

Abstract
Compared to traditional experimental approaches, computational modeling is a promising strategy to efficiently prioritize new candidates with low cost. In this study, we developed a novel data mining and computational modeling workflow proven to be applicable by screening new analgesic opioids. To this end, a large opioid data set was used as the probe to automatically obtain bioassay data from the PubChem portal. There were 114 PubChem bioassays selected to build quantitative structure–activity relationship (QSAR) models based on the testing results across the probe compounds. The compounds tested in each bioassay were used to develop 12 models using the combination of three machine learning approaches and four types of chemical descriptors. The model performance was evaluated by the coefficient of determination (R2) obtained from 5-fold cross-validation. In total, 49 models developed for 14 bioassays were selected based on the criteria and were identified to be mainly associated with binding affinities to different opioid receptors. The models for these 14 bioassays were further used to fill data gaps in the probe opioids data set and to predict general drug compounds in the DrugBank data set. This study provides a universal modeling strategy that can take advantage of large public data sets for computer-aided drug design (CADD).
Keywords: Opioid, Big data, QSAR, Data mining, Machine learning, Profiling
Graphical Abstract
INTRODUCTION
Opioids are commonly used medicines for the treatment of severe acute and chronic pain.1–3 However, opioid misuse may cause drug resistance,4 severe side effects,5 and opioid use disorder.6 In 2017, around two-thirds of the drug overdose deaths in the United States involved opioids and were mainly caused by opioid-induced respiratory depression.7 Opioid drugs mediate the analgesic effect through binding to opioid receptors, which are classified into four major subtypes: μ, δ, and κ opioid receptors (MOR, DOR, KOR) and a nociceptin receptor (NOR).8 Activations of different opioid receptors may display different pharmacological activities in vivo. MOR agonists inhibit severe acute pain by modulating thermal nociception.9 Although KOR agonists are potently analgesic, activation of KOR is associated with sedation and hallucinogenic effects.10,11 DOR activation weakly modulates acute nociception and has antidepressant effects,9 and NOR, while less understood, has been shown to be involved in pain response and development of tolerance to MOR agonists.12 Among these receptors, MOR is the primary target for analgesics with its agonist morphine being a prototype for opioid analgesics. To minimize the side effects of morphine, including physical dependence and respiratory depression, and to produce more efficacious analgesics, many morphine-based semisynthetic (e.g., heroin, oxycodone) and fully synthesized opioids (e.g., fentanyl) have been developed; however, none of these opioids has been shown to be a safe and efficacious analgesic.13
In recent years, many efforts have been made to reduce known opioid side effects by designing new opioid drugs.14–16 Developing a new drug is expensive and time-consuming. It costs nearly $2.7 billion and more than 10 years to bring a new drug to market,17 and the development of new opioid drugs is no exception. Computational modeling approaches, especially those used for computer-aided drug design (CADD), are promising to reduce the drug development cost by rapidly analyzing data and evaluating new compounds.18 The quantitative structure–activity relationship (QSAR) is a classic CADD modeling strategy used to identify the contributions of chemical structures to target biological activities.19 With the development of advanced machine learning algorithms and chemical descriptors, QSAR models have been widely used in the early stage of drug research and development to identify drug candidates with desired therapeutic activities and exclude compounds with potential side effects or toxicity before experimental testing.20–22 In previous studies, several QSAR models have been developed to predict the binding activity of compounds to various opioid receptors. For example, one 3D-QSAR model and two k nearest neighbor (kNN) models using ECFP6 and FCFP6 descriptors were developed for the prediction of MOR binding affinity using 115 fentanyl-like compounds.23 Pan et al. performed 3D-QSAR modeling based on a training set of 46 compounds to predict DOR binding activity.24 However, these models were developed using small in-house congeneric data sets, which limit their applicability. Moreover, existing opioid QSAR models were developed to predict a single endpoint, so it is not feasible to make a comprehensive evaluation of the new compounds of interest as potential analgesics based on these models. Benefiting from the high-throughput screening (HTS) technique development and many associated data-sharing projects, modern drug discovery has stepped into a big data era.25 For example, PubChem is a publicly available big data resource with over 96 million compounds, including many drugs and drug-like compounds, tested against over 1 million bioassays.26,27 Thus data-driven modeling studies, fueled by newly developed machine learning algorithms, have become popular in the current CADD procedure.
The challenge raised by big data is how to efficiently identify useful data for modeling purposes and develop numerous models using these data. In this study, we developed a novel data mining and computational modeling workflow that can be used to prioritize new opioid candidates with analgesic activities of interest for future experimental testing. The utility of this workflow was shown by automatically developing predictive models that can be used to identify new opioids. A large data set of human MOR ligands was used as a probe data set to search PubChem for all the available bioassay data. If a probe compound was tested against one or more PubChem assays, the associated responses obtained from these assays were extracted. Due to an enormous amount of data available for the probe data set in PubChem, an automatic data mining approach was used to extract and compile critical bioassay data relevant to opioid binding targets. Then, the curated PubChem data were used as training sets for combinatorial QSAR modeling using various machine learning approaches and molecular descriptors. This procedure resulted in thousands of QSAR models for various opioid-binding endpoints. These models were filtered by model performance and activity distribution. Finally, the selected models were used to fill the data gaps of all probe opioid compounds and prioritize untested compounds with the desired activities for safe pain treatments.
METHODS
Data Sets and Profiling.
A large data set was used as a probe data set to search PubChem for all the available bioassay data. This probe data set consisting of 3656 active ligands of human MOR was retrieved from the ChEMBL database (https://www.ebi.ac.uk/chembl/target_report_card/CHEMBL233, accessed April 14, 2020). Structures in the probe data set were curated and standardized using the CASE-Ultra DataKurator 1.8.0.0 software (MulitCASE Inc., Beachwood, OH). This includes the removal of duplicates, mixtures, and inorganics, and the correction of structural errors. The curated probe data set consisted of 3656 unique compounds, which were used for public bioactivity data curation and modeling.
Using an in-house automatic profiling tool,28,29 the public in vitro bioassay data of the 3656 probe compounds were extracted from the PubChem portal. The hypothesis of public data profiling of the probe compounds is that the opioids were likely studied for their analgesic activities in previous studies, and thus, the extracted PubChem assays are likely somewhat associated with analgesic activities. The extracted data were selected based on (1) the number of probe compounds in a PubChem assay and (2) the distribution of activities in tested compounds to ensure the modelability for model developments. The resulting profile was first screened by removing those PubChem bioassays with limited activity data among probe compounds (i.e., less than 17 testing results across the compounds in the probe data set). The compounds tested by each of the selected bioassays were retrieved from PubChem. Duplicate compounds with different activity values were removed. Activity data that are shown as a range (e.g., Ki > 1000 nM) instead of exact activity values were excluded as well. We then further examined the activity distribution of the remaining compounds in each bioassay. Bioassays were excluded if their activity distributions had large gaps (i.e., any two consecutive activities with a difference larger than 20% of the entire activity range in the training set).30 After applying each of the above criteria, all compounds that remained in a bioassay were used as the training set of that bioassay for QSAR model development.
An external validation set that consisted of 20 approved opioid drugs, which were not in the training set for model development, was collected from ChEMBL.31 The opioid receptor binding activities of these compounds were collected from literatures to be compared to the model predictions.32–41 External general drugs were retrieved from the DrugBank database42 for prediction and comparison purposes. After removing overlap compounds in the probe data set and external validation set, the remaining 2042 DrugBank compounds were also used for external predictions. Details of compounds in each data set are given in the Supporting Information, Data sets.
Chemical Descriptors.
Four types of chemical descriptors, including chemical fingerprints and molecular descriptors, were used in this study. Extended-connectivity fingerprints (ECFPs) and functional-class fingerprints (FCFPs), which both contain 1024 fragments, have been used successfully in similarity search and QSAR modeling.43 Additionally, Molecular ACCess system (MACCS) keys consisting of 166 substructure fingerprints were used in this study.44 These three types of fingerprints were calculated using the RDKit (www.rdkit.org) package. Furthermore, 200 RDKit molecular descriptors, including topological, compositional, and electro-topological states, were calculated for the training set compounds using the RDKit package. All the molecular descriptor values were normalized to the range from zero to one for the training set compounds before model development.
QSAR Modeling.
