Variants were annotated against COSMIC cancer gene list (v82)78, and filtered if gene does not occur in COSMIC database. Filtered variants were used to generate an oncoprint with cBioPortal’s Oncoprinter (https://www.cbioportal.org/oncoprinter). RNA FASTQs were aligned using STAR 2.3.1z 79.
Histology
Frozen tissue cores were embedded in Optimal Cutting Temperature (OCT) compound and 8μm sections were cut onto plain glass microscope slides. One section was stained and archived. The tissue section was fixed in 70% ethanol, stained with hematoxylin and eosin, dehydrated in graded ethanol (70%, 95%, 100%), rinsed in xylene, and a coverslip was applied using Permount mounting medium.
Laser capture microdissection
Laser capture microdissection was used to procure enriched populations of breast tumor cells for proteomic analysis. Frozen tissue sections were fixed in 70% ethanol, stained with hematoxylin only, dehydrated in graded ethanol (70%, 95%, 100%) and rinsed in xylene immediately prior to laser capture microdissection (LCM) using an ArcturusXT or PixCell IIe LCM in infrared capture mode. The staining reagents contained protease inhibitors (CompleteMini tablets, Roche). The tumor compartment was microdissected from each tissue section. Microdissected cells were stored on the LCM cap (CapSure Macro cap) at −80°C prior to lysis with protein extraction buffer (10% (v/v) solution of Tris(2-carboxyethyl)phosphine (TCEP; Pierce, Rockford, IL) in equal volumes of Tissue Protein Extraction Reagent (T-PER™, Pierce) and Novex Tris-glycine 2X SDS buffer (Invitrogen). Lysates were denatured at 95°C for 5 min prior to constructing reverse phase protein arrays.
Reverse phase protein microarray
Proteins can be quantified in a low number of cells using reverse phase protein arrays (RPPA). RPPA allow multi-plex analysis of many different specimens and proteins under identical experimental conditions. We used RPPA to quantify various proteins related to glucose and serine metabolism in TNBCs.
Reverse phase protein arrays (RPPA) were constructed containing microdissected breast tissue lysates diluted to 0.25μg/mL, and commercial cell line lysates and bovine serum albumin as controls and calibrators 80–82 Lysates were printed in technical replicates onto nitrocellulose coated glass slides (Oncyte Avid, Grace Bio-Labs, Bend OR), in serial 2-fold dilution curves, using a Quanterix 2470 arrayer (Billerica, MA) equipped with 350μm solid pins. MOLT-4 (Santa Cruz Biotechnology sc-2233, acute lymphoblastic leukemia), MCF7+EGF+β-estradiol (Santa Cruz Biotechnology sc-24730, breast adenocarinoma), and Jurkat+Calyculin (Santa Cruz Biotechnology sc-2277, human acute T cell leukemia induced with Calyculin A) cell lysates were printed on each array as quality control samples. Bovine Serum Albumin (BSA, Pierce #23209) was printed in an 8 point, 2-fold dilution series, starting at 1.0mg/mL as a calibration curve for total protein 82,83. The total protein within each array spot was determined using Sypro Ruby protein blot stain (Invitrogen/Molecular Probes) per manufacturer’s directions and scanned using a Cy3 laser (Tecan Power Scanner).
RPPA staining and analysis
Immunostaining was performed on a Dako Autostainer per manufacturer’s instructions (CSA kit, Agilent Dako) 84,85. Prior to immunostaining, RPPA slides were incubated in 1X ReBlot Mild solution (Fisher Scientific), washed twice with phosphate buffered saline without calcium or magnesium (PBS), and incubated in I-Block solution (Invitrogen T2015) at room temperature for a minimum of 30 min. Each slide was incubated with a single primary antibody at room temperature for 30 min. Antibody specificity was confirmed by Western blotting as previously described 86. The negative control slide was incubated with antibody diluent. Secondary antibody was gt anti-rabbit IgG H+L (1:10,000) (Vector Labs, Burlingame, CA) or anti-mouse IgG (1:10, Agilent Dako CSA kit). Signal detection was amplified via horseradish peroxidase mediated biotinyl tyramide deposition with chromogenic detection (Diaminobenzidine) per manufacturer’s instructions (Agilent Dako).
Arrays were scanned at 600dpi, grayscale, on a flatbed scanner (UMAX PowerLook). Spot (pixel) intensity was analyzed using ImageQuant ver 5.2 (GE Healthcare), with mean local area background subtraction 80. Signal:background intensity for each spot was calculated using the freely available data reduction algorithm (RAS ver16, www.capmm.gmu.edu) 83. Spot intensities were normalized to total protein/spot.
QUANTIFICATION AND STATISTICAL ANALYSIS
Samples sizes are defined in each figure legend. Unless otherwise specified, results for biological replicates are presented as mean ± SEM. Fractional carbon labeling = [(fraction M+1) + 2*(fraction M+2) + … K*(fraction M+K)]/K, where K is the number of carbon atoms in the metabolite. For “α” calculations, error was propagated from both biopsy and serum measurements using standard error propagation calculations. Correlative gene expression analysis to serum metabolite data was performed using log2-transformed TPM output from Salmon (for RNA) and normalized total protein (for protein) followed by a Pearson correlation test. Statistical significance was calculated using unpaired Student’s t-test when comparing two groups, one-way ANOVA followed by Tukey’s post hoc analysis when comparing three or more, and F-testing for assessing the significance of linear regression. All statistical calculations were performed using the software package GraphPad Prism 7.03.
Supplementary Material
Table S1. Patient characteristics; related to Figure 1
Table S2. TNBC biopsy WES filtered variants; related to Figure 1
Table S5. Mouse tissue metabolite labeling; related to Figure 4
Table S7. TNBC biopsy metabolomics data; related to Figure 7
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies for RPPA | ||
| Secondary antibody, goat anti-rabbit IgG (H+L) | Vector | BA-1000 |
| Secondary antibody, anti-mouse IgG | Agilent | K1500 |
| Aldolase-A | Cell Signaling Technology | 8060 |
| ATP Citrate Lyase | Cell Signaling Technology | 4331 |
| Enolase-1 | Cell Signaling Technology | 3810 |
| Enolase-2 | Cell Signaling Technology | 8171 |
| G6PD | Cell Signaling Technology | 12263 |
| GAPDH | Cell Signaling Technology | 5174 |
| GLUT-4 | Bioss | bs-03848 |
| Hexokinase 1 | Cell Signaling Technology | 2024 |
| Hexokinase2 | Cell Signaling Technology | 2867 |
| LDHA | Cell Signaling Technology | 3582 |
| MCT-1 | Millipore/Sigma | AB3538P |
| MCT-4 | Millipore/Sigma | AB3314P |
| PFKFB2 | Cell Signaling Technology | 13045 |
| PFKFB3 | Cell Signaling Technology | 13123 |
| PGAM1 | Cell Signaling Technology | 12098 |
| PHDGH | Cell Signaling Technology | 13428 |
| Pyruvate Dehydrogenase | Cell Signaling Technology | 3205 |
| Transaldolase | Abcam | ab137629 |
| Biological Samples | ||
| Human blood samples | This study | N/A |
| Human tumor biopsy samples | This study | N/A |
| Mouse tissue samples | This study | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| [1,2-13C]glucose | Sigma-Aldrich | 661422-CONF |
| [U-13C]glucose | Cambridge Isotope Laboratories | CIL-1396 |
| Critical Commercial Assays | ||
| DNeasy Blood & Tissue kit | Qiagen | 69582 |
| SureSelect XT Strexome v2 | Agilent | Custom version based on G9611A |
| RNeasy Mini Kit | Qiagen | 74104 |
| KAPA RNA HyperPrep Kit with RiboErase HMR | Roche | 08098131702 |
| KAPA Unique Dual Index Adapters | Roche | 08861919702 |
| Deposited Data | ||
| N/A | ||
| Experimental Models: Cell Lines | ||
| MOLT-4 | Santa Cruz Biotechnology | sc-2233 |
| MCF7+EGF+β-estradiol | Santa Cruz Biotechnology | sc-24730 |
| Jurkat+Calyculin | Santa Cruz Biotechnology | sc-2277 |
| Experimental Models: Organisms/ Strains | ||
| Mouse: C57BL/6 | Charles River Laboratories | C57BL |
| Oligonucleotides | ||
| N/A | ||
| Software and Algorithms | ||
