

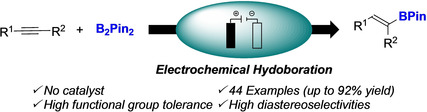
Abstract
Herein we reported the electrochemical hydroboration of alkynes by using B2Pin2 as the boron source. This unprecedented reaction manifold was applied to a broad range of alkynes, giving the hydroboration products in good to excellent yields without the need of a metal catalyst or a hydride source. This transformation relied on the possible electrochemical oxidation of an in situ formed borate. This anodic oxidation performed in an undivided cell allowed the formation of a putative boryl radical, which reacted on the alkyne.
Keywords: alkynes, anodic oxidation, boryl radical, electrochemistry, hydroboration
The electrochemical hydroboration of alkynes by using B2Pin2 as the boron source is reported. This reaction manifold was applied to a broad range of alkynes, giving the hydroboration products in good to excellent yields without the need of a metal catalyst or a hydride source. In addition, the mechanism of this transformation was study.
In the realm of organic synthesis organoboron compounds play an important role to build up complex molecules. [1] In addition, boron‐containing molecules found applications in medicinal chemistry, which culminated in the approval of boron‐containing drugs (e. g. Dutogliptin). [2] These key building blocks showcase various and specific reactivities, which provide them a pivotal role in the arsenal of organic chemists to design retrosynthetic disconnections. [3] For instance, organoboron compounds have been widely employed in allylation, aldolisation, or homologation reactions and it is important to notice that the hydroboration and the Suzuki‐Miyaura cross‐coupling reactions were celebrated by the Nobel prize given to H. C. Brown and A. Suzuki in 1979 and 2010, respectively. Among these synthetically useful reactions, the hydroboration of an alkene or an alkyne is of paramount interest and has been widely studied for almost half a century. [4] These endeavors gave birth to noble and non‐noble metal catalyzed processes. [5] In parallel, the chemistry of boryl radicals was recently highlighted as a straightforward alternative to introduce a boron residue. [6] With that respect, Curran, Lacôte, Fensternbank and Malacria explored the formation and reactivity of these radicals from NHC‐borane. They demonstrated the possibility to form the radical using usual initiators, such as peroxides or AIBN, for instance. [7] Among the studied reactions, the addition of the boryl radical to olefin and alkynes, pioneered by Lalevée and Curran, [8] has been less explored and the development of new protocols is still highly desirable to extend the portfolio of possible reactions. Very recently, the photocatalyzed formation of boryl radicals using iridium catalysts has been reported (Scheme 1). This new method was applied to the addition of NHC‐boron residues on fluorinated olefins and aryls, according to an addition/fluoride elimination sequence [9] and to the hydroboration of imines, [10] alkenes [11] and electron poor aromatic derivatives. [12] Importantly, in contrast to the formation of boryl radicals from NHC‐borane, their formation from diboron compounds remains scarce. [13] Within the last five years organic electrochemistry has known an impressive renewal of interest. Indeed, electrochemical synthesis, by replacing hazardous chemical redox reagents by electrons, avoids the generation of wastes and if electricity is produced from renewable sources, it can be considered as a green process. [14] As part of our program to develop straightforward access to boron‐containing molecules, [15] we sought to use organic electrochemistry to generate boryl radicals that would be used as an unprecedented reaction manifold in the hydroboration of alkynes. Worth of note, during the preparation of this manuscript, Qing and co‐workers reported the electrochemical hydroboration of styrene using HBpin as the boron source and DIPEA as auxiliary. [16] Herein, we report an original electrochemical hydroboration of terminal alkyne formation from the readily available B2Pin2 using electron as a green redox reagent.
Scheme 1.
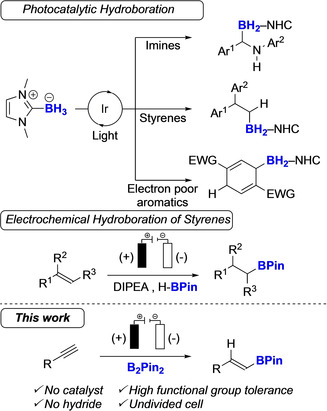
State of the art and present work.
As a model reaction we chose phenylacetylene as the substrate and B2Pin2 as the boron source. After extensive optimization, we found that the open‐air reaction performed in MeOH using n‐Bu4NBF4 as the electrolyte (0.05 M), stainless steel electrodes (SST) at both the cathode and anode along with a constant current of 10 mA and a total charge of 2 F mol−1 were required to ensure the formation of 1 in 84 % yield (Table 1, entry 1).
Table 1.
Optimization of the reaction and sensitivity tests.
|
| ||
|---|---|---|
|
Entry |
Variation from standard conditions |
Yield [%][a] |
|
1 |
none |
84[b] |
|
2 |
EtOH or i‐PrOH instead of MeOH |
<25 |
|
3 |
graphite electrodes instead of SST |
0 |
|
4 |
platinum electrodes instead of SST |
<30 |
|
5 |
1 F mol−1 instead of 2 F mol−1 |
51 |
|
6 |
n‐Bu4NBF4: 0.025 M instead of 0.05 M |
75 |
|
7 |
H‐BPin instead of B2Pin2 |
0 |
|
8 |
no current |
0 |
|
9 |
NaOMe (1 equiv.) & no current |
0 |
|
| ||
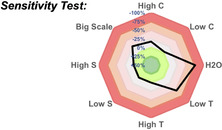
Reaction conditions: phenylacetylene (0.2 mmol), B2Pin2 (0.4 mmol), MeOH (4 mL), r.t. [a] NMR yield. [b] Isolated yield. C: concentration, T: temperature, S: electrodes surface.
Indeed, the use of EtOH or i‐PrOH did not afford 1 with decent yields (entry 2, <25 %), while the use of other electrodes was deleterious for the reaction outcome (entries 3 and 4). Then, a total charge of 1 F mol−1 or a decrease of the electrolyte concentration gave lower yields (entries 5 and 6). Finally, control experiments revealed that i) the reaction did not occurred when B2Pin2 was replaced by H‐BPin (entry 7), ii) the reaction did not occurred in the absence of current (entry 8) and iii) no reaction occurred with MeONa in the absence of current (entry 9), precluding a base assisted hydroboration reaction. [17] Then, to ascertain the versatility and to ensure the reproducibility of our electrochemical hydroboration reaction, a sensitivity assessment regarding the reaction parameters was performed (Table 1). [18] This hydroboration protocol was found sensitive to the presence of water and to the temperature, while the yield slightly decreased (c.a. 25 % less) when the scale of the reaction was increased. [19]
Having optimized the reaction conditions, we explored the scope of this catalyst‐ and hydride‐free hydroboration reaction (Scheme 2). First, a large panel of aryl substituted terminal alkynes was tested. Aryls substituted with a methyl group provided the corresponding vinyl boronate derivatives (2–4) in good to excellent yields, whatever the substitution pattern. Other electron donating groups like methoxy, phenyl, N,N‐dimethyl and non‐protected amines were suitable affording the products (6–11) in good to excellent yields. Interestingly, the reaction was tolerant to the presence of a BPin group, a benzylic alcohol as well as an acetal, furnishing the synthetically useful hydroborated products 12, 13 and 14. Halogens and CF3 substituents on the aromatic did not affect the efficiency of the reaction, offering an access to compounds 15–20 in good to excellent yields. Regarding the presence of electron‐withdrawing on the aromatic, the aldehyde 21 and the ester 22 were synthesized in moderate to good yields, at the cost of an increase of the charge from 2.0 to 2.5 F mol−1 for 22. Similarly, naphthyl derivatives 23 and 24 were readily obtained using 2.5 F mol−1. This methodology was applied to pyridine derivative, giving 25 in 63 % isolated yield. Then, alkyl substituted terminal alkynes were tested. The presence of a primary, secondary and tertiary alcohols, as well as a phthalimide did not affect the outcome of the reaction and the hydroborated products 26–31 were isolated in moderate to excellent yields. The reaction was selective to alkyne, since an enyne was selectively functionalized on the alkyne residue (32). Interestingly, the cyclopropyl derivative 33 was obtained with an excellent 70 % NMR yield and a modest 39 % isolated yield, due to a tedious purification. Surprisingly, the vinyl boronates 34–39 were isolated as a mixture of α‐ and β‐isomers in moderate yields, the β‐isomer being the major one in all cases. [20] Pleasingly, the scope of this electrochemical hydroboration reaction was extended to internal alkynes. Using one equivalent of K2CO3 as an additive, the boronic esters 40 and 41 were isolated in 49 % and 58 % yield, respectively. Note that in the case of 40, a 70 : 30 mixture of α‐ and β‐isomers were obtained. Finally, this methodology was applied to complexes molecules to demonstrate the synthetic utility of the reaction. The steroids derivatives 42 and 43 were obtained in 66 % and 37 % isolated yields. Regarding 43, the low efficiency of the process was explained by the poor solubility of the starting material in the solvent of the reaction. Finally, the α‐tocopherol derivative 44 was isolated in 27 % yield.
Scheme 2.
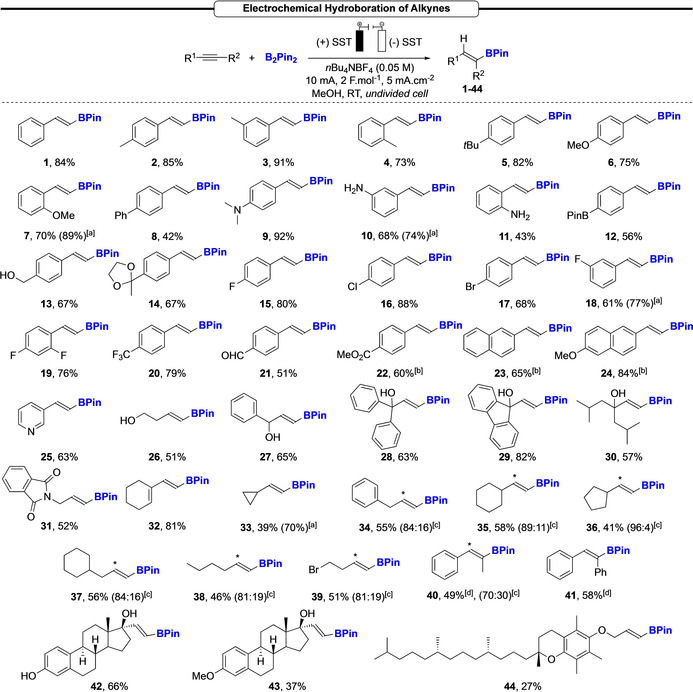
Exploration of the scope of the electrochemical hydroboration of alkynes. All data are the average of two runs. [a] Yield determined by 1H NMR by using DMF as an internal standard. [b] 2.5 F mol−1 instead of 2 F mol−1. [c] Determined by 1H NMR on the crude reaction mixture. [d] 1 equiv. of K2CO3 was added.
To get further insight into the mechanism, the reaction was carried out in the presence of CD3OD instead of CH3OH (Scheme 3, Eq. (1)). [19] This control experiment furnished the deuterated product [D]‐1 in 42 % yield with a complete incorporation of deuterium, which demonstrates that the hydrogen atom comes from the solvent. Then, to determine if the H abstraction results from the cleavage of the C−H or O−H bond of the solvent, reactions were carried out with CD3OH and CH3OD, independently (Scheme 3, Eqs. (2) & (3)). These results clearly highlight that the H results from the cleavage of the O−H bond, despite its higher BDE compared to the C−H bond. Finally, we studied the kinetic isotopic effect (KIE). Parallel reactions in CH3OH or CD3OD revealed a KIE=2, while a KIE of 5.75 was measured in a 1 : 1 mixture of CH3OH/CD3OD, suggesting that the hydrogen abstraction from the solvent after the addition of the boryl radical might be the rate determining step (Scheme 3, Eqs. (4) & (5)). Finally, to ascertain the reaction does not result from the presence of metallic salts released from the electrodes, the electrochemical reaction was stopped after 32 minutes (1 F mol−1) and the reaction mixture was stirred within the electrosynthesis cell for an additional 24 h (Scheme 3, Eq. (6)). Pleasingly, we did not observe an increase of the reaction yield after this additional stirring. This observation precluded a background hydroboration reaction catalyzed by metallic salts released from the SST electrodes. Then, since we surmised a possible oxidation of a transient borate species, we carried out cyclic voltammetry (CV) experiments to depict a possible reaction mechanism. The CV measurements of B2Pin2 (orange), NaOMe (yellow), B2Pin2 and NaOMe (blue), phenylacetylene (grey) and the mixture of B2Pin2, NaOMe and phenylacetylene (green) in MeOH were carried out. [19] To mimic the formation of the alkoxide, which is unlikely during the CV measurement due to the tiny surface of the anode, NaOMe was added to the B2Pin2 species. The CV analysis of the mixture B2Pin2/NaOMe (blue) demonstrated an irreversible oxidation wave at +1.2 V vs. SCE, that is not observed on the CV measurement of the B2Pin2 (orange). Moreover, a similar oxidation wave was observed on the mixture of all reagents (green). Finally, no oxidation nor reduction of B2Pin2 or phenylacetylene was observed, while a completely different oxidation wave was witnessed with NaOMe (yellow). Hence, with these experimental data in hand, we suggested the following plausible mechanism (Scheme 4). First, we surmised the formation of the boryl radical from the oxidation of an in situ formed borate A. [21] Indeed, the coordination of an alkoxide, here generated by the reduction of the solvent at the cathode (i. e. MeOH), to the empty orbital of a boron atom of B2Pin2 might gave birth to this species. [17] Then, the anodic oxidation of the latter would generate B, that would quickly collapse into the boryl radical C (i. e. •BPin) and MeOBPin. This boryl radical is probably a 7‐electrons centered one, rather than a less stable 5‐electrons, thanks to the coordination of the solvent to the empty orbital of the boron atom. [6b] This radical would then undergo an addition reaction onto the alkyne to form the corresponding highly reactive vinyl radical D. Then, taking into account the above‐mentioned results from the labelling experiments, a plausible pathway to explain the abstraction of the H from the stronger O−H bond is suggested. [22] The coordination of a boron species, which can act as a Lewis acid, to MeOH might decrease the O−H bond dissociation energy and allow the H abstraction by the vinyl radical. Then, the resulting O radical could be reduced at the cathode to complete the redox cycle. [23] Alternatively, the methoxide anion, generated at the cathode, might act as a hydrogen donor as well. Although its concentration in the reaction media is much lower than MeOH this pathway can be productive as well. [24]
Scheme 3.
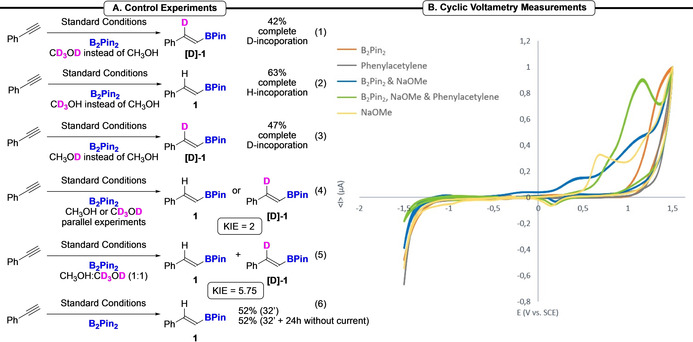
Mechanistic study: control experiments, CV measurements. nBu4NBF4 was used as the electrolyte for the cyclic voltammetry measurements (see Supporting Information for details). All potentials are reported vs. SCE.
Scheme 4.
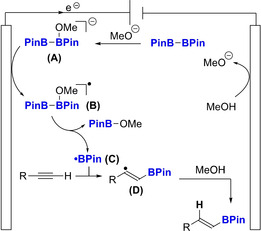
Plausible Mechanism.
In conclusion, we developed the first electrochemical hydroboration of alkynes. This catalyst‐ and hydride‐free methodology provides a greener alternative for the synthesis of borylated alkenes. Furthermore, the use of stainless steel electrodes makes the process cost‐effective. According to a possible anodic oxidation of an in situ formed borate species, a putative boryl radical derived from the cleavage of the B−B bond of B2Pin2 was added to a large panel of alkynes, including complex molecules such as steroids. The resulting products were obtained in good to excellent yields.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was partially supported by Normandie Université (NU), the Région Normandie, the Centre National de la Recherche Scientifique (CNRS), Université de Rouen Normandie (URN), INSA Rouen Normandie, Labex SynOrg (ANR‐11‐LABX‐0029), Innovation Chimie Carnot (I2C) and the Agence National pour la Recherche (ANR‐CE07‐0004‐1). M.A. thanks the Agence National pour la Recherche (ANR‐CE07‐0004‐1) for a doctoral fellowship. M.S. thanks the NSERC‐CREATE Program in Continuous Flow Science for support. P.J. and T.P. thank the Région Normandie for funding (RIN TREMPLIN EFLUX). T.P. thanks the Institut Universitaire de France (IUF) for support. Dr. Muriel Durandetti is gratefully acknowledged for her help regarding the CV measurements.
M. Aelterman, M. Sayes, P. Jubault, T. Poisson, Chem. Eur. J. 2021, 27, 8277.
Contributor Information
Prof. Dr. Philippe Jubault, Email: philippe.jubault@insa-rouen.fr.
Prof. Dr. Thomas Poisson, Email: thomas.poisson@insa-rouen.fr.
References
- 1.
- 1a. Synthesis and Applications of Organoboron Compounds., Eds. E. Fernandez, A. Whiting, Springer 2015; For selected reviews, see:
- 1b. Carreras J., Caballero A., Pérez P. J., Chem. Asian J. 2019, 14, 329–343; [DOI] [PubMed] [Google Scholar]
- 1c. Fyfe J. W. B., Watson A. J. B., Chem 2017, 3, 31–55; [Google Scholar]
- 1d. Miyaura N., Suzuki A., Chem. Rev. 1995, 95, 2457–2483; [Google Scholar]
- 1e. Xu L., Zhang S., Li P., Chem. Soc. Rev. 2015, 44, 8848–8858; [DOI] [PubMed] [Google Scholar]
- 1f. Lennox A. J. J., Lloyd-Jones G. C., Chem. Soc. Rev. 2014, 43, 412–443; [DOI] [PubMed] [Google Scholar]
- 1g. Duret G., Quinlan R., Bisseret P., Blanchard N., Chem. Sci. 2015, 6, 5366–5382; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1h. Ollivier C., Renaud P., Chem. Rev. 2001, 101, 3415–3434; [DOI] [PubMed] [Google Scholar]
- 1i. Namirembe S., Morken J. P., Chem. Soc. Rev. 2019, 48, 3464–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yang F., Zhu M., Zhang J., Zhou H., MedChemComm 2018, 9, 201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Friese F. W., Studer A., Chem. Sci. 2019, 10, 8503–8518; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Neeve E. C., Geier S. J., Mkhalid I. A. I., Westcott S. A., Marder T. B., Chem. Rev. 2016, 116, 9091–9161; [DOI] [PubMed] [Google Scholar]
- 3c. Ishiyama T., Miyaura N., Chem. Rec. 2004, 3, 271–280; [DOI] [PubMed] [Google Scholar]
- 3d. Mkhalid I. A. I., Barnard J. H., Marder T. B., Murphy J. M., Hartwig J. F., Chem. Rev. 2010, 110, 890–931; [DOI] [PubMed] [Google Scholar]
- 3e. Ros A., Fernández R., Lassaletta J. M., Chem. Soc. Rev. 2014, 43, 3229–3243; [DOI] [PubMed] [Google Scholar]
- 3f. Snieckus V., Chem. Rev. 1990, 90, 879–933. [Google Scholar]
- 4.
- 4a. Burgess K., Ohlmeyer M. J., Chem. Rev. 1991, 91, 1179–1191; [Google Scholar]
- 4b. Crudden C. M., Edwards D., Eur. J. Org. Chem. 2003, 2003, 4695–4712; [Google Scholar]
- 4c. Vogels C. M., Westcott S. A., Curr. Org. Chem. 2005, 9, 687–699; [Google Scholar]
- 4d. Chen J.-B., Whiting A., Synthesis 2018, 50, 3843–3861. [Google Scholar]
- 5. Obligacion J. V., Chirik P. J., Nat. Chem. Rev. 2018, 2, 15–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Curran D. P., Solovyev A., Makhlouf Brahmi M., Fensterbank L., Malacria M., Lacôte E., Angew. Chem. Int. Ed. 2011, 50, 10294–10317; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 10476–10500; [Google Scholar]
- 6b. Taniguchi T., Eur. J. Org. Chem. 2019, 6308–6319. [Google Scholar]
- 7.
- 7a. Ueng S.-H., Brahmi M. M., Derat E., Fensterbank L., Lacôte E., Malacria M., Curran D. P., J. Am. Chem. Soc. 2008, 130, 10082–10083; [DOI] [PubMed] [Google Scholar]
- 7b. Ueng S.-H., Solovyev A., Yuan X., Geib S. J., Fensterbank L., Lacôte E., Malacria M., Newcomb M., Walton J. C., Curran D. P., J. Am. Chem. Soc. 2009, 131, 11256–11262. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Lalevée J., Blanchard N., Chany A.-C., Tehfe M.-A., Allonas X., Fouassier J.-P., J. Phys. Org. Chem. 2009, 22, 986–993; [Google Scholar]
- 8b. Lalevée J., Tehfe M.-A., Allonas X., Fouassier J.-P., Macromolecules 2008, 41, 9057–9062; [Google Scholar]
- 8c. Watanabe T., Hirose D., Curran D. P., Taniguchi T., Chem. Eur. J. 2017, 23, 5404–5409; [DOI] [PubMed] [Google Scholar]
- 8d. Watanabe T., Geib S. J., Curran D. P., Taniguchi T., J. Org. Chem. 2017, 82, 13034–13042; [DOI] [PubMed] [Google Scholar]
- 8e. Shimoi M., Watanabe T., Maeda K., Curran D. P., Taniguchi T., Angew. Chem. Int. Ed. 2018, 57, 9485–9490; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 9629–9634; For recent examples, see: [Google Scholar]
- 8f. Jin J.-K., Zheng W.-X., Xia H.-M., Zhang F.-L., Wang Y.-F., Org. Lett. 2019, 21, 8414–8418; [DOI] [PubMed] [Google Scholar]
- 8g. Liu X., Lin E. E., Chen G., Li J.-L., Liu P., Wang H., Org. Lett. 2019, 21, 8454–8458; [DOI] [PubMed] [Google Scholar]
- 8h. Shimoi M., Maeda K., Geib S. J., Curran D. P., Taniguchi T., Angew. Chem. Int. Ed. 2019, 58, 6357–6361; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 6423–6427; [Google Scholar]
- 8i. Huang Y.-S., Wang J., Zheng W.-X., Zhang F.-L., Yu Y.-J., Zheng M., Zhou X., Wang Y.-F., Chem. Commun. 2019, 55, 11904–11907; [DOI] [PubMed] [Google Scholar]
- 8j. Ren S.-C., Zhang F.-L., Xu A.-Q., Yang Y., Zheng M., Zhou X., Fu Y., Wang Y.-F., Nat. Commun. 2019, 10, 1934; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8k. Jin J.-K., Zhang F.-L., Zhao Q., Lu J.-A., Wang Y.-F., Org. Lett. 2018, 20, 7558–7562. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Xu W., Jiang H., Leng J., Ong H. W., Wu J., Angew. Chem. Int. Ed. 2020, 59, 4009–4016; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 4038–4045; [Google Scholar]
- 9b. Qi J., Zhang F.-L., Jin J.-K., Zhao Q., Li B., Liu L. X., Wang Y.-F., Angew. Chem. Int. Ed. 2020, 59, 12876–12884; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 12976–12984; [Google Scholar]
- 9c. Chen G., Wang L., Liu X., Liu P., Adv. Synth. Catal. 2020, 362, 2990–2996. [Google Scholar]
- 10. Zhou N., Yuan X.-A., Zhao Y., Xie J., Zhu C., Angew. Chem. Int. Ed. 2018, 57, 3990–3994; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 4054–4058. [Google Scholar]
- 11. Xia P.-J., Song D., Ye Z.-P., Hu Y.-Z., Xiao J.-A., Xiang H.-Y., Chen X.-Q., Yang H., Angew. Chem. Int. Ed. 2020, 59, 6706–6710; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 6772–6776. [Google Scholar]
- 12. Dai W., Geib S. J., Curran D. P., J. Am. Chem. Soc. 2020, 142, 6261–6267. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Wang G., Zhang H., Zhao J., Li W., Cao J., Zhu C., Li S., Angew. Chem. Int. Ed. 2016, 55, 5985–5989; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 6089–6093; [Google Scholar]
- 13b. Xu R., Lu G.-P., Cai C., New J. Chem. 2018, 42, 16456–16459. [Google Scholar]
- 14.For selected recent reviews, see:
- 14a. Kingston C., Palkowitz M. D., Takahira Y., Vantourout J. C., Peters B. K., Kawamata Y., Baran P. S., Acc. Chem. Res. 2020, 53, 72–83; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14b. Pollok D., Waldvogel S. R., Chem. Sci. 2020, 11, 12386–12400; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14c. Yan M., Kawamata Y., Baran P. S., Chem. Rev. 2017, 117, 13230–13319; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14d. Yamamoto K., Kuriyama M., Onomura O., Acc. Chem. Res. 2020, 53, 105–120; [DOI] [PubMed] [Google Scholar]
- 14e. Mitsudo K., Kurimoto Y., Yoshioka K., Suga S., Chem. Rev. 2018, 118, 5985–5999; [DOI] [PubMed] [Google Scholar]
- 14f. Waldvogel S. R., Lips S., Selt M., Riehl B., Kampf C. J., Chem. Rev. 2018, 118, 6706–6765; [DOI] [PubMed] [Google Scholar]
- 14g. Yuan Y., Lei A., Acc. Chem. Res. 2019, 52, 3309–3324. [DOI] [PubMed] [Google Scholar]
- 15. Nitelet A., Thevenet D., Schiavi B., Hardouin C., Fournier J., Tamion R., Pannecoucke X., Jubault P., Poisson T., Chem. Eur. J. 2019, 25, 3262–3266. [DOI] [PubMed] [Google Scholar]
- 16. Zhang Y., Zhao X., Bi C., Lu W., Song M., Wang D., Qing G., Zhang Y., Green Chem. 2021, 23, 1691–1699. [Google Scholar]
- 17.For a review, see:
- 17a. Kuang Z., Yang K., Zhou Y., Song Q., Chem. Commun. 2020, 56, 6469–6479; [DOI] [PubMed] [Google Scholar]
- 17b. Bonet A., Pubill-Ulldemolins C., Bo C., Gulyás H., Fernandez E., Angew. Chem. Int. Ed. 2011, 50, 7158–7161; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 7296–7299; [Google Scholar]
- 17c. Hong S., Zhang W., Liu M., Yao Z.-J., Deng W., Tetrahedron Lett. 2016, 57, 1–4; [Google Scholar]
- 17d. Deng C. M., Ma Y. F., Wen Y. M., ChemistrySelect 2018, 3, 1202–1204. [Google Scholar]
- 18. Pitzer L., Schäfers F., Glorius F., Angew. Chem. Int. Ed. 2019, 58, 8572–8576; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 8660–8664. [Google Scholar]
- 19.See Supporting Information for details.
- 20.The presence of the α-isomer has not been so far explained and is in contrast to the previous reports of hydroboration reaction of alkyne with boryl radicals, see:
- 20a. Shimoi M., Watanabe T., Maeda K., Curran D. P., Taniguchi T., Angew. Chem. Int. Ed. 2018, 57, 9485–9490; [DOI] [PubMed] [Google Scholar]
- 20b. Zhong M., Gagné Y., Hope T., Pannecoucke X., Frenette M., Jubault P., Poisson T., Angew. Chem. Int. Ed. 2021, 10.1002/anie.202101874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.For selected examples of electrochemical oxidation of borates, see:
- 21a. Geske D. H., J. Phys. Chem. 1959, 63, 1062–1070; [Google Scholar]
- 21b. Geske D. H., J. Phys. Chem. 1962, 66, 1743–1744; [Google Scholar]
- 21c. Beil S. B., Möhle S., Enders P., Waldvogel S. R., Chem. Commun. 2018, 54, 6128–6131; [DOI] [PubMed] [Google Scholar]
- 21d. Music A., Baumann A. N., Spieß P., Plantefol A., Jagau T. C., Didier D., J. Am. Chem. Soc. 2020, 142, 4341–4348; [DOI] [PubMed] [Google Scholar]
- 21e. Baumann A. N., Music A., Dechent J., Müller N., Jagau T. C., Didier D., Chem. Eur. J. 2020, 26, 8382–8387; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21f. Yan H., Chen M., Zhao D., Xu J., Li C., Lu C., Angew. Chem. Int. Ed. 2021, 60, 7838–7844; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 7917–7923. [Google Scholar]
- 22.H-CH2OH, BDE=96 kcal.mol−1 and CH3O−H BDE=104 kcal.mol−1.
- 23.The reduction of the vinyl radical D into the corresponding vinyl anion followed by protonation to afford the hydroborated product is unlikely, since no reduction has been witnessed during the CV measurement with all reaction partners, see Scheme 3B.
- 24. Boyle W. J., Bunnett J. F., J. Am. Chem. Soc. 1974, 96, 1418–1422. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary