

Abstract
Nuclear envelope proteins play an important role in regulating nuclear size and structure in cancer. Altered expression of nuclear lamins are found in many cancers and its expression is correlated with better clinical outcomes. The nucleus is the largest organelle in the cell with a diameter between 10 and 20 μm. Nuclear size significantly impacts cell migration. Nuclear structural changes are predicted to impact cancer metastasis by regulating cancer cell migration. Here we show emerin regulates nuclear structure in invasive breast cancer cells to impact cancer metastasis. Invasive breast cancer cells had 40–50% less emerin than control cells, which resulted in decreased nuclear size. Overexpression of GFP-emerin in invasive breast cancer cells rescued nuclear size and inhibited migration through 3.0 and 8.0 μm pores. Mutational analysis showed emerin binding to nucleoskeletal proteins was important for its regulation of nuclear structure, migration, and invasion. Importantly, emerin expression inhibited lung metastasis by 91% in orthotopic mouse models of breast cancer. Emerin nucleoskeleton-binding mutants failed to inhibit metastasis. These results support a model whereby emerin binding to the nucleoskeleton regulates nuclear structure to impact metastasis. In this model, emerin plays a central role in metastatic transformation, since decreased emerin expression during transformation causes the nuclear structural defects required for increased cell migration, intravasation and extravasation.
Keywords: emerin, breast cancer, nuclear envelope, metastasis, nucleoskeleton
Introduction
The ability for cells to move within their environment is crucial for numerous physiological and pathological processes. During development, cell migration contributes to shaping of the growing embryo and forming nascent tissues [1]. In mature organisms, immune cells, such as neutrophils, are mobilized from the blood to enter sites of infection [1]. Migration of epithelial cells and fibroblasts is vital for proper wound healing and tissue repair [2]. Cancer cell migration is required for metastasis, which is responsible for approximately 90% of all cancer deaths [3]. During metastasis, cancer cells leave the primary tumor by both pushing and pulling their way through surrounding tissues to enter and exit blood vessels and colonize distant sites [1, 4, 5].
Altered nuclear structure remains a hallmark for cancer diagnosis [6]. Histologic features such as nuclear size, shape, number of nucleoli, and chromatin texture are considered clinically relevant, however the functional significance of these alterations in the context of cancer progression remains unclear [6]. Both smaller and larger nuclei have been implicated in increased invasiveness during carcinogenesis, but one thing all of these cells share during metastatic transformation is an increase in nuclear deformability regardless of size [6, 7]. The cytoplasm is very flexible and can undergo large deformations [6]. The cytoskeleton can also actively remodel, allowing the cell to penetrate openings as small as 1 μm [8]. The cell nucleus, on the other hand, is two- to ten-times stiffer than the surrounding cytoplasm and occupies about 10% of the cellular volume, with a typical diameter of 10–20 μm [9]. The nucleus is larger than many of the openings encountered in the extracellular environment. Thus, substantial nuclear deformations [10] are required for cells to move through these small spaces.
Nuclear deformability is affected by several factors, including nuclear envelope protein expression and chromatin structure [4, 11, 12]. The nuclear envelope is comprised of two lipid bilayers, the outer nuclear membrane and the inner nuclear membrane (INM). Underlying the INM is a network of type-V intermediate filament proteins called lamins. This network is composed of overlapping A- and B-type lamin filaments that form along the INM, which along with the integral INM proteins form the nuclear lamina [13]. The relative expression of A- and B-type lamins differentially effects the shape and mechanical properties of the nucleus [14]. Cells with low levels of A-type lamins have irregularly shaped nuclei, while those with high levels of lamin A/C have rounded nuclei [4, 15]. Nuclei of cells with low levels of B-type lamins form blebs, while cells with high levels of lamin B form invaginations [15].
There are more than 130 predicted INM proteins, many of which show cell type-specific expression patterns [16–18]. The importance of the nuclear lamina in providing structural support to the nucleus and controlling nuclear size is well established [19, 20]. Additionally, several studies have reported altered distribution, expression, or both, of INM proteins in various human cancers including ovarian cancer, breast cancer, basal cell carcinoma, colorectal carcinoma, hepatocellular carcinoma, osteosarcoma, and prostate and thyroid cancers [21–27]. Although the impact of lamins in cancer progression and metastasis is becoming clearer [9, 15, 19, 28–30], how INM proteins impact cancer progression and metastasis remains unclear.
Emerin is a ubiquitously expressed integral INM protein [31, 32] with roles in nuclear structure, chromatin architecture, genomic organization, cell signaling, and gene expression [33–37]. Recent evidence supports a role for emerin in cancer [30]. Emerin expression is decreased 7.5-fold (EMBL-EBI; GSE52914) in triple-negative breast cancer [38–40]. Breast cancer prognosis is negatively correlated with emerin expression, as 5-yr and 10-yr survival rates for emerin low-expressors was 75% and 47%, respectively, compared to emerin high-expressors, which were 88% and 63%, respectively (Human Protein Atlas analysis of data from the Cancer Genome Atlas Network). The Cancer Genome Atlas Network also reports at least 102 EMD mutations linked to cancer, many of which are expected to affect emerin expression and protein folding.
Emerin is important for regulating nuclear structure through its interactions with lamins, nuclear actin, the LINC complex, and chromatin [26, 41, 42]. How these functional interactions affect nuclear structure during cancer transformation to impact invasive cancer cell migration has not been investigated. Here we report the functional interactions of emerin with the nucleoskeleton is important for the nuclear structural changes occurring during metastatic transformation and establish a link between increased emerin function and metastasis inhibition.
Materials and Methods
Cell culture
MDA-MB-231 (ATCC cat#: HTB-26, RRID:CVCL_0062), MDA-157 (ATCC cat#: HTB-24, RRID:CVCL_0618), MCF10A (ATCC cat#: CRL-10317, RRID:CVCL_0598), and normal primary mammary epithelial cells (cat#: PCS-600–010) were purchased from American Type Culture Collection (ATCC, Manassas, VA). The MDA-231, and MDA-157 cells were grown in Dulbecco minimum essential medium (DMEM) with high glucose and GlutaMAX supplement (Gibco/LifeTechnologies, Carlsbad, CA, cat#: 10566–016) with 10% fetal bovine serum (FBS, Gibco/LifeTechnologies, cat#: 16000–044) and 1% Penicillin/Streptomycin (Gibco/LifeTechnologies, cat#1: 5140–122). MCF10A cells were grown in Ham’s F-12 media (Lonza, Basel, Switzerland, cat#: 12–615F) with 5% horse serum (Gibco/LifeTechnologies, cat#: 16050–130), 20 ng/ml EGF (LifeTechnologies, cat#: PHG0313), 0.5 mg/mL hydrocortisone (ThermoFisher Scientific, Waltham, MA, cat#: AC35245–0010), 100 ng/mL cholera toxin (MilliporeSigma, Burlington, MA, cat#: 227036), 10 μg/mL Insulin (Sigma-Aldrich, St. Louis, MO, cat#: 10516), and 1% Pennicillin/Streptomycin. Primary mammary epithelial cells were grown in mammary epithelial cell basal medium (ATCC, cat#: PCS-600–030) with all the components from the mammary epithelial cell growth kit (ATCC, cat#: PCS-600–040). All cell lines were maintained at 37°C in 5% CO2. Cell lines are authenticated yearly using ATCC short tandem repeat (STR) profiling through their Cell Line Authentication Service. The latest cell line authentication was done in January 2021. Mycoplasma testing is done regularly by PCR using the Universal Mycoplasma Detection kit (ATCC, cat# 30–1012K).
Proliferation and Cell Cycle Analysis
Cell cycle analysis was done using a Guava Muse Cell Analyzer (Luminex, Austin, TX, cat#: 0500–3115) using the Muse Cell Cycle kit (Luminex, cat#: MCH100106) and Muse Annexin V & Dead Cell kit (Luminex, cat#: MCH100105), per manufacturer instructions. Proliferation was analyzed by plating 1×105 cells of each cell line in a 12-well plate. Cells were trypsinized at 24, 48, 72, 96, and 120 hours after plating and counted using a Countess II FL Automated Cell Counter (ThermoFisher Scientific).
Trans-well migration and invasion assays
Falcon (Santa Clara, CA) trans-well inserts with 3.0 μm (cat#: 353096), or 8.0 μm (cat#: 353097) pores were used for the cell migration assays. For the cell invasion assays, 8.0 μm pores coated with Matrigel (cat #354483) were placed in 24-well plates (Falcon, cat#: 353504). Cells were plated at 2×105 cells/insert in serum-free media. Complete growth media was placed in the well beneath the insert. Cells were allowed to migrate for 24 hours. Cells that did not migrate remained on top of the porous membrane. These cells were wiped off using PBS and a sterile cotton swab. The cells on the bottom of the membrane were fixed for 15 minutes using 3.7% formaldehyde in PBS, washed three times with PBS, and permeabilized with 0.5% Triton X-100 in PBS for 20 minutes. Membranes were cut away from the inserts and placed cell-side down on a drop of Prolong Diamond Antifade with DAPI (LifeTechnologies, cat#: P36971) on a slide (VWR, Radnor, PA, cat#: 48311–703). A coverslip (VWR, cat#: 48366–205) was placed on top and allowed to solidify in the dark overnight.
Western Blot
Tissue protein lysates were obtained from Origene (Rockville, MD). Samples used were CP565563, CP531533, CP544778, CP552484, CP627491, CP529617, CP531866, CP537436, CP537799, CP541360, CP542781, CP603959, CP607640, CP626552, CP8115065. Samples were suspended in NuPAGE LDS buffer (LifeTechnologies, cat#: NP0008) containing reducing agent (LifeTechnologies, cat#: NP0009). MDA-231 cells stably expressing GFP-emerin, GFP-M45A, GFP-M151, GFP-S54F, and GFP-M196 were collected and resuspended in NuPAGE LDS buffer containing reducing agent. Samples were resolved on by SDS-PAGE (LifeTechnolgies, cat#: NP323BOX) and transferred to nitrocellulose membranes (GE Healthcare, Chicago, IL cat#: 10600004). Membranes were blocked using 3% BSA (Sigma-Aldrich, cat#: A7906) in PBS + 0.1% Tween-20 (PBST) for 2 hours. Appropriate primary and secondary antibodies were diluted in 3% BSA in PBST and incubated on the membrane for two hours each. Signal intensity was measured using the ChemiDoc Imaging system (Bio-Rad, Hercules, CA). Densitometric assessment of bands was done using the Image Lab software (Bio-Rad, Inc.). The content of each protein was normalized to the proper control band (either actin or gamma-tubulin). Patient samples were normalized to the CP565563 control patient without cancer. MDA-231 cells expressing GFP-emerin or each GFP-emerin mutant were normalized to MDA-231 cells. Primary antibodies used for blotting included rabbit emerin polyclonal antibody (1:5,000 dilution; Proteintech, Rosemont, IL, cat#: 10351-I-AP), mouse gamma-tubulin monoclonal antibody (1:10,000 dilution; Sigma-Aldrich, cat#: T6557, RRID:AB_532292), mouse actin monoclonal antibody (1:2,500 dilution; Sigma-Aldrich, cat#: A5441, RRID:AB_476744), rabbit H3K9me2 polyclonal antibody (1:5,000 dilution; Active Motif, cat#: 39239, RRID:AB_2793199), rabbit H3K9me3 polyclonal antibody (1:10,000 dilution; Abcam, Cambridge, UK, cat#: ab8898, RRID:AB_306848), and rabbit H3K27me3 polyclonal antibody (1:10,000 dilution; MilliporeSigma, cat#: 07–449, RRID:AB_310624). Secondary antibodies included horseradish peroxidase-conjugated rabbit (1:10,000 dilution; Invitrogen, Carlsbad, CA, cat#: 31462) and mouse (1:10,000 dilution; Invitrogen, cat#: 31432) antibodies.
qPCR
Total RNA from tumors and normal breast tissue controls were purchased from Origene Technologies, Inc. The RNAs purchased were matched to protein samples. 1-step qPCR was performed on 10 ng of total RNA from each sample using the Verso SYBR Green 1-Step RT-qPCR ROX Mix (ThermoFisher Scientific, Inc.) per manufacturer instructions, except that 500 nm of each primer were added, using the ABI StepOne Plus Real Time PCR Machine (ThermoFisher Scientific, Inc.). Relative expression was determined by comparing each experimental to the normal control breast RNA samples. Primer sequences used were purchased from Integrated DNA Technologies and are: GAPDH, 5’-ACATCGCTCAGACACCATG-3’ and 5’-TGTAGTTGAGGTCAATGAAGGG-3’; and EMD, 5’-AGCTCTCTTCATAGTAGTCGTCA and 5’-CTCCTCTTATAGCTTCTCTGACTTG.
Orthotopic Mouse Models
This study was conducted according to the guidelines of the National Institutes of Health (NIH) and the University of the Sciences’ Institutional Animal Care and Use Committee (IACUC). 6–8-week-old athymic NCR-nu/nu female nude mice were obtained from Charles River (Wilmington MA). Mice were injected intraperitoneally with 0.1–0.6 mg/kg buprenorphine 1 hr prior to surgery. Mice were anesthetized with 2–5% isoflurane by inhalation. Using the orthotopic injection technique as previously described [43], a ventral incision was performed to expose the fourth inguinal right mammary gland. MDA-231 cells (5 × 106) stably expressing infrared fluorescent protein 713 (iRFP713) and GFP-emerin or each GFP-emerin mutant were injected into the right mammary fat pad using a sterile syringe with a 26-gauge needle (n=14–25 for each group). The incision was subsequently closed with 4–5 surgical staples and mice recovered from surgery on a 37°C heated pad and were closely monitored post-surgery for the entire protocol.
In vivo Imaging
Mice were anesthetized with 2–5% isoflurane and imaged with the LICOR Pearl Trilogy small animal system (LICOR, Lincoln, NE). Images were analyzed using the LICOR Image Studio software auto-selection tool with 5.5 standard deviations. The background reference circle was standardized for all image analyses and was placed on the contralateral mammary fat pad that was not injected. Eight weeks after fat pad injections, mice were euthanized by CO2 inhalation and tumors were harvested, weighed, measured, and imaged. The tumors collected were cut in half and each half was either frozen in liquid nitrogen or fixed in 4% paraformaldehyde (PFA) for western blotting and immunohistochemical analyses, respectively. Lungs were inflated with PFA, excised and imaged on the LICOR Pearl Trilogy small animal system. Lungs were preserved in PFA for immunohistochemical analyses.
Transduction
Wells were coated with poly-L-lysine (Advanced Biomatrix, San Diego, CA, cat#: 5048) prior to seeding cells. Cells were seeded at confluency and pretreated with 10 μg/mL polybrene (Vector Builder). Cells were transduced using a multiplicity of infection of 100 with LV-iRFP713-P2A-Puro (Imanis Life Sciences, Rochester, MN, cat#: LV032-L). Media was changed four, 24, and 48 hours after treatment with the lentivirus. Selection with 4 ng/mL of puromycin dihydrochloride (Corning, Corning, NY, cat#: 61–385-RA) started 72 hours after transduction.
Transfection
Wells were coated with poly-L-lysine prior to seeding cells. MDA-231 or MCF10A cells were plated at 70% confluency. The Lipofectamine3000 (Invitrogen, cat#: L3000–015) protocol was used to transfect cells with GFP-emerin or each GFP-emerin mutant plasmid. Media was changed 24 hours after transfection. Selection with 3.6 mg/mL of G418 (VWR, cat#: J847) was started 72 hours after transfection.
Electroporation
The Neon Transfection System (Invitrogen, cat#: MPK1096) kit was used to electroporate cells. MDA-231, MDA-157, or MCF10A cells (5 × 106 cells/ml) were resuspended in the 100 μl tip. Cells were electroporated using protocols per manufacturer instructions. The MDA-231 and MDA-157 parameters used were 1350V, 20 ms, 2 pulses. The MCF10A parameters used were 1250V, 20 ms, 2 pulses. Media was changed 24 hours after electroporation. Selection with 3.6 mg/ml of G418 started 72 hours after electroporation.
Immunofluorescence and Nuclear Area and Nuclear Volume Analysis
Invasive breast cancer cells lines containing GFP-emerin or each GFP-emerin mutant were plated on coverslips at equal densities. Cells were rinsed with PBS, fixed with 3.7% formaldehyde in PBS, washed three times with PBS, treated with wheat germ agglutinin 594 conjugate (ThermoFisher Scientific, cat#: W11262) at 5.0 μg/mL for 10 minutes, and permeabilized with a 0.5% Triton X-100 in PBS for 20 minutes. Cells were rinsed three times with PBS and mounted with Prolong Diamond Antifade with DAPI (LifeTechnologies, cat#: P36971) and visualized using the DAPI fluorescent light cube (ThermoFisher Scientific, EVOS, cat#: amep4650), Texas Red fluorescent light cube (ThermoFisher Scientific, EVOS, cat#: amep4655), and GFP fluorescent light cube (ThermoFisher Scientific, EVOS, cat#: amep4651) on the EVOS FL Auto using a 40X objective. Nuclear area was analyzed with the NII_Plugin on Image J (ImageJ, RRID:SCR_003070) using these DAPI images [44]. Nuclei intensity segmentation threshold was standardized at 30 pixels and a minimum nuclear area was set at 200 pixels for all images. Images were manually checked to ensure one nucleus was selected in each box and debris or apoptosing nuclei were avoided. Cytoplasmic area was analyzed with the NII_Plugin on Image J using the Texas Red images. The intensity segmentation threshold was standardized at 30 pixels and a minimum area was set at 300 pixels for all images. The cytoplasmic and nuclear area was converted from pixels to microns prior to analysis. Volume analysis was performed using 60X images from a Nikon Ti2-E confocal microscope using the DAPI and GFP lasers (Nikon, Tokyo, Japan). The NIS Elements AR system was used to measure volume.
Results
To examine if emerin expression is correlated with breast cancer invasiveness, we measured the protein expression levels of emerin in two invasive breast cancer cell lines. MDA-231 and MDA-157 cells had 1.5- to 1.8-fold less emerin protein expression compared to primary mammary epithelial cells and MCF10A cells (Figure 1A,B). MDA-231 (109.5 μm2 ± 2.9 μm2) and MDA-157 (115.6 μm2 ± 2.5 μm2) nuclei each had a 1.6- to 1.7-fold decrease in nuclear area when compared to the MCF10A nuclei (183.5 μm2 ± 5.9 μm2 Figure 1A,C). The DAPI images of cells plated at the same density highlight the extensive nuclear deformations seen in the invasive breast cancer cell lines when compared to the size, regularity, and circularity of the MCF10A nuclei (Figure 1D, Supplemental Figure 1A). The circularity of the nuclei was analyzed with 1.0 being a perfect, uniform circle. The MDA-231 (0.89 ± 0.08) and MDA-157 (0.87 ± 0.07) cells had more irregularly shaped nuclei when compared to the MCF10A cells (0.96 ± 0.06; Figure 1E). Similar results were seen in emerin-null myogenic progenitor cells (Supplemental figure 1B,C).
Figure 1: Invasive breast cancer cells have decreased protein expression and smaller nuclear area.
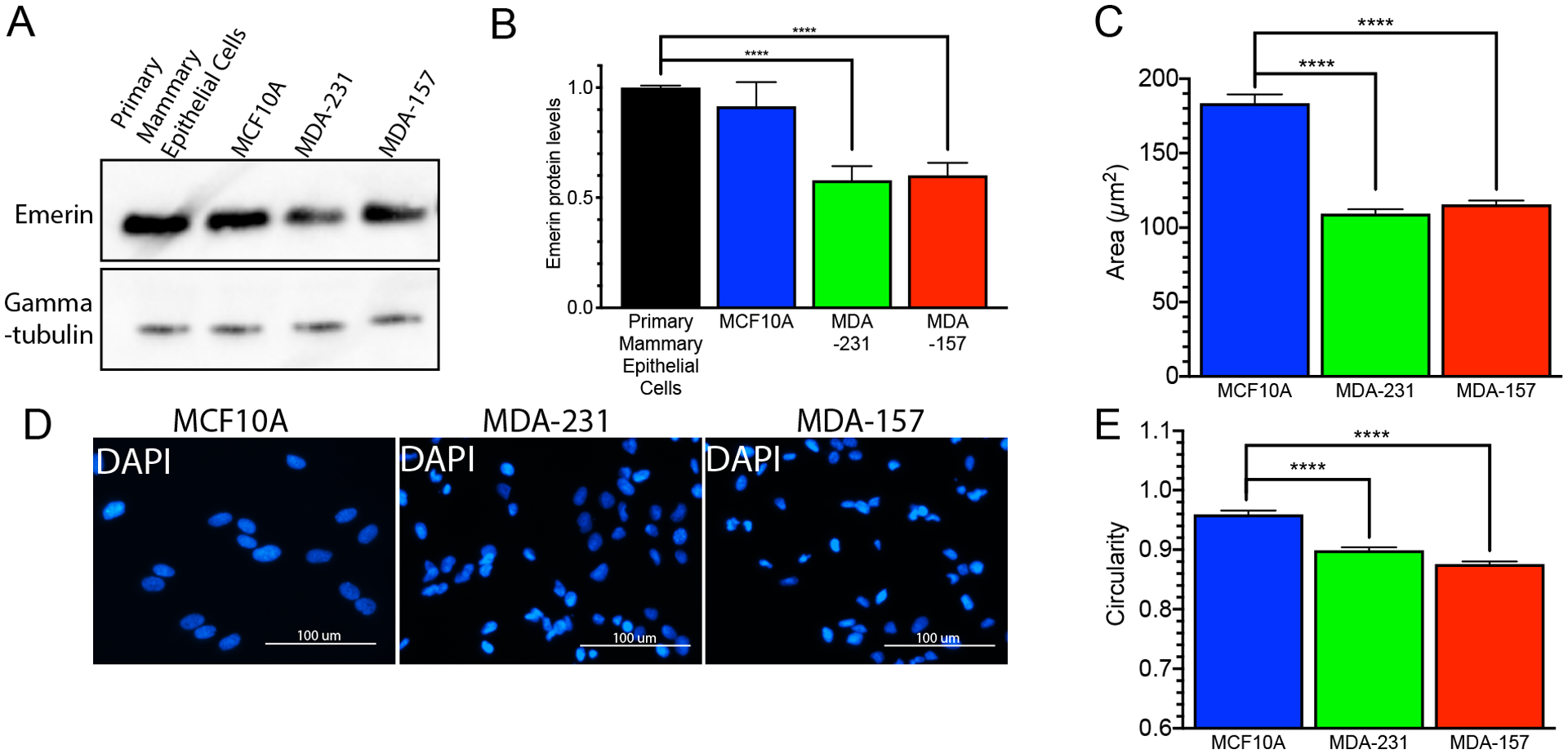
(A) Western Blot analysis of emerin in primary mammary epithelial cells, MCF10A cells, MDA-231 cells and MDA-157 cells. (B) Western Blot quantification of emerin expression for each cell line. Emerin expression was normalized to tubulin and primary mammary epithelial cells. Error bars represent standard error. (n=3) ****p-value < 0.0001, unpaired t-test. (C) Nuclear area for MCF10A (n=116, blue), MDA-231 (n=237, green) and MDA-157 (n=342, red) cells. Error bars represent standard error. ****p-value < 0.0001, unpaired t-test (D) Representative DAPI (blue) images of MCF10A, MDA-231, and MDA-157 cells. Scale bars: 100 μm. (E) Nuclear circularity for MCF10A (n=75, blue), MDA-231 (n=301, green), and MDA-157 (n=237, red) cells. Error bars represent standard error. ****p-value < 0.0001, unpaired t-test
GFP-emerin was expressed in MDA-231 cells to investigate whether emerin regulates nuclear morphology in invasive breast cancer cells. The nuclear area of cells expressing GFP or GFP-emerin was measured and compared to untransfected MDA-231 cells. There was a significant increase in nuclear area of GFP-emerin-expressing cells compared to untransfected cells (Figure 2A,B). GFP-emerin was expressed at similar levels as endogenous emerin, which increases total emerin expression to levels similar to endogenous emerin levels in MCF10A cells (Figure 3A,B). Similar results were seen in MDA-157 cells (Supplemental Figure 2A,B). GFP-emerin expression in MCF10A cells had no significant effect on nuclear area (Figure 2C,D).
Figure 2: GFP-emerin increases nuclear area in invasive breast cancer cells.
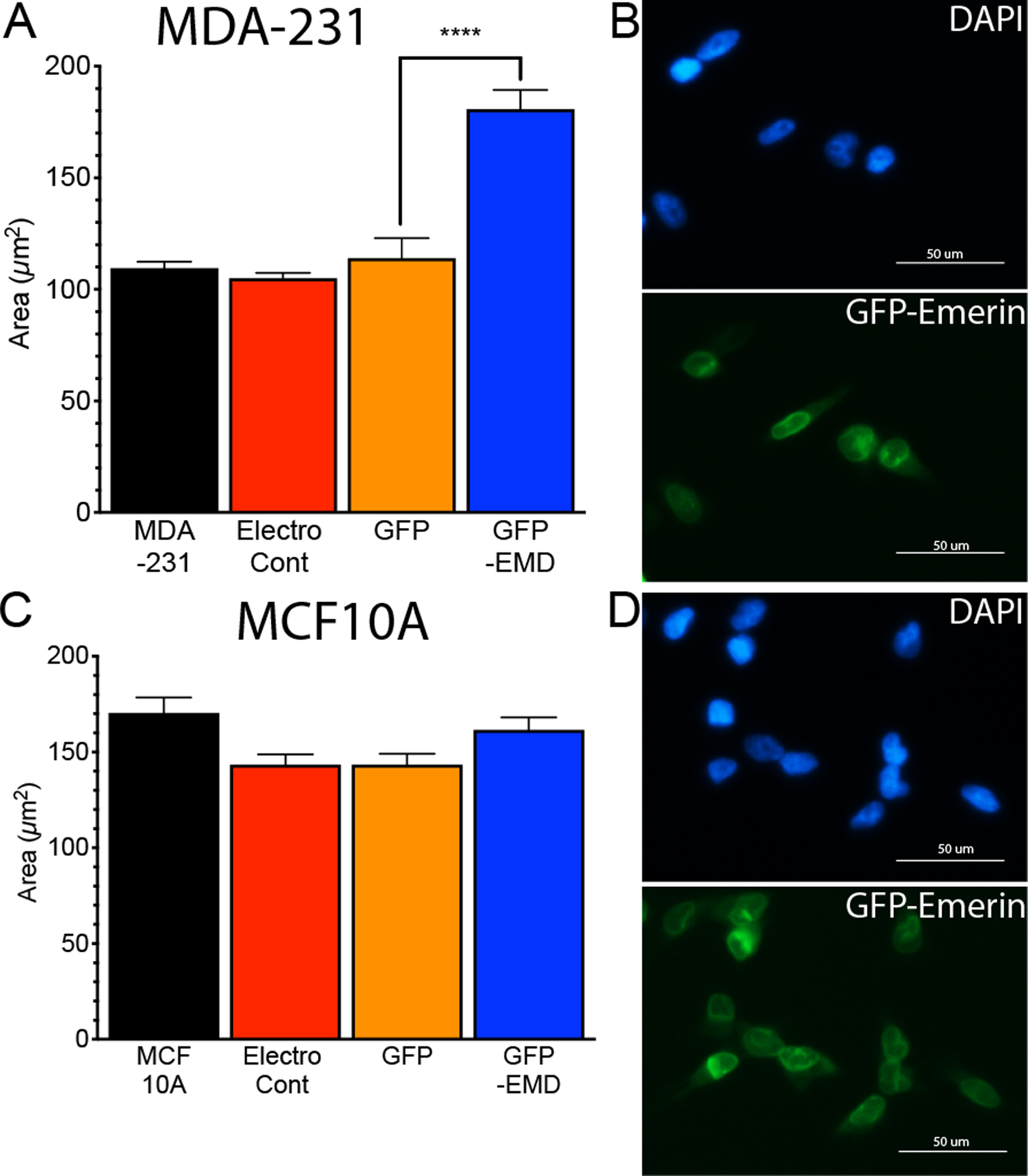
(A) Nuclear area of MDA-231 cells (n=237, black), MDA-231 cells with no plasmid, but electroporated (electroporation control; n=195, red), and MDA-231 cells expressing GFP (n=53, orange) or GFP-emerin (n=157, blue). Error bars represent standard error. ****p-value < 0.0001, unpaired t-test (B) Representative DAPI (blue) and GFP-emerin (green) images of MDA-231 cells. Scale bar: 50 μm. (C) Nuclear area of MCF10A cells (n=65, black), MCF10A cells with no plasmid, but electroporated (electroporation control; n=45, red), and MCF10A cells expressing GFP (n=90, orange) or GFP-emerin (n=76, blue). Error bars represent standard error. All comparisons had p-values > 0.05. (D) Representative DAPI (blue) and GFP-emerin (green) images of MCF10A cells. Scale bar: 50 μm.
Figure 3: GFP-emerin mutants that disrupt binding to nucleoskeletal partners fail to rescue nuclear area.
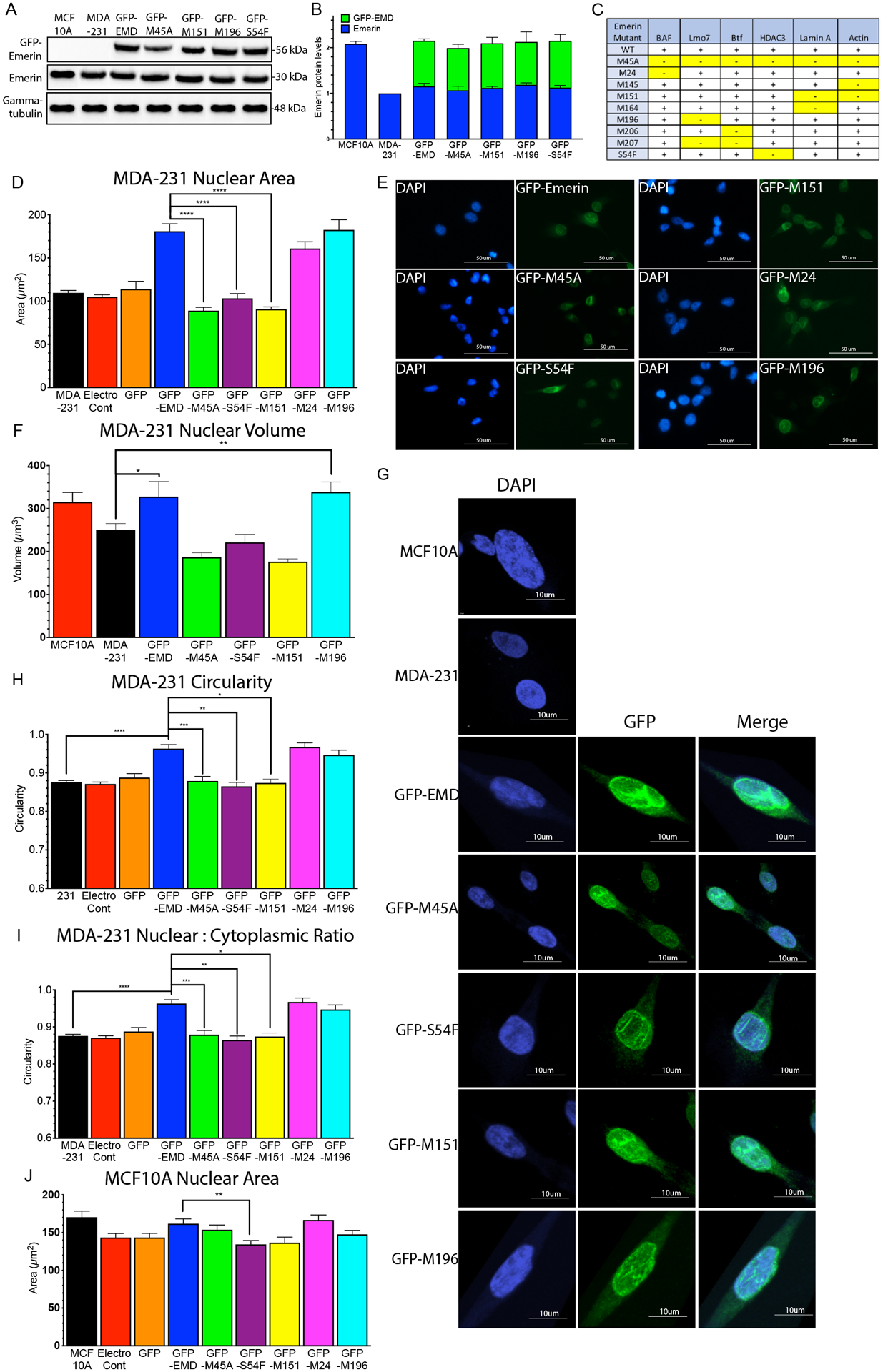
(A) Western blot of MCF10A cells, MDA-231 cells and MDA-231 cells stably expressing GFP-emerin or each GFP-emerin mutant. (B) Endogenous emerin, GFP-emerin and total emerin protein expression was normalized to gamma-tubulin and wildtype emerin in MDA-231 cells. Error bars represent standard deviation. (C) Panel showing known emerin-binding proteins and their disruption by the indicated emerin mutant protein. +, indicates emerin mutant binds to specific binding partner; -, indicates emerin mutant disrupts binding to the protein [58]. Disruptions are highlighted in yellow for clarity. (D) Nuclear area of MDA-231 cells (n=237, black), MDA-231 cells with no plasmid, but electroporated (electroporation control; n=195, black), and MDA-231 cells expressing GFP (n=53, orange), GFP-emerin (n=157, blue), GFP-M45A (n=142, green), GFP-S54F (n=145, purple), GFP-M151 (n=155, yellow), GFP-M24 (n=158, pink) or GFP-M196 (n=149, cyan). Error bars represent standard error. ****p-value < 0.0001, one-way ANOVA. (E) Representative DAPI (blue) and GFP (green) images for GFP-emerin and each GFP-emerin mutant analyzed in D. Scale bars: 50 μm. (F) Nuclear volume of MCF10A cells (n=25, red), MDA-231 cells (n=29, black), and MDA-231 cells expressing GFP-emerin (n=32, blue), GFP-M45A (n=22, green), GFP-S54F (n=28, purple), GFP-M151 (n=26, yellow), and GFP-M196 (n=19, cyan). Error bars represent standard error. **p-value = 0.0030, *p-value = 0.041, one-way ANOVA. (G) Representative DAPI (blue), GFP (green), and merged images for MCF10A cells, MDA-231 cells and MDA-231 cells expressing GFP-emerin or each GFP-emerin mutant analyzed in F. Scale bars: 10 μm. (H) Nuclear circularity of MDA-231 cells (n=55, black) and MDA-231 cells expressing GFP-emerin (n=52, blue), GFP-M45A (n=53), green), GFP-S54F (n=50, purple), GFP-M151 (n=49, yellow), GFP-M24 (n=44, pink), or GFP-M196 (n=59, cyan). Error bars represent standard error. *p-value = 0.0035, **p-value = 0.0005, ***p-value = 0.0225, ****p-value < 0.0001, one-way ANOVA (I) Ratio of nuclear to cytoplasmic area in MDA-231 cells (n=61, black) and MDA-231 cells expressing GFP-emerin (n=63, blue), GFP-M45A (n=45, green), GFP-S54F (n=65, purple), GFP-M151 (n=55, yellow) or GFP-M196 (n=54, cyan). Error bars represent standard error. *p-value = 0.051, **p-value = 0.035, ****p-value < 0.0001, one-way ANOVA. (J) Nuclear area of MCF10A cells (n=65, black), MCF10A cells with no plasmid, but electroporated (electroporation control; n=45, red), and MCF10A cells expressing GFP (n=90, orange), GFP-emerin (n=76, blue), GFP-M45A (n=90, green), GFP-S54F (n=91, purple), GFP-M151 (n=72, yellow), GFP-M24 (n=72, pink) or GFP-M196 (n=72, cyan). Error bars represent standard error. **p-value = 0.0101, one-way ANOVA.
Selected GFP-emerin mutants (Figure 3C) were over-expressed in MDA-231 cells to examine the functional interactions important for emerin modulating nuclear size. Each emerin mutant disrupts the binding of only one or two binding partners (Figure 3C). GFP-M45A (negative control) disrupts binding to all emerin partners. We successfully created MDA-231 cells stably expressing each GFP-emerin mutant at wildtype levels without altering the expression of endogenous emerin in these cells. This resulted in a 2-fold increase of total emerin expression (Figure 3A,B), which equals emerin expression in MCF10A cells (Figures 1A, 3A,B) and normal primary mammary epithelial cells (Figure 1A,B). GFP-emerin rescued nuclear area of MDA-231 cells (180.8 ± 8.5 μm2; Figure 3D,E), as it was comparable to MCF10A nuclear area (183.5 ± 2.8 μm2; Figure 2C). GFP-M45A failed to rescue nuclear area (88.8 ± 3.9 μm2), as expected. GFP-S54F (103.2 ± 5.5 μm2) and GFP-M151 (90.7 ± 2.6 μm2) also failed to rescue nuclear area. GFP-M24 (160.8 +/− 7.8 μm2) and GFP-M196 (182.4 ± 11.7 μm2) rescued nuclear area (Figure 3D,E). Nuclear volume was measured in MDA-231 cells expressing each GFP-emerin mutant to confirm changes in nuclear area correlated with changes in nuclear volume. The nuclear volume of MDA-231 cells was 250.7 ± 14.57 μm3 and the nuclear volume of MCF10A cells was 314.9 ± 12.72 μm3 (Figure 3F,G). GFP-emerin rescued nuclear volume (327 ± 10.24 μm3; Figure 3F,G). GFP-M45A failed to rescue nuclear volume (186 ± 10.65 μm3), as expected. GFP-S54F (221.2 ± 18.91 μm3) and GFP-M151 (176.3 ± 6.45 μm3) also failed to rescue nuclear volume. GFP-M196 (338.1 ± 23.71 μm3) rescued nuclear volume (Figure 3F,G).
Nuclear circularity was measured in MDA-231 cells expressing GFP-emerin and each GFP-emerin mutant. GFP-emerin rescued nuclear circularity (0.94 ± 0.08, Figure 3H), as it was comparable to MCF10A cells (0.96 ± 0.05, Figure 1E). GFP-M45A (0.81 ± 0.05), GFP-S54F (0.82 ± 0.03), and GFP-M151 (0.87 ± 0.05) failed to rescue nuclear circularity (Figure 3H). GFP-M24 (0.96 ± 0.05) and GFP-M196 (0.94 ± 0.06) rescued nuclear circularity (Figure 3H). Nuclear area was compared to cytoplasmic area to test if changes in nuclear size were due to changes in cell size. MDA-231 cells are known to be smaller than MCF10A cells, so it was no surprise the cytoplasmic area of MDA-231 cells was 50% smaller than MCF10A cells (Supplemental figure 2C). Importantly, nuclear size was reduced even further than expected, as the nuclear to cytoplasmic ratio of MDA-231 cells was reduced by 25% (0.476 ± 0.022) compared to MCF10A cells (0.631 ± 0.016, Figure 3I). Expression of GFP-emerin rescued both the cytoplasmic area of MDA-231 cells (Supplemental figure 2A) and the nuclear to cytoplasmic ratio (0.578 ± 0.021; Figure 3I). GFP-M45A (0.456 ± 0.022) failed to rescue the nuclear to cytoplasmic ratio, as expected. GFP-S54F (0.466 ± 0.027) and GFP-M151 (0.480 ± 0.024) also failed to rescue the nuclear to cytoplasmic ratio. Only GFP-M196 (0.573 ± 0.017) rescued the nuclear to cytoplasmic ratio (Figure 3I) and cytoplasmic area of MDA-231 cells. Thus, although expression of emerin alters cytoplasmic area, its expression has a larger effect on nuclear size. The result of this inappropriate scaling is a much smaller nucleus in the absence of emerin. Expression of GFP-emerin or each GFP emerin mutant in MCF10A cells caused little change in their nuclear area (Figure 3J).
Recent research has shown variations in chromatin compaction plays an important role in nuclear mechanics for small (one- to two-micron) deformations, while lamins are more important for larger deformations [12]. To better understand the effects of emerin expression on heterochromatin formation, the levels of histone posttranslational modifications indicative of heterochromatin were analyzed (Figure 4A,B). The levels of H3K9me2, H3K9me3 and H3K27me3 were unchanged upon expression of GFP-emerin, GFP-M45A, GFP-S54F, GFP-M151 and GFP-M196 (Figure 4A,B).
Figure 4: GFP-emerin expression decreases cell migration in invasive breast cancer cells.
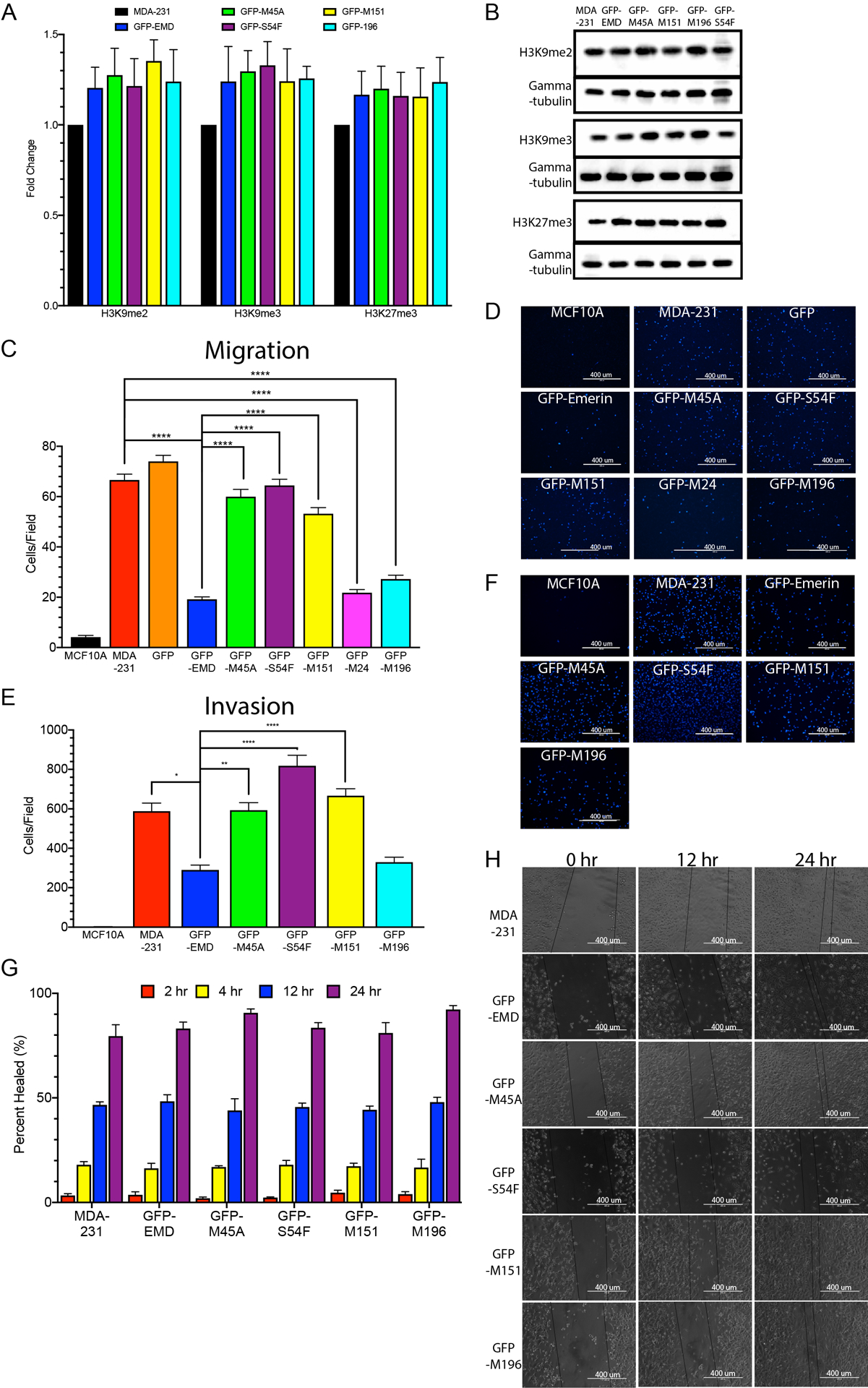
(A) Western blot quantification of H3K9me2, H3K9me3, and H3K27me3 expression in MDA-231 cells, and MDA-231 cells expressing GFP-emerin, GFP-M45A, GFP-S54F, GFP-M151, and GFP-M196. Expression was normalized to gamma-tubulin and MDA-231 cells. Error bars represent standard error. All comparisons had p-value > 0.05. (B) Representative Western blot of MDA-231 cells and MDA-231 cells expressing GFP-emerin, GFP-M45A, GFP-S54F, GFP-M151, and GFP-M196. (C) The number of cells migrating through 3.0 μm transwell pores is shown for MCF10A cells, MDA-231 cells, and MDA-231 cells expressing GFP, GFP-emerin, GFP-M45A, GFP-S54F, GFP-M151, GFP-M24 or GFP-M196. Error bars represent standard deviation. ****p-value < 0.0001, one-way ANOVA (D) Representative DAPI (blue) images for each of the cell lines analyzed in C. Scale bars: 400 μm. (E) The number of cells invading through 8.0 μm transwell pores with a Matrigel® coating was measured for MCF10A cells, MDA-231 cells, and MDA-231 cells expressing GFP-emerin, GFP-M45A, GFP-S54F, GFP-M151, or GFP-M196. Error bars represent standard deviation. *p-value = 0.0009, **p-value = 0.0005, ****p-value < 0.0001, one-way ANOVA (F) Representative DAPI (blue) images used for E. Scale Bars: 400 μm. (G) Scratch-wound healing assay. MCF10A cells, MDA-231 cells, and MDA-231 cells expressing GFP-emerin, GFP-M45A, GFP-S54F, GFP-M151, or GFP-M196 were plated, scratched with a pipette tip, and migration of cells into the wound area was monitored for 2, 4, 12 and 24 hours. Percent healed (%) refers to the ability of cells to migrate into the wound area. Error bars represent standard deviation. (H) Representative phase images for each cell line analyzed in G. Scale bars: 400 μm.
The effect of changes in nuclear area on migration of MDA-231 cells through 3.0 μm pores was studied using transwell assays. A 3.0 μm pore size was chosen because this pore size requires significant nuclear deformation for cells to successfully migrate. Transwell assays were performed on MDA-231 cells stably expressing GFP-emerin or each GFP-emerin mutant. GFP-emerin expression decreased migration by 71 ± 3.2% (Figure 4C,D). GFP-M45A (10 ± 6.3% decrease), GFP-S54F (4.3 ± 8.6% decrease) and GFP-M151 (16 ± 4.4% decrease) failed to impair migration (Figure 4C,D). GFP-M24 (66 ± 4.5% decrease) and GFP-M196 (57 ± 2.3% decrease) both impaired migration (Figure 4C,D). Similar results were seen in transwell inserts with 8.0 μm pores (Supplemental Figure 4), which is the size routinely used for studying impeded migration of breast cancer cells. Thus, GFP-emerin mutants that rescued nuclear size also impaired migration through small pores.
Cell invasion assays were done using 8.0 μm transwell inserts coated with Matrigel as the extracellular matrix. GFP-emerin expression decreased invasion by 51 ± 4.2% (Figure 4E,F). GFP-M45A (1.0 ± 6.3% increase), GFP-S54F (39 ± 9.2% increase), and GFP-M151 (13 ± 6.0% increase) failed to impair invasion (Figure 4E,F). GFP-M196 impaired invasion (56 ± 4.3% decrease). Thus, GFP-emerin mutants that rescued migration also rescued invasion to a similar extent. To ensure these affects were not caused by a general migration failure or an inability to respond to a signaling cue, migration was examined using scratch-wound healing assays. The scratch-wound closed at similar rates in MDA-231 cells and MDA-231 cells expressing GFP-emerin and each GFP-emerin mutant over 2-, 4-, 12-, and 24-hour periods (Figure 4G,H).
Emerin inhibition of cancer cell migration in vitro was predicted to affect metastasis in vivo. Whether emerin expression impaired metastasis was tested in orthotopic mouse models of breast cancer. MDA-231 cells expressing iRFP713 (to follow implanted cells) and GFP-emerin or each GFP-emerin mutant were injected into the mammary fat pad of nude mice. Mice were imaged every week for eight weeks. Mice were euthanized after eight weeks and the tumors and lungs were removed and imaged. There was a 61 ± 12% decrease in primary tumor size and a 50 ± 9% decrease in tumor growth rate of GFP-emerin cells (66,637 fluorescent units [FU], 8,610 FU/week; Figure 5A,B) compared to MDA-231 cells expressing only iRFP713 (control cells; 171,822 FU, 17,205 FU/week). GFP-M45A (184,376 FU, 22,722 FU/week), GFP-S54F (157,809 FU, 14,806FU/week), and GFP-M151 (240,951 FU, 26,731 FU/week) failed to rescue primary tumor size and growth rate (Figure 5A,B). GFP-M196 also significantly decreased primary tumor size and tumor growth rate (93,166 FU, 10,590 FU/week; Figure 5A,B). Surprisingly, there was no correlation between tumor growth and the proliferation rate of MDA-231 cells or MDA-231 cells expressing GFP-emerin or each GFP-emerin mutant (Supplemental figure 3D). Neither cell cycle dynamics nor apoptosis was affected by expression of emerin or any of the emerin mutants (Supplemental figure 3A–C).
Figure 5: GFP-emerin expression decreased primary tumor growth and blocked lung metastasis.
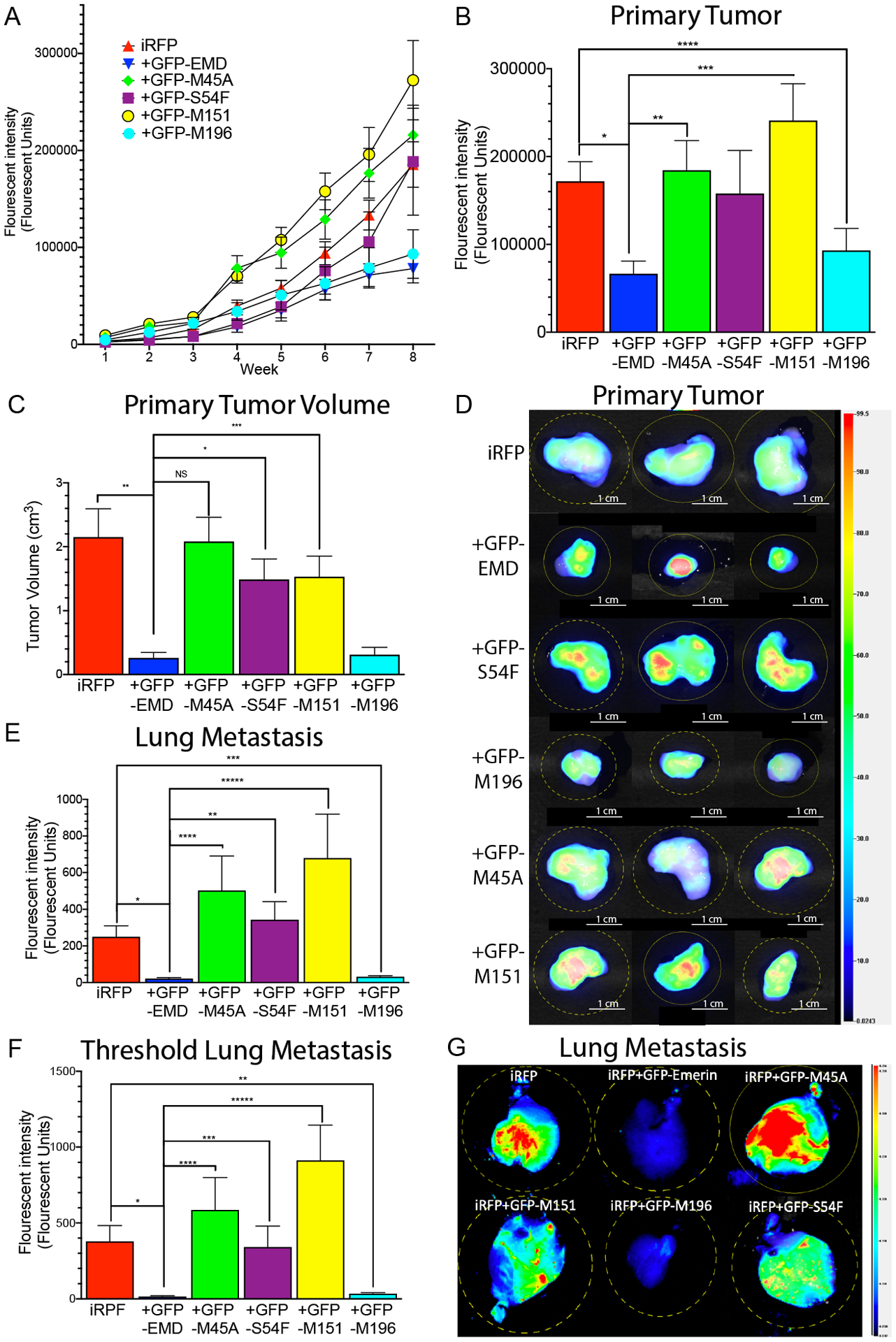
(A) Fluorescence intensity of the primary tumor was measured weekly for eight weeks. (B) Primary tumor size after excision at week eight. n=38 for iRFP713, n=30 for GFP-emerin, n=28 for each GFP-emerin mutant. Error bars represent standard error. *p-value = 0.0004, **p-value = 0.0050, ***p-value = 0.0006, ****p-value= 0.0266, one-way ANOVA. C) Volume of excised tumors at eight weeks post-injection with MDA-231 cells expressing iRFP713 (n=38), and GFP-emerin (n=30), GFP-S54F (n=28), GFP-M196 (n=28), GFP-M45A (n=28), or GFP-M151 (n=28) cells. Error bars represent standard error. *p-value = 0.0005, **p-value = 0.0015, ***p-value = 0.0239, one-way ANOVA (D) Representative fluorescent images of excised primary tumors at eight weeks post-injection. Heat-map shows iRFP713 fluorescence intensity. Scale bars: 1 cm (E) Fluorescence intensity of lung metastasis at eight weeks post-injection. Data was normalized to control lungs with no metastasis. n=38 for iRFP713 alone, n=30 for GFP-emerin, n=28 for each GFP-emerin mutant. Error bars represent standard error. *p-value = 0.0027, **p-value = 0.0040, ***p-value = 0.0034, ****p-value = 0.0156, *****p-value = 0.0107, one-way ANOVA. (F) Lung metastasis fluorescence intensity from mice whose primary tumors were greater than 50,000 FU is shown. Data was normalized to control lungs with no metastasis. n=28 for iRFP713, n=12 for GFP-emerin, n=20 for GFP-S54F, n=16 for GFP-M196, n=24 for GFP-M45A, n=28 for GFP-M151. Error bars represent standard error. *p-value = 0.0023, **p-value = 0.0035, ***p-value = 0.0331, ****p-value = 0.0136, *****p-value = 0.0006, one-way ANOVA. (G) Representative fluorescent images of excised lungs at eight weeks post-injection. Heat-map shows iRFP713 fluorescent intensity.
The volume of excised tumors was measured after eight weeks. There was an 88 ± 4.0% decrease in the primary tumor volume of GFP-emerin cells (0.26 cm3 ± 0.09 cm3) compared to MDA-231 cells expressing only iRFP713 (2.15 cm3 ± 0.4 cm3, Figure 5C,D). GFP-M45A (2.08 cm3 ± 0.38 cm3), GFP-S54F (1.49 cm3 ± 0.32 cm3), and GFP-M151 (1.53 cm3 ± 0.32 cm3) failed to rescue primary tumor volume (Figure 5C,D). GFP-M196 also significantly decreased primary tumor volume (0.31 cm3 ± 0.11 cm3; Figure 5C,D).
GFP-emerin expression completely blocked lung metastasis (21.7 ± 5.3 FU) as compared to iRFP713 control cells (250 ± 59.8 FU; Figure 5E,G). GFP-M196 also blocked lung metastasis (32.6 ± 5.9 FU; Figure 5E,G). GFP-M45A (503 ± 186.2 FU), GFP-S54F (343 ± 98.1 FU) and GFP-M151 (679 ± 233.7 FU) failed to rescue lung metastasis. Only lungs from mice whose primary tumors grew over 50,000 FU were then analyzed to confirm the failure to metastasize in GFP-emerin and GFP-M196 cells was not caused by reduced tumor burden. 50,000 FU was selected because primary tumors >50,000 FU metastasized to the lungs in 100% of control cases. Using this threshold, GFP-emerin (15.4 ± 5.5 FU) and GFP-M196 (34.8 ± 7.3 FU) still eliminated lung metastasis (Figure 5F). iRFP713 MDA-231 cells had 370 ± 55.2 FU in the lungs using this threshold (Figure 5F). This demonstrates the function of emerin in metastasis formation is independent of its role in primary tumor growth.
To determine if emerin expression correlates with cancer aggressiveness in patients, emerin protein expression was measured in patient tissues. A significant decrease in emerin expression was seen in fourteen different patients with a variety of receptor expression profiles, stages, and metastases (Figure 6A,B,D). qPCR analysis of RNA samples matching ten of the protein samples was done to determine emerin mRNA expression; RNA was unavailable for the other four patient samples. There was no discernable pattern between emerin protein expression and emerin mRNA expression (Figure 6C,D). Interestingly, there is a correlation between HER2-negative cancers having significantly less emerin when compared to healthy normal controls (Figure 6D). Emerin expression was similar in multiple normal controls (Supplemental Figure 5).
Figure 6: Patients with invasive breast cancer have decreased emerin expression.
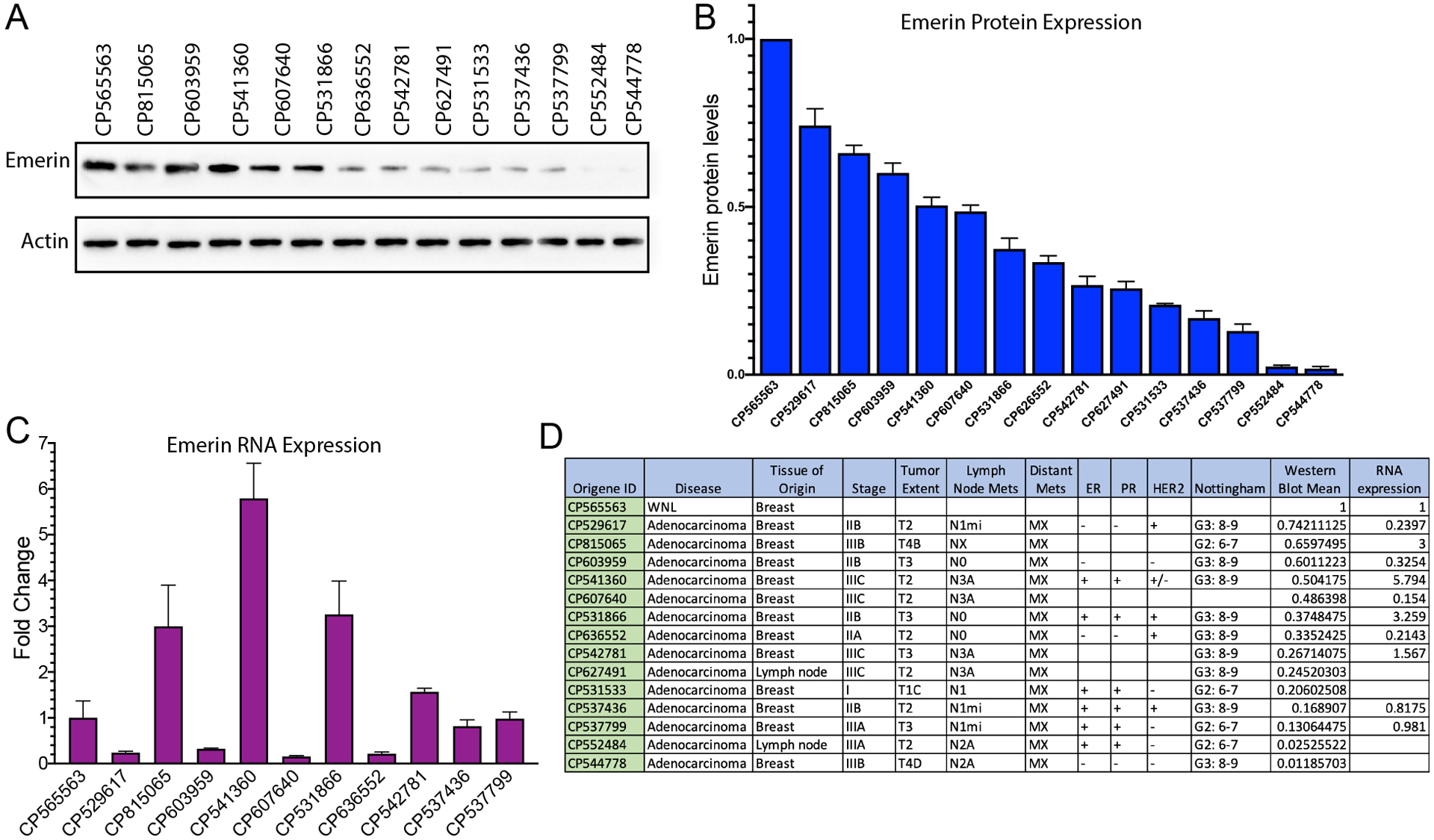
(A) Western blot analysis of protein lysates from a patient without breast cancer (control; CP565563) or from breast cancer patients. (B) Quantitation of protein lysates. Bands were normalized to actin and a patient without breast cancer (n=3). Error bars represent standard deviation. (C) qPCR analysis of matched RNA samples from one normal control patient sample and ten breast cancer patients. Samples were normalized to GAPDH and control patient sample without breast cancer. Error bars represent standard deviation. (D) Summary panel of patient tumor information.
Discussion
In this study, we showed emerin plays a direct and significant role in breast cancer migration and metastasis. The dynamics of cell migration and adhesion are particularly important for the process of metastasis, in which cancer cells escape from the primary tumor and enter the vasculature (intravasation); travel to a distant site; and extravasate to invade a new niche [5]. The deformability of the nucleus limits the cell’s ability to pass through tight spaces, with decreased compliance reducing or even stalling migration as the pore size decreases [15, 28]. Alterations in nuclear structure and mechanics directly contributes to cancer cell progression and metastasis [15, 20, 28, 45].
The importance of the nuclear lamina in providing structural support to the nucleus and controlling nuclear size is well established [19, 20, 28, 45]. Additionally, several studies have reported altered distribution, expression, or both, of INM proteins in various human cancers [21–27]. Many types of cancer, including breast cancer, present with lamina alterations [24]. Aberrant localization and reduced expression of A-type lamins frequently correlate with cancer subtypes and cancer aggressiveness, proliferative capacity, and differentiation state [9, 15, 19, 28–30]. Previous experiments showed nuclei lacking lamin A/C are significantly more compliant than wild-type controls [4, 46]. Nuclei in cells lacking lamins A and C deform significantly more under strain application, whereas lamin B1 deficient cells have normal nuclear mechanics [4]. This increased compliance in the absence of lamin A/C and other INM proteins, including emerin, is predicted to increase confined cell migration, cell invasion and metastasis (Figure 7). The nuclear lamina also participates in cytoskeletal and nucleoskeleton coupling, so mutations in lamins or other nuclear envelope proteins could alter force transmission between the nucleus and the cytoskeleton to affect nuclear deformation under externally applied loads [20, 47].
Figure 7: Model for emerin regulation of breast cancer metastasis.
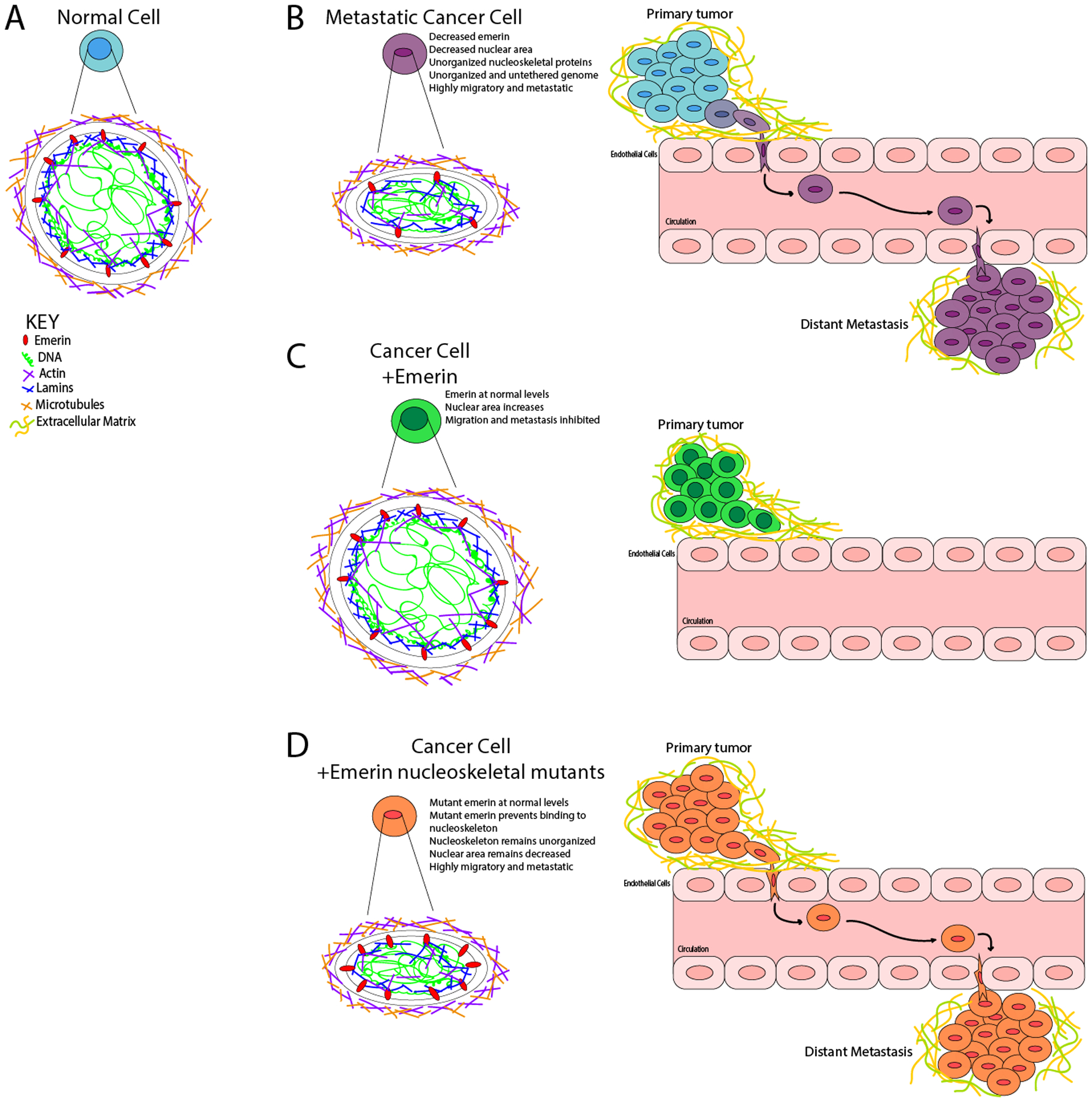
(A) Normal, non-cancerous nuclei are uniformly shaped with an organized nuclear lamina. The forces exerted on the nucleus from the cytoskeleton is countered by the forces within the nucleus, resulting in no deformations. The chromatin is compacted properly with heterochromatin tethered to the periphery and euchromatin centrally localized. (B) In a metastatic cancer cell, there is disruption of nuclear lamina proteins and significantly less emerin. This results in a disorganized nuclear lamina structure. The forces exerted on the nucleus from the cytoskeleton cannot be countered by the nucleoskeleton, resulting in a smaller, deformed nucleus that can easily migrate and metastasize. (C) When emerin is added to an invasive breast cancer cell, the nucleus can now properly organize the nucleoskeleton, which causes the nucleus to increase in size and shape. These changes block intravasation from occurring. (D) When emerin mutants that fail to bind the nucleoskeleton are added to invasive breast cancer cells, the nuclei are unable to reorganize the nuclear lamina properly. This fails to alter the nuclear morphology or size, and thus these cells can still intravasate and metastasize.
In addition to lamins, other nuclear envelope proteins have recently been implicated in a variety of cancers. Emerin expression is altered in osteosarcoma [26], which scales with cancer aggressiveness. Emerin was also recently implicated in prostate cancer aggressiveness, as treatment of prostate cancer cells with shRNA to downregulate emerin increased nuclear deformation and migration [42]. The data presented here is the first to show emerin expression is decreased in invasive breast cancer cells and that increasing emerin expression rescued their nuclear size and structure and decreased their migration, invasion and metastasis in mice.
There is ample evidence for emerin maintaining nuclear architecture. These include biophysical studies showing emerin-null cells have decreased elasticity [48] and the nuclear envelope is more compliant [48, 49]. These cells also have altered nuclear morphology, increased nuclear deformability, impaired viability under mechanical strain and defective mechanotransduction [46, 49]. Emerin binds actin and caps the pointed end of actin filaments to stabilize these short nuclear actin polymers [34]. Emerin co-purifies with nuclear protein 4.1R, which binds both nuclear spectrin and nuclear actin [50]. Thus, emerin is predicted to anchor cortical nuclear actin networks near the nuclear envelope to increase nuclear rigidity. Our results support the role of emerin stabilization of the nucleoskeleton in maintaining proper nuclear rigidity, since disruption of emerin’s interactions with the nucleoskeleton results in cells with smaller, deformed nuclei that migrate and invade more easily and metastasize more readily.
These nuclear size and structural changes may also result from disruption of its interactions with the linker of nucleoskeleton and cytoskeleton (LINC) complex. The LINC complex physically connects the cytoskeleton to the nucleus [51]. LINC complex proteins and lamin A/C directly transmit mechanical force from the plasma membrane and cytoplasm to the nucleus [49, 51]. Application of force to the plasma membrane or cytoskeleton deforms the nucleus and activates mechanosensitive genes [52]. Emerin and lamin A directly bind nesprins and SUN-domain proteins [53], which likely serves to relay the cytoskeletal signals to the nucleoskeleton and chromatin. Thus, nucleoskeleton organization and the structure of the LINC complex are key factors in the transmission of mechanical stress to the nucleus [54]. As wildtype nuclei undergo mechanical force, the nuclear envelope adapts by increasing in stiffness. Force strongly induces the phosphorylation of emerin, mediated by the SRC family kinases (SFKs) [54]. Interestingly, emerin depletion prevented nuclear adaptation to force. Nuclei expressing emerin mutants that prevented phosphorylation, failed to adapt to force [54]. These results indicate application of force to nesprin 1 activates SFKs, which phosphorylate emerin to mediate the mechanical adaptation of isolated nuclei [54].
Besides the nuclear lamina, chromatin is an important contributor to nuclear stiffness [11, 12, 15, 20, 28, 55]. Unlike the loadbearing, elastic shell of the nuclear lamina, chromatin exhibits a more viscoelastic behavior (i.e., it flows when subjected to forces and undergoes plastic deformations) [14]. Chromatin exists in two configurations: euchromatin, which is less compact and typically transcriptionally active, and heterochromatin, which is more tightly compacted and associated with transcriptionally silent genes [56]. A large portion of heterochromatin interacts with the nuclear lamina at the nuclear periphery [56]. Treatment of mouse embryonic fibroblasts (MEFs) with a histone methyltransferases inhibitor (HMTi) caused ~20% larger nuclei with increased compliance and increased blebbing [12]. These increases were attributed to decreased heterochromatin formation, as H3K9me2/3 and H3K27me3 levels were decreased. However, we showed emerin-null myoblasts have decreased H3K9me2/3 and H3K27me3 and increased H3K9ac and H4K5ac [33] yet have smaller nuclei (Supplemental Figure 1). This suggests heterochromatic marks may affect nuclear size and structure differently in a cell-type dependent manner. The results reported here show there is no change in heterochromatin formation upon expression of GFP-emerin or GFP-emerin mutants, as defined by H3K9me2/3 and H3K27me3 levels (Figure 4A,B). The reduction of emerin by 50% in invasive breast cancer cells, as opposed to its complete loss, may also explain this apparent inconsistency. It is also possible that the many mutations accompanying the transformation to invasive breast cancer cells may have affected chromatin architecture independent of emerin or other nuclear envelope proteins [57]. It is also possible the functional interaction of emerin with HDAC3 may affect chromatin compaction independent of histone methylation. Regardless of the mechanism, our data show chromatin minimally contributes to the decreased nuclear size and increased compliance in invasive breast cancer cells. We propose the functional interaction of emerin with the nucleoskeleton is the major contributor to the nuclear structural changes seen in invasive breast cancer cells.
Conclusion
Collectively, our data shows the functional interaction of emerin with the nucleoskeleton is important for the nuclear structural changes seen in invasive breast cancer cells. These smaller, softer nuclei are predicted to migrate more easily and to metastasize more readily (Figure 7).
Loss of emerin protein expression during metastatic transformation would have similar effects to the loss of nucleoskeletal proteins, including lamins [1, 6, 15, 20, 28]. Thus, we predict loss of emerin expression is an important driver of metastatic transformation.
Supplementary Material
Implications:
Modulating emerin expression and function represents new targets for therapeutic interventions of metastasis, since increased emerin expression rescued cancer metastasis.
Acknowledgments:
We thank the Department of Pharmaceutical Sciences at the University of the Sciences and the Department of Biomedical Sciences at Cooper Medical School of Rowan University for providing laboratory space and funding. This work was supported by a predoctoral grant from the New Jersey Commission on Cancer Research (A. Liddane), a grant from the W. W. Smith Charitable Trust (J. Holaska) and a grant from the National Institute of Arthritis, and Musculoskeletal and Skin Diseases (R15AR069935 to J. Holaska). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, the W.W. Smith Charitable Trust or the New Jersey Commission on Cancer Research. This work was also supported by Rowan University under the Camden Health Research Initiative. We thank Chris Janetopoulos, Darren Boehning, and Goutham Kodakandla for their helpful insights with volumetric analysis. We thank the members of the Holaska laboratory for the many helpful discussions regarding these studies and preparation of the manuscript.
Footnotes
Conflicts of interests: The authors declare no conflicts of interest.
References
- 1.Wirtz D, Konstantopoulos K, and Searson PC, The physics of cancer: the role of physical interactions and mechanical forces in metastasis. Nat Rev Cancer, 2011. 11(7): p. 512–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leoni G, et al. , Wound repair: role of immune-epithelial interactions. Mucosal Immunol, 2015. 8(5): p. 959–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chaffer CL and Weinberg RA, A perspective on cancer cell metastasis. Science, 2011. 331(6024): p. 1559–64. [DOI] [PubMed] [Google Scholar]
- 4.Lammerding J, et al. , Lamins A and C but not lamin B1 regulate nuclear mechanics. J Biol Chem, 2006. 281(35): p. 25768–80. [DOI] [PubMed] [Google Scholar]
- 5.Chow KH, Factor RE, and Ullman KS, The nuclear envelope environment and its cancer connections. Nat Rev Cancer, 2012. 12(3): p. 196–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zink D, Fischer AH, and Nickerson JA, Nuclear Structure in Cancer Cells. Nature Reviews, 2004. 4. [DOI] [PubMed] [Google Scholar]
- 7.Mueller JL, et al. , Rapid staining and imaging of subnuclear features to differentiate between malignant and benign breast tissues at a point-of-care setting. J Cancer Res Clin Oncol, 2016. 142(7): p. 1475–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolf K, et al. , Physical limits of cell migration: control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J Cell Biol, 2013. 201(7): p. 1069–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lammerding J, Mechanics of the nucleus. Compr Physiol, 2011. 1(2): p. 783–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolf K, et al. , Collagen-based cell migration models in vitro and in vivo. Semin Cell Dev Biol, 2009. 20(8): p. 931–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schreiner SM, et al. , The tethering of chromatin to the nuclear envelope supports nuclear mechanics. Nat Commun, 2015. 6: p. 7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stephens AD, et al. , Chromatin histone modifications and rigidity affect nuclear morphology independent of lamins. Mol Biol Cell, 2018. 29(2): p. 220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shimi T, et al. , Structural organization of nuclear lamins A, C, B1, and B2 revealed by superresolution microscopy. Mol Biol Cell, 2015. 26(22): p. 4075–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rowat AC, et al. , Towards an integrated understanding of the structure and mechanics of the cell nucleus. Bioessays, 2008. 30(3): p. 226–36. [DOI] [PubMed] [Google Scholar]
- 15.Friedl P, Wolf K, and Lammerding J, Nuclear mechanics during cell migration. Curr Opin Cell Biol, 2011. 23(1): p. 55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Korfali N, et al. , The nuclear envelope proteome differs notably between tissues. Nucleus, 2012. 3(6): p. 552–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilkie GS, et al. , Several novel nuclear envelope transmembrane proteins identified in skeletal muscle have cytoskeletal associations. Mol Cell Proteomics, 2011. 10(1): p. M110 003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Worman HJ and Schirmer EC, Nuclear membrane diversity: underlying tissue-specific pathologies in disease? Curr Opin Cell Biol, 2015. 34: p. 101–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ho CY and Lammerding J, Lamins at a glance. J Cell Sci, 2012. 125(Pt 9): p. 2087–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lammerding J, Mechanics of the Nucleus Compr Physiol, 2011. 1(2): p. 783–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Capo-chichi CD, et al. , Loss of GATA6 leads to nuclear deformation and aneuploidy in ovarian cancer. Mol Cell Biol, 2009. 29(17): p. 4766–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coradeghini R, et al. , Differential expression of nuclear lamins in normal and cancerous prostate tissues. Oncol Rep, 2006. 15(3): p. 609–13. [PubMed] [Google Scholar]
- 23.Kim HJ, et al. , LAP2 is widely overexpressed in diverse digestive tract cancers and regulates motility of cancer cells. PLoS One, 2012. 7(6): p. e39482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsumoto A, et al. , Global loss of a nuclear lamina component, lamin A/C, and LINC complex components SUN1, SUN2, and nesprin-2 in breast cancer. Cancer Med, 2015. 4(10): p. 1547–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun S, et al. , Circulating Lamin B1 (LMNB1) biomarker detects early stages of liver cancer in patients. J Proteome Res, 2010. 9(1): p. 70–8. [DOI] [PubMed] [Google Scholar]
- 26.Urciuoli E, et al. , Nuclear Lamins and Emerin Are Differentially Expressed in Osteosarcoma Cells and Scale with Tumor Aggressiveness. Cancers (Basel), 2020. 12(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J, et al. , Expression of nuclear membrane proteins in normal, hyperplastic, and neoplastic thyroid epithelial cells. Virchows Arch, 2015. 467(4): p. 427–36. [DOI] [PubMed] [Google Scholar]
- 28.Denais C and Lammerding J, Nuclear Mechanics in Cancer. Adv Exp Med Biol, 2014. 773: p. 435–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davidson PM and Lammerding J, Broken nuclei--lamins, nuclear mechanics, and disease. Trends Cell Biol, 2014. 24(4): p. 247–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lammerding J and Lee RT, The nuclear membrane and mechanotransduction: impaired nuclear mechanics and mechanotransduction in lamin A/C deficient cells. Novartis Found Symp, 2005. 264: p. 264–73; discussion 273–8. [PubMed] [Google Scholar]
- 31.Nagano A, et al. , Emerin deficiency at the nuclear membrane in patients with Emery-Dreifuss muscular dystrophy. Nat Genet, 1996. 12(3): p. 254–9. [DOI] [PubMed] [Google Scholar]
- 32.Tunnah D, et al. , The apparent absence of lamin B1 and emerin in many tissue nuclei is due to epitope masking. J Mol Histol, 2005. 36(5): p. 337–44. [DOI] [PubMed] [Google Scholar]
- 33.Demmerle J, Koch AJ, and Holaska JM, The nuclear envelope protein emerin binds directly to histone deacetylase 3 (HDAC3) and activates HDAC3 activity. J Biol Chem, 2012. 287(26): p. 22080–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holaska JM, Kowalski AK, and Wilson KL, Emerin caps the pointed end of actin filaments: evidence for an actin cortical network at the nuclear inner membrane. PLoS Biol, 2004. 2(9): p. E231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holaska JM, Rais-Bahrami S, and Wilson KL, Lmo7 is an emerin-binding protein that regulates the transcription of emerin and many other muscle-relevant genes. Hum Mol Genet, 2006. 15(23): p. 3459–72. [DOI] [PubMed] [Google Scholar]
- 36.Holaska JM and Wilson KL, Multiple roles for emerin: implications for Emery-Dreifuss muscular dystrophy. Anat Rec A Discov Mol Cell Evol Biol, 2006. 288(7): p. 676–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holaska JM and Wilson KL, An emerin “proteome”: purification of distinct emerin-containing complexes from HeLa cells suggests molecular basis for diverse roles including gene regulation, mRNA splicing, signaling, mechanosensing, and nuclear architecture. Biochemistry, 2007. 46(30): p. 8897–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eswaran J, et al. , Transcriptomic landscape of breast cancers through mRNA sequencing. Sci Rep, 2012. 2: p. 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eswaran J, et al. , RNA sequencing of cancer reveals novel splicing alterations. Sci Rep, 2013. 3: p. 1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horvath A, et al. , Novel insights into breast cancer genetic variance through RNA sequencing. Sci Rep, 2013. 3: p. 2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ranade D, et al. , Lamin A/C and Emerin depletion impacts chromatin organization and dynamics in the interphase nucleus. BMC Mol Cell Biol, 2019. 20(1): p. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reis-Sobreiro M, et al. , Emerin Deregulation Links Nuclear Shape Instability to Metastatic Potential. Cancer Res, 2018. 78(21): p. 6086–6097. [DOI] [PubMed] [Google Scholar]
- 43.Mercier I, et al. , CAPER, a novel regulator of human breast cancer progression. Cell Cycle, 2014. 13(8): p. 1256–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Filippi-Chiela EC, et al. , Nuclear morphometric analysis (NMA): screening of senescence, apoptosis and nuclear irregularities. PLoS One, 2012. 7(8): p. e42522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zwerger M, Ho CY, and Lammerding J, Nuclear mechanics in disease. Annu Rev Biomed Eng, 2011. 13: p. 397–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lammerding J, et al. , Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest, 2004. 113(3): p. 370–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maurer M and Lammerding J, The Driving Force: Nuclear Mechanotransduction in Cellular Function, Fate, and Disease. Annu Rev Biomed Eng, 2019. 21: p. 443–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rowat AC, Lammerding J, and Ipsen JH, Mechanical properties of the cell nucleus and the effect of emerin deficiency. Biophys J, 2006. 91(12): p. 4649–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lammerding J, et al. , Abnormal nuclear shape and impaired mechanotransduction in emerin-deficient cells. J Cell Biol, 2005. 170(5): p. 781–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ohanian V, et al. , Analysis of the ternary interaction of the red cell membrane skeletal proteins spectrin, actin, and 4.1. Biochemistry, 1984. 23(19): p. 4416–20. [DOI] [PubMed] [Google Scholar]
- 51.Lombardi ML and Lammerding J, Keeping the LINC: the importance of nucleocytoskeletal coupling in intracellular force transmission and cellular function. Biochem Soc Trans, 2011. 39(6): p. 1729–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang N, Tytell JD, and Ingber DE, Mechanotransduction at a distance: mechanically coupling the extracellular matrix with the nucleus. Nat Rev Mol Cell Biol, 2009. 10(1): p. 75–82. [DOI] [PubMed] [Google Scholar]
- 53.Mislow JM, et al. , Nesprin-1alpha self-associates and binds directly to emerin and lamin A in vitro. FEBS Lett, 2002. 525(1–3): p. 135–40. [DOI] [PubMed] [Google Scholar]
- 54.Guilluy C, et al. , Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat Cell Biol, 2014. 16(4): p. 376–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stephens AD, et al. , Physicochemical mechanotransduction alters nuclear shape and mechanics via heterochromatin formation. Mol Biol Cell, 2019. 30(17): p. 2320–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eissenberg JC and Wallrath LL, Heterochromatin, position effects, and the genetic dissection of chromatin. Prog Nucleic Acid Res Mol Biol, 2003. 74: p. 275–99. [DOI] [PubMed] [Google Scholar]
- 57.Demmerle J, Koch AJ, and Holaska JM, Emerin and histone deacetylase 3 (HDAC3) cooperatively regulate expression and nuclear positions of MyoD, Myf5, and Pax7 genes during myogenesis. Chromosome Res, 2013. 21(8): p. 765–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Berk JM, Tifft KE, and Wilson KL, The nuclear envelope LEM-domain protein emerin. Nucleus, 2013. 4(4): p. 298–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.