| El-Maven | Elucidata | https://resources.elucidata.io/elmaven |
| Accucor | Github | https://github.com/lparsons/accucor |
| GraphPad Prism 7 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Pegasus | TGen | 72 |
| BCL Converter | Illumina | https://www.illumina.com/informatics/sequencing-data-analysis/sequence-file-formats.html |
| BWA-MEM (bwa v0.7.8) | Wellcome Trust Sanger Institute | 73 |
| GATK v3.1-1 | The Broad Institute of Harvard and MIT | 74 |
| GENCODE (v3) | Ensembl | https://www.gencodegenes.org/ |
| Oncoprinter | cBioPortal | https://www.cbioportal.org/oncoprinter |
| STAR 2.3.1z | Cold Spring Harbor Laboratory | 79 |
| ImageQuant ver 5.2 | GE Healthcare | https://www.cytivalifesciences.com |
| RAS ver16 | GMU | www.capmm.gmu.edu |
| Biorender | Biorender | https://biorender.com/ |
| Other | ||
| N/A | ||
Highlights:
Breast cancer patients were infused with [1,2-13C]glucose to study tumor metabolism
Glycolytic flux far exceeds pentose phosphate pathway flux
Certain highly glycolytic tumor cells make most tumor lactate
Tumors make substantial serine and glutamine from glucose
Context and Significance:
Triple negative breast cancer (TNBC) is an aggressive disease with limited treatment options. To fuel their proliferation, tumor cells require energy and biopolymer building blocks. Glucose is a major tumor fuel, and can be catabolized by glycolysis or the pentose phosphate pathway. Here, a 13C-labeled form of glucose that distinguishes these two catabolic routes was infused into patients with TNBC prior to tumor biopsy. Subsequent metabolic analyses revealed that TNBCs catabolize glucose primarily via glycolysis, releasing lactate into the tumor microenvironment (Warburg metabolism). In parallel, tumors use glucose as an oxidative and anabolic substrate, making metabolites required for DNA, RNA, and protein synthesis. Targeting these pathways may help slow tumor growth or facilitate antitumor immune responses.
Acknowledgements
We thank all of the patients who participated in this study. We also thank Ralph DeBerardinis for generously providing plasma lactate enrichment data from the studies described in Courtney et al. 47, and Shawn Davidson for the mouse circulating glucose and lactate data described in Davidson et al. 50. We thank members of the Rabinowitz laboratory for helpful comments and suggestions. The infusion scheme, red blood cell imagery, and Graphical abstract were created using BioRender. The results published here are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. The clinical trial, genomic, and proteomic studies were funded by a grant from Baylor Scott & White Dallas Foundation, Dallas, Texas. Metabolite analysis was supported by NIH grants 1DP1DK113643, P30 CA072720 (Metabolomics, Biomedical Informatics, and Biospecimen Repository Service Shared Resource, Rutgers Cancer Institute of New Jersey) and R01 CA163591.
Funding:
The clinical trial, genomics, and proteomics were funded by the Baylor Scott & White Dallas Foundation, Dallas, Texas. Metabolic analyses were supported by NIH grants 1DP1DK113643, R01CA163591, and P30CA072720.
Footnotes
Declaration of interests
J.D.R. is a co-founder and stockholder in Toran and Serien Therapeutics, and advisor and stockholder in Agios Pharmaceuticals, Kadmon Pharmaceuticals, Bantam Pharmaceuticals, Colorado Research Partners, Rafael Pharmaceuticals, Barer Institute, and L.E.A.F. Pharmaceuticals. D.D.V.H. is a consultant for L.E.A.F Pharmaceuticals. V.E. is a consultant for OncBioMune d/b/a Theralink Technologies, Inc and is listed on George Mason University owned patents for laser capture microdissection and breast cancer treatment. J.D.R. and J.M.G. are listed as inventors on a Princeton University patent application concerning G6PD inhibitors.
ADDITIONAL RESOURCES
Clinical trial information
Patients were enrolled in the study “Pilot Study To Investigate Targetable Metabolic Pathways Sustaining Triple Negative Breast Cancer,” registered as NCT03457779.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bray F et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians 68, 394–424, doi: 10.3322/caac.21492 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Waks AG & Winer EP Breast Cancer Treatment. Jama 321, 288, doi: 10.1001/jama.2018.19323 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Foulkes WD, Smith IE & Reis-Filho JS Triple-Negative Breast Cancer. New England Journal of Medicine 363, 1938–1948, doi: 10.1056/nejmra1001389 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Harbeck N et al. Breast cancer. Nature Reviews Disease Primers 5, doi: 10.1038/s41572-019-0111-2 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Hanahan D & Weinberg, Robert A. Hallmarks of Cancer: The Next Generation. Cell 144, 646–674, doi: 10.1016/j.cell.2011.02.013 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Fletcher JW et al. Recommendations on the use of 18F-FDG PET in oncology. J Nucl Med 49, 480–508, doi: 10.2967/jnumed.107.047787 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Warburg O On the Origin of Cancer Cells. Science (New York, N.Y.) 123, 309, doi: 10.1126/science.123.3191.309 (1956). [DOI] [PubMed] [Google Scholar]
- 8.Lunt SY & Vander Heiden MG Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annual review of cell and developmental biology 27, 441–464, doi: 10.1146/annurev-cellbio-092910-154237 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Liberti MV & Locasale JW The Warburg Effect: How Does it Benefit Cancer Cells? Trends in biochemical sciences 41, 211–218, doi: 10.1016/j.tibs.2015.12.001 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Osthus RC et al. Deregulation of Glucose Transporter 1 and Glycolytic Gene Expression by c-Myc. Journal of Biological Chemistry 275, 21797–21800, doi: 10.1074/jbc.C000023200 (2000). [DOI] [PubMed] [Google Scholar]
- 11.Shen L et al. Metabolic reprogramming in triple-negative breast cancer through Myc suppression of TXNIP. Proceedings of the National Academy of Sciences 112, 5425–5430, doi: 10.1073/pnas.1501555112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morrish F, Isern N, Sadilek M, Jeffrey M & Hockenbery DM c-Myc activates multiple metabolic networks to generate substrates for cell-cycle entry. Oncogene 28, 2485–2491, doi: 10.1038/onc.2009.112 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang R et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882, doi: 10.1016/j.immuni.2011.09.021 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shim H et al. c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proceedings of the National Academy of Sciences of the United States of America 94, 6658–6663, doi: 10.1073/pnas.94.13.6658 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Menssen A & Hermeking H Characterization of the c-MYC-regulated transcriptome by SAGE: Identification and analysis of c-MYC target genes. Proceedings of the National Academy of Sciences 99, 6274–6279, doi: 10.1073/pnas.082005599 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doherty JR et al. Blocking lactate export by inhibiting the Myc target MCT1 Disables glycolysis and glutathione synthesis. Cancer research 74, 908–920, doi: 10.1158/0008-5472.Can-13-2034 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun L et al. cMyc-mediated activation of serine biosynthesis pathway is critical for cancer progression under nutrient deprivation conditions. Cell Research 25, 429–444, doi: 10.1038/cr.2015.33 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nikiforov MA et al. A functional screen for Myc-responsive genes reveals serine hydroxymethyltransferase, a major source of the one-carbon unit for cell metabolism. Molecular and cellular biology 22, 5793–5800, doi: 10.1128/mcb.22.16.5793-5800.2002 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morrish F et al. Myc-dependent mitochondrial generation of acetyl-CoA contributes to fatty acid biosynthesis and histone acetylation during cell cycle entry. The Journal of biological chemistry 285, 36267–36274, doi: 10.1074/jbc.M110.141606 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wise DR et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proceedings of the National Academy of Sciences of the United States of America 105, 18782–18787, doi: 10.1073/pnas.0810199105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao P et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458, 762–765, doi: 10.1038/nature07823 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bott Alex J. et al. Oncogenic Myc Induces Expression of Glutamine Synthetase through Promoter Demethylation. Cell metabolism 22, 1068–1077, doi: 10.1016/j.cmet.2015.09.025 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu W et al. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proceedings of the National Academy of Sciences of the United States of America 109, 8983–8988, doi: 10.1073/pnas.1203244109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cappelletti V et al. Metabolic Footprints and Molecular Subtypes in Breast Cancer. Disease Markers 2017, 1–19, doi: 10.1155/2017/7687851 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Basu S et al. Comparison of triple-negative and estrogen receptor-positive/progesterone receptor-positive/HER2-negative breast carcinoma using quantitative fluorine-18 fluorodeoxyglucose/positron emission tomography imaging parameters: a potentially useful method for disease characterization. Cancer 112, 995–1000, doi: 10.1002/cncr.23226 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Jadvar H, Alavi A & Gambhir SS 18F-FDG uptake in lung, breast, and colon cancers: molecular biology correlates and disease characterization. J Nucl Med 50, 1820–1827, doi: 10.2967/jnumed.108.054098 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palaskas N et al. 18F-fluorodeoxy-glucose positron emission tomography marks MYC-overexpressing human basal-like breast cancers. Cancer research 71, 5164–5174, doi: 10.1158/0008-5472.Can-10-4633 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hussein YR et al. Glut-1 Expression Correlates with Basal-like Breast Cancer. Transl Oncol 4, 321–327, doi: 10.1593/tlo.11256 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang X et al. High expressions of LDHA and AMPK as prognostic biomarkers for breast cancer. The Breast 30, 39–46, doi: 10.1016/j.breast.2016.08.014 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Dong T et al. Tumor LDH-A expression and serum LDH status are two metabolic predictors for triple negative breast cancer brain metastasis. Scientific Reports 7, doi: 10.1038/s41598-017-06378-7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson JM et al. MCT1 in Invasive Ductal Carcinoma: Monocarboxylate Metabolism and Aggressive Breast Cancer. Front Cell Dev Biol 5, 27, doi: 10.3389/fcell.2017.00027 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pinheiro C et al. Monocarboxylate transporter 1 is up-regulated in basal-like breast carcinoma. Histopathology 56, 860–867, doi: 10.1111/j.1365-2559.2010.03560.x (2010). [DOI] [PubMed] [Google Scholar]
- 33.Whitaker-Menezes D et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors. Cell Cycle 10, 1772–1783, doi: 10.4161/cc.10.11.15659 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sonveaux P et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. The Journal of Clinical Investigation 118, 3930–3942, doi: 10.1172/JCI36843 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patgiri A et al. An engineered enzyme that targets circulating lactate to alleviate intracellular NADH:NAD+ imbalance. Nature Biotechnology 38, 309–313, doi: 10.1038/s41587-019-0377-7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rabinowitz JD & Enerbäck S Lactate: the ugly duckling of energy metabolism. Nature Metabolism 2, 566–571, doi: 10.1038/s42255-020-0243-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Possemato R et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476, 346–350, doi: 10.1038/nature10350 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim SK, Jung WH & Koo JS Differential expression of enzymes associated with serine/glycine metabolism in different breast cancer subtypes. PloS one 9, e101004–e101004, doi: 10.1371/journal.pone.0101004 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ngo B et al. Limited Environmental Serine and Glycine Confer Brain Metastasis Sensitivity to PHGDH Inhibition. Cancer discovery, CD-19–1228, doi: 10.1158/2159-8290.CD-19-1228 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen J et al. Phosphoglycerate dehydrogenase is dispensable for breast tumor maintenance and growth. Oncotarget 4, 2502–2511, doi: 10.18632/oncotarget.1540 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beckonert O et al. Metabolic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts. Nature protocols 2, 2692–2703, doi: 10.1038/nprot.2007.376 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Jang C, Chen L & Rabinowitz JD Metabolomics and Isotope Tracing. Cell 173, 822–837, doi: 10.1016/j.cell.2018.03.055 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maher EA et al. Metabolism of [U-13 C]glucose in human brain tumors in vivo. NMR Biomed 25, 1234–1244, doi: 10.1002/nbm.2794 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Faubert B et al. Lactate Metabolism in Human Lung Tumors. Cell 171, 358–371.e359, doi: 10.1016/j.cell.2017.09.019 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hensley Christopher T. et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 164, 681–694, doi: 10.1016/j.cell.2015.12.034 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hui S et al. Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115–118, doi: 10.1038/nature24057 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Courtney KD et al. Isotope Tracing of Human Clear Cell Renal Cell Carcinomas Demonstrates Suppressed Glucose Oxidation In Vivo. Cell metabolism 28, 793–800.e792, doi: 10.1016/j.cmet.2018.07.020 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johnston K et al. Isotope tracing reveals glycolysis and oxidative metabolism in childhood tumors of multiple histologies. Med, doi: 10.1016/j.medj.2021.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee W-NP et al. Mass isotopomer study of the nonoxidative pathways of the pentose cycle with [1,2–13C2]glucose. American Journal of Physiology-Endocrinology and Metabolism 274, E843–E851, doi: 10.1152/ajpendo.1998.274.5.E843 (1998). [DOI] [PubMed] [Google Scholar]
- 50.Davidson SM et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell metabolism 23, 517–528, doi: 10.1016/j.cmet.2016.01.007 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.TeSlaa T et al. The Source of Glycolytic Intermediates in Mammalian Tissues. Cell metabolism, doi: 10.1016/j.cmet.2020.12.020 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pederson BA et al. Glucose Metabolism in Mice Lacking Muscle Glycogen Synthase. Diabetes 54, 3466, doi: 10.2337/diabetes.54.12.3466 (2005). [DOI] [PubMed] [Google Scholar]
- 53.Yudilevich DL, De Rose N & Sepulveda FV Facilitated transport of amino acids through the blood-brain barrier of the dog studied in a single capillary circulation. Brain Research 44, 569–578, doi: 10.1016/0006-8993(72)90319-8 (1972). [DOI] [PubMed] [Google Scholar]
- 54.Newgard CB et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell metabolism 9, 311–326, doi: 10.1016/j.cmet.2009.02.002 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mullen JL et al. Protein synthesis dynamics in human gastrointestinal malignancies. Surgery 87, 331–338 (1980). [PubMed] [Google Scholar]
- 56.Pacold ME et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nature Chemical Biology 12, 452, doi:10.1038/nchembio.207010.1038/nchembio.2070https://www.nature.com/articles/nchembio.2070#supplementary-informationhttps://www.nature.com/articles/nchembio.2070#supplementary-information (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ngo B et al. Limited Environmental Serine Confers Sensitivity to PHGDH Inhibition in Brain Metastasis. bioRxiv, 2020.2003.2003.974980, doi: 10.1101/2020.03.03.974980 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ducker GS et al. Human SHMT inhibitors reveal defective glycine import as a targetable metabolic vulnerability of diffuse large B-cell lymphoma. Proceedings of the National Academy of Sciences of the United States of America 114, 11404–11409, doi: 10.1073/pnas.1706617114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.García-Cañaveras JC et al. SHMT inhibition is effective and synergizes with methotrexate in T-cell acute lymphoblastic leukemia. Leukemia, doi: 10.1038/s41375-020-0845-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Birsoy K et al. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 162, 540–551, doi: 10.1016/j.cell.2015.07.016 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sullivan Lucas B. et al. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 162, 552–563, doi: 10.1016/j.cell.2015.07.017 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gross MI et al. Antitumor Activity of the Glutaminase Inhibitor CB-839 in Triple-Negative Breast Cancer. Molecular cancer therapeutics 13, 890, doi: 10.1158/1535-7163.MCT-13-0870 (2014). [DOI] [PubMed] [Google Scholar]
- 63.Kamphorst JJ et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer research 75, 544–553, doi: 10.1158/0008-5472.can-14-2211 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chiu M, Taurino G, Bianchi MG, Kilberg MS & Bussolati O Asparagine Synthetase in Cancer: Beyond Acute Lymphoblastic Leukemia. Frontiers in Oncology 9, doi: 10.3389/fonc.2019.01480 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Awada AH et al. 381TiP - TRYbeCA-2: A randomized phase II/III study of eryaspase in combination with gemcitabine and carboplatin chemotherapy versus chemotherapy alone as first-line treatment in patients with metastatic or locally recurrent triple-negative breast cancer. Annals of Oncology 30, v138, doi: 10.1093/annonc/mdz242.076 (2019). [DOI] [Google Scholar]
- 66.Knott SRV et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 554, 378–381, doi: 10.1038/nature25465 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lin HH et al. Autophagic reliance promotes metabolic reprogramming in oncogenic KRAS-driven tumorigenesis. Autophagy 14, 1481–1498, doi: 10.1080/15548627.2018.1450708 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Banerjee I et al. Assessing treatment response in triple-negative breast cancer from quantitative image analysis in perfusion magnetic resonance imaging. Journal of Medical Imaging 5, 1, doi: 10.1117/1.jmi.5.1.011008 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tasdogan A et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature, doi: 10.1038/s41586-019-1847-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lu W et al. Metabolomic analysis via reversed-phase ion-pairing liquid chromatography coupled to a stand alone orbitrap mass spectrometer. Analytical chemistry 82, 3212–3221, doi: 10.1021/ac902837x (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Su X, Lu W & Rabinowitz JD Metabolite Spectral Accuracy on Orbitraps. Analytical chemistry 89, 5940–5948, doi: 10.1021/acs.analchem.7b00396 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nasser S et al. An integrated framework for reporting clinically relevant biomarkers from paired tumor/normal genomic and transcriptomic sequencing data in support of clinical trials in personalized medicine. Pac Symp Biocomput, 56–67 (2015). [PubMed] [Google Scholar]
- 73.Li H & Durbin R Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760, doi: 10.1093/bioinformatics/btp324 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McKenna A et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303, doi: 10.1101/gr.107524.110 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Christoforides A et al. Identification of somatic mutations in cancer through Bayesian-based analysis of sequenced genome pairs. BMC Genomics 14, 302, doi: 10.1186/1471-2164-14-302 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Saunders CJ et al. Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci Transl Med 4, 154ra135, doi: 10.1126/scitranslmed.3004041 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cibulskis K et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31, 213–219, doi: 10.1038/nbt.2514 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tate JG et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res 47, D941–d947, doi: 10.1093/nar/gky1015 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, doi: 10.1093/bioinformatics/bts635 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Holmes FA et al. Pathologic complete response after preoperative anti-HER2 therapy correlates with alterations in PTEN, FOXO, phosphorylated Stat5, and autophagy protein signaling. BMC Res Notes 6, 507, doi: 10.1186/1756-0500-6-507 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Paweletz CP et al. Reverse phase protein microarrays which capture disease progression show activation of pro-survival pathways at the cancer invasion front. Oncogene 20, 1981–1989, doi: 10.1038/sj.onc.1204265 (2001). [DOI] [PubMed] [Google Scholar]
- 82.Petricoin EF 3rd et al. Phosphoprotein pathway mapping: Akt/mammalian target of rapamycin activation is negatively associated with childhood rhabdomyosarcoma survival. Cancer Res 67, 3431–3440, doi: 10.1158/0008-5472.CAN-06-1344 (2007). [DOI] [PubMed] [Google Scholar]
- 83.Chiechi A et al. Improved data normalization methods for reverse phase protein microarray analysis of complex biological samples. Biotechniques 0, 1–7, doi: 10.2144/000113926 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gallagher RI, Silvestri A, Petricoin EF 3rd, Liotta LA & Espina V Reverse phase protein microarrays: fluorometric and colorimetric detection. Methods Mol Biol 723, 275–301, doi: 10.1007/978-1-61779-043-0_18 (2011). [DOI] [PubMed] [Google Scholar]
- 85.VanMeter AJ et al. Laser capture microdissection and protein microarray analysis of human non-small cell lung cancer: differential epidermal growth factor receptor (EGPR) phosphorylation events associated with mutated EGFR compared with wild type. Mol Cell Proteomics 7, 1902–1924, doi: 10.1074/mcp.M800204-MCP200 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Signore M, Manganelli V & Hodge A Antibody Validation by Western Blotting. Methods Mol Biol 1606, 51–70, doi: 10.1007/978-1-4939-6990-6_4 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Patient characteristics; related to Figure 1
Table S2. TNBC biopsy WES filtered variants; related to Figure 1
Table S5. Mouse tissue metabolite labeling; related to Figure 4
Table S7. TNBC biopsy metabolomics data; related to Figure 7
Data Availability Statement
There are IRB restrictions regarding next generation sequencing data sharing for this study and patients were not consented for public genomic data sharing. Select somatic variants and processed RNA-Seq data (TPM), RPPA data, metabolomics and TNBC isotope labeling data are available in Tables S2–S7. Isotope labeling data for NSCLC were obtained from Faubert et al 44. Isotope labeling data for RCC and brain tumors were obtained from Courtney et al47. Further access to data may be made by contacting the corresponding author.