

Abstract
Coronaviruses (CoVs) represent enveloped, ss RNA viruses with the ability to infect a range of vertebrates causing mainly lung, CNS, enteric, and hepatic disease. While the infection with human CoV is commonly associated with mild respiratory symptoms, the emergence of SARS‐CoV, MERS‐CoV, and SARS‐CoV‐2 highlights the potential for CoVs to cause severe respiratory and systemic disease. The devastating global health burden caused by SARS‐CoV‐2 has spawned countless studies seeking clinical correlates of disease severity and host susceptibility factors, revealing a complex network of antiviral immune circuits. The mouse hepatitis virus (MHV) is, like SARS‐CoV‐2, a beta‐CoV and is endemic in wild mice. Laboratory MHV strains have been extensively studied to reveal coronavirus virulence factors and elucidate host mechanisms of antiviral immunity. These are reviewed here with the aim to identify translational insights for SARS‐CoV‐2 learned from murine CoVs.
Keywords: Animal models, Host/pathogen interaction, Immune responses, Mouse Hepatitis Virus, SARS‐CoV‐2
Common properties of the infection etiology, host‐pathogen interactions, and immune responses shared between the mouse hepatitis virus (MHV‐A59) and SARS‐CoV‐2. A main distinguisher of MHV from other preclinical animal models of COVID‐19 is the fully adapted host replication machinery that recapitulates a multiorgan disease.
Introduction
Coronaviridae are a family of enveloped, ss RNA viruses, consisting of species‐restricted strains that can infect humans, mice, pigs, cows, chickens, cattle, cats, and bats [1]. Many of these viruses induce a respiratory and/or enteric infection, with murine strains additionally causing neurological and hepatic infections [1]. Human coronaviruses (CoVs) typically induce mild respiratory infections caused by the common cold coronaviruses (HCoV‐229E, HCoV‐OC43), however, highly virulent strains can cause severe acute respiratory syndromes and multiorgan involvement as seen by SARS‐CoV [2], MERS‐CoV [3], and SARS‐CoV‐2 [4, 5]. As of March 2021, SARS‐CoV‐2 has caused over 2.65 million deaths worldwide, a number expected to grow until vaccines will be widely distributed. While inflammatory correlates help to identify pathways dysregulated in severe cases, preclinical models are needed to resolve causative mechanisms of viral spread, antiviral immunity, and multiorgan involvement. Here, we provide an overview of viral pathogenicity and host immune defence mechanisms that has been elucidated from studies using murine CoVs and discuss the suitability of these viruses as a preclinical model for SARS‐CoV‐2.
Coronavirus genome and replication cycle
The CoV genome and replication cycle have been reviewed extensively elsewhere [1, 6] and are summarized briefly here. The ss RNA genome is stabilised by the nucleocapsid protein, and surrounded by a viral membrane envelope containing the spike, transmembrane, and envelope glycoproteins. The spike glycoprotein mediates viral attachment to host receptors and is cleaved by host proteases permitting fusion with the cell membrane. Following release of the viral genome into the host cytoplasm, two viral polypeptides encoding nonstructural genes are translated. The nonstructural proteins encoding the replicase–transcriptase complex — RNA polymerase, helicase, exoribonuclease, and methyltransferase are translated first as a polyprotein and subsequently cleaved creating the machinery for viral replication and translation of the structural genes. Encapsidated viral genomes and translated structural proteins are inserted into the ER–Golgi intermediate compartment, and mature virions are released via exocytosis. In addition to the species and tissue tropism determined by the spike protein, the requirement of the host cell's translation machinery generally restricts the efficient replication of CoVs across a species barrier.
Mouse hepatitis virus (MHV) strains and mutants reveal coronavirus virulence factors
Murine CoVs, also referred to as mouse hepatitis viruses (MHV), consist of diverse strains that cause varying degrees of respiratory, gastrointestinal, hepatic, and neuronal symptoms [1, 7, 8]. All MHV strains belong to the genus of beta‐CoVs, as do certain human CoVs (HCoV‐OC43, HCoV‐HKU1, SARS‐CoV, MERS‐CoV, and SARS‐CoV‐2) [9]. Distinct MHV strains exhibit differences in tropism and virulence, and studies of recombinant MHV variants have revealed host and viral factors that determine viral spread or circumvent immune recognition.
The spike glycoprotein confers viral tropism
Studies comparing different MHV strains or recombinant variants illuminate the importance of the spike protein for tissue tropism. The MHV spike protein binds to the host cellular receptor carcinoembryonic antigen‐related cell adhesion molecule 1 (CEACAM‐1), which is expressed on epithelial cells in the liver, intestines, respiratory tract and pancreas, on proximal tubules of the kidneys, to a low extent on glial cells in the CNS, and broadly expressed on endothelial and hematopoietic cells [10, 11]. This host receptor–viral ligand pair is conserved across MHV strains, however, strain‐specific differences in the spike protein confer differences in cellular tropism. While the MHV‐A59 strain is neurotropic, hepatotropic, and mildly pneumotropic, the MHV‐JHM and MHV‐4 (an isolate of MHV‐JHM) strains are highly neurotropic and weakly hepato‐ and pneumotropic, while MHV‐2 and ‐3 strains are strongly hepatotropic, and the MHV‐1 strain is strongly pneumotropic. A series of studies using chimeric MHV viruses identify the spike protein as the key determinant of tissue tropism. For instance, swapping the MHV‐A59 spike protein with that of MHV‐2 prevents the demyelinating consequences of oligodendrocyte infection in the CNS [12], while swapping in the spike protein of MHV‐1 confers increased pneumotropism to MHV‐A59 [8]. Similarly, replacement of the MHV‐A59 spike glycoprotein with that of MHV‐4, a neurotropic strain, attenuated hepatotropism [13]. However, introduction of the MHV‐A59 spike protein onto the MHV‐JHM background failed to introduce a hepatotropic phenotype [14], suggesting that additional viral genes may influence tropism.
Structural and nonstructural genes promote virulence
Recombinant MHV variants have also revealed the contribution of other structural and nonstructural genes in conferring virulence. Increased neurovirulence associated with MHV‐JHM correlates with elevated cytokine production (IFN‐β, IL‐1β, IL‐6, CCL3, CCL4), but not CD8 T‐cell responses compared to MHV‐A59 infection [15]. Recombinant MHV‐JHM viruses further reveal that the spike glycoprotein promotes CCL3/CCL4‐driven macrophage immunopathology [16], while the magnitude of the T‐cell response is orchestrated by other viral genes [17]. Perhaps the most relevant example in the context of SARS‐CoV‐2 infection, is the observation that MHV‐1 structural proteins account for increased pneumovirulence associated with exacerbated interstitial pneumonia and elevated levels of the type I and II IFNs and TNF, but not viral titres [8]. A similar disconnect between viral titres and inflammatory cytokines and histopathology was reported by Thiel and colleagues, who showed that a single amino acid mutation in the MHV‐A59 ADP‐ribose‐1”‐phosphatase encoded by the nonstructural protein (Nsp) 3 resulted in reduced liver pathology and IL‐6 production, despite having no effect on viral titres in the liver [18].
Mechanisms of viral evasion
In addition to modulating host cytokine responses, CoVs have evolved mechanisms to evade host antiviral strategies including inhibition of type I IFN signalling cascades or circumventing recognition by pattern recognition receptors [19, 20]. For example, deletion of Nsp1 that encodes the 5’ end of the viral replicase in the MHV‐A59 genome resulted in attenuation of viral replication and impaired type I IFN responses in professional APCs [21]. Moreover, the coronavirus ribose 2’‐O‐methyltransferase encoded by Nsp16 serves to disguise viral mRNA as eukaryotic mRNA that is methylated at the 5’ cap, thereby, avoiding recognition by the cytoplasmic PRR, melanoma differentiation‐associated gene 5 (MDA5) and downstream type I IFN responses [19]. Notably, a similar Nsp16/10‐dependent mechanism or RNA cap modification has been reported for SARS‐CoV‐2 [22]. Moreover, SARS‐CoV‐2 nonstructural proteins may inhibit splicing‐, translational‐, and protein trafficking processes involved in the host type I IFN response to viral infection [23, 24]. Although strain‐specific differences exist among CoVs, recombinant MHV variants provide a platform for resolving host‐viral interactions and virulence factors conferred by CoV structural and nonstructural genes.
Mechanistic insight into antiviral mechanisms to Coronaviruses learned from MHV
Host innate defence mechanisms contain early viral infection
Early control of MHV critically depends on IFN‐mediated host defence. In vitro studies reveal that plasmacytoid DCs (pDCs) produce the first wave of type I IFNs within 12 h of exposure [25]. As a ss RNA virus that is temporarily in a ds form during viral genome replication, infected cells can sense the coronavirus via either the toll‐like receptor 7 (TLR7) or MDA5, respectively [25, 26]. Studies using Tlr7‐deficient pDCs [25] or Mda5‐deficient macrophages [19] suggest that both sensors contribute to type I IFN‐mediated attenuation of MHV titres in vitro. Similarly, a compensatory role for TLR7 in type I IFN production is observed in Mda5‐deficient mice, although these mice exhibit reduced survival, higher viral titres and increased serum cytokines (IL‐6, TNF, IFN‐γ) compared to MDA5‐sufficient counterparts [26]. Following early type I IFN production by pDCs, signalling via the type I IFN‐α/β receptor (IFNAR) by macrophages, and to a lesser extent by CD11c‐expressing cells, is required to control viral replication and secure host survival [27]. The inverse relationship between the magnitude of the type I IFN responses (or pDC number) and viral load has been mathematically modelled and reveals the importance for tissues bearing a high number of pDCs, such as the spleen, to act as a sink for viral replication, protecting peripheral organs [28] (Fig. 1A and B).
Figure 1.
