

Significance Statement
Hypertension and elevated levels of LDL cholesterol (LDLc) are vital risk factors for cardiovascular disease (CVD) in patients with CKD. Although statins indisputably reduce plasma cholesterol levels, not all patients with renal disease benefit from them, making CVD the leading cause of CKD-related mortality. CKD induces hyperelongation of hepatic heparan sulfate (HS) chains from HS proteoglycans (HSPG), thereby increasing the HSPG–proprotein convertase subtilisin kexin type-9 (PCSK9) interaction. These changes associate with elevated LDLc levels, without affecting LDL-receptor expression. This study, using rat models, discloses a novel mechanism behind hypercholesterolemia in CKD with prospects for future investigation of the HSPG-PCSK9 interaction and development of novel heparin-related glycomimetics targeting interruption of HSPG-PCSK9 binding.
Keywords: chronic kidney disease, dyslipidemia, PCSK9, heparan sulfate, syndecan-1
Visual Abstract

Abstract
Background
Dyslipidemia is an important risk factor in CKD. The liver clears triglyceride-rich lipoproteins (TRL) via LDL receptor (LDLR), LDLR-related protein-1 (LRP-1), and heparan sulfate proteoglycans (HSPGs), mostly syndecan-1. HSPGs also facilitate LDLR degradation by proprotein convertase subtilisin/kexin type 9 (PCSK9). Progressive renal failure affects the structure and activity of hepatic lipoprotein receptors, PCSK9, and plasma cholesterol.
Methods
Uninephrectomy- and aging-induced CKD in normotensive Wistar rats and hypertensive Munich-Wistar-Frömter (MWF) rats.
Results
Compared with 22-week-old sex- and strain-matched rats, 48-week-old uninephrectomized Wistar-CKD and MWF-CKD rats showed proteinuria, increased plasma creatinine, and hypercholesterolemia (all P<0.05), which were most apparent in hypertensive MWF-CKD rats. Hepatic PCSK9 expression increased in both CKD groups (P<0.05), with unusual sinusoidal localization, which was not seen in 22-week-old rats. Heparan sulfate (HS) disaccharide analysis, staining with anti-HS mAbs, and mRNA expression of HS polymerase exostosin-1 (Ext-1), revealed elongated HS chains in both CKD groups. Solid-phase competition assays showed that the PCSK9 interaction with heparin-albumin (HS-proteoglycan analogue) was critically dependent on polysaccharide chain length. VLDL binding to HS from CKD livers was reduced (P<0.05). Proteinuria and plasma creatinine strongly associated with plasma cholesterol, PCSK9, and HS changes.
Conclusions
Progressive CKD induces hepatic HS elongation, leading to increased interaction with PCSK9. This might reduce hepatic lipoprotein uptake and thereby induce dyslipidemia in CKD. Therefore, PCSK9/HS may be a novel target to control dyslipidemia.
Hypertension and dyslipidemia are imperative risk factors for cardiovascular disease (CVD) and CKD.1–4 Dyslipidemia—associated with elevated total cholesterol (TC), LDL cholesterol (LDLc), lipoprotein-a, plasma triglycerides (TGs), and reduced HDL cholesterol (HDLc) levels—is implicated to increase the incidence of CVD in CKD.1,3 CVD-related death is the primary cause of mortality in patients with CKD and is highly prevalent in all age groups and in all stages of CKD.3,5–7 Therefore, the American Heart Association and the National Kidney Foundation categorized patients with renal disease as having a very high risk for developing CVD.1,8
The severity of dyslipidemia in CKD depends on the level of kidney function and proteinuria.1,2,9,10 The pathophysiology behind dyslipidemia in CKD involves reduced hepatic lipase, lipoprotein lipase, and lecithin-cholesterol acyltransferase, along with increased 3-hydroxy-3-methyl-glutaryl-CoA reductase and angiopoietin-like protein-4.9,11 Recently, hepatic loss of syndecan-1 and increased LDL-receptor (LDLR) degradation induced by proprotein convertase subtilisin/kexin type 9 (PCSK9) have been shown to increase cholesterol levels.12–15 The loss of hepatic syndecan-1 was found to be strongly, positively associated with increased TGs, suggesting hepatic syndecan-1 shedding could hamper lipoprotein uptake by the liver and could thus contribute to dyslipidemia.16
The liver is the primary organ for lipoprotein uptake and clearance via LDLRs, LDLR-related protein-1 (LRP-1), and heparan sulfate (HS) proteoglycan (HSPG). Lipoproteins bound to these receptors are endocytosed for lysosomal hydrolysis, whereas receptors are recycled back to the membrane.17–22 Changes in the structure of the HS polysaccharide side chains (chain length and/or degree of sulfation) highly affects the functionality of HSPG,23–28 and such changes have previously been reported in renal HSPGs in various CKD models and in cases of high dietary salt intake.24,25,29,30
In the liver, syndecan-1/HS acts as a lipoprotein receptor, independent of LDLR and LRP-1.17,31–33 Recently, Gustafsen et al. 34 showed hepatic HSPGs at the surface of hepatocytes act as liver-specific coreceptors for PCSK9. Via their HS chains, these HSPGs present PCSK9 to the LDLR. PCSK9 then binds to the EGF-like A domain of LDLR and becomes internalized into hepatocytes together with LDLR, where it directs the LDLR for lysosomal degradation.22,35,36 In animal models of CKD, marked elevation of plasma PCSK9 has been observed with a reduction in hepatic LDLR expression and increase in plasma LDLc levels,37–39 whereas PCSK9 knockout improved dyslipidemia.37 These studies suggest an important role of PCSK9 in CKD-related dyslipidemia.
Recent studies in patients with CKD have reported a 50%–60% increase in plasma PCSK9 levels.40–44 Longitudinal human studies showed a decrease in plasma PCSK9 levels in patients with nephrotic syndrome after effective treatment of the disorder,37 suggesting PCSK9-mediated LDLR degradation is increased in patients with CKD, which hampers the normal lipoprotein clearance.
In this study, we investigated the effects of CKD on hepatic lipoprotein receptors, including HSPG, and the interaction of HSPG with PCSK9 using two models of CKD: (1) normotensive, aged Wistar rats with uninephrectomy; and (2) hypertensive Munich-Wistar-Frömter (MWF) rats with uninephrectomy. We observed progressive loss of renal function induces hepatic HS elongation, leading to increased interaction with PCSK9. These changes might cause dyslipidemia by reducing hepatic lipoprotein clearance. Our study reveals potential novel targets for future treatment strategies in CKD-related dyslipidemia.
Methods
Animals and Treatments
Male MWF rats (n=14) were obtained from Harlan (Indianapolis, IN), and male Wistar rats (n=13) were obtained from Harlan (Melderslo, The Netherlands). The MWF rat is a model for CKD, which is induced by progressive hypertension.45 All animals received care in compliance with the Principles of Laboratory Animal Care (National Institutes of Health publication number 86-23, revised 1985), the University of Groningen guidelines for animal husbandry, and the Dutch law on experimental care, and experiments were approved by the ethical committee on animal experiments at the University of Groningen. All rats underwent unilateral nephrectomy at 22 weeks. Uninephrectomy accelerates the development of CKD due to glomerular hyperfiltration. Uninephrectomy in normal Wistar rats also causes CKD upon aging; however, the CKD is less severe compared with the MWF rat, because of the absence of hypertension. Proteinuria and BP (tail-cuff method) were assessed at 24 and 25 weeks (Wistar-baseline and MWF-baseline), and the mean of those two measurements was taken as the baseline value. Blood was drawn for baseline measurements at 25 weeks of age. At 26 weeks after the unilateral nephrectomy (i.e., the age of 48 weeks; Wistar-CKD and MWF-CKD) proteinuria and BP were measured again and the rats were euthanized. A young Wistar control group was euthanized at 22 weeks of age (Wistar-22W; n=6) without any intervention. Plasma and livers were obtained and cryopreserved for serologic, molecular, and histologic analysis.
Urine and Plasma Analysis
Creatinine in plasma and urine was measured using an enzymatic ultraviolet (UV) assay (Roche Modular P; Roche, Mannheim, Germany). The total protein in urine was determined using the turbidimetric assay (Roche Modular P). PCSK9 levels in the plasma of Wistar-22W, MWF-baseline, and MWF-CKD were determined using the rat PCSK9 ELISA kit (SEK80005; Sino Biologic Inc., Wayne, PA). No plasma from Wistar-CKD were available for this analysis. Methods and procedures were followed according to the those provided in the data sheet of the manufacturer.
Lipid Measurements
TC levels were measured with a colorimetric assay (11489232; Roche) with Cholesterol Standard FS (DiaSys Diagnostic Systems, Holzheim, Germany) as reference. TG levels were measured using the Trig/GB kit (Roche) with Roche Precimat Glycerol standard (Roche) as reference.
Fast-Performance Liquid Chromatography
Plasma samples from the young Wistar-22W control group, MWF-baseline, and MWF-CKD rats were individually fractionated for VLDL cholesterol (VLDLc), LDLc, and HDLc by fast-performance liquid chromatography (FPLC) for lipoprotein profiling as previously described,46 with minor modifications. No plasma from Wistar-CKD rats was available for this analysis. In brief, the system contained a PU-980 ternary pump with an LG-980-02 linear degasser and a UV-975 UV/VIS detector (Jasco). EDTA plasma was diluted 1:1 with Tris-buffered saline (TBS), and 300 μl sample/buffer mixture was loaded onto a Superose 6 HR 10/300 column (GE Healthcare, Life Sciences Division, Chicago, IL) for lipoprotein separation at a flow rate of 0.5 ml/min.
RNA Isolation, cDNA Synthesis, and Quantitative RT-PCR
Total RNA was isolated from liver tissues of young Wistar control rats, Wistar-CKD rats, and MWF-CKD rats using the RNeasy Mini Kit (Favorgen Biotech Corp.). The concentration and purity of RNA was determined using Nanodrop. RNA (1 µg) was reverse transcribed to cDNA using QuantiTect Reverse Transcription Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Endogenous Gapdh was used as a housekeeping gene along with the transcripts for the following genes: Pcsk9, Ldlr, Lrp-1, Syndecan-1, and Ext1. Primers were purchased from Sigma-Aldrich (St Louis, MO), except for Syndecan-1 and Ldlr, which were from Qiagen (QuantiTect Primer Assay; Qiagen on-demand primers). Primer sequences are provided in Supplemental Table 1. The cycle procedure was as follows: 10 minutes at 95°C, followed by 40 repeats of a 15-second denaturation step at 95°C, a 15-second extension and annealing step at 60°C, and then a final 5-second extension step at 72°C. Real-time PCR was performed by CFX384 Touch Real-Time PCR (Bio-Rad, Hercules, CA) with SYBR Green I dye (Bioline, Dublin, Ireland) according to the manufacturer’s instructions. Fluorescent data were converted to cycle threshold (CT) values. Relative mRNA levels were calculated as 2 to the power of −ΔCT, in which ΔCT is the CT gene of interest minus the CT housekeeping gene. We tested the melt curves and efficiency of the PCR reaction. Negative controls were included using the samples without cDNA (PCR-grade water and mRNA sample mix in which no reverse transcriptase was added while making cDNA). Measurements were performed in triplicate.
Immunofluorescence Staining
Details on Immunofluorescence procedures are given in Table 1. Liver stainings by immunofluorescence were evaluated using the Leica DM4000B equipped with the DFC345FX camera and LAS software package. For quantification, we obtained six to ten random pictures per section at a magnification of 200×, with standardized magnification, exposure, and threshold settings, using the Mac Biophotonics ImageJ program (US National Institutes of Health, Bethesda, MD). Data are expressed as percentage positive surface area. No primary antibodies were added in the negative control sections; double staining protocols were controlled by cross-checking the absence of binding of conjugates with the other primary antibody.
Table 1.
Immunofluorescence staining protocols a
| Target | Antibodies | Conjugates | Visualization |
|---|---|---|---|
| Syndecan-1 | Goat polyclonal anti–syndecan-1 (1:25 in PBS with 1% BSA; N‐18, SC 7100; Santa Cruz Biotechnology) | Rabbit anti-goat Ig HRP and goat anti-rabbit Ig-HRP (both 1:100 in PBS with 1% BSA; DAKO Diagnostics) | Tetramethylrhodamine system (1:50; PerkinElmer LAS Inc.) |
| LDLR | Rabbit polyclonal anti-LDLR (1:300 in PBS with 1% BSA; Pab8804; Abnova) | Goat anti-rabbit Ig-HRP (1:100 in PBS with 1% BSA; DAKO) | Tetramethylrhodamine system (1:50; PerkinElmer LAS Inc.) |
| LRP1 | Rabbit mAb anti–LRP-1 (1:7500 in PBS with 1% BSA; 92544; Abcam) | Goat anti-rabbit Ig-HRP (1:100 in PBS with 1% BSA; DAKO Diagnostics) | Tetramethylrhodamine system (1:50; PerkinElmer LAS Inc.) |
| PCSK9 b | Rabbit anti-mouse PCSK9 (1:300 in PBS with 1% BSA; 552C kindly provided by Jayson D. Horton, University of Texas Southwestern Medical Center, Dallas, TX) | Goat anti-rabbit Ig-HRP (1:100 in PBS with 1% BSA; DAKO Diagnostics) | Tetramethylrhodamine system (1:50; PerkinElmer LAS Inc.) |
| Heparan sulfate motif: 10E4 c | Mouse anti-HS mAb biotinylated 10E4 (1:50 in PBS with 1% BSA; AMS Biotech, Abingdon, United Kingdom) | Streptavidine FITC (1:200 in PBS with 1% BSA; Invitrogen) | Streptavidine FITC (1:200; Invitrogen) |
| Heparan sulfate motif: 3G10 c # | Mouse anti-HS mAb 3G10 (1:1600 in PBS with 1% BSA; AMS Biotech) | Goat anti-mouse IgG2b-HRP (1:100 in PBS with 1% BSA; Southern Biotech, Birmingham, AL) | Tetramethylrhodamine system (1:50; PerkinElmer LAS Inc.) |
| LYVE1 | Sheep anti-rat (1:300 in PBS with 1% BSA; R&D Systems, Minneapolis, MN) | Donkey anti-sheep Alexa 488 (1:100 in PBS with 1% BSA; A11015; Invitrogen) | Tetramethylrhodamine system (1:50; PerkinElmer LAS Inc.) |
| LYVE1-3G10 | Sheep anti-rat (1:300 in PBS with 1% BSA; R&D Systems) and mouse anti-HS mAb 3G10 (1:1600 in PBS with 1% BSA; AMS Biotech) | Donkey anti-sheep Alexa 488 (1:100 in PBS with 1% BSA; A11015; Invitrogen) and goat anti-mouse IgG2b-HRP (1:100 in PBS with 1% BSA; Southern Biotech) | Tetramethylrhodamine system (1:50; PerkinElmer LAS Inc.) |
| 3G10-PCSK9 b | Mouse anti-HS mAb 3G10 (1:1600 in PBS with 1% BSA; AMS Biotech) and rabbit anti-mouse PCSK9 (1:300 in PBS with 1% BSA; 552C) | Goat anti-mouse IgG2b (1:100 in PBS with 1% BSA; Southern Biotech) and goat anti-rabbit Ig-HRP (1:100 in PBS with 1% BSA; DAKO Diagnostics) | Streptavidine FITC (1:200 in PBS with 1% BSA; Invitrogen) and tetramethylrhodamine system (1:50; PerkinElmer LAS Inc.) |
No primary antibodies were used in negative controls. In double staining protocols, cross contamination controls were also added. Liver staining was quantified in 5-10 randomly taken photomicrographs using standardized settings at 200× magnification by using Mac Biophotonics ImageJ program (Rasband, W.S., ImageJ, U.S. National Institute of Health, Bethesda, MD). Data are expressed as % positive surface area.
Described in the work by: Rashid et al.
Anti-Hs mAb 10E4 recognizes an epitope characterized by mixed N-acetylated/N-sulfated disaccharide units. Intensity of staining is reduced by hypersulfation. Anti-HS mAb 3G10 recognizes HS stubs after heparitinase digestion and stains all HS chains.
VLDL Binding Assay
Liver cryosections were fixed in acetone, followed by incubation with DiI-labeled human VLDL (Kalen Biomedical LLC, Germantown, MD) for 2 hours. HS specificity of this in situ binding assay has been shown by the lack of VLDL binding after heparitinase treatment of the sections.16 Washing steps were performed in TBS. We performed fluorescence microscopy using the Leica DM4000B equipped with the DFC345FX camera, using the LAS software package. For quantification, we obtained six to ten pictures per section at a magnification of 200×. This quantification was performed using the Mac Biophotonics ImageJ program, and scored as mean percentage positive area per field.
Dot‐Blot Assay
Proteins were isolated from liver cryosections of Wistar-22W rats (n=6), Wistar-CKD rats (n=13), and MWF-CKD rats (n=11) using radioimmunoprecipitation buffer with protease inhibitors (sc-24948; Santa Cruz Biotechnology, Dallas, TX). Tissue was lysed on ice, resuspended, and undissolved material was spun down. We determined the protein concentration using the Pierce BCA Protein Assay Kit (product number 23227; Thermo Scientific, Waltham, MA). The polyvinylidene difluoride (PVDF) membrane was activated using methanol. We blotted 100 μl of 1.25 µg/µl of protein on an activated PVDF membrane using a Bio‐Dot (Bio‐Rad, Hercules, CA) device. After blotting, the membrane was dried, incubated with methanol for 1 minute, and washed three times with demineralized water. Thereafter, the membrane was blocked for endogenous peroxidase activity with 3% hydrogen peroxide in water for 10 minutes, followed by overnight blocking with 5% skimmed milk in TBS with approximately 0.05% Tween 20. The membrane was incubated for 1 hour with anti–syndecan‐1 N18 antibody (sc7100; Santa Cruz Biotechnology), followed by rabbit anti‐goat IgG–horseradish peroxidase (HRP) (Dako, Glostrup, Denmark) secondary antibody. Detection and quantification were performed using the Western Lightning Ultra (NEL112001EA; PerkinElmer, Waltham, MA). Data are expressed as fold increase compared with the control group. As a positive control, we spotted recombinant syndecan‐1 on the membrane. Wells without any spotted sample, but incubated with anti–syndecan‐1 and HRP‐labeled secondary antibody, served as negative control.
Western Blotting
For Western blotting, liver homogenates from Wistar-22W rats (n=6), Wistar-CKD rats (n=4), and MWF-CKD rats (n=5) were obtained using nonidet P-40 buffer (0.1% nonidet P-40, 0.4 M sodium chloride [NaCl], 10 mM Tris–hydrogen chloride [pH 8.0], 1 mM EDTA) supplemented with protease and phosphatase inhibitors (Roche). We determined the protein concentration using the Bradford assay (Bio-Rad). Protein (30 μg) was separated by SDS-PAGE and transferred to Amersham Hybond-P PVDF Transfer Membrane (RPN303F; GE Healthcare). The transfer membrane was activated with methanol beforehand. Membranes were blocked in 5% milk in TBS with 0.01% Tween 20 (MilliporeSigma, Burlington, MA) and incubated with rabbit polyclonal anti‐LDLR (1:300 in PBS with 1% BSA; Pab8804; Abnova, Taipei City, Taiwan), rabbit mAb anti–LRP-1 (1:7500 in PBS with 1% BSA; 92544; Abcam, Cambridge, United Kingdom), and rabbit anti-mouse PCSK9 (1:300 in PBS with 1% BSA; 552C; kindly provided by Jayson D. Horton, University of Texas Southwestern Medical Center, Dallas, TX) antibodies. Detection and quantification were performed using the Western Lightning Ultra (NEL112001EA; PerkinElmer). Proteins were visualized using a ChemiDoc XRS+ System using Image Lab software version 5.2.1 (both from Bio-Rad). Data are expressed as fold increase compared with the Wistar-22W control group.
Extraction and Purification of HS from Liver Samples
We collected approximately 30 mg frozen liver tissue from each rat. Five liver samples per group were pooled, resulting in three pooled samples (Wistar-22W, Wistar-CKD, and MWF-CKD). Extraction and purification of HS was performed as described previously.24 Briefly, hepatic tissues were resuspended in 50 mM TBS, 2 mM EDTA, and 6 M urea, and mechanically disrupted with a Potter grinder. After recovery of the supernatant, the pellet was washed again in 50 mM TBS, 2 mM EDTA, and 6 M urea and centrifuged. Both supernatants were pooled and dialyzed against 25 mM Tris, and 5 mM EDTA, pH 7.8. Proteins were then degraded by pronase digestion (2 mg/ml of pronase, final concentration, incubation for 24 hours at 37°C), precipitated by addition of ice-cold TCA (5% vol/vol final concentration), and incubated at 4°C for 1 hour. The samples were centrifuged, and the pellets were treated again with TCA. Supernatants from both TCA treatments were collected, pooled, and supplemented with diethyl ether (50% vol/vol final concentration). After shaking, we discarded the organic upper phase and repeated the diethyl ether washing four times. The pH from the recovered aqueous phase was then adjusted to 7 by addition of 1 M sodium carbonate, and residual diethyl ether was eliminated by leaving the samples overnight in a low-pressure environment. The samples were then applied to a DEAE Sephacel column (2 ml), equilibrated in 20 mM phosphate, pH 6.5. After extensive washing with 20 mM phosphate, 0.3 M NaCl, pH 6.5, glycosaminoglycan (GAG) chains were step eluted with 20 mM phosphate, 1 M NaCl, pH 6.5. Recovered samples were desalted over a PD-10 column, lyophilized, and stored at −20°C before analysis.
HS Disaccharide Analysis
Disaccharide analysis of HS was performed by reverse-phase, ion-pair high-performance liquid chromatography (HPLC). Samples were dissolved in 100 mM sodium acetate, 0.5 mM calcium chloride, pH 7.1, and HS was exhaustively digested into disaccharides by incubation with heparinase I (10 mU; Grampian Enzymes, Orkney, United Kingdom) overnight at 30°C, followed by a second incubation with heparinase II and heparinase III (10 mU each; Grampian Enzymes) for 24 hours at 37°C. Compositional analysis was performed by reverse-phase, ion-pair HPLC, as described previously.24,47 Samples were applied to a Luna 5 μm C18 reverse-phase column (4.6×150 mm; Phenomenex), equilibrated at 0.5 ml/minute in 1.2 mM tetra-N-butylammonium hydrogen sulfate and 8.5% acetonitrile, and then resolved using an NaCl gradient (0–8 mM in 10 minutes, 8–30 mM in 1 minute, 30–56 mM in 11.5 minutes, 56–106 mM in 1.5 minutes, and 106 mM for 6 minutes) calibrated with disaccharide standards (Iduron, Alderley Edge, United Kingdom). We achieved on-line postcolumn disaccharide derivatization by adding 2-cyanoacetamide (0.25%) in sodium hydroxide (0.5%) at a flow rate of 0.16 ml/min, followed by fluorescence detection (excitation, 346 nm; emission, 410 nm). Disaccharide isolation and analysis of each pool was performed twice.
PCSK9 ELISA Competition Assay
We used ELISA to evaluate whether different heparins compete with the binding of PCSK9 to heparin-albumin. For this purpose, MaxiSorp 96-well, flat-bottomed microtiter plates (U96; VWR International, Amsterdam, The Netherlands) were coated overnight in PBS with 1 μg/ml heparin-albumin (HSPG analogue). Heparin-albumin was from Sigma-Aldrich. According to the data sheet, this artificial proteoglycan comprised 4.8 mol heparin per mole of albumin, yielding a protein content of about 55%. After washing in PBS, wells were blocked with 5% BSA in PBS for 1 hour. In separate microtubes, 4 μg/ml recombinant PCSK9 (PC9-H5223; ACROBiosystems, Newark, DE) was prepared. A dilution range of unfractionated heparin (15 kDa; Sigma, Zwijndrecht, The Netherlands) and two different low-molecular-mass variants of heparin, Enoxaparin (Clexane [Rhone-Poulenc Rorer], Paris, France; a low-molecular-mass heparin with molecular mass of approximately 4500 Da) and sulodexide (Alfa Wassermann SpA, Bologna, Italy; composed of 80% fast-moving heparin with molecular mass of approximately 7500 Da and 20% dermatan sulfate), were also prepared in separate Eppendorf tubes. Equal parts of human recombinant PCSK9 with equal parts of heparins (separately for each heparin) were added to the wells and incubated for 1.5 hours. The final concentration of PCSK9 (2 μg/ml) was chosen to achieve an ELISA signal of around 1.5–2.0 OD. The wells were washed again, and rabbit polyclonal anti-human PCSK9 antibody (1:2000 in PBS with 1% BSA; 125251; Abcam) was added for 1 hour. After washing, HRP-labeled goat anti-rabbit Ig (1:1000; DAKO) was added and incubated for 1 hour. The substrate reaction was achieved using 3,3′,5,5′-tetramethylbenzidine substrate (Sigma) for 10 minutes in the dark, and the reaction was stopped by adding 1.5 N sulfuric acid. Absorbance was measured at 450 nm in a microplate reader. All incubations were performed at room temperature at a volume of 100 μl per well. Results are expressed as percentage inhibition: (1−ELISA signal with inhibitor/ELISA signal without inhibitor)×100%.
Statistical Analyses
Analyses were performed using GraphPad version 5 (GraphPad software). One way ANOVA (Kruskal–Wallis test) was used to compare Wistar-22W, Wistar-CKD, and MWF-CKD rats. When significant differences were observed between the means, the Dunn multiple comparison test was as used as post-test to identify which specific means were significant from the others. Data are given as mean±SEM. The paired t test was used to compare clinical characteristics between the groups for normally distributed data. The Wilcoxon, matched-pairs, signed-rank test was used to compare clinical characteristics between the groups for non-normally distributed data. Nonparametric Spearman correlation was used to analyze the association between parameters. For all experiments, a P value of <0.05 was considered statistically significant. The calculation of the P value takes into account the number of comparisons made.
Results
Clinical Parameters in Rat Models for CKD
CKD was induced by uninephrectomy followed by aging in normal, healthy, male Wistar and male MWF rats. Clinical parameters were assessed at baseline (25 weeks) and at the end of the study (48 weeks), which are presented in Table 2. Wistar-CKD and MWF-CKD rats showed an increase in body weight due to normal aging. No changes were observed in the mean arterial pressure (MAP) of Wistar-CKD compared with Wistar-baseline rats, whereas MWF-CKD rats showed increase in MAP at 48 weeks as compared with MWF-baseline rats. Wistar-baseline and MWF-baseline rats did not show differences in MAP. Plasma creatinine was increased in Wistar-CKD and MWF-CKD rats compared with their respective baseline values. Similarly, Wistar-CKD rats showed increased proteinuria compared with Wistar-baseline rats due to aging and unilateral nephrectomy. MWF-baseline rats were proteinuric, which was further raised significantly upon uninephrectomy and aging at 48 weeks. Nonfasted plasma TGs seemed to increase in Wistar-CKD rats upon aging and nephrectomy; however, this did not reach statistical significance. MWF animals showed increased plasma TGs at baseline and these remained high upon aging. Both Wistar-CKD and MWF-CKD rats had significant increases in plasma cholesterol compared with their baseline values. By combining all four groups (Wistar-baseline, Wistar-CKD, MWF-baseline, and MWF-CKD) both plasma creatinine and proteinuria correlated with TC values (r=0.57; 95% CI, 0.34 to 0.73; P≤0.0001; and r=0.73; 95% CI, 0.57 to 0.84; P≤0.0001; respectively). Moreover, both plasma creatinine and proteinuria were positively correlated with TC values when only Wistar-baseline (25 weeks) and Wistar-CKD (48 weeks) measurements were taken into account (r=0.64; 95% CI, 0.33 to 0.83; P=0.0004; and r=0.51; 95% CI, 0.14 to 0.76; P=0.007; respectively). Also, both plasma creatinine and proteinuria positively correlated with TC values when only MWF-baseline and MWF-CKD measurements were taken into account (r=0.84; 95% CI, 0.67 to 0.92; P≤0.0001; and r=0.61; 95% CI, 0.28 to 0.81; P=0.001; respectively), indicating loss of renal function induces hypercholesterolemia in both CKD rat models.
Table 2.
Clinical characteristics of Wistar-baseline (25 weeks n=13), Wistar-CKD (48 weeks, n=14), MWF-baseline (25 weeks, n=13), and MWF-CKD (48 weeks, n=14) rats
| Parameters | Wistar-Baseline (25 weeks) | Wistar End Point; Wistar-CKD (48 Weeks) | P Value | MWF-Baseline (25 weeks) | MWF End Point; MWF-CKD (48 weeks) | P Value | P Value (Wistar-baseline versus MWF-baseline) |
|---|---|---|---|---|---|---|---|
| Body weight (g) | 453±9 | 567±13 | <0.0001 | 365±20 | 454±14 | <0.0001 | <0.005 |
| Systolic BP (mm Hg) | 143±4 | 151±3 | NS | 141±7 | 194±9 | 0.005 | NS |
| Diastolic BP (mm Hg) | 102±4 | 106±3 | NS | 108±5 | 143±9 | 0.01 | NS |
| Mean BP (mm Hg) | 115±4 | 121±3 | NS | 126±2 | 160±9 | 0.003 | NS |
| Proteinuria (mg/24 h) | 9±3 | 57±20 | 0.0002 | 60±7 | 103±7 | 0.002 | <0.0001 |
| Plasma creatinine (μmol/L) | 20±3 | 42±1 | <0.0001 | 17±1 | 118±25 | 0.0005 | NS |
| TGs (mmol/L) | 1.31±0.12 | 1.76±0.34 | NS | 1.66±0.24 | 1.64±0.17 | NS | NS |
| TC (mmol/L) | 2.34±0.13 | 3.59±0.41 | 0.002 | 2.71±0.11 | 3.83±0.19 | 0.008 | NS |
Data are presented as mean±SEM. NS represents nonsignificant differences between the groups.
Plasma Lipid Profiling
To get more insight into cholesterol profiles, we separated plasma lipoproteins in young Wistar-22W, MWF-baseline, and MWF-CKD rats by FPLC. Plasma samples of Wistar-baseline and Wistar-CKD rats were lost and could not be measured. No significant differences were found in VLDLc, LDLc, and HDLc levels when compared between Wistar-22W and MWF-baseline rats. VLDLc, LDLc, and non-HDLc (VLDLc and LDLc) were found to be significantly increased in MWF-CKD compared with MWF-baseline rats (Figure 1), which might indicate an increased amount of remnant lipoproteins, likely due to impaired hepatic clearance. Association analyses revealed VLDLc associated (borderline) with proteinuria (r=0.45; 95% CI, −0.02 to 0.76; P=0.05), and LDLc and non-HDLc associated with proteinuria and plasma creatinine (for LDLc, r=0.51; 95% CI, 0.06 to 0.79; P=0.03; and r=0.67; 95% CI, 0.3 to 0.87; P=0.002; and for non-HDLc, r=0.49; 95% CI, 0.03 to 0.78; P=0.03; and r=0.66; 95% CI, 0.29 to 0.86; P=0.002; respectively).
Figure 1.

Cholesterol profiling by FPLC in Wistar-22W (n=6), MWF-baseline (25 weeks, n=14), and MWF-CKD (48 weeks, n=14) rats. (A) FPLC profiles for plasma cholesterol for Wistar-22W, MWF-baseline, and MWF-CKD rats. The dark lines indicate the mean; the light shades indicate SEM. (B) Fold increase in VLDLc, (C) in LDLc, (D) in HDLc, and (E) in non-HDLc. Non-HDLc was calculated as the sum of VLDLc and LDLc. Data shown as mean±SEM. *P<0.05, **P<0.005, ***P<0.0001.
Expression of Hepatic Lipoprotein Receptors
We next investigated the expression of major hepatic lipoprotein receptors (LDLR, LRP-1, and syndecan-1) by quantitative RT-PCR, immunofluorescence staining, and Western blotting or dot blotting (Supplemental Figure 1). mRNA analyses of Ldlr and Syndecan-1 showed no changes in Wistar-CKD and MWF-CKD rats, whereas Lrp-1 was significantly upregulated in Wistar-CKD and MWF-CKD rats compared with Wistar-22W rats. Immunofluorescence staining of livers for LDLR, LRP-1, and syndecan-1 showed a sinusoidal staining pattern for basal hepatocyte expression, without any change in intensity or distribution of all three groups. Similarly, Western blotting showed no significant changes in total LDLR protein, but significant upregulation of LRP-1 total protein in Wistar-CKD and MWF-CKD rats. Syndecan-1 was analyzed by dot blot, because the presence of HS chains hampered proper electrophoresis migration and transfer in Western blotting, and this also showed no changes between the groups. Altogether, hepatic lipoprotein receptors were not reduced by mild (Wistar-CKD) or more severe (MWF-CKD) progressive renal failure.
Changes in Hepatic PCSK9 Expression Is Associated with Renal Function Loss and TC
Plasma PCSK9 was not significantly different in MWF-CKD compared with MWF-baseline and Wistar-22W rats (Figure 2A), although half of the CKD rats in both groups show plasma PCSK9 levels above control values. Univariate correlation analysis suggested plasma PCSK9 correlated with non-HDLc (r=0.46; 95% CI, −0.02 to 0.77; P=0.05); however, this did not reach statistical significance.
Figure 2.

Plasma and hepatic PCSK9 expression. (A) Measurement of plasma PCSK9 in healthy, untreated Wistar-22W (n=6), MWF-baseline (25 weeks, n=14), and MWF-CKD (48 weeks, n=14) plasma. (B) Fold change in mRNA expression of PCSK9 in livers of Wistar-22W (n=6), Wistar-CKD (48 weeks, n=7), and MWF-CKD (48 weeks, n=7) rats. (C) Measurement of PCSK9 protein by Western blot. (D) Quantification of Western blots. (E) Immunofluorescence staining of hepatic PCSK9. (F) Quantification of immunofluorescence staining of hepatic PCSK9. (G) Univariate correlation analysis of PCSK9 immunofluorescence staining with plasma creatinine. Scale bars in photomicrographs represent 150 µm. *P<0.05.
No significant differences were observed in Pcsk9 mRNA expression and total protein expression in Wistar-CKD and MWF-CKD rats compared with Wistar-22W rats, although increasing tendencies were observed (Figure 2, B–D). However, PCSK9 protein surface expression, measured by immunofluorescence staining, was significantly upregulated in both Wistar-CKD and MWF-CKD rats as compared with Wistar-22W rats (Figure 2, E and F). Interestingly, the distribution of PCSK9 protein in the livers of Wistar-CKD and MWF-CKD rats was strikingly different from Wistar-22W rats (Figure 2E). In Wistar-22W rats, PCSK9 staining was uniform and weak in a cytoplasmic pattern, whereas PCSK9 was increased and especially accumulated in the sinusoids in both CKD groups. We found positive correlations of PCSK9 staining (thus representing the degree of accumulation in the sinusoids) with plasma creatinine (r=0.53; 95% CI, 0.07 to 0.8; P=0.02) (Figure 2G), LDLc (r=0.75; 95% CI, 0.24 to 0.93; P=0.009), and non-HDLc (r=0.75; 95% CI, 0.24 to 0.93; P=0.009). These data might suggest sinusoidal PCSK9 hampers lipoprotein clearance, but without affecting LDLR protein expression.
Profiling of HS GAG Side Chains
Because both PCSK9 and lipoproteins are functional ligands of HS, we evaluated structural changes in hepatic HS by immunofluorescence staining using monoclonal anti-HS antibodies, by HS disaccharide analysis, and by mRNA expression of Ext1 (the major enzyme for HS polymerization). As reported in previous studies, anti-HS mAb-10E4 binds to HS domains containing mixed N-acetylated and N-sulfated disaccharide units.48 The binding of anti-HS mAb-10E4 weakens with both an increase (hypersulfation) and decrease (hyposulfation) in sulfation of HS. Anti-HS mAb-10E4 was found to be expressed in the sinusoids of the liver, most likely the basal cell membranes of the hepatocytes facing the sinusoids, in all three groups. This staining pattern resembled syndecan-1 protein expression, suggesting anti-HS mAb-10E4 recognized the HS chains of syndecan-1. We observed no differences in the expression of 10E4 in between Wistar-22W rats and both CKD groups (Figure 3, A and C).
Figure 3.

Expression of HS by anti-HS mAbs 10E4 and 3G10. Expression and quantification of HS by anti-HS mAbs (A and C) 10E4 and (B and D) 3G10 in livers of Wistar-22W, Wistar-CKD, and MWF-CKD rats. Univariate correlation analysis of HS-3G10 with (E) plasma creatinine and (F) TC. Scale bar represents 150 µm. **P<0.005, ***P<0.0001.
The anti-HS mAb-3G10 reacts with HS neoepitopes containing unsaturated uronic acid residues on HS fragments generated by digesting HS with heparitinase I. Therefore, staining intensity of anti-∆HS reflects the number of heparitinase-digestible cleavage sites in HS.49 Intensity of 3G10 staining was significantly increased in both CKD models compared with Wistar-22W rats (Figure 3, B and D), indicating more heparitinase-digestible cleavage sites. Interestingly, 3G10 expression was positively associated with plasma creatinine (r=0.52; 95% CI, 0.18 to 0.76; P=0.004) and TC levels (r=0.53; 95% CI, 0.19 to 0.76; P=0.003) (Figure 3, E and F).
In a double staining of anti-HS mAb-3G10 with sinusoidal endothelial marker LYVE1, we demonstrate HS to be present on the outer margins of the sinusoids, most likely the basolateral membrane of the hepatocytes, where syndecan-1 is also localized (Figure 4). These data indicate the proteinuric state changed the structure of HS, but not its distribution, in hepatic tissues.
Figure 4.
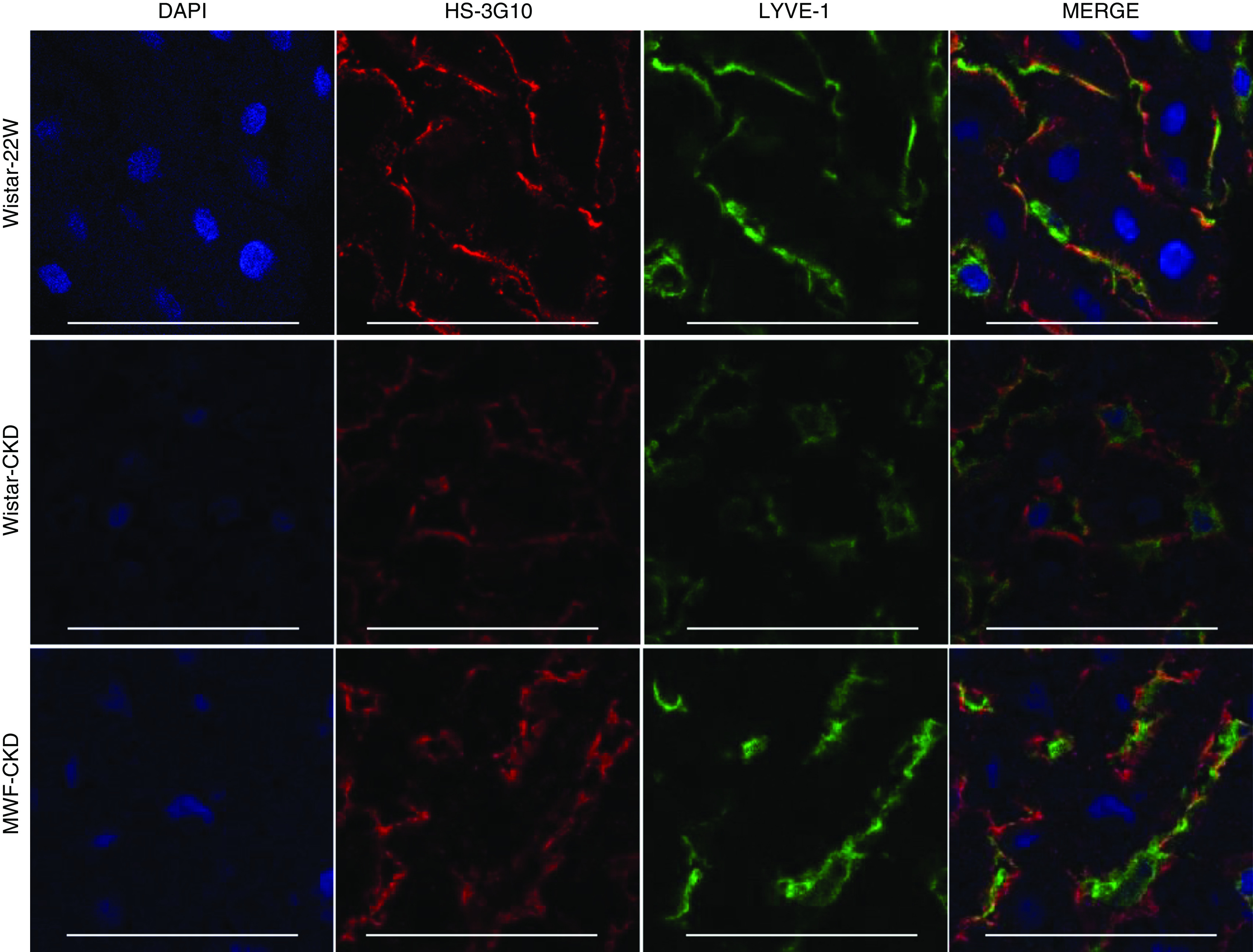
Double staining of HS with sinusoidal endothelial marker LYVE1. Confocal immunofluorescence images of hepatic HS using anti-HS mAb 3G10 with sinusoidal endothelial marker LYVE1. DAPI represents nuclear staining. Scale bars in photomicrographs represent 50 µm. *P<0.05, **P<0.01. DAPI, 4′,6-diamidino-2-phenylindole.
To investigate the structural changes in HS side chains in detail, disaccharide analysis of hepatic HS was performed by HPLC after exhaustive heparinase/heparitinase digestion. The results did not highlight any gross changes in the disaccharide composition of hepatic HS in both CKD groups compared with Wistar-22W group (Figure 5A). However, the amount of HS-derived uronic acid was found to be higher in both CKD groups compared with Wistar-22W rats (Figure 5B), in line with 3G10 immunofluorescence staining data suggesting longer HS chains. Finally, we measured mRNA transcript coding for Ext1, the major enzyme for HS polymerization. Interestingly, EXT-1 mRNA expression was significantly increased in both CKD groups (Figure 5C). Altogether, unchanged syndecan-1 and anti-HS mAb-10E4 staining, together with enhanced anti-∆HS mAb-3G10 staining, increased HS uronic acid content and increased Ext1 expression, strongly indicating a longer HS chain length in the livers of CKD rats, without major changes in HS sulfation patterns. In addition, Ext1 mRNA expression positively associated with plasma creatinine (r=0.52; 95% CI, 0.06 to 0.8; P=0.03), TC (r=0.66; 95% CI, 0.27 to 0.87; P=0.003) (Figure 5D), and PCSK9 total protein (r=0.84; 95% CI, 0.55 to 0.95; P=0.0001) (Figure 5E) and PCSK9 mRNA levels (r=0.53; 95% CI, 0.08 to 0.8; P=0.02), suggesting a relationship between HS chain elongation and PCSK9 sinusoidal expression.
Figure 5.

Elongated hepatic HS in both CKD models correlates with total cholesterol, PCSK9 plasma levels and PCSK9 binding affinity. (A) Hepatic HS disaccharides are expressed as the percentage of total disaccharides. (B) Total quantity of HS in the livers of healthy Wistar-22W, Wistar-CKD, and MWF-CKD rats measured by HPLC and expressed in nanograms per milligram of tissue. (C) Fold change in mRNA expression of EXT1 in the livers of Wistar-22W (n=6), Wistar-CKD (n=7), and MWF-CKD (n=6) rats. (D and E) Univariate correlation analysis showing associations between (D) EXT1 mRNA with TC, and (E) EXT1 mRNA with PCSK9 total protein. *P<0.05. (F) Competition of heparinoids with PCSK9 binding to immobilized heparin-albumin is critically dependent on polysaccharide chain length. LMW, low molecular weight.
PCSK9 Interaction with Heparins Depends on Chain Length
To investigate the eventual effect of HS chain length on the binding with PCSK9, we performed a PCSK9 competition experiment to immobilized heparin-albumin. Competition was done using unfractionated heparin (15 kDa), Sulodexide (7.5 kDa) and low-molecular-mass heparin (Enoxaparin; 4.5 kDa). We observed that decreasing heparin chain length from 15 to 7.5 and to 4.5 kDa decreased its ability to compete with heparin-albumin for an interaction with PCSK9 (Figure 5F). Thus, longer heparin (substitute for HS) chains bind PCSK9 with higher efficiency.
To investigate an association of PCSK9 with elongated HS in vivo, we performed confocal immunofluorescence staining of PCSK9 and anti-HS mAb-3G10. As shown in Figure 6, PCSK9 partially colocalizes to HS in the hepatic sinusoidal regions. These observations were exclusively seen in both CKD groups endowed with hyperelongated HS and not in Wistar-22W rats lacking these hyperelongated modifications (Figure 6).
Figure 6.
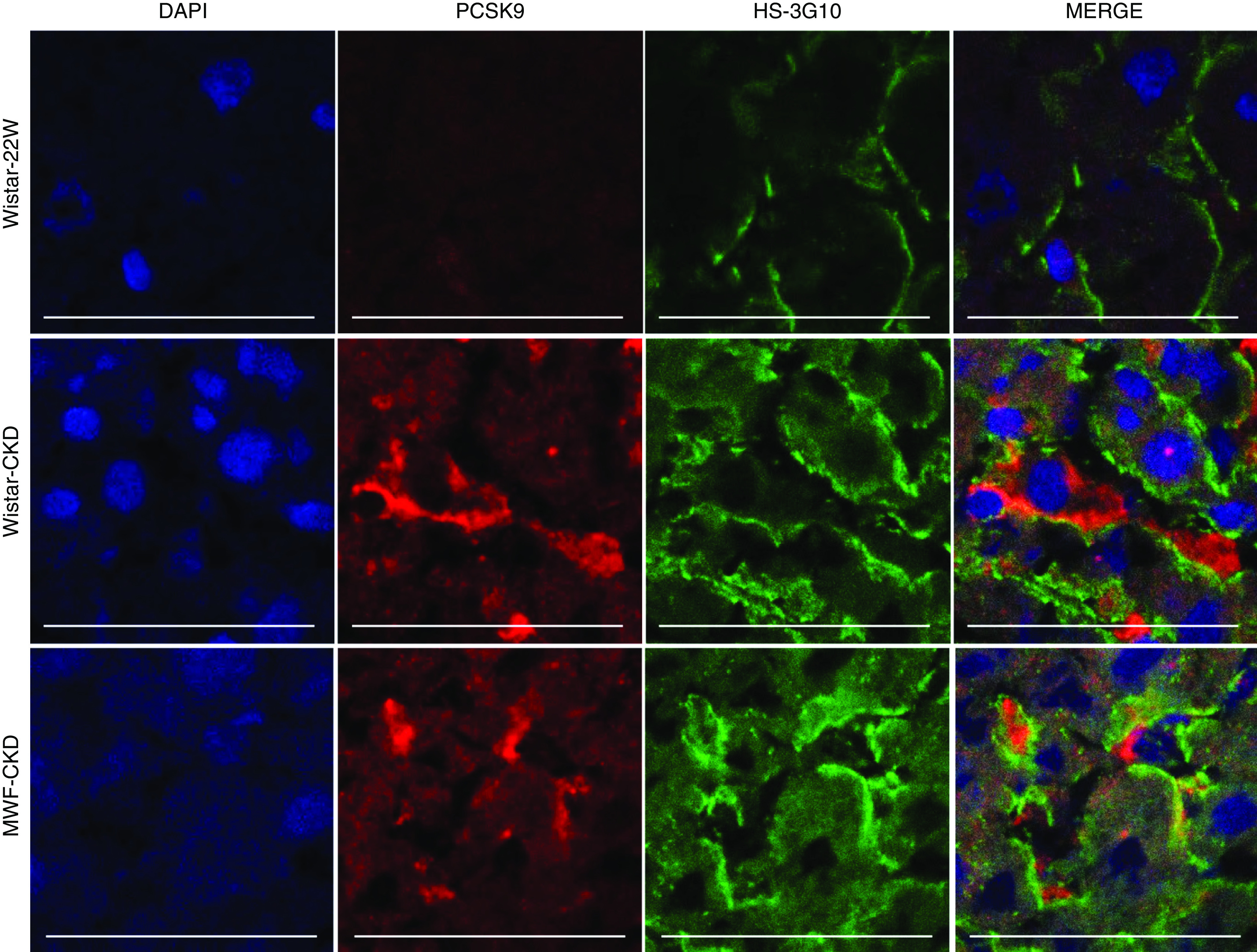
Confocal immunofluorescence double stainings of PCSK9 (red) with anti-HS mAb 3G10 (green). PCSK9 partly colocalizes with HS at the hepatic sinusoids in Wistar-CKD and MWF-CKD rats, but this is absent in Wistar-22W rats. Scale bars represent 50 μm. DAPI, 4′,6-diamidino-2-phenylindole.
Lipoprotein Binding Capacity Is Reduced in CKD Conditions
Lastly, because the HS chains of syndecan-1 are crucially important in hepatic lipoprotein clearance, we evaluated the binding capacity of DiI-labeled VLDL particles with liver sections from the Wistar-22W group and both CKD groups. In the Wistar-22W group, binding was seen in a sinusoidal pattern (Figure 7), comparable to the distribution of syndecan-1 and the HS mAbs 10E4 and 3G10 (Figure 3). Heparitinase pretreatment of the sections abolished all DiI-VLDL binding, demonstrating the HS-dependent binding of DiI-VLDL to the liver sections (not shown). Importantly, the DiI-VLDL binding capacity of hepatic HS was reduced in Wistar-CKD and MWF-CKD rats compared with Wistar-22W rats; this reached significance in Wistar-CKD rats. In addition, VLDL binding was negatively associated with LDLc (r=−0.5; 95% CI, −0.8 to −0.06; P=0.03) and non-HDLc levels (r=−0.5; 95% CI, −0.79 to −0.04; P=0.03), strongly suggesting that HS-dependent lipoprotein binding and hepatic clearance is reduced as renal disease progresses.
Figure 7.

Reduced VLDL binding in liver sections of CKD rats. (A) VLDL binding assay on liver sections of Wistar-22W, Wistar-CKD, and MWF-CKD rats. (B) Quantification of VLDL binding. Scale bars represent 150 µm. *P<0.05.
Discussion
The major findings of this study are the sinusoidal accumulation of PCSK9 and production of hyperelongated HS chains in both models of CKD. Furthermore, the interaction of PCSK9 with HS/heparin critically depends on the polysaccharide chain length. Lipoprotein binding capacity of hepatic HSPGs was reduced in both CKD groups, and associations with renal function were observed with TC, PCSK9, and HS size changes. Altogether, these data indicate a novel mechanism behind hypercholesterolemia in progressive CKD, which involves PCSK9 and elongated hepatic HS chains.
Our study shows, for the very first time, that CKD can modify (elongate) hepatic HS chains, and that elongated HS chains can bind to PCSK9 more effectively. Because HSPGs facilitate PCSK9-mediated LDLR degradation, we initially assumed that increased HS-PCSK9 interaction would promote LDLR degradation in both CKD models causing hypercholesterolemia. However, LDLR protein levels remained unchanged. Interestingly, we found an accumulation of PCSK9 in the sinusoids, along with the production of hyperelongated HS chains in both CKD models. On the basis of these intriguing observations, we designed a model suggesting a potential novel mechanism behind dyslipidemia in CKD.
PCSK9 can self-associate to dimers and multimers via their catalytic domain in a concentration-, pH-, and temperature-dependent manner, and also at concentrations close to that in human plasma.50,51 Altogether, we hypothesize that PCSK9 homeostasis is affected by the structural changes taking place on HS in CKD. As shown in Figure 8B, elongated HS chains (via their trisulfated domains) provide a platform to effectively bind several PCSK9 molecules at a time (via the PCSK9 prodomain), increasing PCSK9 concentrations at the HS site and facilitating PCSK9 dimerization (via their catalytic domains). This results in the formation of complexes involving HSPG and PCSK9 dimers at the surface of hepatocytes. It is well recognized that GAGs, especially HS through their sulfated domains, efficiently bind to protein dimers/oligomers.52–54 Furthermore, many studies have shown GAGs could actually induce ligand oligomerization, with significant consequences for their biologic activities.55–57
Figure 8.
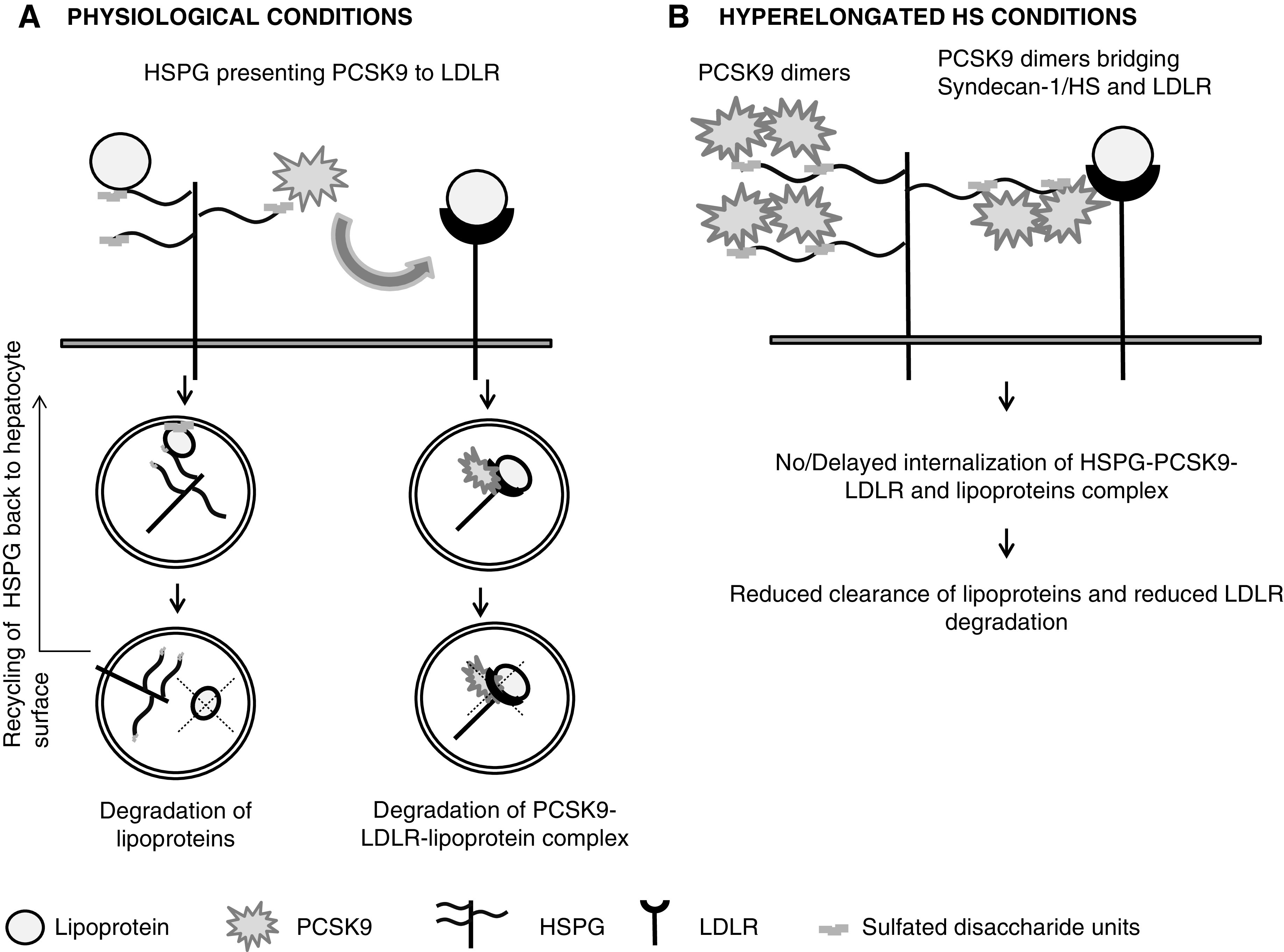
Proposed model representing potential novel mechanism for dyslipidemia in progressive CKD. (A) HSPG presenting PCSK9 to LDLR in normal conditions. (B) Formation of HSPG-PCSK9 dimer–LDLR complex due to hyperelongated state of HS in CKD.
Therefore, we propose the PCSK9 dimer, attached at two sulfated domains of HS, becomes efficiently attached or immobilized with the HS side chain. Immobilization of HSPG-bound ligands has been previously observed with basic fibroblast growth factor.58–60 Because the HS binding domain in PCSK9 differs from the LDLR binding domain,34,61 PCSK9 dimers attached to HS bind to a neighboring LDLR, forming LDLR-PCSK9- HSPG (most likely syndecan-1) complexes. Such complex formations have been previously reported for other ligands.62–64 These complexes might either become immobilized on the surface of hepatocytes or their internalization might be very delayed. The hypothesis of complex formation is further supported by the observation of PCSK9 positivity in liver cryosections, even after successful treatment with heparitinase (Figure 6) or heparin competition (data not shown). These data suggest endogenous PCSK9 bound to hepatic HS remains attached to other membrane receptors, such as LDLR.
These phenomena compromise the functionality of HSPGs and LDLR and prevent lipoprotein clearance, causing dyslipidemia in CKD. This might explain the absence of PCSK9-mediated LDLR degradation and PCSK9 clusters in the hepatic sinusoids in both CKD groups. In line with our findings, Meyers et al. 65 reported that proteoglycans secreted from statin-exposed monkey aortic smooth muscle cells showed longer HS chains with reduced LDL binding.66 Reduced HS binding to (V)LDL, in this case, could be due to steric hindrance by the immobilized PCSK9 dimer.
Significant upregulation of LRP-1 mRNA and protein levels, without improvements in lipoprotein values, is another intriguing finding from our study. The increase in LRP-1 could be due to the effects of LDLc, as observed in the study by Llorente-Cortés et al. 67–69 However, no beneficial effects on lipoprotein levels suggests a dysfunctional LRP-1. Canuel et al. 70 previously reported that PCSK9 can also induce LRP-1 degradation, similarly to LDLR degradation, however, only in the absence of LDLR. Therefore, in our models of CKD where hepatic HSPGs are hyperelongated, PCSK9-HSPG forms a complex with LDLR first. When all LDLRs are complexed with PCSK9-HSPG, then LRP-1-PCSK9-HSPG complexes are formed. This phenomenon might make LRP-1 nonfunctional despite its significant upregulation.
Clinical trials with the PCSK9 mAbs alirocumab and evolocumab, in both CKD and non-CKD populations, showed a reduction in LDLc of 45%–60% and 60%–70% when used as monotherapy or in combination with statins and ezetimibe, respectively, along with a reduction in TG of 35%–45%.71–79 Similarly, treatment with small-interfering RNA against PCSK9 (inclisiran) reduced LDLc levels by >50%.80 Such an effective reduction in LDLc and TG is possible when the complex formation of both LDLR and HSPG by PCSK9 is prevented, which is in line with our model/hypothesis. Heparinoids might be an economic alternative to expensive PCSK9 inhibitors. Nevertheless, further studies are essential to validate specificity, long-term use, and safety of heparinoids.81,82 Furthermore, serum PCSK9 has been reported to be a key player in metabolic dyslipidemia (diabetes, metabolic conditions, and in the obese population).83–87 Therefore, blocking PCSK9, irrespective of changes in hepatic HSPG, might be beneficial in these populations as well. The extent of LDLc modification in metabolic conditions, however, needs further investigation to understand if these populations are equally benefited by blocking of PCSK9.
Although statins have proved unequivocally effective in reducing plasma cholesterol levels, not all patients with renal disorders benefit from them.3,9,88–90 Further, statins have been recently reported to increase PCSK9 levels and LDLR levels by suppression of IDOL (inducible degrader of LDLR) in two independent studies.91,92 Our study suggests the statin-insensitive population might have hyperelongated hepatic HPSGs. In such conditions, regardless of the beneficial effects of statins, the complex formation between HSPG, PCSK9, and LDLR makes both lipoprotein receptors nonfunctional. Targeting PCSK9 might also provide an additional benefit in the statin-insensitive population by rescuing both HSPGs and LDLRs from lipoprotein clearance.
In this study, we did not investigate the factor(s) modifying hepatic HSPGs. TGF-β and angiotensin II, important renal factors, are known to hyperelongate GAG chains in vascular smooth muscle cells and human SV40-transformed podocytes, respectively.66,93–95 Similarly, oxidative stress is known to enhance total proteoglycan synthesis and GAG chain elongation in vascular smooth muscle cells.66,96 Therefore, effects of these factors on hepatic HSPGs and molecules targeting their reduction/inhibition need to be further studied, because proteoglycan metabolism in vascular tissues is markedly different from that in hepatic tissues.97,98 Further, inhibition of the TGF-β signaling pathway requires vigilant consideration because it can disrupt normal physiologic functions.66,94
This study provides novel insight into the biologic mechanism of reduced hepatic lipoprotein clearance leading to hypercholesterolemia in CKD. It also reveals prospects for future investigation on the HS-PCSK9 interaction and development of novel heparin mimetics targeting modification of this HS-PCSK9 interaction.
Disclosures
S.J.L. Bakker reports receiving research funding from Astellas Pharma and Chiesi; and being a scientific advisor for or member of the Dutch Health Council and the scientific board of the Dutch Kidney Foundation. R.R. Vivès reports being a member of the Centre National de la Recherche Scientifique (CNRS) scientific network Groupement De Recherche Structure, Fonction et Régulation des Glycosaminoglycanes (GDR GAG) 3739 (GDR GAG 3739). All remaining authors have nothing to disclose.
Funding
This work was supported by a Universitair Medisch Centrum Groningen, Graduate School of Medical Sciences personal talent grant (to P. Shrestha), a Jan Kornelis de Cock Stichting personal grant (to P. Shrestha), by CNRS grants, Groupement De Recherche Structure, Fonction et Régulation des Glycosaminoglycanes (GDR GAG) 3739, Investissements d’avenir grant ANR-15-IDEX-02 through the Glyco@Alps program, and Agence Nationale de la Recherche grant ANR-17-CE11-0040.
Supplementary Material
Acknowledgments
The authors are grateful to Dr. Jayson D. Horton (University of Texas Southwestern Medical Center, Dallas, TX) for providing rabbit polyclonal antibody against mouse PCSK9 (552C). The authors thank Mr. Albert Gerding for helping with FPLC.
Dr. J. van den Born, Dr. B. van de Sluis, and Ms. P. Shrestha designed the study; Dr. A. Klooster carried out animal experiments; Ms. P. Shrestha, Dr. S. Adepu, Dr. R.R. Vivès, Ms. R. El Masri, Dr. F. Kaptein, and Ms. W. Dam carried out the majority of experiments; Ms. P. Shrestha and Dr. S. Adepu made the figures; Ms. P. Shrestha prepared the paper; Dr. J. van den Born, Dr. S.J.L. Bakker, Dr. H. van Goor, and Dr. B. van de Sluis revised the paper; and all authors approved the final version of the manuscript.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020091376/-/DCSupplemental.
Supplemental Figure 1. Expression of hepatic lipoprotein receptors: LDLR, LRP-1 and syndecan-1 in Wistar-22W, Wistar-CKD and MWF-CKD rats.
Supplemental Table 1. Primer sequences used for qRT-PCR.
References
- 1. Weiner DE, Tighiouart H, Amin MG, Stark PC, MacLeod B, Griffith JL, et al.: Chronic kidney disease as a risk factor for cardiovascular disease and all-cause mortality: A pooled analysis of community-based studies. J Am Soc Nephrol 15: 1307–1315, 2004. [DOI] [PubMed] [Google Scholar]
- 2. Cases A, Coll E: Dyslipidemia and the progression of renal disease in chronic renal failure patients. Kidney Int Suppl 68: S87–S93, 2005. [DOI] [PubMed] [Google Scholar]
- 3. Tsimihodimos V, Mitrogianni Z, Elisaf M: Dyslipidemia associated with chronic kidney disease. Open Cardiovasc Med J 5: 41–48, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tedla FM, Brar A, Browne R, Brown C: Hypertension in chronic kidney disease: Navigating the evidence. Int J Hypertens 2011: 132405, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. GBD Chronic Kidney Disease Collaboration: Global, regional, and national burden of chronic kidney disease, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 395: 709–733, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thompson S, James M, Wiebe N, Hemmelgarn B, Manns B, Klarenbach S, et al.; Alberta Kidney Disease Network: Cause of death in patients with reduced kidney function. J Am Soc Nephrol 26: 2504–2511, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shrestha P, van de Sluis B, Dullaart RPF, van den Born J: Novel aspects of PCSK9 and lipoprotein receptors in renal disease-related dyslipidemia. Cell Signal 55: 53–64, 2019. [DOI] [PubMed] [Google Scholar]
- 8. Collins AJ, Li S, Gilbertson DT, Liu J, Chen SC, Herzog CA: Chronic kidney disease and cardiovascular disease in the Medicare population. Kidney Int Suppl 64: S24–S31, 2003. [DOI] [PubMed] [Google Scholar]
- 9. Mikolasevic I, Žutelija M, Mavrinac V, Orlic L: Dyslipidemia in patients with chronic kidney disease: Etiology and management. Int J Nephrol Renovasc Dis 10: 35–45, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gofman JW, Lindgren F: The role of lipids and lipoproteins in atherosclerosis. Science 111: 166–171, 1950. [DOI] [PubMed] [Google Scholar]
- 11. Kaysen GA: Dyslipidemia in chronic kidney disease: Causes and consequences. Kidney Int 70: 55–58, 2006. [Google Scholar]
- 12. Maxwell KN, Breslow JL: Proprotein convertase subtilisin kexin 9: The third locus implicated in autosomal dominant hypercholesterolemia. Curr Opin Lipidol 16: 167–172, 2005. [DOI] [PubMed] [Google Scholar]
- 13. Maxwell KN, Fisher EA, Breslow JL: Overexpression of PCSK9 accelerates the degradation of the LDLR in a post-endoplasmic reticulum compartment. Proc Natl Acad Sci U S A 102: 2069–2074, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lagace TA: PCSK9 and LDLR degradation: Regulatory mechanisms in circulation and in cells. Curr Opin Lipidol 25: 387–393, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Poirier S, Hamouda HA, Villeneuve L, Demers A, Mayer G: Trafficking dynamics of PCSK9-induced LDLR degradation: Focus on human PCSK9 mutations and C-terminal domain. PLoS One 11: e0157230, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Adepu S, Katta K, Tietge UJ, Kwakernaak AJ, Dam W, van Goor H, et al.: Hepatic syndecan-1 changes associate with dyslipidemia after renal transplantation. Am J Transplant 14: 2328–2338, 2014. [DOI] [PubMed] [Google Scholar]
- 17. Stanford KI, Bishop JR, Foley EM, Gonzales JC, Niesman IR, Witztum JL, et al.: Syndecan-1 is the primary heparan sulfate proteoglycan mediating hepatic clearance of triglyceride-rich lipoproteins in mice. J Clin Invest 119: 3236–3245, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Christianson HC, Belting M: Heparan sulfate proteoglycan as a cell-surface endocytosis receptor. Matrix Biol 35: 51–55, 2014. [DOI] [PubMed] [Google Scholar]
- 19. Wilsie LC, Gonzales AM, Orlando RA: Syndecan-1 mediates internalization of apoE-VLDL through a low density lipoprotein receptor-related protein (LRP)-independent, non-clathrin-mediated pathway. Lipids Health Dis 5: 23, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fuki IV, Kuhn KM, Lomazov IR, Rothman VL, Tuszynski GP, Iozzo RV, et al.: The syndecan family of proteoglycans. Novel receptors mediating internalization of atherogenic lipoproteins in vitro . J Clin Invest 100: 1611–1622, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van Kerkhof P, Lee J, McCormick L, Tetrault E, Lu W, Schoenfish M, et al.: Sorting nexin 17 facilitates LRP recycling in the early endosome. EMBO J 24: 2851–2861, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bartuzi P, Billadeau DD, Favier R, Rong S, Dekker D, Fedoseienko A, et al.: CCC- and WASH-mediated endosomal sorting of LDLR is required for normal clearance of circulating LDL. Nat Commun 7: 10961, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sarrazin S, Lamanna WC, Esko JD: Heparan sulfate proteoglycans. Cold Spring Harb Perspect Biol 3: 1–33, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hijmans RS, Shrestha P, Sarpong KA, Yazdani S, El Masri R, de Jong WHA, et al.: High sodium diet converts renal proteoglycans into pro-inflammatory mediators in rats. PLoS One 12: e0178940, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Celie JWAM, Rutjes NW, Keuning ED, Soininen R, Heljasvaara R, Pihlajaniemi T, et al.: Subendothelial heparan sulfate proteoglycans become major L-selectin and monocyte chemoattractant protein-1 ligands upon renal ischemia/reperfusion. Am J Pathol 170: 1865–1878, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thomas G, Clayton A, Thomas J, Davies M, Steadman R: Structural and functional changes in heparan sulfate proteoglycan expression associated with the myofibroblastic phenotype. Am J Pathol 162: 977–989, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gandhi NS, Mancera RL: The structure of glycosaminoglycans and their interactions with proteins. Chem Biol Drug Des 72: 455–482, 2008. [DOI] [PubMed] [Google Scholar]
- 28. Stopschinski BE, Holmes BB, Miller GM, Manon VA, Vaquer-Alicea J, Prueitt WL, et al.: Specific glycosaminoglycan chain length and sulfation patterns are required for cell uptake of tau versus α-synuclein and β-amyloid aggregates. J Biol Chem 293: 10826–10840, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Katta K, Boersema M, Adepu S, Rienstra H, Celie JWAM, Mencke R, et al.: Renal heparan sulfate proteoglycans modulate fibroblast growth factor 2 signaling in experimental chronic transplant dysfunction. Am J Pathol 183: 1571–1584, 2013. [DOI] [PubMed] [Google Scholar]
- 30. Celie JWAM, Reijmers RM, Slot EM, Beelen RHJ, Spaargaren M, Ter Wee PM, et al.: Tubulointerstitial heparan sulfate proteoglycan changes in human renal diseases correlate with leukocyte influx and proteinuria. Am J Physiol Renal Physiol 294: F253–F263, 2008. [DOI] [PubMed] [Google Scholar]
- 31. Foley EM, Esko JD: Hepatic heparan sulfate proteoglycans and endocytic clearance of triglyceride-rich lipoproteins. Prog Mol Biol Transl Sci 93: 213–233, 2010. [DOI] [PubMed] [Google Scholar]
- 32. Foley EM, Gordts PLSM, Stanford KI, Gonzales JC, Lawrence R, Stoddard N, et al.: Hepatic remnant lipoprotein clearance by heparan sulfate proteoglycans and low-density lipoprotein receptors depend on dietary conditions in mice. Arterioscler Thromb Vasc Biol 33: 2065–2074, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Al-Haideri M, Goldberg IJ, Galeano NF, Gleeson A, Vogel T, Gorecki M, et al.: Heparan sulfate proteoglycan-mediated uptake of apolipoprotein E-triglyceride-rich lipoprotein particles: A major pathway at physiological particle concentrations. Biochemistry 36: 12766–12772, 1997. [DOI] [PubMed] [Google Scholar]
- 34. Gustafsen C, Olsen D, Vilstrup J, Lund S, Reinhardt A, Wellner N, et al.: Heparan sulfate proteoglycans present PCSK9 to the LDL receptor. Nat Commun 8: 503, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. van de Sluis B, Wijers M, Herz J: News on the molecular regulation and function of hepatic low-density lipoprotein receptor and LDLR-related protein 1. Curr Opin Lipidol 28: 241–247, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lambert G, Sjouke B, Choque B, Kastelein JJP, Hovingh GK: The PCSK9 decade. J Lipid Res 53: 2515–2524, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Haas ME, Levenson AE, Sun X, Liao WH, Rutkowski JM, de Ferranti SD, et al.: The role of proprotein convertase subtilisin/kexin type 9 in nephrotic syndrome-associated hypercholesterolemia. Circulation 134: 61–72, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sucajtys-Szulc E, Szolkiewicz M, Swierczynski J, Rutkowski B: Up-regulation of liver Pcsk9 gene expression as a possible cause of hypercholesterolemia in experimental chronic renal failure. Mol Cell Biochem 411: 281–287, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sucajtys-Szulc E, Szolkiewicz M, Swierczynski J, Rutkowski B: Up-regulation of Hnf1α gene expression in the liver of rats with experimentally induced chronic renal failure - a possible link between circulating PCSK9 and triacylglycerol concentrations. Atherosclerosis 248: 17–26, 2016. [DOI] [PubMed] [Google Scholar]
- 40. Konarzewski M, Szolkiewicz M, Sucajtys-Szulc E, Blaszak J, Lizakowski S, Swierczynski J, et al.: Elevated circulating PCSK-9 concentration in renal failure patients is corrected by renal replacement therapy. Am J Nephrol 40: 157–163, 2014. [DOI] [PubMed] [Google Scholar]
- 41. Kwakernaak AJ, Lambert G, Slagman MC, Waanders F, Laverman GD, Petrides F, et al.: Proprotein convertase subtilisin-kexin type 9 is elevated in proteinuric subjects: Relationship with lipoprotein response to antiproteinuric treatment. Atherosclerosis 226: 459–465, 2013. [DOI] [PubMed] [Google Scholar]
- 42. Jin K, Park BS, Kim YW, Vaziri ND: Plasma PCSK9 in nephrotic syndrome and in peritoneal dialysis: A cross-sectional study. Am J Kidney Dis 63: 584–589, 2014. [DOI] [PubMed] [Google Scholar]
- 43. Sabatine MS: PCSK9 inhibitors: Clinical evidence and implementation. Nat Rev Cardiol 16: 155–165, 2019. [DOI] [PubMed] [Google Scholar]
- 44. Shen H, Feng S, Lu Y, Jiang L, Yang T, Wang Z: Correlation between plasma proprotein convertase subtilisin/kexin type 9 and blood lipids in patients with newly diagnosed primary nephrotic syndrome. Ren Fail 42: 405–412, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lim BJ, Yang HC, Fogo AB: Animal models of regression/progression of kidney disease. Drug Discov Today Dis Models 11: 45–51, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wijers M, Zanoni P, Liv N, Vos DY, Jäckstein MY, Smit M, et al.: The hepatic WASH complex is required for efficient plasma LDL and HDL cholesterol clearance. JCI Insight 4: e126462, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ledin J, Staatz W, Li JP, Götte M, Selleck S, Kjellén L, et al.: Heparan sulfate structure in mice with genetically modified heparan sulfate production. J Biol Chem 279: 42732–42741, 2004. [DOI] [PubMed] [Google Scholar]
- 48. van den Born J, Salmivirta K, Henttinen T, Ostman N, Ishimaru T, Miyaura S, et al.: Novel heparan sulfate structures revealed by monoclonal antibodies. J Biol Chem 280: 20516–20523, 2005. [DOI] [PubMed] [Google Scholar]
- 49. David G, Bai XM, Van der Schueren B, Cassiman JJ, Van den Berghe H: Developmental changes in heparan sulfate expression: In situ detection with mAbs. J Cell Biol 119: 961–975, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fan D, Yancey PG, Qiu S, Ding L, Weeber EJ, Linton MF, et al.: Self-association of human PCSK9 correlates with its LDLR-degrading activity. Biochemistry 47: 1631–1639, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lagace TA, Curtis DE, Garuti R, McNutt MC, Park SW, Prather HB, et al.: Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J Clin Invest 116: 2995–3005, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Collins LE, Troeberg L: Heparan sulfate as a regulator of inflammation and immunity. J Leukoc Biol 105: 81–92, 2019. [DOI] [PubMed] [Google Scholar]
- 53. Liang WG, Triandafillou CG, Huang TY, Zulueta MM, Banerjee S, Dinner AR, et al.: Structural basis for oligomerization and glycosaminoglycan binding of CCL5 and CCL3. Proc Natl Acad Sci U S A 113: 5000–5005, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Esko JD, Linhardt RJ: Proteins that bind sulfated glycosaminoglycans. In: Essentials of Glycobiology, edited by Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, et al., Cold Spring Harbor, NY, Cold Spring Harbor Laboratory Press, 2009. [PubMed] [Google Scholar]
- 55. Vivès RR, Sadir R, Imberty A, Rencurosi A, Lortat-Jacob H: A kinetics and modeling study of RANTES(9-68) binding to heparin reveals a mechanism of cooperative oligomerization. Biochemistry 41: 14779–14789, 2002. [DOI] [PubMed] [Google Scholar]
- 56. Proudfoot AEI, Handel TM, Johnson Z, Lau EK, LiWang P, Clark-Lewis I, et al.: Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc Natl Acad Sci U S A 100: 1885–1890, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Salanga CL, Handel TM: Chemokine oligomerization and interactions with receptors and glycosaminoglycans: The role of structural dynamics in function. Exp Cell Res 317: 590–601, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jackson RL, Busch SJ, Cardin AD: Glycosaminoglycans: Molecular properties, protein interactions, and role in physiological processes. Physiol Rev 71: 481–539, 1991. [DOI] [PubMed] [Google Scholar]
- 59. Koenig A, Norgard-Sumnicht K, Linhardt R, Varki A: Differential interactions of heparin and heparan sulfate glycosaminoglycans with the selectins. Implications for the use of unfractionated and low molecular weight heparins as therapeutic agents. J Clin Invest 101: 877–889, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li M, Yang S, Xu D: Heparan sulfate regulates the structure and function of osteoprotegerin in osteoclastogenesis. J Biol Chem 291: 24160–24171, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kwon HJ, Lagace TA, McNutt MC, Horton JD, Deisenhofer J: Molecular basis for LDL receptor recognition by PCSK9. Proc Natl Acad Sci U S A 105: 1820–1825, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Park PW, Reizes O, Bernfield M: Cell surface heparan sulfate proteoglycans: Selective regulators of ligand-receptor encounters. J Biol Chem 275: 29923–29926, 2000. [DOI] [PubMed] [Google Scholar]
- 63. Faham S, Hileman RE, Fromm JR, Linhardt RJ, Rees DC: Heparin structure and interactions with basic fibroblast growth factor. Science 271: 1116–1120, 1996. [DOI] [PubMed] [Google Scholar]
- 64. Kan M, Wang F, To B, Gabriel JL, McKeehan WL: Divalent cations and heparin/heparan sulfate cooperate to control assembly and activity of the fibroblast growth factor receptor complex. J Biol Chem 271: 26143–26148, 1996. [DOI] [PubMed] [Google Scholar]
- 65. Meyers CD, Tannock LR, Wight TN, Chait A: Statin-exposed vascular smooth muscle cells secrete proteoglycans with decreased binding affinity for LDL. J Lipid Res 44: 2152–2160, 2003. [DOI] [PubMed] [Google Scholar]
- 66. Yang SNY, Burch ML, Getachew R, Ballinger ML, Osman N, Little PJ: Growth factor-mediated hyper-elongation of glycosaminoglycan chains on biglycan requires transcription and translation. Arch Physiol Biochem 115: 147–154, 2009. [DOI] [PubMed] [Google Scholar]
- 67. Llorente-Cortés V, Royo T, Otero-Viñas M, Berrozpe M, Badimon L: Sterol regulatory element binding proteins downregulate LDL receptor-related protein (LRP1) expression and LRP1-mediated aggregated LDL uptake by human macrophages. Cardiovasc Res 74: 526–536, 2007. [DOI] [PubMed] [Google Scholar]
- 68. Llorente-Cortés V, Costales P, Bernués J, Camino-Lopez S, Badimon L: Sterol regulatory element-binding protein-2 negatively regulates low density lipoprotein receptor-related protein transcription. J Mol Biol 359: 950–960, 2006. [DOI] [PubMed] [Google Scholar]
- 69. Llorente-Cortés V, Otero-Viñas M, Sánchez S, Rodríguez C, Badimon L: Low-density lipoprotein upregulates low-density lipoprotein receptor-related protein expression in vascular smooth muscle cells: Possible involvement of sterol regulatory element binding protein-2-dependent mechanism. Circulation 106: 3104–3110, 2002. [DOI] [PubMed] [Google Scholar]
- 70. Canuel M, Sun X, Asselin MC, Paramithiotis E, Prat A, Seidah NG: Proprotein convertase subtilisin/kexin type 9 (PCSK9) can mediate degradation of the low density lipoprotein receptor-related protein 1 (LRP-1). PLoS One 8: e64145, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Koren MJ, Lundqvist P, Bolognese M, Neutel JM, Monsalvo ML, Yang J, et al.; MENDEL-2 Investigators: Anti-PCSK9 monotherapy for hypercholesterolemia: The MENDEL-2 randomized, controlled phase III clinical trial of evolocumab. J Am Coll Cardiol 63: 2531–2540, 2014. [DOI] [PubMed] [Google Scholar]
- 72. Farnier M, Gaudet D, Valcheva V, Minini P, Miller K, Cariou B: Efficacy of alirocumab in high cardiovascular risk populations with or without heterozygous familial hypercholesterolemia: Pooled analysis of eight ODYSSEY Phase 3 clinical program trials. Int J Cardiol 223: 750–757, 2016. [DOI] [PubMed] [Google Scholar]
- 73. Robinson JG, Farnier M, Krempf M, Bergeron J, Luc G, Averna M, et al.; ODYSSEY LONG TERM Investigators: Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med 372: 1489–1499, 2015. [DOI] [PubMed] [Google Scholar]
- 74. Langslet G, Emery M, Wasserman SM: Evolocumab (AMG 145) for primary hypercholesterolemia. Expert Rev Cardiovasc Ther 13: 477–488, 2015. [DOI] [PubMed] [Google Scholar]
- 75. Kereiakes DJ, Robinson JG, Cannon CP, Lorenzato C, Pordy R, Chaudhari U, et al.: Efficacy and safety of the proprotein convertase subtilisin/kexin type 9 inhibitor alirocumab among high cardiovascular risk patients on maximally tolerated statin therapy: The ODYSSEY COMBO I study. Am Heart J 169: 906–915.e13, 2015. [DOI] [PubMed] [Google Scholar]
- 76. Cannon CP, Cariou B, Blom D, McKenney JM, Lorenzato C, Pordy R, et al.; ODYSSEY COMBO II Investigators: Efficacy and safety of alirocumab in high cardiovascular risk patients with inadequately controlled hypercholesterolaemia on maximally tolerated doses of statins: The ODYSSEY COMBO II randomized controlled trial. Eur Heart J 36: 1186–1194, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zheng-Lin B, Ortiz A: Lipid management in chronic kidney disease: Systematic review of PCSK9 targeting. Drugs 78: 215–229, 2018. [DOI] [PubMed] [Google Scholar]
- 78. Charytan DM, Sabatine MS, Pedersen TR, Im K, Park JG, Pineda AL, et al.; FOURIER Steering Committee and Investigators: Efficacy and safety of evolocumab in chronic kidney disease in the FOURIER trial. J Am Coll Cardiol 73: 2961–2970, 2019. [DOI] [PubMed] [Google Scholar]
- 79. Tuñón J, Steg PG, Bhatt DL, Bittner VA, Díaz R, Goodman SG, et al.; ODYSSEY OUTCOMES Investigators: Effect of alirocumab on major adverse cardiovascular events according to renal function in patients with a recent acute coronary syndrome: Prespecified analysis from the ODYSSEY OUTCOMES randomized clinical trial. Eur Heart J 41: 4114–4123, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wang N, Tall AR: A new approach to PCSK9 therapeutics. Circ Res 120: 1063–1065, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gajic-Veljanoski O, Phua CW, Shah PS, Cheung AM: Effects of long-term low-molecular-weight heparin on fractures and bone density in non-pregnant adults: A systematic review with meta-analysis. J Gen Intern Med 31: 947–957, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bengalorkar GM, Sarala N, Venkatrathnamma PN, Kumar TN: Effect of heparin and low-molecular weight heparin on serum potassium and sodium levels. J Pharmacol Pharmacother 2: 266–269, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Elewa U, Fernández-Fernández B, Mahillo-Fernández I, Martin-Cleary C, Sanz AB, Sanchez-Niño MD, et al.: PCSK9 in diabetic kidney disease. Eur J Clin Invest 46: 779–786, 2016. [DOI] [PubMed] [Google Scholar]
- 84. Baass A, Dubuc G, Tremblay M, Delvin EE, O’Loughlin J, Levy E, et al.: Plasma PCSK9 is associated with age, sex, and multiple metabolic markers in a population-based sample of children and adolescents. Clin Chem 55: 1637–1645, 2009. [DOI] [PubMed] [Google Scholar]
- 85. Eisenga MF, Zelle DM, Sloan JH, Gaillard CAJM, Bakker SJL, Dullaart RPF: High serum PCSK9 is associated with increased risk of new-onset diabetes after transplantation in renal transplant recipients. Diabetes Care 40: 894–901, 2017. [DOI] [PubMed] [Google Scholar]
- 86. Filippatos TD, Liberopoulos E, Georgoula M, Tellis CC, Tselepis AD, Elisaf M: Effects of increased body weight and short-term weight loss on serum PCSK9 levels - a prospective pilot study. Arch Med Sci Atheroscler Dis 2: e46–e51, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kawanami D, Matoba K, Utsunomiya K: Dyslipidemia in diabetic nephropathy. Renal Replacement Therapy 2: 16, 2016. [Google Scholar]
- 88. Tannock L: Dyslipidemia in Chronic Kidney Disease. In Endotext, edited by Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dungan K, et al., South Dartmouth, MA, MDText.com, Inc., 2008. [Google Scholar]
- 89. Vaziri ND: Dyslipidemia of chronic renal failure: The nature, mechanisms, and potential consequences. Am J Physiol Renal Physiol 290: F262–F272, 2006. [DOI] [PubMed] [Google Scholar]
- 90. Nozue T, Yamamoto S, Tohyama S, Fukui K, Umezawa S, Onishi Y, et al.; TRUTH Investigators: Comparison of arterial remodeling and changes in plaque composition between patients with progression versus regression of coronary atherosclerosis during statin therapy (from the TRUTH study). Am J Cardiol 109: 1247–1253, 2012. [DOI] [PubMed] [Google Scholar]
- 91. Mayne J, Dewpura T, Raymond A, Cousins M, Chaplin A, Lahey KA, et al.: Plasma PCSK9 levels are significantly modified by statins and fibrates in humans. Lipids Health Dis 7: 22, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Dong B, Wu M, Cao A, Li H, Liu J: Suppression of Idol expression is an additional mechanism underlying statin-induced up-regulation of hepatic LDL receptor expression. Int J Mol Med 27: 103–110, 2011. [DOI] [PubMed] [Google Scholar]
- 93. Nangaku M, Fujita T: Activation of the renin-angiotensin system and chronic hypoxia of the kidney. Hypertens Res 31: 175–184, 2008. [DOI] [PubMed] [Google Scholar]
- 94. López-Hernández FJ, López-Novoa JM: Role of TGF-β in chronic kidney disease: An integration of tubular, glomerular and vascular effects. Cell Tissue Res 347: 141–154, 2012. [DOI] [PubMed] [Google Scholar]
- 95. Brinkkoetter PT, Holtgrefe S, van der Woude FJ, Yard BA: Angiotensin II type 1-receptor mediated changes in heparan sulfate proteoglycans in human SV40 transformed podocytes. J Am Soc Nephrol 15: 33–40, 2004. [DOI] [PubMed] [Google Scholar]
- 96. Chang MY, Potter-Perigo S, Tsoi C, Chait A, Wight TN: Oxidized low density lipoproteins regulate synthesis of monkey aortic smooth muscle cell proteoglycans that have enhanced native low density lipoprotein binding properties. J Biol Chem 275: 4766–4773, 2000. [DOI] [PubMed] [Google Scholar]
- 97. MacArthur JM, Bishop JR, Stanford KI, Wang L, Bensadoun A, Witztum JL, et al.: Liver heparan sulfate proteoglycans mediate clearance of triglyceride-rich lipoproteins independently of LDL receptor family members. J Clin Invest 117: 153–164, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Williams KJ, Fuki IV: Cell-surface heparan sulfate proteoglycans: Dynamic molecules mediating ligand catabolism. Curr Opin Lipidol 8: 253–262, 1997. [DOI] [PubMed] [Google Scholar]
- 99. Rashid S, Curtis DE, Garuti R, Anderson NN, Bashmakov Y, Ho YK, et al.: Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9. Proc of the Natl Acad Sci 102: 5374–5379, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.