

Abstract
Spinocerebellar ataxia type 7 (SCA7) is an inherited neurodegenerative disease mainly characterized by motor incoordination because of progressive cerebellar degeneration. SCA7 is caused by polyglutamine expansion in ATXN7, a subunit of the transcriptional coactivator SAGA, which harbors histone modification activities. Polyglutamine expansions in specific proteins are also responsible for SCA1-SCA3, SCA6, and SCA17; however, the converging and diverging pathomechanisms remain poorly understood. Using a new SCA7 knock-in mouse, SCA7140Q/5Q, we analyzed gene expression in the cerebellum and assigned gene deregulation to specific cell types using published datasets. Gene deregulation affects all cerebellar cell types, although at variable degree, and correlates with alterations of SAGA-dependent epigenetic marks. Purkinje cells (PCs) are by far the most affected neurons and show reduced expression of 83 cell-type identity genes, including these critical for their spontaneous firing activity and synaptic functions. PC gene downregulation precedes morphologic alterations, pacemaker dysfunction, and motor incoordination. Strikingly, most PC genes downregulated in SCA7 have also decreased expression in SCA1 and SCA2 mice, revealing converging pathomechanisms and a common disease signature involving cGMP-PKG and phosphatidylinositol signaling pathways and LTD. Our study thus points out molecular targets for therapeutic development, which may prove beneficial for several SCAs. Furthermore, we show that SCA7140Q/5Q males and females exhibit the major disease features observed in patients, including cerebellar damage, cerebral atrophy, peripheral nerves pathology, and photoreceptor dystrophy, which account for progressive impairment of behavior, motor, and visual functions. SCA7140Q/5Q mice represent an accurate model for the investigation of different aspects of SCA7 pathogenesis.
SIGNIFICANCE STATEMENT Spinocerebellar ataxia 7 (SCA7) is one of the several forms of inherited SCAs characterized by cerebellar degeneration because of polyglutamine expansion in specific proteins. The ATXN7 involved in SCA7 is a subunit of SAGA transcriptional coactivator complex. To understand the pathomechanisms of SCA7, we determined the cell type-specific gene deregulation in SCA7 mouse cerebellum. We found that the Purkinje cells are the most affected cerebellar cell type and show downregulation of a large subset of neuronal identity genes, critical for their spontaneous firing and synaptic functions. Strikingly, the same Purkinje cell genes are downregulated in mouse models of two other SCAs. Thus, our work reveals a disease signature shared among several SCAs and uncovers potential molecular targets for their treatment.
Keywords: SCA7, cell type-specific gene deregulation, Purkinje cells, polyglutamine expansion, cerebellum, retinopathy
Introduction
Spinocerebellar ataxia type 7 (SCA7) is an inherited neurodegenerative disorder caused by expansion of CAG repeats encoding polyglutamine (polyQ) in ATXN7 (David et al., 1997), a subunit of the SAGA transcriptional coactivator complex (Helmlinger et al., 2004). SCA7 expansions range from 34 to >200 repeats (Niewiadomska-Cimicka and Trottier, 2019), and the length of the expanded repeats correlates inversely with the onset of symptoms and positively with disease severity.
SCA7 patients show difficulty to coordinate balance, gait, and speech and present a variety of additional clinical features, including visual impairment (David et al., 1998; Martin, 2012). The pathology arises primarily from progressive damage of the cerebellum and its associated tracts, and the retina, leading to important loss of Purkinje cells (PCs) and cone and rod photoreceptors (Martin et al., 1994; Aleman et al., 2002; Michalik et al., 2004; Horton et al., 2013). Alterations of other structures of CNS and PNS are observed and may contribute to additional symptoms (van de Warrenburg et al., 2004; Rub et al., 2008; Martin, 2012; Salas-Vargas et al., 2015).
The visual impairment in SCA7 mouse models revealed a progressive loss of photoreceptor-specific gene expression, resulting in shortage of proteins necessary for renewal of outer segments (La Spada et al., 2001; Abou-Sleymane et al., 2006; Helmlinger et al., 2006; Yefimova et al., 2010). The results suggest that ATXN7 mutation compromises the maintenance of photoreceptor terminal differentiation (Niewiadomska-Cimicka and Trottier, 2019). Interestingly, knockdown of atxn7 in zebrafish altered the differentiation of photoreceptor and PC during development (Yanicostas et al., 2012; Carrillo-Rosas et al., 2018). Whether PCs or other neurons also progressively lose their differentiation identity in SCA7 remains to be investigated.
In contrast to the retinopathy, the pathomechanisms underlying SCA7 motor incoordination remain to be determined. Different transgenic and knock-in mouse models showed different vulnerability of PCs to mutant ATXN7 (mATXN7) expression (Yvert et al., 2000, 2001; Yoo et al., 2003; Custer et al., 2006; Mookerjee et al., 2009; Chou et al., 2010; Furrer et al., 2013; Karam and Trottier, 2018) and little overlap of cerebellar gene deregulation (Gatchel et al., 2008; Chou et al., 2010; Friedrich et al., 2012; Stoyas et al., 2020). Interestingly, mice overexpressing mATXN7 in the whole brain, except PCs or specifically in Bergmann glia, developed motor dysfunction (Custer et al., 2006; Mookerjee et al., 2009), questioning the contribution of different cerebellar cell types to the pathology. A deeper understanding of the SCA7 cerebellar pathology at a single cell type resolution is thus warranted. Furthermore, the contribution of SAGA dysfunction to cerebellar pathology remains unclear as conditional deletion of the GCN5 histone acetyl-transferase, a subunit of SAGA, in mouse PCs led to very mild ataxia (Chen et al., 2012).
SCA7 is part of a group comprising 5 other SCAs (SCA1-SCA3, SCA6, and SCA17) that are also caused by polyQ expansion in different proteins. PolyQ SCAs cause damage to the cerebellum, often with high vulnerability of PCs (Klockgether, 2011). Despite shared genetic, clinical, and neuropathological features, more is needed to identify points of intersection and divergence of the pathophysiology of polyQ SCAs (Niewiadomska-Cimicka et al., 2020). While SCA proteins do not share any domain and have different cellular functions, changes in gene expression are central features in most polyQ SCAs. Therefore, the comparison of differentially expressed genes should provide insight into converging disease mechanisms.
Yet, the broad spectrum of SCA7 pathology has not been fully recapitulated in the current models. We describe here a new SCA7140Q/5Q knock-in mouse line that combines the major clinical features observed in patients, including retinal, cerebellar, cerebral, and peripheral nerve pathologies, which account for progressive impairment of behavior, motor, and vision functions. Transcriptome profiling allowed to identify cell type-specific gene deregulation in the cerebellum and to highlight PCs as the most affected neurons. Strikingly, PCs show a profound downregulation of neuronal identity genes, which are also downregulated in other polyQ SCAs, revealing converging pathomechanisms.
Materials and Methods
Mouse information
The SCA7140Q/5Q knock-in mouse line, which is derived from the SCA7266Q/5Q mouse model (Yoo et al., 2003) by CAG repeat contraction, harbors an expansion ranging from 140 to 150 CAG repeats in exon1 of the Atxn7 locus. Mice are kept on C57Bl/6J background and bred in the Mouse Clinical Institute with a 12 h light/dark cycle and free access to food and water. Genotyping was performed by PCR as described previously (Yoo et al., 2003). WT and SCA7140Q/5Q males and females were analyzed, unless indicated.
Ethics approval and consent to participate
All animal procedures were conducted in strict accordance with the French and German national laws for laboratory animal welfare and the guidelines of the Federation of European Laboratory Animal Science Associations, based on European Union Legislation (Directive 2010/63/EU). Experiments were approved by local Ethics Committees under the supervision of the Ministry of Higher Education, Research and Innovation in France (agreement numbers 14199-2018032117483097, #1803-2015072215481972, and #4354-2016030212155187-v3; CREMEAS; CEEA35) and the Regierungspraesidium Tuebingen in Germany.
Behavioral and motor performance analysis
To ensure the replicability of SCA7 mouse phenotype, two mouse colonies were raised in the husbandry of different research centers and were analyzed using standardized protocols by independent experimenters and instruments. The experiments were all performed during the light phase of a 12 h light/dark cycle.
Open field test
The open fields were placed in a room homogeneously illuminated at 150 lux. Mice of the first colony were tested in automated open fields (44.3 × 44.3 × 16.8 cm, Panlab), virtually divided into central and peripheral regions. Each mouse was placed in the periphery of the open field and allowed to explore freely for 30 min, with the experimenter out of the animal's sight. The distance traveled, the number of rears, average speeds, and resting time were automatically recorded and reflect the general locomotor or motor activity phenotype. The number of entries and the time spent in the central and peripheral regions were used as index of emotionality/anxiety. Mice of the second colony were tested in an open field (46 × 46 cm). Each mouse was tracked over 30 min with EthoVision XT 11 and 12 (Noldus Information Technology) and automatically analyzed for the same parameters, except for the number of rears that were manually scored.
Rotarod
To assess the ability to maintain balance, mice of the first colony were tested on a rotating rod of 5 cm in diameter covered by gray rubber foam (Bioseb). Mice were given three testing trials during which the rotation speed accelerated from 4 to 40 rpm in 5 min. Trials were separated by a minimum of 5-10 min interval. The average latency before falling from the rod was used as index of motor coordination performance. Mice of the second colony were assessed using an apparatus from TSE Systems with 3.5 cm rod diameter without rubber foam, using the same parameters as for the first colony. The floor of the rotarod was covered with soft material to avoid animal discomfort when falling. Light intensity was set to ∼115 lux.
Notched bar
The test was used to test hindlimb coordination. On the first day, mice were trained on a 2-cm-wide and 110-cm-long natural wooden notched bar made of 26 platforms of 2-cm-long spaced by 27 gaps of 2 cm and connected to the goal box (12 × 12 × 14 cm), under 115 lux. The second day, mice were tested in the same conditions and time to cross the notched bar was measured. A back paw going through a gap was considered as an error.
Beam walking test
The test consisted of a 110-cm-long wooden beam (2 cm diameter), positioned 50 cm above the floor. One extremity of the beam was used as starting position while the opposite extremity was connected to a goal box (of 12 × 12 × 14 cm for the first colony, and 8.5 × 8.5 × 12 cm for the second colony). The floor of the goal boxes was covered with clean sawdust. The floor under the beam was covered with soft material. During the whole protocol, the light intensity in proximity of the beam was maintained at ∼115 Lux. Each mouse was first habituated to the goal box for 1 min before three training and three testing trials. On the first training trial, mice were placed on the beam near and facing the goal box and allowed to access it. On the second and third training trials, mice were placed further away (33 and 66 cm, respectively) facing the box and were allowed to reach and enter the box. On the testing sections, mice were placed at the beam extremity (100 cm, starting position), facing the box and were allowed to reach the goal box. After each training and testing trial, mice were left for 15 s in the goal box. The latency to reach the box (cut off 60 s) and the number of errors (slips) while crossing the beam were recorded.
Footprint
After coating the front and hind feet with two different colors of nontoxic inks, mice walked through a tunnel (length 50 cm, width 9 cm, height 6 cm) covered with paper. The following five footprint patterns were manually measured: matching between paws, stride length, stance width, distance between forelimbs and hindlimbs.
Grip strength test
This test measures the maximal muscle strength (g) using an isometric dynamometer connected to a grid (Bioseb). Mice were held by the tail and allowed to grip the metal grid with the four paws; then they were pulled backwards until they released it. Each mouse was submitted to three consecutive trials, and the maximal strength developed by the mouse before releasing the grid was recorded. The average value of the three trials adjusted to body weight was used as index of the muscle strength.
CatWalk
Gait analyses were performed using CatWalk XT 8.1 (Noldus Information Technology). The CatWalk apparatus consists of a closed corridor resting on a glass plate. The walkway (20 cm) is illuminated with red light from the ceiling and green light from the edges of the plate (green intensity threshold: 0.30; camera gain: 20). When an animal enters the corridor, the light reflected by the paws is detected by a camera placed 25 cm below the walkway. Each mouse was placed on the walkway at one extremity and allowed to move in the corridor in both directions. Only runs lasting a maximum of 10 s with 60% maximal speed variation were counted. Five successful runs per animal were recorded. The swing (s) defined as the duration of no contact of a paw with the glass plate was analyzed separately for forelimbs and hindlimbs and averaged between left and right sides.
Optical coherence tomography (OCT) and electroretinography (ERG)
OCT
Pupils were dilated with a bilateral topical application of atropine (1%, Virbac). Animals were then anesthetized using isoflurane (4% for induction, 2% for maintenance in a 0.4 L/min flow of 50%-50% mix of air and O2) and placed on the stage of a Bioptigen R2200 spectral domain OCT (Bioptigen). Cornea was kept moisturized with a thin layer of ocular gel (Ocry-gel, TVM) applied with surgical eye spears. For each eye, a rectangular scan was obtained, corresponding to a region of 1.4 × 1.4 mm centered on the optic nerve. The thicknesses of the whole retina (including the pigmentary epithelium), the segments, the outer nuclear layer (ONL), the inner nuclear layer (INL), and the inner plexiform layer (IPL) for a given eye were calculated as the average of 2 measures performed manually at 0.5 mm from the optic nerve center using the Bioptigen InVivoVue 2.4.33 software.
ERG
Pupils were dilated and cornea moisturized as described above. Mice were then anesthetized with an intraperitoneal injection of a mixture of ketamine (100 mg/kg) and medetomidine (1.2 mg/kg, Domitor). Mice were placed on a platform on top of a chemical heating pad (Aquapack, China). A DTL thread was used as the active electrode and placed across the cornea. To minimize invasiveness, we replaced the subcutaneous needles traditionally used as reference and ground electrodes, respectively, by a gold loop placed in the mouth (around the incisors), and a finger clip electrode placed on the tail with conductive paste. The platform was then moved forward to position the mouse head inside a Ganzfeld with flashes controlled with a Visiosystem (SIEM Bio-médicale). The background light was set at 25 Cd·m−2. Single-flash photopic responses were recorded for intensities of 1, 3, and 10 Cd·s·m−2. and flicker responses were recorded at frequencies of 2, 5, 10, 20, and 25 Hz with three Cd·s·m−2 flashes. The b-wave amplitude was measured offline with the Visiosystem software. At the end of the test, mice received a subcutaneous injection of atipamezole (10 mg/kg, Antisedan) to counteract the action of medetomidine, and were placed on a heating plate set at 37°C until awakening.
Electrophysiology
Electromyography
Recordings were performed using the Natus UltraProS100 apparatus (Mag2Health). Mice were anesthetized using intraperitoneal injection with ketamine (130 mg/kg)/xylazine (13 mg/kg). Animals were maintained at 37°C during the whole experiment until awakening. Latency and amplitude of M and H waves were recorded in the plantar hind paw muscle after sciatic nerve stimulation (0.1 ms and 8 mA intensity).
Electrophysiology of PCs
Following anesthesia by isoflurane inhalation, cerebellum extraction was done in ice-cold (∼1°C) ACSF bubbled with carbogen (95% O2, 5% CO2) containing the following (in mm): 120 NaCl, 3 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2.5 CaCl2, 2 MgCl2, 10 D-glucose, and 0.05 minocyclin. Cerebella were sectioned (Microm HM650V) in an ice-cold low-sodium and zero-calcium slicing buffer containing the following (in mm): 93 NMDG, 2.5 KCl, 0.5 CaCl2, 10 MgSO4, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 3 Na-pyruvate, 2 thiourea, 5 Na-ascorbate, 25 D-glucose, and 1 kynurenic acid. Sagittal slices 300 µm thick were immediately transferred in bubbled ACSF for 30 min at 34°C and maintained at room temperature (∼25°C) in bubbled ACSF for at least 1 h before transfer in a recording chamber, continuously perfused with bubbled ACSF. In order to block inhibitory and excitatory synaptic transmission, blockers of GABAA receptors (100 μm picrotoxin) and ionotropic glutamate receptors (1 mm kynurenic acid) were added in ACSF. PCs located in lobules VI or IX/X were visually identified under infrared illumination. All recordings were performed at near physiological temperature (32 ± 0.5°C) using an AxoPatch 200A (Molecular Devices). PC discharges were monitored using extracellular recordings with pipettes filled with 0.5 m NaCl (resistance: 15-30 mΩ). Data were acquired using WinWCP 4.5.4 freeware (John Dempster, University of Strathclyde, Glasgow; RRID:SCR_014713). Recordings were sampled at 50 kHz and filtered at 2 kHz. In extracellular recordings, action potentials were detected using OpenElectrophy, an open source software (http://neuralensemble.org/OpenElectrophy/; RRID:SCR_000819) (Garcia and Fourcaud-Trocmé, 2009). Data were stored in SQL databases (Oracle). Spike trains were analyzed using Python software written in-house (SynaptiQs developed by Antoine Valera) and custom-based routines. discharge irregularity was assessed by two parameters: (1) the mean of interspike intervals (ISIs); and (2) coefficient of variation for adjacent ISIs (CV2) calculated as follows:
where ISIn and ISIn+1 are two consecutive ISIs. An average of CV2 over all consecutive pairs of ISIs estimates the intrinsic variability of PC pacemaking activity. This parameter is independent of slow variations in the firing pattern.
Neuroanatomical study
Neuroanatomical study was conducted using 10 mice at 33 weeks (n = 5 per group) and 12 mice at 50 weeks (n = 6 per group). Brain samples were fixed in 4% buffered formalin for 48 h. Seventy-eight brain parameters, made of area and length measurements, were taken blind to the genotype across three coronal sections as described previously (Mikhaleva et al., 2016). Data were analyzed using a t test with equal variances to determine whether a brain region is associated with neuroanatomical defect or not.
MRI
After PFA perfusion of 25-week-old mice, brains were removed and stained for 1 week in a solution of gadolinium (Dotarem) in PBS at 2.5 mm. This protocol enhances the signal- and contrast-to-noise ratios on MR images of fixed brains (Dhenain et al., 2006). Ex vivo MRI experiments were performed on a horizontal 11.7 T Bruker scanner (Bruker). A quadrature cryoprobe (Bruker) was used for radio frequency transmission and reception. High-resolution anatomic images were acquired using EPI sequence (Resolution = 100 × 100 × 200 µm3, TR = 3000 ms, TE = 28 ms). In order to preserve integrity to avoid deformations, brains were kept inside skulls for ex vivo MRI experiments. Anatomical images were coregistered in the same space to create a study template. Transformations to match the study template to an atlas composed of a high-resolution template, and a map of region's labels adapted from the Allen Mouse Brain Atlas (Lein et al., 2007) was calculated. Finally, transformations to match the study template and labels to each individual anatomic image were calculated. The automated segmentation pipeline was performed using an in-house Python library (Sammba-MRI; https://github.com/sammba-mri/sammba-mri).
Immunofluorescence
Cryosections
Mice were transcardially perfused with 1× PBS followed by 4% PFA/1× PBS. Brains and eyecups were immediately dissected and incubated in 4% PFA/1× PBS overnight or for 2 h, respectively. They were washed 3 times in 1× PBS before a 24 h incubation in 30% (brain) or 20% (eyecup) sucrose for cryoprotection and then mounted in Shandon Cryomatrix embedding resin (Thermo Fisher Scientific) and immediately frozen; 14-20 μm brain sagittal and 12 μm eyecup cryosections (Leica Microsystems, CM 3050S) were collected and stored at −80°C.
Vibratome sections
Brains were fixed as described above, washed 3 times in 1× PBS, and mounted in 2% agarose/1× PBS. Vibratome sagittal sections (Leica Microsystems, VT 1000S) of 50 μm were collected, washed in 1× PBS, next in 20% glycerol/1× PBS, and stored in 60% glycerol/1× PBS at −20°C. The 60% glycerol/1× PBS was washed out with 1 wash of 20% glycerol/PBS and three washes of 1× PBS before immunofluorescence.
Cryosections were permeabilized in PBSTx (1× PBS/0.1% Triton X-100) and blocked for 1 h at room temperature with PBSTx supplemented with 7% deactivated FCS. Sections were incubated overnight at 4°C with primary antibodies in a blocking buffer using the following dilutions: 1:20,000 (cryosections) or 1:5,000 (floating sections) anti-CALB1 (rabbit, D28k, Swant, CB38a); 1:5000 (cryosection) or 1:1000 (floating section) anti-CALB1 (mouse, D28k, Sigma Millipore, C9848); 1:1000 (floating sections) vGLUT2 (guinea pig, Merck, AB2251-I); 1:10,000 anti-vGLUT1 (guinea pig, Millipore, AB5905); 1:1000 anti-ATXN7 (rabbit, 1261; Institut de Génétique et de Biologie Moléculaire et Cellulaire [IGBMC]); 1:700 anti-GFAP (mouse, Sigma Millipore, G3893). Secondary goat anti-rabbit, goat anti-mouse, goat anti-guinea pig IgG antibodies (Thermo Fisher Scientific, dilution 1:1000) conjugated with Alexa-555, -488, and -488 fluorophores, respectively, were applied for 1 h at room temperature and subsequently washed 3 times in PBSTx. Nuclei were counterstained with 1 μg/ml DAPI (4′,6-diamidino-2-phenylindole dihydrochloride). Controls without primary antibodies were performed in parallel. Slides were mounted with Aqua Poly Mount (Polysciences), dried and stored at 4°C protected from light.
Floating cerebellar sections were processed as described above with the following modifications. Sections were incubated in Eppendorf tubes for 3 d at 4°C with primary antibodies in 300 μl of blocking buffer. Secondary antibodies were applied for 2 h at room temperature. Sections were positioned on slides directly before mounting.
Retina images were acquired on a Leica DM 4000B with a Cool SNAP HQ2 camera with 40× objective with zoom factor 2.5 and processed with Fiji. Cerebellum images were acquired either on a confocal microscope (SP8, Leica Microsystems) with 40×/1.3 oil objective with a zoom factor 1 or on a Spinning disk with Leica DMI 8 inverted microscope equipped with Hamamatsu Orca flash 4.0 Camera 5 (Gataca Sytem) connected with MetaMorph software (Molecular Devices). For the acquisitions, HC PL APO 40×/1.3 oil objective and three diode lasers (405, 488, and 561 nm) were used. Image brightness and contrast were equivalently adjusted in Fiji for display purposes when necessary.
PC size and circularity measurements
Sagittal floating sections (50 µm) of central vermis were immunostained with anti-calbindin 1 antibody and acquired using confocal SP8 (Leica Microsystems) microscope. The obtained z stacks were subdivided into three groups of 15 slices using Fiji. After background subtraction, the PC layer was outlined manually in the vicinity of PC somas. Next, a customized Fiji macro allowing an automatic detection and measurement of the PC area and circularity was applied. Edge PCs were excluded from measurements.
Morphometry of cerebellar vermis sections
Sagittal 20 µm sections of central vermis were immunostained with anti-CALB1 antibody and counterstained with DAPI. Sections were scanned with Digital Slide Scanner Nanozoomer 2.0 HT (Hamamatsu) using 40× magnification. Images of the cerebellum were visualized using NDPI viewer and exported with 5× magnification into tiff format. Length and width of cerebellar sections as well as the thickness of the molecular cell layer (MCL) were measured using Fiji software.
vGLUT2 density, volume, and extension in the MCL
Sagittal floating sections (50 µm) of central vermis were immunostained with anti-calbindin 1 and anti-vGLUT2 antibodies, counterstained with DAPI. Sections were acquired using YoKogawa CSU W1 Spinning disk with Leica DMI 8 inverted microscope and Hamamatsu Orca flash 4.0 Camera (Gataca Sytem) and a driver by the MetaMorph software (Molecular Devices). For the acquisitions, HC PL APO 40×/1.3 oil objective and three diode lasers (405, 488, and 561 nm) were used. Stacks consisted of 225 z slices with 0.2 µm z step using 6.5 µm × 6.5 µm pixel size.
Images were saved in tiff format. Subsequently, RGB channels were split in Fiji. Channel with vGLUT2 acquisition (.ims format) was exported to Imaris version 9.3.1 (Bitplane AG) for 3D reconstruction. The ROI, namely, the MCL, was selected and 3D volumes were created with the surface module of Imaris. Next, vGLUT2 was analyzed only on this region with the Imaris spot module. The detection of objects in three-dimensional space was based on both intensity and size (with the estimated XY diameter of 1 µm). Only spots ranging from 0.52-30 µm3 were selected to eliminate background. Density and volume measurements of spots were analyzed in Excel and GraphPad Prism 6. The extension of vGLUT2-positive climbing fiber (CF) terminals along the dendrites of PC was measured using Fiji. Namely, the ratio of the extension of the 3D reconstructed vGLUT2 spots in the MCL over the total length of the MCL was calculated for the central part of lobule VI and for the side part of lobule X.
Histology and electron microscopy analysis
Eyes were prepared for histology and electron microscopy as in Yefimova et al. (2010). Semi-thin (2 μm) sagittal sections were cut with an ultramicrotome (Leica Ultracut UCT), stained with toluidine blue, and histologically analyzed by light microscopy. For sciatic nerve preparation, animals were perfused with 5 ml of PBS and 10 ml 4% PFA in saline, and tissues were fixed in 2.5% PFA/2.5% glutaraldehyde in cacodylate buffer. Ultra-thin (70 nm) sections were cut and contrasted with uranyl acetate and lead citrate and examined at 70 kV with a Morgagni 268D electron microscope. Images were captured digitally by Mega View III camera (Soft Imaging System), and contrast was adjusted for display purposes.
Western blot analysis
Cerebellum and retina were dissected and snap frozen in liquid nitrogen. Their whole-cell extracts were obtained by lysis and sonication of the tissue in, respectively, 500 µl and 150 µl of the buffer containing 50 mm HEPES-KOH, pH 7.5, 140 mm NaCl, 1 mm EDTA, 0.1% Na-deoxycholate, 1% SDS, 1% Triton-X100, 1× Protease Inhibitor Cocktail (Roche), 20 mm N-ethylmaleide, and 20 mm nicotinamide. The last two compounds were not used for ATXN7 detection. Protein concentrations were measured using Bradford protein assay; 10 µg (histone analysis) and 30 µg (other analysis) of total proteins were used for electrophoresis. Electrophoresis was run in 4%-12% Bis-Tris Protein Gels in NuPAGE MOPS SDS Running Buffer (Thermo Fisher Scientific) for histone analysis and in NuPAGE 3%-8% Tris-Acetate Protein Gels in NuPAGE Tris-Acetate SDS Running Buffer (Thermo Fisher Scientific) for ATXN7 detection. Following electrophoresis, proteins were transferred on nitrocellulose membrane for 1.5 h at 200 mA or overnight at 30 mA. Antibodies were diluted in 1× PBSTM (1× PBS with 0.1% Tween, and 5% nonfat milk). The following primary antibodies were applied overnight at 4°C: anti-ATXN7, 1:1000 (Thermo Fisher Scientific, PA1-749); 1:20,000 anti-vGLUT1 (Millipore, AB5905), 1: 2000 anti-vGLUT2 (Millipore, AB2251), anti-RGS8, 1:1000 (Novus, NBP2-20 153), anti-FAM107b, 1:500 (Abcam, ab122566), 1:10,000 anti-IP3R1 (Novus, NB120-5908), anti-CALB1, 1:5000 (D28k, Swant, CB38a), anti-ALDOC, 1:10,000 (#69075; TMDU, lab Sugihara), anti-H2B, 1:10,000 (IGBMC, clone H2-2A8); anti-H2Bub 1:2000 (MediMabs, MM-0029); anti-H3K9ac 1:2000 (Abcam, ab4441); anti-GAPDH 1:10,000 (Merck Millipore, MAB374); anti-TUBB 1:2000 (IGBMC, 2A2), anti-histone H3, 1:20,000 (IGBMC). Secondary antibodies are as follows: Peroxidase AffiniPure F(ab′)2 Fragment Goat Anti-Rabbit IgG (H + L) (GARPO; 1:10,000, Jackson ImmunoResearch Laboratories), Peroxidase AffiniPure Goat Anti-Mouse IgG (H + L) (GAMPO; 1:10,000, Jackson ImmunoResearch Laboratories), and Donkey anti-Goat IgG (H + L) HRP (DAGPO, 1:10,000, Thermo Fisher Scientific) were applied in 1× PBSTM for 2 h at room temperature, washed 3 times in 1× PBST, and proteins were detected using SuperSignal West Pico PLUS Chemiluminescent kit (Thermo Fisher Scientific). Chemiluminescent signal was acquired on GE HealthcareImager 600. Images were analyzed using Fiji.
RNA-Seq library preparation and sequencing
RNA-Seq libraries (3 independent samples/genotype) were prepared from 300 ng of total cerebellar RNA using TruSeq Stranded mRNA Library Prep Kit and TruSeq RNA Single Indexes kits A and B (Illumina), according to the manufacturer's instructions. Briefly, following purification with poly-T oligo-attached magnetic beads, mRNAs were fragmented using divalent cations at 94°C for 2 min. RNA fragments were reverse-transcribed into first-strand cDNA using random primers. Strand specificity was achieved by replacing dTTP with dUTP for the second-strand cDNA synthesis using DNA Polymerase I and RNase H. Following addition of a single A base and subsequent ligation of the adapter on double-stranded cDNA fragments, products were purified and amplified by PCR (30 s at 98°C [10 s at 98°C, 30 s at 60°C, 30 s at 72°C] × 12 cycles; 5 min at 72°C) to create the cDNA library. Surplus PCR primers and adapter dimers were removed by purification using AMPure XP beads (Beckman-Coulter) and the final cDNA libraries were checked for quality and quantified using capillary electrophoresis. Libraries were sequenced on Illumina Hiseq 4000 Sequencer as Single-Read 50 base reads following Illumina's instructions. Image analysis and base calling were performed using RTA 2.7.3 and bcl2fastq 2.17.1.14. Adapter dimer reads were removed using DimerRemover (https://sourceforge.net/projects/dimerremover/). The RNA-seq data have been submitted to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under GSE138527 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE138527).
RNA-Seq data analysis
Image analysis and base calling were performed using RTA 2.7.3 and bcl2fastq 2.17.1.14. Reads were mapped onto the mm10 assembly of mouse genome using TopHat version 2.0.14 (Kim et al., 2013) with bowtie version 2.1.0 aligner. Gene expression was quantified using HTSeq version 0.6.1 (Anders et al., 2015) and gene annotations from Ensembl release 86. Statistical analysis was performed using R and DESeq2 version 1.6.3 Bioconductor library (Love et al., 2014). Gene functional annotations were performed using Ingenuity Pathway Analysis (IPA) and String version 10.5 software (Kramer et al., 2014; Szklarczyk et al., 2015).
Analysis of public datasets
For SCA1 dataset (Gene Expression Omnibus: GSE75778), FASTQ files were retrieved from Sequence Read Archive using fastq-dump (SRA Toolkit version 2.8.0, BioProject PRJNA305316, samples: GSM1967518-GSM1967520; GSM1967524-GSM1967529; GSM1967533-GSM1967538; GSM1967542-GSM1967544), and only the first 50 bases of read 1 sequences were retained for further analysis. For SCA2 dataset (Array Express: E-MTAB-6293), FASTQ files were retrieved from Sequence Read Archive using fastq-dump (SRA Toolkit version 2.8.0, BioProject PRJEB24319, samples ERR2237861-ERR2237880, ERR2237882-ERR2237893). Reads were then mapped onto the mm10 assembly of mouse genome using TopHat version 2.0.14 (Kim et al., 2013) with bowtie version 2.1.0 aligner. Gene expression was quantified using HTSeq version 0.6.1 (Anders et al., 2015) and gene annotations from Ensembl release 90. Statistical analysis was performed using R and DESeq2 version 1.18.1 (Love et al., 2014).
Analysis of cell type-specific gene deregulation in SCA7 cerebellum
To gain insight into the cell type-specific distribution of differentially expressed genes in SCA7 cerebella, we used published datasets of Doyle et al. (2008). In this study, 9 BacTRAP transgenic mice were engineered to selectively purify 9 different cerebellar cell-type populations (PCs, granule cells (GCs), Golgi cells, stellate and basket cells, unipolar brush cells, oligodendrocytes expressing Olig2 or Cmtm5, Bergmann glia, and astrocytes) from the whole cerebellum. For each cell type, the actively translated mRNA profile (translatome) was determined using Affymetrix microarray analysis.
We first reannotated gene names according to the updated mm10 annotation using the Affymetrix NetAffx Annotation File (Mouse430_2 Annotations, Release 36). The reannotation showed that a total of 9671 genes are expressed in the 9 cell types. From these, we identified that 5058 genes are exclusively present in a single cell type and that 4613 genes are present in more than one cell type dataset. Then, using this information, we could assign that, among the 676 deregulated genes in SCA7 mouse cerebellum, 240 were specific to a single cerebellar cell type while another 230 were present in more than one cell type; 206 genes had no assignment because they were not reported in the datasets of Doyle et al. (2008).
qRT-PCR
Total RNA samples were extracted from 1 cerebellum/sample or 2 retinas/animal/sample using TriReagent (MRC) following the manufacturer's instruction. For the subregional qRT-PCR analysis, anterior (lobules II-V) and posterior (lobules VI-IX) vermis, as well as lobule X were cut apart from freshly dissected cerebellum under a binocular, and total RNA samples were also extracted using TriReagent; 500 ng of purified RNA was then reverse-transcribed using QuantiTect Reverse Transcription kit (QIAGEN) according to the manufacturer's protocol. The qRT-PCRs were performed on obtained cDNAs (1/20 dilution) using QuantiTect SYBR Green PCR Master Mix (QIAGEN), a Light Cycler 480 (Roche) and specific primers as follows: Calb1 (forward (F): GCAGAGTACACAGACCTCATG, reverse (R): ATGTATCCGTTGCCATCCTG); Grid2 (F: TGTGGTTTGTGTATGGGTCC; R: GGGTGATAGTGAGGAAAGCG); Rora (F: CGCTAGAGGTGGTGTTTATTAGG; R: ATTTCGTCTTCGGTCAGGTG); Rgs8 (F: GAAAGGTGGACAAGGTGGAG, R:TTGACAGACTGAAGGCAAGG); Fam21 (F: GTGACCTCTTTGCTGACTCTG, R: ATCTGTTTGTCCCTCTTTCCG); Fam107b (F: AAATCACCAAGACCTCCACAG, R:TGCTTTATCACTTGGTCTCGTC); Pcp4 (F: TGCCTTCTGAGCTGTTCTG, R:TCTGAACTGAGACTGAATGGC); 36B4 (Rplp0): (F: ACCCTGAAGTGCTCGACATC, R: AGGAAGGCCTTGACCTTTTC); Rho (F: ACCCTCTACACATCACTCCA, R: AAGACCACACCCATGATAGC); Opn1sw (F: CACCTGGTTCCTCTTCATCTTC, R: GTTCAGCCTTTTGTGTCGTAG); Opn1mw (F: GCTACTTCGTTCTGGGACAC; R: CAAATCTCACATTGCCAAAGGG); Hprt (F: GTAATGATCAGTCAACGGGGGAC, R: CCAGCAAGCTTGCAACCTTAACCA); Foxp4 (F: CCAGTTCCCACCCTCTTTATG, R: CAAAGCGTGTTCTGTGTTGAG); Orai2 (F: CTGGAACTCGTCACGTCTAAC, R: AGGGTACTGGTACTTGGTCTC); Kctd12 (F: CCAGAGCGAGGACAAGATC, R: AGAAGGAATCGGGAAACAGG); Prkg1 (F: AAGGGAGATTGGTTTGGAGAG, R: TGCTTCTGCGTCCTCATATG); Gng13 (F: TCTCTGCTTTTGCTGTCTCC, R: TCAATCCACTTGAGAAGCTCG); Itpr1 (F: GATGCTAAAGAGGGACAGAAGG, R: GCAGCGGAGAATGAGATCAAC); Plcb3 (F: CTGCCCTGCTTATCTACACTG, R: GGTCTCCTGATATGTCTCTGTG); Plcb4 (F: CAAACCCCAGATTTAGCGATG, R: GGAACTGCCCTGATATAACCTG); Aldoc (F: TATTGTGGAGCCTGAGATTCTG, R: CTTCTGGGCTATACTTGATGGG); Gabbr1 (F: CGTTATATCCAGAACTCCCAGC, R: GAACATAGAGCCATAGCCCAG). Gene expressions were normalized based on the expression of Hprt or 36B4 (Rplp0).
Experimental design and statistical analysis
Information on number (n) values and what n represents (e.g., animals, sex, biological replicates) and definition of center and dispersion and precision measures (e.g., mean, median, SEM) can be found in the figure legends. Data were analyzed using GraphPad Prism 7 or 8 or Stat view. Differences between means were assessed with unpaired Student's t test, ANOVA, or ordinary two-way ANOVA or two-way ANOVA with repeated measurement, followed by post hoc testing of pairwise comparisons among genotypes (Bonferroni correction), as indicated. Differences between median were analyzed using Mann–Whitney test. Survival of mice with different genotype and sex was plotted using the Kaplan–Meier method, and statistical comparisons were performed using the Log Mantel-Cox test. Distribution of PC size and circularity was analyzed using χ2. Further information is indicated in the figure legends. Significance was established at p < 0.05. Data are mean ± SEM unless indicated. Datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Results
General phenotypic features of SCA7 mice carrying 140 CAG repeats
We performed the phenotypic characterization of the SCA7140Q/5Q mouse line, which is derived from the SCA7266Q/5Q knock-in mouse model by CAG repeat contraction (Yoo et al., 2003). SCA7266Q/5Q mice show a very severe infantile-like pathology and early death. SCA7140Q/5Q heterozygous mice have a median lifespan of 54 weeks for both males and females and SCA7140Q/140Q homozygous mice have a much shorter lifespan than heterozygotes (Fig. 1A). Age-related gain in body weight of SCA7140Q/5Q and SCA7140Q/140Q mice significantly differed from their WT littermates. SCA7140Q/5Q females and males gained weight at the same rate as WT mice up to 12 and 14 weeks, respectively, and then plateaued until 24 weeks with an underweight of 18% for females and 20% for males compared with WT of the same sex and age (Fig. 1B). Subsequently, the weight of SCA7140Q/5Q decreased, while remaining stable for WT mice. The body weight of SCA7140Q/140Q mice plateaued earlier at 10 weeks for males and 12 weeks for females, and strongly decreased after 20 weeks. Compared with age-matched WT male and female littermates, the body lengths of adult SCA7140Q/5Q and SCA7140Q/140Q males were significantly shorter by 6.6% (p = 0.0121) and 23% (p < 0.0001), respectively, and those of adult SCA7140Q/5Q and SCA7140Q/140Q females by 6.5% (p < 0.0034) and 21% (p < 0.0001), respectively (Fig. 1C). SCA7140Q/5Q mice, which represent the genetic status of most SCA7 patients, were then used for a detailed phenotypic characterization.
Figure 1.

Lifespan, and body size and composition of SCA7 mice. A, Kaplan–Meier survival curves indicate that SCA7140Q/5Q heterozygous (HTZ) and SCA7140Q/140Q homozygous (HMZ) males (M) and females (F) have shorter lifespan than WT littermates (n = 11 WT F, 5 WT M, 9 HTZ F, 4 HTZ M, 6 HMZ F, 5 HMZ M). B, Gain in body weight of males and females of SCA7 HTZ and SCA7 HMZ mice differs from WT littermates. Two-way ANOVA with repeated measurement (males-genotypes: F(2,20) = 62.06, p < 0.0001; females-genotypes: F(2,22) = 13.92, p = 0.0001) and post hoc Bonferroni test (*p < 0.05; **p < 0.01; ***p < 0.001) (n = 11 WT F, 5 WT M, 8 HTZ F, 9 HTZ M, 6 HMZ F, 9 HMZ M). C, Body length of 38-week-old SCA7 HTZ and SCA7 HMZ mice differs from WT littermates. One-way ANOVA (males: F(2,17) = 62.45, p < 0.0001; females: F(2,20) = 50.07, p < 0.0001) and post hoc Bonferroni test (n = 11 WT F, 5 WT M, 8 HTZ F, 9 HTZ M, 4 HMZ F, 6 HMZ M). D, Analysis of proportion of lean and fat tissues normalized to animal body weight, bone area, bone mineral density (BMD), bone mineral content (BMC), and BMC normalized on body weight. SCA7 HTZ mice show no difference in body composition compared with WT mice, except for the BMC of females and bone area for females and males. Two-tailed Student's t test (n = 4 mice/genotype). B-D, Data are mean ± SEM.
Examination of general physical appearance and body shape of SCA7140Q/5Q mice did not reveal gross morphologic abnormalities at 28 weeks of age. The proportion of lean, fat, and bone tissues was similar to their WT littermates, when normalizing to body weight (Fig. 1D). The only exceptions were the total bone area that was smaller in SCA7140Q/5Q males and females compared with WT mice, and the bone mineral content that was smaller in SCA7140Q/5Q females. In contrast to the severely affected SCA7266Q/5Q mice, SCA7140Q/5Q mice presented no complication of male fertility and offspring delivery (data not shown). From ∼40 weeks of age, SCA7140Q/5Q mice progressively showed severe motor deficit and were easily distinguishable from control animals because of their smaller body size and hypoactivity, and some with a hunched posture (kyphosis) and tremors. Because of the poor health status and following recommendation of our animal health regulator, SCA7140Q/5Q mice were generally killed at this stage.
Degeneration of photoreceptors, pigmented epithelium, and Bruch's membrane
Previous studies of SCA7 mouse retinopathy mainly focused on photoreceptor pathology. To get a broader insight into the SCA7140Q/5Q mouse retinopathy, longitudinal analysis was made by correlating noninvasive OCT and ERG, and histologic and molecular analysis. The OCT measurement of 8 different morphologic parameters indicated that the overall structure of SCA7140Q/5Q retina was normal at 9 weeks (Fig. 2A–I). However, from 15 weeks, photoreceptor segment layers of SCA7 mice progressively got thinner with age compared with WT littermates (Fig. 2C). The similar thinning was observed for the ONL and the retina thickness of SCA7 mice but occurred later (23 weeks) and not as severely (Fig. 2B,D). There was no clear scalable alteration of thickness of the INL, the IPL, or any other eye structures in SCA7 mice (Fig. 2E–H), except for the anterior chamber that showed a significant reduction from 23 weeks onward (Fig. 2I).
Figure 2.

Retina alterations in SCA7 mice. A, Representative OCT B-scan showing reduced thickness of the segment layers of SCA7140Q/5Q mice compared with WT at 29 weeks. OS, outer segment; IS, inner segment. B-I, Longitudinal OCT measurements at 9, 15, 23, and 29 weeks show progressive thinning of retina thickness (genotypes: F(1,32) = 63.11, p < 0.0001; B), photoreceptor segment layers (genotypes: F(1,32) = 86.03, p < 0.0001; C), ONL (genotypes: F(1,32) = 17.93, p = 0.0002; D), and anterior (Ant.) chamber (genotypes: F(1,30) = 6.688, p = 0.0148; I) in SCA7140Q/5Q mice relative to WT littermates. The INL (E), IPL (F), vitreous (G), and cornea (H) show no steady difference between WT and mutant mice. Data are analyzed using two-way ANOVA with repeated measurement and post hoc Bonferroni test (n = both eyes of 8 WT and 9 SCA7 mice): *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. J, K, Longitudinal cone-derived b-wave amplitudes of photopic response to 1 Cd·s·m−2 flashes (J) and 5 Hz flicker responses to 3 Cd·s·m−2 flashes (K) show progressive reduction of cone function in SCA7140Q/5Q retina relative to WT retina. Background illumination was 25 Cd·m−2. Data are mean ± SEM and analyzed with two-way ANOVA with repeated measurement and post hoc Bonferroni test (n = single eyes of 5 WT and 5 SCA7 mice). L, qRT-PCR analysis shows early and progressive downregulation of Opn1mw and Opn1sw cone genes and Rhodopsin (Rho) rod gene in SCA7140Q/5Q retina, as shown by normalization to WT gene expression levels. Two-tailed Student's t test (n = 3-6/genotype).
ERG studies in SCA7 patients showed that cone dysfunction precedes rod dysfunction (Michalik et al., 2004). We evaluated cone function of WT and SCA7140Q/5Q mice by measuring the cone-derived b-wave amplitudes of photopic (single flashes ERG of 1, 3, and 10 Cd·s·m−2) and flicker responses (5 frequencies between 2 and 25 Hz) over time. At 5 weeks, b-wave amplitudes of both photopic and flicker responses of SCA7140Q/5Q mice were similar to WT littermates. However, at 10 weeks, the b-wave amplitudes of photopic and flicker responses of SCA7140Q/5Q mice significantly decreased relative to WT mice, between 32% (p = 0.035) and 36% (p = 0.045) for photopic flashes and between 37% (p < 0.012) and 45% (p = 0.007) depending on flicker frequency (Fig. 2J,K; Table 1). The loss of b-wave amplitudes of photopic responses and flicker further worsened with age, with reductions between 81% and 100% at 23 weeks.
Table 1.
Electroretinograph analysis of SCA7 mice
| 5 weeks (b-wave, µV) |
10 weeks (b-wave, µV) |
15 weeks (b-wave, µV) |
23 weeks (b-wave, µV) |
Two-way repeated-measures ANOVAa |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | SCA7 | p | WT | SCA7 | p | %b | WT | SCA7 | p | %b | WT | SCA7 | p | %b | G | A | GxA | ||
| Photopic (Cd·s·m-2) | 1 | 66.2 ± 22.8 | 65.8 ± 24.5 | 0,971 | 66.4 ± 16.4 | 42.4 ± 17.1 | 0,035 | 36% | 75.6 ± 16.8 | 28.2 ± 13.6 | <0.001 | 63% | 39.8 ± 13.3 | 6.4 ± 6 | 0,004 | 84% | <0.001 | < 0.001 | 0,046 |
| 3 | 132 ± 31.9 | 150 ± 41.5 | 0,384 | 132.4 ± 20.5 | 90.6 ± 42.7 | 0,049 | 32% | 134 ± 28.3 | 52.8 ± 34.9 | <0.001 | 61% | 90.4 ± 32 | 14.8 ± 16.9 | <0.001 | 84% | 0,002 | < 0.001 | 0,009 | |
| 10 | 167.6 ± 41.3 | 174.8 ± 25.2 | 0,776 | 164.8 ± 26.1 | 131.4 ± 58.1 | 0,194 | 20% | 171.4 ± 45.1 | 83.6 ± 53 | 0,001 | 51% | 124.4 ± 28.9 | 20.8 ± 23.8 | <0.001 | 83% | 0,005 | < 0.001 | 0,011 | |
| Flicker (Hz) | 2 | 135.8 ± 24.2 | 138 ± 35.1 | 0,910 | 137.6 ± 12 | 86.4 ± 50.9 | 0,012 | 37% | 136.4 ± 30.3 | 47.2 ± 32.3 | <0.001 | 65% | 96.6 ± 26.4 | 12.4 ± 14 | <0.001 | 87% | <0.001 | < 0.001 | 0,008 |
| 5 | 109.8 ± 27 | 135 ± 52.5 | 0,211 | 125.4 ± 13.5 | 68.4 ± 39 | 0,007 | 45% | 124.4 ± 28.1 | 43 ± 29.6 | <0.001 | 65% | 83.2 ± 27.5 | 11.6 ± 13.5 | 0,001 | 86% | 0,003 | < 0.001 | 0,002 | |
| 10 | 78.6 ± 22.3 | 104.2 ± 49.9 | 0,128 | 85.4 ± 9.3 | 52 ± 28.9 | 0,050 | 39% | 89.4 ± 18.7 | 37 ± 25 | 0,003 | 59% | 59 ± 16.7 | 11.2 ± 13.7 | 0,007 | 81% | 0,026 | < 0.001 | 0,004 | |
| 20 | 33 ± 14.8 | 48.8 ± 22.5 | 0,101 | 34.6 ± 11.8 | 23.6 ± 15.3 | 0,248 | 32% | 48 ± 17.6 | 23.4 ± 13.8 | 0,013 | 51% | 27.4 ± 12.1 | 0 | 0,006 | 100% | 0,067 | 0,001 | 0,006 | |
| 25 | 24.4 ± 12 | 39.2 ± 29.6 | 0,080 | 23.8 ± 6.7 | 15.2 ± 11.1 | 0,301 | 36% | 36 ± 10.4 | 15.8 ± 4.2 | 0,019 | 56% | 13 ± 4.9 | 0 | 0,122 | 100% | 0,169 | 0,001 | 0,027 | |
aG, Genotype; A, age; GxA, genotype × age. p values, post hoc Bonferroni test. Single eye of n = 5 mice.
bPercentage of decrease in SCA7.
The green and blue opsin (OPN1MW, OPN1SW, respectively) of cones and the rhodopsin of rods are light-sensitive pigments that initiate vision and represent major components of photoreceptor outer segments. qRT-PCR showed that expression of these photoreceptor identity genes progressively decreased with age in SCA7140Q/5Q mice (Fig. 2L). Notably, the level of Opn1mw and Opn1sw mRNAs already showed ∼65% (p < 0.0036) and 90% (p < 0.0043) reduction, respectively, in 10-week-old mutant mice, when cone-derived ERG responses began to decrease. The decrease of cone opsin mRNAs was earlier and more drastic than for the Rho mRNA, consistent with the primary alteration of cone function in SCA7 patients. Therefore, the loss of cone ERG of SCA7140Q/5Q mice closely correlates with the loss of cone opsins expression, and both occur before a significant shortening of segments detected by OCT.
Histology and electron microscopy indicated that photoreceptor outer segments are almost completely lost at 44 weeks in SCA7140Q/5Q mice and at 20 weeks in SCA7140Q/140Q mice, whereas inner segments showed only a slight reduction in both SCA7 genotypes, compared with WT retina (Fig. 3A,B). Additional degenerative signs of SCA7 photoreceptors included disorganization of the remnant outer segments, swollen mitochondria and vesicular membrane accumulation in inner segments, several dark-stained inner segments, and photoreceptor nuclei (Fig. 3C). Interestingly, we found striking anomalies of the retinal pigmented epithelium (RPE) and Bruch's membrane of SCA7140Q/5Q retina, never reported in previous models to our knowledge. First, the thickness of RPE was clearly increased in SCA7 mice compared with controls (Fig. 3D). Second, on electron microscopy, while the normal RPE (Fig. 3E) contains translucent infolding membranes in contact all along the basement membrane, SCA7 RPE showed discontinuous and opaque infolding membranes, with homogeneous deposits between the plasma membrane and the basement membrane (Fig. 3E). The Bruch's membrane of SCA7 retina showed enlargement and disorganization compared with WT in which the Bruch's membrane was thin and regular (Fig. 3E).
Figure 3.
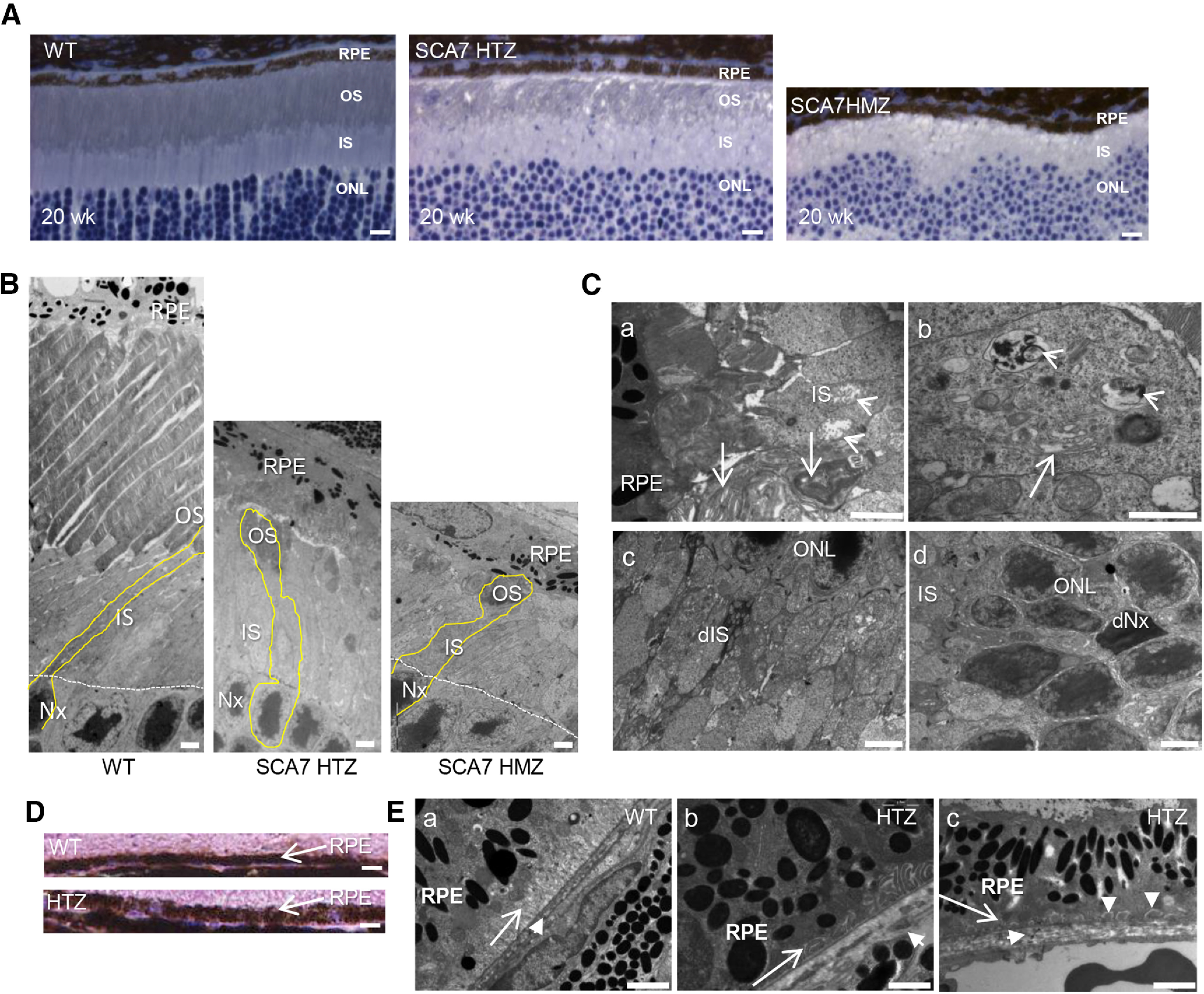
Tissular and cellular alterations of SCA7 mouse eye. A, Histologic sections of retina at 20 weeks represent the progressive thinning of outer segment (OS) layer in the SCA7140Q/5Q heterozygous (HTZ) retina and the absence of OS in the more severe SCA7140Q/140Q homozygous (HMZ) retina compared with WT retina. IS, Inner segment layer. Scale bar, 15 µm. B, Electron micrographs represent the progressive disappearance of OS and the shortening of IS of 44-week-old SCA7 HTZ and 20-week-old SCA7 HMZ, compared with WT mice. Photoreceptor nuclei (Nx). Scale bar, 10 µm. C, Electron micrographs of 20-week-old SCA7 HMZ retina. The remnant OSs have lost their parallel organization (arrows), and the IS contains swollen and disrupted mitochondria (arrowheads) (Ca). The IS shows dilated and vesiculated endoplasmic reticulum (arrow) and accumulation of vesicular membranes (arrowhead) (Cb). Dark degenerating IS (dIS) with numerous abnormal mitochondria (Cc). Dark photoreceptor nuclei (dNx) (Cd). Scale bars: Ca, Cb, 2 μm; Cc, Cd, 5 µm. D, Histologic sections comparing the thickness of WT and SCA7 RPE at 20 weeks. Scale bar, 15 µm. E, Electron micrographs comparing the basal infolding membrane of RPE and Bruch's membrane of WT and SCA7 mice. In WT (Ea), the basal infolding membrane of RPE (arrow) shows a typical lamellar organization with translucid lumen, and a regular Bruch's membrane (short arrow). In contrast, in SCA7 retina, the infolding membrane (long arrow) is either opaque (Eb) or completely absent (Ec), whereas the Bruch's membrane (short arrow) is enlarged and disorganized (Eb, Ec), and occasionally interrupted (Eb). In addition, homogeneous deposits are found between the basement membrane and plasma membrane of RPE (vertical arrowheads in Ec). Scale bar, 2 µm.
Deficits in motor activity and coordination
We performed longitudinal motor performance analysis of SCA7140Q/5Q and WT male littermates using a variety of motor activity paradigms. The spontaneous activity measured with the open field showed that SCA7140Q/5Q mice become significantly hypoactive at 16 weeks, and hypoactivity worsened with age (Fig. 4A–D). Specifically, they traveled shorter distances in the arena, ran slower, rested longer times, and made less rears than WT littermates. However, SCA7140Q/5Q mice did not show anxiety behavior, since they explored the central area similarly to WT mice (Fig. 4E).
Figure 4.

Behavioral and motor deficits of SCA7 mice. A-E, Open field analyses show that SCA7140Q/5Q males (n = 6) progressively become hypoactive relative to WT male littermates (n = 6), according to the distance traveled (genotypes: F(1,5) = 82.21, p = 0.0001; A), the resting time (genotypes: F(1,5) = 43.32, p = 0.0001; B), the average speed (genotypes: F(1,5) = 64.12, p = 0.0005; C), and the number (Nb) of rears (genotypes: F(1,5) = 11.71, p = 0.0188; D). However, SCA7 mice show no difference with WT mice as for the time spent in the arena center (genotypes: F(1,5) = 0.26, p = 0.6307) (E). Accelerating rotarod (mean latency to fall) shows progressive motor deficit of SCA7140Q/5Q males compared with WT males (genotypes: F(1,5) = 6.61, p = 0.0499; F). Notched bar [time to cross the bar (genotypes: F(1,123) = 27.96, p < 0.0001; G) and number of mistakes (genotypes: F(1,123) = 29.78, p < 0.0001; H)] indicates that SCA7140Q/5Q males (n = 10) progressively have altered motor coordination and balance compared with WT males (n = 12). Beam walking also shows that motor performances of SCA7140Q/5Q males (n = 10) mice are affected compared with WT males (n = 12) (genotypes: F(1,123) = 13.60, p = 0.0015; I). Spatial gait analysis using footprint patterns indicates that SCA7140Q/5Q males (n = 9-11) progressively have shorter stride length of the four limbs [only right forelimb shown (genotypes: F(1,74) = 4.75, p = 0.0325; J) and shorter stance width (only forelimbs shown (genotypes: F(1,74) = 10.11, p = 0.0021; K))] compared with WT littermates (n = 9-12). Temporal gait analysis using Catwalk shows that swing times of forelimbs [average of right and left forelimbs (genotypes: F(1,26) = 19.17, p = 0.0004; L), but not hindlimbs (average of right and left hindlimbs (genotypes: F(1,26) = 0.92, p = 0.3458; M)] of SCA7140Q/5Q males (n = 15) progressively become slower than those of WT males (n = 10). Grip strength performance of SCA7140Q/5Q males (n = 6) normalized to mouse body weight is similar to WT males (n = 6) (genotypes: F(1,40) = 0.19, p = 0.6613; N). O-S, Sixteen-week-old SCA7140Q/5Q females (SCA7-F) (n = 7) are hypoactive relative to WT female littermates (WT-F) (n = 11) in open field test, according to the distance traveled (O), resting time (P), average speed (Q), and Nb of rears (R), and 25-week-old SCA7-F (n = 7) show motor defect in rotarod relative to WT-F (n = 11) (S). Data are mean ± SEM. Two-way ANOVA with repeated measurement (A-F,I,L-N) or ordinary two-way ANOVA (G,H,J,K), followed by post hoc Bonferroni test: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; two-tailed Student's t test (O-S).
When comparing the mean latency to fall from the accelerated rotarod, SCA7140Q/5Q mice fell earlier than WT mice with statistically significant differences from the age of 24 weeks (Fig. 4F). On the notched bar test, SCA7140Q/5Q mice performed similarly to WT littermates up to 17 weeks (Fig. 4G,H). From 20 weeks, performance worsened relative to age-matched WT mice for the time required and the number of mistakes done when crossing the bar. For beam walking test, SCA7140Q/5Q mice took longer time to cross the bar from 20 weeks (Fig. 4I), while the number of mistakes was not significantly different between genotypes (data not shown).
The forward gait patterns were quantitatively assessed for spatial and temporal gait parameters, both for the forelimbs and hindlimbs, using, respectively, footprint and Catwalk, an automated gait analysis system. On footprint, SCA7 and WT mice performed similarly in all gait parameters until 17 weeks. From 21 weeks, SCA7140Q/5Q mice made significantly shorter steps (stride length) with forelimbs and hindlimbs of each body side (Fig. 4J). Similarly, the stance width between forelimbs or between hindlimbs was consistently shorter in SCA7140Q/5Q mice from 21 weeks (Fig. 4K). Temporal gait analysis as recorded by Catwalk showed that the swing duration of right and left forelimbs was significantly affected in SCA7140Q/5Q mice from 25 weeks, while the swing duration of hindlimbs was not affected (Fig. 4L,M). Finally, the grip strength test showed that the muscle strength of SCA7140Q/5Q mice normalized to animal body weight was similar to WT mice (Fig. 4N). Similar to males, SCA7140Q/5Q females showed initial motor defects at 16 weeks on open field test and at 25 weeks on rotarod test (Fig. 4O–S), and their motor performances worsened with age (data not shown).
Replicability of mouse phenotyping across laboratories has important implications in fundamental research and preclinical studies and is often hampered by methodological issues. To assess the replicability of SCA7 mouse phenotype, a second SCA7140Q/5Q mouse colony was raised in the husbandry of a different research center and was analyzed by independent experimenters and instruments. Mice from the second colony were tested using the standardized protocols established for the first colony (for further details, see Materials and Methods). As in the first colony, SCA7140Q/5Q males gained less weight than WT littermates, reaching a plateau at 14 weeks (Fig. 5A). However, the average weights of adult SCA7140Q/5Q and WT males from the second colony were 6.5% (p < 0.0001) lighter than the mice of the first colony (Fig. 5B), indicating a significant environment effect regardless of the genotype. As for the first colony, open field analysis revealed the hypoactivity of SCA7140Q/5Q mice from 16 weeks of age, with a decreased total distance traveled in the arena, a decreased average speed, an increased resting time, and a decreased number of rears (Fig. 5C–G). In contrast to the first colony, the exploration of the open central area suggested a tendency of SCA7 mice to anxiety behavior in the second environment. As for the first colony, SCA7140Q/5Q mice of the second colony took significantly longer time to cross the beam walking bar (Fig. 5H). However, accelerated rotarod, which had a smaller rod diameter than the previous apparatus used for the first colony, led to short latency to fall even for WT at baseline and hence did not reveal any difference in the latency between the two genotypes (Fig. 5I).
Figure 5.

Motor phenotypes of a second colony of SCA7 mice. A, Gain in body weight of a second colony of SCA7140Q/5Q males (SCA7-SCA2) (n = 10) is lower than their WT (WT-2) male littermates (n = 10) (genotypes: F(1,26) = 15.3, p = 0.0006). B, Comparison of body weight of adult males of the first colony [WT-1 (n = 5), SCA7-1 (n = 9)] and the second colony (WT-2, SCA7-SCA2) indicates that animals of the second colony have lower body weight, regardless of the genotype. Two-tailed Student's t test. C-F, Open field analyses show that SCA7-SCA2 mice are hypoactive relative to their WT-2 littermates from 16 weeks of age (distance: genotypes: F(1,26) = 14.3, p = 0.0008; resting time: genotypes: F(1,26) = 15.29, p = 0.0006; average speed: genotypes: F(1,26) = 14.34, p = 0.0008; nb of rear: genotypes: F(1,26) = 8.48, p = 0.0073). G, Open field indicates that SCA7-SCA2 mice spend less time in the arena center, compared with WT-2 littermates (genotypes: F(1,26) = 8.21, p = 0.0081). H, Motor performances on beam walking of SCA7-SCA2 mice are affected compared with WT-2 littermates (genotypes: F(1,26) = 9.41, p = 0.0050). I, Motor performance on accelerating rotarod (mean latency to fall) of SCA7-SCA2 mice is comparable with WT-2 littermates (genotypes: F(1,26) = 0.78, p = 0.3860). A, C-I, *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; two-way ANOVA with repeated measurement, followed by post hoc Bonferroni test. Data are mean ± SEM.
In summary, the hypoactivity of SCA7140Q/5Q mice revealed by open field tests appeared at an early time point (onset at 16 weeks), while defects in motor performances (as shown in rotarod of the first colony) and in specific proprioceptive abilities (as reflected by gait alteration in notched bar, beam walking, footprint, and catwalk) appeared later between 20 and 25 weeks, depending on the test. Except for rotarod, the onset and progression of motor phenotypes of SCA7140Q/5Q were replicable in two different research centers.
Morphologic and functional alterations of the PNS
A peripheral neuropathy was reported in some, but not all, neurophysiological studies in patients (van de Warrenburg et al., 2004; Horton et al., 2013; Salas-Vargas et al., 2015) and has never been explored in mouse models. To understand the cause of motor alterations in SCA7140Q/5Q mice, the activity of the PNS was first assessed over time by measuring the sciatic nerve function by electromyography. At 15 weeks, median amplitudes of plantar M-wave and H-wave of SCA7140Q/5Q mice were similar to WT littermates (Fig. 6A,B). However, both plantar M-wave and H-wave amplitudes were reduced by 21% (p = 0.0238) and by 33% (p = 0.0226), respectively, at 32 weeks in SCA7 mice. At 43 weeks, the plantar M-wave amplitude was reduced by 31% (p < 0.0153), while the H-wave was almost absent (95%, p < 0.0001). To correlate these functional abnormalities with structural status, sciatic nerve sections were analyzed by histology and electron microscopy. Compared with WT mice, a cross section of sciatic nerve of SCA7 mice showed clear alterations of myelinated fibers and decreased number of small fibers (Fig. 6C). On electron microscopy (Fig. 6D), several structural alterations were observed in myelinated fibers of SCA7140Q/5Q mice, including myelin degeneration and infolding-like structures as well as axonal modifications characterized by inner swelling tongue and autophagy. Nonmyelinated fibers displayed abnormal structures of Remak bundles. In conclusion, motor components of the sciatic nerve morphology and activity are affected in SCA7140Q/5Q mice.
Figure 6.

Peripheral nerve alterations in SCA7 mice. Electromyograph activities of the sciatic nerve. Amplitude of motor M-wave (A) and sensory H-wave (B) in SCA7140Q/5Q mice relative to WT littermates (15 weeks: n = 16 WT and 15 SCA7; 32 week: n = 22 WT and 21 SCA7; 43 week: n = 22 WT and 21 SCA7). Scatter plot represents median with interquartile range. Mann–Whitney test. C, Comparison of semithin sections of sciatic nerve of SCA7140Q/5Q and WT mice. The mutant shows irregular and degenerated myelinated fibers and loss of small fibers. Scale bars, 50 µm. D, Electron microscopy analyses of the sciatic nerve. Compared with WT littermates (Da-Dd), sciatic nerve section of SCA7140Q/5Q mice (De-Dl) at 43 week shows severe abnormalities in both myelinated and nonmyelinated fibers, including myelin degeneration (* in De, Dg, Dl), infolding-like structure (I in De, Dg), abnormal Remak bundle (∧ in De-Dg, Di-Dk), autophagy ($ in Df, Dh), and inner swelling tongue (ist in Dj). Scale bars: Da, Dj, Dk, 5 µm; Db-Dd, Df-Di, Dl, 2 µm; De, 10 µm.
Distinct morphologic alterations in SCA7 mouse brain
Brains of SCA7 patients show prominent neurodegeneration in the cerebellum and atrophy of other brain regions. To establish a systematic survey of brain damage of SCA7 mice, the morphology of three different coronal brain sections of SCA7140Q/5Q and WT mice was studied using neuroanatomical measurement of 78 anatomy parameters across 20 distinct brain subregions (Mikhaleva et al., 2016). At 33 weeks, there was no major surface difference between SCA7140Q/5Q mice and WT littermates (Fig. 7A,C). However, end-stage SCA7140Q/5Q mice (50 weeks) showed significant reduction of brain areas in the three coronal sections (from −6.7% to −8%) with both white and gray matter alterations. Atrophy of gray matter structures included the somatosensory cortex, the pons, whereas affected white matter regions comprised the corpus callosum, the anterior commissure, the internal capsule, and the fimbria of the hippocampus (Fig. 7B,C).
Figure 7.
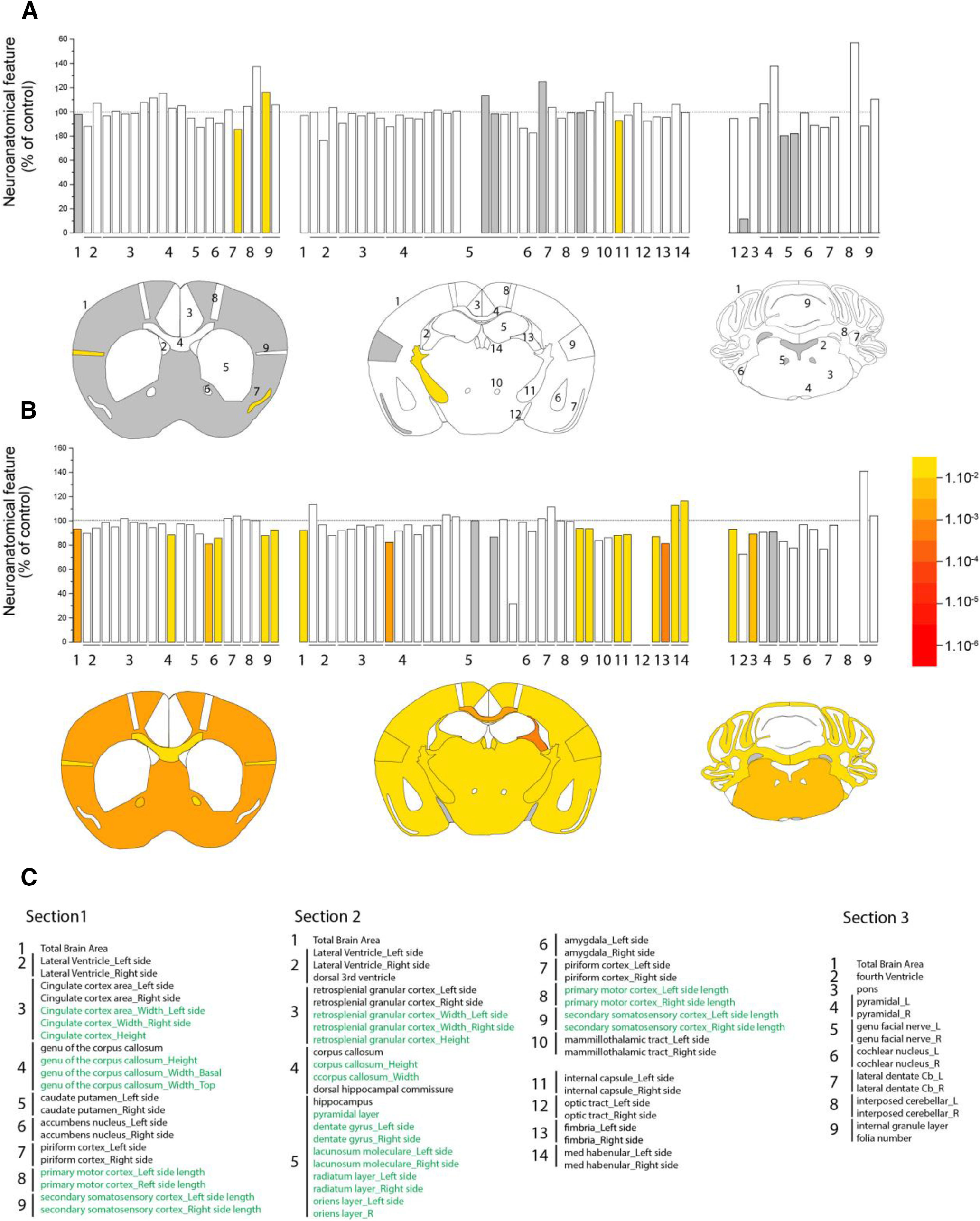
Neuroanatomical alterations in SCA7 mice. The figure summarizes systematic surface measurement of 78 anatomy parameters across 20 distinct brain subregions with a precision of 30 μm and down to 5 μm resolution. A, B, Top, Histograms represent the percentage increase or decrease of measured brain regions of SCA7 mice relative to the controls (100%) at 33 weeks (A) and 50 weeks (B). A, B, Bottom, Schematic representation of affected brain regions plotted in coronal planes according to p values in SCA7140Q/5Q mice versus WT littermates (5 or 6 mice per genotype). Left, Striatum section (bregma 0.98 mm). Middle, Hippocampus section (bregma −1.34 mm). Right, Cerebellum section (bregma −5.80 mm). White represents a p value >0.05. Gray represents missing data. Numbers indicate a total of 78 assessed brain regions (listed in C). C, List of assessed parameters and associated numbers across the three coronal sections. Green represents a length measurement. Black represents an area measurement. At 33 weeks (A), there is no major surface difference between SCA7140Q/5Q mice and WT littermates. At 50 weeks (B), the three coronal sections of SCA7 mouse brain show striking reduction (section 1: −6.7%, p = 0.0017; section 2: −8.1%, p = 0.0145; section 3: −7%, p = 0.0238) with both white and gray matter atrophy. Affected white matter regions include the corpus callosum (section 1: −11.6%, p = 0.0226; section 2: −17.8%, p = 0.0049, for the area of the soma of the corpus callosum), the anterior commissure (−17.8%, p = 0.0070, based on average of left and right hemispheres), the internal capsule (−11.9%, p = 0.0099, based on an average between the left and the right hemispheres), and the fimbria of the hippocampus (−13.9%, p = 0.0288). Altered gray matter structures are confined to the somatosensory cortex across two sections (section 1: −9.8%, p = 0.0055, based on an average between the left and the right hemispheres; section 2: −6.6%, p = 0.0004) and the pons of the cerebellum section (−10.9%, p = 0.0054). Data were analyzed using a Student's t test.
We then aimed to capture SCA7 mouse brain alterations at earlier disease stage using high-resolution MRI. MRI measurement clearly showed a global atrophy of the SCA7 mouse brains compared with WT at 25 weeks (p < 0.05) (Fig. 8A). Moreover, the decrease of whole-brain volume of SCA7140Q/140Q mice was 2 times higher than SCA7140Q/5Q (p < 0.05), confirming the stronger severity of the disease in homozygous mice. Interestingly, our automated segmentation pipeline highlighted specific brain regions particularly atrophied in the mouse model, such as the corpus callosum, and subparts of the hippocampus and cortex (Fig. 8B,C). No significant atrophy of the cerebellar cortex was measured in SCA7 mice, which can be because of strong variability of volume measurement in this structure.
Figure 8.
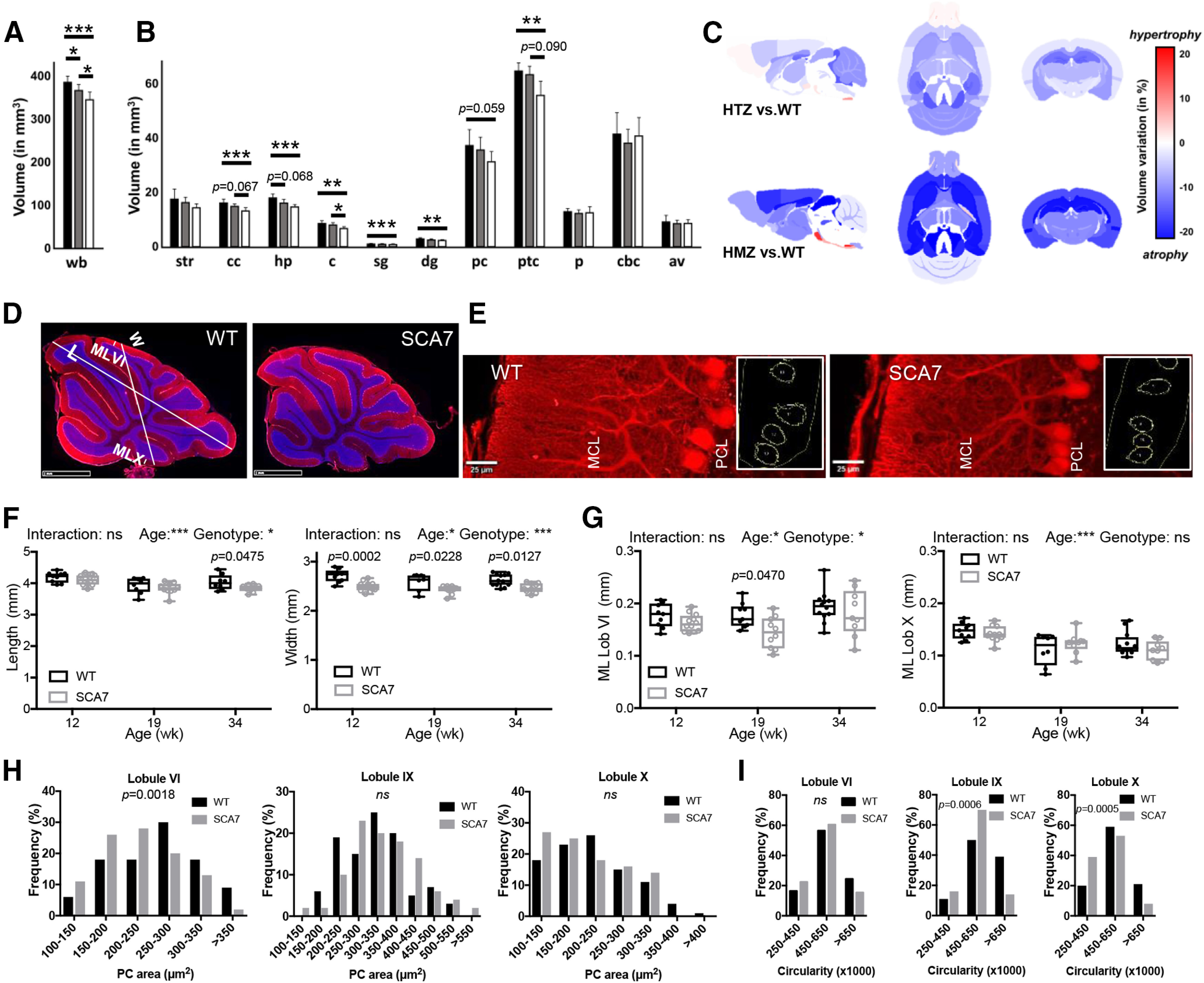
Brain region atrophy and morphologic alteration of PCs in SCA7. A-C, Ex vivo MRI measurement of whole-brain volume (A) and individual brain structures (B) represents differences between WT (n = 8), SCA7140Q/5Q (n = 9), and SCA7140Q/140Q (n = 6) mice at 24 weeks (black, gray and white bars, respectively). wb, Whole brain; str, striatum; cc, corpus callosum; hp, hippocampus; c, colliculus; sg, stratum granulosum; dg, dentate gyrus; pc, prefrontal cortex; ptc, parietotemporal cortex; p, pons; cbc, cerebellar cortex; av, arbor vitae. A, B, Data are mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; one-way ANOVA with post hoc Bonferroni test. Variation maps (C) of brain structure volumes of MRI that compare WT versus SCA7140Q/5Q heterozygous (HTZ) mice (top) and WT versus SCA7140Q/140Q homozygous (HMZ) mice (bottom). Variation of brain region volume was calculated in each ROI as follows: Variation = 100 × [Volume(SCA7) – Volume(WT))/Volume(WT)]. Statistical analysis was performed by a nonparametric Kruskal–Wallis test with correction for multiple comparisons. Probability levels are indicated above vertical bars when trends of difference between groups are observed. D, E, CALB 1 immunostaining (red) of cerebellar vermis section (D) and PCs (E) in lobule VI of the vermis of WT and SCA7 mice (DAPI in blue). Axis from lobule V to IX and axis from lobule VI to X in D are used to measure the vermis length (L) and vermis width (W). The CALB 1 staining in E is used to evaluate the PC soma size and circularity (right). PCL, PC layer. Scale bar, 20 µm. F, Longitudinal analysis of the cerebellar vermis length shows shortening at late disease stage (34 weeks) in SCA7140Q/5Q relative to WT mice, while the width has reduced size from 12 weeks onward in SCA7140Q/5Q mice (n = 8-12 tissue sections/age-genotype). G, Longitudinal analysis of the thickness of molecular layer (ML) of lobules (Lob) VI and X of cerebellar vermis sections shows no steady difference between SCA7140Q/5Q and WT mice (9-12 sections/mouse). F, G, Data points illustrated with interleaved box and whiskers represent minimal and maximal values. Ordinary two-way ANOVA with post hoc Bonferroni test. H, I, Frequency of PC soma area (H) and frequency of PC circularity index (I) as measured in lobules VI, IX, and X of the vermis of 34-week-old WT (n = 3) and SCA7140Q/5Q mice (n = 3) (33-51 PC/lobule/mouse were measured). SCA7140Q/5Q mice show different distribution of PC area in lobule VI and different distribution of PC circularity in lobules IX and X, relative to WT mice. Statistical analysis was performed using χ2.
To further correlate cerebellar neuropathology and motor alterations, morphometric parameters were measured on sagittal sections of the central vermis at different disease stages. The vermis length (axis from lobule V to IX) of SCA7140Q/5Q mice was smaller to WT at 34 weeks (Fig. 8D,F). Interestingly, the vermis width (axis from lobule VI to X) was significantly shorter in SCA7140Q/5Q mice at 12 weeks already, and this difference remained at later stages but did not worsen with the motor phenotype (Fig. 8D,F). Strikingly, there was no major or progressive alteration of the MCL thickness in any of the lobules analyzed (Fig. 8G; and data not shown).
Morphologic and functional alterations of PCs
Since PCs are highly vulnerable neurons in inherited ataxias, their morphology, connectivity, and function were investigated. The area of PC soma (Fig. 8E) was significantly smaller in lobule VI (p = 0.0018) of 34 week SCA7140Q/5Q compared with age-matched WT mice, while in lobules IX and X PC soma did not differ (Fig. 8H). In contrast, PC circularity index was affected in lobules IX and X (p = 0.0006 and p = 0.0005, respectively), but not in lobule VI (Fig. 8I).
We then analyzed synapse integrity between PC and CFs of inferior olivary neurons as well as between PC and parallel fibers (PFs) of GCs using vGLUT2 and vGLUT1 markers, respectively. By Western blot analysis, the bulk level of vGLUT2 showed a 20% decrease (p = 0.0022) in SCA7 cerebellum compared with WT, while the level of vGLUT1 was not affected (Fig. 9A). Consistently, vGLUT1 immunostaining of PC-PF contact points in the MCL of cerebellar sections showed no significant change (Fig. 9B). In contrast, vGLUT2 immunolabeling of PC-CF contact points showed that the extension of CF terminals along the PC dendritic tree was significantly reduced in lobule VI and X, respectively, by 15.8% (p = 0.029) and 23% (p = 0.034) in SCA7140Q/5Q compared with WT mice (Fig. 9B,C). Further quantification did not show abnormal average density and average volume of vGLUT2 contact points in SCA7 mice (data not shown). However, coimmunolabeling of vGLUT2 and CALB1 revealed frequent aggregation of vGLUT2 contact points along large dendritic arborization of PC in SCA7 cerebellum (Fig. 9C, left). Therefore, despite different morphologic alterations of PC soma of lobule VI and X, synaptic contacts between PC and CF were affected in both lobules.
Figure 9.
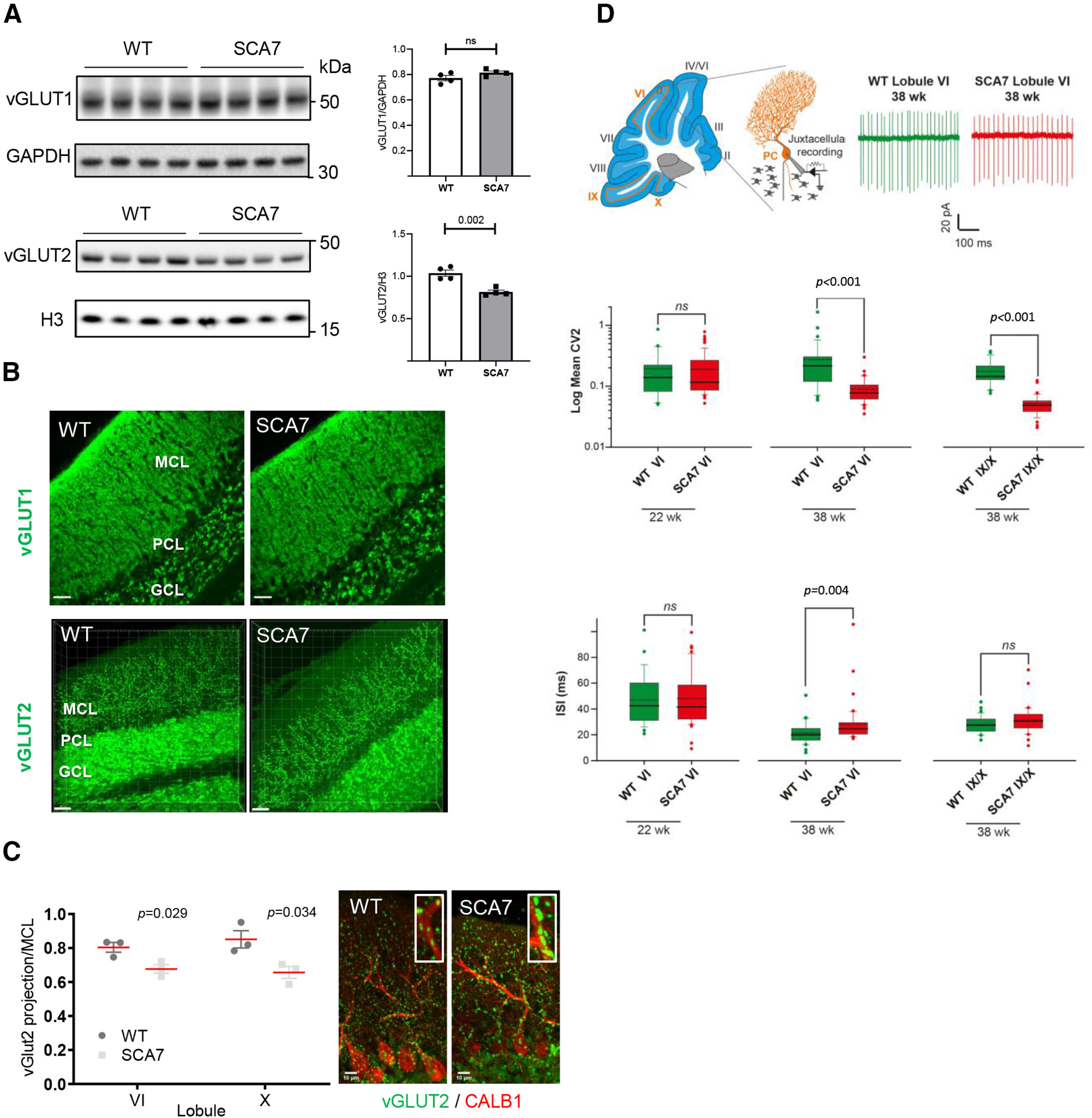
Alteration of synaptic contact and intrinsic activity of SCA7 PCs. A, Western blot analyses of SCA7140Q/5Q and WT cerebella show decreased level of vGLUT2 in SCA7 mice at 34 weeks, whereas the vGLUT1 is not affected. GAPDH and histone H3 protein levels are used as loading controls. Molecular weight in kDa. B, vGLUT1 and vGLUT2 immunostaining of lobule VI of the vermis in WT and SCA7 mice. GCL, GC layer; PCL, PC layer. Scale bar, 30 µm. C, Left, Measurement of vGLUT2-labeled area in the MCL of lobule VI and X of the vermis shows reduced width size in SCA7140Q/5Q (n = 3) compared with WT mice (n = 3). Data are mean ± SEM. Two-tailed Student's t test. Right, Coimmunostaining of vGLUT2 and CALB1 showing aggregation of vGLUT2 contact points located along large dendritic arborization of PC in SCA7 cerebellum. Scale bar, 10 µm. D, Top, Schematic of a sagittal cerebellar section where cerebellar lobules are numbered from I to X and orange lines indicate the locations of PC intrinsic activity recording sites (left). Middle, Schematic of juxtacellular recording of PC pacemaking activity. Representative traces of spontaneous firing in PCs located in lobule VI from WT or SCA7140Q/5Q mice (right). Middle, Box plots represent the values of CV2 of adjacent ISI in lobule VI and IX/X in WT and SCA7 mice. Dashed and solid lines indicate mean and median values, respectively: 22-week-old WT (n = 27) and SCA7 (n = 65); 38-week-old WT (n = 33 in lobule VI and 34 in IX/X) and SCA7 (n = 41 in lobule VI and 36 in IX/X). Bottom, Same representation as in middle panel for ISI values. Statistical analysis was performed using Mann–Whitney rank-sum test. ns. not significant.
Intrinsic membrane properties of PC determine their spontaneous firing pattern even in the absence of synaptic inputs (Gruol and Franklin, 1987; Hausser and Clark, 1997). Any alteration in the precision of their pacemaking activity may be the result of PC cellular dysfunctions and may affect cerebellar functions (Hoxha et al., 2018). To investigate the correlation between PC function, morphologic phenotypes, and motor deficits, we assessed whether the intrinsic properties of PC spontaneous discharge were altered in SCA7140Q/5Q mice at 22 and 38 weeks. We recorded spontaneous activity of PCs located in lobule VI and IX/X in WT and SCA7 littermates using juxtacellular recordings of PCs in acute cerebellar slices at a near physiological temperature (32°C) (Fig. 9D). To focus on PC intrinsic excitability and to rule out network contributions, excitatory and inhibitory synaptic transmissions were pharmacologically blocked. PC discharges were evaluated using the mean firing frequency (measurement of the mean ISI) and the CV2, which estimates the variability of the firing pattern between two consecutive ISIs (Holt et al., 1996; Arancillo et al., 2015). At 22 weeks, no difference in the firing properties of PCs located in lobule VI was detected between WT and SCA7 mice. In contrast, in symptomatic SCA7 mice of 38 weeks, PCs exhibited a higher regularity of discharge in both lobules VI (p < 0.001) and IX/X (p < 0.001) (Fig. 9D, middle) and a decrease in the mean firing rate in lobule VI (p = 0.004) (Fig. 9D, bottom). Our results show that intrinsic properties of PC excitability are altered in symptomatic SCA7 mice.
Accumulation of mutant ATXN7 in retina and cerebellum
Previous studies showed that mATXN7 tends to misfold and form intracellular protein aggregates (for review, see Niewiadomska-Cimicka and Trottier, 2019). To determine the dynamic of mATXN7 aggregation in SCA7 mice, we performed immunofluorescence and Western blot analysis of retina and cerebellar sections. In WT retina and cerebellum, ATXN7 immunolabeling is barely detectable (Fig. 10A,B). In contrast, the immunolabeling progressively increased with age in SCA7 mouse tissues, indicating that mATXN7 accumulates over time. This resulted in different labeling profiles depending on the neuronal population and disease stage. mATXN7 aggregates were readily detected in many photoreceptor nuclei at 12 weeks and became widespread in all retinal neuron nuclei at later stages (Fig. 10A). In SCA7 cerebellum, PCs showed the earliest mATXN7 accumulation that progressively increased between 12 and 19 weeks, leading to the formation of large nuclear aggregates at later stages (Fig. 10B). mATXN7 nuclear aggregates were also observed in the MCL and GC layer. All cerebellar lobules showed the accumulation of mATXN7 in PC nuclei (Fig. 10C).
Figure 10.
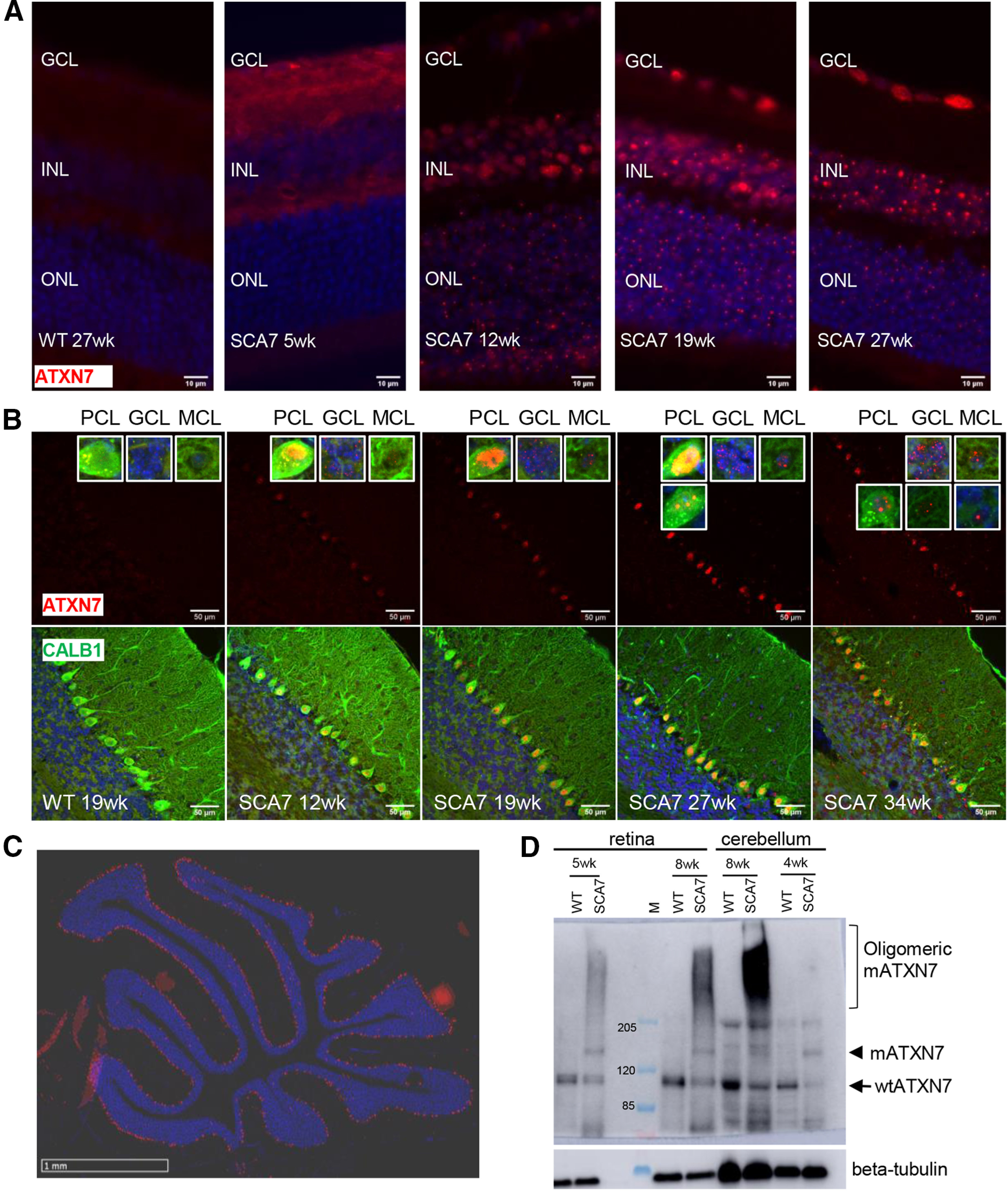
Accumulation of mutant ATXN7 in SCA7 mouse retina and cerebellum. A, Immunostaining of ATXN7 (red) on retinal sections of WT and SCA7140Q/5Q mice at different ages, showing the progressive accumulation of mutant ATXN7 (mATXN7) aggregates in retinal cells. DAPI-stained nuclei (blue). GCL, Ganglion cell layer. Scale bar, 10 µm. B, Coimmunostaining of ATXN7 (red) and CALB1 (green) of cerebellar sections, showing the progressive accumulation of mATXN7 in different cerebellar cells. DAPI-stained nuclei (blue). PCL, PC layer; GCL, GC layer. Scale bar, 50 µm. C, Immunostaining of ATXN7 (red) showing the global distribution of mATXN7 aggregates in the PCL of SCA7 cerebellum. DAPI-stained nuclei (blue). Scale bar, 1 mm. D, Western blot analysis of whole protein extract of retina and cerebellum of WT and SCA7140Q/5Q mice at different ages, probed with ATXN7 and β-tubulin antibodies. Arrows and arrowhead indicate the WT and mutant forms of ATXN7, respectively. Bracket indicates the oligomeric high molecular weight mATXN7 material in SCA7 samples. Molecular weight (M) is given in kDa.
Western blot analysis showed the expression of normal ATXN7 as a 110 kDa band in control retina and cerebellar samples (Fig. 10D). In SCA7 samples, the mutant form of ATXN7 migrated at ∼140 kDa. From 5 to 8 weeks of age, mATXN7 was also detected as high molecular weight smear in these tissues, indicating that mATXN7 accumulates as oligomeric forms, much before the formation of visible nuclear aggregates as detected by immunolabeling of tissues.
Cerebellar gene expression changes in SCA7 mice
To gain molecular insight into the cerebellar dysfunctions of SCA7140Q/5Q mice, gene expression profiles of cerebellar samples of symptomatic SCA7 mice and their WT littermates were analyzed by RNA-seq. Analysis of differentially expressed genes indicated that 406 genes are downregulated in SCA7 cerebella and 270 genes are upregulated (p < 0.05) (Fig. 11A). IPA (Kramer et al., 2014) of the 676 differentially expressed genes revealed that the most significantly affected functions (p ≤ 0.0002) concern major neuronal signaling pathways involving glutamate, G-protein coupled receptors, calcium, CREB, and cAMP, and LTD (Fig. 11B; Extended Data Fig. 11-1), most of which have previously been implicated in the pathogenesis of other ataxias (Hoxha et al., 2018).
Figure 11.
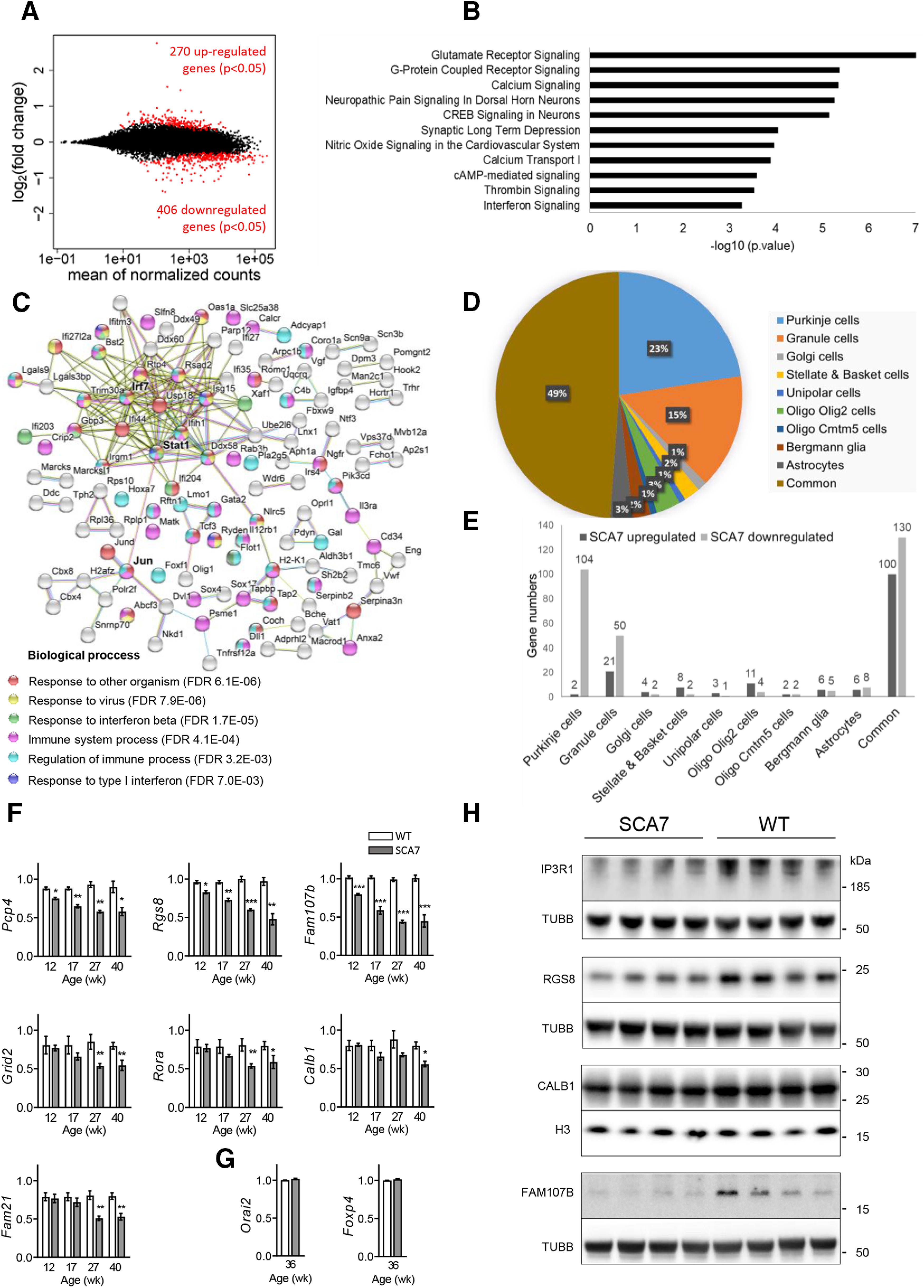
Gene deregulation in SCA7 cerebellum. A, MA plot of transcriptomic dataset between SCA7140Q/5Q and WT mice cerebellum at 40 weeks of age. Red points represent 676 genes deregulated in SCA7140Q/5Q (p < 0.05). B, Functional annotations of the deregulated genes performed using IPA. The most significantly affected pathways (p ≤ 0.0002) in SCA7140Q/5Q are indicated (for additional information, see Extended Data Fig. 11-1). C, The STRING version 10.5 software was used to generate the protein network based on known functional partnerships and interactions: light blue represents curated databases; pink represents experimentally determined; light green represents text mining; black represents coexpression. The assigned GO Biological Processes of upregulated genes are indicated and show significant enrichment in immune response and interferon signaling pathways, with IRF7, STAT1, and JUN at the center a large network of 189 edges. D, Cerebellar cell-type distribution of the genes deregulated in SCA7140Q/5Q mice based on curated dataset of Doyle et al. (2008) (for additional information, see Extended Data Fig. 11-2). E, Cell-type classification of upregulated and downregulated genes indicates PCs as the predominant cell type affected by gene downregulation in SCA7 cerebellum. Common means genes deregulated in more than one cell type. F, G, qRT-PCR analyses of SCA7140Q/5Q and WT cerebella point out the downregulation of Purkinje specific genes Pcp4, Rgs8, and Fam107b at presymptomatic stage and Grid2, Rora, Calb1, and Fam21 at symptomatic stage in SCA7 mice (n = 3 WT and 3 SCA7 mice per age) (F). qRT-PCR analyses confirm that two Purkinje-specific genes nonderegulated in RNA-seq (Orai2, Foxp4) have the same expression level in SCA7 and WT cerebella at 36 weeks (G). All data are normalized to the 36B4 level and are expressed as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; two-tailed Student's t test. H, Western blot analyses of SCA7140Q/5Q and WT cerebella show the strong decreased level of IP3R1, RGS8, and FAM107B in SCA7 mice at 26 weeks, while the CALB1 is not affected. Tubulin (TUBB) and histone H3 protein levels are used as loading controls.
IPA analysis and top 10 most predicted upstream regulators of genes deregulated in SCA7140Q/5Q mice cerebellum. Download Figure 11-1, XLSX file (12.8KB, xlsx) .
Analysis of cell-type specific gene deregulation in SCA7140Q/5Q mice cerebellum. Download Figure 11-2, XLSX file (129.6KB, xlsx) .
IPA also pointed out the impairment of interferon signaling pathway (Fig. 11B). Interestingly, among the 10 most predicted upstream regulators (p ≤ 7 × 10−9) of the differentially expressed genes, 8 are implicated in the modulation of interferon signaling pathways (Extended Data Fig. 11-1). Moreover, functional annotations of the 270 upregulated genes using STRING (Szklarczyk et al., 2015) further highlighted a strong enrichment (false discovery rate [FDR] <2 × 10−6) of Type 1 interferon pathway (Fig. 11C). The interferon regulator factor 7 gene (Irf7), which is the second most significantly upregulated protein-coding gene (2.7-fold; p = 2.28 × 10−10) in SCA7 mouse cerebella, composed with high confidence (0.700) a protein-protein association network of 19 direct edges that includes signal transducer and activator of transcription 1 (Stat1), which further interconnects with the protooncogene Jun. Together, Irf7, Stat1, and Jun constituted the center a large network of 189 edges involving multiple upregulated genes coding for proteins involved in immune response (Fig. 11C), further emphasizing the important activation of interferon signaling pathway in SCA7 cerebella. Chort et al. (2013) previously reported the increased level of interferon-β and its receptor in the cerebellum of the SCA7266Q/5Q model. Our transcriptome data provide additional evidence of the activation of a regulatory network involving interferon signaling.
Downregulation of a large subset of PC identity genes
To gain insight into the cell type-specific distribution of differentially expressed genes in SCA7 cerebella, we took advantage of the publicly available translatome datasets of each major cerebellar cell type established using BacTRAP transgenic mouse lines (Doyle et al., 2008). Among a total of 9671 genes present in these datasets, 5058 genes were exclusively expressed in one cell type, including 801 genes in PC, 2357 in GCs, 156 in Golgi cells, 340 in stellate and basket cells, 144 in unipolar brush cells, 563 in oligodendrocytes expressing Olig2, 133 in oligodendrocytes expressing Cmtm5, 279 in Bergmann glia, and 285 in astrocytes (Extended Data Fig. 11-2). We compared the list of genes deregulated in SCA7 cerebellum and identified that 240 are expressed in a single cerebellar cell type and 230 are present in more than one cell type (Extended Data Fig. 11-2). The analysis revealed that all the cerebellar cell types presented differentially expressed genes (Fig. 11D), indicating that SCA7 pathology is widespread across cerebellar cell types. However, despite the rarity of PC in the cerebellum estimated to 0.1% of cerebellar cells, 23% (106 of 470) of SCA7 deregulated genes were assigned to PC. In contrast, 15% of SCA7 deregulated genes were assigned to GCs, while this cell type represents 90% of cerebellar cells. Importantly, while GC genes were both upregulated and downregulated, almost all PC genes (104 of 106 genes) were downregulated (Fig. 11E), indicating that repression of PC genes is a major component of SCA7 cerebellar pathology. Another publicly available dataset from Kratz et al. (2014) allowed us to identify 5 additional PC genes that were downregulated in SCA7 mouse cerebella (Extended Data Fig. 11-2), resulting in a total of 109 downregulated PC genes.
To ascertain the specificity of these 109 genes in PC, their pattern of expression was analyzed using ISH datasets reported in the Brain Transcriptome Database (BrainTx; CDT) (Sato et al., 2008) and in the Allen Brain Atlas (Lein et al., 2007). We established three categories of genes according to the level of enriched expression in PC, as specifically expressed in PC compared with other cerebellar cells, enriched in PC or nonenriched in PC (e.g., equally present in other cell types). The analysis showed that 83 genes had specific enrichment in PC, and are thus considered as PC identity genes that showed reduced expression in SCA7 mice. The remaining genes were expressed in wider range of cerebellar cell types or were not present in ISH datasets and were then excluded from further analysis (Extended Data Fig. 11-2).
qRT-PCR was used to study the time course of repression of a subset of PC identity genes in SCA7140Q/5Q cerebella (Fig. 11F). The Grid2, Calb1, Fam21, and Rora genes showed decreased expression in SCA7 mice from 27 weeks, coinciding with the onset of motor alterations. Interestingly, the 3 PC genes Pcp4, Fam107b, and Rgs8 showed downregulation already at 12 weeks, a presymptomatic stage of the disease, suggesting a strong implication in PC pathology of SCA7 mice. The PC genes Orai2 and Foxp4, which were not deregulated in RNA-seq data, were also not affected by qRT-PCR in SCA7 mice compared with WT, indicating that PC gene deregulation is not a consequence of cell loss (Fig. 11G). Consistently, Western blot analysis showed that FAM107B and RGS8 as well as IP3R1 proteins were strongly decreased in SCA7140Q/5Q cerebella compared with WT samples at 26 weeks, while CALB1 level was not affected (Fig. 11H).
A signature of downregulated PC identity genes in SCA1, SCA2, and SCA7
To further explore whether PC genes downregulated in SCA7 have pathologic relevance in cerebellar ataxia, we looked at publicly available transcriptomic datasets of SCA1 and SCA2 mouse models in which the mutant genes were specifically expressed in PC and caused spontaneous firing dysfunction (Dansithong et al., 2015; Ingram et al., 2016). In these two models, expression level of a subset genes was also shown to be altered compared with their respective WT littermates (Dansithong et al., 2015; Ingram et al., 2016). Before proceeding with the comparative analysis, we have reanalyzed the RNA-seq raw data of SCA1 mouse cerebella at 5, 12, and 28 weeks and SCA2 cerebella at 6 weeks with the same software that was used for SCA7. We then compared the differentially expressed genes in the cerebellum of the three SCA models. The analysis revealed that 136 genes common to the three datasets were downregulated and only 5 were upregulated (Fig. 12A,B). Interestingly, of the 136 downregulated genes, 67 were PC identity genes, including 10 genes previously associated with genetic ataxias (Table 2; Extended data Table 2-1). These 67 genes were downregulated already at 5 weeks in SCA1 and at 6 weeks in SCA2 mice, which are disease onset stages of these models (Hansen et al., 2013; Ingram et al., 2016), and constitute a signature of early pathologic processes of PC. The comparison performed with the SCA1 dataset at later disease stages revealed 13 additional downregulated PC identity genes in common with SCA2 and SCA7 mice (Table 2), suggesting that these 13 genes were associated with the progression of PC pathology.
Figure 12.
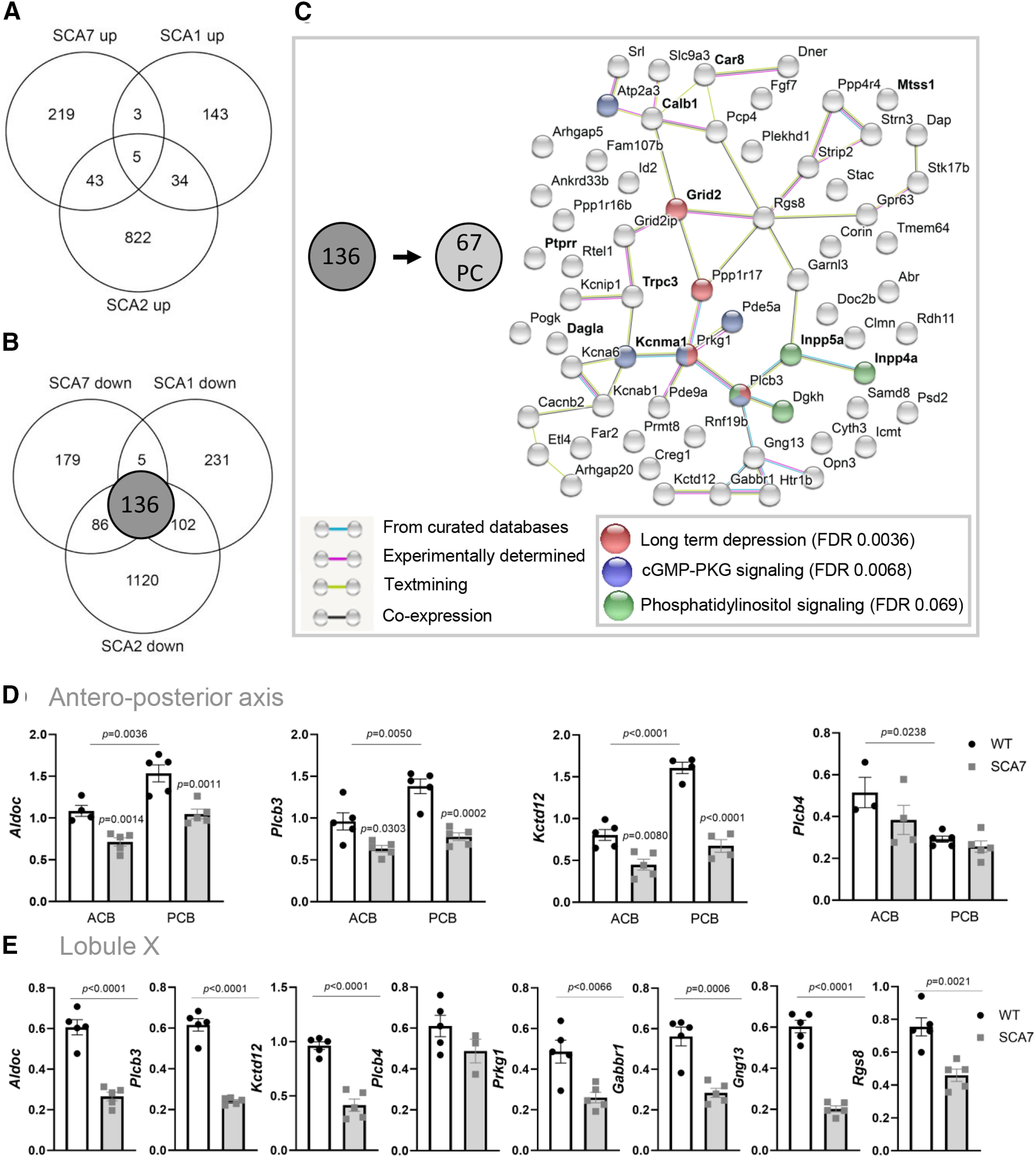
Downregulation of PC identity genes in SCA1, SCA2, and SCA7 and network analysis. A, B, Venn diagrams show overlap between genes upregulated (A) and downregulated (B) in SCA1 (5-week-old) (Ingram et al., 2016), SCA2 (6-week-old) (Dansithong et al., 2015), and SCA7140Q/5Q cerebellum. C, Genetic network showing functional partnerships and interactions between proteins encoded by the 67 PC genes downregulated in SCA1, SCA2, and SCA7 mouse models, as determined using the STRING software. Bold type represents known ataxia genes. Bottom right, Functional annotations of the 67 PC-enriched genes downregulated in SCA1, SCA2, and SCA7 mouse models, using KEGG pathway analysis. D, qRT-PCR analyses of ACB (ZII– enriched lobules II-V) and PCB (ZII+ enriched lobule VI-IX) vermal cerebella of SCA7140Q/5Q and WT mice show strong downregulation of ZII+ associated genes (Aldoc, Plcb3, Kctd12) in PCB of SCA7 mice compared with WT, whereas the expression of ZII– associated gene Plcb4 is not affected in ACB and PCB. Data are normalized to the 36B4 level and are expressed as mean ± SEM. Ordinary two-way ANOVA, followed by post hoc Bonferroni test. E, qRT-PCR analyses of ZII+ enriched lobule X show strong downregulation of ZII+ associated genes (Aldoc, Plcb3, Kctd12) and 4 other genes (Prkg1, Gabbr1, Gng13, Rgs8) from the downregulated network (C), in SCA7 mice compared with WT. The ZII– associated gene Plcb4 is not affected in SCA7 lobule X. Data are normalized to the 36B4 level and are expressed as mean ± SEM. Two-tailed Student's t test.
Table 2.
PC specific or enriched genes commonly downregulated in the cerebellum of SCA1, SCA2, and SCA7 mice models
| Gene symbol | Gene names | SCA7 log2FC (SCA7/WT) 40 weeks | SCA1 |
SCA2 log2FC (SCA2/WT) 6 weeks | ||
|---|---|---|---|---|---|---|
| log2FC (SCA1/WT) 5 weeks | log2FC (SCA1/WT) 12 weeks | log2FC (SCA1/WT) 28 weeks | ||||
| Abr r | active BCR-related gene | −0.32 | −0.27 | −0.02 | −0.20 | −0.46 |
| Ankrd33b | ankyrin repeat domain 33B | −0.52 | −0.49 | −1.28 | −1.58 | −0.73 |
| Arhgap20 | Rho GTPase activating protein 20 | −0.73 | −0.47 | −0.79 | −1.22 | −0.70 |
| Arhgap26 | Rho GTPase activating protein 26 | −0.62 | NS | −0.68 | −0.84 | −0.94 |
| Arhgap31 | Rho GTPase activating protein 31 | −0.37 | NS | −0.64 | −1.12 | −0.70 |
| Arhgap32 | Rho GTPase activating protein 32 | −0.39 | NS | −0.49 | −1.03 | −0.69 |
| Arhgap5 | Rho GTPase activating protein 5 | −0.49 | −0.26 | −1.22 | −1.05 | −0.56 |
| Atp2a3 | ATPase, Ca++ transporting, ubiquitous | −0.38 | −0.79 | −1.50 | −1.84 | −1.26 |
| B4galnt3 | beta-1,4-N-acetyl-galactosaminyl transferase 3 | −0.55 | NS | −4.15 | −0.56 | −1.77 |
| Cacnb2 r | calcium channel, voltage-dependent, beta 2 subunit | −0.42 | −0.51 | −0.92 | −1.05 | −0.49 |
| Calb1 ar | calbindin 1 | −0.47 | −0.61 | −1.17 | −1.42 | −0.87 |
| Car8 ar | carbonic anhydrase 8 | −0.51 | −0.73 | −1.59 | −1.51 | −1.13 |
| Casq2 | calsequestrin 2 | −0.42 | NS | −0.73 | −1.03 | −0.58 |
| Clmn | calmin | −0.65 | −0.70 | −1.32 | −1.84 | −1.34 |
| Corin | corin | −0.53 | −0.86 | −2.11 | −2.83 | −1.22 |
| Creg1 | cellular repressor of E1A-stimulated genes 1 | −0.49 | −0.78 | −0.85 | −0.97 | −0.31 |
| Cyth3 | cytohesin 3 | −0.37 | −0.42 | −0.79 | −0.65 | −0.56 |
| Dagla a | diacylglycerol lipase, alpha | −0.45 | −0.90 | −1.12 | −1.44 | −1.58 |
| Dap r | death-associated protein | −0.35 | −1.09 | −0.94 | −1.31 | −0.64 |
| Dgkh r | diacylglycerol kinase, eta | −0.77 | −1.06 | −1.80 | −2.37 | −1.14 |
| Dlg2 | discs large MAGUK scaffold protein 2 | −0.43 | NS | −0.93 | −0.98 | −0.43 |
| Dner | δ/notch-like EGF repeat containing | −0.36 | −0.56 | −1.13 | −1.08 | −1.20 |
| Doc2b | double C2, beta | −0.64 | −1.01 | −1.43 | −1.87 | −1.53 |
| Etl4 | enhancer trap locus 4 | −0.50 | −0.35 | −0.51 | −0.72 | −0.82 |
| Fam107b r | family with sequence similarity 107, member B | −1.03 | −1.08 | −1.92 | −1.65 | −1.87 |
| Fam117a | family with sequence similarity 117, member A | −0.68 | NS | −0.82 | −0.60 | −1.18 |
| Fam78b r | family with sequence similarity 78, member B | −0.41 | NS | −0.45 | −0.85 | −0.60 |
| Far2 | fatty acyl CoA reductase 2 | −0.54 | −0.58 | −1.36 | −1.22 | −0.70 |
| Fgf7 | fibroblast growth factor 7 | −0.51 | −1.55 | −3.26 | −2.59 | −1.41 |
| Gabbr1 | GABA B receptor, 1 | −0.37 | −0.57 | −0.69 | −0.76 | −0.53 |
| Garnl3 | GTPase activating RANGAP domain-like 3 | −0.67 | −0.65 | −1.12 | −1.41 | −0.96 |
| Gm5083 | predicted gene 5083 | −0.47 | −0.68 | −0.90 | −0.98 | −1.08 |
| Gng13 | guanine nucleotide binding protein (G protein), γ 13 | −0.74 | −1.30 | −1.92 | −1.97 | −1.54 |
| Gpr63 | GPCR 63 | −0.90 | −1.07 | −1.93 | −2.58 | −2.17 |
| Grid2 a | glutamate receptor, ionotropic, δ 2 | −0.50 | −0.42 | −1.18 | −1.32 | −0.80 |
| Grid2ip | glutamate receptor, ionotropic, δ 2 (Grid2) interacting protein 1 | −0.66 | −1.09 | −1.66 | −2.95 | −1.68 |
| Htr1b | 5-hydroxytryptamine (serotonin) receptor 1B | −0.45 | −0.52 | −1.36 | −1.21 | −1.16 |
| Icmt r | isoprenylcysteine carboxyl methyltransferase | −0.54 | −0.63 | −1.41 | −1.56 | −0.88 |
| Id2 r | inhibitor of DNA binding 2 | −0.32 | −0.46 | −0.59 | −0.52 | −0.49 |
| Inpp4a a | inositol polyphosphate-4-phosphatase, Type I | −0.29 | −0.35 | −0.72 | −0.90 | −1.00 |
| Inpp5a a | inositol polyphosphate-5-phosphatase A | −0.41 | −0.56 | −0.89 | −1.07 | −1.00 |
| Itpr1 ar | inositol 1,4,5-trisphosphate receptor 1 | −0.56 | NS | −1.43 | −2.00 | −1.00 |
| Kcna6 r | potassium voltage-gated channel, shaker-related, subfamily, member 6 | −0.45 | −0.49 | −0.77 | −0.47 | −0.70 |
| Kcnab1 r | potassium voltage-gated channel, shaker-related subfamily, beta member 1 | −0.34 | −0.51 | −1.30 | −1.07 | −0.74 |
| Kcnip1 | Kv channel-interacting protein 1 | −0.34 | −0.62 | −0.97 | −1.10 | −0.38 |
| Kcnma1 a | potassium large conductance calcium-activated channel, subfamily M, alpha member 1 | −0.67 | −0.37 | −0.97 | −1.62 | −0.83 |
| Kctd12 | potassium channel tetramerization domain containing 12 | −0.95 | −0.87 | −1.96 | −1.44 | −1.23 |
| Mtss1 ar | metastasis suppressor 1 | −0.36 | −0.27 | −0.59 | −0.88 | −0.93 |
| Nexn | nexilin | −0.36 | NS | −0.67 | −0.81 | −0.62 |
| Opn3 | opsin 3 | −1.03 | −1.05 | −1.74 | −0.76 | −1.94 |
| Pcp4 r | PC protein 4 | −0.57 | −0.85 | −1.14 | −1.30 | −0.81 |
| Pde5a | phosphodiesterase 5A, cGMP-specific | −0.70 | −0.45 | −1.50 | −1.83 | −0.89 |
| Pde9a | phosphodiesterase 9A | −0.46 | −0.37 | −0.12 | −0.47 | −0.47 |
| Plcb3 r | phospholipase C, beta 3 | −0.72 | −0.91 | −0.92 | −1.10 | −0.98 |
| Plekhd1 | pleckstrin homology domain containing family D (with coiled-coil domains) member 1 | −0.34 | −0.39 | −0.74 | −0.78 | −0.61 |
| Pogk | pogo transposable element with KRAB domain | −0.45 | −0.32 | −0.75 | −0.67 | −0.54 |
| Ppp1r16b | protein phosphatase 1, regulatory subunit 16B | −0.72 | −0.74 | −1.31 | −1.44 | −1.25 |
| Ppp1r17 | protein phosphatase 1, regulatory subunit 17 | −0.34 | −0.45 | −1.08 | −0.93 | −0.65 |
| Ppp4r4 | protein phosphatase 4, regulatory subunit 4 | −0.49 | −0.46 | −0.98 | −1.26 | −0.70 |
| Prkg1 | protein kinase, cGMP-dependent, Type I | −0.80 | −0.48 | −1.05 | −1.20 | −0.90 |
| Prmt8 | protein arginine N-methyltransferase 8 | −0.52 | −0.35 | −0.49 | −0.50 | −0.70 |
| Psd2 | pleckstrin and Sec7 domain containing 2 | −0.31 | −0.38 | −0.57 | −0.38 | −0.49 |
| Ptprr ar | protein tyrosine phosphatase, receptor type, R | −0.36 | −0.36 | −1.14 | −0.83 | −0.50 |
| Rdh11 | retinol dehydrogenase 11 | −0.46 | −0.40 | −0.90 | −0.60 | −0.45 |
| Rgs7bp | regulator of G-protein signaling 7 binding protein | −0.32 | NS | −0.87 | −0.64 | −0.33 |
| Rgs8 | regulator of G-protein signaling 8 | −0.91 | −0.97 | −2.06 | −2.13 | −1.50 |
| Rnf19b | ring finger protein 19B | −0.45 | −0.49 | −0.70 | −0.68 | −0.90 |
| Rora ar | RAR-related orphan receptor alpha | −0.39 | NS | −0.87 | −1.25 | −0.63 |
| Rtel1 | regulator of telomere elongation helicase 1 | −0.32 | −0.55 | −0.79 | −0.71 | −0.41 |
| Samd8 | sterile alpha motif domain containing 8 | −0.33 | −0.43 | −0.90 | −0.92 | −0.60 |
| Slc9a3 r | solute carrier family 9 (sodium/hydrogen exchanger), member 3 | −0.60 | −0.82 | −1.66 | −2.00 | −1.22 |
| Srl | sarcalumenin | −0.77 | −0.91 | −1.24 | −1.04 | −0.78 |
| Stac | src homology three (SH3) and cysteine-rich domain | −0.66 | −0.92 | −1.26 | −1.83 | −1.22 |
| Stk17b | serine/threonine kinase 17b (apoptosis-inducing) | −0.71 | −0.84 | −1.86 | −1.74 | −1.02 |
| Strip2 | striatin interacting protein 2 | −0.49 | −0.45 | −1.37 | −1.24 | −0.73 |
| Strn3 | striatin, calmodulin binding protein 3 | −0.34 | −0.42 | −0.75 | −0.87 | −0.37 |
| Tmem64 | transmembrane protein 64 | −0.33 | −0.27 | −0.85 | −0.58 | −0.37 |
| Trpc3 a | transient receptor potential cation channel subfamily C member 3 | −0.50 | −0.65 | −1.57 | −1.58 | −1.26 |
| 2410124H12Rik | RIKEN cDNA 2410124H12 gene | −0.88 | −1.75 | −3.11 | −3.67 | −2.08 |
| 3632451O06Rik | –– | −0.40 | NS | −0.84 | −0.53 | −0.25 |
a, genes associated with genetic ataxias;
r, RORa genetic targets. See also Extended Data Table 2-1.
NS. Not Significant
136 transcripts commonly downregulated in cerebellum of SCA1, SCA2 and SCA7 mice models. Download Table 2-1, XLSX file (14.6KB, xlsx) .
The biological significance of the early downregulation of the 67 PC identity genes was researched using three analyses. First, the KEGG pathway analysis indicated that this set was enriched for genes involved in LTD (FDR 0.0036), cGMP-PKG signaling pathway (FDR 0.0068), and phosphatidylinositol signaling system (FDR 0.0069) (Fig. 12C). Second, the STRING analysis (Szklarczyk et al., 2015) was used to search for functional partnerships and interactions between proteins encoded by the 67 PC genes. It revealed a network of 45 edges (p = 1.2e−13) (Fig. 12C) interconnecting a large number of proteins encoded by genes showing mutations in human or mouse model ataxias, such as Grid2, Calb1, Car8, Trpc3, Kcnma1, Inpp5a, and Inpp4a. The network also showed central positions to the regulator of G-protein signaling 8 (RGS8), cGMP-dependent kinase PRKG1, and its phosphorylation target phospholipase C β 3 (PLCB3); the latter two are related to LTD and synapse functions of PC (Feil et al., 2003; Hoxha et al., 2018). Finally, our list of 67 PC identity genes was compared with a published list of genes composing the SCA1 Magenta gene module (Ingram et al., 2016). This module was established using the Weighted Gene Coexpression Network Analysis of SCA1 cerebellar transcriptomes and reported to be enriched in PC genes involved in LTD and glutamate signaling and to be of high relevance for SCA1 pathogenesis (Ingram et al., 2016). We found that 48 of 67 PC-enriched genes were present in the Magenta module. Furthermore, two major hub genes described in the module, namely, Fam107b and Rgs8, were within the top strongest downregulated genes in SCA1, SCA2, and SCA7 models, and were already downregulated at 12 weeks in SCA7 mice, hence before onset of motor symptoms (Fig. 11F). Together, these analyses suggest that the dysfunction of synaptic LTD of PC constitutes a common pathway in the early pathogenesis of SCA1, SCA2, and SCA7.
The heterogeneity of PCs in the cerebellum is characterized at the topographical and molecular level by striped arrangement of Zebrin II (ALDOC) positive (ZII+) or negative (ZII–) PCs (Sillitoe and Hawkes, 2002; Fujita et al., 2014; Cerminara et al., 2015). ZII+ PCs express Aldoc and Plcb3 and are enriched in the posterior cerebellum (lobules VI-X), whereas ZII– PCs express Plcb4 and are enriched in the anterior part (lobules I-V). The presence of the Plcb3 at a central position in the downregulated network (Fig. 12C) suggested that ZII+ PCs are particularly vulnerable in SCA1, SCA2, and SCA7. We tested this possibility in SCA7 mice by analyzing the related markers in the anterior (ACB) and posterior vermal cerebellum (PCB) by qRT-PCR. As expected, Plcb4 showed a significantly higher expression in the ACB of WT mice, and Aldoc and Plcb3 were significantly more expressed in the PCB (Fig. 12D). Interestingly, Plcb4 expression was not affected in SCA7 cerebellum, whereas Aldoc and Plcb3 were downregulated and showed a stronger decreased expression in the PCB than in the ACB. Similarly, the gene coding for KCTD12, another marker expressed in ZII+ PCs (Metz et al., 2011) and present in the downregulated network (Fig. 12C), showed a strong decreased expression in the PCB. As shown in Figure 9, SCA7 PCs showed firing impairment in the vermal lobules IX/X, two lobules strongly enriched in ZII+ PCs. Consistently, all markers of ZII+ PCs (Aldoc, Plcb3, and Kctd12) were strongly downregulated in SCA7 lobule X compared with WT, whereas the marker of ZII– PCs (Plcb4) was not affected (Fig. 12E). Four other central genes of the downregulated network (Prgk1, Gabbr1, Gng13, and Rgs8) also showed decreased expression in SCA7 lobule X. Together, the data indicate a particular vulnerability of ZII+ PCs in SCA7 mouse cerebellum.
Finally, to identify mechanisms that could account for the downregulation of the 67 PC identity genes, we first looked at RORa (RAR-related orphan receptor α), a key transcription factor involved in PC differentiation and survival (Gold et al., 2003) and showing downregulation in SCA1, SCA2, and SCA7 mice (Table 2). Among the 67 PC genes, 19 genes were known RORa-target genes (Gold et al., 2003; Sarachana and Hu, 2013) (Table 2), suggesting that RORa dysfunction accounts for PC gene deregulation. We then performed in silico analysis of 2 kb promoter sequence of the 67 PC genes to search for de novo motifs and transcription factors binding sites using MEME, TOMTOM, AME, or FIMO (Bailey et al., 2009). The analysis did not reveal major enrichment of other transcription factor binding motifs in the promoter of the 67 PC identity genes affected, compared with control sequences (e.g., shuffled promoter sequences or promoter sequences of 67 PC-enriched nonderegulated genes) (data not shown).
Deregulation of gene expression may depend on alteration of diverse processes, including chromatin structure, nucleosomal occupancy, and histone modifications, which play important roles in transcriptional regulation. Given the presence of ATXN7 in the histone-modifying SAGA complex, we determined whether the bulk level of histone marks regulated by SAGA were altered in SCA7 cerebellum. The level of H2Bub was significantly increased (p = 0.0072) in SCA7140Q/5Q cerebellum compared with WT, whereas the level of H3K9ac showed no significant difference (Fig. 13A). However, a significant decrease (p = 0.0003) of H3K9ac was observed in the more severely affected SCA7140Q/140Q homozygous mice (Fig. 13B). Together, this suggests that mutant ATXN7 differentially alters the histone-modifying activities of SAGA complex along the progression of cerebellar pathology and could impact on transcription of cerebellar genes.
Figure 13.

Alteration of SAGA-dependent histone marks in SCA7 cerebellum. A, Western blot analyses showing that the bulk level of monoubiquitinated histone H2B (H2Bub) is significantly increased in the cerebellum of SCA7140Q/5Q heterozygous mice (23 weeks) compared with WT, whereas the bulk level of acetylated lysine 9 of histone H3 (H3K9ac) is not affected. B, Western blot analyses showing the increased bulk level of H2Bub and the decreased bulk level of H3K9ac in the cerebellum of SCA7140Q/140Q homozygous mice (12 weeks) compared with WT littermates. Immunodetected signals were quantified in Fiji and normalized using the level of H2B and GAPDH. Data are mean ± SEM. Two-tailed Student's t test.
Discussion
We characterized the behavioral, molecular, and cellular phenotypes of a new SCA7 knock-in mice. By analyzing cell type-specific gene deregulation as well as morphologic and functional alterations of the cerebellum, PCs came out as the most affected cerebellar cell type by decreased expression of identity genes, critical for their spontaneous firing and synaptic functions. These observations suggest disease mechanisms primarily altering the maintenance of PC differentiation, and are consistent with similar pathologic features of SCA7 mouse photoreceptors in this and earlier studies. The same PC identity genes were also affected in presymptomatic SCA1 and SCA2 mice, indicating converging pathomechanisms of PC vulnerability in these SCAs.
Retinal, PNS, and CNS features of SCA7 mice
The SCA7140Q/5Q line combines cardinal features observed in adult SCA7 patients, including retinal, cerebral, cerebellar, and peripheral nerve pathologies (Fig. 14). Many of these phenotypes were not reported in previous models, such as alterations of RPE, Bruch's membrane, peripheral nerve, and different cerebral and brainstem regions. SCA7140Q/5Q photoreceptor dysfunction correlates with progressive reduced expression of photoreceptor identity genes, such as opsins and the disappearance of outer segments. In zebrafish, knockdown of atxn7 impedes the formation of outer segments during photoreceptor differentiation (Yanicostas et al., 2012; Carrillo-Rosas et al., 2018). Together, this suggests that mATXN7 toxicity alters a normal function of ATXN7 in the maintenance of outer segments, a cell type-specific structure characteristic of photoreceptor differentiation (Swaroop et al., 2010). Alterations of RPE and Bruch's membrane warrant further investigation as they are essential for photoreceptor visual function. Interestingly, homogeneous deposits observed between RPE and basement membrane resemble sub-RPE deposits reported in Malattia Leventinese mouse model (Marmorstein et al., 2007), a dominantly inherited macular degenerative disease.
Figure 14.
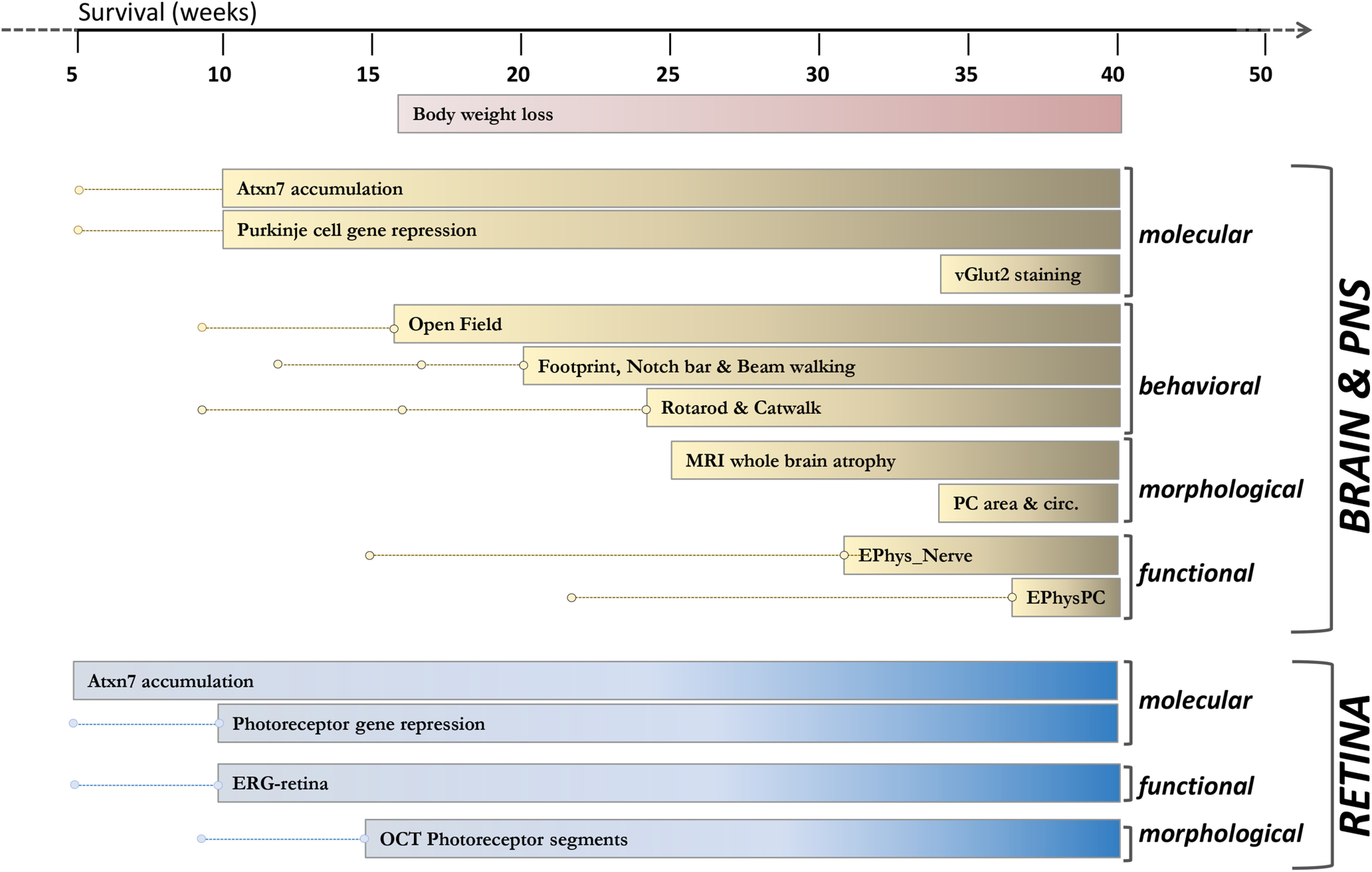
Summary of onset and progression of SCA7 mouse phenotypes. Dots and lines indicate analytic time points before phenotype onset. Bars represent phenotype onset and progression until 40 weeks of age.
Peripheral pathologies account for SCA7140Q/5Q neurologic phenotype. The reduced motor peripheral activities observed at 32 weeks are because of lesions of myelin and myelinated fibers of sciatic nerve. Nonmyelinated fibers are also altered and might affect a sensory component. These findings are consistent with abnormal motor and sensory nerve conductions observed in SCA7 patients (Salas-Vargas et al., 2015).
SCA7140Q/5Q mice present decreased volume of selected brain regions at 24 weeks, including corpus callosum, hippocampus, and cortical regions, which parallels observations of human SCA7 (Rub et al., 2008; Horton et al., 2013; Adanyeguh et al., 2018). Recent studies showed altered functional connectivity between patients' cerebellum and hippocampus or parahippocampal areas that may account for specific memory dysfunctions (Alcauter et al., 2011; Hernandez-Castillo et al., 2013, 2016; Chirino et al., 2018). Interestingly, hippocampal neurons showed very early accumulation of mATXN7 (data not shown) and alteration of short-term synaptic plasticity in SCA7266Q/5Q mice (Yoo et al., 2003). The cerebellar cortex presented no significant atrophy, which can be because of variability of MRI volume measurement in our imaging setup. Morphometric analysis of cerebellar sections showed that the vermis width is slightly, but significantly, smaller before the onset of the motor symptoms. At end-disease stage (52 weeks), SCA7140Q/5Q brain showed additional atrophy of the pons and somatosensory cortex, anterior commissure, internal capsule, and the fimbria of the hippocampus. Overall, brain alterations in the SCA7140Q/5Q mice recapitulate many characteristics of human SCA7.
Consistent with alterations of CNS and PNS, SCA7140Q/5Q mice show progressive motor dysfunctions in a variety of experimental paradigms. At motor dysfunction onset, body composition, day and night food intake (data not shown), and normalized muscle strength were normal. The progressive visual impairment might affect the motor performance. However, to perceive their environment, mice primarily use on their excellent olfactory system, vomeronasal organ, and highly developed whiskers (Ache and Young, 2005; Diamond et al., 2008). The motor tests used in this study do not rely much on vision and are common first line tests for preclinical trials in mice.
PC pathology of SCA7 mice
PCs in SCA7140Q/5Q mice show morphologic alterations of soma and synapses, impairment of spontaneous firing, and early and progressive repression of genes involved in firing and synaptic functions. PC pacemaking activities are essential for normal cerebellar functions and are shaped by a complex interplay between Ca2+, Na+, and K+ conductance (Raman and Bean, 1999). In several ataxia models mutated in ion channel genes, PCs present a lower rate of spontaneous firing and/or an irregular firing pattern (Hansen et al., 2013; Hoxha et al., 2018; Bushart and Shakkottai, 2019). PCs in SCA7 mice fire with a higher regularity (low CV2) in lobules VI and IX/X, and a lower frequency in lobule VI, than in WT. The downregulation of PC genes associated with potassium channels (Table 2) may account for alterations of pacemaking activity. The absence of major morphologic alteration of MCL associated with low CV2 of PCs was reported in ataxic waddles mice mutated in Car8 gene (White et al., 2016). Interestingly, Car8 is one of the 83 PC genes downregulated in SCA7 mice. The PC CV2 of waddles mice was improved by chlorzoxazone, a pharmacological agent increasing the probability of opening of Ca2+-dependent K+ channels (White et al., 2016).
SCA7 PCs in lobule VI and IX/X present different excitability impairment that may be linked to different morphologic alteration (smaller soma in VI and altered circularity in IX/X). Based on the topographical organization of the olivo-cortico-cerebellar network (Apps and Hawkes, 2009), lobule-specific differences in PC firing rate between WT and SCA7 may suggest that defects are associated to specific cerebellar tasks.
Synaptic functions integrating afferent signals that arrive from CF, PF, or interneurons are essential to control the output of PCs, and mutations affecting the function of synaptic circuits lead to ataxia (Hoxha et al., 2018). SCA7140Q/5Q dysfunction at CF-PC synapses is revealed by the reduced vGLUT2 immunolabeling in the MCL, whereas downregulation of PC genes encoding postsynaptic membrane-associated proteins involved in glutamate signaling and LTD, such as GLURδ2 (coded by Grid2), GRID2ip, GABBR1, and TRPC3, indicates functional alteration of PF-PC synapses (Kashiwabuchi et al., 1995; Tabata et al., 2004; Kato et al., 2012; Hawkes, 2014).
There is strong interest in identifying converging pathways of PC pathology across SCAs to improve our understanding of polyQ pathogenesis and to identify common potential targets for therapeutic intervention (Smeets and Verbeek, 2014). We identified 67 PC identity genes downregulated in SCA7 and in early symptomatic SCA1 and SCA2 mice (Hansen et al., 2013; Ingram et al., 2016) (Table 2), suggesting that they are primary targets of the pathogenesis in these SCAs. These PC genes compose a downregulated network involving hub proteins, such as RGS8, PLCB3, and PRKG1 and point out toward alteration of LTD, cGMP-PKG signaling, and phosphatidylinositol signaling pathways. This downregulated network further suggests that PCs located in ZII+ stripes are particularly vulnerable in SCA1, SCA2, and SCA7. The dysfunction of ZII+ PC subclass in SCA7 mice is further supported by the concomitant decreased expression of ZII+ associated genes (Aldoc, Plcb3, Kctd12) and PC firing impairment in IX/X of the vermis, which are enriched in ZII+ PCs. MRI analysis indicated that lobule X present atrophy in most SCA7 patients (Chirino et al., 2018).
Loss of PC identity gene expression
Our study determined cerebellar cell type-specific gene deregulation in SCA7 mouse cerebellum. PCs differ from other cell types by a high number of cell identity genes that are downregulated. Many of these genes are essential for major PC functions (spontaneous firing and synaptic functions involving glutamate signaling and LTD) and are mutated in other genetic ataxias (Table 2). One emerging concept is that selective transcription factors that specify the acquisition of neuron-type identity during development continue to function later to maintain identity of postmitotic neuronal type (Holmberg and Perlmann, 2012; Deneris and Hobert, 2014). Dysfunction of such key transcription factors is proposed to account for neurodegenerative disorders. RORa is highly expressed in fetal PC and is essential for their maturation (Boukhtouche et al., 2006) and maintaining adult PC characteristics (Chen et al., 2013). RORa expression is decreased in the cerebellum of SCA1, SCA2, and SCA7, together with many RORa-regulated PC identity genes, suggesting that RORa transcriptional program of PC maintenance is disrupted in these SCAs. Previous studies showed that mATXN7 alters the transactivation activity of RORa in cultured cells (Strom et al., 2005), and that postnatal loss of Rora expression worsened neurodegeneration of PCs in SCA1 mice (Serra et al., 2006), while increasing RORa level (through partial loss of TIP60) delayed cerebellar pathology (Gehrking et al., 2011).
Studies established that epigenetic regulation at super-enhancers is involved in the expression of cell identity genes (Hnisz et al., 2013; Whyte et al., 2013). In Huntington's disease, neuronal identity genes regulated by super-enhancers are preferentially downregulated in the disease striatum (Achour et al., 2015). Our study revealed that mATXN7 alters the level of both epigenetic marks (H3K9ac and H2Bub) dependent of SAGA function in SCA7 mouse cerebellum. Previous studies showed that mATXN7 aggregates can sequester components of SAGA as well as CBP, which is responsible for H3K27 acetylation on active enhancers (Yvert et al., 2001; Lan et al., 2015). Therefore, further genome-wide investigation of SAGA- and CBP-dependent epigenetic marks might provide insights into mechanisms leading to loss neuron identity gene expression in SCA7.
Footnotes
This work was supported by European Union Joint Program–Neurodegenerative Disease Research Project ModelPolyQ Grant Agreement 643417 jointly funded by national funding organizations: ANR-15-JPWG-0008-03 to Y.T. and BMBF 01ED1606 to H.H.P.N.; E-Rare Research Program, which is a transnational R&D program jointly funded by national funding organizations within the framework of the ERA-NET E-Rare 3 to Y.T, H.H.P.N., and E.B.; French foundation Connaître les Syndromes Cérébelleux to Y.T.; ANR-17-RAR3-0008-04_ACT and ANR-18-CE12-0026-02 to Y.T.; ANR-CE37-2015 (CeMod) and Fondation pour la Recherche Médicale (Equipes FRM DEQ20140329514) to P.I.; and ANR-10-LABX-0030-INRT under the frame program Investissements d'Avenir labeled ANR-10-IDEX-0002-02 (to Institut de Génétique et de Biologie Moléculaire et Cellulaire). The 11.7T MRI scanner was funded by a grant from NeurATRIS: A Translational Research Infrastructure for Biotherapies in Neurosciences (Investissements d'Avenir, ANR-11-INBS-0011). We thank Dr. Huda Zoghbi for providing the SCA7 knock-in mice; the Mouse Clinical Institute of Illkirch for mouse phenotyping support; the GenomEast Platform at Institut de Génétique et de Biologie Moléculaire et Cellulaire, a member of the France Genomique consortium (ANR-10-INBS-0009) for processing Illumina libraries; Stephan Collins for macro allowing PC morphological analysis; Filiz Kavla, Raphaël Bour, and Christel Wagner for technical assistance; Tao Ye for fruitful discussion regarding motif enrichments; Doulaye Dembele for suggestions regarding statistical analysis; and Nathalie Daigle and Hélène Puccio for critical reading of the manuscript and fruitful discussion.
The authors declare no competing financial interests.
References
- Abou-Sleymane G, Chalmel F, Helmlinger D, Lardenois A, Thibault C, Weber C, Merienne K, Mandel JL, Poch O, Devys D, Trottier Y (2006) Polyglutamine expansion causes neurodegeneration by altering the neuronal differentiation program. Hum Mol Genet 15:691–703. 10.1093/hmg/ddi483 [DOI] [PubMed] [Google Scholar]
- Ache BW, Young JM (2005) Olfaction: diverse species, conserved principles. Neuron 48:417–430. 10.1016/j.neuron.2005.10.022 [DOI] [PubMed] [Google Scholar]
- Achour M, Le Gras S, Keime C, Parmentier F, Lejeune FX, Boutillier AL, Neri C, Davidson I, Merienne K (2015) Neuronal identity genes regulated by super-enhancers are preferentially down-regulated in the striatum of Huntington's disease mice. Hum Mol Genet 24:3481–3496. 10.1093/hmg/ddv099 [DOI] [PubMed] [Google Scholar]
- Adanyeguh IM, Perlbarg V, Henry PG, Rinaldi D, Petit E, Valabregue R, Brice A, Durr A, Mochel F (2018) Autosomal dominant cerebellar ataxias: imaging biomarkers with high effect sizes. Neuroimage Clin 19:858–867. 10.1016/j.nicl.2018.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcauter S, Barrios FA, Diaz R, Fernandez-Ruiz J (2011) Gray and white matter alterations in spinocerebellar ataxia type 7: an in vivo DTI and VBM study. Neuroimage 55:1–7. 10.1016/j.neuroimage.2010.12.014 [DOI] [PubMed] [Google Scholar]
- Aleman TS, Cideciyan AV, Volpe NJ, Stevanin G, Brice A, Jacobson SG (2002) Spinocerebellar ataxia type 7 (SCA7) shows a cone-rod dystrophy phenotype. Exp Eye Res 74:737–745. 10.1006/exer.2002.1169 [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W (2015) HTSeq: a Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169. 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apps R, Hawkes R (2009) Cerebellar cortical organization: a one-map hypothesis. Nat Rev Neurosci 10:670–681. 10.1038/nrn2698 [DOI] [PubMed] [Google Scholar]
- Arancillo M, White JJ, Lin T, Stay TL, Sillitoe RV (2015) In vivo analysis of Purkinje cell firing properties during postnatal mouse development. J Neurophysiol 113:578–591. 10.1152/jn.00586.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37:W202–W208. 10.1093/nar/gkp335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukhtouche F, Doulazmi M, Frederic F, Dusart I, Brugg B, Mariani J (2006) RORalpha, a pivotal nuclear receptor for Purkinje neuron survival and differentiation: from development to ageing. Cerebellum 5:97–104. 10.1080/14734220600750184 [DOI] [PubMed] [Google Scholar]
- Bushart DD, Shakkottai VG (2019) Ion channel dysfunction in cerebellar ataxia. Neurosci Lett 688:41–48. 10.1016/j.neulet.2018.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo-Rosas S, Weber C, Fievet L, Messaddeq N, Karam A, Trottier Y (2018) Loss of zebrafish Ataxin-7, a SAGA subunit responsible for SCA7 retinopathy, causes ocular coloboma and malformation of photoreceptors. Hum Mol Genet 28:912–927. [DOI] [PubMed] [Google Scholar]
- Cerminara NL, Lang EJ, Sillitoe RV, Apps R (2015) Redefining the cerebellar cortex as an assembly of non-uniform Purkinje cell microcircuits. Nat Rev Neurosci 16:79–93. 10.1038/nrn3886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XR, Heck N, Lohof AM, Rochefort C, Morel MP, Wehrle R, Doulazmi M, Marty S, Cannaya V, Avci HX, Mariani J, Rondi-Reig L, Vodjdani G, Sherrard RM, Sotelo C, Dusart I (2013) Mature Purkinje cells require the retinoic acid-related orphan receptor-alpha (RORalpha) to maintain climbing fiber mono-innervation and other adult characteristics. J Neurosci 33:9546–9562. 10.1523/JNEUROSCI.2977-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YC, Gatchel JR, Lewis RW, Mao CA, Grant PA, Zoghbi HY, Dent SY (2012) Gcn5 loss-of-function accelerates cerebellar and retinal degeneration in a SCA7 mouse model. Hum Mol Genet 21:394–405. 10.1093/hmg/ddr474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirino A, Hernandez-Castillo CR, Galvez V, Contreras A, Diaz R, Beltran-Parrazal L, Fernandez-Ruiz J (2018) Motor and cognitive impairments in spinocerebellar ataxia type 7 and its correlations with cortical volumes. Eur J Neurosci 48:3199–3211. 10.1111/ejn.14148 [DOI] [PubMed] [Google Scholar]
- Chort A, Alves S, Marinello M, Dufresnois B, Dornbierer JG, Tesson C, Latouche M, Baker DP, Barkats M, El Hachimi KH, Ruberg M, Janer A, Stevanin G, Brice A, Sittler A (2013) Interferon beta induces clearance of mutant ataxin 7 and improves locomotion in SCA7 knock-in mice. Brain 136:1732–1745. 10.1093/brain/awt061 [DOI] [PubMed] [Google Scholar]
- Chou AH, Chen CY, Chen SY, Chen WJ, Chen YL, Weng YS, Wang HL (2010) Polyglutamine-expanded ataxin-7 causes cerebellar dysfunction by inducing transcriptional dysregulation. Neurochem Int 56:329–339. 10.1016/j.neuint.2009.11.003 [DOI] [PubMed] [Google Scholar]
- Custer SK, Garden GA, Gill N, Rueb U, Libby RT, Schultz C, Guyenet SJ, Deller T, Westrum LE, Sopher BL, La Spada AR (2006) Bergmann glia expression of polyglutamine-expanded ataxin-7 produces neurodegeneration by impairing glutamate transport. Nat Neurosci 9:1302–1311. 10.1038/nn1750 [DOI] [PubMed] [Google Scholar]
- Dansithong W, Paul S, Figueroa KP, Rinehart MD, Wiest S, Pflieger LT, Scoles DR, Pulst SM (2015) Ataxin-2 regulates RGS8 translation in a new BAC-SCA2 transgenic mouse model. PLoS Genet 11:e1005182. 10.1371/journal.pgen.1005182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G, Abbas N, Stevanin G, Durr A, Yvert G, Cancel G, Weber C, Imbert G, Saudou F, Antoniou E, Drabkin H, Gemmill R, Giunti P, Benomar A, Wood N, Ruberg M, Agid Y, Mandel JL, Brice A (1997) Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet 17:65–70. 10.1038/ng0997-65 [DOI] [PubMed] [Google Scholar]
- David G, Durr A, Stevanin G, Cancel G, Abbas N, Benomar A, Belal S, Lebre AS, Abada-Bendib M, Grid D, Holmberg M, Yahyaoui M, Hentati F, Chkili T, Agid Y, Brice A (1998) Molecular and clinical correlations in autosomal dominant cerebellar ataxia with progressive macular dystrophy (SCA7). Hum Mol Genet 7:165–170. 10.1093/hmg/7.2.165 [DOI] [PubMed] [Google Scholar]
- Deneris ES, Hobert O (2014) Maintenance of postmitotic neuronal cell identity. Nat Neurosci 17:899–907. 10.1038/nn.3731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhenain M, Delatour B, Walczak C, Volk A (2006) Passive staining: a novel ex vivo MRI protocol to detect amyloid deposits in mouse models of Alzheimer's disease. Magn Reson Med 55:687–693. 10.1002/mrm.20810 [DOI] [PubMed] [Google Scholar]
- Diamond ME, von Heimendahl M, Knutsen PM, Kleinfeld D, Ahissar E (2008) 'Where' and 'what' in the whisker sensorimotor system. Nat Rev Neurosci 9:601–612. 10.1038/nrn2411 [DOI] [PubMed] [Google Scholar]
- Doyle JP, Dougherty JD, Heiman M, Schmidt EF, Stevens TR, Ma G, Bupp S, Shrestha P, Shah RD, Doughty ML, Gong S, Greengard P, Heintz N (2008) Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell 135:749–762. 10.1016/j.cell.2008.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil R, Hartmann J, Luo C, Wolfsgruber W, Schilling K, Feil S, Barski JJ, Meyer M, Konnerth A, De Zeeuw CI, Hofmann F (2003) Impairment of LTD and cerebellar learning by Purkinje cell-specific ablation of cGMP-dependent protein kinase I. J Cell Biol 163:295–302. 10.1083/jcb.200306148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich B, Euler P, Ziegler R, Kuhn A, Landwehrmeyer BG, Luthi-Carter R, Weiller C, Hellwig S, Zucker B (2012) Comparative analyses of Purkinje cell gene expression profiles reveal shared molecular abnormalities in models of different polyglutamine diseases. Brain Res 1481:37–48. 10.1016/j.brainres.2012.08.005 [DOI] [PubMed] [Google Scholar]
- Fujita H, Aoki H, Ajioka I, Yamazaki M, Abe M, Oh-Nishi A, Sakimura K, Sugihara I (2014) Detailed expression pattern of aldolase C (Aldoc) in the cerebellum, retina and other areas of the CNS studied in Aldoc-Venus knock-in mice. PLoS One 9:e86679. 10.1371/journal.pone.0086679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furrer SA, Waldherr SM, Mohanachandran MS, Baughn TD, Nguyen KT, Sopher BL, Damian VA, Garden GA, La Spada AR (2013) Reduction of mutant ataxin-7 expression restores motor function and prevents cerebellar synaptic reorganization in a conditional mouse model of SCA7. Hum Mol Genet 22:890–903. 10.1093/hmg/dds495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia S, Fourcaud-Trocmé N (2009) OpenElectrophy: an electrophysiological data- and analysis-sharing framework. Front. Neuroinform 3:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatchel JR, Watase K, Thaller C, Carson JP, Jafar-Nejad P, Shaw C, Zu T, Orr HT, Zoghbi HY (2008) The insulin-like growth factor pathway is altered in spinocerebellar ataxia type 1 and type 7. Proc Natl Acad Sci USA 105:1291–1296. 10.1073/pnas.0711257105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrking KM, Andresen JM, Duvick L, Lough J, Zoghbi HY, Orr HT (2011) Partial loss of Tip60 slows mid-stage neurodegeneration in a spinocerebellar ataxia type 1 (SCA1) mouse model. Hum Mol Genet 20:2204–2212. 10.1093/hmg/ddr108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold DA, Baek SH, Schork NJ, Rose DW, Larsen DD, Sachs BD, Rosenfeld MG, Hamilton BA (2003) RORalpha coordinates reciprocal signaling in cerebellar development through sonic hedgehog and calcium-dependent pathways. Neuron 40:1119–1131. 10.1016/S0896-6273(03)00769-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruol DL, Franklin CL (1987) Morphological and physiological differentiation of Purkinje neurons in cultures of rat cerebellum. J Neurosci 7:1271–1293. 10.1523/JNEUROSCI.07-05-01271.1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen ST, Meera P, Otis TS, Pulst SM (2013) Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum Mol Genet 22:271–283. 10.1093/hmg/dds427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausser M, Clark BA (1997) Tonic synaptic inhibition modulates neuronal output pattern and spatiotemporal synaptic integration. Neuron 19:665–678. 10.1016/S0896-6273(00)80379-7 [DOI] [PubMed] [Google Scholar]
- Hawkes R (2014) Purkinje cell stripes and long-term depression at the parallel fiber-Purkinje cell synapse. Front Syst Neurosci 8:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmlinger D, Hardy S, Abou-Sleymane G, Eberlin A, Bowman AB, Gansmuller A, Picaud S, Zoghbi HY, Trottier Y, Tora L, Devys D (2006) Glutamine-expanded ataxin-7 alters TFTC/STAGA recruitment and chromatin structure leading to photoreceptor dysfunction. PLoS Biol 4:e67. 10.1371/journal.pbio.0040067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmlinger D, Hardy S, Sasorith S, Klein F, Robert F, Weber C, Miguet L, Potier N, Van-Dorsselaer A, Wurtz JM, Mandel JL, Tora L, Devys D (2004) Ataxin-7 is a subunit of GCN5 histone acetyltransferase-containing complexes. Hum Mol Genet 13:1257–1265. 10.1093/hmg/ddh139 [DOI] [PubMed] [Google Scholar]
- Hernandez-Castillo CR, Alcauter S, Galvez V, Barrios FA, Yescas P, Ochoa A, Garcia L, Diaz R, Gao W, Fernandez-Ruiz J (2013) Disruption of visual and motor connectivity in spinocerebellar ataxia type 7. Mov Disord 28:1708–1716. 10.1002/mds.25618 [DOI] [PubMed] [Google Scholar]
- Hernandez-Castillo CR, Galvez V, Diaz R, Fernandez-Ruiz J (2016) Specific cerebellar and cortical degeneration correlates with ataxia severity in spinocerebellar ataxia type 7. Brain Imaging Behav 10:252–257. 10.1007/s11682-015-9389-1 [DOI] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, Young RA (2013) Super-enhancers in the control of cell identity and disease. Cell 155:934–947. 10.1016/j.cell.2013.09.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg J, Perlmann T (2012) Maintaining differentiated cellular identity. Nat Rev Genet 13:429–439. 10.1038/nrg3209 [DOI] [PubMed] [Google Scholar]
- Holt GR, Softky WR, Koch C, Douglas RJ (1996) Comparison of discharge variability in vitro and in vivo in cat visual cortex neurons. J Neurophysiol 75:1806–1814. 10.1152/jn.1996.75.5.1806 [DOI] [PubMed] [Google Scholar]
- Horton LC, Frosch MP, Vangel MG, Weigel-DiFranco C, Berson EL, Schmahmann JD (2013) Spinocerebellar ataxia type 7: clinical course, phenotype-genotype correlations, and neuropathology. Cerebellum 12:176–193. 10.1007/s12311-012-0412-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoxha E, Balbo I, Miniaci MC, Tempia F (2018) Purkinje cell signaling deficits in animal models of ataxia. Front Synaptic Neurosci 10:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram M, Wozniak EA, Duvick L, Yang R, Bergmann P, Carson R, O'Callaghan B, Zoghbi HY, Henzler C, Orr HT (2016) Cerebellar transcriptome profiles of ATXN1 transgenic mice reveal SCA1 disease progression and protection pathways. Neuron 89:1194–1207. 10.1016/j.neuron.2016.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karam A, Trottier Y (2018) Molecular mechanisms and therapeutic strategies in spinocerebellar ataxia type 7. Adv Exp Med Biol 1049:197–218. [DOI] [PubMed] [Google Scholar]
- Kashiwabuchi N, Ikeda K, Araki K, Hirano T, Shibuki K, Takayama C, Inoue Y, Kutsuwada T, Yagi T, Kang Y, Aizawa S, Mishina M (1995) Impairment of motor coordination, Purkinje cell synapse formation, and cerebellar long-term depression in GluR delta 2 mutant mice. Cell 81:245–252. 10.1016/0092-8674(95)90334-8 [DOI] [PubMed] [Google Scholar]
- Kato AS, Knierman MD, Siuda ER, Isaac JT, Nisenbaum ES, Bredt DS (2012) Glutamate receptor delta2 associates with metabotropic glutamate receptor 1 (mGluR1), protein kinase Cgamma, and canonical transient receptor potential 3 and regulates mGluR1-mediated synaptic transmission in cerebellar Purkinje neurons. J Neurosci 32:15296–15308. 10.1523/JNEUROSCI.0705-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14:R36. 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klockgether T (2011) Update on degenerative ataxias. Curr Opin Neurol 24:339–345. 10.1097/WCO.0b013e32834875ba [DOI] [PubMed] [Google Scholar]
- Kramer A, Green J, Pollard J Jr, Tugendreich S (2014) Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30:523–530. 10.1093/bioinformatics/btt703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratz A, Beguin P, Kaneko M, Chimura T, Suzuki AM, Matsunaga A, Kato S, Bertin N, Lassmann T, Vigot R, Carninci P, Plessy C, Launey T (2014) Digital expression profiling of the compartmentalized translatome of Purkinje neurons. Genome Res 24:1396–1410. 10.1101/gr.164095.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Spada AR, Fu YH, Sopher BL, Libby RT, Wang X, Li LY, Einum DD, Huang J, Possin DE, Smith AC, Martinez RA, Koszdin KL, Treuting PM, Ware CB, Hurley JB, Ptacek LJ, Chen S (2001) Polyglutamine-expanded ataxin-7 antagonizes CRX function and induces cone-rod dystrophy in a mouse model of SCA7. Neuron 31:913–927. 10.1016/S0896-6273(01)00422-6 [DOI] [PubMed] [Google Scholar]
- Lan X, Koutelou E, Schibler AC, Chen YC, Grant PA, Dent SY (2015) Poly(Q) expansions in ATXN7 affect solubility but not activity of the SAGA deubiquitinating module. Mol Cell Biol 35:1777–1787. 10.1128/MCB.01454-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chi Chin M, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, et al. (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445:168–176. 10.1038/nature05453 [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein LY, McLaughlin PJ, Peachey NS, Sasaki T, Marmorstein AD (2007) Formation and progression of sub-retinal pigment epithelium deposits in Efemp1 mutation knock-in mice: a model for the early pathogenic course of macular degeneration. Hum Mol Genet 16:2423–2432. 10.1093/hmg/ddm199 [DOI] [PubMed] [Google Scholar]
- Martin JJ (2012) Spinocerebellar ataxia type 7. Handb Clin Neurol 103:475–491. [DOI] [PubMed] [Google Scholar]
- Martin JJ, Krols L, Ceuterick C, Van Broeckhoven C, Van Regemorter N, Hayer-Delatte F, Brucher JM, de Barsy T, Szliwowski H, Evrard P, Tassignon MJ, Smet-Dieleman H, Willems PJ (1994) On an autosomal dominant form of retinal-cerebellar degeneration: an autopsy study of five patients in one family. Acta Neuropathol 88:277–286. 10.1007/BF00310370 [DOI] [PubMed] [Google Scholar]
- Metz M, Gassmann M, Fakler B, Schaeren-Wiemers N, Bettler B (2011) Distribution of the auxiliary GABAB receptor subunits KCTD8, 12, 12b, and 16 in the mouse brain. J Comp Neurol 519:1435–1454. 10.1002/cne.22610 [DOI] [PubMed] [Google Scholar]
- Michalik A, Martin JJ, Van Broeckhoven C (2004) Spinocerebellar ataxia type 7 associated with pigmentary retinal dystrophy. Eur J Hum Genet 12:2–15. 10.1038/sj.ejhg.5201108 [DOI] [PubMed] [Google Scholar]
- Mikhaleva A, Kannan M, Wagner C, Yalcin B (2016) Histomorphological phenotyping of the adult mouse brain. Curr Protoc Mouse Biol 6:307–332. 10.1002/cpmo.12 [DOI] [PubMed] [Google Scholar]
- Mookerjee S, Papanikolaou T, Guyenet SJ, Sampath V, Lin A, Vitelli C, DeGiacomo F, Sopher BL, Chen SF, La Spada AR, Ellerby LM (2009) Posttranslational modification of ataxin-7 at lysine 257 prevents autophagy-mediated turnover of an N-terminal caspase-7 cleavage fragment. J Neurosci 29:15134–15144. 10.1523/JNEUROSCI.4720-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewiadomska-Cimicka A, Trottier Y (2019) Molecular targets and therapeutic strategies in spinocerebellar ataxia type 7. Neurotherapeutics 16:1074–1096. 10.1007/s13311-019-00778-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewiadomska-Cimicka A, Hache A, Trottier Y (2020) Gene deregulation and underlying mechanisms in spinocerebellar ataxias with polyglutamine expansion. Front Neurosci 14:571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Bean BP (1999) Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J Neurosci 19:1663–1674. 10.1523/JNEUROSCI.19-05-01663.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rub U, Brunt ER, Seidel K, Gierga K, Mooy CM, Kettner M, Van Broeckhoven C, Bechmann I, La Spada AR, Schols L, den Dunnen W, de Vos RA, Deller T (2008) Spinocerebellar ataxia type 7 (SCA7): widespread brain damage in an adult-onset patient with progressive visual impairments in comparison with an adult-onset patient without visual impairments. Neuropathol Appl Neurobiol 34:155–168. 10.1111/j.1365-2990.2007.00882.x [DOI] [PubMed] [Google Scholar]
- Salas-Vargas J, Mancera-Gervacio J, Velázquez-Pérez L, Rodrígez-Labrada R, Martínez-Cruz E, Magaña JJ, Durand-Rivera A, Hernández-Hernández O, Cisneros B, Gonzalez-Piña R (2015) Spinocerebellar ataxia type 7: a neurodegenerative disorder with peripheral neuropathy. Eur Neurol 73:173–178. 10.1159/000370239 [DOI] [PubMed] [Google Scholar]
- Sarachana T, Hu VW (2013) Genome-wide identification of transcriptional targets of RORA reveals direct regulation of multiple genes associated with autism spectrum disorder. Mol Autism 4:14. 10.1186/2040-2392-4-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato A, Sekine Y, Saruta C, Nishibe H, Morita N, Sato Y, Sadakata T, Shinoda Y, Kojima T, Furuichi T (2008) Cerebellar development transcriptome database (CDT-DB): profiling of spatio-temporal gene expression during the postnatal development of mouse cerebellum. Neural Netw 21:1056–1069. 10.1016/j.neunet.2008.05.004 [DOI] [PubMed] [Google Scholar]
- Serra HG, Duvick L, Zu T, Carlson K, Stevens S, Jorgensen N, Lysholm A, Burright E, Zoghbi HY, Clark HB, Andresen JM, Orr HT (2006) RORalpha-mediated Purkinje cell development determines disease severity in adult SCA1 mice. Cell 127:697–708. 10.1016/j.cell.2006.09.036 [DOI] [PubMed] [Google Scholar]
- Sillitoe RV, Hawkes R (2002) Whole-mount immunohistochemistry: a high-throughput screen for patterning defects in the mouse cerebellum. J Histochem Cytochem 50:235–244. 10.1177/002215540205000211 [DOI] [PubMed] [Google Scholar]
- Smeets CJ, Verbeek DS (2014) Cerebellar ataxia and functional genomics: identifying the routes to cerebellar neurodegeneration. Biochim Biophys Acta 1842:2030–2038. 10.1016/j.bbadis.2014.04.004 [DOI] [PubMed] [Google Scholar]
- Stoyas CA, Bushart DD, Switonski PM, Ward JM, Alaghatta A, Tang MB, Niu C, Wadhwa M, Huang H, Savchenko A, Gariani K, Xie F, Delaney JR, Gaasterland T, Auwerx J, Shakkottai VG, La Spada AR (2020) Nicotinamide pathway-dependent Sirt1 activation restores calcium homeostasis to achieve neuroprotection in spinocerebellar ataxia type 7. Neuron 105:630–644.e639. 10.1016/j.neuron.2019.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom AL, Forsgren L, Holmberg M (2005) A role for both wild-type and expanded ataxin-7 in transcriptional regulation. Neurobiol Dis 20:646–655. 10.1016/j.nbd.2005.04.018 [DOI] [PubMed] [Google Scholar]
- Swaroop A, Kim D, Forrest D (2010) Transcriptional regulation of photoreceptor development and homeostasis in the mammalian retina. Nat Rev Neurosci 11:563–576. 10.1038/nrn2880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, von Mering C (2015) STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43:D447–D452. 10.1093/nar/gku1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata T, Araishi K, Hashimoto K, Hashimotodani Y, van der Putten H, Bettler B, Kano M (2004) Ca2+ activity at GABAB receptors constitutively promotes metabotropic glutamate signaling in the absence of GABA. Proc Natl Acad Sci USA 101:16952–16957. 10.1073/pnas.0405387101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Warrenburg BP, Notermans NC, Schelhaas HJ, van Alfen N, Sinke RJ, Knoers NV, Zwarts MJ, Kremer BP (2004) Peripheral nerve involvement in spinocerebellar ataxias. Arch Neurol 61:257–261. 10.1001/archneur.61.2.257 [DOI] [PubMed] [Google Scholar]
- White JJ, Arancillo M, King A, Lin T, Miterko LN, Gebre SA, Sillitoe RV (2016) Pathogenesis of severe ataxia and tremor without the typical signs of neurodegeneration. Neurobiol Dis 86:86–98. 10.1016/j.nbd.2015.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA (2013) Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153:307–319. 10.1016/j.cell.2013.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanicostas C, Barbieri E, Hibi M, Brice A, Stevanin G, Soussi-Yanicostas N (2012) Requirement for zebrafish ataxin-7 in differentiation of photoreceptors and cerebellar neurons. PLoS One 7:e50705. 10.1371/journal.pone.0050705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yefimova MG, Messaddeq N, Karam A, Jacquard C, Weber C, Jonet L, Wolfrum U, Jeanny JC, Trottier Y (2010) Polyglutamine toxicity induces rod photoreceptor division, morphological transformation or death in Spinocerebellar ataxia 7 mouse retina. Neurobiol Dis 40:311–324. 10.1016/j.nbd.2010.06.005 [DOI] [PubMed] [Google Scholar]
- Yoo SY, Pennesi ME, Weeber EJ, Xu B, Atkinson R, Chen S, Armstrong DL, Wu SM, Sweatt JD, Zoghbi HY (2003) SCA7 knockin mice model human SCA7 and reveal gradual accumulation of mutant ataxin-7 in neurons and abnormalities in short-term plasticity. Neuron 37:383–401. 10.1016/S0896-6273(02)01190-X [DOI] [PubMed] [Google Scholar]
- Yvert G, Lindenberg KS, Picaud S, Landwehrmeyer GB, Sahel JA, Mandel JL (2000) Expanded polyglutamines induce neurodegeneration and trans-neuronal alterations in cerebellum and retina of SCA7 transgenic mice. Hum Mol Genet 9:2491–2506. 10.1093/hmg/9.17.2491 [DOI] [PubMed] [Google Scholar]
- Yvert G, Lindenberg KS, Devys D, Helmlinger D, Landwehrmeyer GB, Mandel JL (2001) SCA7 mouse models show selective stabilization of mutant ataxin-7 and similar cellular responses in different neuronal cell types. Hum Mol Genet 10:1679–1692. 10.1093/hmg/10.16.1679 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
IPA analysis and top 10 most predicted upstream regulators of genes deregulated in SCA7140Q/5Q mice cerebellum. Download Figure 11-1, XLSX file (12.8KB, xlsx) .
Analysis of cell-type specific gene deregulation in SCA7140Q/5Q mice cerebellum. Download Figure 11-2, XLSX file (129.6KB, xlsx) .
136 transcripts commonly downregulated in cerebellum of SCA1, SCA2 and SCA7 mice models. Download Table 2-1, XLSX file (14.6KB, xlsx) .