

Abstract
Resistance to platinum‐based anticancer drugs represents an important limit for their clinical effectiveness and one of the most important field of investigation in the context of platinum compounds. From our previous studies, PtII complexes containing the triphenylphosphino moiety have been emerging as promising agents, showing significant cytotoxicity to resistant ovarian carcinoma cells. Two brominated triphenylphosphino trans‐platinum derivatives were prepared and evaluated on human tumor cell lines, sensitive and resistant to cisplatin. The new complexes exert a notable antiproliferative effect on resistant ovarian carcinoma cells, showing a remarkable intracellular accumulation and the ability to interact with different intracellular targets. The interaction with DNA, the collapse of mitochondrial transmembrane potential, and the impairment of intracellular redox state were demonstrated. Moreover, a selectivity towards the selenocysteine of thioredoxin reductase was observed. The mechanism of action is discussed with regard to the resistance phenomenon in ovarian carcinoma cells.
Keywords: cytotoxicity, mitochondria, platinum, thiol redox regulation, thioredoxin reductase
Increase targets, decrease resistance: Two new brominated trans‐PtII complexes show an interesting cytotoxicity on ovarian carcinoma resistant cells. Study of the mechanism of action reveals the ability to affect different biomolecular targets: interaction with DNA, inhibition of thioredoxin reductase and impairment of mitochondria were demonstrated.
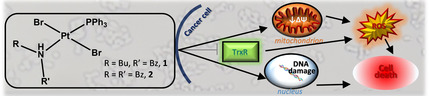
Introduction
The approval of cisplatin by FDA in 1978, for the treatment of advanced testicular, ovarian and bladder cancer, represented a milestone inside the field of metal‐based anticancer drugs. Nowadays, cisplatin acts also as the keystone of combination therapy for a wide range of solid tumors, including lung, gastric, head and neck cancers, and mesothelioma. [1] Notwithstanding it is one of the most compelling anticancer drugs, significant challenges remain with regard to drug resistance and undesired side effects.[ 2 , 3 ] With the aim to enhance the clinical outcome of cisplatin‐based therapy, thousands of analogues have been synthesized and tested and, up to now, two additional PtII complexes have been approved, carboplatin [4] and oxaliplatin, [5] while three others, nedaplatin, heptaplatin and lobaplatin, achieved clinical approval in some countries. [6] Although carboplatin and cisplatin possess similar mechanisms of action, their clinical efficacy is not always the same for all types of tumor, and many data suggest that the mechanisms of resistance are very similar.[ 7 , 8 ] As concerning oxaliplatin, many data point to a limited activity against cisplatin resistant cancer, especially as single agent, although it was initially regarded as active in cisplatin‐resistant cells. [9] Actually, resistance phenomenon remains one of the most important limiting factors in platinum‐based anticancer therapy. Therefore, the myriad and complexity of events concurring to the development of the resistant phenotype are very difficult hurdles to overcome.[ 3 , 10 ]
The overexpression of enzymes belonging to the thiol redox systems has been reported to play a role in the process of tumor chemoresistance.[ 11 , 12 ] The thioredoxin and glutathione systems are the major thiol‐dependent reducing systems involved in the regulation of the redox balance in cells. The thioredoxin system is formed by NADPH, thioredoxin reductase (TrxR) and thioredoxin (Trx), the glutathione system is composed of NADPH, glutathione reductase (GR), glutathione (GSH), glutathione peroxidases (GPxs) and glutaredoxins (Grxs). [13] Both thioredoxin and glutathione systems transfer the reducing equivalents from NADPH downstream to the other components of the system. Cellular glutathione is mainly found in its reduced form. This highly reduced condition is maintained by GR, which, in turn, uses reducing equivalents deriving from NADPH. Although GR effectively reduces GSSG in basal conditions, any marked alteration in GSSG/GSH ratio during oxidative stress may lead to a disruption in redox homeostasis. [14] TrxR is the central enzyme of the thioredoxin system and is a selenoenzyme. [14] Its exposed and highly reactive selenocysteine present in the second active site, renders TrxR efficient in its reducing activity but it is also easily targeted by electrophilic compounds. [12]
In this scenario, we moved our interest toward new transplatinum complexes containing the triphenylphosphino moiety and indeed, in our previous papers, we reported for some new complexes the ability to exert an interesting antiproliferative activity on human tumor cell lines and to affect mitochondrial functions, leading to apoptosis.[ 15 , 16 ] More recently, we synthesized analogues carrying N‐(butyl),N‐(arylmethyl)amino ligands that demonstrated the capacity to overcome cisplatin resistance in ovarian cancer cells. [17]
Considering that in the latest years interesting studies were published about the effects of changing the halogen on cisplatin analogues,[ 18 , 19 ] some of us have developed a useful procedure to prepare brominated triphenylphosphino platinum derivatives. [20]
In this paper we describe the synthesis and the characterization of two new trans‐triphenylphosphinodialkylamino platinum(II) complexes, where the chlorido substituents have been replaced by two bromido groups (complexes 1 and 2).
The antiproliferative effect was evaluated on a panel of human tumor cell lines (HeLa, cervix adenocarcinoma; HT29, colorectal adenocarcinoma and A549, non‐small cell lung cancer) and on A2780 and A2780cis, sensitive and resistant ovarian carcinoma cell lines, respectively. The uptake in resistant cell line and the ability to interact with DNA were assayed to highlight possible differences in behavior with respect to the reference drug, cisplatin. Moreover, the capacity to affect mitochondria and the intracellular redox balance were in depth investigated with the aim to point out the mechanism(s) of action responsible for the biological profile in resistant cells.
Results and Discussion
Chemistry
The synthesis of trans‐[PtBr2(NHRR′)(PPh3)] complexes (R=Bu, R′=Bz, 1; R=R′=Bz, 2) was carried out according to a well consolidated procedure, consisting in the bridge splitting of dinuclear trans‐[Pt(μ‐Br)Br(PPh3)]2 by the suitable dialkylamine (Scheme 1). The reaction is guided by the strong trans effect exerted by triphenylphosphine and affords the trans products in generally good yields. Complexes 1 and 2 are structural analogues of the corresponding chlorinated derivatives, for which interesting antiproliferative properties had been previously described.[ 15 , 17 ]
Scheme 1.

Synthesis of complexes 1 and 2.
Both complexes were characterized spectroscopically (for NMR spectra, see Figures S1–S8 in the Supporting Information) and showed features in good agreement with their chlorinated counterparts,[ 15 , 17 ] except for 195Pt NMR signals, which were shifted downfield (ca. −4000 ppm), due to the presence of two bromide substituents, as expected. [21] Crystals of good quality were obtained from recrystallization of 2 from a chloroform solution and the structure was determined by single crystal X‐ray diffraction (Figure 1). The coordination around platinum is square planar, with small deviations from ideality and the geometry around the metal center is trans. The Pt−N and Pt−P bond lengths are in good agreement with those of the chlorinated analogues; [16] the Pt−Br bond lengths are similar to those measured in cis‐[PtBr2(NCMe)(PPh3)]. [20]
Figure 1.
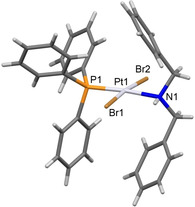
Structure of complex 2. Selected bond lengths: Pt(1)‐N(1)=2.145(3) Å; Pt(1)‐P(1)=2.2291(8) Å; Pt(1)‐Br(1)=2.4296(4) Å; Pt(1)‐Br(2)=2.4360(4) Å. Selected bond angles: N(1)‐Pt(1)‐P(1)=174.64(8)°; N(1)‐Pt(1)‐Br(1)=84.69(8)°; P(1)‐Pt(1)‐Br(1)=90.77(2)°; N(1)‐Pt(1)‐Br(2)=90.86(8)°; P(1)‐Pt(1)‐Br(2)=93.43(2)°; Br(1)‐Pt(1)‐Br(2)=173.500(15)°.
Both complexes 1 and 2 are stable in solid state in air. They are not soluble in water nor ethanol, but they are well soluble in DMSO. The stability in DMSO/water (92 : 8, v/v) over time was checked by 31P NMR experiments (Figure S9)
Cell growth inhibition
The effect of the new complexes on cell growth was investigated by an in vitro assay on a panel of human tumor cell lines. HeLa (cervix adenocarcinoma), HT29 (colorectal adenocarcinoma), A549 (non‐small‐cell lung cancer) and the ovarian carcinoma cisplatin sensitive parent line (A2780) and cisplatin resistant subline (A2780cis), were considered.
Data were expressed as IC50 values, that is, the concentration of the complex [μm] required for 50 % inhibition of cell growth with respect to the control culture, and the resistance factor (RF; IC50 resistant line/IC50 parent line) was calculated for each complex (Table 1).
Table 1.
Effect of test complexes (1 and 2) on cell growth after 72 h of incubation. Stock solutions of 1 and 2 were made in DMSO and the amount of added solvent was always lower than 0.5 %. Cisplatin, dissolved in 0.9 % NaCl, was used as reference.
|
Complex |
|
IC50 [μm][a] |
RF[b] |
|||
|---|---|---|---|---|---|---|
|
A2780 |
A2780cis |
HeLa |
HT29 |
A549 |
||
|
1 |
2.19±0.06 |
2.15±0.19 |
7.93±0.71 |
4.85±0.99 |
6.60±1.90 |
1.0 |
|
2 |
2.99±0.19 |
3.45±0.34 |
7.05±0.72 |
4.40±1.10 |
7.80±1.90 |
1.1 |
|
cisplatin[c] |
0.91±0.13 |
6.61±0.61 |
1.51±0.09 |
2.57±0.67 |
2.10±0.25 |
7.3 |
[a] IC50 values are the mean±SD of at least three independent experiments in duplicate. [b] IC50(A2780cis)/IC50(A2780); [c] Ref. [22].
The new complexes induce a significant antiproliferative effect in all tested cell lines, showing IC50 values in the low micromolar range. In detail, both 1, bearing a n‐buthyl,benzylamino substituent and 2, where two benzyl groups characterize the coordinated amine, demonstrate the same effect, thus indicating a similar role for both substituents. Interestingly, notwithstanding the cytotoxicity exerted by the new complexes appears lower with respect to the reference drug in all cell lines, the effect on resistant ovarian carcinoma cells, A2780cis, is comparable to that on the parent line A2780. Indeed, the RF is about 1 for both 1 and 2, while for cisplatin a value of about 7 is calculated. Moreover, it is noteworthy that on the resistant cell line, the IC50 values obtained for the new complexes are 3 (complex 1) and 1.9 (complex 2) times lower than that obtained for cisplatin, pointing to a higher antiproliferative effect.
The notable cytotoxicity on resistant cells prompted us to investigate the mechanism responsible for such effect, focusing our interest on intracellular molecular and/or macromolecular targets.
Cell uptake
It is well recognized that an inadequate drug content can contribute to the resistance phenotype, which in most cases was related to a reduced drug uptake instead of an increased drug efflux.[ 3 , 10 , 23 ] To investigate the uptake of 1 in resistant cells, the amount of platinum and phosphorus (ppb) in A2780cis, incubated for different times in the presence of the test complex and cisplatin, as reference, was analyzed by ICP‐AES. The measurement of phosphorus in each sample was considered as internal standard. The obtained results, expressed as [Pt]/[P], are shown in Figure 2.
Figure 2.
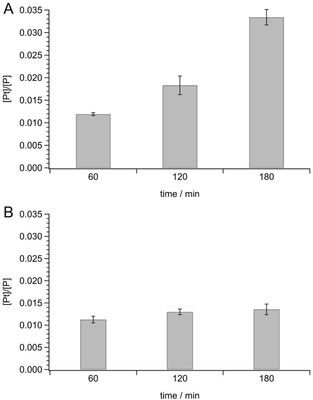
Platinum (ppb)/Phosphorus (ppb) ratio ([Pt]/[P]) in A2780cis cells incubated in the presence of 100 μm A) 1 or B) cisplatin for 60, 120 or 180 min. Mean values ±SD of four experiments are reported.
The comparison between the uptake of cisplatin and 1 reveals significant differences. In detail, the total amount of both complexes accumulated in cells after 60 min of incubation is comparable. Nevertheless, by increasing the incubation time up to 180 min, the uptake of cisplatin remains practically unchanged, also as consequence of a possible increased efflux, while the new complex shows a time‐dependent accumulation that at 180 min reaches a value about three times higher with respect to that detected at 60 min. The remarkable difference observed after 180 min of incubation clearly indicates for 1 a greater capacity to cross plasma membrane than the reference drug. It is to note that this ability could contribute to the higher antiproliferative effect on the drug resistant cell line A2780cis.
Interaction with DNA
A great number of studies pointed to DNA as the primary target of cisplatin, demonstrating a preferential platination at the most nucleophilic sites, that are the N7 atoms of purine bases guanine and adenine. In particular, cisplatin forms monofunctional adducts, followed by crosslinks both on the same strand (intrastrand crosslinks) and on the two opposite strands (interstrand crosslinks) of DNA. The covalent addition to the macromolecule significantly affects the tridimensional structure, giving rise to bending and unwinding of the double helix, and ultimately leading to impairment of DNA functions and cell death.[ 24 , 25 ] In this connection, we investigated the capacity of 1 to platinate DNA, in comparison with cisplatin. In detail, salmon testes DNA was incubated in the presence of test complexes at [DNA]/[complex]=10 for different times (0–48 h), and the amount of platinum (ppb) covalently bound was determined by ICP‐AES (Figure 3A). The analysis of phosphorus as internal standard in each sample was also performed, according to a previous study. [26]
Figure 3.
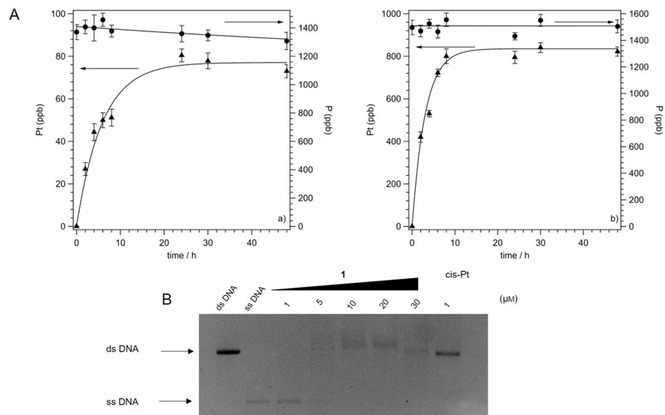
A) Platinum (ppb, ▴) and phosphorus (ppb, •) bound to salmon testes DNA incubated with Left: 1 or right: cisplatin for different incubation times (0–48 h). [DNA]=9×10−4 m, [DNA]/[complex]=10. The data represent the mean±SD of four experiments. B) Formation of interstrand crosslinks in the presence of increasing concentrations of 1 (1–30 μm) or 1 μm cisplatin (cis‐Pt). The linearized untreated ds‐DNA and urea/heat‐denatured ss‐DNA are also shown for reference.
The platination capacity of complex 1 and cisplatin appeared significantly different. In detail, the amount of platinum (ppb) bound to DNA following the incubation with complex 1 increases in a time‐dependent manner up to 24 h of treatment, and reaches a maximum of about 80 ppb after 48 h. The reference drug appears able to bind to the macromolecule with a greater efficiency, because an amount about ten times higher (800 ppb) with respect to that of the new complex, is determined at 48 h. Of note, 1 is a molecule where the small size and planar geometry of cisplatin is lacking, and rather a bulky triphenylphosphino hydrophobic moiety along with a trans geometry of the bromido groups, are present. Interestingly, both these structural differences could contribute to the observed different binding ability to DNA.
Although this result points to DNA as a possible molecular target for 1, it is likely that the interaction with the macromolecule can contribute only partially to the cytotoxic effect exerted by the new complex. Indeed, the differences in IC50 values between 1 and cisplatin, reported in Table 1 for all cell lines taken into consideration, are less pronounced than the platination ability and, more importantly, the lower platination ability of 1 cannot account for the high cytotoxicity on resistant cells.
In this connection, to further investigate the interactions between 1 and DNA, the occurrence of interstrand crosslinks in linearized plasmid DNA was assayed. Indeed, a correlation between this molecular lesion and cytotoxicity has been reported and for both cisplatin and transplatin the ability to give rise to this diadduct was demonstrated.[ 27 , 28 ] Actually, both isomers form interstrand crosslinks, although of distinct natures and causing different distortions in DNA structure.[ 29 , 30 ] In detail, supercoiled plasmid pBR322 DNA was linearized with restriction enzyme, treated with increasing concentrations of 1 (1–30 μm) or 1 μm cisplatin, and then urea/heat denatured. The results are shown in Figure 3B. Linear untreated double‐stranded (ds) DNA and linear denatured single‐stranded (ss) DNA are also shown.
The results in Figure 3B demonstrate the ability of complex 1 to induce crosslinks between the two DNA strands, as revealed by the appearance of the band corresponding to ds‐DNA. Such damage appears at 5 μm concentration, nevertheless, the total amount of interstrand crosslinks appears practically unaffected by a further increase in concentration and, in any case, significantly lower with respect to that induced by 1 μm cisplatin. This result is in agreement with literature, reporting a lower ability of the trans isomer, transplatin, to give rise to the interstrand diadduct.[ 28 , 29 ]
The lower capacity of 1 to bind and to damage DNA with respect to the reference drug, notwithstanding the higher cell uptake and cytotoxicity on resistant A2780cis cell line, prompted us to investigate other intracellular targets that could contribute to the biological effects.
Effect on mitochondria
Mitochondria are intracellular organelles that play a crucial role for cell life as well as for cell death. The inner mitochondrial membrane, thanks to the electron transport chain components and ATP synthase, allows the energy supply generating ATP by oxidative phosphorylation. Otherwise, some extrinsic and intrinsic stimuli can induce the permeabilization of mitochondrial outer membrane, thus promoting a signaling cascade that can lead to apoptosis, the major form of regulated cell death. [31] Owing to their fundamental role in the regulation of important cell functions and of programmed cell death, mitochondria have been proposed as interesting targets for anticancer therapy and, in particular, mitochondria‐targeted anticancer agents emerged as potential drugs to circumvent some mechanisms of resistance.[ 32 , 33 ] In previous studies we described several PtII complexes able to promote cell apoptosis, affecting mitochondrial functions[ 15 , 34 ] and for some of them the ability to overcome drug‐resistance was also demonstrated. [22]
Starting from these results, we tested the effect of complex 1 on mitochondria and in particular, on mitochondrial transmembrane potential in resistant A2780cis cells. Indeed, intact mitochondria are characterized by a high mitochondrial transmembrane potential (ΔΨ), negative inside and generated by the respiratory chain. Damaged mitochondria can undergo membrane depolarization losing the ΔΨ. The variations in ΔΨ can be measured by using the green monomeric fluorescent cationic probe JC‐1, which selectively enters into mitochondria driven by the transmembrane potential, negative inside, leading to the formation of aggregates characterized by red fluorescence emission. In detail, A2780cis cells were treated with 1 and cisplatin, loaded with JC‐1, and fluorescence was detected by cytofluorimetric analysis. The results are reported in Figure 4A as percentage of cells with high ΔΨ (high red fluorescence) and low ΔΨ (low red fluorescence).
Figure 4.
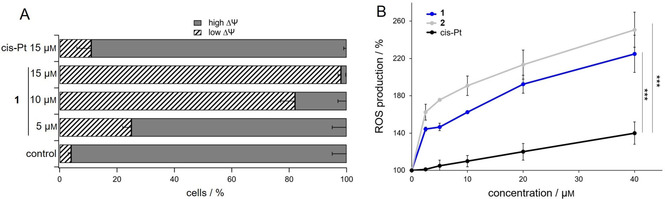
Analysis of the effect of the platinum complexes on mitochondria. A) Cytofluorimetric analysis of mitochondrial transmembrane potential (ΔΨ) in A2780cis cells in the presence of 1 or cisplatin (cis‐Pt) at the indicated concentrations for 40 h. B) Measurement of ROS production in cells upon incubation with the platinum complexes. A2780cis cells (10 000/well) were treated with increasing concentrations of 1, 2 or cis‐Pt, and short‐term ROS production was assessed by means of the CM‐DCFDA probe. Results are reported as percentage ROS production with respect to the control. For both experiments mean values ±SD of four experiments are reported.
Complex 1 causes a remarkable dose‐dependent increase of cells with depolarized mitochondria. In fact, the very low percentage of A2780cis with low ΔΨ in the control condition (ca. 4 %) becomes significantly high at 5 μm concentration (ca. 25 %), getting more than 90 % at the maximum tested concentration (15 μm). Interestingly, the incubation of resistant cells with cisplatin leads to a completely different result. In fact, cisplatin at the highest considered concentration, 15 μm, induces a percentage of about 10 % of cells with depolarized ΔΨ, a value notably lower than that obtained for 1 in the same experimental conditions. These results clearly point to mitochondria as a possible intracellular target of complex 1 and, more interestingly, the difference in mitochondria damaging effect between 1 and cisplatin could account for the more pronounced cytotoxicity exerted on resistant cells.
As mitochondria are the major reactive oxygen species (ROS)‐producing organelles in the cell, and cancer cells are particularly susceptible to a redox imbalance, we assessed whether the treatment of A2780cis cells with the platinum complexes could induce ROS production.
As shown in Figure 4B, the platinum complexes 1 and 2 determine a great and concentration‐dependent ROS production in A2780cis cells. This induction of ROS species is similar between the two complexes and significantly larger than that observed with cisplatin. This result is in accordance with the data obtained by measuring the ΔΨ, where 1 was far more potent than cisplatin at the same concentration, and raises the hypothesis that the impairment in mitochondrial function induced by 1 and 2 may be responsible for the increase in ROS production. This large amount of ROS could determine the high cytotoxicity especially in A2780cis.
Consequences on cellular redox state
At this point, it is apparent that the new complexes are able to induce an effect on the cellular redox state. Therefore, we evaluated the impact of cell treatment with the platinum complexes on the thiol antioxidant system.
Interestingly, compounds 1 and 2 determined a considerable decrease of total thiols at the concentration of 10 μm in both cell lines (Figure 5A), while cisplatin was effective although to a lower extent, in decreasing the free thiols only in the cisplatin sensitive A2780 cell line. This result suggests that the complexes are able to overcome the cisplatin resistance and are more effective than the reference drug to induce protein oxidation.
Figure 5.
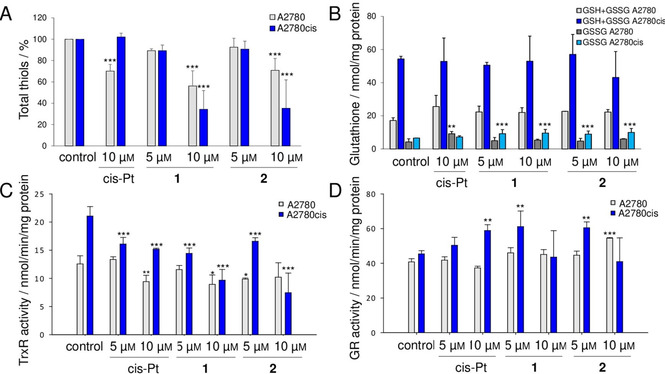
Effects of the platinum complexes on the cellular thiol redox systems. Cells (250 000/well) were treated with 5 and 10 μm 1 and 2 or cis‐Pt for 48 h with a refresh after 24 h. A) Total free thiols titrated with DTNB as described in the Experimental Section and expressed as a percentage with respect to the control. B) Total glutathione (GSH+GSSG) and oxidized glutathione (GSSG) levels in A2780 and A2780cis cells reported as nmol/mg protein. C) Total TrxR activity and D) total GR activity estimated spectrophotometrically and reported as nmol/min/mg protein. * p<0.05, ** p<0.01, *** p<0.001. Mean values ±SD of four experiments are reported.
