

Abstract
Emerging evidence supports the notion that inflammation fosters the development of common benign gynecologic disorders, including uterine leiomyoma, endometriosis, and adenomyosis. Numerous cytokines, chemokines, and growth and transcription factors have indisputable roles in the establishment and maintenance of benign gynecologic disorders by initiating complex cascades that promote proliferation, angiogenesis, and lesion progression. The interaction between inflammation and benign gynecologic disorders is orchestrated by a plethora of factors, including sex steroids, genetics, epigenetics, extracellular matrix, stem cells, cardiometabolic risk factors, diet, vitamin D, and the immune system. The role of inflammation in these disorders is not limited to local pathobiology but also extends to involve clinical sequelae that range from those confined to the reproductive tract, such as infertility and gynecologic malignancies, to systemic complications such as cardiovascular disease. Enhanced understanding of the intricate mechanisms of this association will introduce us to unvisited pathophysiological perspectives and guide future diagnostic and therapeutic implications aimed at reducing the burden of these disorders. Utilization of inflammatory markers, microRNA, and molecular imaging as diagnostic adjuncts may be valuable, noninvasive techniques for prompt detection of benign gynecologic disorders. Further, use of novel as well as previously established therapeutics, such as immunomodulators, hormonal treatments, cardiometabolic medications, and cyclooxygenase-2 and NF-κB inhibitors, can target inflammatory pathways involved in their pathogenesis. In this comprehensive review, we aim to dissect the existing literature on the role of inflammation in benign gynecologic disorders, including the proposed underlying mechanisms and complex interactions, its contribution to clinical sequelae, and the clinical implications this role entails.
Keywords: adenomyosis, chemokine, cytokine, endometriosis, growth factor, infertility, inflammation, prostaglandin, uterine leiomyoma
Reconceptualizing common benign gynecologic disorders as having inherent inflammatory pathobiology will introduce us to previously unvisited perspectives of their development and devise novel clinical implications aimed at reducing their burden.
Introduction
Inflammation has been historically known as one of the most ancient and extensively studied pathophysiological phenomena. Once defined by the five cardinal signs of rubor, calor, tumor, dolor, and functio laesa, it now represents a far more complex cascade of intra- and extracellular events in response to injury that culminate in tissue healing, repair, or occasionally, disease [1]. Various immunologic, neoplastic, infectious, and even atherosclerotic conditions driven and maintained by inflammation have been recognized, where aberrant inflammatory processes perpetuate tissue injury, and resolution does not occur, leading a chronic disease state [2].
Considerable evidence supports the notion that chronic inflammation fosters the development of benign gynecologic disorders (BGDs), including uterine leiomyoma, endometriosis, and adenomyosis; conditions that were once merely considered neoplastic or metastatic in nature. Whereas multiple experimental studies have explored the interplay of inflammatory mediators in uterine leiomyoma development [3], others imply that adenomyotic and endometriotic lesions may also induce local and systemic inflammatory milieus, respectively [4, 5]. A plethora of cytokines, chemokines, growth factors, and prostaglandins are found to be key promoters of BGD initiation, maintenance, and progression. In addition, not only does inflammation contribute to BGD pathogenesis but also to their manifestations that cover a wide array of local and systemic clinical sequelae [6, 7].
While these findings represent breakthroughs in BGD pathogenesis, the majority of evidence remains obscure and is rarely presented comprehensively. Therefore, this exhaustive review aims to explore various and unique perspectives of the association between inflammation and BGDs, debate the underlying mechanisms and complex interactions, and shed the light on the role of inflammation in their clinical sequelae. Lastly, we introduce novel and previously established diagnostic and therapeutic implications tailored to the context of this association.
Ethics statement
An ethical approval was not required for this work as no human participants or animal models were included and no new data were generated during its production.
Pathophysiologic considerations
Local and systemic inflammation in benign gynecologic disorders
Uterine leiomyoma
As we explore the role of inflammation in BGDs, it remains largely elusive how and where the inciting inflammatory events occur, and whether they start locally then progress systemically, vice versa, or develop in parallel. In uterine leiomyoma, for example, it is demonstrated that CD68+ macrophages exist in abundance as compared with distant autologous myometrium, supporting a role for local inflammation in leiomyoma pathogenesis [3]. Local macrophages then secrete various growth factors, including transforming growth factor β (TGF-β), a chemoattractant of more macrophages and established key player in fibrosis and development of leiomyoma, where it is overexpressed alongside other proinflammatory cytokines, including tumor necrosis factor ⍺ (TNF-⍺), interleukin (IL)-11, and IL-13 [8–10]. Chronic myometrial inflammation can be induced by a plethora of culprits, including infection, ovulation, menstruation, implantation, foreign bodies, surgery, male reproductive proteins, and even stress [11]. Chronic uterine inflammation, regardless of the cause, leads to uncontrolled production of extracellular matrix (ECM) by myofibroblasts and failure of their apoptosis (Figure 1) [12]. Systemic evidence of inflammation was also noted in women with uterine leiomyoma, who are found to have twice as high serum TNF-⍺ levels compared with controls, with levels correlating with tumor size. In addition, not only is TNF-⍺ thought to contribute to leiomyoma development but also to its clinical manifestations [13].
Figure 1 .
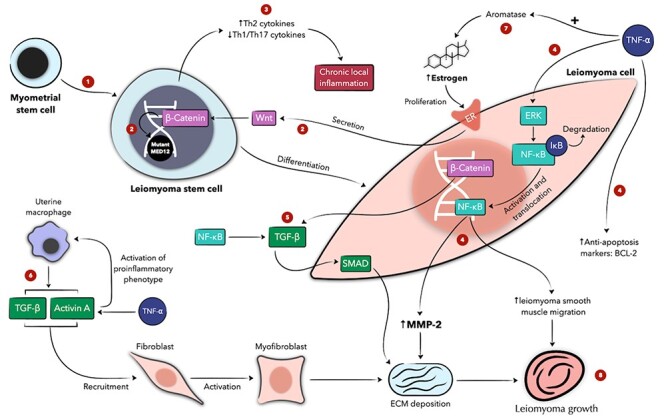
Schematic presentation of the role of inflammation in uterine leiomyoma development and its interaction with hormones, extracellular matrix, and stem cells. In myometrial stem cells, Wnt, whose secretion is mediated by estrogen, stimulates β-Catenin, whose function is regulated by the MED12 gene. In leiomyoma stem cells with mutated MED12, β-Catenin can result in uncontrolled proliferation of leiomyoma cells (1, 2) [244]. Leiomyoma stem cells also have increased expression of proinflammatory cytokines, favoring local inflammation and leiomyoma development (3). TNF-⍺ exerts various effects on leiomyoma cells and estrogen metabolism that culminate in increased proliferation and ECM deposition (4, 7, 8). TGF β and activin A, also modulated by inflammation, mediate ECM deposition and leiomyoma growth (5, 6, 8). Numeric labels are meant to guide the reader through the figure; concurrent or subsequent steps may carry the same label. + denotes an activating function. ECM denotes extracellular matrix, ER estrogen receptor, ERK extracellular signal–regulated kinase, MMP matrix metalloproteinase, NF-κB nuclear factor kappa-light chain enhancer of activated B cells, TGF-β transforming growth factor β, and TNF-⍺ tumor necrosis factor ⍺.
Endometriosis
Endometriosis is now largely recognized for being an inflammatory disease and here, the process of local inflammation is better comprehended knowing that ectopic endometrial fragments are the likely culprit. Upon implantation, macrophages and neutrophils are first recruited, secreting numerous cytokines and growth factors with proinflammatory, chemotactic, and angiogenic properties, including TNF-⍺, IL-1, IL-6, IL-8, and vascular endothelial growth factor (VEGF) (Figure 2) [14, 15]. In addition, excessive amount of retrograde menstruation and subsequent iron overload appear to overwhelm macrophage physiology, triggering aberrant inflammatory signaling and impaired phagocytic potential [16]. This local inflammatory environment favors lesion establishment, progression, and angiogenesis [17]. Women with stage III/IV endometriosis have significantly higher peritoneal TNF-⍺ levels compared with stage I/II patients, which alongside IL-8, is found to correlate with size and number of active lesions, reflecting increased activation of peritoneal macrophages [18–20]. On the other hand, emerging evidence shows that endometriosis establishes a state of systemic inflammation as evidenced by high serum levels of TNF-⍺, IL-1β, and IL-6 in women with the disease (Figure 3) [17, 21]. While it is not entirely clear how peritoneal inflammation progresses systemically, microRNAs (miRNA) are hypothesized to alter cytokine expression of serum macrophages distant from the peritoneal lesions [21]. Specifically, miRNA 125b-5p upregulation and miRNA Let-7b-5p downregulation correlate with increased serum levels of proinflammatory cytokines in endometriosis [21]. The exact mechanism underlying this correlation remains largely unknown, but Let-7b is thought to (1) target C/EBP-δ, a transcription factor that sustains responsiveness to toll-like receptor (TLR) signaling, and (2) regulate the nuclear factor kappa-light chain enhancer of activated B cells (NF-κB) inflammatory pathway, a major transcription factor of inflammatory cascades [22]. Thus, endometriosis is no longer perceived as an exclusively local disease of the peritoneum but one of extensive systemic effects [23].
Figure 2 .
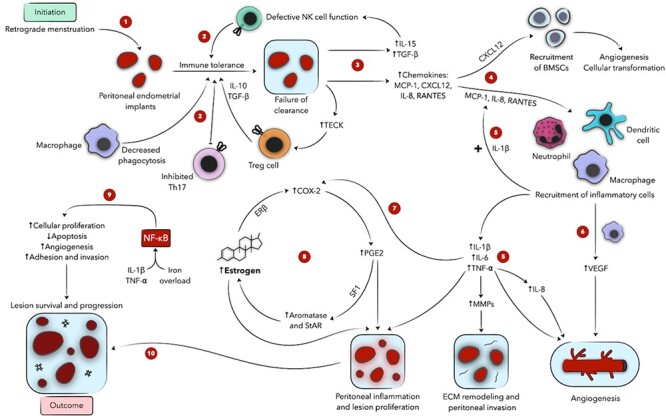
Schematic presentation of the role of inflammation and immune cells in endometriosis. Ectopic endometrial implants evade the immune system due to defective immunosurveillance (1, 2) and secrete various chemokines, recruiting BMSCs and inflammatory cells to the proendometriotic niche (3, 4). Inflammatory cells, in turn, express proinflammatory cytokines, activating transcription factors and accentuating lesional proliferation, angiogenesis, and invasion (5, 6, 9, 10). These mediators also drive the local estradiol-PGE2 cycle, fueling peritoneal inflammation and disease progression (7, 8). Numeric labels are meant to guide the reader through the figure; concurrent or subsequent steps may carry the same label. + denotes an activating function. BMSC denotes bone marrow-derived stem cell, COX-2 cyclooxygenase-2, CXCL12 CXC motif chemokine 12 ECM extracellular matrix, ER estrogen receptor, IL interleukin, MMP matrix metalloproteinase, MCP monocyte chemoattractant protein, NF-κB nuclear factor kappa-light chain enhancer of activated B cells, NK natural killer, PGE2 prostaglandin E2, RANTES regulated-on-activation, normal-T-cell-expressed and -secreted, SF1 steroidogenic factor-1, StAR steroidogenic acute regulatory protein, TECK thymus-excreted chemokine, TGF-β transforming growth factor β, TNF-⍺ tumor necrosis factor ⍺, Treg regulatory T cells, VEGF vascular endothelial growth factor.
Figure 3 .
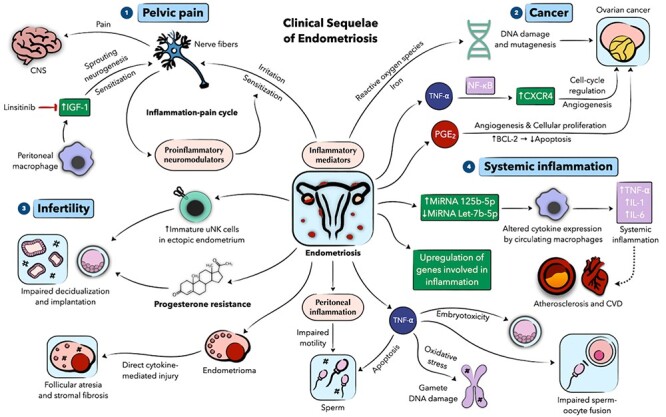
Schematic presentation of the role of inflammation in the clinical sequalae of endometriosis. (1) Proinflammatory mediators in the endometriotic environment induce irritation and sensitization of local nerve fibers, which in turn release neuromodulators that further augment this cycle, predisposing to pelvic pain development. Local macrophages contribute to pelvic pain via insulin growth factor 1 (IGF-1) that induces sprouting neurogenesis and nerve fiber sensitization. (2) Through actions of inflammatory mediators and reactive oxygen species, endometriosis may predispose to ovarian malignancy. (3) Aberrant immune cells, progesterone resistance, and peritoneal inflammation can impact fertility in endometriosis patients. (4) Endometriosis can induce a systemic inflammatory state, potentially contributing to CVD. Numeric labels are meant to guide the reader through the figure. Dotted lines denote hypothesized associations. CNS denotes central nervous system, CVD cardiovascular disease, IL interleukin, MiRNA microRNA, PGE2 prostaglandin E2, TNF-⍺ tumor necrosis factor ⍺, and uNK uterine natural killer.
Adenomyosis
Inflammation is hypothesized to actively participate in the pathogenesis of adenomyosis. The theory of tissue injury and repair (TIAR) has particularly received a great deal of attention as a possible mechanism (Figure 4). This theory encompasses the notion that mechanical strain in the form of chronic myometrial contractions may invoke microtrauma to the junctional zone, creating an inflammatory microenvironment that stimulates basal endometrial proliferation into the uterine wall, eventually forming the classical adenomyotic lesions [24]. Chronic inflammation at the endometrial–myometrial interface preceding active disease may be characterized by an interplay of macrophages, platelets, and the cytokines they secrete, promoting endometrial attachment and infiltration. In fact, this theory is also proposed to explain, at least partially, the migration of endometrial implants outside the uterus and endometriosis establishment [25]. Likewise, extrinsic adenomyosis, i.e., endometriotic lesions invading from outside the uterus, is more common in patients with deep infiltrating endometriosis, the most clinically severe form of endometriosis, and is associated with increased levels of TNF-⍺, IL-1β, and IL-33 [26, 27].
Figure 4 .
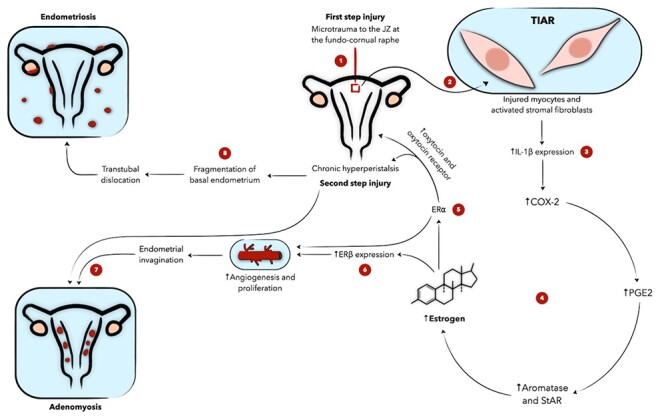
Schematic presentation of the role of inflammation in the theory of tissue injury and repair (TIAR) involved in the pathophysiology of adenomyosis and endometriosis as proposed by Leyendecker. JZ injury activates local TIAR (1, 2), leading to upregulation of inflammatory mediators and local estrogen (3, 4). Upon binding both of its receptors, estrogen perpetuates myometrial contractions and endometrial proliferation (5, 6). This amplifies the effect of TIAR and predisposes to endometrial invagination and fragmentation, leading to adenomyosis and endometriosis, respectively (7, 8). Numeric labels are meant to guide the reader through the figure. COX-2 denotes cyclooxygenase-2, ER estrogen receptor, IL interleukin, JZ junctional zone, PGE2 prostaglandin E2, StAR steroidogenic acute regulatory protein.
Extracellular and intracellular inflammatory signaling
Inflammation in BGDs constitutes an intricate network of various mediators that regulate a chain of extra- and intracellular signals, creating microenvironments that foster disease maintenance and progression. These mediators comprise cytokines, chemokines, prostaglandins, growth factors, peptides, angiogenic factors, hormones, and transcription factors [28].
Uterine leiomyoma
In uterine leiomyoma, several proinflammatory markers are found to be implicated in tumor development (Table 1). Among others, TNF-⍺ may have a particularly notable role (Figure 1). TNF-⍺ has been found to be highly expressed in leiomyoma cells compared with the normal myometrium [10]. The exact mechanism by which TNF-⍺ mediates its downstream signaling in uterine leiomyoma remains largely unknown, but a few hypotheses are recognized. It has been established that TNF-⍺ contributes to reproductive physiology through stimulating trophoblastic proliferation and differentiation soon after implantation [29], which raises the prospect that it may function similarly in uterine pathologies. Indeed, in vitro studies have demonstrated enhanced human leiomyoma cell proliferation in the presence of TNF-⍺, with increased expression of pro-proliferative and antiapoptotic makers such as BCL-2, effects that were reversed on adding anti-TNF-⍺ antibodies [30]. Lending support to these findings, Matsuo et al. reported an abundant cellular expression of BCL-2 in leiomyomas relative to the normal myometrium [31], a feature by which TNF-⍺ may block apoptotic cascades and promote tumor development. By contrast, TNF-⍺ is recognized for its proapoptotic actions elsewhere that are in fact blocked by the BCL-2 protein [32]. These observations highlight the crosstalk between TNF-⍺ and BCL-2 in regulating apoptotic and tumorigenic pathways in leiomyoma cells that are complex and not entirely comprehended, emphasizing the need to explore other in vivo factors participating in this crosstalk. Given the abundance of myometrial macrophages in uterine leiomyoma [3], it may be intuitive to assume that they mediate local TNF-⍺ production [33]; however, as leiomyoma cells are found to express TNF-⍺ as well, the exact source needs to be accurately investigated.
Table 1.
Expression and function of cytokines, chemokines, growth and transcription factors, and miRNA in uterine leiomyoma
| Mediator | Expression/activity in uterine leiomyoma | Special remarks |
|---|---|---|
| TNF-⍺ | ↑ | |
| NF-κB | ↑ | |
| Wnt/β-Catenin | ↑ | |
| Activin | ↑ | |
| TGF-β | ↑ |
|
| CXC motif chemokine 12 (CXCL12) | ↑ |
|
| MiRNA-200c | ↓ |
|
BMSC denotes bone marrow–derived stem cell, ECM extracellular matrix, IL interleukin, MiRNA microRNA, MMP matrix metalloproteinase, NF-κB nuclear factor kappa-light chain enhancer of activated B cells, TGF-β transforming growth factor β, and TNF-⍺ tumor necrosis factor ⍺.
Another experiment has demonstrated that TNF-⍺ promotes cultured leiomyoma smooth muscle cell migration through upregulating the NF-κB and extracellular signal–regulated kinase (ERK) pathways (Figure 1). Using PD98059, a specific ERK inhibitor, the same study shows that ERK signaling is required for NF-κB activation and is therefore a critical step in TNF-⍺–induced smooth muscle cell migration in uterine leiomyoma [34]. Indeed, the Ras/Raf/MEK/ERK pathway has an established role in cellular proliferation and is particularly implicated in leiomyoma pathobiology as shown by other studies [35, 36]. On the other hand, NF-κB is known for upregulating tumorigenic cytokines, such as IL-1, IL-6, and TNF-⍺ itself, and the BCL-2 protooncogene in certain tumors [37], whereas the mitogen-activated protein kinase (MAPK) pathway, also upregulated by TNF-⍺, is shown to promote cellular proliferation and differentiation [2, 38–40]. In the context of leiomyoma, both NF-κB and MAPK p38 mediate the downstream signaling of the focal adhesion kinase pathway activated by TGF-β [41]. Although TNF-⍺ seems to be a chief regulator of the aforementioned signaling cascades, whether such interconnections between inflammation and tumorigenesis occur in uterine leiomyoma remains to be confirmed.
The convergence of inflammatory and tumorigenic pathways in uterine leiomyoma may be further exemplified by the crosstalk between NF-κB and Wnt/β-Catenin signaling. Wnt/β-Catenin regulates NF-κB, both positively and negatively, with the latter shown to potentiate the mitogenic properties of Wnt/β-Catenin in some cancers [42]. Of interest, constitutive activation of Wnt/β-Catenin in mice is shown to induce mesenchymal tumors and myometrial hyperplasia that are histologically and molecularly similar to uterine leiomyoma [43], modulate leiomyoma stem cell–mature leiomyoma cell interaction, and mediate, at least partially, sex steroid-induced leiomyomatous proliferation (Figure 1) [44].
Endometriosis
Endometriosis is characterized by aberrant upregulation of cytokines, chemokines, and prostaglandins in ectopic lesions and peritoneal fluid, of which TNF-⍺, IL-1β, IL-6, macrophage migration inhibitory factor (MIF), IL-8, regulated-on-activation, normal-T-cell-expressed and -secreted (RANTES), and monocyte chemotactic protein 1 (MCP-1) are master regulators (Figure 2) [45]. Their specific roles in endometriotic inflammation have been extensively evaluated by various studies and are presented in Table 2. RANTES, MCP-1, and IL-8, which are regulated by IL-1β, function as in situ chemokines recruiting macrophages, natural killer (NK) cells, and granulocytes to endometriotic lesions and appear to contribute to angiogenesis, proliferation, and lesion remodeling by sustaining an inflammatory niche [46]. Macrophage-secreted TNF-⍺ promotes endometriotic proliferation by inducing IL-8 and IL-6 [47, 48] as well as prostaglandin (PG) E2 by means of cyclooxygenase-2 (COX-2) overexpression upon canonically activating NF-κB [49]. As will be discussed below, PGE2 is recognized as an essential mediator in endometriosis and together with estrogen, it creates a self-sustaining cycle of locally overproduced estradiol and inflammatory mediators that ensure lesion viability and proliferation. In addition, the local pool of PGE2 is further augmented by IL-1β, MIF, and VEGF [50–52]. MIF, a proinflammatory cytokine that regulates innate immune responses, is also increasingly recognized for its role in endometriosis pathogenesis, including angiogenesis, lesion proliferation, and PGE and estradiol synthesis [53, 54]. Indeed, peritoneal, peripheral, and lesional MIF levels are higher in women with endometriosis [55–57].
Table 2.
Expression and function of cytokines, chemokines, miRNA, and growth, angiogenic, and transcription factors in endometriosis
| Mediator | Expression/activity in endometriosis | Special remarks |
|---|---|---|
| IL-1β | ↑ | |
| IL-6 | ↑ | |
| IL-8 (CXCL8) | ↑ | |
| IL-9 | – |
|
| IL-10 | ↑ | |
| IL-13 | ↓ |
|
| IL-15 | ↑ |
|
| TNF-⍺ | ↑ | |
| PGE2 | ↑ | |
| MCP-1 | ↑ | |
| TGF-β | ↑ | |
| RANTES (CCL5) | ↑ |
|
| TECK | ↑ |
|
| C-C motif ligand 17 (CCL17) | ↑ |
|
| CXC motif chemokine 12 (CXCL12) | ↑ |
|
| MIF | ↑ | |
| VEGF | ↑ | |
| NF-κB | ↑ |
|
| CD200 | ↑ | |
| MiRNA | ↑ (miR-106b-3p, -451a, -486-5p, and -185-5p) | |
| ↓ (miRNA-Let-7b) |
BMSC denotes bone marrow–derived stem cell, COX-2 cyclooxygenase-2, ESC endometriotic stromal cell, E2 estradiol, ER estrogen receptor, GM-CSF granulocyte macrophage colony stimulating factor, ICAM-1 intercellular adhesion molecule-1, IL interleukin, MCP monocyte chemoattractant protein, MIF macrophage migration inhibitory factor, MiRNA microRNA, MMP matrix metalloproteinase, NF-κB nuclear factor kappa-light chain enhancer of activated B cells, NK natural killer, PGE2 prostaglandin E2, RANTES regulated-on-activation, normal-T-cell-expressed and -secreted, TECK thymus-excreted chemokine, TGF-β transforming growth factor β, TNF-⍺ tumor necrosis factor ⍺, Treg regulatory T cells, and VEGF vascular endothelial growth factor.
Although inflammatory cells secrete many of these mediators, it is now thought that endometriotic lesions themselves participate in priming the local peritoneal environment, the proendometriotic niche, by secreting a plethora of cytokines and chemokines. In fact, not only does this phenomenon contribute to local inflammation but also to creating an immunosuppressed milieu that favors initial lesion persistence and progression (Figure 2) [58]. For example, it has been shown that endometriotic stromal cell (ESC)-derived IL-15, alongside platelet-derived TGF-β, plays a role in suppressing NK cell cytotoxicity [59, 60] whereas ESC-derived thymus-expressed chemokines upregulate T-regulatory (Treg) cells, promoting local immune tolerance and lesion invasiveness [61]. In addition, Wang et al. have demonstrated that the crosstalk between ESCs and immune cells augments the inflammatory and immunosuppressive effects produced by each player alone. On the one hand, ESC-monocyte (MO) co-culture stimulates Treg chemotaxis by upregulating CCL22 and CCL17 as well as Treg-mediated TGF-β production, promoting immune tolerance and angiogenesis, respectively. On the other hand, ESC-MO co-culture induces local expression of IL-1β and TNF-⍺, which synergistically with TGF-β, are also angiogenic [62]. Intriguingly, endometriosis-associated haptoglobin (ENDO-1), an extrahepatic haptoglobin expressed by endometriotic lesions, is postulated to alter peritoneal macrophage function, possibly contributing to the disease [63], but its role beyond these speculations warrants further investigation.
Intracellularly, NF-κB is a crucial transcription factor in endometriotic inflammation and manifests increased constitutive activation in peritoneal macrophages and red (active) peritoneal lesions compared with black lesions [64]. Not only has NF-κB been implicated in early development of in vivo endometriosis but also in promoting ongoing lesion proliferation, adhesion, angiogenesis, and invasion (Figure 2) [65]. TNF-⍺ and IL-1β, on the one hand, and iron overload, on the other hand, seem to activate the canonical and atypical NF-κB pathways, respectively, in macrophages and ectopic endometrial cells, amplifying local cytokine expression and generating a positive feedback loop of autocrine signaling that facilitates lesion progression [64, 65]. Although may differ in other cell types, NF-κB confers resistance against apoptosis and promotes survival of endometriotic cells, an effect when blocked shown to reverse these phenomena, both in vivo and in vitro, through downregulating IL-8, MIF, BCL-2, and Bcl-XL and activating caspases [65, 66]. Nevertheless, NF-κB role in endometriosis is far more complex and involves the interaction of various cofactors, depicting the unique functionality of NF-κB in this disorder.
Adenomyosis
Expanding on the aforementioned TIAR theory of adenomyosis development, microtrauma to the junctional zone at the fundo-cornual raphe generates myocyte and fibroblast injury, upregulating inflammatory cascades, mainly involving IL-1β, which induce COX-2 and in turn PGE2 overexpression (Figure 4). Inflammatory cascades then go on to activate local hormonal production as will be explained in the next section, perpetuating the injurious cycle and lesion development as proposed by Leyendecker et al. [24]. Indeed, intramural lesions are also found to increasingly express COX-2 and lipoxygenase-5 (LOX-5) that correlate with local IL-6 and IL-8 levels, lending support to the inflammatory hypothesis of adenomyosis pathogenesis [4]; however, the exact roles of these cytokines in regulating adenomyotic inflammation merit further investigation. Another study concluded that TNF-⍺ and VEGF can induce CXCL1 chemokine release in human endometrial epithelial cells derived from the endometrium of adenomyosis patients. This effect is more robust with VEGF and mediated by several pathways, including NF-κB activation and IκB phosphorylation. In turn, CXCL1 attracts vascular endothelial cell migration, promoting local neovascularization and adenomyosis progression [67].
Sex steroids
Female sex steroids have inflammation-modulating properties that may explain, at least in part, the link inflammation has with BGDs (Figure 5). Estrogen regulates immune responses and influences, both genomically and nongenomically, proinflammatory cytokine production [68]. Nevertheless, there is a conflicting body of evidence as to whether estrogen promotes or ameliorates inflammation. Whereas estrogen is known for its anti-inflammatory properties and suppressive effects on major proinflammatory transcription factors such as NF-κB [68] that are known contributors to some BGDs [64], it can alternatively promote inflammatory milieus of the same array of diseases [69]. This intriguing paradox was thoroughly addressed by Straub in his review, where the effects of estrogen on inflammation have been demonstrated to be reliant on many factors, including immune stimuli, cell types involved, afflicted organ and surrounding microenvironment, as well as variable expression of estrogen receptors ⍺ and β [70].
Figure 5 .
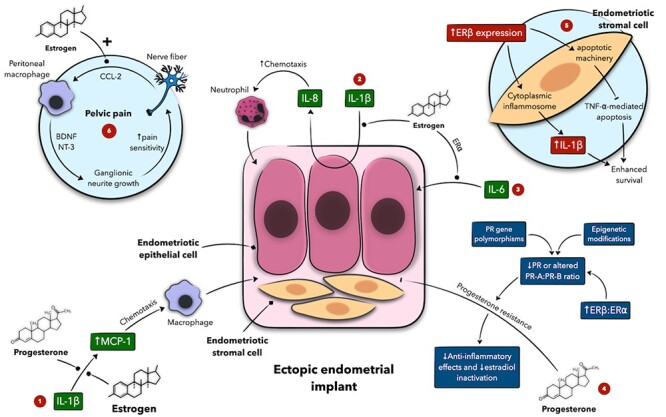
Schematic presentation of the interplay between sex steroids and inflammation in the pathogenesis of endometriosis and associated pelvic pain. Estrogen and progesterone modulate IL-β-mediated MCP-1 expression, which acts as a macrophage chemoattractant (1) whereas estrogen also enhances IL-β-mediated IL-8 secretion by endometriotic cells (2) and modulates IL-6 actions on these cells (3). Progesterone resistance deprives the local peritoneal environment of progesterone anti-inflammatory actions and facilitates endometriosis progression (4). (5) Represents the crosstalk between estrogen receptor (ER) β and inflammatory mediators in endometriotic stromal cells. (6) Estrogen stimulates nerve fibers to produce CCL-2, attracting macrophages, which release BDNF and NT-3. These mediators promote neurite growth, increasing pain sensitivity. Estrogen and progesterone actions shown here are largely modulatory in nature as hormone-mediated activation or inhibition can be receptor-dependent. Numeric labels are meant to guide the reader through the figure. + denotes an activating function. BDNF denotes brain-derived neurotrophic factor, CCL-2 C-C motif ligand 2, IL interleukin, MCP monocyte chemoattractant protein, NT-3 neurotrophin-3, and PR progesterone receptor.
Endometriosis
In women with endometriosis, peritoneal fluid macrophages manifest higher expression of IL-6, IL-1β, and TNF-⍺ that significantly correlate with macrophage estrogen receptor (ER) expression. For ERβ, however, this correlation was also seen in women without the disease, whereas for ER⍺, it was exclusively noted in endometriosis. This may suggest a particular role for ER⍺ in mediating inflammation in endometriosis as opposed to ERβ that may rather maintain a baseline inflammatory status in the nondiseased peritoneum [71]. ER⍺ is known to modulate IL-6 through CEBPβ and NF-κB pathways [72, 73], and Burns et al. have indeed shown the importance of this crosstalk in endometriotic lesion development (Figure 5) [74]. However, this demarcation between the inflammatory properties of ER⍺ and ERβ is not always clear-cut in endometriosis. For instance, ER expression in ESCs is unique in that ERβ expression is markedly higher compared with ER⍺ at a ratio of almost 16:1, which may point to a possible role for ERβ in endometriotic inflammation. ERβ participates in the local estradiol-PG production cycle by mediating estrogen induction of COX-2 and contributes to a state of progesterone resistance seen in endometriosis, aggravating the imbalance between pro- and anti-inflammatory pathways [75]. In addition, ERβ overexpression in ESCs appears to inhibit TNF-⍺–mediated apoptosis through interaction with the cytoplasmic apoptotic machinery and induce IL-1β expression, enhancing cell survival through interaction with the cytoplasmic inflammasome [76].
Intriguingly, local estradiol may be indirectly linked to neutrophil chemotaxis through enhancing epithelial and stromal endometriotic cell responsiveness to IL-1, thus increasing the secretion of the chemokine IL-8 [77]. Estradiol is also shown to enhance IL-1β–mediated production of MCP-1 in endometriotic lesions, in turn promoting chemoattraction of immune mediators, particularly macrophages (Figure 5) [15, 78]. Nevertheless, Arici et al. has shown that estradiol rather inhibits MCP-1 expression in ESC possibly via a receptor-mediated mechanism [79]. This discrepancy in the estrogenic effect on MCP-1 may be attributed to various in vivo factors, including aberrant estrogen receptor expression, that may modulate this interaction and would be difficult to account for in vitro. In line with this observation, cotransfection of endometrial epithelial cells with ER⍺ and ERβ dampens ERβ-mediated IL-1β production, suggesting an inhibitory role for ER⍺ that may attenuate ERβ-promoted inflammation and MCP-1 production [80].
Progesterone resistance is hypothesized to additionally contribute to a proinflammatory phenotype in the setting of endometriosis (Figure 5). Together with estrogen, progesterone can exert anti-inflammatory properties by inhibiting MCP-1 [79]. However, endometriotic lesions are limited in progesterone receptor (PR) expression, possess progesterone receptor (PR) gene polymorphisms and altered miRNA expression, and harbor epigenetic modifications to PRs and downstream targets. Thus, deviant progesterone signaling may be an additional driver of endometriotic inflammation [15, 81]. The interplay of hormones and inflammation in endometriosis does not only involve cytokines and extracellular signaling but also extends to influence transcriptional function. On the one hand, ERs in ESCs inhibit the assembly of NF-κB subunits, whereas the latter, once activated, decreases ER⍺ responsiveness to estrogen in these cells [82], which may be counterintuitive knowing that both NF-κB and estrogen have crucial roles in endometriosis. On the other hand, Shen et al. have concluded that increased NF-κB activation and decreased PR-B immunoreactivity, in the context of their close relationship, jointly constitutes a good biomarker to predict recurrence of ovarian endometrioma [83]. This observation may imply an inverse association between progesterone action and NF-κB activation that is accentuated in endometriosis due to progesterone resistance.
On the contrary, the reciprocal effects of inflammation on estrogen are similarly elucidated in endometriosis. As previously discussed, cytokine-driven production of PGE2 plays a central role in lesional inflammation. Unlike the eutopic endometrium, endometriotic stromal cells possess exceptionally high levels of P450arom (aromatase) and steroidogenic acute regulatory protein (StAR) that are induced by PGE2 to produce estradiol de novo [84]. The latter, in turn induces PGE2 production, which goes on to produce more estradiol [85]. This positive feedback loop of local estradiol overproduction is now recognized as one of the hallmarks of endometriosis pathobiology, characterizing the complex interplay of estrogen and inflammation in this disorder (Figure 2).
Uterine leiomyoma
The role of estrogen and progesterone in inflammation is just as complex in uterine leiomyoma. Similar to endometriosis, differential expression of ERs may play a role in myometrial inflammation. Uterine ERβ is shown to modulate the proinflammatory actions of ER⍺ by inhibiting its stimulatory effect on IL-1 secretion [86]. This role-reversal of ER function as compared to what happens in ectopic endometrial implants of endometriosis is not fully understood. It has been demonstrated that ER⍺ mRNA is highly expressed in leiomyoma cells but further studies are necessary to verify its role in promoting a downstream inflammatory pathway in these tumors [87]. Besides its proven tumorigenic properties in uterine leiomyoma [88], progesterone is found to exert anti-inflammatory actions by decreasing TNF-⍺ expression [10]. Nevertheless, high TNF-⍺ levels in leiomyomas may imply the intrusion of other mediators that counteract progesterone action [40]. On the other hand, TNF-⍺ induces aromatase activity and influences estrogen metabolism, rendering the latter more available, potent, and tumorigenic (Figure 1) [89, 90].
Adenomyosis
In adenomyosis, intramural chronic inflammation potentiates local production of estrogen by estrone sulfatase and aromatase [91]. Aromatase and StAR upregulation is mediated by COX-2, and the locally overproduced estrogen induces ERβ overexpression, which alongside ER⍺, stimulates local angiogenesis and endometrial proliferation (Figure 4) [24]. Estrogen, through actions of ER⍺, upregulates oxytocin and oxytocin receptor expression, inducing a state of persistent myometrial hyperperistalsis likely originating from the junctional zone. This hormone-driven action further augments autotraumatization, creating a self-perpetuating vicious cycle that culminates in endometrial invagination into the uterine wall [24, 92].
Genetics
Uterine leiomyoma
A genetic link between inflammation and BGDs has been documented in the literature. Multiple studies show that uterine leiomyomas are associated with polymorphisms in IL-6, IL-1β, and TNF-⍺ genes [93–95], which may be used to predict susceptibility to myoma development [94]. In addition, somatic gain-of-function mutations in the mediator complex subunit 12 (MED12) gene are found to play a critical role in leiomyoma pathogenesis [96]. Al-Hendy et al. have demonstrated that MED12-knockdown human leiomyoma cells have less proliferative activity, fibrogenesis, and sex steroid receptor expression, highlighting the tumorigenic properties of MED12 [97]. Interestingly, MED12 is additionally involved in NF-κB signaling [98], suggesting a mechanism by which mutated MED12 may link tumorigenesis and inflammation.
Endometriosis
Antiñolo et al. have postulated that gene variants encoding for CCR5 and CCR2, the receptors for RANTES and MCP-1, respectively, may alter the transcription and expression of these receptors in the endometrium and, hence, the interaction with their ligands, contributing to endometriosis; however, no association has been detected in their study [99]. On the other hand, Ahn et al. have explored gene signatures involved in inflammation in endometriosis patients. They have concluded that endometriotic lesions are molecularly distinct compared with the eutopic endometrium of healthy women and detected 396 genes significantly different in ectopic tissues, including those involved in cytokine–cytokine receptor interaction (IL-18, CCL5, CCR2), NK cell cytotoxicity (NFIL3, GNLY, IL-15), and TNF and MAPK signaling (TRAF4, MAPK1, DUSP4) [100].
Epigenetics
Epigenetic phenomena have been reported between inflammation and BGDs. As previously discussed, MED12 may be implicated in both leiomyoma and inflammatory pathways, and mutations in this gene may be, in part, subsequent to epigenetic alterations in myometrial cell DNA [101]. Trimethylated histone 3 at lysine residue 27 (H3K27me3), on the other hand, has been studied for its role in endometriosis pathogenesis. This repressive mark is created by epigenetic methylation, a process remarkably noted in endometriosis, and found to halt endometrial progression from a proliferative to a decidual phenotype [102]. However, Colón-Caraballo et al. have not detected significantly different H3K27me3 levels between healthy women and those with endometriosis but detected higher levels in the secretory eutopic endometrium of both cohorts [103]. Of particular importance, H3K27me3 can be induced by inflammatory environments, such as colitis, as demonstrated by Takeshima et al. [104], possibly implying inflammation-induced epigenetic changes in endometriosis that merit future investigation. Another inflammatory mediator shown to epigenetically modulate expression of genes involved in endometriotic pathogenesis is PGE2, as part of the local estradiol-PG cycle (Figure 2). PGE2 mediates these actions via the transcription factor steroidogenic factor-1 (SF1), which binds to P450arom and StAR promoters, upregulating their expression and fueling local estradiol synthesis [84]. This further symbolizes the intricately regulated cointeraction between inflammation and hormones that exists on many levels in endometriosis.
Intriguingly, endometriosis is shown to reciprocally induce gene expression in peripheral leukocytes already delineated in nongynecologic inflammatory diseases (Figure 3). Gentilini et al. have demonstrated that endometriosis upregulates 26 genes involved in inflammation, including pre–B-cell colony enhancing factor 1 (PBEF1) and dual specificity phosphatase 1 (DUSP1), which are linked to rheumatoid arthritis and insulin resistance, disease states known for their core inflammatory components. Notably, this gene expression is ameliorated after surgical removal of endometriosis [105]. These observations depict endometriosis as having epigenetic modulating properties and suggest a causative, lesion-autonomous role in systemic disease.
Extracellular matrix and stromal elements
Uterine leiomyoma
A growing body of evidence suggests an impact of chronic inflammation on ECM remodeling in BGDs. Inciting myometrial events may induce a microinflammatory state that promotes fibrogenesis and leiomyoma development [28]. Tissue macrophages appear to actively participate in attempting tissue repair and activating myofibroblasts, a key event in leiomyomatous fibrogenesis (Figure 1) [106]. Myofibroblasts are collagen-producing fibroblasts recruited and transformed by actions of growth factors, of which macrophage-secreted TGF-β is chief [8]. Being a pleiotropic cytokine and growth factor, TGF-β is involved in various processes in uterine leiomyoma, including tumorigenesis, ECM synthesis, and inflammation [40]. In fact, the aforementioned NF-κB–regulated Wnt/β-catenin pathway is shown to stimulate TGF-β3 expression and fibronectin deposition [43, 107], implying another mechanism by which inflammation and fibrogenesis are cross-regulated.
Activin A is another central profibrotic mediator in the TGF-β family secreted by proinflammatory macrophages and upregulated by TNF-⍺ in leiomyomas [108]. Not only is activin A postulated to induce myofibroblast transition and fibrogenesis but also shown to inhibit the anti-inflammatory macrophage phenotype and activate the proinflammatory phenotype in a positive feedback mechanism that perpetuates local inflammation and ECM deposition [108]. TNF-⍺ modulating role in ECM may be mediated, at least partially, via ERKs, which exert mitogenic properties through the Ras/Raf/MEK/ERK signaling cascade, a proposed pathway in uterine leiomyoma pathogenesis [35, 41].
Endometriosis
Inflammation has demonstrated the ability to alter ECM physiology in endometriosis, facilitating central disease processes, such as invasion and lesion progression. Locally produced TNF-⍺, TGF-β, and IL-1 stimulate matrix metalloproteinases (MMPs) 1, 2, and 3 expression, which are essential for ECM remodeling and peritoneal invasion (Figure 2) [109]. Further, NF-κB has an additional role in promoting cell adhesion and invasion of endometriotic cells into the submesothelial space by positively modulating MMP-9 expression [47]. Indeed, increased activation of NF-κB and expression of MMPs and activins in deep infiltrating endometriosis as compared with ovarian endometriosis and superficial peritoneal endometriosis correlate with a more invasive phenotype [110]. Although NF-κB seems to also positively regulate plasminogen activators involved in ECM remodeling, this particular effect is yet to be proven in endometriosis [111]. As local inflammation intensifies, mesothelial-to-mesenchymal transition of the peritoneal lining can also be seen, a process that triggers peritoneal fibrosis, adhesion, and angiogenesis [112]. Angiogenesis, although beyond the scope of this review, is inarguably an essential process in the progression of early disease and is similarly regulated by proangiogenic cytokines, including TNF-⍺, IL-1, IL-6, and IL-8, to name a few [113, 114], which are in turn modulated by NF-κB as well. For example, NF-κB–regulated MIF manifests proproliferative properties in endothelial cells in vitro [115, 116] whereas in vivo, it is more expressed in red lesions compared with black lesions [57], emphasizing the versatile yet central role NF-κB plays in endometriosis.
Stem cells
Uterine leiomyoma
Undifferentiated myometrial stem cells are proposed as potential culprits in uterine leiomyoma development [117]. Of importance, they are postulated to maintain a chronic inflammatory microenvironment that favors tumor sustainability by means of immune cell modulation and cytokine regulation (Figure 1). Indeed, Orciani et al. [118] have documented a significant differential expression of Th2 and Th1/Th17 pathways in myometrial progenitor cells (MPCs) and leiomyoma progenitor cells (LPCs). In LPCs, greater expression of Th2 pathway cytokines is noted, including IL-4, IL-5, IL-10, and IL-13, as opposed to lower expression of Th1/Th17 pathway cytokines, TGF-β in particular. The latter mediates differentiation of naïve T cells to Treg cells, which are essentially involved in suppressing inflammation whereas Th2 cells are implicated in promoting chronic inflammation. Therefore, these observations suggest that LPCs may actively promote leiomyoma development and maintenance of a proinflammatory state. Embryologically, it is still unclear as to whether LPCs intrinsic to the myometrial stroma are responsible for the initial generation of leiomyomas, although the evidence above suggests that LPCs participate in at least the maintenance phase of tumor growth. It would be interesting to determine whether LPCs could be identified during development well before their clinical manifestation in adulthood. Besides local stem cells, uterine leiomyoma is capable of secreting the chemokine CXCL12, which recruits bone marrow–derived stem cells (BMSCs) to the tumor, perhaps facilitating tumor growth through engrafting stem cells into the leiomyoma [119].
Endometriosis
A role for BMSCs is also observed in endometriosis (Figure 2). Particularly, CXCL12 is found to promote BMSC recruitment to the peritoneal cavity where they undergo cellular transformation under the influence of various in situ cytokines [58, 120]. This process is further enhanced by estradiol, which is shown to upregulate CXCL12 expression [121]. Additionally, BMSCs are found to secrete VEGF, MCP-1, as well as other proinflammatory cytokines and MMPs, accentuating angiogenesis and lesion migration and contributing to local inflammation and disease progression [122].
Cardiometabolic risk factors
Our group has previously reviewed the association of cardiometabolic risk factors (CMRFs) with BGDs and addressed the role of inflammation in this association [123]. Briefly, CMRFs, including obesity, hypertension, hyperlipidemia, and diabetes mellitus, may result from or contribute to local and systemic inflammatory milieus [124]. In obese individuals, for example, adipose tissue secretes various proinflammatory cytokines and adipokines, introducing the concept of metabolic inflammation or metaflammation, i.e., low-grade, chronic inflammation regulated by metabolically active cells in response to excess nutrients and energy [125]. Relating to BGDs, it is found that human leiomyoma cells demonstrate increased proliferation when cultured in adipocyte-conditioned media or co-cultured with human adipocytes, observations attributed to TNF-⍺ [30]. In particular, visceral adipose tissue is the major contributor to obesity-induced inflammation and insulin resistance [126]. Indeed, Sun et al. have concluded that increased visceral adipose tissue correlates relatively weakly, however, with the risk and size of uterine leiomyoma in women [127].
Adipokines are specific cytokines secreted by adipose tissue. Evidence suggests that a dysregulated adipokine profile manifested by obesity leads to chronic inflammation, promotes angiogenesis and cellular proliferation, and alters antitumor immune responses [128]. Key adipokines, including leptin and adiponectin, are implicated in leiomyoma pathophysiology. Evidence reveals that the leptin gene and protein, along with the leptin receptor, are preferentially expressed in uterine leiomyoma but absent in the normal myometrium [129], whereas serum adiponectin is significantly lower among women with the tumor [130]. Besides its proinflammatory properties, leptin is found to promote cellular proliferation and angiogenesis while suppressing apoptosis in leiomyoma cells [126]. Conversely, adiponectin manifests anti-inflammatory and antineoplastic properties. With the former being elevated in obesity and the latter being reduced, aberrant adipokine alterations may in fact be linked to inflammation and subsequent leiomyoma tumorigenesis [126].
Addressing the association between hypertension and uterine leiomyoma, multiple hypotheses are raised to explain a role for inflammation via the renin–angiotensin–aldosterone system. First, angiotensin II and aldosterone contribute to cellular proliferation, fibrosis, and inflammation by inducing proinflammatory cytokine production. In particular, angiotensin II is proposed to activate the NF-κB signaling pathway via angiotensin II receptor 2 [131]. Isobe et al. have documented that angiotensin II can additionally induce leiomyomatous proliferation, however, via angiotensin II receptor 1 [132]. Reciprocally, proinflammatory cytokines are also hypothesized to regulate hepatic and renal production of angiotensinogen, predisposing to angiotensin II-dependent hypertension [133]. Regardless of angiotensin II direct actions, hypertension may induce smooth muscle injury and proinflammatory milieus, promoting leiomyomatous development possibly through TGF-β [134], a mechanism reminiscent of atheromatous plaque formation. From a clinical perspective, Faerstein et al. have concluded that hypertensive women are more likely to have uterine leiomyoma compared with their nonhypertensive counterparts, and that women with the tumor have longer durations of hypertension [135]. Further support has come from genetic analysis showing that angiotensin converting enzyme (ACE) gene polymorphisms, specifically, the I/D and D/D genotypes, confer a 2–3 times higher risk of leiomyoma development as compared with the I/I genotype [136]. Nevertheless, further studies are warranted to verify these observations and elucidate a causal role for inflammation.
Diet and vitamin D
A role for diet is implicated in both inflammation and BGDs. The literature reveals that nutritional deficiencies, such as folic acid, vitamin B12, zinc, and choline deficiencies, may result in epigenetic changes that predispose to endometriotic inflammation. CpG hypomethylation, for example, can upgrade the expression of SF1 and ERβ, and therefore, estradiol and PGE2, fueling local estrogen production and disease progression [137]. Parazzini et al. have demonstrated an increased risk of endometriosis with trans-unsaturated fat and palmitic acid consumption but not with other components of animal fats, including saturated and monosaturated fats [138]. From a pathophysiological perspective, trans fatty acids increase serum levels of proinflammatory cytokines, such as TNF-⍺ and IL-6 [139], which are established players in endometriosis. In addition, trans fatty acids downregulate peroxisome proliferator–activated receptor 𝛾 (PPAR 𝛾) expression, a nuclear receptor that modulates anti-inflammatory functions. Notably, PPAR 𝛾 can promote the regression of surgically induced endometriosis in rats, possibly conferring a protective mechanism against the disease [140].
Vitamin D can downregulate proinflammatory cytokine production, such as TNF-⍺ and IL-6 [141], and suppress endometrial COX-2 expression and prostaglandin synthesis [137]. Women with endometriosis have lower vitamin D levels [141], possibly implying that vitamin D deficiency may allow proliferative and inflammatory processes in endometriosis to progress uninhibited. Similarly, vitamin D deficiency is associated with increased incidence of uterine leiomyoma. Elhusseini et al. have provided evidence that vitamin D deficiency may promote a chronic inflammatory state in the murine myometrium and induce DNA damage, facilitating tumorigenesis [142]. Moreover, vitamin D ameliorates the effects of TGF-β3 and collagen 1 expression and modulates MMPs, suggesting a role for vitamin D deficiency in inducing ECM deposition in leiomyomatous growth [143].
Immune mechanisms
Endometriosis
Immune cells are key masters of inflammation, and dysregulation of the immune system has been implicated in BGD pathobiology. Aberrancies in NK and Treg cell function are of particular interest (Figure 2). Decreased NK cell cytotoxicity may result in impaired immunosurveillance and defective lesion clearance [144]. The lymphocyte function–associated antigen-1 (LFA1)–intercellular adhesion molecule-1 (ICAM-1) pathway is another mechanism by which NK function is altered. Normally, lymphocytes expressing LFA1 bind to peritoneal endometrial cells expressing ICAM-1, facilitating their clearance by NK cells; however, endometriotic cells shed a soluble form of ICAM-1 that competes for LFA1 binding, rendering endometriotic lesions less recognizable by NK cells [145]. While their function may be impaired, some studies found an increased number of NK cells peripherally [146] and higher expression of chemokines, namely CXCL12 and CX3CL, involved in NK chemotaxis locally [144]. This may be explained by an unsuccessful attempt of the immune system to eliminate ectopic implants leading to lesion persistence. Further, it is proposed that dominance of Foxp3+ Treg cells in the eutopic endometrium of women with endometriosis may dampen the ability of immune cells to recognize and mount a response against shed endometrial cells, facilitating ectopic implantation. Treg cells mediate this effect by secreting inhibitory cytokines that suppress immunosurveillance by NK cells, macrophages, and CD4+ and CD8+ lymphocytes [147].
Uterine leiomyoma and adenomyosis
In uterine leiomyoma, local chronic inflammatory microenvironments are hypothesized to occur due to systemic dysfunction of the immune system. In contrast to endometriosis, this dysfunction is characterized by low levels of Treg cells, which essentially inhibit improper inflammatory signals that would otherwise contribute to leiomyoma development [11]. In adenomyosis, a role for the immune system was reported, but the exact biological mechanisms are yet to be fully explored. Gui et al. were first to investigate Th17 and Treg cell involvement in patients with adenomyosis. They found a significantly higher level of Th17 cells in women with focal and diffuse adenomyosis as opposed to a significantly lower level of Treg cells, suggesting a Th17–Treg cell imbalance [148]. Although the contribution of this finding to adenomyosis pathophysiology remains undefined, it resembles what has been previously seen in autoimmune diseases [149], providing plausible analogies that may ground future experimental studies.
Clinical manifestations and sequelae
Pelvic pain
Pelvic pain is a common symptom and presentation of BGDs, and inflammation may contribute to its complex biology. In uterine leiomyoma, TNF-⍺ may be a key regulator given its well-documented role as a pain inducer [150]. In addition, TNF-⍺ facilitates production of prostaglandins, including PGE2 and PGF2⍺ [151], which are strongly implicated in dysmenorrhea. In endometriosis, local inflammation mediates pain through nerve ending irritation, and in turn nerve fibers modulate release of proinflammatory neuromodulators, further contributing to pelvic pain in a process known as neurogenic inflammation (Figure 3) [152]. Various proinflammatory cytokines, chemokines, and growth factors directly induce peripheral nerve sensitization and participate in feedback loops, perpetuating the inflammation–pain cycle [7]. In addition, chronic inflammatory milieus, including those of endometriosis, may induce nerve fiber redistribution, wherein local sensory fibers increase and sympathetic fibers are lost, possibly to preserve local inflammatory microenvironments while increasing pain signaling [153].
Intriguingly, disease-modified macrophages appear to assume a role in pelvic pain pathophysiology in endometriosis mediated by insulin-like growth factor 1 (IGF-1), a possible neurotrophic and sensitizing agent (Figure 3) [154]. Indeed, peritoneal IGF-1 levels are found to be elevated in women with endometriosis and positively correlated with their pain scores. IGF-1 mechanistic role in endometriotic pain seems to occur through sprouting neurogenesis and nerve sensitization as shown in vitro by Forster et al. Further, pain behaviors in mice with endometriosis were reversed by linsitinib, an IGF-1 receptor inhibitor [154]. As shown by another study, macrophage–nerve crosstalk in endometriosis is modulated by locally produced estradiol. Estradiol induces nerve fiber production of the chemokine C-C motif ligand 2 (CCL-2), recruiting macrophages, which in turn express brain-derived neurotrophic factor (BDNF) and neurotrophin-3 (NT-3) (Figure 5) [155]. These mediators promote ganglionic neurite outgrowths, potentially contributing to increased pain sensitivity and highlighting, once again, the complex interaction between hormones and inflammation in endometriosis. On the other hand, gene expression of TLRs is found to be increased in the eutopic endometrium of adenomyosis patients reporting debilitating dysmenorrhea and heavy menstrual bleeding [156]. TLRs are key players in activating the endometrial innate immune system and inflammatory pathways and may therefore be implicated in modulating pelvic pain severity in adenomyosis [156]. Nonetheless, this association remains largely hypothetical, and additional research is needed to elucidate its validity.
Abnormal uterine bleeding
Abnormal uterine bleeding (AUB) is a prevalent and bothersome symptom of BGDs, such as uterine leiomyoma and adenomyosis, that may be associated with inflammation [157]. Women with AUB have increased endometrial levels of TNF-⍺, an angiogenic cytokine [158]. In vivo and in vitro studies show that transmembrane expression of TNF-⍺ exerts permissive properties by sensitizing endothelial cells to VGEF, increasing their vascular permeability [159]. Tabibzadeh et al. suggest that TNF-⍺ may be a key mediator of endometrial shedding and bleeding through inhibition of endometrial proliferation and apoptosis induction, promotion of epithelial cell–cell dissociation, and disruption of vascular integrity. Epithelial dyscohesion is mediated by leukocyte infiltration and trafficking, which are greatly facilitated by TNF-⍺ [160]. In fact, Shalaby et al. have demonstrated that TNF-⍺ administration in mice leads to vascular damage and endometrial hemorrhage, an event reminiscent of human menstrual bleeding [161]. As stated previously, TNF-⍺, alongside IL-1, promotes release of PGE2 and PGF2⍺ in cultured human luteal phase endometrial cells, thus facilitating vasoconstriction and endometrial shedding [151]. Although these observations point to a notable role for inflammation in AUB, whether it truly occurs in BGDs remains to be investigated.
Infertility
Infertility afflicts a significant proportion of women with endometriosis, uterine leiomyoma, and adenomyosis, and inflammation may contribute to its occurrence. Although mechanisms of endometriosis-associated infertility (EAI) remain largely unknown, a few hypotheses are suggested (Figure 3). Infertile women with endometriosis have increased density of immature CD16+ cytotoxic uterine NK cells in their eutopic endometrium [162]. These cells can influence the development of placental vasculature and secrete proinflammatory cytokines implicated in trophoblastic invasion [163]. Aberrancies in these cells may create inflammatory environments that hinder decidualization and successful implantation and are observed in recurrent pregnancy loss and EAI [162]. In addition, progesterone resistance in endometriosis deprives the endometrium of progesterone anti-inflammatory properties and may induce production of proinflammatory cytokines, prostaglandins, and reactive oxygen species, leading to implantation failure [164].
In addition, inflammation in endometriosis is shown to upregulate ER and aromatase expression, altering estrogen metabolism. This combination of progesterone resistance and estrogen dominance, if proven to occur in the eutopic endometrium, can be detrimental to implantation [165]. The effect of inflammation in endometriosis is also observed on gametes. Peritoneal inflammation can be directly toxic to oocytes and sperm by means of proinflammatory cytokines. For example, IL-1 and MIF, alongside other cytokines, can impair sperm mobilization [166, 167] whereas TNF-⍺ can interfere with sperm–oocyte fusion and induce sperm apoptosis and DNA damage through oxidative stress [167, 168]. Endometriomas have similar effects on adjacent ovarian follicles. Cystic fluid of an endometrioma is rich with proinflammatory cytokines and can contribute to follicular atresia and decreased density as well as stromal fibrosis, decreasing ovarian reserve and possibly fertility [169, 170]. Of note, surgical ovarian endometrioma excision does not positively impact antral follicle count, although it is associated with improvement in spontaneous conception rates of up to 30–50% among women with EAI [171], implying that endometrioma may have permanent effects on fertility possibly through inflammation and reactive oxygen species.
Local inflammation in uterine leiomyoma may similarly establish an endometrial milieu that disfavors sperm transport and zygote implantation. TNF-⍺ and IL-1 seem to play a substantial role in inhibiting decidualization of endometrial stromal cells by inhibiting cAMP-stimulated prolactin production [172]. Leiomyomatous and adenomyotic uteri have significantly higher expression of cytokines, including TGF-β1, and MMPs, which may influence the uterine decidua and endometrial receptivity [173]. Obesity, regardless of a BGD diagnosis, may also induce ovarian inflammation and reduce oocyte quality and fertility potential. Innate immune cells populate the ovaries of obese women and animal models, with evidence of inflammatory signaling and oxidative stress. TNFα, IL-6, and IL-8 expression and NF-κB signal transduction are increased in ovaries from obese women and mice that may occur on similar pathophysiological grounds in BGDs [174]. Nevertheless, research is needed to further explore the mechanisms, implications, and treatment strategies addressing the role of inflammation in infertility.
Cardiovascular disease
Systemic inflammation in endometriosis is suggested to predispose to cardiovascular disease, and endometriosis may be perceived as a cardiovascular risk factor [5, 123]. In a large cohort study designed to examine the prospective association between laparoscopically confirmed endometriosis and subsequent coronary heart disease, women with endometriosis had higher risk of various adverse cardiovascular outcomes, including angina, myocardial infarction, coronary angioplasty, and coronary artery bypass graft surgery [175]. Whether these observations are causal or merely confounded by common genetics, therapeutic agents used for, or stress associated with endometriosis remains to be determined; nevertheless, underlying biological mechanisms are suggested. It has become well established that atherosclerosis is largely driven by inflammation either directly by induction and maintenance of endothelial injury or indirectly by contributing to insulin resistance and lipid derangements [176]. It is shown that European women with endometriosis have significantly higher rates of subclinical atherosclerosis, increasing their risk of cardiovascular disease, and are more likely to have endothelial dysfunction even in the absence of structural atherosclerotic lesions [177]. In another study, regression of endothelial dysfunction was documented in women after surgical resection of endometriotic lesions [178], reinforcing the plausibility of this hypothesis.
Gynecologic cancer
Some BGDs are now recognized as precursors to gynecologic malignancies, and inflammation may mediate part of this association. Whereas endometriosis has long been known as a benign disease, evidence shows its contribution to low-grade serous, endometrioid, and clear cell ovarian carcinoma development (Figure 3) [179]. Experimental studies have, in fact, detected mutations in endometriosis-associated cancers that are also found in adjacent endometriotic lesions [180]. It is postulated that inflammation in endometriosis may allow tissue exposure to reactive oxygen species and iron, inducing DNA damage and mutagenesis [16, 181]. In particular, ovarian clear cell carcinoma is often attributed to microsatellite instability with the mismatch repair system being particularly susceptible to oxidative stress-induced damage in the setting of chronic inflammation [182, 183]. In addition, ovarian endometrioid adenocarcinoma is commonly found to have KRAS mutations, correlating with similar mutations in deep infiltrating endometriosis lesions in the same patients, suggesting a common mechanism. Although it is unclear whether these mutations are inflammation induced, NF-κB dysfunction evident in this tumor may be another mechanism that links inflammation to carcinogenesis [182, 183]. The relationship between ovarian low-grade serous carcinoma and endometriosis can often be linked via BRAF mutations often found in cancer cells, eutopic endometrium, and ectopic endometrium from these patients, although a potential role for inflammation in this particular cancer subtype remains less clear [183].
Proinflammatory cytokines are also demonstrated to promote epithelial ovarian cancer development and progression [184], and TNF-⍺ involvement in particular was studied in vitro. TNF-⍺ mRNA is found to be expressed up to 1000 times more in cultured ovarian cancer cells compared with normal ovarian epithelium [185]. Additionally, TNF-⍺ gene expression appears to take part in cell cycle regulation, adhesion, and angiogenesis possibly through NF-κB–mediated induction of CXCR4, a chemokine receptor expressed in epithelial ovarian cancer [186]. PGE2, another crucial inflammatory mediator in endometriosis, can also influence cellular proliferation and angiogenesis and suppress apoptosis in ovarian cancer by increasing expression of the antiapoptotic protein BCL-2 [187]. Nevertheless, it remains an unsolved mystery as to why certain inflammatory states found in BGDs lead to carcinogenesis while others lead to benign growths without significant risk of malignant transformation.
Clinical implications
Diagnostic implications
Inflammatory markers
Various inflammatory markers have been evaluated in the search for potential noninvasive diagnostic adjuncts for BGDs. To begin with, serum TNF-⍺ levels may help diagnose uterine leiomyoma and differentiate it from other pathologies, such as smooth muscle tumors of uncertain malignant potential (STUMP lesions) and leiomyosarcoma, using a level cutoff point [13, 40]. In addition, TNF-⍺ levels may assist in evaluating the risk of clinical symptoms and tumor occurrence as well as treatment effectiveness [40]. While using TNF-⍺ solely may not be of sufficient specificity, addition of other markers, including 25-hydroxyvitamin D or TGF-β3, may enhance test specificity [188]. In endometriosis, serum IL-6 and peritoneal fluid TNF-⍺ levels are suggested as reliable diagnostic indicators given their high sensitivity and specificity [189]. Other advantages include lack of variation with the menstrual cycle or disease stage, allowing for their use as qualitative tests. A minimally invasive approach by peritoneal fluid sampling can be achieved through transvaginal ultrasound-guided aspiration and may ideally avoid the need to confirm the disease laparoscopically [189] whereas for early stages, elevated serum levels of soluble TNF-⍺ receptor II and IL-1β may be potential beneficial indicators [190]. Another marker that may be valuable in infertile women with endometriosis is the neutrophil:lymphocyte (N:L) ratio measured as the ratio of absolute neutrophil to absolute lymphocyte counts in peripheral blood. An elevated N:L ratio is associated with worse overall survival in several solid tumors likely due to (1) neutrophilic response suppressing tumor-infiltrating cytotoxic T cells or (2) increased neutrophilic activity signifying increased cytokine/chemokine release by the tumor [191]. Further, N:L ratio is found to be an independent risk factor for infertility and positively correlates with disease stage, tubal adhesion, and diameter of ovarian endometrioma. Furthermore, it may differentiate between endometriosis and other benign ovarian tumors by increasing diagnostic sensitivity when used with CA-125 [6]. Another study showed that quantitative detection of endometrial cell autoantibodies may serve as a novel technique for noninvasive diagnosis of endometriosis in infertile women [192]. Prognostically, elevated levels of anti-inflammatory cytokines, including IL-4, IL-10, and IL-1Ra, may be possible indictors of endometriosis outcomes [193]. MIF is also showing promising results as a potential biomarker and severity indicator as women with endometriosis, especially if infertile, have more than 3-fold increase in serum MIF levels compared with healthy women, especially as disease advances [56]. Given that the current gold standard diagnosis of endometriosis is surgical, it is imperative to conduct further studies to verify and validate clinically relevant noninvasive testing methods to facilitate improved diagnosis and treatment in women with this disease.
MicroRNA
MiRNAs are small noncoding RNAs that inhibit gene translation by binding to transcribed mRNA [194]. This post-transcriptional regulation happens in various physiological and pathophysiological states, including endometriosis [195]. MiRNAs seem to influence the inflammatory peritoneal environment and fertility potential in women with the disease and may therefore be potential predictors of fertility outcome [196]. In addition, their role in intercellular communication and systemic inflammation may enhance our understanding of the pathogenesis of endometriosis and its sequelae [21]. Significantly higher miR-106b-3p, -451a, and -486-5p levels are detected in women with endometriosis, correlating with disease stage whereas miR-185-5p, whose targets are implicated in angiogenesis and inflammation, is upregulated in sterile cases and controls, implying a possible role in EAI [196]. Further, as miRNA Let-7b downregulation is linked to the inflammatory pathobiology of endometriosis, its role as a local therapeutic agent has been experimented (Table 3) [197].
Table 3.
Medications with inflammation-modulating properties for potential use in benign gynecologic disorders
| Medication | BGD | Experimental model/study design | Effects |
|---|---|---|---|
| Anti-TNF-⍺ | |||
| Pentoxifylline [220] | Endometriosis | In vivo, rat; in vivo, human (data inconsistent). |
|
| Etanercept [221, 264] | Endometriosis | In vivo, baboon; in vitro, rat. |
|
| Adalimumab [265] | Endometriosis | In vitro, mouse embryo and peritoneal fluid, human. |
|
| Other immunomodulators | |||
| IL-2 [266, 267] | Endometriosis | In vivo, rat; in vivo, human. |
|
| Interferon-⍺-2b [268] | Endometriosis | In vivo, rat. |
|
| Lipoxin A4 [269] | Endometriosis | In vivo, rat. |
|
| IL-37b [270] | Endometriosis | In vivo, mouse. |
|
| Niclosamide [271] | Endometriosis | In vitro, human ESC. |
|
| MicroRNA [197, 272, 273] | Endometriosis; uterine leiomyoma. | In vivo, mouse; in vitro, human leiomyoma smooth muscle cells. |
|
| Hormonal treatment | |||
| Aromatase inhibitors [40, 274] | Uterine leiomyoma; endometriosis |
|
|
| Paclitaxel and 2-methylestradiol [226] | Not tested in BGDs | In vitro, human normal or malignant breast tissue. |
|
| GnRH analogs [227, 229] | Uterine leiomyoma; endometriosis; adenomyosis | In vitro, endometrial cells and ESCs. |
|
| Danazol and progesterone [275] | Endometriosis | In vitro, ESCs. |
|
| Cardiometabolic medications | |||
| Statins [233, 234, 276] | Endometriosis; uterine leiomyoma | In vivo, baboon; in vivo, mouse; in vitro, human leiomyoma cells. |
|
| Metformin [232] | Uterine leiomyoma | Retrospective cohort study. |
|
| Thiazolidinediones [140, 277] | Endometriosis | In vivo, rat; in vitro, ESCs. |
|
| ACE inhibitors [235, 238] | Uterine leiomyoma | Nested case–control study. |
|
| Cyclooxygenase-2 inhibitors | |||
| Celecoxib [239, 241, 242] | Uterine leiomyoma; endometriosis | In vitro, leiomyoma cells; in vitro, ESCs; clinical trial. |
|
| Dexketoprofen trometamol [243] | Endometriosis | In vivo, rat. |
|
| Parecoxib [278] | Endometriosis | In vivo, rat. |
|
| Rofecoxib [242] | Endometriosis | In vivo, human. |
|
| NF-κB inhibitors | |||
| BAY 11-7085 and SN-50 [66] | Endometriosis | In vivo, mouse. |
|
| IKK-2 inhibitor [47] | Endometriosis | In vitro, ESCs. |
|
| NF-κB decoy oligonucleotides [279] | Endometriosis | In vitro, endometriotic epithelial cells. |
|
| Dietary phytochemicals, e.g., indole-3-carbinol, lycopene, ursolic acid [216] | Uterine leiomyoma | Not experimentally tested. |
|
ACE denotes angiotensin converting enzyme, AMPK AMP-activated protein kinase, BGD benign gynecologic disorder, COX-2 cyclooxygenase-2, ECM extracellular matrix, ESC endometriotic stromal cell, ER estrogen receptor, GnRH gonadotrophin releasing hormone, ICAM-1 intercellular adhesion molecule-1, IFN-𝛾 interferon 𝛾, IKK IκB kinase, IL interleukin, MCP monocyte chemoattractant protein, MIF macrophage migration inhibitory factor, MiRNA microRNA, MMP matrix metalloproteinase, NF-κB nuclear factor kappa-light chain enhancer of activated B cells, PGE2 prostaglandin E2, RANTES regulated-on-activation, normal-T-cell-expressed and -secreted, TLR toll-like receptor, TNF-⍺ tumor necrosis factor ⍺, and VEGF vascular endothelial growth factor.
MiRNA can also regulate inflammation in uterine leiomyoma. In leiomyoma smooth muscle cells, MiR-200c represses IκB⍺ phosphorylation, increasing its activity as an NF-κB inhibitor, i.e., miR-200c indirectly downregulates NF-κB signal transduction. Uterine leiomyoma have low levels of miR-200c, which in turn disinhibit NF-κB signaling and increase IL-8 expression when compared with the normal myometrium [198]. IL-8 can have antiapoptotic effects through upregulating Bcl-xL and Bcl-2, promoting cell survival in endothelial cells [199], but whether this occurs in uterine leiomyoma needs further investigation.
Molecular imaging
In vivo molecular imaging has been revolutionizing diagnostic medicine and showed great promise in disease screening and personalized medicine [200]. In fact, molecular imaging aids in diagnosing BGDs, such as uterine leiomyoma and endometriosis, by detecting altered biochemical and metabolic reactions [201, 202]. Although not yet used to characterize the inflammatory processes in BGDs, molecular imaging has been used to visualize inflammation elsewhere, including the central nervous system and blood vessels [203]. Using nanoparticles, such as small and ultrasmall paramagnetic iron oxide, macrophages and monocytes can be targeted in tissues. Other modalities to molecularly image macrophages include PET and SPECT ligands, optical imaging, and computed tomography [203, 204]. Given the abundance of macrophages in uterine leiomyoma and endometriosis, these imaging techniques may theoretically be of use in screening and diagnosis, but their actual effectiveness in clinical settings remains to be seen. Similarly, lymphocyte imaging can be achieved using labeled cytokines and chemokines, such as IL-1, IL-2, and IL-8 [205], many of which are established mediators in BGDs. Molecular imaging of cytokines has also been reported in the literature. For example, scintigraphic detection of TNF-⍺ via 99mTc-labelled infliximab or 99mTc-adalimumab is employed in rheumatoid arthritis patients to identify foci of active inflammation and monitor response to therapy [206]. This technique may show benefit in early, noninvasive detection of uterine leiomyoma knowing how important a role TNF-⍺ plays in its pathogenesis. Nevertheless, despite their high resolution, substantial sensitivity, and noninvasiveness, molecular imaging of inflammation is yet to be evaluated as a diagnostic adjunct for BGDs.
Therapeutic implications
Behavioral modification
Lifestyle modifications represent a convenient, nonpharmacological initial approach that may combat the role of inflammation in BGDs, possibly decreasing their incidence. Knowing that obesity induces a systemic inflammatory state that may influence leiomyoma pathogenesis, measures, such as exercise and dietary restriction, may be of benefit. In fact, reduction of IL-6 and CRP serum levels is documented in subjects who lose weight through these measures [207]. In addition, high-intensity interval training and moderate-intensity continuous training are shown to immunomodulate IL-6 and IL-10. Dietary modifications can also help downregulate inflammation [208]. Whereas decreased richness of gut microbiota is implicated in obesity and systemic inflammation [209], this can be largely modulated by dietary factors. For example, high-fat diets reduce colonic colonization of beneficial species of commensal microbiota, increasing serum lipopolysaccharides and hence inflammatory milieus [210]. In contrast, oleic acid and omega-3-polyunsaturated fatty acids can counteract the harmful effects on gut microbiota [211]. Women with endometriosis may be advised to decrease red meat consumption as it contains arachidonic acid (omega 6), which if taken in excess, can contribute to inflammation [212, 213]. Alternatively, increased intake of omega 3 is proposed to decrease growth of endometrial implants and improve pain and inflammation in endometriosis [213]. Further, dietary phytochemicals, which are nonnutritive compounds with disease-preventing properties found in different foods, possess anti-inflammatory actions that have been evaluated for potential use in antifibrotic and anti-inflammatory medications against uterine leiomyoma and endometriosis. For example, curcumin, a dietary supplement found in turmeric, inhibits NF-κB protein expression in leiomyoma cells [214] whereas apigenin inactivates NF-κB and hence suppresses TNF-⍺–induced cellular proliferation and PGE2 synthesis in endometriotic stromal cells [215]. An excellent review has explored in depth the possible therapeutic roles of dietary phytochemicals in uterine leiomyoma [216]. Lastly, vitamins A, C, and E are potent antioxidants through decreasing lipid peroxidation in inflammatory diseases and are, in fact, shown to reduce markers of oxidative stress in endometriosis [217, 218]. Whether diets high in antioxidants or low in saturated fats can decrease chronic pelvic pain or reduce other inflammation-related sequelae including ovarian carcinoma remains to be seen.
Medical therapy
This paper introduces novel as well as previously established therapeutics that can be integrated in the management practices of BGDs. An extensive list of experimentally and clinically tested medications is shown in Table 3.
Immunomodulators
Given the important role of TNF-⍺ in uterine leiomyoma pathogenesis, it may be beneficial to utilize anti–TNF-⍺ agents to treat leiomyoma-related symptoms; nevertheless, there is no current consensus on the efficacy of these medications in leiomyoma management. Future potential studies could start with observational approaches to patients taking anti–TNF-⍺ agents for other indications, such as rheumatoid arthritis and inflammatory bowel disease, to examine rates of fibroids and fibroid growth and involution as compared with demographically matched controls without exposure to such agents. In women with endometriosis, pentoxifylline was shown to reduce in vitro production and proinflammatory actions of TNF-⍺ as well as reduce pain postoperatively [219, 220] but as additional trials showed no significant improvement, its true benefits in humans is yet to be seen. Etanercept, a TNF-⍺ blocker, significantly decreased red lesion surface area in a randomized controlled trial using baboon models [221] while infliximab has not been shown to reduce pain scores in patients with endometriosis-related chronic pelvic pain in a Cochrane Review of the evidence [222], and overall, there is still no enough evidence that supports the use of anti–TNF-⍺ agents to relieve pelvic pain in women with the disease. Other immunomodulatory medications have shown promise in treating endometriosis by targeting key players in disease pathogenesis. For example, glucosaminyl muramyl dipeptide was shown to prevent peritoneal macrophage hyperactivation [223] whereas loxoribine induced epithelial and stromal regression and reduced number of NK cells in endometriotic lesions [224]. Kotlyar et al. have suggested in their review a plethora of immunomodulatory medications that can be potentially used in endometriosis, including disease modifying antirheumatic drugs, cytokines, mTOR inhibitors, nucleotide inhibitors, and miRNAs (Table 3); however, their efficacy in humans is yet to be tested [225]. As evidence shows a growing role for macrophages in the pathogenesis in endometriosis and its clinical manifestations, therapies that specifically target aberrant macrophage function and attenuate their disease-promoting properties could serve as potential options for treating endometriosis and its associated pain [154]. Nevertheless, while diverse immunomodulators appear promising in targeting inflammation in BGDs, further experimental studies and human randomized controlled trials are warranted to establish the full efficacy of these medications.
Hormonal treatment
Besides their inhibitory actions on aromatase and decreasing estrogen availability in patients with BGDs, aromatase inhibitors may have anti-inflammatory properties through TNF-⍺–dependent pathways. Notably, as TNF-⍺ stimulates aromatase activity, increasing the estrogenic load in patients with BGDs, aromatase inhibitors may interrupt the downstream effects of TNF-⍺ on estrogen metabolism [40]. Targeting the same pathway, paclitaxel, a microtubule-stabilizing agent, and 2-methylestradiol, an endogenous estrogen metabolite, were found to inhibit TNF-⍺–mediated aromatase activity in stromal fibroblasts and downregulate TNF-⍺ receptors in human macrophages in breast tissues [226] and may be of benefit in patients with BGDs. While GnRH analogs have been classically used as hormonal treatments of BGDs, experimental evidence shows their ability to attenuate inflammatory pathways in these disorders. For example, they influence TNF-⍺–induced cellular proliferation in endometrial stromal cells but not endometriotic stromal cells [227] and attenuate IL-8 expression by reducing TNF-⍺–induced NF-κB activation in endometriosis [228]. In another study, GnRH analogs increased the apoptotic index of eutopic endometria, lesions, and myometria of women with uterine leiomyoma, endometriosis, and adenomyosis and reduced CD68+ macrophage infiltration in the endometria of women with these diseases compared with nontreated groups, reconceptualizing these traditionally used medications as having anti-inflammatory properties [229].
Cardiometabolic medications
Medications originally used to target CMRFs were demonstrated to influence several inflammatory pathways in BGDs. To begin with, metformin can change the composition of gut microbiota by increasing the Akkermansia species, which in turn improves insulin resistance and reduces tissue inflammation [230]. In addition, metformin influences inflammation both indirectly by improving hyperglycemia, hyperlipidemia, and insulin resistance and directly by inhibiting TNF-⍺ and NF-κB signaling via AMPK-dependent and independent pathways [231]. Indeed, metformin has been reported to confer a lower risk of uterine leiomyoma in diabetic women [232]. On the other hand, Lebovic et al. have found that ciglitazone, a thiazolidinedione and a PPAR 𝛾 ligand, reduces the size of endometriotic implants in rat models and inferred that these medications, originally used for type 2 diabetes mellitus, may be potentially used in women with endometriosis [140].
The antihyperlipidemic medications statins were also experimented for their anti-inflammatory effect in endometriosis. In a study by Taylor et al. on baboon endometriosis models, statins downregulated ERβ, which is known to activate inflammatory responses and increase IL-1β levels in the disease [233] whereas in another study, statins reduced the expression of MCP-1 in vitro and in mouse models [234]. Multiple studies demonstrated that ACE inhibitors and angiotensin II receptor blockers can reduce inflammation and proinflammatory mediators [235–237]. Additionally, Fischer et al. have concluded that ACE inhibitors reduce the incidence of uterine leiomyoma in hypertensive patients, rendering these medications possible therapeutic candidates [238] that may or may not target the inflammatory pathways in these tumors.
Cyclooxygenase-2 and NF-κB inhibitors
The COX-2 inhibitor celecoxib was found to inhibit leiomyoma cell proliferation by decreasing gene expression of proinflammatory cytokines and transcription factors, including IL-1β, IL-6, TNF-⍺, and NF-κB. Furthermore, it showed inhibitory actions on production of ECM elements, such as collagen A and fibronectin, and reduced the expression of several growth factors [239]. This suggests that COX-2 inhibitors may attenuate the inflammatory microenvironment harboring leiomyoma growth, but more experimental and clinical studies are needed to confirm its beneficial effects. As previously documented, COX-2 and PGE2 are key inflammatory players in endometriosis, and theoretically, COX-2 inhibitors may be substantially beneficial, although data on their effectiveness have been controversial in the past. For example, in a study published more than 15 years back, nimesulide, a COX-2 inhibitor, has shown no effect on lesion size or number in a mouse model of endometriosis [240]. However, more recent studies, including clinical trials, show that celecoxib [241], rofecoxib [242], and a new COX-2 inhibitor, dexketoprofen trometamol [243], have promising therapeutic effects as shown in Table 3. On the other hand, as evidence accumulates on the various mechanisms by which NF-κB participates in endometriotic pathogenesis, it may serve as an attractive potential target to halt endometriosis progression. Among others, the NF-κB inhibitors BAY 11-7085 and SN-50 have shown remarkable lesion inhibitory effects in vivo through boosting apoptosis and decreasing ICAM-1, an adhesion molecule, as well as reducing proliferation in the BAY 11-7085 group [66]. NF-κB inhibitors cover a wide spectrum of medications that involve hormonal agents, herbal compounds, and oligonucleotides, to name a few [65]. However, these agents have been mostly tested in vitro and in animal models, and clinical trials are therefore needed to evaluate their effectiveness in humans.
Conclusion and future directions
BGDs have been heavily studied over the decades uncovering unique perspectives of their pathophysiology. This exhaustive paper enriches our understanding of how inflammation represents an inherent feature of BGDs, contributing remarkably to their development. As we become increasingly aware of this association, curiosity to learn its mechanistic aspects will lead us to previously unexplored pathobiological avenues and introduce novel frameworks that will ground future diagnostic and therapeutic implications. Therefore, while this paper comprehensively explores this association, it also emphasizes the need for further experimental and clinical research to delve into the cellular and molecular mechanisms involved and elucidate a casual role for inflammation in these chronic, costly, and debilitating disorders.
Conflict of interest
The authors have no conflict of interest to declare.
Data availability statement
No new data were generated or analyzed in support of this research.
Author contribution statement
AA, LR, and GK searched the literature, wrote the manuscript, and approved the final version. MB wrote the manuscript and approved the final version.
Grant Support: This work was supported, in part, by NIH grant 1R01HD094380 (MAB).
Contributor Information
Abdelrahman AlAshqar, Department of Gynecology and Obstetrics, Johns Hopkins University, Baltimore, MD, USA; Department of Obstetrics and Gynecology, Kuwait University, Kuwait City, Kuwait.
Lauren Reschke, Department of Gynecology and Obstetrics, Johns Hopkins University, Baltimore, MD, USA.
Gregory W Kirschen, Department of Gynecology and Obstetrics, Johns Hopkins University, Baltimore, MD, USA.
Mostafa A Borahay, Department of Gynecology and Obstetrics, Johns Hopkins University, Baltimore, MD, USA.
References
- 1. Punchard NA, Whelan CJ, Adcock I. The journal of inflammation. J Inflamm (Lond) 2004; 1:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen L, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018; 9:7204–7218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Protic O, et al. Possible involvement of inflammatory/reparative processes in the development of uterine fibroids. Cell Tissue Res 2016; 364:415–427. [DOI] [PubMed] [Google Scholar]
- 4. Li C, et al. Correlation of LOX5 and COX2 expression with inflammatory pathology and clinical features of adenomyosis. Mol Med Rep 2019; 19:727–733. [DOI] [PubMed] [Google Scholar]
- 5. Alderman MH 3rd, Yoder N, Taylor HS. The systemic effects of endometriosis. Semin Reprod Med 2017; 35:263–270. [DOI] [PubMed] [Google Scholar]
- 6. Jing X, et al. Systemic inflammatory response markers associated with infertility and endometrioma or uterine leiomyoma in endometriosis. Ther Clin Risk Manag 2020; 16:403–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McKinnon BD, et al. Inflammation and nerve fiber interaction in endometriotic pain. Trends Endocrinol Metab 2015; 26:1–10. [DOI] [PubMed] [Google Scholar]
- 8. Fallowfield JA, et al. Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J Immunol 2007; 178:5288–5295. [DOI] [PubMed] [Google Scholar]
- 9. Yang Q, et al. The emerging spectrum of early life exposure-related inflammation and epigenetic therapy. Cancer Stud Mol Med 2018; 4:13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kurachi O, et al. Tumor necrosis factor-alpha expression in human uterine leiomyoma and its down-regulation by progesterone. J Clin Endocrinol Metab 2001; 86:2275–2280. [DOI] [PubMed] [Google Scholar]
- 11. Wegienka G. Are uterine leiomyoma a consequence of a chronically inflammatory immune system? Med Hypotheses 2012; 79:226–231. [DOI] [PubMed] [Google Scholar]
- 12. Islam MS, et al. Extracellular matrix in uterine leiomyoma pathogenesis: a potential target for future therapeutics. Hum Reprod Update 2018; 24:59–85. [DOI] [PubMed] [Google Scholar]
- 13. Ciebiera M, et al. TNF-alpha serum levels are elevated in women with clinically symptomatic uterine fibroids. Int J Immunopathol Pharmacol 2018; 32: 2058738418789805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McLaren J, et al. Vascular endothelial growth factor is produced by peritoneal fluid macrophages in endometriosis and is regulated by ovarian steroids. J Clin Invest 1996; 98:482–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ahn SH, et al. Pathophysiology and immune dysfunction in endometriosis. Biomed Res Int 2015; 2015:795976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kobayashi H, et al. The role of iron in the pathogenesis of endometriosis. Gynecol Endocrinol 2009; 25:39–52. [DOI] [PubMed] [Google Scholar]
- 17. Pizzo A, et al. Behaviour of cytokine levels in serum and peritoneal fluid of women with endometriosis. Gynecol Obstet Invest 2002; 54:82–87. [DOI] [PubMed] [Google Scholar]
- 18. Bullimore DW. Endometriosis is sustained by tumour necrosis factor-alpha. Med Hypotheses 2003; 60:84–88. [DOI] [PubMed] [Google Scholar]
- 19. Rana N, et al. Basal and stimulated secretion of cytokines by peritoneal macrophages in women with endometriosis. Fertil Steril 1996; 65:925–930. [PubMed] [Google Scholar]
- 20. Harada T, Iwabe T, Terakawa N. Role of cytokines in endometriosis. Fertil Steril 2001; 76:1–10. [DOI] [PubMed] [Google Scholar]
- 21. Nematian SE, et al. Systemic inflammation induced by microRNAs: endometriosis-derived alterations in circulating microRNA 125b-5p and let-7b-5p regulate macrophage cytokine production. J Clin Endocrinol Metab 2018; 103:64–74. [DOI] [PubMed] [Google Scholar]
- 22. Teng GG, et al. Let-7b is involved in the inflammation and immune responses associated with Helicobacter pylori infection by targeting toll-like receptor 4. PLoS One 2013; 8:e56709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Taylor HS, Kotlyar AM, Flores VA. Endometriosis is a chronic systemic disease: clinical challenges and novel innovations. Lancet 2021; 397:839–852. [DOI] [PubMed] [Google Scholar]
- 24. Leyendecker G, Wildt L, Mall G. The pathophysiology of endometriosis and adenomyosis: tissue injury and repair. Arch Gynecol Obstet 2009; 280:529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bodur S, et al. The clinical significance of classical and new emerging determinants of adenomyosis. Int J Clin Exp Med 2015; 8:7958–7964. [PMC free article] [PubMed] [Google Scholar]
- 26. Khan KN, et al. Biological differences between intrinsic and extrinsic adenomyosis with coexisting deep infiltrating endometriosis. Reprod Biomed Online 2019; 39:343–353. [DOI] [PubMed] [Google Scholar]
- 27. Koninckx PR, Martin DC. Deep endometriosis: a consequence of infiltration or retraction or possibly adenomyosis externa? Fertil Steril 1992; 58:924–928. [DOI] [PubMed] [Google Scholar]
- 28. Chegini N. Proinflammatory and profibrotic mediators: principal effectors of leiomyoma development as a fibrotic disorder. Semin Reprod Med 2010; 28:180–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Terranova PF, et al. Tumor necrosis factor-alpha in the female reproductive tract. Proc Soc Exp Biol Med 1995; 209:325–342. [DOI] [PubMed] [Google Scholar]
- 30. Nair S, Al-Hendy A. Adipocytes enhance the proliferation of human leiomyoma cells via TNF-alpha proinflammatory cytokine. Reprod Sci 2011; 18:1186–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Matsuo H, Maruo T, Samoto T. Increased expression of Bcl-2 protein in human uterine leiomyoma and its up-regulation by progesterone. J Clin Endocrinol Metab 1997; 82:293–299. [DOI] [PubMed] [Google Scholar]
- 32. Haviv R, Stein R. Nerve growth factor inhibits apoptosis induced by tumor necrosis factor in PC12 cells. J Neurosci Res 1999; 55:269–277. [DOI] [PubMed] [Google Scholar]
- 33. Mantovani A, et al. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 2004; 25:677–686. [DOI] [PubMed] [Google Scholar]
- 34. Wang Y, et al. Differential effects of tumor necrosis factor-alpha on matrix metalloproteinase-2 expression in human myometrial and uterine leiomyoma smooth muscle cells. Hum Reprod 2015; 30:61–70. [DOI] [PubMed] [Google Scholar]
- 35. Borahay MA, et al. Signaling pathways in leiomyoma: understanding pathobiology and implications for therapy. Mol Med 2015; 21:242–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yu L, et al. Differential expression of receptor tyrosine kinases (RTKs) and IGF-I pathway activation in human uterine leiomyomas. Mol Med 2008; 14:264–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Terzic J, et al. Inflammation and colon cancer. Gastroenterology 2010; 138:2101–2114 e5. [DOI] [PubMed] [Google Scholar]
- 38. Riches DW, Chan ED, Winston BW. TNF-alpha-induced regulation and signalling in macrophages. Immunobiology 1996; 195:477–490. [DOI] [PubMed] [Google Scholar]
- 39. Sun SC. Non-canonical NF-kappaB signaling pathway. Cell Res 2011; 21:71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ciebiera M, et al. The role of tumor necrosis factor alpha in the biology of uterine fibroids and the related symptoms. Int J Mol Sci 2018; 19:3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ciebiera M, et al. Role of transforming growth factor beta in uterine fibroid biology. Int J Mol Sci 2017; 18:2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ma B, Hottiger MO. Crosstalk between Wnt/beta-catenin and NF-kappaB signaling pathway during inflammation. Front Immunol 2016; 7:378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tanwar PS, et al. Constitutive activation of Beta-catenin in uterine stroma and smooth muscle leads to the development of mesenchymal tumors in mice. Biol Reprod 2009; 81:545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ono M, et al. Paracrine activation of WNT/beta-catenin pathway in uterine leiomyoma stem cells promotes tumor growth. Proc Natl Acad Sci USA 2013; 110:17053–17058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Burney RO, Giudice LC. Pathogenesis and pathophysiology of endometriosis. Fertil Steril 2012; 98:511–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kayisli UA, Mahutte NG, Arici A. Uterine chemokines in reproductive physiology and pathology. Am J Reprod Immunol 2002; 47:213–221. [DOI] [PubMed] [Google Scholar]
- 47. Grund EM, et al. Tumor necrosis factor-alpha regulates inflammatory and mesenchymal responses via mitogen-activated protein kinase kinase, p38, and nuclear factor kappaB in human endometriotic epithelial cells. Mol Pharmacol 2008; 73:1394–1404. [DOI] [PubMed] [Google Scholar]
- 48. Iwabe T, et al. Tumor necrosis factor-alpha promotes proliferation of endometriotic stromal cells by inducing interleukin-8 gene and protein expression. J Clin Endocrinol Metab 2000; 85:824–829. [DOI] [PubMed] [Google Scholar]
- 49. Garcia-Gomez E, et al. Regulation of inflammation pathways and inflammasome by sex steroid hormones in endometriosis. Front Endocrinol (Lausanne) 2019; 10:935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tamura M, et al. Interleukin-1beta elevates cyclooxygenase-2 protein level and enzyme activity via increasing its mRNA stability in human endometrial stromal cells: an effect mediated by extracellularly regulated kinases 1 and 2. J Clin Endocrinol Metab 2002; 87:3263–3273. [DOI] [PubMed] [Google Scholar]
- 51. Tamura M, et al. Vascular endothelial growth factor up-regulates cyclooxygenase-2 expression in human endothelial cells. J Clin Endocrinol Metab 2002; 87:3504–3507. [DOI] [PubMed] [Google Scholar]
- 52. Nothnick W, Alali Z. Recent advances in the understanding of endometriosis: the role of inflammatory mediators in disease pathogenesis and treatment. F1000Res 2016; 5:F1000 Faculty Rev-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Veillat V, et al. Macrophage migration inhibitory factor elicits an angiogenic phenotype in human ectopic endometrial cells and triggers the production of major angiogenic factors via CD44, CD74, and MAPK signaling pathways. J Clin Endocrinol Metab 2010; 95:E403–E412. [DOI] [PubMed] [Google Scholar]
- 54. Carli C, et al. Up-regulation of cyclooxygenase-2 expression and prostaglandin E2 production in human endometriotic cells by macrophage migration inhibitory factor: involvement of novel kinase signaling pathways. Endocrinology 2009; 150:3128–3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kats R, et al. Marked elevation of macrophage migration inhibitory factor in the peritoneal fluid of women with endometriosis. Fertil Steril 2002; 78:69–76. [DOI] [PubMed] [Google Scholar]
- 56. Morin M, et al. Elevated levels of macrophage migration inhibitory factor in the peripheral blood of women with endometriosis. Fertil Steril 2005; 83:865–872. [DOI] [PubMed] [Google Scholar]
- 57. Kats R, Metz CN, Akoum A. Macrophage migration inhibitory factor is markedly expressed in active and early-stage endometriotic lesions. J Clin Endocrinol Metab 2002; 87:883–889. [DOI] [PubMed] [Google Scholar]
- 58. Liang Y, et al. Pro-endometriotic niche in endometriosis. Reprod Biomed Online 2019; 38:549–559. [DOI] [PubMed] [Google Scholar]
- 59. Yu JJ, et al. IL15 promotes growth and invasion of endometrial stromal cells and inhibits killing activity of NK cells in endometriosis. Reproduction 2016; 152:151–160. [DOI] [PubMed] [Google Scholar]
- 60. Guo SW, Du Y, Liu X. Platelet-derived TGF-beta1 mediates the down-modulation of NKG2D expression and may be responsible for impaired natural killer (NK) cytotoxicity in women with endometriosis. Hum Reprod 2016; 31:1462–1474. [DOI] [PubMed] [Google Scholar]
- 61. Li MQ, et al. CD4+Foxp3+ regulatory T cell differentiation mediated by endometrial stromal cell-derived TECK promotes the growth and invasion of endometriotic lesions. Cell Death Dis 2014; 5:e1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wang XQ, et al. Synergistic effect of regulatory T cells and proinflammatory cytokines in angiogenesis in the endometriotic milieu. Hum Reprod 2017; 32:1304–1317. [DOI] [PubMed] [Google Scholar]
- 63. Sharpe-Timms KL, et al. Differential expression and localization of de-novo synthesized endometriotic haptoglobin in endometrium and endometriotic lesions. Hum Reprod 2000; 15:2180–2185. [DOI] [PubMed] [Google Scholar]
- 64. Gonzalez-Ramos R, et al. Nuclear factor-kappa B is constitutively activated in peritoneal endometriosis. Mol Hum Reprod 2007; 13:503–509. [DOI] [PubMed] [Google Scholar]
- 65. Gonzalez-Ramos R, et al. Involvement of the nuclear factor-kappaB pathway in the pathogenesis of endometriosis. Fertil Steril 2010; 94:1985–1994. [DOI] [PubMed] [Google Scholar]
- 66. Gonzalez-Ramos R, et al. Agents blocking the nuclear factor-kappaB pathway are effective inhibitors of endometriosis in an in vivo experimental model. Gynecol Obstet Invest 2008; 65:174–186. [DOI] [PubMed] [Google Scholar]
- 67. Lai TH, Wu PH, Wu WB. Involvement of NADPH oxidase and NF-kappaB activation in CXCL1 induction by vascular endothelial growth factor in human endometrial epithelial cells of patients with adenomyosis. J Reprod Immunol 2016; 118:61–69. [DOI] [PubMed] [Google Scholar]
- 68. Murphy AJ, Guyre PM, Pioli PA. Estradiol suppresses NF-kappa B activation through coordinated regulation of let-7a and miR-125b in primary human macrophages. J Immunol 2010; 184:5029–5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jiang L, et al. Inflammation and endometriosis. Front Biosci (Landmark Ed) 2016; 21:941–948. [DOI] [PubMed] [Google Scholar]
- 70. Straub RH. The complex role of estrogens in inflammation. Endocr Rev 2007; 28:521–574. [DOI] [PubMed] [Google Scholar]
- 71. Montagna P, et al. Peritoneal fluid macrophages in endometriosis: correlation between the expression of estrogen receptors and inflammation. Fertil Steril 2008; 90:156–164. [DOI] [PubMed] [Google Scholar]
- 72. Liu H, Liu K, Bodenner DL. Estrogen receptor inhibits interleukin-6 gene expression by disruption of nuclear factor kappaB transactivation. Cytokine 2005; 31:251–257. [DOI] [PubMed] [Google Scholar]
- 73. Stein B, Yang MX. Repression of the interleukin-6 promoter by estrogen receptor is mediated by NF-kappa B and C/EBP beta. Mol Cell Biol 1995; 15:4971–4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Burns KA, et al. Early endometriosis in females is directed by immune-mediated estrogen receptor alpha and IL-6 cross-talk. Endocrinology 2018; 159:103–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bulun SE. Endometriosis. N Engl J Med 2009; 360:268–279. [DOI] [PubMed] [Google Scholar]
- 76. Chantalat E, et al. Estrogen receptors and endometriosis. Int J Mol Sci 2020; 21:2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Akoum A, et al. Ectopic endometrial cells express high concentrations of interleukin (IL)-8 in vivo regardless of the menstrual cycle phase and respond to oestradiol by up-regulating IL-1-induced IL-8 expression in vitro. Mol Hum Reprod 2001; 7:859–866. [DOI] [PubMed] [Google Scholar]
- 78. Boucher A, et al. Ovarian hormones modulate monocyte chemotactic protein-1 expression in endometrial cells of women with endometriosis. Mol Hum Reprod 2000; 6:618–626. [DOI] [PubMed] [Google Scholar]
- 79. Arici A, et al. Regulation of monocyte chemotactic protein-1 expression in human endometrial stromal cells by estrogen and progesterone. Biol Reprod 1999; 61:85–90. [DOI] [PubMed] [Google Scholar]
- 80. Simmen RC, Kelley AS. Reversal of fortune: estrogen receptor-beta in endometriosis. J Mol Endocrinol 2016; 57:F23–F27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Patel BG, et al. Progesterone resistance in endometriosis: origins, consequences and interventions. Acta Obstet Gynecol Scand 2017; 96:623–632. [DOI] [PubMed] [Google Scholar]
- 82. Guzeloglu-Kayisli O, et al. DNA-binding ability of NF-kappaB is affected differently by ERalpha and ERbeta and its activation results in inhibition of estrogen responsiveness. Reprod Sci 2008; 15:493–505. [DOI] [PubMed] [Google Scholar]
- 83. Shen F, et al. Immunoreactivity of progesterone receptor isoform B and nuclear factor kappa-B as biomarkers for recurrence of ovarian endometriomas. Am J Obstet Gynecol 2008; 199:486 e1–486 e10. [DOI] [PubMed] [Google Scholar]
- 84. Attar E, et al. Prostaglandin E2 via steroidogenic factor-1 coordinately regulates transcription of steroidogenic genes necessary for estrogen synthesis in endometriosis. J Clin Endocrinol Metab 2009; 94:623–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bulun SE, et al. Role of aromatase in endometrial disease. J Steroid Biochem Mol Biol 2001; 79:19–25. [DOI] [PubMed] [Google Scholar]
- 86. Weihua Z, et al. Estrogen receptor (ER) beta, a modulator of ERalpha in the uterus. Proc Natl Acad Sci USA 2000; 97:5936–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Asada H, et al. Potential link between estrogen receptor-alpha gene hypomethylation and uterine fibroid formation. Mol Hum Reprod 2008; 14:539–545. [DOI] [PubMed] [Google Scholar]
- 88. Ishikawa H, et al. Progesterone is essential for maintenance and growth of uterine leiomyoma. Endocrinology 2010; 151:2433–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Macdiarmid F, et al. Stimulation of aromatase activity in breast fibroblasts by tumor necrosis factor alpha. Mol Cell Endocrinol 1994; 106:17–21. [DOI] [PubMed] [Google Scholar]
- 90. Kamel M, et al. Effect of tumour necrosis factor-alpha on estrogen metabolic pathways in breast cancer cells. J Cancer 2012; 3:310–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yamamoto T, et al. Evidence for estrogen synthesis in adenomyotic tissues. Am J Obstet Gynecol 1993; 169:734–738. [DOI] [PubMed] [Google Scholar]
- 92. Orazov MR, et al. Immune-inflammatory predictors of the pelvic pain syndrome associated with adenomyosis. Gynecol Endocrinol 2017; 33:44–46. [DOI] [PubMed] [Google Scholar]
- 93. Hsieh YY, et al. Tumor necrosis factor-alpha-308 promoter and p53 codon 72 gene polymorphisms in women with leiomyomas. Fertil Steril 2004; 82:1177–1181. [DOI] [PubMed] [Google Scholar]
- 94. Litovkin KV, et al. Interleukin-6 -174G/C polymorphism in breast cancer and uterine leiomyoma patients: a population-based case control study. Exp Oncol 2007; 29:295–298. [PubMed] [Google Scholar]
- 95. Pietrowski D, et al. Uterine leiomyoma is associated with a polymorphism in the interleukin 1-beta gene. Am J Reprod Immunol 2009; 62:112–117. [DOI] [PubMed] [Google Scholar]
- 96. Makinen N, et al. MED12, the mediator complex subunit 12 gene, is mutated at high frequency in uterine leiomyomas. Science 2011; 334:252–255. [DOI] [PubMed] [Google Scholar]
- 97. Al-Hendy A, et al. Silencing Med12 gene reduces proliferation of human leiomyoma cells mediated via Wnt/beta-catenin signaling pathway. Endocrinology 2017; 158:592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Mittal P, et al. Med12 gain-of-function mutation causes leiomyomas and genomic instability. J Clin Invest 2015; 125:3280–3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Antinolo G, et al. Analysis of the involvement of CCR5-Delta32 and CCR2-V64I variants in the development of endometriosis. Mol Hum Reprod 2004; 10:155–157. [DOI] [PubMed] [Google Scholar]
- 100. Ahn SH, et al. Immune-inflammation gene signatures in endometriosis patients. Fertil Steril 2016; 106:1420–1431 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. El Andaloussi A, et al. Uterine fibroids: bridging genomic defects and chronic inflammation. Semin Reprod Med 2017; 35:494–498. [DOI] [PubMed] [Google Scholar]
- 102. Grimaldi G, et al. Down-regulation of the histone methyltransferase EZH2 contributes to the epigenetic programming of decidualizing human endometrial stromal cells. Mol Endocrinol 2011; 25:1892–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Colon-Caraballo M, Monteiro JB, Flores I. H3K27me3 is an epigenetic mark of relevance in endometriosis. Reprod Sci 2015; 22:1134–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Takeshima H, et al. Induction of aberrant trimethylation of histone H3 lysine 27 by inflammation in mouse colonic epithelial cells. Carcinogenesis 2012; 33:2384–2390. [DOI] [PubMed] [Google Scholar]
- 105. Gentilini D, et al. Gene expression profiling of peripheral blood mononuclear cells in endometriosis identifies genes altered in non-gynaecologic chronic inflammatory diseases. Hum Reprod 2011; 26:3109–3117. [DOI] [PubMed] [Google Scholar]
- 106. Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis 2010; 30:245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Arici A, Sozen I. Transforming growth factor-beta3 is expressed at high levels in leiomyoma where it stimulates fibronectin expression and cell proliferation. Fertil Steril 2000; 73:1006–1011. [DOI] [PubMed] [Google Scholar]
- 108. Protic O, et al. Activin a in inflammation, tissue repair, and fibrosis: possible role as inflammatory and fibrotic mediator of uterine fibroid development and growth. Semin Reprod Med 2017; 35:499–509. [DOI] [PubMed] [Google Scholar]
- 109. Pitsos M, Kanakas N. The role of matrix metalloproteinases in the pathogenesis of endometriosis. Reprod Sci 2009; 16:717–726. [DOI] [PubMed] [Google Scholar]
- 110. Tosti C, et al. Pathogenetic mechanisms of deep infiltrating endometriosis. Reprod Sci 2015; 22:1053–1059. [DOI] [PubMed] [Google Scholar]
- 111. Novak U, Cocks BG, Hamilton JA. A labile repressor acts through the NFkB-like binding sites of the human urokinase gene. Nucleic Acids Res 1991; 19:3389–3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Aguilera A, et al. Effects of rapamycin on the epithelial-to-mesenchymal transition of human peritoneal mesothelial cells. Int J Artif Organs 2005; 28:164–169. [DOI] [PubMed] [Google Scholar]
- 113. Sheveleva T, et al. Innovative approach in assessing the role of neurogenesis, angiogenesis, and lymphangiogenesis in the pathogenesis of external genital endometriosis. Gynecol Endocrinol 2016; 32:75–79. [DOI] [PubMed] [Google Scholar]
- 114. Tariverdian N, et al. Neuroendocrine-immune disequilibrium and endometriosis: an interdisciplinary approach. Semin Immunopathol 2007; 29:193–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Veillat V, et al. Involvement of nuclear factor-kappaB in macrophage migration inhibitory factor gene transcription up-regulation induced by interleukin-1 beta in ectopic endometrial cells. Fertil Steril 2009; 91:2148–2156. [DOI] [PubMed] [Google Scholar]
- 116. Yang Y, et al. Identification of macrophage migration inhibitory factor as a potent endothelial cell growth-promoting agent released by ectopic human endometrial cells. J Clin Endocrinol Metab 2000; 85:4721–4727. [DOI] [PubMed] [Google Scholar]
- 117. Carneiro MM. Stem cells and uterine leiomyomas: what is the evidence? JBRA Assist Reprod 2016; 20:33–37. [DOI] [PubMed] [Google Scholar]
- 118. Orciani M, et al. Chronic inflammation may enhance leiomyoma development by the involvement of progenitor cells. Stem Cells Int 2018; 2018:1716246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Moridi I, et al. CXCL12 attracts bone marrow-derived cells to uterine leiomyomas. Reprod Sci 2020; 27:1724–1730. [DOI] [PubMed] [Google Scholar]
- 120. Hopman RK, DiPersio JF. Advances in stem cell mobilization. Blood Rev 2014; 28:31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Wang X, et al. Chemoattraction of bone marrow-derived stem cells towards human endometrial stromal cells is mediated by estradiol regulated CXCL12 and CXCR4 expression. Stem Cell Res 2015; 15:14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Koippallil Gopalakrishnan Nair AR, et al. Endometriotic mesenchymal stem cells exhibit a distinct immune phenotype. Int Immunol 2015; 27:195–204. [DOI] [PubMed] [Google Scholar]
- 123. AlAshqar A, et al. Cardiometabolic risk factors and benign Gynecologic disorders. Obstet Gynecol Surv 2019; 74:661–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Sverdlov AL, et al. Interplay between oxidative stress and inflammation in cardiometabolic syndrome. Mediators Inflamm 2016; 2016:8254590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol 2011; 29:415–445. [DOI] [PubMed] [Google Scholar]
- 126. Renehan AG, Zwahlen M, Egger M. Adiposity and cancer risk: new mechanistic insights from epidemiology. Nat Rev Cancer 2015; 15:484–498. [DOI] [PubMed] [Google Scholar]
- 127. Sun K, et al. A case-control study of the relationship between visceral fat and development of uterine fibroids. Exp Ther Med 2019; 18:404–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Sanchez-Jimenez F, et al. Obesity and breast cancer: role of leptin. Front Oncol 2019; 9:596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Markowska A, et al. Further studies on leptin and leptin receptor expression in myometrium and uterine myomas. Eur J Gynaecol Oncol 2005; 26:517–525. [PubMed] [Google Scholar]
- 130. Chen HS, et al. Aberrant serum adiponectin levels in women with uterine leiomyomas. Gynecol Obstet Invest 2004; 58:160–163. [DOI] [PubMed] [Google Scholar]
- 131. Benigni A, Cassis P, Remuzzi G. Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO Mol Med 2010; 2:247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Isobe A, et al. Dual repressive effect of angiotensin II-type 1 receptor blocker telmisartan on angiotensin II-induced and estradiol-induced uterine leiomyoma cell proliferation. Hum Reprod 2008; 23:440–446. [DOI] [PubMed] [Google Scholar]
- 133. Satou R, Penrose H, Navar LG. Inflammation as a regulator of the renin-angiotensin system and blood pressure. Curr Hypertens Rep 2018; 20:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Kimura C, et al. Development of vascular smooth muscle contractility by endothelium-derived transforming growth factor beta proteins. Pflugers Arch 2014; 466:369–380. [DOI] [PubMed] [Google Scholar]
- 135. Faerstein E, Szklo M, Rosenshein NB. Risk factors for uterine leiomyoma: a practice-based case-control study. II. Atherogenic risk factors and potential sources of uterine irritation. Am J Epidemiol 2001; 153:11–19. [DOI] [PubMed] [Google Scholar]
- 136. Keshavarzi F, et al. Association of ACE I/D and AGTR1 A1166C gene polymorphisms and risk of uterine leiomyoma: a case-control study. Asian Pac J Cancer Prev 2019; 20:2595–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Halpern G, Schor E, Kopelman A. Nutritional aspects related to endometriosis. Rev Assoc Med Bras (1992) 2015; 61:519–523. [DOI] [PubMed] [Google Scholar]
- 138. Parazzini F, et al. Selected food intake and risk of endometriosis. Hum Reprod 2004; 19:1755–1759. [DOI] [PubMed] [Google Scholar]
- 139. Mozaffarian D, et al. Trans fatty acids and systemic inflammation in heart failure. Am J Clin Nutr 2004; 80:1521–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Lebovic DI, Kir M, Casey CL. Peroxisome proliferator-activated receptor-gamma induces regression of endometrial explants in a rat model of endometriosis. Fertil Steril 2004; 82:1008–1013. [DOI] [PubMed] [Google Scholar]
- 141. Norman PE, Powell JT. Vitamin D and cardiovascular disease. Circ Res 2014; 114:379–393. [DOI] [PubMed] [Google Scholar]
- 142. Elhusseini H, et al. Diet-induced vitamin D deficiency triggers inflammation and DNA damage profile in murine myometrium. Int J Womens Health 2018; 10:503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Skowronska P, et al. The role of vitamin D in reproductive dysfunction in women - a systematic review. Ann Agric Environ Med 2016; 23:671–676. [DOI] [PubMed] [Google Scholar]
- 144. Bellelis P, et al. Transcriptional changes in the expression of chemokines related to natural killer and T-regulatory cells in patients with deep infiltrative endometriosis. Fertil Steril 2013; 99:1987–1993. [DOI] [PubMed] [Google Scholar]
- 145. Somigliana E, et al. Human endometrial stromal cells as a source of soluble intercellular adhesion molecule (ICAM)-1 molecules. Hum Reprod 1996; 11:1190–1194. [DOI] [PubMed] [Google Scholar]
- 146. Dias JA Jr, et al. Patients with endometriosis of the rectosigmoid have a higher percentage of natural killer cells in peripheral blood. J Minim Invasive Gynecol 2012; 19:317–324. [DOI] [PubMed] [Google Scholar]
- 147. Berbic M, et al. The role of Foxp3+ regulatory T-cells in endometriosis: a potential controlling mechanism for a complex, chronic immunological condition. Hum Reprod 2010; 25:900–907. [DOI] [PubMed] [Google Scholar]
- 148. Gui T, et al. The disturbance of TH17-Treg cell balance in adenomyosis. Fertil Steril 2014; 101:506–514. [DOI] [PubMed] [Google Scholar]
- 149. Stromnes IM, et al. Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nat Med 2008; 14:337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Sommer C, Kress M. Recent findings on how proinflammatory cytokines cause pain: peripheral mechanisms in inflammatory and neuropathic hyperalgesia. Neurosci Lett 2004; 361:184–187. [DOI] [PubMed] [Google Scholar]
- 151. Chen DB, et al. Stimulation of prostaglandin (PG) F2 alpha and PGE2 release by tumour necrosis factor-alpha and interleukin-1 alpha in cultured human luteal phase endometrial cells. Hum Reprod 1995; 10:2773–2780. [DOI] [PubMed] [Google Scholar]
- 152. Morotti M, Vincent K, Becker CM. Mechanisms of pain in endometriosis. Eur J Obstet Gynecol Reprod Biol 2017; 209:8–13. [DOI] [PubMed] [Google Scholar]
- 153. Pongratz G, Straub RH. Role of peripheral nerve fibres in acute and chronic inflammation in arthritis. Nat Rev Rheumatol 2013; 9:117–126. [DOI] [PubMed] [Google Scholar]
- 154. Forster R, et al. Macrophage-derived insulin-like growth factor-1 is a key neurotrophic and nerve-sensitizing factor in pain associated with endometriosis. FASEB J 2019; 33:11210–11222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Greaves E, et al. Estradiol is a critical mediator of macrophage-nerve cross talk in peritoneal endometriosis. Am J Pathol 2015; 185:2286–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Jiang C, et al. The expression of toll-like receptors in eutopic and ectopic endometrium and its implication in the inflammatory pathogenesis of adenomyosis. Sci Rep 2017; 7:7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Berbic M, Ng CH, Fraser IS. Inflammation and endometrial bleeding. Climacteric 2014; 17:47–53. [DOI] [PubMed] [Google Scholar]
- 158. Malik S, et al. Reduced levels of VEGF-A and MMP-2 and MMP-9 activity and increased TNF-alpha in menstrual endometrium and effluent in women with menorrhagia. Hum Reprod 2006; 21:2158–2166. [DOI] [PubMed] [Google Scholar]
- 159. Clauss M, et al. A permissive role for tumor necrosis factor in vascular endothelial growth factor-induced vascular permeability. Blood 2001; 97:1321–1329. [DOI] [PubMed] [Google Scholar]
- 160. Tabibzadeh S. The signals and molecular pathways involved in human menstruation, a unique process of tissue destruction and remodelling. Mol Hum Reprod 1996; 2:77–92. [DOI] [PubMed] [Google Scholar]
- 161. Shalaby MR, et al. Tumor necrosis factor-alpha-associated uterine endothelial injury in vivo. Influence of dietary fat. Lab Invest 1989; 61:564–570. [PubMed] [Google Scholar]
- 162. Giuliani E, et al. Characterization of uterine NK cells in women with infertility or recurrent pregnancy loss and associated endometriosis. Am J Reprod Immunol 2014; 72:262–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Dosiou C, Giudice LC. Natural killer cells in pregnancy and recurrent pregnancy loss: endocrine and immunologic perspectives. Endocr Rev 2005; 26:44–62. [DOI] [PubMed] [Google Scholar]
- 164. Lin YH, et al. Chronic niche inflammation in endometriosis-associated infertility: current understanding and future therapeutic strategies. Int J Mol Sci 2018; 19:2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Lessey BA, Kim JJ. Endometrial receptivity in the eutopic endometrium of women with endometriosis: it is affected, and let me show you why. Fertil Steril 2017; 108:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Carli C, et al. Direct effect of macrophage migration inhibitory factor on sperm function: possible involvement in endometriosis-associated infertility. Fertil Steril 2007; 88:1240–1247. [DOI] [PubMed] [Google Scholar]
- 167. Faber BM, et al. Macrophage secretory products and sperm zona pellucida binding. Obstet Gynecol 2001; 98:668–673. [DOI] [PubMed] [Google Scholar]
- 168. Mansour G, et al. The impact of peritoneal fluid from healthy women and from women with endometriosis on sperm DNA and its relationship to the sperm deformity index. Fertil Steril 2009; 92:61–67. [DOI] [PubMed] [Google Scholar]
- 169. Sanchez AM, et al. The distinguishing cellular and molecular features of the endometriotic ovarian cyst: from pathophysiology to the potential endometrioma-mediated damage to the ovary. Hum Reprod Update 2014; 20:217–230. [DOI] [PubMed] [Google Scholar]
- 170. Kitajima M, et al. Endometriomas as a possible cause of reduced ovarian reserve in women with endometriosis. Fertil Steril 2011; 96:685–691. [DOI] [PubMed] [Google Scholar]
- 171. Leone Roberti Maggiore U, Gupta JK, Ferrero S. Treatment of endometrioma for improving fertility. Eur J Obstet Gynecol Reprod Biol 2017; 209:81–85. [DOI] [PubMed] [Google Scholar]
- 172. Inoue T, et al. Tumour necrosis factor alpha inhibits in-vitro decidualization of human endometrial stromal cells. Hum Reprod 1994; 9:2411–2417. [DOI] [PubMed] [Google Scholar]
- 173. Inagaki N, et al. Uterine cavity matrix metalloproteinases and cytokines in patients with leiomyoma, adenomyosis or endometrial polyp. Eur J Obstet Gynecol Reprod Biol 2003; 111:197–203. [DOI] [PubMed] [Google Scholar]
- 174. Snider AP, Wood JR. Obesity induces ovarian inflammation and reduces oocyte quality. Reproduction 2019; 158:R79–R90. [DOI] [PubMed] [Google Scholar]
- 175. Mu F, et al. Endometriosis and risk of coronary heart disease. Circ Cardiovasc Qual Outcomes 2016; 9:257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Wilson PW. Evidence of systemic inflammation and estimation of coronary artery disease risk: a population perspective. Am J Med 2008; 121:S15–S20. [DOI] [PubMed] [Google Scholar]
- 177. Santoro L, et al. Endothelial dysfunction but not increased carotid intima-media thickness in young European women with endometriosis. Hum Reprod 2012; 27:1320–1326. [DOI] [PubMed] [Google Scholar]
- 178. Santoro L, et al. Regression of endothelial dysfunction in patients with endometriosis after surgical treatment: a 2-year follow-up study. Hum Reprod 2014; 29:1205–1210. [DOI] [PubMed] [Google Scholar]
- 179. Dawson A, et al. Endometriosis and endometriosis-associated cancers: new insights into the molecular mechanisms of ovarian cancer development. Ecancermedicalscience 2018; 12:803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Anglesio MS, et al. Multifocal endometriotic lesions associated with cancer are clonal and carry a high mutation burden. J Pathol 2015; 236:201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Kobayashi H, et al. Understanding the role of epigenomic, genomic and genetic alterations in the development of endometriosis (review). Mol Med Rep 2014; 9:1483–1505. [DOI] [PubMed] [Google Scholar]
- 182. Kobayashi H, et al. Molecular pathogenesis of endometriosis-associated clear cell carcinoma of the ovary (review). Oncol Rep 2009; 22:233–240. [PubMed] [Google Scholar]
- 183. Wendel JRH, Wang X, Hawkins SM. The Endometriotic tumor microenvironment in ovarian cancer. Cancers (Basel) 2018; 10:261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Clendenen TV, et al. Circulating inflammation markers and risk of epithelial ovarian cancer. Cancer Epidemiol Biomarkers Prev 2011; 20:799–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Szlosarek PW, et al. Expression and regulation of tumor necrosis factor alpha in normal and malignant ovarian epithelium. Mol Cancer Ther 2006; 5:382–390. [DOI] [PubMed] [Google Scholar]
- 186. Kulbe H, et al. The inflammatory cytokine tumor necrosis factor-alpha regulates chemokine receptor expression on ovarian cancer cells. Cancer Res 2005; 65:10355–10362. [DOI] [PubMed] [Google Scholar]
- 187. Munkarah AR, et al. Effects of prostaglandin E(2) on proliferation and apoptosis of epithelial ovarian cancer cells. J Soc Gynecol Investig 2002; 9:168–173. [PubMed] [Google Scholar]
- 188. Halder S, Al-Hendy A. Hypovitaminosis D and high serum transforming growth factor beta-3: important biomarkers for uterine fibroids risk. Fertil Steril 2016; 106:1648–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Bedaiwy MA, et al. Prediction of endometriosis with serum and peritoneal fluid markers: a prospective controlled trial. Hum Reprod 2002; 17:426–431. [DOI] [PubMed] [Google Scholar]
- 190. Mu F, et al. A prospective study of inflammatory markers and risk of endometriosis. Am J Epidemiol 2018; 187:515–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Faria SS, et al. The neutrophil-to-lymphocyte ratio: a narrative review. Ecancermedicalscience 2016; 10:702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Inagaki J, et al. An association of IgG anti-laminin-1 autoantibodies with endometriosis in infertile patients. Hum Reprod 2003; 18:544–549. [DOI] [PubMed] [Google Scholar]
- 193. Malutan AM, et al. Serum anti-inflammatory cytokines for the evaluation of inflammatory status in endometriosis. J Res Med Sci 2015; 20:668–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009; 136:215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Mari-Alexandre J, et al. miRNAs regulation and its role as biomarkers in endometriosis. Int J Mol Sci 2016; 17:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Mari-Alexandre J, et al. Micro-RNA profile and proteins in peritoneal fluid from women with endometriosis: their relationship with sterility. Fertil Steril 2018; 109:675–684 e2. [DOI] [PubMed] [Google Scholar]
- 197. Sahin C, et al. microRNA let-7b: a novel treatment for endometriosis. J Cell Mol Med 2018; 22:5346–5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Chuang TD, Khorram O. miR-200c regulates IL8 expression by targeting IKBKB: a potential mediator of inflammation in leiomyoma pathogenesis. PLoS One 2014; 9:e95370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Li A, et al. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J Immunol 2003; 170:3369–3376. [DOI] [PubMed] [Google Scholar]
- 200. Pysz MA, Gambhir SS, Willmann JK. Molecular imaging: current status and emerging strategies. Clin Radiol 2010; 65:500–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Feider CL, et al. Molecular imaging of endometriosis tissues using desorption electrospray ionization mass spectrometry. Sci Rep 2019; 9:15690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Al-Zaghal A, et al. The detection of uterine leiomyoma (fibroid) calcifications on 18F-NaF PET/CT. Clin Nucl Med 2018; 43:e287–e288. [DOI] [PubMed] [Google Scholar]
- 203. Hammoud DA. Molecular imaging of inflammation: current status. J Nucl Med 2016; 57:1161–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Weissleder R, Nahrendorf M, Pittet MJ. Imaging macrophages with nanoparticles. Nat Mater 2014; 13:125–138. [DOI] [PubMed] [Google Scholar]
- 205. Malviya G, et al. Imaging T-lymphocytes in inflammatory diseases: a nuclear medicine approach. Q J Nucl Med Mol Imaging 2014; 58:237–257. [PubMed] [Google Scholar]
- 206. Chianelli M, et al. New radiopharmaceuticals for imaging rheumatoid arthritis. Q J Nucl Med Mol Imaging 2006; 50:217–225. [PubMed] [Google Scholar]
- 207. Park J, et al. Obesity and cancer—mechanisms underlying tumour progression and recurrence. Nat Rev Endocrinol 2014; 10:455–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Barry JC, et al. Short-term exercise training reduces anti-inflammatory action of interleukin-10 in adults with obesity. Cytokine 2018; 111:460–469. [DOI] [PubMed] [Google Scholar]
- 209. Le Chatelier E, et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013; 500:541–546. [DOI] [PubMed] [Google Scholar]
- 210. Cani PD, Delzenne NM. The gut microbiome as therapeutic target. Pharmacol Ther 2011; 130:202–212. [DOI] [PubMed] [Google Scholar]
- 211. Candido FG, et al. Impact of dietary fat on gut microbiota and low-grade systemic inflammation: mechanisms and clinical implications on obesity. Int J Food Sci Nutr 2018; 69:125–143. [DOI] [PubMed] [Google Scholar]
- 212. Seah JY, et al. Consumption of red meat, but not cooking oils high in polyunsaturated fat, is associated with higher arachidonic acid status in Singapore Chinese adults. Nutrients 2017; 9:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Khanaki K, et al. Evaluation of the relationship between endometriosis and omega-3 and omega-6 polyunsaturated fatty acids. Iran Biomed J 2012; 16:38–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Malik M, et al. Curcumin, a nutritional supplement with antineoplastic activity, enhances leiomyoma cell apoptosis and decreases fibronectin expression. Fertil Steril 2009; 91:2177–2184. [DOI] [PubMed] [Google Scholar]
- 215. Suou K, et al. Apigenin inhibits tumor necrosis factor alpha-induced cell proliferation and prostaglandin E2 synthesis by inactivating NFkappaB in endometriotic stromal cells. Fertil Steril 2011; 95:1518–1521. [DOI] [PubMed] [Google Scholar]
- 216. Islam MS, et al. Use of dietary phytochemicals to target inflammation, fibrosis, proliferation, and angiogenesis in uterine tissues: promising options for prevention and treatment of uterine fibroids? Mol Nutr Food Res 2014; 58:1667–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Fusco D, et al. Effects of antioxidant supplementation on the aging process. Clin Interv Aging 2007; 2:377–387. [PMC free article] [PubMed] [Google Scholar]
- 218. Mier-Cabrera J, et al. Women with endometriosis improved their peripheral antioxidant markers after the application of a high antioxidant diet. Reprod Biol Endocrinol 2009; 7:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Deree J, et al. Insights into the regulation of TNF-alpha production in human mononuclear cells: the effects of non-specific phosphodiesterase inhibition. Clinics (Sao Paulo) 2008; 63:321–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Kamencic H, Thiel JA. Pentoxifylline after conservative surgery for endometriosis: a randomized, controlled trial. J Minim Invasive Gynecol 2008; 15:62–66. [DOI] [PubMed] [Google Scholar]
- 221. Barrier BF, et al. Efficacy of anti-tumor necrosis factor therapy in the treatment of spontaneous endometriosis in baboons. Fertil Steril 2004; 81:775–779. [DOI] [PubMed] [Google Scholar]
- 222. Lu D, Song H, Shi G. Anti-TNF-alpha treatment for pelvic pain associated with endometriosis. Cochrane Database Syst Rev 2013; 3. CD008088. [DOI] [PubMed] [Google Scholar]
- 223. Antsiferova Y, Sotnikova N, Parfenyuk E. Different effects of the immunomodulatory drug GMDP immobilized onto aminopropyl modified and unmodified mesoporous silica nanoparticles upon peritoneal macrophages of women with endometriosis. Biomed Res Int 2013; 2013:924362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Keenan JA, et al. Regression of endometrial explants in a rat model of endometriosis treated with the immune modulators loxoribine and levamisole. Fertil Steril 1999; 72:135–141. [DOI] [PubMed] [Google Scholar]
- 225. Kotlyar A, Taylor HS, D'Hooghe TM. Use of immunomodulators to treat endometriosis. Best Pract Res Clin Obstet Gynaecol 2019; 60:56–65. [DOI] [PubMed] [Google Scholar]
- 226. Purohit A, et al. Inhibition of tumor necrosis factor alpha-stimulated aromatase activity by microtubule-stabilizing agents, paclitaxel and 2-methoxyestradiol. Biochem Biophys Res Commun 1999; 261:214–217. [DOI] [PubMed] [Google Scholar]
- 227. Taniguchi F, et al. Gonadotropin-releasing hormone analogues reduce the proliferation of endometrial stromal cells but not endometriotic cells. Gynecol Obstet Invest 2013; 75:9–15. [DOI] [PubMed] [Google Scholar]
- 228. Sakamoto Y, et al. Tumor necrosis factor-alpha-induced interleukin-8 (IL-8) expression in endometriotic stromal cells, probably through nuclear factor-kappa B activation: gonadotropin-releasing hormone agonist treatment reduced IL-8 expression. J Clin Endocrinol Metab 2003; 88:730–735. [DOI] [PubMed] [Google Scholar]
- 229. Khan KN, et al. Changes in tissue inflammation, angiogenesis and apoptosis in endometriosis, adenomyosis and uterine myoma after GnRH agonist therapy. Hum Reprod 2010; 25:642–653. [DOI] [PubMed] [Google Scholar]
- 230. Hur KY, Lee MS. New mechanisms of metformin action: focusing on mitochondria and the gut. J Diabetes Investig 2015; 6:600–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Saisho Y. Metformin and inflammation: its potential beyond glucose-lowering effect. Endocr Metab Immune Disord Drug Targets 2015; 15:196–205. [DOI] [PubMed] [Google Scholar]
- 232. Tseng CH. Metformin use is associated with a lower risk of uterine leiomyoma in female type 2 diabetes patients. Ther Adv Endocrinol Metab 2019; 10:2042018819895159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Taylor HS, et al. Effect of simvastatin on baboon endometriosis. Biol Reprod 2017; 97:32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Cakmak H, et al. Statins inhibit monocyte chemotactic protein 1 expression in endometriosis. Reprod Sci 2012; 19:572–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Kortekaas KE, et al. ACE inhibitors potently reduce vascular inflammation, results of an open proof-of-concept study in the abdominal aortic aneurysm. PLoS One 2014; 9:e111952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Montecucco F, Pende A, Mach F. The renin-angiotensin system modulates inflammatory processes in atherosclerosis: evidence from basic research and clinical studies. Mediators Inflamm 2009; 2009:752406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Silva IVG, Figueiredo RC, Rios DRA. Effect of different classes of antihypertensive drugs on endothelial function and inflammation. Int J Mol Sci 2019; 20:3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Fischer NM, et al. Angiotensin-converting enzyme inhibitors reduce uterine fibroid incidence in hypertensive women. J Clin Endocrinol Metab 2020; 106(2):e650–e659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Park SB, et al. Cyclooxygenase-2 inhibitor, celecoxib, inhibits leiomyoma cell proliferation through the nuclear factor kappaB pathway. Reprod Sci 2014; 21:1187–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Hull ML, et al. Nimesulide, a COX-2 inhibitor, does not reduce lesion size or number in a nude mouse model of endometriosis. Hum Reprod 2005; 20:350–358. [DOI] [PubMed] [Google Scholar]
- 241. Olivares C, et al. Effects of a selective cyclooxygenase-2 inhibitor on endometrial epithelial cells from patients with endometriosis. Hum Reprod 2008; 23:2701–2708. [DOI] [PubMed] [Google Scholar]
- 242. Cobellis L, et al. The treatment with a COX-2 specific inhibitor is effective in the management of pain related to endometriosis. Eur J Obstet Gynecol Reprod Biol 2004; 116:100–102. [DOI] [PubMed] [Google Scholar]
- 243. Kilico I, et al. Regression of experimentally induced endometriosis with a new selective cyclooxygenase-2 enzyme inhibitor. Gynecol Obstet Invest 2014; 77:35–39. [DOI] [PubMed] [Google Scholar]
- 244. Bulun SE. Uterine fibroids. N Engl J Med 2013; 369:1344–1355. [DOI] [PubMed] [Google Scholar]
- 245. Wu MH, et al. Prostaglandin E2: the master of endometriosis? Exp Biol Med (Maywood) 2010; 235:668–677. [DOI] [PubMed] [Google Scholar]
- 246. Arici A, et al. Interleukin-8 concentration in peritoneal fluid of patients with endometriosis and modulation of interleukin-8 expression in human mesothelial cells. Mol Hum Reprod 1996; 2:40–45. [DOI] [PubMed] [Google Scholar]
- 247. Topley N, et al. Human peritoneal mesothelial cells synthesize interleukin-6: induction by IL-1 beta and TNF alpha. Kidney Int 1993; 43:226–233. [DOI] [PubMed] [Google Scholar]
- 248. Kang YJ, et al. An increased level of IL-6 suppresses NK cell activity in peritoneal fluid of patients with endometriosis via regulation of SHP-2 expression. Hum Reprod 2014; 29:2176–2189. [DOI] [PubMed] [Google Scholar]
- 249. Harada T, et al. Increased interleukin-6 levels in peritoneal fluid of infertile patients with active endometriosis. Am J Obstet Gynecol 1997; 176:593–597. [DOI] [PubMed] [Google Scholar]
- 250. Mukaida N, Harada A, Matsushima K. Interleukin-8 (IL-8) and monocyte chemotactic and activating factor (MCAF/MCP-1), chemokines essentially involved in inflammatory and immune reactions. Cytokine Growth Factor Rev 1998; 9:9–23. [DOI] [PubMed] [Google Scholar]
- 251. Garcia-Velasco JA, Arici A. Chemokines and human reproduction. Fertil Steril 1999; 71:983–993. [DOI] [PubMed] [Google Scholar]
- 252. Ryan IP, et al. Interleukin-8 concentrations are elevated in peritoneal fluid of women with endometriosis. Fertil Steril 1995; 63:929–932. [PubMed] [Google Scholar]
- 253. Tarumi Y, et al. Interleukin-9 produced by helper T cells stimulates interleukin-8 expression in endometriosis. Am J Reprod Immunol 2020; e13380. [DOI] [PubMed] [Google Scholar]
- 254. Somigliana E, et al. Modulation of NK cell lytic function by endometrial secretory factors: potential role in endometriosis. Am J Reprod Immunol 1996; 36:295–300. [DOI] [PubMed] [Google Scholar]
- 255. Suen JL, et al. Serum level of IL-10 is increased in patients with endometriosis, and IL-10 promotes the growth of lesions in a murine model. Am J Pathol 2014; 184:464–471. [DOI] [PubMed] [Google Scholar]
- 256. McLaren J, et al. Decreased levels of the potent regulator of monocyte/macrophage activation, interleukin-13, in the peritoneal fluid of patients with endometriosis. Hum Reprod 1997; 12:1307–1310. [DOI] [PubMed] [Google Scholar]
- 257. Cameron MJ, Kelvin DJ. Cytokines and chemokines—their receptors and their genes: an overview. Adv Exp Med Biol 2003; 520:8–32. [DOI] [PubMed] [Google Scholar]
- 258. Arici A, et al. Monocyte chemotactic protein-1 concentration in peritoneal fluid of women with endometriosis and its modulation of expression in mesothelial cells. Fertil Steril 1997; 67:1065–1072. [DOI] [PubMed] [Google Scholar]
- 259. Liu YG, et al. Induction of endometrial epithelial cell invasion and c-fms expression by transforming growth factor beta. Mol Hum Reprod 2009; 15:665–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Hornung D, et al. Immunolocalization and regulation of the chemokine RANTES in human endometrial and endometriosis tissues and cells. J Clin Endocrinol Metab 1997; 82:1621–1628. [DOI] [PubMed] [Google Scholar]
- 261. Zhou WJ, et al. The CCL17-CCR4 axis between endometrial stromal cells and macrophages contributes to the high levels of IL-6 in ectopic milieu. Am J Reprod Immunol 2017; 78:e12644. [DOI] [PubMed] [Google Scholar]
- 262. Thiruchelvam U, Wingfield M, O'Farrelly C. Natural killer cells: key players in endometriosis. Am J Reprod Immunol 2015; 74:291–301. [DOI] [PubMed] [Google Scholar]
- 263. Weng LC, et al. Estrogen-regulated CD200 inhibits macrophage phagocytosis in endometriosis. J Reprod Immunol 2020; 138:103090. [DOI] [PubMed] [Google Scholar]
- 264. Zulfikaroglu E, et al. Efficacy of anti-tumor necrosis factor therapy on endometriosis in an experimental rat model. Arch Gynecol Obstet 2011; 283:799–804. [DOI] [PubMed] [Google Scholar]
- 265. Chen YJ, et al. A tumor necrosis factor-alpha inhibitor reduces the embryotoxic effects of endometriotic peritoneal fluid. Fertil Steril 2013; 100:1476–1485. [DOI] [PubMed] [Google Scholar]
- 266. Velasco I, et al. Intraperitoneal recombinant interleukin-2 activates leukocytes in rat endometriosis. J Reprod Immunol 2007; 74:124–132. [DOI] [PubMed] [Google Scholar]
- 267. Acien P, et al. Treatment of endometriosis with transvaginal ultrasound-guided drainage under GnRH analogues and recombinant interleukin-2 left in the cysts. Gynecol Obstet Invest 2005; 60:224–231. [DOI] [PubMed] [Google Scholar]
- 268. Ingelmo JM, Quereda F, Acien P. Effect of human interferon-alpha-2b on experimental endometriosis in rats: comparison between short and long series of treatment. Eur J Obstet Gynecol Reprod Biol 2013; 167:190–193. [DOI] [PubMed] [Google Scholar]
- 269. Xu Z, et al. Lipoxin A4 inhibits the development of endometriosis in mice: the role of anti-inflammation and anti-angiogenesis. Am J Reprod Immunol 2012; 67:491–497. [DOI] [PubMed] [Google Scholar]
- 270. He Y, et al. Interleukin-37b inhibits the growth of murine endometriosis-like lesions by regulating proliferation, invasion, angiogenesis and inflammation. Mol Hum Reprod 2020; 26:240–255. [DOI] [PubMed] [Google Scholar]
- 271. Sekulovski N, et al. Niclosamide suppresses macrophage-induced inflammation in endometriosis dagger. Biol Reprod 2020; 102:1011–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Wu M, Zhang Y. MiR-182 inhibits proliferation, migration, invasion and inflammation of endometrial stromal cells through deactivation of NF-kappaB signaling pathway in endometriosis. Mol Cell Biochem 2021; 476:1575–1588. [DOI] [PubMed] [Google Scholar]
- 273. Chuang TD, Rehan A, Khorram O. Tranilast induces MiR-200c expression through blockade of RelA/p65 activity in leiomyoma smooth muscle cells. Fertil Steril 2020; 113:1308–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Ebert AD, Bartley J, David M. Aromatase inhibitors and cyclooxygenase-2 (COX-2) inhibitors in endometriosis: new questions—old answers? Eur J Obstet Gynecol Reprod Biol 2005; 122:144–150. [DOI] [PubMed] [Google Scholar]
- 275. Horie S, et al. Progesterone and progestational compounds attenuate tumor necrosis factor alpha-induced interleukin-8 production via nuclear factor kappa B inactivation in endometriotic stromal cells. Fertil Steril 2005; 83:1530–1535. [DOI] [PubMed] [Google Scholar]
- 276. Afrin S, et al. Simvastatin ameliorates altered mechanotransduction in uterine leiomyoma cells. Am J Obstet Gynecol 2020; 223:733 e1–733 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Ohama Y, et al. Peroxisome proliferator-activated receptor-gamma ligand reduced tumor necrosis factor-alpha-induced interleukin-8 production and growth in endometriotic stromal cells. Fertil Steril 2008; 89:311–317. [DOI] [PubMed] [Google Scholar]
- 278. Machado DE, et al. A selective cyclooxygenase-2 inhibitor suppresses the growth of endometriosis with an antiangiogenic effect in a rat model. Fertil Steril 2010; 93:2674–2679. [DOI] [PubMed] [Google Scholar]
- 279. Xiu-li W, et al. NF-kappaB decoy oligonucleotides suppress RANTES expression and monocyte chemotactic activity via NF-kappaB inactivation in stromal cells of ectopic endometrium. J Clin Immunol 2009; 29:387–395. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new data were generated or analyzed in support of this research.