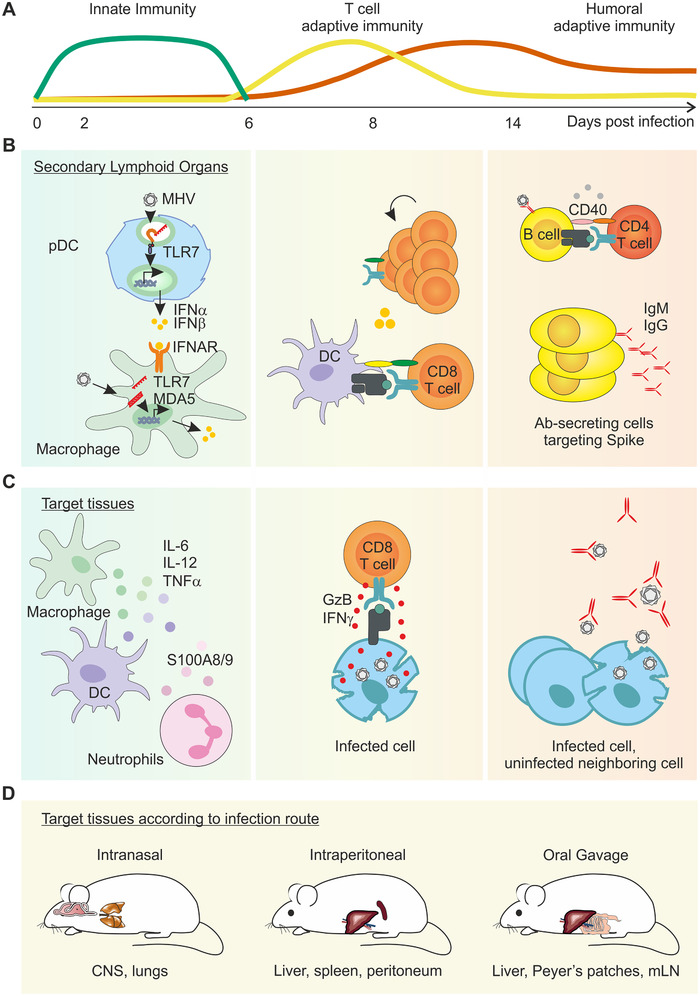
Schematic diagram of the overlapping layers of protective immunity against murine coronaviruses. (A) Schematic diagram of the timely overlap of innate and adaptive immune responses to MHV‐A59. (B) Virus‐infected cells rapidly produce type I IFN as the first line of innate immune defence. Plasmacytoid dendritic cells (pDC) residing in secondary lymphoid organs are a dominant source of type I IFNs, and signalling via the IFN‐α/β receptor (IFNAR) on macrophages triggers downstream innate mechanisms to limit viral spread. Uptake of viral antigen and activation by dendritic cells (DC) instigates cytotoxic CD8 T‐cell priming. Germinal centre B‐cell responses also take place in secondary lymphoid organs, promoting the expansion and maturation of high affinity, antibody‐secreting cells. (C) In the target tissue, cytokine‐secreting, activated myeloid cells help contain viral spread until primed, cytotoxic CD8 T cells migrate to the site of infection and critically eliminate virus‐infected cells. Long‐term humoral immunity is mediated by neutralizing antibodies (Ab). (D) Schematic diagram of the known target organs following MHV‐A59 infection according to distinct infection routes.
Type I IFN sensing leads to induction of an antiviral state and upregulation of chemokines and cytokines that activate sequential arms of the innate immune response. Studies elaborating the kinetics of early immune cell infiltration reveal that neutrophils are among the first cell type to accumulate in infected organs [29, 30, 31], promoting subsequent mononuclear cell infiltration and viral containment [32]. A recent study demonstrated that lung‐infiltrating neutrophils upregulate the bacterial alarmins S100A8/A9 in response to CoVs, including MHV‐A59, but not other viruses [33]. Pharmacological blockade of S100A9 reduced lung pathology and protected mice against fatal MHV infection in IFNAR‐deficient mice, suggesting an important second line of innate immune defence by neutrophils following MHV‐infection.
Proinflammatory cytokines are induced in response to CoVs, and genetic studies have begun to dissect the roles of inflammatory cytokines in MHV infection. Inflammasome‐related cytokines IL‐1 and IL‐18 both contribute to controlling viral replication, and IL‐18 signalling is additionally associated with proficient IFN‐gamma (IFN‐γ) production by T cells and host survival [34]. Earlier studies of IFN‐γ‐deficient mice have highlighted the critical role for this cytokine to protect from fatal peritonitis following intraperitoneal MHV infection [35]. Thus, diverse innate immune cell types and inflammatory mediators orchestrate the early innate immune response to MHV (Fig. 1A).
Adaptive immunity mediates viral clearance and protective immunity to MHV
Following the first line of innate immune defence, MHV is critically controlled by cytotoxic T cells (Fig. 1B). Studies tracking virus‐specific T cells, demonstrate that CD4+ and CD8+ T cells accumulate in the target organ from day 6 following infection [36], and that their CCR7‐dependent cell recruitment is required to restrict viral replication and ensure host survival, at least in the CNS [37]. A series of adoptive transfer and T‐cell depletion experiments in WT mice have shown that both CD8+ and CD4+ T cells are required to contain viral spread in the target tissue [38, 39]. While CD8 T cells eliminate virus‐infected cells, CD4+ T cells have been shown to enhance the peripheral activation and local cytotoxic activity of CD8+ T cells [40], and promote T cell‐dependent humoral immunity [41]. Notably, long‐term control of MHV depends on neutralizing antibodies (Fig. 1C). B‐cell deficient mice, or mice unable to secrete IgM experience viral recrudescence following initial viral clearance [41, 42], a manifestation that can be prevented by the transfer of convalescent serum [42] or spike‐targeted neutralizing antibodies [43]. Longitudinal tracking of serum immunoglobulins following intranasal MHV infection demonstrates that neutralizing antibodies remain stable for at least 60 days following intranasal infection [41], however, further studies are warranted to explore later time points and functional protection following reinfection.
Parallel immune phenotypes in MHV and SARS‐CoV‐2
Plasma levels of proinflammatory mediators, such as IL‐2, IL‐6, IL‐7, IL‐10, CXCL10, CCL3, and TNF, are significantly elevated in patients experiencing severe COVID‐19 compared to a mild or moderate disease course [4]. Moreover, several reports suggest that type I IFN responses are attenuated in COVID‐19 patients [44, 45], including observations of severe disease progression in individuals with loss of function TLR7 variants (who exhibit further impaired type I IFN responses) [46], and genetic variants in components of the IFN‐signalling pathway [47]. MHV infection also induces increased serum levels of proinflammatory cytokines, including TNF‐α, IL‐6, IL‐1β, and IFN‐γ [29, 34, 48], which may be further elevated in virulent MHV variants [15, 16, 18, 19, 21], or older mice [49]. Furthermore, the critical role of type I IFNs in containing MHV replication has been dissected in genetic models as discussed above. Thus, MHV and SARS‐CoV‐2 induce similar soluble antiviral and proinflammatory mediators, which are correlated with virulence, age, and survival.