Next, we determined the amount of total cellular glutathione and its redox state. Glutathione is a very abundant tripeptide in the cell environment and, in homeostatic conditions, the ratio between its reduced and oxidized forms (GSH/GSSG) is elevated whereas oxidative stimuli can lower it. [14] In addition, glutathione possesses detoxifying properties thanks to the activity of the enzymes glutathione S‐transferases (GSTs) and glutathione peroxidases (GPx) which utilize the reduced glutathione to inactivate electrophiles and peroxides, respectively.
After treatment of the two cell lines with the complexes at the concentration of 5 and 10 μm for 48 h, total glutathione amount was estimated. As shown in Figure 5B, none of the compounds was able to affect the total glutathione pool. From these data, it is interesting to note that A2780cis cells showed a greater basal amount of glutathione in comparison to A2780. This finding is in accordance with previous papers linking the cisplatin resistance to an increased expression of detoxification systems.[ 35 , 36 ] Regarding the analysis of the glutathione redox state, cisplatin elicited an increase of oxidized glutathione only in A2780 cells, while the two platinum compounds 1 and 2 were able to increase GSSG amount specifically in A2780cis cells. These data further support the idea that the two complexes are able to overcome the mechanisms involved in cisplatin resistance and emerge as potent stressors of the cisplatin‐resistant tumor cell type.
As ROS can damage not only proteins but also other cellular components such as lipid bilayers impairing the cell membrane functioning, we investigated the effect of the complexes by measuring lipid peroxidation. Intriguingly, 1 and 2 did not significantly increase lipid peroxidation in both cell lines (Figure S10) fostering a major effect on specific protein targets more than an unspecific oxidizing action.
The control of thiol redox state in the cell is regulated by the thioredoxin and the glutathione systems. For both, the reducing equivalents derive from NADPH and two pyridine nucleotide oxidoreductases mediate their utilization: thioredoxin reductase (TrxR) for the thioredoxin system and glutathione reductase (GR) for the glutathione network. The two proteins are homologous, but TrxR possesses a second active site bearing a peculiar selenocysteine in a flexible arm at the C‐ter [14] which confers to the enzyme an affinity to a broad range of substrates. Given the key role of TrxR and GR in the whole process of redox regulation, we explored the effect of the platinum complexes on these two enzymes.
Cells treated with 1 and 2 at the indicated concentrations, were lysed and the total TrxR and GR activities were estimated. Cisplatin was able to partially affect TrxR activity only in A2780 cells. Its inhibitory effect towards TrxR was already reported in literature. [37] As apparent in Figure 5C, concerning the complexes 1 and 2, we observed a good inhibitory action on thioredoxin reductase in A2780cis cells, while in the sensitive cell line, TrxR was less responsive. In addition, the compounds did not affect glutathione reductase activity (Figure 5D), which on the contrary is rather stimulated in A2780cis cells suggesting a sort of backup response to the stress induced to the cell.
Altogether these findings highlight that 1 and 2 are able to induce oxidative stress especially in the cisplatin‐resistant cancer cell line and that the activity of TrxR, one of the key enzymes for the modulation of the cellular antioxidant response, is principally affected. As reported in Figure 5C, A2780cis exhibited a high level of TrxR, that constitutes one of the characteristics of cisplatin resistance, and for this reason its inhibition may be a crucial mechanism in the observed redox imbalance in the cancer cell.
To further investigate the inhibition of the complexes toward thioredoxin reductase activity, we analyzed the effect of the platinum compounds on the isolated isoforms of the enzyme. In particular, we tested their inhibitory activity on the two mammalian isoforms of TrxR: the cytosolic TrxR1 and the mitochondrial TrxR2, on the homolog GR and on the TrxR from Escherichia coli, both lacking the second active site containing the selenocysteine.
As shown in Figures 6A and B, 1 and 2 exhibited a remarkable inhibitory effect on the two mammalian thioredoxin reductases, especially on TrxR1, the cytosolic isoform. Indeed, in comparison with cisplatin, both compounds showed a strong inhibitory activity in the nanomolar range of concentrations. In detail, for TrxR1, the IC50 was 2.27 nm and 2.59 nm for 1 and 2 respectively, while for cisplatin was 22.5 nm. For the mitochondrial isoform (TrxR2), the compounds showed an IC50 of 206 nm (1), 132 nm (2) and 405 nm (cis‐Pt). These results highlight the different action of 1 and 2 toward the two different isoforms of TrxR, but remarkably more efficient with respect to cisplatin. However, when the complexes were incubated with GR (Figure 6C) or TrxR from E. coli (Figure 6D), no inhibition was observed even at higher concentrations, in the micromolar range. The fact that these two latter enzymes both lack the selenocysteine in their structure, makes this amino acid a strong candidate for the site of inhibition of the two new transplatinum complexes on mammalian TrxR, probably involving coordination of platinum to the Sec‐containing redox center, similarly to cisplatin.[ 38 , 39 ] In addition, the ligands present in 1 and 2 might have a crucial role in the binding with selenol group which is essential for the enzyme activity.
Figure 6.
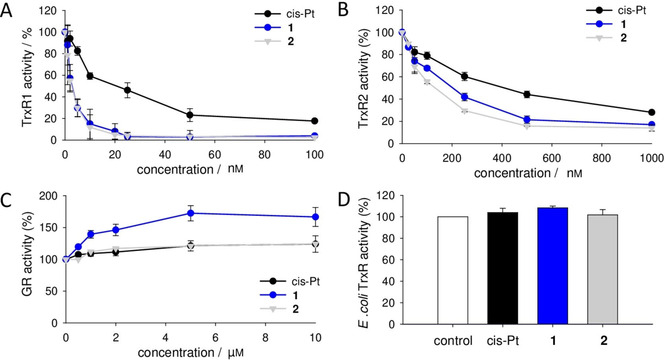
Effect of the platinum complexes on isolated enzymes in vitro. A) Purified rat liver TrxR1, B) rat liver TrxR2, and C) yeast GR were incubated with increasing concentrations of 1, 2 or cisplatin for 5 min. Then, their enzymatic activity was assessed spectrophotometrically, as described in the Experimental Section. D) E. coli TrxR was incubated with 10 μm of the three platinum complexes for 5 min prior to the determination of its enzymatic activity. Mean values ±SD of four experiments are reported.
Conclusion
The synthesis of trans‐[PtBr2(NHRR′)(PPh3)] complexes (R=Bu, R′=Bz, 1; R=R′=Bz, 2) is described. The complexes were characterized and the molecular structure of 2 was confirmed by single crystal X‐ray diffraction. The complexes show an interesting antiproliferative activity on human tumor cell lines and, more remarkably, overcome cisplatin resistance in ovarian carcinoma cells. The investigation of the mechanism of action reveals a cell uptake significantly higher with respect to the reference drug and the ability to act on several intracellular targets. The interaction with DNA was demonstrated and a significant depolarization of mitochondrial membrane also occurs. Moreover, 1 and 2 are able to induce a notable oxidative stress on resistant cells. In this connection, the effect on isolated in vitro TrxRs, key enzymes for the modulation of the cellular antioxidant response, allowed to propose selenocysteine as a possible target for the inhibitory activity exerted by the new complexes.
Taking into consideration that cisplatin resistance is the result of a myriad of phenotypic changes and that an impairment of drug accumulation represents one of the most prominent features, the multiple intracellular targets, and the remarkable intracellular accumulation of the new complexes, render the trans‐[PtBr2(NHRR′)(PPh3)] structure a compelling scaffold for the design of more effective anticancer agents.
Experimental Section
Chemistry
All manipulations were carried out under a dinitrogen atmosphere, if not otherwise stated. Solvents were purified according to reported procedures. [40] 1H‐, 13C‐, 31P‐ and 195Pt NMR spectra were recorded with a Bruker ‘‘Avance DRX400’’ spectrometer, in CDCl3 solution if not otherwise stated. Chemical shifts were measured in ppm (δ) from TMS by residual solvent peaks for 1H and 13C, from aqueous (D2O) H3PO4 (85 %) for 31P and from aqueous (D2O) H2PtCl6 for 195Pt. A sealed capillary containing C6D6 was introduced in the NMR tube to lock the spectrometer to the deuterium signal when non‐deuterated solvents were used. FTIR spectra in solid phase were recorded with a Perkin–Elmer ‘‘Spectrum One’’ spectrometer, equipped with an ATR accessory. The following abbreviations are used to describe band intensities: w (weak), m (medium), s (strong). Elemental analyses (C, H, N) were performed at Dipartimento di Chimica e Chimica Industriale, Università di Pisa. Trans‐[Pt(μ‐Br)Br(PPh3)]2 [20] was prepared according to a reported procedure. A sample of benzylbutylamine (BzBuNH) was prepared according to a reported procedure [17] and purified by distillation. A sample of dibenzylamine (Aldrich™) was passed over a short package of dry Al2O3 immediately before use.
General procedure for the synthesis of trans‐[PtBr2(NHRR’)(PPh3)]
A Schlenk tube equipped with a magnetic stirrer was charged with a suspension of [Pt(μ‐Br)Br(PPh3)]2 (0.200–0.400 g), in 1,2‐DCE (10–12 mL). A solution of the suitable dialkylamine (amine/Pt=1.15 molar ratio) in the same solvent (2 mL) was added under stirring at room temperature. The suspension turned into a yellow solution in few minutes. A sample of the solution was analyzed (31P NMR) and showed the formation of a single product. The liquid phase was concentrated under vacuum up to one fourth of the original volume and then treated with heptane. The yellow solid precipitated was recovered by filtration, washed with heptane and dried under vacuum.
trans‐[PtBr2(NHBzBu)(PPh3)] (1). From 0.2200 g of [Pt(μ‐Br)Br(PPh3)]2 a sample of 0.1650 g of 2 was obtained (59 % yield). C29H32Br2NPPt requires C 44.63, H 4.13, N 1.79 %. Experimental: C 44.38, H 4.23, N 1.85 %. FT‐I.R. (ATR, cm−1): 3192 m, 3053w, 2966w, 2927w, 2860w, 2197w, 2001w, 1958w, 1588w, 1480w, 1432 m, 1385w, 1311w, 1098 m, 999w, 963w, 912w, 743 s. 1H‐ NMR: 7.68 (m, 6H, Harom),7.55–7.35 (m, 14H, Harom) 4.55 (m, 1H, CHHPh), 4.30 (m, 1H, NH), 3.97 (m, 1H, CHHPh), 3.42 (m, 1H, NHCHH), 2.77 (m, 1H, NHCHH), 2.26 (m, 1H, CH2CHH), 1.84(m, 1H, CH2CHH), 1.46 (m, 1H, CHHCH3), 1.38 (m, 1H, CHHCH3), 0.96 (t, 3H, CH3). 13C‐NMR: 135.9, 135.0 (d, J=11 Hz), 130.6 (d, J=3 Hz), 130.2 (d, 1JC‐P=60 Hz), 130.1, 128.7, 128.2, 127.6 (d, J=12 Hz), 58.8, 51.3, 31.0, 20.1, 13.9. 31P‐NMR: 2.91 (1JP‐Pt=3486 Hz). 195 Pt NMR: −4081 (1JP‐Pt=3486 Hz).
trans‐[PtBr2(NHBz2)(PPh3)] (2). From 0.3560 g of [Pt(μ‐Br)Br(PPh3)]2 a sample of 0.3410 g of 2 was obtained (73 % yield). C32H30Br2NPPt requires C 47.19, H 3.71, N 1.72 %. Experimental: C 47.28, H 3.62, N 1.82 %. FT‐IR (ATR, cm−1): 3222w; 3198w; 3059w; 3030w; 2934w; 2877w; 1497w; 1481w; 1435 m; 1389w; 1187w;1154w; 1125w; 1097 m; 1072w; 997w. 1H NMR: 7.54 (m, 10H, Harom), 7.37 (m, 15H, Harom),4.60 (m, 3H, PhCHH+NH), 4.03 (m, 2H, PhCHH). 13C NMR: 135.7, 135.0 (d, J=10 Hz), 130.6, 130.4, 130.1 (d, 1JC‐P=63 Hz), 128.7, 128.3, 127.7, 127.6, 55.3. 31P NMR: 2.83 (1JP‐Pt=3516 Hz). 195 Pt‐ NMR: −4086 (1JP‐Pt=3516 Hz). A sample of 2 was recrystallized from CHCl3 and afforded crystals suitable for single crystal X‐ray diffraction, which confirmed the trans geometry of the complex.
X‐ray determination
Crystals of 2 were selected at room temperature (296 K), glued to glass fibers and analyzed with a Bruker Smart Breeze CCD diffractometer. Table 2 summarizes the lattice parameters and the space group. Intensity data were collected in the range of 2θ angles reported in Table 2. After correction for Lorentz and polarization effects and for absorption, the structure solution was obtained using the direct methods contained in SHELXS program. [41] The final reliability factors of the refinement procedure, done using SHELXL program, [42] are listed in Table 2. Other control calculations were performed with the programs contained in the WINGX suite. [43] Deposition number 2057464 (for 2) contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
Table 2.
Crystal data and structure refinement for complex 2.
|
Compound |
1 |
|
|---|---|---|
|
empirical formula |
C32H30Br2NPPt |
|
|
formula weight |
814.45 |
|
|
temperature |
296(2) K |
|
|
wavelength |
0.71073 Å |
|
|
crystal system |
Monoclinic |
|
|
space group |
P21/c |
|
|
unit cell dimensions |
a=11.7011(10) Å b=20.6928(17) Å c=12.3442(10) Å |
α=90° β=91.0630(10)° γ=90° |
|
volume |
2988.4(4) Å3 |
|
|
Z |
4 |
|
|
density (calculated) |
1.810 Mg/m3 |
|
|
absorption coefficient |
7.444 mm−1 |
|
|
F(000) |
1568 |
|
|
crystal size |
0.339×0.317×0.228 mm3 |
|
|
θ range for data collection |
3.121 to 28.020°. |
|
|
index ranges |
−15<=h<=13, 27<=k<=27, 15<=l<=16 |
– – – |
|
reflections collected |
41164 |
|
|
independent reflections |
7231 [R(int)=0.0321] |
|
|
completeness to θ=25.242° |
99.8 % |
|
|
refinement method |
Full‐matrix least‐squares on F2 |
|
|
data/restraints/parameters |
7231/0/334 |
|
|
goodness‐of‐fit on F 2 |
1.116 |
|
|
final R indices [I>2σ(I)] |
R1=0.0260, wR2=0.0627 |
|
|
R indices (all data) |
R1=0.0342, wR2=0.0652 |
|
|
extinction coefficient |
n/a |
|
|
largest diff. peak and hole |
1.962 and −0.597 e.Å−3 |
Biological assays
Cell cultures: A2780 (human ovarian carcinoma), A2780cis (human ovarian carcinoma cisplatin‐resistant) and HT29 (colorectal cancer) were grown in RPMI 1640 (Sigma–Aldrich R6504). HeLa (human cervix adenocarcinoma) and A549 (non‐small‐cell lung cancer) were grown in Ham's F12 Nutrient Mixture (Sigma‐Aldrich N6760 and N3540, respectively). 1.5 g/L NaHCO3, 10 % heat‐inactivated fetal bovine serum (Biowest), 100 U/mL penicillin, 100 μg/mL streptomycin, and 0.25 μg/mL amphotericin B (Sigma‐Aldrich A5955) were added to the media. To maintain drug‐resistance, A2780cis cells were supplemented with sub‐lethal concentration 1 μm cisplatin (Sigma–Aldrich P4394) every 2–3 passages. All cell lines were cultured at 37 °C in a humidified atmosphere under 5 % carbon dioxide in air.
Inhibition growth assay: Cells were seeded at a density of 2.5–3×104/well to a 24‐well cell culture plate and allowed to grow for 24 h before addition of different concentrations (from 0.1 to 20 μm) of the test complexes to the complete medium. Stock solutions of the new complexes were made in dimethylsulfoxide at 20 mm concentration and then diluted with complete medium in such a way that the final amount of solvent in each well did not exceed 0.5 %. Cisplatin was dissolved in 0.9 % NaCl at 4 mm concentration. After 72 h exposure, the drug‐containing medium was removed, the cells washed with phosphate buffered saline (PBS, 0.1 m NaCl, 2 mm KCl, 8 mm Na2HPO4 ⋅ 2 H2O, 1.5 mm KH2PO4) and harvested. A Trypan blue assay was performed to determine cell viability. Cytotoxicity data were expressed as IC50 values, i. e., the concentration of the test agent inducing 50 % reduction in cell number compared with control cultures.
Cell uptake: A2780cis cells (2×106) were seeded into 6‐well cell culture plate in complete medium and allowed to grow for 24 h. Then, the test agents were added at 100 μm concentration. At fixed incubation time (0–180 min), the cells were harvested, washed twice with 0.9 % NaCl and the pellet was added with 195 μL of HNO3 (65 %) and heated 90 °C for 1 h. Finally the samples were diluted in 37 % (v/v) HCl (3 volumes), and milliQ® water was added up to 5 mL. The content of P and Pt were detected by Inductively Coupled Plasma Atomic Emission Spectrometry (ICP‐AES) at emission lines λ (P)=178.290 nm and λ (Pt)=214.423 nm. A Spectroflame Modula sequential and simultaneous ICP‐spectrometer (ICP SPECTRO Arcos with EndOnPlasma torch) equipped with a capillary cross‐flow Meinhard nebulizer was used (Spectro Analytical). Analytical determinations were performed using a plasma power of 1.2 kW, a radiofrequency generator of 27.12 MHz and an argon gas flow with nebulizer, auxiliary, and coolant set at 1, 0.5 and 14 L/min, respectively. Calibration was carried out by preparing 15 multielement standard solutions containing platinum and phosphorous in the concentration range 0–800 mg L−1 (ppm). Standard solutions were prepared by diluting phosphorous and platinum stock solutions (1000 mg L−1, Spectrascan standards Teknolab, Norway) with 1 % HCl‐water solution.
Nucleic acids: Salmon testes DNA was purchased from Sigma‐Aldrich (D1626) The DNA concentration was determined using the molar absorption coefficient ϵ=6600 m −1 cm−1 at 260 nm. Plasmid pBR322 DNA was purchased from Fermentas Life Sciences.
Binding to DNA: The platinum and phosphorus analyses were performed according to a previously established method. [34] Briefly, salmon testes DNA dissolved in ultra‐pure water (9×10−4 m) was incubated at 37 °C with test complexes at a [DNA]/[drug]=10. At fixed incubation times (0–48 h) aliquots of exact volume were collected and DNA was precipitated twice with sodium acetate (up to 0.3 m concentration) and ice‐cold absolute ethanol (2 vol), washed with ice‐cold 70 % ethanol, dried at room temperature and solubilized in 250 μL of milliQ® water. All samples were then mineralized by the addition 195 μL of HNO3 (65 %) and heating at 90 °C for 20 min. Finally, each sample was diluted in 3 volumes of HCl (37 %) and milliQ® water, up to 5 mL, was added. The amount (ppb) of P and Pt was analyzed by ICP‐AES at emission lines λ (P)=178.290 nm and λ (Pt)=214.423 nm, as described in cell uptake section.
Interstrand‐crosslink assay: Supercoiled pBR322 plasmid DNA (1 μg) was incubated at 37 °C for 2 h with 1 U EcoRI restriction enzyme (Thermo Fisher Scientific) in 50 μL of reaction buffer (50 mm Tris, pH 7.5, 10 mm MgCl2, 100 mm NaCl, 0.02 % Triton X‐100, 0.1 mg/mL bovine serum albumin). The linearized DNA was then precipitated by the addition of Na‐acetate (up to 0.3 m concentration) and cold absolute ethanol (2 vol), washed with 70 % ethanol, dried at room temperature and dissolved in milliQ® water to obtain 0.1 μg/μL final concentration. The concentration was determined by Genova Nano Spectrophotometer (Jenway, Cole‐Parmer Ltd.). Linearized DNA (80 ng) was incubated with the complexes at indicated concentrations for 18 h at 37 °C in 10 μL final volume of milliQ® water. After incubation, 5 μL of each sample was added to 25 μL of denaturation buffer (8 m urea, 1 mm TRIS, 1 % Igepal (v/v)) and heated at 95 °C for 10 min. Then, 6 μL of loading buffer (0.125 % Bromophenol Blue, 0.125 % xylene cyanol and 50 % glycerol) were added and 30 μL of each sample were loaded into a 1.4 % agarose gel in TAE‐Urea buffer (0.04 m TRIS, 0.02 m glacial acetic acid, 0.001 m EDTA, 1 m urea). A non‐denatured sample was included as control. Electrophoresis at 40 V for 6 h was performed. Finally, the gel was stained with ethidium bromide solution (1 μg/mL) in TAE buffer, transilluminated by UV light, and the fluorescence emission was visualized by a CCD camera coupled to a Gel Doc XR apparatus (Bio‐Rad).
Mitochondrial transmembrane potential measurement: The mitochondrial transmembrane potential was measured by BD™ MitoScreen Kit (BD Pharmigen), according to Cossarizza et al. [44] Briefly, A2780cis cells (3×105) were seeded into 60 mm culture cell plates in complete medium and allowed to grow for 24 h. Cells were then treated with different concentrations of the test complexes and incubated for a further 40 h. The cells were harvested, centrifuged and resuspended in JC‐1 Working Solution. After incubation for 30 min at 37 °C in the dark, cells were washed in Assay buffer, resuspended and promptly analyzed by a FACSCanto II flow cytometer (Becton‐Dickinson, Mountain View, CA).
Formation of reactive oxygen species: A2780cis cells (1×104 per well) were seeded into a 96 well culture plate in complete growth medium. After incubation for 24 h, the medium was removed, and the cells washed with phosphate buffered saline (PBS). Cells were incubated with 10 μm of the fluorogenic probe CM‐H2DCFDA (Molecular Probes, Thermo Fisher Scientific) in PBS‐glucose 10 mm at 37 °C in the dark for 20 min, washed with PBS, and treated with increasing concentrations of the platinum complexes in PBS/glucose 10 mm. Fluorescence increase was detected by a plate reader (Tecan Infinite® M200 PRO, Männedorf, CH) at λ ex=485 nm and λ ex=527 nm.
Estimation of total thiol groups: A2780 and A2780cis cells (2.5×105 per well) were treated with the complexes (5–10 μm) for 48 h, with refresh after 24 h. Then, total thiols were measured with the Ellman's assay. [45] Briefly, at the end of incubation, cells were washed with PBS and then 1 mL of ice‐cold 0.2 m Tris ⋅ HCl buffer (pH 8.1), containing 7.2 M guanidine was added. The titration of free thiols after addition of 30 mm 5,5’‐dithiobis(2‐nitrobenzoic acid) (DTNB) was monitored spectrophotometrically at 412 nm on a Lambda 2 spectrophotometer (PerkinElmer). Protein quantifications were performed using the Bradford assay. [46]
Determination of glutathione concentration and redox state in cell lysates: A2780 and A2780cis cells (2.5×105 per well) were incubated with the complexes (5–10 μm) for 48 h, with refresh after 24 h. Then, cells were washed with cold PBS, and directly deproteinized with 6 % meta‐phosphoric acid. After 20 min at 4 °C cells were scraped and centrifuged at 15 800 g for 10 min at 4 °C. Supernatants were neutralized with 15 % Na3PO4 and utilized for total glutathione estimation. [47] Aliquots of the samples were incubated with 0.2 mM NADPH and 0.4 units of glutathione reductase from baker's yeast (Sigma‐Aldrich) in 0.2 M Na‐K‐Pi buffer (pH 7.4) added of 5 mm EDTA. The reaction was started by the addition of 0.25 mm DTNB and the absorbance was monitored spectrophotometrically at 412 nm for about 10 min on a Lambda 2 spectrophotometer. The nanomoles of total glutathione were calculated using a standard curve. For measurement of oxidized glutathione, sample aliquots were treated with 2 % 2‐vinylpyridine for 40 min before performing the assay. [48] For protein estimation, cell pellets were washed with 1 mL of ice‐cold acetone, centrifuged at 11 000 g, dried, then dissolved in 62.5 mm Tris ⋅ HCl buffer (pH 8.1) containing 1 % SDS and analyzed by the Lowry assay. [49]
TrxR and GR activities in cell lysates: Cells (1×106) were incubated for 48 h, with refresh after 24 h, in the presence of the compounds (5–10 μm) and then harvested and washed with PBS. Each sample was lysed with a modified radioimmunoprecipitation assay (RIPA) buffer: NaCl (150 mm), Tris ⋅ HCl (50 mm), EDTA (1 mm), 1 % Triton X‐100, 0.1 % SDS, 0.5 % sodium deoxycholate, NaF (1 mm) and an antiprotease cocktail (Complete, Roche) containing phenylmethylsulfonyl fluoride (PMSF, 0.1 mm). After 40 min at 4 °C, the lysates were centrifuged at 12 000 g, to discard the debris, and tested for total TrxR and GR activities. The determination of TrxR activity was performed in 0.2 m Na‐K‐Pi buffer (pH 7.4) added of 5 mm EDTA and NADPH (0.25 mm) and started by the addition of DTNB (0.1 m). The absorbance of the DTNB reduction product was monitored spectrophotometrically at 412 nm at 25 °C. GR activity of cell lysates was measured in Tris ⋅ HCl buffer (0.2 m, pH 8.1) containing EDTA (1 mm) and NADPH (0.25 mm). The assay was initiated by addition of glutathione disulfide (1 mm), and monitored spectrophotometrically at 340 nm at 25 °C.
Determination of lipid peroxidation: Lipid peroxidation was quantified by measuring MDA production. A2780 and A2780cis cells (2.5×105 per well) were incubated with the complexes (5–10 μM) for 48 h, with refresh after 24 h. Afterwards, cells were washed with PBS and treated with 1 mL of 0.1 N H2SO4 and 150 μL of 10 % phosphotungstic acid (PTA) for 10 min at room temperature. Then, cells were centrifuged at 15 800 g for 10 min at 4 °C. Discarded the supernatants, the pellets were resuspended in 1 mL of 0.1 N H2SO4 and 150 μL of PTA and again centrifuged as described above. Afterwards, the pellets were dissolved with 350 μL of a medium composed by 0.25 % NONIDET P‐40, 0.01 % BHT, 0.17 % thiobarbituric acid and incubated at 95 °C for 60 min. At the end, samples were ice‐cooled for 5 min and centrifuged at 15800 g for 10 min. Supernatants were treated with 400 μL of n‐butanol, vigorously mixed and centrifuged at 15 800 g for 15 min. The fluorescence of the upper phase was estimated (λ ex=530 nm, λ em=590 nm) using a Tecan Infinites M200 PRO plate reader. For protein determination, the pellets were washed with 500 μL of acetone/1 m HCl solution (98 : 2) for 10 min at 4 °C, centrifuged at 15 800 g for 10 min at 4 °C, dissolved in 75 μL of RIPA buffer and subjected to the Lowry assay. [49]
Enzymatic activity estimation of isolated rat TrxR1, rat TrxR2, yeast GR and E. coli TrxR: Highly purified cytosolic (TrxR1) and mitochondrial (TrxR2) thioredoxin reductases were prepared from rat liver according to Luthman and Holmgren [50] and Rigobello and Bindoli, [51] respectively. E. coli thioredoxin reductase was prepared following the procedure of Williams et al. [52] The protein content of the purified enzyme preparations was measured according to Lowry et al. [49] Glutathione reductase from baker's yeast was purchased from Sigma‐Aldrich. Thioredoxin reductase activity was determined by estimating the DTNB reducing properties of the enzymes in the presence of NADPH. Aliquots of highly purified TrxR in 0.2 m Na‐K‐Pi buffer (pH 7.4) added of 5 mm EDTA and 0.25 mm NADPH were preincubated for 10 min with various compounds. Afterwards, the reaction was initiated with 1 mm DTNB and monitored spectrophotometrically at 412 nm for about 10 min. GR activity was measured in 0.2 m Tris ⋅ HCl buffer (pH 8.1), 1 mm EDTA, and 0.25 mm NADPH in the presence of the various compounds. After an incubation of 10 min, the assay was started by the addition of 1 mm GSSG and followed spectrophotometrically at 340 nm.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
M.P.R. and V.S. acknowledge BIRD187299/18 granted by University of Padova (Italy). L. D. V. is grateful to the financial support provided by Dipartimento di Scienze del Farmaco‐Università di Padova‐Progetti di Ricerca di Dipartimento PRID 2017 – “An in depth investigation on novel Pt‐based agents to shed light on cancer resistance mechanims”. L.L., F.M. and S.S. ackowledge University of Pisa (Fondi di Ateneo 2020 and Progetti di Ricerca di Ateneo 2020‐PRA_2020_39). Consorzio Interuniversitario di Ricerca in Chimica dei Metalli nei Sistemi Biologici (CIRCMSB) is gratefully acknowledged.
M. Hyeraci, V. Scalcon, A. Folda, L. Labella, F. Marchetti, S. Samaritani, M. P. Rigobello, L. Dalla Via, ChemMedChem 2021, 16, 1956.
Contributor Information
Prof. Maria Pia Rigobello, Email: mariapia.rigobello@unipd.it.
Prof. Lisa Dalla Via, Email: lisa.dallavia@unipd.it.
References
- 1. Dasari S., Tchounwou P. B., Eur. J. Pharmacol. 2014, 740, 364–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kelland L., Nat. Rev. Cancer 2007, 7, 573–584. [DOI] [PubMed] [Google Scholar]
- 3. Rottenberg S., Disler C., Perego P., Nat. Rev. Cancer 2021, 21, 37–50. [DOI] [PubMed] [Google Scholar]
- 4. Wagstaff A. J., Ward A., Benfield P., Heel R. C., Drugs 1989, 37, 162–190. [DOI] [PubMed] [Google Scholar]
- 5. Ibrahim A., Hirschfeld S., Cohen M. H., Griebel D. J., Williams G. A., Pazdur R., Oncologist 2004, 9, 8–12. [DOI] [PubMed] [Google Scholar]
- 6. Dilruba S., Kalayda G. V., Cancer Chemother. Pharmacol. 2016, 77, 1103–1124. [DOI] [PubMed] [Google Scholar]
- 7. Ho G. Y., Woodward N., Coward J. I. G., Crit. Rev. Oncol. Hematol. 2016, 102, 37–46. [DOI] [PubMed] [Google Scholar]
- 8. Stewart D. J., Crit. Rev. Oncol. Hematol. 2007, 63, 12–31. [DOI] [PubMed] [Google Scholar]
- 9. Stordal B., Pavlakis N., Davey R., Cancer Treat. Rev. 2007, 33, 347–357. [DOI] [PubMed] [Google Scholar]
- 10. Shen D. W., Pouliot L. M., Hall M. D., Gottesman M. M., Pharmacol. Rev. 2012, 64, 706–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Arnér E. S. J., Holmgren A., Semin. Cancer Biol. 2006, 16, 420–426. [DOI] [PubMed] [Google Scholar]
- 12. Scalcon V., Bindoli A., Rigobello M. P., Free Radical Biol. Med. 2018, 127, 72–79. [DOI] [PubMed] [Google Scholar]
- 13. Lu J., Holmgren A., Free Radical Biol. Med. 2014, 66, 75–87. [DOI] [PubMed] [Google Scholar]
- 14. Bindoli A., Rigobello M. P., Antioxid. Redox Signaling 2013, 18, 1157–1193. [DOI] [PubMed] [Google Scholar]
- 15. Dalla Via L., Garcia-Argaez A. N., Adami A., Grancara S., Martinis P., Toninello A., Belli Dell'Amico D., Labella L., Samaritani S., Bioorg. Med. Chem. 2013, 21, 6965–6972. [DOI] [PubMed] [Google Scholar]
- 16. Belli Dell'Amico D., Dalla Via L., Garcia-Argaez A. N., Labella L., Marchetti F., Samaritani S., Polyhedron 2015, 85, 685–689. [Google Scholar]
- 17. Dalla Via L., Garcia-Argaez A. N., Agostinelli E., Belli Dell'Amico D., Labella L., Samaritani S., Bioorg. Med. Chem. 2016, 24, 2929–2937. [DOI] [PubMed] [Google Scholar]
- 18. Marzo T., Bartoli G., Gabbiani C., Pescitelli G., Severi M., Pillozzi S., Michelucci E., Fiorini B., Arcangeli A., Quiroga A. G., Messori L., BioMetals 2016, 29, 535–542. [DOI] [PubMed] [Google Scholar]
- 19. Tolbatova I., Marzo T., Cirri D., Gabbiani C., Coletti C., Marrone A., Paciotti R., Messori L., Re N., J. Inorg. Biochem. 2020, 209, 111096. [DOI] [PubMed] [Google Scholar]
- 20. Fioco D., Belli Dell′Amico D., Labella L., Marchetti F., Samaritani S., Eur. J. Inorg. Chem. 2019, 36, 3970–3974. [Google Scholar]
- 21. Pregosin P. S., Coord. Chem. Rev. 1982, 44, 247–291. [Google Scholar]
- 22. Hyeraci M., Colalillo M., Labella L., Marchetti F., Samaritani S., Scalcon V., Rigobello M. P., Dalla Via L., ChemMedChem 2020, 15, 1464–1472. [DOI] [PubMed] [Google Scholar]
- 23. Holohan C., Van Schaeybroeck S., Longley D. B., Johnston P. G., Nat. Rev. 2013, 13, 714–726. [DOI] [PubMed] [Google Scholar]
- 24. Fichtinger-Schepman A. M. J., van der Veer J. L., den Hartog J. H. J., Lohman P. H. M., Reedijk J., Biochemistry 1985, 24, 707–713. [DOI] [PubMed] [Google Scholar]
- 25. Cohen G. L., Bauer W. R., Barton J. K., Lippard S. J., Science 1979, 203, 1014–1016. [DOI] [PubMed] [Google Scholar]
- 26. Dalla Via L., Di Noto V., Gia O., Marciani Magno S., J. Photochem. Photobiol. B 2005, 79, 59–65. [DOI] [PubMed] [Google Scholar]
- 27. Deans A. J., West S. C., Nat. Rev. Cancer 2011, 11, 467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Noll D. M., McGregor Mason T., Miller P. S., Chem. Rev. 2006, 106, 277–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brabec V., Leng M., Proc. Natl. Acad. Sci. USA 1993, 90, 5345–5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brabec V., Prog. Nucleic Acid Res. Mol. Biol. 2002, 71, 1–68. [DOI] [PubMed] [Google Scholar]
- 31. Bock F. J., Tait S. W. G., Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [DOI] [PubMed] [Google Scholar]
- 32. Fulda S., Galluzzi L., Kroemer G., Nat. Rev. Drug Discovery 2010, 9, 447–464. [DOI] [PubMed] [Google Scholar]
- 33. Pathania D., Millard M., Neamati N., Adv. Drug Delivery Rev. 2009, 61, 1250–1275. [DOI] [PubMed] [Google Scholar]
- 34. Dalla Via L., Santi S., Di Noto V., Venzo A., Agostinelli E., Calcabrini A., Condello M., Toninello A., J. Biol. Inorg. Chem. 2011, 16, 695–713. [DOI] [PubMed] [Google Scholar]
- 35. Galluzzi L., Senovilla L., Vitale I., Michels J., Martins I., Kepp O., Castedo M., Kroemer G., Oncogene 2012, 31, 1869–1883. [DOI] [PubMed] [Google Scholar]
- 36. Citta A., Schuh E., Mohr F., Folda A., Massimino M. L., Bindoli A., Casini A., Rigobello M. P., Metallomics 2013, 5, 1006–1015. [DOI] [PubMed] [Google Scholar]
- 37. Prast-Nielsen S., Cebula M., Pader I., Arnér E. S. J., Free Radical Biol. Med. 2010, 49, 1165–1178. [DOI] [PubMed] [Google Scholar]
- 38. Rackham O., Shearwood A. M., Thyer R., McNamara E., Davies S. M., Callus B. A., Miranda-Vizuete A., Berners-Price S. J., Cheng Q., Arnér E. S. J., Filipovska A., Free Radical Biol. Med. 2011, 50, 689–699. [DOI] [PubMed] [Google Scholar]
- 39. Cheng P., Liu H., Li Y., Pi P., Jiang Y., Zang S., Li X., Fu A., Ren X., Xu J., Holmgren A., Lu J., Biochem. Pharmacol. 2020, 117, 113873. [DOI] [PubMed] [Google Scholar]
- 40.W. L. F. Armarego, D. D. Perrin in Purification of Laboratory Chemicals, Butterworth-Heinemann, 1996.
- 41.G. M. Sheldrick, SHELXS, version 2014/7, 2013, Georg-August-Universität of Göttingen (Germany).
- 42.G. M. Sheldrick, SHELXL (Release 97–2), 1998, University of Göttingen (Germany).
- 43. Farrugia L. J., J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar]
- 44. Cossarizza A., Baccarani-Contri M., Kalashnikova G., Franceschi C., Biochem. Biophys. Res. Commun. 1993, 197, 40–45. [DOI] [PubMed] [Google Scholar]
- 45. Ellman G. L., Arch. Biochem. Biophys. 1959, 82, 70–77. [DOI] [PubMed] [Google Scholar]
- 46. Bradford M. M., Anal. Biochem. 1976, 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 47. Tietze F., Anal. Biochem. 1969, 27, 502–522. [DOI] [PubMed] [Google Scholar]
- 48. Anderson M. E., Methods Enzymol. 1985, 113, 548–555. [DOI] [PubMed] [Google Scholar]
- 49. Lowry O. H., Rosebrough N. J., Farr A. L., Randall R. J., J. Biol. Chem. 1951, 193, 265–275. [PubMed] [Google Scholar]
- 50. Luthman M., Holmgren A., Biochemistry 1982, 21, 6628–6633. [DOI] [PubMed] [Google Scholar]
- 51. Rigobello M. P., Bindoli A. in Methods in Enzymology: Thiol Redox Transitions in Cell Signaling, Pt B: Cellular Localization and Signaling, Vol. 474 (Ed.: Cadenas and L. Packer E.), 2010, pp. 109–122. [Google Scholar]
- 52. Williams C. H. Jr., Zanetti G., Arscott L. D., McAllister J. K., J. Biol. Chem. 1967, 242, 5226–5231. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary