

Abstract
Wound healing is a complex sequence of tissue protection, replacement, and reorganization leading to regenerated tissue. Disruption of any of these steps results in the process being incomplete as an ulcer or over-exuberant as a hypertrophic scar. Over the past decade, it has become evident that the extracellular matrix and associated components orchestrate this process. However, the cellular events that are induced by the extracellular matrix to accomplish wound healing remain to be defined. Herein we propose that matrix-regulated cellular macro-autophagy is key to both the tissue replacement and resolution stages of healing by directing cellular function or apoptosis. Further, disruptions in matrix turnover alter autophagic function leading to chronic wounds or scarring. While the literature that directly investigates autophagy during wound healing is sparse, the emerging picture supports our proposing a model of the centrality of the matrix-autophagy modulation as central to physiologic and pathologic healing.
Keywords: Matricellular proteins, Tenascin-C, Decorin, Macro-autophagy, Mitophagy, Chronic wounds, Scars
Introduction
Wound healing is a highly dynamic and complex process that involves three major phases of repair: hemostasis/inflammation, tissue replacement, and resolution. The modulation, structure, and signaling of extracellular matrix (ECM) through all these phases is key to obtaining a successful wound resolution. The dynamic influential nature of the ECM not only directs the transitions through the various phases of wound healing1, 2, but also determines the survival and fate of cells in the wound bed. Early in the inflammatory phase, the provisional matrix includes many pro-migratory and promitogenic matricellular components that then provide for the rapid tissue replacement phase. These include not just matrix-associated growth factors, but also bioactive matricellular elements such as thrombospondin, fibronectin, and tenascin-C1, 2.These support the expansion of the cell numbers not only by driving proliferation but also by protecting against death signals. As an example, tenascin-C protects stem cells and others from death signals via cryptic matrikine domains activating the epidermal growth factor (EGF) receptor in a tonic manner to signal via the Akt survival pathway3. This is critical during the very early stages before viable vasculature can provide both nutrients and oxygen, in addition to the removal of toxic cell fragments and death signals released by innate immune cells. An open question relates to how the cells that provide for the tissue replacement can withstand the lack of nutrients; this reveals an avenue for macro-autophagy4.
Macro-autophagy (hereafter, autophagy) is an evolutionarily conserved catabolic process that utilizes double membraned vacuoles called autophagosomes to transport cellular cargo to the lysosomal degradation machinery to maintain homeostasis within the cell5. Output metabolites of the system (amino acids, nucleotides, etc) can then be reused to make other macromolecules desired by the cell. The autophagic process functions in two conditions: selective and non-selective. Under normal circumstances, this process is discriminatory and only selects cargo such as a dysfunctional organelle or aggregate prone proteins6. However, under situations where a cell is under an extreme amount of stress such as nutrient deprivation or hypoxia, this process becomes non-selective and all cellular components can be randomly metabolized to keep the cell alive7. It is the responsiveness of autophagy to both extracellular and intracellular cues that lends it to contribute to a variety of physiologic pathways from cell survival to differentiation and signaling5. This has led to further investigations examining how the ECM interacts and influences autophagic activity involved in wound healing (Table 1).
Table 1.
Extracellular matrix – autophagic relationships during wound healing in the literature.
| Article Reference | Phase of Wound Healing | Key Finding | Implications in wound healing |
|---|---|---|---|
| Ouseph et al.19 | Hemostasis/Inflammation | Autophagy is induced upon platelet activation via LC3 turnover and is essential for platelet function. | ECM platelet activators such as Col I and fibrinogen may act as key regulators of autophagy within the activated platelets. |
| Soto-Pantoja et al.23 | Hemostasis/Inflammation | Thrombospondin 1 signals through CD47 to downregulate autophagic flux subsequently limiting cell survival of cells in ischemic conditions. | Thrombospondin 1 acts as a negative regulator of angiogenesis early in hemostasis to prevent leaky vasculature and hemorrhaging in the wound microenvironment. |
| Poluzzi et al. 42 | Hemostasis/Inflammation | Biglycan is an SLRP protein that will induce autophagic flux within M1 macrophages by signaling through the TLR-4/CD44 receptor. | Biglycan’s induction of autophagy in M1 macrophages enhances their survival under the ischemic conditions of the wound bed. |
| Castagnaro et al.49 | Tissue Replacement | Fibroblasts from a COLVI null mouse model exhibit an impaired autophagic response resulting in increased apoptosis when subjected to nutrient-stringent conditions. | Collagen VI is a key regulator of fibroblasts survival and function through regulation of autophagic flux. This is key for migrating fibroblasts entering the wound bed during the early phases of repair when the wound is still ischemic. |
| Wang et al.51 | Tissue Replacement | Tenascin-C upregulates Beclin 1 through 13–3-3t and is required for induction of autophagy in immortalized cells under nutrient limited conditions. | Tenascin-C is a vital protein for the tissue replacement phase of wound healing. Its ability to induce autophagy may add additional insight to its pro-survival capacity of MSCs and other cell types and their regulation during the early phases of wound repair. |
| Zheng et al.57 | Tissue Replacement | Osteopontin stimulates autophagosome formation and expression of autophagic flux of vascular smooth muscle cells through signaling integrin/CD44 and MAPK pathways. This enhancement of autophagy has been linked to increased cell death in diseases such as abdominal aortic aneurysm. | The timing of osteopontin in wound healing is critical, as it had been shown to both improve wound healing outcomes as well as lead to increased fibrosis. It acts as a major chemokine for inflammatory cells and MSCs entering the wound bed, with additional observations of key signaling through the CD44 pathway. The downstream autophagic regulations on smooth muscles cells and other cells types may be a determining feature for healthy wound healing and abnormal wound healing. |
| Buraschi et al.61 | Resolution | Decorin suppresses endothelial cell angiogenesis via binding to VEGF receptor 2 and inducing a pro-autophagic response through the Beclin 1 and Peg3 convergence. | Decorin is a major stop signal in the resolution phase of wound healing with one of the key functions being the pruning of unnecessary vasculature. Decorin’s ability to initiate an autophagic response in endothelial cells actively leads to the suppression of angiogenesis within the wound bed. |
| Poluzzi et al.62 | Resolution | Endorepellin suppresses endothelial cell angiogenesis by signaling through VEGF receptor 2 and α2β1 integrin to modulate the expression of Beclin 1 and Peg3 to enhance autophagic flux. | Endorepellin is another SLRP that supports decorin in the resolution phase of wound healing to prune excess vasculature and help return the wound to homeostasis. |
As the wound progresses towards resolution, the matrix changes to a suppressive phenotype, dominated by collagen I fibrils8. The stimulatory matricellular proteins are diminished, with a near-complete absence of thrombospondin and tenascin-C1. In their stead appear a family of small leucine-rich proteoglycans (SLRPs), including lumican and decorin1, 9. The latter molecule acts as a non-competitive inhibitor of multiple growth factors, preventing activation of the receptors for EGF, IGF-1, HGF, and even VEGF10, 11. This change is commensurate with the skin epidermis quiescing to reduce basal proliferation with fewer transitional keratinocytes and an epidermis returning to its paucicellular state. Over the weeks of wound resolution, approximately 90% of the neovessels involute and over half of the fibroblasts disappear, returning the skin to a resolved homeostatic state8.
The above wound healing process is found in near regenerative healing, with the biphasic matrix-induce survival and death signals leading to an orderly tissue regeneration and subsequent quiescent resolution. However, disruptions of this orderly progression leads to pathologies of healing, including chronic wounds/ulcers and hypertrophic scars1. Scarring ensues when the tissue replacement phase persists with a weakened resolution. In these circumstances, the matrix remains rich in tenascin-C, fibronectin, and thrombospondin while being relatively devoid of the SLRPs, such as decorin12, 13. We will discuss below, how this matrix may increase one mode of autophagy to pathologically enhance cell numbers while suppressing the autophagy that triggers cellular apoptosis.
The second wound pathology occurs when the tissue replacement phase of healing does not cover the wound bed and stalls. There are a number of situations that impact and block tissue replacement. Repeated injury, as in pressure ulcers, activates excessive matrix breakdown by matrix metalloproteinases (MMP)14. Insufficient vascular supply, due to diabetic arteriolosclerosis or pressure as in pressure ulcers, limits cellular replacement14, 15. Insufficient drainage, as in venous stasis ulcers, leaves toxic components and increased MMP activity that drives premature cell apoptosis and dysfunctional matrix14, 15. Wound infection, even without biofilms, also leads to matrix breakdown secondary to chronic inflammatory infiltrates16. In all these situations, the pathological wound beds impact the survival of the parenchymal cells, in situations of nutrient limitations that regulate autophagy.
Thus, we are confronted with complex processes leading to near regenerative healing when the notes are played in proper sequence, but ulcers or scarring when the tone is discordant. This is orchestrated by matrix and matrix-associated signals directing cells to play their assigned roles. Central to the melodious outcome is the sequencing of cell differentiation and then involution of over-exuberant tissues. An aspect that is not fully appreciated is the intracellular processes that enact these changes. Herein, we explore the supposition that autophagy, as regulated by matrix and matricellular components, and how dysfunctional autophagy can lead to chronic wounds or scarring. As direct studies are few in number (Table 1), the following posits a testable model wherein matrix components regulate the autophagic flux in physiologic and pathologic skin wound healing.
Extracellular matrix regulation of autophagy in the normal wound healing response.
Hemostasis and inflammation phase of wound healing.
Hemostasis is the first reaction in an activated wound healing response (Fig.1). It is initiated when an injury to the skin results in damaging the underlying vasculature and serves as the primary mechanism to prevent extensive hemorrhaging. This is accomplished through the formation of a fibrin clot; where circulating platelets adhere to newly exposed collagen I matrix and subsequently become activated through the glycoprotein VI receptor17. These activated platelets undergo morphological changes to increase their surface area along with secreting additional pro-coagulation factors in the local environment. The activated platelets also begin to express binding receptors for fibrinogen. This allows for the capture of circulating plasma fibrinogen, which then acts as a tether for additional circulating platelet cells allowing for their aggregation and activation18. Fibrinogen can also activate platelets through glycoprotein VI receptor similar to the earlier exposed collagen I fragments17. Similar to other mammalian cells, platelets have been found to express key autophagic related markers such as Beclin-1, LC3 and ATG 719, 20. Further investigation revealed that resting platelets undergo a basal level of autophagic activity. Where activated platelets exhibited an agonist-induced loss of LC3-II demonstrating a change in autophagic activity19. In vitro autophagic manipulations of MTOR and in vivo murine knock out models of ATG7 and Beclin-1 demonstrated that disruption to the autophagic mechanism resulted in decreased platelet aggregation, adhesion, and activation19, 20. Though not directly linked to a full thickness cutaneous wound model, these studies suggest that collagen I and fibrinogen may be associated with inducing and regulating the autophagic response of activated platelets.
Figure 1:
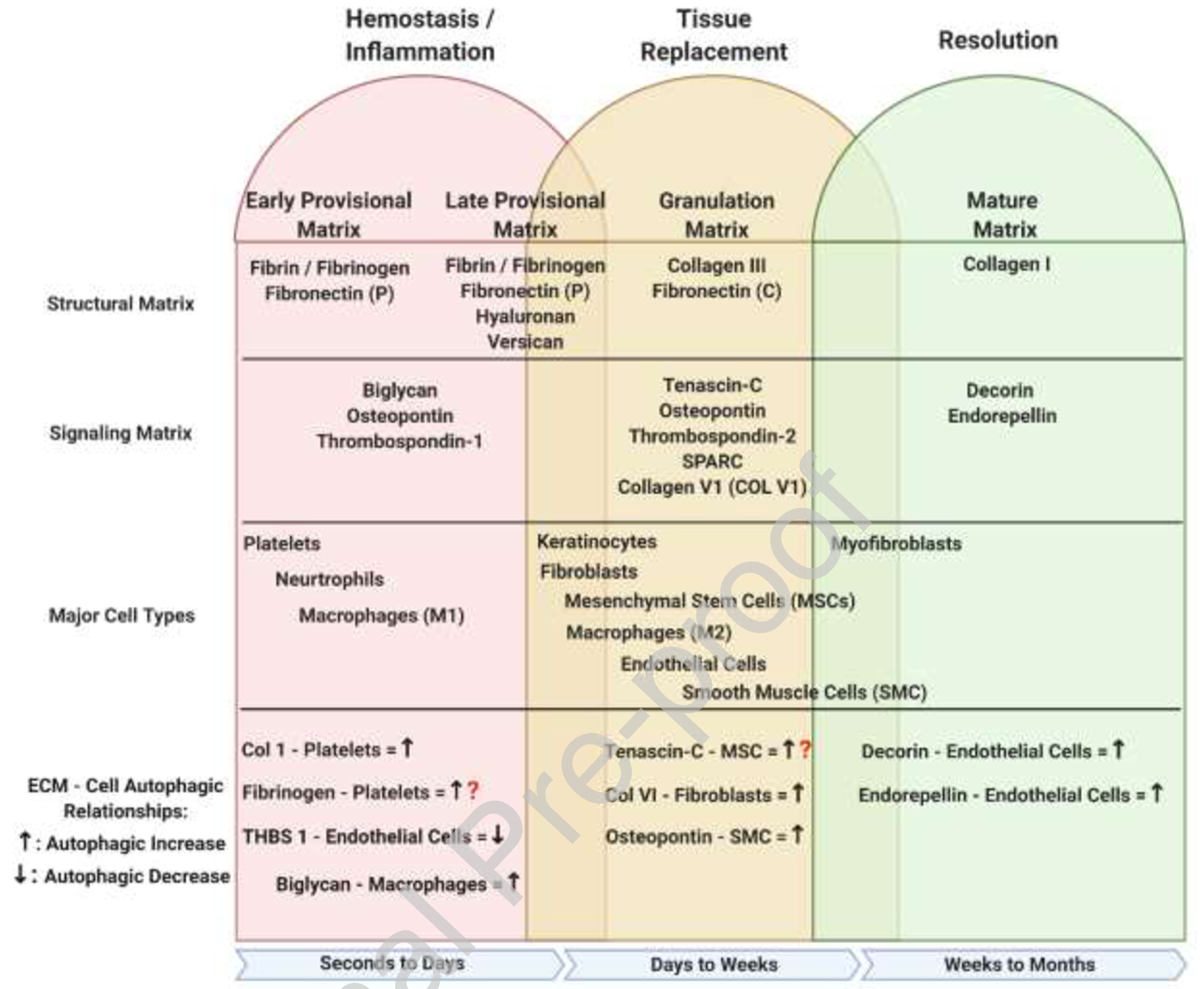
Overview of ECM-Autophagic relationships during wound healing. During the initial stages of the hemostasis/inflammation phase of repair, a provisional matrix barrier known as the fibrin clot is generated to suppress further hemorrhaging from the wound site. Concurrently with the formation of the clot, activated platelets signal to the surrounding tissue to initiate a hasty immune response recruiting neutrophils and monocyte derived M1 macrophages to sterilize the wound and remove all damaged ECM debris; this is driven by cell-specific autophagy to elicit the cell behaviors. The increase of inflammatory cell manipulations along with early fibroblasts ECM secretions further modify the provisional matrix changing it from an early to late form. The late provisional matrix further manipulates the inflammatory response, preparing it to transition from anti-inflammatory to pro-repair. During the tissue replacement phase, further modification by fibroblasts and M2 macrophages to the late provisional matrix transforms it to a granulation matrix. The granulation matrix is meant to provide easier cell movement and function as it mostly made from a loose collagen 3 base with interspersed signaling ECM proteins. The last phase of repair is the resolution phase where the ECM is recognized to its final form, vasculature is pruned, and the spare cells are removed, again involving autophagy-initiated apoptosis of endothelial cells and autophagy-triggered phenotypic shifts of fibroblasts. This figure was created with BioRender.com (on 10 October 2020). ECM – Extracellular Matrix, Fibronectin (P) – Plasma, Fibronectin (C) – Cellular, THBS1 – Thrombospondin 1, MSC – Mesenchymal Stem Cell, SMC – Smooth Muscle Cell, Col V1 – Collagen Type VI, SPARC – Secreted Protein Acidic and Cysteine Rich
As circulating platelets and fibrinogen continue to build up at the injury site, they eventually form what is known as the platelet plug. As more and more platelets become activated within the platelet plug, they proceed to enhance the coagulation signaling cascade. This involves a final step where the structure of the platelet plug is further reinforced and transformed into a fibrin clot. Within this clot, additional plasma fibrinogen is enzymatically modified into fibrin and crosslinked together with plasma fibronectin to form a structural mesh that solidifies the entire structure21.This newly formed barrier is now termed the “Early Provisional Matrix” and is the first major ECM framework within the activated wound healing response22. In addition to creating a barrier to stop the bleeding, the body also releases the matrix glycoprotein thrombospondin-1 that acts as an endogenous angiogenic inhibitor. Thrombospondin-1 adheres to the CD47 receptor on endothelial cells and subsequently downregulates their ability to undergo enhanced autophagy; thus further limiting their ability to survive and respond to any pro-angiogenic signals coming from the early wound bed23–25.
Once the primary goal of stopping the localized hemorrhage is accomplished, the early provisional matrix priorities shift to augment a robust inflammatory response to counter the breach in the anti-microbial barrier. The main goal of the immune system is to sterilize the wound bed against foreign pathogens and to remove all damaged tissue to prepare for a smooth transition into the tissue replacement phase. Neutrophils are one of the first immune cells to respond, sensing chemical cues in the local microenvironment from the fibrin clot and transmigrating through the vascular wall to reach the site of injury within 24 hours after injury26, 27. They begin the fight against infection and combat foreign pathogens through a variety of measures including the phagocytosis of small microbes, the release of antimicrobial granules and reactive oxygen species, and the distribution of neutrophil extracellular traps (NETs)28. In recent findings, autophagy is a major regulating factor in all three of these defense mechanisms29, 30. A unique form of LC3-associated phagocytosis (LAP) has been exhibited by neutrophils as a selective type of autophagy that is induced upon activation of pathogen associated molecular pattern (PAMPs) sensing pathways and internalization of foreign pathogens, ultimately enhancing the cells phagocytosis capabilities31–33. It has also been shown that a deficiency in autophagic machinery can lead to reduced degranulation and reactive oxygen species (ROS) generation in neutrophils29, 31. Furthermore, autophagy is thought to be linked to the second step of NET preparation by providing the necessary bioactive proteins31, 34. The link between these key autophagic regulation events and the ECM's ability to modulate these responses in neutrophils during wound healing awaits elucidation. Neutrophil functions have been shown to be enhanced and altered through ECM attachment via β2 integrin35. Overall, there is much to still investigate as to how the ECM dynamics influence neutrophile form and function.
Monocytes are the next major immune cell to emigrate into the wound bed. They are recruited by pro-inflammatory cytokines (IL-6, TNF-α, CCL2, CCL5) produced by activated platelets from the fibrin clot or activated neutrophils already on site36, 37. Additionally, ECM- derived damage associated modifying proteins (DAMPS) can also recruit monocytes as smaller ECM fragments can break from the wound area and into the localized vasculature38. Once monocytes arrive at the site of injury, they extravasate into the wound bed relying on β1 integrins to adhere to ECM proteins such as fibronectin to navigate around39. Once in the wound bed they receive additional inflammatory chemical cues, such as IFN-γ and lipopolysaccharide, which stimulate their polarization into the M1 -pro-inflammatory macrophage phenotype40. The M1 macrophage relies heavily on its phagocytosing ability, eradicating additional remnants of pathogen fragments missed by neutrophils, and debriding the rest of the wound bed of damaged tissue40.
Similar to neutrophils, M1 macrophages rely on autophagic machinery for most inflammatory processes from activation to phagocytosis to regulating the inflammasome41. Yet, little information has connected ECM regulation of natural autophagic function within macrophages of the skin. In recent findings in the kidney however, a small leucine-rich proteoglycan called biglycan, which is also expressed in the skin, was able to induce a pro-autophagic response in M1 macrophages through interactions with a TLR-4/CD44 receptor complex during a renal ischemia/reperfusion injury. Interestingly this increase in autophagic ability in M1 macrophages was associated with an increase of M2 macrophage recruitment and polarization in the later stages of repair42.
As the composition of the original early provisional matrix becomes altered through debridement and release of ECM modifying enzymes, macrophages and early migrating fibroblasts start to synthesize and deposit versican and hyaluronan into the matrix43. This is a second major modification to the wound ECM, which will be termed the “Late Provisional Matrix”. Versican is a large proteoglycan that can be cleaved by a disintegrin and metalloproteinase with thrombospondin motifs(ADAMTS) proteases to elicit different responses throughout wound healing. It has the ability to attach to numerous inflammatory cell receptors and is considered a key molecule in regulating the inflammation response44. Hyaluronan is a glycosaminoglycan that can also play a part in all aspects of wound healing. For the immune response, iťs size dictates whether it will have pro-inflammatory or anti-inflammatory effects45. Together these two ECM proteins mark the end of the hemostatic/inflammation phase of repair as the wound transitions away from the provisional matrix to the next stage of wound healing where it will need to support the survival, proliferation, and migration of numerous cell types.
Tissue replacement phase of wound healing.
As the wound reaches its final stages of sterilization and most of the damaged ECM has been removed, the late provisional matrix starts to transform into a more modular granulation tissue; ultimately setting the stage for the tissue replacement phase to begin. The overall goal of this phase of wound healing is to repopulate and restore the tissue to its original state. This involves various cell types proliferating around the edges of the wound and then migrating within to rebuild the tissue.
Within the epidermal layer, keratinocytes begin to proliferate and migrate into the edges of the provisional matrix under the eschar, creating what is known as the epithelial tongue. The keratinocytes will continue to bore through the provisional matrix until they converge with keratinocytes migrating in from the adjacent side. Once this connection is made in the epidermis, dermal fibroblasts arrive and begin to reconstruct the basement membrane below, further separating the epidermis/dermis of the skin. A recent study discovered that the keratinocytes rely upon TNF induced autophagy through AMPK-MAPK1/3-AP1 signaling pathway to regulate the expression of CCL2. This increase in CCL2 expression could then be used to activate keratinocytes behind the leading edge for their proliferation and migration into the wound bed. Even more interesting was that this CCL2 expression was also critical for dermal fibroblast activation as well46.
In the underlying dermis, fibroblasts all along the wound margin proliferate and migrate into the wound bed. They are tasked with creating new stroma that will replace the existing fibrin/platelet dense provisional matrix. The structure of the new matrix called the “granulation matrix” is made from a more accommodating collagen III environment. Collagen VI is also produced during the tissue repair phase as it helps to regulate dermal fibroblast motility and ECM assembly47, 48. Studies have shown fibroblasts from knockout collagen VI models exhibited increased apoptosis when subjected to nutrient-stringent conditions similar to those seen at the initiation of the tissue replacement phase. This increase in cell death was due to a deficiency in autophagic flux while also increasing negative regulation pathways like the Akt/mTOR signaling pathway49. In addition to collagen secretion, other matricellular ECM proteins such as tenascin-C, osteopontin, and fibronectin are also present and aid the migration and survival of incoming cells. In particular, tenascin-C has been shown to enhance the survival of mesenchymal stem cells (MSCs) post transplantation into a cutaneous wound healing model. As stated earlier, the EGF-like repeat domain of tenascin-C will bind to the EGF receptor and promote survival via the Akt survival pathway3, 50. Tenascin-C also has the capability to regulate and induce Beclin-1 expression via 14–3-3T in immortalized cell lines51. More work needs to be done exploring the relationship between autophagy and tenascin-C in the context of wound healing, but this may provide added insight to tenascin-C's survival influence overs MSCs during the early stages of wound repair. . These new ECM proteins are important as prior to this point the provisional matrix was an acellular space devoid of supporting vasculature. Hence, any cells migrating into the granulation tissue early on will face ischemic conditions and be challenged both in survival and in function.
Macrophages also play a large role in the early part of the tissue replacement stage as newly recruited monocytes and M1 macrophages start to polarize towards the M2 anti-inflammatory/pro-repair phenotype. The macrophage polarization is triggered by a culmination of a few signaling pathways, with the major signals of CCL2 and IL-6 pathways triggering an autophagic induction response. These signals help macrophages improve their survival and help fibroblasts with granulation tissue formation52. M2 macrophages will also start to release growth factors, such as VEGF and PDGF, to help in the initiation of angiogenesis36. In addition to the construction of a new matrix, angiogenesis plays just as vital of a role in the tissue replacement phase as a substantial amount of energy (e.g. nutrients and oxygen) is required to rebuild the wound bed to its proper form. In conjunction with the M2 macrophages, the ischemic/hypoxic conditions of the wound bed will also trigger MSCs to secrete higher levels of VEGF through enhanced autophagic flux53, 54. An additional ECM protein, osteopontin, further aids in the angiogenic response by acting as a major chemokine for macrophages and MSCs to migrate to the wound bed, while also promoting the differentiation of MSCs into endothelial cells55, 56. Osteopontin has also been observed to induce an autophagic response in smooth muscle cells (SMCs) via integrin/CD44 and p38 MAPK57. However, osteopontin's autophagic increase in SMCs was also linked to enhanced cell death57. Additional wound healing studies have shown that higher levels of osteopontin could be linked to chronic ulcers or fibrosis56, 58. Thus, timing of osteopontin and its regulation of autophagy in various cell types will be a key area to explore to further understand why it can lead to both normal and abnormal wound healing events.
Resolution phase of wound healing
The final phase of the wound healing mechanism is the resolution phase. Up to this point both the provisional matrix and granulation matrix focused heavily on recruiting cells into the wound bed and supporting their additive functions. The focus of the “mature matrix” is to quell all superfluous tissue repair mechanisms and reorganize into a more mature matrix that restores the local microenvironment back to a homeostatic state.
The reorganization of the granulation matrix into the mature matrix starts with the turnover of collagen III fibrils to collagen I. The collagen III network provided a flexible environment that allowed for an abundant amount of cell migration and function in and around the wound bed. Turnover into collagen I fibrils enhances the tensile strength of the tissue and offers the additional support needed to return to normal function. This is achieved through fibroblasts differentiating into myofibroblasts which acquire higher amounts of intracellular alpha smooth muscle actin, allowing for stronger manipulation and contraction of the underlying ECM via integrin interactions59. The myofibroblasts along with remaining M2 macrophages release a series of ECM modulating enzymes such as MMPs, transglutaminases, and lysyl oxidases to strategically crosslink ECM back to its collagen-I state59. Subsequently after this conversion, majority of the myofibroblasts and M2 macrophages undergo apoptotic events and are removed from the restored wound bed.
A second component of the wound bed that is largely manipulated during the resolution phase is the underlying vasculature. The tissue replacement phase required a large amount of cellular traffic, nutrients, and cargo to come in and out of the wound bed. Consequently, these systems are not necessary anymore resulting in vascular pruning to regress the density of blood vessels back to normal levels and allowing for the remaining vasculature to fully mature. One of the key regulators during this step is a small leucine rich proteoglycan called decorin. Decorin is considered an important shut off switch for the pro-reparative mechanism of the tissue replacement phase, as it binds to growth factor receptors such as EGFR and VEGFR260; and limits the occurrence of hypertrophic scarring through preventing excessive repair. Upon binding VEGFR2 on endothelial cells, decorin induces a pro-autophagic response through the AMPKa/Vps34/Peg3 signaling pathway and concurrently inhibits angiogenesis by suppressing VEGFA levels10, 60, 61. In conjunction with the decorin finding, the same group of researchers have since looked at other ECM proteins as they relate to the modification of endothelial cells. They identified endorepellin, a matrikine subdomain protein of perlecan, which was also found to induce a pro-autophagic response via the same signaling cascade as decorin 62. Similarly, endorepellin actively contributes to blocking the angiogenic capability of endothelial cells63. Like the myofibroblasts and macrophages, the autophagic signaling of these SLRPs eventually lead to an apoptotic clearing of all leftover endothelial cells in the final transition from resolution to homeostasis.
Extracellular matrix regulation of autophagy in the pathological wound healing response.
The pathologies of healing fall into two large groups – failure to regenerate the lost tissue (stalled healing or ulcer) and excessive dermal/matrix expansion (scarring)(Fig. 2). Since these groups relate to either too few or too many cells, the interactions of autophagy with cell number may underlie these pathologies.
Figure 2:
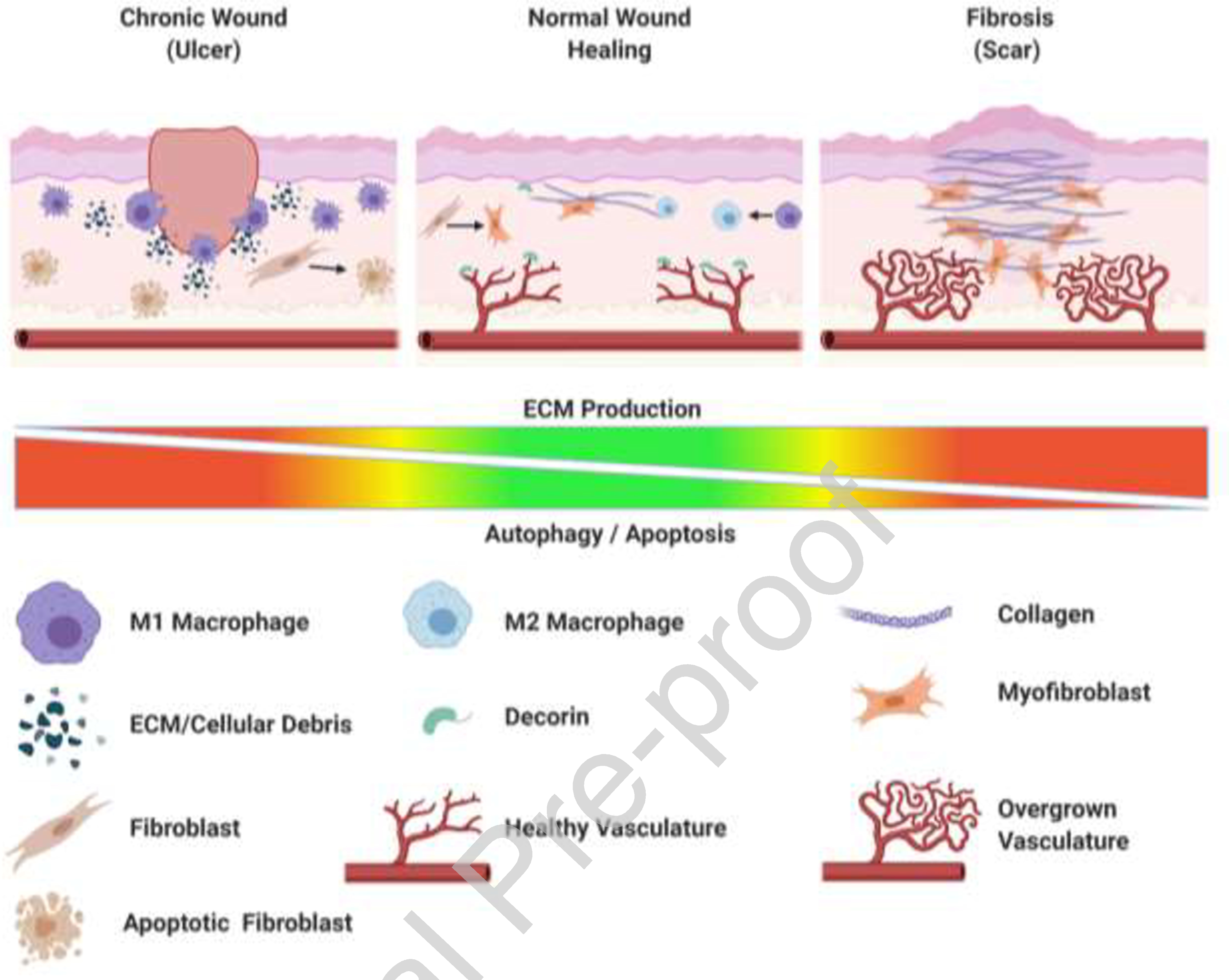
Disruption of wound healing balance leads to chronic ulcers or scars. Wounds that do not heal properly result in two categories: chronic wounds or fibrotic wounds. Chronic wounds occur most often due to an over-activate turnover of ECM within the wound bed. The persistent exposure to debris, heightened inflammatory cytokines, and ischemic conditions places an extraordinary amount of stress on surrounding cells. This leads to activation of internal pro-survival responses including autophagy within these cells, but the longevity of stress will eventually lead to apoptotic induction and a failure of tissue replacement. This results in a 'stalled' healing response or open chronic wound. In Fibrosis or scarring the wound fails to successfully complete the resolution phase of healing. Often the key stop signal regulators such as decorin or other SLRPs, are under expressed, with reduced autophagy-triggered apoptosis of the excess endothelial cells, and limited autophagy leading to excess myofibroblasts that produce bio-active replacement matrix, rather than suppressive collagen-1 rich resolution matrix. This figure was created with BioRender.com (on 15 October 2020).
Skin ulcers ensue usually from underlying co-morbidities or extreme age. The age-related failures to heal can be traced to limited replicative capacity of the dermal fibroblasts, the Hayflick number64, and/or lack of stem cells65,66. but successful and stable coverage requires a mature dermis. Even though this cause of ulceration does not involve autophagy, the other main comorbidities likely impact by healing via dysregulated autophagy.
Diabetic ulcers occur in areas of the skin with limited revascularization and hyperglycemia in the extracellular fluids. The resultant prolonged hypoxia and limited nutrients push the balance of autophagy past the tipping point and into apoptosis of the MSCs67. The hyperglycemia of diabetes contributes to the healing dysfunction by nonenzymatic glycating matrix proteins resulting in advanced glycation end products (AGEs) that impact multiple aspects of healing. Fibroblasts are driven to apoptosis via AGE-induced autophagy68, and macrophages are stuck in the inflammatory M1 polarization69. The authors showed autophagy to be a key regulating mechanism for impaired wound healing as iťs inhibition restored normal healing in the their diabetic mouse model.
Venous stasis ulcers and infected wounds prevent completion of the tissue replacement phase by the same final mechanisms of increased matrix turnover. In venous stasis ulcers the sterile edematous fluids polarize macrophages towards M1 with secretion of MMPs. This mirrors the leukocyte activation by infecting bacteria and also increases the presence of MMPs and other matrix-degrading proteases. It is the unrelenting degradation of the matrix and produced matrix fragments that increase the autophagic flux in the otherwise stressed fibroblasts and induces MSCs to push the cells towards apoptosis and away from differentiation.
Scarring can be viewed as a dysregulation of the transition to resolution. Here the main issue is that by delaying and decreasing the levels of decorin and other SLRP, the autophagy-mediated involution is limited, leading to increased deposition of matrix. This is compounded by the lower levels of autophagy not reaching the threshold for apoptosis but rather redirecting the fibroblasts into myofibroblasts70. Thus, insufficient autophagic activity leads to a higher population of synthetic and fibrotic cells in the dermis in a feed forward manner.
Conclusion
The matrix is emerging as the main regulator of wound healing. A procession of ECM components directs a diverse array of cells in the wound bed to in-migrate, expand, form tissue and then mostly involute to return to a pauci-cellular tissue. However, the key cellular processes induced by the ECM components are only now being deciphered. Within, we presented a strong case that autophagy is critical to every phase of wound healing, and changes in the relative balance of autophagy leads to either unhealed chronic wounds or excessive ECM deposition and scarring.
Throughout the discussion of this model of healing, we often have alluded to apoptosis, or rescue from, in relationship to autophagy modulated by matrix components. It must be borne in mind that autophagy is just one inducer of apoptosis, and apoptosis is just a single outcome of autophagy, though a major one. We have tried to link this event to the physiologic (such as vascular involution and resolution) or pathologic (for instance, persistence of excess fibroblast in scarring) healing phenomenon. However, these causative links remain to be directly tested
What makes this proposed model of matrix-modulated autophagy during wound healing especially attractive is that autophagy would fit into a physiological process that is characterized by cellular starvation to initiate the wound healing. As autophagy can rescue cells from both nutrient or energy deprivation by recycling cellular elements, it is centrally placed to be a trigger for stem cell recruitment at the beginning of the tissue replacement phase. Meanwhile during the resolution phase, autophagy may be able to induce the cell death of involution. Thus, by reinstating autophagy, the cells would survive or die, and result in the pathologies of dysfunctional healing if unbalanced. Should this centrality of autophagy be supported, it opens avenues for direct interventions to enhance or restore wound healing and subsequently treat hypertrophic scars and ulcerations.
Highlights.
We propose that autophagy plays a major role in wound healing.
The procession of the ECM drives the extent of autophagic flux.
Alterations in timing or extent of autophagy results in pathologies of healing.
Acknowledgements
Funding: The concepts reported on herein were supported by grants from the NIH (GM69668, GM63569 and NR016436) and the VA Merit Program.
We thank members of the Wells, Roy (Univ Pittsburgh, Bioengineering), and Wang (Cornell University, Bioengineering) laboratories for helpful discussions and feedback.
Both authors conceived of, wrote and edited the manuscript. A.W. provided the funding.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
There are no conflicts of interest to disclose related to the work herein. There are no germane human subjects or vertebrate animal considerations specific for this review.
References.
- [1].Wells A, Nuschke A, Yates CC: Skin tissue repair: Matrix microenvironmental influences. Matrix Biol 2016, 49:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Olczyk P, Mencner L, Komosinska-Vassev K: The role of the extracellular matrix components in cutaneous wound healing. Biomed Res Int 2014, 2014:747584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rodrigues M, Yates CC, Nuschke A, Griffith L, Wells A: The matrikine tenascin-C protects multipotential stromal cells/mesenchymal stem cells from death cytokines such as FasL. Tissue Eng Part A 2013, 19:1972–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Nuschke A, Rodrigues M, Stolz DB, Chu CT, Griffith L, Wells A: Human mesenchymal stem cells/multipotent stromal cells consume accumulated autophagosomes early in differentiation. Stem Cell Res Ther 2014, 5:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yang Z, Klionsky DJ: Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 2010, 22:124–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Klionsky DJ, Cuervo AM, Dunn WA Jr., Levine B, van der Klei I, Seglen PO: How shall I eat thee? Autophagy 2007, 3:413–6. [DOI] [PubMed] [Google Scholar]
- [7].Mizushima N, Levine B, Cuervo AM, Klionsky DJ: Autophagy fights disease through cellular self-digestion. Nature 2008, 451:1069–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Krafts KP: Tissue repair: The hidden drama. Organogenesis 2010, 6:225–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pang X, Dong N, Zheng Z: Small Leucine-Rich Proteoglycans in Skin Wound Healing. Front Pharmacol 2019, 10:1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Buraschi S, Neill T, Iozzo RV: Decorin is a devouring proteoglycan: Remodeling of intracellular catabolism via autophagy and mitophagy. Matrix Biol 2019, 75–76:260–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Neill T, Schaefer L, Iozzo RV: Decorin: a guardian from the matrix. Am J Pathol 2012, 181:380–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yates CC, Bodnar R, Wells A: Matrix control of scarring. Cell Mol Life Sci 2011, 68:1871–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yates CC, Hebda P, Wells A: Skin wound healing and scarring: fetal wounds and regenerative restitution. Birth Defects Res C Embryo Today 2012, 96:325–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rohani MG, Parks WC: Matrix remodeling by MMPs during wound repair. Matrix Biol 2015, 44–46:113–21. [DOI] [PubMed] [Google Scholar]
- [15].Davis FM, Kimball A, Boniakowski A, Gallagher K: Dysfunctional Wound Healing in Diabetic Foot Ulcers: New Crossroads. Curr Diab Rep 2018, 18:2. [DOI] [PubMed] [Google Scholar]
- [16].Krishnaswamy VR, Mintz D, Sagi I: Matrix metalloproteinases: The sculptors of chronic cutaneous wounds. Biochim Biophys Acta Mol Cell Res 2017, 1864:2220–7. [DOI] [PubMed] [Google Scholar]
- [17].Mangin PH, Onselaer MB, Receveur N, Le Lay N, Hardy AT, Wilson C, Sanchez X, Loyau S, Dupuis A, Babar AK, Miller JL, Philippou H, Hughes CE, Herr AB, Ariens RA, Mezzano D, Jandrot-Perrus M, Gachet C, Watson SP: Immobilized fibrinogen activates human platelets through glycoprotein VI. Haematologica 2018, 103:898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zuliani-Alvarez L, Midwood KS: Fibrinogen-Related Proteins in Tissue Repair: How a Unique Domain with a Common Structure Controls Diverse Aspects of Wound Healing. Adv Wound Care (New Rochelle) 2015, 4:273–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ouseph MM, Huang Y, Banerjee M, Joshi S, MacDonald L, Zhong Y, Liu H, Li X, Xiang B, Zhang G, Komatsu M, Yue Z, Li Z, Storrie B, Whiteheart SW, Wang QJ: Autophagy is induced upon platelet activation and is essential for hemostasis and thrombosis. Blood 2015, 126:1224–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Feng W, Chang C, Luo D, Su H, Yu S, Hua W, Chen Z, Hu H, Liu W: Dissection of autophagy in human platelets. Autophagy 2014, 10:642–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Laurens N, Koolwijk P, de Maat MP: Fibrin structure and wound healing. J Thromb Haemost 2006, 4:932–9. [DOI] [PubMed] [Google Scholar]
- [22].Barker TH, Engler AJ: The provisional matrix: setting the stage for tissue repair outcomes. Matrix Biol 2017, 60–61:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Soto-Pantoja DR, Miller TW, Pendrak ML, DeGraff WG, Sullivan C, Ridnour LA, Abu-Asab M, Wink DA, Tsokos M, Roberts DD: CD47 deficiency confers cell and tissue radioprotection by activation of autophagy. Autophagy 2012, 8:1628–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Isenberg JS, Romeo MJ, Maxhimer JB, Smedley J, Frazier WA, Roberts DD: Gene silencing of CD47 and antibody ligation of thrombospondin-1 enhance ischemic tissue survival in a porcine model: implications for human disease. Ann Surg 2008, 247:860–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kyriakides TR, Maclauchlan S: The role of thrombospondins in wound healing, ischemia, and the foreign body reaction. J Cell Commun Signal 2009, 3:215–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kim MH, Liu W, Borjesson DL, Curry FR, Miller LS, Cheung AL, Liu FT, Isseroff RR, Simon SI: Dynamics of neutrophil infiltration during cutaneous wound healing and infection using fluorescence imaging. J Invest Dermatol 2008, 128:1812–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Park JE, Barbul A: Understanding the role of immune regulation in wound healing. Am J Surg 2004, 187:11S-6S. [DOI] [PubMed] [Google Scholar]
- [28].Wang J: Neutrophils in tissue injury and repair. Cell Tissue Res 2018, 371:531–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bhattacharya A, Wei Q, Shin JN, Abdel Fattah E, Bonilla DL, Xiang Q, Eissa NT: Autophagy Is Required for Neutrophil-Mediated Inflammation. Cell Rep 2015, 12:1731–9. [DOI] [PubMed] [Google Scholar]
- [30].Shrestha S, Lee JM, Hong CW: Autophagy in neutrophils. Korean J Physiol Pharmacol 2020, 24:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Skendros P, Mitroulis I, Ritis K: Autophagy in Neutrophils: From Granulopoiesis to Neutrophil Extracellular Traps. Front Cell Dev Biol 2018, 6:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Deretic V: Autophagy in immunity and cell-autonomous defense against intracellular microbes. Immunol Rev 2011, 240:92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Martinez J, Malireddi RK, Lu Q, Cunha LD, Pelletier S, Gingras S, Orchard R, Guan JL, Tan H, Peng J, Kanneganti TD, Virgin HW, Green DR: Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol 2015, 17:893–906. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [34].Stakos DA, Kambas K, Konstantinidis T, Mitroulis I, Apostolidou E, Arelaki S, Tsironidou V, Giatromanolaki A, Skendros P, Konstantinides S, Ritis K: Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur Heart J 2015, 36:1405–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].O'Brien XM, Reichner JS: Neutrophil Integrins and Matrix Ligands and NET Release. Front Immunol 2016, 7:363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Krzyszczyk P, Schloss R, Palmer A, Berthiaume F: The Role of Macrophages in Acute and Chronic Wound Healing and Interventions to Promote Pro-wound Healing Phenotypes. Front Physiol 2018, 9:419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Prame Kumar K, Nicholls AJ, Wong CHY: Partners in crime: neutrophils and monocytes/macrophages in inflammation and disease. Cell Tissue Res 2018, 371:551–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Frevert CW, Felgenhauer J, Wygrecka M, Nastase MV, Schaefer L: Danger-Associated Molecular Patterns Derived From the Extracellular Matrix Provide Temporal Control of Innate Immunity. J Histochem Cytochem 2018, 66:213–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Muller WA: Getting leukocytes to the site of inflammation. Vet Pathol 2013, 50:7–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Mahdavian Delavary B, van der Veer WM, van Egmond M, Niessen FB, Beelen RH: Macrophages in skin injury and repair. Immunobiology 2011, 216:753–62. [DOI] [PubMed] [Google Scholar]
- [41].Wu MY, Lu JH: Autophagy and Macrophage Functions: Inflammatory Response and Phagocytosis. Cells 2019, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Poluzzi C, Nastase MV, Zeng-Brouwers J, Roedig H, Hsieh LT, Michaelis JB, Buhl EM, Rezende F, Manavski Y, Bleich A, Boor P, Brandes RP, Pfeilschifter J, Stelzer EHK, Munch C, Dikic I, Brandts C, Iozzo RV, Wygrecka M, Schaefer L: Biglycan evokes autophagy in macrophages via a novel CD44/Toll-like receptor 4 signaling axis in ischemia/reperfusion injury. Kidney Int 2019, 95:540–62. [DOI] [PubMed] [Google Scholar]
- [43].Wight TN: Provisional matrix: A role for versican and hyaluronan. Matrix Biol 2017, 60–61:38–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wight TN, Kang I, Evanko SP, Harten IA, Chang MY, Pearce OMT, Allen CE, Frevert CW: Versican-A Critical Extracellular Matrix Regulator of Immunity and Inflammation. Front Immunol 2020, 11:512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Garantziotis S, Savani RC: Hyaluronan biology: A complex balancing act of structure, function, location and context. Matrix Biol 2019, 78–79:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Qiang L, Yang S, Cui YH, He YY: Keratinocyte autophagy enables the activation of keratinocytes and fibroblasts and facilitates wound healing. Autophagy 2020:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Theocharidis G, Drymoussi Z, Kao AP, Barber AH, Lee DA, Braun KM, Connelly JT: Type VI Collagen Regulates Dermal Matrix Assembly and Fibroblast Motility. J Invest Dermatol 2016, 136:74–83. [DOI] [PubMed] [Google Scholar]
- [48].Cescon M, Gattazzo F, Chen P, Bonaldo P: Collagen VI at a glance. J Cell Sci 2015, 128:3525–31. [DOI] [PubMed] [Google Scholar]
- [49].Castagnaro S, Chrisam M, Cescon M, Braghetta P, Grumati P, Bonaldo P: Extracellular Collagen VI Has Prosurvival and Autophagy Instructive Properties in Mouse Fibroblasts. Front Physiol 2018, 9:1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yates CC, Nuschke A, Rodrigues M, Whaley D, Dechant JJ, Taylor DP, Wells A: Improved Transplanted Stem Cell Survival in a Polymer Gel Supplemented With Tenascin C Accelerates Healing and Reduces Scarring of Murine Skin Wounds. Cell Transplant 2017, 26:103–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wang B, Ling S, Lin WC: 14–3-3Tau regulates Beclin 1 and is required for autophagy. PLoS One 2010, 5:e10409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Chen P, Cescon M, Bonaldo P: Autophagy-mediated regulation of macrophages and its applications for cancer. Autophagy 2014, 10:192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].An Y, Liu WJ, Xue P, Ma Y, Zhang LQ, Zhu B, Qi M, Li LY, Zhang YJ, Wang QT, Jin Y: Autophagy promotes MSC-mediated vascularization in cutaneous wound healing via regulation of VEGF secretion. Cell Death Dis 2018, 9:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sylakowski K, Bradshaw A, Wells A: Mesenchymal Stem Cell/Multipotent Stromal Cell Augmentation of Wound Healing: Lessons from the Physiology of Matrix and Hypoxia Support. Am J Pathol 2020, 190:1370–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wang W, Li P, Li W, Jiang J, Cui Y, Li S, Wang Z: Osteopontin activates mesenchymal stem cells to repair skin wound. PLoS One 2017, 12:e0185346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Chimento S, Billero V, Cavallin L, Romanelli M, Nadji M, Romanelli P: Evaluation of osteopontin expression in chronic wounds: a potential prognostic and therapeutic biomarker. J Wound Care 2017, 26:S4–S8. [DOI] [PubMed] [Google Scholar]
- [57].Zheng YH, Tian C, Meng Y, Qin YW, Du YH, Du J, Li HH: Osteopontin stimulates autophagy via integrin/CD44 and p38 MAPK signaling pathways in vascular smooth muscle cells. J Cell Physiol 2012, 227:127–35. [DOI] [PubMed] [Google Scholar]
- [58].Mori R, Shaw TJ, Martin P: Molecular mechanisms linking wound inflammation and fibrosis: knockdown of osteopontin leads to rapid repair and reduced scarring. J Exp Med 2008, 205:43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Klingberg F, Hinz B, White ES: The myofibroblast matrix: implications for tissue repair and fibrosis. J Pathol 2013, 229:298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Gubbiotti MA, Buraschi S, Kapoor A, Iozzo RV: Proteoglycan signaling in tumor angiogenesis and endothelial cell autophagy. Semin Cancer Biol 2020, 62:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Buraschi S, Neill T, Goyal A, Poluzzi C, Smythies J, Owens RT, Schaefer L, Torres A, Iozzo RV: Decorin causes autophagy in endothelial cells via Peg3. Proc Natl Acad Sci U S A 2013, 110:E2582–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Poluzzi C, Casulli J, Goyal A, Mercer TJ, Neill T, Iozzo RV: Endorepellin evokes autophagy in endothelial cells. J Biol Chem 2014, 289:16114–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Goyal A, Gubbiotti MA, Chery DR, Han L, Iozzo RV: Endorepellin-evoked Autophagy Contributes to Angiostasis. J Biol Chem 2016, 291:19245–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Tran KT, Rusu SD, Satish L, Wells A: Aging-related attenuation of EGF receptor signaling is mediated in part by increased protein tyrosine phosphatase activity. Exp Cell Res 2003, 289:359–67. [DOI] [PubMed] [Google Scholar]
- [65].Akamatsu H, Hasegawa S, Yamada T, Mizutani H, Nakata S, Yagami A, Matsunaga K: Age-related decrease in CD271(+) cells in human skin. J Dermatol 2016, 43:311–3. [DOI] [PubMed] [Google Scholar]
- [66].Zobiri O, Deshayes N, Rathman-Josserand M: Evolution of the clonogenic potential of human epidermal stem/progenitor cells with age. Stem Cells Cloning 2012, 5:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Nuschke A, Rodrigues M, Wells AW, Sylakowski K, Wells A: Mesenchymal stem cells/multipotent stromal cells (MSCs) are glycolytic and thus glucose is a limiting factor of in vitro models of MSC starvation. Stem Cell Res Ther 2016, 7:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Dai J, Chen H, Chai Y: Advanced Glycation End Products (AGEs) Induce Apoptosis of Fibroblasts by Activation of NLRP3 Inflammasome via Reactive Oxygen Species (ROS) Signaling Pathway. Med Sci Monit 2019, 25:7499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Guo Y, Lin C, Xu P, Wu S, Fu X, Xia W, Yao M: AGEs Induced Autophagy Impairs Cutaneous Wound Healing via Stimulating Macrophage Polarization to M1 in Diabetes. Sci Rep 2016, 6:36416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Bernard M, Dieude M, Yang B, Hamelin K, Underwood K, Hebert MJ: Autophagy fosters myofibroblast differentiation through MTORC2 activation and downstream upregulation of CTGF. Autophagy 2014, 10:2193–207. [DOI] [PMC free article] [PubMed] [Google Scholar]