

Abstract
SARS‐CoV‐2 has caused a significant ongoing pandemic worldwide. A number of studies have examined the T cell mediated immune responses against SARS‐CoV‐2, identifying potential T cell epitopes derived from the SARS‐CoV‐2 proteome. Such studies will aid in identifying targets for vaccination and immune monitoring. In this study, we applied tandem mass spectrometry and proteomic techniques to a library of ∼40,000 synthetic peptides, in order to generate a large dataset of SARS‐CoV‐2 derived peptide MS/MS spectra. On this basis, we built an online knowledgebase, termed virusMS (https://virusms.erc.monash.edu/), to document, annotate and analyse these synthetic peptides and their spectral information. VirusMS incorporates a user‐friendly interface to facilitate searching, browsing and downloading the database content. Detailed annotations of the peptides, including experimental information, peptide modifications, predicted peptide‐HLA (human leukocyte antigen) binding affinities, and peptide MS/MS spectral data, are provided in virusMS.
Keywords: COVID19, database, LC–MS, SARS‐CoV‐2, synthetic peptides, tandem mass spectrometry
1. INTRODUCTION
The SARS (severe acute respiratory syndrome)‐CoV‐2 virus was first identified in Wuhan, China in December 2019 and has gone on to cause a global pandemic [1, 2]. The pneumonia and other related syndromes caused by SARS‐CoV‐2 infection was further defined as COVID‐19 (i.e., coronavirus disease 2019) by the WHO [3, 4]. To date, a number of studies using diverse biological techniques, including mass spectrometry, have explored and characterised the human proteome‐wide functional disruptions and immune responses upon SARS‐CoV‐2 infection [5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21].
T cell mediated immunity plays a crucial role in controlling and eliminating viral disease [22, 23, 24, 25]. Antigen processing and presentation are two of the most important steps of T cell mediated immunity, where peptides derived from viral antigens are generated and presented at the cell surface by MHC (major histocompatibility complex or human leukocyte antigen; i.e., HLA) class I and class II molecules. These peptide‐MHC complexes are scrutinised by the clonally distributed T cell receptors (TCRs) expressed on the surface of T cells, with recognition of foreign peptides triggering immune responses [26, 27, 28]. A number of studies have been dedicated to the discovery of the T cell epitopes derived from SARS‐CoV‐2 [29, 30, 31, 32, 33]. However, the accurate identification of SARS‐CoV‐2 peptides presented to the immune system by MHC molecules (collectively termed the immunopeptidome) remains challenging and is critical for a better understanding of human immune responses to SARS‐CoV‐2, vaccine design and clinical monitoring of COVID‐19.
Given its complexity (particularly with the host antigen‐derived peptide background), the task of mapping the viral immunopeptidome can be hampered by ambiguous spectral assignments, often requiring extensive validation of peptide spectra using synthetic peptides [26]. In order to facilitate rapid SARS‐CoV‐2 peptide validation, herein we describe the generation of an interactive and comprehensive online database of SARS‐CoV‐2 peptides, termed virusMS, which harbours in total 39,650 synthetic peptides generated by extensive and diverse proteolytic digestion of peptide precursors derived from the viral proteome. We have synthesized the SARS‐COV‐2 15mers peptides and digested by four proteases to generate a large dataset of SARS‐COV‐2‐derived peptides without synthesising each peptide. We used two non‐specific proteases (pepsin and elastase) and also trypsin and chymotrypsin for mimicking the activity of β2 (trypsin like) and β5 (chymotrypsin‐like) proteasome subunits. To the best of our knowledge, virusMS is the first database offering MS/MS information for SARS‐CoV‐2 synthetic peptides. For each peptide, virusMS provides comprehensive annotation regarding the experimental MS/MS information, predicted peptide‐HLA (human leukocyte antigen) binding affinity for HLA class I supertypes, and the full MS/MS spectral data. The implementation of a user‐friendly interface significantly boosts the user experience when searching and browsing the entries from virusMS. In addition, all data documented in virusMS is publicly available for customised and bulk download. We anticipate that virusMS will facilitate hypothesis generation for immunological studies and provide foundational data for the validation of immunopeptidomics studies related to SARS‐CoV‐2.
Significance statement
SARS‐CoV‐2 has caused an ongoing severe global pandemic since 2020. It is therefore crucial to understand the mechanisms of T cell mediated immune responses for effective therapies against COVID‐19. To aid the immunopeptidomics and other immunological studies of SARS‐CoV‐2 and COVID‐19, we have proposed a user‐friendly knowledgebase of synthetic peptides of SARS‐CoV‐2, termed virusMS, by applying tandem mass spectrometry and proteomic techniques. So far, two datasets harbouring in total 39,650 synthetic peptides have been harvested, documented, annotated and analysed in virusMS. Detailed annotations of these synthetic peptides, including basic experimental information, peptide‐HLA‐I binding prediction, third‐party database cross‐referencing, and detailed spectral data, are freely available at virusMS. VirusMS offers multiple functions to facilitate smooth user experience, including database search, browsing, download, with the help of multiple plug‐ins. Altogether, with the experimental data documented, virusMS is able to contribute to the generation of immunological hypothesis and the validation of the immunopeptidomic data associated with SARS‐CoV‐2 and COVID‐19.
2. MATERIAL AND METHODS
2.1. Sample preparation
A peptide library (1809 total peptides, comprising 15 amino acid length each, overlapping by nine amino acids) was synthesized from the entire SARS‐CoV‐2 proteome (Mimotopes, Australia). Peptides were dissolved in 100 μL of 5% DMSO (Figure 1A). Each sample was individually digested with four proteases, including trypsin, chymotrypsin, elastase and pepsin. For trypsin, chymotrypsin and elastase digestion, ∼100 μg (at 1 μg/μL) of each peptide sample was mixed with 5 μL of 1 M Tris and pH adjusted to ∼8 prior to addition of 4 μg of enzyme. Tryptic digestions were incubated for 60 min and elastase and chymotrypsin digest were incubated for 20 min, with all digestions at 37°C. After digestion, 4 μL of formic acid was added to terminate the reaction. For pepsin digestion, 1 μL of 50% formic acid in water was added to each sample (final pH = 3). Fifty units of pepsin was added to each sample, prior to incubation for 5 min at room temperature. All samples were centrifuged for 2 min at 13,000 rpm and peptide‐containing supernatant was concentrated and cleaned up using OMIX C18 Mini‐Bed tips (Agilent Technologies). Vacuum concentrated peptide samples were reconstituted to 12 μL with 0.1% formic acid in water.
FIGURE 1.
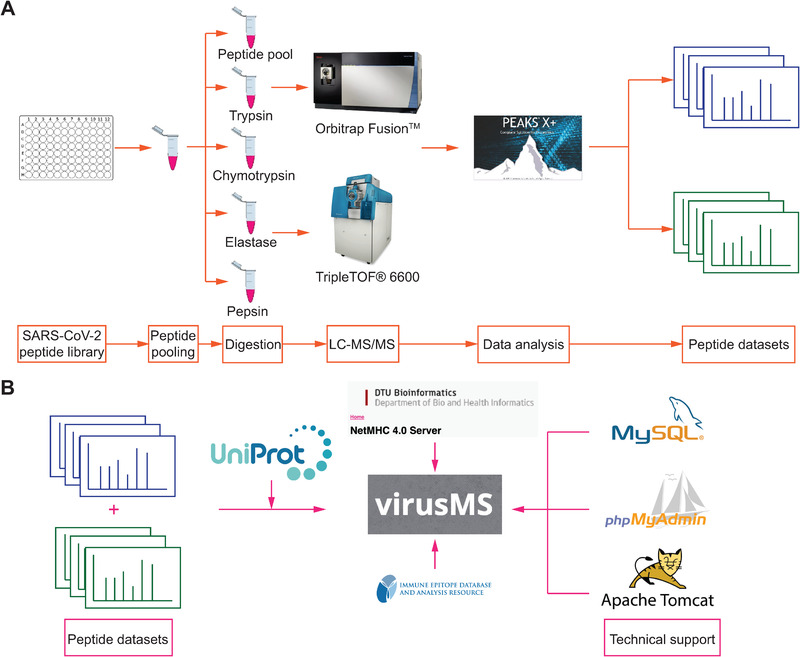
A graphical illustration demonstrating (A) the process SARS‐CoV‐2 synthetic peptide dataset generation and (B) the construction of virusMS database
2.2. LC–MS/MS analysis
Five samples were generated from each pool (one without digestion and four after digestion, the composition of the pools is available in the supplementary data, each synthesis rack being equivalent to one pool) (Figure 1A). Each sample was run in duplicate on two different instruments. We used a Dionex UltiMate 3000 RSLCnano System equipped with a Dionex UltiMate 3000 RS Autosampler. The samples were loaded via an Acclaim PepMap 100 Trap Column (100 mm × 2 cm, nanoViper, C18, 5 mm, 100 Å; Thermo Fisher Scientific) onto an Acclaim PepMap RSLC Analytical Column (75 mm × 50 cm, nanoViper, C18, 2 mm, 100 Å; Thermo Fisher Scientific). The peptides were separated by increasing concentrations of 80% acetonitrile/0.1% formic acid at a flow of 250 nL/min for 158 min and analysed with an Orbitrap Fusion Tribrid Mass Spectrometer (Thermo Fisher Scientific). Six microlitre of each sample fraction was loaded onto the trap column at a flow rate of 15 μL/min.
Orbitrap Fusion Tribrid Mass Spectrometer (Thermo Fisher Scientific) was set to data‐dependent acquisition (DDA) mode with the following settings: all MS spectra (MS1) profiles were recorded from full ion scan mode 375 to 1800 m/z in the Orbitrap at 120,000 resolution with AGC target of 400,000 and dynamic exclusion of 15 s. The top 12 precursor ions were selected using top speed mode at a cycle time of 2 s. For tandem mass spectrometry (MS/MS), a decision tree was utilised to help select peptides of charge state 1 and 2 to 6 separately. For single charged analytes, only ions falling within the range of m/z 800 to 1800 were selected. For +2 to +6 charge states, no such parameter was set. The c‐trap was loaded with a target of 200,000 ions with an accumulation time of 120 ms and isolation width of 1.2 amu. Normalized collision energy was set to 32 (high energy collisional dissociation) and fragments were analysed in the Orbitrap at 30,000 resolution.
Peptide acquisition was also carried out using a SCIEX TripleTOF 6600 equipped with an on‐line Eksigent Ekspert nanoLC 415 (SCIEX, Canada). 4 μL of each sample was directly loaded onto a trap column (ChromXP C18, 5 μm 120 Å, 300 μm × 10 mm [SCIEX]) maintained at an isocratic flow of buffer A (2% v/v acetonitrile in water supplemented with 0.1% v/v formic acid) at 10 μL/min for 5 min and then separated using an analytical column (ChromXP C18, 3 μm 120 Å, 0.3 mm × 150 mm [SCIEX]) by increasing linear concentrations of buffer B (0.1% v/v formic acid, 80% v/v acetonitrile) at a flow rate of 5 μL/min for 57 min. Up to 30 MS/MS spectra were acquired per cycle using an IDA strategy with accumulation times of 250 and 100 ms for MS1 and MS2, respectively. The MS1 scan range was set to 370–1250 m/z and MS2 set to 60–1500 m/z. To prevent multiple sequencing of the same peptide, MS1 masses were excluded for sequencing after two occurrences for 10 s.
MS/MS data were searched against the SARS‐CoV‐2 proteome by PEAKS Studio X plus (PEAKS Studio 10.5 build 20200219; Bioinformatics Solutions) using the UniProt database for SARS CoV‐2 (29 entries, dated 04/20). MS data files were imported into PEAKS Studio subjected to default data refinement. For SCIEX generated MS data, the parent mass error tolerance was set to 15 ppm and the fragment mass error tolerance to 0.1 Da. For Fusion MS data, the parameters were set to 10 ppm and the fragment mass error tolerance to 0.02 Da. For all the searches, enzyme specificity was turned off. Oxidation of Methionine, deamidation of Asn or Gln, and cysteinylation of Cys were included as variable modifications in the database peptide searches. A –10logP > 15 threshold was applied to allow selection of high‐confidence peptides. The raw data are available via ProteomeXchange [34] with identifier PXD022191. For generating the database, we further applied several criteria to select high‐quality peptides for the construction of virusMS, including (i) only peptides between 8 and 14 amino acids in length were considered, (ii) only peptides where more than 30% of theoretical b and y ions were retained.
2.3. Peptide‐HLA class I binding prediction
In this study, we conducted two major predictions for peptide‐HLA binding affinity, including (i) using peptides from experiment and (ii) proteome‐wide binding prediction. NetMHC 4.0 [35, 36] was employed for the binding affinity between peptides and HLA class I molecule (Figure 1B). We selected 12 HLA class I supertype representatives to carry out the binding affinity prediction, including HLA‐A*01:01 (A1), HLA‐A*02:01 (A2), HLA‐A*03:01 (A3), HLA‐A*24:02 (A24), HLA‐A*26:01 (A26), HLA‐B*07:02 (B7), HLA‐B*08:01 (B8), HLA‐B*27:05 (B27), HLA‐B*39:01 (B39), HLA‐B*40:01 (B44), HLA‐B*58:01 (B58), and HLA‐B*15:01 (B62). We used the peptides as direct input for NetMHC 4.0 to predict. For proteome‐wide binding prediction, we first collected all protein sequences of SARS‐CoV‐2 database from UniProt database (as described above). When conducting binding prediction using the proteome data, the peptide length for was set as 8 to 14 amino acids to reflect typical ligand lengths of HLA class I molecules. We used all default settings for the parameter configuration. A strong binder is defined as one with prediction ranking equal to or lower than 0.5%; while a weak binder has the predicted ranking between 0.5% and 2%. Any peptides with predicted ranking higher than 2% are regarded as non‐binders.
2.4. Cross‐referencing third‐party databases
We cross‐referenced two additional databases to help users further interpret the data documented in virusMS, namely IEDB (Immune Epitope Database and Analysis Resource) [37] (data compared on 8th October 2020) and UniProt [38] (downloaded on 23rd September 2020) (Figure 1B). We mapped the peptides documented in virusMS to IEDB using a wide range of coronavirus‐related filters, including Severe acute respiratory syndrome‐related coronavirus (ID: 694009), SARS‐CoV2 (ID: 2697049), SARS coronavirus Tor2 (ID: 227984), SARS coronavirus P2 (ID: 627442), SARS coronavirus TJF (ID: 284672), Bat SARS CoV Rp3/2004 (ID: 349344), SARS coronavirus BJ01 (ID: 228407), SARS coronavirus Urbani (ID: 228330), Coronavirus (ID: 11118), Betacoronavirus (ID: 694002), Alphacoronavirus (ID: 693996), Human coronavirus 229E (ID: 11137), Gammacoronavirus (ID: 694013), Middle East respiratory syndrome‐related coronavirus (ID: 1335626), Avian coronavirus (ID: 694014), Betacoronavirus 1 (ID: 694003), Coronavirus HKU15 (ID: 1965089), Alphacoronavirus 1 (ID: 693997), Bovine coronavirus (ID: 11128), Feline coronavirus (ID: 12663), Murine coronavirus (ID: 694005), and Turkey coronavirus (ID: 11152). For any peptide that can be mapped to IEDB, the related information of the peptide, including IEDB accession, the protein name and species are provided. The UniProt database was mainly utilised to provide coronavirus‐related protein sequences for peptide mapping and proteome‐wide peptide‐HLA binding prediction.
2.5. Presenting data in virusMS
Three open‐source JavaScript plugins were applied in virusMS to provide interactive data presentation, including the NeXtProt sequence viewer (part of the NeXtProt project [39]), Lorikeet (https://github.com/UWPR/Lorikeet), and Vue.js (https://vuejs.org/). The NeXtProt sequence viewer is used to present the peptide and its parent protein sequences. Lorikeet is applied to demonstrate interactive presentation of mass spectrometry data for peptides in virusMS; while Vue.js is employed to illustrate the predicted peptide‐HLA class I binding affinity.
2.6. Database construction
VirusMS resides on the Nectar (the National eResearch Collaboration Tools and Resources project) cloud server, which is managed by Monash University eResearch Centre and equipped with 12 virtual CPU cores and 500 GB disk space. We used JSP (Java Server Page), Apache Tomcat (version 9) to build and manage the backstage server of virusMS. The database was constructed using MySQL workbench (version 8.0.19) and managed by phpMyAdmin (version 5.0.4) (Figure 1B).
3. RESULTS AND DISCUSSION
3.1. Brief statistics of virusMS
To generate a comprehensive dataset of as many possible SARS‐CoV‐2 peptides, we first synthesised an overlapping peptide library (15mers, overlapping by nine amino acids) of the SARS‐CoV‐2 viral proteome. Next, we subjected peptide pools to proteolytic digestion using four different proteases (trypsin, chymotrypsin, elastase and pepsin) to generate a heterogeneous mix of peptides relevant to potential HLA‐I ligands. Pooled peptides were analysed by two common LC–MS/MS instruments with different mass analysers (orbitrap vs. TOF‐MS) and analysed by PEAKS X plus software (refer to Section 2 for more details). After filtering the original datasets, a total of 39,650 synthetic peptides were retained and used to construct virusMS. 5264 peptide entries have at least one modification identified. From our experiments, we identified two common types of peptide modifications, including deamidation (NQ) and oxidation (M). Based on the peptide‐HLA binding prediction, 10,108 peptides (25.5%) were predicted to bind at least one of the HLA class I supertypes and 672 synthetic peptides were predicted to bind to at least three HLA class I supertypes. Note that the statistics is subject to change upon the database updates. Detailed breakdowns of peptide‐HLA binding prediction can be viewed at and downloaded from virusMS.
3.2. The utility of virusMS
VirusMS provides several major functionalities as demonstrated in Figure 2, including database search (Figure 2A), peptide‐HLA binding prediction results (Figure 2B), browse (Figure 2C), interactive presentations of peptide information (Figure 2D), and bulk and customisable download (Figure 2E). All documented peptide datasets are summarised and categorised based on the types of virus on the browse webpage (Figure 3). Users can click on the experiment ID of interest (Figure 3A) directly to extract all the peptides harvested from the experiment (Figure 3B).
FIGURE 2.

A schematic illustration of the functionalities in virusMS, including (A) database keyword search, (B) peptide‐HLA binding prediction summary, (C) database browsing, (D) detailed presentation of peptide information, and (E) bulk and customisable download from virusMS
FIGURE 3.
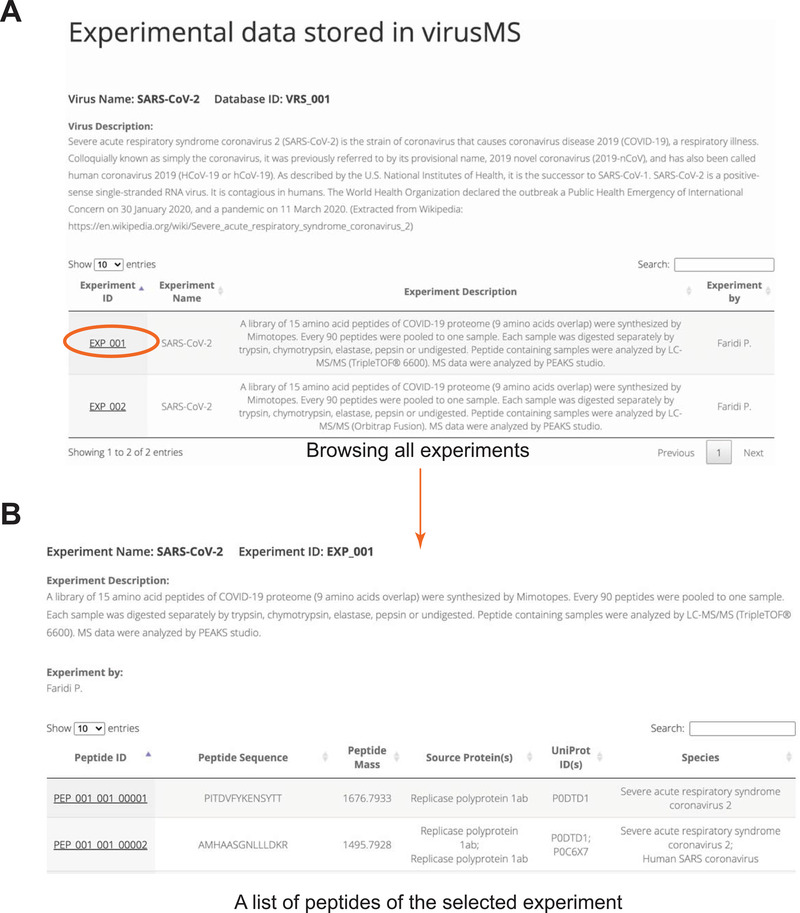
Extracting all peptides of an experiment documented in virusMS. (A) Browsing all experiments documented in virusMS. (B) Extracting all peptides by clicking the experiment ID
It is fairly straightforward and easy to search virusMS. Four types of keywords have been provided to facilitate efficient searching against virusMS, including peptide sequence, peptide source protein, peptide modification type, and peptide‐HLA binding (Figure 4). VirusMS uses a “fuzzy search” strategy when processing the user‐provided peptide sequences (Figure 4A). As such, users do not need to provide a full peptide sequence—only part of the sequence is sufficient for virusMS to identify all possible hits in the database that contain the partial sequence information provided by the users. The source proteins of all peptides have been summarised and provided for users. All the peptides derived from the same source protein can be easily extracted using the source protein search (Figure 4B). Users can select deamidation (NQ) or oxidation (M) as modifications to quickly locate all modified peptides (Figure 4C). More importantly, virusMS provides convenient searches for users to extract binders to HLA class I supertype representatives, based on our peptide‐HLA binding prediction results (Figure 4D). Two binding types (i.e., strong binder and weak binder) and all 12 HLA class I supertypes (refer to Section 2 for more details) are listed for users to perform combinatory searches. Users can perform binding searches within a total of 24 combinations to directly extract the peptides of interest.
FIGURE 4.
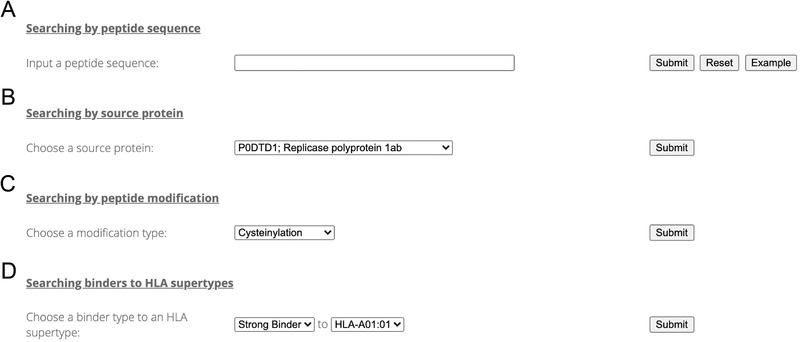
Searching virusMS using a variety of keywords, including (A) peptide sequence, (B) peptide source protein, (C) peptide modification, and (D) peptide‐HLA binding prediction
We have designed an informative webpage (Figure 5) to present the experimental data (including mass spectrometry data, peptide modification), prediction information (such peptide‐HLA binding affinity), and other identity information by cross‐referencing third‐party databases including IEDB and UniProt. For each peptide, we extracted a variety of experimental information, including basic peptide information (e.g., mass, ppm, m/z, normalised retention time, and ‐10logP) (Figure 5A), peptide‐HLA‐I binding prediction (Figure 5B), and the MS data displayed using the Lorikeet JavaScript plugin (https://github.com/jmchilton/lorikeet), which allows users to investigate the peptide spectral data interactively, including ions, mass type, peak alignment and labels (Figure 5C). In addition, users have an option to compare the spectral data of the same peptide with different modifications by clicking the “Compare the spectral data with other identical peptides” link. All MS data will be displayed using the Lorikeet plug‐in on a newly opened webpage. Meanwhile, for peptides with modification(s), the modified residue(s) are highlighted by the Lorikeet plugin. We further mapped the peptide sequence to the IEDB platform and extracted the possible IEDB accession for this peptide. Note that the accession ID was obtained by sequence match to a range of coronavirus species (see Section 2 for more details); therefore, one should bear in mind that the matching just aims to provide relevant information regarding the peptides with identical sequences. In addition to the experimental information, virusMS provides predicted peptide‐HLA binding affinities (Figure 5B). The detailed predicted results including binding affinity (nM) and rank of binding level (%) are illustrated using two interactive radar charts powered by Vue.js. Detailed definitions of strong and weak binders are provided on the webpage. Based on the definitions, all HLA supertypes that the peptide binds with are listed in the table below the radar charts.
FIGURE 5.
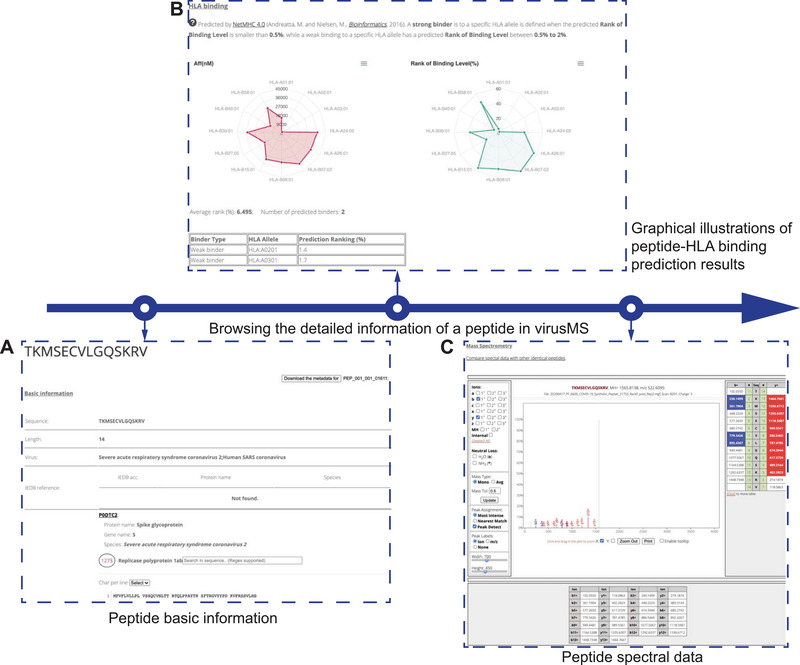
A graphical illustration for the detailed information of a peptide presented in virusMS, including (A) peptide basic information (experimental information and cross‐references), (B) prediction affinities of the peptide binding to 12 HLA class I supertype representatives, and (C) the spectral data of the peptide using the Lorikeet plug‐in
VirusMS allows both bulk and customisable download of the meta‐data in the database. Several ways have been offered to download the database content (Figure 6). First, users can download either selected or all the search results using keywords in virusMS, by clicking the “Download Selected” or the “Download All” button, respectively (Figure 6A). Alternatively, for the peptide of interest, users can click the “Download the metadata” button on the webpage for the detailed peptide information (Figure 6B). The downloaded file is in TSV (Tab Separated Values) format and contains all the information for the selected peptide(s), including the virus, experimental description, peptide basic information, modification, spectral data, and peptide‐HLA binding predictions (Figure 6C). In addition, we have exported the entire database to a SQL file, which can be easily imported to the MySQL database workbench or the phpMyAdmin management system (Figure 6D). The peptide spectral data is also available for MS users to generate libraries. To keep up with the research progress, we endeavour to incorporate state‐of‐the‐art literature to virusMS in a timely manner. Individual submission is also welcome, but users need to contact us for next steps.
FIGURE 6.
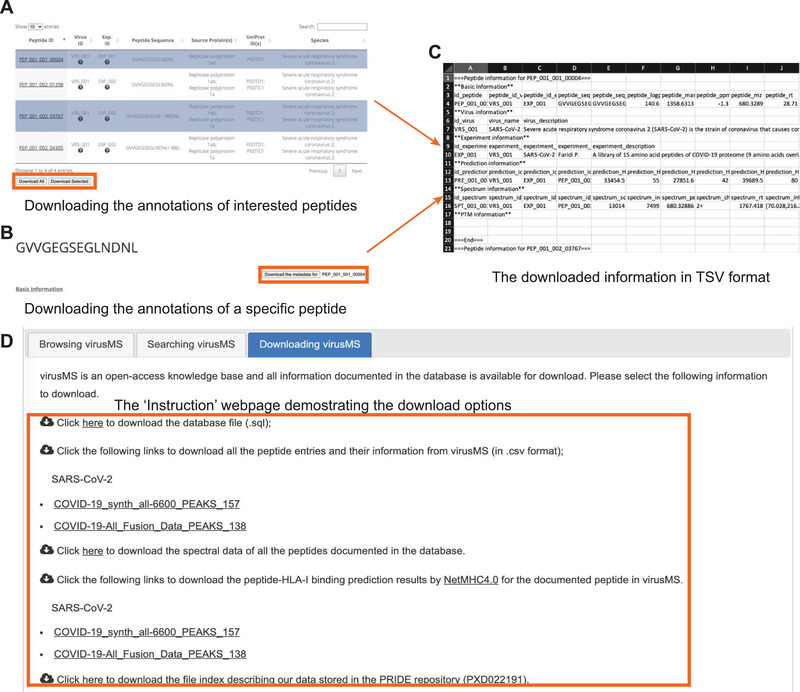
Bulk and customisable download from virusMS. Users can either (A) download the search results by selecting peptides of interests, or (B) download the information of a specific peptide on the detailed information page. The downloaded file is in TSV format and can be opened by any text editor or Microsoft Excel (C). The “Instruction” webpage provides fouroptions for downloading the entire database (D), including the database SQL file, data tables, peptide spectral data and peptide‐HLA‐I binding predictions
4. CONCLUSION
In this study, two large‐scale synthetic peptide datasets from the SARS‐CoV‐2 proteome have been generated using a SCIEX TripleTOF 6600 or an Orbitrap Fusion LC–MS. With the two datasets, we have constructed a user‐friendly database, termed virusMS, for resourcing the synthetic peptides Using virusMS, users are able to search, extract and download their peptides of interest together with the related MS/MS data. In addition, virusMS documents predicted peptide‐HLA binding affinity for all peptides stored in the database, thereby shedding light on the potential immunogenicity of these peptides. VirusMS is fully open‐access and all the information in the database is freely available. We hope virusMS will serve as an essential data resource for immunological and vaccine studies of SARS‐CoV‐2 and COVID‐19. As more data becomes available, we will continue to expand the content of the virusMS database, including further coverage of the viral proteome and additional MS platforms such as the Bruker TIMS TOF Pro.
[dataset] VirusMS is accessible at https://virusms.erc.monash.edu/. The mass spectrometry proteomics data of SARS‐CoV‐2 is available at the ProteomeXchange Consortium with the dataset identifier PXD022191.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Supporting information.
ACKNOWLEDGMENTS
The authors acknowledge the donation of a peptide library spanning the entire sequence of the SARS‐CoV‐2 proteome by Mimotopes Pty Ltd (Mulgrave, Victoria, Australia). The authors acknowledge instrumentation, training and technical support by the Monash Biomedical Proteomics Facility. Computational resources were supported by the R@CMon/Monash Node of the Nectar Research Cloud, an initiative of the Australian Government's Super Science Scheme and the Education Investment Fund. This work was financially supported by a project grant from the National Health and Medical Research Council of Australia (NHMRC) (1165490). AWP is supported by a NHMRC Principal Research Fellowship (1137739). C.L. is supported by an NHMRC CJ Martin Early Career Research Fellowship (1143366).
Li C., Revote J., Ramarathinam Sri. H., Chung S. Z., Croft N. P., Scull K. E., Huang Z., Ayala R., Braun A., Mifsud N. A., Illing P. T., Faridi P., Purcell A. W., (2021). Resourcing, annotating, and analysing synthetic peptides of SARS‐CoV‐2 for immunopeptidomics and other immunological studies. Proteomics, 21, e2100036. 10.1002/pmic.202100036
Contributor Information
Pouya Faridi, Email: Pouya.Faridi@monash.edu.
Anthony W. Purcell, Email: Anthony.Purcell@monash.edu.
DATA AVAILABILITY STATEMENT
VirusMS is freely available for academic purposes at https://virusms.erc.monash.edu. The peptide datasets (including from TripleTOF 6600 and Orbitrap Fusion) are available for download at http://virusms.erc.monash.edu/instruction.jsp. The peptide‐HLA class I binding prediction results using proteome data and peptide data in virusMS are available at http://virusms.erc.monash.edu/HLA.jsp and http://virusms.erc.monash.edu/instruction.jsp. The mass spectrometry proteomics data of SARS‐CoV‐2 in this study have been deposited to the ProteomeXchange Consortium [34] via the PRIDE [40] partner repository with the dataset identifier PXD022191.
REFERENCES
- 1. Khan, S. , Siddique, R. , Shereen, M. A. , Ali, A. , Liu, J. , Bai, Q. , Bashir, N. , & Xue, M. , (2020). Emergence of a novel coronavirus, severe acute respiratory syndrome coronavirus 2: Biology and therapeutic options. Journal of Clinical Microbiology, 58(5). 10.1128/JCM.00187-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mo, P. , Xing, Y. , Xiao, Y. , Deng, L. , Zhao, Q. , Wang, H. , Xiong, Y. , Cheng, Z. , Gao, S. , Liang, K. , Luo, M. , Chen, T. , Song, S. , Ma, Z. , Chen, X. , Zheng, R. , Cao, Q. , Wang, F. , & Zhang, Y. , (2020). Clinical characteristics of refractory COVID‐19 pneumonia in Wuhan, China. Clinical Infectious Diseases, 1.23. 10.1093/cid/ciaa270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Phelan, A. L. , Katz, R. , & Gostin, L. O. , (2020). The Novel Coronavirus Originating in Wuhan, China. JAMA, the Journal of the American Medical Association, 323(8), 709–710. 10.1001/jama.2020.1097 [DOI] [PubMed] [Google Scholar]
- 4. Zhou, F. , Yu, T. , Du, R. , Fan, G. , Liu, Y. , Liu, Z. , Xiang, J. , Wang, Y. , Song, B. , Gu, X. , Guan, L. , Wei, Y. , Li, H. , Wu, X. , Xu, J. , Tu, S. , Zhang, Y. , Chen, H. , & Cao, B. , (2020). Clinical course and risk factors for mortality of adult inpatients with COVID‐19 in Wuhan, China: A retrospective cohort study. Lancet, 395(10229), 1054–1062. 10.1016/S0140-6736(20)30566-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bojkova, D. , Klann, K. , Koch, B. , Widera, M. , Krause, D. , Ciesek, S. , Cinatl, J. , & Münch, C. , (2020). Proteomics of SARS‐CoV‐2‐infected host cells reveals therapy targets. Nature, 583(7816), 469–472. 10.1038/s41586-020-2332-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bouhaddou, M. , Memon, D. , Meyer, B. , White, K. M. , Rezelj, V. V. , Correa Marrero, M. , Polacco, B. J. , Melnyk, J. E. , Ulferts, S. , Kaake, R. M. , Batra, J. , Richards, A. L. , Stevenson, E. , Gordon, D. E. , Rojc, A. , Obernier, K. , Fabius, J. M. , Soucheray, M. , Miorin, L. , …, Krogan, N. J. , (2020). The global phosphorylation landscape of SARS‐CoV‐2 infection. Cell, 182(3), 685–712. e619. 10.1016/j.cell.2020.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gordon, D. E. , Jang, G. M. , Bouhaddou, M. , Xu, J. , Obernier, K. , White, K. M. , O'meara, M. J. , Rezelj, V. V. , Guo, J. Z. , Swaney, D. L. , Tummino, T. A. , Hüttenhain, R. , Kaake, R. M. , Richards, A. L. , Tutuncuoglu, B. , Foussard, H. , Batra, J. , Haas, K. , Modak, M. , …, Krogan, N. J. , (2020). A SARS‐CoV‐2 protein interaction map reveals targets for drug repurposing. Nature, 583(7816), 459–468. 10.1038/s41586-020-2286-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shen, B. , Yi, X. , Sun, Y. , Bi, X. , Du, J. , Zhang, C. , Quan, S. , Zhang, F. , Sun, R. , Qian, L. , Ge, W. , Liu, W. , Liang, S. , Chen, H. , Zhang, Y. , Li, J. , Xu, J. , He, Z. , Chen, B. , …, Guo, T. , (2020). Proteomic and metabolomic characterization of COVID‐19 patient sera patient sera. Cell, 182(1), 59–72.e15. 10.1016/j.cell.2020.05.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Whetton, A. D. , Preston, G. W. , Abubeker, S. , & Geifman, N. , (2020). Proteomics and informatics for understanding phases and identifying biomarkers in COVID‐19 disease. Journal of Proteome Research, 19(11), 4219–4232, 10.1021/acs.jproteome.0c00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang, Y. , Zhao, W. , Mao, Y. , Chen, Y. , Wang, S. , Zhong, Y. , Su, T. , Gong, M. , Du, D. , Lu, X. , Cheng, J. , & Yang, H. , (2020). Site‐specific N‐glycosylation characterization of recombinant SARS‐CoV‐2 spike proteins. Molecular and Cellular Proteomics. 20, 100058, 10.1074/mcp.RA120.002295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Agarwal, S. , & June, C. H. , (2020). Harnessing CAR T‐cell insights to develop treatments for hyperinflammatory responses in patients with COVID‐19. Cancer discovery, 10(6), 775–778. 10.1158/2159-8290.CD-20-0473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen, Z. , & John Wherry, E. , (2020). T cell responses in patients with COVID‐19. Nature Reviews Immunology, 20(9), 529–536. 10.1038/s41577-020-0402-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grifoni, A. , Weiskopf, D. , Ramirez, S. I. , Mateus, J. , Dan, J. M. , Moderbacher, C. R. , Rawlings, S. A. , Sutherland, A. , Premkumar, L. , Jadi, R. S. , Marrama, D. , De Silva, A. M. , Frazier, A. , Carlin, A. F. , Greenbaum, J. A. , Peters, B. , Krammer, F. , Smith, D. M. , Crotty, S. , & Sette, A. , (2020). Targets of T cell responses to SARS‐CoV‐2 coronavirus in humans with COVID‐19 disease and unexposed individuals. Cell, 181(7), 1489–1501.e1415. 10.1016/j.cell.2020.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Habel, J. R. , Nguyen, T. H. O. , Van De Sandt, C. E. , Juno, J. A. , Chaurasia, P. , Wragg, K. , Koutsakos, M. , Hensen, L. , Jia, X. , Chua, B. , Zhang, W. , Tan H.‐X., Flanagan, K. L. , Doolan, D. L. , Torresi, J. , Chen, W. , Wakim, L. M. , Cheng, A. C. , Doherty, P. C. , …, Kedzierska, K. , (2020). Suboptimal SARS‐CoV‐2‐specific CD8 + T cell response associated with the prominent HLA‐A*02:01 phenotype. PNAS, 117(39), 24384–24391. 10.1073/pnas.2015486117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Juno, J. A. , Tan, H.‐X. , Lee, W. S. , Reynaldi, A. , Kelly, H. G. , Wragg, K. , Esterbauer, R. , Kent, H. E. , Batten, C. J. , Mordant, F. L. , Gherardin, N. A. , Pymm, P. , Dietrich, M. H. , Scott, N. E. , Tham, W.‐H. , Godfrey, D. I. , Subbarao, K. , Davenport, M. P. , Kent, S. J. , & Wheatley, A. K. , (2020). Humoral and circulating follicular helper T cell responses in recovered patients with COVID‐19. Nature Medicine, 26(9), 1428–1434. 10.1038/s41591-020-0995-0 [DOI] [PubMed] [Google Scholar]
- 16. Kroemer, M. , Spehner, L. , Vettoretti, L. , Bouard, A. , Eberst, G. , Pili Floury, S. , Capellier, G. , Lepiller, Q. , Orillard, E. , Mansi, L. , Clairet, A.‐L. , Westeel, V. , Limat, S. , Dubois, M. , Malinowski, L. , Bohard, L. , Borg, C. , Chirouze, C. , & Bouiller, K. , (2020). COVID‐19 patients display distinct SARS‐CoV‐2 specific T‐cell responses according to disease severity. Journal of Infection, 4816, 282–327. 10.1016/j.jinf.2020.08.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Le Bert, N. , Tan, A. T. , Kunasegaran, K. , Tham, C. Y. L. , Hafezi, M. , Chia, A. , Chng, M. H. Y. , Lin, M. , Tan, N. , Linster, M. , Chia, W. N. , Chen, M. I.‐C. , Wang, L.‐F. , Ooi, E. E. , Kalimuddin, S. , Tambyah, P. A. , Low, J. G.‐H. , Tan, Y.‐J. , & Bertoletti, A. (2020). SARS‐CoV‐2‐specific T cell immunity in cases of COVID‐19 and SARS, and uninfected controls. Nature, 584(7821), 457–462. 10.1038/s41586-020-2550-z [DOI] [PubMed] [Google Scholar]
- 18. Mateus, J. , Grifoni, A. , Tarke, A. , Sidney, J. , Ramirez, S. I. , Dan, J. M. , Burger, Z. C. , Rawlings, S. A. , Smith, D. M. , Phillips, E. , Mallal, S. , Lammers, M. , Rubiro, P. , Quiambao, L. , Sutherland, A. , Yu, E. D. , Da Silva Antunes, R. , Greenbaum, J. , Frazier, A. , …, Weiskopf, D. , (2020). Selective and cross‐reactive SARS‐CoV‐2 T cell epitopes in unexposed humans. Science, 370(6512), 89–94. 10.1126/science.abd3871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mazzoni, A. , Maggi, L. , Capone, M. , Spinicci, M. , Salvati, L. , Colao, M. G. , Vanni, A. , Kiros, S. T. , Mencarini, J. , Zammarchi, L. , Mantengoli, E. , Menicacci, L. , Caldini, E. , Romagnani, S. , Liotta, F. , Morettini, A. , Rossolini, G. M. , Bartoloni, A. , Cosmi, L. , & Annunziato, F. , (2020). Cell‐mediated and humoral adaptive immune responses to SARS‐CoV‐2 are lower in asymptomatic than symptomatic COVID‐19 patients. European Journal of Immunology, 50(12), 2013–2024. 10.1002/eji.202048915. [DOI] [PubMed] [Google Scholar]
- 20. Oja, A. E. , Saris, A. , Ghandour, C. A. , Kragten, N. A. M. , Hogema, B. M. , Nossent, E. J. , Heunks, L. M. A. , Cuvalay, S. , Slot, E. , Linty, F. , Swaneveld, F. H. , Vrielink, H. , Vidarsson, G. , Rispens, T. , Schoot, E. , Lier, R. A. W. , Ten Brinke, A. , & Hombrink, P. , P. (2020). Divergent SARS‐CoV‐2‐specific T and B cell responses in severe but not mild COVID‐19 patients. European Journal of Immunology.1998–2012, 10.1002/eji.202048908. [DOI] [PubMed] [Google Scholar]
- 21. Zhang, J.‐Y. , Wang, X.‐M. , Xing, X. , Xu, Z. , Zhang, C. , Song, J.‐W. , Fan, X. , Xia, P. , Fu, J.‐L. , Wang, S.‐Y. , Xu, R.‐N. , Dai, X.‐P. , Shi, L. , Huang, L. , Jiang, T.‐J. , Shi, M. , Zhang, Y. , Zumla, A. , Maeurer, M. , …, Wang, F.‐S. , (2020). Single‐cell landscape of immunological responses in patients with COVID‐19. Nature Immunology, 21(9), 1107–1118. 10.1038/s41590-020-0762-x [DOI] [PubMed] [Google Scholar]
- 22. Bain, C. , Parroche, P. , Lavergne, J. P. , Duverger, B. , Vieux, C. , Dubois, V. , Komurian‐Pradel, F. , Trépo, C. , Gebuhrer, L. , Paranhos‐Baccala, G. , Penin, F. , & Inchauspé, G. , (2004). Memory T‐cell‐mediated immune responses specific to an alternative core protein in hepatitis C virus infection. Journal of Virology, 78(19), 10460–10469. 10.1128/JVI.78.19.10460-10469.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Channappanavar, R. , Zhao, J. , & Perlman, S. , (2014). T cell‐mediated immune response to respiratory coronaviruses. Immunologic Research, 59(1‐3), 118–128. 10.1007/s12026-014-8534-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cox, R. J. , & Brokstad, K. A. , (2020). Not just antibodies: B cells and T cells mediate immunity to COVID‐19. Nature Reviews Immunology, 20(10), 581–582. 10.1038/s41577-020-00436-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. La Gruta, N. L. , & Turner, S. J. , (2014). T cell mediated immunity to influenza: Mechanisms of viral control. Trends in Immunology, 35(8), 396–402. 10.1016/j.it.2014.06.004 [DOI] [PubMed] [Google Scholar]
- 26. Purcell, A. W. , Ramarathinam, S. H. , & Ternette, N. , (2019). Mass spectrometry‐based identification of MHC‐bound peptides for immunopeptidomics. Nature Protocols, 14(6), 1687–1707. 10.1038/s41596-019-0133-y [DOI] [PubMed] [Google Scholar]
- 27. Vyas, J. M. , Van Der Veen, A. G. , & Ploegh, H. L. , (2008). The known unknowns of antigen processing and presentation. Nature Reviews Immunology, 8(8), 607–618. 10.1038/nri2368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Watts, C. , & Powis, S. , (1999). Pathways of antigen processing and presentation. Reviews in Immunogenetics, 1(1), 60–74. [PubMed] [Google Scholar]
- 29. Gouveia, D. , Grenga, L. , Gaillard, J.‐C. , Gallais, F. , Bellanger, L. , Pible, O. , & Armengaud, J. , (2020). Shortlisting SARS‐CoV‐2 peptides for targeted studies from experimental data‐dependent acquisition tandem mass spectrometry data. Proteomics, 20(14), 2000107. 10.1002/pmic.202000107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nelde, A. , Bilich, T. , Heitmann, J. S. , Maringer, Y. , Salih, H. R. , Roerden, M. , Lübke, M. , Bauer, J. , Rieth, J. , Wacker, M. , Peter, A. , Hörber, S. , Traenkle, B. , Kaiser, P. D. , Rothbauer, U. , Becker, M. , Junker, D. , Krause, G. , Strengert, M. , …, Walz, J. S. , (2020). SARS‐CoV‐2‐derived peptides define heterologous and COVID‐19‐induced T cell recognition. Nature Immunology 74–85, 10.1038/s41590-020-00808-x. [DOI] [PubMed] [Google Scholar]
- 31. Poh, C. M. , Carissimo, G. , Wang, B. , Amrun, S. N. , Lee, C. Y.‐P. , Chee, R. S.‐L. , Fong, S.‐W. , Yeo, N. K.‐W. , Lee, W.‐H. , Torres‐Ruesta, A. , Leo, Y.‐S. , Chen, M. I.‐C. , Tan, S.‐Y. , Chai, L. Y. A. , Kalimuddin, S. , Kheng, S. S. G. , Thien, S.‐Y. , Young, B. E. , Lye, D. C. , …, Ng, L. F. P. (2020). Two linear epitopes on the SARS‐CoV‐2 spike protein that elicit neutralising antibodies in COVID‐19 patients. Nature communications, 11(1), 2806. 10.1038/s41467-020-16638-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vanpatten, S. , He, M. , Altiti, A. F. , Cheng, K. , Ghanem, M. H. , & Al‐Abed, Y. , (2020). Evidence supporting the use of peptides and peptidomimetics as potential SARS‐CoV‐2 (COVID‐19) therapeutics. Future medicinal chemistry, 12(18), 1647–1656. 10.4155/fmc-2020-0180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Weingarten‐Gabbay, S. , Klaeger, S. , Sarkizova, S. , Pearlman, L. R. , Chen, D. Y. , Bauer, M. R. , Taylor, H. B. , Conway, H. L. , Tomkins‐Tinch, C. H. , Finkel, Y. , Nachshon, A. , Gentili, M. , Rivera, K. D. , Keskin, D. B. , Rice, C. M. , Clauser, K. R. , Hacohen, N. , Carr, S. A. , Abelin, J. G. , … Sabeti, P. C. , (2020). SARS‐CoV‐2 infected cells present HLA‐I peptides from canonical and out‐of‐frame ORFs. bioRxiv. 10.1101/2020.10.02.324145 [DOI] [Google Scholar]
- 34. Deutsch, E. W. , Bandeira, N. , Sharma, V. , Perez‐Riverol, Y. , Carver, J. J. , Kundu, D. J. , García‐Seisdedos, D. , Jarnuczak, A. F. , Hewapathirana, S. , Pullman, B. S. , Wertz, J. , Sun, Z. , Kawano, S. , Okuda, S. , Watanabe, Y. , Hermjakob, H. , Maclean, B. , Maccoss, M. J. , Zhu, Y. , … Vizcaíno, J. A. , (2020). The ProteomeXchange consortium in 2020: Enabling 'big data' approaches in proteomics. Nucleic Acids Research, 48(D1), D1145–D1152. 10.1093/nar/gkz984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Andreatta, M. , & Nielsen, M. , (2016). Gapped sequence alignment using artificial neural networks: Application to the MHC class I system. Bioinformatics, 32(4), 511–517. 10.1093/bioinformatics/btv639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nielsen, M. , Lundegaard, C. , Worning, P. , Lauemøller, S. L. , Lamberth, K. , Buus, S. , Brunak, S. , & Lund, O. , (2003). Reliable prediction of T‐cell epitopes using neural networks with novel sequence representations. Protein Science, 12(5), 1007–1017. 10.1110/ps.0239403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vita, R. , Mahajan, S. , Overton, J. A. , Dhanda, S. K. , Martini, S. , Cantrell, J. R. , Wheeler, D. K. , Sette, A. , & Peters, B. , (2019). The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Research, 47(D1), D339–D343. 10.1093/nar/gky1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. UniProt Consortium . (2019). UniProt: A worldwide hub of protein knowledge. Nucleic Acids Research, 47(D1), D506–D515. 10.1093/nar/gky1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zahn‐Zabal, M. , Michel, P.‐A. , Gateau, A. , Nikitin, F. , Schaeffer, M. , Audot, E. , Gaudet, P. , Duek, P. D. , Teixeira, D. , Rech De Laval, V. , Samarasinghe, K. , Bairoch, A. , & Lane, L. , (2020). The neXtProt knowledgebase in 2020: Data, tools and usability improvements. Nucleic Acids Research, 48(D1), D328–D334. 10.1093/nar/gkz995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Perez‐Riverol, Y. , Csordas, A. , Bai, J. , Bernal‐Llinares, M. , Hewapathirana, S. , Kundu, D. J. , Inuganti, A. , Griss, J. , Mayer, G. , Eisenacher, M. , Pérez, E. , Uszkoreit, J. , Pfeuffer, J. , Sachsenberg, T. , Yılmaz, Ş. , Tiwary, S. , Cox, J. , Audain, E. , Walzer, M. , …, Vizcaíno, J. A. , (2019). The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Research, 47(D1), D442–D450. 10.1093/nar/gky1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Data Availability Statement
VirusMS is freely available for academic purposes at https://virusms.erc.monash.edu. The peptide datasets (including from TripleTOF 6600 and Orbitrap Fusion) are available for download at http://virusms.erc.monash.edu/instruction.jsp. The peptide‐HLA class I binding prediction results using proteome data and peptide data in virusMS are available at http://virusms.erc.monash.edu/HLA.jsp and http://virusms.erc.monash.edu/instruction.jsp. The mass spectrometry proteomics data of SARS‐CoV‐2 in this study have been deposited to the ProteomeXchange Consortium [34] via the PRIDE [40] partner repository with the dataset identifier PXD022191.