Further parallels between murine CoVs and SARS‐CoV‐2 are drawn in the nature of the humoral immune response. As in SARS‐CoV‐2, neutralizing antibodies are directed against the spike glycoprotein in MHV‐infected mice. While IgM, IgG, and IgA antibody responses are generated following SARS‐CoV‐2 infection, near‐germline antibodies directed at the receptor‐binding domain of the spike glycoprotein have been shown to exert potent neutralizing activity [50], akin to observed importance of germline IgM antibodies in MHV‐A59‐infected mice [41]. Moreover, the clinical benefit of convalescent serum transfer or spike‐targeted antibody therapy in COVID‐19 patients recapitulates earlier proof‐of‐principle experiments elaborated in MHV‐infected mice [42, 43]. Thus, despite differences in tropism or receptor usage, CoVs induce potent, neutralizing antibodies against the spike glycoprotein in mice and men.
Murine coronaviruses as a model for SARS‐COV‐2
Selection of MHV strains and infection routes for preclinical modelling of diverse facets of SARS‐CoV‐2 infection
Distinct MHV strains exhibit differences in virulence and tissue tropism, which should be taken into account when modelling SARS‐CoV‐2 infection. For instance, the MHV‐JHM strain induces a high mortality in mice, superseding the virulence of SARS‐CoV‐2. The MHV‐1 strain, although not widely used, is the most pneuomotropic MHV strain and recapitulates moderate to severe pneumonia [31]. In turn, the prototypical laboratory variant is the MHV‐A59 strain, which induces a lung, CNS, gastrointestinal, or hepatic infection according to the infection route and dose, and may be best suited to recapitulate the multiorgan involvement of SARS‐CoV‐2. Intracerebral and intranasal inoculation results in a CNS infection [51], and intranasal administration of MHV‐A59 also induces an acute, self‐resolving respiratory infection that recapitulates the acute pneumonia experienced by the majority of SARS‐CoV‐2‐infected individuals. However, mice exposed to a sufficiently high dose [52], or aged [49], experience increased lung pathology, but succumb to the infection. Intraperitoneal or intravenous infection leads to liver disease [51], whereas oral application of the virus precipitates gastrointestinal symptoms and hepatitis at higher doses [53]. The known affected organs following different infection routes are recapitulated in Fig. 1D, however, further studies are needed to fully understand the dynamics of viral spread. In sum, MHV‐A59 is well‐suited as a preclinical model recapitulating an acute pulmonary and extrapulmonary coronavirus infection in a natural host.
Comparison of MHV, humanized mice, and mouse‐adapted viruses as SARS‐CoV‐2 models
As described above, several features of the antiviral immune response to MHV‐A59 are shared with SARS‐CoV‐2 (early type I IFN responses, elevated serum cytokines, neutralizing antibodies against the spike glycoprotein). However, MHV is distinct to SARS‐CoV‐2 in its virulence factors and host cell entry receptors, and much effort has gone into generating humanized, transgenic, or knock‐in mice expressing hACE2 [54, 55, 56, 57, 58] as well as mouse‐adapted SARS‐CoV‐2 strains [59, 60]. These models recapitulate the pneumotropism of SARS‐CoV‐2 in human hosts, the neutralizing capacity of Spike‐directed antibodies [58, 59] and requirement of type I IFN signalling and CD8 T cells to attenuate viral titres [57]. Nevertheless, it has been similarly difficult to recapitulate the spectrum of COVID‐19 pathogenicity in hACE2 or mouse‐adapted SARS‐CoV‐2 mouse models. In many of these studies, a very high infection dose is needed, or very young or old mice are used for sufficient infection efficacy (Table 1). Aged hACE2‐transgenic mice infected with SARS‐CoV‐2 [54] and aged WT mice infected with mouse‐adapted SARS‐CoV‐2 [60] exhibit exacerbated lung pathology compared to young counterparts, but do not readily succumb to viral infection suggesting that these models may be suitable to examine long‐term consequences of CoV infection. Nevertheless, none of these models recapitulate the prolonged clinical pneumonia exhibited in COVID‐19 patients. Pulmonary viral titres are detected for up to 5 or 6 days [57, 58] and viral transcripts for 7‐10 days [54, 55, 56] in hACE2 models, while viral titres are cleared within 4 days of intranasal infection in mouse‐adapted SARS‐CoV‐2 mice [59].
Table 1.
Overview of preclinical models of SARS‐CoV‐2
| Virus | Dose, Route | Host strain | Host age | Clinical features | Survival | Reported duration of viral pneumonia | Ref. |
|---|---|---|---|---|---|---|---|
| MHV‐A59 | 5 × 104 PFU, i.n. | C57Bl/6 | 6‐9 weeks | Acute neuroinflammation | 100% survival | n.d. | [37] |
| 1.5 × 104–5 PFU, i.n. | C57Bl/6 | 4 weeks | Acute pneumonia | 97% survival | <11 days (infectious viral particles) | [48] | |
| 5 × 103 PFU, i.n. | A/J | 6‐8 weeks | Acute pneumonia | 20% survival by day 10 | 7 days (infectious viral particles) | [8] | |
| 7 × 102 – 3 PFU, i.n. | C57Bl/6 | 20‐24 months | Pneumonia, weight loss, reduced O2 saturation, and elevated serum proinflammatory cytokines in older mice. | 0% survival in old mice by day 7 | n.d. | [49] | |
| Mouse‐adapted SARS‐CoV‐2 1 | 1.6 × 104 PFU, i.n. | BALB/c | 6 weeks (young), 9 months (old) | Acute pneumonia in young mice, accompanying lung pathology in older mice. No clinical symptoms or weight loss | 60% survival | <4 days (infectious viral particles) | [59] |
| Mouse‐adapted SARS‐CoV‐2 2 | 105 PFU, i.n. | BALB/c | 12 weeks (young), 12 months (old) | Acute pneumonia in young mice, accompanying lung pathology in older mice. Weight loss and impaired viral clearance in older mice. | 100% survival | 7 days (viral RNA) | [60] |
| SARS‐CoV‐2 | 3 × 104 PFU, i.n. | hACE2 transgenic (HFH4 promoter) C3H and C57Bl/6 | 8‐10 weeks | Interstitial pneumonia, lymphopenia in mice with severe disease | 33‐50% survival by day 7 | 7 days (viral RNA) | [54] |
| 105 PFU, i.n. | hACE2 transgenic (mACE2 promoter) ICR | 6‐11 months | Lung pathology, weight loss, and viral titres in lungs | 100% survival | 7 days (viral RNA) | [58] | |
| 4 × 105 PFU, i.n. |
hACE2 (CRISPR/Casp9) C57Bl/6 |
4.5 weeks (young) 30 weeks (old) | Interstitial pneumonia and elevated cytokines in old mice, no clinical symptoms of weight loss in young mice | 100% survival | 6 days (viral RNA) | [56] | |
| 105 PFU, i.n./i.v. | AdV/hACE2 BALB/c | 8‐10 weeks | Lung pathology, weight loss and viral titres in lungs | 100% survival | 10 days (viral RNA) | [55] | |
| 105 PFU, i.n. | Ad5‐hACE2 BALB/c and C57Bl/6 | 6‐8 weeks | Lung pathology, weight loss and viral titres in lungs | 100% survival | 5 days C57Bl/6 6 days BALB/c (infectious viral particles) | [57] |
Ad5, Adenovirus type 5; AdV, Adenovirus; HFH4, hepatocyte nuclear factor 3/forkhead homologue 4; i.n., intranasal; i.v., intravenous; n.d., not determined.
1Six passages in BALB/c mice (MASCp6).
2 Spike Q498T/P499Y mutant binds mACE‐2.
Additionally, marked differences exist between models in terms of recapitulating hypercytokinemia and multiorgan involvement. Similar to MHV infection [49, 52], Gu et al., report elevated serum proinflammatory cytokines (including IL‐1β, TNF‐α, IL‐2, IL‐4, IL‐5, IL‐6, IL‐10, G‐CSF, GM‐CSF, and CCL2) in young and old mice infected with a mouse‐adapted SARS‐CoV‐2 [60], and Sun et al., report elevated serum levels of IFN‐γ, IL‐9, G‐CSF, and Eotaxin in old but not young SARS‐CoV‐2‐infected hACE2‐transgenic mice [56]. In other models, serum cytokines were found to be unchanged compared to uninfected controls [59]. Finally, in terms of multiorgan involvement, with the exception of the heart [55], infection of hACE2‐transgenic mice with SARS‐CoV‐2 does not recapitulate the multiorgan viral spread seen in COVID‐19 [5]. Some studies, albeit greatly underpowered, report detectable viral titres in the brain [54, 56], and others report the absence of viral titres outside of the respiratory tract [58]. Moreover, intragastric SARS‐CoV‐2 infection of humanized mice did not result in detectable viral titres in the spleen, kidneys, liver, or intestine [56], in contrast to MHV‐A59 [53]. In a mouse‐adapted SARS‐CoV‐2 strain, detectable viral transcript was reported in the lung, heart, liver, and faeces, but not the intestines or kidneys [60], recapitulating only a limited spectrum of the multiorgan involvement in COVID‐19 patients.
None of the available models recapitulate the prolonged detection of viral titres and pneumonia exhibited in severe COVID‐19 patients. Considering the absence of a fully‐adapted host replication machinery in hACE2 mice, these models are advantageous for preclinical testing of therapeutic interventions at the level of Spike — hACE2 interaction. MHV‐A59 is best suited to elucidate CoV virulence mechanisms and multiorgan involvement, and mouse‐adapted SARS‐CoV‐2 and MHV‐A59 strains are most adept for the mechanistic dissection of the mediators of antiviral immunity in the upper and lower airways.
Conclusions
MHV is a murine coronavirus that causes acute respiratory, CNS, gastrointestinal, and/or liver disease depending on the infection route and viral strain. The study of recombinant and natural MHV strains has elucidated the function of CoV structural and nonstructural genes, revealing determinants of tropism, virulence, and evasion of host immunity. Moreover, genetic and pharmacological studies in mice have helped to identify innate and adaptive mechanisms of antiviral immunity to CoVs. Immunological correlates of disease severity in COVID‐19 patients underscore the cross‐species conservation of antiviral mechanisms to CoVs. In addition to MHV, humanized ACE2‐expressing mice and mouse‐adapted SARS‐CoV‐2 strains have recently been developed. While none of the currently available models recapitulate the prolonged pneumonia exhibited in patients with severe COVID‐19, the native murine CoV represents a well‐suited model to study CoV virulence factors, multiorgan involvement, and antiviral immunity. Detailed histopathological studies following viral clearance are required to determine to what extent these models recapitulate long‐term complications of CoV infection (“long COVID”), a condition facing many COVID‐19 patients globally [61].
Conflict of Interest
The authors declare no commercial or financial conflict of interest.
Abbreviations
- IFNAR
IFN‐α/β receptor
- MDA5
melanoma differentiation‐associated gene 5
- Nsp
nonstructural protein
- pDCs
plasmacytoid DCs
Acknowledgments
This study received financial support from the Swiss National Science Foundation (Grant 180011 to N.B.P. and Grants 177208 and 182583 to B.L.) and Novartis Foundation for Biomedical Research to N.B.P. The funders had no role in preparation of the manuscript.
References
- 1. Weiss, S. R. and Navas‐Martin, S. , Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol Mol Biol Rev 2005. 69: 635–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lee, N. , Hui, D. , Wu, A. , Chan, P. , Cameron, P. , Joynt, G. M. , Ahuja, A. et al., A major outbreak of severe acute respiratory syndrome in Hong Kong. N Engl J Med 2003. 348: 1986–1994. [DOI] [PubMed] [Google Scholar]
- 3. Arabi, Y. M. , Balkhy, H. H. , Hayden, F. G. , Bouchama, A. , Luke, T. , Baillie, J. K. , Al‐Omari, A. et al., Middle east respiratory syndrome. N Engl J Med 2017. 376: 584–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huang, C. , Wang, Y. , Li, X. , Ren, L. , Zhao, J. , Hu, Y. , Zhang, L. et al., Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020. 395: 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gupta, A. , Madhavan, M. V. , Sehgal, K. , Nair, N. , Mahajan, S. , Sehrawat, T. S. , Bikdeli, B. et al., Extrapulmonary manifestations of COVID‐19. Nat Med 2020. 26: 1017–1032. [DOI] [PubMed] [Google Scholar]
- 6. Fehr, A. R. and Perlman, S. , Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol 2015. 1282: 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bender, S. J. and Weiss, S. R. , Pathogenesis of murine coronavirus in the central nervous system. J Neuroimmune Pharmacol 2010. 5: 336–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Leibowitz, J. L. , Srinivasa, R. , Williamson, S. T. , Chua, M. M. , Liu, M. , Wu, S. , Kang, H. et al., Genetic determinants of mouse hepatitis virus strain 1 pneumovirulence. J Virol 2010. 84: 9278–9291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gorbalenya, A. E. , Snijder, E. J. and Spaan, W. J. , Severe acute respiratory syndrome coronavirus phylogeny: toward consensus. J Virol 2004. 78: 7863–7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Coutelier, J. P. , Godfraind, C. , Dveksler, G. S. , Wysocka, M. , Cardellichio, C. B. , Noel, H. and Holmes, K. V. , B lymphocyte and macrophage expression of carcinoembryonic antigen‐related adhesion molecules that serve as receptors for murine coronavirus. Eur J Immunol 1994. 24: 1383–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Godfraind, C. , Langreth, S. G. , Cardellichio, C. B. , Knobler, R. , Coutelier, J. P. , Dubois‐Dalcq, M. and Holmes, K. V. , Tissue and cellular distribution of an adhesion molecule in the carcinoembryonic antigen family that serves as a receptor for mouse hepatitis virus. Lab Invest 1995. 73: 615–627. [PubMed] [Google Scholar]
- 12. Sarma Das, J., Fu , L., Tsai , C. J., Weiss, S. R. and Lavi, E. , Demyelination determinants map to the spike glycoprotein gene of coronavirus mouse hepatitis virus. J Virol 2000. 74: 9206–9213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Navas, S. , Seo, S. H. , Chua, M. M. , Das Sarma, J. , Lavi, E. , Hingley, S. T. and Weiss, S. R. , Murine coronavirus spike protein determines the ability of the virus to replicate in the liver and cause hepatitis. J Virol 2001. 75: 2452–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Navas, S. and Weiss, S. R. , Murine coronavirus‐induced hepatitis: JHM genetic background eliminates A59 spike‐determined hepatotropism. J Virol 2003. 77: 4972–4978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rempel, J. D. , Murray, S. J. , Meisner, J. and Buchmeier, M. J. , Differential regulation of innate and adaptive immune responses in viral encephalitis. Virology 2004. 318: 381–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rempel, J. D. , Murray, S. J. , Meisner, J. and Buchmeier, M. J. , Mouse hepatitis virus neurovirulence: evidence of a linkage between S glycoprotein expression and immunopathology. Virology 2004. 318: 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Iacono, K. T. , Kazi, L. and Weiss, S. R. , Both spike and background genes contribute to murine coronavirus neurovirulence. J Virol 2006. 80: 6834–6843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eriksson, K. K. , Cervantes‐Barragan, L. , Ludewig, B. and Thiel, V. , Mouse hepatitis virus liver pathology is dependent on ADP‐ribose‐1''‐phosphatase, a viral function conserved in the alpha‐like supergroup. J Virol 2008. 82: 12325–12334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zust, R. , Cervantes‐Barragan, L. , Habjan, M. , Maier, R. , Neuman, B. W. , Ziebuhr, J. , Szretter, K. J. et al., Ribose 2'‐O‐methylation provides a molecular signature for the distinction of self and non‐self mRNA dependent on the RNA sensor Mda5. Nat Immunol 2011. 12: 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fehr, A. R. , Channappanavar, R. , Jankevicius, G. , Fett, C. , Zhao, J. , Athmer, J. , Meyerholz, D. K. et al., The conserved coronavirus macrodomain promotes virulence and suppresses the innate immune response during severe acute respiratory syndrome coronavirus infection. mBio 2016. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zust, R. , Cervantes‐Barragan, L. , Kuri, T. , Blakqori, G. , Weber, F. , Ludewig, B. and Thiel, V. , Coronavirus non‐structural protein 1 is a major pathogenicity factor: implications for the rational design of coronavirus vaccines. PLoS Pathog 2007. 3: e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Viswanathan, T. , Arya, S. , Chan, S. H. , Qi, S. , Dai, N. , Misra, A. , Park, J. G. et al., Structural basis of RNA cap modification by SARS‐CoV‐2. Nat Commun 2020. 11: 3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Banerjee, A. K. , Blanco, M. R. , Bruce, E. A. , Honson, D. D. , Chen, L. M. , Chow, A. , Bhat, P. , Ollikainen, N. et al., SARS‐CoV‐2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell 2020. 183:1325–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xia, H. , Cao, Z. , Xie, X. , Zhang, X. , Chen, J. Y. , Wang, H. , Menachery, V. D. , Rajsbaum, R. and Shi, P. Y. , Evasion of type I interferon by SARS‐CoV‐2. Cell Rep 2020. 33: 108234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cervantes‐Barragan, L. , Zust, R. , Weber, F. , Spiegel, M. , Lang, K. S. , Akira, S. , Thiel, V. and Ludewig, B. , Control of coronavirus infection through plasmacytoid dendritic‐cell‐derived type I interferon. Blood 2007. 109: 1131–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zalinger, Z. B. , Elliott, R. , Rose, K. M. and Weiss, S. R. , MDA5 is critical to host defense during infection with murine coronavirus. J Virol 2015. 89: 12330–12340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cervantes‐Barragan, L. , Kalinke, U. , Zust, R. , Konig, M. , Reizis, B. , Lopez‐Macias, C. , Thiel, V. and Ludewig, B. , Type I IFN‐mediated protection of macrophages and dendritic cells secures control of murine coronavirus infection. J Immunol 2009. 182: 1099–1106. [DOI] [PubMed] [Google Scholar]
- 28. Bocharov, G. , Zust, R. , Cervantes‐Barragan, L. , Luzyanina, T. , Chiglintsev, E. , Chereshnev, V. A. , Thiel, V. and Ludewig, B. , A systems immunology approach to plasmacytoid dendritic cell function in cytopathic virus infections. PLoS Pathog 2010. 6: e1001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bleau, C. , Filliol, A. , Samson, M. and Lamontagne, L. , Mouse hepatitis virus infection induces a toll‐like receptor 2‐dependent activation of inflammatory functions in liver sinusoidal endothelial cells during acute hepatitis. J Virol 2016. 90: 9096–9113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Templeton, S. P. , Kim, T. S. , O'Malley, K. and Perlman, S. , Maturation and localization of macrophages and microglia during infection with a neurotropic murine coronavirus. Brain Pathol 2008. 18: 40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. De Albuquerque, N. , Baig, E. , Ma, X. , Zhang, J. , He, W. , Rowe, A. , Habal, M. et al., Murine hepatitis virus strain 1 produces a clinically relevant model of severe acute respiratory syndrome in A/J mice. J Virol 2006. 80: 10382–10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhou, J. , Stohlman, S. A. , Hinton, D. R. and Marten, N. W. , Neutrophils promote mononuclear cell infiltration during viral‐induced encephalitis. J Immunol 2003. 170: 3331–3336. [DOI] [PubMed] [Google Scholar]
- 33. Guo, Q. , Zhao, Y. , Li, J. , Liu, J. , Yang, X. , Guo, X. , Kuang, M. et al., Induction of alarmin S100A8/A9 mediates activation of aberrant neutrophils in the pathogenesis of COVID‐19. Cell Host Microbe 2020. 29: 222–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zalinger, Z. B. , Elliott, R. and Weiss, S. R. , Role of the inflammasome‐related cytokines Il‐1 and Il‐18 during infection with murine coronavirus. J Neurovirol 2017. 23: 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kyuwa, S. , Tagawa, Y. , Shibata, S. , Doi, K. , Machii, K. and Iwakura, Y. , Murine coronavirus‐induced subacute fatal peritonitis in C57BL/6 mice deficient in gamma interferon. J Virol 1998. 72: 9286–9290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Haring, J. S. , Pewe, L. L. and Perlman, S. , High‐magnitude, virus‐specific CD4 T‐cell response in the central nervous system of coronavirus‐infected mice. J Virol 2001. 75: 3043–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cupovic, J. , Onder, L. , Gil‐Cruz, C. , Weiler, E. , Caviezel‐Firner, S. , Perez‐Shibayama, C. , Rulicke, T. et al., Central nervous system stromal cells control local CD8(+) T cell responses during virus‐induced neuroinflammation. Immunity 2016. 44: 622–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Williamson, J. S. and Stohlman, S. A. , Effective clearance of mouse hepatitis virus from the central nervous system requires both CD4+ and CD8+ T cells. J Virol 1990. 64: 4589–4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yamaguchi, K. , Goto, N. , Kyuwa, S. , Hayami, M. and Toyoda, Y. , Protection of mice from a lethal coronavirus infection in the central nervous system by adoptive transfer of virus‐specific T cell clones. J Neuroimmunol 1991. 32: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Phares, T. W. , Stohlman, S. A. , Hwang, M. , Min, B. , Hinton, D. R. and Bergmann, C. C. , CD4 T cells promote CD8 T cell immunity at the priming and effector site during viral encephalitis. J Virol 2012. 86: 2416–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gil‐Cruz, C. , Perez‐Shibayama, C. , Firner, S. , Waisman, A. , Bechmann, I. , Thiel, V. , Cervantes‐Barragan, L. and Ludewig, B. , T helper cell‐ and CD40‐dependent germline IgM prevents chronic virus‐induced demyelinating disease. Proc Natl Acad Sci USA 2012. 109: 1233–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Matthews, A. E. , Weiss, S. R. , Shlomchik, M. J. , Hannum, L. G. , Gombold, J. L. and Paterson, Y. , Antibody is required for clearance of infectious murine hepatitis virus A59 from the central nervous system, but not the liver. J Immunol 2001. 167: 5254–5263. [DOI] [PubMed] [Google Scholar]
- 43. Ramakrishna, C. , Bergmann, C. C. , Atkinson, R. and Stohlman, S. A. , Control of central nervous system viral persistence by neutralizing antibody. J Virol 2003. 77: 4670–4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mudd, P. A. , Crawford, J. C. , Turner, J. S. , Souquette, A. , Reynolds, D. , Bender, D. , Bosanquet, J. P. et al., Targeted immunosuppression distinguishes COVID‐19 from influenza in moderate and severe disease. medRxiv 2020. 10.1101/2020.05.28.20115667 [DOI] [Google Scholar]
- 45. Hadjadj, J. , Yatim, N. , Barnabei, L. , Corneau, A. , Boussier, J. , Smith, N. , Pere, H. et al., Impaired type I interferon activity and inflammatory responses in severe COVID‐19 patients. Science 2020. 369: 718–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. van der Made, C. I. , Simons, A. , Schuurs‐Hoeijmakers, J. , van den Heuvel, G. , Mantere, T. , Kersten, S. , van Deuren, R. C. et al., Presence of genetic variants among young men with severe COVID‐19. JAMA 2020. 324: 663–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang, Q. , Bastard, P. , Liu, Z. , Le Pen, J. , Moncada‐Velez, M. , Chen, J. , Ogishi, M. et al., Inborn errors of type I IFN immunity in patients with life‐threatening COVID‐19. Science 2020. 370: eabd4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yang, Z. , Du, J. , Chen, G. , Zhao, J. , Yang, X. , Su, L. , Cheng, G. and Tang, H. , Coronavirus MHV‐A59 infects the lung and causes severe pneumonia in C57BL/6 mice. Virol Sin 2014. 29: 393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ryu, S. , Shchukina, I. , Youm, Y. H. , Qing, H. , Hilliard, B. K. , Dlugos, T. , Zhang, X. et al., Ketogenesis restrains aging‐induced exacerbation of COVID in a mouse model. bioRxiv 2020. 10.1101/2020.09.11.294363 [DOI] [Google Scholar]
- 50. Kreer, C. , Zehner, M. , Weber, T. , Ercanoglu, M. S. , Gieselmann, L. , Rohde, C. , Halwe, S. et al., Longitudinal isolation of potent near‐germline SARS‐CoV‐2‐neutralizing antibodies from COVID‐19 patients. Cell 2020. 182: 843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lavi, E. , Gilden, D. H. , Highkin, M. K. and Weiss, S. R. , The organ tropism of mouse hepatitis virus A59 in mice is dependent on dose and route of inoculation. Lab Anim Sci 1986. 36: 130–135. [PubMed] [Google Scholar]
- 52. Qing, H. , Sharma, L. , Hilliard, B. K. , Peng, X. , Swaminathan, A. , Tian, J. , Israni‐Winger, K. et al., Type I interferon limits viral dissemination‐driven clinical heterogeneity in a native murine betacoronavirus model of COVID‐19. bioRxiv 2020: 2020.2009.2011.294231. [Google Scholar]
- 53. Gil‐Cruz, C. , Perez‐Shibayama, C. , Onder, L. , Chai, Q. , Cupovic, J. , Cheng, H. W. , Novkovic, M. et al., Fibroblastic reticular cells regulate intestinal inflammation via IL‐15‐mediated control of group 1 ILCs. Nat Immunol 2016. 17: 1388–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jiang, R. D. , Liu, M. Q. , Chen, Y. , Shan, C. , Zhou, Y. W. , Shen, X. R. , Li, Q. et al., Pathogenesis of SARS‐CoV‐2 in transgenic mice expressing human angiotensin‐converting enzyme 2. Cell 2020. 182: 50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hassan, A. O. , Case, J. B. , Winkler, E. S. , Thackray, L. B. , Kafai, N. M. , Bailey, A. L. , McCune, B. T. et al., A SARS‐CoV‐2 infection model in mice demonstrates protection by neutralizing antibodies. Cell 2020. 182: 744–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sun, S. H. , Chen, Q. , Gu, H. J. , Yang, G. , Wang, Y. X. , Huang, X. Y. , Liu, S. S. et al., A mouse model of SARS‐CoV‐2 infection and pathogenesis. Cell Host Microbe 2020. 28: 124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sun, J. , Zhuang, Z. , Zheng, J. , Li, K. , Wong, R. L. , Liu, D. , Huang, J. et al., Generation of a broadly useful model for COVID‐19 pathogenesis, vaccination, and treatment. Cell 2020. 182: 734–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bao, L. , Deng, W. , Huang, B. , Gao, H. , Liu, J. , Ren, L. , Wei, Q. et al., The pathogenicity of SARS‐CoV‐2 in hACE2 transgenic mice. Nature 2020. 583: 830–833. [DOI] [PubMed] [Google Scholar]
- 59. Dinnon, K. H., 3rd , Leist, S. R. , Schafer, A. , Edwards, C. E. , Martinez, D. R. , Montgomery, S. A. , West, A. et al., A mouse‐adapted model of SARS‐CoV‐2 to test COVID‐19 countermeasures. Nature 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gu, H. , Chen, Q. , Yang, G. , He, L. , Fan, H. , Deng, Y. Q. , Wang, Y. et al., Adaptation of SARS‐CoV‐2 in BALB/c mice for testing vaccine efficacy. Science 2020. 369: 1603–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Meeting the challenge of long COVID. Nat Med 2020. 26: 1803. [DOI] [PubMed] [Google Scholar]