

Summary
Increasing evidence suggests that free haem and iron exert vasculo‐toxic and pro‐inflammatory effects by activating endothelial and immune cells. In the present retrospective study, we compared serum samples from transfusion‐dependent patients with β‐thalassaemia major and intermedia, hereditary spherocytosis and sickle cell disease (SCD). Haemolysis, transfusions and ineffective erythropoiesis contribute to haem and iron overload in haemolytic patients. In all cohorts we observed increased systemic haem and iron levels associated with scavenger depletion and toxic ‘free’ species formation. Endothelial dysfunction, oxidative stress and inflammation markers were significantly increased compared to healthy donors. In multivariable logistic regression analysis, oxidative stress markers remained significantly associated with both haem‐ and iron‐related parameters, while soluble vascular cell adhesion molecule 1 (sVCAM‐1), soluble endothelial selectin (sE‐selectin) and tumour necrosis factor α (TNFα) showed the strongest association with haem‐related parameters and soluble intercellular adhesion molecule 1 (sICAM‐1), sVCAM‐1, interleukin 6 (IL‐6) and vascular endothelial growth factor (VEGF) with iron‐related parameters. While hereditary spherocytosis was associated with the highest IL‐6 and TNFα levels, β‐thalassaemia major showed limited inflammation compared to SCD. The sVCAM1 increase was significantly lower in patients with SCD receiving exchange compared to simple transfusions. The present results support the involvement of free haem/iron species in the pathogenesis of vascular dysfunction and sterile inflammation in haemolytic diseases, irrespective of the underlying haemolytic mechanism, and highlight the potential therapeutic benefit of iron/haem scavenging therapies in these conditions.
Keywords: heme, haemoglobinopathies, haemolysis, iron, transfusion, haemopexin
Introduction
Blood diseases such as sickle cell disease (SCD), β‐thalassaemia (β‐thal) and hereditary spherocytosis (SPH) are hallmarked by haemolysis, leading to cell‐free haemoglobin (Hb) release in the circulation. 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 Mechanistically haemolysis can be extra‐ or intra‐vascular. While extra‐vascular haemolysis involves red blood cell (RBC) phagocytosis by hepatic and splenic macrophages of the reticulo‐endothelial system, and accounts for most haemolytic events in β‐thal and hereditary SPH, intravascular haemolysis occurs in the form of erythrocyte destruction within the blood vessel lumen and accounts for one‐third of the haemolytic process in SCD. 5
Previous work showed that circulating free Hb levels are increased in these diseases and haptoglobin (Hp), the Hb scavenger is reduced. Hp is part of a detoxification system in mammals that scavenges haemolysis by‐products in the circulation and promotes intracellular metabolism. 1 , 2 , 9 This detoxification system also includes the haem scavenger haemopexin (Hx) and the cellular enzymes haem‐oxygenases (HO‐1 and ‐2). 9 , 10 Hp and Hx, by binding with high affinity Hb and haem, respectively, deliver Hb to macrophages and haem to hepatocytes, thus preventing their pro‐oxidant action in the circulation and non‐specific uptake in non‐target cells. 1 , 2 , 11 , 12 , 13 , 14 , 15 Within cells, HOs degrade the haem porphyrin into iron, carbon monoxide and biliverdin, thus exerting primary anti‐inflammatory, anti‐oxidant and anti‐apoptotic effects. 10 , 16 , 17 , 18 , 19 Once iron is released, the ferritin heavy chain (H‐Ferritin) oxidises ferrous iron (Fe2+) to catalytically inactive ferric iron (Fe3+) through its ferroxidase activity and allows iron storage inside ferritin light chain (L‐Ferritin). 20 , 21 , 22 Due to a high rate of haemolysis, haem detoxification systems are often saturated in haemolytic diseases. 1 , 11 , 23 , 24 , 25 While Hp is used as a clinical marker for haemolysis and its reduction in SCD, β‐thal and SPH is known; the modulation and comparison of haem‐related parameters across different haemolytic diseases is less clear.
Anaemia in haemolytic diseases is often treated with transfusions. While regular transfusions cause iron overload, exchange transfusions are expected to limit iron accumulation. In transfused patients, the iron binding capacity of the plasma carrier transferrin (Tf) is often exceeded, resulting in non‐Tf‐bound iron (NTBI) formation and iron overload. Low hepcidin levels in patients with β‐thal in response to inefficient erythropoiesis, are an additional cause of iron overload. 7 , 26 , 27 , 28 , 29 , 30 The hepatic hormone hepcidin controls iron absorption and recycling through the occlusion and proteolytic degradation of the iron exporter ferroportin on the surface of duodenal enterocytes and macrophages. 31 , 32 Hepcidin levels are reduced in response to hypoxia and increased erythropoietic activity in patients with haemoglobinopathies, promoting dietary iron uptake for erythropoiesis despite sufficient iron availability. 31 , 32 Previous work showed a reduction of hepcidin in patients with β‐thal, whereas data on hepcidin in SCD remain controversial. Differences between patients with SCD receiving simple versus exchange transfusions have not explored to date.
A vasculo‐toxic and pro‐inflammatory role has been attributed to unbound haem and iron. 1 , 33 , 34 , 35 , 36 due to their ability to induce endothelial and immune cell activation. 37 , 38 , 39 , 40 , 41 , 42 , 43 , 44 Through these mechanisms, haem and iron promote adhesion of leucocytes and RBCs to the vessel wall and trigger vasculopathy 45 , 46 and sterile inflammation, which contributes to a dysfunctional vasculature and highly inflammatory environment typical of SCD and β‐thal. 36 , 47 , 48 , 49 , 50 Overall, haemolysis and related mechanisms likely contribute to a number of complications associated with these diseases, including vaso‐occlusive crisis, pulmonary hypertension, stroke, acute chest syndrome, leg ulceration and thrombosis. 46 , 62 Whereas haem‐induced toxicity has been explored in patients with SCD, the association between haem/iron parameters and markers of vascular dysfunction and inflammation has been poorly investigated in patients with β‐thal and SPH, and comparisons across these diseases are not available.
In the present study, we analysed cohorts of transfusion‐dependent patients with β‐thal major, β‐thal intermedia, SPH and SCD to shed light on the relationship between the formation of free haem and iron species and vascular dysfunction, as well as inflammation across haemolytic diseases. Specifically, the aims of the present study were: (i) to systematically compare haem‐ and iron‐related parameters and vascular/inflammatory markers across different haemolytic diseases; (ii) to correlate markers of vascular alterations, oxidative stress and inflammation with haem‐ and iron‐related parameters; (iii) to compare iron‐related parameters in patients with SCD receiving simple versus exchange transfusions; (iv) to assess the impact of splenectomy on anaemia and haemolysis, as well as vascular/inflammatory markers.
Patients and methods
Details on patients and methods are included in Data S1.
Results
Patients’ characteristics
We analysed (i) a cohort of patients with β‐thal major (n = 59) and β‐thal intermedia (n = 7). These patients were transfusion‐dependent (one transfusion every 3–4 weeks); however, transfusions were unable to fully correct the anaemia (average Hb 78 g/l for β‐thal major). 95% of the patients received iron chelation (75% patients receiving either deferoxamine or deferiprone alone; 20% a combination of deferoxamine or deferiprone with deferasirox; 5% non‐chelated) (Tables SI and SII). We compared patients with β‐thal to healthy subjects (n = 17). The β‐thal cohorts predominantly included children (average age 11–14, range 2–36 years); therefore, it was not possible to obtain a perfectly age‐matched control population of healthy donors (average age 27, range 8–42 years). Nevertheless, throughout the study we did not observe major differences between younger (aged 8–15 years) and older (aged >25 years) control subjects for the parameters analysed; (ii) a cohort of patients with hereditary SPH (n = 14; mean [SD] age 8·35 [3·89] years), receiving one transfusion every 3 up to 8 weeks (average Hb 84 g/l). Only 30% of these patients were treated by chelation (deferoxamine) (Tables SIII and SIV); (iii–iv) two cohorts of patients with SCD (genotype SS), whereby the first cohort received simple transfusions (SCD 1, n = 16; mean [SD] 19·18 [5·75] years) and the second automated exchange transfusions (SCD 2, n = 19; mean [SD] age 22·5 [6·17] years). The SCD 1 patients were sporadically transfused every 3–6 weeks (average Hb 82.5 g/l) and did not receive chelation. 25% of SCD 1 patients were treated with hydroxyurea. The SCD 2 patients received exchange transfusion every 4 weeks (average Hb 100.8 g/l) and only 21% required iron chelation therapy (deferasirox). The SPH and SCD cohorts were compared to a control population of healthy subjects (34; aged ≥25 years) (Tables SV–SVII). The SCD 2 patients were mainly of African American origin, 63 whereas all the other cohorts of patients analysed in the present study (β‐thal, SPH, SCD 1) were of Palestinian origin. 64 Compliance with iron chelation therapy was poor, especially in the patients with β‐thal and SPH.
All parameters reported in our study were measured as long as a sufficient sample was available. Lack of sample availability prevented us from measuring all parameters in the entire cohorts and controls. The β‐thal cohorts and the SCD and SPH cohorts were analysed at different times, therefore data are presented in separate graphs. To directly compare between the results from the patients with β‐thal with patients with SPH and SCD, we included a few β‐thal serum samples still available at the time of the analysis of SCD and SPH serum samples. Comparisons are presented in Figs 1H, I, 2F, G, 4E, F, 5E; Figure S4.
Systemic haem and iron are elevated in transfusion‐dependent patients with haemolytic diseases
In all patient cohorts serum levels of cell‐free haem, which include both bound and free Hb and haem, were significantly elevated compared to healthy subjects, without significant differences among the three haemolytic diseases (Fig 1A, G, H). However, most patients with SPH had lower Hb/haem levels compared to patients with β‐thal and SCD (Fig 1H). Consistent with the increase in circulating haem, Hx was severely reduced in patients with β‐thal, both major and intermedia, as well as in patients with SCD, reaching almost undetectable levels (Fig 1C, D, G, I, J). Hx levels were reduced to a lesser extent in patients with SPH (Fig 1G, I). Additionally, we observed reduced Hp and increased bilirubin levels in patients with β‐thal (Fig 1B; Figure S1). Hb/haem scavenger saturation and depletion likely result in the formation of ‘free’ unbound Hb and haem species, which we estimated in the patients with β‐thal based on Hp and Hx levels. While in control subjects the total serum Hp and Hx levels accounted for an average Hb/haem binding capacity of 20·66 (9·83–31·5) µmol/l, in patients with β‐thal major this capacity was as low as 4·53 (0·59–8·47) µmol/l (Fig 1E). However, serum total Hb/haem levels were increased from an average of 34·82 (22·77–46·87) µmol/l in healthy subjects up to 80 (41·68–118·52) µmol/l in patients with β‐thal major (Fig 1A). Therefore, the average amount of ‘free’ non‐Hp‐bound Hb (NHBHb) and non‐Hx‐bound haem (NHBH) was about 16·9 (3·88–29·9) µmol/l in healthy subjects and elevated up to 76 (36·4–115·5) µmol/l in patients with β‐thal major (Fig 1F). Patients with β‐thal carried at least an average 50 µmol/l NHBHb and NHBH in excess compared to healthy individuals.
Fig 1.
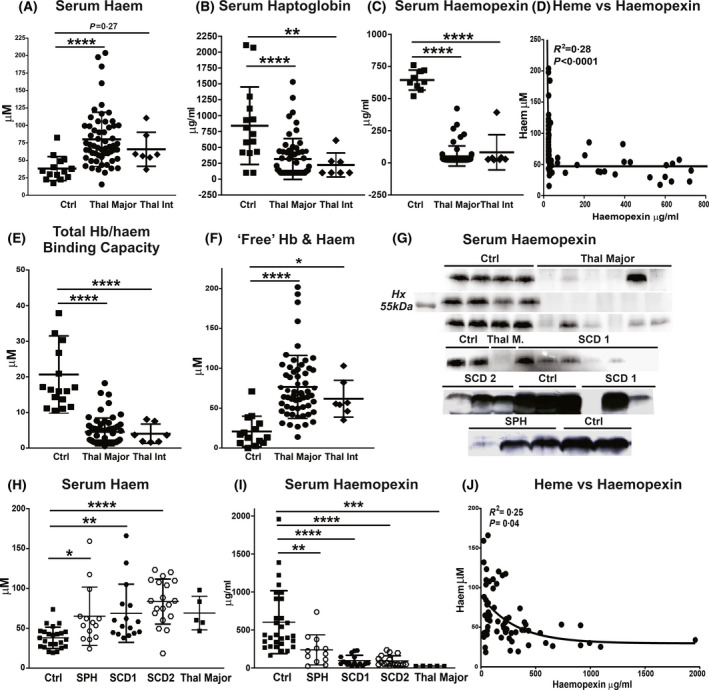
Transfusion‐dependent patients with β‐thalassaemia (β‐thal), sickle cell disease (SCD) and spherocytosis (SPH) are hallmarked by haemolysis and haem scavenger depletion. (A–C) Measurement of total cell‐free Hb/haem, haptoglobin and haemopexin in sera of healthy subjects (Ctrl), and patients with β‐thal major and intermedia (int). (D) Correlation between haemopexin and haem includes data from Ctrls and patients with β‐thal. (E,F) Total µmol/l Hb and haem binding capacity of haptoglobin and haemopexin and ‘free’ unbound Hb/haem (NHBHb, NHBH) in patients with β‐thal major and int (calculated as described in Methods). (G) Representative Western blot for haemopexin in sera of healthy subjects (Ctrl), as well as patients with β‐thal major, SCD (SCD 1 and 2), and SPH; (H,I) Measurement of total cell‐free Hb/haem and haemopexin in sera of Ctrls and patients with SPH and SCD (SCD 1 and 2). Few samples of patients with β‐thal major were included for comparison. (J) Correlation of haemopexin with haem includes data from Ctrls, and patients with SCD1 and 2. Values represent mean ± SD. *P < 0·05; **P < 0·01; ***P < 0·001; ****P < 0·0001.
Furthermore, serum iron, Tf saturation and NTBI were highly elevated in β‐thal major and intermedia and, to a lower extent in SCD and SPH (Fig 2A–C, E–G; Figure S1). Patients with β‐thal major had the highest levels of serum iron, Tf saturation (average 80%) and NTBI, whereas patients with β‐thal intermedia and SCD had a more moderate Tf saturation (average 55–60%) and NTBI increase (Fig 2A–C). Tf saturation and NTBI were only mildly elevated in SPH and more pronounced in SCD (Fig 2E–G). Markers for iron overload were increased to similar levels in patients with SCD receiving simple and exchange transfusions (Tf saturation average 55% in SCD 1 vs. 50% in SCD 2) (Fig 2E–G). In all cohorts, NTBI showed a direct correlation with Tf saturation (Fig 2D, H).
Fig 2.
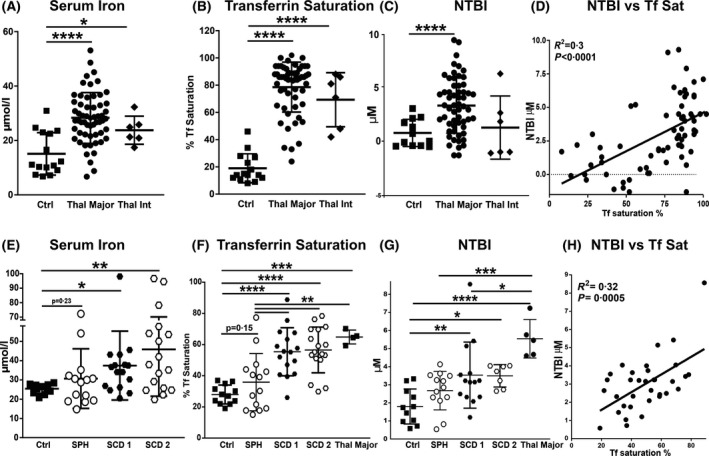
Transfusion‐dependent patients with haemolytic diseases are hallmarked by elevated transferrin (Tf) saturation and non‐Tf‐bound iron (NTBI) formation. (A–C) Measurement of iron, Tf saturation and NTBI in sera of healthy subjects (Ctrl), as well as patients with β‐thalassaemia (β‐thal) intermedia (int) and major. (D) Correlation between NTBI and Tf saturation includes data from Ctrls and patients with β‐thal. (E–G) Measurement of iron, Tf saturation and NTBI in sera of Ctrls, as well as patients with sickle cell disease (SCD1 and 2) and spherocytosis (SPH). A few samples of patients with β‐thal major were included for comparison. (H) Correlation between NTBI and Tf saturation includes data from Ctrls and patients with SCD (SCD 1 and 2). Values represent mean ± SD. *P < 0·05; **P < 0·01; ***P < 0·001; ****P < 0·0001.
In patients with β‐thal the Hb levels were decreased and the erythropoietin (EPO) as well as hepcidin levels increased (Fig 3A–C). However, hepcidin induction by iron appears inadequate when compared to the elevated iron burden, monitored as serum ferritin, in these patients. This is indicated by the hepcidin–ferritin ratio, which was significantly decreased in patients with β‐thal compared to healthy subjects (Fig 3D).
Fig 3.
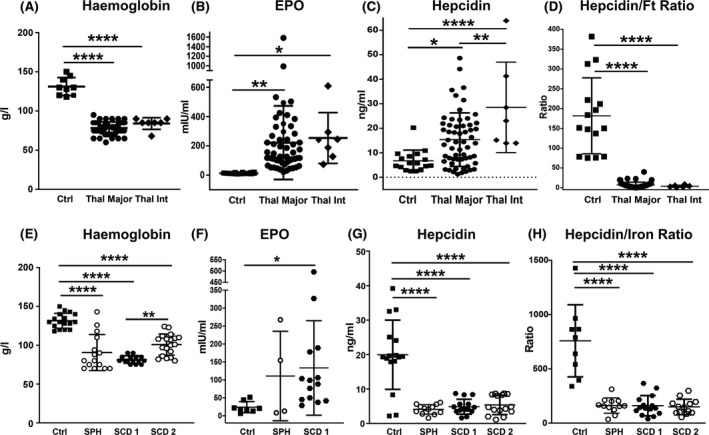
Iron overload in transfusion‐dependent patients with haemolytic diseases is associated with inappropriate hepcidin levels. (A–D) Measurement of haemoglobin, erythropoietin (EPO), hepcidin and hepcidin–serum ferritin (Ft) ratio in blood samples and sera of healthy subjects (Ctrl), and patients with β‐thalassaemia (β‐thal) intermedia (int) and major; (E–H) Measurement of haemoglobin, EPO, hepcidin and hepcidin–Ft ratio in sera of Ctrls, as well as patients with sickle cell disease (SCD 1 and 2) and spherocytosis (SPH). EPO is shown only in the SPH an SCD 1 groups. Values represent mean ± SD. *P < 0·05; ***P < 0·001; ****P < 0·0001.
Anaemia was also seen in patients with SCD and SPH (Fig 3E). Conversely to the β‐thal cohorts, in the SCD and SPH cohorts hepcidin levels were decreased (Fig 3G), together with a less severe iron burden (Tf saturation 35–60%, Fig 2E–G) and increased erythropoietic activity (demonstrated by EPO induction, Fig 3F). The modulation of the hepcidin–serum iron ratio reflected that of absolute hepcidin levels (Fig 3H). While haem and Hx levels, as well the hepcidin–ferritin ratio, were similarly altered across patients with different haemolytic diseases, Tf saturation, NTBI and EPO were significantly higher in patients with β‐thal, reflecting higher transfusional iron burden and more severe ineffective erythropoiesis (Figs 1H, I, 2F, Gand 3B, C, F, G).
Collectively, these data indicate that haemolytic diseases are hallmarked by elevated systemic haem and iron levels, associated with scavenger saturation and formation of unbound Hb, haem and iron species. Hepcidin levels are reduced or inappropriately low in these disease conditions, likely contributing to NTBI formation together with transfusions.
Markers of vasculopathy are increased in transfusion‐dependent patients with haemolytic diseases
We recently showed that in mouse models of haemolytic (SCD and β‐thal) and iron‐overload disease (hereditary haemochromatosis) unbound haem and iron are drivers of vascular damage. 15 , 33 , 39 , 65 We thus analysed whether markers of vascular activation and dysfunction are altered in the serum of patients with haemolytic anaemias. Specifically, we monitored circulating soluble adhesion molecules, whose increase indicates endothelial activation, and nitrotyrosine, whose reduced levels indicate poor availability of the key endothelial homeostatic regulator nitric oxide (NO) and thus vascular dysfunction. Consistent with our observations in disease models, patients with β‐thal, SCD and SPH had an increase of markers of vascular activation and dysfunction. Soluble adhesion molecules, including soluble vascular cell adhesion molecule 1 (sVCAM‐1), soluble intercellular adhesion molecule 1 (sICAM‐1), soluble platelet and endothelial selectins (sP‐selectin; sE‐selectin), were increased in the serum of patients with β‐thal major and intermedia (Fig 4A). sE‐selectin and sVCAM‐1 were significantly elevated also in patients with SCD and SPH (Fig 4E). While sE‐selectin levels were similar across the cohorts, sVCAM‐1 levels were higher in patients with β‐thal and SCD receiving simple transfusions (SCD 1) compared to patients with SPH and SCD receiving exchange transfusions (SCD 2) (Fig 4A, E). Most circulating adhesion molecules were directly correlated with haem‐ and iron‐related parameters (Fig 4B–D, F). Nitrotyrosine levels were almost unchanged in patients with SPH and reduced in patients with β‐thal and SCD compared to healthy donors (Fig 4G, I), indicating reduced NO production. Nitrotyrosine levels were directly correlated with Hx and inversely correlated with Tf saturation (Fig 4H).
Fig 4.
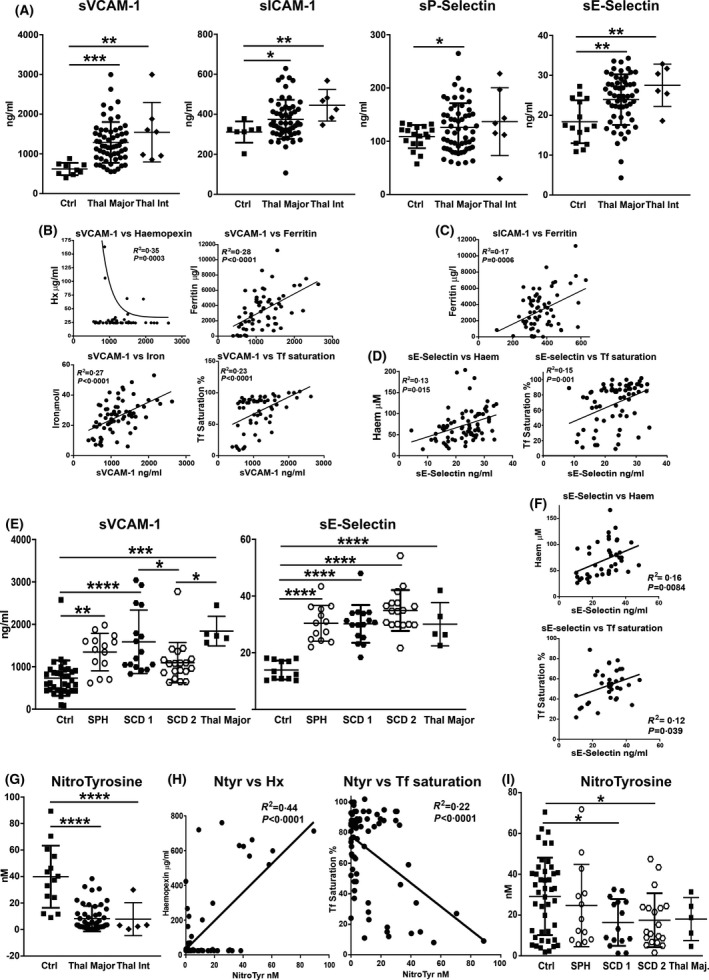
Transfusion‐dependent patients with haemolytic diseases are characterised by elevated biomarkers of vascular dysfunctions. (A) Measurement of soluble vascular cell and intercellular adhesion molecules 1 (sVCAM‐1, sICAM‐1), soluble endothelial and platelet selectins (sE‐selectin, sP‐selectin) in sera of healthy subjects (Ctrls), as well as patients with β‐thalassaemia (β‐thal) intermedia (int) and major. (B–D) Significant correlations between soluble adhesion molecules and haem/iron‐related parameters including data from Ctrls and patients with β‐thal major are shown. (E) Measurement of sVCAM‐1 and sE‐selectin in sera of Ctrls, as well as patients with sickle cell disease (SCD 1 and 2), and spherocytosis (SPH). (F) Correlations between sE‐selectin and haem or transferrin (Tf) saturation including data from Ctrls and SCD 1 and 2 are shown. (G, I) Nitrotyrosine measurement in sera of Ctrls, as well as patients with β‐thal int and major, SCD 1 and 2, and SPH. A few samples of patients with β‐thal major were included for comparison. (H) Correlations between nitrotyrosine (Ntyr) and haemopexin (Hx) or Tf saturation including data from Ctrls and patients with β‐thal are shown. Values represent mean ± SD. *P < 0·05; **P < 0·01; ***P < 0·001; ****P < 0·0001.
To determine the relationship between haem/iron‐related parameters and damage biomarkers, we performed both uni‐ and multivariate regression analysis. In Figs 4, 5 and 6 the univariate correlations shown are those that remain significant in the multivariate regression analysis reported in Table I. Furthermore, we generated correlation matrices with Spearman correlations, which are visualised through correlation dot plots in Fig 8A–C.
Fig 5.
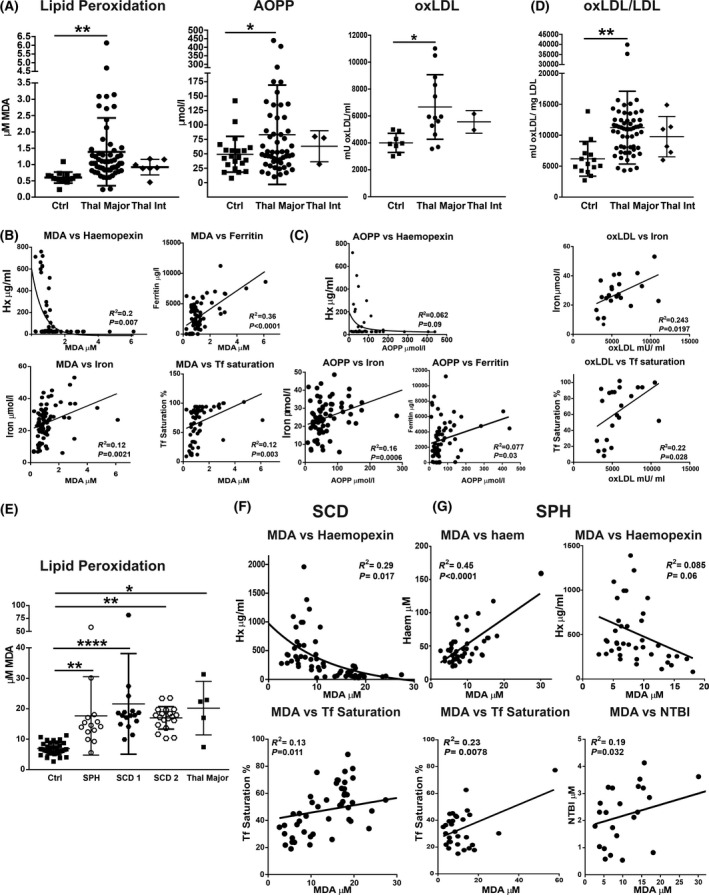
Transfusion‐dependent patients with haemolytic diseases are hallmarked by oxidative stress. (A) Measurement of malondialdehyde (MDA), advanced oxidised protein products (AOPP) and oxidised low‐density lipoproteins (oxLDL) in sera of healthy subjects (Ctrls), as well as patients with β‐thalassaemia (β‐thal) intermedia (int) and major. (B, C) Significant correlations of MDA and AOPP with haem/iron‐related parameters including data from Ctrls and patients with β‐thal major are shown. (D) oxLDL levels in Ctrls as well as patients with β‐thal major who show an average level of circulating LDLs of about 0·8 mg/ml (See Figure S3). Correlations between oxLDL and free haem or iron levels in this subgroup of patients and Ctrls is shown. (E) Measurement of MDA in sera of Ctrls, as well as patients with sickle cell disease (SCD) 1 and 2, and spherocytosis (SPH). Few samples of patients with β‐thal major were included for comparison. (F, G) Significant correlations between MDA and haem/iron‐related parameters including data from Ctrls, and patients with SCD (SCD 1 and 2) and SPH are shown. Values represent mean ± SD. *P < 0·05; **P < 0·01; ****P < 0·0001.
Fig 6.
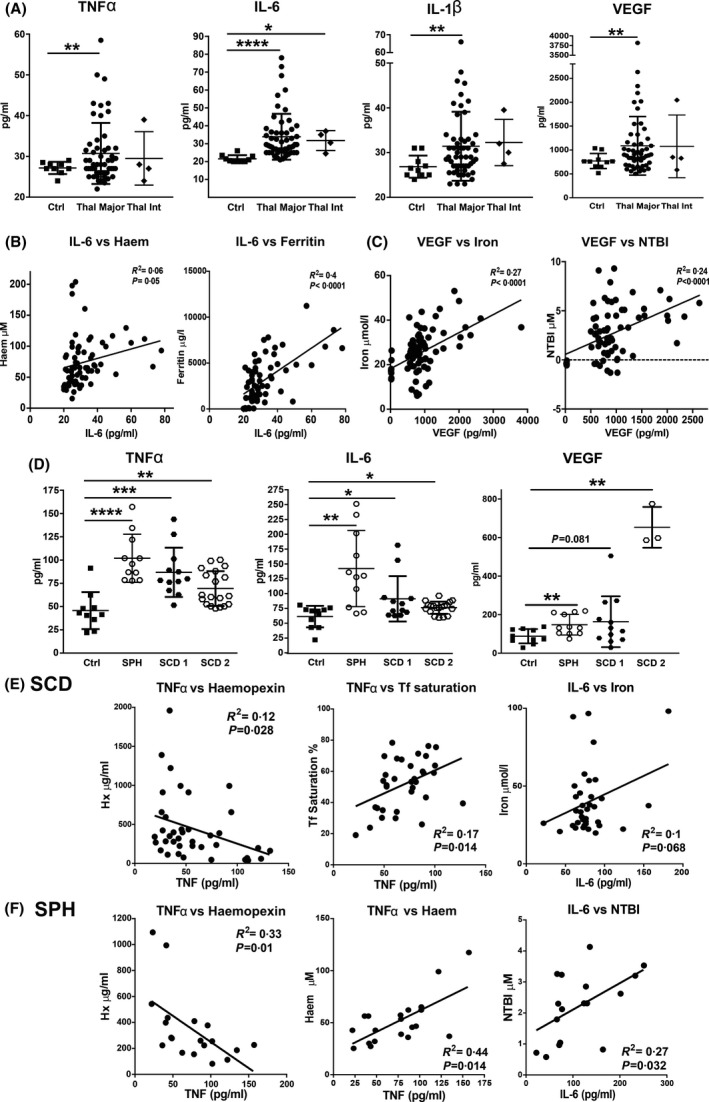
Transfusion‐dependent patients with haemolytic diseases are hallmarked by elevated circulating inflammatory cytokines. (A) Measurement of tumour necrosis factor α (TNFα), interleukin 6 (IL‐6), IL‐1β and vascular endothelial growth factor (VEGF) in sera of healthy subjects (Ctrls), as well as patients with β‐thalassaemia (β‐thal) intermedia (int) and major. (B, C) Significant correlations of IL‐6 and VEGF with haem/iron‐related parameters including data from Ctrls and patients with β‐thal major are shown. (D) Measurement of TNFα, IL‐6 and VEGF in sera of Ctrls, as well as patients with sickle cell disease (SCD 1 and 2), and spherocytosis (SPH). (E, F) Significant correlations of TNFα and IL‐6 with haem/iron‐related parameters including data from Ctrls and patients with SCD 1 and 2 or SPH are shown. Values represent mean ± SD. *P < 0·05; **P < 0·01; ***P < 0·001; ****P < 0·0001.
Table I.
Multivariable linear regression models in transfusion‐dependent patients with β‐thal, SCD and SPH.
| Endothelial dysfunction | Oxidative stress | Inflammation | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variable | Parameter | Beta coeff. | SE | P value | Parameter | Beta coeff. | SE | P value | Parameter | Beta coeff. | SE | P value |
| β‐thal Major | ||||||||||||
| Serum Heme | (Intercept) | 12·27 | 20·22 | 0·547 | (Intercept) | 86·72 | 15·48 | <0·001 | (Intercept) | 77·779 | 26·28 | 0·0045 |
| sVCAM1 | 0·012 | 0·01 | 0·249 | MDA | 5·33 | 5·184 | 0·309 | IL‐6 | 1·0611 | 0·565 | 0·066 | |
| sE‐selectin | 2·601 | 0·945 | 0·009** | oxLDL | −0·004 | 0·002 | 0·104 | |||||
| Ntyr | −0·432 | 0·272 | 0·12 | |||||||||
| Haemopexin | (Intercept) | 254·89 | 90·61 | 0·007 | (Intercept) | 11·31 | 50·183 | 0·8227 | (Intercept) | 189·23 | 94·23 | 0·05 |
| sVCAM1 | −0·118 | 0·047 | 0·015* | MDA | −23·697 | 15·36 | 0·13 | IL‐6 | −2·523 | 1·956 | 0·203 | |
| sE‐selectin | −11·17 | 4·233 | 0·012* | oxLDL | 0·021 | 0·008 | 0·013* | |||||
| Ntyr | 6·71 | 1·22 | <0·001*** | AOPP | −0·383 | 0·196 | 0·057 | |||||
| Serum Iron | (Intercept) | 3·23 | 5·49 | 0·56 | (Intercept) | 18·31 | 3·22 | <0·001 | (Intercept) | 13·71 | 6·084 | 0·028 |
| sVCAM1 | 0·0066 | 0·003 | 0·025* | MDA | 3·429 | 1·09 | 0·003** | VEGF | 0·008 | 0·003 | 0·002** | |
| sE‐selectin | 0·399 | 0·257 | 0·128 | AOPP | 0·04 | 0·014 | 0·012* | |||||
| Ntyr | −0·171 | 0·074 | 0·026* | |||||||||
| Serum Ferritin | (Intercept) | −396·92 | 1217·83 | 0·746 | (Intercept) | 644·8 | 698·4 | 0·3598 | (Intercept) | −399·4 | 1277·69 | 0·756 |
| sVCAM1 | 1·746 | 0·625 | 0·008** | MDA | 1440 | 236·1 | <0·001*** | TNF | −90·286 | 67·44 | 0·186 | |
| sICAM1 | 12·33 | 4·025 | 0·004** | AOPP | 8·184 | 3·017 | 0·009** | IL‐6 | 110·52 | 27·251 | 0·0002*** | |
| Ntyr | −25·48 | 16·384 | 0·127 | |||||||||
| NTBI | (Intercept) | 0·443 | 1·77 | 0·804 | (Intercept) | 3·262 | 0·978 | 0·002 | (Intercept) | 0·177 | 1·81 | 0·922 |
| sVCAM1 | 0·001 | 0·001 | 0·182 | MDA | 0·51 | 0·33 | 0·132 | TNF | 0·107 | 0·092 | 0·25 | |
| oxLDL | −0·00002 | 0·0001 | 0·191 | VEGF | 0·002 | 0·001 | 0·015* | |||||
| Tf Saturation | (Intercept) | 48·15 | 13·35 | <0·001 | (Intercept) | 73·15 | 8·77 | <0·001 | (Intercept) | 32·69 | 16·38 | 0·051 |
| sVCAM1 | 0·019 | 0·007 | 0·008** | MDA | 8·43 | 2·93 | 0·006** | VEGF | 0·011 | 0·007 | 0·102 | |
| sE‐selectin | 1·205 | 0·624 | 0·06 | oxLDL | −0·003 | 0·001 | 0·046* | |||||
| Ntyr | −0·645 | 0·178 | 0·0008*** | AOPP | 0·051 | 0·037 | 0·176 | |||||
| sP‐selectin | −0·087 | 0·078 | 0·272 | |||||||||
| Sickle cell disease | ||||||||||||
| Serum Heme | (Intercept) | 33·2 | 19·21 | 0·092 | (Intercept) | 50·15 | 6·41 | <0·001 | ||||
| sE‐selectin | 1·53 | 0·61 | 0·017* | MDA | 0·7 | 0·38 | 0·07 | |||||
| Haemopexin | (Intercept) | 538·21 | 151·78 | 0·001 | (Intercept) | 487·61 | 74·54 | <0·001 | (Intercept) | 510·966 | 118·464 | <0·001 |
| sE‐selectin | −11·75 | 4·96 | 0·024* | MDA | −12·14 | 4·35 | 0·007** | TNF | −4·31 | 1·52 | 0·008** | |
| Ntyr | 4·28 | 3 | 0·163 | |||||||||
| Serum Iron | (Intercept) | 22·3 | 17·11 | 0·202 | (Intercept) | 31·97 | 4·93 | <0·001 | (Intercept) | 17·276 | 12·4505 | 0·175 |
| sE‐_selectin | 0·78 | 0·51 | 0·132 | MDA | 0·33 | 0·26 | 0·204 | IL_6 | 0·19 | 0·15 | 0·207 | |
| Tf Saturation | (Intercept) | 37·4 | 12·01 | 0·004 | (Intercept) | 39·97 | 3·87 | <0·001 | (Intercept) | 37·2726 | 9·382 | <0·001 |
| sVCAM1 | −0·006 | 0·005 | 0·212 | MDA | 0·54 | 0·21 | 0·012* | TNF | 0·2 | 0·12 | 0·107 | |
| sE‐_selectin | 0·77 | 0·36 | 0·038* | |||||||||
| NTBI | (Intercept) | 2·53 | 0·4 | <0·001 | (Intercept) | 2 | 0·84 | 0·029 | ||||
| MDA | 0·03 | 0·02 | 0·198 | TNF | 0·01 | 0·01 | 0·276 | |||||
| Spherocytosis | ||||||||||||
| Serum Heme | (Intercept) | 25·57 | 21·58 | 0·251 | (Intercept) | 37·93 | 5·96 | <0·001 | (Intercept) | 22·01 | 10·19 | 0·045 |
| sE‐_selectin | 1·04 | 0·82 | 0·223 | MDA | 0·89 | 0·44 | 0·050* | TNF | 0·42 | 0·12 | 0·003** | |
| Haemopexin | (Intercept) | 692·51 | 220·29 | 0·006 | (Intercept) | 631·28 | 91·319 | <0·001 | (Intercept) | 712·05 | 125·07 | <0·001 |
| sE‐_selectin | −13·37 | 8·64 | 0·14 | MDA | −14·44 | 6·62 | 0·035* | TNF | −3·19 | 1·54 | 0·055 | |
| Ntyr | 4·01 | 2·53 | 0·132 | IL_6 | −1·209 | 0·9928 | 0·241 | |||||
| Serum Iron | (Intercept) | 18·65 | 2·6 | <0·001 | ||||||||
| MDA | 0·76 | 0·16 | <0·001*** | |||||||||
| Tf Saturation | (Intercept) | 26·32 | 3·43 | <0·001 | ||||||||
| MDA | 0·633 | 0·22 | 0·008** | |||||||||
| NTBI | (Intercept) | 1·76 | 0·3018 | <0·001 | (Intercept) | 0·94 | 0·57 | 0·123 | ||||
| MDA | 0·04 | 0·018 | 0·032* | IL_6 | 0·01 | 0·004 | 0·111 | |||||
Complete case regression analysis for β‐thal, SCD and SPH patients with models generated for each domain of variables: endothelial dysfunction, oxidative stress and inflammation. Significant positive and negative associations between haem and iron parameters (serum haem, haemopexin, iron, Tf saturation, NTBI) and markers of endothelial dysfunction(soluble adhesion molecules, nytotyrosine), oxidative stress (MDA, oxLDL, AOPP) and inflammation (IL‐1β, TNFα, IL‐6, VEGF) are shown. Analysis includes parameters of patients’ group and corresponding healthy donors.
The beta estimates, standard errors and parameter P‐values are reported for variables with a P‐value
P < 0·05;
P < 0·01;
P < 0·001.
Fig 8.
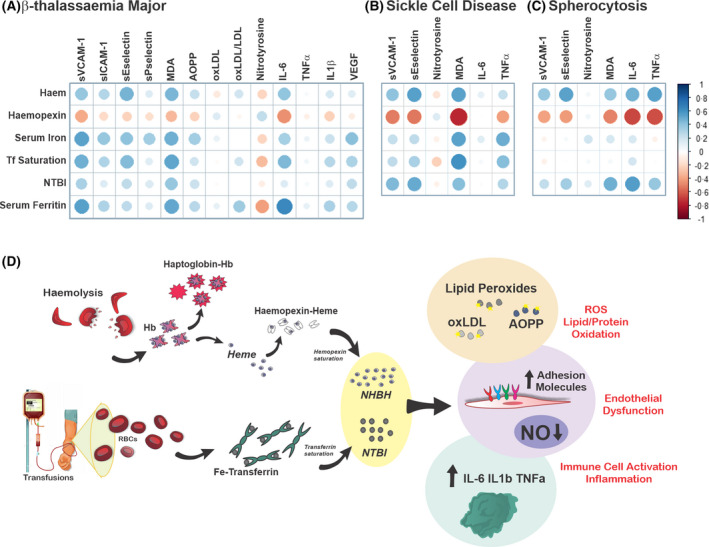
Working model: haem‐ and iron‐mediated toxicity in haemolytic diseases. (A) Correlation plots between haem/iron‐related parameters [serum haem, iron, transferrin (Tf) saturation, non‐Tf‐bound iron (NTBI), haemopexin (Hx)] and damage‐related parameters [malondialdehyde (MDA), soluble vascular cell and intercellular adhesion molecules 1 (sVCAM1, sICAM‐1), soluble endothelial and platelet selectins (sP‐selectin, sE‐selectin), oxidation of low‐density lipoprotein (oxLDL), nitrotyrosine, advanced oxidised protein product (AOPP), interleukin 1β (IL‐1β), tumour necrosis factor α (TNFα), IL‐6, vascular endothelial growth factor (VEGF)] in patients with β‐thalassaemia (β‐thal) major. Corresponding Spearman correlation coefficients and P values are reported in Table SIII. (B, C) Correlation plots between iron‐related parameters (haem, iron, Tf saturation, NTBI, Hx) and damage‐related parameters [sVCAM1, sE‐selectin, nitrotyrosine, MDA, IL‐6, TNFα, VEGF, monocyte chemoattractant protein‐1 (MCP‐1)] in patients with sickle cell disease (SCD; SCD 1 and 2)and spherocytosis (SPH). Corresponding Spearman correlation coefficients and P values are reported in Tables SIX and SX. The size of the dot suggests the strength of the correlation; the colour of the dot indicate the direction of the correlation (blue dot: positive; red dot: negative). (D) In haemolytic diseases, haemolysis leads to the release of elevated haemoglobin (Hb) and haem levels in the circulation. Saturation of the Hb and haem scavengers haptoglobin and haemopexin triggers the formation of ‘free’ non‐haptoglobin‐bound Hb (NHBHb), non‐haemopexin‐bound haem (NHBH). In parallel, transfusions increase systemic iron levels and transferrin saturation. This generates ‘free’ NTBI. NHBHb, NHBH and NTBI exert a synergistic detrimental action on the vasculature and immune cells. While the pro‐oxidant potential of NHBH and NTBI is reflected by elevated lipid and proteins oxidation (lipid peroxides, oxLDL, AOPP), their vasculo‐toxic and pro‐inflammatory action is reflected by elevated soluble adhesion molecules (e.g. E‐selectin, P‐selectin, ICAM‐1, VCAM‐1), VEGF and inflammatory cytokines (e.g. IL‐6, TNFα, IL‐1β) and reduced nitric oxide (NO) levels. These alterations likely contribute to NHBH‐ and NTBI‐triggered vasculopathy and chronic sterile inflammation, typical hallmarks of several haemolytic diseases. [Colour figure can be viewed at wileyonlinelibrary.com]
Multivariate linear regression analysis indicated that serum haem and Hx were correlated with sE‐selectin in all cohorts. Moreover, serum iron and Tf saturation showed a positive correlation with sE‐selectin in the β‐thal major and SCD cohorts (Table I; Fig 4D, F). sVCAM‐1 showed a positive correlation with most haem and iron parameters, particularly in the β‐thal major cohort (Table I; Fig 4B). Serum ferritin was positively correlated with sICAM‐1 in the β‐thal major cohort (Table I; Fig 4C). Nitrotyrosine was negatively correlated with Hx in all cohorts and also with most haem and iron parameters in the β‐thal major cohort (Table I; Fig 4H).
These findings indicate a direct correlation between the haem/iron status and vascular alterations (Fig 7A–C) and suggest that elevated free haem and iron contribute to vascular dysfunction in patients with haemolytic diseases.
Fig 7.
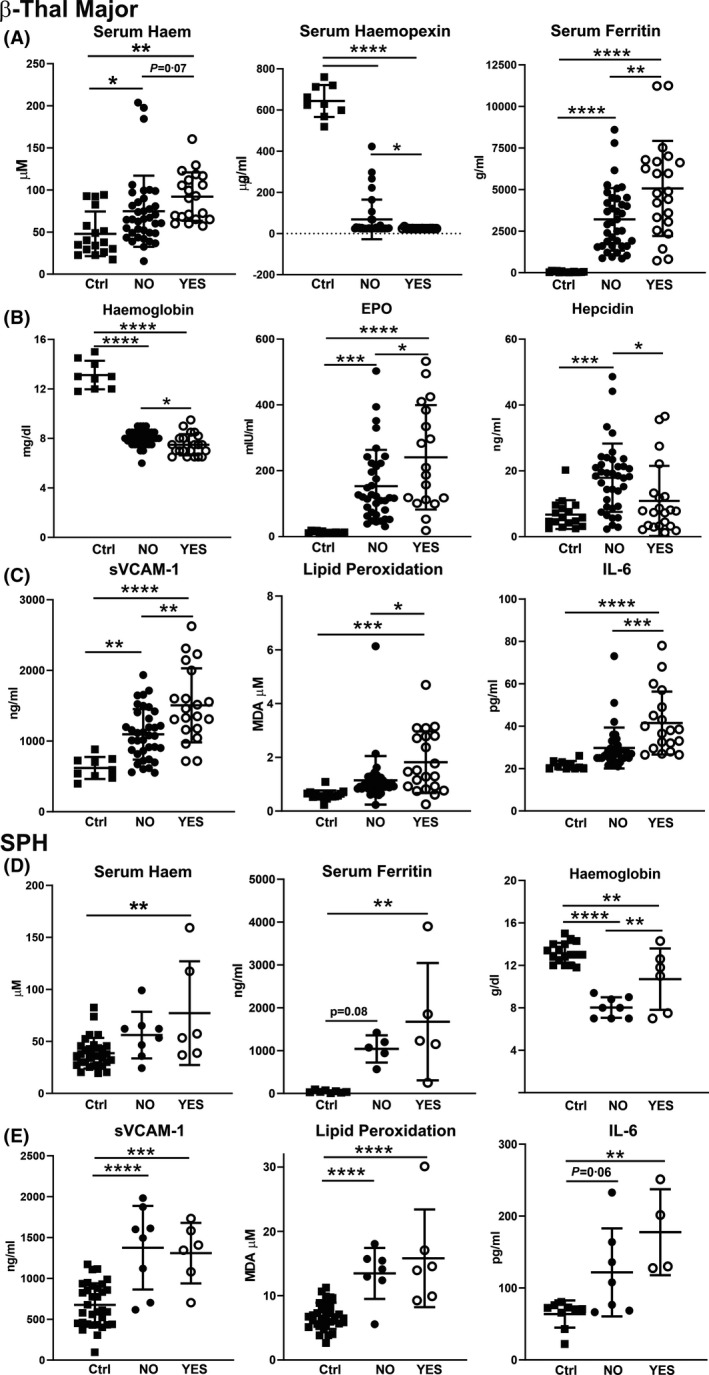
Effect of splenectomy on anaemia, haem/iron parameters and markers of vascular dysfunction, oxidative stress and inflammation in transfusion‐dependent patients with β‐thalassaemia (β‐thal) major and spherocytosis (SPH). (A) Measurement of total cell‐free haemoglobin/haem, haemopexin and ferritin, (B) haemoglobin, erythropoietin (EPO) and hepcidin, (C) soluble vascular cell adhesion molecule 1 (sVCAM‐1), lipid peroxidation (malondialdehyde) and interleukin 6 (IL‐6) in sera and blood samples of healthy subjects (Ctrls) and patients with β‐thal major, non‐splenectomised (NO; n = 38) and splenectomised (YES; n = 21). (D) Cell‐free Hb/haem, ferritin and haemoglobin, (E) sVCAM‐1, lipid peroxidation (MDA) and IL‐6 in sera and blood samples of Ctrls and patients with SPH, non‐splenectomised (NO; n = 8) and splenectomised (YES; n = 6). Values represent mean ± SD. *P < 0·05; **P < 0·01; ***P < 0·001; ****P < 0·0001.
Markers of oxidative stress and inflammation are increased in transfusion‐dependent patients with haemolytic diseases
Due to their pro‐oxidant and inflammatory properties, unbound haem and iron trigger oxidative stress and inflammation. 14 , 15 , 38 , 66 , 67 Therefore, we analysed serum markers of oxidative stress as well as cytokines in our patient cohorts. Compared to healthy subjects, patients with β‐thal had increased levels of lipid peroxidation, assessed by malondialdehyde (MDA) measurement, and protein oxidation, assessed by advanced oxidised protein product (AOPP) levels (Fig 5A). The MDA increase was higher in β‐thal major compared to intermedia. Lipid peroxidation was also elevated in the SCD and SPH cohorts compared to healthy subjects (Fig 5E). While lipid peroxidation was similarly increased in the two SCD cohorts, patients with SPH and β‐thal major had a trend towards lower and higher MDA levels respectively (Fig 5A, E). Patients with β‐thal major further had increased oxidation of low‐density lipoprotein (oxLDL) per mg LDL (Fig 5A). Despite overall reduced LDL levels in the β‐thal population (Figure S2A, B), those patients that had LDL levels comparable to healthy controls (average 0·8 mg/ml; Figure S2C) had higher oxLDL, correlating with increased serum iron and Tf saturation (Fig 5D). Uni‐ and multivariable regression linear analysis showed a strong correlation between MDA and most haem as well as iron parameters in all cohorts (Table I; Fig 5B, F, G). Similarly, AOPP showed a correlation with Hx, serum iron and ferritin in β‐thal major (Table I; Fig 5C).
Circulating levels of interleukin 1β (IL‐1β), IL‐6, tumour necrosis factor α (TNFα) and vascular endothelial growth factor (VEGF) were elevated in patients with β‐thal major and intermedia (Fig 6A). By contrast, monocyte chemoattractant protein‐1 (MCP‐1) levels were significantly decreased in these patients (Figure S3). Patients with SCD and SPH had increased IL‐6, TNFα, MCP‐1 and VEGF levels, whereby IL‐6 and TNFα levels were higher in patients with SPH (Fig 6D), and VEGF and MCP‐1 levels were higher in patients with SCD receiving exchange transfusions (SCD 2) (Fig 6D; Figure S3). Patients with β‐thal had lower TNFα, VEGF and MCP‐1 levels compared to patients with SPH and SCD, indicating overall more moderate inflammation (Figure S4). Regression analysis showed that IL‐6 was positively correlated with serum haem and ferritin in the β‐thal major cohort (Table I; Fig 6B); a good correlation was also seen between IL‐6 and Hx, serum iron and NTBI in the SCD and SPH cohorts (Table I; Fig 6E, F). Interestingly, TNFα had a positive correlation with serum haem and Hx in the SCD and SPH cohorts, but not the β‐thal major one (Table I; Fig 6E, F). VEGF showed a specific correlation with iron parameters, serum iron, Tf saturation and NTBI (Table I; Fig 6C). Hydroxyurea treatment reduced IL‐6 and TNFα levels in patients with SCD receiving simple transfusions (SCD 1) (Figure S5).
Splenectomy aggravates haemolysis in transfusion‐dependent patients with β‐thal major and spherocytosis
As the spleen is frequently removed in haemolytic disorders, we performed a sub‐analysis by comparing splenectomised and non‐splenectomised patients with transfusion‐dependent β‐thal major, SCD (only cohort SCD 2) and SPH in order to evaluate the effect of splenectomy on haem‐ and iron‐related parameters, as well as vascular and inflammatory markers (Fig 7, full analysis: Figures S6–S11). The most severe effect was seen in patients with β‐thal major. Compared to non‐splenectomised patients (n = 38), splenectomised patients with β‐thal major (n = 21) had more severe anaemia and haemolytic disease, hallmarked by lower total Hb, higher cell‐free haem, and lower serum Hp and Hx levels (Fig 7A; Figure S6A, C). Moreover, they developed a more pronounced iron overload, as suggested by significantly higher serum iron and ferritin levels (NTBI showed a trend to higher levels) (Fig 7A; Figure S6B). EPO was further elevated and hepcidin lower, highlighting a higher degree of ineffective erythropoiesis in splenectomised patients with β‐thal (Fig 7B; Figure S6C). Consistent with the more severe haem/iron overload, splenectomised patients had elevated circulating markers of endothelial dysfunction (sVCAM‐1, sICAM‐1, sP‐Selectin, sE‐Selectin, VEGF), oxidative stress (MDA and AOPP) and inflammation (IL‐6, white blood cell count) (Fig. 7C; Figure S7). Interestingly, nitrotyrosine levels were higher in splenectomised patients. Similar to patients with β‐thal, splenectomised patients with SPH (six patients) had a more pronounced haem and iron phenotype compared to non‐splenectomised ones (eight). However, in contrast to β‐thal, anaemia was improved by splenectomy, as indicated by higher Hb levels (Fig. 7D; Figure S8). Splenectomised patients with SPH had a mild elevation of vascular (sE‐selectin, VEGF) and inflammatory markers (IL‐6, TNFα), as well as lipid peroxidation (Fig. 7E; Figure S9). Likely due to limited sample numbers, patients with SCD with exchange transfusions did not show significant differences when they underwent splenectomy (6 vs. 9 non‐splenectomised), besides a mild drop in Hb and increase in iron and IL‐6 (Figures S10 and S11).
Overall, the present study highlights a direct correlation of the haem/iron status with vascular and oxidative stress markers and inflammatory mediators, as shown by the dot plots and relative Spearman correlation analysis (Fig 8A–C; Tables SVIII–SX), and support a causal link between haem/iron excess and vascular dysfunction, oxidative damage and inflammation (Fig 8D). Splenectomy amplifies these detrimental effects by aggravating haemolysis.
Discussion
Haemolysis and free iron accumulation as a cause for vascular dysfunction, inflammation and oxidative stress was mainly demonstrated by us and others in in vitro systems and in vivo mouse models. 14 , 15 , 37 , 39 , 41 , 42 , 65 , 66 , 68 In the present study, we analysed and compared cohorts of transfusion‐dependent patients with three different haemolytic diseases and found that free haem and iron excess were correlated with vasculo‐toxicity, oxidative stress and inflammation. While the present study does not inform about the direct influence of transfusions on haemolysis and inflammatory markers due to the lack of a non‐transfused patient group, we believe our observations are of significance for patients who present with haemolytic anaemias and transfusion dependency.
Elevated levels of circulating Hb and haem lead to the saturation of Hp and Hx in mouse models of Hb/haem overload as well as murine disease models for SCD and β‐thal. 15 , 38 , 69 Our present data show that this is fully recapitulated in transfused patients with β‐thal major and intermedia, SCD and hereditary SPH and confirm previous observations. 5
A first result was that circulating Hb/haem levels increased in all three haemolytic diseases independently of the underlying haemolytic mechanism. This implies that irrespective whether haemolysis occurs extra‐ (within tissues) or intra‐vascularly, Hb and haem reach the circulation, saturating plasma scavengers and giving rise to ‘free’ NHBHb and NHBH (Fig 8D). Most patients with SPH had lower circulating Hb/haem levels, suggesting a reduced haemolysis rate compared to patients with β‐thal and SCD (Fig 1).
We further show that systemic iron levels, Tf saturation and NTBI are increased according to transfusion burden, being more elevated in patients with β‐thal than in patients with SCD and SPH (Fig 2). The majority of previous studies showed a reduction of hepcidin in β‐thal. 70 , 71 , 72 , 73 , 74 , 75 , 76 , 77 , 78 In the present study, we observed increased hepcidin levels, suggesting that transfusional iron overload (Fig 3) dominates over hepcidin regulatory signals induced by anaemia and ineffective erythropoiesis (EPO, erythroferrone) in these β‐thal cohorts. The discrepancy is likely explained by the severe transfusional iron overload indicated by very high serum ferritin levels (average 3763 ng/ml) in the present β‐thal cohort compared to most β‐thal cohorts analysed previously where ferritin levels were lower (<1000 ng/ml). Similarly to our present study, Rashidy et al. 79 observed high hepcidin in patients with β‐thal associated with elevated ferritin levels (average 2451 ng/ml). Indeed, transfusions acutely induce hepcidin expression in patients with β‐thal major by triggering a temporary suppression of erythropoiesis and by causing iron accumulation. Persistent hepcidin elevation likely reflects iron overload due to inadequate chelation and poor compliance in our present cohort. Despite absolute high hepcidin levels, these are inadequately low when compared to the iron burden, as suggested by the low hepcidin–ferritin ratio in patients with β‐thal compared to healthy subjects (Fig 3D). Elevated erythropoietic activity and iron demand likely account for inappropriately low hepcidin induction. Overall, these data indicate that in transfused patients with β‐thal hepcidin levels result from the combined effect of ineffective erythropoiesis and transfusional iron overload. In SCD, hepcidin modulation is controversial: previous studies showed either increased or reduced hepcidin levels. Kroot et al. 70 showed that in four of nine patients with HbSS SCD hepcidin levels were decreased compared to control subjects, which was attributed to increased erythropoietic activity. In agreement with this study Karafin et al. 71 has proven that while erythropoietic drive, iron status, and inflammation all contribute to the control of hepcidin levels, the strongest contributor is the erythropoietic drive. Therefore, low hepcidin values were found in patients with high reticulocyte percentage and EPO. Consistently, hepcidin levels were reduced and EPO was increased in our present SCD cohorts, suggesting that enhanced erythropoietic activity overrides iron burden and inflammation in the regulation of hepcidin and has a key role in triggering iron overload in SCD (Fig 3). The observation of comparable NTBI levels in the two SCD cohorts suggests that NTBI may not only result from iron overload due to simple transfusions, but are likely explained by reduced hepcidin levels in patients with SCD receiving exchange transfusions.
Another important result of our present study was the appearance of soluble adhesion molecules and elevated VEGF levels in the circulation, reflecting upon vascular activation and damage correlates with both haem‐ and iron‐related parameters in all the haemolytic cohorts (Fig 4). This finding supports a connection between haemolysis and vasculopathy. Consistent with previous analyses in SCD, our present data show that sVCAM‐1 and sE‐selectin are linked to haemolysis markers also in β‐thal major. 80 Moreover, sVCAM‐1 and sICAM‐1 are associated with the iron status, suggesting a synergistic effect of free haem and iron in vascular dysfunction.
Free haem and iron have the ability to accumulate in endothelial cells, where they cause vascular oxidative stress and endothelial activation by promoting reactive oxygen species (ROS) formation and cell apoptosis, and reduce NO availability, thereby triggering vascular permeabilisation and dysfunction. 15 , 38 , 39 , 41 , 42 , 72 , 81 In addition, haem and iron exert toxic effects on smooth muscle and cardiac cells. 82 , 83 , 84 , 85 These findings are of relevance for patients with SCD and β‐thal, who in the long term may develop vascular‐related complications, including vaso‐occlusive crisis, acute chest syndrome, increased blood pressure, altered cardiac function, stroke and atherosclerosis. 54 , 56 , 58 , 86 , 87 An increased risk of vascular complications has also been observed in SPH. 6 , 40 Moreover, the presence of elevated serum sP‐selectin also reflects upon enhanced haem‐driven platelet activation during clotting formation, as recently demonstrated. 88
NO availability is severely reduced in patients with SCD and β‐thal major, as suggested by reduced nitrotyrosine levels that likely reflect upon reduced NO production by endothelial NO synthase (Fig 4). NO plays a major role in vascular homeostasis and is a critical regulator of vasomotor tone, endothelial adhesion molecule expression and platelet activation. 89 , 90 , 91 Free Hb‐mediated NO scavenging, haem/iron‐driven NO oxidative inactivation and decreased production likely lead to an overall reduction in the active NO fraction and contribute to vascular dysfunction in haemolytic conditions. 1 , 5 , 58
Besides inducing vasculo‐toxicity, free haem and iron behave as ‘alarmin’ by triggering the activation of immune cells, including macrophages and neutrophils, and promoting sterile inflammation. 36 , 48 , 50 , 92 Haem stimulates neutrophil extracellular trap (NET) formation and inflammatory neutrophil activation, contributing to SCD pathogenesis. 93 , 94 , 95 Macrophages activated by free haem and iron undergo a pro‐inflammatory phenotypic switching hallmarked by elevated IL‐6, TNFα, IL‐1β synthesis and are involved in tissue damage and chronic inflammation in SCD. 14 , 66 , 68 , 96 , 97 , 98 , 99 , 100 Increased circulating levels of inflammatory cytokines were observed in all haemolytic patient cohorts analysed in the present study (Fig 6). Lower TNFα, VEGF and MCP‐1 levels in patients with β‐thal compared to patients with SCD and SPH might be explained by a different underlying disease pathophysiology, but also by the chelation therapy that only patients with β‐thal received (Figures S3 and S4). The TNFα and MCP‐1 levels in SCD are highest and thus may serve as markers for chronic sterile inflammation. The trend to higher TNFα and IL‐6 levels in the SCD 1 versus SCD 2 cohort, support the concept that exchange transfusions suppress pro‐inflammatory mechanisms by reducing the number of sickling cells. 37 , 41 , 66 , 67 , 101 The administration of exogenous Hx in SCD and β‐thal mice, by promoting hepatic haem detoxification and limiting haem‐iron loading in cardiovascular and immune cells, prevents haem‐induced vasculo‐toxicity and inflammation. 14 , 15 , 60 , 72 In the present study, we show that a severe drop in Hb and haem scavengers is a signature of human haemolytic diseases and underlies vascular dysfunction and sterile inflammation in these cohorts of patients. The elevation of oxidative stress markers in patients with haemolytic diseases also indicates a saturation of the anti‐oxidant systems, resulting in ROS production and related sequelae 102 , 103 (Fig 5).
Recently we have shown that NTBI and vascular iron accumulation contribute to atherosclerosis through a multifactorial action involving vascular oxidative stress and dysfunction, lipid and protein oxidation, inflammation and elevated VEGF. 39 , 87 Likewise, unbound haem exerts a pro‐oxidant, pro‐atherogenic and pro‐thrombotic effects due to its oxidant and inflammatory action. 25 , 112 These findings can be translated to patients with hereditary haemochromatosis 39 and haemolytic disorders, where the same markers are increased and can represent valuable biomarkers for iron‐aggravated risk of cardiovascular disease in these pathological conditions. Patients with β‐thal have an increased incidence of cardiovascular complications, whereby congestive heart failure is the main cause of death, especially in transfusion‐dependent β‐thal major. 113 , 114 Furthermore, these patients are also prone to arterial and venous thromboembolic events, stroke, cardiovascular abnormalities and premature atherosclerosis. 87 , 115 , 116 , 117 , 118 , 119 , 120 , 121 The low LDL cholesterol levels in β‐thal, which we and others observed (Figure S2), may protect against an atherogenic risk, explaining the higher incidence of cardiac rather than atherosclerotic mortality in these patients. 122 , 123 , 124 , 125 In patients with β‐thal and SCD, accelerated erythropoiesis and enhanced cholesterol consumption have been suggested as a dominant mechanism of low LDL levels. 126 Conversely, children with β‐thal may be at an increased risk of premature atherosclerosis because of dyslipidaemia with elevated triglycerides. 121 , 124 , 127 , 128 Interestingly, we found that patients with β‐thal with LDL levels in the same range as the control populations had a clear increase in oxLDL levels (Fig 5), correlating with free haem and iron levels. Therefore, iron might superimpose a pro‐atherosclerotic risk through increased serum LDL oxidation in patients with β‐thal who carry average LDL levels at least comparable to the normal population. Independently of cholesterol levels, iron overload in β‐thal has been implicated in early atherosclerotic changes, including endothelial dysfunction, structural arterial alterations (e.g. re‐modelling, increased stiffness) 117 , 129 and carotid thickness. 117 , 130 , 131 Overall, our present data suggest that patients with β‐thal bear a vascular and inflammatory phenotype together with an athero‐protective lipid pattern, the interplay of which eventually determine the development of cardiovascular dysfunction and/or atherosclerotic disease. While SCD is a less common risk factor for atherosclerosis probably due to the overall lower extent of iron overload, in recent decades the longer life expectancy and the tendency to transfuse significantly increased such risk in this patient population. 132
Previous reports suggested that splenectomy aggravates haemolysis in β‐thal, leading to higher circulating levels of free Hb as well as sE‐ and P‐Selectin. 133 , 134 In the present study, we found that also sVCAM‐1 and sICAM‐1, as well as oxidative stress markers and cytokines, were more elevated after splenectomy. More importantly, we found that the effect of splenectomy was not limited to aggravated haemolysis, but extends to a more severe iron overload (Fig 7; Figures S6 and S7). This is likely triggered by a more severe ineffective erythropoiesis due to the lack of extra‐medullary splenic erythropoiesis contribution. Therefore, in β‐thal, anaemia is worsened by the combined effect of suppressed splenic erythropoiesis and accelerated intravascular haemolysis, as consequence of spleen removal. 133 , 135 By contrast, the improved anaemia and increased cell‐free haem in splenectomised patients with SPH suggests that the lack of spleen‐mediated RBC clearance on the one hand moderately enhances intravascular haemolysis, and on the other prolongs RBC half‐life (Fig 7; Figures S8 and S9). 135 , 136 Thus, while splenectomy accelerates intravascular haemolysis in both β‐thal and SPH, its effect on anaemia is opposite due to a major role of suppressed splenic erythropoiesis in β‐thal and reduced extra‐vascular haemolysis in SPH. The increased intravascular haemolysis is associated with aggravated inflammation and oxidative stress in both β‐thal and SPH.
Overall, the present study supports the concept that (i) haemolytic conditions are hallmarked by the presence of free haem and iron species that are generated in the circulation independently of the haemolytic mechanisms and also upon transfusions; (ii) free haem and iron are involved in the pathogenesis of chronic inflammation and cardiovascular‐related complications in haemolytic patients through the induction vascular dysfunction and immune cell activation; 87 , 105 , 137 (iii) β‐thal and SCD show more severe vascular alterations compared to SPH, consistent with higher levels of free Hb, haem and iron; (iv) splenectomy accelerates intravascular haemolysis, thus aggravating vascular dysfunction and inflammation. We believe that the markers analysed in the present study can serve as valuable early biomarkers for haem‐ and iron‐driven vasculo‐toxicity and inflammation in iron overload and haemolytic conditions, potentially reflecting predisposition to disease‐related complications. Finally, our present data highlight the potential therapeutic benefit of Hp/Hx replacement treatment, as well as anti‐oxidant and anti‐inflammatory strategies to counteract vascular toxicity and inflammation in haemolytic and iron‐overload conditions. 3 , 143
Author contributions
Francesca Vinchi designed and supervised the study, analysed data, wrote the manuscript and prepared the Figs; Richard Sparla performed experiments and measurements on samples from the thalassaemia cohort; Sara T. Passos, Richa Sharma and S.Zebulon Vance performed experiments and measurements on samples from the SCD and SPH cohorts; Hala S. Zreid, Hesham Juaidi, Deepa Manwani, Karina Yazdanbakhsh and Eitan Fibach collected and provided the samples and contributed to manuscript preparation; Vijay Nandi performed statistical analysis of the data; André M. N. Silva and Anand R. Agarvas performed NTBI measurements on patients’ samples; John D. Belcher and Gregory M. Vercellotti contributed to study design and manuscript preparation; Husam Ghoti provided the samples, supervised the study and contributed to manuscript preparation; Husam Ghoti and Martina U. Muckenthaler designed and supervised the study and wrote the manuscript.
Conflict of interest
Francesca Vinchi: research funding from Novartis, Silence Therapeutics, Vifor Pharma and PharmaNutra; John D. Belcher and Gregory M. Vercellotti: research funding from CSL Behring and Mitobridge/Astellas. Martina U. Muckenthaler: research funding from Novartis and Silence Therapeutics.
Funding statement
This work was supported by: research funding from the Dietmar Hopp‐Stiftung, the Deutsche Forschungsgemeinschaft (SFB 1118/1036) and the Federal Ministry of Education and Research (NephrESA project Nr 031L0191C) (to Martina U. Muckenthaler); fellowships from the Olympia Morata Programme of the Medical Faculty of Heidelberg and National Blood Foundation (NBF 19‐08) and National Institutes of Health (NIH) research grant HL149626 (to Francesca Vinchi); NIH research grant HL114567‐01 (to John D. Belcher and Gregory M. Vercellotti).
Supporting information
Figure S1. Serum ferritin and transferrin analysed in patients with β‐thalassaemia.
Figure S2. Serum LDL and oxLDL in patients with β‐thalassaemia.
Figure S3. MCP‐1 levels in patients with haemolytic diseases.
Figure S4. Cytokines in patients with haemolytic diseases.
Figure S5. Effect of hydroyurea treatment in sickle cell disease.
Figure S6. Effect of splenectomy on haem and iron parameters and anaemia in transfusion‐dependent patients with β‐thalassaemia major.
Figure S7. Effect of splenectomy on markers of vascular dysfunction, oxidative stress and inflammation in transfusion‐dependent patients with β‐thalassaemia major.
Figure S8. Effect of splenectomy on haem and iron parameters and anaemia in transfusion‐dependent patients with spherocytosis.
Figure S9. Effect of splenectomy on markers of vascular dysfunction, oxidative stress and inflammation anaemia in transfusion‐dependent patients with spherocytosis.
Figure S10. Effect of splenectomy on haem and iron parameters and anaemia in patients with sickle cell disease receiving exchange transfusions.
Figure S11. Effect of splenectomy on markers of vascular dysfunction, oxidative stress and inflammation in patients with sickle cell disease receiving exchange transfusions.
Table SI. Clinical parameters analysed in patients with β‐thalassaemia major and intermedia.
Table SII. Clinical parameters analysed in patients with β‐thalassaemia major and intermedia.
Table SIII. Clinical parameters analysed in patients with spherocytosis.
Table SIV. Clinical parameters analysed in patients with spherocytosis.
Table SV. Clinical parameters analysed in patients with sickle cell disease.
Table SVI. Clinical parameters analysed in patients with sickle cell disease treated with simple transfusions.
Table SVII. Clinical parameters analysed in patients with sickle cell disease treated with exchange transfusions.
Table SVIII. Pairwise comparisons between heme‐/iron parameters and biomarkers in patients with β‐thalassaemia major.
Table SIX. Pairwise comparisons between heme‐/iron parameters and biomarkers in patients with sickle cell disease.
Table SX. Pairwise comparisons between heme‐/iron parameters and biomarkers in patients with spherocytosis.
Acknowledgements
The authors dedicate the current manuscript to Eliezer Rachmilewitz who made this work possible by promoting synergism and interaction among scientists and medical doctors.
Contributor Information
Francesca Vinchi, Email: fvinchi@nybc.org.
Martina U. Muckenthaler, Email: martina.muckenthaler@med.uni-heidelberg.de.
References
- 1. Vinchi F, Tolosano E. Therapeutic approaches to limit hemolysis‐driven endothelial dysfunction: scavenging free heme to preserve vasculature homeostasis. Oxid Med Cell Longev. 2013;2013:396527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tolosano E, Fagoonee S, Morello N, Vinchi F, Fiorito V. Heme scavenging and the other facets of hemopexin. Antioxid Redox Signal. 2010;12:305–20. [DOI] [PubMed] [Google Scholar]
- 3. Schaer DJ, Buehler PW, Alayash AI, Belcher JD, Vercellotti GM. Hemolysis and free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood. 2013;121:1276–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schaer DJ, Vinchi F, Ingoglia G, Tolosano E, Buehler PW. Haptoglobin, hemopexin, and related defense pathways‐basic science, clinical perspectives, and drug development. Front Physiol. 2014;5:415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kato GJ, Steinberg MH, Gladwin MT. Intravascular hemolysis and the pathophysiology of sickle cell disease. J Clin Invest. 2017;127:750–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet. 2008;372:1411–26. [DOI] [PubMed] [Google Scholar]
- 7. Mariani R, Trombini P, Pozzi M, Piperno A. Iron metabolism in thalassemia and sickle cell disease. Mediterr J Hematol Infect Dis. 2009;1:e2009006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Iolascon A, De Franceschi L, Muckenthaler M, Taher A, Rees D, de Montalembert M, et al. EHA research roadmap on hemoglobinopathies and thalassemia: an update. Hemasphere. 2019;3:e208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chiabrando D, Vinchi F, Fiorito V, Mercurio S, Tolosano E. Heme in pathophysiology: a matter of scavenging, metabolism and trafficking across cell membranes. Front Pharmacol. 2014;5:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Belcher JD, Mahaseth H, Welch TE, Otterbein LE, Hebbel RP, Vercellotti GM. Heme oxygenase‐1 is a modulator of inflammation and vaso‐occlusion in transgenic sickle mice. J Clin Invest. 2006;116:808–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Smith A, McCulloh RJ. Hemopexin and haptoglobin: allies against heme toxicity from hemoglobin not contenders. Front Physiol. 2015;6:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Smith A, Morgan WT. Hemopexin‐mediated transport of heme into isolated rat hepatocytes. J Biol Chem. 1981;256:10902–9. [PubMed] [Google Scholar]
- 13. Smith A, Morgan WT. Hemopexin‐mediated heme uptake by liver. Characterization of the interaction of heme‐hemopexin with isolated rabbit liver plasma membranes. J Biol Chem. 1984;259:12049–53. [PubMed] [Google Scholar]
- 14. Vinchi F, Costa da Silva M, Ingoglia G, Petrillo S, Brinkman N, Zuercher A, et al. Hemopexin therapy reverts heme‐induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood. 2016;127:473–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vinchi F, De Franceschi L, Ghigo A, Townes T, Cimino J, Silengo L, et al. Hemopexin therapy improves cardiovascular function by preventing heme‐induced endothelial toxicity in mouse models of hemolytic diseases. Circulation. 2013;127:1317–29. [DOI] [PubMed] [Google Scholar]
- 16. Belcher JD, Vineyard JV, Bruzzone CM, Chen C, Beckman JD, Nguyen J, et al. Heme oxygenase‐1 gene delivery by Sleeping Beauty inhibits vascular stasis in a murine model of sickle cell disease. J Mol Med. 2010;88:665–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Belcher JD, Young M, Chen C, Nguyen J, Burhop K, Tran P, et al. MP4CO, a pegylated hemoglobin saturated with carbon monoxide, is a modulator of HO‐1, inflammation, and vaso‐occlusion in transgenic sickle mice. Blood. 2013;122:2757–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gomperts E, Belcher JD, Otterbein LE, Coates TD, Wood J, Skolnick BE, et al. The role of carbon monoxide and heme oxygenase in the prevention of sickle cell disease vaso‐occlusive crises. Am J Hematol. 2017;92:569–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gozzelino R, Jeney V, Soares MP. Mechanisms of cell protection by heme oxygenase‐1. Annu Rev Pharmacol Toxicol. 2010;50:323–54. [DOI] [PubMed] [Google Scholar]
- 20. Arosio P, Elia L, Poli M. Ferritin, cellular iron storage and regulation. IUBMB Life. 2017;69:414–22. [DOI] [PubMed] [Google Scholar]
- 21. Vercellotti GM, Khan FB, Nguyen J, Chen C, Bruzzone CM, Bechtel H, et al. H‐ferritin ferroxidase induces cytoprotective pathways and inhibits microvascular stasis in transgenic sickle mice. Front Pharmacol. 2014;5:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Balla J, Vercellotti GM, Jeney V, Yachie A, Varga Z, Jacob HS, et al. Heme, heme oxygenase, and ferritin: how the vascular endothelium survives (and dies) in an iron‐rich environment. Antioxid Redox Signal. 2007;9:2119–37. [DOI] [PubMed] [Google Scholar]
- 23. Santiago RP, Guarda CC, Figueiredo CV, Fiuza LM, Aleluia MM, Adanho CSA, et al. Serum haptoglobin and hemopexin levels are depleted in pediatric sickle cell disease patients. Blood Cells Mol Dis. 2018;72:34–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wochner RD, Spilberg I, Iio A, Liem HH, Muller‐Eberhard U. Hemopexin metabolism in sickle‐cell disease, porphyrias and control subjects–effects of heme injection. N Engl J Med. 1974;290:822–6. [DOI] [PubMed] [Google Scholar]
- 25. Yalamanoglu A, Deuel JW, Hunt RC, Baek JH, Hassell K, Redinius K, et al. Depletion of haptoglobin and hemopexin promote hemoglobin‐mediated lipoprotein oxidation in sickle cell disease. Am J Physiol Lung Cell Mol Physiol. 2018;315:L765–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morton DJ, Smith A, Ren Z, Madore LL, VanWagoner TM, Seale TW, et al. Identification of a haem‐utilization protein (Hup) in Haemophilus influenzae. Microbiology. 2004;150:3923–33. [DOI] [PubMed] [Google Scholar]
- 27. Fibach E, Rachmilewitz EA. Pathophysiology and treatment of patients with beta‐thalassemia ‐ an update. F1000Res. 2017;6:2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rivella S. Iron metabolism under conditions of ineffective erythropoiesis in beta‐thalassemia. Blood. 2019;133:51–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Porter JB, Garbowski MW. Interaction of transfusion and iron chelation in thalassemias. Hematol Oncol Clin North Am. 2018;32:247–59. [DOI] [PubMed] [Google Scholar]
- 30. Coffey R, Ganz T. Erythroferrone: an erythroid regulator of hepcidin and iron metabolism. Hemasphere. 2018;2:e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Muckenthaler MU, Rivella S, Hentze MW, Galy B. A red carpet for iron metabolism. Cell. 2017;168:344–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hentze MW, Muckenthaler MU, Galy B, Camaschella C. Two to tango: regulation of mammalian iron metabolism. Cell. 2010;142:24–38. [DOI] [PubMed] [Google Scholar]
- 33. Belcher JD, Nath KA, Vercellotti GM. Vasculotoxic and proinflammatory effects of plasma heme: cell signaling and cytoprotective responses. ISRN Oxidative Med. 2013;2013:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jeney V. Pro‐inflammatory actions of red blood cell‐derived DAMPs. Exp Suppl. 2018;108:211–33. [DOI] [PubMed] [Google Scholar]
- 35. Jeney V, Balla J, Yachie A, Varga Z, Vercellotti GM, Eaton JW, et al. Pro‐oxidant and cytotoxic effects of circulating heme. Blood. 2002;100:879–87. [DOI] [PubMed] [Google Scholar]
- 36. Soares MP, Bozza MT. Red alert: labile heme is an alarmin. Curr Opin Immunol. 2016;38:94–100. [DOI] [PubMed] [Google Scholar]
- 37. Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso‐occlusion in murine sickle cell disease. Blood. 2014;123:377–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vinchi F, Gastaldi S, Silengo L, Altruda F, Tolosano E. Hemopexin prevents endothelial damage and liver congestion in a mouse model of heme overload. Am J Pathol. 2008;173:289–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vinchi F, Porto G, Simmelbauer A, Altamura S, Passos ST, Garbowski M, et al. Atherosclerosis is aggravated by iron overload and ameliorated by dietary and pharmacological iron restriction. Eur Heart J. 2020;41(28):2681–95. [DOI] [PubMed] [Google Scholar]
- 40. Frei AC, Guo Y, Jones DW, Pritchard KA Jr, Fagan KA, Hogg N, et al. Vascular dysfunction in a murine model of severe hemolysis. Blood. 2008;112:398–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Erdei J, Toth A, Balogh E, Nyakundi BB, Banyai E, Ryffel B, et al. Induction of NLRP3 inflammasome activation by heme in human endothelial cells. Oxid Med Cell Longev. 2018;2018:4310816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Balla G, Vercellotti GM, Muller‐Eberhard U, Eaton J, Jacob HS. Exposure of endothelial cells to free heme potentiates damage mediated by granulocytes and toxic oxygen species. Lab Invest. 1991;64:648–55. [PubMed] [Google Scholar]
- 43. Carlini RG, Alonzo E, Bellorin‐Font E, Weisinger JR. Apoptotic stress pathway activation mediated by iron on endothelial cells in vitro. Nephrol Dial Transplant. 2006;21:3055–61. [DOI] [PubMed] [Google Scholar]
- 44. Balla J, Jacob HS, Balla G, Nath K, Eaton JW, Vercellotti GM. Endothelial‐cell heme uptake from heme proteins: induction of sensitization and desensitization to oxidant damage. Proc Natl Acad Sci USA. 1993;90:9285–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kalambur VS, Mahaseth H, Bischof JC, Kielbik MC, Welch TE, Vilback A, et al. Microvascular blood flow and stasis in transgenic sickle mice: utility of a dorsal skin fold chamber for intravital microscopy. Am J Hematol. 2004;77:117–25. [DOI] [PubMed] [Google Scholar]
- 46. Kato GJ, Taylor JG. Pleiotropic effects of intravascular haemolysis on vascular homeostasis. Br J Haematol. 2010;148:690–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Belcher JD, Mahaseth H, Welch TE, Vilback AE, Sonbol KM, Kalambur VS, et al. Critical role of endothelial cell activation in hypoxia‐induced vasoocclusion in transgenic sickle mice. Am J Physiol Heart Circ Physiol. 2005;288:H2715–25. [DOI] [PubMed] [Google Scholar]
- 48. Belcher JD, Marker PH, Weber JP, Hebbel RP, Vercellotti GM. Activated monocytes in sickle cell disease: potential role in the activation of vascular endothelium and vaso‐occlusion. Blood. 2000;96:2451–9. [PubMed] [Google Scholar]
- 49. Solovey A, Somani A, Belcher JD, Milbauer L, Vincent L, Pawlinski R, et al. A monocyte‐TNF‐endothelial activation axis in sickle transgenic mice: therapeutic benefit from TNF blockade. Am J Hematol. 2017;92:1119–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dutra FF, Bozza MT. Heme on innate immunity and inflammation. Front Pharmacol. 2014;5:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Adisa OA, Hu Y, Ghosh S, Aryee D, Osunkwo I, Ofori‐Acquah SF. Association between plasma free haem and incidence of vaso‐occlusive episodes and acute chest syndrome in children with sickle cell disease. Br J Haematol. 2013;162:702–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kato GJ, Hebbel RP, Steinberg MH, Gladwin MT. Vasculopathy in sickle cell disease: Biology, pathophysiology, genetics, translational medicine, and new research directions. Am J Hematol. 2009;84:618–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kato GJ. Sickle cells and sickle trait in thrombosis. Blood. 2019;133:2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kato GJ. Sickle cell vasculopathy: vascular phenotype on fire! J Physiol. 2019;597:993–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kato GJ, Hsieh M, Machado R, Jt T, Little J, Butman JA, et al. Cerebrovascular disease associated with sickle cell pulmonary hypertension. Am J Hematol. 2006;81:503–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Minniti CP, Delaney KM, Gorbach AM, Xu D, Lee CC, Malik N, et al. Vasculopathy, inflammation, and blood flow in leg ulcers of patients with sickle cell anemia. Am J Hematol. 2014;89:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gladwin MT, Kato GJ. Hemolysis‐associated hypercoagulability in sickle cell disease: the plot (and blood) thickens! Haematologica. 2008;93:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gladwin MT, Kato GJ. Cardiopulmonary complications of sickle cell disease: role of nitric oxide and hemolytic anemia. Hematology Am Soc Hematol Educ Program. 2005;2005:51–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rother RP, Bell L, Hillmen P, Gladwin MT. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA. 2005;293:1653–62. [DOI] [PubMed] [Google Scholar]
- 60. Ghosh S, Adisa OA, Chappa P, Tan F, Jackson KA, Archer DR, et al. Extracellular hemin crisis triggers acute chest syndrome in sickle mice. J Clin Invest. 2013;123:4809–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ofori‐Acquah SF, Hazra R, Orikogbo OO, Crosby D, Flage B, Ackah EB, et al. Hemopexin deficiency promotes acute kidney injury in sickle cell disease. Blood. 2020;135:1044–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Roumenina LT, Rayes J, Lacroix‐Desmazes S, Dimitrov JD. Heme: modulator of plasma systems in hemolytic diseases. Trends Mol Med. 2016;22:200–13. [DOI] [PubMed] [Google Scholar]
- 63. Liu Y, Jing F, Yi W, Mendelson A, Shi P, Walsh R, et al. HO‐1(hi) patrolling monocytes protect against vaso‐occlusion in sickle cell disease. Blood. 2018;131:1600–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ghoti H, Fibach E, Rachmilewitz EA, Jeadi H, Filon D. New Insights on beta‐Thalassemia in the Palestinian population of Gaza: high frequency and milder phenotype among homozygous IVS‐I‐1 (HBB: c.92+1G>A) patients with high levels of Hb F. Hemoglobin. 2017;41:144–6. [DOI] [PubMed] [Google Scholar]
- 65. Ghosh S, Flage B, Weidert F, Ofori‐Acquah SF. P‐selectin plays a role in haem‐induced acute lung injury in sickle mice. Br J Haematol. 2019;186:329–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Dutra FF, Alves LS, Rodrigues D, Fernandez PL, de Oliveira RB, Golenbock DT, et al. Hemolysis‐induced lethality involves inflammasome activation by heme. Proc Natl Acad Sci USA. 2014;111:E4110–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Figueiredo RT, Fernandez PL, Mourao‐Sa DS, Porto BN, Dutra FF, Alves LS, et al. Characterization of heme as activator of Toll‐like receptor 4. J Biol Chem. 2007;282:20221–9. [DOI] [PubMed] [Google Scholar]
- 68. Costa da Silva M, Breckwoldt MO, Vinchi F, Correia MP, Stojanovic A, Thielmann CM, et al. Iron induces anti‐tumor activity in tumor‐associated macrophages. Front Immunol. 2017;8:1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Belcher JD, Chen C, Nguyen J, Zhang P, Abdulla F, Nguyen P, et al. Control of oxidative stress and inflammation in sickle cell disease with the Nrf2 activator dimethyl fumarate. Antioxid Redox Signal. 2017;26:748–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kroot JJ, Laarakkers CM, Kemna EH, Biemond BJ, Swinkels DW. Regulation of serum hepcidin levels in sickle cell disease. Haematologica. 2009;94:885–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Karafin MS, Koch KL, Rankin AB, Nischik D, Rahhal G, Simpson P, et al. Erythropoietic drive is the strongest predictor of hepcidin level in adults with sickle cell disease. Blood Cells Mol Dis. 2015;55:304–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Belcher JD, Chen C, Nguyen J, Abdulla F, Zhang P, Nguyen H, et al. Haptoglobin and hemopexin inhibit vaso‐occlusion and inflammation in murine sickle cell disease: Role of heme oxygenase‐1 induction. PLoS One. 2018;13:e0196455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jenkins ZA, Hagar W, Bowlus CL, Johansson HE, Harmatz P, Vichinsky EP, et al. Iron homeostasis during transfusional iron overload in beta‐thalassemia and sickle cell disease: changes in iron regulatory protein, hepcidin, and ferritin expression. Pediatr Hematol Oncol. 2007;24:237–43. [DOI] [PubMed] [Google Scholar]
- 74. Gomez S, Diawara A, Gbeha E, Awadalla P, Sanni A, Idaghdour Y, et al. Comparative analysis of iron homeostasis in Sub‐Saharan African children with sickle cell disease and their unaffected siblings. Front Pediatr. 2016;4:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Piperno A, Mariani R, Trombini P, Girelli D. Hepcidin modulation in human diseases: from research to clinic. World J Gastroenterol. 2009;15:538–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mangaonkar AA, Thawer F, Son J, Ajebo G, Xu H, Barrett NJ, et al. Regulation of iron homeostasis through the erythroferrone‐hepcidin axis in sickle cell disease. Br J Haematol. 2020;189:1204–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. El Beshlawy A, Alaraby I, Abdel Kader MS, Ahmed DH, Abdelrahman HE. Study of serum hepcidin in hereditary hemolytic anemias. Hemoglobin. 2012;36:555–70. [DOI] [PubMed] [Google Scholar]
- 78. Ezeh C, Ugochukwu CC, Weinstein J, Okpala I. Hepcidin, haemoglobin and ferritin levels in sickle cell anaemia. Eur J Haematol. 2005;74:86–8. [DOI] [PubMed] [Google Scholar]
- 79. Rashidy F, Abo Elghar H, Kamal Eldin S, Taha M. Hepcidin and iron regulation in chronic hemolytic anemia. Menoufia Med J. 2015;28:463–70. [Google Scholar]
- 80. Kato GJ, Martyr S, Blackwelder WC, Nichols JS, Coles WA, Hunter LA, et al. Levels of soluble endothelium‐derived adhesion molecules in patients with sickle cell disease are associated with pulmonary hypertension, organ dysfunction, and mortality. Br J Haematol. 2005;130:943–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Petrillo S, Chiabrando D, Genova T, Fiorito V, Ingoglia G, Vinchi F, et al. Heme accumulation in endothelial cells impairs angiogenesis by triggering paraptosis. Cell Death Differ. 2018;25:573–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Alvarado G, Jeney V, Toth A, Csosz E, Kallo G, Huynh AT, et al. Heme‐induced contractile dysfunction in human cardiomyocytes caused by oxidant damage to thick filament proteins. Free Radic Biol Med. 2015;89:248–62. [DOI] [PubMed] [Google Scholar]
- 83. Ingoglia G, Sag CM, Rex N, De Franceschi L, Vinchi F, Cimino J, et al. Data demonstrating the anti‐oxidant role of hemopexin in the heart. Data Brief. 2017;13:69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ingoglia G, Sag CM, Rex N, De Franceschi L, Vinchi F, Cimino J, et al. Hemopexin counteracts systolic dysfunction induced by heme‐driven oxidative stress. Free Radic Biol Med. 2017;108:452–64. [DOI] [PubMed] [Google Scholar]
- 85. Gall T, Petho D, Nagy A, Hendrik Z, Mehes G, Potor L, et al. Heme induces endoplasmic reticulum stress (HIER stress) in human aortic smooth muscle cells. Front Physiol. 2018;9:1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hebbel RP, Belcher JD, Vercellotti GM. The multifaceted role of ischemia/reperfusion in sickle cell anemia. J Clin Invest. 2020;130:1062–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Vinchi F, Muckenthaler MU, Da Silva MC, Balla G, Balla J, Jeney V. Atherogenesis and iron: from epidemiology to cellular level. Front Pharmacol. 2014;5:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Bourne JH, Colicchia M, Di Y, Martin E, Slater A, Roumenina LT, et al. Heme induces human and mouse platelet activation through C‐type‐lectin‐like receptor‐2. Haematologica. 2020. [Online ahead of print]. 10.3324/haematol.2020.246488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. De Caterina R, Libby P, Peng HB, Thannickal VJ, Rajavashisth TB, Gimbrone MA Jr, et al. Nitric oxide decreases cytokine‐induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest. 1995;96:60–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Space SL, Lane PA, Pickett CK, Weil JV. Nitric oxide attenuates normal and sickle red blood cell adherence to pulmonary endothelium. Am J Hematol. 2000;63:200–4. [DOI] [PubMed] [Google Scholar]
- 91. Spiecker M, Darius H, Kaboth K, Hubner F, Liao JK. Differential regulation of endothelial cell adhesion molecule expression by nitric oxide donors and antioxidants. J Leukoc Biol. 1998;63:732–9. [PubMed] [Google Scholar]
- 92. Sohn YS, Ghoti H, Breuer W, Rachmilewitz E, Attar S, Weiss G, et al. The role of endocytic pathways in cellular uptake of plasma non‐transferrin iron. Haematologica. 2012;97:670–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Porto BN, Alves LS, Fernandez PL, Dutra TP, Figueiredo RT, Graca‐Souza AV, et al. Heme induces neutrophil migration and reactive oxygen species generation through signaling pathways characteristic of chemotactic receptors. J Biol Chem. 2007;282:24430–6. [DOI] [PubMed] [Google Scholar]
- 94. Chen G, Zhang D, Fuchs TA, Manwani D, Wagner DD, Frenette PS. Heme‐induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood. 2014;123:3818–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Graca‐Souza AV, Arruda MA, de Freitas MS, Barja‐Fidalgo C, Oliveira PL. Neutrophil activation by heme: implications for inflammatory processes. Blood. 2002;99:4160–5. [DOI] [PubMed] [Google Scholar]
- 96. Fortes GB, Alves LS, de Oliveira R, Dutra FF, Rodrigues D, Fernandez PL, et al. Heme induces programmed necrosis on macrophages through autocrine TNF and ROS production. Blood. 2012;119:2368–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Tangudu NK, Alan B, Vinchi F, Worle K, Lai D, Vettorazzi S, et al. Scavenging reactive oxygen species production normalizes ferroportin expression and ameliorates cellular and systemic iron disbalances in hemolytic mouse model. Antioxid Redox Signal. 2018;29:484–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hao K, Hanawa H, Ding L, Ota Y, Yoshida K, Toba K, et al. Free heme is a danger signal inducing expression of proinflammatory proteins in cultured cells derived from normal rat hearts. Mol Immunol. 2011;48:1191–202. [DOI] [PubMed] [Google Scholar]
- 99. Simoes RL, Arruda MA, Canetti C, Serezani CH, Fierro IM, Barja‐Fidalgo C. Proinflammatory responses of heme in alveolar macrophages: repercussion in lung hemorrhagic episodes. Mediators Inflamm. 2013;2013:946878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Sindrilaru A, Peters T, Wieschalka S, Baican C, Baican A, Peter H, et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J Clin Invest. 2011;121:985–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Nyakundi BB, Toth A, Balogh E, Nagy B, Erdei J, Ryffel B, et al. Oxidized hemoglobin forms contribute to NLRP3 inflammasome‐driven IL‐1beta production upon intravascular hemolysis. Biochim Biophys Acta Mol Basis Dis. 2019;1865:464–75. [DOI] [PubMed] [Google Scholar]
- 102. Amer J, Ghoti H, Rachmilewitz E, Koren A, Levin C, Fibach E. Red blood cells, platelets and polymorphonuclear neutrophils of patients with sickle cell disease exhibit oxidative stress that can be ameliorated by antioxidants. Br J Haematol. 2006;132:108–13. [DOI] [PubMed] [Google Scholar]
- 103. Fibach E, Rachmilewitz E. The role of oxidative stress in hemolytic anemia. Curr Mol Med. 2008;8:609–19. [DOI] [PubMed] [Google Scholar]
- 104. Balla G, Jacob HS, Eaton JW, Belcher JD, Vercellotti GM. Hemin: a possible physiological mediator of low density lipoprotein oxidation and endothelial injury. Arterioscler Thromb. 1991;11:1700–11. [DOI] [PubMed] [Google Scholar]
- 105. Belcher JD, Marker PH, Geiger P, Girotti AW, Steinberg MH, Hebbel RP, et al. Low‐density lipoprotein susceptibility to oxidation and cytotoxicity to endothelium in sickle cell anemia. J Lab Clin Med. 1999;133:605–12. [DOI] [PubMed] [Google Scholar]
- 106. Cheng C, Noordeloos AM, Jeney V, Soares MP, Moll F, Pasterkamp G, et al. Heme oxygenase 1 determines atherosclerotic lesion progression into a vulnerable plaque. Circulation. 2009;119:3017–27. [DOI] [PubMed] [Google Scholar]
- 107. Jeney V, Balla G, Balla J. Red blood cell, hemoglobin and heme in the progression of atherosclerosis. Front Physiol. 2014;5:379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Nagy E, Eaton JW, Jeney V, Soares MP, Varga Z, Galajda Z, et al. Red cells, hemoglobin, heme, iron, and atherogenesis. Arterioscler Thromb Vasc Biol. 2010;30:1347–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Nath KA, Grande JP, Belcher JD, Garovic VD, Croatt AJ, Hillestad ML, et al. Antithrombotic effects of heme‐degrading and heme‐binding proteins. Am J Physiol Heart Circ Physiol. 2020;318:H671–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Potor L, Banyai E, Becs G, Soares MP, Balla G, Balla J, et al. Atherogenesis may involve the prooxidant and proinflammatory effects of ferryl hemoglobin. Oxid Med Cell Longev. 2013;2013:676425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Mehta NU, Grijalva V, Hama S, Wagner A, Navab M, Fogelman AM, et al. Apolipoprotein E‐/‐ mice lacking hemopexin develop increased atherosclerosis via mechanisms that include oxidative stress and altered macrophage function. Arterioscler Thromb Vasc Biol. 2016;36:1152–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Merle NS, Grunenwald A, Rajaratnam H, Gnemmi V, Frimat M, Figueres ML, et al. Intravascular hemolysis activates complement via cell‐free heme and heme‐loaded microvesicles. JCI Insight. 2018;3:e96910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Gammella E, Recalcati S, Rybinska I, Buratti P, Cairo G. Iron‐induced damage in cardiomyopathy: oxidative‐dependent and independent mechanisms. Oxid Med Cell Longev. 2015;2015:230182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Porter JB, Wood J, Olivieri N, Vichinsky EP, Taher A, Neufeld E, et al. Treatment of heart failure in adults with thalassemia major: response in patients randomised to deferoxamine with or without deferiprone. J Cardiovasc Magn Reson. 2013;15:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Aessopos A, Farmakis D, Tsironi M, Diamanti‐Kandarakis E, Matzourani M, Fragodimiri C, et al. Endothelial function and arterial stiffness in sickle‐thalassemia patients. Atherosclerosis. 2007;191:427–32. [DOI] [PubMed] [Google Scholar]
- 116. Chen YG, Lin CL, Ho CL, Chen YC, Kao CH. Risk of coronary artery disease in transfusion‐naive thalassemia populations: a nationwide population‐based retrospective cohort study. Eur J Intern Med. 2015;26:250–4. [DOI] [PubMed] [Google Scholar]
- 117. Cheung YF, Chan GC, Ha SY. Arterial stiffness and endothelial function in patients with beta‐thalassemia major. Circulation. 2002;106:2561–6. [DOI] [PubMed] [Google Scholar]
- 118. Gursel O, Kurekci AE, Tascilar E, Ileri T, Altun D, Tapan S, et al. Premature atherosclerosis in children with beta‐thalassemia major. J Pediatr Hematol Oncol. 2012;34:630–4. [DOI] [PubMed] [Google Scholar]
- 119. Hahalis G, Kalogeropoulos A, Terzis G, Tselepis AD, Kourakli A, Mylona P, et al. Premature atherosclerosis in non‐transfusion‐dependent beta‐thalassemia intermedia. Cardiology. 2011;118:159–63. [DOI] [PubMed] [Google Scholar]
- 120. Hahalis G, Zacharioglou E, Xanthopoulou I, Koniari I, Kalogeropoulou C, Tsota I, et al. Coronary atherosclerosis burden is not advanced in patients with beta‐thalassemia despite premature extracardiac atherosclerosis: a coronary artery calcium score and carotid intima‐media thickness study. J Geriatr Cardiol. 2016;13:158–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Sherief LM, Dawood O, Ali A, Sherbiny HS, Kamal NM, Elshanshory M, et al. Premature atherosclerosis in children with beta‐thalassemia major: new diagnostic marker. BMC Pediatr. 2017;17:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Ricchi P, Ammirabile M, Spasiano A, Costantini S, Di Matola T, Cinque P, et al. Hypocholesterolemia in adult patients with thalassemia: a link with the severity of genotype in thalassemia intermedia patients. Eur J Haematol. 2009;82:219–22. [DOI] [PubMed] [Google Scholar]
- 123. Ricchi P, Ammirabile M, Spasiano A, Costantini S, Cinque P, Di Matola T, et al. Combined chelation therapy in thalassemia major with deferiprone and desferrioxamine: a retrospective study. Eur J Haematol. 2010;85:36–42. [DOI] [PubMed] [Google Scholar]
- 124. Amendola G, Danise P, Todisco N, D'Urzo G, Di Palma A, Di Concilio R. Lipid profile in beta‐thalassemia intermedia patients: correlation with erythroid bone marrow activity. Int J Lab Hematol. 2007;29:172–6. [DOI] [PubMed] [Google Scholar]
- 125. Ho PJ, Hiwase D, Ramakrishna R, Viiala N, Solterbeck A, Traficante R, et al. Cardiac and hepatic siderosis in myelodysplastic syndrome, thalassemia and diverse causes of transfusion‐dependent anemia: the TIMES study. Hemasphere. 2019;3:e224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Shalev H, Kapelushnik J, Moser A, Knobler H, Tamary H. Hypocholesterolemia in chronic anemias with increased erythropoietic activity. Am J Hematol. 2007;82:199–202. [DOI] [PubMed] [Google Scholar]
- 127. Khera R, Singh M, Goel G, Gupta P, Singh T, Dubey AP. Hypertriglyceridemia thalassemia syndrome: a report of 4 cases. Indian J Hematol Blood Transfus. 2014;30(Suppl 1):288–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Boudrahem‐Addour N, Izem‐Meziane M, Bouguerra K, Nadjem N, Zidani N, Belhani M, et al. Oxidative status and plasma lipid profile in beta‐thalassemia patients. Hemoglobin. 2015;39:36–41. [DOI] [PubMed] [Google Scholar]
- 129. Tantawy AA, Adly AA, El Maaty MG, Amin SA. Subclinical atherosclerosis in young beta‐thalassemia major patients. Hemoglobin. 2009;33:463–74. [DOI] [PubMed] [Google Scholar]
- 130. Celletti FL, Waugh JM, Amabile PG, Brendolan A, Hilfiker PR, Dake MD. Vascular endothelial growth factor enhances atherosclerotic plaque progression. Nat Med. 2001;7:425–9. [DOI] [PubMed] [Google Scholar]
- 131. Fatkhullina AR, Peshkova IO, Koltsova EK. The role of cytokines in the development of atherosclerosis. Biochemistry. 2016;81:1358–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Elsharawy MA, Moghazy KM, Shawarby MA. Atherosclerosis in sickle cell disease ‐ a review. Int J Angiol. 2009;18:62–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Atichartakarn V, Chuncharunee S, Archararit N, Udomsubpayakul U, Aryurachai K. Intravascular hemolysis, vascular endothelial cell activation and thrombophilia in splenectomized patients with hemoglobin E/beta‐thalassemia disease. Acta Haematol. 2014;132:100–7. [DOI] [PubMed] [Google Scholar]
- 134. Conran N. Intravascular hemolysis: a disease mechanism not to be ignored. Acta Haematol. 2014;132:97–9. [DOI] [PubMed] [Google Scholar]
- 135. Chapman RG, McDonald LL. Red cell life span after splenectomy in hereditary spherocytosis. J Clin Invest. 1968;47:2263–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Sakamoto TM, Canalli AA, Traina F, Franco‐Penteado CF, Gambero S, Saad ST, et al. Altered red cell and platelet adhesion in hemolytic diseases: hereditary spherocytosis, paroxysmal nocturnal hemoglobinuria and sickle cell disease. Clin Biochem. 2013;46:1798–803. [DOI] [PubMed] [Google Scholar]
- 137. Switzer JA, Hess DC, Nichols FT, Adams RJ. Pathophysiology and treatment of stroke in sickle‐cell disease: present and future. Lancet Neurol. 2006;5:501–12. [DOI] [PubMed] [Google Scholar]
- 138. Belcher JD, Gomperts E, Nguyen J, Chen C, Abdulla F, Kiser ZM, et al. Oral carbon monoxide therapy in murine sickle cell disease: beneficial effects on vaso‐occlusion, inflammation and anemia. PLoS One. 2018;13:e0205194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Fibach E, Rachmilewitz EA. The role of antioxidants and iron chelators in the treatment of oxidative stress in thalassemia. Ann N Y Acad Sci. 2010;1202:10–6. [DOI] [PubMed] [Google Scholar]
- 140. Kaul DK, Liu XD, Choong S, Belcher JD, Vercellotti GM, Hebbel RP. Anti‐inflammatory therapy ameliorates leukocyte adhesion and microvascular flow abnormalities in transgenic sickle mice. Am J Physiol Heart Circ Physiol. 2004;287:H293–301. [DOI] [PubMed] [Google Scholar]
- 141. Louie JE, Anderson CJ, Fayaz MF, Henry A, Killeen T, Mohandas N, et al. Case series supporting heme detoxification via therapeutic plasma exchange in acute multiorgan failure syndrome resistant to red blood cell exchange in sickle cell disease. Transfusion. 2018;58:470–9. [DOI] [PubMed] [Google Scholar]
- 142. Vercellotti GM, Zhang P, Nguyen J, Abdulla F, Chen C, Nguyen P, et al. Hepatic overexpression of hemopexin inhibits inflammation and vascular stasis in murine models of sickle cell disease. Mol Med. 2016;22:437–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Matte A, Zorzi F, Mazzi F, Federti E, Olivieri O, De Franceschi L. New therapeutic options for the treatment of sickle cell disease. Mediterr J Hematol Infect Dis. 2019;11:e2019002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Serum ferritin and transferrin analysed in patients with β‐thalassaemia.
Figure S2. Serum LDL and oxLDL in patients with β‐thalassaemia.
Figure S3. MCP‐1 levels in patients with haemolytic diseases.
Figure S4. Cytokines in patients with haemolytic diseases.
Figure S5. Effect of hydroyurea treatment in sickle cell disease.
Figure S6. Effect of splenectomy on haem and iron parameters and anaemia in transfusion‐dependent patients with β‐thalassaemia major.
Figure S7. Effect of splenectomy on markers of vascular dysfunction, oxidative stress and inflammation in transfusion‐dependent patients with β‐thalassaemia major.
Figure S8. Effect of splenectomy on haem and iron parameters and anaemia in transfusion‐dependent patients with spherocytosis.
Figure S9. Effect of splenectomy on markers of vascular dysfunction, oxidative stress and inflammation anaemia in transfusion‐dependent patients with spherocytosis.
Figure S10. Effect of splenectomy on haem and iron parameters and anaemia in patients with sickle cell disease receiving exchange transfusions.
Figure S11. Effect of splenectomy on markers of vascular dysfunction, oxidative stress and inflammation in patients with sickle cell disease receiving exchange transfusions.
Table SI. Clinical parameters analysed in patients with β‐thalassaemia major and intermedia.
Table SII. Clinical parameters analysed in patients with β‐thalassaemia major and intermedia.
Table SIII. Clinical parameters analysed in patients with spherocytosis.
Table SIV. Clinical parameters analysed in patients with spherocytosis.
Table SV. Clinical parameters analysed in patients with sickle cell disease.
Table SVI. Clinical parameters analysed in patients with sickle cell disease treated with simple transfusions.
Table SVII. Clinical parameters analysed in patients with sickle cell disease treated with exchange transfusions.
Table SVIII. Pairwise comparisons between heme‐/iron parameters and biomarkers in patients with β‐thalassaemia major.
Table SIX. Pairwise comparisons between heme‐/iron parameters and biomarkers in patients with sickle cell disease.
Table SX. Pairwise comparisons between heme‐/iron parameters and biomarkers in patients with spherocytosis.