

Summary
Tumor multiregion sequencing reveals intratumor heterogeneity (ITH) and clonal evolution playing a key role in tumor progression and metastases. Large-scale high-depth multiregional sequencing of colorectal cancer, comparative analysis among patients with right-sided colon cancer (RCC), left-sided colon cancer (LCC), and rectal cancer (RC), as well as the study of lymph node metastasis (LN) with extranodal tumor deposits (ENTDs) from evolutionary perspective remain weakly explored. Here, we recruited 68 patients with RCC (18), LCC (20), and RC (30). We performed high-depth whole-exome sequencing of 206 tumor regions including 176 primary tumors, 19 LN, and 11 ENTD samples. Our results showed ITH with a Darwinian pattern of evolution and the evolution pattern of LCC and RC was more complex and divergent than RCC. Genetic and evolutionary evidences found that both LN and ENTD originated from different clones. Moreover, ENTD was a distinct entity from LN and evolved later.
Subject area: Evolutionary Biology, Cancer Systems Biology, Genomics
Graphical abstract
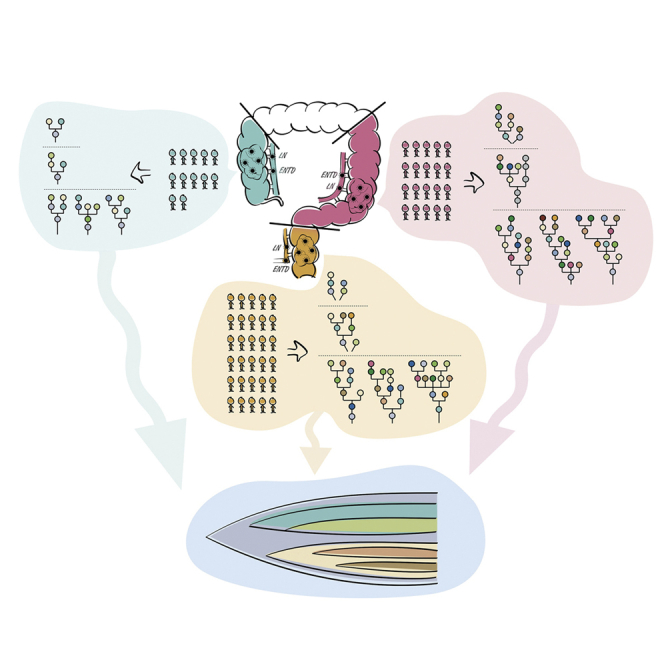
Highlights
-
•
Clonal evolution of colorectal tumors followed Darwinian pattern of evolution
-
•
Intratumor heterogeneity in patients with right-sided and left-sided colon and rectal cancer
-
•
Evolution of left-sided colon cancer and rectal cancer was more complex and divergent
-
•
Lymph node metastasis and extranodal tumor deposits were of polyclonal in origin
Evolutionary biology; Cancer systems biology; Genomics
Introduction
Colorectal cancer (CRC) is the third most common malignancy and the second leading cause of cancer death worldwide (Cancer Fact Sheet, World Health Organization [WHO]). As per the WHO GLOBOCAN database, there were 1,849,518 estimated new CRC cases and 880,792 CRC-related deaths in 2019 (Colorectal Cancer Facts & Figures 2020-2022). In China, CRC is the second most common neoplasia, occupying the fifth position in mortality, accounting for an incidence of 521,490 new cases and 248,400 deaths in 2019.
Tumor multiregion sequencing reveals intratumor heterogeneity (ITH) and clonal evolution which play a key role in progression and metastases of the tumor (Alizadeh et al., 2015). The development of effective target-based precision medicine and personalized cancer therapy is based on ITH and the pattern of clonal evolution in colorectal tumors (Turner and Reis-Fiho, 2012). Therefore, patients with CRC may respond variably to the same treatment, owing to ITH and differences in clonal evolution, despite there being no significant differences identified in the tumor histopathology (Waddell et al., 2015). Hence, study of ITH and comparative analysis of clonal evolution is highly significant from both clinical and biological perspective, to understand the genomic changes driving the malignant process, which is fundamental for developing an effective personalized cancer therapy.
Recently, tumor multiregion sequencing studies of colorectal cancer have demonstrated ITH (Sottoriva et al., 2015; Roerink et al., 2018; Uchi et al., 2016; Saito et al., 2018; Wei et al., 2017; Alves et al., 2019; Hu et al., 2019; Zhang et al., 2020). This multiregional sequencing approach, sequencing DNA samples from geographically separated regions of a single tumor, explores ITH and cancer evolution. Large-scale multiregional sequencing studies have systematically revealed ITH as well as cancer evolution in patients with non-small-cell lung cancer and renal cancer (Jamal-Hanjani et al., 2017; Turajlic et al., 2018; Turajlic et al., 2018, 2018). However, large-scale multiregional sequencing studies of CRC have not been well reported. In addition, multiregional sequencing studies in CRC were performed at relatively shallow sequencing depths (Sottoriva et al., 2015; Roerink et al., 2018; Uchi et al., 2016; Saito et al., 2018; Wei et al., 2017), making it difficult to assess ITH, owing to inability to detect somatic mutations with low frequencies.
CRC is no longer regarded as a single disease with increasing knowledge of the molecular mechanisms of carcinogenesis. The location of the primary tumor, with respect to the right side or left side of the splenic flexure and rectum, is an important prognostic factor of CRC (Loupakis et al., 2015; Petrelli et al., 2017). Patients with left-sided colon cancer (LCC) and rectal cancer (RC) (originating from splenic flexure, descending colon, sigmoid colon, and rectum) survive longer than patients with right-sided colon cancer (RCC) (originating from hepatic flexure, ascending colon, and cecum). Clinical symptoms are also different between patients with RCC and LCC/RC (Missiaglia et al., 2014; Lee et al., 2017). Patients with RCC tend to be older, female, and have advanced stage of tumors with frequent metastasis to the peritoneum compared with metastasis to the lung and liver in patients with LCC/RC. In addition, patients with RCC and LCC/RC exhibit different treatment outcomes toward antiepidermal growth factor receptor therapy (Lee et al., 2017). Many studies have been conducted to explore the possible reasons for clinical heterogeneity between RCC and LCC/RC and found differences in their CpG island methylator phenotype (CIMP) status, embryonic origin, blood supplies, genetic mutations, genomic expression profiles, immunological composition, and bacterial population in tumor microenvironment (Advani et al., 2018; Cheng et al., 2008; Missiaglia et al., 2014; Lee et al., 2017; Hu et al., 2018; Imperial et al., 2018; Baek, 2019). For example, CIMP cases, comprising 20% of CRCs, tend to occur in RCC (Advani et al., 2018; Cheng et al., 2008). However, the understanding of the ITH and clonal evolution that determine the pathogenesis of RCC and LCC/RC is still unclear.
Among patients with CRC, the stage of the disease is one of the most important prognostic factors which is correlated with the disease survival rate (O’Connell et al., 2014). Tumor node metastasis (TNM)/American Joint Committee on Cancer cancer staging system is the gold standard for determining the correct cancer stage, helping to make appropriate treatment plans. Among patients with CRC, the presence of cancer cells in lymph nodes is defined as stage III disease (NIH consensus conference, 1990). In the seventh and eighth editions of TNM staging system, a separate entity, entitled extranodal tumor deposits (ENTDs), was included as “N1c” subcategory (Weiser, 2018). However, inclusion of ENTDs within nodal staging has worldwide debates in CRC because of lack of significant improvement of prognostic value (Ueno et al., 2012; Lord et al., 2017; Nagtegaal et al., 2017). Although, many ITH and evolution studies of CRC focus on spreading routes of lymphatic metastases by sampling paired primary tumors and lymph node metastasis (LN), none of them included ENTD samples (Saito et al., 2018; Wei et al., 2017; Alves et al., 2019; Hu et al., 2019; Zhang et al., 2020). Therefore, the molecular signature and evolutionary relationship between LN and ENTD has not been clear till now. Hence, the characterization of the molecular signature and evolution of the primary tumor, LN and ENTD, is very significant for TNM staging and therapeutic interventions for the patients with CRC.
To overcome the drawbacks of previous studies, we have comprehensively studied the ITH and clonal evolution of CRC, using high-depth (median depth of 395×) whole-exome sequencing (WES) of 206 multiregion tumor samples and 68 matched germline samples from 68 CRC tumors, determined the differences of ITH, and the clonal evolution of CRC in patients with RCC, LCC, and RC.
Results
Comprehensive clinical descriptions of these 68 patients with CRC were provided in Table S1. Tumor multiregion high-depth (median depth of 395×, range 179–596) WES was performed with 206 tumor regions (2–7 regions/tumor) including 176 primary tumor regions, 19 LN regions, and 11 ENTD regions, as well as 68 matched germline samples from 68 patients with CRC. WES identified 6 hypermutated (mutation burden of each tumor region were >10 mutations/1 Mb bases) patients with CRC; of these, four patients were identified with microsatellite instability (MSI). The remaining 62 patients with CRC were microsatellite stable (MSS), and of these, 12 are patients with RCC, 20 are patients with LCC, and 30 are patients with RC. Hypermutated patients were analyzed separately.
ITH in colorectal tumors
WES of 62 tumors with 188 tumor regions identified 19,454 somatic mutations including 17,560 single-nucleotide variants (SNVs) (14,361 nonsilent SNVs) and 1894 insertions/deletions (INDELs) (Table S2). Identified somatic mutations were divided into clonal and subclonal mutations. Mutations with cancer cell fraction (CCF) > 0.9 across all regions of a tumor were considered as clonal mutations, otherwise they were considered as subclonal mutations. The mutation burden identified by the multiregion WES was significantly higher than single sample sequencing due owing to detection of subclonal mutations (median number of mutations/1-MB bases, 4.61 vs. 3.23; P = 8.9 × 10−9) (Figure S3). In our study, the mutation burden of single sample sequencing was significantly higher than single CRC sample sequencing data from The Cancer Genome Atlas (Cancer Genome Atlas Network, 2012) (TCGA) (median number of mutations/1-MB bases, 3.23 vs. 2.07; P = 1.7 × 10−22) (Figure S3).
It is worth noting that 2 patients (CRC32 and CRC36) with LCC and 6 patients (CRC49, CRC42, CRC51, CRC48, CRC52, and CRC60) with RC had not identified with clonal mutations (Figure 1A), suggesting the existence of coexisting tumor-initiating events and widespread of branched evolution of mutations during tumor progression in patients with LCC and RC. In addition, patients with RCC had significantly more clonal mutations than patients with RC (median number, 160 vs. 119; P = 0.035) (Figure S4).
Figure 1.

Overview of genomic heterogeneity in CRC tumors
(A) Heterogeneity of mutations and somatic copy-number alterations (SCNAs). Tumors were sorted by location and stage. (1) Number of all SNV and INDEL mutations (including coding and noncoding mutations) in CRC tumors. (2) The percentages of clonal mutations in CRC tumors. (3) Quantification of SCNAs in CRC tumors. (4) The percentages of clonal SCNAs in CRC tumors. (5) Demographic and clinical characteristics of the 62 patients with CRC in this study (divided by histology; stage; number of regions; tumor size; age and tumor location).
(B) Mutation frequency of driver genes (driver mutations occurred in not less than 10 patients) and comparison with TCGA data. (C) Frequency of SCNAs in CRC tumors. The dotted lines were frequency of SCNAs in TCGA CRC samples.
Somatic copy number alterations (SCNAs) were measured as length of segments affected by either gains or losses (detailed copy number data are given in Table S3). Any segment of gain or loss that spanned across all the regions was defined as clonal, and all other segments of SCNA were defined as subclonal. We summarized the total length of the genome that subjected to SCNAs and calculated the percentage of clonal and subclonal SCNAs (Figure 1A). Interestingly, in a patient with RC (CRC43), all the identified SCNAs were subclonal. There were no significant differences in the length and percentage of SCNAs among patients with RCC, LCC and RC (Figure S5).
In our study, we identified that the mutation frequency of 14 driver genes (APC, TP53, KRAS, LZTR1, LRP1B, FBXW7, TCF7L2, FAT4, ARID1A, ATM, PIK3CA, AMER1, CSMD3, and SMAD4) were higher at the patient level than at the sample level, except SMAD4 gene (Figure 1B). Consistent with previous study that loss of SMAD4 promote metastasis in CRC (Itatani et al., 2013; Voorneveld et al., 2014), mutations in SMAD4 were frequently identified in LN/ENTD and primary samples of patients with advanced tumors. Therefore, SMAD4 mutations appeared more frequently at the sample level than at the patient level. In addition, we also found that the mutation frequency was higher at the patient level than that in the TCGA data (Cancer Genome Atlas Network, 2012) except CSMD3 gene (Figure 1B). Notably, the mutation frequency of the LZTR1 gene was much higher than TCGA data (Cancer Genome Atlas Network, 2012) (Figure 1B), which deserved further study. Our study also identified higher frequency of SCNAs than TCGA (Cancer Genome Atlas Network, 2012) data, probably owing to the identification of subclonal SCNAs (Figure 1C).
Clonal architecture in colorectal tumors
All the mutations (SNVs and INDELs) were clustered as per their CCF values to understand the clonal architecture and evolutionary history of 62 colorectal tumors. Each colored circle in the phylogenetic tree represented one cluster of the tumor (Figure 2). Phylogenetic trees for 62 tumors and 188 regions together with schematic diagram of 100 tumor cells representing distribution of clusters in each tumor region were shown in Data S1. Driver mutations, driver SCNAs and their clusters were annotated beside the phylogenetic trees (Data S1). Detailed information of cluster numbers for each tumor was listed in Table S4, with a median of 6 clusters per tumor (range, 1 to 13). Our study showed that patients with LCC possessed significantly more cluster numbers than patients with both RCC (median number, 7.5 vs. 6; P = 0.028) and RC (median number, 7.5 vs. 5.5; P = 0.025) (Figure S6), which potentially reflected that patients with LCC were structurally more complex than patients with RCC in evolutionary perspective.
Figure 2.

Phylogenetic trees
Phylogenetic trees for each CRC tumor were shown. The trees were ordered by overall stage (I, Ⅱ, Ⅲ, IV) and position (right-sided colon, left-sided colon and rectum). The cluster number corresponding to the color was displayed in the upper right corner with largest cluster labeled “1.” The lines connecting clusters does not contain any information.
Driver event alterations in CRC evolution
Identifying cancer driver events and their clonality is highly significant to understand the driving force underlying the transformation of a benign tumor to a malignant one. Therefore, driver mutations, driver SCNAs, arm-level SCNAs, and their clonality were analyzed for colorectal tumors (Figure 3). All genes enlisted in the COSMIC Cancer Gene Census (v88) (Forbes et al., 2015) were considered as driver genes. Variants of no less than 3 matches with COSMIC in oncogene or deleterious variants in tumor-suppressor gene (TSG) were classified as driver mutations. The copy number of oncogene >2 × ploidy or copy number of TSG = 0 was classified as driver SCNAs.
Figure 3.

Summary of driver events in CRC evolution
Mutations and SCNAs were shown as frequency in patients indicating whether the events are clonal (blue) or subclonal (red). Only genes that were mutated in at least five patients in total or two patients in right-sided colon/left-sided colon/rectum were shown. For SCNAs, driver SCNAs in at least 20% of the patients were shown, while all the arm-level SCNAs were shown. A driver event (driver mutation, driver SCNA, or arm-level SCNA) was classified as a late event if it appeared more often as subclonal in patients than as clonal, otherwise it was an early event. In the arm-level SCNA part, “G” represented gain, “L” represented loss, and the numbers in parentheses represented the time of occurrence in tumors.
We identified 1373 driver events (405 driver mutations, 707 driver SCNAs, and 261 arm-level SCNAs) among 62 colorectal tumors. Among these events, 605 of 1373 driver events (44%) were subclonal (41% of driver mutations, 40% of driver SCNAs, and 60% of arm-level SCNAs). Significantly lower percentage of clonal driver events were identified in patients with RC than in patients with both RCC (median percentage, 56% vs. 72%; P = 0.031) and LCC (median percentage, 56% vs. 74%; P = 0.047) (Figures S7 and S8). Moreover, ITH index was calculated as the numbers of subclonal driver events divided by the numbers of clonal driver events. In a multivariate logistic regression analysis, patients with RC were associated with high ITH index compared with patients with RCC after adjustment for age, gender, tumor purity, number of sampling regions, tumor size, and stage (odds ratio, 6.04; 95% confidence interval [CI], 1.19 to 37.94; P = 0.037) (see Table S5). Hence, our study showed increased diversity in driver events existed in patients with RC.
In addition, no driver events were found consistently clonal among 62 patients (Figure 3), suggesting high ITH status and evolutionary diversity existed among colorectal tumors. A driver event was classified as a late event if it appeared more often as subclonal in patients than as clonal, otherwise it was an early event. All the driver SCNAs and most of the driver mutations were identified as “early events,” while very few arm-level SCNAs were identified as “early events,” suggesting that the genomic instability process occurred firstly at the driver SCNA level, then at the driver mutations level, and finally at the arm-level SCNA level.
Driver mutations in APC, TP53, and KRAS were mostly identified in all these 62 patients, which were predominantly clonal and identified as “early event,” suggesting their significance and key roles in tumor initiation. Except for driver mutations in APC, TP53, and KRAS, other identified driver mutations were completely different between patients with RCC and LCC, while 11 driver mutations identified were the same in patients with LCC and RC (Figure 3). The difference between RCC and LCC in driver mutations in Wnt-signaling pathway gene AMER1, RTK/RAS pathway gene BRAF, and TGFβ pathway gene ACVR2A was confirmed in the TCGA CRC cohort. In the TCGA data set, driver mutations in AMER1 were identified in 19% of RCC, 3% of LCC, and 5% of RC; driver mutations in BRAF occurred in 9% of RCC, 3% of BRAF, and 2% of RC; driver mutations in ACVR2A were found in 7% of RCC, 2% of LCC, and 0% of RC. The genes of driver SCNAs identified were the same in patients with LCC and RC, whereas only 3 of 24 genes of driver SCNAs (CYSLTR2, FLT3, and FOXO1) were same in patients with RCC and LCC (Figure 3). These huge differences in both driver mutations and driver SCNAs between the patients with RCC and LCC suggested that patients with LCC were evolutionary closer to the patients with RC than patients with RCC.
Conserved evolutionary features in CRC
To understand the constraints and features of CRC evolution, we analyzed conserved patterns of driver events by REVOLVER (Caravagna et al., 2018) (Figure 4). Evolutionary trajectories were clustered by the CCF and cluster information of all the driver events in 62 patients and four clusters (cluster red, blue, green, and purple) were found (Figure 4). Repeated evolutionary trajectories in four clusters were summarized (Figure S9). To understand whether conserved patterns of CRC evolution correlated to distinct clinical phenotypes, clinical and genomic metrics were shown under four clusters (Figure 4).
Figure 4.

Evolutionary subtypes
Evolutionary trajectories were clustered based on CCF value and cluster information of driver mutations, driver SCNAs and arm-level SCNAs. Heat maps showed the most recurrent evolution for the most recurrent driver mutations, driver SCNAs and arm-level SCNAs. Alterations were ordered by their frequencies in CRC tumors. CRC tumors are annotated by the following parameters: ITH index (high: half of the largest ITH index value; low: the other half), TMB (high > median, low ≤ median), SCNA index (high > median, low ≤ median), tumor location, histology, stage, number of regions, tumor size, and age.
We found that the red and blue clusters had relatively fewer driver events than green and purple clusters. There were no specific genomic or clinical features for the tumors in red cluster. The green and purple clusters had similar clinical features, which were enriched in patients with LCC and RC. In a multivariate logistic regression analysis, LCC patients were associated with green and purple clusters compared with RCC patients after adjustment for age, gender, tumor purity, number of sampling regions, tumor size, and stage (odds ratio, 11.65; 95% CI, 1.39 to 268.18; P = 0.049) (Table S6). Taken together, these findings suggested that patients with LCC and RC were functionally more divergent than patients with RCC in evolutionary perspective.
Phylogenetic distance between LN and ENTD
We analyzed 10 stage III patients to understand the phylogenetic distance and evolutionary relationship amongs primary tumor, LN, and ENTD. CRC21, CRC28, CRC43, and CRC48 were identified with both LN and ENTD samples which were sequenced (Figure 5). In CRC21, we identified that the clonal evolution of LN and ENTD was similar, while ENTD appeared evolutionarily later than LN (Data S1). In CRC28, two ENTD samples were clustered together (Figure 5). In CRC43 and CRC48, we identified that the ENTDs were not clustered together with LN and evolved separately (Figure 5 and Data S1). In tumors with more than one LN sequenced (CRC01, CRC11, CRC29, and CRC33), some LNs were clustered together, while some LNs were not (Figure 5). In tumors with two ENTDs sequenced (CRC60), these two ENTDs were far away from each other in the phylogenetic tree (Figure 5). These findings suggested that for one thing, not necessarily all LNs/ENTDs derived at the same time and from the same population. For another, not necessarily all ENTDs derived from a corresponding LN. In short, both LN and ENTD originated from different clones.
Figure 5.

Phylogenetic distance between primary tumor, LN, and ENTD
Heatmap showed the presence (blue) and absence (white) of all the mutations (SNVs and INDELs) among different tumor regions of the patients with lymph node metastasis or ENTD.
Phylogeny reconstruction using maximum parsimony based on mutational presence or absence of all the mutations were shown beside heatmap. Genes with driver mutations were labeled in the phylogenetic trees.
Evolutionary process at the mutational level
Convergent features in CRC
Evidence of convergent mutations in tumor driver genes may shed light on evolutionary selection, which may provide therapeutic targets for treatment. APC, TP53, and KRAS were the most frequently mutated driver genes identified in our study, with a mutation frequency of 80.6% (50/62), 80.6% (50/62), and 51.6% (32/62) respectively (Figure S10). Among these three genes, APC was the most frequent mutated gene in tumor samples. Among these 50 patients with APC mutations, 19 (38%) had 2 mutations, consistent with the two-hit hypothesis of APC genes in CRC tumorigenesis (Rowan et al., 2000) (Figure S11).
Mutation signature
We analyzed mutational processes based on previously published mutational signatures (Alexandrov et al., 2020). We found that the clock-like signature SBS1 was the predominant mutational process for all these 62 patients, with a median percentage of age-related mutations of 23% (Figure S12).
The median percentage of clock-like signature SBS1 for clonal mutations was 28%, whereas it dropped to 19% for subclonal mutations (Figure S12). This finding suggested that except for age, other mutational processes played more important roles in subclonal than clonal mutations in tumors, which accounted for ITH of CRC. Except for clock-like signature SBS1, other main mutational processes were thiopurine-chemotherapy-treatment-related signature SBS87 and defective DNA-mismatch-repair-related signature SBS15.
Evolutionary process at copy number alteration level
Chromosome instability
Previously, we analyzed the length and clonality of SCNAs (Figure 1A), and we then measured the SCNAs frequency pattern in patients with RCC, LCC, and RC. The SCNA frequency pattern in patients with LCC and RC was similar with each other, whereas that of patients with RCC was very different (Figure S13). As shown in Figure Supplement S14, patients with RCC had more 9p gain, 3q gain, and 19p loss and less 20q gain, 18p loss, and 8p loss than both patients with LCC and RC.
Mirrored subclonal allelic imbalance
Recent studies identified parallel evolution of SCNAs in NSCLC and renal cancer through mirrored subclonal allelic imbalance (MSAI) (Jamal-Hanjani et al., 2017; Turajlic et al., 2018). We identified MSAI events in 23 of 62 patients (37%, found in 5 patients with RCC, 6 patients with LCC, and 12 patients with RC) (Data S2). MSAI parallel gain or loss events found in this study were summarized (Figure 6A). Interestingly, patients with RCC had 42% MSAI events, more compared with both patients with LCC (30%) and RC (40%). We also analyzed parallel evolution of driver SCNAs, 5 tumors (4 tumors with parallel amplification and 1 tumor with parallel deletion) were found to have driver SCNAs which overlapped with MSAI events (Figures 6B and 6C). Interestingly, 2 of 5 patients (CRC12 and CRC59) were identified with parallel amplification of FLT3 gene in chromosome 13 (Figure 6C).
Figure 6.

Parallel evolution
(A) Genomic position and size of all mirrored subclonal allelic imbalance (MSAI) parallel gain or loss events found in this study. This included genome-wide copy number gains and losses which were subjected to MSAI events and their occurrence in CRC tumors.
(B) Parallel evolution of driver SCNAs observed in 5 CRC tumors, indicted by the depth ratio and B-allele frequency values of the same chromosome on which the driver SCNAs were located.
(C) Phylogenetic trees that indicated parallel evolution of driver amplifications (Amp) or deletions (Del) (Driver SCNAs) detected through the observation of MSAI (arrows).
Evolution landscape of hypermutated CRC tumors
All 6 (CRC04, CRC05, CRC09, CRC13, CRC15 and CRC17) patients with hypermutated patients were identified with RCC; of these, two patients (CRC09 and CRC13) were with MSS and remaining four patients (CRC04, CRC05, CRC15, CRC17) were with MSI tumors (Figure S14A). All the 6 hypermutated patients had mutations in mismatch-repair genes, or in POLE or POLD gene family (Figure S14A). CRC09 had one missense mutation and one nonsense mutation of POLE. CRC13 had one missense mutation of POLE (Figure S14A). These findings were consistent with the predominant mutational process in these two patients with MSS tumors was POLE exonuclease-domain-mutation-related signatures SBS10a and SBS10b (Figure S14B). Defective-DNA-mismatch-repair-related signature SBS6, SBS15, or SBS26 contributed to the mutational process of 4 patients with MSI tumors (Figure S14B). We also analyzed the evolution landscape of hypermutated tumors in the SCNA level. The absolute SCNAs of hypermutated patients with CRC occurred less compared with nonhypermutated ones (Figures S14C and S15), which suggested that these hypermutated patients with CRC were mainly having low chromosomal instability and mutation-driven tumors. Interestingly, CRC04 had MSAI events in X chromosome (Figure S16).
Discussion
In this present study, we performed high-depth WES and analyzed 206 multiregion tumor samples from 68 patients with CRC. Our result showed that patients with LCC were structurally and functionally more complex and divergent than patients with RCC in terms of evolutionary perspective. Our result showed ENTD were later events in the evolution of the tumor than LN. In addition, all patients with CRC followed the Darwinian pattern of evolution.
Patients with RCC, LCC, and RC: In the light of clonal evolution
Previous studies have shown remarkable differences among RCC, LCC, and RC based on CIMP status, genetic mutations (hypermutation and MSI status), genomic expression profiles, immunological composition, and bacterial population in the tumor microenvironment (Advani et al., 2018; Cheng et al., 2008; Lee et al., 2017; Hu et al., 2018; Imperial et al., 2018; Baek, 2019; O’Connell et al., 2014).
However, almost no research has been conducted till date for understanding the differences between different locations of CRC from evolutionary perspective, which is the key to explore the differences among RCC, LCC, and RC in tumor initiation and progression. As summarized in Table S7, our study demonstrated that ITH and evolution among patients with LCC, RCC, and RC were different in the following aspects: mutations, SCNAs, polygenetic tree, and driver events. First, patients with RC had shown fewer clonal mutations than patients with RCC, indicating higher ITH in patients with RC at the mutational level. Second, the SCNA frequency pattern in patients with RCC was different from that in patients with LCC and RC patients, which addressed the evolutionary difference between them at the SCNA level. Third, the structure of phylogenetic trees in patients with LCC and RC were more complicated and branched than that of patients with RCC. Specifically, patients with LCC were identified with the most complicated structure of the phylogenetic tree, reflected by more cluster numbers. In addition, only patients with LCC and RC were polyclonal in origin. Fourth, patients with LCC and RC were enriched in clusters (green and purple clusters) which had more driver events, indicating that patients with LCC and RC showed more functional diversity in evolution. Moreover, patients with RC were identified with less percentage of clonal driver events than both patients with LCC and RCC, suggested that more functional diversity occurred in the process of evolution of patients with RC.
In conclusion, our data showed that patients with LCC and RC were more divergent and complicated in terms of evolution than patients with RCC, not only structurally but also functionally, which indicated that the evolutionary diversity might play an important role in the initiation and progression of CRC among patients with LCC and RC. This is probably owing to the fact that patients with RCC are more susceptible to genetic (hypermutation and MSI status) and epigenetic instability (CIMP status) in the evolutionary process.
Primary tumor, LN, and ENTD: In evolutionary perspective
To date, no systematic research studies have been conducted to understand the similarities and differences between ENTD and LN. In this study, we found that ENTDs were later events in the evolution of the tumor than LN as per the clonal evolution history in CRC21. LN and ENTD could not be clustered together in the polygenetic tree as per the occurrence of mutations. In CRC21 and CRC28, mutations in driver genes of TSC1, CASP8 or FAT4 were identified in LN samples but were not found in ENTD samples of the same tumor (Figure 5). The biological significance of TSC1, CASP8, and FAT4 in promoting the formation of LN instead of ENTD was still unknown. This question is worthy of further study. Unlike in previous studies (Wei et al., 2017; Hu et al., 2019), different LN or ENTD in the same tumor did not cluster together in all cases, indicating their origin from different clones. In conclusion, ENTD was a distinct entity from LN and evolved later.
Evolution pattern: Darwinian pattern of evolution and neutral evolution
In this present study, we found predominantly Darwinian pattern of evolution (59 of 62 patients) as well as linear evolution (3 of 62 tumors). Previous studies proposed neutral evolution model for CRCs (Sottoriva et al., 2015; Williams et al., 2016; Loeb et al., 2019), while our conclusion was different from them, based on three reasons. First, clonal events of both mutations (SNVs and INDELs) and SCNAs were widespread, with a median percentage of 47% and 43%, respectively. Second, 59% of driver mutations were clonal, whereas only 41% of nondriver mutations were clonal, which indicated the enrichment of clonal driver mutations in course of evolution. Finally, convergent events were present for driver genes in both mutational and SCNA level, especially for genes APC, TP53, and KRAS. Previous studies also showed Darwinian pattern of evolution for the patients with colorectal cancer followed by neutral evolution (Uchi et al., 2016; Saito et al., 2018). In our study, we identified that 28% of subclonal mutations were shared by tumor regions (either branch or trunk mutations), which suggested the importance of branches in phylogenetic trees.
Limitation of the study
First, we recruited a relatively small cohort, 68 patients with CRC including RCC (18), LCC (20), and RC (30). Although, comparative analysis usually considers equal number of patients from different groups but owing to time constraints, the recruited and the studied number of patients with RCC, LCC and RC were not equal. Second, single-cell sequencing technology is usually performed in recent research or studies of ITH and clonal evolution. However, single-cell sequencing technology was not used in our present study. Third, in this present study, we focused on ITH and clonal evolution in primary CRC tumors. Now, we are following up the survival rate and metastasis. We will address these questions in our future studies.
STAR★Methods
Key resources table
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Prof. Santasree Banerjee (santasree.banerjee@genomics.cn)
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate code. The published article contains all data sets generated or analyzed during this study. The sequencing data has been deposited at the CNGB Nucleotide Sequence Archive (CNSA: https://db.cngb.org/cnsa), under accession number CNP0000594.
Experimental model and subject details
Study approval
The study was approved by the Ethics Committee of the Affiliated Hospital of Qingdao University. All the samples were collected after obtaining written informed consent from the patients.
Patient recruitment
The study was approved by the Ethics Committee of the Affiliated Hospital of Qingdao University. All the samples were collected after obtaining written informed consent from the patients. Patients were recruited based on the following criteria. (i) age greater than 18 years and (ii) patients clinically diagnosed with CRC by enteroscopy, imaging, biopsy and followed by surgery, and histopathology performed with the resected tumor tissues. Patients with sufficient tissue were available for the study.
In this present studied and analyzed cohort, the comprehensive and detailed information of patients (gender, age range, tumor location, pTNM, disease stage, histology, tumor size, tumor form, HER2, Ki-67+, MSI status, vascular invasion, perineural invasion, adjuvant therapy before surgery) has given in Table S1.
Sample collection
A pathologist performed macroscopic examination of all surgically resected specimens to guide the multiregion sampling in this study. First, the pathologist performed routine pathological sampling for clinical diagnosis, and then, multiregion sampling was performed by using the remaining samples. At least 2 regions of each tumor, which were at least 3 mm apart, were collected. Areas with significant necrosis, fibrosis, or hemorrhage were avoided to maximize the viability of tumor cells. Normal colorectal mucosa tissues were also sampled from areas remote from the primary tumor (at least 2 cm distant from the tumor edge). Peri-intestinal nodules including lymph nodes present in the resected specimen were sampled. If there was malignancy appearance (the cut section appeared tan-gray and hard), after confirming the malignancy, a portion of the lymph nodes was sampled for diagnostic requirements. The remaining part was taken for this study. Each selected tissue block was split into two for snap freezing and formalin fixing, respectively (mirrored FFPE sample). Fresh samples were placed in a 2-mL cryotube and snap-frozen with immediate immersion into liquid nitrogen before transferred to a -80°C freezer for storage. Peripheral blood was collected and processed into EDTA anticoagulation tube. The tumor tissue samples from 68 patients were sequenced and analyzed after filtering as per the filtering pipeline, schematically presented in the CONSORT diagram (CONSORT flowchart, Figure S1). The workflow summarizing experiments and data analysis in our study was shown in Figure S2.
Sample processing
Approximately 50 mm3 of tumor tissue from each region was used for genomic DNA extraction using the QIAamp DNA Mini Kit (Qiagen, Germany) as per the manufacturer’s instructions. Two milliliter of peripheral blood was used for germline DNA extraction using the QIAamp DNA Blood midi kit (Qiagen, Germany) as per the manufacturer’s instructions. DNA was quantified by the Qubit Fluorometric Quantitation (Thermo Fisher Scientific, USA), and the quality of DNA was assessed by agarose gel electrophoresis.
Pathology diagnoses and review
Pathological diagnoses were established as per the WHO classification and independently reviewed by two pathologists. Clinical details were summarized in Table S1. Hematoxylin-eosin sections of mirrored FFPE samples for each region in every case (387 sections from 70 patients) were evaluated. Only primary tumor regions with more than 30% tumor component and pathological heterogeneity were considered for sequencing. For example, as shown in Figure S17, the pathological conditions of tumor regions TR1, TR2, and TR3 in CRC09 are similar, which are glandular adenocarcinoma. The pathological condition of TR5 is quite different from them, which is mucinous adenocarcinoma. Therefore, we selected tumor regions TR1 and TR5 for sequencing. In addition, pathologist distinguished LN and ENTD by reviewing hematoxylin-eosin sections of their mirrored FFPE samples in this study which were also sent for sequencing.
Method details
Whole-exome library construction and sequencing
Tumor tissues and matched germline tissues were subjected to whole-exome sequencing. Exome capture was performed on 1 μg of genomic DNA. Covaris (LE220) was used to randomly fragmented DNA into 150–250 bp. These fragments were purified and connected through a PE Index Adaptor designed by BGI and then captured by using the the MGIeasy Exome Capture V4 probe set (~ 59 Mb; MGI Tech Co., Ltd, China). All constructed libraries were loaded onto BGISEQ-500 (MGI Tech Co., Ltd, China), and the sequences were generated as 100-bp paired-end reads.
Sequencing reads containing sequencing adapters, more than 10% of unknown bases and low-quality bases (> 50% bases with quality <5) were removed by SOAPnuke (v1.5.6) (Chen et al., 2018). The processed sequencing reads were then aligned to UCSC human reference genome (hg19) using BWA-MEM (v0.7.12) (Li et al., 2009). Picard (v1.137) (https://broadinstitute.github.io/picard/) was used to generate chromosomal coordinate-sorted bam files to remove PCR duplicates. Then, the median sequencing depth of the generated data for the tumor area was reached 391 (range 179-537), and the matched germline tissues were reached 414.5 (range 243-596). We then used the Genomic Analysis Toolkit (GATK v3.8.0) (McKenna et al., 2010) to perform base quality score recalibration and local realignment of the aligned reads to improve alignment accuracy.
Quality control to prevent contamination, interpatient sample swaps, and removal of regions with extremely low mutation occurrence
ContEst (Cibulskis et al., 2011), a GATK module, was used to estimate the cross-individual contamination level. Samples with contamination level more than 1% were deleted (3 samples failed the QC owing to contamination as shown in Figure S1. To avoid sample swaps between patients, we used BAM-matcher (Wang et al., 2016).
The number of mutations in each tumor region was called independently. The median number of mutations across all regions for each tumor was calculated. A region in one tumor was removed if less than 20% of the median mutation count of that tumor was identified in that region.
Somatic mutation detection and filtering
After processed the sequencing data, SAMtools (v1.2) (Li et al., 2009) mpileup was used to locate nonreference locations in tumor and germline samples. Bases with phred scores less than 20 or reads with mapping quality (MAPQ) values less than 20 were deleted. Base-alignment quality computation was disabled with adjust mapping quality coefficient set of 50. Both VarScan 2 (v2.4.3) (Koboldt et al., 2012) and MuTect (v1.1.7) (Cibulskis et al., 2013) were used to call somatic mutations. The somatic variants called by VarScan 2 were filtered, and the minimum coverage of the germline sample was set to 10, the minimum variant frequency was changed to 0.01, and tumor purity was set to 0.5. We further filtered the resulting single-nucleotide variant (SNV) calls for false positives using VarScan-2-associated fpfilter.pl script. We used bam-readcount (v0.8.0) (https://github.com/genome/bam-readcount) to prepare input files for fpfilter and min-var-freq was set to 0.02. All insertions/deletions (INDELs) called in reads that VarScan 2 processSomatic classified as "high confidence" were recorded for further downstream filtering. MuTect was used to detect SNVs using annotation files contained in GATK bundle (v2.8) and variants were filtered as per the filter parameter “PASS.”
Additional filtering was performed to reduce false positive mutation calls. If the variant allele frequency (VAF) is greater than 2%, and both VarScan 2 (with a somatic p-value <= 0.01) and MuTect called the mutation, then a SNV was considered as truly positive. Alternatively, if an SNV was called only in VarScan 2 with a somatic p-value <=0.01, a frequency of 5% was required. In addition, the sequencing depth supporting the variant call in each region required >= 30, and the sequence reads required >= 5. In contrast, the VAF value of the variant in the germline should be <= 1%. We filtered the INDEL using the same parameters as aforementioned, except that reads >= 10 were required to support mutation calls, somatic p-values <= 0.001 and sequencing depth >= 50.
ANNOVAR (Wang et al., 2010) was used to annotate mutations with COSMIC (v88) (Forbes et al., 2015), SIFT (Kumar et al., 2009), PolyPhen-2 (Adzhubei et al., 2013), and MutationTaster (Schwarz et al., 2010) databases. All mutations used in the analysis can be found in Table S2. Mutations were classified as clonal or subclonal using PyClone (v0.13.1) (Roth et al., 2014). The PyClone cancer cell fraction (CCF) value was calculated as described in the subclonal deconstruction section. Mutations with CCF > 0.9 across all regions of a tumor were considered as clonal mutations, otherwise they were considered as subclonal mutations.
Driver mutation identification
All variants were compared with all genes identified and enlisted in the COSMIC Cancer Gene Census (v88) (Forbes et al., 2015). Then, three types of mutations were classified as a driver mutation as per the following criteria. First, if the gene was annotated as TSG (tumor suppressor gene) by COSMIC, and the nonsilent variant was considered deleterious: either loss of function (stop-gain/stop-loss, frameshift deletion/insertion or nonframeshift insertion/deletion) or predicted deleterious in two of these three computational approaches applied – SIFT (Kumar et al., 2009), PolyPhen-2 (Adzhubei et al., 2013), and MutationTaster (Schwarz et al., 2010), then the specific variant would be classified as a driver mutation. Second, if the variant was annotated as oncogene by COSMIC, then we tried to identify exact matches to nonsilent variants in COSMIC. If an exact match was found ≥ 3 times, the variant was categorized as a driver mutation. Third, if the gene was annotated as TSG by COSMIC, and the variant is located at the canonical splice site, then the specific variant would be classified as a driver mutation. Finally, we compared all these three types of driver mutations to the CpG island location file on UCSC Genome Bioinformatics website (http://genome.ucsc.edu). We then deleted all mutations that occurred on the CpG island and finally got all driver mutations.
Copy number analysis
Sequenza (v3.0.0) (Favero et al., 2015) was used to detect the somatic copy number alterations (SCNAs) and evaluate the purity and ploidy of tumor cells as follows. First, we used SAMtools (v1.2) (Li et al., 2009) mpileup to convert the Bam file to Pileup format. Second, paired tumors and normal Pileup files were processed by sequenza-utils to extract the sequencing depth, determine the homozygous and heterozygous positions of variants in normal samples, and calculate the variant alleles and allelic frequencies from tumor samples. The sequenza-utils output was further processed by using Sequenza R package to provide segmented copy number data, cellularity, and estimated ploidy for each sample. All segmented copy number data have been given in Table S3. Heatmap of genome-wide SCNAs is visualized by R package copynumber (v1.24.0) (Nilsen et al., 2012).
The driver gene copy number variations (driver SCNAs) of all genes enlisted in the COSMIC cancer gene census were analyzed as follows. First, if the gene was annotated as oncogene by COSMIC, gene-level amplification was called if gene copy number >2 × ploidy of that sample. Second, if the gene was annotated as TSG by COSMIC, gene-level deletion was called if gene copy number = 0. To determine the ITH status of driver SCNAs, we called driver SCNAs across all regions from each tumor. If at least one region showed an amplified SCNA, we called a gene as clonal amplification if all other regions of this gene showed copy number > ploidy + 1. If at least one region showed a deleted SCNA, we called a gene as clonal deletion if all other regions of this gene showed copy number < ploidy -1. All other driver SCNAs were defined as subclonal amplification or deletion. In 8 polyclonally originated tumors (CRC32, CRC36, CRC42, CRC48, CRC49, CRC51, CRC52, and CRC60) without founder clusters (cluster with CCF > 0.9 across all regions of a tumor), all their driver SCNAs were subclonal. To correlate driver SCNAs with specific mutation clusters of PyClone, we first identified all clusters where >= 50% CCF was present in each tumor region. We then identified all the clusters present in the same regions as a given driver SCNAs. We called a gene as clonally amplified if all the regions of this gene showed copy number >2 × ploidy, while we called a gene as clonally deleted if all the regions of this gene showed copy number = 0. Then, we repeated the association test mentioned previously. If an SCNA still could not be associated with a mutant cluster, it was annotated as a subclone associated with no known cluster (NA cluster).
To determine the ITH status of global SCNA, all parts of the genome were considered independently and divided into the smallest contiguous segments that overlap in all the regions within each tumor. The gains and losses of segment were determined as follows. First, copy number data for each segment were divided by the sample mean ploidy and then converted to log2. Second, gain and loss were defined as log2 (2.5/2) and log2 (1.5/2), respectively. Third, any segment of gain or loss that spanned across all the regions was defined as clonal, and all other segments of SCNA were defined as subclonal. Within each tumor, we summarized the length of the genome that subjected to SCNA in any region (total SCNA), the length of the genome that subjected to clonal SCNA (clonal gain or clonal loss), and the length of the genome that subjected to subclonal SCNA (subclonal gain, subclonal loss or subclonal undetermined). The proportion of subclonal SCNAs were then defined as the percentage of genomes subjected to subclonal SCNA divided by the percentage of genomes subjected to total SCNAs.
Chromosomal arm-level SCNAs were determined if at least one region has shown an increase or decrease of at least 97% in chromosomal arm. To determine the ITH status of chromosome arm gain and loss, we called clonal arm gain or loss if the same chromosomal arm showed at least 75% gain or loss in all the remaining regions. We called subclonal arm gain or loss if at least one of the remaining regions showed less than 75% gain or loss. In 8 polyclonally originated tumors, all their arm-level SCNAs were subclonal. As previously described in the driver SCNA part, we correlated arm-level SCNAs with specific mutation clusters of PyClone in the same way.
Subclonal deconstruction
To estimate whether mutations were clonal or subclonal, and the phylogenetic trees of each tumor, the following formula were used (Jamal-Hanjani et al., 2017; Zhang et al., 2019):
where vaf is the mutated allele frequency of the mutated base; p is the estimated tumor purity; CNt is tumor-locus-specific copy number; CNn is normal-locus-specific copy number, assuming 2 for autosomal chromosomes; CCF is the fraction of tumor cells carrying mutations. Considering that CNmut is the copy number of the chromosome harboring the mutation, the possible CNmut range is from 1 to CNt (integer). We then assigned one of the possible values to CCF: 0.01, 0.02, ..., 1, together with every possible CNmut to find the best fit CCF using maximum likelihood. In detail, for point mutations with alternative reads as “a” and sequencing coverage as “N,” we used Bayesian probability theory and binomial distribution to estimate the probability of a given CCF:
Then, the distribution of CCF was obtained by calculating P (CCF) on 100 uniform grids with CCF values from 0.01 to 1 and dividing by their sum.
Then, we used PyClone (v0.12.9) (Roth et al., 2014) Dirichlet process clustering to cluster all the mutations (SNVs and INDELs). For each mutation, we used the observed mutation count and set the reference count so that vaf equal to half of the CCF value calculated by maximum likelihood previously. We set the major allele copy number to 2, the minor allele copy number to 0, and the purity to 0.5 because they had been modified.
Because the vaf values of INDELs were potentially unreliable, we multiplied each estimated INDEL CCF with a region-specific correction factor, which was calculated by dividing the median mutation CCF of the ubiquitous mutations (mutations presented in all regions) in that region by the median INDEL CCF of the ubiquitous INDELs (INDELs presented in all regions) in that region. We ran PyClone with 10,000 iterations and a burn-in of 1000.
Phylogenetic tree construction
Phylogenetic trees were constructed using the published tool CITUP (v0.1.0) (Malikic et al., 2015). As input, CITUP requires mutation clusters and their mean cancer cell prevalence values which were collected from PyClone. All clusters with at least 5 mutations were used as input to CITUP. Clusters for phylogenetic tree construction were summarized in Table S4. The optimal phylogenetic trees for each patient from CITUP were illustrated using MapScape (v1.8.0) (Smith et al., 2017).
Evolution subtype analysis
Evolutionary subtypes were clustered and visualized by REVOLVER (v0.2.0) (Caravagna et al., 2017). CCF values and cluster information of driver events were processed as previously described, which were used as input to REVOLVER. REVOLVER requires a founder cluster for all the input tumors. Therefore, we artificially defined a founder cluster for 8 polyclonally originated tumors. The ITH index was calculated as the numbers of subclonal driver events divided by the numbers of clonal driver events, and the SCNA index was indicated by the length of total SCNA.
Phylogenetic analysis
Phylogenetic distance between primary tumor, LN, and ENTD was analyzed by using the binary matrix of mutations present or absent in each region of tumors with LN or ENTD. ITH could be overestimated owing to false-negative calls of low-frequency mutations. Therefore, we collected all the mutations from all samples of a given patient and then assigned a “1” in the binary to all the samples with any sequencing reads supporting the corresponding mutation.
Private mutations of each region were discarded from phylogenetic tree building owing to lack of information. Fake outgroups with no mutations were generated for each individual as a root. Phylogenies were constructed using the PHYLIP (v3.697) (Falenstein, 1989) suite of tools. For each tumor, we used seqboot to generate 100 bootstrap replicates by resampling of the mutations with replacement.
Phylogenetic trees were then constructed for each bootstrap replicate by maximum parsimony using the Mix programme in Wagner method. The jumble = 10 option was used and the order of the input samples was randomized 10 times for each bootstrap replicate. Finally, the Consense program was used to build a consensus of all the phylogenetic trees by using the majority rule (extended) option. Phylogenetic trees were redrawn by FigTree (v1.4.4) (Rambaut, 2007) with the length of trunks and branches, proportional to the number of mutations.
Mutation signature analysis
Mutation signatures were estimated by using the DeconstructSigs (v1.8.0) (Rosenthal et al., 2016) package in R. Mutational signature analysis was applied only in the presence of at least 15 mutations. Signatures in COSMIC v3.1 (Alexandrov et al., 2020) were used in mutation signature analysis.
Mirrored sub-clonal allelic imbalance analysis
Single-nucleotide polymorphisms (SNPs) were called by using Platypus (v0.8.1) (Rimmer et al., 2014), and only SNPs with a minimum coverage of 20× were analyzed. The B allele frequency (BAF) of each SNP was calculated as the ratio of reads of reference base to variant. Heterozygous SNPs and BAFs were used as input and mirror subclone allelic imbalances (MSAIs) were analyzed and visualized by RECUR (Jakubek et al., 2019).
Parallel evolution events for driver SCNAs were identified as follows. First, driver SCNAs were identified as described in the "copy number analysis" section. Secondly, we annotated the regions of MSAI events in each tumor to the events of driver SCNAs. If two events coincided with each other, then these driver SCNAs have undergone parallel evolution.
Statistical analysis
All analyses were performed in R statistical environment, version >= 3.5.0. All statistical comparisons of two distributions used the Wilcoxon test (wilcox.test function in R). Multivariate logistic regression was performed with ITH index/evolutionary clusters versus tumor position, with age, gender, tumor purity, number of sampling regions, tumor size, and stage included in the model.
Acknowledgments
We are thankful to the proband and all the family members for participating in our study and we are thankful to the China National GeneBank. The study was supported by the grants from Guangdong Provincial Key Laboratory of Genome Read and Write (No. 2017B030301011), National Natural Science Foundation of China (No. 81802473), Key Research and Development Plan of Shandong Province (No. 2018GSF118206) and "Clinical medicine + X″ project from Medical College of Qingdao University.
Author contributions
Conception and design: Santasree Banerjee, Shan Kuang, Junnian Liu, Yun Lu, Xin Liu; Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): Xianxiang Zhang, Qingyao Wu, Shujian Yang, Jigang Wang, Xiaobin Ji, Peng Han, Yong Li, Xiaofen Tian, Zhiwei Wang; Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): Lei Li, Santasree Banerjee, Shan Kuang, Shui Shun, Li Deng, Yue Zhang; Writing, review, and/or revision of the manuscript: Santasree Banerjee, Shan Kuang, Lei Li, Xianxiang Zhang, Jigang Wang; Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): Huanming Yang, Lars Bolund, Yonglun Luo, Kui Wu, Shida Zhu, Guangyi Fan, Xun Xu; Study supervision: Santasree Banerjee, Shan Kuang, Junnian Liu, Yun Lu, Xin Liu.
Declarations of interests
The authors declare no competing interests.
Published: July 23, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102718.
Contributor Information
Junnian Liu, Email: chris.liu@genomics.cn.
Yun Lu, Email: cloudylucn@126.com.
Xin Liu, Email: liuxin@genomics.cn.
Supporting citations
The following reference appears in the Supplemental Information: International Agency for Research on Cancer, 2019; Li and Durbin, 2009; World Health Organization, 2019.
Supplemental information
References
- Adjuvant therapy for patients with colon and rectal cancer. JAMA. 1990;264:1444–1450. [PubMed] [Google Scholar]
- Advani S.M., Advani P., DeSantis S.M., Brown D., VonVille H.M., Lam M., Loree J.M., Sarshekeh A.M., Bressler J., Lopez D.S. Clinical, pathological, and molecular characteristics of CpG island methylator phenotype in colorectal cancer: a systematic review and meta-analysis. Transl. Oncol. 2018;11:1188–1201. doi: 10.1016/j.tranon.2018.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei I., Jordan D.M., Sunyaev S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013;7:7–20. doi: 10.1002/0471142905.hg0720s76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov L.B., Kim J., Haradhvala N.J., Huang M.N., Ng A.W.T., Wu Y., Boot A., Covington K.R., Gordenin D.A., Bergstrom E.N. The repertoire of mutational signatures in human cancer. Nature. 2020;578:94–101. doi: 10.1038/s41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alizadeh A.A., Aranda V., Bardelli A., Blanpain C., Bock C., Borowski C., Caldas C., Califano A., Doherty M., Elsner M. Toward understanding and exploiting tumor heterogeneity. Nat. Med. 2015;21:846. doi: 10.1038/nm.3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves J.M., Prado-López S., Cameselle-Teijeiro J.M., Posada D. Rapid evolution and biogeographic spread in a colorectal cancer. Nat. Commun. 2019;10:5139. doi: 10.1038/s41467-019-12926-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek S.K. Laterality: immunological differences between right-sided and left-sided colon cancer. Ann. Coloproctol. 2019;35:291–293. doi: 10.3393/ac.2019.11.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caravagna G., Giarratano Y., Ramazzotti D., Tomlinson I., Graham T.A., Sanguinetti G., Sottoriva A. Detecting repeated cancer evolution from multi-region tumor sequencing data. Nat. Methods. 2018;15:707–714. doi: 10.1038/s41592-018-0108-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Chen Y., Shi C., Huang Z., Zhang Y., Li S., Li Y., Ye J., Yu C., Li Z. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience. 2018;7:1–6. doi: 10.1093/gigascience/gix120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y.W., Pincas H., Bacolod M.D., Schemmann G., Giardina S.F., Huang J., Barral S., Idrees K., Khan S.A., Zeng Z. CpG island methylator phenotype associates with low-degree chromosomal abnormalities in colorectal cancer. Clin. Cancer Res. 2008;14:6005–6013. doi: 10.1158/1078-0432.CCR-08-0216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K., McKenna A., Fennell T., Banks E., DePristo M., Getz G. ContEst: estimating cross-contamination of human samples in next-generation sequencing data. Bioinformatics. 2011;27:2601–2602. doi: 10.1093/bioinformatics/btr446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K., Lawrence M.S., Carter S.L., Sivachenko A., Jaffe D., Sougnez C., Gabriel S., Meyerson M., Lander E.S., Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falenstein J. PHYLIP—phylogeny inference packages (version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- Favero F., Joshi T., Marquard A.M., Birkbak N.J., Krzystanek M., Li Q., Szallasi Z., Eklund A.C. Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann. Oncol. 2015;26:64–70. doi: 10.1093/annonc/mdu479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes S.A., Beare D., Gunasekaran P., Leung K., Bindal N., Boutselakis H., Ding M., Bamford S., Cole C., Ward S. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43:D805–D811. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W., Yang Y., Li X., Huang M., Xu F., Ge W., Zhang S., Zheng S. Multi-omics approach reveals distinct differences in left-and right-sided Colon Cancer. Mol. Cancer Res. 2018;16:476–485. doi: 10.1158/1541-7786.MCR-17-0483. [DOI] [PubMed] [Google Scholar]
- Hu Z., Ding J., Ma Z., Sun R., Seoane J.A., Shaffer J.S., Suarez C.J., Berghoff A.S., Cremolini C., Falcone A. Quantitative evidence for early metastatic seeding in colorectal cancer. Nat. Genet. 2019;51:1113–1122. doi: 10.1038/s41588-019-0423-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imperial R., Ahmed Z., Toor O.M., Erdoğan C., Khaliq A., Case P., Case J., Kennedy K., Cummings L.S., Melton N. Comparative proteogenomic analysis of right-sided colon cancer, left-sided colon cancer and rectal cancer reveals distinct mutational profiles. Mol. Cancer. 2018;17:177. doi: 10.1186/s12943-018-0923-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Agency for Research on Cancer Cancer today. 2019. https://gco.iarc.fr/today/
- Itatani Y., Kawada K., Fujishita T., Kakizaki F., Hirai H., Matsumoto T., Iwamoto M., Inamoto S., Hatano E., Hasegawa S. Loss of SMAD4 from colorectal cancer cells promotes CCL15 expression to recruit CCR1+ myeloid cells and facilitate liver metastasis. Gastroenterology. 2013;145:1064–1075. e1011. doi: 10.1053/j.gastro.2013.07.033. [DOI] [PubMed] [Google Scholar]
- Jakubek Y.A., San Lucas F.A., Scheet P. Directional allelic imbalance profiling and visualization from multi-sample data with RECUR. Bioinformatics. 2019;35:2300–2302. doi: 10.1093/bioinformatics/bty885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamal-Hanjani M., Wilson G.A., McGranahan N., Birkbak N.J., Watkins T.B., Veeriah S., Shafi S., Johnson D.H., Mitter R., Rosenthal R. Tracking the evolution of non–small-cell lung cancer. N. Engl. J. Med. 2017;376:2109–2121. doi: 10.1056/NEJMoa1616288. [DOI] [PubMed] [Google Scholar]
- Koboldt D.C., Zhang Q., Larson D.E., Shen D., McLellan M.D., Lin L., Miller C.A., Mardis E.R., Ding L., Wilson R.K. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Lee M.S., Menter D.G., Kopetz S. Right versus left colon cancer biology: integrating the consensus molecular subtypes. J. Natl. Compr. Canc. Netw. 2017;15:411–419. doi: 10.6004/jnccn.2017.0038. [DOI] [PubMed] [Google Scholar]
- Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb L.A., Kohrn B.F., Loubet-Senear K.J., Dunn Y.J., Ahn E.H., O’Sullivan J.N., Salk J.J., Bronner M.P., Beckman R.A. Extensive subclonal mutational diversity in human colorectal cancer and its significance. Proc. Natl. Acad. Sci. U S A. 2019;116:26863–26872. doi: 10.1073/pnas.1910301116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord A.C., D'Souza N., Pucher P.H., Moran B.J., Abulafi A.M., Wotherspoon A., Rasheed S., Brown G. Significance of extranodal tumour deposits in colorectal cancer: a systematic review and meta-analysis. Eur. J. Cancer. 2017;82:92–102. doi: 10.1016/j.ejca.2017.05.027. [DOI] [PubMed] [Google Scholar]
- Loupakis F., Yang D., Yau L., Feng S., Cremolini C., Zhang W., Maus M.K., Antoniotti C., Langer C., Scherer S.J. Primary tumor location as a prognostic factor in metastatic colorectal cancer. J. Natl. Cancer Inst. 2015;107:dju427. doi: 10.1093/jnci/dju427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malikic S., McPherson A.W., Donmez N., Sahinalp C.S. Clonality inference in multiple tumor samples using phylogeny. Bioinformatics. 2015;31:1349–1356. doi: 10.1093/bioinformatics/btv003. [DOI] [PubMed] [Google Scholar]
- McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missiaglia E., Jacobs B., D'ario G., Di Narzo A., Soneson C., Budinska E., Popovici V., Vecchione L., Gerster S., Yan P. Distal and proximal colon cancers differ in terms of molecular, pathological, and clinical features. Ann. Oncol. 2014;25:1995–2001. doi: 10.1093/annonc/mdu275. [DOI] [PubMed] [Google Scholar]
- Nagtegaal I.D., Knijn N., Hugen N., Marshall H.C., Sugihara K., Tot T., Ueno H., Quirke P. Tumor deposits in colorectal cancer: improving the value of modern staging-a systematic review and meta-analysis. J. Clin. Oncol. 2017;35:1119–1127. doi: 10.1200/JCO.2016.68.9091. [DOI] [PubMed] [Google Scholar]
- Nilsen G., Liestøl K., Van Loo P., Moen Vollan H.K., Eide M.B., Rueda O.M., Chin S.F., Russell R., Baumbusch L.O., Caldas C. Copynumber: Efficient algorithms for single- and multi-track copy number segmentation. BMC. Genomics. 2012;13:591. doi: 10.1186/1471-2164-13-591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell J.B., Maggard M.A., Ko C.Y. Colon cancer survival rates with the new American Joint Committee on Cancer sixth edition staging. J. Natl. Cancer Inst. 2014;96:1420–1425. doi: 10.1093/jnci/djh275. [DOI] [PubMed] [Google Scholar]
- Petrelli F., Tomasello G., Borgonovo K., Ghidini M., Turati L., Dallera P., Passalacqua R., Sgroi G., Barni S. Prognostic survival associated with left-sided vs right-sided colon cancer: a systematic review and meta-analysis. JAMA. Oncol. 2017;3:211–219. doi: 10.1001/jamaoncol.2016.4227. [DOI] [PubMed] [Google Scholar]
- Rambaut A. 2007. FigTree, a Graphical Viewer of Phylogenetic Trees.http://tree.bio.ed.ac.uk/software/figtree/ [cited 2015 Jul 27] [Google Scholar]
- Rimmer A., Phan H., Mathieson I., Iqbal Z., Twigg S.R.F., Wilkie A.O.M., McVean G., Lunter G. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat. Genet. 2014;46:912–918. doi: 10.1038/ng.3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roerink S.F., Sasaki N., Lee-Six H., Young M.D., Alexandrov L.B., Behjati S., Mitchell T.J., Grossmann S., Lightfoot H., Egan D.A. Intra-tumour diversification in colorectal cancer at the single-cell level. Nature. 2018;556:457–462. doi: 10.1038/s41586-018-0024-3. [DOI] [PubMed] [Google Scholar]
- Rosenthal R., McGranahan N., Herrero J., Taylor B.S., Swanton C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17:31. doi: 10.1186/s13059-016-0893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth A., Khattra J., Yap D., Wan A., Laks E., Biele J., Ha G., Aparicio S., Bouchard-Côté A., Shah S.P. PyClone: statistical inference of clonal population structure in cancer. Nat. Methods. 2014;11:396–398. doi: 10.1038/nmeth.2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan A., Lamlum H., Ilyas M., Wheeler J., Straub J., Papadopoulou A., Bicknell D., Bodmer W., Tomlinson I. APC mutations in sporadic colorectal tumors: a mutational “hotspot” and interdependence of the “two hits”. Proc. Natl. Acad. Sci. U S A. 2000;97:3352–3357. doi: 10.1073/pnas.97.7.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T., Niida A., Uchi R., Hirata H., Komatsu H., Sakimura S., Hayashi S., Nambara S., Kuroda Y., Ito S. A temporal shift of the evolutionary principle shaping intratumor heterogeneity in colorectal cancer. Nat. Commun. 2018;9:2884. doi: 10.1038/s41467-018-05226-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz J.M., Rödelsperger C., Schuelke M., Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- Smith M.A., Nielsen C.B., Chan F.C., McPherson A., Roth A., Farahani H., Machev D., Steif A., Shah S.P. E-scape: interactive visualization of single-cell phylogenetics and cancer evolution. Nat. Methods. 2017;14:549–550. doi: 10.1038/nmeth.4303. [DOI] [PubMed] [Google Scholar]
- Sottoriva A., Kang H., Ma Z., Graham T.A., Salomon M.P., Zhao J., Marjoram P., Siegmund K., Press M.F., Shibata D. A Big Bang model of human colorectal tumor growth. Nat. Genet. 2015;47:209–216. doi: 10.1038/ng.3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turajlic S., Xu H., Litchfield K., Rowan A., Chambers T., Lopez J.I., Nicol D., O’Brien T., Larkin J., Horswell S. Tracking cancer evolution reveals constrained routes to metastases: TRACERx Renal. Cell. 2018;173:581–594. doi: 10.1016/j.cell.2018.03.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner N.C., Reis-Filho J.S. Genetic heterogeneity and cancer drug resistance. Lancet Oncol. 2012;13:e178–e185. doi: 10.1016/S1470-2045(11)70335-7. [DOI] [PubMed] [Google Scholar]
- Uchi R., Takahashi Y., Niida A., Shimamura T., Hirata H., Sugimachi K., Sawada G., Iwaya T., Kurashige J., Shinden Y. Integrated multiregional analysis proposing a new model of colorectal cancer evolution. PLoS. Genetics. 2016;12:e1005778. doi: 10.1371/journal.pgen.1005778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno H., Mochizuki H., Akagi Y., Kusumi T., Yamada K., Ikegami M., Kawachi H., Kameoka S., Ohkura Y., Masaki T. Optimal colorectal cancer staging criteria in TNM classification. J. Clin. Oncol. 2012;30:1519–1526. doi: 10.1200/JCO.2011.39.4692. [DOI] [PubMed] [Google Scholar]
- Voorneveld P.W., Kodach L.L., Jacobs R.J., Liv N., Zonnevylle A.C., Hoogenboom J.P., Biemond I., Verspaget H.W., Hommes D.W., De Rooij K. Loss of SMAD4 alters BMP signaling to promote colorectal cancer cell metastasis via activation of Rho and ROCK. Gastroenterology. 2014;147:196–208. e113. doi: 10.1053/j.gastro.2014.03.052. [DOI] [PubMed] [Google Scholar]
- Waddell N., Pajic M., Patch A.M., Chang D.K., Kassahn K.S., Bailey P., Johns A.L., Miller D., Nones K., Quek K. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501. doi: 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P.P., Parker W.T., Branford S., Schreiber A.W. BAM-matcher: a tool for rapid NGS sample matching. Bioinformatics. 2016;32:2699–2701. doi: 10.1093/bioinformatics/btw239. [DOI] [PubMed] [Google Scholar]
- Wei Q., Ye Z., Zhong X., Li L., Wang C., Myers R., Palazzo J., Fortuna D., Yan A., Waldman S. Multiregion whole-exome sequencing of matched primary and metastatic tumors revealed genomic heterogeneity and suggested polyclonal seeding in colorectal cancer metastasis. Ann. Oncol. 2017;28:2135–2141. doi: 10.1093/annonc/mdx278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiser M.R. AJCC 8th edition: colorectal cancer. Ann. Surg. Oncol. 2018;25:1454–1455. doi: 10.1245/s10434-018-6462-1. [DOI] [PubMed] [Google Scholar]
- Williams M.J., Werner B., Barnes C.P., Graham T.A., Sottoriva A. Identification of neutral tumor evolution across cancer types. Nat. Genet. 2016;48:238–244. doi: 10.1038/ng.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization Cancer. 2019. https://www.who.int/news-room/fact-sheets/detail/cancer
- Zhang C., Zhang L., Xu T., Xue R., Yu L., Zhu Y., Wu Y., Zhang Q., Li D., Shen S. Mapping the spreading routes of lymphatic metastases in human colorectal cancer. Nat. Commun. 2020;11:1993. doi: 10.1038/s41467-020-15886-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Liao J., Zhang X., Zhao E., Liang X., Luo S., Shi J., Yu F., Xu J., Shen W. Sex difference of mutation clonality in diffuse glioma evolution. Neuro. Oncol. 2019;21:201–213. doi: 10.1093/neuonc/noy154. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate code. The published article contains all data sets generated or analyzed during this study. The sequencing data has been deposited at the CNGB Nucleotide Sequence Archive (CNSA: https://db.cngb.org/cnsa), under accession number CNP0000594.