

Abstract
Background and Purpose
Inflammation associated with the tumour microenvironment (TME) is critical for cancer development, and immunotherapeutic strategies modulating the immune response in cancer have been crucial. In this study, a methotrexate‐loaded (MTX) poly(lactic‐co‐glycolic acid)‐based (PLGA) drug nanocarrier covered with polyethyleneimine (Pei) and hyaluronic acid (HA) was developed and combined with an PD‐L1 antibody to investigate anti‐cancer and immunomodulatory effects in breast cancer TME.
Experimental Approach
Naked or HA‐coated PeiPLGA‐MTX nanoparticles (NPs) were assessed on 4T1 breast cancer cells grown in culture and in a mouse model of orthotopic tumour growth. Tumours were evaluated by qRT‐PCR and immunohistochemistry. The cell death profile and cell migration were analysed in vitro in 4T1 cells. Polarization of murine macrophages (RAW cells) was also carried out.
Key Results
Naked or HA‐coated PeiPLGA‐MTX NPs used alone or combined with PD‐L1 antibody modified the tumourigenic course by TME immunomodulation, leading to reduction of primary tumour size and metastases. STAT3 and NF‐κB were the major genes downregulated by NPs. In tumor‐associated macrophages (TAM) such regulation switched M2 phenotype (CD163) towards M1 (CD68) and reduced levels of IL‐10, TGF‐β and CCL22. Moreover, malignant cells showed overexpression of FADD, APAF‐1, caspase‐3 and E‐cadherin, and decreased expression of Bcl‐2, MDR‐1, survivin, vimentin, CXCR4 and PD‐L1 after treatment with NPs.
Conclusion and Implications
NPs‐mediated STAT3/NF‐κB signalling axis suppression disrupted crosstalk between immune and malignant cells, reducing immunosuppression and critical pro‐tumour events. These findings provide a promising therapeutic approach capable of guiding the immune TME to suppress the development of breast cancer.
Keywords: Immunomodulation, M2‐like macrophages, NF‐κB, PD‐L1 antibody, PLGA nanoparticles, STAT3, tumour microenvironment
Abbreviations
- EMT
epithelial–mesenchymal transition
- HA
hyaluronic acid
- MTX
methotrexate
- Pei
polyethyleneimine
- TAM
tumour‐associated macrophage
- TME
tumour microenvironment
What is already known
NF‐κB and STAT3 are constitutively expressed in immune and malignant cells and promote tumourigenesis.
M2‐like tumour‐associated macrophages favour an immunosuppressive tumour microenvironment and establishment of the epithelial–mesenchymal transition.
What this study adds
Nanoparticles containing methotrexate mediate STAT3/NF‐κB downregulation, disrupting crosstalk between immune and cancer cells.
Suppression of IL‐10/STAT3/NF‐κB signalling in M2‐like tumour‐associated macrophages blocked development of the immunosuppressive tumour microenvironment.
What is the clinical significance
Our results provide a better understanding of immunosuppressive mechanisms for effective development of immunomodulatory therapies.
They also suggest combining new immunotherapeutic strategies to treat triple negative breast cancer.
1. INTRODUCTION
Tumour‐associated inflammation is an emerging hallmark of cancer (Fouad & Aanei, 2017). The role played by the immune system in tumourigenesis has become widely recognized over the years (Lin et al., 2018). According to the cellular and molecular immune pattern produced in the tumour microenvironment (TME), immune cells develop an antagonistic action that not only protects the host but also prevents tumour growth by direct elimination or inhibition of malignant cell growth. Immune cells often paradoxically promote cancer development and progression through selecting tumour cells to escape from immunosurveillance, or by establishing immunosuppression in the TME, which favours malignant cell growth and spread (Criscitiello et al., 2014). Such a process is known as “cancer immunoediting” and has been separated into three phases, namely, elimination, equilibrium, and escape (Tower, Ruppert, & Britt, 2019).
The establishment of immune cell‐mediated pro‐tumour inflammatory conditions is complex and mechanistically involves several cellular interactions, as well as the activation of various intracellular signalling pathways by many types of chemical mediators. Among these, transcription factors such as NF‐κB and signal transducer activator of transcription‐3 (STAT3) have been particularly involved (Colotta, Allavena, Sica, Garlanda, & Mantovani, 2009). NF‐κB and STAT3 are constitutively expressed in both immune and malignant cells and are important mediators of immunosuppressive inflammatory response in the TME and the tumour immune escape process, as well as controlling the expression of critical genes for tumourigenesis such as anti‐apoptotic, pro‐proliferative, survival, angiogenesis, and tissue remodelling genes (Grivennikov & Karin, 2010).
The immune cell‐mediated inflammatory TME is associated with effective tumour cell‐induced leukocyte recruitment through the release of chemoattractants, via NF‐κB and STAT3 activation. Resident macrophage mobilization and circulating monocyte recruitment support the accumulationof tumour‐associated macrophages (TAMs), which comprise the most abundant and important inflammatory cell population in the TME (Qiu et al., 2018). Influenced by malignant cells through the coordination of NF‐κB and STAT3, tumour‐associated immune cells, mainly TAMs, are often conditioned to change their cancer‐fighting phenotypes towards a cooperative tumour development profile (Szala, Jarosz‐Biej, Cichoń, Smolarczyk, & Sochanik, 2015). In this context, the majority of TAMs are activated to present an immunosuppressive M2 phenotype that secretes IL‐10 (J. Wang, Li, Cang, & Guo, 2019).
As a result of the numerous chemical mediators released by M2‐like TAMs, an immunosuppressive TME is established promoting optimal conditions for cancer development and spread. In this context, the establishment of the epithelial–mesenchymal transition (EMT) process, one major mechanisms of metastasis in solid tumours whose main characteristic is the loss of E‐cadherin expression, is likely to be favoured in the immunosuppressive TME (Huang, Cao, & Tang, 2017). Meanwhile, immunosuppressive M2‐like TAMs may also favour immunosurveillance tumour escape through the expression of the immune checkpoint protein, programmed cell death‐1 ligand 1 (PD‐L1) (Kitamura, Qian, & Pollard, 2015).
Despite considerable effort and successive improvements in anti‐cancer therapy, the management of breast cancer is still challenging and much remains to be done. Therefore, immune‐based therapies that aim to affect the development of immunosuppression in the TME, as well as reducing tumour escape from immunosurveillance, and the use of immune checkpoint inhibitors have been extensively investigated in recent years.
Here we describe the development of a drug delivery system based on poly(lactic‐co‐glycolic acid) (PLGA) polymeric nanoparticles (NPs) carrying methotrexate (MTX), a drug known to exert anti‐neoplastic and anti‐inflammatory effects (Nedelcu et al., 2019; Ochaion et al., 2006). However, its failure to differentiate healthy from malignant cells can lead to systemic toxicity and severe side effects. Thus, in order to improve the MTX delivery efficiency towards malignant breast cells, which overexpress hyaluronic acid (HA) receptors (CD44) (S. Wang, Zhang, Wang, & Chen, 2016), the MTX‐loaded PLGA NPs were coated with polyethyleneimine (Pei) followed by HA externally. Thus, an anti‐PD‐L1 immune checkpoint inhibitor was used in combination with HA‐coated PeiPLGA‐MTX NPs in order to target tumour escape. The effects of such therapeutic approaches on tumourigenesis and immunomodulation of the microenvironment in breast cancer were analysed, using cultures of a murine mammary carcinoma cell (4T1 cells) and and orthotopic injection in mice.
2. METHODS
2.1. Cell cultures
Human embryonic kidney epithelial cell (HEK‐293) [RRID:CVCL_0045], murine mammary carcinoma cell (4T1) [RRID:CVCL_0125], and murine macrophage (RAW 264.7) [RRID:CVCL_0493] were purchased from Banco de Células do Rio de Janeiro (BCRJ, Brazil). Cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 0.15% penicillin and streptomycin (10,000 U·ml−1) (Gibco) in a 5% CO2 humid atmosphere at 37°C. The culture medium replaced every 3 days, and trypsin/EDTA (Gibco) was used to remove the cells, when necessary.
2.2. Animals
All animal care and experimental procedures were approved by the Ethics Committee of the Federal University of Rio Grande do Norte (number 063/2016). Animal studies are reported in compliance with the ARRIVE guidelines (Percie du Sert et al., 2020) and with the recommendations made by the British Journal of Pharmacology (Lilley et al., 2020).
Female BALB/c mice [RRID:IMSR_CRL:028] approximately 7–9 weeks old weighing between 21 and 28 g were used for experimental orthotopic breast cancer model and toxicity studies. In this lineage, tumours from 4T1 cell injection are well developed and immunologically tolerated. Animals were purchased from the Keizo Asami Immunology Laboratory (Fiocruz) and treated according to the ethical principles for animal experimentation. Up to five mice were housed in individually ventilated cages (with top filter‐isolator) at 22 °C with an exhaust having a capacity of 30 air exchanges per hour, 12 h light/dark cycle, and with water and food freely accessible. For both experimental models, the mice were blindly and randomly distributed in cages by a different assistant (UI) from the experimenter (RC) for group composition (n = 6). Therapeutic and surgical interventions were preceded by anaesthesia with i.p. xylazine (10 mg kg−1) and ketamine (100 mg kg−1). The animals were killed by cervical dislocation preceded by anaesthesia.
2.3. Preparation and characterization of PeiPLGA‐based NPs
2.3.1. Preparation of NPs
PLGA NPs were prepared using an adjusted solvent displacement method (dos‐Santos‐Silva et al., 2020). Briefly, a solution of PLGA (MW: ~44 kDa; 50:50 lactide:glycolide monomer composition; 63 ml·g−1 at 30°C), final concentration 0.75% w/v, and MTX, final concentration 0.075% w/v, was prepared in 6 ml of acetone:1‐methyl‐2‐pyrrolidone solution (50:50 v/v; Labsynth, Cat#01M1009.01.BJ) to obtain the organic phase at a 1:10 (w/w) MTX:PLGA ratio. This solution was later added dropwise into 14 ml of aqueous phase containing 0.5% w/v poloxamer 407 surfactant (Pluronic F127, MW: 12.6 kDa) and hyperbranched polyethyleneimine (Pei) (1:50 w/w Pei:PLGA; MW: 25 kDa) at an output flux of 1.0 ml·min−1, under 720 rpm of magnetic stirring, which was left overnight for solvent evaporation. The samples were hermetically sealed in glass flasks and stored at 25°C. All the PLGA empty NPs did not contain MTX. All data on the characterisation of the NPs are presented as mean ± SD of five independent assays with at least three replicates.
2.3.2. Preparation of hyaluronic acid‐functionalized PeiPLGA NPs
MTX‐loaded PeiPLGA NPs and empty PeiPLGA NPs were further functionalized with hyaluronic acid by adsorption (S. Wang, Zhang, Wang, & Chen, 2016). For this, hyaluronic acid (HA) was dissolved in a colloidal solution of NPs at a concentration of 2 mg·ml−1 and mixed for 1 h at room temperature. Then, HA‐PeiPLGA and HA‐PeiPLGA‐MTX NPs were filtered (pore size, 0.45 μm) and stored at 4°C.
2.3.3. Drug loading efficiency and efficiency encapsulation
The MTX loading was assessed using the indirect method, in which dispersions were centrifuged at 16.9 x g for 60 min, at 4°C using the ultra‐centrifugal filter (Sartorius®, Vivaspin 2, Ultra‐15 MWCO 10 kDa). The filtered supernatant was diluted in purified water 1:20 (v/v) and measured at 303 nm, using a UV Thermo Fisher Scientific, 60S Evolution. The drug concentration was determined according to a previously validated method, and the drug loading efficiency (DL) was calculated as follows: DL% = [(Total mass of drug added − Free drug mass)/Mass of the nanoparticle] × 100. The encapsulation efficiency (EE) was indirectly calculated from the equation: EE% = [(Total added drug − Drug determined in the supernatant)/Total drug] × 100.
2.3.4. Physicochemical aspects and colloidal stability
The measurements of mean diameter and particle size distribution were performed by dynamic light scattering in a ZetaPlus (Brookhaven Instruments), equipped with a 90Plus/BI‐MAS apparatus, at a wavelength of 659 nm with a scattering angle of 90°. Zeta potential of NPs was measured by laser Doppler anemometry using the same equipment. All analyses were performed at 25°C.
To investigate the colloidal stability of the NPs, the different formulations ‐ empty PeiPLGA, HA‐PeiPLGA, PeiPLGA‐MTX, and HA‐PeiPLGA‐MTX ‐ were stored for 6 weeks in hermetically sealed vials and conditioned at 5°C. Each day, the samples were collected and evaluated for diameter size. Measurements were performed at 25°C using dynamic light scattering method. The samples containing HA had the substance added before the measurements. Experimental values are shown as the mean ± SD of five independent assays with at least three replicates.
2.4. Cell viability
Cell viability was estimated by the resazurin reduction method. Four thousand cells were cultured in each well of 96‐well plate in DMEM with 10% FBS for 24 h. HEK‐293 and 4T1 cells were treated with different concentrations of empty or MTX‐loaded (0–80 μg·ml−1), PeiPLGA NPs (0–934 μg·ml−1), and naked or HA‐coated (0–584 μg·ml−1). After 24 h, 10 μl of resazurin solution (0.04%; Sigma‐Aldrich, Cat# R7017) was added to each well and the plate incubated again for 4 h. The absorbance was measured at 570 and 600 nm, whose values were applied in the equation [% of resazurin reduction = [AT570 − (AT600 × CF)]/[AC570 − (AC600 × CF)] × 100}, where AT570 and AT600 are absorbances from the treated group, AC570 and AC600 are absorbances from the negative control group, and CF is the correction factor. Results were normalized to express percentage values, so that negative control values were set to 100%. For the following in vitro experiments, the concentrations of each component used in the treatments were as follows: HA, 0.73 μg·ml−1, PeiPLGA, 1.17 μg·ml−1 and MTX, 0.1 μg·ml−1.
2.5. Cell uptake of NPs
The 4T1 cells were seeded (2 × 104 cells per well) on glass coverslips (12 mm) in DMEM with 10% FBS. After adhesion, the cells were treated for 4 h at 37°C with naked or HA‐coated PeiPLGA‐MTX NPs previously adsorbed with 10 μl propidium iodide (PI, 100 μg·ml−1; Invitrogen, Cat#P1304MP) under agitation for 12 h. Then, the cells were washed and fixed with PBS and 4% buffered paraformaldehyde (PFA; Sigma‐Aldrich Cat#158127), respectively. The coverslips were removed and mounted on a glass slide over DAPI‐containing mounting medium (Abcam, Cat#ab104139) and then analysed by fluorescence microscopy (Axio Observer Z1, Zeiss).
2.6. Macrophage polarization towards M2
The polarization of RAW cells towards the M2 phenotype was carried out as described by de Araujo Junior et al. (2020). Briefly, 5 × 105 RAW cells were cultured in DMEM supplemented with 10% FBS and IL‐4 (20 ng·ml−1; PeproTech, Cat#214‐14). After 24 h, cells were washed with FBS‐free DMEM and maintained in FBS‐free DMEM supplemented with IL‐4 for 48 h. The cells were used as M2‐polarized TAMs, and the collected supernatant was used in other experiments as M2‐polarized TAMs enriched medium.
2.7. M2‐polarized TAM‐induced 4T1 cell migration
The TAMs‐induced 4T1 malignant cells promigratory potential was observed by indirect co‐culture with M2‐polarized RAW cells. In a 12‐well plate, 5 × 104 M2‐polarized RAW cells per well were cultured in DMEM with 10% FBS. At the same time, 4T1 cells (1 × 104 cells per insert) were added to the Transwell insert chamber (8 μm pore size; Corning) in DMEM with 10% FBS. After 24 h, 4T1 cells were treated with HA‐PeiPLGA NPs in the absence or presence of IL‐10 receptor antibody (1.2 μg·ml−1; Invitrogen, Cat#PA5‐109852, RRID:AB_2855263) and maintained for another 48 h. Then, 4T1 cells on the inner surface of the insert were washed with PBS and removed using a swab. Migratory 4T1 cells outside the membrane were fixed with 4% PFA and rested under glass slide DAPI‐containing mounting medium (Abcam). The cells were observed by fluorescence microscopy (Zeiss) and the images analysed using ImageJ software (NIH, RRID:SCR_003070).
2.8. Wound healing assay
The wound healing test was performed as described by Tanaka et al. (2009). Briefly, in a 12‐well plate, a north/south scar was performed in a 4T1 cell monolayer using a tip (200 μl). After successive washes with PBS and proliferation inhibition with Mitomycin C (10 μg·ml−1; Fisher Scientific, Cat#BP25312), the cells were treated with HA‐PeiPLGA NPs in the absence or presence of IL‐10 receptor antibody (1.2 μg·ml−1) in FBS‐free DMEM and enriched culture medium (M2‐polarized TAMs supernatant) at a 1:1 ratio. After 24 h, images were collected using Olympus IX70 light microscope (Olympus) equipped with a Leica DFC340 FX digital camera (Leica).
2.9. Enzyme‐linked immunosorbent assay
The supernatant from RAW cultures, previously treated for 48 h with IL‐4 (20 ng·ml−1) and HA‐PeiPLGA NPs in the absence or presence of IL‐10 receptor antibody (1.2 μg·ml−1), was collected and the levels of murine IL‐10 and IL‐12 measured using a LEGEND MAX™ Mouse IL‐10/IL‐12 ELISA Kit (BioLegend, Cat#430607 and Cat#430707, respectively) according to the manufacturer's instructions. Five independent assays with at least three replicates were performed for each experiment.
2.10. Flow cytometry
Apoptotic 4T1 cells were analysed from Annexin V/FITC and PI double labelling. Briefly, 2 × 104 cells/well were seeded in a six‐well plate containing DMEM with 10% FBS. After adhesion, the cells were treated with naked or HA‐coated PeiPLGA‐MTX NPs or MTX for 24 h. The cells were double labelled with Annexin V/FITC and PI (Invitrogen, Cat#V13242) according to the manufacturer's instructions and analysed by flow cytometer FACS Calibur (BD Biosciences).
The M2‐polarized TAMs evaluation was performed on RAW cells cultured previously as described above. The cells were treated with IL‐4 (20 ng·ml−1) and HA‐PeiPLGA in the absence or presence of IL‐10 receptor antibody (1.2 μg·ml−1) for 48 h. RAW cells were harvested and blocked with 0.5% BSA/PBS for 45 min and then labelled with anti‐CD163‐PerCP (1:1000; eBioscience, Cat#46‐1631‐82, RRID:AB_2716956) or anti‐CD68‐FITC (1:1000; Invitrogen, Cat#MA5‐16676, RRID:AB_2538170). For intracellular IL‐10 labelling, the cells were fixed in 4% PFA, washed, and permeabilized with 0.2% TritonX‐100 (Sigma‐Aldrich, Cat#T8787). The cells were incubated with blocking solution (0.5% BSA/PBS) for 45 min and subsequently incubated with anti‐IL10‐FITC antibody (1:1000; eBioscience, Cat#11‐7101‐82, RRID:AB_465403) for 60 min at 4°C. The cells were protected from light until analysed by flow cytometer FACSCalibur (BD Biosciences, USA). FlowJo software (NIH, RRID:SCR_008520) was used for data analysis.
2.11. Immunofluorescence
4T1 cells were seeded (2 × 104 cells per well) on glass coverslips (12 mm) in DMEM with 10% FBS. For apoptosis evaluation, 4T1 cells were treated with naked or HA‐coated PeiPLGA‐MTX for 24 h. HA‐PeiPLGA and IL‐10 receptor antibody (1.2 μg·ml−1) in FBS‐free DMEM and enriched medium (M2‐polarized TAMs supernatant) (1:1 ratio) were used for EMT evaluation for 48 h. The cells were washed, fixed, and permeabilized as previously described and incubated with anti‐Bcl‐2 (1:200; Abcam, Cat#ab59348, RRID:AB_2064155), anti‐caspase‐3 (1:200; Invitrogen, Cat#43‐7800, RRID:AB_2533540), anti‐Fadd (1:200; Abcam, Cat# ab24533, RRID:AB_448126), anti‐Apaf‐1 (1:200; Abcam, Cat# ab2001, RRID:AB_302753), anti‐vimentin (1:100; Invitrogen, Cat# MA5‐16409, RRID:AB_2537928), and anti‐E‐cadherin (1:100; Invitrogen, Cat# MA5‐12547, RRID:AB_10982676) antibodies overnight in a humid chamber. Antibodies were diluted in Diamond antibody diluent (Cell Marque, Cat#938B‐09). Primary antibodies were captured with Alexa Fluor 488 anti‐rabbit (Invitrogen, Cat# A32731, RRID:AB_2633280) or anti‐mouse (Invitrogen, Cat# A32723, RRID:AB_2633275), and Alexa Fluor 555 anti‐rabbit (Invitrogen, Cat# A‐21428, RRID:AB_141784) or anti‐mouse (Invitrogen, Cat# A‐21422, RRID:AB_141822) secondary antibodies. After washes, the glass coverslips were removed and rested on mounting medium containing DAPI (Abcam) on glass slides for labelling the cores. The cells were analysed under a fluorescence microscope (Zeiss), and digital images were collected to determine the mean fluorescence intensity using Black ZEN software (Zeiss, RRID:SCR_018163). Five independent assays with at least three replicates were performed for each experiment.
2.12. NPs toxicity in vivo
A similar orthotopic tumour growth model (Liu et al., 2015) was adapted and used to investigate the toxicity of NPs. BALB/c mice were randomized into six groups with n = 6 each; 24 h later, the animals were treated i.p. with 100 μl of sterile saline (negative control group), MTX (0.1 mg·kg−1, positive control), empty or MTX‐loaded PeiPLGA NPs, naked or HA‐coated (HA 0.73 mg·kg−1, PeiPLGA 1.17 mg·kg−1, and MTX 0.1 mg·kg−1; test groups) after previous anaesthesia with xylazine and ketamine solution (2:8 ratio). The drugs were administered again every 5 days (three treatments). On the 15th day, the animals were killed by cervical dislocation and had blood, liver, and lungs collected for systemic toxicity analysis by serum biochemical measurements (liver and renal toxicity indicators) and histopathological analysis by H&E staining, respectively. A double‐blind histopathological analysis was done by non‐operator analysts (RA & RC). Three histological sections per animal tissue (n = 6) were analysed in each group.
2.13. Orthotopic tumour growth
Orthotopic breast tumour growth in an allographic model was performed as described (Paschall & Liu, 2016) with adaptations. 4T1 cells were inoculated (1 × 104 cells per 100 μl FBS‐free DMEM) immediately below the fourth left breast in previously anaesthetized BALB/c mice (xylazine and ketamine ratio 2:8). Then, mice were randomized into six groups with n = 6 animals each and tumour growth monitored daily. When tumours had reached 3 mm in diameter, the mice were treated peritumourally with sterile saline (negative control group), MTX (0.1 mg·kg−1; positive control), PeiPLGA‐MTX (1.17: 0.1 mg·kg−1), HA‐PeiPLGA‐MTX (0.73: 1.17: 0.1 mg·kg−1), and the PD‐L1 antibody (2.16 mg·kg−1, administered 30 min before) followed by HA‐PeiPLGA‐MTX. Treatments were repeated every 5 days (three treatments). PD‐L1 antibody was administered in a single dose in the first treatment, with the others being administered only HA‐PeiPLGA‐MTX. On the 21st day, the animals were anaesthetized and killed by cervical dislocation and the blood (from the heart), tumour, lungs, and liver collected for biochemical and histopathological analysis. Results were expressed as a growth curve from the average tumour volume (volume mm3 = (width × length2) × 0.52). Metastatic niches in the liver and lung were assessed semiquantitatively as described (Van den Eynden et al., 2012). Scores representing percentage of tumour cells in the tissue parenchyma were applied (1, <5%; 2, 5% to 25%; 3, 26% to 50%; 4, 51% to 75%; 5, >75%). The scores were applied to histological images captured in 20 random fields of all the extension the organs. Histopathological analysis of tumour tissue, liver, and lungs were independently assessed by two non‐operator analysts (RA and RC). Three histological sections per animal tissue (n = 6) were analysed in each group.
2.14. Biochemical and haematological analysis
Biochemical and haematological assessments were performed using whole blood collected with EDTA, and standard techniques were performed manually. Erythrocyte and leukocyte count, as well as haemoglobin quantification and haematocrit determination were observed. AST (Cat#MS80022230083), ALT (Cat#MS80022230086), creatinine (Cat#MS80022230143), and urea (Cat#MS80022230075) levels were evaluated according to the manufacturer's instructions (Gold Analisa). Blood samples from all animals in each group (n = 6) were analysed in triplicate.
2.15. qRT‐PCR
Gene expression analysis was performed on 4T1 cells and partial orthotopic tumour from BALB/c. 4T1 cells were cultured in DMEM with 10% FBS (2 × 105 cells per 100 mm plate) and subsequently treated with naked or HA‐coated PeiPLGA‐MTX NPs and MTX for 24 h. The total RNA of both 4T1 cells and orthotopic tumour were obtained with Trizol reagent (Invitrogen, Cat#15596026) and purified with SV Total RNA Isolation System (Promega, Cat#Z3105) according to the manufacturer's instructions. Then, cDNA was synthesized using high‐capacity RNA‐to‐cDNA kit (Applied Biosystems, Cat#4387406). PowerUp SYBR Green Master Mix (Applied Biosystems, Cat#A25742) was used for real‐time amplification. The used forward and reverse primers sequences (Thermo Fisher Scientific) are listed in Table S1. The experiments were performed in triplicate. Gene expression data were normalized relative to the housekeeping gene β‐actin using the 2−ΔΔCt. Five independent assays with at least two replicates were performed for the 4T1 experiment. All animals in each group (n = 6) were analysed in duplicate.
2.16. Immunohistochemistry
The immunohistochemistry of partial orthotopic tumour from BALB/c mice was performed as described (Araujo Junior et al., 2016) in accordance with the BJP immunohistochemistry guide (Alexander et al., 2018). Briefly, after deparaffinization, rehydration, and antigenic recovery (11 μM sodium citrate solution at 90°C for 30 min) steps, the paraffin‐embedded tumour tissue sections were incubated overnight at 4°C with anti‐Bcl‐2 (Abcam, Cat#ab59348, RRID:AB_2064155), anti‐caspase‐3 (Invitrogen, Cat#43‐7800, RRID:AB_2533540), anti‐Fadd (Abcam, Cat# ab24533, RRID:AB_448126), anti‐Apaf‐1 (Abcam, Cat# ab2001, RRID:AB_302753), anti‐NF‐κB (Santa Cruz Biotechnology, Cat#sc‐8008, RRID:AB_628017), anti‐STAT3 (Santa Cruz Biotechnology, Cat#sc‐8019, RRID:AB_628293), anti‐CD163 (Proteintech, Cat#16646‐1‐AP, RRID:AB_2756528), anti‐CD25 (Invitrogen, Cat#MA5‐12680, RRID:AB_10979943), anti‐TGF‐β (Santa Cruz Biotechnology Cat#sc‐17799, RRID:AB_628348), anti‐E‐cadherin (Invitrogen, Cat#MA5‐12547, RRID:AB_10982676), anti‐IL‐10 (Invitrogen, Cat# PA5‐85660, RRID:AB_2792799), and anti‐PD‐L1 (Proteintech, Cat#66248‐1‐Ig, RRID:AB_2756526) primary antibodies (1:300). Biotinylated pan‐specific universal secondary antibody (Vector, Cat#PK‐7800) followed by streptavidin/HRP‐conjugated (Vector, Cat#PK‐7800) incubation and diaminobenzidine (DAB; DAKO, Cat#GV825) were used to reveal the markings. Sections were counterstained with haematoxylin and analysed under a Nikon E200 LED light microscope (Minato) coupled to a digital camera (Moticam). For immunoreactivity classification, the product between estimated percentage of positively stained cells (from undetectable, equivalent to 0%, to completely stained, 100%) and estimated intensity of positive cells immunostaining (0, negative; 1, weak; 2, moderate; and 3, strong staining) was calculated according to Charafe‐Jauffret et al. (2004). Immunohistochemical analyses of tumour tissues were independently assessed by two non‐operator analysts (RA and RC). For each tumour tissue (n = 6 per group), three histological sections were evaluated for each antibody.
2.17. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). All experiments in vitro were presented as mean ± SD of five independent assays with at least three replicates. The in vitro experiments were analysed by two authors (RA & AA) in a randomised and double‐blind manner. In vivo data (tumour growth) as well as ex vivo data (histopathology/immunohistochemistry) were analysed in a double‐blind manner by RA & AA and RC & RA, respectively. Experiments were designed to obtain randomized groups of equal sizes. Independent values with less than 20% variance were chosen to compose the groups. Statistical analysis was only undertaken for experiments whose group size was n ≥ 5 independent values. No data were removed, and all outliers were included in data analysis. All analyses were performed using Prism 6.01 software (GraphPad Software, RRID:SCR_002798) and SPSS Statistics 20 (IBM Corp., RRID:SCR_002865). Data evaluation for normal distribution was assessed using Shapiro–Wilk normality test, and the Levene test was used to verify the homogeneity of variances. Comparison between groups was performed using Mann–Whitney U test for variables with non‐normal distribution, whereas parametric data for pairwise comparison was analysed using the Student t‐test. For multiple comparisons between groups, one‐way ANOVA was used, followed by post hoc Bonferroni tests only if F achieved the necessary level of statistical significance (P < .05) and there is no significant variance inhomogeneity. When necessary, data underwent logarithmic transformation to enable group comparison by ANOVA. Kruskal–Wallis test with Dunn's correction was used for multiple comparison of non‐normal distribution data. P < .05 values were considered indicative of statistical significance. The results of cell viability, by resazurin reduction, and qRT‐PCR analyses were normalized to control for unwanted sources of variation (negative controls values were set to 100%).
2.18. Materials
Methotrexate was supplied by DEG (São Paulo, Brazil) and HA (Cat#600‐01‐01) was supplied by Contipro (Dolní Dobrouč, Czech Rep). Pei (Cat#408727) and Pluronic F127 (Cat#P2443) was supplied by Sigma‐Aldrich (St. Louis, MO, USA). PLGA (Cat#B6010‐2) was supplied by LACTEL Absorbable Polymers (Birmingham, AL, USA). The IL‐10 receptor antibody (Cat#PA5‐109852, RRID:AB_2855263) was supplied by Invitrogen (Carlsbad, CA, USA) and the PD‐L1 antibody (Cat#BE0101) was from Bio X Cell (Lebanon, NH, USA).
2.19. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in the IUPHAR/BPS Guide to Pharmacology (http://www.guidetopharmacology.org) and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander, Fabbro et al., 2019; Alexander, Kelly et al., 2019).
3. RESULTS
3.1. Characterization of NPs
Figure 1 shows the physico‐chemical characterization of NP formulations. Empty PeiPLGA NPs had a size around 100 nm (±0.9), with polydispersity index of 0.117 ± 0.01 and zeta potential of 8.51 ± 3.2 mV (Figure 1b–d). The MTX incorporation of MTX into the NPs (PeiPLGA‐MTX) resulted in a slight increase in size (137.3 ± 1.2 nm) and zeta potential (2.38 ± 0.3 mV). On the other hand, the HA‐coating of empty or MTX‐loaded NPs caused considerable increases in size and zeta potential (230.8 ± 6.3 nm and −24.60 ± 1.1 mV for HA‐PeiPLGA, as well as 617.0 ± 37.7 nm and −30.5 ± 1.5 mV for HA‐PeiPLGA‐MTX, respectively). Despite this, the encapsulation efficiency (EE) of both naked and HA‐coated PeiPLGA‐MTX NPs remained the same at around 41%, and the MTX loading efficiency of NPs was approximately 4.02% (data not shown). In addition, despite the complexity‐dependent changes, the nanoformulations remained stable in terms of size variability over 3 weeks (Figure 1e).
FIGURE 1.
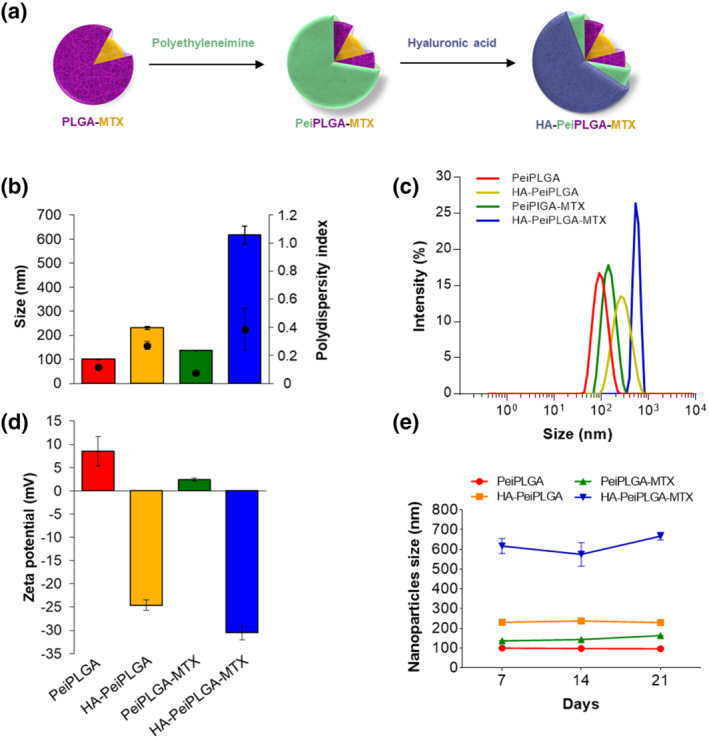
Physicochemical characterization of cationic PLGA nanoparticles (NPs). Schematic representation of PeiPLGA NPs coating steps (a). PLGA nanoparticles were evaluated for size (b) and size distribution (DLS; c), zeta potential (d), as well as size variability over the days of storage (e). Pei, polyethyleneimine; PLGA, poly(lactic‐co‐glycolic acid); MTX, methotrexate; HA, hyaluronic acid. All data are presented as mean ± SD of five independent assays with at least three replicates
3.2. Cell viability
The cytotoxic potential of the NPs is shown in Figure S1. Empty or MTX‐loaded PeiPLGA NPs, naked or HA‐coated, significantly reduced cell viability by up to 100% at the highest concentrations for both evaluated cell lines (Figure S1A–D and G–J). Such effects were more pronounced in 4T1 tumour cells treated with MTX‐loaded PLGA NPs, which had cell viability affected by up to 42% at low concentrations (0.73: 1.17: 0.1 μg·ml−1; Figure S1C,D). Under these same conditions, empty PLGA NPs were not toxic (Figure S1A,B). The PLGA NPs were not cytotoxic at concentrations lower than 116.79 μg·ml−1 (of the polymer) in HEK‐293 cells, except for HA‐PeiPLGA‐MTX, which reduced cell viability by 21% at 7.3:11.68:1 μg·ml−1 (HA:PeiPLGA:MTX; Figure S1J). Free MTX decreased the viability of 4T1 cells by an average of 46% (0.1–80 μg·ml−1), with no significant statistical difference between these concentrations (Figure S1E). On the other hand, MTX did not have a significant cytotoxicity in HEK‐293 cells under the same conditions (Figure S1K). Similarly, free HA did not promote any cytotoxic effect on the evaluated cell lines (Figure S1F,L). Thus, concentrations of MTX (0.1 μg·ml−1) and of PeiPLGA (1.17 μg·ml−1) were the lowest concentrations causing cytotoxic effects in tumour cells (but not in healthy cells), and were selected to be used in subsequent tests.
3.3. Cell uptake of NPs
The incorporation rate of MTX‐loaded PeiPLGA NPs by 4T1 tumour cells was assessed by fluorescence intensity quantification. The results shown in Figure 2 indicate that MTX‐loaded PeiPLGA NPs were internalized by tumour cells after 4 h of treatment. However, HA‐PeiPLGA‐MTX NPs were significantly better internalized (Figure 2g–i) than those without HA.
FIGURE 2.
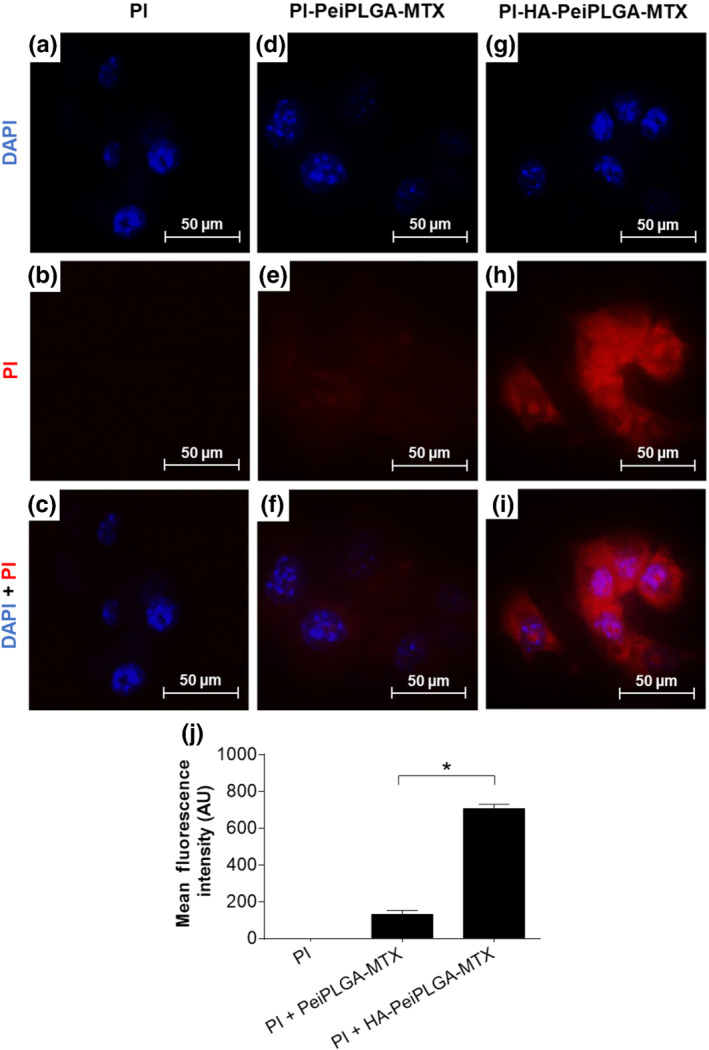
Cellular uptake of PeiPLGA NPs. The naked or HA‐coated PeiPLGA‐MTX NPs PI‐adsorbed were incubated with 4T1 tumour cells for 4 h. The images were captured by fluorescence microscopy (a–i). Graphical representation of mean intensity of fluorescence (j). PI, propidium iodide (red); DAPI (blue). All data are presented as mean ± SD of five independent assays with at least three replicates. *, P < .05, significantly different as indicated; Student's t‐test
3.4. Cell death profile
Flow cytometer analysis revealed that treatment with free MTX increased the apoptotic cell population by 255.3% compared with the DMEM‐treated group, whereas PeiPLGA‐MTX and HA‐PeiPLGA‐MTX increased by 204.7% and 236.2%, respectively. However, there was no statistically significant difference between the three treatments (Figure 3a). The gene expression profile of intrinsic and extrinsic apoptosis pathway revealed that, unlike free MTX, both PeiPLGA‐MTX and HA‐PeiPLGA‐MTX NPs promoted overregulation of APAF‐1 and FADD genes (Figure 3b). Immunofluorescence analysis (Figure 3c) showed a significant increase in Caspase‐3 and FADD proapoptotic proteins expression, as well as decrease of BCL‐2 anti‐apoptotic protein for all treatments compared with DMEM. However, these effects were more pronounced for HA‐PeiPLGA‐MTX. No significant differences were found for APAF‐1 expression after treatments.
FIGURE 3.
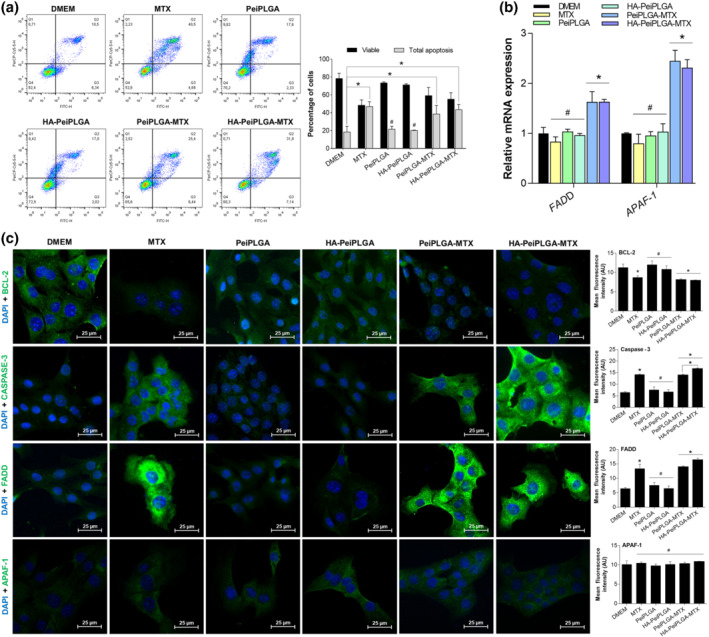
Effect of naked or HA‐coated PeiPLGA‐MTX NPs on apoptosis. The cell death profile was analysed by flow cytometry (a), whose graphical representation expresses the percentage of viable and apoptosis cells. The gene expression profile (b), as well as immunofluorescence immunostaining (c) of key markers in apoptosis were also evaluated. All data are presented as mean ± SD of five independent assays with at least three replicates. *, P < .05, significantly different as indicated; #not significantly different; one‐way ANOVA with post hoc Bonferroni correction or Kruskal–Wallis with post hoc Dunn correction
3.5. RAW cell polarization modulation
Figure 4 shows that population of CD68+ (M1 phenotype indicator) IL‐4‐induced M2‐polarized TAMs decreased by 38% compared with non‐polarized RAW cells. On the other hand, a significant increase in the CD163+ (M2 phenotype indicator) and IL‐10‐positive cell population was observed for IL‐4‐induced M2‐polarized TAMs (190% and 13%, respectively). However, HA‐PeiPLGA NPs, IL‐10 receptor antibody and both combined treatments increased the population of CD68+ M2‐polarized TAMs by 86%, 247%, and 88% (Figure 4a–e) as well as decreasing CD163+ (16% to 23%; Figure 4f–j) and IL‐10+ (6.5% to 21.5%; Figure 4k–o) M2‐polarized TAMs.
FIGURE 4.
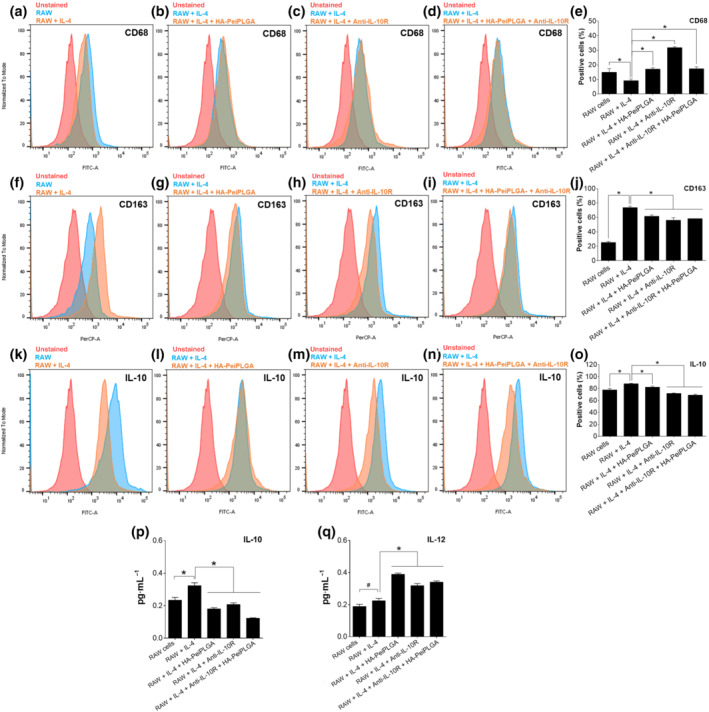
Modulation of M2‐polarized RAW cells by PeiPLGA NPs. After treatment with HA‐coated PeiPLGA NPs and the IL‐10 receptor antibody (anti‐IL‐10R), the IL‐4‐induced M2‐polarized RAW cells were evaluated for labelling for CD68 (a,d), CD163 (b,e), and IL‐10 (c,f) by flow cytometry. The levels of IL‐10 (g) and IL‐12 (h) secreted by RAW cells were also assessed. All data are presented as mean ± SD of five independent assays with at least three replicates. *, P < .05, significantly different as indicated; #not significantly different; one‐way ANOVA with post hoc Bonferroni correction
Cytokine quantification by ELISA revealed an increased level of IL‐10 secreted by IL‐4‐induced M2‐polarized TAMs compared with non‐polarized RAW cells (Figure 4p). On the other hand, HA‐PeiPLGA NPs, IL‐10 receptor antibody and both combined treatments reduced the levels of IL‐10 secreted compared with untreated group (36% to 62%). Low IL‐12 levels were secreted by untreated M2‐polarized TAMs, while IL‐12 secretion was increased after HA‐PeiPLGA NPs, IL‐10 receptor antibody and both combined treatments (42% to 73%, Figure 4q).
3.6. M2‐polarized TAM‐induced migration of4T1 cells
The transwell migration test (Figure 5a) showed that only a few 4T1 tumour cells migrated in the presence of DMEM. However, 4T1 cell migration was significantly enhanced in the RAW cells' presence and mainly M2‐polarized TAMs, suggesting a migratory influence of these on tumour cells. On the other hand, 4T1 cell migration was greatly reduced when treated with HA‐PeiPLGA NPs (77%), IL‐10 receptor antibody (58%), or both combined treatments (84%), even co‐cultured with M2‐polarized TAMs. Similarly, migration of 4T1 cells to fill the scar (over the cell monolayer) was also reduced by treatments. However, this anti‐migratory effect was more pronounced in response to combined treatment (HA‐PeiPLGA + IL‐10 receptor antibody; Figure 5b). Furthermore, the expression of E‐cadherin and vimentin proteins by 4T1 cells fed with M2‐polarized TAMs‐enriched medium was analysed by immunofluorescence. The results presented in Figure 5c show that the constituents present in the M2‐enriched medium in contact with 4T1 tumour cells, decreased expression of E‐cadherin by 46%. The addition of treatments (HA‐PeiPLGA NPs, IL‐10 receptor antibody, and both combined) to M2‐enriched medium considerably increased the E‐cadherin expression in 4T1 tumour cells (439%, 551%, and 863%, respectively), compared with cells with only M2‐enriched medium. On the other hand, under similar conditions, vimentin expression was stimulated in 4T1 tumour cells treated with M2‐enriched medium (96%), whereas HA‐PeiPLGA NPs, IL‐10 receptor antibody, and both combined treatments significantly reduced its expression (68%, 78%, and 80%, respectively), even in the presence of M2‐enriched medium (Figure 5d).
FIGURE 5.
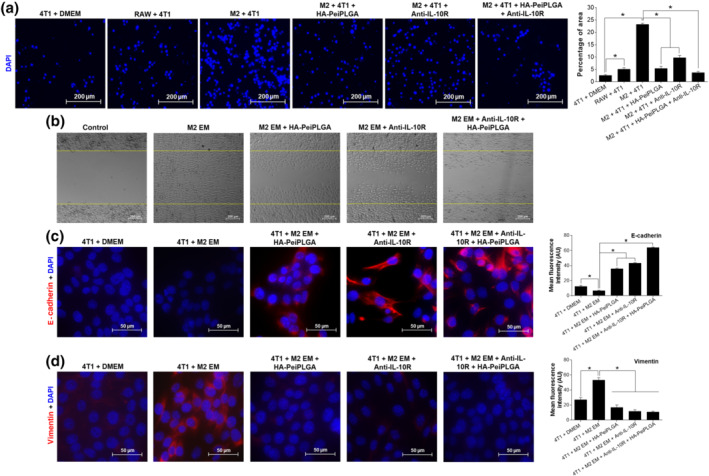
Anti‐migratory effect of HA‐coated PeiPLGA NPs and IL‐10 receptor antibody. The 4T1 tumour cell migration induced by M2‐polarized RAW cells was performed by transwell assay (a) and wound healing assay (b). The control image represents the scar demarcation 5 min after the treatments addition. The expression of E‐cadherin (c) and vimentin (d) adhesion proteins were also evaluated. M2, IL‐4‐induced M2‐polarized macrophages; EM, enriched medium produced by M2 (supernatant). All data are presented as mean ± SD of five independent assays with at least three replicates. * P < .05, significantly different as indicated; one‐way ANOVA with post hoc Bonferroni correction
3.7. In vivo toxicity analysis
The toxicity of the different PeiPLGA formulations were performed prior to allograft tumour growth in female BALB/c mice. The histopathological analysis revealed that none of the PeiPLGA NPs changed the hepatic and pulmonary tissue structures, suggesting that these NO Ps were nontoxic, as used here (Figure S2C–J). On the other hand, free MTX treatment promoted mild hepatocyte vacuolation, followed by mononuclear inflammatory cell infiltrate (arrowhead; Figure S2K), as well as slight interalveolar septa thickening (Figure S2L). No changes in AST/ALT liver enzymes, creatinine, or urea were observed in blood samples from animals treated with different PLGA formulations; however, a subtle increase in AST/ALT was found for MTX alone (Figure S2M‐P).
3.8. Antitumour effect of NPs in vivo
The tumour growth curve reveals that different PeiPLGA formulations (including pretreatment with PD‐L1 antibody) and MTX similarly decreased tumour volume by approximately 45% compared with saline‐treated animals (Figure 6). Except for the saline group, there were no statistically significant differences between treatments.
FIGURE 6.
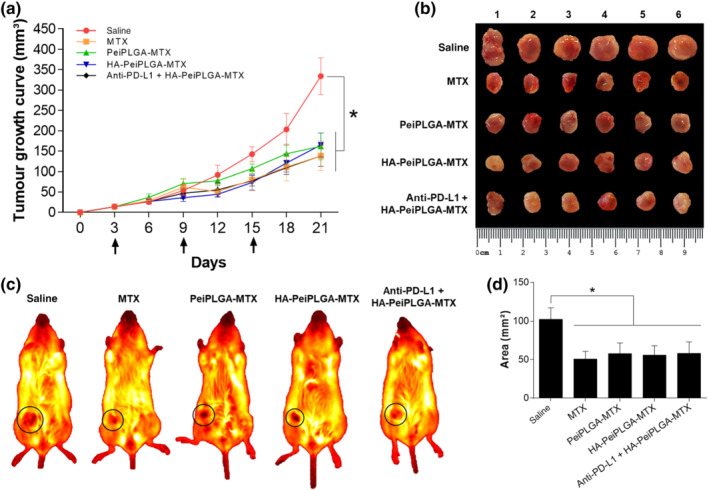
Inhibition of tumour growth. The growth curve of orthotopic breast tumours in BALB/c mice shows a reduction in tumour volumes after treatment with naked and HA‐coated MTX‐loaded PLGA NPs, as well as with free MTX (a). Arrows indicate therapeutic interventions via peritumour administration. The excised tumours were presented on a panel for comparative analysis of tumour size (b). The exact growth area location of orthotopic tumours in mice is illustrated with black circles (c). From these images, the areas occupied by tumours were measured for graphical representation in mm2 (d). All data are presented as mean ± SD of n = 6 per group, * P < .05, significantly different as indicated; one‐way ANOVA with post hoc Bonferroni correction or Kruskal–Wallis with post hoc Dunn correction
The histopathological analysis of liver showed the presence of tumour cells infiltrated throughout the organ, in all groups tested (arrowheads; Figure 7a–e). However, the number and diameter of metastatic niches appear to be smaller when treated with naked or HA‐coated MTX‐loaded PeiPLGA NPs and PD‐L1 antibody pretreatment, than the saline or MTX groups (Figure 7f–j,u). Massive metastatic niches were also found in the lungs of saline‐ and MTX‐treated animals (Figure 7k–o). On the other hand, the MTX‐loaded PeiPLGA NPs, with or without PD‐L1 antibody pretreatment, clearly reduced metastases in lungs (Figure 7k–u).
FIGURE 7.
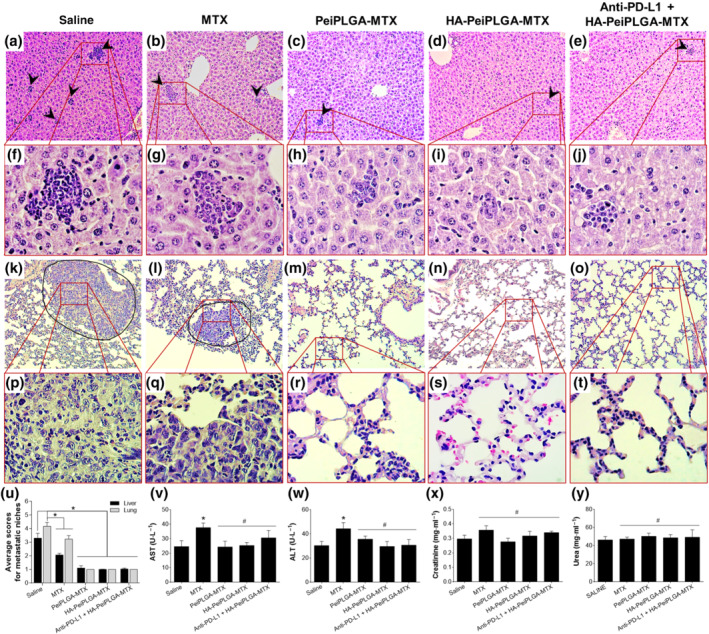
Evaluation of metastatic niches. The evaluation of tumour cell migration to secondary sites was observed in the liver (a–e, 100×, and f–j, 400×) and lung (k–o, 100×, and p–t, 400×). The arrowheads indicate the presence of metastatic niches (a–e), which are best seen in greater magnification (f–j). The surrounding areas (k,l) show tumour masses in the lungs, observed in greater detail in (p) and (q). Metastatic niches in the liver and lung were assessed semiquantitatively (u) by applying scores representing the tumour cells percentage in the tissue parenchyma. Biochemical analyses were carried out in addition to histopathological evaluation (v–y). All data are presented as mean ± SD of six independent values (n = 6) per group or five independent assays with at least two replicates (biochemical analyses). * P < .05, significantly different as indicated; #not significantly different; one‐way ANOVA with post hoc Bonferroni correction or Kruskal–Wallis with post hoc Dunn correction
No other morphological changes were found in any organs or treatment groups (data not shown). Nevertheless, MTX treatment caused subtle increases in liver AST/ALT enzymes (Figure 7v,w). Moreover, a haematological analysis of the animals did not show an anaemic process initiated by any treatment (data not shown). However, the major finding was the significant total leukocyte increase in all treated groups compared with healthy tumour‐free animals (approximately seven times more, except for the goup receiving treatment with PD‐L1 antibody + HA‐PeiPLGA‐MTX, which was five times more). There was a reversed leukocyte profile with neutrophil increase and lymphocyte decrease in all treated groups including saline, compared with tumour‐free animals (data not shown).
3.9. Gene and protein expression profile in TME
The apoptosis, cell survival, drug resistance, tumour immunology, and EMT pathways in the TME were assessed. Tables S2 and S3 summarize how the different treatments modulated gene and protein expression in relative numbers.
The naked and HA‐coated PeiPLGA‐MTX NPs positively regulated the expression of FADD and APAF‐1 apoptosis‐related genes while downregulating the survival and drug resistance genes Survivin and MDR1, respectively, compared with saline group. Among these genes, only APAF‐1 was upregulated by MTX (Figure 8a–d) as well as its protein product APAF‐1 (Figure 8e). Similarly, both NPs increased the expression of APAF‐1 and FADD proapoptotic proteins approximately three and four times, respectively. Expression of another pro‐apoptotic protein caspase‐3 was also increased with MTX and PeiPLGA NPs treatments (approximately two and three times, respectively), which contrastingly reduced expression of the anti‐apoptotic protein Bcl‐2.
FIGURE 8.
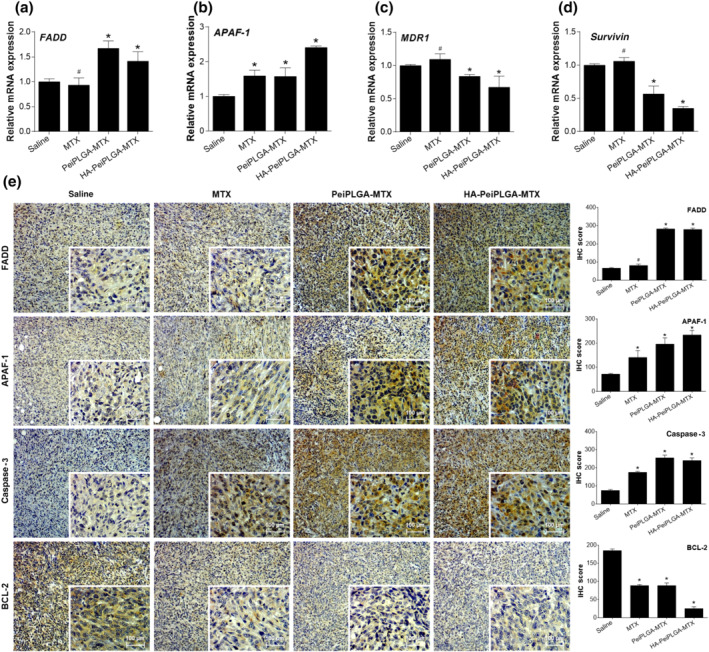
Evaluation of cell death and survival in the TME. The tumours excised from the previously treated female BALB/c were analysed for relative gene expression (a–d) and immunostaining by immunohistochemistry (e). The immunohistological panel presents images with 100× magnification; 400× magnification is shown in the lower right corner of each image. Scores were applied to each image to represent them graphically. All data are presented as mean ± SD of six independent values (n = 6) per group with at least two replicates. * P < .05, significantly different as indicated; #not significantly different; one‐way ANOVA with post hoc Bonferroni correction or Kruskal–Wallis with post hoc Dunn correction
The expression of genes coding for transcription factors (NF‐κB and STAT3) as well as EMT and metastasis regulators (SNAIL and CXCR4) in the TME were markedly downregulated by PeiPLGA‐MTX and HA‐PeiPLGA‐MTX NPs (Figure 9a–d). On the other hand, only NF‐κB and SNAIL were downregulated by MTX. Following gene downregulation, NF‐κB and STAT3 had their protein levels reduced by NPs and MTX in the TME, according to immunostaining tests. Similarly, in line with SNAIL downregulation, an increase in the expression of tumoural E‐cadherin adhesion protein was observed after treatments with NPs (~75%) and MTX (38%; Figure 9e).
FIGURE 9.
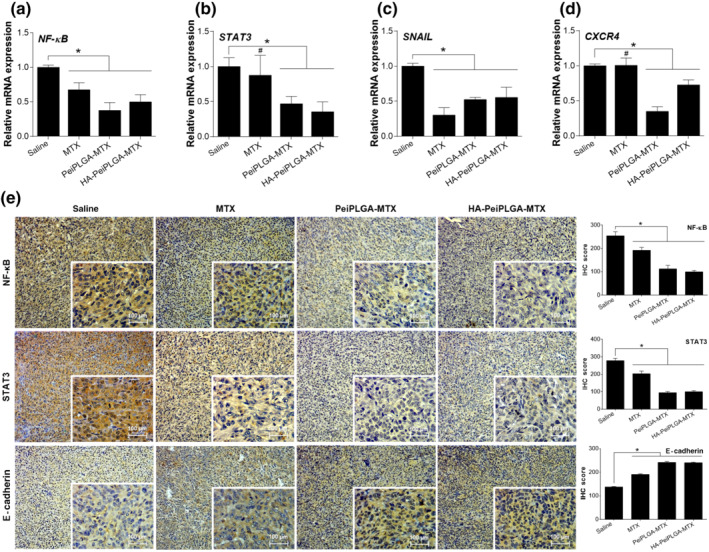
Intracellular signalling modulation in TME. The modulating effect of naked or HA‐coated PeiPLGA‐MTX NPs on key element expression from different signalling routes was evaluated (a–d). The consequences of such modulations on E‐cadherin expression were also assessed (e). The immunohistological panel presents images with 100× magnification; 400× magnification is shown in the lower right corner of each image. Scores were applied to each image to represent them graphically. All data are presented as mean ± SD of six independent values (n = 6) per group with at least two replicates. * P < .05, significantly different as indicated; #not significantly different; one‐way ANOVA with post hoc Bonferroni correction or Kruskal–Wallis with post hoc Dunn correction
PD‐L1 antibody combined with HA‐PeiPLGA‐MTX NPs was employed for tumour inflammatory profile evaluation. This combined treatment upregulated CD8 and IL‐10 genes (160% and 21%, respectively; Figure 10a,d), as well as downregulating the CD68 gene (36%; Figure 10b). In turn, NPs alone downregulated the genes encoding Treg‐recruiting chemokines (CCL22) and those for markers of immunosuppressive activity in the TME (CD163 and IL‐10). On the other hand, the expression of anti‐tumour activity indicators (CD8 and CD68) was highly upregulated (Figure 10a–e). MTX downregulated only CD8, CD68, and CCL22 genes. Immunohistochemical evaluations showed a significant reduction in PD‐L1 protein expression for all evaluated treatments, as well as an increase in CD25. Both the combination of PD‐L1 antibody + HA‐PeiPLGA‐MTX, MTX NPs, given alone, increased the immunosuppressive protein TGF‐β. On the other hand, treatment with the combination also reduced CD163 and IL‐10 protein levels, as did both PeiPLGA formulations. In addition to these proteins, PeiPLGA‐MTX and HA‐PeiPLGA‐MTX also reduced TGF‐β. The evaluation of such markers indicates a reduction in the number of inflammatory cells and their immunosuppressive products, as well as an increase in cancer‐fighting effector cells in the TME.
FIGURE 10.
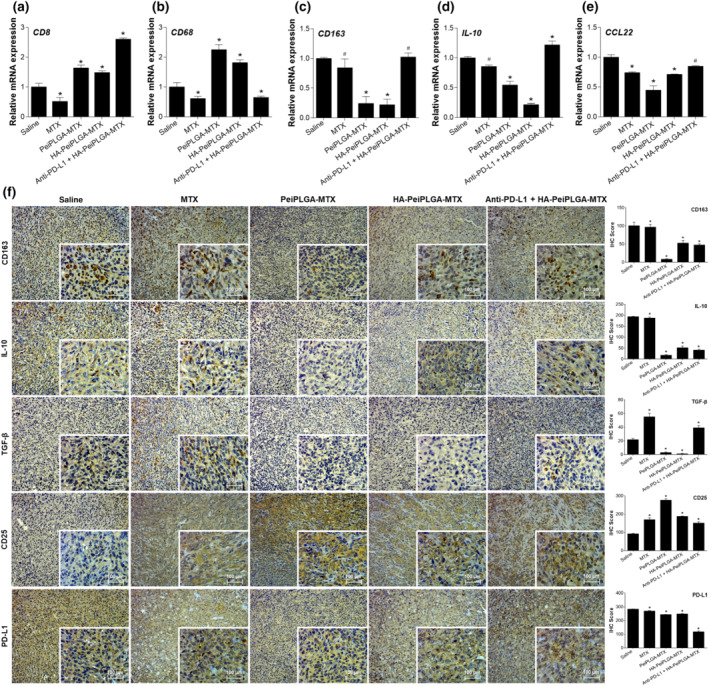
Reduction of the immunosuppressive TME. In combination with PD‐L1 antibody (anti‐PD‐L1), HA‐coated PeiPLGA‐MTX NPs, or only PeiPLGA‐MTX NPs triggered several inflammatory effects in the TME (a–f). The immunohistological panel presents images with 100× magnification; 400× magnification is shown in the lower right corner of each image. Scores were applied to each image to represent them graphically. All data are presented as mean ± SD of six independent values (n = 6) per group with at least two replicates. * P < .05, significantly different as indicated; #not significantly different; One‐way ANOVA with post hoc Bonferroni correction or Kruskal–Wallis with post hoc Dunn correction
4. DISCUSSION
The modulation of breast cancer TME behaviour by naked and HA‐coated PeiPLGA‐MTX NPs has been demonstrated. It was observed that the expression profile of genes involved in important pathways, which favour tumour development and progression, was significantly altered. NF‐κB and STAT3 were the most importantly downregulated genes due to their crucial role in the crosstalk between malignant cells and tumour‐associated immune cells (Fan, Mao, & Yang, 2013). Thus, a range of signalling pathways in both immune and malignant cells were disrupted.
In this context, NF‐κB and STAT3 play an important role in modulating cancer‐associated inflammation‐promoting TAMs polarization towards M2 (Xia, Shen, & Verma, 2014; Zhang et al., 2013). Therefore, NP‐mediated NF‐κB and STAT3 downregulation may be directly related to the inversion of TAMs phenotype in the TME with M1‐polarized TAM increase (CD68 cells). Disruption of TAMs polarization course towards M2 seems to have triggered the decreased expression of CCL22, a chemokine released by M2‐polarized TAM. CCL22 acts as chemoattractant for regulatory T cell (Treg) recruitment (Martinenaite et al., 2016) and creates an immunosuppressive TME through IL‐10 and TGF‐β release, in turn responsible for inhibiting NK cells, CD4 and CD8 T cell recruitment to the tumour (Jiang & Shapiro, 2014; Sakaguchi, Miyara, Costantino, & Hafler, 2010). Moreover, M2‐polarized TAMs can directly reduce the cytotoxic activity of CD8 T cells by IL‐10 and TGF‐β release (Szala, Jarosz‐Biej, Cichoń, Smolarczyk, & Sochanik, 2015), as well as through the expression of PD‐L1 (Pedoeem, Azoulay‐Alfaguter, Strazza, Silverman, & Mor, 2014). In this context, NF‐κB and STAT3 increase the expression of immunosuppression‐related genes in tumour‐associated immune cells, including CCL22, IL‐10, and TGF‐β (Grivennikov & Karin, 2010; Qin, Yan, Zhang, & Zhang, 2019), as well as PD‐L1 expression in cancer cells (Sasidharan Nair, Toor, Ali, & Elkord, 2018). Such regulations help tumour cells to escape from immune elimination (Qin, Yan, Zhang, & Zhang, 2019). Therefore, the significant reduction of IL‐10, TGF‐β levels, and PD‐L1 expression, as well as the increase of CD8+ T cell population in the TME, here may also be related to NF‐κB and STAT3 downregulation by naked or HA‐coated PeiPLGA‐MTX NPs. In addition, the treatment with PD‐L1 antibody combined with HA‐coated PeiPLGA‐MTX NPs induced the highest levels of CD8 T cells according to gene expression analysis. Such cells are important immune prognostic markers and are especially and closely associated with improved survival in patients with breast cancer (Savas et al., 2018).
Our in vitro studies also give us indications that HA‐PeiPLGA NPs appear to be affecting the switch of M2‐polarized phenotype to M1‐like, triggering, for example, the reduction of IL‐10 levels. Moreover, experiments using the IL‐10 receptor antibody showed the role of this cytokine in promoting autocrine activation of M2‐polarized macrophages via IL‐10/STAT3 (Chuang, Hung, Cangelose, & Leonard, 2016; Martinez & Gordon, 2014). Such NP‐mediated inhibition of self‐activation may have triggered the increased release of IL‐12 via NF‐κB and STAT3 downregulation (downregulation > switch of M2 towards M1 > reduced IL‐10 levels > inhibition of M2 autocrine activation by IL‐10 > high levels of IL‐12). IL‐12 is an important Th1 immune response mediator that exerts anti‐tumour activity through increased proliferation, survival, and/or cytotoxicity of NK cells, CD8, and CD4 T cells, as well as suppression of Treg activity (Garcia Paz et al., 2014; Zagozdzon & Lasek, 2016).
As in tumour‐associated immune cells, NF‐κB and STAT3 orchestrate several signalling pathways in malignant cells. In this study, we show how NP‐mediated downregulation of such transcription factors disrupted a complex communication network, crucial to tumour development and progression. Two branches of this network, apoptosis resistance and drug resistance, are major obstacles for chemotherapy of malignant diseases and were significantly reduced by naked and HA‐coated PeiPLGA‐MTX NPs. Both intrinsic and extrinsic apoptotic pathways as well as increased pro‐apoptotic molecule expression were greatly stimulated in malignant cells. NF‐κB/STAT3 signalling disruption may be closely associated with observed decrease in apoptosis resistance, mainly by decreasing of anti‐apoptotic gene downregulation such as BCL‐2 (Fathi, Rashidi, Khodadadi, Shahi, & Sharifi, 2018; Xia, Shen, & Verma, 2014). In this same way, NP‐mediated Survivin downregulation was also observed and may have contributed to unlocking apoptosis (Cui et al., 2017). Survivin is a downstream mediator of NF‐κB and STAT3 signalling and member of the inhibitors of apoptosis (IAP) protein family, which acts in the cell division regulation and suppresses apoptosis by inhibiting caspase (Cui et al., 2017; Gritsko et al., 2006). Such findings may also reflect the NP‐mediated MDR1 downregulation via suppression of NF‐κB/STAT3, which in turn may sensitize malignant cells to death‐inducing stimuli (including chemotherapeutic agents) through MDR1 downregulation (Bentires‐Alj et al., 2003; Zhang, Xiao, Wang, Tian, & Zhang, 2011).
Suppression of NF‐κB/STAT3 signalling also considerably affected tumour progression. The consequent regulation loss of SNAIL and CXCR4 transcription allowed a reduced expression of these genes in our study. Thus, an increased E‐cadherin expression was observed, given that SNAIL gene family directly influences the repression of E‐cadherin transcription (Barrallo‐Gimeno & Nieto, 2005; Y. Wang, Shi, Chai, Ying, & Zhou, 2013). In addition, TGF‐β (downregulated in the TME) can directly or indirectly upregulate SNAIL genes (Y. Wang, Shi, Chai, Ying, & Zhou, 2013; Wendt, Balanis, Carlin, & Schiemann, 2014). CXCR4 overexpression in turn is positively stimulated in response to NF‐κB and STAT3 activity, favouring the tumour spread to sites of greater CXCL12 production, such as lung, liver, and bones (Xu, Zhao, Chen, & Yao, 2015). Furthermore, in vitro studies using the IL‐10 receptor antibody revealed the involvement of immune cells, especially M2‐polarized macrophages, in the malignant cells spread (antimigratory activity assay) supposedly through IL‐10 release (with STAT3 activation) and establishment of an immunosuppressive microenvironment. However, HA‐coated PeiPLGA NPs intervention has interestingly reduced this migratory effect, also helped by reduced vimentin expression, and increased E‐cadherin expression. All this regulation triggered a size and quantity reduction of metastatic niches in the liver and lung in BALB/c mice.
Notably, naked and HA‐coated PeiPLGA‐MTX NPs significantly modulated several cellular responses in the TME. However, the exact mechanisms by which NPs achieve their effects are still unclear. Nevertheless, based on previous studies and physicochemical properties of nanosystems developed here, some suggestions about the intracellular behaviour of NPs may be made. PLGA NPs have been widely used as drug delivery systems. However, anionic drugs such as MTX may have their spontaneous encapsulation hampered by repulsive electrostatic forces due to the negatively charged PLGA surface. Thus, polycations such as Pei have been used as an alternative (Abolmaali, Tamaddon, Yousefi, Javidnia, & Dinarvand, 2014; Shau et al., 2012). Pei gives a positive charge to NPs, favouring internalization and intracellular accumulation of NPs through non‐specific electrostatic interactions with cell membranes and other slightly anionic macromolecules (S. Wang et al., 2017). However, despite that, Pei itself has high cytotoxic potential (Fischer, Li, Ahlemeyer, Krieglstein, & Kissel, 2003) and its use was limited. Thus, both MTX‐loaded PLGA formulations, naked or HA‐coated, showed less than 50% encapsulation efficiency.
To further reduce the Pei‐mediated toxicity, and to improve drug delivery to a specific site, NPs were coated with low molecular weight HA. HA is a negatively charged polysaccharide whose receptors (CD44) are overexpressed in breast cancer cells (S. Wang, Zhang, Wang, & Chen, 2016). The NPs size followed their complexity, especially when loading MTX. Therefore, HA‐mediated neutralization of the positively charged Pei led to a zeta potential reduction of NPs. Similar results using HA‐coated docetaxel‐loaded PeiPLGA NPs were found (Maiolino et al., 2015). Despite the reduced zeta potential among HA‐coated NPs, no evaluated formulation changed its stability over 21 days. Even with reduced zeta potential, HA‐coated PeiPLGA‐MTX NPs did not have their cell uptake impaired. Interestingly, HA‐uncoated PeiPLGA‐MTX NPs did not show the same performance. Consequently, we can suggest that the HA/CD44 interaction in tumour cells may have favoured internalizing the HA‐coated NPs (Gotte & Yip, 2006), whereas HA‐devoid NPs had their uptake delayed.
Although no internalization mechanism of HA‐PeiPLGA‐MTX NPs (and their variations) has been evaluated by us, a study developed with doxorubicin‐loaded mesoporous silica NPs covered with HA and Pei reveal that both coatings seem to play a successful partnership, in the internalization process of such NPs. via CD44‐mediated endocytosis (Fortuni et al., 2019). After endocytosis, NP‐loaded endosomal vesicles fuse with lysosomes to form endolysosomes. Within the endolysosomes, the HA‐coating layer is initially digested by lysosomal hyaluronidases exposing the naked NPs (internally covered with Pei). The “proton sponge effect” supports the escape hypothesis of Pei‐linked NPs from endolysosomes towards cytoplasm. Such an escape protects NPs from degradation and any loaded drugs from inactivation. Due to the protonation of tertiary amines at a low pH, Pei with its high buffering degree stimulates the accumulation of protons into endolysosomes, causing its rupture by osmotic imbalance (Akinc, Thomas, Klibanov, & Langer, 2005). Until their complete degradation, cationic NPs loaded with doxorubicin can interact with several slightly anionic cytoplasmic and nuclear macromolecules, thereby affecting diverse intracellular events. Despite the numerous differences between mesoporous silica NPs and those NPs used in this study, the possibility that similar mechanisms to those described above may also occur during internalization of empty or MTX‐loaded HA‐PeiPLGA NP cannot be ruled out.
A complex and surprising communication network established between immune and malignant cells, and orchestrated by TAMs was disrupted by both naked and HA‐coated PeiPLGA‐MTX NPs, including those NPs associated with anti‐PD‐L1 inhibitor. We have suggested that NF‐κB and STAT3 downregulation affected and reduced the tumour‐promoting inflammation, immunosuppressive and pro‐tumour signalling through blocking the polarization of macrophages towards the M2 phenotype. Furthermore, downregulation of the IL‐10/STAT3/NF‐κB signalling axis reduced tumour escape from immunosurveillance, thus decreasing growth and the spread of breast cancer. The combined administration of anti‐PD‐L1 and HA‐coated PeiPLGA‐MTX proved to be very interesting, because it considerably increases the anti‐tumour cytotoxic immune response. Together, these findings are promising and encourage further tests using the PD‐L1 antibody as a immune checkpoint inhibitor to improve efficient NP drug delivery systems.
AUTHOR CONTRIBUTIONS
R.C. and U.I. conducted the biological experiments. E.S. and A.S. synthesized and characterized the nanoparticles. I.U. prepared the histological and immunohistochemistry slides. R.A., A.A., and R.C. analysed the data. R.C. and E.S. interpreted the data and wrote the manuscript. A.C., L.C., and A.A. kindly provided access to equipment and reagents. A.C., L.C., A.A., and R.A. carefully reviewed the manuscript. All authors approved the final version of the manuscript.
CONFLICT OF INTEREST
The authors declare no potential conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, and Animal Experimentation, and as recommended by funding agencies, publishers, and other organizations engaged with supporting research.
Supporting information
Table S1: Mouse primers sequence, amplicon size and annealing temperature used in this study.
Table S2: Percentage of gene expression modulation by different treatments
Table S3: Modulation of protein expression by different treatments expressed in IHC score units.
Fig. S1: Cell viability after treatment with PLGA nanoparticles. Different formulations of PeiPLGA NPs, as well as their isolated constituents, were performed in 4T1 breast 3 cancer cells (A‐F) and HEK‐293 kidney embryonic cells (G‐L). IC50 of each test was assessed. One‐way ANOVA with post‐hoc Bonferroni correction; *, P < .05. All data are presented as mean ± SD of five independent assays with at least three replicates.
Fig. S2: In vivo toxicity of NPs. The different formulations of PeiPLGA NPs were tested in female BALB/c mice after intraperitoneal administration of NPs every five days for fifteen days. Hepatic (A, C, E, G, I, K) and pulmonary (B, D, F, H, J, L) tissues were evaluated histologically. The arrowheads indicate inflammatory sites. In the lower right corner of each image a 400x magnification of is shown. Systemic toxicity by measuring biochemical markers of liver and kidney injury (M‐P). One‐way ANOVA with post‐hoc Bonferroni correction or Kruskal‐Wallis with post‐hoc Dunn correction; *, P < .05; #, no significant difference. All data are presented as mean ± SD of six independent values (n = 6) per group.
ACKNOWLEDGEMENT
This work was supported by CNPq (425786/2016‐1 Universal project) and EU Programs H2020‐MSCA‐2015‐RISE PRISAR (grant number 644373). We acknowledge support by doctoral fellowship from RSC by CAPES. LJC and AC received funding from the H2020‐MSCA‐2017‐RISE CANCER (grant number 777682). AC received funding from the EU Programs H2020‐WIDESPREAD‐2017‐Twinning ACORN (grant number 807281) and H2020‐WIDESPREAD‐2018‐Twinning SIMICA (grant number 852985). The authors would like to thank Dr. Aldo C Medeiros and MSc. Italo M Azevedo from Departamento de Cirurgia ‐ UFRN for the images acquired in the Kodak In‐Vivo FX image station.
Cavalcante RS, Ishikawa U, Silva ES, et al. STAT3/NF‐κB signalling disruption in M2 tumour‐associated macrophages is a major target of PLGA nanocarriers/PD‐L1 antibody immunomodulatory therapy in breast cancer. Br J Pharmacol. 2021;178:2284–2304. 10.1111/bph.15373
DATA AVAILABILITY STATEMENT
Data available on request from the authors.
REFERENCES
- Abolmaali, S. S. , Tamaddon, A. , Yousefi, G. , Javidnia, K. , & Dinarvand, R. (2014). Sequential optimization of methotrexate encapsulation in micellar nano‐networks of polyethyleneimine ionomer containing redox‐sensitive cross‐links. International Journal of Nanomedicine, 9, 2833–2848. 10.2147/IJN.S61614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akinc, A. , Thomas, M. , Klibanov, A. M. , & Langer, R. (2005). Exploring polyethylenimine‐mediated DNA transfection and the proton sponge hypothesis. The Journal of Gene Medicine, 7(5), 657–663. 10.1002/jgm.696 [DOI] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , … CGTP Collaborators . (2019). The Concise Guide to PHARMACOLOGY 2019/20: Catalytic receptors. British Journal of Pharmacology, 176, S247–S296. 10.1111/bph.14751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , … Collaborators, C . (2019). The Concise Guide to PHARMACOLOGY 2019/20: Other protein targets. British Journal of Pharmacology, 176(Suppl 1), S1–S20. 10.1111/bph.14747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Roberts, R. E. , Broughton, B. R. S. , Sobey, C. G. , George, C. H. , Stanford, S. C. , … Ahluwalia, A. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology. British Journal of Pharmacology, 175(3), 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araujo Junior, R. F. , Garcia, V. B. , Leitao, R. F. , Brito, G. A. , Miguel Ede, C. , Guedes, P. M. , & de Araujo, A. A. (2016). Carvedilol improves inflammatory response, oxidative stress and fibrosis in the alcohol‐induced liver injury in rats by regulating Kuppfer cells and hepatic stellate cells. PLoS ONE, 11(2), e0148868. 10.1371/journal.pone.0148868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrallo‐Gimeno, A. , & Nieto, M. A. (2005). The Snail genes as inducers of cell movement and survival: Implications in development and cancer. Development, 132(14), 3151–3161. 10.1242/dev.01907 [DOI] [PubMed] [Google Scholar]
- Bentires‐Alj, M. , Barbu, V. , Fillet, M. , Chariot, A. , Relic, B. , Jacobs, N. , … Bours, V. (2003). NF‐κB transcription factor induces drug resistance through MDR1 expression in cancer cells. Oncogene, 22(1), 90–97. 10.1038/sj.onc.1206056 [DOI] [PubMed] [Google Scholar]
- Charafe‐Jauffret, E. , Tarpin, C. , Bardou, V. J. , Bertucci, F. , Ginestier, C. , Braud, A. C. , … Viens, P. (2004). Immunophenotypic analysis of inflammatory breast cancers: Identification of an 'inflammatory signature'. The Journal of Pathology, 202(3), 265–273. 10.1002/path.1515 [DOI] [PubMed] [Google Scholar]
- Chuang, Y. , Hung, M. E. , Cangelose, B. K. , & Leonard, J. N. (2016). Regulation of the IL‐10‐driven macrophage phenotype under incoherent stimuli. Innate Immunity, 22(8), 647–657. 10.1177/1753425916668243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colotta, F. , Allavena, P. , Sica, A. , Garlanda, C. , & Mantovani, A. (2009). Cancer‐related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis, 30(7), 1073–1081. 10.1093/carcin/bgp127 [DOI] [PubMed] [Google Scholar]
- Criscitiello, C. , Esposito, A. , Gelao, L. , Fumagalli, L. , Locatelli, M. , Minchella, I. , … Curigliano, G. (2014). Immune approaches to the treatment of breast cancer, around the corner? Breast Cancer Research, 16(1), 204–211. 10.1186/bcr3620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, X. , Shen, D. , Kong, C. , Zhang, Z. , Zeng, Y. , Lin, X. , & Liu, X. (2017). NF‐κB suppresses apoptosis and promotes bladder cancer cell proliferation by upregulating survivin expression in vitro and in vivo. Scientific Reports, 7, 40723. 10.1038/srep40723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175(7), 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Araujo Junior, R. F. , Eich, C. , Jorquera, C. , Schomann, T. , Baldazzi, F. , Chan, A. B. , & Cruz, L. J. (2020). Ceramide and palmitic acid inhibit macrophage‐mediated epithelial–mesenchymal transition in colorectal cancer. Molecular and Cellular Biochemistry, 468(1–2), 153–168. 10.1007/s11010-020-03719-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos‐Santos‐Silva, E. , Alves‐Silva, M. F. , de Medeiros, J. S. , dos Santos‐Cavalcante, R. , Cornélio, A. M. , Fernandes‐Pedrosa, M. F. , … da Silva‐Júnior, A. A. (2020). Colloidal properties of self‐assembled cationic hyperbranched‐polyethyleneimine covered poly lactide‐co‐glycolide nanoparticles: Exploring modified release and cell delivery of methotrexate. Journal of Molecular Liquids, 315, 113721. 10.1016/j.molliq.2020.113721 [DOI] [Google Scholar]
- Fan, Y. , Mao, R. , & Yang, J. (2013). NF‐κB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein & Cell, 4(3), 176–185. 10.1007/s13238-013-2084-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fathi, N. , Rashidi, G. , Khodadadi, A. , Shahi, S. , & Sharifi, S. (2018). STAT3 and apoptosis challenges in cancer. International Journal of Biological Macromolecules, 117, 993–1001. 10.1016/j.ijbiomac.2018.05.121 [DOI] [PubMed] [Google Scholar]
- Fischer, D. , Li, Y. , Ahlemeyer, B. , Krieglstein, J. , & Kissel, T. (2003). In vitro cytotoxicity testing of polycations: Influence of polymer structure on cell viability and hemolysis. Biomaterials, 24(7), 1121–1131. 10.1016/s0142-9612(02)00445-3 [DOI] [PubMed] [Google Scholar]
- Fortuni, B. , Inose, T. , Ricci, M. , Fujita, Y. , Van Zundert, I. , Masuhara, A. , … Uji, I. H. (2019). Polymeric engineering of nanoparticles for highly efficient multifunctional drug delivery systems. Scientific Reports, 9(1), 2666. 10.1038/s41598-019-39107-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouad, Y. A. , & Aanei, C. (2017). Revisiting the hallmarks of cancer. American Journal of Cancer Research, 7(5), 1016–1036. [PMC free article] [PubMed] [Google Scholar]
- Garcia Paz, F. , Madrid Marina, V. , Morales Ortega, A. , Santander Gonzalez, A. , Peralta Zaragoza, O. , Burguete Garcia, A. , … Bermudez Morales, V. (2014). The relationship between the antitumor effect of the IL‐12 gene therapy and the expression of Th1 cytokines in an HPV16‐positive murine tumor model. Mediators of Inflammation, 2014, 510846. 10.1155/2014/510846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotte, M. , & Yip, G. W. (2006). Heparanase, hyaluronan, and CD44 in cancers: A breast carcinoma perspective. Cancer Research, 66(21), 10233–10237. 10.1158/0008-5472.CAN-06-1464 [DOI] [PubMed] [Google Scholar]
- Gritsko, T. , Williams, A. , Turkson, J. , Kaneko, S. , Bowman, T. , Huang, M. , … Jove, R. (2006). Persistent activation of Stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clinical Cancer Research, 12(1), 11–19. 10.1158/1078-0432.CCR-04-1752 [DOI] [PubMed] [Google Scholar]
- Grivennikov, S. I. , & Karin, M. (2010). Dangerous liaisons: STAT3 and NF‐κB collaboration and crosstalk in cancer. Cytokine & Growth Factor Reviews, 21(1), 11–19. 10.1016/j.cytogfr.2009.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, A. , Cao, S. , & Tang, L. (2017). The tumor microenvironment and inflammatory breast cancer. Journal of Cancer, 8(10), 1884–1891. 10.7150/jca.17595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, X. , & Shapiro, D. J. (2014). The immune system and inflammation in breast cancer. Molecular and Cellular Endocrinology, 382(1), 673–682. 10.1016/j.mce.2013.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura, T. , Qian, B. Z. , & Pollard, J. W. (2015). Immune cell promotion of metastasis. Nature Reviews. Immunology, 15(2), 73–86. 10.1038/nri3789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley, E. , Stanford, S. C. , Kendall, D. E. , Alexander, S. P. H. , Cirino, G. , Docherty, J. R. , … Ahluwalia, A. (2020). ARRIVE 2.0 and the British Journal of Pharmacology: Updated guidance for 2020. British Journal of Pharmacology, 177(16), 3611–3616. 10.1111/bph.15178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Q. , Jin, S. , Han, M. , Zheng, W. , Liu, J. , & Wei, X. (2018). Inflammation in the tumor microenvironment. Journal of Immunology Research, 2018, 1965847. 10.1155/2018/1965847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, H. , Xu, H. W. , Zhang, Y. Z. , Huang, Y. , Han, G. Q. , Liang, T. J. , … Qin, C. K. (2015). Ursodeoxycholic acid induces apoptosis in hepatocellular carcinoma xenografts in mice. World Journal of Gastroenterology, 21(36), 10367–10374. 10.3748/wjg.v21.i36.10367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiolino, S. , Russo, A. , Pagliara, V. , Conte, C. , Ungaro, F. , Russo, G. , & Quaglia, F. (2015). Biodegradable nanoparticles sequentially decorated with polyethyleneimine and hyaluronan for the targeted delivery of docetaxel to airway cancer cells. J Nanobiotechnology, 13, 29–41. 10.1186/s12951-015-0088-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinenaite, E. , Munir Ahmad, S. , Hansen, M. , Met, O. , Westergaard, M. W. , Larsen, S. K. , … Andersen, M. H. (2016). CCL22‐specific T cells: Modulating the immunosuppressive tumor microenvironment. Oncoimmunology, 5(11), e1238541. 10.1080/2162402X.2016.1238541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez, F. O. , & Gordon, S. (2014). The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep, 6, 13. 10.12703/P6-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedelcu, R. I. , Balaban, M. , Turcu, G. , Brinzea, A. , Ion, D. A. , Antohe, M. , … Zurac, S. A. (2019). Efficacy of methotrexate as anti‐inflammatory and anti‐proliferative drug in dermatology: Three case reports. Experimental and Therapeutic Medicine, 18(2), 905–910. 10.3892/etm.2019.7511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochaion, A. , Bar‐Yehuda, S. , Cohn, S. , Del Valle, L. , Perez‐Liz, G. , Madi, L. , … Fishman, P. (2006). Methotrexate enhances the anti‐inflammatory effect of CF101 via up‐regulation of the A3 adenosine receptor expression. Arthritis Research & Therapy, 8(6), R169. 10.1186/ar2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschall, A. V. , & Liu, K. (2016). An orthotopic mouse model of spontaneous breast cancer metastasis. Journal of Visualized Experiments, 2016(114), e54040. 10.3791/54040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedoeem, A. , Azoulay‐Alfaguter, I. , Strazza, M. , Silverman, G. J. , & Mor, A. (2014). Programmed death‐1 pathway in cancer and autoimmunity. Clinical Immunology, 153(1), 145–152. 10.1016/j.clim.2014.04.010 [DOI] [PubMed] [Google Scholar]
- Percie du Sert, N. , Hurst, V. , Ahluwalia, A. , Alam, S. , Avey, M. T. , Baker, M. , … Würbel, H. (2020). The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. PLoS Biol, 18, e3000410. 10.1371/journal.pbio.3000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, J. J. , Yan, L. , Zhang, J. , & Zhang, W. D. (2019). STAT3 as a potential therapeutic target in triple negative breast cancer: A systematic review. Journal of Experimental & Clinical Cancer Research, 38(1), 195–210. 10.1186/s13046-019-1206-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu, S. Q. , Waaijer, S. J. H. , Zwager, M. C. , de Vries, E. G. E. , van der Vegt, B. , & Schroder, C. P. (2018). Tumor‐associated macrophages in breast cancer: Innocent bystander or important player? Cancer Treatment Reviews, 70, 178–189. 10.1016/j.ctrv.2018.08.010 [DOI] [PubMed] [Google Scholar]
- Sakaguchi, S. , Miyara, M. , Costantino, C. M. , & Hafler, D. A. (2010). FOXP3+ regulatory T cells in the human immune system. Nature Reviews. Immunology, 10(7), 490–500. 10.1038/nri2785 [DOI] [PubMed] [Google Scholar]
- Sasidharan Nair, V. , Toor, S. M. , Ali, B. R. , & Elkord, E. (2018). Dual inhibition of STAT1 and STAT3 activation downregulates expression of PD‐L1 in human breast cancer cells. Expert Opinion on Therapeutic Targets, 22(6), 547–557. 10.1080/14728222.2018.1471137 [DOI] [PubMed] [Google Scholar]
- Savas, P. , Virassamy, B. , Ye, C. , Salim, A. , Mintoff, C. P. , Caramia, F. , … Loi, S. (2018). Single‐cell profiling of breast cancer T cells reveals a tissue‐resident memory subset associated with improved prognosis. Nature Medicine, 24(7), 986–993. 10.1038/s41591-018-0078-7 [DOI] [PubMed] [Google Scholar]
- Shau, M. D. , Shih, M. F. , Lin, C. C. , Chuang, I. C. , Hung, W. C. , Hennink, W. E. , & Cherng, J. Y. (2012). A one‐step process in preparation of cationic nanoparticles with poly (lactide‐co‐glycolide)‐containing polyethylenimine gives efficient gene delivery. European Journal of Pharmaceutical Sciences, 46(5), 522–529. 10.1016/j.ejps.2012.04.006 [DOI] [PubMed] [Google Scholar]
- Szala, S. , Jarosz‐Biej, M. , Cichoń, T. , Smolarczyk, R. , & Sochanik, A. (2015). Polarization of tumor milieu: Therapeutic implications. In Rezaei N. (Ed.), Cancer immunology (pp. 401–408). Berlin, Heidelberg: Springer. [Google Scholar]
- Tanaka, K. , Arao, T. , Maegawa, M. , Matsumoto, K. , Kaneda, H. , Kudo, K. , … Nishio, K. (2009). SRPX2 is overexpressed in gastric cancer and promotes cellular migration and adhesion. International Journal of Cancer, 124(5), 1072–1080. 10.1002/ijc.24065 [DOI] [PubMed] [Google Scholar]
- Tower, H. , Ruppert, M. , & Britt, K. (2019). The immune microenvironment of breast cancer progression. Cancers (Basel), 11(9), 1375–1389. 10.3390/cancers11091375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Eynden, G. G. , Bird, N. C. , Majeed, A. W. , Van Laere, S. , Dirix, L. Y. , & Vermeulen, P. B. (2012). The histological growth pattern of colorectal cancer liver metastases has prognostic value. Clinical & Experimental Metastasis, 29(6), 541–549. 10.1007/s10585-012-9469-1 [DOI] [PubMed] [Google Scholar]
- Wang, J. , Li, D. , Cang, H. , & Guo, B. (2019). Crosstalk between cancer and immune cells: Role of tumor‐associated macrophages in the tumor microenvironment. Cancer Medicine, 8(10), 4709–4721. 10.1002/cam4.2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S. , Shao, M. , Zhong, Z. , Wang, A. , Cao, J. , Lu, Y. , … Zhang, J. (2017). Co‐delivery of gambogic acid and TRAIL plasmid by hyaluronic acid grafted PEI‐PLGA nanoparticles for the treatment of triple negative breast cancer. Drug Delivery, 24(1), 1791–1800. 10.1080/10717544.2017.1406558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S. , Zhang, J. , Wang, Y. , & Chen, M. (2016). Hyaluronic acid‐coated PEI‐PLGA nanoparticles mediated co‐delivery of doxorubicin and miR‐542‐3p for triple negative breast cancer therapy. Nanomedicine, 12(2), 411–420. 10.1016/j.nano.2015.09.014 [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Shi, J. , Chai, K. , Ying, X. , & Zhou, B. P. (2013). The role of snail in EMT and tumorigenesis. Current Cancer Drug Targets, 13(9), 963–972. 10.2174/15680096113136660102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt, M. K. , Balanis, N. , Carlin, C. R. , & Schiemann, W. P. (2014). STAT3 and epithelial–mesenchymal transitions in carcinomas. JAKSTAT, 3(1), e28975. 10.4161/jkst.28975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, Y. , Shen, S. , & Verma, I. M. (2014). NF‐κB, an active player in human cancers. Cancer Immunology Research, 2(9), 823–830. 10.1158/2326-6066.CIR-14-0112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, C. , Zhao, H. , Chen, H. , & Yao, Q. (2015). CXCR4 in breast cancer: Oncogenic role and therapeutic targeting. Drug Design, Development and Therapy, 9, 4953–4964. 10.2147/DDDT.S84932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagozdzon, R. , & Lasek, W. (2016). Interleukin 12: Antitumor activity and immunotherapeutic potential in oncology. Switzerland: Springer International Publishing. [Google Scholar]
- Zhang, X. , Tian, W. , Cai, X. , Wang, X. , Dang, W. , Tang, H. , … Chen, T. (2013). Hydrazinocurcumin encapsuled nanoparticles “re‐educate” tumor‐associated macrophages and exhibit anti‐tumor effects on breast cancer following STAT3 suppression. PLoS ONE, 8(6), e65896. 10.1371/journal.pone.0065896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. , Xiao, W. , Wang, L. , Tian, Z. , & Zhang, J. (2011). Deactivation of signal transducer and activator of transcription 3 reverses chemotherapeutics resistance of leukemia cells via down‐regulating P‐gp. PLoS ONE, 6(6), e20965. 10.1371/journal.pone.0020965 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Mouse primers sequence, amplicon size and annealing temperature used in this study.
Table S2: Percentage of gene expression modulation by different treatments
Table S3: Modulation of protein expression by different treatments expressed in IHC score units.
Fig. S1: Cell viability after treatment with PLGA nanoparticles. Different formulations of PeiPLGA NPs, as well as their isolated constituents, were performed in 4T1 breast 3 cancer cells (A‐F) and HEK‐293 kidney embryonic cells (G‐L). IC50 of each test was assessed. One‐way ANOVA with post‐hoc Bonferroni correction; *, P < .05. All data are presented as mean ± SD of five independent assays with at least three replicates.
Fig. S2: In vivo toxicity of NPs. The different formulations of PeiPLGA NPs were tested in female BALB/c mice after intraperitoneal administration of NPs every five days for fifteen days. Hepatic (A, C, E, G, I, K) and pulmonary (B, D, F, H, J, L) tissues were evaluated histologically. The arrowheads indicate inflammatory sites. In the lower right corner of each image a 400x magnification of is shown. Systemic toxicity by measuring biochemical markers of liver and kidney injury (M‐P). One‐way ANOVA with post‐hoc Bonferroni correction or Kruskal‐Wallis with post‐hoc Dunn correction; *, P < .05; #, no significant difference. All data are presented as mean ± SD of six independent values (n = 6) per group.
Data Availability Statement
Data available on request from the authors.