Three machine learning approaches implemented by scikit-learn were used to develop QSAR models for each bioassay endpoint: k-nearest neighbors (kNN), random forest (RF), and support vector machines (SVM). kNN uses the average of its k chemical nearest neighbors in the training set as its prediction and employs the variable selection procedure to define neighbors.45 RF is an ensemble algorithm that constructs several randomized decision trees and outputs the mean prediction of forest trees to improve the prediction accuracy and avoid overfitting.46 SVM regression models try to find a function of descriptor–activity space that has a limited deviation from the actual activity value for all the training data and at the same time is as flat as possible.47 These machine learning approaches were tuned to identify the optimal parameters for model performance, as described previously.48,49
Individual regression models for each bioassay endpoint were developed using the combination of one type of descriptors (ECFP6, FCFP6, MACCS, rdkit) and one of the modeling approaches (kNN, RF, SVM), resulting in 12 individual models. The consensus QSAR model, which was generated by averaging predictions of various individual models, was also used in this study.19,20,48 All models were evaluated using a standard 5-fold cross-validation procedure, with 20% of the training set compounds left out for testing purposes during each iteration, as described in previous studies.20,48,50 Each bioassay training set was randomly split into five equal subsets. Four subsets (80% of the total compounds) were used for model training, and the remaining 20% was used to test the resulted model. This procedure was repeated five times so that every compound was used for prediction once.
Model Selections.
Two criteria were used to select models and corresponding bioassays: (1) model performance and (2) prediction distribution. First, models were selected based on the 5-fold cross-validation procedure with satisfactory predictivity (R2 ≥ 0.5). Then, each remaining model was used to predict the remaining probe compounds that were not experimentally tested against the corresponding assays and were therefore not also in the training set of that model. The distribution of the resulted predictions was evaluated by a Kurtosis parameter. Kurtosis is a measurement of the combined weight of the tails relative to the rest of the prediction distribution. Kurtosis of the standard normal distribution is equal to zero, and values between −2 and +2 are considered acceptable to prove normal univariate distribution.51,52 In this study, models with poor prediction distributions, such as heavy tails and/or uniform distribution, were excluded if their predictions for new opioids had |Kurtosis| ≥ 2.51,52
Applicability Domain.
Two different ways of implementing applicability domains (ADs) were defined for chemical fingerprints and molecular descriptors. For the RDKit molecular descriptors, the AD was calculated based on the distribution of Euclidean distances between each test set compound and its nearest neighbor in the training set. The threshold value to define the AD for a QSAR model using RDKit descriptors was set as the mean plus one-half of the standard deviation of the distribution of distances between each training set compound and its nearest neighbor in the training set, as described in previous studies.53,54 If the Euclidean distance of the test compound from its nearest neighbor in the training set exceeded this threshold, the prediction was considered unreliable. For ECFP6, FCFP6, and MACCS fingerprints, a similarity-based approach was used to define AD, and the threshold was set at a Jaccard similarity of 0.5.48 If the Jaccard similarity of the test compound and its nearest neighbor in the training set was less than 0.5, it was considered to be out of AD.
Statistical Analysis.
In this study, the coefficient of determination (R2) and mean absolute error (MAE) were used to evaluate model performance. The coefficient of determination represents the proportion of variance in responses that have been explained by the feature variables in a regression model, and MAE measures the average prediction error in the same units of the variable.55 R2 and MAE are defined as follows
| (1) |
| (2) |
where n is the total number of compounds, yi is the prediction value of the ith compound, Yi is the corresponding experimental value of the ith compound, and is the averaged experimental value of all compounds.
Another parameter, Kurtosis, was used to assess the distribution of predictions. In Fisher’s definition, Kurtosis is zero for normal distribution.51,52 The equation for calculating Kurtosis is as follows
| (3) |
where n is the total number of compounds, yi is the prediction value of the ith compound, is the mean value, and s is the standard deviation of all the predictions.
RESULTS AND DISCUSSION
Workflow in This Study.
The workflow designed for this study is summarized in Figure 1. The curated probe compounds were first profiled against PubChem for all the bioactivity data. Critical bioassays were then automatically identified using several criteria based on the testing results across the probe compounds. Automatic QSAR modeling was performed for the selected bioassays using all PubChem compounds and their associated bioactivity data, including both probe compounds and other compounds being tested. The resulted models were then selected based on model performance and prediction distributions. The final selected models were used to predict new compounds, including approved opioid drugs, probe compounds without experimental data, and nonopioid drugs obtained from the DrugBank data set.
Figure 1.
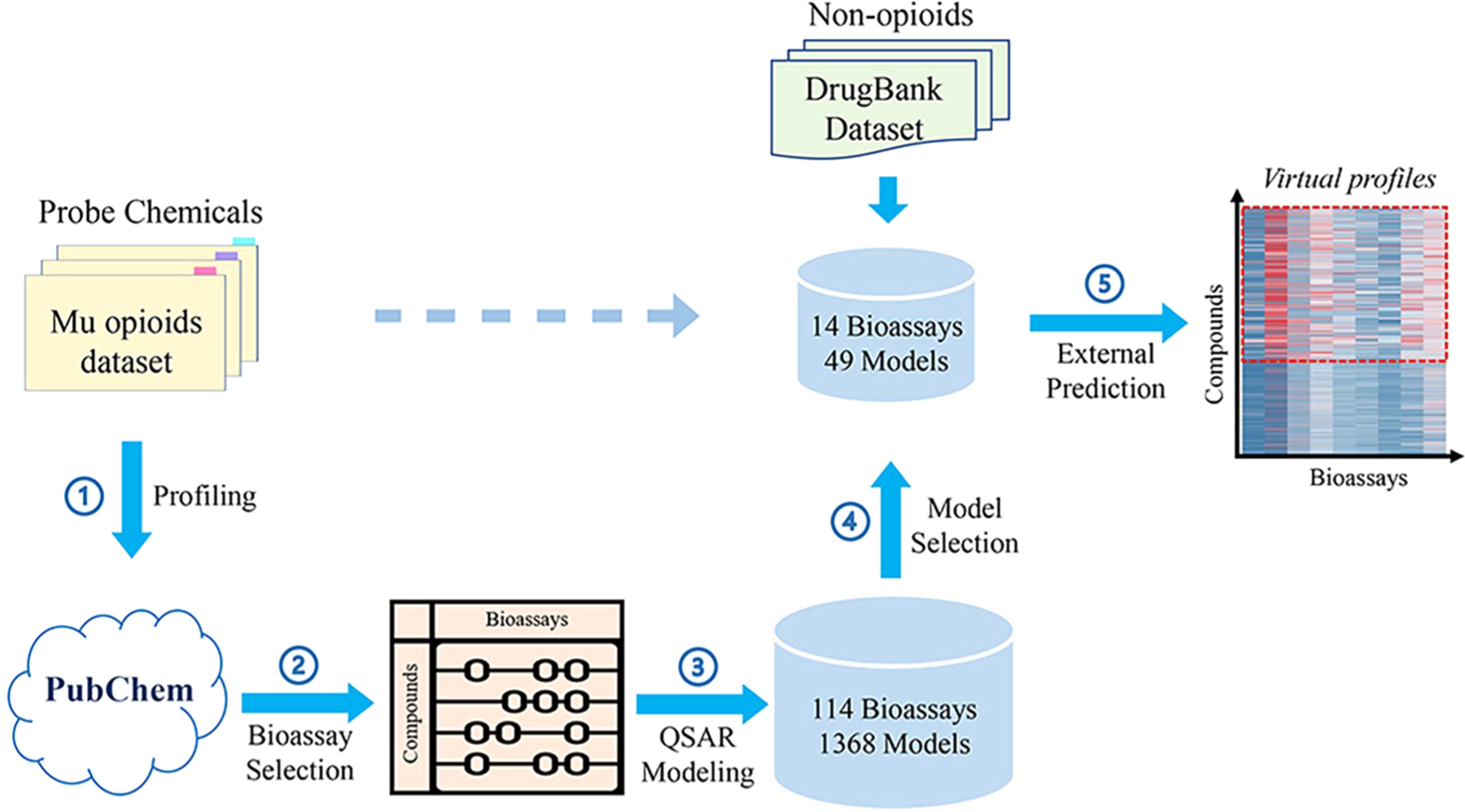
Diagrammatic depiction of the workflow in this study, including (1) profiling probe compounds against PubChem bioassays, (2) identifying critical bioassays, (3) automatic QSAR modeling, (4) model selection, and (5) external prediction.
Profiling and Resulted Training Sets.
The probe data set consisted of the ligands of MOR, which is the primary site of action for most analgesic opioids.56 After removing duplicates, and chemicals that could not be processed by our descriptor generator (salts, mixtures, and inorganics), 3656 unique compounds remained in the final probe data set. The probe data set was then searched against the PubChem portal to extract all the biological data, generating the profiles of their bioactivities. In PubChem, compounds’ responses in bioassays were classified into active, inactive, and inconclusive. Most of the actives also showed quantitative response results such as Ki, KD, EC50, and IC50. The initial bioprofile for probe compounds consisted of 19,256 PubChem bioassays, most of which were sparse, consisted of little data, and needed further curations.
It is important to select critical bioassays based on their relationships to probe compounds. First, a bioassay was selected if it had quantitative testing results for at least 17 probe compounds. This criterion is to ensure the modelability of the training set with enough compounds and was based on previous studies.54 This effort resulted in 200 critical bioassays from the originally extracted 19,256 PubChem assays. After examining the activity distributions of these 200 bioassays, 114 bioassays were selected by removing bioassays that had large gaps in the activity distribution.30 All the compounds tested in these bioassays, including probe compounds and the other PubChem compounds, were used as the training sets for automatic QSAR model development. There was a total of 2152 unique compounds included in the training sets for these 114 bioassays, and the training set size ranged from 17 to 223. Here, all compounds with testing results against individual assays were included to expand the chemical space of the training set and to ensure the coverage of the resulted models. Some compounds were tested multiple times in these assays. For example, morphine (PubChem Compound ID (CID) 5288826) was the most frequently tested compound and showed activities in 15 bioassays. A total of 1609 training set compounds were also in the 3656 probe opioid compounds, indicating the potential relationships of these bioassays to opioid receptors. Details of the 114 bioassays and all the training set compounds are listed in the Supporting Information, Training sets and models.
QSAR Modeling and Model Selections.
For each bioassay, 12 combinatorial QSAR models were developed using the combination of three machine learning approaches (kNN, RF, SVM) and four types of chemical descriptors (ECFP6, FCFP6, MACCS, and RDKit). In total, 1368 models were developed for the selected 114 PubChem bioassays. All the models were evaluated by the 5-fold cross-validation process, which was described previously.57 The resulting models were selected if they (1) had an acceptable model performance for 5-fold cross-validation and (2) were able to predict the remaining probe opioid compounds within a reasonable prediction range. Among the 1368 models generated by automatic QSAR modeling, 184 models showed acceptable predictivity for 5-fold cross-validation (R2 ≥ 0.5), which were developed for 47 bioassays (Figure 2A). The number of acceptable models for a specific bioassay ranged from 1 to 12. Models for the 47 bioassays were then used to predict the remaining probe compounds that were not included in the training set for modeling. Models with poor prediction distributions (|Kurtosis| ≥ 2) of these probe compounds were removed. If two bioassays had the same target and endpoint type, the one that has fewer predicted probe compounds within the AD was removed. This process resulted in 49 models, which were developed for 14 unique bioassays. The R2 and MAE values of the best performing models (model with the highest R2) for the 14 selected bioassays are shown in Figure 2B, and the details of these models are provided in the Supporting Information, Training sets and models. The combination of different types of descriptors (e.g., combining ECFP6 and MACCS as a new descriptor set) was also used to model selected assays, but the resulting models did not show improvement (results not shown here). Therefore, when the diversity of descriptors is high enough, simply increasing the number of descriptors used for modeling will not improve the resulting models in this study.
Figure 2.
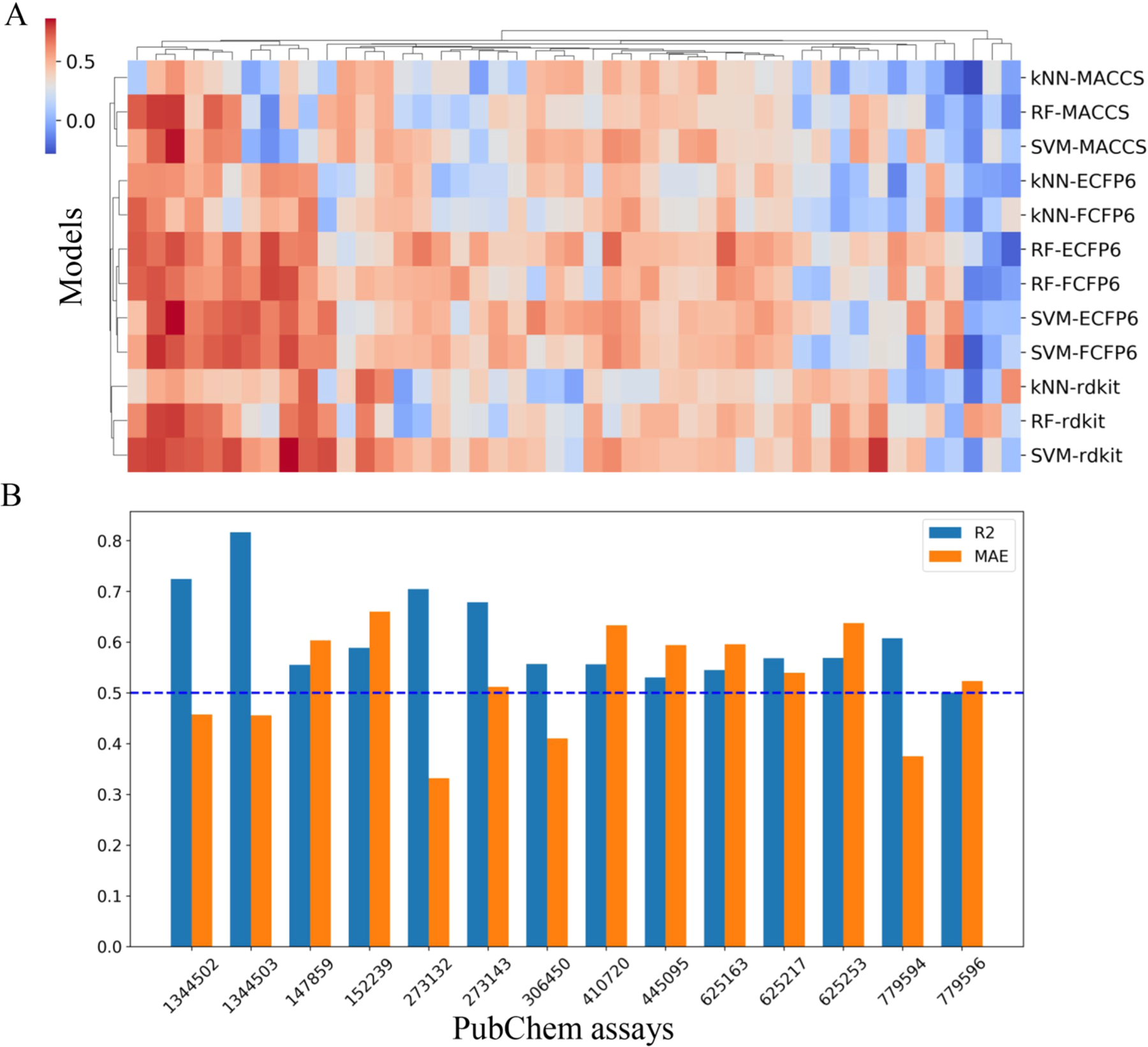
Performance of QSAR models. (A) Heatmap of 5-fold cross-validation R2 values of models developed for 47 bioassays. (B) R2 and MAE values of the best performance models (model with highest R2) for the 14 selected bioassays.
As listed in Table 1, 12 selected bioassays were related to four types of opioid receptors, including MOR, DOR, KOR, and NOR. The other two bioassays (PubChem Assay ID (AID) 625217 and 625253) were related to a serotonin receptor (5-HT2B) and dopamine receptor (D2L), respectively. These bioassays can be classified into two types: binding affinity assays and functional assays. Among the five functional assays, three assays tested GTPγS signaling as an assay of opioid agonism/antagonism at MOR (AID 1344502), KOR (AID 1344503), and DOR (AID 273143). The other two functional assays (AID 779594, 779596) tested the opioids’ activation of MOR downstream intracellular signaling pathways.
Table 1.
Details of Selected 14 Bioassays
| Targets | PubChem AID | No. compounds | Assay typesa | Descriptions |
|---|---|---|---|---|
| MOR | 152239 | 35 | B, IC50 | Binding affinity against MOR using [3H] DAMGO |
| 445095 | 40 | B, Ki | Displacement of [3H] DAMGO from MOR | |
| 625163 | 36 | B, Ki | DrugMatrix: MOR binding using [3H] diprenorphine | |
| 779594 | 18 | F, EC50 | Functional activity at MOR: β-arrestin2 recruitment | |
| 779596 | 31 | F, EC50 | Functional activity at MOR: forskolin-stimulated cAMP accumulation | |
| 1344502 | 37 | F, EC50 | [35S] GTPγS functional assay at MOR | |
| KOR | 147859 | 39 | B, IC50 | Binding affinity against KOR using [3H] U69593 |
| 410720 | 27 | B, Ki | Displacement of [3H] U69593 form KOR | |
| 1344503 | 43 | F, EC50 | [35S] GTPγS functional assay at KOR | |
| DOR | 273132 | 19 | B, Ki | Displacement of [3H]Cl-DPDPE from DOR |
| 273143 | 22 | F, IC50 | [35S] GTPγS functional assay at DOR | |
| NOR | 306450 | 40 | B, Ki | Displacement of [125I] nociceptin from nociceptin receptor |
| Serotonin | 625217 | 118 | B, Ki | DrugMatrix: Serotonin 5-HT2B binding using [3H] LSD |
| Dopamine | 625253 | 58 | B, Ki | DrugMatrix: Dopamine D2L binding using [3H] spiperone |
B: binding affinity assays, F: functional assays.
External Validation Using Approved Opioid Drugs.
External validation was performed to evaluate the predictivity of the selected QSAR models. A total of 20 approved opioid drugs that were not in the training set were collected as an external validation set.20,48 Since the experimental data of NOR, serotonin 5-HT2B and the dopamine D2L receptor binding, as well as some functional bioassays were not available for most opioid drugs, only nine opioid drugs with experimental data could be used for validation purposes. In Table 2, we listed nine representative opioid drugs that had binding affinity data (Ki) to three opioid receptors (MOR, DOR, KOR), which were compared to the model predictions.
Table 2.
External Predictions of Nine Opioid Drugs for Their Binding Affinities
| Name (CIDa) | Structure | Bioassays (AIDb) | Predicted log Ki (nM) | Experimental log Ki (nM) | Target |
|---|---|---|---|---|---|
|
Oxycodone (5284603) |
 |
445095 | 1.31 | 1.2532 | MOR |
| 273132 | 2.24 | 2.9832 | DOR | ||
| 410720 | 1.22 | 2.8332 | KOR | ||
|
Levorphanol (5359272) |
 |
445095 | −0.30 | −1.0633 | MOR |
| 273132 | 0.73 | 0.7033 | DOR | ||
| 410720 | 0.74 | 0.8133 | KOP | ||
|
Sufentanil (41693) |
 |
445095 | 1.19* | −0.8233 | MOR |
| 273132 | 1.64 | 1.7033 | DOR | ||
| 410720 | 1.01 | 1.8733 | KOR | ||
|
Alvimopan (5488548) |
 |
445095 | 0.95* | −0.1134 | MOR |
| 273132 | 0.63 | 0.6434 | DOR | ||
| 410720 | 0.90 | 1.6034 | KOR | ||
|
Methadone (4095) |
 |
445095 | 1.31* | 0.1635 | MOR |
| 273132 | 1.62 | 2.1236 | DOR | ||
| 410720 | 1.36 | 1.1935 | KOR | ||
|
Fentanyl (3345) |
 |
445095 | −0.13 | −0.1536 | MOR |
| 273132 | 1.64 | 2.1836 | DOR | ||
| 410720 | 0.90* | 1.9036 | KOR | ||
|
Morphine (5288826) |
 |
445095 | 1.20 | 1.1533 | MOR |
| 273132 | 0.60 | 0.3037 | DOR | ||
| 410720 | 0.29 | 1.5333 | KOR | ||
|
Loperamide (3955) |
 |
445095 | 0.64 | −0.2738 | MOR |
| 273132 | 1.62 | 1.7039 | DOR | ||
| 410720 | 1.39* | - | KOR | ||
|
Levallorphan (5359371) |
 |
445095 | −0.44 | −0.3240 | MOR |
| 273132 | 0.75 | 0.3441 | DOR | ||
| 410720 | 0.20 | 1.5441 | KOR |
CID: PubChem CID.
AID: PubChem AID.
Cannot find relevant data.
Prediction out of AD.
The AD was implemented to evaluate whether the new compounds were confidently predicted. If the similarity between a new compound and its nearest neighbor in the training set exceeded the AD threshold, the prediction was considered to be unreliable.48 As shown in Table 2, most of the in-AD predictions were accurate, compared to the literature-reported Ki values (i.e., the difference of predicted and experimental log Ki values is less than one unit). Several poor predictions may be caused by the factor of “activity cliffs”, (i.e., structurally similar compounds exhibit vastly different activities).58 For example, nalorphine (CID 5284595) is a derivative of morphine (CID 5288826) in which the n-methyl group was replaced by an allyl group. Nalorphine was the nearest neighbor of morphine in the training set of the KOR binding bioassay; however, their binding affinities to KOR were significantly different (i.e., log Ki values of nalorphine and morphine are −0.42 and 1.53, respectively) (Supporting Information, Table S1). Similarly, oxycodone (CID 5284603) and its nearest neighbor naloxone (CID 5284596) also had a large difference in binding affinities to KOR (i.e., log Ki values of oxycodone and naloxone are 2.83 and 0.08, respectively). The model for the KOR binding bioassay (AID 410720) was based on a limited number of compounds (27 compounds), so this issue is expected to be addressed when there are more compounds to be tested to increase the size of the training set in the future.
Potential Applications of the Resulting Models.
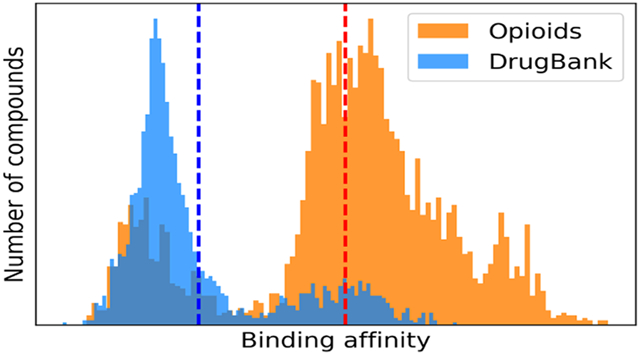
For each of the 14 selected bioassays, including nine binding assays and five functional assays, the model with the highest R2 was used to fill the data gaps for the probe opioid compounds and, as a comparison, to predict DrugBank compounds.42 DrugBank compounds consist of thousands of common drug molecules. Most of these are not likely to be opioids and thus can be used as negative controls of the external prediction. After removing overlaps with the probe and external validation sets, the final DrugBank data set of 2042 drug compounds was used for prediction and comparison purposes. Figure 3 shows the prediction results of opioids and normal drug compounds using the models of the nine binding assays. Opioids and general drug molecules from the DrugBank showed different prediction results. The opioids were predicted to have significantly higher binding affinities than DrugBank molecules (Supporting Information, Table S2). The average binding affinity predictions of each compound against four opioid receptors in these two data sets are shown in Figure 4. Most opioids showed higher predicted binding affinities than the DrugBank compounds, especially for MOR and NOR receptors.
Figure 3.
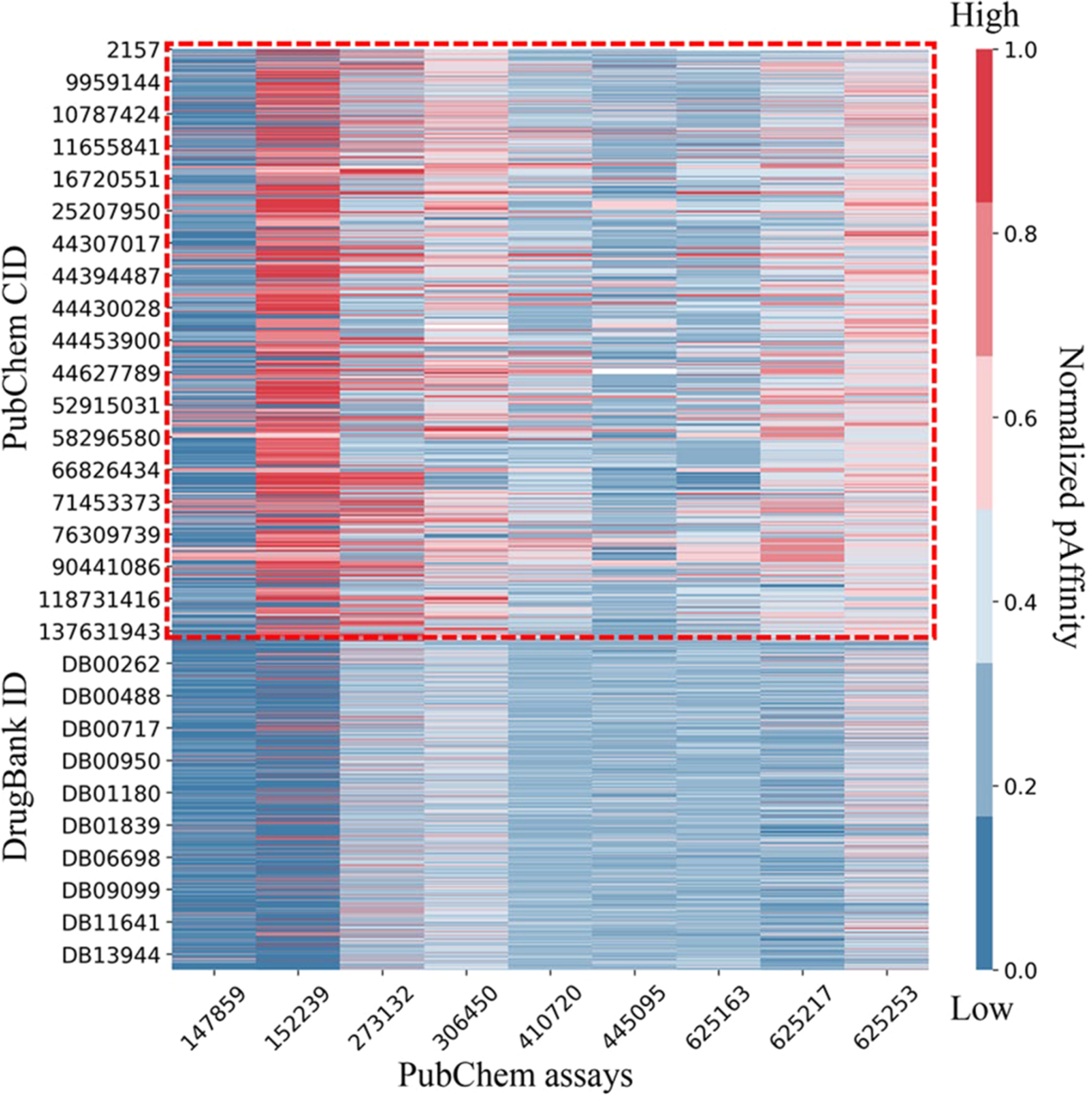
Virtual bioprofile of nine PubChem binding assays for the opioids and DrugBank compounds. The probe compound bioprofile is circled with red dots. For each bioassay, the predicted pKi (−log Ki) or pIC50 (−log IC50) values were normalized to [0,1] so that a higher value means a higher binding affinity.
Figure 4.
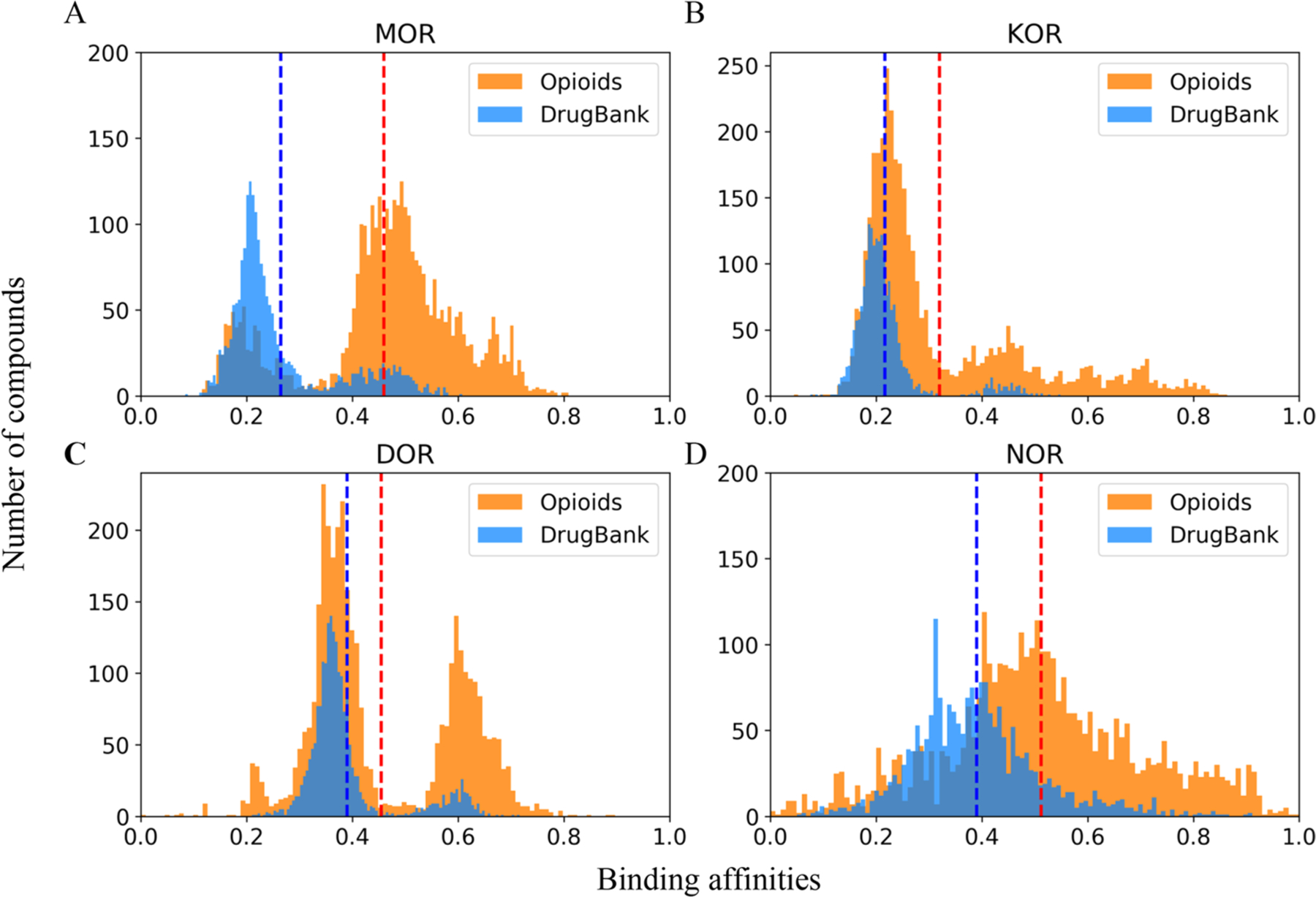
Distribution of the predicted averaged affinities of opioids and DrugBank molecules to four opioid receptors: (A) MOR, (B) KOR, (C) DOR, and (D) NOR. For compounds in the probe data set and DrugBank data set, the predicted pKi or pIC50 range was normalized to [0,1]. Normalized predictions for the same receptor were averaged, and their distributions are shown. Red and blue dashed lines indicate the mean prediction values of opioids and DrugBank molecules, respectively.
Several strategies have been proposed for the development of safer opioids.15,59–61 For example, the development of the biased MOR agonist showed an improved therapeutic index (analgesia vs adverse effects), which preferentially stimulates G protein-dependent signaling over the β-arrestin2 recruitment pathway.15,59 In this way, the model results we identify here have important implications for prioritizing the development of potential alternative opioids. Potentially biased compounds can be identified by calculating the difference of the activities between the bioassays that test the activation of G protein-dependent signaling (AID 779596) and the β-arrestin2 recruitment pathway (AID 779594).62 In the Supporting Information, Table S3, we prioritize five opioids that are predicted to preferentially activate the G protein-dependent signaling pathway (i.e., cAMP activation potential is larger than that of β-arrestin2). Of these five candidates, the best compound (CID 24822630) is a high-affinity MOR agonist that showed a potent analgesic effect,63 and its predicted log EC50 values for β-arrestin2 recruitment versus the G protein-dependent signaling pathway are 4.30 and 0.84, respectively. Our model results could also exclude some opioids with undesired activities. For example, potent activation of KOR can produce critical side effects.11 Nalorphine can antagonize morphine, but its potent activation of KOR can cause anxiety and hallucinations. Thus, this molecule is no longer being used medically.64–66 A similar approach to that described above could be used to identify candidates with unacceptably high actions at KOR, thus minimizing the risk of KOR-mediated off-target effects.
As shown in Figures 3 and 4, a small number of nonopioid compounds showed modest binding to opioid receptors. These molecules may represent potential nonopioid drugs that can bind to the opioid receptors. To select potential nonopioid drugs from DrugBank molecules, a consensus model was applied based on the resulting models to extend predictions to a larger chemical diversity. The individual models developed using different chemical descriptors could provide multiple diverse chemical spaces. The consensus prediction was generated by averaging the predictions within the AD of individual models, which can merge their chemical spaces. The advantage of using consensus predictions was described in previous studies.19,20,48 On the basis of consensus predictions, we identified eight drugs as being in the top five compounds in models of more than one bioassay (Supporting Information, Table S4). Somewhat surprisingly, these eight drugs were reported to mainly target two types of receptors: dopamine receptors and muscarinic acetylcholine receptors (mAChR).67,68 Similar to opioid receptors, they all belong to G protein-coupled receptors.
It is possible that opioids could interact with other G protein-coupled receptors, indicating potential drug interactions.69,70 Knowing such information could help us comprehensively evaluate the activity profile and prioritize optimal compounds with desired activities. In Figure 3, some probe opioids were predicted to potentially bind to serotonin receptors. Previous studies showed that opioids like methadone and tramadol could target the serotonin receptor and provoke serotonin toxicity when combined with certain serotonergic drugs, e.g., anti-depressants.71–73 Tramadol can also inhibit a mAChR-induced response.74,75 Several dopamine receptor ligands (e.g., haloperidol) were predicted to bind to opioid receptors MOR and KOR (Supporting Information, Table S4). Haloperidol is a high affinity dopamine D2 receptor antagonist that is used as an antipsychotic drug, which was reported to bind MOR (Ki = 633 nM) and inhibit the development of morphine tolerance and dependence following chronic administration.76–78 The interaction between brain opioid and dopamine systems is important in the addictive effects of opioids, although the exact molecular mechanism is still unknown.79 In Figure 3, some probe opioids also predicted to bind dopamine D2 receptors, potentially reflecting a functional similarity between these two types of ligands, although this possibility should be tested further.
CONCLUSIONS
In this study, a large data set of 3656 opioids was used as the probe to profile all the public bioactivity data from PubChem, and 114 critical bioassays were selected for automatic QSAR modeling. A total of 12 QSAR models were developed for each bioassay and sufficiently explored the chemical space and modeling ability of the training set compounds. Finally, 14 bioassays were further selected for profiling and prediction purposes by examining the model performance and prediction distributions. The resulting models of these bioassays not only showed good model performance but were also shown to make reasonable predictions to differentiate opioids and nonopioid drug compounds. These models can generate a comprehensive bioprofile for unknown compounds that can make it feasible to extensively study the binding mechanisms to opioid receptors and other relevant receptors, leading to safer analgesic medicine design. The computational modeling workflow developed in this study can automatically retrieve useful biological data from publicly available resources and perform predictive model development. This effort thus contributes to a universal data mining and modeling strategy that can take advantage of large public data sets to accelerate drug discovery and other related fields.
Supplementary Material
ACKNOWLEDGMENTS
This project was partially supported by the National Institute of Environmental Health Sciences (Grant R01ES031080, R15ES023148, and P30ES005022). M.H.J. is supported by a K99/R00 grant (K99DA045765) from the National Institute on Drug Abuse.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssuschemeng.0c09139.
Possible activity cliffs in QSAR model of KOR binding (PubChem AID 410720) (Table S1). Comparisons between predictions of probe opioids and DrugBank compounds for nine PubChem assays (Table S2). Possible G protein signaling biased MOR ligands prioritized from probe compounds (Table S3). Eight general drugs with potential opioid receptor binding activity (Table S4). (PDF)
Probe data set compounds, DrugBank data set compounds, and external validation compounds (Data sets). (XLSX)
Details of 114 bioassays, compounds of 114 training sets, and models for the selected 14 bioassays (Training sets and models). (XLSX)
Complete contact information is available at: https://pubs.acs.org/10.1021/acssuschemeng.0c09139
The authors declare no competing financial interest.
Contributor Information
Morgan H. James, Department of Psychiatry, Robert Wood Johnson Medical School, Rutgers University and Rutgers Biomedical Health Sciences, Piscataway, New Jersey 08854, United States; Brain Health Institute, Rutgers University and Rutgers Biomedical and Health Sciences, Piscataway, New Jersey 08854, United States
Hao Zhu, The Rutgers Center for Computational and Integrative Biology, Joint Health Sciences Center, Camden, New Jersey 08103, United States; Department of Chemistry, Rutgers University, Camden, New Jersey 08102, United States;.
REFERENCES
- (1).Eccleston C; Fisher E; Thomas KH; Hearn L; Derry S; Stannard C; Knaggs R; Moore RA Interventions for the reduction of prescribed opioid use in chronic non-cancer pain. Cochrane Database Syst. Rev 2017, 11, CD010323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Reuben DB; Alvanzo AA; Ashikaga T; Bogat GA; Callahan CM; Ruffing V; Steffens DC National Institutes of Health Pathways to Prevention Workshop: the role of opioids in the treatment of chronic pain. Ann. Intern. Med 2015, 162 (4), 295–300. [DOI] [PubMed] [Google Scholar]
- (3).Wiffen PJ; Wee B; Derry S; Bell RF; Moore RA Opioids for cancer pain - an overview of Cochrane reviews. Cochrane Database Syst. Rev 2017, 7, CD012592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Volkow ND; McLellan AT Opioid Abuse in Chronic Pain–Misconceptions and Mitigation Strategies. N. Engl. J. Med 2016, 374 (13), 1253–1263. [DOI] [PubMed] [Google Scholar]
- (5).Shim H; Gan TJ Side effect profiles of different opioids in the perioperative setting: are they different and can we reduce them? Br. J. Anaesth 2019, 123 (3), 266–268. [DOI] [PubMed] [Google Scholar]
- (6).Darcq E; Kieffer BL Opioid receptors: drivers to addiction? Nat. Rev. Neurosci 2018, 19 (8), 499–514. [DOI] [PubMed] [Google Scholar]
- (7).Scholl L; Seth P; Kariisa M; Wilson N; Baldwin G Drug and Opioid-Involved Overdose Deaths - United States, 2013–2017. Morb. Mortal. Wkly. Rep 2018, 67 (5152), 1419–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Dietis N; Rowbotham DJ; Lambert DG Opioid receptor subtypes: fact or artifact? Br. J. Anaesth 2011, 107 (1), 8–18. [DOI] [PubMed] [Google Scholar]
- (9).Madariaga-Mazon A; Marmolejo-Valencia AF; Li Y; Toll L; Houghten RA; Martinez-Mayorga K Mu-Opioid receptor biased ligands: A safer and painless discovery of analgesics? Drug Discovery Today 2017, 22 (11), 1719–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Bruchas MR; Roth BL New Technologies for Elucidating Opioid Receptor Function. Trends Pharmacol. Sci 2016, 37 (4), 279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Stein C; Schafer M; Machelska H Attacking pain at its source: new perspectives on opioids. Nat. Med 2003, 9 (8), 1003–1008. [DOI] [PubMed] [Google Scholar]
- (12).Toll L; Bruchas MR; Calo G; Cox BM; Zaveri NT Nociceptin/Orphanin FQ Receptor Structure, Signaling, Ligands, Functions, and Interactions with Opioid Systems. Pharmacol. Rev 2016, 68 (2), 419–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Pasternak GW; Pan Y-X Mu opioids and their receptors: evolution of a concept. Pharmacol. Rev 2013, 65 (4), 1257–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Schuckit MA Treatment of Opioid-Use Disorders. N. Engl. J. Med 2016, 375 (4), 357–368. [DOI] [PubMed] [Google Scholar]
- (15).Wadman M ‘Biased’ opioids could yield safer pain relief. Science 2017, 358 (6365), 847–848. [DOI] [PubMed] [Google Scholar]
- (16).Cunningham CW; Elballa WM; Vold SU Bifunctional opioid receptor ligands as novel analgesics. Neuropharmacology 2019, 151, 195–207. [DOI] [PubMed] [Google Scholar]
- (17).Goozner M A Much-Needed Corrective on Drug Development Costs. JAMA Int. Med 2017, 177 (11), 1575–1576. [DOI] [PubMed] [Google Scholar]
- (18).Osakwe O The Significance of Discovery Screening and Structure Optimization Studies. In Social Aspects of Drug Discovery, Development and Commercialization; Academic Press: Cambridge, 2016; pp 109–128, DOI: 10.1016/B978-0-12-802220-7.00005-3. [DOI] [Google Scholar]
- (19).Golbraikh A; Wang XS; Zhu H; Tropsha A Predictive QSAR Modeling: Methods and Applications in Drug Discovery and Chemical Risk Assessment. In Handbook of Computational Chemistry; Springer: Dordrecht, 2017; pp 2303–2340, DOI: 10.1007/978-3-319-27282-5_37. [DOI] [Google Scholar]
- (20).Wang W; Kim MT; Sedykh A; Zhu H Developing Enhanced Blood-Brain Barrier Permeability Models: Integrating External Bio-Assay Data in QSAR Modeling. Pharm. Res 2015, 32 (9), 3055–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kim MT; Huang R; Sedykh A; Wang W; Xia M; Zhu H Mechanism Profiling of Hepatotoxicity Caused by Oxidative Stress Using Antioxidant Response Element Reporter Gene Assay Models and Big Data. Environ. Health Perspect 2016, 124 (5), 634–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zhao L; Russo DP; Wang W; Aleksunes LM; Zhu H Mechanism-Driven Read-Across of Chemical Hepatotoxicants Based on Chemical Structures and Biological Data. Toxicol. Sci 2020, 174 (2), 178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Floresta G; Rescifina A; Abbate V Structure-Based Approach for the Prediction of Mu-opioid Binding Affinity of Unclassified Designer Fentanyl-Like Molecules. Int. J. Mol. Sci 2019, 20 (9), 2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Pan C; Meng H; Zhang S; Zuo Z; Shen Y; Wang L; Chang KJ Homology modeling and 3D-QSAR study of benzhydrylpiperazine delta opioid receptor agonists. Comput. Biol. Chem 2019, 83, 107109. [DOI] [PubMed] [Google Scholar]
- (25).Zhu H Big Data and Artificial Intelligence Modeling for Drug Discovery. Annu. Rev. Pharmacol. Toxicol 2020, 60, 573–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kim S; Thiessen PA; Bolton EE; Chen J; Fu G; Gindulyte A; Han L; He J; He S; Shoemaker BA; Wang J; Yu B; Zhang J; Bryant SH PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44 (D1), D1202–D1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Wang Y; Bryant SH; Cheng T; Wang J; Gindulyte A; Shoemaker BA; Thiessen PA; He S; Zhang J PubChem BioAssay: 2017 update. Nucleic Acids Res. 2017, 45 (D1), D955–D963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Russo DP; Kim MT; Wang W; Pinolini D; Shende S; Strickland J; Hartung T; Zhu H CIIPro: a new read-across portal to fill data gaps using public large-scale chemical and biological data. Bioinformatics 2017, 33 (3), 464–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zhang J; Hsieh JH; Zhu H Profiling animal toxicants by automatically mining public bioassay data: a big data approach for computational toxicology. PLoS One 2014, 9 (6), e99863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Tropsha A Best Practices for QSAR Model Development, Validation, and Exploitation. Mol. Inf 2010, 29 (6–7), 476–488. [DOI] [PubMed] [Google Scholar]
- (31).Davies M; Nowotka M; Papadatos G; Dedman N; Gaulton A; Atkinson F; Bellis L; Overington JP ChEMBL web services: streamlining access to drug discovery data and utilities. Nucleic Acids Res. 2015, 43 (W1), W612–W620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Monory K; Greiner E; Sartania N; Sallai L; Pouille Y; Schmidhammer H; Hanoune J; Borsodi A Opioid binding profiles of new hydrazone, oxime, carbazone and semicarbazone derivatives of 14-alkoxymorphinans. Life Sci. 1999, 64 (22), 2011–2020. [DOI] [PubMed] [Google Scholar]
- (33).Raynor K; Kong H; Chen Y; Yasuda K; Yu L; Bell GI; Reisine T Pharmacological characterization of the cloned kappa-, delta-, and mu-opioid receptors. Mol. Pharmacol 1994, 45 (2), 330–334. [PubMed] [Google Scholar]
- (34).Wentland MP; Lou R; Lu Q; Bu Y; VanAlstine MA; Cohen DJ; Bidlack JM Syntheses and opioid receptor binding properties of carboxamido-substituted opioids. Bioorg. Med. Chem. Lett 2009, 19 (1), 203–208. [DOI] [PubMed] [Google Scholar]
- (35).Standifer KM; Cheng J; Brooks AI; Honrado CP; Su W; Visconti LM; Biedler JL; Pasternak GW Biochemical and pharmacological characterization of mu, delta and kappa 3 opioid receptors expressed in BE (2)-C neuroblastoma cells. J. Pharmacol. Exp. Ther 1994, 270 (3), 1246–1255. [PubMed] [Google Scholar]
- (36).Toll L; Berzetei-Gurske IP; Polgar WE; Brandt SR; Adapa ID; Rodriguez L; Schwartz RW; Haggart D; O’Brien A; White A; Kennedy JM; Craymer K; Farrington L; Auh JS Standard binding and functional assays related to medications development division testing for potential cocaine and opiate narcotic treatment medications. NIDA Res. Monogr 1998, 178, 440–466. [PubMed] [Google Scholar]
- (37).Molenveld P; Bouzanne des Mazery R; Sterk GJ; Storcken RPM; Autar R; van Oss B; van der Haas RNS; Frohlich R; Schepmann D; Wunsch B; Soeberdt M Conformationally restricted κ-opioid receptor agonists: Synthesis and pharmacological evaluation of diastereoisomeric and enantiomeric decahydroquinoxalines. Bioorg. Med. Chem. Lett 2015, 25 (22), 5326–5330. [DOI] [PubMed] [Google Scholar]
- (38).Chen Z; Davies E; Miller WS; Shan S; Valenzano KJ; Kyle DJ Design and synthesis of 4-phenyl piperidine compounds targeting the mu receptor. Bioorg. Med. Chem. Lett 2004, 14 (21), 5275–5279. [DOI] [PubMed] [Google Scholar]
- (39).Breslin HJ; Miskowski TA; Rafferty BM; Coutinho SV; Palmer JM; Wallace NH; Schneider CR; Kimball ES; Zhang S-P; Li J; Colburn RW; Stone DJ; Martinez RP; He W Rationale, design, and synthesis of novel phenyl imidazoles as opioid receptor agonists for gastrointestinal disorders. J. Med. Chem 2004, 47 (21), 5009–5020. [DOI] [PubMed] [Google Scholar]
- (40).Majumdar S; Burgman M; Haselton N; Grinnell S; Ocampo J; Pasternak AR; Pasternak GW Generation of novel radiolabeled opiates through site-selective iodination. Bioorg. Med. Chem. Lett 2011, 21 (13), 4001–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Codd EE; Shank RP; Schupsky JJ; Raffa RB Serotonin and norepinephrine uptake inhibiting activity of centrally acting analgesics: structural determinants and role in antinociception. J. Pharmacol. Exp. Ther 1995, 274 (3), 1263–1270. [PubMed] [Google Scholar]
- (42).Wishart DS; Feunang YD; Guo AC; Lo EJ; Marcu A; Grant JR; Sajed T; Johnson D; Li C; Sayeeda Z; Assempour N; Iynkkaran I; Liu Y; Maciejewski A; Gale N; Wilson A; Chin L; Cummings R; Le D; Pon A; Knox C; Wilson M DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46 (D1), D1074–D1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Rogers D; Hahn M Extended-connectivity fingerprints. J. Chem. Inf. Model 2010, 50 (5), 742–754. [DOI] [PubMed] [Google Scholar]
- (44).Durant JL; Leland BA; Henry DR; Nourse JG Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Comput. Sci 2002, 42 (6), 1273–1280. [DOI] [PubMed] [Google Scholar]
- (45).Zheng W; Tropsha A Novel variable selection quantitative structure–property relationship approach based on the k-nearest-neighbor principle. J. Chem. Inf. Comput. Sci 2000, 40 (1), 185–194. [DOI] [PubMed] [Google Scholar]
- (46).Breiman L Random forests. Mach. Learn 2001, 45 (1), 5–32. [Google Scholar]
- (47).Smola AJ; Schölkopf B A tutorial on support vector regression. Stat Comput. 2004, 14 (3), 199–222. [Google Scholar]
- (48).Ciallella HL; Russo DP; Aleksunes LM; Grimm FA; Zhu H Predictive modeling of estrogen receptor agonism, antagonism, and binding activities using machine- and deep-learning approaches. Lab Invest. 2020, 2020, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Russo DP; Zorn KM; Clark AM; Zhu H; Ekins S Comparing Multiple Machine Learning Algorithms and Metrics for Estrogen Receptor Binding Prediction. Mol. Pharmaceutics 2018, 15 (10), 4361–4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Russo DP; Strickland J; Karmaus AL; Wang W; Shende S; Hartung T; Aleksunes LM; Zhu H Nonanimal Models for Acute Toxicity Evaluations: Applying Data-Driven Profiling and Read-Across. Environ. Health Perspect 2019, 127 (4), 047001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Sharma C; Ojha C Statistical Parameters of Hydrometeorological Variables: Standard Deviation, SNR, Skewness and Kurtosis. In Advances in Water Resources Engineering and Management; Springer: Singapore, 2020; pp 59–70, DOI: 10.1007/978-981-13-8181-2_5. [DOI] [Google Scholar]
- (52).Arnett AB; Pennington BF; Willcutt EG; DeFries JC; Olson RK Sex differences in ADHD symptom severity. J. Child Psychol. Psychiatry 2015, 56 (6), 632–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Zhao L; Wang W; Sedykh A; Zhu H Experimental Errors in QSAR Modeling Sets: What We Can Do and What We Cannot Do. ACS Omega 2017, 2 (6), 2805–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Tropsha A, Golbraikh A Predictive Quantitative Structure-Activity Relationships Modeling. In Handbook of Chemoinformatics Algorithms; CRC Press: Boca Raton, FL, 2010; pp 173–210, DOI: 10.1201/9781420082999-c7. [DOI] [Google Scholar]
- (55).Willmott CJ; Matsuura K Advantages of the mean absolute error (MAE) over the root mean square error (RMSE) in assessing average model performance. Clim. Res 2005, 30, 79–82. [Google Scholar]
- (56).Ehrlich AT; Kieffer BL; Darcq E Current strategies toward safer mu opioid receptor drugs for pain management. Expert Opin. Ther. Targets 2019, 23 (4), 315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Tropsha A; Gramatica P; Gombar VK The importance of being earnest: validation is the absolute essential for successful application and interpretation of QSPR models. QSAR Comb. Sci 2003, 22 (1), 69–77. [Google Scholar]
- (58).Maggiora GM On outliers and activity cliffs–why QSAR often disappoints. J. Chem. Inf. Model 2006, 46 (4), 1535–1535. [DOI] [PubMed] [Google Scholar]
- (59).Chen XT; Pitis P; Liu G; Yuan C; Gotchev D; Cowan CL; Rominger DH; Koblish M; Dewire SM; Crombie AL; Violin JD; Yamashita DS Structure-activity relationships and discovery of a G protein biased mu opioid receptor ligand, [(3-methoxythiophen-2-yl)methyl]({2-[(9R)-9-(pyridin-2-yl)-6-oxaspiro-[4.5]decan-9-yl]-ethyl})amine (TRV130), for the treatment of acute severe pain. J. Med. Chem 2013, 56 (20), 8019–8031. [DOI] [PubMed] [Google Scholar]
- (60).Majumdar S; Devi LA Strategy for making safer opioids bolstered. Nature 2018, 553 (7688), 286–288. [DOI] [PubMed] [Google Scholar]
- (61).Service RF New pain drugs may lower overdose and addiction risk. Science 2018, 361 (6405), 831–832. [DOI] [PubMed] [Google Scholar]
- (62).Winpenny D; Clark M; Cawkill D Biased ligand quantification in drug discovery: from theory to high throughput screening to identify new biased mu opioid receptor agonists. Br. J. Pharmacol 2016, 173 (8), 1393–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Ghirmai S; Azar MR; Polgar WE; Berzetei-Gurske I; Cashman JR Synthesis and biological evaluation of alpha- and beta-6-amido derivatives of 17-cyclopropylmethyl-3, 14beta-dihydroxy-4, 5alpha-epoxymorphinan: potential alcohol-cessation agents. J. Med. Chem 2008, 51 (6), 1913–1924. [DOI] [PubMed] [Google Scholar]
- (64).Glatt MM Alcohol dependence: The ‘lack of control’ over alcohol and its implications. In The Dependence Phenomenon; Springer: Dordrecht, 1982; pp 119–155, DOI: 10.1007/978-94-011-7457-2_6. [DOI] [Google Scholar]
- (65).Aggrawal A APC Essentials of Forensic Medicine and Toxicology; Avichal Publishing Company: New Delhi, 2014; pp 553–554, DOI: 10.13140/2.1.2702.4967. [DOI] [Google Scholar]
- (66).Satoskar RS; Rege N; Bhandarkar S Pharmacology and Pharmacotherapeutics; Elsevier: Chennai, India, 2017; pp 165–166, DOI: 10.4103/0250-474X.29672. [DOI] [Google Scholar]
- (67).Kruse AC; Kobilka BK; Gautam D; Sexton PM; Christopoulos A; Wess J Muscarinic acetylcholine receptors: novel opportunities for drug development. Nat. Rev. Drug Discovery 2014, 13 (7), 549–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Beaulieu JM; Espinoza S; Gainetdinov RR Dopamine receptors - IUPHAR Review 13. Br. J. Pharmacol 2015, 172 (1), 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Liu R; Huang XP; Yeliseev A; Xi J; Roth BL Novel molecular targets of Dezocine and their clinical implications. Anesthesiology 2014, 120 (3), 714–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Olson KM; Duron DI; Womer D; Fell R; Streicher JM Comprehensive molecular pharmacology screening reveals potential new receptor interactions for clinically relevant opioids. PLoS One 2019, 14 (6), e0217371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Schenk M; Wirz S Serotonin syndrome and pain medication: What is relevant for practice? Schmerz 2015, 29 (2), 229–251. [DOI] [PubMed] [Google Scholar]
- (72).Rickli A; Liakoni E; Hoener MC; Liechti ME Opioid-induced inhibition of the human 5-HT and noradrenaline transporters in vitro: link to clinical reports of serotonin syndrome. Br. J. Pharmacol 2018, 175 (3), 532–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Baldo BA; Rose MA The anaesthetist, opioid analgesic drugs, and serotonin toxicity: a mechanistic and clinical review. Br. J. Anaesth 2020, 124 (1), 44–62. [DOI] [PubMed] [Google Scholar]
- (74).Shiraishi M; Minami K; Uezono Y; Yanagihara N; Shigematsu A Inhibition by tramadol of muscarinic receptor-induced responses in cultured adrenal medullary cells and in Xenopus laevis oocytes expressing cloned M1 receptors. J. Pharmacol. Exp. Ther 2001, 299 (1), 255–260. [PubMed] [Google Scholar]
- (75).Nakamura M; Minami K; Uezono Y; Horishita T; Ogata J; Shiraishi M; Okamoto T; Terada T; Sata T The effects of the tramadol metabolite O-desmethyl tramadol on muscarinic receptor-induced responses in Xenopus oocytes expressing cloned M1 or M3 receptors. Anesth. Analg 2005, 101 (1), 180–186. [DOI] [PubMed] [Google Scholar]
- (76).Titeler M; Lyon RA; Kuhar MJ; Frost JF; Dannals RF; Leonhardt S; Bullock A; Rydelek LT; Price DL; Struble RG Mu opiate receptors are selectively labelled by [3H]carfentanil in human and rat brain. Eur. J. Pharmacol 1989, 167 (2), 221–228. [DOI] [PubMed] [Google Scholar]
- (77).Bower CM; Hyde TM; Zaka M; Hamid EH; Baca SM; Egan MF Decreased mu-opioid receptor binding in the globus pallidus of rats treated with chronic haloperidol. Psychopharmacology (Berl) 2000, 150 (3), 260–263. [DOI] [PubMed] [Google Scholar]
- (78).Yang C; Chen Y; Tang L; Wang ZJ Haloperidol disrupts opioid-antinociceptive tolerance and physical dependence. J. Pharmacol. Exp. Ther 2011, 338 (1), 164–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Galaj E; Newman AH; Xi ZX Dopamine D3 receptor-based medication development for the treatment of opioid use disorder: Rationale, progress, and challenges. Neurosci. Biobehav. Rev 2020, 114, 38–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.