

Abstract
The regulation of enzyme activity is a method to control biological function. We report two systems enabling the ultrasound‐induced activation of thrombin, which is vital for secondary hemostasis. First, we designed polyaptamers, which can specifically bind to thrombin, inhibiting its catalytic activity. With ultrasound generating inertial cavitation and therapeutic medical focused ultrasound, the interactions between polyaptamer and enzyme are cleaved, restoring the activity to catalyze the conversion of fibrinogen into fibrin. Second, we used split aptamers conjugated to the surface of gold nanoparticles (AuNPs). In the presence of thrombin, these assemble into an aptamer tertiary structure, induce AuNP aggregation, and deactivate the enzyme. By ultrasonication, the AuNP aggregates reversibly disassemble releasing and activating the enzyme. We envision that this approach will be a blueprint to control the function of other proteins by mechanical stimuli in the sonogenetics field.
Keywords: Enzymes, Nucleic acids, Polymer mechanochemistry, Sonogenetics, Ultrasound
The precise regulation of protein activity is important to understand complex biological signaling networks and to devise new therapeutic approaches. Here, we use either polyaptamers or split aptamer‐conjugated AuNPs to inhibit the activity of thrombin. Mechanical force generated by ultrasound is exploited to activate thrombin, thereby catalyzing the formation of fibrin from fibrinogen.
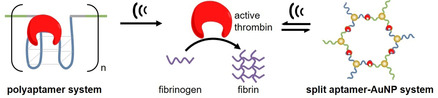
Introduction
Proteins fulfill many functions within the body and are involved in nearly all biological processes. [1] The dysregulation of such processes is associated to a number of pathological conditions, such as cancer and neurological disorders, and can have multiple causes. [2] Hence, the precise regulation of protein activity is important to understand complex biological signaling networks and to devise new therapeutic approaches.[ 3 , 4 ] Over the last decades, various internal and external stimuli have been used for this purpose.[ 5 , 6 , 7 ] Among these, light is popular [8] due to its non‐invasive character and high spatiotemporal resolution. [9] However, light application can be accompanied by UV‐toxicity, thermal irradiation damage to healthy cells, and low tissue penetration depths. [10] Other external stimuli used are temperature, [11] electric fields, [12] and small molecules, [13] yet with a lack of spatiotemporal control.
Mechanical force exerted by ultrasound (US) [14] is only sparsely used in this context. While seminal discoveries made use of protein activity regulation indirectly by manipulating gene transcription and protein expression with mechanosensitive ion channels in cells,[ 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 ] we genetically engineered proteins with supercharged polypeptide chains rendering them intrinsically US‐responsive in vitro. [23] The lack of further examples is surprising as US has been used as stimulus to release small molecules from microbubbles, [24] liposomes,[ 25 , 26 ] and nanoparticles [27] underlining its biocompatibility, excellent tissue penetration depth, and capability for spatiotemporally remote‐controlled release. [28]
In polymer mechanochemistry, force‐reactive functional molecular motifs (mechanophores) are designed such that they undergo transformations on the molecular level by rearranging or cleaving bonds with reasonable specificity.[ 29 , 30 , 31 , 32 , 33 , 34 ] Mechanophore activation can occur not only in bulk material by exposure to mechanical stress and strain but also in solution via the collapse of US‐induced cavitation bubbles generating shear stress. [35] Non‐invasive US is used for biomedical applications as it combines spatial and temporal dosage with easy regulation of tissue penetration depth by varying frequency and energy through exposure time. [36] Though polymer mechanochemistry mainly employs low frequency, high energy US (20 kHz), [35] and compatibilization with clinically employed high frequency, low energy US will take considerable efforts, [36] the first examples of site‐selective bond‐scission for the release of drug molecules[ 37 , 38 , 39 , 40 ] underline the potential of this direction of research.
Using the principles of mechanochemistry, we here demonstrate the activation of a protein inhibited by specific binding to high molar mass polynucleotide aptamers (polyaptamers) through ultrasonication. We selected the enzyme thrombin that catalyzes the formation of fibrin from fibrinogen as a model protein, since its activity can be easily monitored by light scattering. [8] The deactivating polyaptamers (pTBA15) were prepared by rolling circle amplification (RCA) (Figure 1 a).[ 41 , 42 , 43 ] Based on the robust recognition and protein‐loading ability of the TBA15 aptamer, thrombin was captured and its activity was inhibited through the formation of a well‐defined aptamer‐protein complex.[ 7 , 8 ] Upon irradiation with US, the polyaptamer loaded with thrombin was stretched and the specific non‐covalent interactions of thrombin and the polyaptamer (hydrogen bonds, hydrophobic interactions, etc.) within the loop structures were cleaved. In addition, non‐specific chain fragmentation along the phosphodiester backbone was induced. Both resulted in the release of the protein and subsequent turn‐on of its catalytic activity (Figure 1 b). [39] Notably, this approach circumvents the limitations of single mechanophore architectures [44] suffering from low loading ratios [45] to release sufficient amounts of functional species [46] or the difficulty to accurately control the chain‐centered position of the mechanophore. [47]
Figure 1.

Schematic depiction of two approaches to US activation of thrombin. a) pTBA15 formation by RCA. b) Thrombin loading and deactivation by pTBA15 and subsequent release and activation via US to catalyze the formation of fibrin from fibrinogen. c) Split aptamer‐enabled aggregation of AuNPs in the presence of thrombin. The AuNPs are functionalized with TBA15‐L or TBA15‐R. US treatment induces reversible disaggregation leading to thrombin release.
To further expand the scope of the method introduced here, we prepared an aptamer‐nanoparticle system to deactivate and subsequently release thrombin (Figure 1 c). Therefore, we prepared two types of gold nanoparticles (AuNPs) functionalized on the surfaces with the thiolated split aptamer TBA15‐L or TBA15‐R. In the presence of thrombin, the two split aptamer parts assembled into the intact aptamer tertiary structure and at the same time induced the aggregation of the two AuNP types. Upon the application of US, these aggregates disassembled, and thrombin was released and hence activated. Notably, this process could be repeated for several cycles without obvious fatigue rendering this hybrid structure reversible adding new characteristics to US‐responsive protein systems. [23]
Results and Discussion
Initially we evaluated the binding quality of the monoaptamer mTBA15 to thrombin. Gel electrophoresis of thrombin@mTBA15 revealed that binding was near stoichiometric and no binding was observed using a random DNA sequence (Ran‐mDNA, Figure S1a) highlighting the selectivity of mTBA15. Subsequently, the propensity of mTBA15 to inhibit thrombin activity was investigated. While both free thrombin and thrombin with Ran‐mDNA induced fibrin formation, thrombin@mTBA15 showed greatly diminished activity of ca. 10 % compared to pure thrombin, which is consistent with the low dissociation constant (K d) around 0.6 nM of mTBA15 reported in the literature (Figure S1a and Table S1). [48]
Then, the circular template for pTBA15 synthesis was prepared by ligation of 5′‐phosphorylated linear DNA with T4 DNA ligase (Figure S2a) and the successful synthesis was confirmed by agarose gel electrophoresis with the circularized template exhibiting reduced mobility compared to the linear template. pTBA15 received from the subsequent RCA process was then analyzed by gel electrophoresis. The RCA product remained within the wells indicating its high molar mass (Figure S2b). We then determined the binding quality of thrombin@pTBA15 using gel electrophoresis by staining DNA with ROTI®GelStain and labeling thrombin with the fluorescent dye Cy3 (Figure S3a). The absence of fluorescence originating from free thrombin together with the superposition of Cy3 and ROTI®GelStain fluorescence in the thrombin@pTBA15 lane showed that all thrombin was bound to pTBA15 (Figure 2 a). Again, non‐specific binding was ruled out as random polynucleotides (Ran‐pDNA) did not show such features (Figure S3b). Light scattering was used to prove that thrombin@pTBA15 showed considerably diminished catalytic activity of ca. 21 % compared to pristine thrombin for the formation of fibrin (Figure 2 b and S4).
Figure 2.
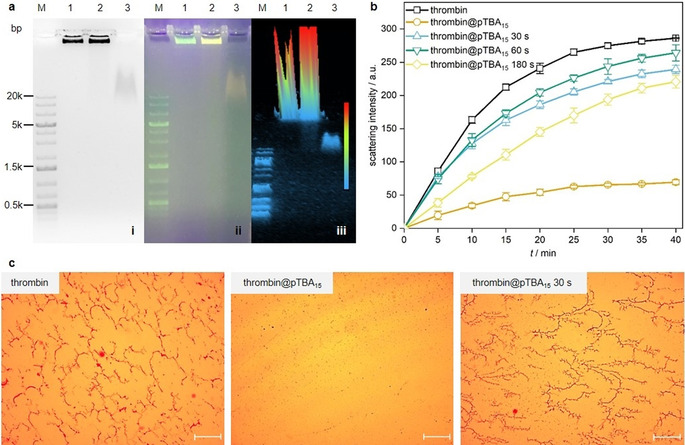
Activation of thrombin@pTBA15 by US (20 kHz). a) Agarose gel (1 %) for binding test of pTBA15 with Cy3‐thrombin. (i) Fluorescence image in grayscale; (ii) corresponding fluorescence image in color scale; (iii) corresponding 3D gel image. Lane M: 1 kb plus DNA marker; Lane 1: pTBA15; Lane 2: Cy3‐thrombin@pTBA15; Lane 3: Cy3‐thrombin. b) Real‐time light scattering spectra of fibrinogen solution with thrombin (control) and thrombin@pTBA15, treated with US for 0, 30, 60, and 180 s, respectively. c) Optical microscope images of fibrinogen treated with thrombin, thrombin@pTBA15, and thrombin@pTBA15 treated with US for 30 s. Scale bar: 50 μm. Mean values ± SD from the mean, N=3 independent experiments.
Subsequently, we investigated the mechanochemical activation of thrombin@pTBA15 by US (Figure S5 and S6). After 30–60 s US application, near‐complete recovery of the original thrombin catalytic activity (80–90 %) for fibrin formation was observed (Figure S6). Longer US exposition reduced the activity, as thrombin itself is a biomacromolecule denaturing under shear force in solution (Figure S4). Conversely, thrombin@mTBA15 did not show sensitivity towards US leading to no fibrin formation even after 180 s ultrasonication.
Optical microscopy confirmed the above results: Combining fibrinogen and thrombin@pTBA15 treated with US for 30–60 s resulted in dense fibrin fiber networks similar to pristine thrombin (Figure 2 c). Conversely, only small fibrin fibers were observed when thrombin@pTBA15 was not subjected to US. Underlining the results obtained by light scattering, thrombin activity was diminished after 180 s treatment of thrombin@pTBA15 with US (Figure S7). This was in line with the activity profile of pure thrombin with different ultrasonication times (Figure S8). Importantly, thrombin@mTBA15 did not show any fiber formation with or without US (Figure S9), once more highlighting the mechanochemical origin of the observed effects.
In addition to the visual inspection of fibrin formation, we verified the activation of thrombin@pTBA15 using fibrinogen‐modified gold nanoparticles (F‐AuNPs) in a catalytic colorimetric assay. While free F‐AuNPs catalyzed the reduction of the yellow‐colored 4‐nitrophenol with NaBH4, the active thrombin led to aggregation of F‐AuNPs into fibrin meshes, rendering the AuNPs inaccessible for the reduction of 4‐nitrophenol. The remaining yellow color of 4‐nitrophenol is hence an indicator for the presence of free thrombin, while loss of color indicated the presence of free AuNPs (Scheme S1). We employed 13 nm F‐AuNPs (2 nM) (Figure S10) that catalyzed the reduction of 4‐nitrophenol (Figure S11), which was inhibited by the presence of thrombin. While thrombin@pTBA15 and thrombin@mTBA15 allowed the reduction to proceed, application of US for 30–60 s led to thrombin release from pTBA15, subsequent fibrin formation, and inhibition of the reduction reaction cascade. Note here that this process was irreversible as thrombin release was accompanied by non‐specific chain scission along the polyaptamer backbone (Figure S5). [39]
For broadening the scope of thrombin activation by US and to introduce reversibility, we used AuNPs as building blocks to develop a nanoparticle‐based US‐responsive system. Using AuNPs as platform had several advantages, such as easy synthesis in aqueous solution, uniform sizes and shapes, low biotoxicity, and efficient functionalization with oligonucleotides.[ 49 , 50 ] Using split aptamers was inspired by previous research showing that these can reassemble into an intact G‐quadruplex structure in the presence of their target molecules.[ 51 , 52 ] Hence, we split the thrombin aptamer into TBA15‐L and TBA15‐R according to literature protocols. [53] The successful functionalization of AuNPs with the split aptamers was firstly confirmed by light scattering where the hydrodynamic diameters of AuNPs increased after conjugation with TBA15‐L or TBA15‐R from around 13 to 25 nm (Figure S12a). Moreover, successful functionalization was confirmed via agarose gel (3 %) electrophoresis where the TBA15‐L‐ or TBA15‐R‐conjugated AuNPs exhibited reduced mobility compared to the pristine AuNPs (Figure S12b).
The aggregation process in the presence of thrombin was followed by UV‐vis absorption spectroscopy (Figure 3 a). With increasing thrombin concentration, the surface plasmon resonance (SPR) bands of Au@TBA15 shifted to longer wavelengths indicating aggregation. This observation was supported by transmission electron microscopy (TEM) where AuNP clusters could easily be discerned (Figure S13).
Figure 3.
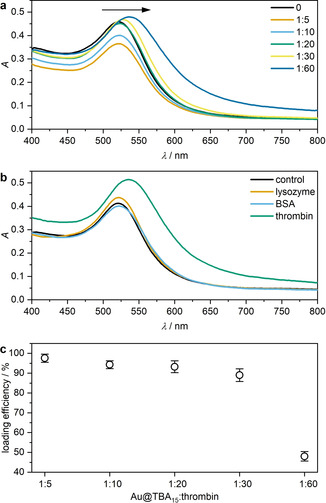
Aggregation behavior of split aptamer‐functionalized AuNPs in the presence of thrombin. a) UV‐vis spectra of different molar ratios Au@TBA15:thrombin. b) UV‐vis spectra of Au@TBA15 reacted with lysozyme, BSA, and thrombin individually to test specific aggregation behavior at a molar ratio Au@TBA15:protein=1:30. c) Thrombin loading efficiency as a function of the Au@TBA15:thrombin ratio. Mean values ± SD from the mean. N=3 independent experiments.
To assess the selectivity and specificity of the system, control experiments were performed with the proteins lysozyme and bovine serum albumin (BSA). Therefore, Au@TBA15 was incubated with the proteins and aggregation only observed when these acted as NP crosslinkers by binding to the aptamers.
Indeed, UV‐vis spectra showed that only in the presence of thrombin, but neither lysozyme nor BSA, the SPR band was shifted bathochromically thus confirming above hypothesis (Figure 3 b). This result was again confirmed by TEM (Figure S14). Optimization of the thrombin loading efficiency was performed using a thrombin activity assay kit. This was based on free thrombin cleaving a substrate bearing a Förster resonance energy transfer (FRET) pair into two fragments resulting in the increase in fluorescence intensity of the FRET donor 5‐carboxyfluorescein (5‐FAM) in linear correlation to the thrombin concentration (Figure S15). We found an optimal loading efficiency of ca. 30 thrombin molecules per Au@TBA15 (Figure 3 c), which is a reasonable value given that a single AuNP is functionalized with ca. 40 split aptamers (Figure S16).
Subsequently, we applied US to induce the disassembly of thrombin@Au@TBA15. With progressing sonication time, the SPR band shifted hypsochromically (Figure 4 a,c) and dynamic light scattering (DLS) revealed decreasing hydrodynamic diameters (Figure 4 b,c) both indicating the successful US‐induced disassembly. While the hypsochromic shift was attributed to the decrease of the plasmonic resonator size, the hypochromic effect likely had its origin in the dissolution and degradation of the AuNPs into Au atoms over the course of the sonication. Both the final SPR peak maximum and the hydrodynamic diameter obtained after 300 s sonication resembled those of single, non‐assembled Au@TBA15 and the disassembly process could even be followed by the naked eye (Figure 4 d). Most notably, this US‐induced transition was reversible. The sample was transformed to its disassembled state simply by US irradiation for 15 s and returned to its assembled state by equilibrating at room temperature for 30 min, as indicated by the SPR band in UV‐vis measurements (Figure 5 a), DLS (Figure 5 b), and TEM (Figure 5 c). The hysteresis visible in Figure 5 a was possibly caused by the interaction of denatured thrombin and AuNPs.
Figure 4.
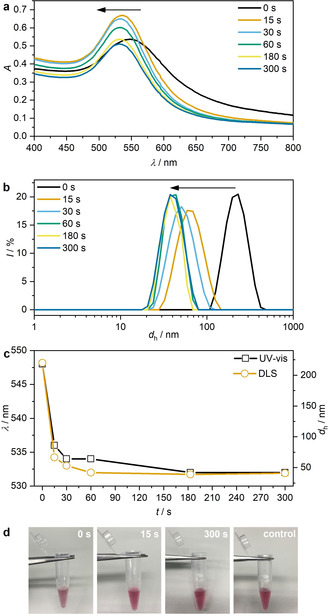
US‐induced disassembly of thrombin@Au@TBA15 for different sonication times (20 kHz). a) UV‐vis spectra, b) hydrodynamic diameter d h measured by DLS, c) evolution of UV‐vis and DLS peak maxima, and d) photographs of the solutions.
Figure 5.
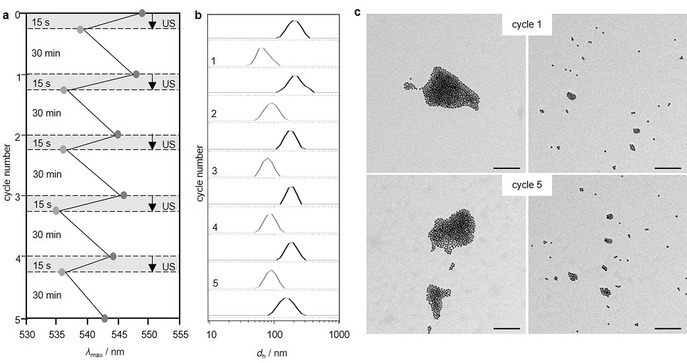
Reversibility of the US‐induced disassembly of thrombin@Au@TBA15 (20 kHz). Five complete disassembly‐assembly cycles (15 s US and 30 min recovery in each cycle) as followed by a) UV‐vis spectroscopy and b) DLS. c) TEM images of samples that contained aggregated (left) and dispersed (right) AuNPs with US in cycle 1 and cycle 5. Scale bar: 100 nm.
Eventually, we investigated whether the catalytic activity of thrombin@Au@TBA15 could be turned on by US. Already 15 s of US irradiation resulted in 50 % of pristine thrombin activity and more than 70 % activity was obtained after 30 s (Figure 6 a). Similar to thrombin@pTBA15, prolonged exposure of the system to US led to a reduced catalytic activity due to irreversible denaturation of thrombin. However, Au@TBA15 efficiently deactivated thrombin while its US‐induced release enabled the catalytic transformation of fibrinogen to fibrin as shown by light scattering (Figure 6 b). Optical microscopy further confirmed these results (Figure 6 c and S17).
Figure 6.
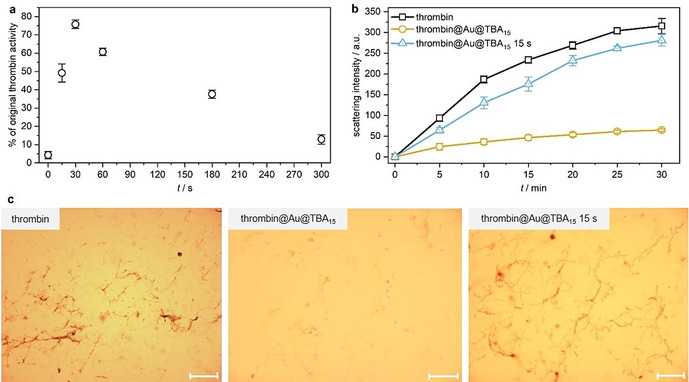
Thrombin activation from thrombin@Au@TBA15 over the course of US irradiation (20 kHz). a) Quantification of thrombin activity. b) Real‐time light scattering of fibrinogen solution with thrombin, thrombin@Au@TBA15, and the latter treated with US for 15 s. c) Optical micrographs of fibrinogen treated with thrombin, thrombin@Au@TBA15, and the latter treated with US for 15 s. Scale bar: 50 μm. Mean values ± SD from the mean. N=3 independent experiments.
To underline the applicability of our developed systems using clinically established US techniques, we performed sonication experiments for 6 min using a focused ultrasound setup at a frequency of 5 MHz. The sonication conditions had little influence on the activity of thrombin (Figure S18). For both the polyaptamer‐ and the AuNP‐based systems more than ≈80 % and ≈75 % of the catalytic activity compared to unaltered thrombin were restored for thrombin@pTBA15 (Figure S19) and thrombin@Au@TBA15 (Figure S20), respectively. We used the catalytic colorimetric assay described above to visualize the activation process using either thrombin@pTBA15 (Figure 7 a) or thrombin@Au@TBA15 (Figure 7 b) per in situ sonication with spatial resolution. Only the wells located on the cross shape were irradiated with US resulting in the activation of thrombin and thus inhibition of the catalytic reduction and discoloration (Figure S21). Untreated wells placed on the vertices of the squares were unaffected and discoloration proceeded with the reduction.
Figure 7.
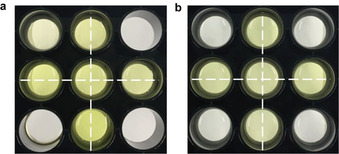
Colorimetric assay for thrombin activation by focused US (5 MHz) in well plates containing solutions of F‐AuNPs, 4‐nitrophenol, NaBH4, and a) thrombin@pTBA15 or b) thrombin@Au@TBA15. US was applied to only those wells within the crossed pattern. Single well diameter: 1.5 cm.
Conclusion
In summary, we presented the first molecular design to activate proteins from their deactivated aptamer‐bound parent form by using mechanical force in the form of ultrasound. We showed that the aptamer‐based deactivation of thrombin was possible both irreversibly by using long polyaptamers or reversibly by using split aptamers conjugated to gold nanoparticles. Thereby, we showed that the formation of fibrin from fibrinogen could be controlled by ultrasound as a trigger. Importantly, cleaving bonds site‐specifically using the mechanochemical approach, our method compares favorably to previous ultrasound‐based methods, where local heating induced by high‐intensity focused ultrasound (HIFU) released proteins from nanodroplets, [54] microcapsules, [55] or hydrogels. [56] In addition and considering other mechanochemical release systems that only hold a single mechanophore within a large polymer chain, [44] our system allows for a considerably higher mechanophore loading along the polymer chain. Another remarkable feature presented here in the context of mechanical activation of enzymes is the reversibility of the gold nanoparticle system involving split aptamers. Importantly, we demonstrated the working principle both with ultrasound producing inertial cavitation and clinically established therapeutic focused ultrasound.
Using external stimuli to control protein function has become extremely relevant, as evidenced by the growing field of optogenetics and its impact on neuroscience and related areas.[ 57 , 58 ] We hence believe that the approaches presented here hold great promise for future applications that may lie in the remote control of protein activity in cells and in vivo leading to sonogenetic technologies.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by The European Union (European Research Council Advanced Grant SUPRABIOTICS No. 694610) and The China Scholarship Council to P.Z. and J.F. Moreover, R.G. is grateful for support by a Freigeist‐Fellowship of the Volkswagen Foundation (No. 92888). Parts of the analytical investigations were performed at the Center for Chemical Polymer Technology CPT, which was supported by the European Commission and the federal state of North Rhine‐Westphalia (No. 300088302). Open access funding enabled and organized by Projekt DEAL.
P. Zhao, S. Huo, J. Fan, J. Chen, F. Kiessling, A. J. Boersma, R. Göstl, A. Herrmann, Angew. Chem. Int. Ed. 2021, 60, 14707.
Contributor Information
Prof. Shuaidong Huo, Email: huosd@xmu.edu.cn.
Prof. Andreas Herrmann, Email: herrmann@dwi.rwth-aachen.de.
References
- 1. Qin X., Yu C., Wei J., Li L., Zhang C., Wu Q., Liu J., Yao S. Q., Huang W., Adv. Mater. 2019, 31, 1902791. [DOI] [PubMed] [Google Scholar]
- 2. Le Quesne J. P., Spriggs K. A., Bushell M., Willis A. E., J. Pathol. 2010, 220, 140–151. [DOI] [PubMed] [Google Scholar]
- 3. Gorostiza P., Isacoff E. Y., Science 2008, 322, 395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kramer R. H., Mourot A., Adesnik H., Nat. Neurosci. 2013, 16, 816–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lu Y., Sun W., Gu Z., J. Controlled Release 2014, 194, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhao P., Liu Y., Xiao L., Deng H., Du Y., Shi X., J. Mater. Chem. B 2015, 3, 7577–7584. [DOI] [PubMed] [Google Scholar]
- 7. Wang J., Wei Y., Hu X., Fang Y.-Y., Li X., Liu J., Wang S., Yuan Q., J. Am. Chem. Soc. 2015, 137, 10576–10584. [DOI] [PubMed] [Google Scholar]
- 8. Xie S., Qiu L., Cui L., Liu H., Sun Y., Liang H., Ding D., He L., Liu H., Zhang J., Chen Z., Zhang X., Tan W., Chem 2017, 3, 1021–1035. [Google Scholar]
- 9. Vázquez-González M., Willner I., Angew. Chem. Int. Ed. 2020, 59, 15342–15377; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 15458–15496. [Google Scholar]
- 10. Karimi M., Sahandi Zangabad P., Baghaee-Ravari S., Ghazadeh M., Mirshekari H., Hamblin M. R., J. Am. Chem. Soc. 2017, 139, 4584–4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stayton P. S., Shimoboji T., Long C., Chilkoti A., Ghen G., Harris J. M., Hoffman A. S., Nature 1995, 378, 472–474. [DOI] [PubMed] [Google Scholar]
- 12. Kwon I. C., Bae Y. H., Kim S. W., Nature 1991, 354, 291–293. [DOI] [PubMed] [Google Scholar]
- 13. Kim H., Kang Y. J., Kang S., Kim K. T., J. Am. Chem. Soc. 2012, 134, 4030–4033. [DOI] [PubMed] [Google Scholar]
- 14. Leinenga G., Langton C., Nisbet R., Götz J., Nat. Rev. Neurol. 2016, 12, 161–174. [DOI] [PubMed] [Google Scholar]
- 15. Pan Y., Yoon S., Sun J., Huang Z., Lee C., Allen M., Wu Y., Chang Y.-J., Sadelain M., Shung K. K., Chien S., Wang Y., Proc. Natl. Acad. Sci. USA 2018, 115, 992–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Heureaux J., Chen D., Murray V. L., Deng C. X., Liu A. P., Cell. Mol. Bioeng. 2014, 7, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ibsen S., Tong A., Schutt C., Esener S., Chalasani S. H., Nat. Commun. 2015, 6, 8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Prieto M. L., Firouzi K., Khuri-Yakub B. T., Maduke M., Ultrasound Med. Biol. 2018, 44, 1217–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maresca D., Lakshmanan A., Abedi M., Bar-Zion A., Arash F., Lu G. J., Szablowski J. O., Wu D., Yoo S., Shapiro M. G., Annu. Rev. Chem. Biomol. Eng. 2018, 9, 229–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang T., Wang H., Pang G., He T., Yu P., Cheng G., Zhang Y., Chang J., ACS Appl. Mater. Interfaces 2020, 12, 56692–56700. [DOI] [PubMed] [Google Scholar]
- 21. Ye J., Tang S., Meng L., Li X., Wen X., Chen S., Niu L., Li X., Qiu W., Hu H., Jiang M., Shang S., shu Q., Zheng H., Duan S., Li Y., Nano Lett. 2018, 18, 4148–4155. [DOI] [PubMed] [Google Scholar]
- 22. Farhadi A., Ho G. H., Sawyer D. P., Bourdeau R. W., Shapiro M. G., Science 2019, 365, 1469–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou Y., Huo S., Loznik M., Göstl R., Boersma A. J., Herrmann A., Angew. Chem. Int. Ed. 2021, 60, 1493–1497; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 1515–1519. [Google Scholar]
- 24. Kauscher U., Holme M. N., Björnmalm M., Stevens M. M., Adv. Drug Delivery Rev. 2019, 138, 259–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Graham S. M., Carlisle R., Choi J. J., Stevenson M., Shah A. R., Myers R. S., Fisher K., Peregrino M.-B., Seymour L., Coussios C. C., J. Controlled Release 2014, 178, 101–107. [DOI] [PubMed] [Google Scholar]
- 26. Nele V., Schutt C. E., Wojciechowski J. P., Kit-Anan W., Doutch J. J., Armstrong J. P. K., Stevens M. M., Adv. Mater. 2020, 32, 1905914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Paris J. L., Cabañas M. V., Manzano M., Vallet-Regí M., ACS Nano 2015, 9, 11023–11033. [DOI] [PubMed] [Google Scholar]
- 28. Ibsen S., Schutt C. E., Esener S., Drug Des. Dev. Ther. 2013, 7, 375–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brown C. L., Craig S. L., Chem. Sci. 2015, 6, 2158–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. De Bo G., Chem. Sci. 2018, 9, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Willis-Fox N., Rognin E., Aljohani T. A., Daly R., Chem 2018, 4, 2499–2537. [Google Scholar]
- 32. Stratigaki M., Göstl R., ChemPlusChem 2020, 85, 1095–1103. [DOI] [PubMed] [Google Scholar]
- 33. O′Neill R. T., Boulatov R., Nat. Rev. Chem. 2021, 5, 148–167. [DOI] [PubMed] [Google Scholar]
- 34. Chen Y., Mellot G., van Luijk D., Creton C., Sijbesma R. P., Chem. Soc. Rev. 2021, 50, 4100–4140. [DOI] [PubMed] [Google Scholar]
- 35. Cravotto G., Gaudino E. C., Cintas P., Chem. Soc. Rev. 2013, 42, 7521–7534. [DOI] [PubMed] [Google Scholar]
- 36. Mitragotri S., Nat. Rev. Drug Discovery 2005, 4, 255–260. [DOI] [PubMed] [Google Scholar]
- 37. Shi Z., Wu J., Song Q., Göstl R., Herrmann A., J. Am. Chem. Soc. 2020, 142, 14725–14732. [DOI] [PubMed] [Google Scholar]
- 38. Shi Z., Song Q., Göstl R., Herrmann A., Chem. Sci. 2021, 12, 1668–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huo S., Zhao P., Shi Z., Zou M., Yang X., Warszawik E., Loznik M., Göstl R., Herrmann A., Nat. Chem. 2021, 13, 131–139. [DOI] [PubMed] [Google Scholar]
- 40. Küng R., Pausch T., Rasch D., Göstl R., Schmidt B. M., Angew. Chem. Int. Ed. 2021, 60, 10.1002/anie.202102383; Angew. Chem. 2021, 133, . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mohsen M. G., Kool E. T., Acc. Chem. Res. 2016, 49, 2540–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhao W., Ali M. M., Brook M. A., Li Y., Angew. Chem. Int. Ed. 2008, 47, 6330–6337; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 6428–6436. [Google Scholar]
- 43. Ali M. M., Li F., Zhang Z., Zhang K., Kang D.-K., Ankrum J. A., Le X. C., Zhao W., Chem. Soc. Rev. 2014, 43, 3324–3341. [DOI] [PubMed] [Google Scholar]
- 44. Potisek S. L., Davis D. A., Sottos N. R., White S. R., Moore J. S., J. Am. Chem. Soc. 2007, 129, 13808–13809. [DOI] [PubMed] [Google Scholar]
- 45. Bowser B. H., Craig S. L., Polym. Chem. 2018, 9, 3583–3593. [Google Scholar]
- 46. Akbulatov S., Boulatov R., ChemPhysChem 2017, 18, 1422–1450. [DOI] [PubMed] [Google Scholar]
- 47. Kean Z. S., Gossweiler G. R., Kouznetsova T. B., Hewage G. B., Craig S. L., Chem. Commun. 2015, 51, 9157–9160. [DOI] [PubMed] [Google Scholar]
- 48. Bai Y., Li Y., Zhang D., Wang H., Zhao Q., Anal. Chem. 2017, 89, 9467–9473. [DOI] [PubMed] [Google Scholar]
- 49. Wang H., Yang R., Yang L., Tan W., ACS Nano 2009, 3, 2451–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Seeman N. C., Nature 2003, 421, 427–431. [DOI] [PubMed] [Google Scholar]
- 51. Kashefi-Kheyrabadi L., Mehrgardi M. A., Biosens. Bioelectron. 2012, 37, 94–98. [DOI] [PubMed] [Google Scholar]
- 52. Zheng X., Peng R., Jiang X., Wang Y., Xu S., Ke G., Fu T., Liu Q., Huan S., Zhang X., Anal. Chem. 2017, 89, 10941–10947. [DOI] [PubMed] [Google Scholar]
- 53. Lin Z., Chen L., Zhu X., Qiu B., Chen G., Chem. Commun. 2010, 46, 5563–5565. [DOI] [PubMed] [Google Scholar]
- 54. Lee J. Y., Carugo D., Crake C., Owen J., de S. Victor M., Seth A., Coussios C., Stride E., Adv. Mater. 2015, 27, 5484–5492. [DOI] [PubMed] [Google Scholar]
- 55. Pavlov A. M., Saez V., Cobley A., Graves J., Sukhorukov G. B., Mason T. J., Soft Matter 2011, 7, 4341–4347. [Google Scholar]
- 56. Yamaguchi S., Higashi K., Azuma T., Okamoto A., Biotechnol. J. 2019, 14, 1800530. [DOI] [PubMed] [Google Scholar]
- 57. Repina N. A., Rosenbloom A., Mukherjee A., Schaffer D. V., Kane R. S., Annu. Rev. Chem. Biomol. Eng. 2017, 8, 13–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kim C. K., Adhikari A., Deisseroth K., Nat. Rev. Neurosci. 2017, 18, 222–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary