

Abstract
Isolated methylmalonic acidaemia (MMA) and propionic acidaemia (PA) are rare inherited metabolic diseases. Six years ago, a detailed evaluation of the available evidence on diagnosis and management of these disorders has been published for the first time. The article received considerable attention, illustrating the importance of an expert panel to evaluate and compile recommendations to guide rare disease patient care. Since that time, a growing body of evidence on transplant outcomes in MMA and PA patients and use of precursor free amino acid mixtures allows for updates of the guidelines. In this article, we aim to incorporate this newly published knowledge and provide a revised version of the guidelines. The analysis was performed by a panel of multidisciplinary health care experts, who followed an updated guideline development methodology (GRADE). Hence, the full body of evidence up until autumn 2019 was re‐evaluated, analysed and graded. As a result, 21 updated recommendations were compiled in a more concise paper with a focus on the existing evidence to enable well‐informed decisions in the context of MMA and PA patient care.
Keywords: diagnosis and management, guidelines, inherited metabolic disease, methylmalonic acidaemia, propionic acidaemia
Synopsis.
Structured evidence evaluation of the present literature resulted in 21 recommendations on how to diagnose and manage patients with methylmalonic acidaemia and propionic acidaemia.
1. INTRODUCTION
The first guidelines on isolated methylmalonic acidaemia (MMA) and propionic acidaemia (PA) were published in September 2014 and accessed over 53 000 times on the journal's website and cited 182 times according to Web of Science (July 2020). 1 The attention the guidelines received signifies the interest and utilisation by health care professionals of this evidence evaluation and deduced recommendations as provided by an expert panel. Especially in the rare disease field, structured evaluation of the present evidence is of great value, as the process involves specific considerations related to the rarity of these types of disorders. High‐quality trials in large samples which would produce firm and reliable evidence are scarce given the small patient populations, often rendering the present evidence low quality. As a result, evidence collected from case series or smaller studies needs to be carefully evaluated.
The advances in the field and the success of the first MMA/PA guidelines prompted us to provide an update 6 years later. The guidelines presented here use a different and more up‐to‐date system to evaluate evidence than the first version, the ‘Grading of Recommendations, Assessment, Development and Evaluation’ (GRADE) approach, 2 which is more suitable for the rare disease field, as it partially accounts for the above described challenges. Due to the change of methods, the whole body of literature on MMA and PA referenced on PubMed (https://pubmed.ncbi.nlm.nih.gov/) was newly evaluated. Since the publication of the initial guidelines, new advances have been made, and more data have been published on MMA and PA. For example, more patients have been organ transplanted and their outcomes were published. Several novel studies investigated the impact of using precursor free amino acids (PFAAs) as part of the dietary regimen of MMA and PA patients. The updated guidelines presented here aim to include this new data, while at the same time attempt to streamline and summarise sections for which the evidence has not significantly changed. The reader is provided with the full recommendations for MMA and PA diagnosis and management but is referred to the initial guidelines for more details regarding certain aspects.
This first revision of the MMA and PA guidelines covers the same subtypes of isolated MMA and PA as before. MMA is caused by a deficiency of the methylmalonyl‐CoA mutase enzyme (MMUT), either by a direct defect of the enzyme, or by a deficient synthesis of its cofactor adenosylcobalamin. Precursors of this pathway are derived from specific amino acids (valine, isoleucine, threonine, methionine), propionate produced by gut bacteria and odd chain fatty acids as well as the cholesterol side chain (Figure 1). Under isolated MMA, the following gene defects are included: deficiencies of MMUT (OMIM #251000), MMAA (OMIM #251100), MMAB (OMIM #251110) and truncating mutations towards the N‐terminal in the MMADHC gene (OMIM #277410). 3 PA (OMIM #606054) is caused by mutations in the PCCA (OMIM #232000) or PCCB gene (OMIM #232050), resulting in deficiency of the propionyl‐CoA carboxylase enzyme function. 4
FIGURE 1.
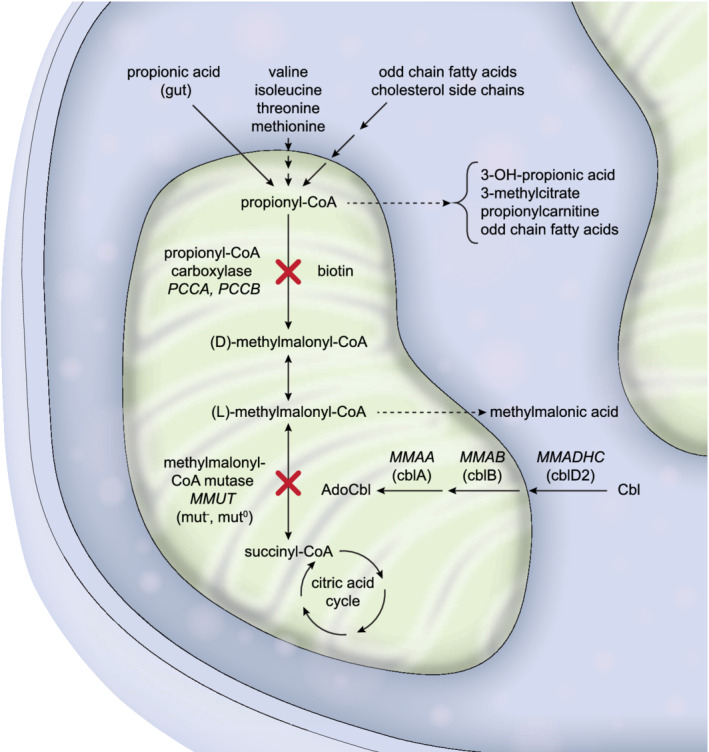
Propionyl‐CoA is metabolised in the mitochondria by specific enzymes. Defects in the genes MMUT, MMAA, MMAB or MMADHC (more specifically variant 2) lead to isolated MMA, whereas defects in PCCA or PCCB lead to PA. Propionyl‐CoA is derived from various sources and is metabolised to alternative products in the case of accumulation in the above‐described defects. In addition, accumulating methylmalonyl‐CoA in isolated MMA is hydrolysed to methylmalonic acid, the main biomarker of this disease. Gene names are italicised, complementation groups are in parenthesis. MMA, methylmalonic acidaemia; PA, propionic acidaemia
2. METHODOLOGY AND OBJECTIVES
The presented revision of the guidelines for MMA and PA was undertaken by a panel of 21 health care professionals who work in the field of metabolic medicine. P. F. (secretary), M. R. B., F. H. (co‐chairs) and M. Huemer (methodology and moderation) formed a core panellist group to coordinate organisational and content aspects. The panel further included paediatric metabolic specialists (D. B., A. C., K. A. C., C. D.‐V., S. C. G., S. G., T. H., D. K., S. S.‐B., G. T., M. W.), a metabolic adult physician (M. Hochuli), a paediatric metabolic dietitian (M. D.), paediatric neurologists (G. H., D. M.), a medical doctor in training and PhD student in inherited metabolic diseases (F. M.) and a clinical biochemist (J. O. S.). The panel was supported by external reviewers for review of nephrology, cardiology, neurology and dietetic recommendations and two patient representatives. The panel met in person in September 2019 and November 2019).
2.1. Literature search and evidence grading
In the initial guidelines, the method to collect the evidence was based on SIGN. 5 In the revised guidelines, the GRADE methodology was used.
To gather the initial evidence base, a PubMed search was performed on September 29, 2019 with the following search term: (“propionic acidemia” OR “propionic acidaemia” OR “propionic aciduria” OR “methylmalonic acidemia” OR “methylmalonic acidaemia” OR “methylmalonic aciduria” OR “propionic acidemias” OR “propionic acidaemias” OR “propionic acidurias” OR “methylmalonic acidemias” OR “methylmalonic acidaemias” OR “methylmalonic acidurias”) AND (“"1900/01/01"[Date ‐ Publication]: "2019/09/05"[Date ‐ Publication]) AND (english[Language]) NOT (animals[mh] NOT humans[mh]). This search yielded 1488 entries. Articles were manually sorted to 194 clinical reports on patients with MMA and/or PA (any lab‐based studies and case reports with less than three patients excluded) that were stratified into categories (P. F., M. B.) (Figure S1). Case reports were considered in bulk for specific questions. Eight articles published past the cut‐off date and seven articles cited within the initial clinical reports were considered by the panel to be of specific importance (total of 209 articles). Articles were allocated to the categories ‘high’, ‘moderate’ and ‘low’ quality of evidence to a certain outcome by at least two panellists.
2.2. Development process
In a first face‐to‐face meeting, panellists were informed about and received guidance on how to apply the GRADE methodology.
The panellists agreed that the guidelines should address all types of isolated MMA and PA in patients of all ages. Panellists and patient representatives selected and rated outcome parameters as critical, important, or not important using an online tool. The literature was assigned to the critical and important outcomes and discussed and evaluated in working groups (email or online/phone meetings), which formulated first drafts for recommendations. A consensus‐oriented, moderated discussion of the evidence and wording of the recommendations was held during the second face‐to‐face meeting. Based on this meeting, the evidence profile was created, and recommendations were formulated. Since during the COVID‐19 pandemic personal in‐person meetings were impossible, the panellists rated the quality of the evidence for each outcome using an online tool. Final rating of the quality of evidence was based on at least 50% of votes for the category ‘high’, ‘moderate’ or ‘low’. If two categories received equal numbers of votes, the quality of the evidence was downgraded to the lower category (Figure S2A).
FIGURE 2.
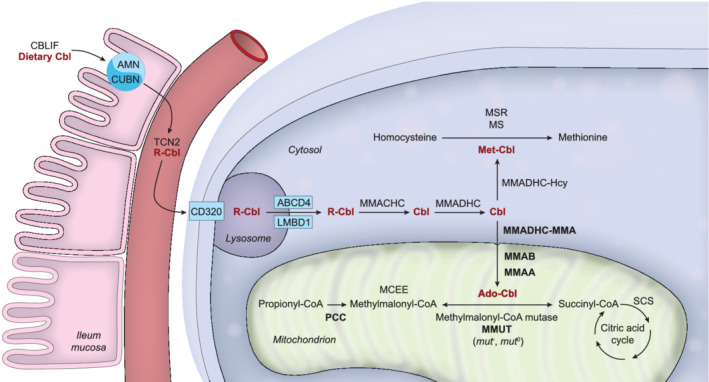
Scheme depicting the extra‐ and intracellular transport and processing of the cobalamin (Cbl) molecule and the involved proteins. Methylmalonyl‐CoA mutase (MMUT), methylmalonic aciduria type A protein (MMAA), methylmalonic aciduria type B protein (MMAB), methylmalonic aciduria and homocystinuria type D protein variant 2 (MMADHC‐MMA), and propionyl‐CoA carboxylase (PCC) defects and their related diseases are discussed in these guidelines, hence these proteins are depicted in bold print. The other proteins are depicted using official protein nomenclature: amnionless (AMN) and cubulin (CUBN) form the cubam receptor to absorb cobalamin bound to cobalamin binding intrinsic factor (CBLIF); haptocorrin (TCN1; not shown), transcobalamin (TCN2) and transcobalamin receptor (CD320) facilitate cobalamin transport and uptake into the cell; lysosomal cobalamin transporter (ABCD4) and lysosomal cobalamin transport escort protein (LMBD1) export cobalamin from the lysosome; methylmalonic aciduria and homocystinuria type C protein (MMACHC) cleaves the R group of cobalamin (upper‐axial ligand); methylmalonic aciduria and homocystinuria type D protein (MMADHC) targets cobalamin towards further processing in the cytosol or the mitochondrion; methionine synthase (MS), kept in its active form by methionine synthase reductase (MSR), uses cobalamin in its methylated form; methylmalonyl‐CoA epimerase (MCEE) converts (R)‐methylmalonyl‐CoA to (S)‐methylmalonyl‐CoA; succinyl‐CoA synthetase (SCS) also called succinate‐CoA ligase forms succinate from succinyl‐CoA in the citric acid cycle
The strength of each recommendation was finally rated by the panellists based on the quality of the evidence as well as the balance of benefits and harms, values, preferences and resources. Recommendations were downgraded to the lower category (weak) if the distribution of votes was exactly equal (Figure S2B).
Strong recommendations are indicated by ‘we recommend’; weak recommendations by ‘we suggest’. Some recommendations were considered extremely well based on clinical grounds or would have serious consequences if not followed; others were clearly supported by biochemical facts and well‐established medical knowledge. In these cases, the wording of the recommendation was strengthened (‘we strongly recommend’). For approval ratings for each recommendation, see Figure S3.
FIGURE 3.

Proposed management flowchart for MMA and PA patients during an acute metabolic decompensation, based on expert opinion. All drugs are given intravenously (IV). MMA, methylmalonic acidaemia; PA, propionic acidaemia
2.3. Guidelines objective
The purpose of these guidelines is to serve health care professionals working with MMA and PA patients as an evidence‐based reference to provide optimal patient care. When clear‐cut recommendations could not be reached because of poor evidence quality, the presented collation of the body of the evidence may serve as a reference, when considering specific aspects of MMA and PA patient care. However, the guidelines do not replace comprehensive clinical reflection and assessment of each patient's individual situation. These guidelines attempt to provide guidance where there is sufficient evidence, while at the same time identifying knowledge gaps in the evidence.
3. GUIDELINES
3.1. Outcome parameters
An outcome parameter reflects a specific entity by which a patient suffering from MMA or PA can be affected. This can be a clinical sign (eg, kidney dysfunction or cardiomyopathy) or a complex mix of several factors [eg, health‐related quality of life (HrQoL) or metabolic stability]. Eleven outcome parameters were initially proposed by the core panellist group. After a round of open feedback from the panel and the patient representatives, four further outcomes were added. The importance of the different outcomes was gradually rated from 1 (of lowest importance) to 9 (most critical for decision‐making) (Table 1). Survival was the most highly rated outcome parameter, followed by HrQoL and metabolic stability. The lowest rated outcomes were haematological abnormalities and bone health. Importantly, none of the outcomes were underestimated by the panel when compared to the patient representatives' ratings (Figure S4).
TABLE 1.
Outcome parameters and their ratings of importance
| Outcome parameter | Median rating of importance of outcome |
|---|---|
| Survival | 9 |
| Health‐related quality of life | 9 |
| Metabolic stability | 8 |
| Cognitive development | 8 |
| Epilepsy | 8 |
| Metabolic stroke | 7 |
| Vision and hearing | 7 |
| Early diagnosis | 7 |
| Cardiomyopathy | 7 |
| Kidney dysfunction | 7 |
| Pancreatitis | 6 |
| Normal growth | 6 |
| Neutropenia | 6 |
| Anaemia | 5.5 |
| Bone health | 5 |
Note: Each outcome parameter was rated by the panellists and the patient representatives from 0 (lowest importance) to 9 (most critical for decision‐making). The second column represents the median value of all the ratings.
FIGURE 4.
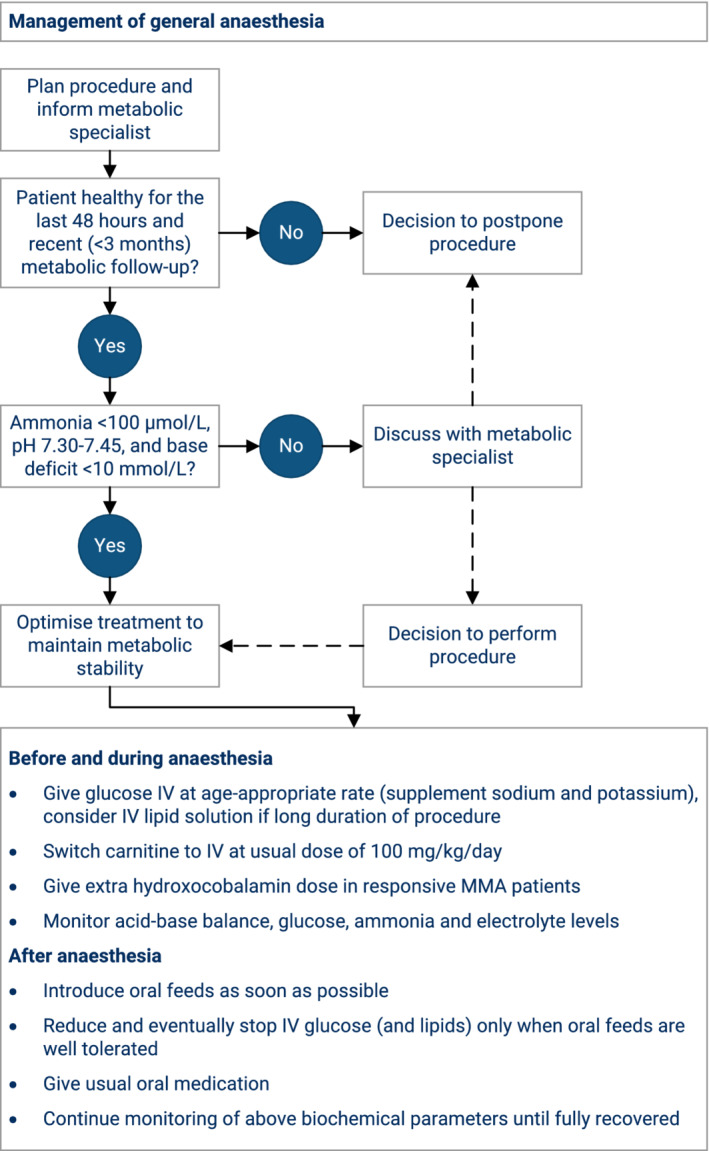
Proposed management flowchart for MMA and PA patients undergoing general anaesthesia, based on expert opinion. MMA, methylmalonic acidaemia; PA, propionic acidaemia
The following recommendations are structured according to a patient's natural pathway from initial presentation and diagnostic procedures to management, long‐term complications and monitoring. The above‐defined outcomes guide this path and each of the recommendations directly refers to an outcome.
3.2. Diagnostic challenges
3.2.1. Clinical presentation
A patient can only be diagnosed with MMA or PA when the physician recognises the clinical picture suggestive of MMA or PA, which can be a challenging process as many of the clinical signs are non‐specific. MMA and PA can present with acute, chronic or intermittent symptoms, which can often aggravate or even occur for the first time following a ‘trigger’ event. Symptoms include vomiting, weight loss, hypoglycaemia, neurological deterioration with hypotonia, irritability and lethargy eventually leading to coma in the case of an acute early onset neonatal presentation. This clinical presentation can be accompanied by the biochemical findings of metabolic acidosis, ketosis, hyperlactataemia and hyperammonaemia. Triggers of an acute episode include but are not limited to post‐partum neonatal stress and other forms of catabolic states induced by infection or prolonged fasting. Late‐onset patients present with a wider range of signs, involving failure to thrive, a range of neurological symptoms (encephalopathy, developmental delay), cardiac and kidney manifestations. We provide a slightly updated table of main and rare signs and symptoms occurring during acute or chronic presentation of MMA or PA (Table 2).
TABLE 2.
Potential signs and symptoms at presentation observed in MMA and PA patients
| Acute presentation | Chronic presentation |
|---|---|
| Nervous system | |
| Acute encephalopathy | Hypotonia |
| Seizures | Developmental delay |
| Movement disorders (more frequent in PA) | Seizures |
| Stroke‐like episodes (more frequent in MMA) | Movement disorders/dystonia |
| Gastrointestinal system | |
| Vomiting | Recurrent vomiting with ketoacidosis |
| Feeding difficulties | Failure to thrive |
| Pancreatitis | |
| Haematopoietic system | |
| Neutropenia, pancytopenia | Neutropenia, pancytopenia |
| Heart (mostly in PA) | |
| Acute cardiac failure (mostly based on cardiomyopathy) | Cardiomyopathy |
| Arrhythmias | Prolonged QTc in ECG |
| Kidney | |
| Chronic renal failure (almost exclusively in MMA) | |
Abbreviations: ECG, electrocardiogram; MMA, methylmalonic acidaemia; PA, propionic acidaemia; QTc, corrected QT interval.
Recommendation #1
We strongly recommend considering MMA and PA in the differential diagnosis of an acute or intermittent neurological deterioration, and in the differential diagnosis of neonatal sepsis.
Outcome: Survival.
Quality of evidence: moderate.
Strength of recommendation: very strong (supported by medical knowledge).
3.2.2. Laboratory diagnosis
When, based on clinical assessment and basic laboratory parameters or on an abnormal result from newborn screening, an organic acidaemia is suspected, the diagnosis of MMA or PA is achieved by measurements of organic acids in urine (Table 3). Elevations of methylmalonic acid together with 3‐hydroxypropionate and presence of 2‐methylcitrate confirm a diagnosis of MMA, while the same pattern without abnormal levels of methylmalonic acid is present in PA. Often, tiglylglycine and propionylglycine can be found in PA. The diagnostic work‐up may be complemented with the measurement of acylcarnitines in dried blood spots or plasma. In this test, a striking elevation of C3 acylcarnitine (propionylcarnitine) can be found in both MMA and PA. In addition, plasma amino acid measurements show elevated glycine levels in MMA and PA.
TABLE 3.
Biochemical presentation of PA and conditions with raised methylmalonic acid
| Organic acids in urine | Acylcarnitines in dried blood or plasma | Plasma | |||||
|---|---|---|---|---|---|---|---|
| Methylmalonic acid | 3‐hydroxy‐propionate | 2‐methylcitrate | Propionylcarnitine | Homocysteine | Vitamin B12 | Holotranscobalamin | |
| Diseases discussed in these guidelines | |||||||
| MMA a | ↑‐↑↑↑ | ↑ | ↑ | ↑↑ | n | n | n |
| PA | n | ↑ | ↑(↑) | ↑↑ (↑) | n | n | n |
| Other defects and deficiencies causing raised methylmalonic acid | |||||||
| MCEE deficiency | ↑ | (↑) | (↑) | (↑) | n | n | n |
| ACSF3 deficiency b | ↑ | n | n | n | n | n | n |
| Adenosyl‐ and methylcobalamin synthesis defects c | ↑ ‐ ↑↑↑ | ↑ | ↑ | ↑‐↑↑ | ↑ ‐ ↑↑↑ | n | n |
| Transcobalamin deficiency | ↑ | n‐↑ | n‐↑ | n‐↑ | ↑ | n‐↓ | ↓ |
| Transcobalamin receptor deficiency | ↑ | n/a | n/a | n/a | n‐↑ | n/a | n/a |
| IF deficiency and Imerslund‐Najman‐Gräsbeck syndrome | ↑ ‐ ↑↑ | n‐↑ | n‐↑ | n‐↑ | ↑ ‐ ↑↑ | ↓↓ | ↓ |
| Nutritional vitamin B12 deficiency | ↑ ‐ ↑↑ | n‐↑ | n‐↑ | n‐↑ | ↑ ‐ ↑↑ | ↓‐↓↓ | ↓‐↓↓ |
Note: Pathognomonic biochemical findings for MMA and PA in urine and blood compared to other related diseases, causing raised methylmalonic acids levels. Isolated defects in the methylcobalamin pathway are not displayed.
Abbreviations: ACSF3, acyl‐CoA synthetase family member 3; IF, intrinsic factor; MCEE, methylmalonyl CoA epimerase; MMA, methylmalonic acidaemia; n, normal; n/a, no data available; PA, propionic acidaemia.
Subtypes mut, cblA, cblB, cblD‐MMA.
In addition to methylmalonic acid, raised malonic acid can be found in urine; biochemical parameters based on Reference 6.
Subtypes cblC, cblD‐MMA/HC, cblF, cblJ.
Recommendation #2
We strongly recommend measurement of organic acids in urine to achieve a reliable diagnosis of MMA or PA.
Outcome: Early diagnosis.
Quality of evidence: high.
Strength of recommendation: very strong (supported by biochemical knowledge).
3.2.3. Differential diagnosis
As illustrated (Figure 1), the methylmalonyl‐CoA mutase enzyme can be dysfunctional due to several different genetic defects, all of which lead to an elevated level of methylmalonic acid in urine and in blood. The differential diagnosis of raised methylmalonic acid entails a range of diseases. Specifically, methylmalonic acid can be raised due to a defect in MMUT or one of the genes distal in the intracellular cobalamin pathway, causing the MMA subtypes discussed in these guidelines, or due defects more proximal in the intracellular cobalamin pathway as well as nutritional deficiency of cobalamin. To differentiate the diseases discussed in these guidelines from the more proximal ones and impaired cobalamin supply, homocysteine and vitamin B12 levels in blood should be measured, which are expected to be normal in isolated MMA (except in the case of incidental concomitant nutritional vitamin B12 deficiency in isolated MMA). The magnitude of methylmalonic acid elevation can further help the classification of the different defects in the pathway 3 (Table 3). In addition, other rare genetic defects lead to an isolated, albeit milder elevation of methylmalonic acid with normal homocysteine but are not part of these guidelines. These genes include MCEE, encoding methylmalonyl‐CoA epimerase, and SUCLG1, SUCLA2, which encode succinate‐CoA ligase, and others (Figure 2). MCEE deficiency can in very rare cases present with an acute metabolic decompensation, but is clearly clinically less severe compared to isolated MMA. 7
As the propionyl‐CoA carboxylase enzyme requires biotin as a cofactor, nutritional biotin deficiency or a defect of the enzymes biotinidase (BTD gene, OMIM #609019) or holocarboxylase synthetase (HLCS gene, OMIM #609018) must be excluded to finalise the diagnosis of PA. In all these cases, the urinary organic acid profiles would not only include elevated levels of the metabolites pathognomonic for PA (3‐hydroxypropionate and 2‐methylcitrate), but also other metabolites arising from deficiencies of other biotin dependent carboxylases such as methylcrotonylglycine.
Both MMA and PA are inherited in an autosomal recessive fashion. To confirm the biochemical diagnosis, guide management and allow genetic counselling of families, it is necessary to identify the underlying genetic defect. Isolated MMA can be caused by defects in four different genes, including MMUT (mut subtypes), MMAA (cblA), MMAB (cblB), MMADHC (cblD‐MMA). 3 Specific mutations in MMADHC can also lead to a combined phenotype of MMA and homocystinuria or isolated homocystinuria. 8 In the rare case of inconclusive genetic results, complementation studies, the propionate incorporation assayor enzyme activity measurements in fibroblasts can aid in the diagnostic process. 3 , 9 , 10 Many of the known MMUT mutations have been functionally tested with the aforementioned assays, allowing the historic classification as a mut 0 subtype (no response to hydroxocobalamin supplementation in the propionate incorporation assay) or a mut − subtype (increased propionate incorporation upon addition of hydroxocobalamin to the cell culture medium). 11 , 12
In the case of PA, there is no evidence that identification of the underlying gene defect (in PCCA or PCCB) can guide management or prognosis, as no genotype‐phenotype correlation is known. 13 However, molecular genetic confirmation of the disease is useful for genetic counselling and potential prenatal diagnosis.
Recommendation #3
We suggest molecular genetic testing in any patient with a biochemical diagnosis of MMA and PA for confirmation of the diagnosis, allocation to subgroups (mut 0, mut −, cblA, cblB, cblD‐MMA), genetic counselling, and to enable prenatal diagnosis.
Outcome: Early diagnosis.
Quality of evidence: moderate.
Strength of recommendation: weak.
3.2.4. Prenatal testing
Prenatal diagnosis is preferentially performed by molecular analyses in foetal DNA. Biochemical analyses for MMA or PA can be used as an alternative or additional method in cases where the genetic results are inconclusive or not available. Prenatal diagnosis of MMA can be performed by measurement of MMA in dried amniotic fluid 14 and of PA by determination of 2‐methylcitrate with the same method. 15 The combination of two independent methods (biochemical and genetics) increases the reliability of results. 16 These guidelines only recommend on the mode of prenatal diagnosis and do not address individual, cultural and ethical values and considerations.
Recommendation #4
We suggest using genetic testing for prenatal diagnosis of MMA and PA.
Outcome: Early diagnosis.
Quality of evidence: moderate.
Strength of recommendation: weak.
3.2.5. Newborn screening
Propionylcarnitine (C3) is a disease biomarker but can display variable elevations in the context of newborn screening for MMA and PA. From a methodological perspective sensitivity and specificity for diagnosis of MMA and PA from dried blood spots, obtained during the first days of life, can be optimised by measuring C3/C2 ratio, 2‐methylcitrate, 17 , 18 , 19 3‐hydroxypropionate, 20 C3/C0 and C16:1OH/C2 21 and C17, 22 either as part of the initial screening or as second tier testing.
In more recent studies, newborn screening has been shown to increase the chance of early diagnosis of MMA and PA patients, 23 , 24 especially in late‐onset cases. 25 While newborn screening may decrease neonatal mortality, 26 it may not prevent neonatal metabolic decompensation, potentially due to the delay until the newborn screening result is available. Additionally, newborn screening may not improve important outcome parameters, including metabolic stability and cognitive development. 23 , 27 , 28 , 29 , 30 In countries in which MMA and PA is not part of newborn screening but the method is based on obtaining a full acylcarnitine profile, immediate ‘unblinding’ of the profile in children with suggestive clinical signs and symptoms can avoid significant diagnostic delay.
A minority of countries in the European Union (7/27) performed newborn screening for MMA and PA in 2015. 31 As the modality of screening, including confirmatory testing, is variable, and often structured follow‐up programs focussing on patient‐relevant outcomes are absent, systematic evaluation of MMA and PA newborn screening programs is warranted in order to allow for a general recommendation in favour or against MMA and PA newborn screening in the future.
3.3. Management of MMA and PA
3.3.1. Initial treatment
Early diagnosis and timely treatment are essential to improve survival and reduce morbidity in MMA and PA patients. Over time survival of these patients has improved. 23 , 26 , 32 , 33 , 34 , 35 Improved survival seems to be due to improved treatment strategies and patient monitoring. The effect of single treatment variables on survival, however, is not clear from the present evidence. In MMA patients, survival also depends on age of onset and disease subtype; early‐onset MMA patients, in particular mut 0 and cblB patients, have a higher mortality risk. 33 , 34
As soon as the diagnosis of MMA and PA is suspected, specific therapies should be initiated and the patient should be referred to a specialist centre as some of the treatments and specialist knowledge are only available there. 35 Intensive care treatment of patients with a metabolic decompensation can be necessary if there is a severe metabolic acidosis with or without hyperammonaemia, and hyperlactataemia. The specifics of these acute treatment modalities, including extracorporeal detoxification, have not been systematically studied yet, and the contribution of individual treatments to the improved survival over the last decades is unclear. The panel has intensively discussed the available evidence and has come up with principles based on expert opinions of how to approach treatment of an (imminent) acute metabolic decompensation in MMA and PA patients (Figure 3). It seems appropriate to advise against the primary use of sodium phenylbutyrate as ammonia scavenger in MMA and PA as it can lead to decreased glutamine levels, potentially hampering tricarboxylic acid cycle anaplerosis via 2‐ketoglutarate. 36 More recently, carglumic acid was trialled as part of hyperammonaemia treatment strategies in MMA and PA. 37 , 38 , 39 While the medication was so far tolerated with no side effects, 40 further systematic studies are required to prove its efficacy. 41 Further, it has to be noted that extensive administration of glucose intravenously can be associated with lactic acidosis, potentially due to the inhibited pyruvate dehydrogenase enzyme in MMA and PA.
It is not possible to make evidence‐based statements on improvement of survival with liver transplantation, as the published studies focus on the survival shortly after transplantation, but long‐term comparisons to non‐transplanted patients are absent. This applies to liver, kidney and liver‐kidney transplantations, where overall survival after surgery is around 85%. 42 , 43 , 44
Recommendation #5
If the diagnosis of MMA or PA is suspected, we suggest immediate specific treatment to improve survival in MMA and PA.
Outcome: Survival.
Quality of evidence: moderate.
Strength of recommendation: weak.
3.3.2. Metabolic stability
The term metabolic stability is often used in the clinical context, but its meaning is seldom stated explicitly. For the purpose of these guidelines, we defined metabolic stability as the absence of hospitalisation and exacerbation of disease signs and symptoms, especially metabolic acidosis and hyperammonaemia. Per this definition metabolic stability is a complex term, as it is influenced by various factors, some of which are addressed in this chapter.
Levels of biochemical metabolites, such as methylmalonic acid, propionylcarnitine and plasma amino acids, are often used as surrogate markers for metabolic stability (refer to the ‘monitoring’ section for details on measurement frequencies). The present evidence, though, suggests to rely on clinical assessment, including review of medical history, and the measurement of simple biochemical parameters, such as acid‐base balance (decreased pH and base excess, raised anion gap), urinary ketones and plasma ammonia in the acute setting to discriminate between a metabolically stable or a decompensating situation. 45 , 46 More recently, measurement of fibroblast growth factor 21 (FGF21) and of 2‐methylcitrate showed potential to predict the development of long‐term complications. 47 , 48 , 49
Trigger events leading to metabolic decompensation include infections, causing fever and catabolism. Similarly, prolonged fasting periods are leading to catabolism and should be avoided to prevent metabolic instability. 50 In such situations of a mild intermittent illness, it is common practice to apply enteral emergency feeds consisting of a glucose polymer solution (occasionally with fat emulsion) to provide adequate energy and meet raised metabolic demands. The emergency feeds should only be given during a limited time and if they are tolerated, that is, absence of vomiting and diarrhoea. In parallel, protein intake (including PFAA mixtures) is stopped or partly reduced, for example, to 50%, depending on the severity of clinical symptoms. A simplified overview of age‐dependent emergency regimens based on glucose polymer solution is provided (Table 4). Similarly, general anaesthesia requires specific considerations to ensure adequate energy supply and metabolic stability (Figure 4). As vaccinations do not seem to trigger decompensation themselves, as demonstrated in urea cycle disorders, 51 regular vaccination protocols are indicated for MMA and PA patients. Pregnancies have been described in a few cases of MMA and PA without metabolic decompensations. 52 , 53 , 54
TABLE 4.
Emergency regimens
| Age (years) | Glucose polymer concentration (% carbohydrate) | Fat emulsion (% fat) a | Energy (kcal per 100 mL from carbohydrates and fat) |
|---|---|---|---|
| 0‐1 | 10 | 3.5 | 71.5 |
| 1‐2 | 15 | 5 | 105 |
| 2‐9 | 20 | 5 | 125 |
| >10 | 25 | 5 | 145 |
Note: The amount of the glucose polymer solution should be determined according to age‐adequate total daily fluid intake.
Fat emulsion can be added if expected to be well tolerated.
Generally, it is unclear whether resting energy expenditure in MMA and PA is abnormal. 55 , 56 , 57 , 58 There is no evidence in MMA and PA, but it may be considered, that energy requirements might be lower in patients after a metabolic stroke with subsequent reduced mobility. Implementation of tube feeding can ensure adequate nutrition and energy intake in a selected group of MMA and PA patients. 59 , 60
Further, the role of upcoming therapies, such as mRNA therapy or other gene therapy approaches, on metabolic stability and other outcome parameters will need to be defined. 61 , 62 , 63 , 64 , 65
Recommendation #6
We recommend avoiding catabolic metabolism in MMA and PA patients to improve metabolic stability.
Outcome: Metabolic stability.
Quality of evidence: moderate.
Strength of recommendation: strong (supported by experience‐based medical knowledge).
More dietary aspects are also described in the section on normal growth. In general, a diet low in natural protein aims at the reduction of precursor molecules funnelling into the defective pathway in MMA and PA. Current practice of dietary management of MMA and PA patients aims at both metabolic stability and normal growth. It is based on adequate energy supply, avoidance of prolonged fasting and reduced intake of precursor amino acids (methionine, threonine, valine, isoleucine) through a diet restricted in natural protein, and an adequate provision of vitamins, minerals and trace minerals and essential fatty acids for age. The amount of natural protein must be assessed individually and must be based on clinical and biochemical monitoring. Breastfeeding is possible considering the total natural protein intake. However, there is insufficient data to define a detailed evidence‐based dietetic strategy in MMA and PA.
Recommendation #7
We suggest a low natural protein diet under consideration of age‐appropriate total protein requirements to improve metabolic stability.
Outcome: Metabolic stability.
Quality of evidence: low.
Strength of recommendation: weak.
The following specific medications are also used to facilitate metabolic stability. Levocarnitine treatment – usually at a dose of 100 mg/kg/day – is applied with the rationale of restoring depleted to normal carnitine and CoA levels in MMA and PA patients, and based on its ability to bind and eliminate propionyl‐CoA molecules, which are assumed to be responsible for some of the toxic metabolite effects in MMA and PA. 66 At least in PA, levocarnitine might be able to improve metabolic stability and help avoiding hyperammonaemic crises. 36 Levocarnitine is considered a well‐tolerated medication with few side effects. A recent study shows elevated plasma levels of trimethylamine N‐oxide in MMA and PA patients on levocarnitine treatment. Whether this finding is related to an increased risk for cardiovascular disease in MMA and PA patients on levocarnitine medication remains yet to be determined. 67
Metronidazole – at 10 to 20 mg/kg/day in two to three doses – has been part of the standard long‐term management to reduce bacterial propionate production in the gut despite low quality of evidence for a benefit. There is no evidence demonstrating neuropathy as an intrinsic long‐term complication in MMA or PA, but peripheral neuropathy has been observed in PA patients under metronidazole treatment 68 and metronidazole is a known cause of peripheral neuropathy. 69 , 70 Other side effects include QTc prolongation and pancreatitis; hence, metronidazole treatment should be cautiously indicated in MMA and PA patients. If metronidazole is not well tolerated, alternative antibiotics, including amoxicillin or cotrimoxazole, have been applied.
Whether carglumic acid can consistently reduce the number of metabolic decompensations needs to be further studied. 71 It has been tried to anapleurotically support Krebs cycle function by providing citrate, which was associated with reduced number of hospitalisations in three PA patients. 72
Recommendation #8
We suggest supplementation with levocarnitine to improve metabolic stability.
Outcome: Metabolic stability.
Quality of evidence: low.
Strength of recommendation: weak.
Some MMA patients clearly benefit from parenteral cobalamin treatment. Patients with cblA will mostly improve, and cblB have been described to respond in one third of the cases. 73 Among the MMUT‐deficient patients, the mut − subgroup is more likely to show benefits from cobalamin injections 33 although clear evidence of cobalamin responsiveness in mut − patients is lacking. The ideal application route is intramuscular and hydroxocobalamin is the preferred form of the cobalamin molecule. A protocol to evaluate biochemical responsiveness has been proposed previously 3 : (a) The patient should be metabolically stable without recent treatment changes. (b) The patient should be off hydroxocobalamin supplementation for at least 1 month prior to assessment. (c) Collect at least three baseline urine or plasma (use the same body fluid throughout) samples on different days for measurement of methylmalonic acid. (d) Inject 1 mg hydroxocobalamin intramuscularly on three consecutive days. (e) Collect urine or plasma samples over 10 days, every other day. (f) A mean decrease of urine or plasma methylmalonic acid concentration of >50% is regarded as a significant response. A similar cofactor treatment with biotin for PA patients is not recommended, as there has been no description of responsive patients so far.
Recommendation #9
We recommend evaluating responsiveness for parenteral vitamin B12 in every patient with suspected MMA.
Outcome: Metabolic stability.
Quality of evidence: moderate.
Strength of recommendation: strong (supported by basic biochemical knowledge).
Liver transplantation in MMA or PA and combined liver‐kidney as well as kidney transplantation in MMA has been performed in several patients to improve the disease outcome. Despite many transplanted patients, it is difficult to provide detailed recommendations on this topic. The lack of detailed follow‐up studies and natural history data leads to incomplete evidence for many of the relevant outcome parameters. For the purpose of these guidelines, we have investigated all articles of our literature search with regards to the patients who underwent organ transplantation. We have identified nearly 300 MMA and PA patients, most of which were MMA undergoing liver transplantation followed by PA patients with the same procedure. It seems that MMA patients increasingly receive a combined liver and kidney transplant. Yap et al recently performed a similar analysis, concluding that transplantation procedures in MMA and PA patients have potential benefits as well as associated risks. 74 We suggest considering the management flowchart for MMA and PA patients undergoing general anaesthesia provided in Figure 4, when performing any transplantation procedure.
After liver and/or kidney transplantation, the number of metabolic decompensations decreases and some report a shortened hospital admission length during an intermittent illness. 42 , 54 , 75 , 76 , 77 , 78 , 79 , 80 , 81 , 82 , 83 , 84 , 85 , 86 , 87 , 88 , 89 , 90 , 91 , 92 , 93 , 94 , 95 , 96 , 97 , 98 , 99 , 100 , 101 , 102 , 103 Also biochemical markers (C3, C3/C2 ratio, methylmalonic acid, 2‐methylcitrate, FGF21) seem to improve after transplantation. 47 , 49 , 75 , 98 , 104 However, acute metabolic decompensations can still occur after transplantation in MMA and PA patients and some of these decompensations have been reported to be lethal. 47 , 102 , 104 , 105 , 106 , 107 , 108 , 109 Therefore, follow‐up of any transplanted MMA or PA patient by a metabolic clinician is indicated and regular metabolic monitoring seems to be essential. 110 Organ transplantation of patients in acute metabolic decompensation should be avoided, as the procedure itself has the potential to cause further decompensation. 111
In transplanted MMA and PA patients diet normalisation, 42 , 84 , 89 , 90 , 93 , 94 , 103 , 104 , 112 , 113 , 114 , 115 liberalisation or increased protein intake 75 , 76 , 78 , 80 , 83 , 88 , 91 , 96 , 97 , 98 , 100 , 102 , 116 , 117 , 118 or continuation of dietary restrictions 85 , 87 , 119 were all applied following transplantation. Liberalised and continuous diet both led to metabolically stable situations without acute decompensation. 89 , 103 Other patients were described to have metabolic acidosis post‐transplant while being on a continued protein restriction. 86 , 102 Further research is necessary to determine if continuation of dietary restriction is necessary after transplantation. Until the evidence is clearer further care by a metabolic specialist, including biochemical monitoring and regular dietetic review, as well as continuation of levocarnitine seems appropriate.
It is of note, that the guideline panel did not include transplantation surgeons and thus the available evidence was read from the perspective of metabolic physicians. The panel suggests considering patients for liver (MMA and PA) or combined liver‐kidney (MMA) transplantation who suffer from frequent metabolic decompensations, as the strongest effect of transplantations is the improvement of metabolic stability.
Recommendation #10
We suggest considering liver transplantation in MMA and PA, and combined liver kidney transplantation in MMA to improve metabolic stability.
Outcome: Metabolic stability.
Quality of evidence: low.
Strength of recommendation: weak.
3.4. Long‐term complications of MMA and PA
With improved survival, long‐term complications become more apparent, including so far unknown affections, such as liver neoplasms, namely hepatoblastoma and hepatocellular carcinoma. 120 It is essential to acknowledge that despite improved survival, metabolic decompensations are still a major mortality cause, 34 and sudden death may occur due to prolonged QT syndrome or cardiomyopathy. 13 , 121 , 122 Detailed recommendations are provided according to specific outcome parameters below.
3.4.1. Neurological complications
A variety of neurological problems are associated with MMA and PA disease. Due to their complexity, they are split in separate sections according to the outcome parameters cognitive development, metabolic stroke, epilepsy, and vision and hearing impairment.
While some patient show stabilisation and even improvement of neurological symptoms following organ transplantation, 80 , 88 , 98 , 99 , 117 it is essential to acknowledge that after transplantation patients are still at risk of developing neurological complications. Cognitive development often remains stable or improves post liver and liver‐kidney transplantation for MMA, and liver transplantation for PA, 44 , 80 , 81 , 86 , 87 , 92 , 97 , 104 , 105 , 115 , 123 , 124 but in one PA patient cognitive abilities got worse post liver transplantation. 86 In both diseases, however, the available evidence mostly lacks structured assessments of neurodevelopmental outcome. Several (including severe) new neurological sequelae occurred after liver, liver‐kidney and kidney transplantation in MMA and liver transplantation in PA in up to 25% of patients. These included onset of seizures, metabolic stroke, 42 , 113 Leigh syndrome, 44 worsening of vision and focal leukoencephalopathy, 125 generalised motor neuropathy 85 and general neurological deterioration 116 in MMA patients; and metabolic stroke, 86 , 104 epilepsy 86 and temporal mesial sclerosis 126 in PA patients.
Cognitive development and psychiatric complications
Lack of natural history studies in terms of domain‐specific outcomes and longitudinal developmental trajectories, different time points of evaluation, variety of patient populations, use of non‐standardised, variable test batteries and cut‐off values for normal development are among the limitations to evaluate long‐term cognitive and psychiatric outcome of MMA and PA.
In MMA cobalamin non‐responsiveness, early‐onset of disease, presence of hyperammonaemia or seizures at onset and a mut 0 subtype are associated with more severe cognitive involvement, while all patients show selective deficits in processing speed. 33 , 127 In addition, up to 20% of MMA patients are affected by neuropsychiatric problems. 128 , 129 Other evidence illustrates the negative impact of the number of metabolic decompensations on cognitive outcome, indicating that improvement of metabolic stability might ameliorate cognitive problems. 130
On the other hand, PA seems to be generally associated with cognitive impairment, which in most studies exceeds 50% of the evaluated patient population. 28 , 32 , 131 , 132 , 133 In addition, autism spectrum disorder, anxiety and acute psychotic episodes are described. 134 , 135 , 136 , 137 , 138 , 139 The present evidence suggests, that early intervention and treatment has little effect on the cognitive outcome in PA. 23
Recommendation #11
We suggest age‐appropriate, standardised developmental, cognitive and behavioural testing, since developmental delay, intellectual disability and/or behavioural abnormalities are known complications in MMA and PA.
Outcome: Cognitive development.
Quality of evidence: moderate.
Strength of recommendation: weak.
Metabolic stroke
The term metabolic stroke refers to the acute onset of a central neurological deficit, often associated with a decompensation of the underlying inborn error of metabolism, that cannot be accounted for by hypoxaemia or vascular insufficiency. 140 Metabolic strokes, particularly involving the basal ganglia area, were reported in both MMA and PA patients. 28 , 126 , 141 , 142 , 143 , 144 , 145 They often occur during an episode of metabolic decompensation or shortly thereafter. In PA, several cases with metabolic stroke without metabolic decompensation have been published. 143 , 146 , 147 , 148 , 149 , 150 Magnetic resonance imaging (MRI) can be of help to diagnose a metabolic stroke and should be performed based on clinical presentation and assessment. 151 , 152 , 153 One study precisely characterised globus pallidus infarcts and developed a grading method to systematically assess the volume of the lesion. 154 Movement disorders are common in MMA and PA and may arise as a consequence of a metabolic stroke event. 33 , 131 , 155
There is no evidence for a specific management of metabolic stroke. Therefore, standard symptomatic treatment strategies for stroke in addition to the management of the acute metabolic decompensation should apply. As described above regarding the outcome of metabolic stability, liver transplantation can improve metabolic stability and often leads to the absence of metabolic decompensations, 101 but is unable to prevent metabolic strokes. 113 In some patients, brain MRI findings improved after transplantation 87 while also new MRI abnormalities without a clear metabolic stroke have been reported after transplantation. 102
Recommendation #12
We strongly recommend standard appropriate investigation for metabolic stroke, if MMA or PA patients present with symptoms suspicious of a stroke‐like episode.
Outcome: Metabolic stroke.
Quality of evidence: high.
Strength of recommendation: very strong.
Epilepsy
Epilepsy is a common complication in MMA (prevalence of 23%‐43% reported) 33 , 155 and PA (prevalence of 25%‐59% reported). 133 , 143 , 145 , 156 , 157 Seizures have even been reported as the presenting symptom of the disease prior to diagnosis in several cases of PA. 143 , 146 , 148
There is no sufficient evidence to recommend a specific frequency of electroencephalogram recordings in the absence of clinical symptoms of seizures. There is no evidence to change standard epilepsy procedures and treatment regimens in MMA and PA patients. Namely, electroencephalogram evaluation should be performed in the presence of any focal neurological symptoms or clinically suspected seizures, and treatment with antiepileptic drugs should be initiated accordingly. Caution is warranted when valproic acid is used, as it can decrease the concentration of levocarnitine by excretion of valproylcarnitine. 158
Recommendation #13
We strongly recommend monitoring symptoms of epilepsy in MMA and PA patients as seizures are a common complication in MMA and PA.
Outcome: Epilepsy.
Quality of evidence: moderate.
Strength of recommendation: very strong.
Vision and hearing
Optic neuropathy is one of the neurological long‐term complications of both MMA 159 , 160 , 161 , 162 and PA 13 , 28 , 159 , 163 , 164 occurring in infancy or later during the disease course, affecting about 25% of patients. Onset of optic neuropathy can be insidious and subclinical or acute. 165 , 166
Sensorineural hearing loss seems to be less common but was reported in several cases of MMA and PA patients. 23 , 28 , 122 , 162 , 163
Currently, there are no biomarkers or clinical signs which would allow a prediction as to which patients will develop visual or hearing impairment. Despite some interesting pathophysiological considerations, which mainly involve mitochondrial dysfunction, 122 or interference with potassium channels in the case of sensorineural deafness, 167 no specific treatment can be recommended for these complications.
Recommendation #14
We suggest monitoring of visual and hearing function, since optic neuropathy and sensorineural hearing loss are known complications in MMA and PA.
Outcome: Vision and hearing.
Quality of evidence: moderate.
Strength of recommendation: weak.
3.4.2. Kidney dysfunction
Impaired kidney function is a well‐documented long‐term complication in MMA and less frequent in PA. 32 , 34 , 122 , 156 , 162 , 168 , 169 , 170 , 171 , 172 Occurrence of kidney dysfunction is related to the molecular subtype: mut 0 patients are affected earlier in life than cblB patients; cblA and mut − patients seem to be affected even later in life. 32 , 33 , 122 , 173 Despite these differences it is thought that all patients with isolated MMA are at risk of developing renal insufficiency in their long‐term course of the disease. 174 Although kidney disease in PA is only reported in single cases, it can be severe, even requiring transplantation of the dysfunctional organ. 169
While the exact pathomechanisms remain unresolved, two pathological correlates in kidneys of MMA patients have been described as tubulo‐interstitial nephritis and renal tubular acidosis. 175 , 176 , 177 , 178 , 179 Mitochondrial impairment seems to play a key role in the pathomechanisms. 122 , 180 , 181
Kidney growth is predicted by height and correlates negatively with serum cystatin C and serum methylmalonic acid concentrations. 182 Estimation of glomerular filtration rate (GFR) is more accurate using cystatin C instead of the traditional creatinine, as it is independent of muscle mass. 4 , 182 As measuring GFR is a complex procedure and often not available, we suggest using cystatin C based estimated GFR. Regular blood pressure monitoring should be part of kidney function assessments. While FGF21 levels do not seem to correlate with kidney function, 48 plasma lipocalin‐2 can be used as a biomarker. 172 , 180
There is no evidence allowing recommendations of specific treatment strategies for kidney impairment or failure in MMA and PA patients, hence, we suggest following standard protocols.
Recommendation #15
We strongly recommend monitoring of kidney function, as chronic kidney disease is a complication in MMA and PA.
Outcome: Renal dysfunction.
Quality of evidence: high.
Strength of recommendation: very strong.
In MMA, combined liver and kidney transplantation seems to be an efficient treatment of kidney failure, resulting in good renal function, even up to 10 years after transplantation. 95 , 117 , 118 , 183 , 184 Kidney transplant improves renal function shortly after transplantation and in some even for years (1.5 years up to 14 years) after transplantation, 54 , 85 , 88 , 179 but recurrence of nephropathy and renal failure has also been reported after kidney transplant. 47 , 185 After liver transplantation several patients showed normal renal function even up to 15 years after transplantation, 119 , 184 while others have or develop (progressive) renal failure after transplantation. 94 , 101 , 106 , 116 , 119 Liver transplantation seems not to correct an already dysfunctional kidney. 97
In PA, one patient was reported with kidney transplant 17 years after liver transplant. 91
In the context of transplantation, it has to be considered that calcineurin inhibitors can be nephrotoxic, which was also found in MMA 96 , 186 and PA patients. 86
3.4.3. Cardiac disease
Cardiomyopathy occurs in few MMA and in approximately 25% of PA patients. 28 , 34 , 84 , 145 , 162 , 170 , 187 , 188 Cardiomyopathy can present as an isolated clinical finding in previously asymptomatic individuals. 189 , 190 There does not seem to be a correlation between the occurrence of cardiomyopathy and metabolic stability, severity of the phenotype or residual enzymatic activity. 84 Cardiomyopathy can be rapidly progressive leading to cardiac failure or even death. 48
Long QT syndrome was reported in 22% and 33% of PA patients in two large cohorts. 28 , 162 Others observed an even higher incidence of prolonged QTc interval (70%) and arrhythmias (20%) in 10 PA patients with. 191 Prolonged QTc and ventricular arrhythmias can result in sudden cardiac arrest in PA patients. 191 , 192
There is weak evidence that high doses of coenzyme Q10 can improve cardiac function in PA patients with cardiomyopathy. 193 Beyond that, there is not enough evidence to recommend any specific management of cardiomyopathy or long QT syndrome in MMA and PA beyond standard cardiac therapy.
Based on the above‐described cardiac complications, we suggest using electrocardiogram and echocardiogram for regular monitoring (Table 5)
TABLE 5.
Monitoring
| Assessment | Frequency |
|---|---|
| Metabolic follow‐up | |
| Plasma: NH3, acid base balance (via blood gas analysis a ), lactate; urine: ketones | Each clinic visit |
| Quantitative plasma amino acids (3‐4 h of fasting prior to sample collection) | 3‐6 monthly |
| Methylmalonic acid in plasma (and urine if available) | 3‐6 monthly |
| Acylcarnitine profile in dried blood or plasma (propionylcarnitine and free carnitine) | 3‐6 monthly |
| Diet and nutritional status | |
| Diet history | Each clinic visit |
| Growth (weight, length or height, head circumference) | Each clinic visitb |
| Full clinical examination | Each clinic visit |
| Albumin, total protein, transferrin | 6‐monthly |
| Bone health (Ca, P, ALP, Mg, PTH, 25‐OH vitamin D in blood; Ca, P in urine) c | 12‐monthly |
| Full blood count, iron status, folic acid, vitamin B12 | 12‐monthly |
| Long‐term complications | |
| Neurological examination with assessment of developmental milestones | Each clinic visit |
| Kidney function (blood pressure, serum creatinine, electrolytes, cystatin C, uric acid; urinary electrolytes and protein loss; GFR) c , d | 6‐monthly |
| Pancreas function (lipase, pancreatic amylase) | 6‐monthly |
| Cardiac assessment (ECG) | 12‐monthly |
| Formal developmental/cognitive assessment | When clinically indicated |
| EEG, cerebral MRI | When clinically indicated |
| Ophthalmologic assessment | 12‐monthly |
| Formal hearing test | When clinically indicated |
Abbreviations: ALP, alkaline phosphatase; Ca, calcium; ECG, electrocardiogram; EEG, electroencephalogram; GFR, glomerular filtration rate; Mg, magnesium; MRI, magnetic resonance imaging; P, phosphate; PA, propionic acidaemia; PTH, parathyroid hormone.
Venous or capillary blood sample.
bMore frequently in infants.
More frequently in the presence of chronic kidney disease.
GFR: if available 12‐monthly, alternatively use estimated GFR based on cystatin C; in PA 12‐monthly biochemistry is sufficient.
Recommendation #16
We strongly recommend regular cardiac examinations, since cardiomyopathy in MMA and PA and long QT syndrome in PA are potentially life‐threatening complications.
Outcome: Cardiac disease.
Quality of evidence: high.
Strength of recommendation: very strong.
There are several reports showing stabilisation or improvement of cardiomyopathy after liver transplantation. 84 , 89 , 91 , 98 , 102 , 114 , 184 , 185 , 194 However, these results are counterbalanced by studies illustrating the development of cardiomyopathy and prolonged QTc after transplantation and reporting patients to have died as a consequence of heart failure. 91 , 105 , 188 , 195 Cardiac complications may at least partly be attributed to medication and/or immunosuppressants used in the context of post‐transplant treatment, hence more systematic data of cardiac parameters and used medication must be collected to enable specific recommendations regarding organ transplantation in the context of cardiac outcome in MMA and PA. Potentially, heart transplantation can be an option for PA patients with isolated cardiomyopathy. 196
3.4.4. Haematological complications
Haematological abnormalities are common in MMA and PA. 122 , 162 Pancytopenia (especially neutropenia) at initial presentation or during metabolic decompensation as well as during the chronic disease course has been frequently described. 13 , 23 , 28 , 148 , 197 , 198 , 199 , 200 Isolated thrombocytopenia can be observed during acute metabolic decompensations. 13 , 23 , 28 , 148 Anaemia may rather present chronically and depend on other factors such as the presence of chronic kidney disease. Other postulated mechanisms include reversible suppressive effects of toxic metabolites on bone marrow function. 198 , 201
Deficiencies of substrates for blood cell production (vitamin B12, folic acid, iron) should be excluded in patients with haematological abnormalities. Treatment of anaemia with erythropoietin in patients with chronic kidney disease should follow common nephrology standards. Indication for treatment with granulocyte colony stimulating factor is unclear, as there is very limited experience in MMA and PA patients. 13
Recommendation #17
We suggest monitoring of full blood count during regular follow‐up and in case of metabolic decompensation, since anaemia, neutropenia and/or thrombocytopenia can be a complication in MMA and PA patients.
Outcome: Haematological complications.
Quality of evidence: moderate.
Strength of recommendation: weak.
3.4.5. Bone health
Patients with MMA and PA are at risk of low bone density and osteoporosis, 128 , 202 , 203 which may be aggravated by kidney dysfunction in MMA. Risk factors for low bone density and osteoporosis in MMA and PA patients can include chronic acidosis (causing osteoclast activation and osteoblast inhibition), impaired kidney function and inadequate dietary intake of calcium, phosphate and vitamin D. Further it has been hypothesised, that the application of amino acid supplements can decrease bone mineralisation, as it increases the load of extracted acids, leading to buffering of protons in the bone tissue and increased bone resorption. 122 Optimisation of calcium and phosphate intake and vitamin D supplementation might be beneficial for bone health.
The time and frequency of assessment of bone health in MMA and PA should be guided by the risk of the individual patient. If bone density measurements (dual‐energy X‐ray absorptiometry [DEXA] scans) are applied in growing children and adolescents, they should be combined with assessment of bone age (and pubertal status) for adequate interpretation of the results. Osteoporosis can only be diagnosed in the presence of clinically significant fracture history, which has only been scarcely observed in MMA and PA. 204 In patients with renal failure, the diagnosis of osteoporosis should only be made in the absence of renal osteodystrophy.
For osteopenia or osteoporosis, treatment decisions need to be individualised, integrating parameters of bone metabolism, including secondary hyperparathyroidism in patients with chronic renal failure. Once the diagnosis of osteoporosis is established, treatment should follow common recommendations. Antiresorptive drugs (eg, bisphosphonates) may be indicated in patients at increased risk for fractures with relevant bone loss documented in serial DEXA measurements despite optimisation of nutritional support. The role of antiresorptive drugs for treatment or prevention of osteoporosis has not been systematically studied in MMA and PA patients.
Recommendation #18
We suggest assessment of bone health, considering the risk of patients with MMA and PA for impaired bone health.
Outcome: Bone health.
Quality of evidence: low.
Strength of recommendation: weak.
3.4.6. Growth
Poor growth outcomes (weight, linear growth and head circumference) are observed in MMA and PA patients 122 , 162 , 170 , 173 , 205 , 206 with a higher prevalence in MMA. 122 Lower birthweights compared to the normal reference population are seen in newborns with MMA but not in PA. 207
Poor growth is more pronounced in mut 0 and cblB subtypes than cblA and mut − subtypes 33 , 58 , 206 with no reported difference between early and late‐onset patients. 207 Linear growth appears more affected than weight gain which can result in overweight and obesity in MMA and PA patients. 28 , 58 , 59 , 122 , 208 A high percentage of fat mass is reported in MMA, particularly in mut 0 and cblB, 58 , 205 and PA patients. 208 Failure to thrive was associated with intellectual disability and increased mortality in cobalamin non‐responsive MMA patients. 194 In MMA, body length SD score decreased over time in patients with movement disorders compared to those without. 162 Linear growth improves in liver transplanted patients with MMA, when protein intake was relaxed but not unrestricted. 101
The causes for poor growth are considered to be multifactorial. Nutritional reasons are commonly cited and include over‐restriction of natural protein, 209 , 210 , 211 inadequate protein and energy intakes, 212 use of PFAAs, 203 , 206 , 208 , 213 , 214 malnutrition secondary to feeding difficulties 206 and frequent hospitalisations for metabolic decompensations preventing the usual diet and nutrients intake. 215 While total energy intake does not seem to correlate with linear growth a protein to energy ratio of 1.5 to 2.9 g protein/100 kcal/day is positively correlated with growth outcomes in patients with inborn errors of intermediary protein metabolism. 208
Use of PFAAs is common practice in some centres with a variable prescription regarding percentage of total protein intake. 33 , 60 , 203 , 209 , 210 , 211 , 215 However, PFAAs are low in valine and isoleucine but high in leucine content and can result in low plasma isoleucine and valine levels and iatrogenic amino acid deficiencies associated with adverse growth outcomes. 206 The height z‐score seems positively correlated with natural‐protein‐to‐energy prescription ratio and plasma L‐valine and L‐arginine levels, while negatively associated with the amount of PFAAs. 214 Hence, PFAAs should not substitute for an adequate provision of natural protein and the need for PFAAs requires further review. 206 , 208 It is common practice to monitor plasma amino acids on a regular basis and adjust the dietary regimen accordingly.
Recommendation #19
We suggest regular monitoring of anthropometric measurements in MMA and PA patients, as growth outcomes can be poor.
Outcome: Normal growth.
Quality of evidence: moderate.
Strength of recommendation: weak.
Growth can potentially be improved after organ transplantation, but persistent growth delay and even decreased growth rate have also been reported. 44 , 80 , 81 , 97 , 100 , 102 , 104 , 106 , 118 , 216 Factors influencing growth after transplantation need to be determined.
3.4.7. Pancreatitis
Acute, recurrent acute and chronic pancreatitis are possible long‐term complications of MMA 32 , 122 , 217 and PA. 13 , 23 , 28 , 122 , 205 , 218 , 219 Pancreatitis has been observed in around 5% to 10% of patients 13 , 122 , 205 and may develop independently from metabolic decompensations and metabolic control. 13 , 194 There is weak evidence that pancreatitis correlates with mortality. 32 , 194
Like in individuals without MMA or PA, the clinical presentation of pancreatitis is variable. It is of note that pancreatitis can occur without abdominal pain and may therefore be under‐recognised. 220 , 221
There is no evidence that management of acute or chronic pancreatitis in MMA and PA has specific aspects different from general standards on pancreatitis treatment. Adequate energy supply should be maintained throughout an episode of pancreatitis to ensure metabolic stability.
Recommendation #20
We strongly recommend prompt evaluation for pancreatitis in MMA and PA patients if clinically suspected, since both acute and chronic pancreatitis are known complications in PA and MMA.
Outcome: Pancreatitis.
Quality of evidence: moderate.
Strength of recommendation: very strong.
3.5. Health‐related quality of life
HrQoL and psychological adjustment are meaningful outcome parameters that should be considered in the care for children with chronic disease. 222 , 223 The high rating of the outcome HrQoL illustrates the importance of this parameter to health professionals and experts as well as patient representatives. In MMA and PA, HrQoL has so far scarcely been systematically addressed. 224 Most of our knowledge on HrQoL in MMA and PA has been extrapolated from studies in combined samples of inherited metabolic disease patients applying generic or chronic generic instruments. Only very few reports present isolated data on HrQoL in PA or MMA patients.
HrQoL was addressed in 13 PA patients, using a generic instrument (KINDL), and found significantly lower HrQoL for the ‘psychological’ and ‘friends’ domain. Higher HrQoL was reported for the ‘school’ domain. Correlations with disease‐ or family‐related parameters or impact of interventions on HrQoL were not investigated. 28
For 35 MMA mut 0 patients their parents or caregivers completed the PedsQl generic core, transplant as well as family impact modules proxy versions. They reported lower mean PedsQl core scale scores (in comparison to healthy children) and lower scores on the transplant scales (as compared to other indication groups for liver transplantation). Compared to families of children with other complex chronic conditions, MMA families had a lower quality of life. Free comments suggested a favourable effect of liver transplantation on HrQoL. 225
Recently, Zeltner et al have developed a specific questionnaire for the assessment of HrQoL in intoxication‐type metabolic disorders. 226 In contrast to generic instruments, specific tools are tailor‐made for disease groups with patients participating in the process. 227 Specific instruments are more suitable to detect changes over time or following interventions.
If considered, we strongly recommend systematic assessment of HrQoL with standardised instruments. We recommend choosing generic, chronic generic and/or specific instruments in line with the question to be addressed. While there is weak evidence suggesting that MMA and PA patients and their families have impaired HrQoL, there is not enough evidence to give recommendations on interventions to improve HrQoL in MMA and PA.
Recommendation #21
We recommend considering health‐related quality of life a relevant outcome parameter in MMA and PA.
Outcome: Health‐related quality of life.
Quality of evidence: moderate.
Strength of recommendation: strong.
3.6. Monitoring
There is no strong evidence base as to when which monitoring investigations are indicated in MMA and PA patients. The panel recommends a slightly modified monitoring scheme compared to the initial guidelines (Table 5), which can guide clinical management but should be tailored to individual patients' needs. In addition to the items represented in the table, we recommend regular contact to a metabolic dietitian. A full clinical examination should be performed at each patient visit, focusing on the cardiovascular system and neurological aspects but also comprising an assessment of skin, nails and hair as part of the nutritional assessment of the patient.
4. CONCLUDING REMARKS
This update of the MMA and PA guidelines provides a digest of evaluated clinically relevant evidence published on these disorders. Based on the present evidence, 21 recommendations were formulated by a diverse panel of metabolic disease health care professionals. It has to be noted, though, that the participants predominantly had a European/Mediterranean background. For a next revision, it is intended to overcome this limitation and form a more heterogeneous panel. There remain significant knowledge gaps regarding the optimal management of MMA and PA patients. While more and better‐quality evidence is needed in the study of MMA and PA, we will continue to evaluate the literature on a regular basis to enable health care professionals working in the field to make well‐informed decisions, which are not based on dogmatic expert knowledge.
CONFLICT OF INTEREST
D. B. participated in European Metabolic Group meetings sponsored by Nutricia and received research funds from Nutricia. S. C. G. has received a research fund from Nutricia as well as reimbursement for attending a symposium by MetaX and Vitaflo and fees for speaking on educational events from Nutricia and Vitaflo. J. O. S. received institutional honoraria from Swedish Orphan Biovitrum und Immedica Pharma AB. S. S.‐B. has received reimbursement for symposia fees from Nutricia and Dr Schär and research funds from Nutricia. M. R. B. received a research fund from Nutricia and is a member of the clinical advisory boards of Hemoshear and Moderna. A. C. received financial support from Nutricia, Vitaflo and Recordati/Orphan Europe, including research funds, institutional honoraria and consulting fees. M. D. has received reimbursement to attend symposia and fees for speaking at an organised meeting from Vitaflo and Nutricia and consultancy fees from Nutricia, Vitaflo and Recordati Rare Diseases. The other authors do not declare any conflict of interest related to this work.
AUTHOR CONTRIBUTIONS
All authors were involved in the conception of the paper and the evaluation of literature as well as development of recommendations. Patrick Forny, Friederike Hörster, Martina Huemer and Matthias R. Baumgartner coordinated the panel. Patrick Forny is the guarantor of the paper.
Supporting information
Appendix S1: Supporting information
ACKNOWLEDGMENTS
We thank the external reviewers, namely Burkhard Toenshoff (nephrology) and Christoph Aufricht (nephrology), Alexander Kovacevic (cardiology), Angels García‐Cazorla (neurology) and Stefan Koelker (neurology), Alessio Cremonesi (biochemist), Daniela Moor (dietetics) and the patient representatives Marike Groenendijk (chair Stofwisselkracht, Dutch patient organisation) and Hanka Dekker (executive director of Vereniging Volwassenen, Kinderen en Stofwisselingsziekten) for significant input and evaluation of the guidelines. P. F. is supported by the Filling the Gap grant awarded by the Medical Faculty of the University of Zurich, Switzerland. M. R. B. is supported by a grant from the Swiss National Science Foundation (31003A_175779) and by the radiz, Rare Disease Initiative Zurich and ITINERARE University Priority Research Programs of the University of Zurich, Switzerland. The Society for the Study of Inborn Errors of Metabolism (SSIEM) and The German Society of Pediatrics and Adolescent Medicine supported the development process of these guidelines to enable the face‐to‐face meeting in Brussels and cover other administrative costs. T. H. was supported by research grants from the Ministry of Health of the Czech Republic RVO VFN 64165. J. O. S. gratefully acknowledges financial support by the program ‘FH Zeit für Forschung’ (project ‘KETOplus’, 005‐1703‐0016) of the Ministry of Culture and Science of the German State of North Rhine‐Westphalia. The other authors do not declare any specific funding related to this work.
Forny P, Hörster F, Ballhausen D, et al. Guidelines for the diagnosis and management of methylmalonic acidaemia and propionic acidaemia: First revision. J Inherit Metab Dis. 2021;44:566–592. 10.1002/jimd.12370
Patrick Forny and Friederike Hörster contributed equally.
Martina Huemer and Matthias R. Baumgartner are joint last author.
Communicating Editor: Ivo Barić
Funding information The German Society of Pediatrics and Adolescent Medicine; The Society for the Study of Inborn Errors of Metabolism; Ministry of Culture and Science of the German State of North Rhine‐Westphalia, Grant/Award Number: 005‐1703‐0016; Ministry of Health of the Czech Republic, Grant/Award Number: RVO VFN 64165; ITINERARE University Priority Research Programs of the University of Zurich, Switzerland; radiz, Rare Disease Initiative Zurich; Swiss National Science Foundation, Grant/Award Number: 31003A_175779; Faculty of Medicine of the University of Zurich, Filling the Gap Grant, Switzerland
REFERENCES
- 1. Baumgartner MR, Hörster F, Dionisi‐Vici C, et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J Rare Dis 2014;9:130. 10.1186/s13023-014-0130-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guyatt GH, Oxman AD, Vist GE, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ 2008;336(7650):924‐926. 10.1136/bmj.39489.470347.AD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fowler B, Leonard JV, Baumgartner MR. Causes of and diagnostic approach to methylmalonic acidurias. J Inherit Metab Dis 2008;31(3):350‐360. 10.1007/s10545-008-0839-4. [DOI] [PubMed] [Google Scholar]
- 4. Shchelochkov OA, Carrillo N, Venditti C. GeneReviews®: propionic acidemia. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews((R)). Seattle, WA; University of Washington, National Center for Biotechnology Information; 1993. [Google Scholar]
- 5. Harbour R, Miller J. A new system for grading recommendations in evidence based guidelines. BMJ 2001;323(7308):334‐336. 10.1136/bmj.323.7308.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Levtova A, Waters PJ, Buhas D, et al. Combined malonic and methylmalonic aciduria due to ACSF3 mutations: benign clinical course in an unselected cohort. J Inherit Metab Dis 2019;42(1):107‐116. 10.1002/jimd.12032. [DOI] [PubMed] [Google Scholar]
- 7. Heuberger K, Bailey HJ, Burda P, et al. Genetic, structural, and functional analysis of pathogenic variations causing methylmalonyl‐CoA epimerase deficiency. Biochim Biophys Acta Mol Basis Dis 2019;1865(6):1265‐1272. 10.1016/j.bbadis.2019.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stucki M, Coelho D, Suormala T, Burda P, Fowler B, Baumgartner MR. Molecular mechanisms leading to three different phenotypes in the cblD defect of intracellular cobalamin metabolism. Hum Mol Genet 2012;21(6):1410‐1418. 10.1093/hmg/ddr579. [DOI] [PubMed] [Google Scholar]
- 9. Devi ARR, Naushad SM. Targeted exome sequencing for the identification of complementation groups in methylmalonic aciduria: a south Indian experience. Clin Biochem 2017;50(1–2):68‐72. 10.1016/j.clinbiochem.2016.08.016. [DOI] [PubMed] [Google Scholar]
- 10. Abdrabo LS, Watkins D, Wang SR, Lafond‐Lapalme J, Riviere J‐B, Rosenblatt DS. Genome and RNA sequencing in patients with methylmalonic aciduria of unknown cause. Genet Med 2019;22:432‐436. 10.1038/s41436-019-0640-9. [DOI] [PubMed] [Google Scholar]
- 11. Forny P, Froese DS, Suormala T, Yue WW, Baumgartner MR. Functional characterization and categorization of missense mutations that cause methylmalonyl‐CoA mutase (MUT) deficiency. Hum Mutat 2014;35(12):1449, 10.1002/humu.22633‐1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Forny P, Schnellmann A‐S, Buerer C, et al. Molecular genetic characterization of 151 Mut‐type methylmalonic aciduria patients and identification of 41 novel mutations in MUT. Hum Mutat 2016;37(8):745‐754. 10.1002/humu.23013. [DOI] [PubMed] [Google Scholar]
- 13. Pena L, Franks J, Chapman KA, et al. Natural history of propionic acidemia. Mol Genet Metab 2012;105(1):5‐9. 10.1016/j.ymgme.2011.09.022. [DOI] [PubMed] [Google Scholar]
- 14. Inoue Y, Ohse M. Prenatal diagnosis of methylmalonic aciduria by measuring methylmalonic acid in dried amniotic fluid on filter paper using gas chromatography‐mass spectrometry. Anal Bioanal Chem 2011;400(7):1953‐1958. 10.1007/s00216-011-4805-x. [DOI] [PubMed] [Google Scholar]
- 15. Inoue Y, Ohse M, Shinka T, Kuhara T. Prenatal diagnosis of propionic acidemia by measuring methylcitric acid in dried amniotic fluid on filter paper using GC/MS. J Chromatogr B Analyt Technol Biomed Life Sci 2008;870(2):160‐163. 10.1016/j.jchromb.2008.02.022. [DOI] [PubMed] [Google Scholar]
- 16. Morel CF, Watkins D, Scott P, Rinaldo P, Rosenblatt DS. Prenatal diagnosis for methylmalonic acidemia and inborn errors of vitamin B12 metabolism and transport. Mol Genet Metab 2005;86(1–2):160‐171. 10.1016/j.ymgme.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 17. Al‐Dirbashi OY, McIntosh N, Chakraborty P. Quantification of 2‐methylcitric acid in dried blood spots improves newborn screening for propionic and methylmalonic acidemias. J Med Screen 2017;24(2):58‐61. 10.1177/0969141316645824. [DOI] [PubMed] [Google Scholar]
- 18. Al Dhahouri N, Langhans C‐D, Al Hammadi Z, et al. Quantification of methylcitrate in dried urine spots by liquid chromatography tandem mass spectrometry for the diagnosis of propionic and methylmalonic acidemias. Clin Chim Acta 2018;487:41‐45. 10.1016/j.cca.2018.09.017. [DOI] [PubMed] [Google Scholar]
- 19. Al‐Dirbashi OY, Alfadhel M, Al‐Thihli K, et al. Assessment of methylcitrate and methylcitrate to citrate ratio in dried blood spots as biomarkers for inborn errors of propionate metabolism. Sci Rep 2019;9(1):12366. 10.1038/s41598-019-48885-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. La Marca G, Malvagia S, Pasquini E, Innocenti M, Donati MA, Zammarchi E. Rapid 2nd‐tier test for measurement of 3‐OH‐propionic and methylmalonic acids on dried blood spots: reducing the false‐positive rate for propionylcarnitine during expanded newborn screening by liquid chromatography‐tandem mass spectrometry. Clin Chem 2007;53(7):1364‐1369. 10.1373/clinchem.2007.087775. [DOI] [PubMed] [Google Scholar]
- 21. Lindner M, Ho S, Kölker S, Abdoh G, Hoffmann GF, Burgard P. Newborn screening for methylmalonic acidurias—optimization by statistical parameter combination. J Inherit Metab Dis 2008;31(3):379‐385. 10.1007/s10545-008-0892-z. [DOI] [PubMed] [Google Scholar]
- 22. Malvagia S, Haynes CA, Grisotto L, et al. Heptadecanoylcarnitine (C17) a novel candidate biomarker for newborn screening of propionic and methylmalonic acidemias. Clin Chim Acta 2015;450:342‐348. 10.1016/j.cca.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grünert SC, Müllerleile S, de Silva L, et al. Propionic acidemia: neonatal versus selective metabolic screening. J Inherit Metab Dis 2012;35(1):41‐49. 10.1007/s10545-011-9419-0. [DOI] [PubMed] [Google Scholar]
- 24. McCrory NM, Edick MJ, Ahmad A, et al. Comparison of methods of initial ascertainment in 58 cases of propionic acidemia enrolled in the inborn errors of metabolism information system reveals significant differences in time to evaluation and symptoms at presentation. J Pediatr 2017;180:200‐205. 10.1016/j.jpeds.2016.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Heringer J, Valayannopoulos V, Lund AM, et al. Impact of age at onset and newborn screening on outcome in organic acidurias. J Inherit Metab Dis 2016;39(3):341‐353. 10.1007/s10545-015-9907-8. [DOI] [PubMed] [Google Scholar]
- 26. Dionisi‐Vici C, Deodato F, Röschinger W, Rhead W, Wilcken B. ‘Classical’ organic acidurias, propionic aciduria, methylmalonic aciduria and isovaleric aciduria: long‐term outcome and effects of expanded newborn screening using tandem mass spectrometry. J Inherit Metab Dis 2006;29(2–3):383‐389. 10.1007/s10545-006-0278-z. [DOI] [PubMed] [Google Scholar]
- 27. Leonard JV, Vijayaraghavan S, Walter JH. The impact of screening for propionic and methylmalonic acidaemia. Eur J Pediatr 2003;162(suppl 1):S21‐S24. 10.1007/s00431-003-1345-1. [DOI] [PubMed] [Google Scholar]
- 28. Grünert SC, Müllerleile S, de Silva L, et al. Propionic acidemia: clinical course and outcome in 55 pediatric and adolescent patients. Orphanet J Rare Dis 2013;8(6):6. 10.1186/1750-1172-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Han B, Nie W, Sun M, Liu Y, Cao Z. Clinical presentation, molecular analysis and follow‐up of patients with Mut methylmalonic acidemia in Shandong province, China. Pediatr Neonatol 2019;61:148‐154. 10.1016/j.pedneo.2019.07.004. [DOI] [PubMed] [Google Scholar]
- 30. Haijes HA, Molema F, Langeveld M, et al. Retrospective evaluation of the Dutch pre‐newborn screening cohort for propionic acidemia and isolated methylmalonic acidemia: what to aim, expect, and evaluate from newborn screening? J Inherit Metab Dis 2020;43(3):424‐437. 10.1002/jimd.12193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hörster F, Kölker S, Loeber JG, Cornel MC, Hoffmann GF, Burgard P. Newborn screening programmes in Europe, arguments and efforts regarding harmonisation: focus on organic acidurias. JIMD Rep 2017;32:105‐115. 10.1007/8904_2016_537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Baulny HO d, Benoist JF, Rigal O, Touati G, Rabier D, Saudubray JM. Methylmalonic and propionic acidaemias: management and outcome. J Inherit Metab Dis 2005;28(3):415‐423. 10.1007/s10545-005-7056-1. [DOI] [PubMed] [Google Scholar]
- 33. Hörster F, Baumgartner MR, Viardot C, et al. Long‐term outcome in methylmalonic acidurias is influenced by the underlying defect (mut0, mut‐, cblA, cblB). Pediatr Res 2007;62(2):225‐230. 10.1203/PDR.0b013e3180a0325f. [DOI] [PubMed] [Google Scholar]
- 34. Hörster F, Garbade SF, Zwickler T, et al. Prediction of outcome in isolated methylmalonic acidurias: combined use of clinical and biochemical parameters. J Inherit Metab Dis 2009;32(5):630. 10.1007/s10545-009-1189-6. [DOI] [PubMed] [Google Scholar]
- 35. Chapman KA, Gropman A, MacLeod E, et al. Acute management of propionic acidemia. Mol Genet Metab 2012;105(1):16‐25. 10.1016/j.ymgme.2011.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Filipowicz HR, Ernst SL, Ashurst CL, Pasquali M, Longo N. Metabolic changes associated with hyperammonemia in patients with propionic acidemia. Mol Genet Metab 2006;88(2):123‐130. 10.1016/j.ymgme.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 37. Yap S, Leong HY, Abdul Aziz F, et al. N‐Carbamylglutamate is an effective treatment for acute neonatal hyperammonaemia in a patient with methylmalonic aciduria. Neonatology 2016;109(4):303‐307. 10.1159/000443630. [DOI] [PubMed] [Google Scholar]
- 38. Chakrapani A, Valayannopoulos V, Segarra NG, et al. Effect of carglumic acid with or without ammonia scavengers on hyperammonaemia in acute decompensation episodes of organic acidurias. Orphanet J Rare Dis 2018;13(1):97. 10.1186/s13023-018-0840-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Häberle J, Chakrapani A, Ah Mew N, Longo N. Hyperammonaemia in classic organic acidaemias: a review of the literature and two case histories. Orphanet J Rare Dis 2018;13(1):219. 10.1186/s13023-018-0963-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Abacan M, Boneh A. Use of carglumic acid in the treatment of hyperammonaemia during metabolic decompensation of patients with propionic acidaemia. Mol Genet Metab 2013;109(4):397‐401. 10.1016/j.ymgme.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 41. Nashabat M, Obaid A, Al Mutairi F, et al. Evaluation of long‐term effectiveness of the use of carglumic acid in patients with propionic acidemia (PA) or methylmalonic acidemia (MMA): study protocol for a randomized controlled trial. BMC Pediatr 2019;19(1):195. 10.1186/s12887-019-1571-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Morioka D, Kasahara M, Takada Y, et al. Living donor liver transplantation for pediatric patients with inheritable metabolic disorders. Am J Transplant 2005;5(11):2754‐2763. 10.1111/j.1600-6143.2005.01084.x. [DOI] [PubMed] [Google Scholar]
- 43. Kasahara M, Sakamoto S, Horikawa R, et al. Living donor liver transplantation for pediatric patients with metabolic disorders: the Japanese multicenter registry. Pediatr Transplant 2014;18(1):6‐15. 10.1111/petr.12196. [DOI] [PubMed] [Google Scholar]
- 44. Brassier A, Krug P, Lacaille F, et al. Long‐term outcome of methylmalonic aciduria after kidney, liver, or combined liver‐kidney transplantation: the French experience. J Inherit Metab Dis 2020;43(2):234‐243. 10.1002/jimd.12174. [DOI] [PubMed] [Google Scholar]
- 45. Zwickler T, Haege G, Riderer A, et al. Metabolic decompensation in methylmalonic aciduria: which biochemical parameters are discriminative? J Inherit Metab Dis 2012;35(5):797‐806. 10.1007/s10545-011-9426-1. [DOI] [PubMed] [Google Scholar]
- 46. Zwickler T, Riderer A, Haege G, Hoffmann GF, Kölker S, Burgard P. Usefulness of biochemical parameters in decision‐making on the start of emergency treatment in patients with propionic acidemia. J Inherit Metab Dis 2014;37(1):31‐37. 10.1007/s10545-013-9621-3. [DOI] [PubMed] [Google Scholar]
- 47. Manoli I, Sysol JR, Epping MW, et al. FGF21 underlies a hormetic response to metabolic stress in methylmalonic acidemia. JCI Insight 2018;3(23):e124351. 10.1172/jci.insight.124351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Molema F, Jacobs EH, Onkenhout W, Schoonderwoerd GC, Langendonk JG, Williams M. Fibroblast growth factor 21 as a biomarker for long‐term complications in organic acidemias. J Inherit Metab Dis 2018;41(6):1179‐1187. 10.1007/s10545-018-0244-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Maines E, Catesini G, Boenzi S, et al. Plasma methylcitric acid and its correlations with other disease biomarkers: the impact in the follow up of patients with propionic and methylmalonic acidemia. J Inherit Metab Dis 2020;43:1173‐1185. 10.1002/jimd.12287. [DOI] [PubMed] [Google Scholar]
- 50. Thompson GN, Chalmers RA. Increased urinary metabolite excretion during fasting in disorders of propionate metabolism. Pediatr Res 1990;27(4 pt 1):413‐416. 10.1203/00006450-199004000-00021. [DOI] [PubMed] [Google Scholar]
- 51. Morgan TM, Schlegel C, Edwards KM, et al. Vaccines are not associated with metabolic events in children with urea cycle disorders. Pediatrics 2011;127(5):e1147‐e1153. 10.1542/peds.2010-1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Adeyemi OA, Girish T, Mukhopadhyay S, Olczak SA, Ahmed Z. Methylmalonic acidaemia: a rare metabolic disorder in pregnancy. J Obstet Gynaecol 2004;24(8):927‐928. 10.1080/01443610400019070. [DOI] [PubMed] [Google Scholar]
- 53. Langendonk JG, Roos JCP, Angus L, et al. A series of pregnancies in women with inherited metabolic disease. J Inherit Metab Dis 2012;35(3):419‐424. 10.1007/s10545-011-9389-2. [DOI] [PubMed] [Google Scholar]
- 54. Lubrano R, Bellelli E, Gentile I, et al. Pregnancy in a methylmalonic acidemia patient with kidney transplantation: a case report. Am J Transplant 2013;13(7):1918‐1922. 10.1111/ajt.12282. [DOI] [PubMed] [Google Scholar]
- 55. Feillet F, Bodamer OA, Dixon MA, Sequeira S, Leonard JV. Resting energy expenditure in disorders of propionate metabolism. J Pediatr 2000;136(5):659‐663. 10.1067/mpd.2000.104290. [DOI] [PubMed] [Google Scholar]
- 56. Thomas JA, Bernstein LE, Greene CL, Koeller DM. Apparent decreased energy requirements in children with organic acidemias: preliminary observations. J Am Diet Assoc 2000;100(9):1074‐1076. 10.1016/S0002-8223(00)00313-8. [DOI] [PubMed] [Google Scholar]
- 57. van Hagen CC, Carbasius Weber E, van den Hurk TAM, et al. Energy expenditure in patients with propionic and methylmalonic acidaemias. J Inherit Metab Dis 2004;27(1):111‐112. 10.1023/b:boli.0000016678.78134.7d. [DOI] [PubMed] [Google Scholar]
- 58. Hauser NS, Manoli I, Graf JC, Sloan J, Venditti CP. Variable dietary management of methylmalonic acidemia: metabolic and energetic correlations. Am J Clin Nutr 2011;93(1):47‐56. 10.3945/ajcn.110.004341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Daly A, Evans S, Gerrard A, Santra S, Vijay S, MacDonald A. The nutritional intake of patients with organic acidaemias on enteral tube feeding: can we do better? JIMD Rep 2016;28:29‐39. 10.1007/8904_2015_443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pinto A, Evans S, Daly A, et al. Dietary practices in methylmalonic acidaemia: a European survey. J Pediatr Endocrinol Metab 2020;33(1):147‐155. 10.1515/jpem-2019-0277. [DOI] [PubMed] [Google Scholar]
- 61. Chandler RJ, Venditti CP. Pre‐clinical efficacy and dosing of an AAV8 vector expressing human methylmalonyl‐CoA mutase in a murine model of methylmalonic acidemia (MMA). Mol Genet Metab 2012;107(3):617‐619. 10.1016/j.ymgme.2012.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Guenzel AJ, Hillestad ML, Matern D, Barry MA. Effects of adeno‐associated virus serotype and tissue‐specific expression on circulating biomarkers of propionic acidemia. Hum Gene Ther 2014;25(9):837‐843. 10.1089/hum.2014.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wong ESY, McIntyre C, Peters HL, Ranieri E, Anson DS, Fletcher JM. Correction of methylmalonic aciduria in vivo using a codon‐optimized lentiviral vector. Hum Gene Ther 2014;25(6):529‐538. 10.1089/hum.2013.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. An D, Schneller JL, Frassetto A, et al. Systemic messenger RNA therapy as a treatment for methylmalonic acidemia. Cell Rep 2017;21(12):3548‐3558. 10.1016/j.celrep.2017.11.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chandler RJ, Venditti CP. Gene therapy for methylmalonic acidemia: past, present, and future. Hum Gene Ther 2019;30:1236‐1244. 10.1089/hum.2019.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Morath MA, Okun JG, Müller IB, et al. Neurodegeneration and chronic renal failure in methylmalonic aciduria—a pathophysiological approach. J Inherit Metab Dis 2008;31(1):35‐43. 10.1007/s10545-007-0571-5. [DOI] [PubMed] [Google Scholar]
- 67. Miller MJ, Bostwick BL, Kennedy AD, et al. Chronic oral L‐Carnitine supplementation drives marked plasma TMAO elevations in patients with organic acidemias despite dietary meat restrictions. JIMD Rep 2016;30:39‐44. 10.1007/8904_2016_539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Diodato D, Olivieri G, Pro S, et al. Axonal peripheral neuropathy in propionic acidemia: a severe side effect of long‐term metronidazole therapy. Neurology 2018;91(12):565‐567. 10.1212/WNL.0000000000006209. [DOI] [PubMed] [Google Scholar]
- 69. Wilson BT, Strong A, O'Kelly S, Munkley J, Stark Z. Metronidazole toxicity in Cockayne syndrome: a case series. Pediatrics 2015;136(3):e706‐e708. 10.1542/peds.2015-0531. [DOI] [PubMed] [Google Scholar]
- 70. Goolsby TA, Jakeman B, Gaynes RP. Clinical relevance of metronidazole and peripheral neuropathy: a systematic review of the literature. Int J Antimicrob Agents 2018;51(3):319‐325. 10.1016/j.ijantimicag.2017.08.033. [DOI] [PubMed] [Google Scholar]
- 71. Burlina A, Cazzorla C, Zanonato E, Viggiano E, Fasan I, Polo G. Clinical experience with N‐carbamylglutamate in a single‐centre cohort of patients with propionic and methylmalonic aciduria. Mol Genet Metab Rep 2016;8:34‐40. 10.1016/j.ymgmr.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Longo N, Price LB, Gappmaier E, et al. Anaplerotic therapy in propionic acidemia. Mol Genet Metab 2017;122(1‐2):51‐59. 10.1016/j.ymgme.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Matsui SM, Mahoney MJ, Rosenberg LE. The natural history of the inherited methylmalonic acidemias. N Engl J Med 1983;308(15):857‐861. 10.1056/NEJM198304143081501. [DOI] [PubMed] [Google Scholar]
- 74. Yap S, Vara R, Morais A. Post‐transplantation outcomes in patients with pa or mma: a review of the literature. Advances in Therapy. 2020;37(5):1866‐1896. 10.1007/s12325-020-01305-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Schlenzig JS, Poggi‐Travert F, Laurent J, et al. Liver transplantation in two cases of propionic acidaemia. J Inherit Metab Dis 1995;18(4):448‐461. 10.1007/bf00710056. [DOI] [PubMed] [Google Scholar]
- 76. van 't Hoff WG, Dixon M, Taylor J, et al. Combined liver‐kidney transplantation in methylmalonic acidemia. J Pediatr 1998;132(6):1043‐1044. 10.1016/s0022-3476(98)70407-x. [DOI] [PubMed] [Google Scholar]
- 77. Hsui J‐Y, Chien Y‐H, Chu S‐Y, et al. Living‐related liver transplantation for methylmalonic acidemia: report of one case. Acta Paediatr Taiwan 2003;44(3):171‐173. [PubMed] [Google Scholar]
- 78. Huang H‐P, Chien Y‐H, Huang L‐M, et al. Viral infections and prolonged fever after liver transplantation in young children with inborn errors of metabolism. J Formos Med Assoc 2005;104(9):623‐629. [PubMed] [Google Scholar]
- 79. Manzoni D, Spotti A, Carrara B, Gritti P, Sonzogni V. Anaesthesia for liver transplantation in two infants with an organic acidaemia. Pediatr Transplant 2006;10(5):623‐628. 10.1111/j.1399-3046.2006.00536.x. [DOI] [PubMed] [Google Scholar]
- 80. Morioka D, Kasahara M, Horikawa R, Yokoyama S, Fukuda A, Nakagawa A. Efficacy of living donor liver transplantation for patients with methylmalonic acidemia. Am J Transplant 2007;7(12):2782‐2787. 10.1111/j.1600-6143.2007.01986.x. [DOI] [PubMed] [Google Scholar]
- 81. Rela M, Battula N, Madanur M, et al. Auxiliary liver transplantation for propionic acidemia: a 10‐year follow‐up. Am J Transplant 2007;7(9):2200‐2203. 10.1111/j.1600-6143.2007.01899.x. [DOI] [PubMed] [Google Scholar]
- 82. McGuire PJ, Lim‐Melia E, Diaz GA, et al. Combined liver‐kidney transplant for the management of methylmalonic aciduria: a case report and review of the literature. Mol Genet Metab 2008;93(1):22‐29. 10.1016/j.ymgme.2007.08.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Sato S, Kasahara M, Fukuda A, et al. Liver transplantation in a patient with propionic acidemia requiring extra corporeal membrane oxygenation during severe metabolic decompensation. Pediatr Transplant 2009;13(6):790‐793. 10.1111/j.1399-3046.2008.01029.x. [DOI] [PubMed] [Google Scholar]
- 84. Romano S, Valayannopoulos V, Touati G, et al. Cardiomyopathies in propionic aciduria are reversible after liver transplantation. J Pediatr 2010;156(1):128‐134. 10.1016/j.jpeds.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 85. Clothier JC, Chakrapani A, Preece M‐A, et al. Renal transplantation in a boy with methylmalonic acidaemia. J Inherit Metab Dis 2011;34(3):695‐700. 10.1007/s10545-011-9303-y. [DOI] [PubMed] [Google Scholar]
- 86. Vara R, Turner C, Mundy H, et al. Liver transplantation for propionic acidemia in children. Liver Transpl 2011;17(6):661‐667. 10.1002/lt.22279. [DOI] [PubMed] [Google Scholar]
- 87. Kasahara M, Sakamoto S, Kanazawa H, et al. Living‐donor liver transplantation for propionic acidemia. Pediatr Transplant 2012;16(3):230‐234. 10.1111/j.1399-3046.2011.01607.x. [DOI] [PubMed] [Google Scholar]
- 88. Brassier A, Boyer O, Valayannopoulos V, et al. Renal transplantation in 4 patients with methylmalonic aciduria: a cell therapy for metabolic disease. Mol Genet Metab 2013;110(1–2):106‐110. 10.1016/j.ymgme.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 89. Arrizza C, Gottardi A d, Foglia E, Baumgartner M, Gautschi M, Nuoffer J‐M. Reversal of cardiomyopathy in propionic acidemia after liver transplantation: a 10‐year follow‐up. Transpl Int 2015;28(12):1447‐1450. 10.1111/tri.12677. [DOI] [PubMed] [Google Scholar]
- 90. Chan R, Mascarenhas L, Boles RG, Kerkar N, Genyk Y, Venkatramani R. Hepatoblastoma in a patient with methylmalonic aciduria. Am J Med Genet A 2015;167A(3):635‐638. 10.1002/ajmg.a.36925. [DOI] [PubMed] [Google Scholar]
- 91. Charbit‐Henrion F, Lacaille F, McKiernan P, et al. Early and late complications after liver transplantation for propionic acidemia in children: a two centers study. Am J Transplant 2015;15(3):786‐791. 10.1111/ajt.13027. [DOI] [PubMed] [Google Scholar]
- 92. Niemi A‐K, Kim IK, Krueger CE, et al. Treatment of methylmalonic acidemia by liver or combined liver‐kidney transplantation. J Pediatr 2015;166(6):1455‐61.e1. 10.1016/j.jpeds.2015.01.051. [DOI] [PubMed] [Google Scholar]
- 93. Spada M, Calvo PL, Brunati A, et al. Early liver transplantation for neonatal‐onset methylmalonic acidemia. Pediatrics 2015;136(1):e252‐e256. 10.1542/peds.2015-0175. [DOI] [PubMed] [Google Scholar]
- 94. Spada M, Calvo PL, Brunati A, et al. Liver transplantation in severe methylmalonic acidemia: the sooner, the better. J Pediatr 2015;167(5):1173. 10.1016/j.jpeds.2015.08.022. [DOI] [PubMed] [Google Scholar]
- 95. Duclaux‐Loras R, Bacchetta J, Berthiller J, et al. Pediatric combined liver‐kidney transplantation: a single‐center experience of 18 cases. Pediatr Nephrol 2016;31(9):1517‐1529. 10.1007/s00467-016-3324-6. [DOI] [PubMed] [Google Scholar]
- 96. Khanna A, Gish R, Winter SC, Nyhan WL, Barshop BA. Successful domino liver transplantation from a patient with methylmalonic acidemia. JIMD Rep 2016;25:87‐94. 10.1007/8904_2015_480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Sakamoto R, Nakamura K, Kido J, et al. Improvement in the prognosis and development of patients with methylmalonic acidemia after living donor liver transplant. Pediatr Transplant 2016;20(8):1081‐1086. 10.1111/petr.12804. [DOI] [PubMed] [Google Scholar]
- 98. Silva HM, Nassogne MC, Smets F, et al. Liver transplantation for propionic acidemia. J Pediatr Gastroenterol Nutr 2017;64(3):e73‐e76. 10.1097/MPG.0000000000000626. [DOI] [PubMed] [Google Scholar]
- 99. Quintero J, Molera C, Juamperez J, et al. The role of liver transplantation in propionic acidemia. Liver Transpl 2018;24(12):1736‐1745. 10.1002/lt.25344. [DOI] [PubMed] [Google Scholar]
- 100. Celik N, Squires JE, Soltys K, et al. Domino liver transplantation for select metabolic disorders: expanding the living donor pool. JIMD Rep 2019;48(1):83‐89. 10.1002/jmd2.12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Jiang Y‐Z, Sun L‐Y. The value of liver transplantation for methylmalonic acidemia. Front Pediatr 2019;7:87. 10.3389/fped.2019.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Pillai NR, Stroup BM, Poliner A, et al. Liver transplantation in propionic and methylmalonic acidemia: a single center study with literature review. Mol Genet Metab 2019;128(4):431‐443. 10.1016/j.ymgme.2019.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Porta F, Romagnoli R, Busso M, Tandoi F, Spada M. Differential intraoperative effect of liver transplant in different inborn errors of metabolism. J Pediatr Gastroenterol Nutr 2019;69(2):160‐162. 10.1097/MPG.0000000000002354. [DOI] [PubMed] [Google Scholar]
- 104. Yorifuji T, Kawai M, Mamada M, et al. Living‐donor liver transplantation for propionic acidaemia. J Inherit Metab Dis 2004;27(2):205‐210. 10.1023/B:BOLI.0000028778.54210.13. [DOI] [PubMed] [Google Scholar]
- 105. Barshes NR, Vanatta JM, Patel AJ, et al. Evaluation and management of patients with propionic acidemia undergoing liver transplantation: a comprehensive review. Pediatr Transplant 2006;10(7):773‐781. 10.1111/j.1399-3046.2006.00569.x. [DOI] [PubMed] [Google Scholar]
- 106. Kaplan P, Ficicioglu C, Mazur AT, Palmieri MJ, Berry GT. Liver transplantation is not curative for methylmalonic acidopathy caused by methylmalonyl‐CoA mutase deficiency. Mol Genet Metab 2006;88(4):322‐326. 10.1016/j.ymgme.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 107. Kasahara M, Horikawa R, Tagawa M, et al. Current role of liver transplantation for methylmalonic acidemia: a review of the literature. Pediatr Transplant 2006;10(8):943‐947. 10.1111/j.1399-3046.2006.00585.x. [DOI] [PubMed] [Google Scholar]
- 108. Kamei K, Ito S, Shigeta T, et al. Preoperative dialysis for liver transplantation in methylmalonic acidemia. Ther Apher Dial 2011;15(5):488‐492. 10.1111/j.1744-9987.2011.00974.x. [DOI] [PubMed] [Google Scholar]
- 109. Rammohan A, Gunasekaran V, Reddy MS, Rela M. The role of liver transplantation in propionic acidemia. Liver Transpl 2019;25(1):176‐177. 10.1002/lt.25373. [DOI] [PubMed] [Google Scholar]
- 110. Sloan JL, Manoli I, Venditti CP. Liver or combined liver‐kidney transplantation for patients with isolated methylmalonic acidemia: who and when? J Pediatr 2015;166(6):1346‐1350. 10.1016/j.jpeds.2015.03.026. [DOI] [PubMed] [Google Scholar]
- 111. Baba C, Kasahara M, Kogure Y, et al. Perioperative management of living‐donor liver transplantation for methylmalonic acidemia. Paediatr Anaesth 2016;26(7):694‐702. 10.1111/pan.12930. [DOI] [PubMed] [Google Scholar]
- 112. Saudubray JM, Touati G, Delonlay P, et al. Liver transplantation in propionic acidaemia. Eur J Pediatr 1999;158(suppl 2):S65‐S69. 10.1007/pl00014325. [DOI] [PubMed] [Google Scholar]
- 113. Chakrapani A, Sivakumar P, McKiernan PJ, Leonard JV. Metabolic stroke in methylmalonic acidemia five years after liver transplantation. J Pediatr 2002;140(2):261‐263. 10.1067/mpd.2002.121698. [DOI] [PubMed] [Google Scholar]
- 114. Ameloot K, Vlasselaers D, Dupont M, et al. Left ventricular assist device as bridge to liver transplantation in a patient with propionic acidemia and cardiogenic shock. J Pediatr 2011;158(5):866‐867. 10.1016/j.jpeds.2010.12.031. [DOI] [PubMed] [Google Scholar]
- 115. Shanmugam NP, Valamparampil JJ, Reddy MS, et al. Auxiliary partial orthotopic liver transplantation for monogenic metabolic liver diseases: single‐centre experience. JIMD Rep 2019;45:29‐36. 10.1007/8904_2018_137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Nyhan WL, Gargus JJ, Boyle K, Selby R, Koch R. Progressive neurologic disability in methylmalonic acidemia despite transplantation of the liver. Eur J Pediatr 2002;161(7):377‐379. 10.1007/s00431-002-0970-4. [DOI] [PubMed] [Google Scholar]
- 117. Nagarajan S, Enns GM, Millan MT, Winter S, Sarwal MM. Management of methylmalonic acidaemia by combined liver‐kidney transplantation. J Inherit Metab Dis 2005;28(4):517‐524. 10.1007/s10545-005-0517-8. [DOI] [PubMed] [Google Scholar]
- 118. Imbard A, Garcia Segarra N, Tardieu M, et al. Long‐term liver disease in methylmalonic and propionic acidemias. Mol Genet Metab 2018;123(4):433‐440. 10.1016/j.ymgme.2018.01.009. [DOI] [PubMed] [Google Scholar]
- 119. Noone D, Riedl M, Atkison P, et al. Kidney disease and organ transplantation in methylmalonic acidaemia. Pediatr Transplant 2019;23(4):e13407. 10.1111/petr.13407. [DOI] [PubMed] [Google Scholar]
- 120. Forny P, Hochuli M, Rahman Y, et al. Liver neoplasms in methylmalonic aciduria: an emerging complication. J Inherit Metab Dis 2019;42(5):793‐802. 10.1002/jimd.12143. [DOI] [PubMed] [Google Scholar]
- 121. Massoud AF, Leonard JV. Cardiomyopathy in propionic acidaemia. Eur J Pediatr 1993;152(5):441‐445. 10.1007/bf01955907. [DOI] [PubMed] [Google Scholar]
- 122. Haijes HA, Jans JJM, Tas SY, Verhoeven‐Duif NM, van Hasselt PM. Pathophysiology of propionic and methylmalonic acidemias. Part 1: complications. J Inherit Metab Dis 2019;42(5):730‐744. 10.1002/jimd.12129. [DOI] [PubMed] [Google Scholar]
- 123. Yorifuji T, Muroi J, Uematsu A, Nakahata T, Egawa H, Tanaka K. Living‐related liver transplantation for neonatal‐onset propionic acidemia. J Pediatr 2000;137(4):572‐574. 10.1067/mpd.2000.108391. [DOI] [PubMed] [Google Scholar]
- 124. Nagao M, Tanaka T, Morii M, Wakai S, Horikawa R, Kasahara M. Improved neurologic prognosis for a patient with propionic acidemia who received early living donor liver transplantation. Mol Genet Metab 2013;108(1):25‐29. 10.1016/j.ymgme.2012.10.022. [DOI] [PubMed] [Google Scholar]
- 125. Vernon HJ, Sperati CJ, King JD, et al. A detailed analysis of methylmalonic acid kinetics during hemodialysis and after combined liver/kidney transplantation in a patient with mut (0) methylmalonic acidemia. J Inherit Metab Dis 2014;37(6):899‐907. 10.1007/s10545-014-9730-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Davison JE, Davies NP, Wilson M, et al. MR spectroscopy‐based brain metabolite profiling in propionic acidaemia: metabolic changes in the basal ganglia during acute decompensation and effect of liver transplantation. Orphanet J Rare Dis 2011;6:19. 10.1186/1750-1172-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. O'Shea CJ, Sloan JL, Wiggs EA, et al. Neurocognitive phenotype of isolated methylmalonic acidemia. Pediatrics 2012;129(6):e1541‐e1551. 10.1542/peds.2011-1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Martín‐Hernández E, Lee PJ, Micciche A, Grunewald S, Lachmann RH. Long‐term needs of adult patients with organic acidaemias: outcome and prognostic factors. J Inherit Metab Dis 2009;32(4):523‐533. 10.1007/s10545-009-1191-12. [DOI] [PubMed] [Google Scholar]
- 129. Kang L, Liu Y, Shen M, et al. A study on a cohort of 301 Chinese patients with isolated methylmalonic acidemia. J Inherit Metab Dis 2020;43(3):409‐423. 10.1002/jimd.12183. [DOI] [PubMed] [Google Scholar]
- 130. Nizon M, Ottolenghi C, Valayannopoulos V, et al. Long‐term neurological outcome of a cohort of 80 patients with classical organic acidurias. Orphanet J Rare Dis 2013;8:148. 10.1186/1750-1172-8-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Surtees RA, Matthews EE, Leonard JV. Neurologic outcome of propionic acidemia. Pediatr Neurol 1992;8(5):333‐337. 10.1016/0887-8994(92)90085-d. [DOI] [PubMed] [Google Scholar]
- 132. North KN, Korson MS, Gopal YR, et al. Neonatal‐onset propionic acidemia: neurologic and developmental profiles, and implications for management. J Pediatr 1995;126(6):916‐922. 10.1016/s0022-3476(95)70208-3. [DOI] [PubMed] [Google Scholar]
- 133. Sass JO, Hofmann M, Skladal D, Mayatepek E, Schwahn B, Sperl W. Propionic acidemia revisited: a workshop report. Clin Pediatr 2004;43(9):837‐843. 10.1177/000992280404300908. [DOI] [PubMed] [Google Scholar]
- 134. Shuaib T, Al‐Hashmi N, Ghaziuddin M, et al. Propionic acidemia associated with visual hallucinations. J Child Neurol 2012;27(6):799‐803. 10.1177/0883073811426929. [DOI] [PubMed] [Google Scholar]
- 135. Al‐Owain M, Kaya N, Al‐Shamrani H, et al. Autism spectrum disorder in a child with propionic acidemia. JIMD Rep 2013;7:63‐66. 10.1007/8904_2012_143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. La Dejean Bâtie C d, Barbier V, Valayannopoulos V, et al. Acute psychosis in propionic acidemia: 2 case reports. J Child Neurol 2014;29(2):274‐279. 10.1177/0883073813508812. [DOI] [PubMed] [Google Scholar]
- 137. Witters P, Debbold E, Crivelly K, et al. Autism in patients with propionic acidemia. Mol Genet Metab 2016;119(4):317‐321. 10.1016/j.ymgme.2016.10.009. [DOI] [PubMed] [Google Scholar]
- 138. La Bâtie CD d, Barbier V, Roda C, et al. Autism spectrum disorders in propionic acidemia patients. J Inherit Metab Dis 2018;41(4):623‐629. 10.1007/s10545-017-0070-2. [DOI] [PubMed] [Google Scholar]
- 139. Trakadis YJ, Fulginiti V, Walterfang M. Inborn errors of metabolism associated with psychosis: literature review and case‐control study using exome data from 5090 adult individuals. J Inherit Metab Dis 2018;41(4):613‐621. 10.1007/s10545-017-0023-9. [DOI] [PubMed] [Google Scholar]
- 140. Heidenreich R, Natowicz M, Hainline BE, et al. Acute extrapyramidal syndrome in methylmalonic acidemia: "metabolic stroke" involving the globus pallidus. J Pediatr 1988;113(6):1022‐1027. 10.1016/s0022-3476(88)80574-2. [DOI] [PubMed] [Google Scholar]
- 141. Haas RH, Marsden DL, Capistrano‐Estrada S, et al. Acute basal ganglia infarction in propionic acidemia. J Child Neurol 1995;10(1):18‐22. 10.1177/088307389501000104. [DOI] [PubMed] [Google Scholar]
- 142. Hamilton RL, Haas RH, Nyhan WL, Powell HC, Grafe MR. Neuropathology of propionic acidemia: a report of two patients with basal ganglia lesions. J Child Neurol 1995;10(1):25‐30. 10.1177/088307389501000107. [DOI] [PubMed] [Google Scholar]
- 143. Scholl‐Bürgi S, Haberlandt E, Gotwald T, et al. Stroke‐like episodes in propionic acidemia caused by central focal metabolic decompensation. Neuropediatrics 2009;40(2):76‐81. 10.1055/s-0029-1231065. [DOI] [PubMed] [Google Scholar]
- 144. Broomfield A, Gunny R, Prabhakar P, Grunewald S. Spontaneous rapid resolution of acute basal ganglia changes in an untreated infant with propionic acidemia: a clue to pathogenesis? Neuropediatrics 2010;41(6):256‐260. 10.1055/s-0031-1273720. [DOI] [PubMed] [Google Scholar]
- 145. Karall D, Haberlandt E, Schimmel M, et al. Cytotoxic not vasogenic edema is the cause for stroke‐like episodes in propionic acidemia. Neuropediatrics 2011;42(5):210. 10.1055/s-0031-1287772. [DOI] [PubMed] [Google Scholar]
- 146. Kidd JR, Wolf B, Hsia E, Kidd KK. Genetics of propionic acidemia in a Mennonite‐Amish kindred. Am J Hum Genet 1980;32(2):236‐245. [PMC free article] [PubMed] [Google Scholar]
- 147. Sethi KD, Ray R, Roesel RA, et al. Adult‐onset chorea and dementia with propionic acidemia. Neurology 1989;39(10):1343‐1345. 10.1212/wnl.39.10.1343. [DOI] [PubMed] [Google Scholar]
- 148. Ozand PT, Rashed M, Gascon GG, et al. Unusual presentations of propionic acidemia. Brain Dev 1994;16(suppl):46‐57. 10.1016/0387-7604(94)90096-5. [DOI] [PubMed] [Google Scholar]
- 149. Pérez‐Cerdá C, Merinero B, Martí M, et al. An unusual late‐onset case of propionic acidaemia: biochemical investigations, neuroradiological findings and mutation analysis. Eur J Pediatr 1998;157(1):50‐52. 10.1007/s004310050765. [DOI] [PubMed] [Google Scholar]
- 150. Nyhan WL, Bay C, Beyer EW, Mazi M. Neurologic nonmetabolic presentation of propionic acidemia. Arch Neurol 1999;56(9):1143‐1147. 10.1001/archneur.56.9.1143. [DOI] [PubMed] [Google Scholar]
- 151. Brismar J, Ozand PTCT. MR of the brain in disorders of the propionate and methylmalonate metabolism. AJNR Am J Neuroradiol 1994;15(8):1459‐1473. [PMC free article] [PubMed] [Google Scholar]
- 152. Harting I, Seitz A, Geb S, et al. Looking beyond the basal ganglia: the spectrum of MRI changes in methylmalonic acidaemia. J Inherit Metab Dis 2008;31(3):368‐378. 10.1007/s10545-008-0801-5. [DOI] [PubMed] [Google Scholar]
- 153. Kandel A, Amatya SK, Yeh EA. Reversible diffusion weighted imaging changes in propionic acidemia. J Child Neurol 2013;28(1):128‐131. 10.1177/0883073812441057. [DOI] [PubMed] [Google Scholar]
- 154. Baker EH, Sloan JL, Hauser NS, et al. MRI characteristics of globus pallidus infarcts in isolated methylmalonic acidemia. AJNR Am J Neuroradiol 2015;36(1):194‐201. 10.3174/ajnr.A4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Ma X, Zhang Y, Yang Y, et al. Epilepsy in children with methylmalonic acidemia: electroclinical features and prognosis. Brain Dev 2011;33(9):790‐795. 10.1016/j.braindev.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 156. Lehnert W, Sperl W, Suormala T, Baumgartner ER. Propionic acidaemia: clinical, biochemical and therapeutic aspects. Experience in 30 patients. Eur J Pediatr 1994;153(7 suppl 1):S68‐S80. 10.1007/bf02138781. [DOI] [PubMed] [Google Scholar]
- 157. Haberlandt E, Canestrini C, Brunner‐Krainz M, et al. Epilepsy in patients with propionic acidemia. Neuropediatrics 2009;40(3):120‐125. 10.1055/s-0029-1243167. [DOI] [PubMed] [Google Scholar]
- 158. Okamura N, Ohnishi S, Shimaoka H, Norikura R, Hasegawa H. Involvement of recognition and interaction of carnitine transporter in the decrease of L‐carnitine concentration induced by pivalic acid and valproic acid. Pharm Res 2006;23(8):1729‐1735. 10.1007/s11095-006-9002-9. [DOI] [PubMed] [Google Scholar]
- 159. Williams ZR, Hurley PE, Altiparmak UE, et al. Late onset optic neuropathy in methylmalonic and propionic acidemia. Am J Ophthalmol 2009;147(5):929‐933. 10.1016/j.ajo.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 160. Pinar‐Sueiro S, Martínez‐Fernández R, Lage‐Medina S, Aldamiz‐Echevarria L, Vecino E. Optic neuropathy in methylmalonic acidemia: the role of neuroprotection. J Inherit Metab Dis 2010;33(suppl 3):S199‐S203. 10.1007/s10545-010-9084-8. [DOI] [PubMed] [Google Scholar]
- 161. Traber G, Baumgartner MR, Schwarz U, Pangalu A, Donath MY, Landau K. Subacute bilateral visual loss in methylmalonic acidemia. J Neuroophthalmol 2011;31(4):344‐346. 10.1097/WNO.0b013e31822db480. [DOI] [PubMed] [Google Scholar]
- 162. Kölker S, Valayannopoulos V, Burlina AB, et al. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 2: the evolving clinical phenotype. J Inherit Metab Dis 2015;38(6):1059‐1074. 10.1007/s10545-015-9840-x. [DOI] [PubMed] [Google Scholar]
- 163. Stigsby B, Yarworth SM, Rahbeeni Z, et al. Neurophysiologic correlates of organic acidemias: a survey of 107 patients. Brain Dev 1994;16:125‐144. 10.1016/0387-7604(94)90104-X. [DOI] [PubMed] [Google Scholar]
- 164. Ianchulev T, Kolin T, Moseley K, Sadun A. Optic nerve atrophy in propionic acidemia. Ophthalmology 2003;110(9):1850‐1854. 10.1016/S0161-6420(03)00573-6. [DOI] [PubMed] [Google Scholar]
- 165. Noval S, López‐Rodríguez M, González‐Sánchez E, Contreras I, Royo A, Boto‐De‐los‐Bueis A. Late optic neuropathy in propionic acidemia following surgical intervention. J Neuroophthalmol 2013;33(1):90‐91. 10.1097/WNO.0b013e318273c03b. [DOI] [PubMed] [Google Scholar]
- 166. Martinez Alvarez L, Jameson E, Parry NRA, Lloyd C, Ashworth JL. Optic neuropathy in methylmalonic acidemia and propionic acidemia. Br J Ophthalmol 2016;100(1):98‐104. 10.1136/bjophthalmol-2015-306798. [DOI] [PubMed] [Google Scholar]
- 167. Grünert SC, Bodi I, Odening KE. Possible mechanisms for sensorineural hearing loss and deafness in patients with propionic acidemia. Orphanet J Rare Dis 2017;12(1):30. 10.1186/s13023-017-0585-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Cosson MA, Benoist JF, Touati G, et al. Long‐term outcome in methylmalonic aciduria: a series of 30 French patients. Mol Genet Metab 2009;97(3):172‐178. 10.1016/j.ymgme.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 169. Lam C, Desviat LR, Perez‐Cerdá C, Ugarte M, Barshop BA, Cederbaum S. 45‐Year‐old female with propionic acidemia, renal failure, and premature ovarian failure; late complications of propionic acidemia? Mol Genet Metab 2011;103(4):338‐340. 10.1016/j.ymgme.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 170. Vatanavicharn N, Champattanachai V, Liammongkolkul S, et al. Clinical and molecular findings in Thai patients with isolated methylmalonic acidemia. Mol Genet Metab 2012;106(4):424‐429. 10.1016/j.ymgme.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 171. Vernon HJ, Bagnasco S, Hamosh A, Sperati CJ. Chronic kidney disease in an adult with propionic acidemia. JIMD Rep 2014;12:5‐10. 10.1007/8904_2013_237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Shchelochkov OA, Manoli I, Sloan JL, et al. Chronic kidney disease in propionic acidemia. Genet Med 2019;21:2830‐2835. 10.1038/s41436-019-0593-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Baumgartner ER, Viardot C. Long‐term follow‐up of 77 patients with isolated methylmalonic acidaemia. J Inherit Metab Dis 1995;18(2):138‐142. 10.1007/bf00711749. [DOI] [PubMed] [Google Scholar]
- 174. Morath MA, Hörster F, Sauer SW. Renal dysfunction in methylmalonic acidurias: review for the pediatric nephrologist. Pediatr Nephrol 2013;28(2):227‐235. 10.1007/s00467-012-2245-2. [DOI] [PubMed] [Google Scholar]
- 175. Wolff JA, Strom C, Griswold W, et al. Proximal renal tubular acidosis in methylmalonic acidemia. J Neurogenet 1985;2(1):31‐39. [DOI] [PubMed] [Google Scholar]
- 176. Ohura T, Kikuchi M, Abukawa D, et al. Type 4 renal tubular acidosis (subtype 2) in a patient with methylmalonic acidaemia. Eur J Pediatr 1990;150(2):115‐118. 10.1007/bf02072052. [DOI] [PubMed] [Google Scholar]
- 177. D'Angio CT, Dillon MJ, Leonard JV. Renal tubular dysfunction in methylmalonic acidaemia. Eur J Pediatr 1991;150(4):259‐263. 10.1007/bf01955526. [DOI] [PubMed] [Google Scholar]
- 178. Rutledge SL, Geraghty M, Mroczek E, Rosenblatt D, Kohout E. Tubulointerstitial nephritis in methylmalonic acidemia. Pediatr Nephrol 1993;7(1):81‐82. 10.1007/bf00861581. [DOI] [PubMed] [Google Scholar]
- 179. Haarmann A, Mayr M, Kölker S, et al. Renal involvement in a patient with cobalamin A type (cblA) methylmalonic aciduria: a 42‐year follow‐up. Mol Genet Metab 2013;110(4):472‐476. 10.1016/j.ymgme.2013.08.021. [DOI] [PubMed] [Google Scholar]
- 180. Manoli I, Sysol JR, Li L, et al. Targeting proximal tubule mitochondrial dysfunction attenuates the renal disease of methylmalonic acidemia. Proc Natl Acad Sci U S A 2013;110(33):13552‐13557. 10.1073/pnas.1302764110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Luciani A, Schumann A, Berquez M, et al. Impaired mitophagy links mitochondrial disease to epithelial stress in methylmalonyl‐CoA mutase deficiency. Nat Commun 2020;11(1):970. 10.1038/s41467-020-14729-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Kruszka PS, Manoli I, Sloan JL, Kopp JB, Venditti CP. Renal growth in isolated methylmalonic acidemia. Genet Med 2013;15(12):990‐996. 10.1038/gim.2013.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Stevenson T, Millan MT, Wayman K, et al. Long‐term outcome following pediatric liver transplantation for metabolic disorders. Pediatr Transplant 2010;14(2):268‐275. 10.1111/j.1399-3046.2009.01228.x. [DOI] [PubMed] [Google Scholar]
- 184. Critelli K, McKiernan P, Vockley J, et al. Liver transplantation for propionic acidemia and methylmalonic acidemia: perioperative management and clinical outcomes. Liver Transpl 2018;24(9):1260‐1270. 10.1002/lt.25304. [DOI] [PubMed] [Google Scholar]
- 185. Lubrano R, Elli M, Rossi M, et al. Renal transplant in methylmalonic acidemia: could it be the best option? Report on a case at 10 years and review of the literature. Pediatr Nephrol 2007;22(8):1209‐1214. 10.1007/s00467-007-0460-z. [DOI] [PubMed] [Google Scholar]
- 186. Molema F, Williams M, Langendonk J, et al. Neurotoxicity including posterior reversible encephalopathy syndrome after initiation of calcineurin inhibitors in transplanted methylmalonic acidemia patients: two case reports and review of the literature. JIMD Rep 2020;51(1):89‐104. 10.1002/jmd2.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Pérez‐Cerdá C, Merinero B, Rodríguez‐Pombo P, et al. Potential relationship between genotype and clinical outcome in propionic acidaemia patients. Eur J Hum Genet 2000;8(3):187‐194. 10.1038/sj.ejhg.5200442. [DOI] [PubMed] [Google Scholar]
- 188. Prada CE, Al Jasmi F, Kirk EP, et al. Cardiac disease in methylmalonic acidemia. J Pediatr 2011;159(5):862‐864. 10.1016/j.jpeds.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 189. Lee TM, Addonizio LJ, Barshop BA, Chung WK. Unusual presentation of propionic acidaemia as isolated cardiomyopathy. J Inherit Metab Dis 2009;32(suppl 1):S97‐S101. 10.1007/s10545-009-1084-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Laemmle A, Balmer C, Doell C, Sass JO, Häberle J, Baumgartner MR. Propionic acidemia in a previously healthy adolescent with acute onset of dilated cardiomyopathy. Eur J Pediatr 2014;173(7):971‐974. 10.1007/s00431-014-2359-6. [DOI] [PubMed] [Google Scholar]
- 191. Baumgartner D, Scholl‐Bürgi S, Sass JO, et al. Prolonged QTc intervals and decreased left ventricular contractility in patients with propionic acidemia. J Pediatr 2007;150(2):192‐197. 10.1016/j.jpeds.2006.11.043. [DOI] [PubMed] [Google Scholar]
- 192. Mardach R, Verity MA, Cederbaum SD. Clinical, pathological, and biochemical studies in a patient with propionic acidemia and fatal cardiomyopathy. Mol Genet Metab 2005;85(4):286‐290. 10.1016/j.ymgme.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 193. Baruteau J, Hargreaves I, Krywawych S, et al. Successful reversal of propionic acidaemia associated cardiomyopathy: evidence for low myocardial coenzyme Q10 status and secondary mitochondrial dysfunction as an underlying pathophysiological mechanism. Mitochondrion 2014;17:150‐156. 10.1016/j.mito.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 194. Fujisawa D, Nakamura K, Mitsubuchi H, et al. Clinical features and management of organic acidemias in Japan. J Hum Genet 2013;58(12):769‐774. 10.1038/jhg.2013.97. [DOI] [PubMed] [Google Scholar]
- 195. Collins J, Kelly D. Cardiomyopathy in propionic acidaemia. Eur J Pediatr 1994;153(1):53. 10.1007/bf02000791. [DOI] [PubMed] [Google Scholar]
- 196. Genuardi MV, Kagawa H, Minervini M, Mathier MA, Sciortino CA. Case report of cardiac transplantation for isolated cardiomyopathy associated with propionic acidemia. Prog Transplant 2019;29:364‐366. 10.1177/1526924819874390. [DOI] [PubMed] [Google Scholar]
- 197. Sweetman L, Nyhan WL, Cravens J, Zomer Y, Plunket DC. Propionic acidaemia presenting with pancytopaenia in infancy. J Inherit Metab Dis 1980;2(3):65‐69. 10.1007/bf01801721. [DOI] [PubMed] [Google Scholar]
- 198. Stork LC, Ambruso DR, Wallner SF, et al. Pancytopenia in propionic acidemia: hematologic evaluation and studies of hematopoiesis in vitro. Pediatr Res 1986;20(8):783‐788. 10.1203/00006450-198608000-00017. [DOI] [PubMed] [Google Scholar]
- 199. Werlin SL. E. coli sepsis as a presenting sign in neonatal propionic acidemia. Am J Med Genet 1993;46(4):455‐456. 10.1002/ajmg.1320460423. [DOI] [PubMed] [Google Scholar]
- 200. Feliz B, Witt DR, Harris BT. Propionic acidemia: a neuropathology case report and review of prior cases. Arch Pathol Lab Med 2003;127(8):e325‐e328. . [DOI] [PubMed] [Google Scholar]
- 201. Hutchinson RJ, Bunnell K, Thoene JG. Suppression of granulopoietic progenitor cell proliferation by metabolites of the branched‐chain amino acids. J Pediatr 1985;106(1):62‐65. 10.1016/s0022-3476(85)80466-2. [DOI] [PubMed] [Google Scholar]
- 202. Childs B, Nyhan WL, Borden M, Bard L, Cooke RE. Idiopathic hyperglycinemia and hyperglycinuria: a new disorder of amino acid metabolism. I. Pediatrics 1961;27(4):522‐538. [PubMed] [Google Scholar]
- 203. Touati G, Valayannopoulos V, Mention K, et al. Methylmalonic and propionic acidurias: management without or with a few supplements of specific amino acid mixture. J Inherit Metab Dis 2006;29(2‐3):288‐298. 10.1007/s10545-006-0351-7. [DOI] [PubMed] [Google Scholar]
- 204. Inoue F, Terada N, Nukina S, Kodo N, Kinugasa A, Sawada T. Methylmalonic aciduria with pathological fracture. J Inherit Metab Dis 1993;16(6):1052‐1053. 10.1007/bf00711530. [DOI] [PubMed] [Google Scholar]
- 205. Rafique M. Propionic acidaemia: demographic characteristics and complications. J Pediatr Endocrinol Metab 2013;26(5‐6):497‐501. 10.1515/jpem-2013-0031. [DOI] [PubMed] [Google Scholar]
- 206. Manoli I, Myles JG, Sloan JL, Shchelochkov OA, Venditti CP. A critical reappraisal of dietary practices in methylmalonic acidemia raises concerns about the safety of medical foods. Part 1: isolated methylmalonic acidemias. Genet Med 2016;18(4):386‐395. 10.1038/gim.2015.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Kölker S, Garcia‐Cazorla A, Cazorla AG, et al. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 1: the initial presentation. J Inherit Metab Dis 2015;38(6):1041‐1057. 10.1007/s10545-015-9839-3. [DOI] [PubMed] [Google Scholar]
- 208. Evans M, Truby H, Boneh A. The relationship between dietary intake, growth, and body composition in inborn errors of intermediary protein metabolism. J Pediatr 2017;188:163‐172. 10.1016/j.jpeds.2017.05.048. [DOI] [PubMed] [Google Scholar]
- 209. van der Meer SB, Poggi F, Spada M, et al. Clinical outcome of long‐term management of patients with vitamin B12‐unresponsive methylmalonic acidemia. J Pediatr 1994;125(6 pt 1):903‐908. 10.1016/s0022-3476(05)82005-0. [DOI] [PubMed] [Google Scholar]
- 210. van der Meer SB, Poggi F, Spada M, et al. Clinical outcome and long‐term management of 17 patients with propionic acidaemia. Eur J Pediatr 1996;155(3):205‐210. 10.1007/bf01953939. [DOI] [PubMed] [Google Scholar]
- 211. Daly A, Pinto A, Evans S, et al. Dietary practices in propionic acidemia: a European survey. Mol Genet Metab Rep 2017;13:83‐89. 10.1016/j.ymgmr.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Yannicelli S, Acosta PB, Velazquez A, et al. Improved growth and nutrition status in children with methylmalonic or propionic acidemia fed an elemental medical food. Mol Genet Metab 2003;80(1‐2):181‐188. [DOI] [PubMed] [Google Scholar]
- 213. Zwickler T, Lindner M, Aydin HI, et al. Diagnostic work‐up and management of patients with isolated methylmalonic acidurias in European metabolic centres. J Inherit Metab Dis 2008;31(3):361‐367. 10.1007/s10545-008-0804-2. [DOI] [PubMed] [Google Scholar]
- 214. Molema F, Gleich F, Burgard P, et al. Evaluation of dietary treatment and amino acid supplementation in organic acidurias and urea‐cycle disorders: on the basis of information from a European multicenter registry. J Inherit Metab Dis 2019;42:1162‐1175. 10.1002/jimd.12066. [DOI] [PubMed] [Google Scholar]
- 215. Daly A, Evans S, Ashmore C, Chahal S, Santra S, MacDonald A. Refining low protein modular feeds for children on low protein tube feeds with organic acidaemias. Mol Genet Metab Rep 2017;13:99‐104. 10.1016/j.ymgmr.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Coman D, Huang J, McTaggart S, et al. Renal transplantation in a 14‐year‐old girl with vitamin B12‐responsive cblA‐type methylmalonic acidaemia. Pediatr Nephrol 2006;21(2):270‐273. 10.1007/s00467-005-2071-x. [DOI] [PubMed] [Google Scholar]
- 217. Kahler SG, Sherwood WG, Woolf D, et al. Pancreatitis in patients with organic acidemias. J Pediatr 1994;124(2):239‐243. 10.1016/s0022-3476(94)70311-6. [DOI] [PubMed] [Google Scholar]
- 218. Burlina AB, Dionisi‐Vici C, Piovan S, et al. Acute pancreatitis in propionic acidaemia. J Inherit Metab Dis 1995;18(2):169‐172. 10.1007/bf00711758. [DOI] [PubMed] [Google Scholar]
- 219. Bultron G, Seashore MR, Pashankar DS, Husain SZ. Recurrent acute pancreatitis associated with propionic acidemia. J Pediatr Gastroenterol Nutr 2008;47(3):370‐371. 10.1097/MPG.0b013e3181132252. [DOI] [PubMed] [Google Scholar]
- 220. Manoli I, Sloan JL, Venditti CP. GeneReviews®: isolated methylmalonic acidemia. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews((R)). Seattle, WA; University of Washington, National Center for Biotechnology Information; 1993. [Google Scholar]
- 221. Marquard J, El Scheich T, Klee D, et al. Chronic pancreatitis in branched‐chain organic acidurias—a case of methylmalonic aciduria and an overview of the literature. Eur J Pediatr 2011;170(2):241‐245. 10.1007/s00431-010-1313-5. [DOI] [PubMed] [Google Scholar]
- 222. Bullinger M, Quitmann J. Quality of life as patient‐reported outcomes: principles of assessment. Dialogues Clin Neurosci 2014;16(2):137‐145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Hall CA, Donza C, McGinn S, et al. Health‐related quality of life in children with chronic illness compared to parents: a systematic review. Pediatr Phys Ther 2019;31(4):315‐322. 10.1097/PEP.0000000000000638. [DOI] [PubMed] [Google Scholar]
- 224. Zeltner NA, Huemer M, Baumgartner MR, Landolt MA. Quality of life, psychological adjustment, and adaptive functioning of patients with intoxication‐type inborn errors of metabolism ‐ a systematic review. Orphanet J Rare Dis 2014;9:159. 10.1186/s13023-014-0159-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Splinter K, Niemi A‐K, Cox R, et al. Impaired health‐related quality of life in children and families affected by methylmalonic acidemia. J Genet Couns 2016;25(5):936‐944. 10.1007/s10897-015-9921-x. [DOI] [PubMed] [Google Scholar]
- 226. Zeltner NA, Baumgartner MR, Bondarenko A, et al. Development and psychometric evaluation of the MetabQoL 1.0: a quality of life questionnaire for paediatric patients with intoxication‐type inborn errors of metabolism. JIMD Rep 2017;37:27‐35. 10.1007/8904_2017_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Zeltner NA, Landolt MA, Baumgartner MR, et al. Living with intoxication‐type inborn errors of metabolism: a qualitative analysis of interviews with paediatric patients and their parents. JIMD Rep 2017;31:1‐9. 10.1007/8904_2016_545. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting information