

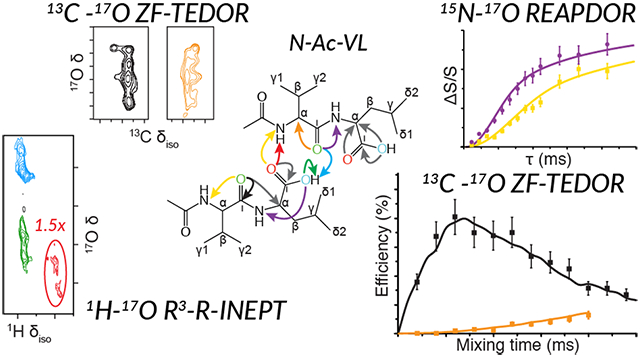
Abstract
The structure of two protected amino acids, FMOC-l-leucine and FMOC-l-valine, and a dipeptide, N-acetyl-l-valyl-l-leucine (N-Ac-VL), were studied via one- and two-dimensional solid-state nuclear magnetic resonance (NMR) spectroscopy. Utilizing 17O magic-angle spinning (MAS) NMR at multiple magnetic fields (17.6–35.2 T/750–1500 MHz for 1H) the 17O quadrupolar and chemical shift parameters were determined for the two oxygen sites of each FMOC-protected amino acids and the three distinct oxygen environments of the dipeptide. The one- and two-dimensional, 17O, 15N─17O, 13C─17O, and 1H─17O double-resonance correlation experiments performed on the uniformly 13C,15N and 70% 17O-labeled dipeptide prove the attainability of 17O as a probe for structure studies of biological systems. 15N─17O and 13C─17O distances were measured via one-dimensional REAPDOR and ZF-TEDOR experimental buildup curves and determined to be within 15% of previously reported distances, thus demonstrating the use of 17O NMR to quantitate interatomic distances in a fully labeled dipeptide. Through-space hydrogen bonding of N-Ac-VL was investigated by a two-dimensional 1H-detected 17O R3-R-INEPT experiment, furthering the importance of 17O for studies of structure in biomolecular solids.
Graphical Abstract
1. INTRODUCTION
The essential role of oxygen in hydrogen bonding in determining the chemistry, structure, and function of peptides and proteins is well known. It is also axiomatic that 17O magic-angle spinning (MAS) nuclear magnetic resonance (NMR) experiments can in principle elucidate the details of these interactions via site-specific measurements of the chemical shift and quadrupole tensors. Furthermore, if a method of dipolar recoupling is included in the experimental protocol, then 13C─17O and 15N─17O and 1H─17O distance measurements and 3D structures are possible.1,2 Nevertheless, the potential of 17O NMR to provide this level of detailed structural information has never been realized primarily for three reasons: (i) 17O is present with a low natural isotopic abundance (0.037%), (ii) it is a low gyromagnetic ratio nuclear spin (γ = −5.774 MHz T−1), and (iii) it is a quadrupolar nucleus (I = 5/2). Thus, at ubiquitous magnetic fields (≤5–14.1 T, ω0H/2π = 200–600 MHz) the sensitivity observed in MAS NMR spectra is inherently low, and the resolution is low due to the residual second-order quadrupolar broadening that is not eliminated by magic-angle spinning.3
The direct solution to the first issue is isotopic enrichment and a relatively new solution to this problem is discussed below. The second and third issues are most easily addressed by using new NMR methods and by performing 17O spectroscopy at high magnetic fields, which we define as greater than 14.1 T.4-13 In particular, we report here spectroscopy at high fields (17.6–35.2 T, ω0H/2π = 750–1500 MHz) which enhances overall sensitivity, but more importantly attenuates the broadening from second-order quadrupole coupling arising from the quadrupole coupling constant (CQ) of 7–9 MHz observed for C═17O chemical environments.14-16 For example, in spectra described below, the second-order powder patterns are narrowed to a few kHz, concurrently increasing the resolution of the spectra. With the emergence of NMR magnets fabricated from high-temperature superconductors that generate 28–35 T on the horizon, these sorts of experimental data should become relatively routine. Thus, the motivation of the results reported here is to demonstrate the potential for significant advances and applications of 17O NMR spectroscopy.
Because of the improved sensitivity and resolution in high-field 17O MAS NMR spectra, it becomes feasible to perform correlation experiments to determine spectral assignments and measure structural parameters with dipole recoupling experiments. Both homonuclear and heteronuclear MAS recoupling experiments are routinely used to study the structure of peptides and proteins,17-20 and provide accurate measurements of 13C─13C,21-23 13C─15N,24,25 and 1H─13C/15N26-28 distances. This class of experiments is also applicable, with suitably modified pulse sequences, to 13C─17O, 15N─17O and 1H─17O distance measurements; for example, heteronuclear recoupling experiments have been reported, although only a few examples of two-dimensional heteronuclear correlation experiments are in the literature.29-35 15N─17O rotational-echo adiabatic-passage double-resonance (REAPDOR)36 experiments were used to examine hydrogen bonding in amyloid fibrils, with the most complete results requiring an isolated spin-pair and a determination of the precise level of 17O enrichment in the sample to precisely measure the 15N─17O dipolar coupling.4,29,37 Dipolar mediated heteronuclear correlation experiments between spin I = 1/2 and quadrupolar nuclei based on J-coupled experiments, such as rotary resonance recoupled (R3) refocused insensitive nuclei enhanced by polarization transfer (R3-R-INEPT), have been shown to produce high-efficiency polarization transfer.30,38 The addition of a multiple-quantum (MQ) or satellite transition (ST) filter to the dipolar meditated heteronuclear experiments was shown to increase the resolution available in these experiments.38 Finally, recent advances in 17O spectroscopy including higher magnetic field experiments (>14.1 T),4-13 application of population transfer techniques,39-41 and dynamic nuclear polarization31-33,42-44 enhance the ability to perform these distance measurements. Thus, successful 17O studies of amino acids, polypeptides, pharmaceutical compounds, and amyloid fibrils have appeared in the literature using one or more of these strategies.4-13,29,31-33,37,39-58
Building on previous 17O NMR experiments, we first describe an efficient procedure for 17O labeling FMOC-protected amino acids using H217O as the source. The approach utilizes a nonequilibrium multiple-turnover reaction under mild and selective conditions.59 Two FMOC-protected amino acid precursors, FMOC-l-valine and FMOC-l-lecuine, were 17O enriched to 40 and 70%, with an efficiency of >95%, and used to prepare [17O/13C/15N]-N-Ac-VL, a model dipeptide used to develop dipole recoupling experiments. We envision that these labeled amino acids can be used in contemporary peptide synthesis60 or in cell-free synthesis protocols61 for 17O labeling of peptides and proteins.4 Second, we recorded 1D MAS spectra at ω0H/2π = 750, 800, 900, and 1500 MHz and simulated the rotational sideband patterns to extract the chemical shift and electric field gradient (EFG) tensors for the three 17O moieties: amide NC═O, carboxyl C═O, and C─OH. The C═17O tensors have a span of 380–450 ppm and the C─17OH span is 310–320; thus, the shift tensors are substantial and the shifts are very sensitive to chemical environment. The quadrupole coupling constants range between 7 and 8.5 MHz. Third, in order to demonstrate the utility of high magnetic fields in 17O spectroscopy we recorded 2D triple-quantum MAS (3QMAS) at 35.2 T (ω0H/2π = 1.5 GHz) for N-Ac-VL using the series-connected hybrid (SCH) magnet at the National High Magnetic Field Laboratory (NHMFL, Tallahassee, FL). At this field strength the second-order quadrupole powder patterns are reduced to ~2 kHz (~10 ppm), yielding excellent sensitivity and resolution. The 3QMAS experiment yields an isotropic dimension with the chemical and second-order quadrupole shift and an anisotropic dimension with the second-order line shapes and chemical shift information. Finally, using a variety of dipole recoupling techniques we measured a total of 14 13C─17O, 15N─17O, and 1H─17O distances in N-acetyl-l-valyl-l-leucine (N-Ac-VL), demonstrating the viability of determining molecular structure with 17O distance constraints.
2. EXPERIMENTAL SECTION
2.1. Materials and Synthesis.
Traditionally 17O enrichment of amino acids is performed via acid-catalyzed exchange at elevated temperature, where the amino acid is heated in the presence of strong acid and 17O enriched water and yield enrichment efficiencies up to 85%.62,63 These conditions prohibit acid-catalyzed exchange to be used for 17O enrichment in protected amino acids or proteins, and therefore we have turned to an alternative procedure utilizing mild conditions and yielding 17O/18O isotopic enrichment efficiencies of >95%.46,59,62-64 Utilizing this approach, illustrated in Scheme 1, FMOC-l-valine and FMOC-l-leucine were prepared using 40% H217O (Cambridge Isotope Laboratories, Andover, MA) for study via 17O NMR spectroscopy. A dipeptide sample, [17O/13C/15N]-N-Ac-VL, was prepared using the same approach with U-13C,15N amino acids and 70% H217O (Cambridge Isotope Laboratories, Andover, MA).
Scheme 1.

Isotopic Labeling of FMOC Amino Acids Using the Multiple-Turnover Reaction with 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Hydrochloride (EDC·HCl)59
The following reagents were purchased from Sigma-Aldrich (St. Louis, MO), 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide hydrochloride (EDC·HCl), dimethylformamide (kept in a septum container), and 3,5-dimethylpyridine, which was then reacted with HBr (33% in acetic acid) to form 3,5-dimethylpyridine·HBr that was lyophilized to remove residual water. All starting materials were dried by preparing a sonicated suspension of each in acetonitrile, and removing residual water as an acetonitrile azeotrope by rotary evaporation. Drying was repeated in triplicate. Flame-dried glassware and stir bars were used and the reactions were carried out under dry Ar gas on a Schlenk line.
First, 430 mg of EDC·HCl (10 equiv) and 850 mg of dry 3,5-dimethylpyridine·HBr (20 equiv) were suspended in ~2–5 mL of DMF and added to 100 mg of FMOC-protected amino acid and 200 μL of 40% H217O (45 equiv) via a syringe. The reaction was stirred at room temperature for 18–24 h after which the reaction mixture was supplemented with 430 mg of EDC·HCl. This was repeated once and the reaction was allowed to stir for an additional 18–24 h. The reaction mixture was diluted with ~25 mL of ethyl acetate and dried with MgSO4, washed three times with 0.1 M citric acid (~15 mL) and once with 0.1 M citric acid in brine (~15 mL) and filtered. The FMOC-protected amino acid products were purified by aqueous extraction with ethyl acetate (~25 mL) three times. The resulting solutions were lyophilized to yield pure FMOC-protected 17O-labeled amino acids.
In addition to the 40% 17O-enriched FMOC-protected amino acids, [U-13C,15N]FMOC-l-valine and [U-13C,15N]FMOC-l-leucine were labeled with 70% 17O H217O (CIL, Andover, MA) using the procedure above. Finally, N-acetyl-[U-13C,15N,70% 17O]-l-Val-l-Leu (N-Ac-VL) was synthesized by New England Peptide (Gardner, MA) using standard solid-phase methods and purified by high-performance liquid chromatography (HPLC).
The products were then recrystallized in dichloromethane (FMOC-l-valine), ethanol (FMOC-l-leucine), or water (N-Ac-VL) by slow evaporation. The 17O labeling of each product was verified by mass spectrometry (Koch Institute MIT, FMOC amino acids; New England Peptide, N-Ac-VL).
2.2. Solid-State Nuclear Magnetic Resonance Spectroscopy.
One- and two-dimensional 17O NMR experiments were performed using a series of magnetic field strengths, all experimental parameters are summarized in Tables 1-3. Experiments at 21.1 T (ω0H/2π = 900 MHz, FBML-MIT), were performed using a Bruker Avance II spectrometer and a 3.2 mm triple resonance (1H, 13C, 17O) probe (Bruker BioSpin, Billerica, MA, USA). Experiments at 18.8 T (ω0H/2π = 800 MHz, FBML-MIT) were performed using a Bruker Avance III spectrometer and a 1.3 mm triple resonance (1H, X, Y) probe (Bruker, BioSpin, Billerica, MA, USA). Experiments at 17.6 T (ω0H/2π = 750 MHz, FBML-MIT) were performed using a home-built spectrometer (courtesy of Dr. David Ruben, FBML-MIT) and a 3.2 mm triple resonance (1H, 17O, 15N) probe with a modified probe head employing a RevolutionNMR (Fort Collins, CO, USA) stator housing within a Bruker probe body. Additional 17O NMR experiments were recorded on the 35.2 T (ω0H/2π = 1500 MHz) series-connected hybrid magnet at the NHMFL65 using a Bruker Avance NEO console and a single-resonance 3.2 mm MAS probe designed and constructed at the NHMFL. All 17O NMR spectra were referenced to liquid water (18% H217O) via the substitution method.66 17O γB1/2π varied between 83 and 156 kHz, and non-spinning experiments were acquired with continuous-wave high-power 1H decoupling during acquisition (γB1/2π = 100 kHz). We note that 1H decoupling during acquisition of 17O MAS NMR spectra did not affect the second-order line shape of the spectrum (Figure S6).54
Table 1.
NMR Acquisition Parameters for One-Dimensional 17O Experiments
| B0 (T) | ω0/2π (MHz) | pulse sequence | sample | ωR/2π (kHz) | recycle delay (s) | scans (×1024) | 1H decouple |
|---|---|---|---|---|---|---|---|
| 17.6 | 101.45 | Hahn-echo | FMOC-l-leucine | 15, 17, 18 | 0.3 | 63 | no |
| 0 | 1.0 | 1340 | yes | ||||
| FMOC-l-valine | 18, 19, 20 | 0.3 | 63 | no | |||
| 0 | 1.0 | 1340 | yes | ||||
| N-Ac-VL | 18, 20 | 0.3 | 63 | no | |||
| 18 | 1.0 | 235 | yes | ||||
| 18.8 | 108.36 | Hahn-echo | FMOC-l-leucine | 60 | 0.3 | 256 | no |
| N-Ac-VL | 60 | 0.15 | 250 | no | |||
| 21.1 | 122.02 | Hahn-echo | FMOC-l-leucine | 24 | 0.25 | 20 | no |
| FMOC-l-valine | 20 | 0.2 | 100 | no | |||
| N-Ac-VL | 24 | 0.3 | 80 | no | |||
| 35.2 | 203.36 | Hahn-echo | N-Ac-VL | 19 | 0.1 | 4 | no |
Table 3.
NMR Acquisition Parameters for One- and Two-Dimensional 1H, 13C, and 15N Experiments
| B0 (T) | nucleusa | ω0/2π (MHz) | pulse sequence | sample | ωR/2π (kHz) | recycle delay (s) | scans | 1H decouple |
|---|---|---|---|---|---|---|---|---|
| 11.7 | 13C | 125.3 | CP MAS | FMOC-l-leucine | 10 | 3 | 21280 | yes |
| 13C | 125.3 | CP MAS | FMOC-l-valine | 10 | 3 | 18560 | yes | |
| 13C | 125.3 | RFDR | N-Ac-VL | 10 | 3 | 16 | yes | |
| 13C─15N | 125.3–50.5 | ZF-TEDOR | N-Ac-VL | 10 | 3 | 256 | yes | |
| 18.8 | 1H | 800 | Bloch decay | N-Ac-VL | 60 | 3 | 32 | no |
| 21.1 | 1H | 900.1 | Bloch decay | N-Ac-VL | 20 | 3 | 16 | no |
| 13C | 226.3 | CP MAS | N-Ac-VL | 20 | 3 | 64 | yes | |
| 15N | 91.2 | CP MAS | N-Ac-VL | 20 | 3 | 256 | yes |
Detection nucleus listed first.
The timing diagrams of the pulse sequences involving 17O (3QMAS, MATPASS, ZF-TEDOR, REAPDOR, and R3-R-INEPT) are illustrated in Figures S15 and S16 with the phase cycling scheme used for each experiment. The two-dimensional 17O shifted-echo 3QMAS spectrum was acquired at 35.2 T with 20 rotor-synchronized t1 increments with an increment of 52.63 μs, and performed with 3Q excitation and conversion pulses of 3 and 1 μs (γB1/2π = 100 kHz), and π/2 and π pulses of 2.5 and 5 μs (γB1/2π = 33.3 kHz). The two-separation (MATPASS)67 spectrum was acquired at 35.2 T with 8 t1 increments with an increment of 26.3 μs and performed with π/2 and π pulses of 1.6 and 3.2 μs with γB1/2π = 52 kHz. Two-dimensional 13C─17O ZF-TEDOR spectra were recorded at 21.1 T with 66 t1 increments with an increment of 25 μs, a mixing time of 2.4 ms, and 13C γB1/2π = 100 kHz, 17O γB1/2π = 100 kHz and 1H γB1/2π = 100 kHz TPPM decoupling.68 A series of one-dimensional 13C─17O ZF-TEDOR experiments with varying mixing times from 0.4 to 4.8 ms was performed to produce buildup curves. Two-dimensional 1H─17O R3-R-INEPT30,38,69 experiments were performed at 17.6 T with 40 t1 increments with an increment of 25 μs, a R3 pulse length of 100 μs at γB1/2π = 20 kHz, and γB1/2π = 100 kHz for hard pulses on 1H and 17O. One-dimensional 15N─17O ZF-TEDOR spectra were recorded at 17.6 T with 15N γB1/2π = 36 kHz, 17O γB1/2π = 100 kHz, and 1H γB1/2π = 100 kHz TPPM decoupling. Mixing times were varied from 2.33 to 20.09 ms to determine the maximum transfer. 15N─17O REAPDOR36 was implemented at 17.6 T with dephasing times varying from 0.222 to 5.55 ms. The adiabatic 17O pulse had a duration of onethird of a rotor period, τR/3, and 17O γB1/2π = 100 kHz; the dipolar dephasing curve (ΔS/S, where ΔS = S – SR) was determined by acquiring spectra with (SR) and without (S) the 17O adiabatic pulse.
13C, 15N cross-polarization (CP),70,71 and 1H MAS NMR experiments were performed at 11.7 T (ω0H/2π = 500 MHz, home-built spectrometer courtesy of Dr. Dave Ruben, FBML-MIT) and 21.1 T, with ωR/2π = 10 and 20 kHz, respectively. Two-dimensional 13C─13C (RFDR)21 and 13C─15N (ZF-TEDOR)72 experiments were performed at 11.7 T.
2.3. Spectral Processing and Simulations.
All spectra were processed with RNMR (Dr. D. Ruben, FBML-MIT) or TOPSPIN (Bruker BioSpin, Billerica, MA, USA) with between 10 and 500 Hz of exponential apodization. Processing of the 3QMAS spectrum was done using home-written scripts in MATLAB (MathWorks Inc., Natick, MA, USA), which was necessary to Q-shear73 the spectrum and “unwind” the spinning sidebands along the indirect dimension. Spectral simulations employed either the WSolids74 or DMFit75 software packages. The SIMPSON76 and SPINEVOLUTION77 software packages were used to simulate one-dimensional 15N─17O REAPDOR and 13C─17O ZF-TEDOR dephasing and buildups curves, respectively.
3. RESULTS AND DISCUSSION
3.1. 17O Labeling of FMOC-Protected Amino Acids and N-Acetyl-l-valyl-l-leucine.
To envisage 17O NMR spectroscopy as a reliable tool for the study of biological solids, efficient enrichment of 17O into biomolecules is paramount due to its low natural abundance (99.76% (16O) vs 0.037% (17O)). Recently, an efficient multiple-turnover labeling reaction has been described by Seyfried et al. that proceeds under mild conditions (i.e., room temperature and neutral pH) for 17O/18O enrichment of protected amino acids. The enrichment of various amino acids with different protecting groups was demonstrated by a reaction utilizing an excess of carbodiimide and H2 18O with a dry proton source to eliminate racemization.59
Utilizing this multiple-turnover exchange reaction, FMOC-l-leucine and FMOC-l-valine were enriched with 40% 17O-labeled H2O (vide supra). The 17O isotopic enrichment of the samples was determined by matrix-assisted laser desorption/ionization mass spectrometry (MALDI MS) (Koch Institute MIT), as shown in Figure S1. While the MALDI matrix appears at similar m/z to that of the FMOC amino acids (Figure S1a) these peaks did not interfere with the ability to determine the 17O labeling of the samples. The MALDI MS results yielded the protected amino acids with a M+ m/z of 376.4 Da for FMOC-l-leucine and 362.4 Da for FMOC-l-valine, the addition of a Na+ ion to the molecule is the cause for the larger than expected m/z. For FMOC-l-leucine the mass spectra yielded peaks at m/z of 376.385, 377.383, and 378.420 Da corresponding to the M+, (M+1)+, and (M+2)+, respectively. Correcting for the 13C natural abundance yields a 16O/16O:16O/17O:17O/17O ratio of 36:38:26 for the FMOC-l-leucine sample, which corresponds to an 17O enrichment of 45 ± 5%. The corrected 16O/16O:16O/17O:17O/17O ratio for the FMOC-l-valine sample was determined to be 37:42:20 that leads to an 17O enrichment of 42 ± 5%. Therefore, the labeling efficiency of the multiple-turnover reaction for the two samples was determined to be 100% (40% 17O, H2 17O) within the error of measuring the enrichment. A new series of FMOC-[U-13C,15N]-l-leucine and FMOC-[U-13C,15N]-l-valine were then labeled with 70% 17O-labeled H2O for the synthesis of the dipeptide, N-acetyl-[U-13C,15N,70% 17O]-l-valyl-l-leucine. N-Acetyl-[U-13C,15N,70% 17O]-l-valyl-l-leucine was synthesized via solid-state peptide synthesis by New England Peptide (Gardner, MA), and the enrichment of 17O was verified by MS (not shown) and 17O MAS NMR (Figures 3 and S5).
Figure 3.

1D and 2D 17O MAS NMR spectra of [U-13C,15N,70% 17O]-N-Ac-VL recorded at 35.2 T (ω0H/2π = 1500 MHz). Line structure (a) of N-Ac-VL illustrating the positions labeled with 17O (NCO, CO, and COH). Experimental (b) and simulated (c) 1D MAS 17O MAS NMR spectra, spinning at 19 kHz; centerbands indicated with asterisks (*). Slices (d) of the anisotropic dimension of the 2D 3QMAS spectrum of N-Ac-VL extracted at the isotropic frequency of the centerbands of each 17O moiety. (e) 2D 3QMAS spectrum of [U-13C, 15N, 70% 17O]-N-Ac-VL at 35.2 T (ω0H/2π = 1500 MHz) with the centerbands indicated with dashed horizontal lines and circles, the remaining peaks in the spectrum are due to spinning sidebands.
3.2. 1D and 2D 3QMAS NMR of FMOC-Protected Amino Acids and N-Acetyl-l-valyl-l-leucine.
The central transition of half-integer spin quadrupolar nucleus, such as 17O (I = 5/2), is subject to residual second-order quadrupolar broadening under MAS that yields a characteristic line shape that can be used as a structural probe. The characteristic quadrupolar line shape can be described by the quadrupole coupling constant and the quadrupole asymmetry parameter, ηQ. In addition to the EFG tensor, the chemical shift tensor elements also influence the 17O NMR spectrum, particularly at high fields due to the linear dependence of the chemical shift anisotropy (CSA) on the external magnetic field. The discussion that follows will employ the IUPAC definitions for chemical shift interactions adopting the Herzfeld–Berger convention.66,78 The chemical shift tensor is described by the isotropic chemical shift, δiso, the breadth of the CSA powder pattern, span (Ω), and the magnitude of the asymmetry of the CSA tensor, skew (κ). More complete explanations of the EFG and CSA tensors in solids can be found in literature.3,79-85
17O NMR at multiple magnetic fields (17.6–35.2 T) under both MAS and non-spinning conditions was utilized in conjunction with spectral simulations to determine the quadrupole and chemical shift tensor parameters for each oxygen site in the samples, as shown in Table 4, Figures 1-3, and Figures S3-S5.
Table 4.
Amino Acid and Dipeptide 17O NMR Parameters
| sample | 17O site | δiso (ppm, ±1) | CQ (MHz, ±0.2) | ηQ (±0.1) | Ω (±75) | κ (±0.25) | α (±20°) | β (±20°) | γ (±20°) |
|---|---|---|---|---|---|---|---|---|---|
| FMOC-l-leucine | CO | 338 | 8.3 | 0.0 | 385 | 0.1 | 30 | 90 | 0 |
| COH | 161 | 7.3 | 0.2 | 320 | −0.8 | 20 | 65 | 70 | |
| FMOC-l-valine | CO | 337 | 8.45 | 0.0 | 340 | 0.2 | 45 | 90 | 0 |
| COH | 169 | 7.25 | 0.15 | 320 | −0.7 | 20 | 65 | 70 | |
| N-Ac-VL | NCO | 286 | 8.1 | 0.4 | 450 | 0.3 | 30 | 50 | 15 |
| CO | 329 | 8.2 | 0.0 | 450 | 0.2 | 30 | 40 | 0 | |
| COH | 165 | 7.2 | 0.2 | 310 | −0.8 | 10 | 65 | 0 |
Figure 1.

Experimental and simulated 17O MAS NMR of FMOC-l-leucine (a–c) and FMOC-l-valine (d–f) at 21.1 T (ω0H/2π = 900 MHz). Experimental 17O MAS NMR (a,d), full spectral simulations (b,e), and simulations of each individual oxygen environment, CO (red) and COH (blue) (c,f) are shown. The line structures of FMOCL-l-leucine (inset above (a)) and FMOC-l-valine (inset above (d)) indicating the oxygen environments of interest are displayed. Spectra were acquired with ωR/2π = 24 (FMOC-l-leucine) or 20 (FMOC-l-valine) kHz, with spinning sidebands noted by asterisks (*). NMR parameters used in spectral simulations are given in Table 4.
The MAS and non-spinning 17O NMR spectra of FMOC-l-leucine are shown in Figures 1a-c and S4, and the 13C─1H CPMAS NMR spectrum is shown in Figure S2a. The 17O CQ of the CO and COH groups were found to be 8.3 and 7.3 MHz, respectively, with ηQ = 0.0 and 0.2. These values are consistent with previously reported studies of FMOC-protected amino acids.10 The chemical shift parameters were found to be δiso = 338 and 161 ppm, Ω = 385 and 320 ppm, and κ = 0.1 and −0.8 for the CO and COH groups, respectively. FMOC-l-valine, as shown in Figures 1d-f and S4, was determined to have chemical shift tensor parameters, δiso = 337 and 169 ppm, Ω = 340 and 320 ppm, and κ = 0.2 and −0.7 for the CO and COH groups, respectively. The CQ and ηQ were determined to be 8.45 and 7.45, and 0.0 and 0.15, respectively. Simulation of the chemical shift tensor parameters was necessary for both MAS and non-spinning spectra due to the large CSA of both oxygen environments and its effect on the intensities of the spinning sidebands present in the 17O MAS spectra at 17.6 and 21.1 T.
Verification of a single crystalline structure of N-Ac-VL was achieved by performing one-dimensional 13C, 15N, and 1H MAS NMR experiments, as shown in Figure S8, and comparing to previous studies.18,72,86 Three crystallographically distinct oxygen environments were found within the structure of N-Ac-VL, one on the valine amino acid and two on the leucine, referred to as NCO, CO, and COH, respectively in the following discussion (as shown on the line structure in Figure 2). The 17O NMR parameters of each 17O site of N-Ac-VL were determined via 17O MAS NMR at multiple magnetic fields, as shown in Figures 2, 3 and S5. Spectral simulations were utilized to determine the quadrupole and chemical shift tensor parameters for each of the three 17O sites (NCO, CO, and COH) in the sample, the results of which are summarized in Table 4. The CQ was found to be 8.1, 8.2, and 7.2 for the NCO, CO, COH sites respectively, with ηQ of 0.4, 0.0, and 0.2. The chemical shift parameters of the NCO site were determined to be δiso = 286 ppm, Ω = 450 ppm and κ = 0.3. For the CO and COH sites these parameters were found to be δiso = 329 and 165, Ω = 450 and 310, and κ = 0.2 and −0.8, respectively (Table 4).
Figure 2.

Experimental 17O MAS NMR of N-Ac-VL (a), full simulation of MAS NMR spectrum (b) and simulation of each individual oxygen environment (c) at 21.1 T (ω0H/2π = 900 MHz). Line structure is shown in the inset indicating the 17O enriched sites: CO (red), NCO (green) and COH (blue). Spectra were acquired with ωR/2π = 24 kHz, spinning sidebands are noted by asterisks (*). NMR parameters used in spectral simulations are given in Table 4.
The relative uncertainty in the EFG tensor fits listed in Table 4 is primarily due to the inability to fully remove the spinning sidebands from the centerbands due to the large span of the second-order quadrupolar line shapes and the chemical shift dispersion in these samples. However, when spinning at ωR/2π = 60 kHz, as shown in Figure S7 for FMOC-l-leucine and N-Ac-VL, the spinning sidebands no longer overlap with the centerband, and the CQ and ηQ are fit with a higher degree of precision. While higher MAS frequencies result in a less ambiguous spectral simulation due to the attenuation of chemical shift tensor interaction and a fully resolved centerband spectrum; a loss of sample volume due to the reduced rotor size (i.e., 30 vs 2.5 μL) required for the increased spinning frequency results in a significant sacrifice in signal-to-noise and increase in acquisition time.
17O MAS NMR experiments were performed at 35.2 T, as shown in Figures 3 and S14, to demonstrate the resolution that is afforded at high magnetic field strengths due to the inverse dependence of the second-order residual quadrupolar interaction. Spectral simulation of the 1D MAS NMR spectrum verified the EFG and CSA tensors that were determined from different magnetic fields. The presence of intense spinning sidebands in the spectra recorded at 35.2 T is due to the spinning frequency that was employed (ωR/2π = 19 kHz) and the increased influence of the CSA on the MAS NMR spectrum at the increased magnetic field. Therefore, the large effect of the CSA tensor on the line shape of each resonance, centerband and spinning sidebands, increased the difficulty in producing an accurate spectral simulation. To isolate the contribution of the 17O MAS NMR spectrum due to the spinning sidebands, 2D MATPASS was performed, as shown in Figure S14. The 2D MATPASS spectrum correlates the anisotropic spinning sideband order and the centerband only spectrum, thus allowing for more precise spectral simulation of the 1D MAS spectrum. To further demonstrate the resolution available at 35.2 T, 2D 3QMAS was performed (Figure 3d,e), yielding an isotropic dimension that does not suffer from the broad resonances of 1D MAS NMR experiments on quadrupolar nuclei. Extracting 1D slices at the isotropic frequency of each oxygen environment demonstrates the isolation of each of the environments and the triple-quantum filtered MAS line shape of the 2D resonances in the 3QMAS experiment (Figure 3d). The resolution that is produced by the two-dimensional 3QMAS experiment at high magnetic field will allow for complex samples to be probed via 17O NMR spectroscopy.
3.3. 17O Heteronuclear Correlation Experiments of N-Ac-VL Using Dipolar Recoupling Methods.
Early studies to validate the use of heteronuclear correlation experiments to measure dipolar couplings in spin I = 1/2 nuclei were performed on small peptides, including N-Ac-VL.18,72,87,88 Jaroniec et al. utilized one-dimensional and two-dimensional heteronuclear correlation experiments to measure 13C─15N dipolar couplings in N-Ac-VL via frequency-selective REDOR, while using RFDR and ZF-TEDOR for resonance assignments. 18,72 To further validate the 17O-enriched N-Ac-VL sample used here, two-dimensional 13C─13C RFDR and 13C─15N ZF-TEDOR were performed (Figures S9 and S10) and compared to the previous results.
Two-dimensional MAS NMR correlation spectroscopy of half-integer quadrupolar nuclei, like 17O, has yet to be utilized routinely for biologically relevant samples.10 However, one-dimensional dephasing experiments such as REAPDOR, TRAPDOR, and REDOR have been shown to measure isolated spin pairs in biological samples, though these experiments require a well resolved one-dimensional spectra and a simple spin system to simulate.10,45,89-91 Despite these limitations, utilizing specific labeling techniques Dupree and co-workers were able to measure 15N─17O interatomic distances in two amyloid-beta peptides using 15N─17O REAPDOR.4,37 Including 17O as a routine nucleus of interest in NMR structural studies of biological systems requires augmenting the known one-dimensional techniques for measuring spin I = 1/2 to 17O distances with the better resolved two-dimensional qualitative and quantitative correlation experiments used for spin I = 1/2 nuclei.
Dipolar dephasing and recoupling experiments of N-Ac-VL were examined to further investigate both the crystal structure of N-Ac-VL and the viability of such techniques for further biological structure studies. Interatomic distances were determined via SIMPSON76 and SPINEVOLUTION77 simulations based on one-dimensional 15N─17O REAPDOR dephasing curves and 13C─17O ZF-TEDOR buildup curves, and are given in Table 5. 15N─17O REAPDOR experiments probed the 15N─17O dipolar couplings in the crystal, as shown in Figure 4. While the REAPDOR results demonstrate the robustness of dipolar dephasing experiments to measure accurate distances involving 17O, to properly fit each dephasing curve the simulation required two 17O nuclei for each 15N resonance. Dipolar dephasing experiments yield accurate measurements of the dipolar coupling between isolated spin pairs; however, when an isolated two-spin system is not present the resulting simulations require multiple dipolar couplings and therefore reducing the accuracy of the resulting distance measurement due to the increase in variables. Despite this limitation, the 15N─17O distances extracted from the simulated dephasing curves were within 12%29 of the distances determined via X-ray diffraction methods.92 The 15N─17O distances extracted from the leucine nitrogen (LN) dephasing curve were found to be 2.36 Å to the NCO oxygen and 3.0 Å to the COH oxygen compared to 2.226 and 2.749 Å from diffraction. Weaker dipolar couplings were not included in the simulations to limit the size of the spin system and the number of matrix variables; however, inclusion of a larger number of dipolar couplings could yield a more accurate distance measurement. This limitation of REAPDOR can be avoided by using either a sample that is specifically labeled to isolate the spin pairs of interest or via a correlation experiment that utilizes a second dimension to increase the resolution of the experiment. The 1D 15N─17O ZF-TEDOR spectrum, as shown in Figure S11, demonstrated the ability to utilize ZF-TEDOR to transfer polarization from 15N to 17O nuclei in biological solids. However, the sensitivity of the experiment prevented 15N─17O distances from being measured via a one-dimensional buildup or a full two-dimensional experiment, an area that dynamic nuclear polarization (DNP) will surely impact.
Table 5.
O–C/N/H Intra- and Intermolecular Distances of Interest within N-Ac-VL
| O | C/N/H | rcryst (Å)a | D (Hz)a | rNMR (Å)b | experiment | description |
|---|---|---|---|---|---|---|
| Intramolecular N-Ac-VL Contacts | ||||||
| O2 | C3 | 2.414 | 291.3 | 2.50 ± 0.15 | TEDOR | NCO-Vα |
| C7 | 1.231 | 2196.4 | 1.23 ± 0.15 | TEDOR | NCO-V′ | |
| C8 | 2.681 | 212.6 | 2.73 ± 0.15 | TEDOR | NCO-Lα | |
| N1 | 2.784 | 76.5 | 2.93 ± 0.2 | REAPDOR | NCO-NV | |
| N2 | 2.226 | 149.8 | 2.36 ± 0.2 | REAPDOR | NCO-NL | |
| O3 | C8 | 2.375 | 305.4 | 2.42 ± 0.15 | TEDOR | CO-Lα |
| C13 | 1.196 | 2394.9 | 1.25 ± 0.15 | TEDOR | CO-L′ | |
| O4 | C8 | 2.384 | 302.4 | 2.43 ± 0.15 | TEDOR | COH-Lα |
| C13 | 1.308 | 1830.9 | 1.45 ± 0.15 | TEDOR | COH-L′ | |
| N2 | 2.749 | 79.5 | 3.0 ± 0.2 | REAPDOR | COH-NL | |
| H24 | 0.859 | 25701.9 | – | R3-R-INEPT | COH-HL′ | |
| Intermolecular N-Ac-VL Contacts | ||||||
| O2 | H24 | 1.784 | 2869.2 | - | R3-R-INEPT | NCO-HL′ |
| O3 | N1 | 2.884 | 68.86 | 3.21 ± 0.2 | REAPDOR | CO-NV |
| H22 | 1.967 | 2140.6 | - | R3-R-INEPT | CO-HNv | |
Distances and couplings taken from ref 92.
Current study.
Figure 4.

One-dimensional 15N–17O REAPDOR dephasing curves of the leucine (blue, circle) and valine (red, squares) nitrogens of N-Ac-VL at 17.6 T (ω0H/2π = 750 MHz). Best-fit simulated REAPDOR dephasing curves (solid lines) and ±0.2 Å error (semi-transparent curve) in simulated dephasing curves generated using SIMPSON.76 15N–17O distances that were determined based on the simulated curves are given in Table 5.
13C─17O ZF-TEDOR was performed, as shown in Figures 5 and 6, to further probe the 17O interatomic distances in the N-Ac-VL crystal structure. The combination of the higher sensitivity of 13C detection with the increase in magnetic field strength (21.1 vs 17.6 T) allowed for both one-dimensional buildup curves and a two-dimensional correlation spectrum to be collected for the N-Ac-VL sample. The one-dimensional buildup curves, performed with mixing times from 0.4 to 4.8 ms, allowed for 13C─17O interatomic distances to be determined via simulation of the curves, as given in Table 5. Due to the insensitivity of the ZF-TEDOR experiment, the buildup curves were not performed by determining the cross peak intensity as a function of mixing time from multiple two-dimensional TEDOR experiments. The 13C─17O distances determined by the fitting of the 13C─17O ZF-TEDOR curves were within 10% of those determined by diffraction methods.92 For the ZF-TEDOR experiments, 13C─17O distances could be probed for both the carbonyl and the alpha carbons of the sample, demonstrating an increase in the number of 17O contacts that can be probed via 13C─17O ZF-TEDOR when compared to 15N─17O REAPDOR. The dependence of the ZF-TEDOR experiment is sensitive to the orientation of the quadrupolar interaction relative to the internuclear vector rendering the simulations of the curves quite sensitive to the atomic-level molecular structure. While it is difficult to precisely measure the orientation of the quadrupolar tensor with respect to the internuclear vector, extraction of the Euler angles between the EFG and CSA tensors were possible by spectral simulations of the 1D 17O MAS NMR spectra of N-Ac-VL, as given in Table 4. These angles were then used for the simulation of the ZF-TEDOR buildup curves to reduce the number of variables within the simulation. Narrowing of the possible orientations of the 17O quadrupolar interaction allowed for the ZF-TEDOR simulations to be completed within an experimental error of ±0.15 Å, as shown in Figure 5. The ZF-TEDOR buildup curves of the carbonyl resonances (V′ and L′) were fit to one and two 13C─17O distances respectively for the valine and leucine peaks. These fits were determined by iteratively simulating the buildup curves starting with the 13C─17O distances from the previously determined crystal structure.92 The distances that were extracted from these fits were found to be 1.23 Å for V′-NCO, 1.25 Å for L′-CO, and 1.45 Å for L′-COH; these 13C─17O distances are slightly longer than the diffraction values of 1.23, 1.196, and 1.308 Å. The Cα resonances were simulated using one and three 13C─17O distances for each 13C resonance. The Cα simulations yielded 13C─17O distances of 2.50 Å for Vα-NCO, 2.42 Å for Lα-CO, 2.43 Å for Lα-COH, and 2.73 Å for Lα-NCO. The 13C─17O distances extracted from the simulations of the ZF-TEDOR experiments were determined to be within 0.15 Å of those determined by diffraction,92 which is within the error present in 13C─15N TEDOR measurements on N-Ac-VL.72 The 13C─17O distances were measured to be slightly longer than the distances determined by X-ray diffraction, which could be due to the fact that additional couplings could be contributing to the buildup curves that were not included in the simulations due to the size of the nuclear spin system. Figure S12 shows the ZF-TEDOR buildup curves with not only the best-fit simulation but also the ±0.15 Å error for each curve, indicating the precision of the simulation method. The ZF-TEDOR simulations show a much larger dependence on the crystallite orientation and thus required a total of 5,702,887 crystal angle combinations to achieve the curves shown in Figures 5 and S12. Simulations with a smaller set of crystallite orientations yielded curves with more pronounced and non-uniform modulations convoluted with the proper dipolar coupling-dependent curve.
Figure 5.

Experimental and simulated one-dimensional 13C─17O ZF-TEDOR buildup curves as a function of mixing for the leucine carbonyl (a), valine carbonyl (b), and Cα (c) sites (leucine, green circles and valine, orange squares) at 21.1 T (ω0H/2π = 900 MHz) with γB1/2π (17O) = 100 kHz, and 100 kHz TPPM 1H decoupling during acquisition. Best fit simulated curves (solid lines) determined using SPINEVOLUTION.77 13C─17O distances that were determined based on the simulated curves are given in Table 5.
Figure 6.

Two-dimensional 13C─17O ZF-TEDOR spectrum recorded at 21.1 T (ω0H/2π = 900 MHz) with 2.4 ms of mixing. Correlations between the NCO and CO oxygen sites with the closest C′ and Cα indicated on the line structure of N-Ac-VL in the inset.
Two-dimensional 13C─17O ZF-TEDOR was performed, as shown in Figure 5, demonstrating the resolution that is available to the ZF-TEDOR experiment. The two-dimensional ZF-TEDOR spectra was collected over the course of 4 days with a mixing time of 2.4 ms, which corresponds to a transfer efficiency of ~1.4 and 0.4% for the carbonyl and alpha carbons. The L′ and Lα correlations to COH are not shown in Figure 5 due to the low intensity of these peaks that likely arises from the imprecise nature of the π pulses on this particular 17O resonance (π pulses for the two-dimensional ZF-TEDOR spectra were calibrated on the NCO resonance). The differing CQ value for the COH oxygen in comparison to the NCO and CO oxygens causes the COH oxygen to have a slightly altered nutation frequency in comparison to the other oxygens. However, the one-dimensional ZF-TEDOR buildup curves required the addition of the COH dipolar coupling and orientation to properly match the experimental shape indicating that magnetization transfer to the COH environment is occurring. As is evident in Figure 6, the two-dimensional ZF-TEDOR spectrum suffers from a lack of sensitivity that is apparent in the low signal-to-noise of the spectrum despite the long acquisition time. Despite the low signal-to-noise, the cross peaks within the spectrum demonstrate the ability of 17O NMR to act as a powerful probe of interactions between 13C and 17O spins within biomolecular solids. While in the current study 13C─17O interactions were limited to couplings equivalent to one- and two-bond distances, with an increased sensitivity the ZF-TEDOR experiment could be extended to a larger range of carbon–oxygen interactions.
To directly examine hydrogen bonding within the N-Ac-VL crystal, 1H─17O correlation spectroscopy was performed via the 1H detected R3-R-INEPT experiment. The use of 1H detection takes advantage of the higher sensitivity and narrower line widths of 1H in comparison to 17O in addition to utilizing the fast relaxation of the quadrupolar nucleus allowing for rapid recycling of the experiment and thus shorter experimental times. The one-dimensional and two-dimensional 1H─17O R3-R-INEPT spectra, as shown in Figure 7, show the correlation of the COH 1H (HL′) to the COH and NCO oxygens. While the expected directly bonded oxygen is seen in this experiment, the more significant finding is the intermolecular correlation between the carbonyl proton and the NCO oxygen. A low signal-to-noise peak for the HL′-CO correlation was seen in a two-dimensional R3-R-INEPT experiment using an R3 pulse length of 100 μs (Figure S13). The two-dimensional 1H-detected 17O correlation experiment demonstrates the ability to directly probe hydrogen bonding, demonstrating the intermolecular contact between the leucine and valine residues of adjacent N-Ac-VL.
Figure 7.

MAS NMR spectroscopy of N-Ac-VL: (a) 1H direct Hahn-echo detection, (b) one-dimensional 1H–17O R3-R-INEPT, (c) and two-dimensional 1H–17O R3-R-INEPT spectrum with R3 = 100 μs. Spectra were acquired with ωR/2π = 20 kHz at 17.6 T (ω0H/2π = 750 MHz). Correlations between the leucine carbonyl proton to its directly bonded oxygen (blue arrow) and its next closest oxygen through space (NCO, green arrow) are indicated on the crystal structure of N-Ac-VL in the inset.92
4. CONCLUSIONS
An efficient multiple-turnover reaction for 17O enrichment was used to 17O enrich two FMOC-protected amino acids, l-leucine and l-valine, which were used as precursors for the synthesis of a uniformly 13C,15N-labeled and 70% 17O-labeled dipeptide, NAc-VL. The 17O quadrupolar and chemical shift parameters for the FMOC-protected amino acid precursors and the dipeptide were determined using multiple magnetic fields and spinning frequencies. The EFG tensor parameters for the FMOC-protected amino acids were found to be similar for each of the two oxygen environments, CQ = 8.4 and 7.3 MHz and ηQ = 0 and 0.2. The leucine residue of the N-Ac-VL was found to have similar quadrupolar and chemical shift parameters to the FMOC-l-leucine sample. However, the oxygen environment of the valine residue was found to have a CQ and ηQ similar to other peptide NCO environments (8.4 MHz and 0.4). 17O NMR experiments were performed at 35.2 T to demonstrate the ability of high magnetic fields to increase the sensitivity and resolution of 17O MAS NMR spectra via 1D (MAS) and 2D (MATPASS and 3QMAS) experiments. One- and two-dimensional correlation experiments between 17O and 15N/13C/1H using dipolar recoupling methods were performed on N-Ac-[U-13C,15N,70% 17O]-VL demonstrating the viability of 17O NMR as a tool for structural studies of biological systems. 15N─17O and 13C─17O interatomic distances were directly measured via dephasing and buildup curves of REAPDOR and ZF-TEDOR experiments and determined to be within 15% of previously reported values. The two-dimensional 17O correlation spectra showed the ability to measure 17O connectivity within a dipeptide and demonstrated the ability to utilize the increased resolution of two-dimensional experiments to study more complex systems in the future. 1H detected 17O NMR was used to directly probe through space hydrogen bonding in the dipeptide; showing the ability to use 17O NMR to directly measure connectivity via hydrogen bonding. The addition of multiple-quantum filters and DNP NMR would allow for the 17O correlation experiments to be performed with reduced experiment times and increased spectral resolution. Thus, enabling ZF-TEDOR buildup curves to be produced from the cross peak intensities of multiple two-dimensional experiments, and for a larger range of mixing times to probe weaker 13C─17O dipolar couplings that were not observed in the one-dimensional ZF-TEDOR buildups. With the enhanced signal-to-noise the R3-R-INEPT experiment could be extended to probe 1H─17O interatomic distances similarly to ZF-TEDOR.
Supplementary Material
Table 2.
NMR Acquisition Parameters for Double Resonance and Two-Dimensional Experiments Involving 17O
| B0 (T) | nucleia | ω0/2π (MHz) | pulse sequence | sample | ωR/2π (kHz) | recycle delay (s) | scans | 1H decouple |
|---|---|---|---|---|---|---|---|---|
| 17.6 | 1H–17O | 749–101.45 | R3-R-INEPT | N-Ac-VL | 20 | 0.1 | 4096 | no |
| 15N–17O | 76–101.45 | REAPDOR | N-Ac-VL | 18 | 3 | 256 | yes | |
| 15N–17O | 76–101.45 | ZF-TEDOR | N-Ac-VL | 18 | 3 | 60416 | yes | |
| 21.1 | 13C─17O | 226.3–122.02 | ZF-TEDOR | N-Ac-VL | 20 | 3 | 512 | yes |
| 35.2 | 17O | 203.36 | MATPASS | N-Ac-VL | 19 | 0.1 | 1200 | no |
| 3QMAS | 19 | 0.2 | 1024 | no |
Detection nucleus listed first.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (NIH) through grant numbers: EB-001960, EB-002804, and EB-002026. The 35.2 T SCH magnet and NMR instrumentation are supported by NSF (DMR-1039938 and DMR-0603042); additional support for user activities on the SCH are provided by NIH P41 GM122698 and the NHMFL DC and NMR/MRI User Facilities supported by NSF DMR-1157490 and the State of Florida. V.K.M. is grateful to the Natural Sciences and Engineering Research Council of Canada and the Government of Canada for a Banting Postdoctoral Fellowship. The authors thank Drs. Graham Sazama and Joseph Walish for helpful discussions and access to the Schlenk line.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b08989.
Additional Figures S1-S16, showing spectra, buildup curves, and pulse sequences (PDF)
REFERENCES
- (1).Glowacki ED; Irimia-Vladu M; Bauer S; Sariciftci NS J. Mater. Chem. B 2013, 1, 3742–3753. [DOI] [PubMed] [Google Scholar]
- (2).Horowitz S; Trievel RC J. Biol. Chem 2012, 287, 41576–41582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Grandinetti PJ; Trease NM; Ash JT Prog. Nucl. Magn. Reson. Spectrosc 2011, 59, 121–196. [DOI] [PubMed] [Google Scholar]
- (4).Antzutkin ON; Iuga D; Filippov AV; Kelly RT; Becker-Baldus J; Brown SP; Dupree R Angew. Chem., Int. Ed 2012, 51, 10289–10292. [DOI] [PubMed] [Google Scholar]
- (5).Aguiar PM; Michaelis VK; McKinley CM; Kroeker SJ Non-Cryst. Solids 2013, 363, 50–56. [Google Scholar]
- (6).Kong X; Shan M; Terskikh V; Hung I; Gan Z; Wu G J. Phys. Chem. B 2013, 117, 9643–9654. [DOI] [PubMed] [Google Scholar]
- (7).Kwan I; Mo X; Wu G J. Am. Chem. Soc 2007, 129, 2398–2407. [DOI] [PubMed] [Google Scholar]
- (8).Zhu J; Ye E; Terskikh V; Wu G Angew. Chem., Int. Ed 2010, 49, 8399–8402. [DOI] [PubMed] [Google Scholar]
- (9).Michaelis VK; Keeler EG; Ong T-C; Craigen KN; Penzel SA; Wren JEC; Kroeker S; Griffin RG J. Phys. Chem. B 2015, 119, 8024–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wong A; Poli F Annu. Rep. NMR Spectrosc 2014, 83, 145–220. [Google Scholar]
- (11).Wu G Solid State Nucl. Magn. Reson 2016, 73, 1–14. [DOI] [PubMed] [Google Scholar]
- (12).Kong XQ; Brinkmann A; Terskikh V; Wasylishen RE; Bernard GM; Duan Z; Wu QC; Wu G J. Phys. Chem. B 2016, 120, 11692–11704. [DOI] [PubMed] [Google Scholar]
- (13).Tang AW; Kong XQ; Terskikh V; Wu G J. Phys. Chem. B 2016, 120, 11142–11150. [DOI] [PubMed] [Google Scholar]
- (14).Chekmenev EY; Waddell KW; Hu J; Gan ZH; Wittebort RJ; Cross TA J. Am. Chem. Soc 2006, 128, 9849–9855. [DOI] [PubMed] [Google Scholar]
- (15).Waddell KW; Chekmenev EY; Wittebort RJ J. Phys. Chem. B 2006, 110, 22935–22941. [DOI] [PubMed] [Google Scholar]
- (16).Zhang QW; Chekmenev EY; Wittebort RJ J. Am. Chem. Soc 2003, 125, 9140–9146. [DOI] [PubMed] [Google Scholar]
- (17).Griffin RG Nat. Struct. Biol 1998, 5, 508–512. [DOI] [PubMed] [Google Scholar]
- (18).Jaroniec CP; Tounge BA; Herzfeld J; Griffin RG J. Am. Chem. Soc 2001, 123, 3507–3519. [DOI] [PubMed] [Google Scholar]
- (19).McDowell LM; Schaefer J Curr. Opin. Struct. Biol 1996, 6, 624–629. [DOI] [PubMed] [Google Scholar]
- (20).Opella SJ Nat. Struct. Biol 1997, 4, 845–848. [PubMed] [Google Scholar]
- (21).Bennett AE; Ok JH; Griffin RG; Vega S J. Chem. Phys 1992, 96, 8624–8627. [Google Scholar]
- (22).De Paepe G; Lewandowski JR; Loquet A; Eddy M; Megy S; Bockmann A; Griffin RG J. Chem. Phys 2011, 134, 095101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).De Paepe G; Lewandowski JR; Loquet A; Bockmann A; Griffin RG J. Chem. Phys 2008, 129, 245101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Gullion T; Schaefer J J. Magn. Reson 1989, 81, 196–200. [Google Scholar]
- (25).Hing AW; Vega S; Schaefer J J. Magn. Reson 1992, 96, 205–209. [Google Scholar]
- (26).Munowitz MG; Griffin RG; Bodenhausen G; Huang TH J. Am. Chem. Soc 1981, 103, 2529–2533. [Google Scholar]
- (27).Munowitz MG; Griffin RG J. Chem. Phys 1982, 76, 2848–2858. [Google Scholar]
- (28).Roberts JE; Harbison GS; Munowitz MG; Herzfeld J; Griffin RG J. Am. Chem. Soc 1987, 109, 4163–4169. [Google Scholar]
- (29).Hung I; Uldry AC; Becker-Baldus J; Webber AL; Wong A; Smith ME; Joyce SA; Yates JR; Pickard CJ; Dupree R; et al. J. Am. Chem. Soc 2009, 131, 1820–1834. [DOI] [PubMed] [Google Scholar]
- (30).Trebosc J; Hu B; Amoureux JP; Gan Z J. Magn. Reson 2007, 186, 220–227. [DOI] [PubMed] [Google Scholar]
- (31).Perras FA; Chaudhary U; Slowing II; Pruski M J. Phys. Chem. C 2016, 120, 11535–11544. [Google Scholar]
- (32).Perras FA; Kobayashi T; Pruski M J. Am. Chem. Soc 2015, 137, 8336–8339. [DOI] [PubMed] [Google Scholar]
- (33).Michaelis VK; Markhasin E; Daviso E; Herzfeld J; Griffin RG J. Phys. Chem. Lett 2012, 3, 2030–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Vogt FG; Yin H; Forcino RG; Wu LM Mol. Pharmaceutics 2013, 10, 3433–3446. [DOI] [PubMed] [Google Scholar]
- (35).Amoureux JP; Trebosc J; Tricot G Magn. Reson. Chem 2007, 45, S187–S191. [DOI] [PubMed] [Google Scholar]
- (36).Gullion T Chem. Phys. Lett 1995, 246, 325–330. [Google Scholar]
- (37).Wei J; Antzutkin ON; Filippov AV; Iuga D; Lam PY; Barrow MP; Dupree R; Brown SP; O’Connor PB Biochemistry 2016, 55, 2065–2068. [DOI] [PubMed] [Google Scholar]
- (38).Martineau C; Bouchevreau B; Taulelle F; Trebosc J; Lafon O; Amoureux JP Phys. Chem. Chem. Phys 2012, 14, 7112–7119. [DOI] [PubMed] [Google Scholar]
- (39).Brinkmann A; Kentgens APM J. Am. Chem. Soc 2006, 128, 14758–14759. [DOI] [PubMed] [Google Scholar]
- (40).Prasad S; Clark TM; Sharma R; Kwak H-T; Grandinetti PJ; Zimmermann H Solid State Nucl. Magn. Reson 2006, 29, 119–124. [DOI] [PubMed] [Google Scholar]
- (41).Prasad S; Kwak HT; Clark T; Grandinetti PJ J. Am. Chem. Soc 2002, 124, 4964–4965. [DOI] [PubMed] [Google Scholar]
- (42).Blanc F; Sperrin L; Jefferson DA; Pawsey S; Rosay M; Grey CP J. Am. Chem. Soc 2013, 135, 2975–2978. [DOI] [PubMed] [Google Scholar]
- (43).Michaelis VK; Corzilius B; Smith AA; Griffin RG J. Phys. Chem. B 2013, 117, 14894–14906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Michaelis VK; Ong TC; Kiesewetter MK; Frantz DK; Walish JJ; Ravera E; Luchinat C; Swager TM; Griffin RG Isr. J. Chem 2014, 54, 207–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Gullion T; Yamauchi K; Okonogi M; Asakura T Macromolecules 2007, 40, 1363–1365. [Google Scholar]
- (46).Yamada K; Yamazaki T; Asanuma M; Hirota H; Yamamoto N; Kajihara Y Chem. Lett 2007, 36, 192–193. [Google Scholar]
- (47).Sefzik TH; Houseknecht JB; Clark TM; Prasad S; Lowary TL; Gan Z; Grandinetti PJ Chem. Phys. Lett 2007, 434, 312–315. [Google Scholar]
- (48).Wong A; Beevers AJ; Kukol A; Dupree R; Smith ME Solid State Nucl. Magn. Reson 2008, 33, 72. [DOI] [PubMed] [Google Scholar]
- (49).Wong A; Howes AP; Yates JR; Watts A; Anupold T; Past J; Samoson A; Dupree R; Smith ME Phys. Chem. Chem. Phys 2011, 13, 12213–12224. [DOI] [PubMed] [Google Scholar]
- (50).Wu G; Dong S; Ida R; Reen N J. Am. Chem. Soc 2002, 124, 1768. [DOI] [PubMed] [Google Scholar]
- (51).Yamauchi K; Okonogi M; Kurosu H; Tansho M; Shimizu T; Gullion T; Asakura T J. Magn. Reson 2008, 190, 327. [DOI] [PubMed] [Google Scholar]
- (52).Zhu J; Lau JYC; Wu GJ Phys. Chem. B 2010, 114, 11681–11688. [DOI] [PubMed] [Google Scholar]
- (53).Wu G Oxygen 17 NMR Studies of Organic and Biological Molecules. In eMagRes, John Wiley & Sons, Ltd.: Chichester, UK; 2011. (DOI: 10.1002/9780470034590.emrstm1212). [DOI] [Google Scholar]
- (54).Keeler EG; Michaelis VK; Griffin RG J. Phys. Chem. B 2016, 120, 7851–7858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Lemaitre V; Pike KJ; Watts A; Anupold T; Samoson A; Smith ME; Dupree R Chem. Phys. Lett 2003, 371, 91–97. [Google Scholar]
- (56).Pike KJ; Lemaitre V; Kukol A; Anupold T; Samoson A; Howes AP; Watts A; Smith ME; Dupree R J. Phys. Chem. B 2004, 108, 9256–9263. [Google Scholar]
- (57).Wong A; Howes AP; Pike KJ; Lemaitre V; Watts A; Anupold T; Past J; Samoson A; Dupree R; Smith ME J. Am. Chem. Soc 2006, 128, 7744–7745. [DOI] [PubMed] [Google Scholar]
- (58).O’Dell LA; Ratcliffe CI; Kong X; Wu G J. Phys. Chem. A 2012, 116, 1008–1014. [DOI] [PubMed] [Google Scholar]
- (59).Seyfried MS; Lauber BS; Luedtke NW Org. Lett 2010, 12, 104–106. [DOI] [PubMed] [Google Scholar]
- (60).Mijalis AJ; Thomas DA 3rd; Simon MD; Adamo A; Beaumont R; Jensen KF; Pentelute BL Nat. Chem. Biol 2017, 13, 464–466. [DOI] [PubMed] [Google Scholar]
- (61).Etzkorn M; Raschle T; Hagn F; Gelev V; Rice AJ; Walz T; Wagner G Structure 2013, 21, 394–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Marecek J; Song B; Brewer S; Belyea J; Dyer RB; Raleigh DP Org. Lett 2007, 9, 4935–4937. [DOI] [PubMed] [Google Scholar]
- (63).Steinschneider A; Burgar MI; Buku A; Fiat D Int. J. Pept. Protein Res 1981, 18, 324–333. [DOI] [PubMed] [Google Scholar]
- (64).Theodorou V; Skobridis K; Alivertis D; Gerothanassis IP J. Labelled Compd. Radiopharm 2014, 57, 481–508. [DOI] [PubMed] [Google Scholar]
- (65).Gan Z; Hung I; Wang X; Paulino J; Wu G; Litvak IM; Gor’kov PL; Brey WW; Lendi P; Schiano JL; et al. J. Magn. Reson 2017, 284, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Harris RK; Becker ED; De Menezes SMC; Granger P; Hoffman RE; Zilm KW Pure Appl. Chem 2008, 80, 59–84. [Google Scholar]
- (67).Hung I; Gan ZH J. Magn. Reson 2010, 204, 150–154. [DOI] [PubMed] [Google Scholar]
- (68).Bennett AE; Rienstra CM; Auger M; Lakshmi KV; Griffin RG J. Chem. Phys 1995, 103, 6951–6958. [Google Scholar]
- (69).Morris GA; Freeman R J. Am. Chem. Soc 1979, 101, 760–762. [Google Scholar]
- (70).Schaefer J; Stejskal EO J. Am. Chem. Soc 1976, 98, 1031–1032. [Google Scholar]
- (71).Pines A; Waugh JS; Gibby MG J. Chem. Phys 1972, 56, 1776–1777. [Google Scholar]
- (72).Jaroniec CP; Filip C; Griffin RG J. Am. Chem. Soc 2002, 124, 10728–10742. [DOI] [PubMed] [Google Scholar]
- (73).Hung I; Trebosc J; Hoatson GL; Vold RL; Amoureux JP; Gan ZH J. Magn. Reson 2009, 201, 81–86. [DOI] [PubMed] [Google Scholar]
- (74).Eichele K WSolids1 NMR Simulation Package, 1.20.21; 2013. [Google Scholar]
- (75).Massiot D; Fayon F; Capron M; King I; Le Calve S; Alonso B; Durand JO; Bujoli B; Gan ZH; Hoatson G Magn. Reson. Chem 2002, 40, 70–76. [Google Scholar]
- (76).Bak M; Rasmussen JT; Nielsen NC J. Magn. Reson 2000, 147, 296–330. [DOI] [PubMed] [Google Scholar]
- (77).Veshtort M; Griffin RG J. Magn. Reson 2006, 178, 248–282. [DOI] [PubMed] [Google Scholar]
- (78).Herzfeld J; Berger AE J. Chem. Phys 1980, 73, 6021–6030. [Google Scholar]
- (79).Ashbrook SE; Smith ME Chem. Soc. Rev 2006, 35, 718–735. [DOI] [PubMed] [Google Scholar]
- (80).Man PP Encyclopedia of Analytical Chemistry; John Wiley and Sons: Chichester, 2000; pp 12224–12265. [Google Scholar]
- (81).Saito H; Ando I; Ramamoorthy A Prog. Nucl. Magn. Reson. Spectrosc 2010, 57, 181–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Slichter CP Principles of magnetic resonance, with examples from solid state physics; Harper & Row: New York, 1963. [Google Scholar]
- (83).Taulelle F NMR of Quadrupolar Nuclei in the Solid State; Kluwer Academic Publishers: London, 1988; Vol. 322, p 476. [Google Scholar]
- (84).Haeberlen U High resolution NMR in solids: selective averaging; Academic Press: New York, 1976; p v. [Google Scholar]
- (85).Mehring M Principles of high-resolution NMR in solids, 2nd ed.; Springer-Verlag: Berlin/ New York, 1983. [Google Scholar]
- (86).Reif B; Jaroniec CP; Rienstra CM; Hohwy M; Griffin RG J. Magn. Reson 2001, 151, 320–327. [DOI] [PubMed] [Google Scholar]
- (87).Jaroniec CP; MacPhee CE; Astrof NS; Dobson CM; Griffin RG Proc. Natl. Acad. Sci. U. S. A 2002, 99, 16748–16753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Rienstra CM; Tucker-Kellogg L; Jaroniec CP; Hohwy M; Reif B; McMahon MT; Tidor B; Lozano-Perez T; Griffin RG Proc. Natl. Acad. Sci. U. S. A 2002, 99, 10260–10265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Chopin L; Vega S; Gullion T J. Am. Chem. Soc 1998, 120, 4406–4409. [Google Scholar]
- (90).Goldbourt A; Vega S; Gullion T; Vega AJ J. Am. Chem. Soc 2003, 125, 11194–11195. [DOI] [PubMed] [Google Scholar]
- (91).Vaneck ERH; Janssen R; Maas WEJR; Veeman WS Chem. Phys. Lett 1990, 174, 428–432. [Google Scholar]
- (92).Carroll PJ; Stewart PL; Opella S J. Acta Crystallogr., Sect. C: Cryst. Struct. Commun 1990, 46, 243–246. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.