

Abstract
Targeting the interaction between the SARS-CoV-2 spike protein and human ACE2, its primary cell membrane receptor, is a promising therapeutic strategy to prevent viral entry. Recent in vitro studies revealed that the receptor binding domain (RBD) of the spike protein plays a prominent role in ACE2 binding, yet a simple and quantitative assay for monitoring this interaction in a cellular environment is lacking. Here, we developed an RBD-ACE2 binding assay that is based on time-resolved FRET, which reliably monitors the interaction in a physiologically relevant and cellular context. Because it is modular, the assay can monitor the impact of different cellular components, such as heparan sulfate, lipids, and membrane proteins on the RBD-ACE2 interaction and it can be extended to the full-length spike protein. The assay is HTS compatible and can detect small-molecule competitive and allosteric modulators of the RBD-ACE2 interaction with high relevance for SARS-CoV-2 therapeutics.
Keywords: SARS-CoV, spike protein, ACE2, TR-FRET, HTRF, COVID-19, 2019-nCoV, HTS, TMPRSS2, heparan sulfate
Graphical abstract
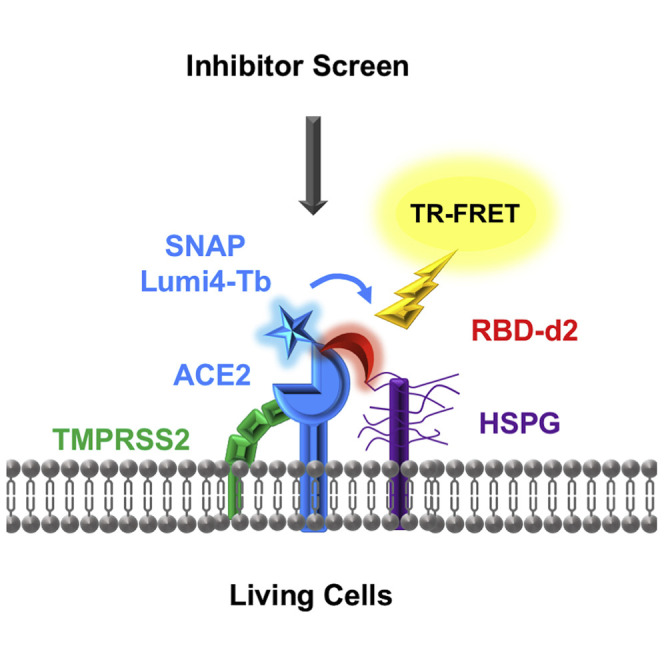
Cecon et al. describe a quantitative, time-resolved FRET assay capable of detecting SARS-CoV-2 spike interactions with ACE2 in living cells. The assay monitors the interaction in a physiologically relevant cellular environment and is suitable for diverse applications, including mechanistic studies, drug screening, and characterization of neutralizing antibodies or vaccine efficacy.
Introduction
The worldwide SARS-CoV-2 pandemic provoked an urgent need for effective therapeutic solutions to prevent and treat the resulting COVID-19 disease. Rapid progress was made on understanding the molecular basis of viral infection, and inhibition of the initial steps of viral-host cell recognition was identified as a promising strategy for therapeutic intervention. The SARS-CoV-2 spike (S) protein is required for virus attachment and entry into target cells through binding to the human angiotensin-converting enzyme 2 (ACE2), the primary SARS-CoV-2 entry receptor (Zhou et al., 2020). Cryoelectron microscopy studies suggest that two S protein trimers bind simultaneously to an ACE2 homodimer (Yan et al., 2020). The S protein is composed of S1 and S2 domains, with the ACE2 receptor binding domain (RBD) located in the S1 region. Successful viral entry requires proteolytic cleavage of the S protein between S1 and S2 domains by the transmembrane protease serine 2 (TMPRSS2) expressed by the host cell (Hoffmann et al., 2020). Other components of the host cell have been suggested to participate in this core SARS-CoV-2/ACE2 complex, such as endogenous heparan sulfate proteoglycans (HSPGs) exposed at the cell surface (Clausen et al., 2020; Zhang et al., 2020) or CD4 (Davanzo et al., 2020). These auxiliary components are likely to confer cell-type specificity but their precise role remains to be determined.
Interfering with the RBD/ACE2 interaction has been proposed as an attractive therapeutic strategy as demonstrated by the inhibitory effect of neutralizing anti-ACE2 antibodies (Baum et al., 2020) and nanobodies (Huo et al., 2020), de novo designed miniprotein inhibitors based on the ACE2 helix interacting with RBD (Cao et al., 2020) or small-molecule FDA-approved drugs (Fu et al., 2020). Small molecules are particularly attractive because they are generally cost-effective, show good stability and a high rate of penetration over biological barriers to easily reach their targets. Identification of such molecules currently relies mainly on acellular in vitro assays suitable for high-throughput screening (HTS) purposes. However, these assays do not take into account the role of the cellular environment, which is likely to have a major impact on the formation of the SARS-CoV-2/ACE2 complex. We developed here a time-resolved fluorescence resonance energy transfer (TR-FRET) assay that probes the molecular proximity and conformational changes of the RBD/ACE2 complex in a cellular context. The assay successfully detects competitive and allosteric modulators of the RBD/ACE2 complex, is suitable for HTS applications and allows validation of candidate compounds identified in in vitro assays. The assay can be easily customized by co-expressing auxiliary components thus providing mechanistic insights in the modulation of the RBD/ACE2 core complex, enabling the design of the most relevant cellular environment for tailored inhibitor screening.
Results
Binding of RBD of the SARS-CoV-2 spike protein to ACE2 monitored by TR-FRET
TR-FRET assays are increasingly used to monitor molecular interactions at the nanometer scale with high signal-to-noise ratio due to the temporal separation between sample excitation and energy transfer measurements (Degorce et al., 2009) (Figure 1 A). The assay is based on the energy transfer between an energy donor (N-terminal SNAP-tagged human ACE2 labeled with terbium [Tb], SNAP-ACE2 in our case) and an energy acceptor (RBD of the SARS-CoV-2 spike protein labeled with the d2 fluorophore, RBD-d2 in our case), which occurs only if both are in close proximity to each other (<10 nm) (Bazin et al., 2002; Degorce et al., 2009; Mathis, 1995) (Figure 1B). SNAP is an O6-alkylguanine-DNA alkyl transferase that catalyzes its own covalent binding to fluorescent derivatives of benzylguanine, such as Lumi4-Tb (Keppler et al., 2003). For the binding assay, the SNAP-ACE2 was expressed in HEK293 cells and labeled with the cell-impermeant Lumi4-Tb. The SNAP-ACE2 fusion protein migrated at an apparent molecular weight of 140 kDa in SDS-PAGE experiments upon expression in HEK293 cells, as expected (Figure S1). Binding of the RBD-d2 tracer to Lumi4-Tb-labeled SNAP-ACE2 was saturable at equilibrium and with nanomolar affinity (Kd = 14.6 ± 2.5 nM; n = 10) (Figure 1C; Table 1 ). Non-specific binding was defined in the presence of an excess of non-labeled RBD (1 μM) and resulted in a signal-to-noise ratio higher than 15 (Figure 1C). RBD-d2 (5 nM) association and dissociation was observed in a time-dependent manner (Figures 1D and 1E), with kon and koff values of 1.3 × 106 ± 5.1 × 105 M−1 s−1 and 2.93 × 10−3 ± 0.5 × 10−3 s−1 (n = 4), respectively, and a calculated Kd of 2.3 ± 1.5 nM (Table 1). The koff was similar to and the kon 6.6 times higher than the mean kon of previous in vitro studies (Table S1). No specific binding of saturating concentration of RBD-d2 (20 nM) was observed in cells expressing similar amounts of the Lumi4-Tb-labeled SNAP-tagged VEGF receptor 2, a single transmembrane control receptor of similar size and topology (Figure 1F). Several hormones, cytokines, chemokines, and lectins (100 nM) found in the extracellular milieu were unable to displace RBD-d2 (5 nM) binding to Lumi4-Tb-labeled SNAP-ACE2, demonstrating the high specificity of the assay (Figure 1G). Fluorescence microscopy experiments showed that RBD-d2 (20 nM) only binds to cells expressing Lumi4-Tb-labeled SNAP-ACE2 but not the control SNAP-LepR (leptin receptor), a single transmembrane protein with a large extracellular domain (Figure 1H). The Lumi4-Tb-label was not observed in the RBD-d2 channel, demonstrating that there was no leakage of the fluorescence signal between the two channels (Figure 1H). Taken together, these results show that fluorescently labeled RBD-d2 binds with high (nanomolar) affinity and high specificity to ACE2 expressed in HEK293 cells.
Figure 1.

Development of SARS-CoV-2 spike protein/ACE2 TR-FRET binding assay
(A) Principle of TR-FRET assay.
(B) Scheme illustrating the TR-FRET-based RBD-d2 binding assay to SNAP-tagged ACE2 labeled with Lumi4-Tb.
(C) Saturation binding curve of RBD-d2 to Lumi4-Tb-labeled SNAP-ACE2 expressed in HEK293 cells (representative curve, expressed as mean ± SD of triplicates; n = 10). Non-specific binding was defined in the presence of an excess of non-labeled RBD (1 μM).
(D and E) Association (D) and dissociation (E) kinetics of RBD-d2 binding (5 nM) to Lumi4-Tb-SNAP-ACE2 expressed in HEK293 cells (representative curve, expressed as mean ± SD of triplicates; n = 4). Dissociation was initiated by adding unlabeled RBD (1 μM).
(F) Binding of RBD-d2 (20 nM) to Lumi4-Tb-SNAP-ACE2 but not Lumi4-Tb-SNAP-VEGFR2. Data are expressed as mean ± SEM of three independent experiments, each performed in triplicate. Insert: representative data of the expression level of Lumi4-Tb-labeled SNAP-ACE2 and SNAP-VEGFR2 assessed by Tb fluorescence measurement (620 nm).
(G) Competition of RBD-d2 (5 nM) binding to Lumi4-Tb-SNAP-ACE2 by non-labeled RBD (1 μM), leptin, IL-6, MCP1, Gal3, insulin, EGF, VEGF (each at 100 nM). Data are expressed as mean ± SEM of three independent experiments, each performed in triplicate; ∗∗∗∗p = 0.0001 by one-way ANOVA. “RBD-d2 Binding” in (F and G) corresponds to the TR-FRET ratio and is expressed as percent of basal (absence of RBD-d2). (H) RBD-d2 (20 nM) interaction with HEK293 cells expressing either SNAP-ACE2 or SNAP-LepR monitored by fluorescence microscopy. Scale bar, 10 μm.
See also Figure S1.
Table 1.
Binding constants of SARS-CoV-2 spike RBD binding to ACE2 in HEK293 cells determined by TR-FRET assay
| Condition | Equilibrium Kd (nM) |
Kon (M−1 s−1) |
Koff (s−1) | Kinetically derived Kd = Koff/Kon (nM) |
RT = 1/Koff (min) |
|---|---|---|---|---|---|
| Control | 14.6 ± 2.5 (n = 10) | 1.3 × 106 ± 5.1 × 105 (n = 4) |
2.93 × 10−3 ± 0.5 × 10−3 (n = 4) |
2.3 ± 1.5 (n = 4) |
7.4 ± 0.5 (n = 4) |
| + TMPRSS2 expression | N/D | 5.9 × 105 ± 6.6 × 104 (n = 3) |
3.4 × 10−3 ± 0.3 × 10−3 (n = 3) |
6.1 ± 1.1 (n = 3) |
5.1 ± 0.5 (n = 3) |
| + Heparin | 25.2 ± 8.7∗ (n = 4) |
3.8 × 105 ± 1.5 × 105 (n = 3) |
2.78 × 10−3 ± 0.49 × 10−3 (n = 3) |
7.3 ± 2.5 (n = 3) |
7.0 ± 1.2 (n = 3) |
| + CD4 expression | 28.9 ± 9.5∗ (n = 4) |
3.9 × 105 ± 4 × 104 (n = 3) |
2.31 × 10−3 ± 0.07 × 10−3 (n = 3) |
6.0 ± 0.2 (n = 3) |
7.2 ± 0.5 (n = 3) |
Data are expressed as mean ± SEM of indicated number (n) of independent experiments. ∗p < 0.05 by paired t test two-tailed compared with respective matched control group performed in the same experimental set. N/D, no data; RT, residence time. See also Figures S3 and S4.
Inhibition of SARS-CoV-2 spike RBD binding to ACE2
Identification of molecules interfering with RBD binding to ACE2 constitutes a major application of our TR-FRET assay. To validate this aspect of the assay we performed competition binding experiments with non-labeled RBD. Competition of the RBD-d2 tracer (5 nM) binding to ACE2 occurred at the nanomolar range (pKi = 7.70 ± 0.03; n = 5), confirming the high-affinity binding of RBD, which is identical to that of the RBD-d2 tracer (Figure 2 A). We then tested the recently developed high-affinity miniprotein LCB1v3, which is based on the ACE2 helix that interacts with RBD (Cao et al., 2020), and we observed a full competition of the RBD-d2 tracer in the subnanomolar range (pKi = 9.42 ± 0.1; n = 5). Similarly, a neutralizing SARS-CoV-1/2 spike RBD Llamabody VHH effectively competed RBD-d2 binding to ACE2, displaying 82% inhibition at 50 μg/mL and displaying an half-maximal inhibitory concentration (IC50) of 5.1 ± 0.04 μg/mL (n = 3) (Figure 2B). To demonstrate the suitability of the assay to identify new inhibitory compounds in an HTS mode, we determined the Z′ that estimates the robustness and reliability of HTS assays (Zhang et al., 1999). A Z′ value of 0.83 was obtained for a decrease in TR-FRET signal of 84% at 5 nM of RBD-d2 and 300 nM of RBD competitor (Figure 2C), which is in the upper range (0.7–1), considered as excellent performance for HTS. Taken together, these results demonstrate the HTS suitability of our TR-FRET assay in the competition mode.
Figure 2.

Detection of competitors of the RBD/ACE2 interaction using the TR-FRET-based assay
(A) Competition of RBD-d2 binding (5 nM) to Lumi4-Tb-SNAP-ACE2 in HEK293 cells by non-labeled RBD or LCB1v3 miniprotein; data are expressed as mean ± SEM of five independent experiments, each performed in triplicate.
(B) Competition of RBD-d2 binding (5 nM) to Lumi4-Tb-SNAP-ACE2 in HEK293 cells by anti-RBD Llamabody VHH (representative curve, n = 3 independent experiments, each performed in triplicate).
(C) Determination of Z′ by collecting TR-FRET signals for RBD-d2 (5 nM) binding to Lumi4-Tb-labeled SNAP-ACE2 expressing HEK293 cells in the absence or presence of RBD (300 nM) from 50 different wells.
(D–H) Competition of RBD-d2 (5 nM) binding to Lumi4-Tb-SNAP-ACE2 by cangrelor (n = 3), elaidic acid (n = 4), fenbendazole (n = 3), enalapril maleate (n = 5), or corilagin (n = 6). “RBD-d2 Binding” corresponds to the TR-FRET ratio and is expressed as percent of control (vehicle-treated group). Data are expressed as mean ± SEM of indicated independent experiments, each performed in triplicate; ∗∗∗∗p < 0.001 by one-way ANOVA.
See also Figure S2.
Several small-molecule compounds have been described to interfere with the RBD-ACE2 interaction in in vitro assays. This is the case of the AlphaLISA proximity assay that was applied to screen 3,384 small-molecule drugs and pre-clinical compounds for drug repurposing (Hanson et al., 2020). We tested 5 out of the 25 high-quality hits identified in this in vitro assay with reported apparent IC50 values in the low micromolar range. Four compounds, cangrelor, elaidic acid, fenbendazole, and enalapril maleate, with reported in vitro IC50 values of around 10–15 μM (Hanson et al., 2020), were unable to compete RBD-d2 (5 nM) at concentrations up to 100 or 1,000 μM (Figures 2D–2G). Corilagin (reported in vitro IC50 of 5.5 μM) (Hanson et al., 2020) showed a statistically significant 30% inhibition at 100 μM (p < 0.001) (Figure 2H). The specificity of this inhibitory effect of corilagin was confirmed in a counter-assay using a similar assay format (Vauthier et al., 2013) in which corilagin did not affected the binding of d2-labeled leptin to its Lumi4-Tb-labeled SNAP-tagged receptor (Figure S2). Of note, corilagin was also the only positive compound in the original article of Hanson et al. (2020) after having performed counter assays. Collectively, these data suggest a significant difference in the apparent IC50 of RBD-ACE2 interaction inhibitors between the in vitro and cellular binding assays, with an estimated loss of affinity of 1–2 logs in the cellular assay. Information obtained with the cellular TR-FRET assay will be crucial to select the most promising compounds to move forward in the drug development process.
Modularity of the SARS-CoV-2 spike RBD/ACE2 binding
The binding of SARS-CoV-2 to ACE2 and cellular entry of the virus occurs in a complex cellular environment that should be taken into consideration when characterizing the spike/ACE2 interaction and when searching for potential inhibitors. To evaluate whether our TR-FRET assay is able to fulfill this requirement we studied the impact of modulating the cellular environment on our binding assay.
We first explored the RBD/ACE2 binding in a transcellular mode. HEK293 cells expressing fluorescently Lumi4-Tb-labeled SNAP-ACE2 were either mixed with mock-transfected cells or cells expressing non-labeled SNAP-ACE2 (1:1 ratio of cells) and the RBD-d2 tracer (5 nM) (Figure 3 A). Only non-labeled SNAP-ACE2 cells inhibited the TR-FRET signal (∼50%) demonstrating the possibility of our TR-FRET assay to detect endogenous transcellular components, such as membrane-anchored proteins and co-factors, that interfere with the interaction.
Figure 3.

Modularity of SARS-CoV-2 spike RBD/ACE2 binding
(A) Competition of RBD-d2 binding (5 nM) to Lumi4-Tb-SNAP-ACE2 by HEK293 cells expressing non-labeled SNAP-ACE2 (transcellular mode) or by mock-transfected cells. Non-specific signal is determined in the presence of excess concentration of non-labeled RBD (1 μM). Data are expressed as mean ± SEM of three independent experiments, each performed in triplicate; ∗∗∗∗p < 0.001 by one-way ANOVA.
(B and C) Binding of RBD-d2 (20 nM) (B) or spike S1-S2-d2 (30 nM) (C) to Lumi4-Tb-labeled SNAP-ACE2 in HEK293 cells with and without TMPRSS2 co-expression. Insert: expression level of Lumi4-Tb-labeled SNAP-ACE2 (energy donor) in both conditions assessed by Tb fluorescence measurement (620 nm). Non-specific signal is determined in the presence of excess concentration of non-labeled RBD (1 μM) or non-labeled S1-S2 (200 nM). Data are expressed as mean ± SEM of three independent experiments, each performed in triplicate; ∗∗p < 0.01, ∗∗∗∗p < 0.001 by one-way ANOVA, or two-way ANOVA when comparing the effect of the presence of TMPRSS2; n.s., not significant.
(D) Competition of RBD-d2 binding (5 nM) to Lumi4-Tb-SNAP-ACE2 by heparin. Data are expressed as mean ± SEM of four independent experiments, each performed in triplicate. HSPGs, heparan sulfate proteoglycans.
(E) Saturation binding curve of RBD-d2 to Lumi4-Tb-SNAP-ACE2 in the absence (black line) and presence (red line) of heparin (representative curve, expressed as mean ± SD of triplicates, 3 mg/mL; n = 4) at equilibrium (2 h incubation time). Non-specific binding was defined by an excess of non-labeled RBD (1 μM).
(F and G) Representative curves of association (F) and dissociation (G) kinetics of RBD-d2 binding (5 nM) to Lumi4-Tb-SNAP-ACE2 in the absence (black line) or presence (red line) of heparin (3 mg/mL; n = 3). Dissociation was initiated by adding non-labeled RBD (1 μM).
(H) ACE2 conformational change assessed by intramolecular TR-FRET between Lumi4-Tb-labeled SNAP-ACE2 and d2-labeled anti-FLAG tag antibody in HEK293 cells, in the presence of non-labeled RBD (5 nM), heparin (3 mg/mL), or RBD + heparin (0.3 and 3 mg/mL). Data are expressed as change of TR-FRET signal (ΔTR-FRET ± SEM) of three independent experiments, each performed in triplicate. ∗p < 0.05, ∗∗p < 0.01 by one-way ANOVA, followed by Dunnett's multiple comparisons test compared with the RBD group.
(I) Saturation binding curve of RBD-d2 to Lumi4-Tb-labeled SNAP-ACE2 in HEK293 cells with and without CD4 co-expression. Non-specific binding was defined by an excess of non-labeled RBD (1 μM). Data are expressed as mean ± SEM of four independent experiments, each performed in triplicate. Insert: data points from 0 to 40 nM of RBD-d2 concentration. “RBD-d2 Binding” in (A, D, H, and I) corresponds to the TR-FRET ratio and is expressed as percent of control group or maximal binding (Bmax).
See also Figures S3 and S4.
TMPRSS2 is known to promote the cellular entry of SARS-CoV-2 by cleaving the spike protein of SARS-CoV-2, a necessary step for viral entry (Hoffmann et al., 2020). Such a mechanism would indicate a molecular proximity between spike/ACE2 complex and TMPRSS2. To elucidate the impact of TMPRSS2 on the RBD/ACE2 interaction, we co-expressed TMPRSS2 and SNAP-ACE2. The presence of TMPRSS2 reduced the TR-FRET signal by 50% at a saturating concentration of the RDB-d2 tracer (20 nM; Figure 3B) and constant amounts of Lumi4-Tb-labeled SNAP-ACE2 (Figure 3B, insert). The effect of TMPRSS2 on the TR-FRET signal was concentration dependent (Figure S3). The kinetic parameters of RBD binding to ACE2 in cells co-expressing TMPRSS2 were not significantly different from control cells (Table 1; Figures S4A and S4B). As the TMPRSS2 cleavage site on the spike protein is located outside of the RBD, between the spike’s S1 and S2 domain, we also investigated the effect of TMPRSS2 on the binding of d2-labeled spike S1-S2 as tracer. Binding of spike S1-S2-d2 (30 nM) to Lumi4-Tb-labeled SNAP-ACE2 was readily observed and competed by non-labeled spike S1-S2 (200 nM; Figure 3C). Reduction of the TR-FRET signal was once again observed, at a constant amount of Lumi4-Tb-labeled SNAP-ACE2. Co-expression of TMPRSS2 did not affect the kinetic parameters or affinity of S1-S2 for ACE2 (Table 2 ; Figures S4C and S4D). S1-S2 displayed a calculated Kd of 3.8 ± 0.9 nM (n = 3) for ACE2 (Table 2), which is similar to the Kd of RBD for ACE2. Collectively, these data indicate that TMPRSS2, by binding to ACE2, modulates the RBD/ACE2 interaction. This effect is independent of the TMPRSS2 cleavage site on the full-length spike protein. TMPRSS2 has no or only a modest impact on the affinity of RBD(S1-S2) for ACE2 and induces a substantial conformational change in the RBD(S1-S2)/ACE2 complex. This latter conclusion was further confirmed by the robust intramolecular conformational change in ACE2 induced by TMPRSS2 (Figure S4F).
Table 2.
Binding constants of SARS-CoV-2 spike S1-S2 binding to ACE2 in cells co-transfected or not with TMPRSS2 determined in TR-FRET assay
| Condition | Kon (M−1 s−1) (n = 3) |
Koff (s−1) (n = 3) |
Kinetically derived Kd = Koff/Kon (nM) (n = 3) |
RT = 1/Koff (min) (n = 3) |
|---|---|---|---|---|
| Control | 6.2 × 105 ± 1.6 × 105 (n = 3) |
2.4 × 10−3 ± 3 × 10−4 (n = 3) |
3.8 ± 0.86 (n = 3) |
6.6 ± 0.9 (n = 3) |
| + TMPRSS2 expression | 3.8 × 105 ± 1.4 × 105 (n = 3) |
2.4 × 10−3 ± 1 × 10−4 (n = 3) |
6.3 ± 3.8 (n = 3) |
5.8 ± 0.5 (n = 3) |
Data are expressed as mean ± SEM of indicated number (n) of independent experiments. ∗p < 0.05 compared with the “control” condition by paired t test two-tailed. RT, residence time. See also Figure S4.
HSPGs are known modulators of SARS-CoV, and SARS-CoV-2 entry and exogenous heparin has been shown to mitigate the binding of RBD to ACE2 (Clausen et al., 2020; Zhang et al., 2020). Consistently, we observed an inhibition of RBD-d2 binding (5 nM) to ACE2 (IC50 = 96 ± 23.4 μg/mL; n = 4) in our TR-FRET assay (Figure 3D). The inhibition was partial (∼50%), which is consistent with the proposed allosteric, rather than competitive, binding mode (Figure 3D). Indeed, heparan sulfate/heparin has been shown to bind to the RBD at a site that is distinct from the ACE2 binding site (Clausen et al., 2020). Binding of heparin is suspected to induce conformational changes in RBD and to stabilize its open conformation prone to ACE2 binding (Clausen et al., 2020). Equilibrium binding experiments show significantly lower affinity of RBD in the presence of heparin (Figure 3E; Table 1), and a similar trend was observed in kinetic binding studies without reaching statistical significance (Figures 3F and 3G; Table 1). To provide further evidence that heparin, by replacing HSPG binding to RBD, induces conformational changes within the ACE2/RBD complex, we assessed conformational changes within ACE2 by intramolecular TR-FRET. TR-FRET was measured between the Lumi4-Tb-labeled SNAP tag and the FLAG tag recognized by d2-labeled anti-FLAG antibodies. Consistent with the close proximity of the two tags at the N terminus of ACE2, a robust TR-FRET signal was observed, which was completely inhibited by non-labeled anti-FLAG antibodies confirming the specificity of the assay (Figure S4E). Addition of RBD (5 nM) induced a significant change of the TR-FRET signal, while the signal was reversed by the addition of heparin (0.3 and 3 mg/mL; n = 3; Figure 3H) consistent with the hypothesis that heparin impacts on the conformation of the RBD/ACE2 complex. Collectively, these results support the allosteric rather than competitive binding mode of heparin on the RBD/ACE2 complex, and clarifies the effect of proximal heparan sulfates present in the extracellular matrix on the RBD/ACE2 interaction, which probably stabilizes a higher-affinity conformation of ACE2/RBD complex.
Another potential modulator of RBD/ACE2 interaction in the cell surface is CD4, which has been recently described as essential for viral entry into CD4+ T helper lymphocytes (Davanzo et al., 2020). Molecular modeling predicted binding of the extracellular N-terminal domain of CD4 to the SARS-CoV-2 spike RBD at a site overlapping with ACE2 binding, suggesting a competition of CD4 and ACE2 for RBD binding. Further functional observations suggest rather a concerted action of ACE2, TMPRSS2, and CD4 to allow the infection of CD4+ T cells by SARS-CoV-2. To clarify this issue, the co-expression of full-length CD4 and SNAP-ACE2 showed a significantly decreased affinity and maximal binding of the RDB-d2 tracer (5 nM) to ACE2 (Figure 3I, Table 1). Kinetic measurements showed a 3.3 times slower association rate (kon) and comparable dissociation rate (koff) than without CD4 (Table 1; Figures S4G and S4H). These results suggest additional allosteric modulation of the binding properties of the RBD/ACE2 interaction by CD4 through conformational changes.
Discussion
In this study, we have developed an innovative binding assay that probes the interaction of ACE2 with the SARS-CoV-2 spike protein at the plasma membrane of living cells. The assay is highly flexible as it is compatible with any cell type of interest that can be transfected/transduced with the SNAP-ACE2 expression vector. This is an advantage compared with in vitro assays, as it can reveal cell-specific membrane components, such as HSPGs and co-factors, such as TMPRSS2 and CD4, that can impact RBD/ACE2 interaction in a cell-dependent manner. The assay is based on the proximity between ACE2 and RBD on cells and displays multiple advantages: rapid and easy to perform, homogeneous without washing steps, compatible with HTS in a physiologically relevant cellular context. The assay will be of valuable importance in the pandemic as a reliable foundation/starting point for drug discovery, being compatible with HTS, and as a research tool for understanding the mechanism of action and pharmacology of the RBD/ACE2 interaction.
In vitro RBD/ACE2 binding assays rely on the immobilization of one of the interacting partners, typically purified ACE2, to a support or sensor chip. Among those assays, the ELISA technique is commonly used to assess RBD binding to purified ACE2 in the presence of putative competitors. However, such assays are not homogenous, require washing steps, and can suffer from detection limits, large variability, and non-negligible false positives as detailed recently in the context of COVID-19 (GeurtsvanKessel et al., 2020). Other more sophisticated in vitro binding techniques include surface plasmon resonance (SPR) and bilayer interferometry (BLI). The range of Kd values of the RBD/ACE2 complex determined by SPR and BLI varied widely from 1.2 to 117 nM (mean 42.5 ± 41.3 nM) across reported in vitro binding studies (Table S1). Likewise, corresponding kon and koff values vary widely with mean values of 1.8 ± 1.1 × 105 M−1 s−1 and 4.9 ± 3.3 × 10−3 s−1, respectively (Table S1). Kinetic parameters from cellular binding assays were lacking. The kon and koff values for RBD binding to ACE2 in HEK293 cells derived from our TR-FRET assay were 1.3 × 106 ± 5.1 × 105 M−1 s−1 and 2.9 × 10−3 ± 0.5 × 10−3 s−1, respectively. Interestingly, the TR-FRET kon was 7.2 times higher compared with in vitro studies indicating that cellular components significantly enhance the association rate of RBD to ACE2.
Available in vitro assays lack the complexity of cellular context, including participation of auxiliary cell surface proteins or cellular elements. Viral entry-associated membrane fusion requires a priming step of the spike protein mediated by host proteases, including TMPRSS2, that cleave the spike protein at a site between the S1 and S2 domains to activate plasma membrane fusion (Hoffmann et al., 2020). Previous studies showed that TMPRSS2 interacts with ACE2 leading to the proteolytic cleavage of ACE2, which was proposed to augment viral infectivity of SARS-CoV-1 (Shulla et al., 2011; Heurich et al., 2014) and potentially of SARS-CoV-2 (Mohammad et al., 2020). In our binding assay, expression of TMPRSS2 decreases the TR-FRET signal between RBD or full-length spike (S1-S2) with ACE2 suggesting a conformational change within the RBD/ACE2 complex. TMPRSS2-induced cleavage of ACE2 is unlikely to account for the decreased TR-FRET signal as the assay is homogeneous and the soluble ACE2 domain remains fully functional in terms of RBD binding, as demonstrated by others (Shang et al., 2020; Toelzer et al., 2020). The hypothesis that TMPRSS2 induces conformational changes within the RBD/ACE2 complex is supported by the induction of intramolecular ACE2 conformational changes by TMPRSS2 and the absence of any striking effect of TMPRSS2 on the binding affinity and kinetics of either RBD or S1-S2 to ACE2.
HSPGs are expressed at the cell surface of many cells and have been shown to be essential for SARS-CoV-2 entry in combination with ACE2, adding a further cell context-dependent component to the RBD/ACE2 interaction (Clausen et al., 2020). HSPGs contain negatively charged heparan sulfates that promote interactions with a variety of positively charged cargos (Christianson and Belting, 2014). The RBD harbors a positively charged cleft suspected to accommodate heparan sulfate in a ternary complex composed of heparan sulfate/RBD/ACE2 (Clausen et al., 2020). Addition of exogenous heparin competes with the heparan sulfate binding to RBD (Clausen et al., 2020; Zhang et al., 2020). By probing the proximity of RBD to ACE2 at the plasma membrane, our TR-FRET assay revealed a 50% decrease of the TR-FRET by heparin and a significant decrease in affinity of RBD binding to ACE2 at equilibrium. Of note, the kon of 3.8 × 105 M−1 s−1 in the presence of heparin was close to the mean kon of in vitro studies of 1.8 × 105 M−1 s−1 (see Table S1). These data are compatible with the suspected function of HSPGs, present in a cellular context, to accelerate and facilitate the interaction of RBD with ACE2 by increasing the affinity and the residential time. The partial displacement of the RBD-d2 tracer by heparin suggests an allosteric effect of heparin on the RBD/ACE2 interaction. Displacement of HSPGs by heparin most likely induces conformational changes within RBD that alter the affinity of RBD for ACE2 in an allosteric manner. This HSPG-induced conformational change would be in favor of the model in which RBD would adopt an “up/open orientation” facilitating endocytosis and viral infection (Wrapp et al., 2020; Walls et al., 2020). By performing an ACE2 intramolecular TR-FRET assay we demonstrated that heparin indeed induces conformational changes in the RBD/ACE2 complex. Alternatively, ACE2 might exist in two conformers, one being heparan sulfate dependent and the other heparan sulfate independent. Heparin would inhibit binding of RBD to one ACE2 conformer and RBD can still bind ACE2 in the second ACE2 conformation, but with a lower affinity. In conclusion, the important impact of HSPGs on the RBD/ACE2 interaction nicely illustrates the importance of the cellular environment for the RBD/ACE2 interaction.
Intensive research on SARS-COV-2 has given rise to the hypothesis that, beside ACE2, other cellular components, including heparan sulfate (Clausen et al., 2020) and alternative secondary receptors, such as neuropilin1 (Cantuti-Castelvetri et al., 2020), CD4 (Davanzo et al., 2020), CD147 (Wang et al., 2020; Ulrich and Pillat, 2020), and GRP78 (Ibrahim et al., 2020), could actively participate in viral binding and entry. Here, we observed that CD4 co-expression with ACE2 systematically decreases the plateau of TR-FRET signal, indicating CD4-mediated modulation of RBD/ACE2 complex. Cellular components, such as membrane proteins, can influence and modulate the RBD/ACE2 interaction, reinforcing the importance of performing the binding studies in cells either for mechanistic investigations, pharmacological characterization, or screening campaigns.
An easy, robust, sensitive and reproducible biochemical interaction assay is essential for implementing HTS campaigns. Our TR-FRET RBD/ACE2 binding assay fulfills these criteria with its homogeneous format, a robust TR-FRET signal (10- to 30-fold over background), an excellent Z′ score of 0.83 and its high sensitivity detecting inhibitors in the subnanomolar range, as well as allosteric modulators. A recent study aimed at identifying inhibitors of the RBD/ACE2 interaction in a drug repurposing strategy through an in vitro AlphaLISA proximity-based assay (Hanson et al., 2020). We picked 5 out of the 25 top hits identified by this in vitro assay for validation in our cellular TR-FRET assay. Only 1 out of 5 compounds, corilagin, the most active compound in the AlphaLISA, showed a partial inhibition at 100 μM, a concentration more than 20 times higher than the IC50 reported in the AlphaLISA assay. The recently described LCB1v3 miniprotein targeting the RBD/ACE2 interaction showed an IC50 in the subnanomolar range (Cao et al., 2020) validating our assay for the screening of RBD/ACE2 interaction inhibitors with high affinity. Collectively, these data show that in vitro assays can overestimate the inhibitory capacity of compounds and highlight the need to perform screening campaigns with cellular assays.
Our results with the anti-spike Llamabody VHH suggests further applications of our TR-FRET assay, including the characterization of therapeutic antibodies in terms of epitope specificity, binding properties, and in structural/functional studies (DeFrancesco, 2020). In addition, performing the TR-FRET assay in a physiological context with cellular complexity can improve the assessment of vaccine efficacy by evaluating whether RBD-specific antisera might block the interaction between RBD to ACE2 at the level of the cell rather than in a restrictive in vitro setting.
In conclusion, the TR-FRET assay allows studying of the interaction of RBD with ACE2 on living cells in a physiological context. Unlike an in vitro binding assay, an additional layer of complexity is observed when using biological membranes containing further relevant components, such as TMPRSS2, HSPG, CD4, etc. Hence the assay can be adapted for a specific purpose via co-expression of membrane receptors. We expect, therefore, that our TR-FRET assay can be applied to boost drug development programs for COVID-19, to characterize neutralizing antibodies or optimize vaccine efficacy, as well as for mechanistic studies of viral spike proteins binding to cells.
Significance
COVID-19 is triggered by infection with the SARS-CoV-2 virus. A critical step in viral infection is the entry of SARS-CoV-2 into host cells, initiated by the interaction between the receptor binding domain (RBD) of the SARS-CoV-2 spike protein, present on the surface of the viral particle, and its receptor present at the cell surface of human cells, the angiotensin I converting enzyme 2 (ACE2). In vitro assays exist to measure the interaction between the purified ACE2 extracellular domain and RBD in a cell-free setting and simple environment. Means to probe the ACE2/RBD interaction in a more complex cellular context were lacking. We have developed here a cellular binding assay measuring the time-resolved FRET signal reflecting the proximity between fluorescent RBD (RBD-d2) and fluorescent SNAP-ACE2 anchored in the plasma membrane. The influence of cellular components, such as proximal membrane proteins (TMPRSS2, HSPG, CD4) on the ACE2/RBD interaction were then investigated and revealed their impact on binding kinetics and conformational changes within ACE2/RBD complex. The cell binding assay not only allows the quantitative detection of the binding of RBD-d2 to SNAP-ACE2 with high sensitivity providing measurements of binding affinity and kinetics at the cell surface, but also allows the detection of inhibitory molecules (small chemical compounds, peptides, nanobodies). Being compatible with high-throughput screening, the SNAP-ACE2/RBD-d2 binding assay is well suited to identify novel inhibitors interfering with the formation of ACE2/RBD complex in living cells. It can also be applied to characterize therapeutic neutralizing antibodies or vaccine efficacy in a cellular environment.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit polyclonal anti-FLAG | Sigma-Aldrich/Millipore | Cat# F7425 |
| rabbit polyclonal anti-TMPRSS2 | Sigma-Aldrich/Millipore | Cat# HPA035787; RRID: AB_2674782 |
| d2-labelled anti-FLAG | Cisbio Bioassays | Cat# 61FG2DLF |
| mouse monoclonal anti-AKT | Cell signaling | Cat# 2920S; RRID: AB_1147620 |
| 680 goat anti-rabbit IgG | LICOR | Cat# 925-68021 RRID: AB_2713919 |
| 800 goat anti-mouse IgG | LICOR | Cat# 926-32210; RRID: AB_621842 |
| Chemicals, peptides, and recombinant proteins | ||
| SARS-CoV-2 spike RBD protein | Sino Biological | Cat# 40592-V08H |
| SARS-CoV-2 spike S1-S2 protein | Sino Biological | Cat# 40589-V08B1 |
| d2-labelled SARS-CoV-2 spike RBD protein | this paper | NA |
| d2-labelled SARS-CoV-2 spike S1-S2 protein | this paper | NA |
| LCB1.v3 mini-protein | Cao et al., 2020 | NA |
| Terbium cryptate Lumi4-Tb | Cisbio Bioassays | Cat# SSNPTBX |
| TagLite labeling medium | Cisbio Bioassays | Cat# LABMED |
| Corilagin | Sigma Aldrich | Cat# G0424 |
| Enalapril Maleate | Sigma Aldrich | Cat# PHR1289 |
| Cangrelor | Sigma Aldrich | Cat# SML2004 |
| Elaidic Acid | Sigma Aldrich | Cat# E4637 |
| Fenbendazole | Sigma Aldrich | Cat# F5396 |
| jetPEI transfection reagent | Polyplus-transfection | Cat# 101-10N |
| Cell Dissociation Solution Non-Enzymatic | Sigma-Aldrich | Cat# C5789 |
| Heparin | Sigma-Aldrich | Cat# H3149 |
| Experimental models: Cell lines | ||
| Human: Human Embryonic Kidney (HEK) 293 cells | Sigma-Aldrich | RRID: CVCL_0063 |
| Recombinant DNA | ||
| SNAP- FLAG-hACE2 plasmid | this paper | N/A |
| hACE2 expression plasmid | Chan et al., 2020 | Addgene, plasmid #141185 |
| CD4 plasmid | Achour et al., 2009 | N/A |
| SNAP-VEGFR2 plasmid | Cisbio Bioassays | N/A |
| SNAP-LepR plasmid | Cisbio Bioassays | N/A |
| TMPRSS2 plasmid | Edie et al., 2018 | Addgene, plasmid #53887 |
| Software and algorithms | ||
| Image J | Schneider et al., 2012 | RRID: SCR_003070 https://imagej.nih.gov/ij/download.html |
| GraphPad Prism 6 | GraphPad Software Inc | RRID: SCR_002798 https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| Tecan F500 | Plate reader | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ralf Jockers (ralf.jockers@inserm.fr).
Materials availability
Plasmids generated in this study are available upon request and MTA agreement.
Data and code availability
-
•
This paper does not report original datasets.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request
Experimental model and subject details
Cell lines
HEK293T (RRID: CVCL 0063) cells were obtained from Sigma-Aldrich and authenticated by the provider. Cell cultures were maintained in Dulbecco's Modified Eagle's Medium (DMEM) Glutamax (Invitrogen) supplemented with 10% fetal bovine serum and 1% streptomycin-penicillin, at 37°C (95% O2, 5% CO2). Cell lines were checked regularly for any mycoplasma contamination.
Method details
Compound sources and preparation
Compounds Corilagin, Enalapril Maleate, Cangrelor, Elaidic Acid and Fenbendazole were purchased from Sigma-Aldrich (Missouri, USA). Compounds were reconstituted in water according to provider's instructions, except for Fenbendazole and Elaidic Acid which were reconstituted in 100% DMSO. DMSO never exceeded 1% in final dilutions for our assay and data for Fenbendazole and Elaidic Acid experiments were normalised to a DMSO vehicle control. Where possible, new stock solutions were made for each repeat. Recombinant RBD and full-length Spike (S1-S2) protein were purchased from SinoBiological (Beijing, China) and reconstituted in water according to provider's instructions.
RBD-d2 and S1-S2-d2
Spike RBD and full-length Spike (S1-S2) proteins were purchased from SinoBiological (Cat#40592-V08H and 40,589-V08B1, respectively) and labeled with d2 by Cisbio Bioassays on lysines with an N-hydroxysuccinimide activated d2 dye in 100mM PO4 buffer (pH8). Molar ratio of d2/protein is calculated according to protein concentration measured at 280 nm and d2 concentration determined at 665 nm.
LCB1.v3 miniprotein
The LCB1.v3 mini-protein was designed using computational methods and produced using standard IPTG expression in BL21pLysS cells (Novagen), as previously described (Cao et al., 2020). The protein was lysed using a microfluidizer and purified via Immobilized Metal Affinity Chromatography (IMAC) using Cytiva IMAC Sepharose FF resin charged with NiS03 and eluted using imidazole. The protein was polished using Size Exclusion Chromatography (SEC) on a Cytiva Superdex 75 Increase column. Binding of the purified protein to RBD was confirmed via Bilayer Interferometry (BLI) using Sartorius Octet96.
Expression vectors and cell transfection
Plasmids encoding for SNAP-tagged proteins (human VEGFR2, human LepR) were obtained from Cisbio Bioassays (Codolet, France). The SNAP-tagged human ACE2 construct was obtained by introducing the ACE2 sequence into the SNAP vector through restriction enzyme cloning. The pCEP4-myc-ACE2 vector was a gift from Erik Procko (Addgene plasmid # 141185; http://n2t.net/addgene:141185; RRID: Addgene_141185) (Chan et al., 2020). The SNAP protein is also fused to an FLAG tag. The TMPRSS2 vector was a gift from Roger Reeves (Addgene plasmid # 53,887; http://n2t.net/addgene:53,887; RRID: Addgene_53,887) (Edie et al., 2018). The CD4 vector was a gift from Dr. Stefano Marullo (Achour et al., 2009). Cells were transfected with 1 μg of SNAP-ACE2 or other SNAP-tagged proteins (when indicated) in 6-well plate using jetPEI reagent according to the supplier's instructions (Polyplus-transfection, New York, NY, USA), and assay was performed 48hr post-transfection.
TR-FRET binding assay
SNAP-tagged proteins (ACE2, LepR, VEGFR2) were fluorescently labeled by incubating cells transfected with the corresponding expression vectors with an SNAP suicide substrate conjugated to the long-lived fluorophore Terbium cryptate (Tb; Lumi4-Tb, 100 nM; Cisbio Bioassays) in Tag-lite labeling medium (1 h, on ice) (Keppler et al., 2003). After several washes, cells were collected using enzyme-free cell dissociation buffer (Sigma-Aldrich), resuspended in Tag-lite buffer and distributed into a 384-well plate. Efficient fluorescent labeling of SNAP was verified by reading fluorescence signal at 620 nm. Experiments were then conducted according to the different modes described below. All reagents were diluted in Tag-lite buffer (final reaction volume of 14 μL) and incubations were performed at room temperature; except in the case of compounds Fenbendazole and Elaidic Acid, where incubation was performed at 37°C to avoid precipitation of compounds. TR-FRET signals were detected using a plate reader (Tecan F500; Tecan, Männedorf, Switzerland) with the following settings: excitation at 340 nm (Tb, energy donor), emission at 665 nm (d2, acceptor) and 620 nm (donor); delay of 150 μs; and integration time of 500 μs. Data is expressed as TR-FRET ratio (acceptor/donor). When indicated, TR-FRET ratio was normalized to % of basal or % of maximal binding (Bmax).
Saturation mode
Cells expressing Lumi4-Tb-labelled SNAP-tagged proteins were incubated with different concentrations of RBD-d2 and TR-FRET ratios were determined after 2 h incubation (at room temperature).
Trans-cellular mode
Cells expressing Lumi4-Tb-labelled SNAP-tagged proteins were mixed with mock-transfected cells or cells expressing non-labelled SNAP-ACE2 (1:1 ratio of cells) and the RBD-d2 tracer (5 nM) and TR-FRET signals were determined after 2 h incubation (at room temperature).
Kinetic mode
Association kinetics were determined by continuously monitoring TR-FRET signal for 1 h after addition of RBD-d2 (5nM) or S1-S2-d2 (20 nM). For dissociation kinetics, cells were incubated for 1 h with RBD-d2 (5 nM) or S1-S2-d2 (20 nM) to reach equilibrium, followed by the addition of saturating concentrations of non-labelled RBD (1 μM) or S1-S2 (200 nM). TR-FRET signals were then immediately and continuously monitored for 1 h at room temperature.
Competition mode
Lumi4-Tb-labelled ACE2 cells were pre-incubated with competitors (either single or increasing concentrations) for 1 h before addition of RBD-d2 (5 nM). Data are expressed as the acceptor/donor ratio or normalized as % when indicated (maximal TR-FRET ratio = 100%, non-specific binding = 0%). Obtained IC50 values from each experiment (performed in triplicates) were converted into Ki using the Cheng-Prussof equation using the mean of the individual Kd values derived from the saturation experiments (Cheng and Prusoff, 1973). The Z′ value (Zhang et al., 1999) of the TR-FRET assay was determined using total and non-specific (excess of non-labelled RBD) TR-FRET ratios as positive and negative controls, respectively, as follow:
where σp = standard deviation of positive control; σn = standard deviation of negative control; μp = mean of positive control; μn = mean of negative control.
Intramolecular TR-FRET assay to assess ACE2 conformational changes
Lumi4-Tb-labelled ACE2 cells, co-expressing TMPRSS2 when indicated, were incubated with d2-labelled anti-FLAG tag antibody (2 μg/mL, 1 h at room temperature; 61FG2DLF, Cisbio Bioassays), followed by addition of non-labelled RBD (5 nM) and heparin (0.3 or 3 mg/mL) when indicated. TR-FRET signal was read after 2 h incubation (room temperature). Non-specific signal was defined in the presence of excess of non-labelled anti-FLAG antibody (20 μg/mL; F7425; Sigma-Aldrich).
SDS-PAGE/Western Blot
Lysates from cells transfected with SNAP-ACE2 were resolved in SDS-PAGE gel (10%), followed by protein transfer to nitrocellulose membranes. Membranes were blocked in 5% non-fat dried milk in TBS (10 mM Tris-HCl, pH 8, 150 mM NaCl), and immunoblotted with primary antibodies against the FLAG tag (1:1,000; F7425, Sigma-Aldrich), or against TMPRSS2 (1:1,000; HPA035787; Sigma-Aldrich) diluted in 0.3% BSA in TBS (overnight, 4°C). Immunoreactivity was revealed using secondary antibodies coupled to 680 or 800 nm fluorophores (1:15,000, diluted in 0.3% non-fat dried milk in TBST; LI-COR Biosciences, Lincoln, NE, USA), and readings were performed with the Odyssey LI-COR IR fluorescent scanner (LI-COR Biosciences).
Fluorescence microscopy
HEK293T cells expressing SNAP-ACE2 were plated in coverslips, labeled with green fluorophore-fused SNAP substrate, and incubated with RBD-d2 (20 nM, 2 h, 37°C). After several washes, cells were fixed with paraformaldehyde 4% solution (15 min), incubated with DAPI (1:1,000, 5 min, Sigma-Aldrich) to stain cell nuclei, and the slides were analyzed under Zeiss Observer.Z1 microscope with an ×40 objective, a laser power of 100% and the following filter settings (exposure time, excitation filter and emission filter) for dapi (100 ms, 392/23, 433/24), labeled SNAP-receptors (800 ms, 509/25, 544/25) and RBD-d2 (500 ms, 635/18, 680/42). Images were analyzed using the ImageJ software.
Quantification and statistical analysis
Data are presented as means ± SEM of the indicated n (number of independent experiments), each performed in triplicate to ensure the reliability of single values. The number (n) of independent experiments was initially set to a minimum of 3–5, each performed in triplicate, and the final number (n) of independent experiments varied due to either technical problems when labeling the receptor and/or due to limited amounts of any of the reagents. Statistical details of experiments can be found in the figure legends and Results section.
TR-FRET signal measurements
TR-FRET data are expressed as the acceptor (fluorescence emission 665 nm)/donor (fluorescence emission 620 nm) ratio or normalized to % of basal or maximal binding (Bmax). Bmax was defined as the maximal TR-FRET ratio obtained in equilibrium, while 0% is defined by the non-specific binding, determined in the presence of an excess of non-labelled RBD (1 μM) or S1-S2 (200 nM). Value of Kd was determined by plotting TR-FRET ratio against RBD-d2 concentration (saturation curves), and data were fitted to the non-linear regression “one-site-total and non-specific binding” (GraphPad Prism software version 6, RRID:SCR_002798). Values of pIC50 (−logIC50) and Imax (maximal inhibition as %, with 100% set as the competition obtained with non-labelled RBD) were obtained following non-linear regression of data from a minimum of eight different concentrations per experiment, repeated at least three times independently using the log (inhibitor) versus response (three parameters) fitting equation (GraphPad Prism). The kon and koff values were calculated from the association and dissociation kinetic experiments by fitting the data to the association kinetic model equation or one phase exponential decay equations, respectively, using the GraphPad Prism software. Statistical analysis was performed using GraphPad Prism software version 6.
Statistical analysis
Comparisons between two groups were performed using Student's t test (paired t test was used when indicated, compared to the corresponding control group in each experimental set), while multiple groups comparisons were performed through ordinary one-way or two-way analysis of variance (ANOVA), followed by Tukey's or Dunnett's multiple comparison post hoc test in the cases where the differences between group means were significantly different (p < 0.05, one-way ANOVA) or when the main effects and interaction effects were detected as statistically significant (p < 0.05, two-way ANOVA). Values of p < 0.05 were considered statistically significant.
Acknowledgments
We thank all the members of the Jockers lab for the discussion at the initial phase of the project. This work was supported by Agence Nationale de la Recherche (ANR-RA-COVID-19 [ANR-20-COV4-0001 to R.J.], [ANR-19-CE16-0025-01 to R.J.], [ANR-16-CE18-0013 to J.D.]), Institut National de la Santé et de la Recherche Médicale (Inserm), Center National de la Recherche Scientifique (CNRS). R.J. was supported by the Fondation pour la Recherche Médicale (Equipe FRM DEQ20130326503) and La Ligue Contre le Cancer N/Ref: RS19/75-127 and E.C. by the Association France Alzheimer (grant no. 2042).
Author contributions
Conceptualization, E.C., J.D., and R.J.; biochemical investigation, E.C., M.B., and J.D.; resources, L.C., R.R., and L.C.; writing – original draft, J.D. and R.J.; writing – review & editing, E.C., J.D., and R.J.; funding acquisition, R.J.; supervision, E.C., J.D., and R.J.
Declaration of interests
The authors declare no competing interests.
Published: October 21, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.chembiol.2021.06.008.
Supplemental information
References
- Achour L., Scott M.G., Shirvani H., Thuret A., Bismuth G., Labbe-Jullie C., Marullo S. CD4-CCR5 interaction in intracellular compartments contributes to receptor expression at the cell surface. Blood. 2009;113:1938–1947. doi: 10.1182/blood-2008-02-141275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum A., Fulton B.O., Wloga E., Copin R., Pascal K.E., Russo V., Giordano S., Lanza K., Negron N., Ni M., et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science. 2020;369:1014–1018. doi: 10.1126/science.abd0831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazin H., Trinquet E., Mathis G. Time resolved amplification of cryptate emission: a versatile technology to trace biomolecular interactions. J. Biotechnol. 2002;82:233–250. doi: 10.1016/s1389-0352(01)00040-x. [DOI] [PubMed] [Google Scholar]
- Cantuti-Castelvetri L., Ojha R., Pedro L.D., Djannatian M., Franz J., Kuivanen S., van der Meer F., Kallio K., Kaya T., Anastasina M., et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science. 2020;370:856–860. doi: 10.1126/science.abd2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L., Goreshnik I., Coventry B., Case J.B., Miller L., Kozodoy L., Chen R.E., Carter L., Walls A.C., Park Y.J., et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science. 2020;370:426–431. doi: 10.1126/science.abd9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K.K., Dorosky D., Sharma P., Abbasi S.A., Dye J.M., Kranz D.M., Herbert A.S., Procko E. Engineering human ACE2 to optimize binding to the spike protein of SARS coronavirus 2. Science. 2020;369:1261–1265. doi: 10.1126/science.abc0870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y., Prusoff W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Christianson H.C., Belting M. Heparan sulfate proteoglycan as a cell-surface endocytosis receptor. Matrix Biol. 2014;35:51–55. doi: 10.1016/j.matbio.2013.10.004. [DOI] [PubMed] [Google Scholar]
- Clausen T.M., Sandoval D.R., Spliid C.B., Pihl J., Perrett H.R., Painter C.D., Narayanan A., Majowicz S.A., Kwong E.M., McVicar R.N., et al. SARS-CoV-2 infection depends on cellular heparan sulfate and ACE2. Cell. 2020;183:1043–1057 e15. doi: 10.1016/j.cell.2020.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davanzo G.G., Codo A.C., Brunetti N.S., Boldrini V.O., Knittel T.L., Monterio L.B., de Moraes D., Ferrari A.J.R., de Souza G.F., Muraro S.P., et al. SARS-CoV-2 uses CD4 to infect T helper lymphocytes. medRxiv. 2020 doi: 10.7554/eLife.84790. 2020.09.25.20200329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFrancesco L. COVID-19 antibodies on trial. Nat. Biotechnol. 2020;38:1242–1252. doi: 10.1038/s41587-020-0732-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degorce F., Card A., Soh S., Trinquet E., Knapik G.P., Xie B. HTRF: a technology tailored for drug discovery—a review of theoretical aspects and recent applications. Curr. Chem. Genomics. 2009;3:22–32. doi: 10.2174/1875397300903010022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edie S., Zaghloul N.A., Leitch C.C., Klinedinst D.K., Lebron J., Thole J.F., McCallion A.S., Katsanis N., Reeves R.H. Survey of human chromosome 21 gene expression effects on early development in Danio rerio. G3 (Bethesda) 2018;8:2215–2223. doi: 10.1534/g3.118.200144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W., Chen Y., Wang K., Hettinghouse A., Hu W., Wang J.Q., Lei Z.N., Chen Z.S., Stapleford K.A., Liu C.J. Repurposing FDA-approved drugs for SARS-CoV-2 through an ELISA-based screening for the inhibition of RBD/ACE2 interaction. Protein Cell. 2020;12:586–591. doi: 10.1007/s13238-020-00803-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GeurtsvanKessel C.H., Okba N.M.A., Igloi Z., Bogers S., Embregts C.W.E., Laksono B.M., Leijten L., Rokx C., Rijnders B., Rahamat-Langendoen J., et al. An evaluation of COVID-19 serological assays informs future diagnostics and exposure assessment. Nat. Commun. 2020;11:3436. doi: 10.1038/s41467-020-17317-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson Q.M., Wilson K.M., Shen M., Itkin Z., Eastman R.T., Shinn P., Hall M.D. Targeting ACE2-RBD interaction as a platform for COVID-19 therapeutics: development and drug-repurposing screen of an AlphaLISA proximity assay. ACS Pharmacol. Transl. Sci. 2020;3:1352–1360. doi: 10.1021/acsptsci.0c00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heurich A., Hofmann-Winkler H., Gierer S., Liepold T., Jahn O., Pohlmann S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014;88:1293–1307. doi: 10.1128/JVI.02202-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Kruger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A., et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280 e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo J., Le Bas A., Ruza R.R., Duyvesteyn H.M.E., Mikolajek H., Malinauskas T., Tan T.K., Rijal P., Dumoux M., Ward P.N., et al. Neutralizing nanobodies bind SARS-CoV-2 spike RBD and block interaction with ACE2. Nat. Struct. Mol. Biol. 2020;27:846–854. doi: 10.1038/s41594-020-0469-6. [DOI] [PubMed] [Google Scholar]
- Ibrahim I.M., Abdelmalek D.H., Elshahat M.E., Elfiky A.A. COVID-19 spike-host cell receptor GRP78 binding site prediction. J. Infect. 2020;80:554–562. doi: 10.1016/j.jinf.2020.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppler A., Gendreizig S., Gronemeyer T., Pick H., Vogel H., Johnsson K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol. 2003;21:86–89. doi: 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- Mathis G. Probing molecular interactions with homogeneous techniques based on rare earth cryptates and fluorescence energy transfer. Clin. Chem. 1995;41:1391–1397. [PubMed] [Google Scholar]
- Mohammad A., Marafie S.K., Alshawaf E., Abu-Farha M., Abubaker J., Al-Mulla F. Structural analysis of ACE2 variant N720D demonstrates a higher binding affinity to TMPRSS2. Life Sci. 2020;259:118219. doi: 10.1016/j.lfs.2020.118219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J., Ye G., Shi K., Wan Y., Luo C., Aihara H., Geng Q., Auerbach A., Li F. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;581:221–224. doi: 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulla A., Heald-Sargent T., Subramanya G., Zhao J., Perlman S., Gallagher T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J. Virol. 2011;85:873–882. doi: 10.1128/JVI.02062-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toelzer C., Gupta K., Yadav S.K.N., Borucu U., Davidson A.D., Kavanagh Williamson M., Shoemark D.K., Garzoni F., Staufer O., Milligan R., et al. Free fatty acid binding pocket in the locked structure of SARS-CoV-2 spike protein. Science. 2020;370:725–730. doi: 10.1126/science.abd3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich H., Pillat M.M. CD147 as a target for COVID-19 treatment: suggested effects of azithromycin and stem cell engagement. Stem Cell Rev. Rep. 2020;16:434–440. doi: 10.1007/s12015-020-09976-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vauthier V., Derviaux C., Douayry N., Roux T., Trinquet E., Jockers R., Dam J. Design and validation of a homogeneous time-resolved fluorescence-based leptin receptor binding assay. Anal. Biochem. 2013;436:1–9. doi: 10.1016/j.ab.2012.12.013. [DOI] [PubMed] [Google Scholar]
- Walls A.C., Park Y.J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181:281–292 e6. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K., Chen W., Zhang Z., Deng Y., Lian J.Q., Du P., Wei D., Zhang Y., Sun X.X., Gong L., et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct. Target. Ther. 2020;5:283. doi: 10.1038/s41392-020-00426-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R., Zhang Y., Li Y., Xia L., Guo Y., Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367:1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.H., Chung T.D., Oldenburg K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Zhang Q., Chen C.Z., Swaroop M., Xu M., Wang L., Lee J., Wang A.Q., Pradhan M., Hagen N., Chen L., et al. Heparan sulfate assists SARS-CoV-2 in cell entry and can be targeted by approved drugs in vitro. Cell Discov. 2020;6:80. doi: 10.1038/s41421-020-00222-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This paper does not report original datasets.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request