

Abstract
Telomeres are regions of repetitive nucleotide sequences capping the ends of eukaryotic chromosomes that protect against deterioration, and whose lengths can be correlated with age and adverse health risk factors. Yet, given their length and repetitive nature, telomeric regions are not easily reconstructed from short-read sequencing, thus making telomere sequencing, mapping, and variant resolution challenging problems. Recently, long-read sequencing, with read lengths measuring in hundreds of kilobase pairs, has made it possible to routinely read into telomeric regions and inspect their sequence structure. Here, we describe a framework for extracting telomeric reads from whole-genome single-molecule sequencing experiments, including de novo identification of telomere repeat motifs and repeat types, and also describe their sequence variation. We find that long, complex telomeric stretches and repeats can be accurately captured with long-read sequencing, observe extensive sequence heterogeneity of human telomeres, discover and localize noncanonical telomere sequence motifs (both previously reported, as well as novel), and validate them in short-read sequence data. These data reveal extensive intra- and inter-population diversity of repeats in telomeric haplotypes, reveal higher paternal inheritance of telomeric variants, and represent the first motif composition maps of multi-kilobase-pair human telomeric haplotypes across three distinct ancestries (Ashkenazi, Chinese, and Utah), which can aid in future studies of genetic variation, aging, and genome biology.
Telomeres are the functional ends of human chromosomes that naturally shorten with cell division and thus with aging (Aubert and Lansdorp 2008). Telomere length is also influenced by a variety of lifestyle factors and environmental exposures (e.g., stress, exercise, air pollution, radiation) (Shammas 2011). Although human telomeres are known to consist largely of a conserved six-nucleotide repeat (5′‐TTAGGG‐3′) (Moyzis et al. 1988), several studies have identified variations of this motif in proximal telomeric regions (Allshire et al. 1989; Coleman et al. 1999; Lee et al. 2018; Bluhm et al. 2019). However, such studies were performed with oligonucleotide hybridization, PCR, immunoprecipitation, and short-read sequencing, requiring prior assumptions about specific target motifs, custom sample preparation, and targeted sequencing and therefore preventing de novo identification of motif variants and their localization. Also, long-range maps of telomeric sequence variation in the human genome are still incomplete or preliminary (Shafin et al. 2020) or have only been completed for a single genome (Jain et al. 2018; Miga et al. 2020). Therefore, completing maps of telomeres and providing new tools for such research (Nurk et al. 2020) can provide new insight into telomere biology and enable novel approaches to analyze the effects of aging, environment, and health status (Lee et al. 2018) on telomere sequence and length.
To improve our understanding of telomere sequence structure and variation, we developed edgeCase, a scalable framework for alignment and de novo telomeric motif discovery from human whole-genome long-read sequencing experiments. We have validated these methods using Genome in a Bottle (GIAB) (Zook et al. 2019) single-molecule real-time (SMRT) sequencing data sets generated with Pacific Biosciences circular consensus sequencing (PacBio CCS) (Eid et al. 2009; Ardui et al. 2018), as well as short-read Illumina (Bentley et al. 2008) and 10x Genomics (Chromium) (www.10xgenomics.com) data sets, as well as with healthy donor peripheral blood mononuclear cells (PBMCs). These results provide evidence for multiple novel, noncanonical telomeric repeats, resolution of multiple chromosome-specific haplotypes with SMRT sequencing, asymmetric inheritance of variants, and a new method for long-range characterization of the structure of telomeric sequences.
Results
A telomere-annotated reference genome enables recovery of telomeric reads from human long-read whole-genome sequencing data sets
We first constructed an extended reference genome, hg38ext, that combines chromosome sequences of the hg38 reference genome (International Human Genome Sequencing Consortium 2001; Schneider et al. 2017) and human subtelomeric assemblies (Stong et al. 2014), resulting in a reference set annotated with boundaries of subtelomeric and telomeric tracts. The layout of this reference set is available in Supplemental File S1, and the set itself can be reproduced with a script available as Supplemental File S2. We then aligned to it PacBio CCS reads of seven GIAB (Zook et al. 2019) human subjects (HG001 through HG007) from three different ancestries (Ashkenazi, Chinese, and Utah), which included two son/father/mother trios (Supplemental Table S1). In total, we observed reads uniquely mapping to the ends of chromosomes and extending into telomeric regions on nine p arms and 14 q arms, with 43–285 such reads on the p arms and 34–250 on the q arms (Supplemental Table S2). Portions of reads contained in the telomeric regions were extracted for further analysis (Fig. 1).
Figure 1.

Mapping of candidate telomeric reads, illustrated with reads from the HG002 data set aligning to Chromosome 12. The chromosome is displayed schematically, centered around the centromere. Vertical red dashed lines denote the position of the boundary of the annotated telomeric tract. Coordinates are given in kilobase pairs, relative to the positions of the telomeric tract boundaries. Statistics for all chromosomes of all seven data sets are provided in Supplemental Table S2.
Telomeric long reads contain variations of the canonical motif
We then performed de novo repeat discovery in the telomeric sequences for motifs of lengths 4 through 16, and we identified motifs in repeat contexts that are statistically enriched in the seven data sets. The majority of motifs were either the canonical TTAGGG/CCCTAA, its variations (e.g., TGAGGG), or a duplet of variants, such as TTAGGGTTAGGGG (Table 1). CG-rich motifs were also observed on the q arms. The top enriched motif (TTAGGG/CCCTAA) explained up to 76.9% of the telomeric repeat content on the p arms and up to 80.1% on the q arms, whereas motifs TGAGGG and TTAGGGG explained up to 8.0% and 6.6% of the repeat content overall, respectively.
Table 1.
Significantly enriched repeating motifs in telomeric regions of GIAB data sets HG001 through HG007

We next visualized the locations of the top three enriched motifs and their reverse complements on the chromosomal ends of the HG002 data set (for p arms, see Fig. 2A; for q arms, see Fig. 2B), as it provided the deepest coverage among the assessed data sets (Supplemental Table S2); plots for the other six data sets are available as Supplemental Figures S2 and S3. Only the chromosomal arms cumulatively covered by at least 25 reads across data sets were plotted. These data showed that the overwhelming majority of the telomeric regions were represented by the canonical repeats, but also novel, chromosome-specific repeat motif patterns could be observed, and they were enriched for the proximal end of the telomere; these data also illustrated the positions of the repeat-rich portions of the genomes in relation to the known subtelomere–telomere boundaries, including deletions/insertions (4p, 8q) and an apparent extension of the 17p subtelomere.
Figure 2.

Densities of the top three enriched motifs at ends of chromosomal p arms (A) and q arms (B) of the HG002 data set. Background represents the remaining sequence content (nonrepeating sequence and not significantly enriched motifs). Reads are shown aligned to the contigs in the hg38ext reference set, and genomic coordinates are given in kilobase pairs. Vertical red dashed lines denote the position of the boundary of the annotated telomeric tract.
To discern if the sequence mapping, read length, or overall coverage had any effect on the discovery or enrichment of these motifs, the motif entropies were examined as a function of their location within reads and coverage across the telomere tracks. When the locations of different motifs were examined within any 10-bp window across the length of the long reads, the entropy data showed consistency among reads and across samples (Fig. 3). Indeed, the coverage-weighted median of normalized Shannon entropy ranged from 0.00 to 0.07 for different data sets, whereas most nonzero values were contained only in the top quartile (between the 75th and the 100th percentile), indicating that locations of the variations are colinear among reads.
Figure 3.

Distribution of motif entropies in 10-bp windows of candidate PacBio CCS reads aligning to the same chromosomal arms in GIAB data sets HG001 through HG007, with respect to per-window coverage, and the coverage-weighted percentiles of the entropy values.
Short-read sequencing validates motif variations observed in long reads
We next validated these findings using short-read sequencing in two ways. First, we extracted telomeric candidate reads with Telomerecat (Farmery et al. 2018) from matching GIAB Illumina data sets and found that they supported a definitive majority of the long-read telomeric candidates, with a median 89% of the p arm sequence and a median 95% of the q arm sequence supported (Supplemental Fig. S1). Second, to ensure these motifs were observed in primary human samples (vs. cell lines), we used human short-read and linked-read (10x Genomics) genomic data sets from donated PBMCs (Garrett-Bakelman et al. 2019; Iosim et al. 2019) to independently confirm 13 of the enriched motifs, with the same three motifs being the most enriched (Supplemental Table S3).
Long-read sequencing uncovers a variety of human telomeric haplotypes
Although reads generally agreed on colinearity of motifs, as evidenced by the low entropy, some rare, nonzero entropy values could be attributable both to sequencing errors and to structural variations within the same subject's data set. To investigate the latter possibility, we clustered reads on each arm of each subject by relative pairwise Levenshtein distances (Levenshtein 1966) and found that hierarchical clustering resulted in high cophenetic correlation between the dendrograms and the pairwise distance matrices (Table 2), as well as in visible structure (Figs. 4, 5).
Table 2.
Measures of cophenetic correlation (Pearson's r and adjusted P-value) between the hierarchical clustering and the pairwise distance matrix on each chromosomal arm
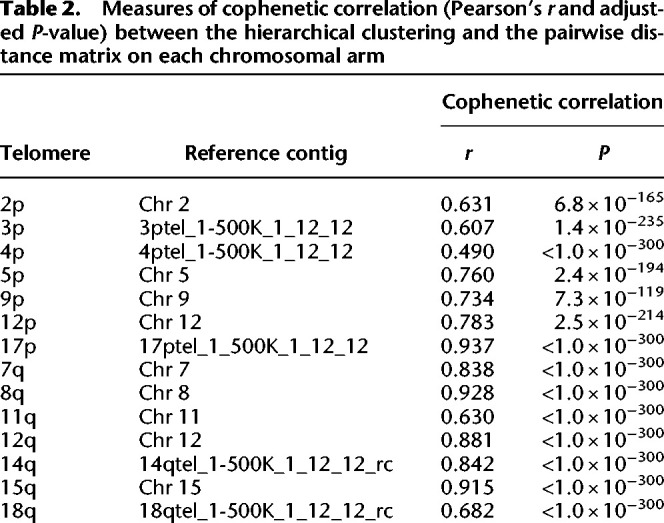
Figure 4.

Clustering of reads by relative pairwise Levenshtein distances (unitless measure) on each chromosomal p arm of data sets HG001 through HG007, as well as densities of the top enriched motifs along each read. Each horizontal line represents an individual read; genomic coordinates are given in kilobase pairs, relative to the positions of the telomeric tract boundaries. Only the chromosomal arms cumulatively covered by at least 25 reads are displayed.
Figure 5.

Clustering of reads by relative pairwise Levenshtein distances (unitless measure) on each chromosomal q arm of data sets HG001 through HG007, and densities of the top enriched motifs along each read. Each horizontal line represents an individual read; genomic coordinates are given in kilobase pairs, relative to the positions of the telomeric tract boundaries. Only the chromosomal arms cumulatively covered by at least 25 reads are displayed.
In this complex clustering, subject- and population-specific variation was evident and quantifiable via relative Levenshtein distances (Methods) (Table 3): Overall, telomeric reads within a subject were more similar than within a population (adjusted Wilcoxon signed-rank test P = 4.2 × 10−56), and telomeric reads within a population were more similar than between populations (P = 2.2 × 10−40).
Table 3.
Adjusted P-values of the Wilcoxon signed-rank tests on relative Levenshtein distances

However, this was true for most, but not all reads: 13.8% of all assessed reads (165 out of 1192) contributed to interpopulation similarity; these reads were twice as close to reads from a different population than they were to any reads of their own subjects. This trend is observable in Figures 4 and 5, with subjects’ and populations’ reads interspersed across multiple clusters. Therefore, the captured reads reflected spectra of haplotypes, generally describing subject- and population-specific similarities, but including a sizable component that described interpopulation similarity. A distinct paternal inheritance of variation was also observed: Each father's telomeric reads were more similar to their son's than to the mother's reads in both the Chinese (adj. P-value = 0.034) and the Ashkenazi (adj. P-value = 3.1 × 10−11) trios.
Discussion
Repeat-rich, low-complexity regions of the human genome such as telomeres have been historically recalcitrant to full mapping and annotation (Miga 2015), mainly owing to the alignment challenge they pose and to the read lengths required to span such areas (Treangen and Salzberg 2012). The advent of long-read, single-molecule methods (third-generation sequencing) has provided new opportunities to map the sequence composition of a previously “dark” area of the human genome, enabling research into the sequence composition and length dynamics (Luxton et al. 2020a, 2020b) of telomeres. Our results not only reaffirm that the canonical repeat (5′-TTAGGG-3′) is certainly the most dominant motif found within telomeres but also reveal a range of diverse, noncanonical repeat variations, which are confirmed by both short- and long-read sequencing technologies. This diversity of repeat sequence includes previously reported variants, as well as novel motifs that are characterized not only by nucleotide substitutions but also by insertions, deletions, and even motif pairing. Repeat patterns were chromosome specific, with different noncanonical repeats being pronounced on different chromosomes, such as TGAGGG on 12q and TTAGGGG on 15q, which may be related to certain biological pathways, such as the telomerase-independent, recombination-mediated alternative lengthening of telomeres (ALT) pathway of telomere length maintenance (Conomos et al. 2012).
Apart from these variations, CG-rich motifs were identified in telomeric regions of q arms, consistent with previously reported findings (Nergadze et al. 2009). Moreover, although short-read sequencing is capable of identifying such variants, it alone cannot reveal the relative locations of these motifs within telomeres, as repetitive short reads can neither be aligned outside of the reference genome nor provide enough overlap variability to be assembled de novo. Long SMRT reads, on the other hand, can be anchored to known subtelomeric sequences of the human genome and extend into the previously unmapped telomeric areas, opening up measures of new types of genetic variation. Furthermore, in contrast to previously published research that used targeted sequencing (Allshire et al. 1989; Coleman et al. 1999; Lee et al. 2018; Bluhm et al. 2019), the method described here allows identification of multiple enriched motifs and their localization de novo, without any bias introduced by prior knowledge about the sequence of target motifs.
These results also highlight the need for better subtelomeric and telomeric annotations in the human genome database: The canonical motif was present on the q arm of Chromosome 8 only 2–3 kbp beyond the annotated boundary in all data sets; the candidate reads on the p arm of Chromosome 17 represented TTAGGG-rich and non-TTAGGG-rich haplotypes, indicating that in multiple subjects and ancestries there exists an extension of the 17p subtelomere, which also contributes to the variation of the percentage of the sequence explained by these repeats (Table 1). For example, the Ashkenazi son (HG002) provided only non-TTAGGG-rich 17p reads, whereas both the father (HG003) and the mother (HG004) had a mixture of apparently telomeric and nontelomeric 17p reads. This supports previous findings (Young et al. 2020) that the existing assemblies do not provide completely accurate subtelomeric annotations, and suggests that methods described herein could help to resolve these areas of reference genomes.
We observed PacBio CCS reads reaching up to 16 kbp beyond the known regions of the genome and resolving the underlying sequence with fidelity, as measured both by the entropy of motif assignment and by pairwise Levenshtein distances between the reads belonging to the same chromosomal arms. Although short reads also provided support for noncanonical motifs, the overlap between the short and the long reads was substantial, but not complete, which can be explained by the necessary bias toward the canonical motif during the selection of short reads. Therefore, telomeric regions with higher content of noncanonical repeats are less likely to be identified through the use of short reads, and so, long reads appear to be more suitable for this purpose as well. Of note, the PacBio CCS read lengths allowed resolution of uniquely mapping reads only on 23 chromosomal arms, and coverage of different arms was uneven. As such, numbers of captured telomeric reads and levels of observed similarity varied from subject to subject; this calls for more sequencing experiments aimed to reconstruct the full picture of this variation. Clustering on a per-subject basis concealed interpopulation similarity but underscored intra-subject variation (Supplemental Figs. S4, S5), suggesting coexistence of two or more telomeric haplotypes per chromosomal arm within a given subject, at least for some chromosomes.
The identified variations in long-range contexts elucidate subject-specific, trio-specific, and population-specific similarities of telomeric sequences, as well as a level of interpopulation similarity, and thus provide a new means of haplotype mapping and reveal the existence and motif composition of haplotype spectra on a multi–kilobase pair scale. Interpopulation similarity, as well as consistently higher paternal inheritance of variation, provided evidence that the observed haplotypes could not be attributed to per–data set batch effects. Moreover, a significant inheritance of telomeric variants was observed in father–son pairs but not in mother–son pairs. This provided a haplotype-based interpretation of an analogous trend previously observed for telomere lengths (Nordfjäll et al. 2005), but no prior study had assessed the heritability of the telomeric repeats themselves.
In sum, these data and methods create new opportunities to map, quantify, and characterize a previously unmappable form of human genetic variation. Given that the reference DNA for the subjects HG001 through HG007 was extracted from culture-derived B lymphoblastoid cells, this suggests that as B cells undergo maturation, distinct clones may gain distinct variations in their telomeric sequence in addition to heterozygosity. This opens up avenues of investigation into the haplotypic variation among not only immune cells but also different cell types overall (e.g., cancer, germline, and developing cells) and, thus, can help delineate the possible mechanisms of selection and propagation of these variants as well as the asymmetric inheritance pattern.
Methods
The extended reference genome
We constructed the extended reference genome by performing an all-to-all alignment of all contigs in the hg38 reference genome (International Human Genome Sequencing Consortium 2001; Schneider et al. 2017) and the subtelomeric assemblies (Stong et al. 2014) with minimap2 (Li 2018) using three settings for assembly-to-reference mapping (asm5, asm10, asm20). Forty subtelomeric contigs mapped to ends of hg38 chromosomes with a mapping quality of 60, one (XpYptel) mapped with the quality of zero and was discarded; one (14qtel) mapped to the ALT version of Chromosome 14 (chr14_KI270846v1_alt) with the quality of 52, which, in turn, mapped to the main Chr 14 contig with the quality score of 60. These data and the exact match and mismatch coordinates were used to create a combined reference (hg38ext) in which subtelomeric contigs informed the locations of the boundaries of the telomeric tracts (tract_anchor). Such contigs that mapped fully within hg38 chromosomes resulted in tract_anchor annotations directly on those hg38 chromosomes; partially mapping contigs were considered as forking from the hg38 sequence and were similarly annotated by themselves. For the purposes of capturing candidate reads that uniquely align to subtelomere–telomere boundaries, subtelomeric contigs that were not previously assembled as extending completely up to the start of the telomere and/or were not precisely localized in relation to the reference genome, such as 1p, 6p, 7p, 8p, 11p, 20p, 3q, 4q, 20q, and Xq (Stong et al. 2014; Young et al. 2020), were masked before downstream analyses.
Detection of telomeric sequences in long-read data sets
Seven subjects were selected for the analysis. The first individual (NA12878/HG001) came from the pilot genome of the HapMap project (The International HapMap Consortium 2003), whereas the other six, including the Ashkenazi Jewish trio (son: NA24385/HG002; father: NA24149/HG003; mother: NA24143/HG004) and the Chinese trio (son: NA24631/HG005; father: NA24694/HG006; mother: NA24695/HG007), are members of the Personal Genome Project, whose genomes are consented for commercial redistribution and reidentification (Zook et al. 2016). These subjects are referred to throughout as HG001 through HG007, respectively.
Multiple GIAB (Zook et al. 2019) PacBio CCS (Eid et al. 2009; Ardui et al. 2018) data sets were available and combined per each subject, with mean coverages of individual data sets ranging from ∼21× to ∼69× (Supplemental Table S1). We mapped these reads to hg38ext with minimap2, allowing secondary mappings, and selected reads that mapped to either end of either chromosome, having an at least 500-bp portion of their sequence mapped to the reference contig and a portion extending beyond the reference (soft- or hard-clipped in the alignment file). As each of such reads can map to multiple subtelomeres owing to paralogy, we considered such multiple mappings and only retained the reads that mapped to a unique subtelomere; furthermore, out of these candidates, we only selected the ones overlapping the subtelomere and the telomere by at least 3 kbp. Sequences past the tract_anchor marker were extracted from the reads that had this marker within their mapped portion (from the 5′ end to the marker on p arms and from the marker to the 3′ end on q arms, accounting for forward and reverse mappings) (Fig. 1).
Evaluation of telomeric content in short- and linked-read data sets
To evaluate the concordance of telomeric reads captured by long- and short-read technologies, we extracted candidate telomeric reads from GIAB Illumina data sets for each subject (Supplemental Table S1) with Telomerecat (Farmery et al. 2018) and mapped the short reads back onto the candidate long reads from the same subject's data set with minimap2, again allowing all secondary mappings. Then, we calculated the fractions of each long read that were supported by the short reads that aligned to them.
To evaluate sequence motifs in independent samples collected from human subjects (as opposed to reference cell lines), we analyzed four whole-genome Illumina data sets (mean coverage ∼104×) and three linked-read 10x data sets (mean coverage ∼28×) for one individual at different time points, as well as one additional linked-read 10x data set (coverage ∼47×) for another individual. These data were originally obtained from astronaut subjects for an unrelated space biology experiment, and the blood samples were collected from the subjects as described in the study (Garrett-Bakelman et al. 2019). For each sample, 1.2 ng of sorted immune cell input was aliquoted for TruSeq PCR-free WGS (short-read) and standard Chromium 10x whole-genome (linked-read) preparation, respectively, and sequenced across one S4 flow cell on an Illumina NovaSeq 6000. From these data sets, candidate telomeric short reads were selected using Telomerecat (Farmery et al. 2018).
Identification of repeat content
Overrepresentation of motifs of lengths k⊂[4…16] was tested within the candidate telomeric regions of the PacBio CCS reads, as well as in the candidate reads from the independently generated Illumina and 10x Chromium data sets. To target motifs in repeat contexts, doubled sequences (e.g., k-mer ACGTACGT for motif ACGT) were counted with Jellyfish (Marçais and Kingsford 2011), and counts of k-mers synonymous with respect to circular shifts (e.g., ACGTACGT and CGTACGTA) were summed together. For each such k-mer, a Fisher's exact test was performed to determine whether its count is significant on the background of counts of other k-mers of the same length. Briefly, we considered k-mers with counts higher than the 1.5 interquartile range above the third quartile of the distribution as potentially classifiable, and a 2 × 2 contingency matrix C for the test was constructed as follows: row 0 contained counts of potentially classifiable k-mers; row 1 contained counts of the remaining (nonclassifiable) k-mers; and columns 0 and 1 contained counts of single and remaining (background) k-mers, respectively; that is, C0,0 = count of target k-mer, C0,1 = sum of counts of other potentially classifiable k-mers, C1,0 = median count of k-mer, C1,1 = sum of counts of other nonclassifiable k-mers. The resultant P-values for each motif among the samples were combined using the Mudholkar–George method (George and Mudholkar 1983) within each technology (PacBio CCS, Illumina, 10x Genomics), and the Bonferroni multiple testing correction was applied. Motifs in the long-read data sets for which k-mers yielded P-values below the cutoff of 0.05 were reported. As even doubled sequences (such as ACGTACGT for motif ACGT) can partially overlap at the boundaries of repeat contexts, we quantified their presence in the telomeric reads in two distinct ways. Consider a sequence such as TTAGGG(TTAGTTAG)GGTTA: The inner (TTAG)×2 repeat can be explained by the repeats of the canonical motif extending into it from either side; the middle part of a similar sequence with a bigger number of the repeats of the 4-mer, TTAGGGTTAG(TTAGTTAG)TTAGGGTTA, can only be explained by the repeats of said 4-mer. On the one hand, the maximum fraction of the sequence that can be explained by any one motif is a useful metric, and it was calculated and reported. On the other hand, the fraction of the k-mers attributable to a specific motif—and not to any others—elucidates the extent of deviation from the background repeat context and identifies motifs that most affect the sequence structure; it was calculated as well and reported as each motif's score. Additionally, motifs that were significantly enriched in the data sets produced by all three technologies (PacBio, Illumina, 10x), with respect to reverse-complemented equivalence, were reported.
Evaluation of sequence concordance in telomeric long reads
As telomeric reads contain long low-complexity regions and present an alignment challenge, we evaluated concordance of their sequences without realignment of their portions that extended past the reference sequence. To that end, for all reads mapping to the same chromosomal arm, we calculated densities of each identified motif in a rolling window starting from the innermost mapped position of each entire read. To evaluate whether the reads on the same arm agree on the positions of different motifs, for each read we calculated motif densities in 10-bp windows with 10-bp smoothing to buffer insertions and deletions. For each window in each read, the motif with the highest density was selected to represent that window. Then, normalized Shannon entropy among all reads was calculated in each window as , where pi is the frequency of each motif in the window, and N is the number of motifs (Minosse et al. 2006). The value of normalized entropy was a metric bounded by [0,1], with 0 describing perfect agreement and 1 describing maximum randomness. As coverage of the windows drops off toward the distal end of the alignment, lower covered windows have less chance to produce entropy; we calculated percentiles of entropy as weighted by coverage minus one (thus prioritizing higher covered windows and removing windows with the coverage of one and no entropy from the calculation). For motif visualization, we performed 1000 rounds of bootstrap of the calculated density values, this time in 100-bp rolling windows to accommodate the scale of multi–kilobase pair plots, and selected the lower and the upper bounds of the 95% confidence interval of bootstrap.
Identification of telomeric haplotypic variation
Within groups of reads mapping to each chromosomal arm, all relative pairwise Levenshtein distances were calculated. In short, Levenshtein distance is a string metric defined as the edit distance between two strings (sequences), equal to the minimum number of single-character insertions, deletions, and substitutions required to make these sequences identical (Levenshtein 1966). For each pair of reads, this metric was calculated and represented absolute edit distance; the relative distance was then computed as the absolute distance divided by the length of the overlap to normalize for the variation of such lengths. Pairwise relative distances were then clustered using Ward's method via the Euclidean metric, resulting in a hierarchical structure describing the extents of similarity among reads. To quantify how accurately hierarchical clustering described this similarity, cophenetic distances (Sokal and Rohlf 1962) between the hierarchies (dendrograms) and the distance matrices were calculated, and their Pearson correlation coefficients and Bonferroni-corrected P-values were reported.
We then traversed the distance matrices and, for each read, tracked the closest reads by category: closest reads from the same subject, from the same trio (population), and from the outgroup (other populations). For the Ashkenazi and the Chinese trios, we also tracked the closest reads between the parents and between each parent and the child. Thus, for each read, we determined whether it locally clustered within its own category (e.g., with other reads of the same subject or with other reads from the same population) or in a different one (e.g., with other reads of a different population), as well as the value of the distances that drove either clustering. Performing the Wilcoxon signed-rank test on these values between either category provided us with P-values that, after a Bonferroni correction, described whether reads tended to cluster in their own category or in a different one. Additionally, we also identified the minority of reads that did not follow the overall trend, and we quantified the extent to which they did so (such as the reads that contributed to interpopulation similarity).
Software availability
The software for identification of telomeric reads, de novo discovery of repeat motifs, haplotype inference, and motif density visualization was implemented in Python and is freely available at GitHub (https://github.com/lankycyril/edgecase), as well as in Supplemental File S3.
Data access
Healthy donor DNA came from a previous study (The NASA Twins Study) (Garrett-Bakelman et al. 2019). The NASA Life Sciences Data Archive (LSDA) is the repository for all human and animal research data, including the whole-genome Illumina and 10x Chromium sequencing data sets from subjects aboard the ISS that were used in this study. These data sets are protected by the terms of the Weill Cornell Medicine internal review board (IRB) and can be made available to be shared upon request. LSDA has a public-facing portal where data requests can be initiated (https://lsda.jsc.nasa.gov/Request/dataRequestFAQ); the LSDA team provides the appropriate processes, tools, and secure infrastructure for archival of experimental data and dissemination while complying with applicable rules, regulations, policies, and procedures governing the management and archival of sensitive data and information. The LSDA team enables data and information dissemination to the public or to authorized personnel either by providing public access to information or by an approved request process for information and data from the LSDA in accordance with NASA Human Research Program and JSC institutional review board direction.
Supplementary Material
Acknowledgments
We thank the Epigenomics Core Facility at Weill Cornell Medicine, the Scientific Computing Unit (SCU), Extreme Science and Engineering Discovery Environment (XSEDE) Supercomputing Resources, as well as the STARR grants I9-A9-071, I13-0052, The Vallee Foundation, The WorldQuant Foundation, The Pershing Square Sohn Cancer Research Alliance, NASA (NNX14AH51G, NNX14AB02G, NNX17AB26G), The National Institutes of Health (R01MH117406, R01NS076465, R01CA249054, R01AI151059, P01HD067244, P01CA214274), Translational Research Institute for Space Health (TRISH) (NNX16AO69A:0107, NNX16AO69A:0061), the Leukemia and Lymphoma Society (LLS) (9238-16, Mak, MCL-982, Chen-Kiang), and the National Science Foundation (1840275).
Author contributions: S.M.B. and C.E.M. conceived the study. K.G., J.F., and C.E.M. developed the framework and analyzed the data. D. Butler, J.J.L., M.J.M., L.T., and K.A.G. participated in the collection and processing of the ISS samples. D. Bezdan, D. Butler, J.J.L, J.R., and C.M. analyzed the data. All authors edited the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.274639.120.
Freely available online through the Genome Research Open Access option.
Competing interest statement
The authors declare no relevant competing interests, although C.E.M. is a cofounder of Onegevity Health.
References
- Allshire RC, Dempster M, Hastie ND. 1989. Human telomeres contain at least three types of G–rich repeat distributed non-randomly. Nucl Acids Res 17: 4611–4627. 10.1093/nar/17.12.4611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardui S, Ameur A, Vermeesch JR, Hestand MS. 2018. Single molecule real-time (SMRT) sequencing comes of age: applications and utilities for medical diagnostics. Nucleic Acids Res 46: 2159–2168. 10.1093/nar/gky066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubert G, Lansdorp PM. 2008. Telomeres and aging. Physiol Rev 88: 557–579. 10.1152/physrev.00026.2007 [DOI] [PubMed] [Google Scholar]
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, et al. 2008. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456: 53–59. 10.1038/nature07517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluhm A, Viceconte N, Li F, Rane G, Ritz S, Wang S, Levin M, Shi Y, Kappei D, Butter F. 2019. ZBTB10 binds the telomeric variant repeat TTGGGG and interacts with TRF2. Nucleic Acids Res 47: 1896–1907. 10.1093/nar/gky1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman J, Baird DM, Royle NJ. 1999. The plasticity of human telomeres demonstrated by a hypervariable telomere repeat array that is located on some copies of 16p and 16q. Hum Mol Genet 8: 1637–1646. 10.1093/hmg/8.9.1637 [DOI] [PubMed] [Google Scholar]
- Conomos D, Stutz MD, Hills M, Neumann AA, Bryan TM, Reddel RR, Pickett HA. 2012. Variant repeats are interspersed throughout the telomeres and recruit nuclear receptors in ALT cells. J Cell Biol 199: 893–906. 10.1083/jcb.201207189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, et al. 2009. Real-time DNA sequencing from single polymerase molecules. Science 323: 133–138. 10.1126/science.1162986 [DOI] [PubMed] [Google Scholar]
- Farmery JHR, Smith ML, Lynch AG. 2018. Telomerecat: a ploidy-agnostic method for estimating telomere length from whole genome sequencing data. Sci Rep 8: 1300. 10.1038/s41598-017-14403-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett-Bakelman FE, Darshi M, Green SJ, Gur RC, Lin L, Macias BR, McKenna MJ, Meydan C, Mishra T, Nasrini J, et al. 2019. The NASA twins study: a multidimensional analysis of a year-long human spaceflight. Science 364: eaau8650. 10.1126/science.aau8650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George EO, Mudholkar GS. 1983. On the convolution of logistic random variables. Metrika 30: 1–13. 10.1007/BF02056895 [DOI] [Google Scholar]
- The International HapMap Consortium. 2003. The International HapMap Project. Nature 426: 789–796. 10.1038/nature02168 [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921. 10.1038/35057062 [DOI] [PubMed] [Google Scholar]
- Iosim S, MacKay M, Westover C, Mason CE. 2019. Translating current biomedical therapies for long duration, deep space missions. Precision Clinical Medicine 2: 259–269. 10.1093/pcmedi/pbz022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, Tyson JR, Beggs AD, Dilthey AT, Fiddes IT, et al. 2018. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol 36: 338–345. 10.1038/nbt.4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Teber ET, Holmes O, Nones K, Patch A-M, Dagg RA, Lau LMS, Lee JH, Napier CE, Arthur JW, et al. 2018. Telomere sequence content can be used to determine ALT activity in tumours. Nucleic Acids Res 46: 4903–4918. 10.1093/nar/gky297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenshtein VI. 1966. Binary codes capable of correcting deletions, insertions, and reversals. Sov Phys Dokl 10: 707–710. [Google Scholar]
- Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100. 10.1093/bioinformatics/bty191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luxton JJ, McKenna MJ, Taylor LE, George KA, Zwart SR, Crucian BE, Drel VR, Garrett-Bakelman FE, Mackay MJ, Butler D, et al. 2020a. Temporal telomere and DNA damage responses in the space radiation environment. Cell Rep 33: 108435. 10.1016/j.celrep.2020.108435 [DOI] [PubMed] [Google Scholar]
- Luxton JJ, McKenna MJ, Lewis A, Taylor LE, George KA, Dixit SM, Moniz M, Benegas W, Mackay MJ, Mozsary C, et al. 2020b. Telomere length dynamics and DNA damage responses associated with long-duration spaceflight. Cell Rep 33: 108457. 10.1016/j.celrep.2020.108457 [DOI] [PubMed] [Google Scholar]
- Marçais G, Kingsford C. 2011. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27: 764–770. 10.1093/bioinformatics/btr011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miga KH. 2015. Completing the human genome: the progress and challenge of satellite DNA assembly. Chromosome Res 23: 421–426. 10.1007/s10577-015-9488-2 [DOI] [PubMed] [Google Scholar]
- Miga KH, Koren S, Rhie A, Vollger MR, Gershman A, Bzikadze A, Brooks S, Howe E, Porubsky D, Logsdon GA, et al. 2020. Telomere-to-telomere assembly of a complete human X chromosome. Nature 585: 79–84. 10.1038/s41586-020-2547-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minosse C, Calcaterra S, Abbate I, Selleri M, Zaniratti MS, Capobianchi MR. 2006. Possible compartmentalization of hepatitis C viral replication in the genital tract of HIV-1–coinfected women. J Infect Dis 194: 1529–1536. 10.1086/508889 [DOI] [PubMed] [Google Scholar]
- Moyzis RK, Buckingham JM, Cram LS, Dani M, Deaven LL, Jones MD, Meyne J, Ratliff RL, Wu JR. 1988. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc Natl Acad Sci 85: 6622–6626. 10.1073/pnas.85.18.6622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nergadze SG, Farnung BO, Wischnewski H, Khoriauli L, Vitelli V, Chawla R, Giulotto E, Azzalin CM. 2009. CpG-island promoters drive transcription of human telomeres. RNA 15: 2186–2194. 10.1261/rna.1748309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordfjäll K, Larefalk Å, Lindgren P, Holmberg D, Roos G. 2005. Telomere length and heredity: indications of paternal inheritance. Proc Natl Acad Sci 102: 16374–16378. 10.1073/pnas.0501724102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk S, Walenz BP, Rhie A, Vollger MR, Logsdon GA, Grothe R, Miga KH, Eichler EE, Phillippy AM, Koren S. 2020. Hicanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res 30: 1291–1305. 10.1101/gr.263566.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider VA, Graves-Lindsay T, Howe K, Bouk N, Chen H-C, Kitts PA, Murphy TD, Pruitt KD, Thibaud-Nissen F, Albracht D, et al. 2017. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res 27: 849–864. 10.1101/gr.213611.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafin K, Pesout T, Lorig-Roach R, Haukness M, Olsen HE, Bosworth C, Armstrong J, Tigyi K, Maurer N, Koren S, et al. 2020. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of 11 human genomes. Nat Biotechnol 38: 1044–1053. 10.1038/s41587-020-0503-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shammas MA. 2011. Telomeres, lifestyle, cancer, and aging. Curr Opin Clin Nutr Metab Care 14: 28–34. 10.1097/MCO.0b013e32834121b1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokal RR, Rohlf FJ. 1962. The comparison of dendrograms by objective methods. Taxon 11: 33–40. 10.2307/1217208 [DOI] [Google Scholar]
- Stong N, Deng Z, Gupta R, Hu S, Paul S, Weiner AK, Eichler EE, Graves T, Fronick CC, Courtney L, et al. 2014. Subtelomeric CTCF and cohesin binding site organization using improved subtelomere assemblies and a novel annotation pipeline. Genome Res 24: 1039–1050. 10.1101/gr.166983.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treangen TJ, Salzberg SL. 2012. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet 13: 36–46. 10.1038/nrg3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young E, Abid HZ, Kwok PY, Riethman H, Xiao M. 2020. Comprehensive analysis of human subtelomeres by whole genome mapping. PLoS Genet 16: e1008347. 10.1371/journal.pgen.1008347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zook JM, Catoe D, McDaniel J, Vang L, Spies N, Sidow A, Weng Z, Liu Y, Mason CE, Alexander N, et al. 2016. Extensive sequencing of seven human genomes to characterize benchmark reference materials. Sci Data 3: 160025. 10.1038/sdata.2016.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zook JM, McDaniel J, Olson ND, Wagner J, Parikh H, Heaton H, Irvine SA, Trigg L, Truty R, McLean CY, et al. 2019. An open resource for accurately benchmarking small variant and reference calls. Nat Biotechnol 37: 561–566. 10.1038/s41587-019-0074-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.